Chapter 421 Acyanotic Congenital Heart Disease
The Obstructive Lesions
421.1 Pulmonary Valve Stenosis with Intact Ventricular Septum
Of the various forms of right ventricular outflow obstruction with an intact ventricular septum, the most common is isolated valvular pulmonary stenosis, which accounts for 7-10% of all congenital heart defects. The valve cusps are deformed to various degrees and, as a result, the valve opens incompletely during systole. The valve may be bicuspid or tricuspid and the leaflets partially fused together with an eccentric outlet. This fusion may be so severe that only a pinhole central opening remains. If the valve is not severely thickened, it produces a dome-like obstruction to right ventricular outflow during systole. Isolated infundibular or subvalvular stenosis, supravalvular pulmonary stenosis, and branch pulmonary artery stenosis are also encountered. In cases where pulmonary valve stenosis is associated with a ventricular septal defect (VSD) but without anterior deviation of the infundibular septum and overriding aorta, this condition is better classified as pulmonary stenosis with VSD rather than as tetralogy of Fallot (Chapter 424.1). Pulmonary stenosis and an atrial septal defect (ASD) are also occasionally seen as associated defects. The clinical and laboratory findings reflect the dominant lesion, but it is important to rule out any associated anomalies. Pulmonary stenosis as a result of valve dysplasia is the most common cardiac abnormality in Noonan syndrome (Chapter 76), and is associated in about 50% of cases with a mutation in the gene PTPN11, encoding the protein tyrosine phosphotase SHP-2 on chromosome 12. The mechanism for pulmonic stenosis is unknown, although maldevelopment of the distal portion of the bulbus cordis and the sequelae of fetal endocarditis have been suggested as etiologies. Pulmonary stenosis, either of the valve or the branch pulmonary arteries, is a common finding in patients with arteriohepatic dysplasia, also known as Alagille syndrome (Chapter 348). In this syndrome and in some patients with isolated pulmonic stenosis, a mutation is present in the Jagged1 gene.
Pathophysiology
The obstruction to outflow from the right ventricle to the pulmonary artery results in increased right ventricular systolic pressure and wall stress, which leads to hypertrophy of the right ventricle (Fig. 421-1). The severity of these abnormalities depends on the size of the restricted valve opening. In severe cases, right ventricular pressure may be higher than systemic arterial systolic pressure, whereas with milder obstruction, right ventricular pressure is only mildly or moderately elevated. Pulmonary artery pressure (distal to the obstruction) is normal or decreased. Arterial oxygen saturation will be normal even in cases of severe stenosis, unless an intracardiac communication such as a VSD or ASD is allowing blood to shunt from right to left. When severe pulmonic stenosis occurs in a neonate, decreased right ventricular compliance often leads to cyanosis due to right-to-left shunting through a patent foramen ovale, a condition termed critical pulmonic stenosis.
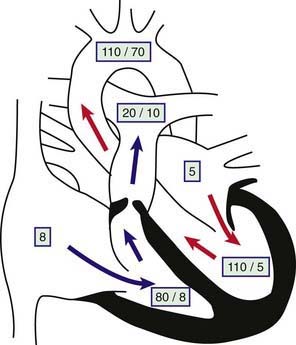
Figure 421-1 Physiology of valvular pulmonary stenosis. Boxed numbers represent pressure in mm Hg. Because of the absence of right-to-left or left-to-right shunting, blood flow through all cardiac chambers is normal at 3 L/min/m2. The pulmonary-to-systemic blood flow ratio (Qp : Qs) is 1 : 1. Right atrial pressure is increased slightly as a result of decreased right ventricular compliance. The right ventricle is hypertrophied, and systolic and diastolic pressure is increased. The pressure gradient across the thickened pulmonary valve is 60 mm Hg. The main pulmonary artery pressure is slightly low, and poststenotic dilatation is present. Left heart pressure is normal. Unless right-to-left shunting is occurring through a foramen ovale, the patient’s systemic oxygen saturation will be normal.
Clinical Manifestations and Laboratory Findings
Patients with mild or moderate stenosis usually do not have any symptoms. Growth and development are most often normal. If the stenosis is severe, signs of right ventricular failure such as hepatomegaly, peripheral edema, and exercise intolerance may be present. In a neonate or young infant with critical pulmonic stenosis, signs of right ventricular failure may be more prominent, and cyanosis is often present because of right-to-left shunting at the foramen ovale.
With mild pulmonary stenosis, venous pressure and pulse are normal. The heart is not enlarged, the apical impulse is normal, and the right ventricular impulse is not palpable. A sharp pulmonic ejection click immediately after the 1st heart sound is heard at the left upper sternal border during expiration. The 2nd heart sound is split, with a pulmonary component of normal intensity that may be slightly delayed. A relatively short, low- or medium-pitched systolic ejection murmur is maximally audible over the pulmonic area and radiates minimally to the lung fields bilaterally. The electrocardiogram is normal or characteristic of mild right ventricular hypertrophy; inversion of the T waves in the right precordial leads may be seen. (Remember that the T wave in lead V1 should normally be inverted until at least 6-8 yr of age. Therefore, a positive T wave in V1 in a young child is a sign of right ventricular hypertrophy.) The only abnormality demonstrable radiographically is usually poststenotic dilatation of the pulmonary artery. Two-dimensional echocardiography shows right ventricular hypertrophy and a slightly thickened pulmonic valve, which domes in systole; Doppler studies demonstrate a right ventricle to pulmonary artery gradient of ≤30 mm Hg.
In moderate pulmonic stenosis, venous pressure may be slightly elevated; in older children, a prominent a wave may be noted in the jugular pulse. A right ventricular lift may be palpable at the lower left sternal border. The 2nd heart sound is split, with a delayed and soft pulmonary component. As valve motion becomes more limited with more severe degrees of stenosis, both the pulmonic ejection click and the pulmonic 2nd sound may become inaudible. With increasing degrees of stenosis, the peak of the systolic ejection murmur is prolonged later into systole, and its quality becomes louder and harsher (higher frequency). The murmur radiates more prominently to both lung fields.
The electrocardiogram reveals right ventricular hypertrophy, sometimes with a prominent spiked P wave. Radiographically, the heart can vary from normal size to mildly enlarged with uptilting of the apex due to prominence of the right ventricle; pulmonary vascularity may be normal or slightly decreased. The echocardiogram shows a thickened pulmonic valve with restricted systolic motion. Doppler examination demonstrates a right ventricle to pulmonary artery pressure gradient in the 30-60 mm Hg range. Mild tricuspid regurgitation may be present and allows Doppler confirmation of right ventricular systolic pressure.
In severe stenosis, mild to moderate cyanosis may be noted in patients with an interatrial communication (atrial septal defect or patent foramen ovale). If hepatic enlargement and peripheral edema are present, they are an indication of right ventricular failure. Elevation of venous pressure is common and is caused by a large presystolic jugular a wave. The heart is moderately or greatly enlarged, and a conspicuous parasternal right ventricular lift is present and frequently extends to the left midclavicular line. The pulmonary component of the 2nd sound is usually inaudible. A loud, long, and harsh systolic ejection murmur, usually accompanied by a thrill, is maximally audible in the pulmonic area and may radiate over the entire precordium, to both lung fields, into the neck, and to the back. The peak of the murmur occurs later in systole as valve opening becomes more restricted. The murmur frequently encompasses the aortic component of the 2nd sound but is not preceded by an ejection click.
The electrocardiogram shows gross right ventricular hypertrophy, frequently accompanied by a tall, spiked P wave. Radiographic studies confirm the presence of cardiac enlargement with prominence of the right ventricle and right atrium. Prominence of the main pulmonary artery segment may be seen due to poststenotic dilatation (Fig. 421-2). Intrapulmonary vascularity is decreased. The two-dimensional echocardiogram shows severe deformity of the pulmonary valve and right ventricular hypertrophy (Fig. 421-3). In the late stages of the disease, systolic dysfunction of the right ventricle may be seen, and in these cases the ventricle may become dilated, with prominent tricuspid regurgitation. Doppler studies demonstrate a high gradient (>60 mm Hg) across the pulmonary valve. The classic findings of severe pulmonary stenosis in older children are rarely seen because of early intervention. Signs of critical pulmonic stenosis, with all of the features of severe pulmonic stenosis plus cyanosis, are usually encountered in the neonatal period.
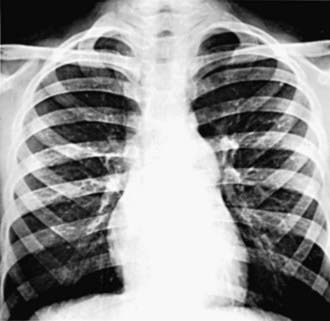
Figure 421-2 Roentgenogram in a patient with valvular pulmonary stenosis and a normal aortic root. The heart size is within normal limits, but poststenotic dilatation of the pulmonary artery is present.
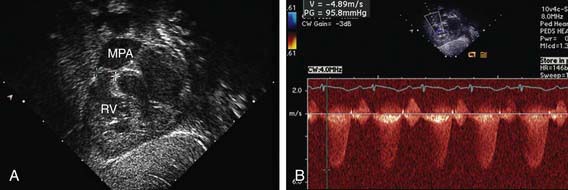
Figure 421-3 Echocardiogram demonstrating valvar pulmonic stenosis. A, Subcostal view showing thickened pulmonary valve leaflets (between crosshatches). B, Doppler study indicating a 95 mm Hg peak pressure gradient across the stenotic valve. MPA, main pulmonary artery; RV, right ventricle.
Cardiac catheterization is not generally required for diagnostic purposes but is undertaken as part of a balloon valvuloplasty procedure. Catheterization demonstrates an abrupt pressure gradient across the pulmonary valve. Pulmonary artery pressure is either normal or low. The severity of the stenosis is graded based on the ratio of right ventricular systolic pressure to systemic systolic pressure or the right ventricle to pulmonary artery pressure gradient: a gradient of 10-30 mm Hg in mild cases, 30-60 mm Hg in moderate cases, and >60 mm Hg or with right ventricular pressure greater than systemic pressure in severe cases. If cardiac output is low or a significant right-to-left shunt exists across the atrial septum, the pressure gradient may underestimate the degree of valve stenosis. Selective right ventriculography demonstrates the thickened, poorly mobile valve. In mild to moderate stenosis, doming of the valve in systole is readily seen. Flow of contrast medium through the stenotic valve in ventricular systole produces a narrow jet of dye that fills the dilated main pulmonary artery. Subvalvular hypertrophy that may intensify the obstruction may be present.
Treatment
Patients with moderate or severe isolated pulmonary stenosis require relief of the obstruction. Balloon valvuloplasty is the initial treatment of choice for the majority of patients (Fig. 421-4). Patients with severely thickened pulmonic valves, especially common in those with Noonan syndrome, may require surgical intervention. In a neonate with critical pulmonic stenosis, urgent treatment by either balloon valvuloplasty or surgical valvotomy is warranted.
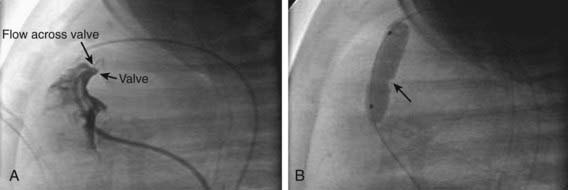
Figure 421-4 Valvar pulmonary stenosis and balloon valvuloplasty. A, Right ventricular angiogram showing severely stenotic pulmonary valve with narrow jet of blood flowing across. B, Inflation of the balloon catheter showing the indentation (arrow) made on the balloon from the stenotic valve.
(Photos courtesy of Dr. Jeffrey Feinstein, Stanford University, Stanford, CA.)
Excellent results are obtained in most instances. The gradient across the pulmonary valve is markedly reduced or abolished. In the early period after balloon valvuloplasty, a small to moderate residual gradient may remain because of muscular infundibular narrowing; it usually resolves with time. A short, early decrescendo diastolic murmur may be heard at the mid to upper left sternal border as a result of pulmonary valvular insufficiency. The degree of insufficiency is not usually clinically significant. No difference in patient status after valvuloplasty or surgery is noted at late follow-up; recurrence is unusual after successful treatment except in those patients with extremely dysplastic valves.
Prognosis and Complications
Heart failure occurs only in severe cases and most often during the 1st mo of life. The development of cyanosis from a right-to-left shunt across a foramen ovale is almost exclusively seen in the neonatal period when the stenosis is severe. Infective endocarditis is a risk but is not common in childhood.
Children with mild stenosis can lead a normal life, but their progress should be evaluated at regular intervals. Patients who have small gradients rarely show progression and do not need intervention, but a significant gradient is more likely to develop in children with moderate stenosis as they grow older. Worsening of obstruction may also be due to the development of secondary subvalvular muscular and fibrous tissue hypertrophy. In untreated severe stenosis, the course may abruptly worsen with the development of right ventricular dysfunction and cardiac failure. Infants with critical pulmonic stenosis require urgent catheter balloon valvuloplasty or surgical valvotomy. Development of right ventricular failure many years after pulmonary balloon valvuloplasty is uncommon. Nonetheless, patients should be followed serially for worsening pulmonary insufficiency and right ventricular dilation.
Crosnier C, Lykavieris P, Meunier-Rotival M, et al. Alagille syndrome. The widening spectrum of arteriohepatic dysplasia. Clin Liver Dis. 2000;4:765-778.
Feinstein JA, Kim N, Reddy VM, Perry SB. Percutaneous pulmonary valve placement in a 10-month-old patient using a hand crafted stent-mounted porcine valve. Catheter Cardiovasc Interv. 2006;67:644-649.
Khambadkone S, Coats L, Taylor A, et al. Percutaneous pulmonary valve implantation in humans: results in 59 consecutive patients. Circulation. 2005;112:1189-1197.
Krantz ID, Smith R, Colliton RP, et al. Jagged 1 mutations in patients ascertained with isolated congenital heart defects. Am J Med Genet. 1999;84:56-60.
Phoon CK. Estimation of pressure gradients by auscultation: an innovative and accurate physical examination technique. Am Heart J. 2001;141:500-506.
Rosales AM, Lock JE, Perry SB, et al. Interventional catheterization management of perioperative peripheral pulmonary stenosis: balloon angioplasty or endovascular stenting. Catheter Cardiovasc Interv. 2002;56:272-277.
Yoshida R, Hasegawa T, Hasegawa Y, et al. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan syndrome. J Clin Endocrinol Metab. 2004;89:3359-3364.
421.2 Infundibular Pulmonary Stenosis and Double-Chamber Right Ventricle
Infundibular pulmonary stenosis is caused by muscular or fibrous obstruction in the outflow tract of the right ventricle. The site of obstruction may be close to the pulmonary valve or well below it; an infundibular chamber may be present between the right ventricular cavity and the pulmonary valve. In many cases, a VSD may have been present initially and later closed spontaneously. When the pulmonary valve is also stenotic, the combined defect is primarily classified as valvular stenosis with secondary infundibular hypertrophy. The hemodynamics and clinical manifestations of patients with isolated infundibular pulmonary stenosis are similar, for the most part, to those described in the discussion of isolated valvular pulmonary stenosis (Chapter 421.1).
A common variation in right ventricular outflow obstruction below the pulmonary valve is that of a double-chambered right ventricle. In this condition, a muscular band is present in the mid-right ventricular region; the band divides the chamber into two parts and creates obstruction between the inlet and outlet portions. An associated VSD that may close spontaneously is often noted. Obstruction is not usually seen early in life but may progress rapidly in a similar manner to the progressive infundibular obstruction observed with tetralogy of Fallot (Chapter 424.1).
The diagnosis of isolated right ventricular infundibular stenosis or double-chambered right ventricle is usually made by echocardiography. The ventricular septum must be evaluated carefully to determine whether an associated VSD is present. The prognosis for untreated cases of severe right ventricular outflow obstruction is similar to that for valvular pulmonary stenosis. When the obstruction is moderate to severe, surgery is indicated. After surgery, the pressure gradient is abolished or markedly reduced and the long-term outlook is excellent.
421.3 Pulmonary Stenosis in Combination with an Intracardiac Shunt
Valvular or infundibular pulmonary stenosis, or both, may be associated with either an ASD or a VSD. In these patients, the clinical features depend on the degree of pulmonary stenosis, which determines whether the net shunt is from left to right or from right to left.
The presence of a large left-to-right shunt at the atrial or ventricular level is evidence that the pulmonary stenosis is mild. These patients have symptoms similar to those of patients with an isolated ASD or VSD. With increasing age, worsening of the obstruction may limit the shunt and result in a gradual improvement in symptoms. Eventually, particularly in patients with pulmonary stenosis and VSD, a further increase in obstruction may lead to right-to-left shunting and cyanosis. When a patient with a VSD has evidence of decreasing heart failure and increased right ventricular forces on the electrocardiogram, one must differentiate between the development of increasing pulmonary stenosis versus the onset of pulmonary vascular disease (Eisenmenger syndrome, Chapter 427.2).
These anomalies are readily repaired surgically. Defects in the atrial or ventricular septum are closed, and the pulmonary stenosis is relieved by resection of infundibular muscle or pulmonary valvotomy, or both, as indicated. Patients with a predominant right-to-left shunt have symptoms similar to those of patients with tetralogy of Fallot (Chapter 424.1).
421.4 Peripheral Pulmonary Stenosis
Single or multiple constrictions may occur anywhere along the major branches of the pulmonary arteries and may range from mild to severe and from localized to extensive. Frequently, these defects are associated with other types of congenital heart disease, including valvular pulmonic stenosis, tetralogy of Fallot, patent ductus arteriosus (PDA), VSD, ASD, and supravalvular aortic stenosis. A familial tendency has been recognized in some patients with peripheral pulmonic stenosis. A high incidence is found in infants with congenital rubella syndrome. The combination of supravalvular aortic stenosis with pulmonary arterial branch stenosis, idiopathic hypercalcemia of infancy, elfin facies, and mental retardation is known as Williams syndrome, a condition associated with deletion of the elastin gene in region 7q11.23 on chromosome 7. Peripheral pulmonary stenosis is also associated with the Alagille syndrome, which may be associated with a mutation in the Jagged1 gene.
A mild constriction has little effect on the pulmonary circulation. With multiple severe constrictions, pressure is increased in the right ventricle and in the pulmonary artery proximal to the site of obstruction. When the anomaly is isolated, the diagnosis is suspected by the presence of murmurs in widespread locations over the chest, either anteriorly or posteriorly. These murmurs are usually systolic ejection in quality but may be continuous. Most often, the physical signs are dominated by the associated anomaly, such as tetralogy of Fallot (Chapter 424.1).
In the immediate newborn period, a mild and transient form of peripheral pulmonic stenosis may be present. Physical findings are generally limited to a soft systolic ejection murmur, which can be heard over either or both lung fields. It is the absence of other physical findings of valvular pulmonic stenosis (right ventricular lift, soft pulmonic 2nd sound, systolic ejection click, murmur loudest at the upper left sternal border) that supports this diagnosis. This murmur usually disappears by 1-2 mo.
If the stenosis is severe, the electrocardiogram shows evidence of right ventricular and right atrial hypertrophy, and the chest radiograph shows cardiomegaly and prominence of the main pulmonary artery. The pulmonary vasculature is usually normal; in some cases, however, small intrapulmonary vascular shadows are seen that represent areas of poststenotic dilatation. Echocardiography is limited in its ability to visualize the distal branch pulmonary arteries. Doppler examination demonstrates the acceleration of blood flow through the stenoses and, if tricuspid regurgitation is present, allows an estimation of right ventricular systolic pressure. MRI and CT are extremely helpful in delineating distal obstructions; if moderate to severe disease is suspected, the diagnosis is usually confirmed by cardiac catheterization.
Severe obstruction of the main pulmonary artery and its primary branches can be relieved during corrective surgery for associated lesions such as the tetralogy of Fallot or valvular pulmonary stenosis. If peripheral pulmonic stenosis is isolated, it may be treated by catheter balloon dilatation, sometimes with placement of an intravascular stent (Fig. 417-29).
421.5 Aortic Stenosis
Pathophysiology
Congenital aortic stenosis accounts for ≈5% of cardiac malformations recognized in childhood; a bicuspid aortic valve, one of the most common congenital heart lesions overall, is identified in up to 1.5% of adults and may be asymptomatic in childhood. Aortic stenosis is more frequent in males (3 : 1). In the most common form, valvular aortic stenosis, the leaflets are thickened and the commissures are fused to varying degrees. Left ventricular systolic pressure is increased as a result of the obstruction to outflow. The left ventricular wall hypertrophies in compensation; as its compliance decreases, end-diastolic pressure increases as well.
Subvalvular (subaortic) stenosis with a discrete fibromuscular shelf below the aortic valve is also an important form of left ventricular outflow tract obstruction. This lesion is frequently associated with other forms of congenital heart disease such as mitral stenosis and coarctation of the aorta (Shone syndrome) and may progress rapidly in severity. It is less commonly diagnosed during early infancy and may develop despite previous documentation of no left ventricular outflow tract obstruction. Subvalvular aortic stenosis may become apparent after successful surgery for other congenital heart defects (coarctation of the aorta, PDA, VSD), may develop in association with mild lesions that have not been surgically repaired, or may occur as an isolated abnormality. Subvalvular aortic stenosis may also be due to a markedly hypertrophied ventricular septum in association with hypertrophic cardiomyopathy (Chapter 433.2).
Supravalvular aortic stenosis, the least common type, may be sporadic, familial, or associated with Williams syndrome, which includes mental retardation (IQ range 41-80), elfin facies (full face, broad forehead, flattened bridge of the nose, long upper lip, and rounded cheeks) (Fig. 421-5), and idiopathic hypercalcemia of infancy. Additional features include loquacious personality, hypersensitivity to sound, spasticity, hypoplastic nails, dental anomalies (partial anodontia, microdontia enamel hypoplasia), joint hypermobility, nephrocalcinosis, hypothyroidism, and poor weight gain. Narrowing of the coronary artery ostia can occur in patients with supravalvar aortic stenosis and should be carefully evaluated. Stenosis of other arteries, in particular, the branch pulmonary arteries, may also be present. Williams syndrome has been shown to be due to a deletion involving the elastin gene on chromosome 7q11.23.
Clinical Manifestations
Symptoms in patients with aortic stenosis depend on the severity of the obstruction. Severe aortic stenosis that occurs in early infancy is termed critical aortic stenosis and is associated with left ventricular failure and signs of low cardiac output. Heart failure, cardiomegaly, and pulmonary edema are severe, the pulses are weak in all extremities, and the skin may be pale or grayish. Urine output may be diminished. If cardiac output is significantly decreased, the intensity of the murmur at the right upper sternal border may be minimal. Most children with less severe forms of aortic stenosis remain asymptomatic and display normal growth and development. The murmur is usually discovered during routine physical examination. Rarely, fatigue, angina, dizziness, or syncope may develop in an older child with previously undiagnosed severe obstruction to left ventricular outflow. Sudden death has been reported with aortic stenosis but usually occurs in patients with severe left ventricular outflow obstruction in whom surgical relief has been delayed.
The physical findings are dependent on the degree of obstruction to left ventricular outflow. In mild stenosis, the pulses, heart size, and apical impulse are all normal. With increasing degrees of severity, the pulses become diminished in intensity and the heart may be enlarged, with a left ventricular apical thrust. Mild to moderate valvular aortic stenosis is usually associated with an early systolic ejection click, best heard at the apex and left sternal edge. Unlike the click in pulmonic stenosis, its intensity does not vary with respiration. Clicks are unusual in more severe aortic stenosis or in discrete subaortic stenosis. If the stenosis is severe, the 1st heart sound may be diminished because of decreased compliance of the thickened left ventricle. Normal splitting of the 2nd heart sound is present in mild to moderate obstruction. In patients with severe obstruction, the intensity of aortic valve closure is diminished, and, rarely in children, the 2nd sound may be split paradoxically (becoming wider in expiration). A 4th heart sound may be audible when the obstruction is severe due to decreased left ventricular compliance.
The intensity, pitch, and duration of the systolic ejection murmur are other indications of severity. The louder, harsher (higher pitch), and longer the murmur, the greater the degree of obstruction is. The typical murmur is audible maximally at the right upper sternal border and radiates to the neck and the left midsternal border. It is usually accompanied by a thrill in the suprasternal notch. In patients with subvalvular aortic stenosis, the murmur may be maximal along the left sternal border or even at the apex. A soft decrescendo diastolic murmur indicative of aortic insufficiency is often present when the obstruction is subvalvular or in patients with a bicuspid aortic valve. Occasionally, an apical short mid-diastolic rumbling murmur is audible; this murmur should raise suspicion of associated mitral valve stenosis.
Laboratory Findings and Diagnosis
The diagnosis can usually be made on the basis of the physical examination and the severity of obstruction confirmed by laboratory tests. If the pressure gradient across the aortic valve is mild, the electrocardiogram is likely to be normal. The electrocardiogram may occasionally be normal even with more severe obstruction, but evidence of left ventricular hypertrophy and strain (inverted T waves in the left precordial leads) is generally present if severe stenosis is long-standing. The chest radiograph frequently shows a prominent ascending aorta, but the aortic knob is normal. Heart size is typically normal. Valvular calcification has been noted only in older children and adults. Echocardiography identifies both the site and the severity of the obstruction. Two-dimensional imaging shows left ventricular hypertrophy and the thickened and domed aortic valve (Fig. 421-6). The echo will also demonstrate the number of valve leaflets and their morphology, and the presence of a subaortic membrane or supravalvar stenosis. Associated anomalies of the mitral valve or aortic arch or a VSD or PDA are present in up to 20% of cases. In the absence of left ventricular failure, the shortening fraction of the left ventricle may be increased because the ventricle is hypercontractile. In infants with critical aortic stenosis, the left ventricular shortening fraction is usually decreased and may be quite poor. The endocardium may appear bright, indicative of the development of endocardial fibrous scarring, known as endocardial fibroelastosis. Doppler studies show the specific site of obstruction and determine the peak and mean systolic left ventricular outflow tract gradients. When severe aortic obstruction is associated with left ventricular dysfunction, the Doppler-derived valve gradient may markedly underestimate the severity of the obstruction because of the low cardiac output across the valve.

Figure 421-6 Echocardiogram showing valvar aortic stenosis with regurgitation. A, In this parasternal long axis view, the stenotic aortic valve can be seen doming in systole. The crosshatch marks delineate the aortic annulus. B, Doppler study shows the presence of aortic regurgitation (arrow). Ao, aorta; LA, left atrium; LV, left ventricle.
Left heart catheterization, usually performed in conjunction with aortic balloon valvuloplasty, demonstrates the magnitude of the pressure gradient from the left ventricle to the aorta. The aortic pressure curve is abnormal if the obstruction is severe. In patients with severe obstruction and decreased left ventricular compliance, left atrial pressure is increased and pulmonary hypertension may be present. When a critically ill infant with left ventricular outflow tract obstruction undergoes cardiac catheterization, left ventricular function is often markedly decreased. As with the echocardiogram, the gradient measured across the stenotic aortic valve may underestimate the degree of obstruction because of low cardiac output. Actual measurement of cardiac output by thermodilution and calculation of the aortic valve area may be helpful.
Treatment
Balloon valvuloplasty is indicated for children with moderate to severe valvular aortic stenosis to prevent progressive left ventricular dysfunction and the risk of syncope and sudden death. It is generally agreed that valvuloplasty should be advised when the peak-to-peak systolic gradient between the left ventricle and aorta exceeds 60-70 mm Hg at rest, assuming normal cardiac output, or for lesser gradients when symptoms or electrocardiographic changes are present. For more rapidly progressive subaortic obstructive lesions, a gradient of 40-50 mm Hg or the presence of aortic insufficiency is considered an indication for surgery. With the development of low-profile balloons and smaller catheters that cause less injury to peripheral arteries, balloon valvuloplasty has become the procedure of choice even in the neonatal period. Surgical treatment is usually reserved for extremely dysplastic aortic valves that are not amenable to balloon therapy or in patients who also have subvalvar or valvar (also known as supravalvar) stenosis.
Discrete subaortic stenosis can be resected without damage to the aortic valve, the anterior leaflet of the mitral valve, or the conduction system. This type of obstruction is not usually amenable to catheter treatment. Relief of supravalvular stenosis is also achieved surgically, and the results are excellent if the area of obstruction is discrete and not associated with a hypoplastic aorta. In association with supravalvular aortic stenosis, one or both coronary arteries may be stenotic at their origins because of a thick supra-aortic fibrous ridge. For patients who have aortic stenosis in association with severe tunnel-like subaortic obstruction, the left ventricular outflow tract can be enlarged by “borrowing” space anteriorly from the right ventricular outflow tract (the Konno procedure).
Regardless of whether surgical or catheter treatment has been carried out, aortic insufficiency or calcification with re-stenosis is likely to occur years or even decades later and eventually require reoperation and often aortic valve replacement. When recurrence develops, it may not be associated with early symptoms. Signs of recurrent stenosis include electrocardiographic signs of left ventricular hypertrophy, an increase in the Doppler echocardiographic gradient, deterioration in echocardiographic indices of left ventricular function, and recurrence of signs or symptoms during graded treadmill exercise. Evidence of significant aortic regurgitation includes symptoms of heart failure, cardiac enlargement on roentgenogram, and left ventricular dilatation on echocardiogram. The choice of reparative procedure depends on the relative degree of stenosis and regurgitation.
When aortic valve replacement is necessary, the choice of procedure often depends on the age of the patient. Homograft valves tend to calcify more rapidly in younger children, but they do not require chronic anticoagulation. Mechanical prosthetic valves are much longer lasting, yet they require anticoagulation, which can be difficult to manage in young children. In adolescent girls who are nearing childbearing age, consideration of the teratogenic effects of warfarin may warrant the use of a homograft valve. None of these options are perfect for a younger child who requires valve replacement because neither homograft nor mechanical valves grow with the patient. An alternative operation is aortopulmonary translocation (Ross procedure); it involves removing the patient’s own pulmonary valve and using it to replace the abnormal aortic valve. A homograft is then placed in the pulmonary position. The potential advantage of this procedure is the possibility for growth of the translocated living “neoaortic” valve and the increased longevity of the homograft valve when placed in the lower pressure pulmonary circulation. The long-term success of this operation, especially in young children, is still being investigated. Stent valves, which are tissue valves sewn into the inside of an expandable metal stent, are currently in clinical trials in adults. These can be implanted in the cardiac catheterization laboratory using a percutaneous approach. Tissue-engineered replacement valves grown in the laboratory from the patient’s own arterial endothelial cells are the best hope for long-term palliation and are currently under development in animal models.
Prognosis
Neonates with critical aortic stenosis may have severe heart failure and deteriorate rapidly to a low-output shock state. Emergency surgery or balloon valvuloplasty is lifesaving, but the mortality risk is not trivial. Neonates who die of critical aortic stenosis frequently have significant left ventricular endocardial fibroelastosis. Those who survive may develop signs of left ventricular diastolic muscle dysfunction (restrictive cardiomyopathy) and require cardiac transplantation (Chapter 433.3).
In older infants and children with mild to moderate aortic stenosis, the prognosis is reasonably good, although disease progression over a period of 5-10 yr is common. Patients with aortic valve gradients <40-50 mm Hg are considered to have mild disease; those with gradients of 40-70 mm Hg have moderate disease. These patients usually respond well to treatment (either surgery or valvuloplasty), although reoperations on the aortic valve are often required later in childhood or in adult life, and many patients eventually require valve replacement. In unoperated patients with severe obstruction, sudden death is a significant risk and often occurs during or immediately after exercise. Aortic stenosis is one of the causes of sudden cardiac death in the pediatric age group.
Patients with moderate to severe degrees of aortic stenosis should not participate in active competitive sports. In those with milder disease, sports participation is less severely restricted. The status of each patient should be reviewed at least annually and intervention advised if progression of signs or symptoms occurs. Prophylaxis against infective endocarditis is no longer recommended unless a prosthetic valve has been inserted.
Older children and adults with isolated bicuspid aortic valve are at increased risk for developing dilation of their ascending aorta, even in the absence of significant stenosis. This risk increases with age, and the rate of increase is greatest in those with the largest aortic roots. In children, this dilation is usually mild and remains stable over many years of observation, but in older patients the aorta can dilate substantially and progressively. Whether these patients have some undiagnosed form of connective tissue disorder remains to be determined (since this form of dilation is similar to that seen in Marfan syndrome). Patients with Turner syndrome and bicuspid aortic valve do have an increased risk of aortic dilation. Although dissection and rupture are described complications of severe aortic root dilation in adults, there is not yet sufficient data to determine these risks in children. Only isolated cases have been reported.
Carabello BA, Paulus WJ. Aortic stenosis. Lancet. 2009;373:956-964.
Harkel ADJT, Berkhout M, Hop WC, et al. Congenital valvular aortic stenosis: limited progression during childhood. Arch Dis Child. 2009;94:531-535.
Hickey EJ, Jung G, Williams WG, et al. Congenital supravalvular aortic stenosis: defining surgical and nonsurgical outcomes. Ann Thorac Surg. 2008;86:1919-1927.
Jahangiri M, Nicholson IA, del Nido PJ, et al. Surgical management of complex and tunnel-like subaortic stenosis. Eur J Cardiothorac Surg. 2000;17:637-642.
Laudito A, Brook MM, Suleman S, et al. The Ross procedure in children and young adults: a word of caution. J Thorac Cardiovasc Surg. 2001;122:147-153.
Lopez L, Arheart KL, Colan SD, et al. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics. 2008;121:e1622-e1627.
Mahle WT, Sutherland JL, Frias PA. Outcome of isolated bicuspid aortic valve in childhood. J Pediatr. 2010;157:445-449.
Marino BS, Bridges ND, Paridon SM. Aortic insufficiency: indications for surgery in children. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 1998;1:147-156.
Morales DL, Carberry KE, Balentine C, et al. Selective application of the pediatric Ross procedure minimizes autograft failure. Congenit Heart Dis. 2008;3:404-410.
Pessotto R, Wells WJ, Baker CJ, et al. Midterm results of the Ross procedure. Ann Thorac Surg. 2001;71(Suppl 5):S336-S339.
Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239-250.
Rocchini A, Beekman RH, Ben Shachar G, et al. Balloon aortic valvuloplasty: Results of the valvuloplasty and angioplasty of congenital anomalies registry. Am J Cardiol. 1990;65:784-789.
Tweddell JS, Pelech AN, Frommelt PC, et al. Complex aortic valve repair as a durable and effective alternative to valve replacement in children with aortic valve disease. J Thorac Cardiovasc Surg. 2005;129:551-558.
Vida VL, Hoehn R, Larrazabal LA, et al. Usefulness of intra-operative epicardial three-dimensional echocardiography to guide aortic valve repair in children. Am J Cardiol. 2009;103:852-856.
421.6 Coarctation of the Aorta
Constrictions of the aorta of varying degrees may occur at any point from the transverse arch to the iliac bifurcation, but 98% occur just below the origin of the left subclavian artery at the origin of the ductus arteriosus (juxtaductal coarctation). The anomaly occurs twice as often in males as in females. Coarctation of the aorta may be a feature of Turner syndrome (Chapters 76 and 580.1) and is associated with a bicuspid aortic valve in more than 70% of patients. Mitral valve abnormalities (a supravalvular mitral ring or parachute mitral valve) and subaortic stenosis are potential associated lesions. When this group of left-sided obstructive lesions occurs together, they are referred to as the Shone complex.
Pathophysiology
Coarctation of the aorta can occur as a discrete juxtaductal obstruction or as tubular hypoplasia of the transverse aorta starting at one of the head or neck vessels and extending to the ductal area (previously referred to as preductal or infantile-type coarctation; Fig. 421-7). Often, both components are present. It is postulated that coarctation may be initiated in fetal life by the presence of a cardiac abnormality that results in decreased blood flow anterograde through the aortic valve (e.g., bicuspid aortic valve, VSD). Alternatively, coarctation may be due to abnormal extension of contractile ductal tissue into the aortic wall.

Figure 421-7 Metamorphosis of coarctation. A, Fetal prototype with no flow obstruction. B, Late gestation. The aortic ventricle increases its output and dilates the hypoplastic segment. Antegrade aortic flow bypasses the shelf via the ductal orifice. C, Neonate. Ductal constriction initiates the obstruction by removing the bypass and increasing antegrade arch flow. D, Mature juxtaductal stenosis. The bypass is completely obliterated, and intimal hypoplasia on the edge of the shelf is aggravating the stenosis. Collaterals develop. E, Persistence of the infantile-type fetal prototype. An intracardiac left-sided heart obstruction precludes an increase in antegrade aortic flow before or after birth. Both isthmus hypoplasia and a contraductal shelf are present. Lower body flow often depends on patency of the ductus.
(From Gersony WM: Coarctation of the aorta. In Adams FH, Emmanouilides GC, Riemenshneider T, editors: Moss heart disease in infants, children, and adolescents, ed 4, Baltimore, 1989, Williams & Wilkins.)
In patients with discrete juxtaductal coarctation, ascending aortic blood flows through the narrowed segment to reach the descending aorta, although left ventricular hypertension and hypertrophy result. In the 1st few days of life, the PDA may serve to widen the juxtaductal area of the aorta and provide temporary relief from the obstruction. Net left-to-right ductal shunting occurs in these acyanotic infants. With more severe juxtaductal coarctation or in the presence of transverse arch hypoplasia, right ventricular blood is ejected through the ductus to supply the descending aorta. Perfusion of the lower part of the body is then dependent on right ventricular output (see Fig. 421-7). In this situation, the femoral pulses are palpable, and differential blood pressures may not be helpful in making the diagnosis. The ductal right-to-left shunting is manifested as differential cyanosis, with the upper extremities being pink and the lower extremities blue.
Such infants may have severe pulmonary hypertension and high pulmonary vascular resistance. Signs of heart failure are prominent. Occasionally, severely hypoplastic segments of the aortic isthmus may become completely atretic and result in an interrupted aortic arch, with the left subclavian artery arising either proximal or distal to the interruption. Coarctation associated with arch hypoplasia was once referred to as infantile type because its severity usually led to recognition of the condition in early infancy. Adult type referred to isolated juxtaductal coarctation, which, if mild, was not usually recognized until later childhood. These terms have been replaced with the more accurate anatomic terms describing the location and severity of the defect.
Blood pressure is elevated in the vessels that arise proximal to the coarctation; blood pressure as well as pulse pressure is lower below the constriction. The hypertension is not due to the mechanical obstruction alone but also involves neurohumoral mechanisms. Unless operated on in infancy, coarctation of the aorta usually results in the development of an extensive collateral circulation, chiefly from branches of the subclavian, superior intercostal, and internal mammary arteries, to create channels for arterial blood to bypass the area of coarctation. The vessels contributing to the collateral circulation may become markedly enlarged and tortuous by early adulthood.
Clinical Manifestations
Coarctation of the aorta recognized after infancy is not usually associated with significant symptoms. Some children or adolescents complain about weakness or pain (or both) in the legs after exercise, but in many instances, even patients with severe coarctation are asymptomatic. Older children are frequently brought to the cardiologist’s attention when they are found to be hypertensive on routine physical examination.
The classic sign of coarctation of the aorta is a disparity in pulsation and blood pressure in the arms and legs. The femoral, popliteal, posterior tibial, and dorsalis pedis pulses are weak (or absent in up to 40% of patients), in contrast to the bounding pulses of the arms and carotid vessels. The radial and femoral pulses should always be palpated simultaneously for the presence of a radial-femoral delay. Normally, the femoral pulse occurs slightly before the radial pulse. A radial-femoral delay occurs when blood flow to the descending aorta is dependent on collaterals, in which case the femoral pulse is felt after the radial pulse. In normal persons (except neonates), systolic blood pressure in the legs obtained by the cuff method is 10-20 mm Hg higher than that in the arms. In coarctation of the aorta, blood pressure in the legs is lower than that in the arms; frequently, it is difficult to obtain. This differential in blood pressures is common in patients with coarctation who are older than 1 yr, about 90% of whom have systolic hypertension in an upper extremity greater than the 95th percentile for age. It is important to determine the blood pressure in each arm; a pressure higher in the right than the left arm suggests involvement of the left subclavian artery in the area of coarctation. Occasionally, the right subclavian may arise anomalously from below the area of coarctation and result in a left arm pressure that is higher than the right. With exercise, a more prominent rise in systemic blood pressure occurs, and the upper-to-lower extremity pressure gradient will increase.
The precordial impulse and heart sounds are usually normal; the presence of a systolic ejection click or thrill in the suprasternal notch suggests a bicuspid aortic valve (present in 70% of cases). A short systolic murmur is often heard along the left sternal border at the 3rd and 4th intercostal spaces. The murmur is well transmitted to the left infrascapular area and occasionally to the neck. Often, the typical murmur of mild aortic stenosis can be heard in the 3rd right intercostal space. Occasionally, more significant degrees of obstruction are noted across the aortic valve. The presence of a low-pitched mid-diastolic murmur at the apex suggests mitral valve stenosis. In older patients with well-developed collateral blood flow, systolic or continuous murmurs may be heard over the left and right sides of the chest laterally and posteriorly. In these patients, a palpable thrill can occasionally be appreciated in the intercostal spaces on the back.
Neonates or infants with more severe coarctation, usually including some degree of transverse arch hypoplasia, initially have signs of lower body hypoperfusion, acidosis, and severe heart failure. These signs may be delayed days or weeks until after closure of the ductus arteriosus. If detected before ductal closure, patients may exhibit differential cyanosis, best demonstrated by simultaneous oximetry of the upper and lower extremities. On physical examination, the heart is large, and a systolic murmur is heard along the left sternal border with a loud 2nd heart sound.
Diagnosis
Findings on roentgenographic examination depend on the age of the patient and on the effects of hypertension and the collateral circulation. Cardiac enlargement and pulmonary congestion are noted in infants with severe coarctation. During childhood, the findings are not striking until after the 1st decade, when the heart tends to be mildly or moderately enlarged because of left ventricular prominence. The enlarged left subclavian artery commonly produces a prominent shadow in the left superior mediastinum. Notching of the inferior border of the ribs from pressure erosion by enlarged collateral vessels is common by late childhood. In most instances, the descending aorta has an area of poststenotic dilatation.
The electrocardiogram is usually normal in young children but reveals evidence of left ventricular hypertrophy in older patients. Neonates and young infants display right or biventricular hypertrophy. The segment of coarctation can generally be visualized by two-dimensional echocardiography (Fig. 421-8); associated anomalies of the mitral and aortic valve can also be demonstrated. The descending aorta is hypopulsatile. Color Doppler is useful for demonstrating the specific site of the obstruction. Pulsed and continuous wave Doppler studies determine the pressure gradient directly at the area of coarctation; in the presence of a PDA, however, the severity of the narrowing may be underestimated. CT and MRI are valuable noninvasive tools for evaluation of coarctation when the echocardiogram is equivocal. Cardiac catheterization with selective left ventriculography and aortography is useful in occasional patients with additional anomalies and as a means of visualizing collateral blood flow. In cases that are well defined by echocardiography, CT, or MRI, diagnostic catheterization is not usually required before surgery.
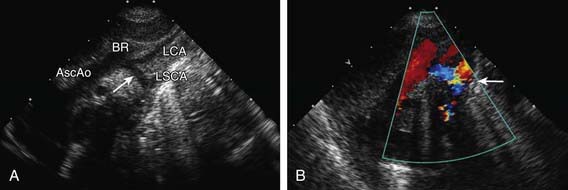
Figure 421-8 Echocardiogram demonstrating coarctation of the aorta with hypoplastic transverse arch. A, Suprasternal notch two-dimensional echocardiogram showing marked narrowing beginning just distal to the brachiocephalic artery. B, Color Doppler demonstrates turbulent flow in the juxtductal area (arrow). AscAo, ascending aorta; BR, brachiocephalic artery; LCA, left carotid artery; LSCA, left subclavian artery.
Treatment
In neonates with severe coarctation of the aorta, closure of the ductus often results in hypoperfusion, acidosis, and rapid deterioration. These patients should be given an infusion of prostaglandin E1 to reopen the ductus and re-establish adequate lower extremity blood flow. Once a diagnosis has been confirmed and the patient stabilized, surgical repair should be performed. Older infants with heart failure but good perfusion should be managed with anticongestive measures to improve their clinical status before surgical intervention. There is usually no reason to delay surgical repair waiting for patient growth; successful repairs have been performed in small premature infants.
Older children with significant coarctation of the aorta should be treated relatively soon after diagnosis. Delay is unwarranted, especially after the 2nd decade of life, when the operation may be less successful because of decreased left ventricular function and degenerative changes in the aortic wall. Nevertheless, if cardiac reserve is sufficient, satisfactory repair is possible well into mid-adult life.
The procedure of choice for isolated juxtaductal coarctation of the aorta is controversial. Surgery remains the treatment of choice at most centers, and several surgical techniques are used. The area of coarctation can be excised and a primary re-anastomosis performed. Often, the transverse aorta is splayed open and an “extended end-to-end” anastomosis performed to increase the effective cross-sectional area of the repair. The subclavian flap procedure, which involves division of the left subclavian artery and incorporation of it into the wall of the repaired coarctation, is used by some, although has grown out of favor due to a higher degree of residual stenosis. Others favor a patch aortoplasty, in which the area of coarctation is enlarged with a roof of prosthetic material. The use of primary angioplasty for native coarctation remains controversial, although is useful in conditions where surgical intervention may be associated with increased risk in patients with severe left ventricular dysfunction.
After surgery, a striking increase in the amplitude of pulsations in the lower extremities is noted. In the immediate postoperative course, “rebound” hypertension is common and requires medical management. This exaggerated acute hypertension gradually subsides and, in most patients, antihypertensive medications can be discontinued. Residual murmurs are common and may be due to associated cardiac anomalies, to a residual flow disturbance across the repaired area, or to collateral blood flow. Rare operative problems include spinal cord injury from aortic cross-clamping if the collaterals are poorly developed, chylothorax, diaphragm injury, and laryngeal nerve injury. If a left subclavian flap is used, the radial pulse and blood pressure in the left arm are diminished or absent.
Postcoarctectomy Syndrome
Postoperative mesenteric arteritis may be associated with acute hypertension and abdominal pain in the immediate postoperative period. The pain varies in severity and may occur in conjunction with anorexia, nausea, vomiting, leukocytosis, intestinal hemorrhage, bowel necrosis, and small bowel obstruction. Relief is usually obtained with antihypertensive drugs (nitroprusside, esmolol, captopril) and intestinal decompression; surgical exploration is rarely required for bowel obstruction or infarction.
Prognosis
Although re-stenosis in older patients after coarctectomy is rare, a significant number of infants operated on before 1 yr of age require revision later in childhood. All patients should be monitored carefully for the development of recoarctation and an aortic anastomotic aneurysm. Should recoarctation occur, balloon angioplasty is the procedure of choice. In these patients, scar tissue from previous surgery may make reoperation more difficult yet makes balloon angioplasty safer because of the lower incidence of aneurysm formation. Relief of obstruction with this technique is usually excellent. Intravascular stents are commonly used, especially in adolescents and young adults, with generally excellent results.
Repair of coarctation in the 2nd decade of life or beyond may be associated with a higher incidence of premature cardiovascular disease, even in the absence of residual cardiac abnormalities. Early onset of adult chronic hypertension may occur, even in patients with adequately resected coarctation.
Abnormalities of the aortic valve are present in most patients. Bicuspid aortic valves are common but do not generally produce clinical signs unless the stenosis is significant. The association of a PDA and coarctation of the aorta is also common. VSDs and ASDs may be suspected by signs of a left-to-right shunt; they are exacerbated by the increased resistance to flow through the left side of the heart. Mitral valve abnormalities are also occasionally seen, as is subvalvular aortic stenosis.
Severe neurologic damage or even death may rarely occur from associated cerebrovascular disease. Subarachnoid or intracerebral hemorrhage may result from rupture of congenital aneurysms in the circle of Willis, rupture of other vessels with defective elastic and medial tissue, or rupture of normal vessels; these accidents are secondary to hypertension. Children with PHACE syndrome (posterior brain fossa anomalies, facial hemangiomas, arterial anomalies, cardiac anomalies and aortic coarctation, eye anomalies) may have strokes (Table 416-2). Abnormalities of the subclavian arteries may include involvement of the left subclavian artery in the area of coarctation, stenosis of the orifice of the left subclavian artery, and anomalous origin of the right subclavian artery.
Untreated, the great majority of older patients with coarctation of the aorta would succumb between the ages of 20 and 40 yr; some live well into middle life without serious disability. The common serious complications are related to systemic hypertension, which may result in premature coronary artery disease, heart failure, hypertensive encephalopathy, or intracranial hemorrhage. Heart failure may be worsened by associated anomalies. Infective endocarditis or endarteritis is a significant complication in adults. Aneurysms of the descending aorta or the enlarged collateral vessels may develop.
Beaton AZ, Nguyen T, Lai WW, et al. Relation of coarctation of the aorta to the occurrence of ascending aortic dilation in children and young adults with bicuspid aortic valves. Am J Cardiol. 2009;103:266-270.
Daniels SR. Repair of coarctation of the aorta and hypertension: does age matter? Lancet. 2001;358:89.
Hager A, Schreiber C, Nutzl S, et al. Mortality and restenosis rate of surgical coarctation repair in infancy: a study of 191 patients. Cardiology. 2009;112:36-41.
Hamdan MA, Maheshwari S, Fahey JT. Endovascular stents for coarctation of the aorta: initial results and intermediate-term follow-up. J Am Coll Cardiol. 2001;38:1518-1523.
Karamlou T, Bernasconi A, Jaeggi E, et al. Factors associated with arch reintervention and growth of the aortic arch after coarctation repair in neonates weighing less than 2.5 kg. J Thorac Cardiovasc Surg. 2009;137:1163-1167.
Markham LW, Knecht SK, Daniels SR, et al. Development of exercise-induced arm-leg blood pressure gradient and abnormal arterial compliance in patients with repaired coarctation of the aorta. Am J Cardiol. 2004;94:1200-1202.
Metry DW, Dowd CF, Barkovich J, et al. The many faces of PHACE syndrome. J Pediatr. 2001;139:117-123.
O’Sullivan JJ, Derrick G, Darnell R. Prevalence of hypertension in children after early repair of coarctation of the aorta: a cohort study using casual and 24 hour blood pressure measurement. Heart. 2002;88:163-166.
Senzaki H, Iwamoto Y, Ishido H, et al. Ventricular-vascular stiffening in patients with repaired coarctation of aorta: integrated pathophysiology of hypertension. Circulation. 2008;118(Suppl 14):S191-S198.
Shone JD, Sellers RD, Anderson RC, et al. The developmental complex of “parachute mitral valve,” supravalvar ring of left atrium, subaortic stenosis, and coarctation of the aorta. Am J Cardiol. 1963;11:714-725.
Vijayalakshmi K, Griffiths A, Hasan A, et al. Late hazards after repair of coarctation of the aorta. BMJ. 2008;336:772-773.
421.7 Coarctation with Ventricular Septal Defect
Coarctation in the presence of a VSD results in both increased preload and afterload on the left ventricle, and patients with this combination of defects will be recognized either at birth or in the 1st mo of life and often have intractable cardiac failure. The magnitude of the left-to-right shunt through a VSD is dependent on the ratio of pulmonary to systemic vascular resistance. In the presence of coarctation, resistance to systemic outflow is enhanced by the obstruction, and the volume of the shunt is markedly increased. The clinical picture is that of a seriously ill infant with tachypnea, failure to thrive, and typical findings of heart failure. Often, the difference in blood pressure between the upper and lower extremities is not very marked because cardiac output may be low. Medical management should be used to stabilize the patient initially; however, it should not be used to delay corrective surgery inordinately.
In most cases, coarctation is the major anomaly causing the severe symptoms, and resection of the coarcted segment results in striking improvement. Many centers routinely repair both the VSD and coarctation at the same operation through a midline sternotomy using cardiopulmonary bypass. Some centers repair the coarctation through a left lateral thoracotomy and, at the same time, place a pulmonary artery band to decrease the ventricular-level shunt. This may be performed when a complicated VSD is present (multiple VSDs, apical muscular VSD), to avoid open heart surgery during infancy for these complex ventricular septal abnormalities.
421.8 Coarctation with Other Cardiac Anomalies and Interrupted Aortic Arch
Coarctation often occurs in infancy in association with other major cardiovascular anomalies, including hypoplastic left heart, severe mitral or aortic valve disease, transposition of the great arteries, and variations of double-outlet or single ventricle. The clinical manifestations depend on the effects of the associated malformations, as well as on the coarctation itself.
Coarctation of the aorta associated with severe mitral and aortic valve disease may have to be treated within the context of the hypoplastic left heart syndrome (Chapter 425.10), even if the left ventricular chamber is not severely hypoplastic. Such patients usually have a long segment of narrow transverse aortic arch in addition to an isolated coarctation at the site of the ductus arteriosus. Coarctation of the aorta with transposition of the great arteries or single ventricle may be repaired alone or in combination with other corrective or palliative measures.
Complete interruption of the aortic arch is the most severe form of coarctation and is usually associated with other intracardiac pathology. Interruption may occur at any level, although it is most commonly seen between the left subclavian artery and the insertion of the ductus arteriosus (type A), followed in frequency by those between the left subclavian and left carotid arteries (type B), or between the left carotid and brachiocephalic arteries (type C). In newborns with an interrupted aortic arch, the ductus arteriosus provides the sole source of blood flow to the descending aorta, and differential oxygen saturations between the right arm (normal saturation) and the legs (decreased saturation) is noted. When the ductus begins to close, severe congestive heart failure, lower extremity hypoperfusion, anuria, and shock usually develops. Patients with an interrupted aortic arch can be supported with prostaglandin E1 to keep the ductus patent before surgical repair. As one of the conotruncal malformations, an interrupted aortic arch, especially type B, can be associated with DiGeorge syndrome (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia). Cytogenetic analysis using fluorescence in situ hybridization demonstrates deletion of a segment of chromosome 22q11 known as the DiGeorge critical region.
421.9 Congenital Mitral Stenosis
Congenital mitral stenosis is a rare anomaly that can be isolated or associated with other defects, the most common being subvalvar and valvar aortic stenosis and coarctation of the aorta (Shone complex). The mitral valve may be funnel-shaped, with thickened leaflets and chordae tendineae that are shortened and deformed. Other mitral valve anomalies associated with stenosis include parachute mitral valve, caused by a single papillary muscle, and double-orifice mitral valve.
If the stenosis is moderate to severe, symptoms usually appear within the 1st yr or 2 of life. These infants have failure to thrive and various degrees of dyspnea and pallor. In some patients, wheezing may be a dominant symptom, and a misdiagnosis of bronchiolitis or reactive airway disease may have been made. Heart enlargement as a result of dilatation and hypertrophy of the right ventricle and left atrium is common. Most patients have rumbling apical diastolic murmurs, but the auscultatory findings may be relatively obscure. The 2nd heart sound is loud and split. An opening snap of the mitral valve may be present. The electrocardiogram reveals right ventricular hypertrophy and may show bifid or spiked P waves indicative of left atrial enlargement. Roentgenograms usually show left atrial and right ventricular enlargement and pulmonary congestion in a perihilar or venous pattern. The echocardiogram is characteristic and shows thickened mitral valve leaflets a significant reduction of the mitral valve orifice, abnormal papillary muscle structure (or a single papillary muscle), and an enlarged left atrium with a normal or small left ventricle. A double orifice may also be visualized. Doppler studies demonstrate a mean pressure gradient across the mitral orifice. Associated anomalies such as aortic stenosis and coarctation can be evaluated. Cardiac catheterization is usually performed to confirm the transmitral pressure gradient before surgery. An increase in right ventricular, pulmonary arterial, and pulmonary capillary wedge pressure can be noted. Angiocardiography shows delayed emptying of the left atrium and the small mitral orifice.
The results of surgical treatment depend on the anatomy of the valve, but if the mitral orifice is significantly hypoplastic, reduction of the gradient may be difficult. In some patients, a mitral valve prosthesis is required, and if the valve orifice is too small, the prosthesis may be placed in the supramitral position. However, whatever prosthesis is used, it must be replaced serially as the child grows. These patients must be managed by anticoagulation with warfarin, and complications of excessive and insufficient anticoagulation are fairly common in infancy. Transcatheter balloon valvuloplasty has been used as a palliative procedure with disappointing results, except in the situation of rheumatic mitral stenosis.
Chandrashekhar Y, Westaby S, Narula J. Mitral stenosis. Lancet. 2009;374:1271-1283.
Marino BS, Kruge LE, Cho CJ, et al. Parachute mitral valve: morphologic descriptors, associated lesions, and outcomes after biventricular repair. J Thorac Cardiovasc Surg. 2009;137:385-393.
Moore P, Adatia I, Spevak PJ, et al. Severe congenital mitral stenosis in infants. Circulation. 1994;89:2099-2106.
Spevak PJ, Bass JL, Ben-Shachar G, et al. Balloon angioplasty for congenital mitral stenosis. Am J Cardiol. 1990;66:472-476.
Toscano A, Pasquini L, Iacobelli R, et al. Congenital supravalvar mitral ring: an underestimated anomaly. J Thorac Cardiovasc Surg. 2009;137:538-542.
421.10 Pulmonary Venous Hypertension
A variety of lesions may give rise to chronic pulmonary venous hypertension, which when extreme may result in pulmonary arterial hypertension and right-sided heart failure. These lesions include congenital mitral stenosis, mitral insufficiency, total anomalous pulmonary venous return with obstruction, left atrial myxomas, cor triatriatum (stenosis of a common pulmonary vein), individual pulmonary vein stenosis, and supravalvular mitral rings. Early symptoms can be confused with chronic pulmonary disease such as asthma because of a lack of specific cardiac findings on physical examination. Subtle signs of pulmonary hypertension may be present. The electrocardiogram shows right ventricular hypertrophy with spiked P waves. Roentgenographic studies reveal cardiac enlargement and prominence of the pulmonary veins in the hilar region, the right ventricle and atrium, and the main pulmonary artery; the left atrium is normal in size or only slightly enlarged.
The echocardiogram may demonstrate left atrial myxoma, cor triatriatum, stenosis of one or more pulmonary veins, or a mitral valve abnormality, especially supravalvar mitral ring. Cardiac catheterization excludes the presence of a shunt and demonstrates pulmonary hypertension with elevated pulmonary arterial wedge pressure. Left atrial pressure is normal if the lesion is at the level of the pulmonary veins, but it is elevated if the lesion is at the level of the mitral valve. Selective pulmonary arteriography usually delineates the anatomic lesion. Cor triatriatum, left atrial myxoma, and supravalvular mitral rings can all be successfully managed surgically.
The differential diagnosis includes pulmonary veno-occlusive disease, an idiopathic process that produces obstructive lesions in 1 or more pulmonary veins. The cause is uncertain and disease that begins in 1 vein can spread to others. Although it is usually encountered in patients after repair of obstructed total anomalous pulmonary venous return (Chapter 425.7), it can occur in the absence of congenital heart disease. The patient initially presents with left-sided heart failure on the basis of congested lungs with apparent pulmonary edema. Dyspnea, fatigue, and pleural effusions are common. Left atrial pressure is normal, but pulmonary arterial wedge pressure is usually elevated. A normal wedge pressure may be encountered if collaterals have formed or the wedge recording is performed in an uninvolved segment. Angiographically, the pulmonary veins return normally to the left atrium, but one or more pulmonary veins are narrowed, either focally or diffusely.
Studies using lung biopsy have demonstrated pulmonary venous and, occasionally, arterial involvement. Pulmonary veins and venules demonstrate fibrous narrowing or occlusion, and pulmonary artery thrombi may be present. Attempts at surgical repair, balloon dilatation, and transcatheter stenting have not significantly improved the generally poor prognosis of these patients. Clinical trials of antiproliferative chemotherapy are currently in progress. Combined heart-lung transplantation (Chapter 437.2) is often the only alternative therapeutic option.