Chapter 395 Cystic Fibrosis
Characterized by obstruction and infection of airways and by maldigestion and its consequences, cystic fibrosis (CF) is an inherited multisystem disorder of children and adults; it is the most common life-limiting recessive genetic trait among white persons. Dysfunction of the cystic fibrosis transmembrane conductance regulator protein (CFTR), the primary defect, leads to a wide and variable array of presenting manifestations and complications.
CF is responsible for most cases of exocrine pancreatic insufficiency in early life and is the major cause of severe chronic lung disease in children. It is also responsible for many cases of salt depletion, nasal polyposis, pansinusitis, rectal prolapse, pancreatitis, cholelithiasis, and insulin-dependent hyperglycemia. CF may manifest as failure to thrive and, occasionally, as cirrhosis or other forms of hepatic dysfunction. Therefore, this disorder enters into the differential diagnosis of many pediatric conditions (Table 395-1).
Table 395-1 COMPLICATIONS OF CYSTIC FIBROSIS
RESPIRATORY
GASTROINTESTINAL
OTHER
Adapted from Silverman FN, Kuhn JP: Essentials of Caffrey’s pediatric x-ray diagnosis, 1990, Chicago, Year Book, p 649.
Genetics
CF occurs most frequently in white populations of northern Europe, North America, and Australia/New Zealand. The prevalence in these populations varies but approximates 1/3,500 live births (1/9,200 individuals of Hispanic descent and 1/15,000 in African Americans). Although less frequent in African, Hispanic, Middle Eastern, South Asian, and eastern Asian populations, the disorder does exist in these populations as well (Fig. 395-1).
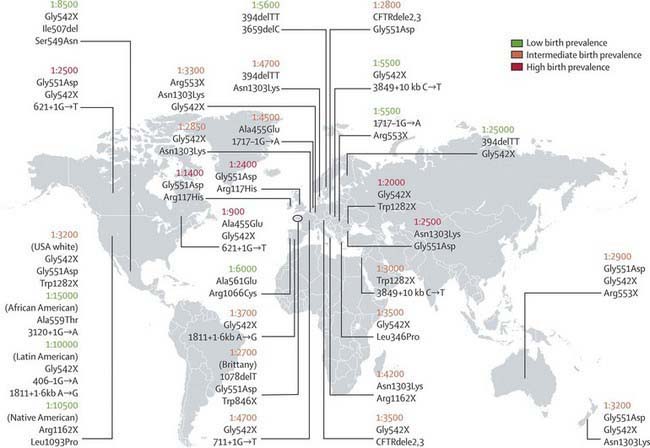
Figure 395-1 Approximate cystic fibrosis birth prevalence and common mutations for selected countries. Birth prevalence is reported as number of live births per case of cystic fibrosis. Common/important mutations in each region are listed below the prevalence figures. The birth prevalence can vary greatly among ethnic groups in a country.
(From O’Sullivan BP, Freedman SD: Cystic fibrosis, Lancet 373:1891–1902, 2009.)
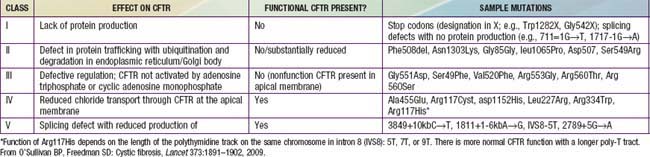
CF is inherited as an autosomal recessive trait. The CF gene codes for the CFTR protein, which is 1,480 amino acids. CFTR is expressed largely in epithelial cells of airways, the gastrointestinal tract (including the pancreas and biliary system), the sweat glands, and the genitourinary system. CFTR is a member of the adenosine triphosphate (ATP)–binding cassette superfamily of proteins. It functions as a chloride channel and has other regulatory functions that are perturbed variably by the different mutations. More than 1,500 CFTR polymorphisms grouped into 5 main classes of mutations that affect protein function are associated with the CF syndrome (Table 395-2). The most prevalent mutation of CFTR is the deletion of a single phenylalanine residue at amino acid 508 (ΔF508). This mutation is responsible for the high incidence of CF in northern European populations and is considerably less frequent in other populations, such as those of southern Europe and Israel. Approximately 50% of individuals with CF who are of northern European ancestry are homozygous for ΔF508, and >80% carry at least one ΔF508 gene. The remainder of patients has an extensive array of mutations, none of which has a prevalence of more than several percentage points, except in circumscribed populations; the W1282X mutation occurs in 60% of Ashkenazi Jews with CF. The relationship between CFTR genotype and clinical phenotype is highly complex and is not predictable for individual patients. Mutations categorized as “severe” are associated almost uniformly with pancreatic insufficiency but only in general with more rapid progression of lung disease. Modifier gene polymorphisms appear to be responsible for much of the variation in the progression of lung disease. The most compelling association with more severe disease is with a single nucleotide change in the transforming growth factor-β1(TGF-β1) gene. Variant alleles of the mannose-binding lectin, a key factor in systemic innate immunity, are associated with more serious lung infections and reduced survival. Polymorphism in the IRFD1 gene, a transcriptional co-regulator that is essential for neutrophil differentiation, has been associated with a more serious CF pulmonary phenotype. Several mutations, such as 3849 + 10kbC→T, are found in patients with normal sweat chloride concentrations. Some individuals with polymorphisms of both CFTR genes have few or no CF manifestations until adolescence or adulthood, when they present with pancreatitis, sinusitis, diffuse bronchiectasis, or male infertility. Whereas CFTR mutations are a sine qua non for CF, two mutations of CFTR can cause disorders that do not meet diagnostic criteria for CF and, occasionally, do not cause discernible clinical problems.
Table 395-2 ONE PROPOSED CLASSIFICATION OF CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR (CFTR) MUTATIONS
Occurrence of liver disease cannot be predicted by CFTR genotype. This finding suggests a major environmental (acquired) component of organ system dysfunction and the presence of other genes that modify the CF phenotype.
Through the use of probes for 40 of the most common mutations, the genotype of 80-90% of Americans with CF can be ascertained. Genotyping using a discreet panel of mutation probes is quick and less costly than more comprehensive sequencing and is commercially available. In special cases, sequencing the entire CFTR gene is necessary to establish the genotype. This procedure is also available commercially although relatively expensive and can identify polymorphisms and unique mutations of unknown clinical importance.
The high frequency of CFTR mutations has been ascribed to resistance to the morbidity and mortality associated with infectious dysenteries through the ages. In support of this hypothesis, cultured CF intestinal epithelial cells homozygous for the ΔF508 mutation are unresponsive to the secretory effects of cholera toxin. The heterozygote (for CFTR) mice experience less mortality when treated with cholera toxin than their unaffected wild type littermates.
Pathogenesis
A number of long-standing observations of CF are of fundamental pathophysiologic importance; they include failure to clear mucous secretions, a paucity of water in mucous secretions, an elevated salt content of sweat and other serous secretions, and chronic infection limited to the respiratory tract. Additionally, there is a greater negative potential difference across the respiratory epithelia of patients with CF than across the respiratory epithelia of control subjects. Aberrant electrical properties are also demonstrated for CF sweat gland duct and rectal epithelia. The membranes of CF epithelial cells are unable to secrete chloride ions in response to cyclic adenosine monophosphate (cAMP)–mediated signals, and at least in the respiratory tract, excessive amounts of sodium are absorbed through these membranes (Fig. 395-2). These defects can be traced to a dysfunction of CFTR (Figs. 395-3 and 395-4).
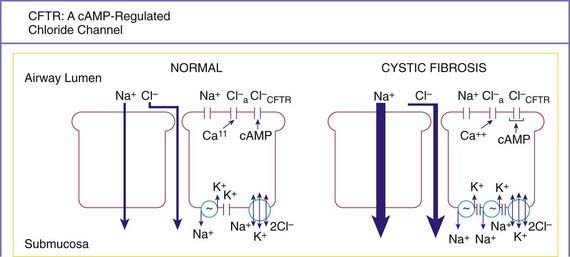
Figure 395-2 The net ion flow across normal and cystic fibrosis (CF) airway epithelia under basal conditions (large arrows). Because water follows salt movement, the predicted net flux of water would be from the airway lumen to the submucosa and would be greater across CF epithelia. The increased Na+ absorption by CF cells is associated with an increased amiloride-sensitive Na+ conductance across the apical (luminal) membrane and increased Na+,K+-ATPase sites at the basolateral membrane. The cyclic adenosine monophosphate (cAMP)–mediated apical membrane conductance of Cl− associated with the CF transmembrane regulator (CFTR) does not function in CF epithelia, but an alternative, calcium (Ca++)−activated Cl− conductance is present in normal and CF cells. It is postulated that CF cells have a limited ability to secrete Cl− and absorb Na+ in excessive amounts, limiting the water available to hydrate secretions and allow them to be cleared from the airways lumen. Cl−a, Ca++–activated Cl− conductance: Cl−CTFR, the CFTR Cl− channel.
(From Knowles MR: Contemporary perspectives on the pathogenesis of cystic fibrosis, New Insights Cystic Fibrosis 1:1, 1993.)
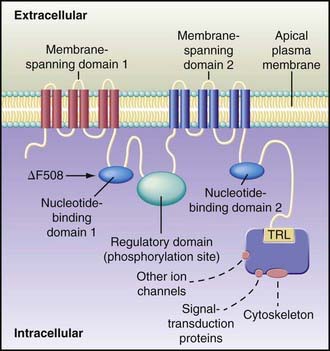
Figure 395-3 Hypothesized structure of cystic fibrosis transmembrane regulator (CFTR). The protein contains 1,480 amino acids and a number of discrete globular and transmembrane domains. Activation of CFTR relies on phosphorylation, particularly through protein kinase A but probably involving other kinases as well. Channel activity is governed by the 2 nucleotide-binding domains, which regulate channel gating. The carboxyl terminal (consisting of threonine, arginine, and leucine [TRL]) of CFTR is anchored through a PDZ-type binding interaction with the cytoskeleton and is kept in close approximation (dashed lines) to a number of important proteins. These associated proteins influence CFTR functions, including conductance, regulation of other channels, signal transduction, and localization at the apical plasma membrane. Each membrane-spanning domain contains 6 membrane-spanning α helixes, portions of which form a chloride-conductance pore. The regulatory domain is a site of protein kinase A phosphorylation. The common δF508 mutation occurs on the surface of nucleotide-binding domain 1.
(From Rowe SM, Miller S, Sorscher EJ: Cystic fibrosis, N Engl J Med 352:1992–2001, 2005.)
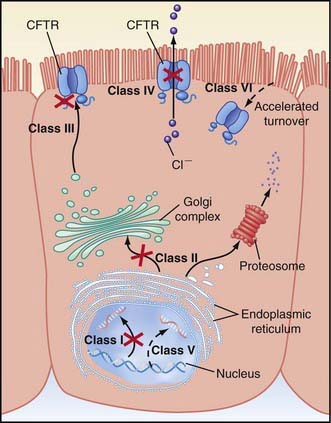
Figure 395-4 Categories of CFTR (cystic fibrosis transmembrane regulator) mutations. Classes of defects in the CFTR gene include the absence of synthesis (class I); defective protein maturation and premature degradation (class II); disordered regulation, such as diminished adenosine triphosphate (ATP) binding and hydrolysis (class III); defective chloride conductance or channel gating (class IV); a reduced number of CFTR transcripts due to a promoter or splicing abnormality (class V); and accelerated turnover from the cell surface (class VI).
(From Rowe SM, Miller S, Sorscher EJ: Cystic fibrosis, N Engl J Med 352:1992–2001, 2005.)
Cyclic AMP–stimulated protein kinase A (PKA) regulation of chloride conductance is the primary function of CFTR; this function is absent in epithelial cells with many different mutations of the CFTR gene. CFTR mutations fall into 6 classes in another classification system, albeit with some overlap (see Fig. 395-4). Individuals with class I, II, and III mutations, on average, have shorter survival than those with “mild” genotypes (class IV or V). The clinical importance of these functional categories is limited because they do not uniformly correlate with specific clinical features or their severity. Rather, clinical features correlate with the residual CFTR activity.
Many hypotheses have been postulated to explain how CFTR dysfunction results in the clinical phenotype. It is likely that no one hypothesis explains the full spectrum of disease. Most believe that the epithelial pathophysiology in airways involves an inability to secrete salt and secondarily to secrete water in the presence of excessive reabsorption of salt and water. The proposed outcome is insufficient water on the airway surface to hydrate secretions. Desiccated secretions become more viscous and elastic (rubbery) and are harder to clear by mucociliary and other mechanisms. In addition it has been suggested that CFTR dysfunction results in an altered microenvironment with low HCO3− and a more acidic pH, thus altering mucus rheology and aggravating poor mucociliary clearance. The result is that these secretions are retained and obstruct airways, starting with those of the smallest caliber, the bronchioles. Airflow obstruction at the level of small airways is the earliest observable physiologic abnormality of the respiratory system.
It is plausible that similar pathophysiologic events take place in the pancreatic and biliary ducts (and in the vas deferens), leading to desiccation of proteinaceous secretions and obstruction. Because the function of sweat gland duct cells is to absorb rather than secrete chloride, salt is not retrieved from the isotonic primary sweat as it is transported to the skin surface; chloride and sodium levels are consequently elevated.
Chronic infection in CF is limited to the airways. A likely explanation for infection is a sequence of events starting with failure to clear inhaled bacteria promptly and then proceeding to persistent colonization and an inflammatory response in airway walls. In addition, it has been proposed that abnormal CFTR creates a proinflammatory state or amplifies the inflammatory response to initial infections (viral or bacterial). Some investigators have identified primary differences in CF-affected immune cells and have suggested that these alterations contribute to this proinflammatory state. It appears that inflammatory events occur first in small airways, perhaps because clearance of altered secretions and microorganisms from these regions is more difficult. Chronic bronchiolitis and bronchitis are the initial lung manifestations (Chapter 383), but after months to years, structural changes in airway walls produce bronchiolectasis and bronchiectasis.
The agents of airway injury include neutrophil products, such as oxidative radicals and proteases, and immune reaction products. With advanced lung disease, infection may extend to peribronchial lung parenchyma.
A finding that is not readily explained by CFTR dysfunction is the high prevalence in patients with CF of airway colonization with Staphylococcus aureus (Chapter 174.1), Pseudomonas aeruginosa (Chapter 197.1), and Burkholderia cepacia (Chapter 197.2), organisms that rarely infect the lungs of other individuals. It has been postulated that the CF airway epithelial cells or surface liquids may provide a favorable environment for harboring these organisms. CF airway epithelium may be compromised in its innate defenses against these organisms, through either acquired or genetic alterations. Antimicrobial activity is diminished in CF secretions; this diminution may be related to hyperacidified surface liquids or other effects on innate immunity. Another puzzle is the propensity for P. aeruginosa to undergo mucoid transformation in the CF airways. The complex polysaccharide produced by these organisms generates a biofilm that provides a hypoxic environment and thereby protects Pseudomonas against antimicrobial agents.
Nutritional deficits, including fatty acid deficiency, have been implicated as predisposing factors for respiratory tract infection. More specifically, concentrations of lipoxins—molecules that suppress neutrophilic inflammation—are suppressed in CF airways. In support of this idea it the observation that the 10-15% of individuals with CF who retain substantial exocrine pancreatic function have delayed onset of colonization with P. aeruginosa and slower deterioration of lung function. It appears that nutritional factors are only contributory because preservation of pancreatic function does not preclude development of typical lung disease.
Pathology
The earliest pathologic lesion in the lung is that of bronchiolitis (mucous plugging and an inflammatory response in the walls of the small airways); with time, mucus accumulation and inflammation extend to the larger airways (bronchitis) (Chapter 383). Goblet cell hyperplasia and submucosal gland hypertrophy become prominent pathologic findings, which is most likely a response to chronic airway infection. Organisms appear to be confined to the endobronchial space; invasive bacterial infection is not characteristic. With long-standing disease, evidence of airway destruction such as bronchiolar obliteration, bronchiolectasis, and bronchiectasis (Chapter 393) becomes prominent. Imaging modalities demonstrate both increased airway wall thickness and luminal cross-sectional area relatively early in lung disease evaluation. Bronchiectatic cysts and emphysematous bullae or subpleural blebs are frequent with advanced lung disease, the upper lobes being most commonly involved. These enlarged air spaces may rupture and cause pneumothorax. Interstitial disease is not a prominent feature, although areas of fibrosis appear eventually. Bronchial arteries are enlarged and tortuous, contributing to a propensity for hemoptysis in bronchiectatic airways. Small pulmonary arteries eventually display medial hypertrophy, which would be expected in secondary pulmonary hypertension.
The paranasal sinuses are uniformly filled with secretions containing inflammatory products, and the epithelial lining displays hyperplastic and hypertrophied secretory elements (Chapter 372). Polypoid lesions within the sinuses and erosion of bone have been reported. The nasal mucosa may form large or multiple polyps, usually from a base surrounding the ostia of the maxillary and ethmoidal sinuses.
The pancreas is usually small, occasionally cystic, and often difficult to find at postmortem examination. The extent of involvement varies at birth. In infants, the acini and ducts are often distended and filled with eosinophilic material. In 85-90% of patients, the lesion progresses to complete or almost complete disruption of acini and replacement with fibrous tissue and fat. Infrequently, foci of calcification may be seen on radiographs of the abdomen. The islets of Langerhans contain normal appearing β cells, although they may begin to show architectural disruption by fibrous tissue in the 2nd decade of life.
The intestinal tract shows only minimal changes. Esophageal and duodenal glands are often distended with mucous secretions. Concretions may form in the appendiceal lumen or cecum. Crypts of the appendix and rectum may be dilated and filled with secretions.
Focal biliary cirrhosis secondary to blockage of intrahepatic bile ducts is uncommon in early life, although it is responsible for occasional cases of prolonged neonatal jaundice. This lesion becomes much more prevalent and extensive with age and is found in 70% of patients at postmortem examination. This process can proceed to symptomatic multilobular biliary cirrhosis that has a distinctive pattern of large irregular parenchymal nodules and interspersed bands of fibrous tissue. Approximately 30-70% of patients have fatty infiltration of the liver, in some cases despite apparently adequate nutrition. At autopsy, hepatic congestion secondary to cor pulmonale is frequently observed. The gallbladder may be hypoplastic and filled with mucoid material and often contains stones. The epithelial lining often displays extensive mucous metaplasia. Atresia of the cystic duct and stenosis of the distal common bile duct have been observed.
Mucus-secreting salivary glands are usually enlarged and display focal plugging and dilatation of ducts.
Glands of the uterine cervix are distended with mucus, copious amounts of which collect in the cervical canal. Endocervicitis may be prevalent in teenagers and young women. In >95% of males, the body and tail of the epididymis, the vas deferens, and the seminal vesicles are obliterated or atretic.
Clinical Manifestations
Mutational heterogeneity and environmental factors appear responsible for highly variable involvement of the lungs, pancreas, and other organs. A list of presenting manifestations is lengthy, although pulmonary and gastrointestinal presentations predominate (Fig. 395-5). With inclusion of CF newborn screening panels, an increasing proportion of children are diagnosed before symptoms appear (Table 395-3).
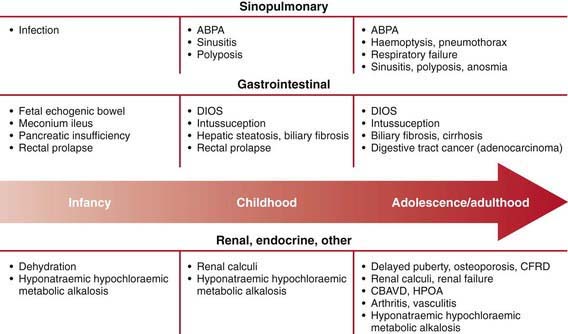
Figure 395-5 Approximate age of onset of clinical manifestations of cystic fibrosis ABPA, allergic bronchopulmonary aspergillosis; CBAVD, congenital bilateral absence of the vas deferens; CFRD, cystic fibrosis-related diabetes mellitus; DIOS, distal intestinal obstruction syndrome; HPOA, hypertrophic pulmonary osteoarthritis.
(From O’Sullivan BP, Freedman SD: Cystic fibrosis, Lancet 373:1891–1902, 2009.)
Table 395-3 PRESENTING FEATURES OF MORE THAN 25,000 PATIENTS WITH CYSTIC FIBROSIS IN THE USA
| FEATURE | % | % of Patients Presenting in 2007 |
|---|---|---|
| Acute or persistent respiratory symptoms | 45.6 | 31.2 |
| Failure to thrive, malnutrition | 37.5 | 18.7 |
| Abnormal stools | 28.8 | 13.8 |
| Meconium ileus, intestinal obstruction | 19.9 | 14 |
| Family history | 16.0 | 12 |
| Newborn Screening | 6.4 | 30.7 |
| Electrolyte, acid-base abnormality | 4.2 | 1.1 |
| Rectal prolapse | 3.3 | 3.3 |
| Nasal polyps, sinus disease | 3.3 | 4.6 |
| Hepatobiliary disease | 1.2 | 1.4 |
| Other* | 3-4 | 6.7 |
* Includes pseudotumor cerebri, azoospermia, acrodermatitis-like rash, vitamin deficiency states, hypoproteinemic edema, hypoprothrombinemia with bleeding, and meconium plug syndrome.
Data from the Patient Registry, Cystic Fibrosis Foundation, Bethesda, MD.
Respiratory Tract
Cough is the most constant symptom of pulmonary involvement. At first, the cough may be dry and hacking, but eventually it becomes loose and productive. In older patients, the cough is most prominent upon arising in the morning or after activity. Expectorated mucus is usually purulent. Some patients remain asymptomatic for long periods or seem to have prolonged but intermittent acute respiratory infections. Others acquire a chronic cough in the first weeks of life, or they have pneumonia repeatedly. Extensive bronchiolitis accompanied by wheezing is a frequent symptom during the first years of life. As lung disease slowly progresses, exercise intolerance, shortness of breath, and failure to gain weight or grow are noted. Exacerbations of lung symptoms, presumably owing to more active airways infection, often require repeated hospitalizations for effective treatment. Cor pulmonale, respiratory failure, and death eventually supervene unless lung transplantation is accomplished. Colonization with B. cepacia and other multidrug-resistant organisms may be associated with particularly rapid pulmonary deterioration and death.
The rate of progression of lung disease is the chief determinant of morbidity and mortality. The course of lung disease is largely independent of genotype. Severe mutations tend to be associated with more rapid progression. A few mutations may substantially or even fully spare the lungs. Male gender and exocrine pancreatic sufficiency are also associated with a slower rate of pulmonary function decline.
Early physical findings include increased anteroposterior diameter of the chest, generalized hyperresonance, scattered or localized coarse crackles, and digital clubbing. Expiratory wheezes may be heard, especially in young children. Cyanosis is a late sign. Common pulmonary complications include atelectasis, hemoptysis, pneumothorax, and cor pulmonale; these usually appear beyond the 1st decade of life.
Even though the paranasal sinuses are virtually always opacified radiographically, acute sinusitis is infrequent. Nasal obstruction and rhinorrhea are common, caused by inflamed, swollen mucous membranes or, in some cases, nasal polyposis. Nasal polyps are most troublesome between 5 and 20 yr of age.
Intestinal Tract
In 15-20% of newborn infants with CF, the ileum is completely obstructed by meconium (meconium ileus). The frequency is greater (≈30%) among siblings born subsequent to a child with meconium ileus and is particularly striking in monozygotic twins, reflecting a genetic contribution from one or more modifying genes. Abdominal distention, emesis, and failure to pass meconium appear in the 1st 24-48 hr of life (Chapters 96.1 and 322.2). Abdominal radiographs (Fig. 395-6) show dilated loops of bowel with air-fluid levels and, frequently, a collection of granular, “ground-glass” material in the lower central abdomen. Rarely, meconium peritonitis results from intrauterine rupture of the bowel wall and can be detected radiographically as the presence of peritoneal or scrotal calcifications. Meconium plug syndrome occurs with increased frequency in infants with CF but is less specific than meconium ileus. Ileal obstruction with fecal material (distal intestinal obstruction syndrome [DIOS]) occurs in older patients, causing cramping abdominal pain and abdominal distention.
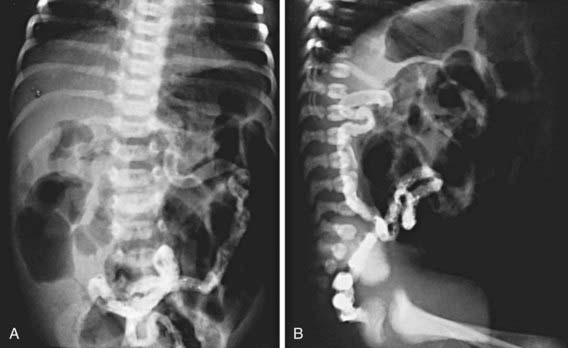
Figure 395-6 A and B, Contrast enema study in a newborn infant with abdominal distention and failure to pass meconium. Notice the small diameter of the sigmoid and ascending colon and dilated, air-filled loops of small intestine. Several air-fluid levels in the small bowel are visible on the upright lateral view.
More than 85% of affected children show evidence of maldigestion from exocrine pancreatic insufficiency. Symptoms include frequent, bulky, greasy stools and failure to gain weight even when food intake appears to be large. Characteristically, stools contain readily visible droplets of fat. A protuberant abdomen, decreased muscle mass, poor growth, and delayed maturation are typical physical signs. Excessive flatus may be a problem. A number of mutations are associated with preservation of some exocrine pancreatic function, including R117H and 3849 + 10kbC→T. Virtually all individuals homozygous for ΔF508 have pancreatic insufficiency.
Less common gastrointestinal manifestations include intussusception, fecal impaction of the cecum with an asymptomatic right lower quadrant mass, and epigastric pain owing to duodenal inflammation. Acid or bile reflux with esophagitis symptoms is common in older children and adults. Subacute appendicitis and periappendiceal abscess have been encountered. Historically a relatively common event, rectal prolapse now occurs much less frequently as the result of earlier diagnosis and initiation of pancreatic enzyme replacement therapy. Occasionally, hypoproteinemia with anasarca appears in malnourished infants, especially if children are fed soy-based preparations. Neurologic dysfunction (dementia, peripheral neuropathy) and hemolytic anemia may occur because of vitamin E deficiency. Deficiency of other fat-soluble vitamins is occasionally symptomatic. Hypoprothrombinemia due to vitamin K deficiency may result in a bleeding diathesis. Clinical manifestations of other fat-soluble vitamin deficiencies, such as decreased bone density and night blindness, have been noted. Rickets is rare.
Biliary Tract
Evidence for liver dysfunction is most often detected in the 1st 15 yr of life and can be found in up to 30% of individuals. Biliary cirrhosis becomes symptomatic in only 5-7% of patients. Manifestations can include icterus, ascites, hematemesis from esophageal varices, and evidence of hypersplenism. A neonatal hepatitis–like picture and massive hepatomegaly owing to steatosis have been reported. Biliary colic secondary to cholelithiasis may occur in the 2nd decade or later. Liver disease occurs independent of genotype but is associated with meconium ileus and pancreatic insufficiency.
Pancreas
In addition to exocrine pancreatic insufficiency, evidence for hyperglycemia and glycosuria, including polyuria and weight loss, may appear, especially in the 2nd decade of life. Eight percent of 11-17 yr old patients and 30% of patients >25 yr of age have CF-related diabetes. Ketoacidosis usually does not occur, but eye, kidney, and other vascular complications have been noted in patients living ≥10 yr after the onset of hyperglycemia. Recurrent, acute pancreatitis occurs occasionally in individuals who have residual exocrine pancreatic function and may be the sole manifestation of two CFTR mutations.
Genitourinary Tract
Sexual development is often delayed but only by an average of 2 yr. More than 95% of males are azoospermic because of failure of development of wolffian duct structures, but sexual function is generally unimpaired. The incidence of inguinal hernia, hydrocele, and undescended testis is higher than expected. Adolescent females may experience secondary amenorrhea, especially with exacerbations of pulmonary disease. Cervicitis and accumulation of tenacious mucus in the cervical canal have been noted. The female fertility rate is diminished. Pregnancy is generally tolerated well by women with good pulmonary function but may accelerate pulmonary progression in those with moderate or advanced lung problems.
Sweat Glands
Excessive loss of salt in the sweat predisposes young children to salt depletion episodes, especially during episodes of gastroenteritis and during warm weather. These children present with hypochloremic alkalosis. Frequently, parents notice salt “frosting” of the skin or a salty taste when they kiss the child. A few genotypes are associated with normal sweat chloride values.
Diagnosis and Assessment
The diagnosis of CF has been based on a positive quantitative sweat test (Cl− ≥ 60 mEq/L) in conjunction with 1 or more of the following features: typical chronic obstructive pulmonary disease, documented exocrine pancreatic insufficiency, and a positive family history. With newborn screening, diagnosis is often made prior to obvious clinical manifestations such as failure to thrive and chronic cough (Table 395-3). Diagnostic criteria have been recommended to include additional testing procedures (Table 395-4).
Table 395-4 DIAGNOSTIC CRITERIA FOR CYSTIC FIBROSIS (CF)
Presence of typical clinical features (respiratory, gastrointestinal, or genitourinary)
OR
A history of CF in a sibling
OR
A positive newborn screening test
PLUS
Laboratory evidence for CFTR (CF transmembrane regulator) dysfunction:
Two elevated sweat chloride concentrations obtained on separate days
OR
Identification of two CF mutations
OR
An abnormal nasal potential difference measurement
Sweat Testing
The sweat test, which involves using pilocarpine iontophoresis to collect sweat and performing chemical analysis of its chloride content, is the standard approach to diagnosis of CF. The procedure requires care and accuracy. An electric current is used to carry pilocarpine into the skin of the forearm and locally stimulate the sweat glands. If an adequate amount of sweat is collected, the specimens are analyzed for chloride concentration. Testing may be difficult in the 1st 2 wk of life because of low sweat rates but is recommended any time after the 1st 48 hr of life. Positive results should be confirmed; for a negative result, the test should be repeated if suspicion of the diagnosis remains.
More than 60 mEq/L of chloride in sweat is diagnostic of CF when 1 or more other criteria are present. Threshold levels of 30-40 mEq/L for infants have been suggested. Borderline (or intermediate) values of 40 to 60 mEq/L have been reported in patients of all ages who have CF with atypical involvement and require further testing. Chloride concentrations in sweat are somewhat lower in individuals who retain exocrine pancreatic function but usually remain within the diagnostic range. Conditions associated with false-negative and false-positive results are noted in Table 395-5.
DNA Testing
Several commercial laboratories test for 30-96 of the most common CFTR mutations. This testing identifies ≥90% of individuals who carry 2 CF mutations. Some children with typical CF manifestations are found to have 1 or no detectable mutations by this methodology. Some laboratories perform comprehensive mutation analysis screening for all of the >1,500 identified mutations.
Other Diagnostic Tests
The finding of increased potential differences across nasal epithelium, the loss of this difference with topical amiloride application, and the absence of a voltage response to a β-adrenergic agonist have been used to confirm the diagnosis of CF in patients with equivocal or frankly normal sweat chloride values.
Pancreatic Function
Exocrine pancreatic dysfunction is clinically apparent in many patients. Documentation is desirable if there are questions about the functional status of the pancreas. Measurement of fat balances with a 3-day stool collection or direct documentation of enzyme secretion after duodenal intubation and pancreozymin-secretin stimulation provides a reliable measure, but the fat collection is cumbersome and duodenal intubation is very invasive and rarely performed. A simpler test that can be used to screen pancreatic function, the quantification of elastase-1 activity in a fresh stool sample, is used routinely. Other indirect measures of pancreatic enzyme secretion are available but have limited or unproven clinical value. Endocrine pancreatic dysfunction may be more prevalent than previously recognized. Many authorities advocate yearly monitoring with a modified 2-hr oral glucose tolerance test (OGTT) after 10 yr of age. This approach is more sensitive than spot checks of blood and urine glucose levels and glycosylated hemoglobin levels.
Radiology
Pulmonary radiologic findings suggest the diagnosis but are not specific. Hyperinflation of lungs occurs early and may be overlooked in the absence of infiltrates or streaky densities. Bronchial thickening and plugging and ring shadows suggesting bronchiectasis usually appear first in the upper lobes. Nodular densities, patchy atelectasis, and confluent infiltrate follow. Hilar lymph nodes may be prominent. With advanced disease, impressive hyperinflation with markedly depressed diaphragms, anterior bowing of the sternum, and a narrow cardiac shadow are noted. Cyst formation, extensive bronchiectasis, dilated pulmonary artery segments, and segmental or lobar atelectasis are often apparent with advanced disease. Typical progression of lung disease is seen in Figure 395-7. Most CF centers obtain chest radiographs (posteroanterior [PA] and lateral) at least annually. Standardized scoring of roentgenographic changes has been used to follow progression of lung disease. CT of the chest can detect and localize thickening of bronchial airway walls, mucous plugging, focal hyperinflation, and early bronchiectasis (Fig. 395-8); it is generally not used for routine evaluation of chest disease. Many children with normal lung function have bronchiectasis on CT, indicating that this imaging modality is sensitive to early lung changes.
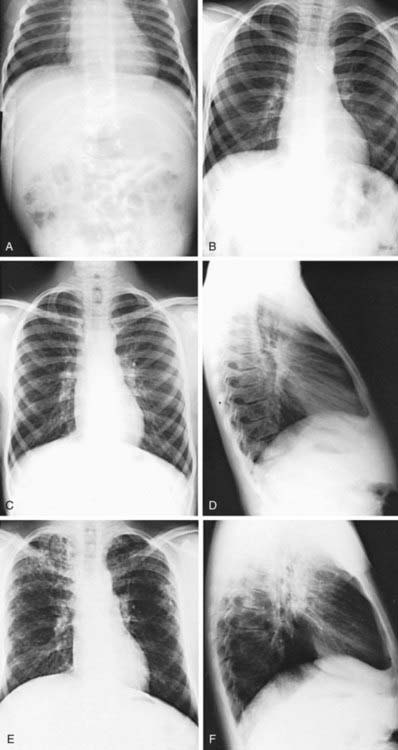
Figure 395-7 Roentgenographic progression of cystic fibrosis lung disease in one patient, from the diagnosis in infancy to 18 yr of age. A, Patient admitted with cough and wheezing at 2 mo of age. Notice the mild increase in bronchovascular markings, especially in the upper lobe areas. B, At age 4 yr, cough was minimal. Bronchovascular markings were mildly increased, and there was some improvement in the upper lobes. The wheeze never recurred. C and D, At age 13 yr, the patient had minimal cough and occasional sputum production. The bronchovascular markings were generally further increased, with early bronchiectatic changes in the right upper lobe. The lateral view does not suggest overinflation. E and F, Age 18 yr. During adolescence, cough and sputum production increased even though outpatient antibiotic therapy was intensified. Small-volume hemoptysis, occasional paroxysms of cough, and weight loss as well as increased nodular infiltrates (especially in the right upper lobe) and hyperinflation (as seen on the lateral view) led to the patient’s 1st hospitalization since infancy. Height and weight were maintained in the 25th-50th percentile range.
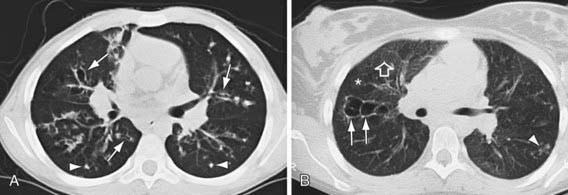
Figure 395-8 CT scans of the chest in cystic fibrosis. A, A 12 yr old boy with moderate lung disease. Airway and parenchymal changes are present throughout both lungs. Multiple areas of bronchiectasis (arrows) and mucous plugging (arrowheads) can be seen. B, A 19 yr old girl has mostly normal lung with 1 area of saccular bronchiectasis in the right upper lobe (arrows) and a focal area of peripheral mucous plugging in the right lower lobe (arrowhead). Lung density is heterogeneous with areas of normal lung (open arrow) and areas of low attenuation reflecting segmental and subsegmental air trapping (asterisk).
Radiographs of paranasal sinuses reveal panopacification and, often, failure of frontal sinus development. CT provides better resolution of sinus changes if this information is required clinically. Fetal ultrasonography may suggest ileal obstruction with meconium early in the 2nd trimester, but this finding is not predictive of meconium ileus at birth.
Pulmonary Function
Standard pulmonary function studies are not obtained until patients are 4-6 yr of age, by which time many patients show the typical pattern of obstructive pulmonary involvement (Chapter 376). Decrease in the midmaximal flow rate is an early functional change, reflecting small airway obstruction. This lesion also affects the distribution of ventilation and increases the alveolar-arterial oxygen difference. The findings of obstructive airway disease and modest responses to a bronchodilator are consistent with the diagnosis of CF at all ages. Residual volume and functional residual capacity are increased early in the course of lung disease. Restrictive changes, characterized by declining total lung capacity and vital capacity, correlate with extensive lung injury and fibrosis and are a late finding. Testing at each clinic visit is recommended to evaluate the course of the pulmonary involvement and allow for early intervention when substantial decrements are documented. Increasing numbers of CF centers are equipped to measure airflow patterns of sedated infants (infant pulmonary function tests). Some patients reach adolescent or adult life with normal pulmonary function and without evidence of overinflation.
Microbiologic Studies
The finding of S. aureus or P. aeruginosa on culture of the lower airways (sputum) strongly suggests a diagnosis of CF. In particular, mucoid forms of P. aeruginosa are often recovered from CF lungs. B. cepacia recovery also suggests CF. A wide range of other organisms are frequently recovered, particularly in advanced lung disease; they include a variety of gram-negative rods, fungi, and nontuberculous mycobacterial species. Failure of respiratory symptom flares to respond to usual antibiotics triggers testing for Mycoplasma and viruses. Fiberoptic bronchoscopy is used to gather lower respiratory tract secretions of infants and young children who do not expectorate.
Heterozygote Detection and Prenatal Diagnosis
Mutation analysis should be fully informative for testing of potential carriers or a fetus, provided that mutations within the family have been previously identified. Testing a spouse of a carrier with a standard panel of probes is ≈90% sensitive, and full CFTR sequence analysis is commercially available if further testing is warranted. Prenatal testing should be offered to all couples planning to have children in addition to individuals with a family history of CF and partners of CF women. The American College of Medical Genetics and the American College of Obstetricians and Gynecologists recommend that CF carrier screening be offered to individuals of Ashkenazi Jewish or white descent and be made available to individuals of other ethnic and racial groups; in one large series, 14% of carrier screening referrals were from Hispanic and African-American individuals, and 12% from individuals with ethnicities other than white or Ashkenazi Jewish. Screening of the siblings of an affected child is also suggested.
Newborn Screening
A variety of newborn screening algorithms are in place to identify infants with CF. Most algorithms utilize a combination of immunoreactive trypsinogen results and limited DNA testing on blood spots, which are then coupled with confirmatory sweat analysis. This screening test is ≈95% sensitive. Newborn diagnoses can prevent early nutritional deficiencies and improve long-term growth and may improve cognitive function. Early diagnosis may improve weight for age; an improved weight for age is associated with better lung function at 6 yr of age. In 2004, the Centers for Disease Control and Prevention recommended the use of newborn screening for early identification of infants with CF. In 2009, newborn screening is mandated in 49 of 50 states, with Texas being the only state that does not require testing. Early diagnosis also has the advantage of genetic counseling for the family and, in some cases, avoids protracted diagnostic efforts.
Treatment
The treatment plan should be comprehensive and linked to close monitoring and early, aggressive intervention.
General Approach to Care
Initial efforts after diagnosis should be intensive and should include baseline assessment, initiation of treatment, clearing of pulmonary involvement, and education of the patient and parents. Follow-up evaluations are scheduled every 1-3 mo, depending on the age at diagnosis, because many aspects of the condition require careful monitoring. An interval history and physical examination should be obtained at each visit. A sputum sample or, if that is not available, a lower pharyngeal swab taken during or after a forced cough is obtained for culture and antibiotic susceptibility studies. Because irreversible loss of pulmonary function from low-grade infection can occur gradually and without acute symptoms, emphasis is placed on a thorough pulmonary history. Table 395-6 lists symptoms and signs that suggest the need for more intensive antibiotic and physical therapy. Protection against exposure to methicillin-resistant S. aureus, P. aeruginosa, B. cepacia, and other resistant gram-negative organisms is essential, including isolation procedures and careful attention to sterilization of inhalation therapy equipment. A nurse, respiratory therapist, social worker, dietitian, and psychologist, as members of the multidisciplinary care team, should evaluate children regularly and contribute to the development of a comprehensive daily care plan. Considerable education and programs to empower families and older children to take responsibility for care are likely to result in the best adherence to daily care programs. Standardization of practice, on the part of both caregivers and families, as well as close monitoring and early intervention for new or increasing symptoms appears to result in the best long-term outcomes.
Table 395-6 SYMPTOMS AND SIGNS ASSOCIATED WITH EXACERBATION OF PULMONARY INFECTION IN PATIENTS WITH CYSTIC FIBROSIS
SYMPTOMS
SIGNS
From Ramsey B: Management of pulmonary disease in patients with cystic fibrosis, N Engl J Med 335:179, 1996.
Because secretions of CF patients are not adequately hydrated, attention in early childhood to oral hydration, especially during warm weather or with acute gastroenteritis, may minimize complications associated with impaired mucus clearance. Intravenous therapy for dehydration should be initiated early.
The goal of therapy is to maintain a stable condition for prolonged periods. This can be accomplished for most patients by interval evaluation and adjustments of the home treatment program. Some children have episodic acute or low-grade chronic lung infection that progresses. For these patients, intensive inhalation and airway clearance and intravenous antibiotics are indicated. Improvement is most reliably accomplished in a hospital setting; selected patients have demonstrated successful outcomes while completing these treatments at home. Intravenous antibiotics may be required infrequently or as often as every 2-3 mo. The goal of treatment is to return patients to their previous pulmonary and functional status.
The basic daily care program varies according to the age of the child, the degree of pulmonary involvement, other system involvement, and the time available for therapy. The major components of this care are pulmonary and nutritional therapies. Because therapy is medication-intensive, iatrogenic problems frequently arise. Monitoring for these complications is also an important part of management (Table 395-7).
Table 395-7 COMPLICATIONS OF THERAPY FOR CYSTIC FIBROSIS*
| COMPLICATION | AGENT |
|---|---|
| Gastrointestinal bleeding | Ibuprofen |
| Hyperglycemia | Corticosteroids (systemic) |
| Growth retardation | Corticosteroids (systemic, inhaled) |
| Renal dysfunction: | |
| Tubular | Aminoglycosides |
| Interstitial nephritis | Semisynthetic penicillins, nonsteroidal anti-inflammatory drugs |
| Hearing loss, vestibular dysfunction | Aminoglycosides |
| Peripheral neuropathy or optic atrophy | Chloramphenicol (prolonged course) |
| Hypomagnesemia | Aminoglycosides |
| Hyperuricemia, colonic stricture | Pancreatic extracts (very large doses) |
| Goiter | Iodine-containing expectorants |
| Gynecomastia | Spironolactone |
| Enamel hypoplasia or staining | Tetracyclines (used in 1st 8 yr of life) |
* Note: Common hypersensitivity reactions to drugs are not included.
Pulmonary Therapy
The object of pulmonary therapy is to clear secretions from airways and to control infection. When a child is not doing well, every potentially useful aspect of therapy should be reconsidered.
Inhalation Therapy
Aerosol therapy is used to deliver medications and hydrate the lower respiratory tract. Metered-dose inhalers can deliver some agents, such as bronchodilators and corticosteroids, with a spacer for younger children. Alternately, these medications can be delivered with a compressor that drives a handheld nebulizer. In some patients β-agonists may decrease PaO2 acutely by increasing ventilation-perfusion mismatch, a concern if the PaO2 is marginal.
Human recombinant DNase (2.5 mg), given as a single daily aerosol dose, improves pulmonary function, decreases the number of pulmonary exacerbations, and promotes a sense of well-being in patients who have moderate disease and purulent secretions. Benefit for those with normal forced expiratory volume in 1 sec (FEV1) values or advanced lung disease has also been documented. Improvement is sustained for ≥12 mo with continuous therapy. Another mucolytic agent, N-acetylcysteine, is toxic to ciliated epithelium, and its repeated administration should be avoided.
Nebulized hypertonic saline, acting as a hyperosmolar agent, is believed to draw water into the airway and rehydrate mucus and the peri ciliary fluid layer, resulting in improved mucociliary clearance. A number of studies have reported 7% hypertonic saline nebulized 2-4 times daily results in increased mucus clearance and improved pulmonary function.
Aerosolized antibiotics are often used when the airways are colonized with Pseudomonas as part of daily therapy. Aerosolized tobramycin, TOBI, used as a suppressive therapy (on 1 month, off 1 month) may reduce symptoms, improve pulmonary function, and alleviate the need for hospitalization (see Aerosolized Antibiotic Therapy).
Airway Clearance Therapy
Airway clearance treatment usually consists of chest percussion combined with postural drainage and derives its rationale from the idea that cough clears mucus from large airways but chest vibrations are required to move secretions from small airways, where expiratory flow rates are low. Chest physical therapy (PT) can be particularly useful for patients with CF because they accumulate secretions in small airways first, even before the onset of symptoms. Although immediate improvement of pulmonary function generally cannot be demonstrated after PT, cessation of chest PT in children with mild to moderate airflow limitation results in deterioration of lung function within 3 wk, and prompt improvement of function occurs when therapy is resumed. Chest PT is recommended 1-4 times a day, depending on the severity of lung dysfunction. Cough, huffing, or forced expirations are encouraged after each lung segment is “drained.” Vest-type mechanical percussors are also useful. Voluntary coughing, repeated forced expiratory maneuvers with and without positive expiratory pressure, patterned breathing, and use of an array of handheld oscillatory devices are additional aids to clearance of mucus. Routine aerobic exercise appears to slow the rate of decline of pulmonary function, and benefit has also been documented with weight training. No one airway clearance technique (ACT) is superior to any other, so all modes should be considered in the development of an airway clearance prescription. Adherence to daily therapy is essential therefore ACT plans are individualized for each patient.
Antibiotic Therapy
Antibiotics are the mainstay of therapy designed to control progression of lung infection. The goal is to reduce the intensity of endobronchial infection and to delay progressive lung damage. The usual guidelines for acute chest infections, such as fever, tachypnea, or chest pain, are often absent. Consequently, all aspects of the patient’s history and examination, including anorexia, weight loss, and diminished activity, must be used to guide the frequency and duration of therapy. Antibiotic treatment varies from intermittent short courses of 1 antibiotic to nearly continuous treatment with 1 or more antibiotics. Dosages for some antibiotics are often 2 to 3 times the amount recommended for minor infections because patients with CF have proportionately more lean body mass and higher clearance rates for many antibiotics than other individuals. In addition, it is difficult to achieve effective drug levels of many antimicrobials in respiratory tract secretions.
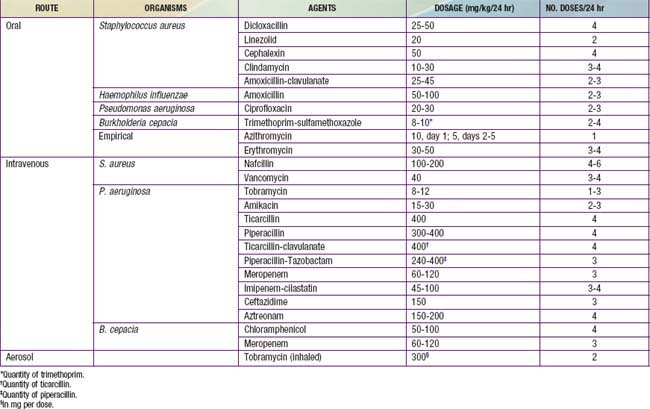
Oral Antibiotic Therapy
Indications for oral antibiotic therapy in a patient with CF include the presence of respiratory tract symptoms and identification of pathogenic organisms in respiratory tract cultures. Whenever possible, the choice of antibiotics should be guided by in vitro sensitivity testing. Common organisms, include S. aureus, nontypable Haemophilus influenzae, P. aeruginosa; B. cepacia and other gram-negative rods, are encountered with increasing frequency. The first 2 can be eradicated from the respiratory tract in CF with use of oral antibiotics, but Pseudomonas is more difficult to treat. The usual course of therapy is ≥2 wk, and maximal doses are recommended. Useful oral antibiotics are listed in Table 395-8. The quinolones are the only broadly effective oral antibiotics for Pseudomonas infection, but resistance against these agents emerges rapidly. Infection with mycoplasmal or chlamydial organisms has been documented, providing a rationale for the use of macrolides on an empirical basis for flare of symptoms. Macrolides may reduce the virulence properties of P. aeruginosa, such as biofilm production, and contribute anti-inflammatory effects. Long-term therapy with azithromycin times a week has been shown to improve lung function in patients with chronic P. aeruginosa infection.
Aerosolized Antibiotic Therapy
P. aeruginosa and other gram-negative organisms are frequently resistant to all oral antibiotics. Aerosol delivery of antibiotics has been used as an option for home delivery of additional agents, such as tobramycin, colistin, and gentamicin. Although these therapies are used, the evidence to support aerosolized antibiotics for an acute pulmonary exacerbation is limited. However, there is good evidence to support the use of inhaled tobramycin as a long-term suppressive therapy in a patient colonized with P. aeruginosa. When tobramycin is given at a dose of 300 mg twice daily on alternate months for 6 mo, Pseudomonas density in sputum decreases, fewer hospitalizations are required, and pulmonary function can improve by ≥10%. Toxicity is negligible. On the basis of available evidence, this therapy is recommended in patients with chronic colonization with P. aeruginosa, to lessen symptoms and/or to improve long-term function in patients with moderate to severe disease. Other antimicrobials, such as colistin (75-150 mg), have been given in aerosolized 2 to 4 times a day; however, their efficacy has not been established. Sensitization and resistance to inhaled antibiotics can occur, but both developments are surprisingly infrequent. Bronchospasm may complicate aerosolized colistin therapy. Another indication for aerosolized antibiotic therapy is the acquisition of P. aeruginosa in the airways. Early infection may be eliminated for months to several years by oral ciprofloxacin with aerosolized colistin or tobramycin. Established infection is rarely eradicated.
Intravenous Antibiotic Therapy
For the patient who has progressive or unrelenting symptoms and signs despite intensive home measures, intravenous antibiotic therapy is indicated. This therapy is usually initiated in the hospital but may be completed on an ambulatory basis. Although many patients show improvement within 7 days, it is usually advisable to extend the period of treatment to at least 14 days. Permanent intravenous access can be provided for long-term or frequent courses of therapy in the hospital or at home. Thrombophilia screening should be considered before the use of totally implantable intravenous devices or for recurring problems with venous catheters.
Commonly used intravenous antibiotics are listed in Table 395-8. In general, treatment of Pseudomonas infection requires 2-drug therapy. A 3rd agent may be required for optimal coverage of S. aureus or other organisms. The aminoglycosides have a relatively short half-life in many patients with CF. The initial parenteral dose, noted in Table 395-8, is generally given every 8 hr. After blood levels have been determined, the total daily dose should be adjusted. Peak levels of 10-15 mg/L are desirable, and trough levels should be kept at <2 mg/L to minimize the risk of ototoxicity and nephrotoxicity. Once- or twice-daily aminoglycoside dosing may have advantages over dosing every 8 hr. Changes in therapy should be guided by lack of improvement and by culture results. If patients do not show improvement, complications such as heart failure and reactive airways or infection with viruses, Aspergillus fumigatus (Chapter 229), nontuberculous mycobacteria (Chapter 209), or other unusual organisms should be considered. B. cepacia is the most frequent of a growing list of gram-negative rods that may be particularly refractory to antimicrobial therapy.
Bronchodilator Therapy
Reversible airway obstruction occurs in many children with CF, sometimes in conjunction with frank asthma or acute bronchopulmonary aspergillosis. Reversible obstruction is defined as improvement of ≥15% in flow rates after inhalation of a bronchodilator. In many patients with CF, flow rates may improve by only 5-10%, however. Nevertheless, subjective benefit is claimed by many following use of a β-adrenergic agonist aerosol. Cromolyn sodium and ipratropium hydrochlorides are alternative agents, but there is no evidence to support their use.
Anti-Inflammatory Agents
Corticosteroids are useful for the treatment of allergic bronchopulmonary aspergillosis and severe reactive airway disease occasionally encountered in children with CF. Prolonged treatment of standard CF lung disease using an alternate-day regimen initially appeared to improve pulmonary function and diminish hospitalization rates. However, a 4-yr double-blind, multicenter study of this regimen for patients with mild to moderate lung disease found only modest efficacy and prohibitive side effects, including growth retardation, cataracts, and abnormalities of glucose tolerance at a dose of 2 mg/kg and growth retardation at 1 mg/kg. Inhaled corticosteroids have theoretical appeal, but there are few data documenting their efficacy and safety; it appears that discontinuing inhaled corticosteroids in patients with CF had no effect on lung function, antibiotic use, or bronchodilator use. Ibuprofen, given long term (dose adjusted to achieve a peak serum concentration of 50-100 µg/mL) for 4 yr, is associated with a slowing of disease progression, particularly in younger patients with mild lung disease. Side effects of nonsteroidal anti-inflammatory drugs have been encountered (see Table 395-7); therefore, this therapy has not gained broad acceptance even though ibuprofen is the only anti-inflammatory agent with documented efficacy in the patient population.
Endoscopy and Lavage
Treatment of obstructed airways sometimes includes tracheobronchial suctioning or lavage, especially if atelectasis or mucoid impaction is present. Bronchopulmonary lavage can be performed by the instillation of saline or a mucolytic agent through a fiberoptic bronchoscope. Antibiotics (usually gentamicin or tobramycin) can also be instilled directly at lavage in order to transiently achieve a much higher endobronchial concentration than can be obtained by using intravenous therapy. There is no evidence for sustained benefit from repeated endoscopic or lavage procedures.
Other Therapies
Expectorants such as iodides and guaifenesin do not effectively assist with the removal of secretions from the respiratory tract. Inspiratory muscle training can enhance maximum oxygen consumption during exercise as well as FEV1.
Emerging Therapies
A number of potential therapies are under development, including promising mutation-specific therapies in clinical trials. For class 1 mutations (nonsense or premature stop-codon mutations) that result in a premature termination of the mRNA that leads to no protein production, PTC124 has been shown to suppress the termination codon, allowing for read through and partial correction of the defect in approximately 30% of subjects with CF. Additional studies are under way to further evaluate PTC124’s efficacy. Several molecules have been identified that allow for proper processing of class 2 mutations; clinical trials are planned to evaluate a number of these substances. Significant progress has been made on a group of small molecules referred to as “potentiators,” including VX-770 (Vertex Pharmaceuticals, Cambridge, MA), that activate CFTR mutants (G551D-CFTR) that traffic to the plasma membrane but do not appropriately activate. Therapies including denufosol and Moli1901 (lancovutide, Lantibio/AOP Orphan Pharmaceuticals AG, Vienna) are directed at bypassing the primary CFTR defect by regulating alternative ion channels and normalizing the ion transport properties of affected tissue, thus correcting the mucociliary abnormality observed in the respiratory tract of the patient with CF.
Treatment of Pulmonary Complications
Atelectasis
Lobar atelectasis occurs relatively infrequently; it may be asymptomatic and noted only at the time of a routine chest radiograph. Aggressive intravenous therapy with antibiotics and increased chest PT directed at the affected lobe may be effective. If there is no improvement in 5-7 days, bronchoscopic examination of the airways may be indicated. If the atelectasis does not resolve, continued intensive home therapy is indicated, because atelectasis may resolve during a period of weeks or months. Persistent atelectasis may be asymptomatic. Lobectomy should be considered only if expansion is not achieved and the patient has progressive difficulty from fever, anorexia, and unrelenting cough (Chapter 402).
Hemoptysis
Endobronchial bleeding usually reflects airway wall erosion secondary to infection. With increasing numbers of older patients, hemoptysis has become a relatively frequent complication. Blood streaking of sputum is particularly common. Small-volume hemoptysis (<20 mL) should not trigger panic and is usually viewed as a need for intensified antimicrobial therapy and chest PT. When the hemoptysis is persistent or increases in severity, hospital admission is indicated. Massive hemoptysis, defined as total blood loss of ≥250 mL in a 24-hr period, is rare in the 1st decade and occurs in <1% of adolescents, but it requires close monitoring and the capability to replace blood losses rapidly. Chest PT is often discontinued until 12-24 hr after the last brisk bleeding episode and is then gradually re-instituted. Patients should receive vitamin K for an abnormal prothrombin time. During brisk hemoptysis, the child and parents require a great deal of reassurance that the bleeding will stop. Blood transfusion is not indicated unless there is hypotension or the hematocrit is significantly reduced. Ticarcillin, salicylates, and nonsteroidal anti-inflammatory drugs interfere with platelet function and may aggravate hemoptysis. Bronchoscopy rarely reveals the site of bleeding. Lobectomy is to be avoided, if possible, because functioning lung should be preserved. Bronchial artery embolization can be useful to control persistent, significant hemoptysis.
Pneumothorax
Pneumothorax (Chapter 405) is encountered in <1% of children and teenagers with CF, although it is more frequently encountered in older patients and may be life-threatening. The episode may be asymptomatic but is often attended by chest and shoulder pain, shortness of breath, or hemoptysis. A small air collection that does not grow can be observed closely. Chest tube placement with or without pleurodesis is often the initial therapy. Intravenous antibiotics are also begun on admission. An open thoracotomy or video-assisted thoracoscopy (VATS) with plication of blebs, apical pleural stripping, and basal pleural abrasion should be considered if the air leak persists. Surgical intervention is usually well tolerated even in cases of advanced lung disease. The thoracotomy tube is removed as soon as possible, usually on the 2nd or 3rd postoperative day. The patient can then be mobilized, and full postural drainage therapy resumed. Previous pneumothorax with or without pleurodesis is not a contraindication to subsequent lung transplantation.
Allergic Aspergillosis
Allergic aspergillosis occurs in 5-10% of patients with CF and may manifest as wheezing, increased cough, shortness of breath, and marked hyperinflation (Chapters 229 and 391). In some patients, a chest radiograph shows new, focal infiltrates. The presence of rust-colored sputum, the recovery of Aspergillus organisms from the sputum, the demonstration of serum precipitating and specific immunoglobulin E (IgE) and IgG antibodies against A. fumigatus, or the presence of eosinophils in a fresh sputum sample supports the diagnosis. The serum IgE level is usually high. Treatment is directed at controlling the inflammatory reaction with oral corticosteroids and preventing central bronchiectasis. For refractory cases, oral antifungals may be required.
Nontuberculous Mycobacteria Infection (Chapter 209)
Injured airways with poor clearance may be colonized by Mycobacterium avium-complex but also Mycobacterium abscessus and Mycobacterium kansasii. Distinguishing endobronchial colonization (frequent) from invasive infection (infrequent) is challenging. Persistent fevers and new infiltrates or cystic lesions coupled with the finding of acid-fast organisms on sputum smear suggest infection. Treatment is prolonged and requires multiple antimicrobial agents. Symptoms may improve, but the nontuberculous mycobacteria (NTM) are not usually cleared from the lungs.
Bone and Joint Complications
Hypertrophic osteoarthropathy causes elevation of the periosteum over the distal portions of long bones and bone pain, overlying edema, and joint effusions. Acetaminophen or ibuprofen may provide relief. Control of lung infection usually reduces symptoms. Intermittent arthropathy unrelated to other rheumatologic disorders occurs occasionally, has no recognized pathogenesis, and usually responds to nonsteroidal anti-inflammatory agents. Back pain or rib fractures from vigorous coughing may require pain management to permit adequate airway clearance. These and other fractures may stem from diminished bone mineralization, the result of reduced vitamin D absorption, corticosteroid therapy, diminished weight-bearing exercises, and, perhaps, other factors. There may be a bone phenotype in CF that is unrelated to therapies or nutritional status and may be due to CFTR dysfunction.
Sleep-Disordered Breathing
Particularly with advanced pulmonary disease and during chest exacerbations, individuals with CF may experience more sleep arousals, less time in rapid eye movement (REM) sleep, nocturnal hypoxemia, hypercapnia, and associated neurobehavioral impairment. Nocturnal hypoxemia may hasten the onset of pulmonary hypertension and right-sided heart failure. Efficacy of specific interventions for this complication of CF has not been systematically assessed. Prompt treatment of airway symptoms and nocturnal oxygen supplementation or bilevel positive airway pressure (BiPAP) support should be considered in selected cases.
Acute Respiratory Failure
Acute respiratory failure (Chapter 65) rarely occurs in patients with mild to moderate lung disease and is usually the result of a severe viral or other infectious illness. Because patients with this complication can regain their previous status, intensive therapy is indicated. In addition to aerosol, postural drainage, and intravenous antibiotic treatment, oxygen is required to raise the arterial PaO2. An increasing PCO2 may require ventilatory assistance. Endotracheal or bronchoscopic suction may be necessary to clear airway inspissated secretions and can be repeated daily. Right-sided heart failure should be treated vigorously. Recovery is often slow. Intensive intravenous antibiotic therapy and postural drainage should be continued for 1-2 wk after the patient has regained baseline status.
Chronic Respiratory Failure
Patients with CF acquire chronic respiratory failure after prolonged deterioration of lung function. Although this complication can occur at any age, it is now seen most frequently in adult patients. Because a long-standing PaO2 <50 mm Hg promotes the development of right-sided heart failure, patients usually benefit from low-flow oxygen to raise arterial PO2 to ≥55 mm Hg. Increasing hypercapnia may prevent the use of optimal FIO2. Most patients improve somewhat with intensive antibiotic and pulmonary therapy measures and can be discharged from the hospital. Low-flow oxygen therapy is needed at home, especially with sleep. Noninvasive ventilatory support can improve gas exchange and has been documented to enhance quality of life. Ventilatory support may be particularly useful for patients awaiting lung transplantation. These patients usually display cor pulmonale and should reduce their salt intake and be given diuretics. Caution should be exercised to avoid ventilation-suppressing metabolic alkalosis that results from CF-related chloride depletion and, in many cases, from diuretic-induced bicarbonate retention. Chronic pain (headache, chest pain, abdominal pain, and limb pain) is frequent at the end of life and responds to judicious use of analgesics, including opioids. Dyspnea has been ameliorated with nebulized fentanyl.
Lung transplantation is an option for end-stage lung disease (Chapter 437) but a topic of vigorous debate. Criteria for referral continue to be a subject of investigation and ideally include estimates of longevity with and without transplant based on lung function and exercise tolerance data. Because of bronchiolitis obliterans (Chapter 386.1) and other complications, transplanted lungs cannot be expected to function for the lifetime of a recipient, and repeat transplantation is increasingly common. The demand for donor lungs exceeds the supply, and waiting lists as well as duration of waits continue to grow. The protocol for matching donor organs with lung transplant recipients has been revised to account for the severity of the patients’ lung disease. In a review of lung transplantation in children with CF between 1992 and 2002, pre-transplantation colonization with B. cepacia, diabetes, and older age decreased post-transplantation survival. The review suggests that transplantation is often associated with many complications and may not prolong life nor significantly improve its quality.
Heart Failure
Some patients experience reversible right-sided heart failure (Chapter 436) as the result of an acute event such as a viral infection or pneumothorax. Individuals with long-standing, advanced pulmonary disease, especially those with severe hypoxemia (PaO2 <50 mm Hg), often acquire chronic right-sided heart failure. The mechanisms include hypoxemic pulmonary arterial constriction and loss of the pulmonary vascular bed. Pulmonary arterial wall changes contribute to increased vascular resistance with time. Evidence for concomitant left ventricular dysfunction is often found. Cyanosis, increased shortness of breath, increased liver size with a tender margin, ankle edema, jugular venous distention, an unusual weight gain, increased heart size seen on chest radiograph, or evidence for right-sided heart enlargement on electrocardiogram or echocardiogram helps to confirm the diagnosis. Diuresis induced by furosemide (1 mg/kg administered intravenously) confirms the suspicion of fluid retention. Repeated doses are often required at 24- to 48-hr intervals to reduce fluid accumulation and accompanying symptoms. Concomitant use of spironolactone may protect against potassium depletion and facilitate long-term diuresis. Hypochloremic alkalosis complicates the long-term use of loop diuretics. Digitalis is not effective in pure right-sided failure but may be useful when there is an associated left-sided dysfunction. The arterial PO2 should be maintained at >50 mm Hg if possible. Intensive pulmonary therapy, including intravenous antibiotics, is most important. Initially, the salt intake should be limited. Volume overload and antibiotics with high sodium content should be avoided. No clear-cut long-term benefit from pulmonary vasodilators has been demonstrated. The prognosis for heart failure is poor, but a number of patients survive for ≥5 yr after the appearance of heart failure. Heart-lung transplantation may be an option (see preceding section).
Nutritional Therapy
Up to 90% of patients with CF have loss of exocrine pancreatic function as well as inadequate digestion and absorption of fats and proteins. They require dietary adjustment, pancreatic enzyme replacement, and supplementary vitamins. In general, children with CF need to exceed the usual required daily caloric intake to grow. The target has been set at 130 kcal per kg of body weight. Daily supplements of the fat-soluble vitamins are required.
Diet
Historically, at the time of diagnosis many infants presented with nutritional deficits; this situation is changing because of newborn screening. Sometimes young infants with a history of wheezy breathing were often started on soy-protein formulas prior to their evaluation; they did not use this protein well and often acquired hypoproteinemia with anasarca. Although in the past a low-fat, high-protein, high-calorie diet was generally recommended for older children, it resulted in deficiencies of essential fatty acids and poor growth. With the advent of improved pancreatic enzyme products, increased amounts of fat in the diet are well tolerated and preferred.
Most individuals with CF have a higher than normal caloric need because of increased work of breathing and perhaps because of increased metabolic activity related to the basic defect. When anorexia of chronic infection supervenes, weight loss occurs. Encouragement to eat high-calorie foods is important, but weight gain is not generally realized unless lung infection is controlled. Weight stabilization or gain sometimes requires nocturnal feeding via nasogastric tube or percutaneous enterostomy or with short-term intravenous hyperalimentation. Not infrequently, parent-child interactions at feeding time are maladaptive, and behavioral interventions can improve caloric intake. Long-term benefits of these interventions include improved quality of life and psychologic well-being. In addition there is good correlation between improved BMI and maintenance of FEV1.
Recombinant growth hormone therapy (3 times per week) has improved nutritional outcomes, including positive effects on nitrogen balance and improved height and weight velocities.
Pancreatic Enzyme Replacement
Extracts of animal pancreas given with ingested food reduce but do not fully correct stool fat and nitrogen losses. Enzyme dosage and product should be individualized for each patient. Enteric-coated, pH-sensitive enzyme microspheres are most often prescribed. Several strengths up to 20,000 IU of lipase/capsule are available. Administration of excessive doses has been linked to colonic strictures requiring surgery. Consequently, enzyme replacement should not exceed 2,500 lipase units/kg/meal in most circumstances. In general, infants need 2,000-4,000 lipase units per feeding, which is most easily given mixed with applesauce. Snacks should also be covered. The dose of enzymes required usually increases with age, but some patients have lower requirements as teenagers and young adults.
Vitamin and Mineral Supplements
Because pancreatic insufficiency results in malabsorption of fat-soluble vitamins (A, D, E, K), vitamin supplementation is recommended. Several vitamin preparations containing all 4 vitamins for patients with CF are available. They should be taken daily. Replacement doses may be required when low serum levels are documented or the patient is symptomatic. Infants with zinc deficiency and an acrodermatitis enteropathica–like rash have been described. In addition, attention should be paid to iron status; in one study, almost 30% of children with CF had low serum ferritin concentrations.
Treatment of Intestinal Complications
Meconium Ileus
When meconium ileus (Chapter 96.1) is suspected, a nasogastric tube is placed for suction and the newborn is hydrated. In many cases, diatrizoate (Gastrografin) enemas with reflux of contrast material into the ileum not only confirm the diagnosis but have also resulted in the passage of a meconium plug and clearing of the obstruction. Use of this hypertonic solution requires careful correction of water losses into the bowel. Children in whom this procedure fails require operative intervention. Children who are successfully treated generally have a prognosis similar to that of other patients with severe CF mutations. Infants with meconium ileus should be treated as if they have CF until adequate diagnostic testing can be carried out.
Distal Intestinal Obstruction Syndrome (Meconium Ileus Equivalent) and Other Causes of Abdominal Symptoms
Despite appropriate pancreatic enzyme replacement, 2-5% of patients accumulate fecal material in the terminal portion of the ileum and in the cecum, which may result in partial or complete obstruction. For intermittent symptoms, pancreatic enzyme replacement should be continued or even increased, and stool softeners (polyethylene glycol [MiraLAX] or docusate sodium [Colace]) given. Increased fluid intake is also recommended. Failure to relieve symptoms signals the need for large-volume bowel lavage with a balanced salt solution containing polyethylene glycol taken by mouth or by nasogastric tube. When there is complete obstruction, a diatrizoate enema, accompanied by large amounts of intravenous fluids, can be therapeutic. Intussusception (Chapter 325.3) and volvulus (Chapter 321.4) must also be considered in the differential diagnosis. Intussusception, usually ileocolic, occurs at any age and often follows a 1- to 2-day history of “constipation.” It can often be diagnosed and reduced via a diatrizoate enema. If a nonreducible intussusception or a volvulus is present, laparotomy is required. Repeated episodes of intussusception may be an indication for cecectomy.
Chronic appendicitis with or without periappendiceal abscess may manifest as recurrent or persistent abdominal pain, raising the question of need for a laparotomy. A lack of acid buffering in the duodenum appears to promote duodenitis and ulcer formation in some children. Other reasons for surgical procedures include carcinoma of the colon or biliary tract and sclerosing colonopathy.
Gastroesophageal Reflux (Chapter 315)
Because several factors raise intra-abdominal pressure, including cough and obstructed airways, pathologic gastroesophageal reflux is not uncommon and may exacerbate lung disease secondary to reflex wheezing and repeated aspiration. Dietary, positional, and medication therapies should be considered. Cholinergic agonists are contraindicated because they trigger mucus secretion and progressive respiratory difficulty. Reduction of stomach acid secretion can help, with proton pump inhibitors being the most effective agents. Fundoplication is a procedure of last resort.
Rectal Prolapse (Chapter 336.5)
Though uncommon, rectal prolapse occurs most often in infants with CF and less frequently in older children with the disease. It is usually related to steatorrhea, malnutrition, and repetitive cough. The prolapsed rectum can usually be replaced manually by continuous gentle pressure with the patient in the knee-chest position. Sedation may be helpful. To prevent an immediate recurrence, the buttocks can be taped closed. Adequate pancreatic enzyme replacement, decreased fat and roughage in the diet, stool softener, and control of pulmonary infection result in improvement. Occasionally, a patient may continue to have rectal prolapse and may require sclerotherapy or surgery.
Hepatobiliary Disease
Liver function abnormalities associated with biliary cirrhosis can be improved by treatment with ursodeoxycholic acid. The ability of bile acids to prevent progression of cirrhosis has not been clearly documented. Portal hypertension with esophageal varices, hypersplenism, or ascites occurs in ≤8% of children with CF (Chapter 359). The acute management of bleeding esophageal varices includes nasogastric suction and cold saline lavage. Sclerotherapy is recommended after an initial hemorrhage. In the past, significant bleeding has also been treated successfully with portosystemic shunting. Splenorenal anastomosis has been the most effective treatment. Pronounced hypersplenism may require splenectomy. Cholelithiasis should prompt surgical consultation. The management of ascites is discussed in Chapter 362.
Obstructive jaundice in newborns with CF needs no specific therapy. Hepatomegaly with steatosis requires careful attention to nutrition and may respond to carnitine repletion. Rarely, biliary cirrhosis proceeds to hepatocellular failure, which should be treated as in patients without CF (Chapters 356 and 359). End-stage liver disease is an indication for liver transplantation in children with CF, especially if pulmonary function is good (Chapter 360).
Pancreatitis
Pancreatitis can be precipitated by fatty meals, alcohol ingestion, or tetracycline therapy. Serum amylase and lipase values may remain elevated for long periods. Treatment of this disorder is discussed in Chapter 343.
Hyperglycemia
Onset of hyperglycemia occurs most frequently after the 1st decade. Approximately 20% of young adults are treated for hyperglycemia, although the incidence of CF-related diabetes (CFRD) may be higher. Prevalence is greater in females and in ΔF508 homozygotes. Ketoacidosis is rarely encountered. The pathogenesis includes both impaired insulin secretion and insulin resistance. Routine screening consists of an annual modified 2-hr oral glucose tolerance test after the child reaches age 8-10 yr. Glucose intolerance without urine glucose losses is usually not treated; glycosylated hemoglobin levels should be followed at least annually. With persistent glycosuria and symptoms, insulin treatment should be instituted. Oral hypoglycemic agents, with or without drugs that reduce insulin resistance, may also be effective. Exocrine pancreatic insufficiency and malabsorption make strict dietary control of hyperglycemia difficult. Corticosteroid therapy should be avoided. The development of significant hyperglycemia favors acquisition of P. aeruginosa and B. cepacia in the airways and may adversely affect pulmonary function. Thus, careful control of blood glucose level is an important goal. Long-term vascular complications of diabetes can occur, providing an additional rationale for good control of blood glucose levels.
Other Therapy
Nasal Polyps
Nasal polyps (Chapter 370) occur in 15-20% of patients with CF and are most prevalent in the 2nd decade of life. Local corticosteroids and nasal decongestants occasionally provide some relief. When the polyps completely obstruct the nasal airway, rhinorrhea becomes constant, or widening of the nasal bridge is noticed, surgical removal of the polyps is indicated; polyps may recur promptly or after a symptom-free interval of months to years. Polyps inexplicably stop developing in many adults.
Rhinosinusitis
Opacification of paranasal sinuses is not an indication for intervention. Acute or chronic sinus-related symptoms are treated initially with antimicrobials, with or without maxillary sinus aspiration for culture. Functional endoscopic sinus surgery has anecdotally provided benefit.
Salt Depletion
Sweat salt losses in patients with CF can be high, especially in warm arid climates. Children should have free access to salt, and precautions against overdressing infants should be observed. Salt supplements are often prescribed to newborns identified through newborn screening and to children who live in hot weather climates. Hypochloremic alkalosis should be suspected in any infant who has had symptoms of gastroenteritis, and prompt fluid and electrolyte therapy should be instituted as needed.
Growth and Maturation
Delayed growth should be vigorously addressed by enhancing nutrition, treating lung disease more vigorously, and, in selected instances, endocrine evaluation and, possibly, growth hormone therapy. The risk : benefit ratio for anabolic steroid therapy does not support its use for undersized children with CF. Delayed sexual maturation, often associated with short stature, occurs fairly frequently in children with CF. Although many patients have severe pulmonary infection or poor nutrition, delayed puberty also occurs in those with otherwise mild disease and is not well explained. Adolescents with CF should receive specific counseling throughout their developing years concerning sexual maturation and reproductive potential.
Surgery
Minor surgical procedures, including dental work, should be performed with the use of local anesthesia if possible in children with CF. Patients with good or excellent pulmonary status can tolerate general anesthesia without any intensive pulmonary measures before the procedure. Those with moderate or severe pulmonary infection usually do better with a 1- to 2-wk course of intensive antibiotic treatment and increased airway clearance before surgery. If this approach is impossible, prompt intravenous antibiotic therapy is indicated once it is recognized that major surgery is required. The total time of anesthesia should be kept to a minimum. After induction, tracheal suctioning is useful and should be repeated. Patients with severe disease require monitoring of blood gas values and may need ventilatory assistance in the immediate postoperative period.
After major surgery, cough should be encouraged and airway clearance treatments should be re-instituted as soon as possible, usually within 24 hr. Adequate analgesia is important if early effective therapy is to be achieved. For those with significant pulmonary involvement, intravenous antibiotics are continued for 7-14 days postoperatively. Early ambulation and intermittent deep breathing are important; an incentive spirometer can also be helpful. After open thoracotomy for treatment of pneumothorax or lobectomy, the chest tube is the greatest single obstacle to effective pulmonary therapy and should be removed as soon as possible so that full postural drainage therapy can resume.
Prognosis
CF remains a life-limiting disorder, although survival has improved dramatically in the past 30-40 yr. Infants with severe lung disease occasionally succumb, but most children survive this difficult period and are relatively healthy into adolescence or adulthood. The slow progression of lung disease eventually reaches disabling proportions, however. Life table data now indicate a median cumulative survival exceeding 35 yr. Male survival is somewhat better than female survival for reasons that are not readily apparent. Children in socioeconomically disadvantaged families have, on average, a poorer prognosis.
Children with CF usually have good school attendance records and should not be restricted in their activities. A high percentage eventually attend and graduate from college. Most adults with CF find satisfactory employment, and an increasing number marry. Transitioning care from pediatric to adult care centers by 21 yr of age is an important objective and requires a thoughtful, supportive approach involving both the pediatric and internal medicine specialists.
With increasing life span for patients with CF, a new set of psychosocial considerations has emerged, including dependence-independence issues, self-care, peer relationships, sexuality, reproduction, substance abuse, educational and vocational planning, medical care costs and other financial burdens, and anxiety concerning health and prognosis. Many of these issues are best addressed in an anticipatory fashion, before the onset of psychosocial dysfunction. With appropriate medical and psychosocial support, children and adolescents with CF generally cope well. Achievement of an independent and productive adulthood is a realistic goal for many.
Aaron SD, Vandemheen KL, Ferris W, et al. Combination antibiotic susceptibility testing to treat exacerbations of cystic fibrosis associated with multiresistant bacteria: a randomized, double-blind, controlled clinical trial. Lancet. 2005;366:463-471.
Aaron SD, Vandemheen KL, Ramotar K, et al. Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA. 2010;304(19):2145-2152.
Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991-2003.
Aldámiz-Echevarria L, Preito JA, Andrade F, et al. Persistence of essential fatty acid deficiency in cystic fibrosis despite nutritional therapy. Pediatr Res. 2009;66:585-589.
Bartlett JR, Friedman KJ, Ling SC, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA. 2009;302:1076-1082.
Boyle MP. Adult cystic fibrosis. JAMA. 2007;298:1787-1793.
Buranawuti K, Boyle MP, Cheng S, et al. Variants in mannose-binding lectin and tumor necrosis factor α affect survival in cystic fibrosis. J Med Genet. 2007;44:209-214.
Casaulta C, Stirnimann A, Schoeni MH, et al. Sweat test in patients with glucose-6-phosphate-1-dehydrogenase deficiency. Arch Dis Child. 2008:93:878-879.
Colombo C, Battezzati A. Growth failure in cystic fibrosis: a true need for anabolic agents? J Pediatr. 2005;146:303-305.
Collaco J, Cutting G. Update on gene modifiers in cystic fibrosis. Curr Opin Pulmon Med. 2008;14:559-566.
Collaco JM, Blackman SM, McGready J, et al. Quantification of the relative contribution of environmental and genetic factors to variation in cystic fibrosis lung function. J Pediatr. 2010;157:802-807.
Collaco JM, Vanscoy L, Bremer L, et al. Interactions between secondhand smoke and genes that affect cystic fibrosis lung disease. JAMA. 2008;299:417-424.
Corey M, Edwards L, Levison H, et al. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr. 1997;131:809-814.
Dasenbrook EC, Checkley W, Merlo CA, et al. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA. 2010;303:2386-2392.
Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335:1255-1259.
Donaldson SH, Bennett WD, Zeman KL, et al. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241-250.
Drumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443-1453.
Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline on patients with cystic fibrosis. N Engl J Med. 2006;354:229-240.
Emerson J, Rosenfeld M, McNamara S, et al. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91-100.
Ferkol T, Rosenfeld M, Milla CE. Cystic fibrosis pulmonary exacerbations. J Pediatr. 2006;148:259-264.
FitzSimmons SC, Burkhart GA, Borowitz D, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med. 1997;336:1283-1289.
Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637-642.
Flume PA, Robinson KA, O’Sullivan BP, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care. 2009;54:522-537.
Flume PA, Robinson KA, O’Sullivan BP, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957-969.
Garcon-Michel N, Roguedas-Contios AM, Rault G, et al. Frequency of aquagenic palmoplantar keratoderma in cystic fibrosis: a new sign of cystic fibrosis? Br J Dermatol. 2010;163:162-166.
Gild R, Clay CD, Morey S. Aquagenic wrinkling of the palms in cystic fibrosis and the cystic fibrosis carrier state: a case-control study. Br J Dermatol. 2010;163:1082-1084.
Grangeia A, Sa R, Carvalho F, et al. Molecular characterization of the cystic fibrosis transmembrane conductance regulator gene in congenital absence of the vas deferens. Genet Med. 2007;9:163-172.
Grasemann H, Stehling F, Brunar H, et al. Inhalation of Moli1901 in patients with cystic fibrosis. Chest. 2007;131:1461-1466.
Green D, Carson K, Leonard A, et al. Current treatment recommendations for correcting vitamin D deficiency in pediatric patients with cystic fibrosis are inadequate. J Pediatr. 2008;153:554-559.
Groman JD, Karczeki B, Sheridan M, et al. Phenotypic and genetic characterization of patients with features of nonclassic forms of cystic fibrosis. J Pediatr. 2005;146:675-680.
Grosse SD, Rosenfeld M, Devine OJ, et al. Potential impact of newborn screening for cystic fibrosis on child survival: a systematic review and analysis. J Pediatr. 2006;149:362-366.
Gu Y, Harley ITW, Henderson LB, et al. Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature. 2009;458:1039-1042.
Hardin DS, Rice J, Ahn C, et al. Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition. J Pediatr. 2005;146:324-328.
Harrison AN, Regelmann WE, Zirbes JM, et al. Longitudinal assessment of lung function from infancy to childhood in patients with cystic fibrosis. Pediatr Pulmonol. 2009;44:330-339.
Hewer SCL, Tyrrell K. Cystic fibrosis and the transition to adult health services. Arch Dis Child. 2008;93:817-820.
Kappler M, Griese M. Nutritional supplements in cystic fibrosis. BMJ. 2006;332:618-619.
Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase 2 trial. Lancet. 2008;372:719-727.
LeGrys VA, Yankaskas JR, Quittell LM, et al. Diagnostic sweat testing: the cystic fibrosis foundation guidelines. J Pediatr. 2007;151:85-89.
Lenaerts C, Lapierre C, Patriqui H, et al. Surveillance for cystic fibrosis-associated hepatobiliary disease: early ultrasound changes and predisposing factors. J Pediatr. 2003;143:343-350.
Levy H, Kalish LA, Cannon CL, et al. Predictors of mucoid Pseudomonas colonization in cystic fibrosis patients. Pediatr Pulmonol. 2008;43:463-467.
Li Z, Kosorok MR, Farrell PM, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA. 2005;293:581-588.
Liou TG, Adler FR, Cox DR, Cahill TGI. Lung transplantation and survival in children with cystic fibrosis. N Engl J Med. 2007;357:2143-2152.
Liou TG, Rubenstein RC. Carrier screening, incidence of cystic fibrosis, and difficult decisions. JAMA. 2009;302:2595-2596.
McColley SA. Cystic fibrosis lung disease: when does it start, and how can it be prevented? J Pediatr. 2004;145:6-7.
Mishra A, Greaves R, Smith K, et al. Diagnosis of cystic fibrosis by sweat testing: age-specific reference intervals. J Pediatr. 2008;153:758-763.
Moran A, Hardin D, Rodman D, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res Clin Pract. 1999;45:61-73.
Moss RB. Infection, inflammation, and the downward spiral of cystic fibrosis lung disease. J Pediatr. 2009;154:162-163.
Moyer K, Balistreri W. Hepatobiliary disease in patients with cystic fibrosis. Curr Opin Gastroenterol. 2009;25:272-278.
Munck A, Houssin E, Rousey M. The importance of sweat testing for older siblings of patients with cystic fibrosis identified by newborn screening. J Pediatr. 2009;155:928-930.
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891-1902.
Poustie VJ, Russell JE, Watling, et al. Oral protein energy supplements for children with cystic fibrosis: CALICO multicentre randomized controlled trial. BMJ. 2006;332:632-635.
Ravzi S, Saiman L. Nontuberculous mycobacteria in cystic fibrosis. Pediatr Infect Dis J. 2007;26:263-264.
Ren CL, Pasta DJ, Rasouliyan L, et al. Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J Pediatr. 2008;153:746-751.
Retsch-Bogart GZ, Quittner AL, Gibson RL, et al. Efficacy and safety of inhaled aztreonam lysine for airway pseudomonas in cystic fibrosis. Chest. 2009;135:1223-1232.
Ross LF. Newborn screening for cystic fibrosis: a lesson in public health disparities. J Pediatr. 2008;153:308-313.
Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749-1756.
Sims EJ, McCormick J, Mehta G, et al. Newborn screening for cystic fibrosis is associated with reduced treatment intensity. J Pediatr. 2005;147:306-311.
Smyth A, Tan KHV, Hyman-Taylor P, et al. Once versus three-times daily regimens of tobramycin treatment for pulmonary exacerbations of cystic fibrosis—the TOPIC study: a randomized controlled trial. Lancet. 2005;365:573-578.
Smyth RL. Daily activity and disease status in cystic fibrosis: an important area for research. J Pediatr. 2005;147:281-283.
Smyth RL. Diagnosis and management of cystic fibrosis. Arch Dis Child. 2005;90:ep1-ep6.
Southern KW, Mérelle MME, Dankert-Roelse JE, et al: Newborn screening for cystic fibrosis (review), Cochrane Database System Rev (1):CD001402, 2009.
Spultan ZN, Foster MM, Newman NB, et al. Sweat chloride testing in infants identified as heterozygote carriers by newborn screening. J Pediatr. 2008;13:857-859.
Sterescu AE, Rhodes B, Jackson R, et al. Natural history of glucose intolerance in patients with cystic fibrosis: ten-year prospective observation program. J Pediatr. 2010;156:613-617.
Stick SM, Brennan S, Murray C, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155:623-628.
Sugarman EA, Rohlfs EM, Silverman LM, et al. CFTR mutation distribution among U.S. Hispanic and African American individuals: evaluation in cystic fibrosis patient and carrier screening populations. Genet Med. 2004;6:392-399.
Texereau J, Marullo S, Hubert D, et al. Nitric oxide synthase 1 as a potential modifier gene of decline in lung function in patients with cystic fibrosis. Thorax. 2004;59:156-158.
Tuchman LK, Schwartz LA, Sawcki GS, et al. Cystic fibrosis and transition to adult medical care. Pediatrics. 2010;125:566-573.
Vandenbussche HL, Klepser ME. Single daily tobramycin dosing in cystic fibrosis: is it better for the patients or the bugs? Lancet. 2005;365:547-548.
Welsch MJ. Targeting the basic defect in cystic fibrosis. N Engl J Med. 2010;363(21):2056-2057.
Wilcken B. Cystic fibrosis: refining the approach to newborn screening. J Pediatr. 2009;155:605-606.
Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433-1441.
Zeitlin P. Emerging drug treatments for cystic fibrosis. Expert Opin Emerg Drugs. 2007;12:329-336.