Chapter 462 The Inherited Pancytopenias
Pancytopenia refers to a reduction below normal values of all 3 peripheral blood lineages: leukocytes, platelets, and erythrocytes. Pancytopenia requires microscopic examination of a bone marrow biopsy specimen and a marrow aspirate to assess overall cellularity and morphology. There are 3 general categories of pancytopenia depending on the marrow findings.
Inherited (“constitutional”) pancytopenia is defined as a decrease in marrow production of the 3 major hematopoietic lineages that occurs on an inherited basis, resulting in anemia, neutropenia, and thrombocytopenia. Any of these conditions (Table 462-1) can be transmitted as a simple mendelian disorder by mutant genes with inherited patterns of autosomal dominant, autosomal recessive, or X-linked types. Modifying genes and acquired factors may also be operative. Inherited pancytopenias account for approximately 30% of cases of pediatric marrow failure. Fanconi anemia is the most common of these disorders.
Fanconi Anemia
Etiology and Epidemiology
Fanconi anemia (FA) is primarily inherited in an autosomal recessive manner (one uncommon form is X-linked recessive). It occurs in all racial and ethnic groups. At presentation, patients with FA may have: (1) typical physical anomalies but normal hematologic findings; (2) normal physical features but abnormal hematologic findings; or (3) physical anomalies and abnormal hematologic findings, which constitute the classic phenotype (39% of cases). There can be sibling discordance in clinical and hematologic findings, even in affected monozygotic twins. Approximately 75% of patients are 3-14 yr of age at the time of diagnosis.
Pathology
Patients have abnormal chromosome fragility, which is seen in metaphase preparations of peripheral blood lymphocytes cultured with phytohemagglutinin and enhanced by adding clastogenic agents such as diepoxybutane (DEB) and mitomycin C. Cell fusion of FA cells with normal cells or with cells from some unrelated patients with FA produces a corrective effect on chromosomal fragility, a process called complementation. This phenomenon allows subtyping of cases of FA into discrete complementation groups. Fourteen separate complementation groups have been identified, and 14 mutant FA (FANC) genes have been cloned so far (A, B, C, D1/BRCA2, D2, E, F, G, I, J, L, M, N, and O) all prefixed with FANC, e.g. FANCA, FANCB and so on); FANCD1 is identical to the breast cancer susceptibility protein BRCA2. The protein products of wild-type FANC genes are involved in the DNA damage recognition and repair biochemical pathways. Therefore, mutant gene proteins lead to genomic instability, chromosome fragility, and FA. An inability of FA cells to remove oxygen-free radicals, resulting in oxidative damage, is a contributing factor in the pathogenesis. Additional factors are also operative. Leukocyte telomere length is significantly shortened but telomerase activity is increased, suggesting a high proliferative rate of marrow progenitors that ultimately leads to their premature senescence. Increased marrow cell apoptosis occurs and is mediated by Fas, a membrane glycoprotein receptor containing an integral death domain. A consistent finding is diminished cellular interleukin-6 production along with markedly heightened tumor necrosis factor-α generation.
Clinical Manifestations
The most common anomaly in FA is hyperpigmentation of the trunk, neck, and intertriginous areas, as well as café-au-lait spots and vitiligo, alone or in combination (Fig. 462-1 and Table 462-2). Half the patients have short stature. Growth failure may be associated with abnormal growth hormone secretion or with hypothyroidism. Absence of radii and thumbs that are hypoplastic, supernumerary, bifid, or absent are common. Anomalies of the feet, congenital hip dislocation, and leg abnormalities are seen. A male patient with FA may have an underdeveloped penis; undescended, atrophic, or absence of the testes; and hypospadias or phimosis. Females can have malformations of the vagina, uterus, and ovary. Many patients have a FA “facies,” including microcephaly, small eyes, epicanthal folds, and abnormal shape, size, or positioning of the ears (see Fig. 462-1). Ectopic, pelvic, or horseshoe kidneys are detected by imaging and may show other organs as duplicated, hypoplastic, dysplastic, or absent kidneys. Cardiovascular and gastrointestinal malformations also occur. Approximately 10% of patients with FA are cognitively delayed.
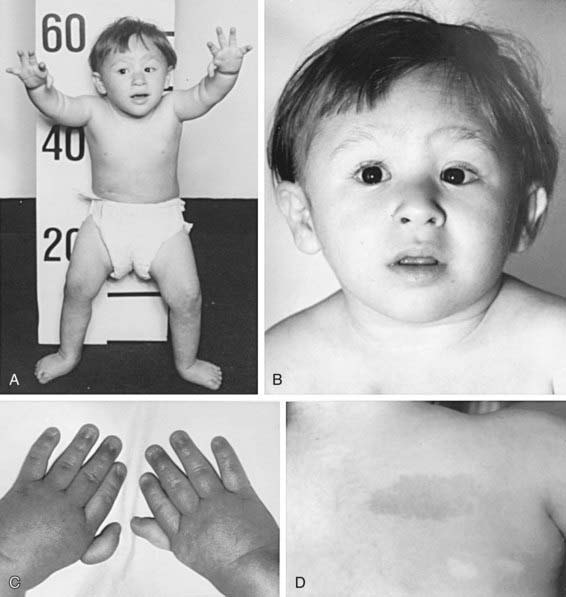
Figure 462-1 A 3 yr old boy with Fanconi anemia who exhibits several classic phenotype features. A, Front view. B, Face. C, Hands. D, Back right shoulder. The features to be noted include short stature, dislocated hips, microcephaly, a broad nasal base, epicanthal folds, micrognathia, thumbs attached by a thread, and café-au-lait spots with hypopigmented areas beneath.
(From Nathan DC, Orkin SH, Ginsburg D, et al, editors: Nathan and Oski’s hematology of infancy and childhood, ed 6, vol I, Philadelphia, 2003, WB Saunders, p 285.)
Table 462-2 CHARACTERISTIC PHYSICAL ANOMALIES IN FANCONI ANEMIA
| ANOMALY | APPROXIMATE FREQUENCY (% OF PATIENTS) |
|---|---|
| Skin pigment changes ± café-au-lait spots | 55 |
| Short stature | 51 |
| Upper limb abnormalities (thumbs, hands, radii, ulnas) | 43 |
| Hypogonadal and genital changes (mostly male) | 35 |
| Other skeletal findings (head/face, neck, spine) | 30 |
| Eye/lid/epicanthal fold anomalies | 23 |
| Renal malformations | 21 |
| Gastrointestinal/cardiopulmonary malformations | 11 |
| Hip, leg, foot, toe abnormalities | 10 |
| Ear anomalies (external and internal), deafness | 9 |
Laboratory Findings
Marrow failure usually ensues in the 1st decade of life. Thrombocytopenia often appears initially, with subsequent onset of granulocytopenia and then macrocytic anemia. Severe aplasia develops in most cases, but its full expression is variable and evolves over a period of months to years. The marrow becomes progressively hypocellular and fatty, like that in severe acquired aplastic anemia. Chromosome fragility is indicated by spontaneously occurring chromatid breaks, rearrangements, gaps, endoreduplications, and chromatid exchanges in blood lymphocytes cultured with phytohemagglutinin as well as in cultured skin fibroblasts, underscoring the constitutional nature of the disorder. With addition of DEB, fragility is strikingly enhanced in lymphocyte cultures of patients with FA in comparison with those of controls. For prenatal diagnosis, abnormal chromosome breakage can be tested for in amniotic fluid cells or in tissue from a chorionic villus biopsy.
Complications
A major feature of the phenotype of FA is the propensity for cancer. The most frequent solid tumors are squamous cell carcinomas of the head, neck, and upper esophagus, followed by carcinomas of the vulva and/or anus, cervix, and lower esophagus. Human papilloma virus is suspected in the pathogenesis. Some patients experience oral cancer after bone marrow transplantation. Benign and malignant liver tumors occur (adenomas, hepatomas) and are usually associated with androgen therapy for aplastic anemia. Androgens are also implicated in the etiology of peliosis hepatis (blood-filled hepatic sinusoids). Peliosis hepatis is reversible when androgen therapy is discontinued, and tumors may regress. Approximately 15% of patients with FA are at risk for acute leukemia or MDS.
Diagnosis
FA should be considered in all children and young adults with unexplained cytopenias. Abnormal hematologic findings and characteristic physical anomalies suggest the diagnosis, which is confirmed with a lymphocyte chromosomal breakage study using DEB. No other inherited pancytopenia is associated with an abnormal DEB chromosomal breakage study result. Ten percent to 15% of patients with suspected FA have “somatic mosaicism” and do not show abnormal lymphocyte chromosomal fragility because of mixed populations of somatic cells, some with 2 abnormal alleles and some with 1. Testing on skin fibroblast cultures instead of lymphocytes confirms the diagnosis.
Most patients have stable elevations of serum α-fetoprotein expressed constitutively, independent of liver complications or androgen therapy. The laboratory measurement of serum α-fetoprotein can be used as a rapid screening diagnostic test.
Specialized laboratories can perform an accurate diagnostic and mutant gene subtyping assay, whereby patient lymphocytes or fibroblasts are studied after exposure to mitomycin C or radiation by immunoblotting for FANCD2. Alternatively, wild-type FANC genes can be transvected into patient T cells by means of retroviral vectors, and if a specific wild-type FANC gene corrects the abnormal T-cell chromosome fragility, the specific mutant gene is deduced.
Treatment
A hematologist and a multidisciplinary team should supervise patients with FA. If the hematologic findings are stable and there are no transfusion requirements, observation is indicated. Subspecialty consultations for anomalies and disabilities can be arranged during this interval. If growth velocity is below expectations, endocrine evaluation is needed to identify growth hormone deficiency or hypothyroidism. Screening for glucose intolerance and hyperinsulinemia should be performed annually or biannually, depending on the degree of hyperglycemia found on initial testing. Blood counts should be performed every 1-3 mo; bone marrow aspiration and biopsy are indicated annually for leukemia and MDS surveillance by means of morphology and cytogenetics. Patients should be assessed for solid tumors at least annually. Beginning at menarche, female patients should be screened annually for gynecologic cancer. Administration of human papilloma virus quadrivalent vaccine to prevent squamous cell carcinoma will likely become a standard intervention.
Hematopoietic stem cell transplantation (HSCT; Chapter 129) is the only curative therapy for the hematologic abnormalities. Patients with FA <10 yr old who undergo transplantation using an HLA–identical sibling donor have a survival rate >80%. Survival rates are lower for patients undergoing the procedure when >10 yr. Preparative regimens are continuously evaluated, refined, and improved worldwide. For patients who do not have a matched sibling donor, a search for a matched unrelated donor (including a search of umbilical cord blood banks) might be initiated. Because of the heightened graft vs host response in patients with FA, the survival and cure rates have not been as good as those for matched sibling donor HSCT (≈ 30% survival). Molecular technology has led to preimplantation genetic diagnosis on parent-derived blastomeres to find an HLA-matched sibling donor without FA.
The potential for recombinant growth factor (cytokine) therapy for FA has not been defined. Granulocyte colony-stimulating factor (G-CSF) can usually induce an increase in the absolute neutrophil count and occasionally may boost platelet counts and hemoglobin levels. However, there may be a heightened risk of marrow cell cytogenetic clonal expansion with monosomy 7. Combination therapy consisting of G-CSF given subcutaneously daily or every 2 days along with erythropoietin given subcutaneously or IV 3 times/wk results in improved neutrophil counts in almost all patients and a sustained rise in platelets and hemoglobin levels in approximately one third of patients, although most patients lose the response after 1 yr owing to progression of marrow failure.
Androgens produce a response in 50% of patients, heralded by reticulocytosis and a rise in hemoglobin within 1-2 mo. White blood cell counts may increase next, followed by platelet counts, but it may take many months to achieve the maximum response. When the response plateaus, androgen dosage can be slowly tapered but not stopped entirely. Oral oxymetholone is used most frequently once a day. Low-dose prednisone orally every 2nd day may be added to counter androgen-induced growth acceleration and prevent thrombocytopenic bleeding by promoting vascular stability. In many patients who are taking androgens, the disease becomes refractory as marrow failure progresses. Potential side effects include masculinization, elevated hepatic enzymes, cholestasis, peliosis hepatis, and liver tumors. Screening for these changes should be performed serially.
The premise for gene therapy in FA is based on the assumption that corrected hematopoietic cells offer a growth advantage. Attempts at gene therapy have been disappointing, possibly because of the type of vector but also because of the chromosomal fragility and impaired proliferative function of the hematopoietic progenitors. Encouraging preclinical data from studies using lentiviral vectors offer hope that gene therapy will be a safe and effective treatment for FA. Transposons are nonviral vectors that have been used successfully for gene delivery in murine models and may hold promise for use in humans.
Prognosis
From FA cases reported in the 1990s, the projected median survival was >30 yr of age, an improvement over that in the previous decade. Successes with HSCT have dramatically improved the outlook. Careful surveillance for known complications, especially cancer, and prompt intervention on their detection has also contributed to the improved survival.
Shwachman-Diamond Syndrome
Etiology and Epidemiology
Shwachman-Diamond syndrome (SDS) is inherited in an autosomal recessive manner; it occurs in all racial and ethnic groups. The two essential diagnostic criteria are exocrine pancreatic insufficiency and variable hematologic cytopenias due to marrow failure (Chapter 341). Chromosomes are normal, and there is no increased chromosomal breakage after DEB testing of SDS lymphocytes.
Pathology
The mutant gene SBDS maps to chromosome 7q11 and in 90% of cases is responsible for the multisystem, pleiotropic phenotype. The wild-type gene protein product is involved in ribosomal biogenesis or function. Pancreatic insufficiency is due to failure of pancreatic acinar development. Fatty replacement of pancreatic tissue is prominent. Bone marrow failure is characterized by dysfunctional marrow stem cells and a defective marrow microenvironment that does not support and maintain normal hematopoiesis.
Clinical Manifestations
Most patients with SDS have symptoms of fat malabsorption from birth that are caused by pancreatic insufficiency, but steatorrhea is not always obvious. Approximately 50% of patients appear to exhibit a modest improvement in pancreatic enzyme secretion as they age. The clinical picture can be dominated by complications from anemia, neutropenia, or thrombocytopenia. Bacterial and fungal infections secondary to neutropenia, neutrophil dysfunction, and immune deficiency can occur. Short stature is a consistent feature of the syndrome; most patients show normal growth velocity yet remain consistently below the 3rd percentile for height and weight. The occasional SDS adult achieves the 25th percentile for height. Although skeletal abnormalities are variable, classic findings are delayed bone maturation, metaphyseal dysplasia, short or flared ribs, thoracic dystrophy, and bifid thumb. Some patients have unexplained hepatomegaly and elevations of liver enzymes. Most patients have dental abnormalities and poor oral health. Many have neurocognitive problems and poor social skills.
Laboratory Findings
Fatty replacement of pancreatic tissue can be visualized by CT scan or ultrasound. Fat malabsorption is proven by assay on a 72-hour stool collection. Pancreatic function tests show markedly impaired enzyme secretion, but with preservation of ductal function. Serum trypsinogen and isoamylase levels are reduced. Neutropenia is present in 100% of patients with SDS on at least 1 occasion. It can be chronic, cyclic, or intermittent. It has been identified in some neonates during an episode of sepsis. Neutrophils may have a defect in mobility, migration, and chemotaxis owing to alterations in neutrophil cytoskeletal or microtubular function. Anemia, thrombocytopenia, and pancytopenia are seen in 66%, 60%, and up to 44% of cases, respectively. Pancytopenia can be severe as a result of full-blown aplastic anemia. Bone marrow biopsy specimens and aspirates usually show varying degrees of marrow hypoplasia and fat infiltration. Patients may also have B-cell defects with 1 or more of the following: low immunoglobulin (Ig) G or IgG subclasses, low percentage of circulating B lymphocytes, decreased in vitro B-cell proliferation, and lack of specific antibody production. Patients may have a low percentage of circulating T cells, subsets, or natural killer cells, and decreased in vitro T-cell proliferation.
Diagnosis
Mutational analysis for SBDS is definitive in 90% of cases. Pearson syndrome (Chapter 443), consisting of refractory sideroblastic anemia, cytoplasmic vacuolization of bone marrow precursors, metabolic acidosis, exocrine pancreatic insufficiency, and a diagnostic mitochondrial DNA mutation is similar to SDS, but the clinical course, morphologic features of the bone marrow, and gene mutation are different. Also, severe anemia requiring transfusion, rather than neutropenia, is present from birth to 1 yr of age. SDS shares some manifestations with Fanconi anemia, such as marrow dysfunction and growth failure, but patients with SDS are readily distinguished because of pancreatic insufficiency with fat malabsorption, fatty changes within the pancreatic body that can be visualized by imaging, characteristic skeletal abnormalities not seen in Fanconi anemia, and a normal chromosomal breakage study with DEB.
Complications
Patients with SDS are predisposed to MDS and leukemic transformation. The crude rate of MDS or acute leukemia in patients with SDS is 8-33%. Marrow cell clonal cytogenetic abnormalities are an isolated finding, occurring in up to 41% of patients. Isochromosome 7 [i(7q)] is particularly common, suggesting that it is a fairly specific clonal marker of SDS and probably related to the presence of mutant SBDS on 7q11. Other clonal chromosome abnormalities include monosomy 7, i(7q) combined with monosomy 7, deletions or translocations involving part of 7q, and deletions of 20q [Del(20q)]. Although i(7q) and Del(20q) are rarely related to leukemic transformation or MDS, the prognostic significance of all marrow clonal changes requires prospective monitoring.
Treatment
Fat malabsorption responds to oral pancreatic enzyme replacement and supplemental fat-soluble vitamins, administered according to guidelines similar to those for cystic fibrosis (Chapter 395). A long-term plan should be initiated to monitor changes in peripheral blood counts that require corrective action and to look for early evidence of malignant myeloid transformation. The latter requires serial bone marrow aspirations for smears and cytogenetics and marrow biopsy. One recommendation is to perform marrow testing annually.
Daily subcutaneous G-CSF for profound neutropenia is effective in inducing a sustained increase in neutrophils. Some patients require transfusion support for management of severe anemia or thrombocytopenia. Experience with erythropoietin is limited. Small numbers of patients have been treated with corticosteroids, and hematologic improvement has been seen in approximately 50%. In some patients who received androgens plus steroids, blood counts have improved. The only curative option for severe marrow failure in SDS is allogeneic HSCT, although experience has been limited. About 50% of patients with SDS who underwent transplantation have died of complications related to the preparative therapy. The risk of cardiotoxicity has been noted. Fludarabine-based protocols using reduced-intensity conditioning appear to be safer and effective for SDS HSCT.
Prognosis
Published literature cites a median survival for patients with SDS as 35 years, but the number of undiagnosed patients with mild or asymptomatic disease is unknown. Hence, overall prognosis may be better than previously thought. Approximately 50% of patients experience spontaneous conversion from pancreatic insufficiency to pancreatic sufficiency as a result of improvement in pancreatic enzyme secretion. Enzyme replacement therapy is then no longer needed. Although all patients have some degree of hematologic cytopenia, the changes in most patients are mild to moderate and do not require therapeutic intervention. Severe neutropenia responds well to G-CSF, but there is concern that the predisposition to MDS and acute leukemia can be heightened by the agent’s powerful growth stimulus on marrow cells. HSCT for severe marrow failure has produced a 50% survival rate, but safer protocols are being introduced. Malignant marrow transformation remains ominous.
Dyskeratosis Congenita
Etiology and Epidemiology
Dyskeratosis congenita (DC) is an inherited multisystem disorder characterized by mucocutaneous abnormalities, bone marrow failure, and a predisposition to cancer and MDS. The diagnostic mucocutaneous (ectodermal) triad is reticulate skin pigmentation of the upper body, mucosal leukoplakia, and nail dystrophy (Fig. 462-2). Skin and nail findings usually become apparent in the 1st 10 yr of life, whereas oral leukoplakia is seen later. These manifestations tend to progress as patients get older. Aplastic anemia occurs in approximately 50% of cases, usually in the 2nd decade of life. About 73% of patients with DC are male, a finding compatible with X-linked recessive inheritance. The remainder has either an autosomal dominant or autosomal recessive mode of inheritance.
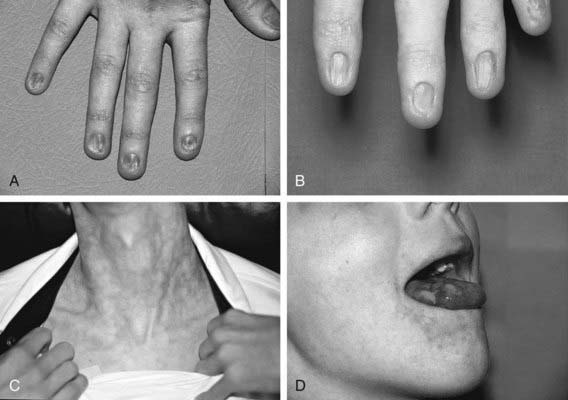
Figure 462-2 Physical findings in patients with dyskeratosis congenita. A and B, Dystrophic fingernails in 2 different patients. C, Lacy reticular pigmentation. D, Leukoplakia on the tongue.
(From Nathan DG, Orkin SH, Ginsburg D, et al, editors: Nathan and Oski’s hematology of infancy and childhood, ed 6, vol I, Philadelphia, 2003, WB Saunders, p 300.)
Pathology
DC is genetically heterogeneous, and patients have mutations in genes that encode components of the telomerase complex (DKC1, TERC, TERT, NOP10, and NHP2), and the telomere shelterin complex (TINF2), all components critical for telomere maintenance. The X-linked recessive form of DC maps to Xq28, and many mutations have been identified in the DKC1 gene, which codes for the nuclear protein dyskerin. The autosomal dominant form is due to mutations in TINF2, or in TERC or TERT, the RNA and enzymatic components of telomerase, respectively. Autosomal recessive DC is linked to mutations in NOP10 or NHP2. Because of impaired telomere maintenance in all 3 inherited forms of DC, short telomeres are demonstrated in the peripheral blood cells of all patients and are a cardinal marker for DC and for marrow failure. The failure is likely due to progressive attrition and depletion of hematopoietic stem cells because of premature senescence, which manifests as pancytopenia.
Clinical Manifestations
Skin pigmentation and nail changes typically appear first, mucosal leukoplakia and excessive ocular tearing appear later, and by the mid-teens, patients with DC have bone marrow failure and malignancy. Many female patients have the same features as male patients. In males, cutaneous findings are the most consistent feature. Lacy reticulated skin pigmentation affecting the face, neck, chest, and arms is a common finding (89%). The degree of pigmentation increases with age and can involve the entire skin surface. There may also be a telangiectatic erythematous component. Nail dystrophy of both hands and feet is the next most common finding (88%). It usually starts with longitudinal ridging, splitting, or pterygium formation and may progress to complete nail loss. Leukoplakia usually involves the oral mucosa (78%), especially the tongue, but may also be seen in the conjunctiva and the anal, urethral, or genital mucosa. Hyperhidrosis of the palms and soles is common, and hair loss is sometimes seen. Eye abnormalities are observed in approximately 50% of cases. Excessive tearing (epiphora) secondary to nasolacrimal duct obstruction is common. Other ophthalmologic manifestations include conjunctivitis, blepharitis, loss of eyelashes, strabismus, cataracts, and optic atrophy. An increased rate of dental decay and early loss of teeth are common. Skeletal abnormalities, such as osteoporosis, avascular necrosis, abnormal bone trabeculation, scoliosis, and mandibular hypoplasia, are seen in approximately 20% of cases. Genitourinary abnormalities include hypoplastic testes, hypospadias, phimosis, urethral stenosis, and horseshoe kidney. Gastrointestinal findings, such as esophageal strictures, hepatomegaly, and cirrhosis, are seen in 10% of cases. A subset of patients has pulmonary complications, with reduced diffusion capacity and/or a restrictive defect. In fatal cases, lung tissue shows pulmonary fibrosis and abnormalities of the pulmonary vasculature.
Laboratory Findings
The initial hematologic change in DC is usually thrombocytopenia, anemia, or both, followed by full-blown pancytopenia and aplastic anemia. The red cells are often macrocytic, and the fetal hemoglobin value can be elevated initially. Initial bone marrow specimens may be hypercellular, but with time, a symmetric depletion of all hematopoietic lineages ensues. Some patients have immunologic abnormalities, including reduced or elevated immunoglobulin values, decreased B- and/or T-lymphocyte count, and reduction of or absence of lymphocyte proliferative responses to phytohemagglutinin. Unlike patients with FA, patients with DC show no abnormal chromosomal breakage in phytohemagglutinin-stimulated lymphocytes upon exposure to DEB. However, primary skin fibroblasts in culture have abnormal morphologic features and doubling rate and show numerous unbalanced chromosome rearrangements, such as dicentrics, tricentrics, and translocations, in the absence of DEB. These findings provide evidence of a defect that predisposes patient cells to chromosomal rearrangements and possibly to DNA damage.
Diagnosis
The following abnormalities are seen in patients with DC but not in those with FA: nail dystrophy, leukoplakia, and tooth abnormalities, hyperhidrosis of the palms and soles, and hair loss. There are overlap syndromes that share some of the features of DC. Hoyeraal-Hreidarsson syndrome is a multisystem disorder comprising aplastic anemia, immunodeficiency, microcephaly, growth retardation, and cerebellar hypoplasia. The syndrome is genetically heterogeneous; some cases are X-linked recessive and caused by mutations in DKC1, and others are autosomal recessive owing to homozygous TERT mutations. Revesz syndrome consists of dystrophic nails, leukoplakia, aplastic anemia, cerebellar hypoplasia, growth retardation, microcephaly, and bilateral exudative retinopathy. TINF2 is mutated in Revesz syndrome, which hence is an autosomal dominant variant of DC.
Complications
Cancer develops in approximately 10-15% of patients with DC, usually in the 3rd and 4th decades of life. Patients with DC are predisposed to MDS as well as to solid tumors. Forty percent of the cancers in such patients are squamous cell carcinomas of the head and neck (tongue, mouth, pharynx). Cancer of the skin and gastrointestinal tract (esophagus, stomach, colon, and especially the anorectal site) is also common.
Treatment
Androgens, usually combined with low-dose prednisone, can induce improvement of marrow function in approximately 50% of patients. When the response is maximal, the androgen dose can be slowly tapered but not stopped. DC can become refractory to androgens as the aplastic anemia progresses. There is no published information on the use of immunosuppressive therapy for this disorder. Although reports are scanty, cytokine therapy with granulocyte-macrophage colony stimulating factor (GM-CSF) or with G-CSF alone or combined with erythropoietin appears to offer potential benefit, at least in the short term, especially for improving neutrophil numbers.
Allogeneic HSCT has been used to correct marrow failure in patients with DC, but with only a 50% survival rate. Inherent telomere shortening in DC may underlie many of the complications. Vascular lesions and fibrosis involving various organs occur early and late after transplantation and carry a high mortality rate. Patients with DC may be more susceptible to endothelial damage that occurs after HSCT as a result of various factors, including the conditioning regimen, infectious disease, and graft versus host disease. Up to 40% of patients with DC experience fatal pulmonary complications after transplantation.
Although the mutated genes for most cases of DC are known, prospects for gene therapy are not imminent.
Prognosis
Considerable heterogeneity exists in DC. Patients with autosomal dominant disease have milder clinical manifestations. Patients with autosomal recessive disease appear to have more physical anomalies and a higher incidence of aplastic anemia and cancer. The mean age of death for patients with DC is approximately 30 yr. The main causes of death are bone marrow failure, complications of HSCT, cancer, and fatal pulmonary problems.
Amegakaryocytic Thrombocytopenia
Etiology and Epidemiology
Congenital amegakaryocytic thrombocytopenia (CAMT) is the rarest of the 4 major inherited pancytopenias. It is transmitted in an autosomal recessive manner. CAMT manifests in infancy as isolated thrombocytopenia due to reduction or absence of marrow megakaryocytes with initial preservation of granulopoietic and erythroid lineages. Pancytopenia due to aplastic anemia often ensues in the first few years of life. The defect in CAMT is directly related to mutations in MPL, the gene for the receptor of thrombopoietin, the growth factor that stimulates megakaryocyte proliferation and maturation. Carriers of the mutant gene have normal hematology; affected individuals have mutations in both alleles. Genotype-phenotype correlations predict disease course and prognosis. Nonsense mutations produce a complete loss of function of the thrombopoietin receptor, causing persistently low platelet counts due to absence of megakaryocytes and a fast progression to pancytopenia and aplastic anemia. Because thrombopoietin also has an anti-apoptotic and cell survival effect on stem cells, impaired stem cell survival with MPL nonsense mutations explains the evolution of CAMT into aplastic anemia. Missense mutations of MPL are associated with a milder course, a transient increase in platelets during the first years of life, and delayed onset, if any, of pancytopenia, indicating residual receptor function. Biologically active plasma thrombopoietin is consistently elevated in all patients with CAMT.
Clinical Manifestations
Patients with CAMT have petechial rash, bruising, or bleeding at birth or in the 1st year of life. Patients with proven MPL mutations appear thus far to have normal physical and imaging features. About 40% of published phenotypic CAMT cases involved physical anomalies, but MPL mutation analyses were not available in these earlier cases, and CAMT was not confirmed genotypically. The most common anomalies in the published cases are neurologic and cardiac. Findings related to cerebellar and cerebral atrophy are frequent, and developmental delay is a prominent feature. Congenital heart disease includes atrial septal defects, ventricular septal defects, patent ductus arteriosus, tetralogy of Fallot, and coarctation of the aorta. Some of these occur in combinations. Other anomalies include abnormal hips or feet, kidney malformations, eye anomalies, and cleft or high-arched palate. Some patients have microcephaly and an abnormal facies.
Laboratory Findings
Thrombocytopenia is the major laboratory finding in CAMT, with normal hemoglobin levels and white blood cell counts initially. Peripheral blood platelets are reduced or totally absent. As in other inherited bone marrow failure syndromes, red blood cells may be macrocytic. Hemoglobin F may be elevated, and there may be increased expression of i antigen. Bone marrow aspirates and biopsy specimens show normal cellularity with marked reduction or absence of megakaryocytes. In patients in whom aplastic anemia develops, marrow cellularity is decreased, with fatty replacement; erythropoietic and granulopoietic lineages are also symmetrically reduced.
Diagnosis
If CAMT manifests beyond the neonatal period, marrow aspirate and biopsy will demonstrate deficient megakaryocytes and suggest the diagnosis; mutational analysis will confirm it. If CAMT occurs at birth or shortly after, it must be distinguished from other causes of inherited and acquired neonatal thrombocytopenia (Chapter 478). Thrombocytopenia with absent radii (TAR syndrome) is distinguished from CAMT because in TAR the radii are absent. CAMT blood lymphocytes do not show increased chromosomal breakage when exposed to DEB, distinguishing the disease it from FA.
Complications
In some patients, clonal marrow cell cytogenetic abnormalities appear such as monosomy 7 and trisomy 8 but without evidence of leukemia. CAMT can evolve into MDS and also acute leukemia, but the true risk cannot be defined because of the rarity of the disease and the paucity of published data.
Therapy and Prognosis
The mortality rate in patients with MPL nonsense mutations from thrombocytopenic bleeding, complications of aplastic anemia, or leukemic transformation has been very close to 100%. Patients with missense mutations have a milder course but may still have serious complications. HSCT is the only curative option. The majority of patients with CAMT who undergo HSCT are cured, especially if the procedure is performed with HLA-matched sibling donors. Before transplantation, platelet transfusion should be used discretely. Platelet count should not always be the sole indication; clinical bleeding is an appropriate trigger. Single-donor filtered platelets are preferred to minimize sensitization, and in a patient for whom HSCT is a possibility, all blood products should be free of cytomegalovirus. Corticosteroids do not appear to be effective for treatment of the thrombocytopenia. For aplastic anemia, androgens in combination with corticosteroids may induce a temporary partial improvement. Interleukin-3 may be an important adjunct to the medical management of CAMT, but it was not adopted broadly and is no longer available. Thrombopoietin has not been tried for the treatment of CAMT and would likely fail, because endogenous thrombopoietin levels are markedly increased and thrombopoietin receptors are either nonfunctional or functioning poorly.
Other Genetic Syndromes
Pancytopenia and bone marrow failure can occur in the context of several nonhematologic syndromes and familial settings that do not exactly correspond to the entities already described.
Down Syndrome
Down syndrome (trisomy 21; Chapter 76) has a unique association with aberrant hematologic findings. In addition to the propensity for acute lymphoblastic and myeloblastic leukemias, especially acute megakaryoblastic leukemia, a few patients with Down syndrome have been reported as having pancytopenia due to aplastic anemia.
Dubowitz Syndrome
Dubowitz syndrome is an autosomal recessive disorder characterized by a peculiar facies, infantile eczema, small stature, and mild microcephaly. The face is small, with a shallow supraorbital ridge, a nasal bridge at the same level as the forehead, short palpebral fissures, variable ptosis, and micrognathia. There is a predilection to cancer as well as to bone marrow dysfunction in these patients. Approximately 10% of patients have hematopoietic disorders, including hypoplastic anemia, moderate pancytopenia, and full-blown aplastic anemia.
Seckel Syndrome
Seckel syndrome, sometimes called “bird-headed dwarfism,” is an autosomal recessive developmental disorder characterized by marked growth failure and mental deficiency, microcephaly, a hypoplastic face with a prominent nose, and low-set and/or malformed ears. Approximately 25% of patients have aplastic anemia or malignancies. One form of Seckel syndrome is caused by a mutant ATR gene, and another is caused by a mutant PCNT2 gene. Different loci for 2 additional forms have also been identified, demonstrating genetic heterogeneity.
Reticular Dysgenesis
Reticular dysgenesis (Chapter 120) is an immunologic deficiency syndrome coupled with congenital agranulocytosis. The mode of inheritance is probably autosomal recessive, but an X-linked mode is also possible in some cases. The disorder is a variant of severe combined immune deficiency in which cellular and humoral immunity are absent and severe lymphopenia and neutropenia are also seen. Anemia and thrombocytopenia may also be present. Bone marrow specimens are hypocellular, with markedly reduced myeloid and lymphoid elements. The only curative therapy is HSCT.
Schimke Immunoosseous Dysplasia
Schimke immunoosseous dysplasia is an autosomal recessive disorder caused by mutations in the chromatin remodeling protein SMARCAL1. Patients have spondyloepiphyseal dysplasia with exaggerated lumbar lordosis and a protruding abdomen. There are pigmentary skin changes and abnormally discolored and configured teeth. Renal dysfunction can be problematic, with proteinuria and nephrotic syndrome. Approximately 50% of patients have hypothyroidism, 50% have cerebral ischemia, and 10% have bone marrow failure with neutropenia, thrombocytopenia, and anemia. Lymphopenia and altered cellular immunity are present in almost all patients. In 1 published case, a patient underwent successful bone marrow transplantation.
Noonan Syndrome
Noonan syndrome is a developmental disorder characterized by the “Noonan facies” (hypertelorism, ptosis, short neck, low-set ears), short stature, congenital heart disease, and multiple skeletal and hematologic abnormalities. It is an autosomal dominant disorder composed of at least 5 genetic types. Heterozygous mutations in PTPN11 cause about 50% of cases of the syndrome; others are caused by a mutant NF1 gene, by a germline KRAS mutation, by mutant SOS1, or by mutant RAF1. In addition to an association with juvenile myelomonocytic leukemia, amegakaryocytic thrombocytopenia, as well as pancytopenia and a hypocellular marrow can develop in patients with Noonan syndrome.
Cartilage-Hair Hypoplasia
Cartilage-hair hypoplasia, an autosomal recessive syndrome seen mostly in Finnish or Amish populations, is characterized by metaphyseal dysostosis, short-limbed dwarfism, and fine, sparse hair. Additional skeletal findings are scoliosis, lordosis, chest deformity, and varus lower limbs. Gastrointestinal abnormalities also occur. Mutations in the RMRP gene cause CHH. Macrocytic anemia is seen in most patients and is sometimes severe and persistent. Neutropenia, lymphopenia, and a predisposition to lymphoma and other cancers are also features.
Familial Aplastic Anemia
Bone marrow failure can cluster in families, but many of these cases cannot be readily classified into discrete diagnostic entities such as FA. The phenotype of these conditions can be complex, with varying combinations of hematologic abnormalities, immunologic deficiency, physical malformations, and predisposition to leukemia. Both autosomal dominant and autosomal recessive inheritances have been observed; both patterns occur with or without associated physical anomalies. An X-linked type is described with physical anomalies; additionally, the X-linked lymphoproliferative syndrome associated with Epstein-Barr virus is associated with pancytopenia.
Unclassified Inherited Bone Marrow Failure Syndromes
Unclassified inherited bone marrow failure syndromes are heterogeneous disorders that may be either atypical presentations of identifiable diseases or new syndromes. Characterized by various cytopenias, with or without physical manifestations, they do not fit into a classic genetic bone marrow failure disease because all features may not be evident at the time of presentation. Compared with classic disorders (presentation ≈ 1 mo of age), infants with unclassified disorders present later (≈ 9 mo) and manifest single or multilineage cytopenia, aplastic anemia, myelodysplasia, or malignancy with variable expression of malformations. Criteria for the diagnosis are seen in Table 462-3. With follow-up, some may demonstrate typical physical features of known syndromes, such as Shwachman-Diamond syndrome, although without obvious mutations in the SBDS gene.
Table 462-3 CANADIAN INHERITED MARROW FAILURE REGISTRY CRITERIA FOR UNCLASSIFIED INHERITED BONE MARROW FAILURE SYNDROMES
FULFILLS CRITERIA 1 AND 2:
FULFILLS AT LEAST 2 OF THE FOLLOWING:
FULFILLS AT LEAST 1 OF THE FOLLOWING:
* The Canadian Inherited Marrow Failure Registry diagnostic guidelines for selected syndromes were adapted from the literature and are available at www.sickkids.ca/cimfr.
† Cytopenia was defined as follows: neutropenia, neutrophil count of <1.5 × 109/L; thrombocytopenia, platelet count of <150 × 109/L; anemia, hemoglobin concentration of <2 standard deviations below mean, adjusted for age.
‡ Hemoglobinopathies with ineffective erythropoiesis and high hemoglobin F should be excluded by clinical or laboratory testing.
Modified from Teo JT, Klaassen R, Fernandez CV, et al: Clinical and genetic analysis of unclassifiable inherited bone marrow failure syndromes. Pediatrics 22:e139–e148, 2008.
Alter BP. Diagnosis, genetics and management of inherited bone marrow failure syndromes. Hematol Am Soc Hematol Educ Program. 2007;2007:29-39.
Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood. 2009;113:6549-6557.
Bagby GC. Constitutional marrow failure. guest editor. Semin Hematol. 2006;43:145-203.
Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23:233-248.
D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362:1909-1918.
De Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res. 2008;668:11-19.
Dokal I, Vulliamy T. Inherited aplastic anemias/bone marrow failure syndromes. Blood Rev. 2008;22:141-153.
Freedman MH. Inherited forms of bone marrow failure. In: Hoffman R, Benz EJ, Shattil SJ, et al, editors. Hematology: basic principles and practice. ed 5. Philadelphia: Churchill Livingstone, Elsevier; 2009:319-357.
Ganapathi KA, Shimamura A. Ribosomal dysfunction and inherited marrow failure. Br J Haematol. 2008;141:376-387.
Geddis AE. Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Hematol Oncol Clin North Am. 2009;23:321-331.
Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am. 2009;23:193-214.
Kerr EN, Ellis L, Dupuis A, et al. The behavioral phenotype of school-age children with Shwachman-Diamond syndrome indicates neurocognitive dysfunction with loss of Shwachman-Bodian-Diamond syndrome gene function. J Pediatr. 2010;156:433-438.
Teo JT, Klaassen R, Fernandez CV, et al. Clinical and genetic analysis of unclassifiable inherited bone marrow failure syndromes. Pediatrics. 2008;122:e139-e148.
Vaz F, Hanenberg H, Schuster B, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42:406-409.
Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol. 2009;145:164-172.