Chapter 652 Diseases of Subcutaneous Tissue
Diseases involving the subcutis are usually characterized by necrosis and/or inflammation; they may occur either as a primary event or as a secondary response to various stimuli or disease processes. The principal diagnostic criteria relates to the appearance and distribution of the lesions, associated symptoms, results of laboratory studies, histopathology, and natural history and exogenous provocative factors of these conditions.
Corticosteroid-Induced Atrophy
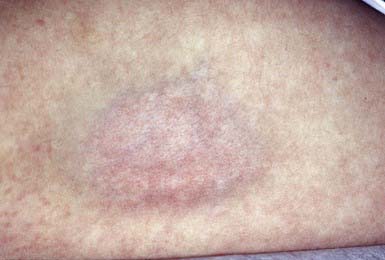
Intradermal injection of a corticosteroid can produce deep atrophy accompanied by surface pigmentary changes and telangiectasia (Fig. 652-1). These changes occur approximately 2 wk after injection and may last for months.
652.1 Panniculitis and Erythema Nodosum
Inflammation of fibrofatty subcutaneous tissue may primarily involve the fat lobule or, alternatively, the fibrous septum that compartmentalizes the fatty lobules. Lobular panniculitis that spares the subcutaneous vasculature includes post-steroid panniculitis, lupus erythematosus profundus, pancreatic panniculitis, α1-antitrypsin deficiency, subcutaneous fat necrosis of the newborn, sclerema neonatorum, cold panniculitis, subcutaneous sarcoidosis, and factitial panniculitis. Lobar panniculitis with vasculitis occurs in erythema induratum and, occasionally, as a feature of Crohn disease (Chapter 328.2). Inflammation predominantly within the septum, sparing the vasculature, may be seen in erythema nodosum (Table 652-1 and Fig. 652-2), necrobiosis lipoidica, progressive systemic sclerosis (Chapter 154), and subcutaneous granuloma annulare (Chapter 649). Septal panniculitis that includes inflammation of the vessels is found primarily in leukocytoclastic vasculitis and polyarteritis nodosa (Chapter 161).
Table 652-1 ETIOLOGY OF ERYTHEMA NODOSUM
VIRUSES
Epstein-Barr, hepatitis B, mumps
FUNGI
Coccidioidomycosis, histoplasmosis, blastomycosis, sporotrichosis
BACTERIA AND OTHER INFECTIOUS AGENTS
Group A streptococcus,* tuberculosis,* Yersinia, cat-scratch disease, leprosy, leptospirosis, tularemia, mycoplasma, Whipple disease, lymphogranuloma venereum, psittacosis, brucellosis
OTHER
Sarcoidosis, inflammatory bowel disease,* estrogen-containing oral contraceptives,* systemic lupus erythematosus, Behçet syndrome, severe acne, Hodgkin disease, lymphoma, sulfonamides, bromides, Sweet syndrome, pregnancy, idiopathic*
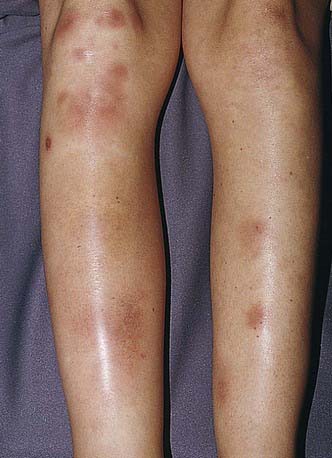
Figure 652-2 Tender red nodules with indistinct borders in a teenage girl with erythema nodosum.
(From Weston AL, Lane AT, Morelli JG: Color textbook of pediatric dermatology, ed 3, St Louis, 2002, Mosby, p 212.)
Erythema Nodosum
Erythema nodosum is a nodular, erythematous hypersensitivity reaction that typically appears with multiple lesions on the exterior surfaces of the arms and legs in the pretibial area (more common) and less often in other cutaneous areas containing subcutaneous fat. The lesions vary in size from 1 to 6 cm, are symmetric, and are oval with the longer axis parallel to the extremity. They initially appear bright or dull red but progress to a brown or purple; they are tense and painful and usually do not ulcerate (see Fig. 652-2). Initial lesions may resolve in 1-2 wk, but new lesions may continue to appear for 2-6 wk. Repeat episodes may occur weeks to months later. Prior to or immediately at the onset of lesions, there may be systemic manifestations that include fever, malaise, arthralgias (50-90%) and rheumatoid factor negative arthritis.
The etiology is unknown in 30-50% of pediatric cases of erythema nodosum; other etiologies are noted in Table 652-1. Group A streptococcal infection and inflammatory disorders (inflammatory bowel disease) are common etiologies in children; sarcoidosis should be considered in young adults.
Treatment includes that of the underlying disease as well as symptomatic relief with nonsteroidal antiinflammatory agents. Salicylates, supersaturated solution of potassium iodide (oral), colchicine, intraintestinal injections of steroidsand, in severe, persistent, or recurrent lesions, oral steroids have been employed. The idiopathic form is a self-limited disorder.
Post-Steroid Panniculitis
Etiology/Pathogenesis
The mechanism of the inflammatory reaction in the fat in post-steroid panniculitis is unknown.
Clinical Manifestations
Fewer than 20 cases of post-steroid panniculitis have been reported, with most of these being in children. The disorder occurs in children who have received high-dose corticosteroids. In 1-2 wk after discontinuation of the drug, multiple subcutaneous nodules usually appear on the cheeks, although other areas may be involved. Nodules range in size from 0.5 to 4 cm, are erythematous or skin colored, and may be pruritic.
Lupus Erythematosus Profundus (Lupus Erythematosus Panniculitis)
Etiology/Pathogenesis
It is unknown what separates those patients in whom lupus erythematosus profundus develops from other patients with systemic lupus erythematosus. This variant of lupus erythematosus is rare in childhood.
Clinical Manifestations
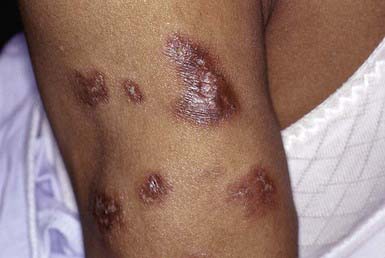
Lupus erythematosus profundus manifests as one to several firm, well-defined, purple plaques or nodules 1 to 3 cm in diameter, most commonly on the face, buttocks, or proximal extremities. This condition may occur in patients with systemic or discoid lupus erythematosus and may precede or follow the development of other cutaneous lesions. The overlying skin is usually normal but may be erythematous, atrophic, poikilodermatous, or hyperkeratotic (Fig. 652-3). Lesions may be painful and may ulcerate. On healing, a shallow depression generally remains or, rarely, soft pink areas of anetoderma result.
Histology
The histopathologic changes in lupus erythematosus profundus are distinctive and may allow the clinician to make the diagnosis in the absence of other cutaneous lesions of lupus erythematosus. The panniculitis is characterized by a mostly lobular dense infiltrate of lymphocytes and plasma cells. A dense perivascular and periappendigeal lymphocytic infiltrate is seen in the dermis. Lichenoid changes may be identified at the epidermal-dermal junction. Histopathologic differentiation from subcutaneous panniculitis–like T-cell lymphoma may be difficult. Results of lupus band and antinuclear antibody tests are usually positive.
Treatment
Nodules tend to be persistent. Hydroxychloroquine (2-5 mg/kg/day) is the treatment of choice for lupus erythematosus profundus. Intralesional corticosteroids may worsen the residual lipoatrophy. Immunosuppressive agents are indicated only for treatment of other severe manifestations of systemic lupus erythematosus. Avoidance of sun exposure and trauma is also important.
α1-Antitrypsin Deficiency
Etiology/Pathogenesis
Individuals with α1-antitrypsin deficiency have severe homozygous deficiency or, rarely, a partial deficiency of the protease inhibitor α1-antitrypsin, which inhibits trypsin activity and the activity of elastase, serine proteases, collagenase, factor VIII, and kallikrein (Chapter 385). Panniculitis occurs with the Z subtype.
Clinical Manifestations
Cellulitis-like areas or tender, red nodules occur on the trunk or proximal extremities (Chapter 385). Nodules tend to ulcerate spontaneously and discharge an oily yellow fluid. Panniculitis may be associated with other manifestations of the disease, such as panacinar emphysema, noninfectious hepatitis, cirrhosis, persistent cutaneous vasculitis, cold contact urticaria, and acquired angioedema. Diagnosis can be substantiated by a decreased level of serum α1-antitrypsin activity.
Treatment
Treatment of the panniculitis in with α1-antitrypsin deficiency is part of the overall treatment of the disease (Chapter 385).
Pancreatic Panniculitis
Etiology/Pathogenesis
Pathogenesis of pancreatic panniculitis appears to be multifactorial, involving liberation of the lipolytic enzymes lipase, trypsin, and amylase into the circulation, causing adipocyte membrane damage and intracellular lipolysis. There is no correlation, however, between the occurrence of panniculitis and the serum concentration of pancreatic enzymes.
Clinical Manifestations
Pancreatic panniculitis manifests most commonly on the pretibial regions, thighs, or buttocks as tender, erythematous nodules that may be fluctuant and occasionally discharge an oily yellowish substance. It appears most often in males with alcoholism but may also occur in patients with pancreatitis as a result of cholelithiasis or abdominal trauma, with rupture of a pancreatic pseudocyst, with pancreatic ductal adenocarcinoma, or with pancreatic acinar cell carcinoma. Associated features may include polyarthritis (PPP [pancreatitis-panniculitis-polyarthritis] syndrome). In almost 65% of patients, abdominal signs are absent or mild, making the diagnosis difficult.
Subcutaneous Fat Necrosis
Etiology/Pathogenesis
The cause of subcutaneous fat necrosis is unknown. The disease in infants may be due to ischemic injury under various circumstances, such as maternal preeclampsia, birth trauma, asphyxia, and prolonged hypothermia; in many affected infants, however, provocative factors are not identified. Susceptibility has been attributed to differences in composition between the subcutaneous tissue of young infants and that of older infants, children, and adults. Neonatal fat solidifies at a relatively high temperature because of its relatively greater concentration of high–melting point saturated fatty acids, such as palmitic and stearic acids.
Clinical Manifestations
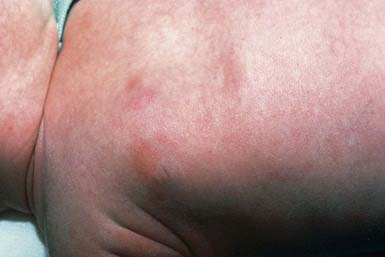
This inflammatory disorder of adipose tissue occurs primarily in the first 4 wk of life in full-term or post-term infants. Typical lesions are asymptomatic, rubbery to firm, erythematous to violaceous plaques or nodules on the cheeks, buttocks, back, thighs, or upper arms (Fig. 652-4). Lesions may be focal or extensive and are generally asymptomatic, although they may be tender during the acute phase. Uncomplicated lesions involute spontaneously within weeks to months, usually without scarring or atrophy. Calcium deposition may occasionally occur within areas of fat necrosis, which may sometimes result in rupture and drainage of liquid material. A rare but potentially life-threatening complication is hypercalcemia. It manifests at 1-6 mo of age as lethargy, poor feeding, vomiting, failure to thrive, irritability, seizures, shortening of the QT interval on electrocardiography, or renal failure. The origin of the hypercalcemia is unknown.
Histology
Histopathologic changes in subcutaneous fat necrosis are diagnostic, consisting of: necrosis of fat; a granulomatous cellular infiltrate composed of lymphocytes, histiocytes, multinucleated giant cells, and fibroblasts; and radially arranged clefts of crystalline triglyceride within fat cells and multinucleated giant cells. Calcium deposits are commonly found in areas of fat necrosis.
Differential Diagnosis
Subcutaneous fat necrosis can be confused with sclerema neonatorum, panniculitis, cellulitis, and hematoma.
Treatment
Because the lesions are self-limited, therapy is not required for uncomplicated cases of subcutaneous fat necrosis. Needle aspiration of fluctuant lesions may prevent rupture and subsequent scarring but is rarely needed. Treatment of hypercalcemia is aimed at enhancing renal calcium excretion with hydration and furosemide (1-2 mg/kg/dose) and at limiting dietary calcium and vitamin D intake. Reduction of intestinal calcium absorption and alteration of vitamin D metabolism may be accomplished by administration of corticosteroids (0.5-1.0 mg/kg/day). Pamidronate (0.25-0.5 mg/kg/day) has been used in severe cases.
Sclerema Neonatorum
Etiology/Pathogenesis
Although the cause remains unknown, four theories of pathogenesis for sclerema neonatorum have been proposed. It is theorized that sclerema neonatorum results from: hardening of the subcutaneous fat due to a decrease in body temperature as a consequence of circulatory shock; a defect in lipolytic enzymes or in lipid transport; association with an underlying severe disease; or a special form of edema affecting the connective tissue that supports the adipocytes.
Clinical Manifestations
This uncommon disorder of adipose tissue manifests abruptly in preterm, gravely ill infants as diffuse, yellowish white woody induration of the skin. Affected skin becomes stony in consistency, cold, and nonpitting. The face assumes a masklike expression, and joint mobility may be compromised because of inflexibility of the skin.
Histology
Histopathologic changes in sclerema neonatorum consist of increases in the size of fat cells and in the width of the fibrous connective tissue septa. In contrast to subcutaneous fat necrosis, with which this disorder is most apt to be confused, fat necrosis, inflammation, giant cells, and calcium crystals are generally absent.
Treatment
Sclerema neonatorum is almost always associated with serious illness, such as sepsis, congenital heart disease, multiple congenital anomalies, or hypothermia. The appearance of sclerema in a sick infant should be regarded as an ominous prognostic sign. The outcome depends on the response of the underlying disorder to treatment.
Cold Panniculitis
Etiology/Pathogenesis
The pathogenic mechanism of cold panniculitis may be similar to that of subcutaneous fat necrosis, involving a greater propensity of fat to solidify in infants than in that in older children and adults as a result of the higher percentage of saturated fatty acids in the subcutaneous fat of infants. Lesions occur in infants after prolonged cold exposure, especially on the cheeks, or after prolonged application of a cold object such as an ice cube, ice bag, or fruit ice pop to any area of the skin.
Clinical Manifestations
Ill-defined, erythematous to bluish, indurated plaques or nodules arise within hours to a couple days of exposure on exposed surfaces (face, arms, legs), persist for 2-3 wk, and heal without residua.
Histology
Histopathologic examination reveals an infiltrate of lymphoid and histiocytic cells around blood vessels at the dermal-subdermal junction and in the fat lobules; by the third day, some of the fat cells in the subcutis may have ruptured and coalesced into cystic structures.
Chilblains (Pernio)
Etiology/Pathogenesis
Vasospasm of arterioles due to damp cold exposure with resultant hypoxemia and localized perivascular mononuclear inflammation appears to be responsible for chilblains. The disease has been associated with cryoglobulins, lupus erythematosus with antiphospholipid antibodies, anorexia nervosa, and thin body habitus.
Clinical Manifestations
The condition is characterized by localized symmetric erythematous to purplish edematous plaques and nodules in areas exposed to cold, typically acral areas (distal hands and feet, ears, face; Chapter 69). Lesions develop 12-24 hr after cold exposure and may be associated with itching, burning, or pain. Blister formation and ulceration are rare.
Histology
Histopathologic examination reveals marked dermal edema and a perivascular and periappendigeal, predominantly T-cell lymphocytic infiltrate in the papillary and reticular dermis.
Factitial Panniculitis
Etiology/Pathogenesis
Factitial panniculitis results from subcutaneous injection by the patient or a proxy of a foreign substance, the most common types of which are organic materials such as milk and feces; drugs such as the opiates and pentazocine; oily materials such as mineral oil and paraffin; and the synthetic polymer povidone.
Clinical Manifestations
Indurated plaques, ulcers, or nodules that liquefy and drain may be noted clinically in factitial panniculitis.
Centers for Disease Control and Prevention. Outbreak of erythema nodosum of unknown cause—New Mexico, November 2007–January 2008. MMWR. 2009;58:1347-1351.
Fraga J, Garcia-Diez A. Lupus erythematosus panniculitis. Dermatol Clin. 2008;26:453-463.
Fregonese L, Stolk J. Hereditary alpa-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis. 2008;19:16.
Kerstan A, Goebeier M, Schmidt E, et al. Lupus erythematosus profundus in an 8-year old child. J Eur Acad Dermatol Venereol. 2007;21:132-133.
Kwon EJ, Emanuel PO, Gribetz CH, et al. Poststeroid panniculitis. J Cutan Pathol. 2007;34(Suppl 1):64-67.
Lombardi G, Cabana R, Bollani L, et al. Effectiveness of pamidronate in severe neonatal hypercalcemia caused by subcutaneous fat necrosis: a case report. Eur J Pediatr. 2009;168:625-627.
Narváez J, Bianchi MM, Santo P, et al. Pancreatitis, panniculitis and polyarthritis. Semin Arthritis Rheum. 2008;47:1814-1819.
Quesada-Cortes A, Campos-Munoz L, Daiz-Diaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:458-459.
Sanmartin O, Requena C, Requena L. Factitial panniculitis. Dermatol Clin. 2008;26:519-527. viii
Simon TD, Soep J, Hollister JR. Pernio in pediatrics. Pediatrics. 2005;116:e472-e475.
Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnosis and management. J Perinatol. 2008;28:453-460.
652.2 Lipodystrophy
Several rare conditions are associated with loss of fatty tissue in a partial or generalized distribution.
Partial Lipodystrophy
Partial lipodystrophy may be familial or acquired. Loss of adipose tissue is not preceded by an inflammatory phase, and histopathologic examination reveals only absence of subcutaneous fat.
There are 3 forms of familial partial lipodystrophy (FPLD):
Type I (FPLD1Köbberlingg) is characterized by loss of adipose tissue confined to the extremities and gluteal region. Fat distribution of the face, neck, and trunk may be normal or increased. Hyperlipidemia, insulin-resistant diabetes mellitus, and eruptive xanthomas may be seen. The gene is unknown, but only females are affected.
Type 2 (FPLD2–Dunnigan) is caused by mutations in the laminin A/C gene. Fat distribution is normal in childhood, but atrophy commences with puberty. Lipodystrophy is seen in the trunk, gluteal region, and extremities. Adipose tissue accumulates in the face and neck and may also be seen in the axillae, back, labia majora, and infra-abdominal region. Insulin-resistant diabetes mellitus and hypertriglyceridemia develop, but high-density lipoprotein and cholesterol levels are low. Both males and females are affected, but the diagnosis may be more difficult in males owing to body habitus.
Type 3 (FPLD3) is caused by mutations in the peroxisome proliferation–activated receptor gamma (PPARG) gene. Lipodystrophy is seen in the limbs and gluteal region. Insulin-resistant diabetes mellitus, primary amenorrhea, acanthosis nigricans, hypertension, and fatty infiltration of the liver are present.
AKT2 and ZMPSTE24 mutations are newly recognized causes of partial lipodystrophy.
Acquired partial lipodystrophy (Barraquer-Simons syndrome) is caused by mutations in the LMNB2 gene. Females are more commonly affected. Fat loss begins in childhood or adolescence and affects the face, neck, arms, thorax, and upper abdomen. Excess fat is seen in the hips and legs, especially in females. Low levels of complement C3 are almost universally seen. C3 nephritic factor is also present. C3 nephritic factor stabilizes C3 convertase, allowing for unopposed activation of the alternate complement pathway and the decreased level of C3. Membranous proliferative glomerulonephritis and other autoimmune diseases may develop. Insulin-resistant diabetes mellitus is rare.
Generalized Lipodystrophy
Generalized lipodystrophy may also be congenital or acquired.
Congenital generalized lipodystrophy is seen in 3 forms:
Type 1 (Berardinelli-Seip congenital lipodystrophy type 1 [BSCL1]) is an autosomal recessive disorder caused by mutations in the 1-acylglycerol-3-phosphate-O-acyltransferase (AGPAT2) gene.
Type 2 (Berardinelli-Seip congenital lipodystrophy type 2 [BSCL2]) is also autosomal recessive and caused by mutations in the seipin gene.
Type 3 (CAV1) is autosomal recessive and caused by mutations in the caveolin 1 gene.
Marked lipodystrophy occurs at birth or in early infancy. Diabetes mellitus, hypertriglyceridemia, hepatic steatosis, acanthosis nigricans, and muscular hypertrophy occur. BSCL2 is a more severe phenotype, with premature death occurring in ≈ 15% of cases.
Acquired generalized lipodystrophy is more common in females. The most common associated disorder is juvenile dermatomyositis (78%). Panniculitis is seen in 17%. More than half of the children may have other complications, including acanthosis nigricans, hyperpigmentation, hepatomegaly, hypertension, protuberant abdomen, and hyperlipidemia.
Localized lipoatrophy is an idiopathic condition that manifests as annular atrophy at the ankles; a bandlike semicircular depression 2-4 cm in diameter on the thighs, abdomen, and/or upper groin or as a centrifugally spreading, depressed, bluish plaque with an erythematous margin.
Insulin lipoatrophy usually occur approximately 6 mo-2 yr after initiation of relatively high doses of insulin. A dimple or well-circumscribed depression at the site of injection is typically seen, although loss of fat may extend beyond the site of injection, leading to an extensive, depressed plaque. Biopsy reveals a marked decrease or absence of subcutaneous tissue, without inflammation or fibrosis. In some patients, hypertrophy occurs clinically. In these cases, the mid-dermal collagen is replaced by hypertrophic fat cells on histopathologic sections. The mechanism of insulin lipoatrophy may be cross-reaction of insulin antibodies with fat cells; the incidence of this condition has decreased since the implementation of widespread use of highly purified insulins. Lesions may also be prevented by frequent alteration of injection sites.
Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009;1791:507-513.
Pope E, Janson A, Khambalia A, et al. Childhood acquired lipodystrophy: a retrospective study. J am Acad Dermatol. 2006;55:947-950.
Serrao VV, Feio AB. Localized abdominal idiopathic lipodystrophy. Dermatol Online. 2008;15:15.