CHAPTER 21 Periodontal Pathogenesis
Understanding periodontal pathogenesis is key to improving management strategies for this common, complex disease. The first challenge is to understand exactly what is meant by the term pathogenesis. According to Merriam Webster’s Collegiate Dictionary, pathogenesis is defined as the origination and development of a disease. Essentially, this means the step-by-step processes that lead to the development of the disease, resulting in a series of changes in the structure and function of, in this case, the periodontium. In broad terms, the pathogenesis of a disease is the mechanism by which an etiologic factor (or factors) causes the disease. The word derives from the Greek pathos (suffering, which is a now obsolete translation of pathos) and genesis (generation/creation).
Our knowledge of periodontal pathogenesis has evolved over the years. It is important to be aware of this, since treatment philosophies have similarly changed in parallel with our improving understanding of disease processes. For example, in the late 1800s, Willoughby D. Miller, the eminent dental researcher who established the important causal role of oral bacteria in the etiology of dental caries, also asserted “during the last few years the conviction has grown continually stronger, among physicians as well as dentists, that the human mouth, as a gathering-place and incubator of diverse pathogenic germs, performs a significant role in the production of varied disorders of the body, and that if many diseases whose origin is enveloped in mystery could be traced to their source, they would be found to have originated in the oral cavity.”115 This marked the beginning of an era of dental treatment strategies that aimed to treat systemic diseases by eliminating so-called foci of infection in the mouth. As a result, many patients underwent dental clearances as a management for their systemic diseases.
By the 1930s, such approaches were beginning to be questioned, as evidenced by a clinical study of 200 patients with rheumatoid arthritis of whom 92 patients had their tonsils removed as treatment for the arthritis (even though only about 15% gave any history of tonsillitis or sore throat) and 52 patients had some or all of their teeth removed.28 Of the 92 who had their tonsils removed, there was no impact on the arthritis in 86 patients (and two got worse), and of the 52 who had teeth removed, there was no benefit in 47 cases (and three patients reported a worsening of their arthritis after the extractions). The authors wrote that “focal infection is a splendid example of a plausible medical theory which is in danger of being converted by its too enthusiastic supporters into the status of an accepted fact.”28 The end of the focal infection era was signalled by an editorial in the Journal of the American Medical Association in 1952 that stated “many patients with diseases presumably caused by foci of infection have not been relieved of their symptoms by removal of the foci, many patients with these same systemic diseases have no evident focus of infection, foci of infection are as common in apparently healthy persons as in those with disease.”163
Advances in the management of periodontitis have been driven by improved knowledge of the epidemiology, etiology, and pathogenesis of the disease.193 In the 1970s, the role of plaque as the sole etiologic factor for periodontitis was unquestioned. In those days, nonsurgical treatment was in its infancy, and most treatment options involved surgery, for example, gingivectomy in the case of shallower pockets or access flap surgery for treatment of deeper sites. When looking back, it becomes clear that treatment strategies used in a given time period are entirely dependent on the prevailing understanding of pathogenesis at that particular point in time. It is therefore very likely that the management options that we take for granted now will change again in the future. This is to be welcomed because a progressive clinical discipline, such as periodontology, that is well founded in science and with patient benefit as its primary value should strive to improve therapeutic strategies in the light of continued discovery.
Periodontal disease results from a complex interplay between the subgingival biofilm and the host immune-inflammatory events that develop in the gingival and periodontal tissues in response to the challenge presented by the bacteria. It is generally accepted that gingivitis precedes periodontitis, but it is clear that not all cases of gingivitis progress to periodontitis. In gingivitis, the inflammatory lesion is confined to the gingiva, but in periodontitis, the inflammatory processes extend to additionally affect the periodontal ligament and alveolar bone. The net result of these inflammatory changes is breakdown of the fibers of the periodontal ligament, resulting in clinical loss of attachment, together with resorption of the alveolar bone.
In the 1970s and 1980s, bacterial plaque was generally considered to be preeminent as the cause of periodontitis. In that era, it was accepted that poor oral hygiene results in increased plaque accumulation, which in turn results in periodontal disease. However, this model failed to take into account observations such as there are many individuals with poor oral hygiene who do not develop advanced periodontal disease, and conversely, there are unfortunate individuals who, despite good oral hygiene and compliance with periodontal treatment protocols, continue to experience progressive periodontal breakdown and would be considered to have aggressive periodontitis. These findings were confirmed by the work of Löe and colleagues who studied Sri Lankan tea laborers who had no access to dental care and who could be divided into three main categories: (1) individuals (≈8% of the population studied) who had rapid progression of periodontal disease, (2) those (≈81%) who had moderate progression, and (3) those (≈11%) who demonstrated no progression of periodontal disease beyond gingivitis.106 All patients in this population displayed abundant plaque and calculus deposits. The etiologic role of plaque bacteria is clear in that the bacteria initiate and perpetuate the inflammatory responses that develop in the gingival tissues. However, the major determinant of susceptibility to disease is the nature of the immune-inflammatory responses themselves. It is paradoxical that these defensive processes, which are protective by intent (to prevent ingress of the bacteria and their products into the tissues), result in the majority of tissue damage leading to the clinical manifestations of disease.
Periodontal disease is therefore a unique clinical entity. It is not an infection in the classic sense of the word. In most infections, a single infective organism causes the disease (e.g., human immunodeficiency virus [HIV], syphilis, or tuberculosis), and identification of that organism provides the basis for the diagnosis. In periodontal disease, a large number of species are identifiable in the periodontal pocket, and many more are, as yet, unknown because they have not been cultured. It is impossible to conclude that a single species, or even a group of species, causes periodontal disease. Many of the species that are considered important in periodontal pathogenesis may simply reside in deep pockets because the pocket is a favorable environment for them to survive (e.g., it is warm, moist, and anaerobic, with a ready supply of nutrients). Many of the unique features of periodontitis derive from the anatomy of the periodontium, in which a hard, nonshedding surface (the tooth) is partly embedded within the body (within connective tissue), crosses an epithelial surface, and is partly exposed to the outside world (within the confines of the mouth). The bacteria that colonize this surface are effectively outside the body (even though they are in the subgingival crevice), yet the inflammatory response that develops is located within the body. These factors add complexity to our understanding of the role of the biofilm and the immune-inflammatory responses in periodontal tissue breakdown.
Histopathology of Periodontal Disease
To better understand periodontal pathogenesis, it is important to have an appreciation of the histologic appearance of clinically healthy tissues, as well as inflamed gingival and periodontal tissues. It is important to note that even in gingival tissues that clinically would be considered to be noninflamed and healthy, there is always evidence of inflammatory responses occurring if they are examined microscopically. This is normal, given that there is a chronic low-grade challenge presented by the subgingival plaque bacteria. The low-grade inflammatory response that results is not detectable macroscopically at the clinical level but is an essential protective mechanism to combat the microbial challenge and to prevent bacteria and their products from infiltrating the tissues and causing tissue damage. Our current understanding of susceptibility to periodontitis suggests that individuals who are more susceptible to the disease mount an excessive, or dysregulated, immune-inflammatory response for a given bacterial challenge, leading to increased tissue breakdown compared to those individuals who have a more normal inflammatory response.
Clinically Healthy Gingival Tissues
Clinically healthy gingival tissues (e.g., those observed in patients with excellent oral hygiene, no visible plaque deposits, and typically who have received regular and meticulous professional cleaning) are pink in appearance, not swollen, not inflamed, and firmly attached to the underlying tooth/bone, with minimal bleeding on probing. The dentogingival junction is a unique anatomic feature whose function is the attachment of the gingiva to the tooth. It comprises an epithelial portion and a connective tissue portion, both of which are of fundamental importance in periodontal pathogenesis. The epithelial portion can be divided into three distinct epithelial structures, the gingival epithelium, sulcular epithelium, and junctional epithelium (Figure 21-1). These epithelial structures are in continuity with each other but have distinct structures and functions, as indicated in Box 21-1.
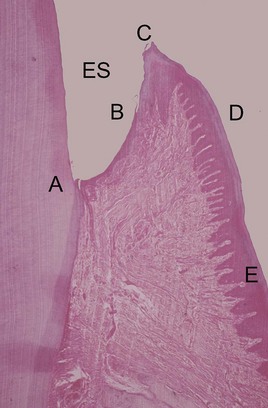
Figure 21-1 Histologic appearance of healthy gingiva. A photomicrograph of a demineralized tooth with the gingival tissues in situ (H&E, low magnification). Amelocemental junction (A). Enamel space (ES). Gingival health is characterized by organization of the epithelium into distinct zones; junctional epithelium (A-B), sulcular epithelium (B-C), free gingiva (C-D) and attached gingiva (D-E). The gingival connective tissue is composed of densely packed, organized, and interlacing collagen bundles. There are a few scattered inflammatory cells, but no significant inflammatory cell infiltrate.
BOX 21-1 Characteristics of the Epithelial Component of the Dentogingival Unit
The junctional epithelium is a particularly unique epithelial structure because the surface cells are specialized for the purpose of attachment to the tooth.11 Therefore, unlike other epithelial tissues elsewhere in the body, there is no opportunity for sloughing of cells from the surface. Instead, cells at the basal layer continually divide and move to within two or three cell layers of the tooth surface and then migrate coronally, parallel to the tooth surface to eventually reach the floor of the sulcus and be sloughed off into the gingival crevice. The extracellular spaces between the junctional epithelium are also greater than other epithelial tissues, with intercellular spaces comprising approximately 18% of the volume of the epithelium. This is a result of a lower density of desmosomes in the junctional epithelium compared to the gingival epithelium, and the junctional epithelium is therefore intrinsically “leaky.” This has great relevance in periodontal pathogenesis, since the widened intercellular spaces in the junctional epithelium permit migration of neutrophils (polymorphonuclear [PMN] leukocytes) and macrophages from the gingival connective tissues to enter the sulcus to phagocytose bacteria, as well as the ingress of bacterial products and antigens.
The connective tissue component of the dentogingival unit contains densely packed collagen fiber bundles (mixture of type I and III collagen fibers) that are arranged in distinct patterns that maintain the functional integrity of the tissues and tight adaptation of the soft tissues to the teeth. These include the following:
It is important to note that even in clinically healthy gingiva, the gingival connective tissue contains at least some inflammatory cells, particularly neutrophils. Neutrophils continually migrate through the connective tissues and pass through the junctional epithelium to enter the sulcus/pocket. These findings were reported in the classic investigations of the histology of periodontal disease reported by Page and Schroeder in 1976.131 This low-grade inflammation occurs in response to the continued presence of bacteria and their products in the gingival crevice. There is a continuous exudate of fluid from the gingival tissues that enters the crevice and flows out as gingival crevicular fluid (GCF). In addition to the continuous migration of neutrophils through the gingival tissues, lymphocytes and macrophages also accumulate. The presence of leukocytes in the connective tissues results from the chemotactic stimulus created by the subgingival biofilm and bacterial products, as well as chemoattractant factors produced by the host.
In clinically healthy tissues, this steady state equilibrium between low-grade inflammation in the tissues and the continual presence of the subgingival microflora may persist for many years or indeed for the lifetime of the individual. Overt clinical signs of gingivitis (redness, swelling, and bleeding on probing) do not develop because of several innate and structural defense mechanisms, including the following:
However, if plaque accumulation increases so that these defense mechanisms are overwhelmed, then inflammation and the classic clinical signs of gingivitis will develop. Even though the development of gingivitis in response to the accumulation of plaque is fairly predictable, research has identified that a spectrum of responses may be observed, with some individuals developing marked gingival inflammation for a given plaque challenge and others developing minimal gingival inflammation.181 These observations underscore the importance of variations in host responses between individuals in terms of gingival inflammatory responses. Furthermore, many individuals may never develop periodontitis despite having widespread gingivitis. The host’s immune-inflammatory response is fundamental in determining which individuals may progress to developing periodontitis, and it is likely that inflammatory responses are markedly different in those individuals who develop periodontitis compared to those who never progress beyond gingivitis. The challenge that this presents clinically is that we do not know (yet) enough about susceptibility to periodontitis to identify these individuals before they actually develop signs of the disease.
Histopathology of Gingivitis and Periodontitis
Development of gingivitis is very clearly observed from a clinical perspective. In addition, the changes that occur within the tissues are very obvious when examined under a microscope. In broad terms, there is infiltration of the connective tissues by numerous defense cells, particularly neutrophils, macrophages, plasma cells, and lymphocytes. As a result of the accumulation of these defense cells and the extracellular release of their destructive enzymes, there is disruption of the normal anatomy of the connective tissues resulting in collagen depletion and subsequent proliferation of the junctional epithelium. Vasodilation and increased vascular permeability lead to increased leakage of fluid out of the vessels and facilitate the passage of defense cells from the vasculature into the tissues, resulting in enlargement of the tissues, which appear erythematous and edematous (i.e., the clinical appearance of gingivitis). These changes are all reversible if the bacterial challenge is substantially reduced by improved oral hygiene.
The landmark studies of Page and Schroeder131 described the histologic changes that occur in the gingival tissues as the initial, early, established, and advanced gingival lesions. In broad terms, the initial lesion corresponds to clinically healthy (but nonetheless slightly inflamed) tissues, the early lesion corresponds to the early stages of (clinically evident) gingivitis, the established lesion corresponds to chronic gingivitis, and the advanced lesion marks the transition to periodontitis, with attachment loss and bone resorption. It is important to note that these are histologic descriptions only, and they should not form part of a clinical diagnosis. It is not possible to make any statements about the histologic status of a patient’s tissues, unless a biopsy is taken and the tissue examined microscopically. It is also important to note that these classic descriptions are primarily based on findings in experimental animals. The histologic stages of gingivitis are described in more detail in Chapter 7, but given their importance for understanding periodontal pathogenesis, these stages are also considered briefly below and summarized in Box 21-2.
BOX 21-2 Adapted from Page RC, Schroeder HE: Lab Invest 33:235-249, 1976 and linked to the clinical stages of gingivitis and periodontitis.
Key Features of the Histologic Stages of Gingivitis and Periodontitis
The Initial Lesion
The initial lesion is typically said to develop within 2 to 4 days of accumulation of plaque at a site that was otherwise plaque-free and at which there was no inflammation evident microscopically. However, this situation is probably never encountered in reality, and the gingival tissues always have characteristics of a low-grade chronic inflammatory response as a result of the continual presence of the subgingival biofilm. In other words, the initial lesion corresponds to the histologic picture that is evident in clinically healthy gingival tissues. This low-grade inflammation is characterized by dilation of the vascular network and increased vascular permeability, permitting the neutrophils and monocytes from the gingival vasculature to migrate through the connective tissues toward the source of the chemotactic stimulus—the bacterial products in the gingival sulcus. Upregulation of adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and E-selectin, in the gingival vasculature facilitates the migration of neutrophils from the capillaries into the connective tissues. Increased leakage of fluid from the vessels increases the hydrostatic pressure in the local microcirculation, and as a result, GCF flow increases. Increased GCF flow has the effect of diluting bacterial products and also potentially has a flushing action to remove bacteria and their products from the crevice, although given the nature of the bacterial biofilm, it is likely that only planktonic (free-floating) bacteria are removed in this way.
The Early Lesion
The early lesion develops after about 1 week of continued plaque accumulation and corresponds to the early clinical signs of gingivitis. The gingiva are erythematous in appearance as a result of proliferation of capillaries, opening up of microvascular beds, and continued vasodilation.104 Increasing vascular permeability leads to increased GCF flow, and transmigrating neutrophils increase significantly in number. The predominant infiltrating cell types are neutrophils and lymphocytes (primarily thymic lymphocytes [T-cells]),135 and the neutrophils migrate through the tissues to the sulcus and phagocytose bacteria. Fibroblasts degenerate, primarily via apoptosis (programmed cell death), which increases the space available for infiltrating leukocytes. Collagen destruction occurs, resulting in collagen depletion in the areas apical and lateral to the junctional and sulcular epithelium. The basal cells of these epithelial structures begin to proliferate to maintain an intact barrier against the bacteria and their products, and as a result the epithelium can be seen proliferating into the collagen depleted areas of the connective tissues (Figure 21-2).151 As a result of edema of the gingival tissues, the gingiva may appear slightly swollen, and accordingly, the gingival sulcus becomes slightly deeper. The subgingival biofilm exploits this ecologic niche and proliferates apically (thereby rendering effective plaque control more difficult). The early gingival lesion may persist indefinitely or may progress further.
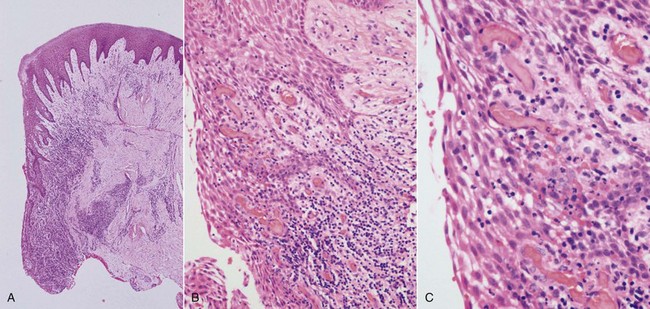
Figure 21-2 Histologic appearance of gingivitis. A series of photomicrographs illustrating gingivitis (H&E). In all cases, the tooth would be to the left side of the image. Low magnification of the gingiva (A) demonstrates hyperplastic junctional and sulcular epithelium with a dense inflammatory cell infiltrate in the adjacent connective tissue. Medium magnification of the epithelial-connective tissue interface (B) shows numerous intraepithelial inflammatory cells along with intercellular edema. The connective tissue contains dilated capillaries (hyperemia), and there is a dense inflammatory cell infiltrate. High magnification (C) shows neutrophils and small lymphocytes transiting the sulcular epithelium.
The Established Lesion
The established lesion roughly corresponds with what clinicians would refer to as chronic gingivitis. The progression from the early lesion to the established lesion depends on many factors, including the plaque challenge (the composition and quantity of the biofilm), host susceptibility factors, and risk factors (both local and systemic). In the initial work by Page and Schroeder, the established lesion was defined as being dominated by plasma cells.131 In human studies, reports have suggested that plasma cells predominate in established gingivitis in older subjects,51 whereas lymphocytes predominate in younger individuals, although the relevance of these findings is not clear.23,51 What is clear from all the studies is that there is a significant inflammatory cell infiltrate in established gingivitis that occupies a considerable volume of the inflamed connective tissues. Large numbers of infiltrating cells can be identified adjacent and lateral to the junctional and sulcular epithelium, around blood vessels, and between collagen fiber bundles.22 Collagen depletion continues, with further proliferation of the epithelium into the connective tissue spaces. Neutrophils accumulate in the tissues and release their lysosomal contents extracellularly (in an attempt to kill bacteria that are not phagocytosed), resulting in the further tissue destruction. Neutrophils are also a major source of MMP-8 (neutrophil collagenase) and MMP-9 (gelatinase B), and these enzymes are produced in large quantities in the inflamed gingival tissues as the neutrophils migrate through the densely packed collagen fiber bundles to enter the sulcus. The junctional and sulcular epithelium form a pocket epithelium that is not firmly attached to the tooth surface and that contains large numbers of neutrophils and is more permeable to the passage of substances into or out of the underlying connective tissue. The pocket epithelium may be ulcerated and is less able to resist the passage of the periodontal probe, so bleeding on probing is a common feature of chronic gingivitis. It is important to remember that these inflammatory changes are still completely reversible if effective plaque control is reinstituted.
The Advanced Lesion
The advanced lesion marks the transition from gingivitis to periodontitis. This transition is determined by many factors, the relative importance of which is, at present, unknown but includes the bacterial challenge (both the composition and the quantity of the biofilm), the host inflammatory response, and susceptibility factors, including environmental and genetic risk factors. Histologic examination reveals continued evidence of collagen destruction (extending now into the periodontal ligament and alveolar bone). Neutrophils predominate in the pocket epithelium and the periodontal pocket, and plasma cells dominate in the connective tissues. The junctional epithelium migrates apically along the root surface into the collagen depleted areas that develop below it to maintain an intact epithelial barrier. Osteoclastic bone resorption commences, and the bone retreats from the advancing inflammatory front as a defense mechanism to prevent spread of bacteria into the bone (Figure 21-3). As the pocket deepens, plaque bacteria proliferate apically into a niche, which is very favorable for many of the species that are regarded as periodontal pathogens. The pocket presents a protected, warm, moist, and anaerobic environment with a ready nutrient supply, and since the bacteria are effectively outside the body (not withstanding that they are in the periodontal pocket), they are not significantly eliminated by the inflammatory response. Thus a cycle develops in which chronic inflammation and associated tissue damage continue; the tissue damage mainly being caused by the inflammatory response, yet the initiating factor, the biofilm, is not eliminated. Destruction of collagen fibers in the periodontal ligament continues, bone resorption progresses, the junctional epithelium migrates apically to maintain an intact barrier, and as a result, the pocket deepens fractionally. This makes it even more difficult to remove the bacteria and disrupt the biofilm through oral hygiene techniques, and thus the cycle perpetuates.
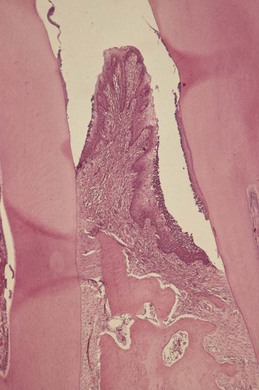
Figure 21-3 Histologic appearance of periodontitis. A photomicrograph of adjacent demineralized teeth with the interproximal gingiva and periodontium in situ (H&E, low magnification). The root of the tooth on the right is coated with a layer of dental plaque/calculus, and there is attachment loss with the formation of a periodontal pocket. The periodontium is densely inflamed and there is alveolar bone loss producing a triangular-shaped defect; vertical bone loss. The base of the pocket is apical to the crest of the alveolar bone and is termed an infrabony periodontal pocket.
(From Soames JV, Southam JC: Oral pathology, ed 4, Oxford, 2005, Oxford University Press.).
Inflammatory Responses in the Periodontium
Now that the histopathology of gingivitis and periodontitis has been reviewed, it is important to consider some of the specific molecules that signal tissue damage as the inflammatory response develops. These can be broadly divided into two main groups: those derived from the subgingival microflora (i.e., microbial virulence factors) and those derived from the host immune-inflammatory response. In terms of the relative importance of each, it is now clear that the great majority of the tissue breakdown results from the host’s inflammatory processes. The bacteria are important because they drive and perpetuate the inflammation but are only responsible directly for a relatively small proportion of the tissue damage that occurs.
Microbial Virulence Factors
The subgingival biofilm initiates and perpetuates inflammatory responses in the gingival and periodontal tissues. The subgingival bacteria also contribute directly to tissue damage by the release of noxious substances, but their primary importance in periodontal pathogenesis is that of activating immune-inflammatory responses that in turn result in tissue damage (which may well be beneficial to the bacteria located within the periodontal pocket by providing nutrient sources). Microbial virulence factors that are important in these processes are now discussed in turn.
Lipopolysaccharide
Lipopolysaccharides (LPS) are large molecules composed of a lipid component (lipid A) and a polysaccharide component. They are found in the outer membrane of gram-negative bacteria, they act as endotoxins (LPS is frequently referred to as endotoxin), and they elicit strong immune responses in animals. LPS is highly conserved in bacterial species, which reflects its importance in maintaining the structural integrity of the bacterial cells. Immune systems in animals have evolved to recognize LPS via toll-like receptors (TLRs), a family of cell surface molecules that are highly conserved in animal species from Drosophila (a genus of fruit flies) to humans, reflecting their importance in innate immune responses. TLRs are also present in lower animals and are in fact more varied than in higher species.26 TLRs are cell surface receptors that recognize microbe-associated molecular patterns (MAMPs), which are conserved molecular structures located on diverse pathogens. TLR-4 recognizes LPS from gram-negative bacteria and functions as part of a complex of cell surface molecules, including CD14 and MD-2 (also known as lymphocyte antigen 96). Interaction of this CD14/TLR-4/MD-2 complex with LPS triggers a series of intracellular events, the net result of which is increased production of inflammatory mediators (most notably cytokines) and the differentiation of immune cells (e.g., dendritic cells) for the development of effective immune responses against the pathogens. It is particularly interesting to the periodontist that the pathogen Porphyromonas gingivalis has an atypical form of LPS and is recognized by both TLR-2 and TLR-4.38,45
It is important to remember that a component of gram-positive cell walls, lipoteichoic acid (LTA), also stimulates immune responses, although less potently than LPS. LTA signals through TLR-2. Both LPS and LTA are released from the bacteria present in the biofilm and stimulate inflammatory responses in the tissues, resulting in increased vasodilation and vascular permeability, recruitment of inflammatory cells by chemotaxis, and release of proinflammatory mediators by the leukocytes that are recruited to the area. LPS in particular is of key importance in initiating and sustaining inflammatory responses in the gingival and periodontal tissues.
Bacterial Enzymes and Noxious Products
Plaque bacteria produce a number of metabolic waste products that contribute directly to tissue damage. These include noxious agents, such as ammonia (NH3) and hydrogen sulfide (H2S), and short-chain carboxylic acids, such as butyric acid and propionic acid. These acids are detectable in GCF and are found in increasing concentrations as the severity of periodontal disease increases. These substances have profound effects on host cells (e.g., butyric acid induces apoptosis in T-cells, B-cells, fibroblasts, and gingival epithelial cells).94,95,164 The short-chain fatty acids may aid P. gingivalis infection through tissue destruction and may also create a nutrient supply for the organism by increasing bleeding into the periodontal pocket. The short-chain fatty acids also influence cytokine secretion by immune cells and may potentiate inflammatory responses after exposure to proinflammatory stimuli such as LPS, interleukin-1 beta (IL-1β), and tumor necrosis factor alpha (TNF-α).122
Plaque bacteria produce proteases, which are capable of breaking down structural proteins of the periodontium such as collagen, elastin, and fibronectin. Bacteria produce these proteases to digest proteins and thereby provide peptides for bacterial nutrition. Bacterial proteases disrupt host responses, compromise tissue integrity, and facilitate microbial invasion of the tissues. P. gingivalis produces two classes of cysteine proteases that have been implicated in periodontal pathogenesis. These are known as gingipains and include the lysine specific gingipain Kgp and the arginine specific gingipains RgpA and RgpB. The gingipains can modulate the immune system and disrupt immune-inflammatory responses, potentially leading to increased tissue breakdown.137 Gingipains can reduce the concentrations of cytokines in cell culture systems7 and they digest and inactivate TNF-α.25 The gingipains can also stimulate cytokine secretion via activation of protease-activated receptors (PARs). For example, RgpB activates two different PARs (PAR-1 and PAR-2), thereby stimulating cytokine secretion107 and both Rgp and Kgp gingipains stimulate IL-6 and IL-8 secretion by monocytes via activation of PAR-1, PAR-2 and PAR-3.182
Microbial Invasion
Microbial invasion of the periodontal tissues has long been a contentious topic. In histologic specimens, bacteria, including cocci, filaments, and rods, have been identified in the intercellular spaces of the epithelium.50 Periodontal pathogens such as P. gingivalis and Aggregatibacter actinomycetemcomitans have been reported to invade the gingival tissues,30,76,146 including the connective tissues.145 Fusobacterium nucleatum can invade oral epithelial cells, and bacteria that routinely invade host cells may facilitate the entry of noninvasive bacteria by coaggregating with them (Figure 21-4).47 It has also been shown that A. actinomycetemcomitans can invade epithelial cells and persist intracellularly.49 The clinical relevance of these findings is unclear, however. Some investigators have suggested that tissue invasion by subgingival bacteria is an active process, whereas others have considered it to be an artifact, or simply a passive translocation process.

Figure 21-4 Invasion of epithelial cells by Fusobacterium nucleatum. In both images, a single epithelial cell is shown, being penetrated by invading F. nucleatum bacteria (3-4 bacteria are evident in A and 1 bacterium is evident in B). The ruffled surface of the epithelial cells (multiple small fingerlike projections, much smaller than the F. nucleatum bacteria) is likely to be an artifact. In B, F. nucleatum may facilitate the colonization of epithelial cells by bacteria unable to adhere or invade directly as evidenced by the single coccoid bacterium (Streptococcus cristatus) that has coaggregated with the F. nucleatum bacterium as it penetrates the epithelial cell.
(Images courtesy Dr. A.E. Edwards, Dr. J.D. Rudney, and Dr. T.J. Grossman, Bath University, United Kingdom, and the University of Minnesota.).
The reports of bacteria present in the tissues have sometimes been used to justify the use of antibiotics in the treatment of periodontitis as a means of attempting to eliminate those organisms that are located in the tissues and that are therefore “protected” from mechanical disruption by root surface debridement. It has also been reported that bacteria in the tissues represent a “reservoir for reinfection” after nonsurgical management. However, until the clinical relevance of bacteria being present in the tissues is better defined, it is inappropriate to make clinical treatment decisions (e.g., whether to use adjunctive systemic antibiotics) on this premise alone.
Fimbriae
The fimbriae of certain bacterial species, particularly P. gingivalis, may also play a role in periodontal pathogenesis. P. gingivalis fimbriae stimulate immune responses, such as IL-6 secretion,96,127 and the major fimbrial structural component of P. gingivalis, FimA, has been shown to stimulate nuclear factor (NF)-κB and IL-8 in a gingival epithelial cell line via TLR-2.5 Monocytes are also stimulated by P. gingivalis FimA, secreting IL-6, IL-8, and TNF-α.48 P. gingivalis fimbriae also interact with complement receptor-3 (CR-3) to activate intracellular signalling pathways that inhibit IL-12 production mediated by TLR-2 signalling.66 This may be of clinical relevance as IL-12 is important in activating natural killer (NK) cells and CD8+ cytotoxic T-cells, which themselves may be important in killing P. gingivalis–infected host cells such as epithelial cells. Indeed, blockade of the CR-3 receptor promotes IL-12–mediated clearance of P. gingivalis and negates its virulence.66 Bacterial fimbriae are therefore important in modifying and stimulating immune responses in the periodontium.
Bacterial Deoxyribonucleic Acid and Extracellular Deoxyribonucleic Acid
Bacterial deoxyribonucleic acid (DNA) stimulates immune cells via TLR-9, which recognizes hypomethylated CpG regions of the DNA.92 CpG sites are regions of DNA at which a cytosine nucleotide is found next to a guanine nucleotide (separated by a phosphate molecule, which links the C and G nucleotides together, hence “CpG”). Extracellular DNA (eDNA) is likely to play a role in the development and structure of the biofilms formed by oral bacteria and has been identified as an important component of the matrix in a number of bacterial biofilms.167,194 eDNA is derived from the chromosomal DNA of bacteria in biofilms, and the majority of eDNA is released after bacterial cell lysis.2,176 However, there is also evidence that eDNA secretion may occur from bacterial cells by mechanisms that are independent of cell lysis.68,140 The significance of this finding is not yet clear, but such “donated” DNA may be used by bacterial species as a means of increasing genetic diversity (if taken up by other bacteria), thereby contributing to antigenic variation and the spread of antibiotic resistance, and it may modulate the host immune response. Thus eDNA may function as a source of genetic information for naturally transformable bacteria in the biofilm189 and/or as a stimulus for host immunity. Little is known about the role of eDNA in oral biofilms, however. It has been demonstrated that DNA isolated from P. gingivalis, A. actinomycetemcomitans, and Peptostreptococcus micros stimulates macrophages and gingival fibroblasts to produce TNF-α and IL-6 in a dose-dependent manner, and therefore immune stimulation by bacterial DNA from subgingival species could contribute to periodontal pathogenesis.123
Host-Derived Inflammatory Mediators
The inflammatory and immune processes that develop in the periodontal tissues in response to the long-term presence of the subgingival biofilm are protective by intent but result in considerable tissue damage. This has sometimes been referred to as bystander damage, denoting that the host response is mainly responsible for the tissue damage that occurs, leading to the clinical signs and symptoms of periodontal disease. It is paradoxical that the host response causes most of the tissue damage, although this is by no means unique to periodontal disease. For example, the tissue damage that occurs in the joints in rheumatoid arthritis results from prolonged and excessive inflammatory responses and is characterized by increased production of many of the cytokines known to be important in periodontal pathogenesis. In the case of rheumatoid arthritis, the initiating factor is an autoimmune response to structural components of the joint, whereas in periodontitis, the initiating factor is the subgingival biofilm. In both cases, however, the destructive inflammatory events are remarkably similar, although the pathogenesis varies as a result of the different anatomy.
Having understood that the majority of the tissue damage in periodontitis derives from the excessive and dysregulated production of a variety of inflammatory mediators and destructive enzymes in response to the presence of the subgingival plaque bacteria, it is important to review the key types of mediators that orchestrate the host responses. These can be broadly divided into the cytokines, the prostanoids, and the matrix metalloproteinases (MMPs).
Cytokines
Cytokines play a fundamental role in inflammation and are key inflammatory mediators in periodontal disease.159 They are soluble proteins and act as messengers to transmit signals from one cell to another. Cytokines bind to specific receptors on target cells and initiate intracellular signalling cascades resulting in phenotypic changes in the cell via altered gene regulation.17,174 Cytokines are effective in very low concentrations, are produced transiently in the tissues, and primarily act locally in the tissues in which they are produced. Cytokines are able to induce their own expression either in an autocrine or paracrine fashion and have pleiotropic effects (i.e., multiple biologic activities) on a large number of cell types. (Autocrine signalling means that the autocrine agent, in this case cytokines, binds to receptors on the cell that secreted the agent, whereas paracrine signalling affects other nearby cells.) Simply, cytokines bind to cell surface receptors, trigger a sequence of intracellular events that lead ultimately to the production of protein by the target cell, which alters that cell’s behavior, and could result in, for example, increased secretion of more cytokines in a positive feedback cycle leading to inflammation.
Cytokines are produced by a large number of cell types, including infiltrating inflammatory cells such as neutrophils, macrophages, and lymphocytes, and also by resident cells in the periodontium, including fibroblasts and epithelial cells.170 Cytokines signal, broadcast, and amplify immune responses and are fundamentally important in regulating immune-inflammatory responses and in combating infections. However, they also have profound biologic effects that lead to tissue damage in chronic inflammation, and prolonged and excessive production of cytokines and other inflammatory mediators in the periodontium leads to the tissue damage that characterizes the clinical signs of the disease. For example, cytokines mediate connective tissue and alveolar bone destruction through the induction of fibroblasts and osteoclasts to produce proteolytic enzymes (i.e., MMPs) that break down structural components of these connective tissues.12
There is significant overlap and redundancy between the function of individual cytokines, and cytokines do not act in isolation, but rather in flexible and complex networks that involve both proinflammatory and antiinflammatory effects and that bring together aspects of both innate and acquired immunity.8 Cytokines play a key role at all stages of the immune response in periodontal disease. Among the most studied (and probably the most important) cytokines in periodontal pathogenesis are the proinflammatory cytokines IL-1β and TNF-α. Both of these cytokines play a key role in the initiation, regulation, and perpetuation of innate immune responses in the periodontium, resulting in vascular changes and migration of effector cells such as neutrophils into the periodontium as part of a normal immune response to the presence of subgingival bacteria.57
Prostaglandins
The prostaglandins (PGs) are a group of lipid compounds derived from arachidonic acid, a polyunsaturated fatty acid found in the plasma membrane of most cells. Arachidonic acid is metabolized by cyclooxygenase-1 and -2 (COX-1 and COX-2) to generate a series of related compounds called the prostanoids, which includes the PGs, thromboxanes, and prostacyclins. PGs are important mediators of inflammation, particularly prostaglandin E2 (PGE2), which results in vasodilation and induces cytokine production by a variety of cell types. COX-2 is upregulated by IL-1β, TNF-α, and bacterial LPS, resulting in increased production of PGE2 in inflamed tissues. PGE2 is produced by various types of cells and most significantly in the periodontium by macrophages and fibroblasts. PGE2 results in induction of MMPs and osteoclastic bone resorption and has a major role in contributing to the tissue damage that characterizes periodontitis.
Matrix Metalloproteinases
MMPs are a family of proteolytic enzymes that degrade extracellular matrix molecules such as collagen, gelatin, and elastin. They are produced by a variety of cell types, including neutrophils, macrophages, fibroblasts, epithelial cells, osteoblasts, and osteoclasts. The names and functions of key MMPs are shown in Table 21-1. The nomenclature of MMPs has been based on the perception that each enzyme has its own specific substrate, for example, MMP-8 and MMP-1 are both collagenases (i.e., they break down collagen). However, it is now appreciated that MMPs usually degrade multiple substrates, with significant substrate overlap between individual MMPs.70 The substrate-based classification is still used, however, and MMPs can be divided into collagenases, gelatinases/type IV collagenases, stromelysins, matrilysins, membrane-type metalloproteinases, and others.
TABLE 21-1 Classification of Matrix Metalloproteinases
| Group | Enzyme | Name |
|---|---|---|
| Collagenases | MMP-1 | Collagenase 1, fibroblast collagenase |
| MMP-8 | Collagenase 2, neutrophil collagenase | |
| MMP-13 | Collagenase 3 | |
| Gelatinases | MMP-2 | Gelatinase A |
| MMP-9 | Gelatinase B | |
| Stromelysins | MMP-3 | Stromelysin 1 |
| MMP-10 | Stromelysin 2 | |
| MMP-11 | Stromelysin 3 | |
| Matrilysins | MMP-7 | Matrilysin 1, pump-1 |
| MMP-26 | Matrilysin 2 | |
| Membrane-type MMPs | MMP-14 | MT1-MMP |
| MMP-15 | MT2-MMP | |
| MMP-16 | MT3-MMP | |
| MMP-17 | MT4-MMP | |
| MMP-24 | MT5-MMP | |
| MMP-25 | MT6-MMP | |
| Others | MMP-12 | Macrophage elastase |
| MMP-19 | — | |
| MMP-20 | Enamelysin |
MMPs, Matrix metalloproteinases; MT, membrane type.
Adapted from Hannas AR, Pereira JC, Granjeiro JM, et al: Acta Odontol Scand 65:1-13, 2007.
MMPs are secreted in a latent form (inactive) and are activated by the proteolytic cleavage of a portion of the latent enzyme. This is achieved by proteases, such as cathepsin G, produced by neutrophils. MMPs are inhibited by proteinase inhibitors, which have antiinflammatory properties. Key inhibitors of MMPs found in the serum include the glycoprotein α1-antitrypsin and α2-macroglobulin, a large plasma protein produced by the liver that is capable of inactivating a wide variety of proteinases. Inhibitors of MMPs that are found in the tissues include the tissue inhibitors of metalloproteinases (TIMPs), which are produced by many cell types; the most important in periodontal disease is TIMP-1.18 MMPs are also inhibited by the tetracycline class of antibiotics, which has led to the development of sub-antimicrobial formulation of doxycycline as a licensed systemic adjunctive drug treatment for periodontitis that exploits the anti-MMP properties of this molecule (see Chapter 48).
Role of Specific Inflammatory Mediators in Periodontal Disease
Interleukin-1 Family Cytokines
The IL-1 family of cytokines comprises at least 11 members, including IL-1α, IL-1β, IL-1 receptor antagonist (IL-1Ra), IL-18, and IL-33.
IL-1β plays a key role in inflammation and immunity, is closely linked to the innate immune response, and induces the synthesis and secretion of other mediators that contribute to inflammatory changes and tissue damage. For example, IL-1β stimulates the synthesis of PGE2, platelet-activating factor (PAF), and nitrous oxide (NO), resulting in vascular changes associated with inflammation, increasing blood flow to the site of infection or tissue injury. IL-1β is mainly produced by monocytes, macrophages, and neutrophils and also by other cell types such as fibroblasts, keratinocytes, epithelial cells, B-cells, and osteocytes.40 IL-1β increases the expression of ICAM-1 on endothelial cells and stimulates secretion of the chemokine CXCL8 (which is IL-8), thereby stimulating and facilitating the infiltration of neutrophils into the affected tissues. IL-1β also synergizes with other proinflammatory cytokines and PGE2 to induce bone resorption. IL-1β has a role in adaptive immunity, regulates the development of antigen-presenting cells, such as dendritic cells, stimulates IL-6 secretion by macrophages (which in turn activates B-cells), and has been shown to enhance antigen-mediated stimulation of T-cells.13 GCF concentrations of IL-1β are increased at sites affected by gingivitis73 and periodontitis,98 and tissue levels of IL-1β correlate with clinical periodontal disease severity.165 Studies in experimental animals have shown that IL-1β exacerbates inflammation and alveolar bone resorption.87 It is clear from the multiplicity of studies that have investigated this cytokine that IL-1β plays a fundamental role in the pathogenesis of periodontal disease.90
IL-1α is primarily an intracellular protein that is not normally secreted and therefore is not usually found in the extracellular environment or in the circulation.43 Unlike IL-1β, biologically active IL-1α is constitutively expressed and likely mediates inflammation only when released from necrotic cells, acting as an “alarmin” to signal the immune system during cell and tissue damage.16 The precise role of IL-1α in periodontal pathogenesis is not well defined, although studies have reported elevated IL-1α levels in GCF and gingival tissues in patients with periodontitis.139 IL-1α is a potent bone resorbing factor involved in the bone loss that is associated with inflammation.172 It is possible that the measured IL-1α in gingival tissues represents intracellular IL-1α that has been released from damaged or necrotic cells. It is probable that IL-1α plays a role in periodontal pathogenesis, possibly as a signalling cytokine (signalling tissue damage) and contributing to bone resorptive activity.
IL-1Ra has structural homology to IL-1β, and binds to the IL-1 receptor (IL-1R1). However, binding of IL-1Ra does not result in signal transduction, therefore IL-1Ra antagonizes the action of IL-1β.42 IL-1Ra is important in regulating inflammatory responses and can be considered to be an antiinflammatory cytokine. IL-1Ra levels have been reported to be elevated in the GCF and tissues of patients with periodontal disease, suggesting a role in immunoregulation in periodontitis.142
IL-18 interacts with IL-1β and shares many of the pro-inflammatory effects of IL-1β. It is mainly produced by stimulated monocytes and macrophages.63 There is increasing evidence to suggest that IL-18 plays a significant role in inflammation and immunity. IL-18 results in proinflammatory responses, including activation of neutrophils.101 It is chemoattractant for T-cells,88 and it interacts with IL-12 and IL-15 to induce interferon gamma (IFN-γ), thereby inducing T-helper (Th1) cells, which activate cell-mediated immunity.197 Interestingly, in the absence of IL-12, IL-18 induces IL-4 and a Th2 response, which regulates humoral (antibody-mediated) immunity.198 There is very limited direct evidence for a role of IL-18 in periodontal pathogenesis. Oral epithelial cells secrete IL-18 in response to stimulation with LPS,143 and a correlation between GCF IL-18 levels and sulcus depth has been reported.82 IL-18 levels have been reported to be higher than those of IL-1β in patients with periodontitis, suggesting that IL-18, along with IL-1β is predominant in periodontitis lesions.129 Since IL-18 has the ability to induce either Th1 or Th2 differentiation, it is likely to play an important role in periodontal disease pathogenesis.130
Other Interleukin-1 Family Cytokines
Six new members of the IL-1 family (IL-1F) of cytokines have been identified on the basis of their sequence homology, structure, gene location, and receptor binding.4,10 Several of these cytokines were identified by different groups, who gave them a variety of names, and proposals were suggested for renaming all of the IL-1F cytokines in a more consistent manner, as indicated in Table 21-2. Our knowledge of the role of these cytokines in inflammation and immunity is very limited at present, and some of these cytokines may be evolutionarily redundant. IL-1F6, IL-1F8, and IL-1F9 are potential agonists (stimulating proinflammatory responses),19,180 whereas IL-1F5 and IL-1F10 are potential antagonists.19,33,102 IL-1F7 appears to have antiinflammatory action.44 It has five splice variants, and one isoform, IL-1F7b, which is highly expressed by monocytes and upregulated by LPS.24 An intracellular mode of action has been suggested for IL-1F7b, and it translocates to the nucleus of macrophages and may act as a transcriptional modulator, reducing the production of LPS-stimulated proinflammatory cytokines, supporting an antiinflammatory role for this cytokine.161
TABLE 21-2 Nomenclature of Interleukin-1 Family (IL-1F) Cytokines
| Cytokine | Systematic Name | Function |
|---|---|---|
| IL-1α | IL-1F1 | Intracellular protein, proinflammatory, contributes to bone resorption, functions as an intracellular transcriptional regulator. |
| IL-1β | IL-1F2 | Key role in inflammation and innate immunity, synergizes with other proinflammatory mediators, major role in adaptive immunity (regulation of T-cells and myeloid cells), stimulates connective tissue breakdown and bone resorption. |
| IL-1Ra | IL-1F3 | Inhibits the action of IL-1α and IL-1β |
| IL-18 | IL-1F4 | Similar proinflammatory profile to IL-1β, activates neutrophils and synergizes with IL-12 to activate Th1 cells. |
| IL-1F5 | IL-1F5 | Antiinflammatory effects via IL-4 induction, antagonizes IL-1F6 action. |
| IL-1F6 | IL-1F6 | Proinflammatory but restricted expression (e.g., localized to skin). |
| IL-1F7 | IL-1F7 | Antiinflammatory, acts as an intracellular regulator, reducing production of LPS-stimulated proinflammatory cytokines. |
| IL-1F8 | IL-1F8 | Proinflammatory but restricted expression (e.g., localized to skin, synovial tissues). |
| IL-1F9 | IL-1F9 | Proinflammatory but restricted expression (e.g., localized to skin, placenta, esophagus). |
| IL-1F10 | IL-1F10 | Putative antagonist with antiinflammatory action. |
| IL-33 | IL-1F11 | Activation of Th2 cells and mast cells, functions as an intracellular transcriptional regulator but restricted expression (e.g., endothelial cells, smooth muscle cells, fibroblasts). |
These novel IL-1F cytokines have limited tissue expression. For example, the agonists IL-1F6, IL-1F8, and IL-1F9 are mainly expressed in skin.180 Therefore, although the primary cellular sources of IL-1β and IL-18 are hematopoietic cells such as neutrophils, macrophages, monocytes, and lymphocytes, IL-1F5-10 are mainly expressed outside these lineages. At present, there are no data to support a role for IL-1F5-10 in periodontal pathogenesis but given that they are expressed mainly by epithelial cells, it will be interesting to learn whether they may play a role in inflammatory responses in the gingiva. This is relevant given the continual exposure of gingival epithelial cells to bacterial challenge, and these cytokines also have properties similar to the primary cytokines such as IL-1β. For example, LPS results in upregulation of IL-1F6, IL-1F8, and IL-1F9, and these cytokines also stimulate the secretion of IL-6 and IL-8.180 P. gingivalis LPS upregulates IL-1F9 mRNA expression in monocytes, although it does not have an effect on IL-1F6, IL-1F7, IL-1F8, or IL-1F10.10
IL-33 (also known as IL-1F11) is of particular interest as, uniquely among the IL-1 cytokines, it stimulates the production of Th2 cytokines such as IL-5 and IL-13, activates Th2 cells, and plays a role in mast cell development and function.1,77,89,116,149 IL-33 is mainly found in nonimmune cells such as bronchial and arterial smooth muscle cells and epithelial cells from the bronchus.149 It is constitutively expressed in endothelial cells of small and large blood vessels, in the fibroblastic reticular cells of lymphoid tissues, and in epithelial cells.27,117 Our knowledge of the expression of IL-33 in myeloid immune cells is very limited, and there are no data to support a role for IL-33 in periodontal pathogenesis. However, it has been reported that IL-33 activates Th2 cells149 and is chemoattractant for these cells.89 Given that Th2 cells are likely to play a role in the destructive phases of periodontal disease and the balance of T-cell subsets is an important factor in determining disease progression,58 then IL-33 may yet prove to play a role in periodontal pathogenesis.
Tumor Necrosis Factor Alpha
TNF-α is a key inflammatory mediator in periodontal disease and shares many of the cellular actions of IL-1β.64 It plays a fundamental role in immune responses, increases neutrophil activity, and mediates cell and tissue turnover by inducing MMP secretion. TNF-α stimulates the development of osteoclasts and limits tissue repair by induction of apoptosis in fibroblasts. TNF-α is secreted by activated macrophages, as well as other cell types, particularly in response to bacterial LPS. Proinflammatory effects of TNF-α include stimulation of endothelial cells to express selectins that facilitate leukocyte recruitment, activation of macrophage IL-1β production, and induction of PGE2 by macrophages and gingival fibroblasts.133 TNF-α, although possessing similar activity to IL-1β, has a less potent effect on osteoclasts, and is present at lower levels in inflamed gingival tissues than IL-1β.166 GCF levels of TNF-α increase as gingival inflammation develops, and higher levels are found in periodontitis.64,73 The importance of TNF-α (and IL-1β) in periodontal pathogenesis is unquestioned and is highlighted, particularly by studies showing that application of antagonists to IL-1β and TNF-α resulted in an 80% reduction in recruitment of inflammatory cells in proximity to the alveolar bone and a 60% reduction in bone loss.6
Interleukin-6 and Related Cytokines
The cytokines in this group, which include IL-6, IL-11, leukemia-inhibitory factor (LIF), and oncostatin M, share common signalling pathways via signal transducers glycoprotein (gp) 130.74 IL-6 is the most extensively studied of this group and has pleiotropic proinflammatory properties.86 IL-6 secretion is stimulated by cytokines such as IL-1β and TNF-α and it is produced by a range of immune cells, including T-cells, B-cells, macrophages, and dendritic cells, as well as resident cells such as keratinocytes, endothelial cells, and fibroblasts.186 IL-6 is also secreted by osteoblasts and stimulates bone resorption and development of osteoclasts.81,93 IL-6 is elevated in the cells, tissues, and GCF of patients with periodontal disease.56,103 IL-6 may have an influence on monocyte differentiation into osteoclasts and a role in bone resorption in periodontal disease.128 IL-6 also has a key role in the regulation of proliferation and differentiation of B-cells and T-cells (in particular the Th17 subset).86 IL-6 therefore has an important role in periodontal pathogenesis, although less than that of IL-1β or TNF-α.
IL-6 also has many activities outside the immune system, for example, the cardiovascular system and the nervous system. It has an important role in hematopoiesis and in signalling the production of C-reactive protein in the liver. Furthermore, IL-6 stimulates T-cell differentiation and function and is important in the regulation of the balance of T-cell subsets, especially the activation of Th17 cells (a subset of T-cells that produce IL-17), and the balance with regulatory T-cells (Treg cells).14
Prostaglandin E2
The cells primarily responsible for PGE2 production in the periodontium are macrophages and fibroblasts. PGE2 levels are increased in the tissues and in GCF at sites undergoing periodontal attachment loss. PGE2 induces the secretion of MMPs and osteoclastic bone resorption and contributes significantly to the alveolar bone loss seen in periodontitis. PGE2 release from monocytes from patients with severe or aggressive periodontitis is greater than that from monocytes from patients who are periodontally healthy.55,125 A large body of evidence has demonstrated the importance of PGE2 in periodontal pathogenesis, and given that prostaglandins are inhibited by nonsteroidal antiinflammatory drugs (NSAIDs), researchers have investigated the use of NSAIDs as potential host-response modulators in the management of periodontal disease.191,192 However, daily administration for extended periods is necessary for the periodontal benefits to become apparent, and NSAIDs are associated with significant unwanted side effects, including gastrointestinal problems, hemorrhage (from impaired platelet aggregation resulting from inhibition of thromboxane formation), and renal and hepatic impairment. NSAIDs are therefore not indicated as adjunctive treatments for the management of periodontitis.
The prostaglandins, including PGE2, are derived from the COX pathway of arachidonic acid metabolism. There are two main isoforms of the COX enzyme, COX-1 and COX-2. COX-1 is constitutively expressed and has antithrombogenic and cytoprotective functions. COX-2 is induced after stimulation with various cytokines, growth factors, and LPS. Inhibition of COX-1 by nonselective NSAIDs results in the majority of the unwanted effects associated with NSAID usage such as gastrointestinal ulceration and impaired hemostasis. Induction of COX-2 results in the production of elevated quantities of prostaglandins, such as PGE2, and therefore inhibition of COX-2 by NSAIDs that selectively inhibit COX-2 results in a reduction of inflammation without the unwanted effects commonly seen after long-term NSAID use. Preliminary studies in animal models showed that selective COX-2 inhibitors slowed alveolar bone loss,15,78 and human studies confirmed that prostaglandin production in the tissues was modified.187 However, in a dramatic and unfortunate development, the selective COX-2 inhibitors were later identified to be associated with significant and life-threatening adverse events, resulting in several of these drugs being withdrawn from the market.46 The selective COX-2 inhibitors cannot therefore be considered as adjunctive treatments for periodontal disease.
Matrix Metalloproteinases
MMPs are a family of zinc-dependent enzymes that are capable of degrading extracellular matrix molecules, including collagens.18,144 MMPs play a key role in periodontal tissue destruction and are secreted by the majority of cell types in the periodontium, including fibroblasts, keratinocytes, endothelial cells, osteoclasts, neutrophils, and macrophages. In healthy tissues, MMPs are mainly produced by fibroblasts, which produce MMP-1 (also known as collagenase-1), and these have a role in the maintenance of the periodontal connective tissues. Transcription of genes coding for MMPs is upregulated by cytokines such as IL-1β and TNF-α.108 MMP activity is regulated by specific endogenous tissue inhibitors of metalloproteinases (TIMPs) and serum glycoproteins such as α-macroglobulins, which form complexes with active MMPs and their latent precursors.141 TIMPs are produced by fibroblasts, macrophages, keratinocytes, and endothelial cells and are specific inhibitors that bind to MMPs in a 1 : 1 stoichiometry.70 MMPs are also produced by some periodontal pathogens, such as A. actinomycetemcomitans and P. gingivalis, but the relative contribution of these bacterially-derived MMPs to periodontal pathogenesis is small. The great majority of MMP activity in the periodontal tissues derives from infiltrating inflammatory cells.
In healthy periodontal tissues, collagen homeostasis is a controlled process that is mediated extracellularly by MMP-1 (expressed by resident cells, primarily fibroblasts) and intracellularly by a variety of lysosomal acid–dependent enzymes. In inflamed periodontal tissues, excessive quantities of MMPs are secreted by resident cells and the large numbers of infiltrating inflammatory cells, particularly neutrophils, as they migrate through the tissues. As a result, the balance between MMPs and their inhibitors is disrupted, resulting in breakdown of the connective tissue matrix,18,175 and leading to the development of collagen depleted areas within the connective tissues, as described earlier. Neutrophils are key infiltrating cells in periodontitis that accumulate in large numbers in inflamed periodontal tissues (see Figure 21-2). Neutrophils have evolved to respond rapidly and aggressively to external stimuli, such as bacterial LPS, and they release large quantities of destructive enzymes very rapidly. The predominant MMPs in periodontitis, MMP-8 and MMP-9, are secreted by neutrophils62 and are very effective at degrading type 1 collagen, the most abundant collagen type in the periodontal ligament.110 MMP-8 and MMP-9 levels increase with increasing severity of periodontal disease and decrease after treatment.61,62,85 The prolonged and excessive release of large quantities of MMPs in the periodontium leads to significant breakdown of structural components of the connective tissues, contributing to the clinical signs of disease.
MMPs play a fundamental role in connective tissue homeostasis and also disease pathogenesis and possess a wide range of biologic effects that are relevant in periodontitis (Table 21-3). MMPs are important in alveolar bone destruction and are expressed by osteoclasts, which also express cathepsin K. Cathepsin K is a lysosomal cysteine protease that is mainly expressed in osteoclasts and plays a key role in bone resorption and remodelling. This enzyme can catabolize collagen, gelatin, and elastin and can therefore contribute to the breakdown of bone and cartilage.
TABLE 21-3 Biologic Activities of Selected MMPs Relevant to Periodontal Disease
| MMP Type | Enzyme | Biologic Activity |
|---|---|---|
| Collagenases | All | Degrade interstitial collagen (type I, II, and III) Digest ECM and non-ECM molecules |
| MMP-1 | Keratinocyte migration and re-epithelialization Platelet aggregation |
|
| MMP-13 | Osteoclast activation | |
| Gelatinases | All | Degrade denatured collagens and gelatin |
| MMP-2 | Differentiation of mesenchymal cells with inflammatory phenotype Epithelial cell migration Increased bioavailability of MMP-9 |
|
| Stromelysins | All | Digest ECM molecules |
| MMP-3 | Activates pro-MMPs Disrupted cell aggregation Increased cell invasion |
|
| Matrilysins | MMP-7 | Disrupted cell aggregation Increased cell invasion |
| Membrane-type MMPs | All | Digest ECM molecules |
| Activate pro-MMP-2 (except MT4-MMP) | ||
| MT1-MMP | Epithelial cell migration Degrade collagen types I, II and III |
MMPs, Matrix metalloproteinases; MT, membrane type; ECM, extracellular matrix.
Adapted from Hannas AR, Pereira JC, Granjeiro JM, et al: Acta Odontol Scand 65:1-13, 2007.
MMPs are critical for osteoclast access to the resorption site, particularly MMP-9 and MMP-14. MMP-14 is located in the ruffled border of osteoclasts, and osteoblasts and osteocytes (but not osteoclasts) express MMP-13, which is present in resorption lacunae, and functions to remove collagen remnants left over by osteoclasts.70 MMPs also contribute to osteoclast recruitment and activity, by releasing cytokines and RANKL (see later section). MMPs are also important in osteoblastic bone formation, including MMP-2, MMP-9, MMP-13, and MMP-14. MMP-14 also contributes to normal bone homeostasis, and MMP-14–activated transforming growth factor beta (TGF-β) inhibits osteoblast apoptosis.
Increased understanding of the importance of MMPs in periodontal pathogenesis has led to the development of systemic drug therapies to modulate the host inflammatory response by inhibiting MMP levels. Doxycycline has been used for this indication, at sub-antimicrobial doses (20 mg twice daily) that have no antibiotic effect but do demonstrate an anticollagenase effect. Doxycycline, like all the tetracyclines, possesses the ability to downregulate MMPs, and this was recognized as representing a novel treatment strategy for the management of periodontitis. The sub-antimicrobial formulation (20 mg twice daily) has been shown to inhibit collagenase activity in the gingival tissues and GCF of patients with chronic periodontitis,61 and a large number of clinical trials have now confirmed the clinical benefits of using this formulation of doxycycline as an adjunct to periodontal therapy.138 Host-response modulation as a treatment concept for periodontitis is discussed further in Chapter 48.
Chemokines
Chemokines are cytokine-like molecules that are characterized by their chemotactic activity. This activity gave rise to the term chemokine (i.e., they are chemotactic cytokines). Chemokines orchestrate leukocyte recruitment in physiologic and pathologic conditions,20 therefore are important in periodontal pathogenesis, resulting in chemotactic migration of neutrophils through the periodontal tissues toward the site of the bacterial challenge in the periodontal pocket.162 Chemokines play a key role in neutrophil recruitment and recruitment of other adaptive and innate immune cells to the site of immune and inflammatory responses. The chemokines are divided into two sub-families according to structural similarity, the CC and CXC sub-families.160 The chemokine, CXCL8, which is more familiarly known as IL-8, has been demonstrated to be localized in the gingival tissues in areas of plaque accumulation, in the presence of neutrophilic infiltration,177 and has also been found in GCF.111 Interaction between bacteria and keratinocytes results in upregulation of IL-8 and ICAM-1 expression in the gingival epithelium and the development of a chemotactic gradient of these molecules in the gingiva, which stimulates neutrophil migration into the epithelial layers and the gingival sulcus.178,179 Similar chemotactic gradients are also present in the gingiva of periodontally healthy individuals, which suggests a role for this process in the maintenance of periodontal health, and supports the findings of infiltrating neutrophils being present even in clinically healthy tissues.179
It is becoming clear that chemokines play an important role in leukocyte migration in periodontal disease. CCL2 and CCL5 (also known as regulation on activation normal T-cell expressed and secreted [RANTES]) play a role in macrophage migration, and CCL3 (also known as macrophage inflammatory protein-1α [MIP-1α]) and CXCL10 play a role in T-cell migration in inflamed periodontal tissues.162 Chemokines play important roles in immune responses, repair, and inflammation and regulate osteoclast activity by influencing myeloid cell differentiation into osteoclasts, which may be of particular importance in the context of periodontitis.
Antiinflammatory Cytokines
The balance between proinflammatory and antiinflammatory events is crucial in determining disease progression, and it is now clear that individual cytokines do not act in isolation but as part of complex networks of mediators that have different functional activities. Antiinflammatory cytokines include IL-10, TGF-β, IL-1Ra, IL-1F5, and possibly IL-1F10.
The IL-10 family of cytokines have multiple pleiotropic effects and of particular interest possess immunosuppressive properties.32,34 IL-10 is produced by Treg cells, monocytes and B-cells and suppresses cytokine secretion from Th1 cells, Th2 cells, monocytes, and macrophages. The role of IL-10 in periodontal disease has been minimally studied, but animal models support that IL-10 downregulates inflammatory responses. For example, IL-10 knock-out mice are more susceptible to alveolar bone loss than wild-type mice.147 IL-10 is also present in GCF and periodontal tissues.79
TGF-β is a growth factor that functions as a cytokine and has immunoregulatory roles, such as the regulation of T-cell subsets and the action of Treg cells, and it also plays a role in repair and regeneration.195 It has multifunctional roles in various cellular functions, including angiogenesis, synthesis of the extracellular matrix, apoptosis, and inhibition of cell growth. TGF-β levels are higher in the GCF and periodontal tissues of patients with periodontitis and gingivitis than those who are periodontally healthy.69
Linking Pathogenesis to Clinical Signs of Disease
Advanced forms of periodontal disease are characterized by the distressing symptoms of tooth mobility and tooth migration. These result from the loss of attachment between the tooth and its supporting tissues following breakdown of the inserting fibers of the periodontal ligament and resorption of alveolar bone. Having reviewed the histopathology and the inflammatory processes that develop in the periodontal tissues as a result of prolonged accumulation of dental plaque, it is now necessary to link these changes to the structural damage that occurs in the periodontium, leading to the well-defined signs of disease.
It is important to note that even clinically healthy tissues demonstrate signs of inflammation when histologic sections are examined. Thus transmigrating neutrophils are evident in the gingival tissues, moving toward the sulcus for the purpose of eliminating bacteria. If the inflammation becomes more extensive, for example, because of an increase in the bacterial challenge, vasodilation and increased vascular permeability lead to edema of the tissues (as well as erythema), causing gingival swelling, a slight deepening of the sulcus, and further compromising plaque removal. Increased infiltration of inflammatory cells, particularly neutrophils, results in development of collagen-depleted areas below the epithelium and as a result, the epithelium proliferates to maintain tissue integrity.
The epithelium provides a physical barrier to impede the ingress of bacteria and their products and disruption of the epithelial barrier can lead to further bacterial invasion and inflammation. Antimicrobial peptides, termed defensins, are expressed by epithelial cells, and gingival epithelial cells express two human β-defensins (hBD-1 and hBD-2). Furthermore, a cathelicidin class antimicrobial peptide, LL-37, which is found in the lysosomes of neutrophils, is also expressed in skin and gingiva. These antimicrobial peptides are important in determining the outcomes of the host-pathogen interactions at the epithelial barrier.190 The epithelium is therefore more than simply a passive barrier, it also has an active role in innate immunity. 37 Epithelial cells in the junctional and sulcular epithelium are in constant contact with bacterial products and respond to these by secreting chemokines (such as IL-8, CXCL8) to attract neutrophils, which migrate up the chemotactic gradient toward the pocket. Epithelial cells are therefore active in responding to infection and signalling further host responses.
If the bacterial challenge persists, the cellular and fluid infiltration continues to develop and neutrophils and other inflammatory cells soon occupy a significant volume of the inflamed gingival tissues. Neutrophils are key components of the innate immune system and play a fundamental role in maintaining periodontal health despite the constant challenge presented by the plaque biofilm. Neutrophils are protective leukocytes that phagocytose and kill bacteria, and deficiencies in neutrophil functioning result in increased susceptibility to infections in general, as well as periodontal disease.99 Neutrophils also release large quantities of destructive enzymes, such as MMPs, as they migrate through the tissues (particularly MMP-8 and MMP-9), resulting in the breakdown of structural components of the periodontium and development of collagen depleted areas. Neutrophils release their potent lysosomal enzymes, cytokines, and reactive oxygen species (ROS) extracellularly, causing further tissue damage.83 Neutrophil hyperactivity in periodontitis has also been suggested, leading to overproduction of damaging ROS and other mediators.53 Patients with periodontitis have been reported to have neutrophils that demonstrate enhanced enzymatic activity and produce increased levels of ROS.112,113 However, it is not yet clear whether enhanced responsiveness of neutrophils is due to an innate properties of the neutrophils in certain individuals or results from priming by cytokines or bacteria, or a combination of these factors.
It is certainly clear, however, that extracellular release of lysosomal enzymes contributes to continued tissue damage and collagen depletion in the periodontal tissues. Degeneration of fibroblasts limits opportunities for repair, and the epithelium continues to proliferate apically, deepening the pocket further, which is rapidly colonized by the subgingival bacteria. The very first steps in the development of the pocket result from a combination of factors, including detachment of cells at the coronal aspect of the junctional epithelium as those at the apical aspect migrate apically into the collagen-depleted areas and intraepithelial cleavage within the junctional epithelium.105,150,171 Epithelial tissues do not have their own blood supply and must rely on diffusion of nutrients from the underlying connective tissues. Thus, as the epithelium proliferates and thickens, necrosis of epithelial cells that are more distant from the connective tissues can lead to intraepithelial clefts and splits, also contributing to the first stages of pocket formation.
A cycle of chronic inflammation is therefore established in which the presence of subgingival bacteria drives inflammatory responses in the periodontal tissues, characterized by infiltration by leukocytes, release of inflammatory mediators and destructive enzymes, connective tissue breakdown, and breakdown and proliferation of the epithelium in an apical direction. The junctional and pocket epithelium becomes thin and ulcerated and bleeds more readily. The bacteria in the pocket are never fully eliminated, as they are effectively outside the body, but their continued presence drives the destructive inflammatory response. Attempts at effective oral hygiene are rendered more difficult by the deepening of the pocket, and the cycle continues.
Alveolar Bone Resorption
As the advancing inflammatory front approaches the alveolar bone, osteoclastic bone resorption commences.31 This is a protective mechanism to prevent bacterial invasion of the bone, but it ultimately leads to tooth mobility and even tooth loss. Resorption of alveolar bone occurs simultaneously with breakdown of periodontal ligament (PDL) in the inflamed periodontal tissues. There are two critical factors that determine whether bone loss occurs: first, the concentration of inflammatory mediators in the gingival tissues must be sufficient to activate pathways that lead to bone resorption, and second, the inflammatory mediators must penetrate to within a critical distance of the alveolar bone.64
Histologic studies have confirmed that the bone resorbs so that there is always a width of noninfiltrated connective tissue of about 0.5 to 1.0 mm overlying the bone.188 It has also been demonstrated that bone resorption ceases when there is at least a 2.5-mm distance between the site of bacteria in the pocket and the bone.132 Osteoclasts are stimulated by proinflammatory cytokines and other mediators of inflammation to resorb the bone, and the alveolar bone “retreats” from the advancing inflammatory front. Osteoclasts are multinucleate cells formed from osteoclast progenitor cells/macrophages, and osteoclastic bone resorption is activated by a variety of mediators such as IL-1β, TNF-α, IL-6, and PGE2.120 Other mediators that also stimulate bone resorption include LIF, oncostatin M, bradykinin, thrombin, and various chemokines.100
Receptor Activator of Nuclear Factor-κB Ligand/Osteoprotegerin
A key system for controlling bone turnover is the receptor activator of nuclear factor-κB (RANK)/RANK ligand (RANKL)/osteoprotegerin (OPG) system (see Figure 25-3). RANK is a cell surface receptor expressed by osteoclast progenitor cells as well as mature osteoclasts. RANKL is a ligand that binds to RANK and is expressed by bone marrow stromal cells, osteoblasts, and fibroblasts. Binding of RANKL to RANK results in osteoclast differentiation and activation and thus bone resorption. Another ligand that binds to RANK is OPG, produced by bone marrow stromal cells, osteoblasts, and PDL fibroblasts. Thus RANKL and OPG are cytokines that bind to RANK, resulting in cellular responses. However, although RANKL promotes activation and differentiation of osteoclasts, OPG has the opposite effect, inhibiting differentiation of osteoclasts. The balance between OPG and RANKL activity can therefore drive bone resorption or bone formation.
IL-1β and TNF-α regulate the expression of RANKL and OPG, and T-cells express RANKL, which binds directly to RANK on the surfaces of osteoclast progenitors and osteoclasts, resulting in cell activation and differentiation to form mature osteoclasts. In periodontitis, elevated levels of proinflammatory cytokines, such as IL-1β and TNF-α, and increasing numbers of infiltrating T-cells result in activation of osteoclasts via RANK, resulting in alveolar bone loss. It has been reported that levels of RANKL are higher and levels of OPG are lower in sites with active periodontal breakdown compared to sites with healthy gingiva,35 and GCF RANKL:OPG ratios are higher in periodontitis than health.21 It is clear that alterations in the relative levels of these key regulators of osteoclasts play a key role in the bone loss that characterizes periodontal disease.
Resolution of Inflammation
Inflammation is an important defense mechanism to combat the threat of bacterial infection, but inflammatory mechanisms are also key in the development and progression of most chronic diseases associated with aging, including periodontal disease.183 It is also becoming evident that resolution of inflammation (i.e., turning off inflammation) is an active process, regulated by specific mechanisms that restore homeostasis. Furthermore, controlling or augmenting these mechanisms may lead to development of novel treatment strategies for managing chronic diseases such as periodontitis. Therefore, although immune-inflammatory responses to infection and injury are necessary for the survival of the host, inflammatory processes can also lead to tissue damage and chronic disease when they persist inappropriately, or are dysregulated or maladaptive.185
Most pharmacologic approaches to control inflammation that have been investigated to date have focussed on inhibiting inflammation. Rather than attempting to inhibit inflammation it is possible that using agonists to stimulate key mechanisms that resolve inflammation may offer new perspectives for controlling chronic diseases. Recently, the mechanisms that regulate inflammation have begun to be identified.84 Resolution of inflammation is an active process that results in a return to homeostasis, and is mediated by specific molecules including a class of endogenous, proresolving lipid mediators, the lipoxins, resolvins, and protectins.155 These molecules are actively synthesized during the resolution phases of acute inflammation, they are antiinflammatory, and they inhibit neutrophil infiltration. They are also chemoattractants but do not cause inflammation. For example, lipoxins stimulate infiltration by monocytes but without stimulating release of inflammatory cytokines.
Lipoxins
The lipoxins include lipoxin A4 (LXA4) and lipoxin B4 (LXB4). The appearance of these molecules signals the resolution of inflammation.153 Lipoxins are lipoxygenase (LO) -derived eicosanoids and are generated from arachidonic acid. They are highly potent, possessing biologic activity at very low concentrations, and inhibit neutrophil recruitment, chemotaxis, and adhesion.169 Lipoxins also signal macrophages to phagocytose the remnants of apoptotic cells at sites of inflammation, without generating an inflammatory response. Pro-inflammatory cytokines such as IL-1β released during acute inflammation can induce expression of lipoxins, which promote resolution of the inflammatory response.114
Resolvins and Protectins
Resolvins (resolution phase interaction products) are derived from the omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) and are classified as the E series resolvins (RvE) and D series resolvins (RvD), respectively.154 Resolvins inhibit neutrophil infiltration and transmigration, they inhibit the production of proinflammatory mediators, and they have potent antiinflammatory and immunoregulatory effects.156 Resolvins are highly potent and have been shown to reduce neutrophil transmigration by around 50% at concentrations as low as 10 nM.168 Protectins are also derived from DHA. They are produced by glial cells and reduce cytokine expression.80 They also inhibit neutrophil infiltration and have been reported to reduce retinal injury118 and stroke damage.109
Neutrophils play a key role in initiating acute inflammation in response to injury or infection, but uncontrolled, or excessive, or persistent inflammatory responses can lead to chronic disease. Release of proinflammatory mediators such as cytokines and prostanoids exacerbates the tissue damage. The release of endogenous proresolving molecules that “switch off” inflammation and act as a braking signal for neutrophil activity indicates that control of inflammation is an active process, rather than a passive dwindling of proinflammatory signals. These molecules could potentially offer benefit in the management of chronic diseases such as periodontitis. This concept has been tested in animal models of periodontitis.184 In a rabbit model of P. gingivalis/ligature-induced experimental periodontitis, periodontal inflammation was clearly evident after 6 weeks, characterized by collagen breakdown and resorption of alveolar bone. As the experiment progressed beyond 6 weeks, topical resolvin E1 (RvE1), 4 µg/tooth, was applied 3 times per week for a further 6 weeks, whereas the control group continued to receive applications of topical P. gingivalis. In the control group, inflammation continued, leading to increased alveolar bone loss, with large increases in the numbers of osteoclasts, infiltrating neutrophils, and significant collagen breakdown. However, in the animals that received the topical RvE1, the progression of periodontitis was prevented, resolution of inflammation occurred, and the bone loss that had occurred in the first 6 weeks of the study was reversed, with evidence of bone gain in the RvE1-treated animals.72 These experiments suggest potential for a new area of research that could have promise for developing exciting new strategies for managing periodontal disease.
Whereas plaque bacteria have a primary etiologic role in initiating and perpetuating periodontal disease, the dysregulated or excessive inflammatory responses that develop as a result are the primary determinants of the progression of the disease and likely explain most of the inter-individual variation that we see in clinical presentation of disease. Inadequate resolution of inflammation is likely to be an important component of the pathogenesis of periodontitis.184 The endogenous proresolving lipid mediators that resolve inflammation could offer potential for the development of powerful and effective new adjunctive treatments for the management of periodontitis. This would represent yet another paradigm shift in the treatment of this complex disease, with a shift in focus toward resolving inflammation rather than inhibiting aspects of the inflammatory response.
Immune Responses in Periodontal Pathogenesis
The immune system is essential for maintenance of periodontal health and is central to the host response to periodontal pathogens. However, if the immune response is dysregulated, inappropriate, persistent, and/or excessive, then damaging chronic inflammatory responses, such as those observed in periodontal disease, can ensue. The immune response to pathogenic microorganisms involves the integration at the molecular, cellular, and organ level of elements often categorized as being part of the innate immune system or the adaptive immune system. Furthermore, host responses in periodontal disease (and other major human diseases) were until recently represented as a linear progression leading from host recognition of microbial pathogens to innate immune responses dominated by the action of phagocytic neutrophils, culminating in the establishment of adaptive immune responses led by antigen specific effector functions such as cytotoxic T-cells and antibodies. Now, it is widely appreciated that immune responses are examples of complex biologic networks in which pathogen recognition, innate immunity, and adaptive immunity are integrated and mutually dependent.52 This complex network is flexible and dynamic with aspects of positive and negative regulation, as well as feedback control; signals are amplified and broadcast leading to diverse effector functions. Furthermore the immune system is integrated with other systems, including the nervous system, hematopoiesis, and hemostasis, as well as elements of tissue repair and regeneration.121
Observational studies of periodontal tissue inform clinical studies and investigations of animal models and isolated cell and tissue systems have allowed us to identify the elements of the immune response relevant to periodontal disease and to relate these to general principles of immunology.90,131 It is important to appreciate that immune responses, which underpin periodontal disease, have unique facets that must be considered before we can truly rationalize the detailed information we have on individual immune cell functions and their responses to specific periodontal pathogens. Thus we need to understand how the polymicrobial plaque biofilm (as opposed to individual species of periodontal pathogens) interacts with host immune defenses. We need also to appreciate specific immunologic properties that relate to the unique anatomy of the periodontium and the oral mucosal tissue that is integrated with it. We also need to understand how immune responses contribute to the dynamic aspects of periodontal disease, the varying clinical courses and presentations thereof, and how elements of host immunity contribute to tissue destruction, resolution, repair, and regeneration.
Innate Immunity
Defenses against infection comprise a wide range of mechanical, chemical, and microbiologic barriers that prevent pathogens invading the cells and tissues of the body. Saliva, GCF, and the epithelial keratinocytes of the oral mucosa all protect the underlying tissues of the oral cavity and in particular the periodontium. The commensal microflora (e.g., in dental plaque) may also be important in providing protection against infection by pathogenic microorganisms through effective competition for resources and ecologic niches and also by stimulating protective immune responses. The complex microanatomy of the periodontium, including the diversity of specialized epithelial tissues, presents many interesting challenges for the study of the immunopathogenesis of periodontal disease.
If these primary defenses are breached, then the cellular and molecular elements of the innate immune response are activated. Innate immunity refers to the elements of the immune response that are determined by inherited factors (and therefore “innate”), have limited specificity, and are “fixed,” in as much as they do not change or improve during an immune response or as the result of previous exposure to a pathogen. Recognition of pathogenic microorganisms and recruitment of effector cells (e.g., neutrophils) and molecules (e.g., the complement system) are central to effective innate immunity. We now have much information about the specific recognition of periodontal pathogens and the events that lead to activation of neutrophils in the periodontium, which are orchestrated by a diverse range of cytokines, chemokines, and cell surface receptors. In pathologic terms, stimulation of innate immunity leads to a state of inflammation, and an important area of periodontal research is to understand the relationship between innate immunity and periodontal disease as a chronic inflammatory disorder.
If innate immune responses fail to eliminate infection (e.g., in the susceptible host), then the effector cells of adaptive immune response (lymphocytes) are activated. It is important to note that whereas historically the adaptive immune system has been the focus of much research in immunology and biomedical science, more recently the innate immune system has enjoyed something of a renaissance, fueled by an explosion in knowledge of pathogen recognition systems such as the TLRs. In particular, how the innate immune response signals adaptive immunity is the subject of intense research, not least in the discipline of periodontology. Thus it is increasingly appreciated that the immune response functions as a network of interacting molecular and cellular elements in which innate immunity and adaptive (antigen-specific) immunity work together toward a common purpose. Aspects of innate immunity that are relevant to periodontal disease are now considered.
Saliva
Saliva secreted from the three major salivary glands (parotid, submandibular, and sublingual), as well as from the numerous minor salivary glands, has an important role in maintaining oral and dental health. The action of shearing forces associated with saliva flow is important in preventing the attachment of bacteria to the dentition and the oral mucosal surfaces. Human saliva also contains numerous molecular components that contribute to host defenses against bacterial colonization and periodontal disease (Table 21-4). These components include molecules that nonspecifically inhibit formation of the plaque biofilm by inhibiting adherence to oral surfaces and promoting agglutination (e.g., mucins), those that inhibit specific virulence factors (e.g., histatins that neutralize LPS), and those that inhibit bacterial cell growth (e.g., lactoferrin) and may induce cell death.60,97 Saliva also contains specific immunoglobulin A (IgA) antibodies to periodontal pathogens that target specific antigens and inhibit bacterial adherence. Patients with periodontal disease have elevated levels of specific IgA, as well as IgG and IgM, antibodies to periodontal pathogens. However, tooth surfaces coated with a salivary pellicle can provide attachment opportunities for plaque bacteria, thus P. gingivalis can attach to the salivary pellicle via fimbriae.
TABLE 21-4 Constituents of Saliva that Contribute to Innate Immunity
| Saliva Constituent | Host Defense Function |
|---|---|
| Antibodies (e.g., IgA) | Inhibit bacterial adherence, promote agglutination |
| Histatins | Neutralize LPS, inhibit destructive enzymes |
| Cystatins | Inhibit bacterial growth |
| Lactoferrin | Inhibits bacterial growth |
| Lysozyme | Lyses bacterial cell walls |
| Mucins | Inhibits bacterial adherence, promotes agglutination |
| Peroxidase | Neutralizes bacterial hydrogen peroxide |
IgA, Immunoglobulin A; LPS, lipopolysaccharides.
Epithelial Tissues
The epithelial tissues play a key role in host defenses as they are the main site of initial interaction between plaque bacteria and the host and also are the site of invasion of microbial pathogens. The keratinized epithelium of the sulcular and gingival epithelial tissues not only provides a protection for the underlying periodontal tissue but also acts as a barrier against bacteria and their products.11,152 In contrast, the unique microanatomic structure of the junctional epithelium has significant intercellular spaces, is not keratinized, and exhibits a higher cellular turnover rate. These properties render the junctional epithelium permeable, allowing for inward movement of microbes and their products and outward movement of GCF and the cells and molecules of innate immunity. Furthermore, the spaces between the cells of the junctional epithelium widen with inflammation, resulting in increased GCF flow.152
Some species of periodontal bacteria invade host epithelial tissues; at the molecular level the processes of adhesion and invasion are coupled. Studies of the invasion of gingival epithelial cells by P. gingivalis have served as a paradigm for the study of this process; infection of host cells by P. gingivalis involves the action of proteases and cell surface fimbriae.3,96 Invasion by P. gingivalis is initiated through signalling via interaction of bacterial components with surface integrins, PAR-1 and PAR-2 and TLR.67,96,196 This in turn activates intracellular signalling pathways (e.g., mitogen-activated protein kinase [MAPK]) and results in the reorganization of actin filaments and microtubules and a modulation of Ca2+ influx. It is thought that inhibition of host cell apoptosis may facilitate survival of intracellular bacteria and that bacteria in this compartment are afforded protection from elements of the host immune response. Invasion of host cells could therefore be relevant in the spread and persistence of certain periodontal bacteria. Also, in vivo analysis and studies of a three-dimensional (3D)-engineered human oral mucosa model demonstrate that P. gingivalis can migrate through the basement membrane of epithelial layers and invade connective tissue.3 Histologic analysis reveals that, in periodontitis, epithelial cells become more rounded and tend to detach from the underlying connective tissue.152 Proteases break down cell-cell junctions in epithelial tissues by digesting transmembrane proteins and adhesion molecules (e.g., E-cadherin). Microanatomic changes associated with the onset of periodontitis, such as the widening of the spaces between the cells of the junctional epithelium and the development of the pocket epithelium, further facilitate bacterial invasion. Infection through the basement membrane and into the underlying connective tissues is facilitated by bacterial-derived proteases and host proteases derived from infiltrating neutrophils.
At the cellular and molecular level, most in vitro studies of epithelial cell responses to periodontal bacteria have been carried out in primary gingival epithelial cells or various immortalized cell lines derived from oral epithelial tissue, and these studies have provided insight into host-cell responses to periodontal bacteria.3,67,69,196 Epithelial cells also constitutively express antimicrobial peptides (e.g., hBDs and LL-37) and the synthesis and secretion of these molecules is upregulated in response to periodontal bacteria. Neutrophils are also a source of antimicrobial peptides (α-defensins). Antimicrobial peptides are small, polycationic peptides that disrupt bacterial cell membranes and thereby directly kill bacteria with broad specificity.
The different categories of antimicrobial peptide are defined on the basis of structural homology. The α-defensins (e.g., human neutrophil peptides 1-4) are expressed by neutrophils and as such are commonly found in GCF. The hBDs, such as hBD1-3, are expressed in gingival epithelial cells, the salivary glands, and in the tongue, as well as in immune cells such as macrophages and dendritic cells; some hBDs are constitutively expressed and others are only expressed in response to cytokines and bacterial products (e.g., gingipains of P. gingivalis).3,37 A third class of antimicrobial peptides are the cathelicidins, of which LL-37 is the only known peptide in this category expressed in human cells. LL-37 is expressed in high levels in the junctional epithelium but like the hBDs has a widespread expression pattern in the mouth and is found in the salivary glands, tongue, and leukocytes, as well as in the connective tissue. Antimicrobial peptides have more recently assumed greater importance because it is recognized that they have a wider role in regulating innate and adaptive immune responses to infection.41 Thus these molecules have chemokine-like activity, stimulating the chemotaxis of a range of leukocytes involved in innate and acquired immunity. Antimicrobial peptides also stimulate mast cell degranulation and cytokine production and likely have a role in wound healing through their effect on keratinocyte differentiation. Furthermore, there is some interest in their possible role in therapy for oral inflammatory diseases.41
Epithelial cells stimulated with bacterial components and cytokines directly produce MMPs, which contribute to loss of connective tissue. Epithelial cells also secrete a range of cytokines in response to periodontal bacteria (such as P. gingivalis, A. actinomycetemcomitans, F. nucleatum, and P. intermedia), which signal immune responses. These include the proinflammatory cytokines IL-1β, TNF-α, and IL-6, as well as the chemokines IL-8 (CXCL8) and monocyte chemoattractant protein-1 (MCP-1), which serve to signal neutrophil and monocyte migration from the vasculature into periodontal tissue.
In some (but not all) experimental systems, P. gingivalis has been shown to inhibit IL-8; it is suggested that in vivo this effect may have a temporary local immune suppression in the periodontium and facilitate the accumulation and invasion of pathogenic periodontal bacteria and the initiation of periodontitis.39,67 P. gingivalis is an example of one periodontal pathogen with a range of virulence factors that affect host immune defenses,67,96 as indicated in Table 21-5.
TABLE 21-5 Virulence Factors of P. gingivalis that Interact with the Immune System
| Virulence Factor | Effect on Immune System |
|---|---|
| Proteases (gingipains) | Degradation of signalling molecules (CD14) and cytokines (IL-1β, IL-6) |
| Cell invasion capability | Inhibition of IL-8 secretion |
| LPS | Antagonism of the stimulatory effects of LPS from other species; no upregulation of E-selectin |
| Fimbriae | Inhibition of IL-12 secretion in macrophages |
| Cell surface polysaccharide | Resistance to complement |
| Short-chain fatty acids | Induction of apoptosis in host cells |
IL, Interleukin; LPS, lipopolysaccharides.
Gingival Crevicular Fluid
GCF originates from the postcapillary venules of the gingival plexus. It has a flushing action in the gingival crevice but also likely functions to bring the blood components (e.g., neutrophils, antibodies, and complement components) of the host defenses into the sulcus.65 The flow of GCF increases in inflammation, and neutrophils are an especially important element of GCF in health and disease.90
Pathogen Recognition and Activation of Cellular Innate Responses
If plaque bacteria and their products penetrate the periodontal tissues, then specialized “sentinel cells” of the immune system can recognize their presence and signal protective immune responses. Thus macrophages and dendritic cells express a range of pattern recognition receptors (PRRs) that interact with specific molecular structures on microorganisms called microbe- associated molecular patterns (MAMPs) to signal immune responses. Thus innate immune responses are activated that provide immediate protection, and adaptive immunity is also activated with the aim of establishing a sustained antigen-specific defense. Excessive and inappropriate immune responses lead to chronic inflammation and the concomitant tissue destruction associated with periodontal disease. A glossary of terms relevant to periodontal immunobiology is presented in Table 21-6.
TABLE 21-6 Glossary of Terms Relevant to Periodontal Immunobiology
| Term | Definition |
|---|---|
| Cluster of differentiation (CD) | Molecules that define different immune cell lineages, functional phenotypes, and stages of development. |
| CD4 | Characteristic coreceptor molecule of T-helper cells. |
| CD8 | Characteristic coreceptor molecule of cytotoxic T-cells. |
| CD14 | Binds to LPS, facilitating interaction with TLR, exists in soluble and membrane-bound forms. |
| Cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) | Enzymes responsible for synthesis of inflammatory mediators (e.g., PGE2) from arachidonic acid. |
| Complement receptor-3 (CR-3) | Cellular receptor for complement that interacts with P. gingivalis fimbriae in signalling cytokines. |
| CXCL and CCL | Two structurally distinct groups of chemokines, which are small proteins that activate and stimulate the movement of leukocytes. |
| Dendritic cell-specific ICAM-3-grabbing nonintegrins (DC-SIGN) | Cell surface molecule on dendritic cells that has a role in cell-cell interactions and antigen presentation. |
| Extracellular DNA (eDNA) | A component of bacterial biofilms that may activate innate immunity. |
| Gingival crevicular fluid (GCF) | Serum exudate that appears in the gingival sulcus (particularly in periodontitis) and that contains molecular and cellular elements of the immune response. |
| GroEl | Bacterial heat shock proteins that stimulate immune responses. |
| Human β-defensins (hBD) | Antimicrobial peptides synthesized by epithelial cells that kill bacterial cells but have other functions in immune responses. |
| Intercellular adhesion molecule-1 (ICAM-1) | Expressed on endothelial molecules during inflammation and interacts with integrins on neutrophils to facilitate neutrophil movement. |
| Interleukin (IL) | Generic term for cytokines produced by leukocytes that are grouped into families based on structural and functional similarity (e.g., IL-1 family). |
| Lysine-specific gingipain A (KgpA) | Enzyme secreted by the periodontal pathogen P. gingivalis, which is an important virulence factor. |
| Leukemia-inhibitory factor (LIF) | Cytokine of the IL-6 family. |
| Peptide cleavage product of human cathelicidin (LL-37) | An antimicrobial peptide widely expressed in tissues, which kills bacterial cells and has other functions in immune responses. |
| Lipopolysaccharide (LPS) | A bacterial cell surface molecule characteristic of gram-negative bacteria which stimulates innate cells via TLR. |
| Lipoteichoic acid (LTA) | Characteristic molecule of gram-positive bacterial cell walls, which activates innate immune cells via TLR. |
| Lipoxin A4 (LXA4), lipoxin B4 (LXB4) | Arachidonic acid derivatives involved in resolution of inflammation. |
| Microbe-associated molecular pattern (MAMP) | Molecules associated with microorganisms, which activate innate immunity through specific signalling pathways. |
| MD-2 | Part of the LPS receptor complex; binds to TLR as a prerequisite for binding LPS. |
| Matrix metalloproteinases (MMPs) | Family of proteolytic enzymes that degrade extracellular matrix molecules. |
| Natural killer (NK) cells | Large granular lymphocytes, distinct from T-cells and B-cells that are important in innate immunity to intracellular pathogens. |
| Nucleotide oligomerization domain receptors 1 (NOD 1) and (NOD 2) 2 | Intracellular PRRs that interact with intracellular bacteria and stimulate innate immunity. |
| Osteoprotegerin (OPG) | Cytokine-like molecule that binds to RANK but antagonizes the action of RANKL. |
| Protease activated receptors 1 (PAR-1) and 2 (PAR-2) | Cellular receptors, which when activated by bacterial enzymes can signal cytokine secretion. |
| Prostaglandin E2 (PGE2) | Inflammatory mediator derived from arachidonic acid. |
| Pattern-recognition receptors (PRRs) | Integrated membrane proteins and intracellular molecules of immune cells that interact with MAMPs and signal innate immunity. |
| Receptor activator of nuclear factor-κB (RANK) | Osteoclast cell surface receptor important in bone regulation. |
| Receptor activator of nuclear factor-κB ligand | Cytokine-like molecule that is the ligand for RANK. |
| Arginine-specific gingipains A (RgpA) and B (RgpB) | Enzymes secreted by the periodontal pathogen P. gingivalis, which are important virulence factors. |
| T-helper 1 subset (Th1 cells) | A subset of CD4+ T-cells that are characterized by their cytokine secretion profile and that activate cellular immunity. |
| T-helper 2 subset (Th2 cells) | A subset of CD4+ T-cells that are characterized by their cytokine secretion profile and that activate humoral (antibody) immunity. |
| T-helper 17 subset (Th17 cells) | A subset of CD4+ T-cells that are characterized by the secretion of IL-17 and that activate inflammatory responses (e.g., neutrophils). |
| Tissue inhibitors of matrix metalloproteinases (TIMPs) | Molecules concerned with the regulation of MMPs. |
| Toll-like receptors (TLRs) | A class of PRRs that recognize MAMPs, such as LPS, and signal cytokine secretion in innate cells. |
| Tumour necrosis factor alpha (TNF-α) | Classic proinflammatory cytokine that activates innate immunity and shares many actions with IL-1β. |
| Regulatory T-cell subset (Treg cells) | A subset of CD4+ T-cells that are characterized by the secretion of TGF-α and IL-10 and that inhibit immune responses. |
The best studied of the signalling systems involved in recognition of plaque bacteria is the interaction of bacterial LPS with TLRs: P. gingivalis, A. actinomycetemcomitans, F. nucleatum all possess LPS molecules that interact with TLR-4 to activate myeloid immune cells. However, individual species of plaque bacteria have a wide variety of MAMPs, which may interact with PRRs. Studies of P. gingivalis have served as a paradigm for investigations of host-bacteria interactions in periodontal disease at the molecular level. Thus P. gingivalis LPS signal via TLR (predominantly TLR-2), and fimbriae, proteases, and DNA from P. gingivalis are all recognized by host cells through interaction with specific PRRs. A number of nonimmune cells in the periodontium (epithelial cells, fibroblasts) also express PRRs and may recognize and respond to MAMPs from plaque bacteria.
Although the signalling pathways activated by PRRs may be diverse, in general terms they converge to elicit similar host-cell responses in the form of upregulation of cytokine secretion and in the case of antigen-presenting cells, such as dendritic cells, cell differentiation leading to enhanced signalling of the adaptive immune response. Dendritic cells also have C-type lectin receptors (e.g., mannose receptor, langerin, and DC-SIGN) which recognize glycans on pathogens but the role of these interactions in periodontal disease is not known.
Signalling of cytokine responses via PRRs influences innate immunity (e.g., neutrophil activity), adaptive immunity (T-cell effector phenotype), and the development of destructive inflammation (e.g., activation of fibroblasts and osteoclasts). A number of cytokines are particularly important in innate immune signalling, and there is now good evidence that these have a role in immune responses in the periodontium. The archetypal proinflammatory cytokine is IL-1β, which exerts its action directly by activating other cells that express the IL-1R1 receptor (e.g., endothelial cells) or by stimulating the synthesis and secretion of other, secondary mediators such as PGE2 and NO. The effect of IL-1β is amplified via a synergistic action with other cytokines such as TNF-α. Upregulation of ICAM-1 and E-selectin on endothelial cells is central to the migration of neutrophils into the periodontium, and this is stimulated by IL-1β and TNF-α. IL-1β also stimulates the secretion of the chemokine IL-8, which stimulates neutrophil chemotaxis (see next section). IL-1β and TNF-α also activate MMP secretion from fibroblasts and osteoclasts; this may facilitate the movement of neutrophils in the connective tissues and thus protective innate responses but may also ultimately contribute to tissue destruction associated with periodontal disease, along with MMPs from neutrophils.
Other cytokines upregulated as a result of activation of PRRs include IL-6, which influences the development of a number of immune cells, including B-cells and dendritic cells, as well as stimulating osteoclast differentiation and thus bone turnover. Other cytokines provide specific signals that contribute to the development of specific CD4+ T-helper cell subsets (e.g., IL-4, IL-12, and IL-18) (see section on Adaptive Immunity). In addition to cytokines that activate immune responses, other cytokines are upregulated that have a role in immune regulation by suppressing the activity of other cytokines; these include IL-1Ra, IL-10, and TGF-β. Cytokines from T-cell subsets feedback and modify innate immune responses; thus IFNγ from Th1 cells activates macrophages, IL-17 from Th17 cells synergizes with IL-1β and TNF-α to reinforce inflammatory reactions, and IL-10 and TGF-β suppress immune responses. The action of many cytokines produced in the periodontium is not limited to one aspect of host immune responses (cytokines are pleiotropic); for example, it is increasingly recognized that IL-1β stimulates dendritic cell differentiation and has a role in activating Th17 cells.
Neutrophil Function
Although macrophages have phagocytic capability, neutrophils are the “professional” phagocytes critical to clearance of bacteria that may invade host tissues. Neutrophils are a feature of healthy gingival tissues, and there is a significant migration of these cells in the absence of clinical signs of inflammation through the intercellular spaces of the junctional epithelium.90,152 This is part of a “low grade defense” against plaque bacteria and is necessary to prevent inflammation and periodontal tissue damage.152 The importance of neutrophils in maintaining periodontal health is demonstrated clinically by the observations of periodontitis in patients with neutrophil defects71 and the association of periodontitis with experimental immunosuppression in animal models.90
Small foci of other leukocytes (lymphocytes, plasma cells, or macrophages) are also found in the healthy gingiva. A small proportion (1% to 2%) of the intercellular spaces in healthy junctional epithelium are occupied by neutrophils (and other leukocytes at various stages of differentiation); but this can increase to 30% with even modest inflammation. In the inflammatory state there are changes to the local vasculature in the gingiva: high endothelial venules develop from the postcapillary venules of the gingival plexus, which facilitates leukocyte emigration and increases flow of GCF into the pocket.152
Neutrophils migrate from the gingival plexus to the extravascular connective tissue and then into the junctional epithelium via the basement membrane. The presence of a layer of neutrophils in the junctional epithelium forms a host defense barrier between subgingival plaque and gingival tissue. At the molecular level, the interaction of adhesion molecules (e.g., ICAM-1 and LFA-3) on endothelial and epithelial cells with β2 integrins on neutrophils facilitates neutrophil migration.
Indeed there is some evidence from immunohistochemistry for the existence of gradients of IL-8 (a “chemotactic gradient”), as well as gradients of ICAM-1, which purportedly direct the neutrophils from the vasculature into the tissues and to the junctional epithelium.179 Increased leukocytes in the periodontium contribute to the disruption of the junctional epithelium by degradation of basement membrane through protease release and the action of reactive oxygen species. Increased expression of IL-8 and adhesion molecules in inflammation may be the result of direct signalling by bacterial products or by signals from cytokines (e.g., IL-1β and TNF-α) upregulated by plaque bacteria. Acute phase proteins, such as α2-macroglobulin, are increased in periodontal tissues as a result of increased vascular leakage in the inflammatory state, resulting in complement and plasmin activation, which also contributes to host defenses.
The neutrophils that infiltrate the periodontium express Fc receptors and complement receptors commensurate with their function in phagocytosis of opsonized bacteria and bacterial antigens. Significantly, the function of neutrophils is not obviously affected by the anaerobic environment within the periodontal pocket. Within the sulcus itself, 95% of the cells are neutrophils, and since neutrophils are present in both health and disease, the transmigration is thought to be a process distinct from that of GCF flow. The remaining 5% of cells are other leukocyte subtypes possibly passively carried into the sulcus by the flow of GCF. The relative proportions of the different leukocytes are highly variable, possibly reflecting the fact that their presence is the result of a passive process and/or a reflection of the natural fluctuations of different cell types during an immune response. Also, the distribution of leukocytes in periodontal tissues is not even; mononuclear cells predominate in the connective tissue and neutrophils in sulcus. This may be the result of selective mechanism mediated by specific chemokine responses and adhesion molecule interactions.
Adaptive Immunity
Adaptive immunity has evolved to provide a focussed and intense defense against infections that overwhelm innate immune responses in the tissues. Adaptive immunity is particularly important as ecologic, social, and demographic changes, which alter susceptibility to existing and emerging infective microorganisms, outpace the natural evolution of biologic systems. Furthermore, the development of effective vaccination is, along with the identification of antibiotics, perhaps one of the greatest triumphs of medical science; this success is based on knowledge of the elements and principles of adaptive immunity. Although the use of effective vaccines against periodontal pathogens remains a topic of research rather than a therapeutic application, it is widely accepted that understanding of immune responses against periodontal pathogens in its widest sense will eventually yield novel therapeutic strategies.
Adaptive immunity contrasts with innate immunity in the dynamic of the underlying cellular and molecular responses: adaptive immunity is slower and reliant on complex interactions between antigen-presenting cells and T- and B-lymphocytes. A key element is the antigen specificity of the responses that facilitates specific targeting of a diverse range of effector elements, including cytotoxic T-cells and antibodies. Another facet is the ability of adaptive immune responses to improve during exposure to antigen and on subsequent infection events. Our current understanding suggests that the cellular and molecular elements of adaptive immunity are more diverse than those of innate immunity, and although a role for many of these factors in periodontal disease has been identified, our knowledge is far from complete. Innate recognition systems interface with periodontal pathogens in the initial stages of pathogenesis, whereas adaptive immune responses predominate in established disease; however, it is widely accepted that the clinical ebb and flow of periodontal disease is a reflection of fluctuations in adaptive immunity.
The importance of adaptive immune responses in periodontal pathogenesis is endorsed by histologic studies of established lesions in periodontal disease.90,131 The population of leukocytes in the periodontium in gingivitis (i.e., the early stages of responses to the plaque biofilm) and in stable periodontal lesions (i.e., those at which tissue destruction is apparently not progressing) is dominated by T-cells, and these cells are clustered mainly around blood vessels. Cell surface marker studies suggest these cells are activated but not proliferating.59 Also, there is a predominance of the helper T-cell subset (i.e., CD4 expressing T-cells) over the cytotoxic T-cell subset (i.e., CD8 expressing T-cells). These T-cells are considered to be proactively maintaining tissue homeostasis in the face of the microbial challenge of the plaque biofilm.59 In contrast, in active periodontitis, B-cells and plasma cells predominate and are associated with pocket formation and progression of disease.
Antigen-Presenting Cells
A central element of the activation and function of T-cells and B-cells is the presentation of antigen by specialized antigen-presenting cells to T-cells and the development of a specific cytokine milieu that influences the development of T-cells with a particular effector function. Antigen-presenting cells are sentinel cells in mucosal tissues such as the periodontium. These cells detect and take up microorganisms and their antigens, after which they may migrate to lymph nodes and interact with T-cells to present antigen. The periodontium is often compared to other mucosal tissues and the skin in terms of its repertoire of immune cells, and it contains a number of “professional” antigen-presenting cells.36 These include B-cells, macrophages, and at least two types of dendritic cells (dermal dendritic cells and Langerhans cells). These cells naturally express major histocompatibility complex (MHC) Class II molecules necessary for antigen presentation to cognate T-cell receptors and may take up specific antigens and transport them to local lymph nodes, thereby facilitating the activation of specific effector T-cells and the generation of an antigen-specific immune response to periodontal pathogens. Although these cells have been identified in periodontal tissues, have been shown to stimulate antigen specific T-cell responses in experimental systems, and are generally increased in periodontitis, their relative contribution to antigen presentation in vivo remains to be determined. Expression of MHC Class II molecules may be induced in other cells present in the periodontium (e.g., fibroblasts and epithelial cells), which then also take up antigen and present antigen locally in the periodontium.
It is increasingly recognized that engagement of PRRs and in particular TLRs by MAMPs from pathogenic microorganisms is not only central to signalling innate immunity in the form of cytokine upregulation but is also a critical element of activation of antigen-presenting cells and the elaboration of T-cell effector function. Thus TLR activation increases the expression of costimulatory molecules on antigen-presenting cells, which are critical in the interaction of these cells with T-cells. Also, TLR activation enhances antigen uptake and processing. Different antigen-presenting cells process and present antigens by different pathways and mechanisms, and this variation is one of the factors, along with the presence of specific combinations of cytokines, that influences the phenotype of T-cell effector function produced during specific immune responses.59
T-Cells
There are a number of different subsets of thymic lymphocytes (T-cells) that develop in the bone marrow and thymus and migrate to the peripheral tissues to participate in adaptive immune responses. The expression of the cell surface molecules (CD4 or CD8) or particular T-cell antigen receptors (αβ or γδ) broadly defines functional T-cell subsets that emerge from the thymus. The role of T-cells in periodontal disease has been established through immunohistologic studies of disease tissues.157 CD4+ helper T-cells are the predominant phenotype in the stable periodontal lesion, and it is thought that alterations in the balance of effector T-cell subsets within the CD4+ population may lead to progression toward a destructive, B-cell–dominated lesion.59 CD4+ T-cell subsets are defined on the basis of their phenotypic characteristics and effector functions. The nature of the antigen-presenting cells, which present antigen to cognate T-cell receptors on T-cells, and the presence of specific combinations of cytokines and chemokines influence the nature of the CD4+ T-cell effector subset, which develops from naive T-cells (Figure 21-5). CD4+ T-cell subsets are defined by the expression of specific transcription factors and their functional characteristics are associated with their cytokine secretion profile.
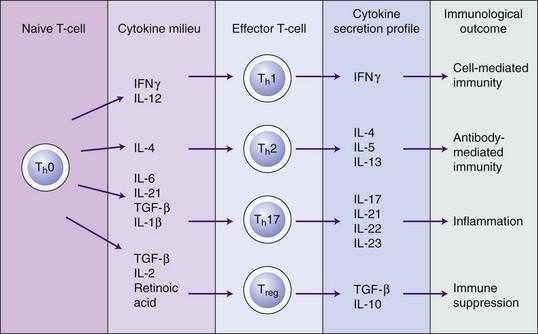
Figure 21-5 CD4+ T-cell subsets and immune responses to microorganisms. Antigen-presenting cells (dendritic cells, macrophages, and B-cells) present specific antigen to naive CD4+ T-cells (Th0 cells), which then differentiate into CD4+ effector T-cells (i.e., Th1, Th2, Th17, and Treg cells) and regulate different aspects of the immune response. Cytokines produced by innate cells in response to microbe-associated molecular patterns (MAMPs) signalling via pattern-recognition receptors (PRRs) influence the nature of the CD4+ effector T-cells that develop from activated Th0 cells. The nature of the antigen-presenting cell and the nature of the antigen itself are also factors that influence effector T-cell function. IL, Interleukin; IFNγ, interferon gamma; TGF-β, transforming growth factor beta.
The best-defined functional subsets of CD4+ T-cells are the Th1 and Th2 cells, and a dynamic interaction between Th1 and Th2 cells may provide an explanation for fluctuations in disease activity and progression of periodontal disease (Box 21-3). Th1 cells secrete IFNγ, which activates cell-mediated immunity (macrophages, natural killer (NK) cells, and CD8+ cytotoxic T-cells) against pathogenic microorganisms. Activation of macrophages promotes phagocytosis and killing of microbial pathogens, whereas NK cells and CD8+ T-cells are cytotoxic T-cells that kill infected host cells. Conversely, Th2 cells regulate humoral (antibody-mediated) immunity and mast cell activity through the secretion of the cytokines IL-4, IL-5, and IL-13. Thus predominance of Th2 cells leads to a B-cell response. The B-cell response may be protective, for example, by the production of specific antibodies that would serve to clear tissue infections through interaction with the complement system and by enhancing neutrophil phagocytosis. However, B-cells are also a source of proinflammatory cytokines that contribute to tissue destruction.
BOX 21-3 The T-Helper 1/T-Helper 2 Concept of Periodontal Disease Progression
A dynamic interaction between T-helper 1 (Th1) and T-helper 2 (Th2) cells represents a possible explanation for aspects of the fluctuations in disease activity and clinical progression seen in periodontal disease. It has been hypothesized that a strong innate response results in interleukin-12 (IL-12) synthesis (e.g., by tissue macrophages), leading to a Th1 response providing protective cell-mediated immunity that would be manifested as a “stable” periodontal lesion. Conversely, a poor innate response would lead to reduced IL-12, which would permit the development of Th2 responses, leading to activation of B-cells that in turn would mediate a destructive lesion possibly through enhanced B-cell–derived IL-1β.59,158 Definitive evidence for the existence of Th1 and Th2 cells and any association with different clinical presentations of periodontal disease has proved difficult to obtain, however. It has been suggested that this is because of the variation in the features of experimental studies, which have differed with respect to the material that has been used (and in particular the definitions of disease stage), the experimental design, and analytic methods employed.54,59,158 Also, in general terms, the Th1/Th2 dichotomy does not explain all aspects of regulation of adaptive immune responses. More recently, other T-cell subsets have been identified and defined. For example, regulatory T-cells (Treg) secrete IL-10 and transforming growth factor beta (TGF-β) and thereby suppress immune responses. Th17 cells have a proinflammatory action via secretion of IL-17, a cytokine that synergizes with IL-1β and tumor necrosis factor alpha (TNF-α). Therefore, although it is widely accepted that Th1 and Th2 cells are likely to be important in the immunopathogenesis of periodontal disease, it is increasingly recognized that the Th1/Th2 model alone is probably inadequate to explain the role of T-cells in this process.
Treg cells have an immunosuppressive action mediated by secretion of TGF-β and are important in preventing autoimmune disease. These cells are increased in periodontitis lesions and may therefore have a role in disease pathogenesis.119 A number of lines of evidence suggest that the pathogenesis of periodontal disease may involve some elements of autoimmunity.59 For example, there is immunologic cross-reactivity between HSP60 expressed on human cells and the GroEL molecule of P. gingivalis, and specific serum antibodies and antigen-specific T-cells to these molecules have been detected in periodontal disease. Similarly autoantibodies and specific T-cells against other host (i.e., self) molecules, such as type I collagen, have been measured in periodontal disease.
Th17 cells are another recently described T-cell subset that has a proinflammatory action important in immune responses against extracellular infections mediated by the cytokine IL-17. Infections with a diverse range of pathogens have been shown to activate strong Th17 cell responses, and Th17 cells are thought to provide a substantial inflammatory response to clear microorganisms that Th1/Th2 cells have failed to eradicate. IL-17 has a number of activities in common with proinflammatory cytokines, such as IL-1β and TNF-α, and has a synergistic activity with these cytokines, particularly TNF-α. IL-17 induces proinflammatory cytokine expression (including IL-1β and TNF-α) in macrophages and stimulates chemokine expression and thereby activates neutrophil infiltration. There is increasing evidence for a role of IL-17 and Th17 cells in periodontal disease.54 IL-17 is detected in periodontal biopsies from disease sites and levels of IL-17 are increased in sites with deep pockets and increased loss of attachment. IL-17 induces IL-6 and IL-8 secretion by gingival fibroblasts and also upregulates MMP-1 and MMP-3 in these cells. IL-17 also induces IL-1β and TNF-α secretion from macrophages and gingival epithelial cells. Significantly, in a mouse model of periodontitis (induced by P. gingivalis infection), IL-17 receptor deficiency (IL-17RA knockouts) resulted in increased susceptibility to alveolar bone loss, suggesting a protective role of IL-17 in bone homeostasis, possibly via an effect on neutrophil function.
A number of other effector CD4+ T-cell subsets have recently been defined based on their cytokine secretion profile: these include Th9 cells and Th22 cells. Also, T-cell subsets have been defined based on their specific anatomic location. For example, Th22 cells home to the skin in which they likely stimulate antimicrobial peptide production and differentiation of keratinocytes. Also, T follicular helper cells (TFH) are located in germinal centers in lymph nodes in which they provide B-cell help and stimulate Ig class switching. The homing of particular T-cell subsets to specific anatomic locations is defined by the expression of specific chemokine receptors which confer responsiveness to specific chemotactic signals which are produced in those locations. However, thus far, the majority of work on these novel T-cell subsets has been carried out in mouse models and in vitro systems; their relevance to human biology in vivo and disease remains to be fully elucidated.
Cytokines produced by differentiated T-cell subsets feedback to stimulate differentiation and sustain the activity of the cells from which they are derived (i.e., in a positive feedback loop). Simultaneously, they inhibit the development of other, competing subsets. For example, IL-4 from Th2 cells inhibits the development of Th1 cells, and IFNγ from Th1 cells inhibits Th2 cells.
It is increasingly appreciated that individual CD4+ T-cell clones, once they have encountered antigen and differentiated in response under the influence of a specific cytokine environment, may not be terminally differentiated cells but rather there is functional flexibility between T-cell subsets and in particular within the memory T-cells population (Figure 21-6). Thus Th17 and Treg cells can interconvert, depending on the local concentrations of IL-6, IL-23, and TGF-β.173 Therefore a picture of functional plasticity in effector T-cells subsets associated with regulating specific immune responses to pathogens is emerging. It is thought that the plethora of functional subsets of T-cells, their anatomic location, and their ability to switch phenotype is a reflection of the requirement for effective responses against diverse pathogens, and this is certainly likely to be a highly relevant facet of immune responses in polymicrobial diseases such as periodontal disease. Other immune cells also have subsets that are defined by expression of cell surface markers and diverse functional and anatomic locations (e.g., myeloid immune cells and NK cells, but these are less well defined than CD4+ T-cell subsets).
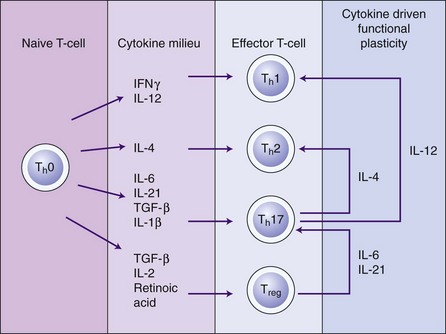
Figure 21-6 Plasticity of T-cell subsets. Although Th1 and Th2 cells have a relatively stable phenotype, other T-cell subsets are functionally “plastic” and can alter phenotype under the influence of different cytokine environments. Thus Th17 cells can develop into Th1 and Th2 cells under the influence of IL-12 and IL-4, respectively. Treg cells can convert into Th17 cells in the presence of IL-6 and IL-21. Other effector T-cell subsets with unique cytokine secretion profiles have been identified (e.g., Th9 and Th22 cells), although these may be transitory and their in vivo relevance remains to be determined. IL, Interleukin; IFNγ, interferon gamma; TGF-β, transforming growth factor beta.
Emerging evidence indicates therefore that periodontal pathogenesis involves complex interactions between a number of interacting T-cell subsets that not only modulate adaptive immune function (Th1, Th2 and Treg cells) but also feedback to modify and enhance innate function (Th17 cells).54 The balance of these different subsets may very well shift and alter longitudinally in periodontal disease. However, the direct relationship (if any) between different T-cell subsets and different clinical presentations remains to be determined.
Antibodies
Specific antibodies are produced in response to an increasing bacterial challenge in periodontal disease and are the endpoint of B-cell activation. Circulating antibodies may be more important than locally produced antibodies, but even so, these generally appear in a high titer but have low biologic activity so there is some doubt as to their effectiveness. Commensurate with the appearance of antibodies against plaque bacterial antigens is the appearance of differentiated plasma cells that characterize the established lesion in periodontal disease. High levels of antibodies appear in GCF (in addition to those in the circulation), and these are produced locally by plasma cells in periodontal tissues.9 Antibodies to periodontal pathogens are primarily IgG with few IgM or IgA types produced.
Many species of oral bacteria elicit a polyclonal B-cell response (with the consequent production of specific antibodies against those bacteria). However, these responses augment responses against nonoral bacteria and may lead to production of autoantibodies (e.g., antibodies against collagen and connective tissue proteins), which may contribute to tissue destruction in periodontal disease.9,59 The incidence and levels of specific serum and GCF IgG antibodies are raised in chronic periodontitis, which suggests that local and peripheral generation of antibodies may be important in the immune response to periodontal pathogens. Antibodies (IgA) to periodontal pathogens are also found in saliva. Variations in the levels of specific antibodies to different species in different clinical presentations suggest differences in pathogenesis. For example, antibodies to A. actinomycetemcomitans of IgG2 subclass predominate in aggressive periodontitis.148
Other P. gingivalis molecules (fimbriae and hemagglutinin) also act as antigens. Specific antibodies are also generated by serotype specific carbohydrate antigens (e.g., capsular polysaccharide of P. gingivalis and carbohydrate of A. actinomycetemcomitans LPS). The subclass distribution of antibodies is influenced by cytokines derived from monocytes.148 For example, IgG2 production is regulated by IL-1α, IL-1β, and PGE2 from monocytes, as well as PAF from neutrophils. PGE2 and PAF indirectly induce Th1 responses and therefore IFNγ, which stimulates IgG2 production. Individuals with aggressive periodontitis have monocytes that are hyper-responsive to LPS and produce elevated quantities of PGE2.9 A. actinomycetemcomitans is commonly associated with aggressive periodontitis, which induces IL-12 production that regulates NK cells and Th1 cells. These cells are a source of IFNγ, which in turn regulates IgG2.
A number of studies have reported an effect of treatment on levels of specific antibodies to periodontal pathogens. For example, plaque removal reduces the titers of antibodies to P. gingivalis and A. actinomycetemcomitans in serum, GCF, and saliva.9 Some studies have observed a transient increase in antibody titers after treatment, which may be due to the release of antigens into the tissue and circulation.
The significance of antibodies in periodontitis is not clear. It is not known if these antibodies have a protective function and whether they participate in disease pathogenesis. Although there is some evidence for a correlation between clinical parameters of disease and titer of specific antibodies to periodontal pathogens, other studies report an inverse correlation of antibody levels and avidity with periodontal destruction. Also, specific antibodies to periodontal pathogens are found in healthy individuals, as well as in those with periodontal disease.
Most research on the analysis of specific antibodies has focussed on antibodies to P. gingivalis, and antigens derived from this organism have been investigated as potential vaccines for periodontal disease.124,136 Thus a significant reduction in disease progression in nonhuman primate and rodent models was observed after immunization with heat-killed P. gingivalis or antigens from P. gingivalis. Also, immunization with P. gingivalis proteases (gingipains) prevents colonization with P. gingivalis and reduces bone loss.124
Concept of Host Susceptibility
The immune and inflammatory processes that result from the challenge presented by the subgingival biofilm are complex, mediated by a large number of proinflammatory and antiinflammatory cytokines and enzymes that function as a network of mediators with overlapping roles and activity. Immune responses to the bacterial challenge do not occur in isolation but take place in the context of other host and environmental factors that influence these responses and thereby determine the progression of disease. A number of risk factors increase susceptibility to periodontal disease, including smoking, systemic diseases such as diabetes, nutritional factors, and stress, and these are considered in detail elsewhere in this book.
A feature of human development and evolution has been that quantitative and qualitative differences exist in immune responses between individuals.75 Indeed, infectious agents (e.g., bacteria) exert evolutionary selection pressures on the species that they infect. This may be relevant in periodontal disease, and a large number of studies have confirmed that immune cells from patients with periodontal disease secrete higher quantities of proinflammatory cytokines than those who are periodontally healthy.174 Cytokine profiles are also different in those individuals with immune-mediated diseases compared to healthy controls.
These observations have led to the development of the concept of the “hyperinflammatory trait,” in which certain individuals possess a hyperinflammatory phenotype and that this accounts for their increased susceptibility to chronic inflammatory conditions such as periodontitis. Such a trait may also underpin shared susceptibility between conditions such as periodontitis and cardiovascular disease or diabetes. Researchers have focused on genetic polymorphisms that may result in this hyperinflammatory trait, and many studies have investigated associations between the presence of single nucleotide polymorphisms (SNPs) in the IL-1 genes and periodontal disease (see Chapter 24). These studies suggest that genetic variants may be identified that confer increased susceptibility to periodontitis. Periodontal disease is a polygenic disease in which many interacting gene variants contribute to disease susceptibility. Given the critical role of cytokines in periodontal pathogenesis, investigation of the cytokine genes and immune regulation continues to be an important area of study.
Therefore, at present, it is not possible to identify with certainty those patients who may possess some form of hyperinflammatory trait. The concept of the periodontal hyper-responder (together with other risk factors) could explain increased susceptibility to periodontal disease.29 The hyper-responder concept was originally proposed in the context of responsiveness of monocytes to LPS challenge, suggesting that patients with disease possess an individual hyper-responsive monocytic trait, characterized by elevated levels of inflammatory mediators released from monocytes in response to bacterial challenge.125 It likely that there are many reasons contributing to disease variation between individuals, such as variations in immune responses, pathogenesis, and the plaque biofilm, resulting in an uneven disease experience in the population.
Figure 21-7 is a schematic illustration of how increasing bacterial (LPS) challenge can result in differing levels of inflammatory response according to the response profile of an individual patient.125 Most individuals would be considered normal and for a given bacterial challenge would produce a certain level of inflammatory mediators in the periodontal tissues. For those who are hyper-responders, the same bacterial challenge results in a greater inflammatory response, which over time would result in increased tissue breakdown, earlier presentation of the clinical signs of disease, and a clinical interpretation of having increased susceptibility to periodontitis. Those individuals who are hypo-responsive produce low levels of inflammatory mediators and are therefore somewhat resistant to developing periodontitis, notwithstanding the fact that plaque may be present and they may have widespread gingivitis. The nature of the inflammatory response will be governed by genetic factors and environmental factors and may vary over time within the same individual (e.g., if environmental factors, such as smoking status, stress, or systemic disease, should change).
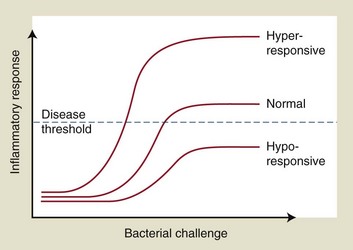
Figure 21-7 Inflammatory response characteristics in relation to bacterial challenge. A given bacterial challenge results in differing levels of inflammatory response according to the response profile of an individual. Most people are close to normal and produce a certain level of inflammatory mediators for a given challenge. Those who are hyper-responders generate an excessive inflammatory response for the same bacterial challenge and cross the threshold into active disease at an earlier stage. Those who are hypo-responsive produce lower levels of inflammatory mediators and despite a significant bacterial challenge, may never develop periodontitis.
(Modified from Champagne CM, Buchanan W, Reddy MS, et al: Periodontol 2000 31:167-180, 2003.)
A similar dose-response curve can also be expressed in the context of stable or progressing disease and as shown in Figure 21-8, a certain level of bacterial challenge results in a moderate release of inflammatory cytokines, mediators, and enzymes. These mediators, together with the infiltrating defense cells have a protective role to eliminate bacteria in the sulcus and do not trigger periodontal disease breakdown. Such a steady-state scenario may persist indefinitely. However, if something changes, such as the quantity or quality of the biofilm alters or the host defenses alter (e.g., as a result of a change in an environmental exposure), then increased secretion of cytokines, prostanoids, MMPs, and other mediators may increase in the tissues, leading to the histopathologic changes described earlier and a transition to periodontitis. There is a threshold therefore between stable and active disease, and this will vary from person to person. The dose-response curve for any individual can shift to the left or the right according to environmental changes. A shift to the left would result in an increase in the quantities of inflammatory mediators produced for a given bacterial challenge and potentially an exacerbation of disease. A shift to the right would have the opposite effect. In all cases, an increase in the LPS challenge would have the tendency to increase the production of inflammatory mediators, which may tip the site from a stable to a progressing periodontal lesion.
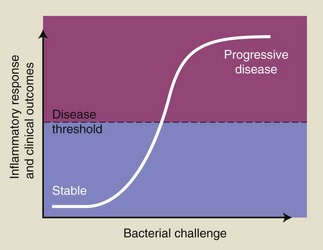
Figure 21-8 Inflammatory response characteristics in relation to threshold for periodontitis. A certain level of bacterial challenge results in a moderate inflammatory response, which is protective by intent, and may not be sufficient to transition to periodontal disease. This stable condition may persist for many years or forever. Changes in the bacterial burden (qualitative and/or quantitative) or changes in the host response (e.g., as a result of change in an environmental exposure), could result in an upregulated inflammatory response characterized by a marked cellular infiltrate and increased secretion of inflammatory mediators leading to tissue damage and a transition from the stable situation to periodontitis. The location of the threshold between stable and active disease varies from person to person. Also, the dose-response curve for any individual can shift to the left or the right according to environmental changes. A shift to the left would result in an increased inflammatory response to a given bacterial challenge and potentially an exacerbation of disease. A shift to the right would have the opposite effect.
(Modified from Champagne CM, Buchanan W, Reddy MS, et al: Periodontol 2000 31:167-180, 2003.)
These are, of course, simplistic models to explain a highly complex phenomenon, and it is clear that cytokines and inflammatory mediators function in complicated networks. Therefore, although increases and decreases in the absolute levels of cytokines have been reported in disease states, it is clear that dysregulation of cytokine networks and other mediators is the key determinant of disease progression. Thus, the relative proportions of mediators within inflammatory networks are fundamental to determining disease progression, and changes in these proportions are driven by the inflammatory challenge and the genetic and environmental factors that govern how the host responds to this challenge. The development and application of techniques to study global gene expression is beginning to provide information to help us understand immune responses on a more holistic basis.69
Schematic illustrations to explain the pathogenesis of periodontal disease are useful, although, given the complexity of the disease processes, they are inevitably simplistic. However, a useful conceptual model of periodontitis was introduced in 1997 (Figure 21-9).134 Earlier models were overly simplistic and were essentially linear, in the first instance suggesting that periodontitis resulted directly from the microbial challenge.91 This concept has influenced periodontology over the decades, resulting in treatment concepts that focussed primarily on the biofilm. Increasing awareness of the importance of host factors in determining inter-individual differences in disease progression led to the model depicted in Figure 21-9, in which, although plaque bacteria initiate the inflammatory response, most of the tissue damage results from the host response, which is influenced by genetic factors and environmental and acquired risk factors. These factors, such as smoking, or genetic risk factors (which are not yet clearly defined) alter the progression of the immune-inflammatory response and shift the balance toward increased periodontal breakdown. This model implies that the presence of plaque bacteria does not inevitably lead to tissue destruction and is supported by a large number of epidemiologic studies, which confirm that more advanced disease is usually confined to a minority of the population.106
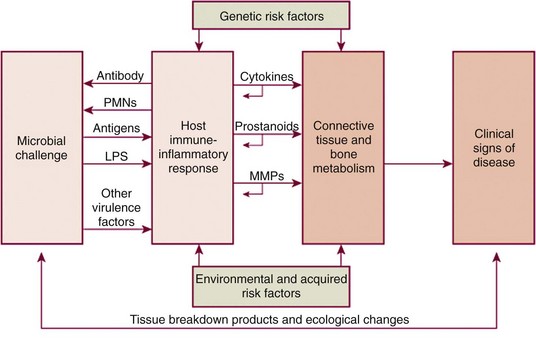
Figure 21-9 Schematic illustration of the pathogenesis of periodontitis. The microbial challenge presented by subgingival plaque bacteria results in an upregulated host immune-inflammatory response in the periodontal tissues that is characterized by the dysregulated and increased production of inflammatory cytokines (e.g., interleukins and tumor necrosis factor alpha), prostanoids (e.g., prostaglandin E2) and enzymes, including the matrix metalloproteinases (MMPs). These upregulated proinflammatory mediators are responsible for the majority of periodontal tissue breakdown that occurs, including alveolar bone resorption via activation of osteoclasts. These changes result over time in the clinical signs of periodontal disease developing. The process is modified by environmental factors such as smoking, and by genetic susceptibility. PMNs, Polymorphonuclear leukocytes; LPS, lipopolysaccharide.
(Modified from Page RC, Kornman KS: Periodontol 2000 14:9-11, 1997.)
The model presented in Figure 21-9 continues to be refined and developed, as new knowledge is obtained about (1) microbial ecologic complexes in the subgingival biofilm and their interactions with the host, (2) links between periodontitis and other complex inflammatory diseases, and (3) increased awareness of the importance of risk factors such as smoking and diabetes. This has led to the development of a biologic systems model for representing periodontal pathogenesis, which involves bacterial components, environmental factors, specific inflammatory mechanisms, and host-genetic variations associated with disease.91 A biologic systems approach provides a framework for viewing the contributions and relative importance of all the components that contribute to the clinical presentation of disease. Thus, in the context of periodontal disease, such a system would include a person level, a genetic/epigenetic level, the biologic phenotype, and ultimately the clinical phenotype (Figure 21-10).126 Such systems will revolutionize conceptual models by creating a more comprehensive view of the disease as a complex regulatory network, in which aspects of the specific genetic factors, environmental exposures, and other modifying factors that an individual is exposed to determine the development of the disease state.
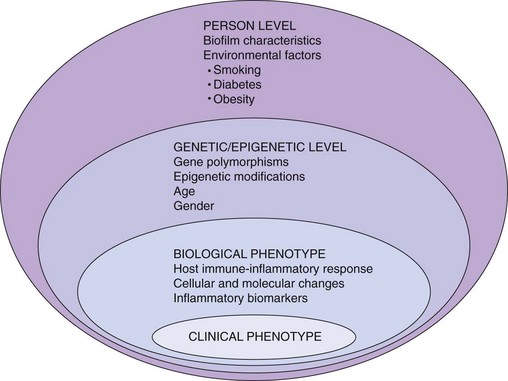
Figure 21-10 A biologic systems model for representing periodontitis. The outermost level of this model is the Person Level, representing an individual’s unique characteristics as they impact on periodontitis. These include the compositional characteristics of the subgingival biofilm, and known risk factors and environmental exposures such as smoking and diabetes. The Person Level characteristics interact with the Genetic/Epigenetic Level characteristics, which include nonmodifiable factors such as age, gender, and genetic composition. Gene polymorphisms are known to be associated with periodontal disease, and epigenetics refers to changes in phenotype (i.e., clinical disease expression) caused by mechanisms other than changes in the underlying DNA sequence. Epigenetics can be defined as all the meiotically and mitotically inherited changes in gene expression that are not encoded in the DNA sequence itself, and epigenetic modifications are important permissive and suppressive factors in controlling the expressed genome via gene transcription. Two major epigenetic mechanisms are posttranslational modification of histone proteins in chromatin and methylation of DNA. The Genetic/Epigenetic Level characteristics influence the Biological Phenotype, which is characterized by the specific immune-inflammatory responses (cellular and molecular events and production of inflammatory mediators) that are associated with the Clinical Phenotype (i.e., the clinical presentation of the disease). This model reflects how different individuals with the same presentation (e.g., periodontitis) may have very different predisposing and risk factors. The model depicts the different biologic factors that underpin the development of periodontal disease in different individuals and ultimately may be used to classify disease according to the contribution provided to the clinical phenotype at each level.
(Modified from Offenbacher S, Barros SP, Beck JD: J Periodontol 79:1577-1584, 2008.)
It is clear that the subgingival bacteria initiate and perpetuate the immune-inflammatory responses in the periodontal tissues. These responses are characterized by classic signs of inflammation that are modified as the result of the unique anatomy of the periodontium and the dentogingival apparatus. The inflammatory events that develop in response to the bacterial challenge are protective by intent but result in the majority of tissue damage and breakdown that lead to the clinical signs of periodontitis. Individuals vary in their susceptibility to periodontal disease and also in the threshold level at which a stable periodontal site progresses to an active site. Such variations are genetically determined and influenced by environmental risk factors, some of which are modifiable and some of which are not. The challenge for the future is to identify at-risk individuals who possess the hyperinflammatory trait so that disease can be prevented by careful management strategies before tissue loss has occurred.
 Science Transfer
Science Transfer
The pathogenesis of gingivitis and periodontitis is a complex molecular phenomenon with many varieties. Thus the exact step-by-step changes that lead from healthy gingiva to gingivitis and periodontitis in any given patient have not been fully elucidated, even though there are accumulated data on many of the theoretical pathways of tissue damage. Gingivitis precedes periodontitis, but not all cases of gingivitis proceed to further destruction of the periodontal tissues, pocket formation, and bone loss.
In the past, the major focus for understanding the pathogenesis of periodontal disease has been the role of anaerobic gram-negative bacteria found in the subgingival biofilms, and periodontal therapy has centered on removing and controlling plaque formation and on using procedures, such as periodontal surgery, to change the milieu so that there are no deep pockets to harbor bacteria and so protect them from oral hygiene plaque removal techniques. Now, it is agreed that the host response involving a variety of cells and inflammatory modulators can be another contributor to the disease process.
Gingival inflammation is based on an initial acute inflammatory reaction coupled with a long-term chronic inflammation. In patients in whom the acute reaction is predominant, there can be a dramatic improvement in redness, swelling, and bleeding on probing and pocket depth when antisubgingival plaque initial therapy is coupled with a high level of oral hygiene. Those patients with more emphasis on chronic inflammatory changes will not show such an obvious clinical improvement with therapy.
Bleeding on probing is often an early sign of gingivitis and continues to be seen as the disease progresses to periodontitis. It is an indication of ongoing connective tissue destruction coupled with vasodilation and ulceration of the gingival lining of the pocket. In the early stages of the disease, plaque control can reverse these inflammatory changes and so patients can reduce and eliminate gingival bleeding in 10 to 14 days. If plaque control becomes inadequate, gingival bleeding can appear as early as 2 days.
New approaches to periodontal treatment couple antibacterial plaque therapy with modulation of the host response. An example of this is the use of low-dose systemic doxycycline coupled with conventional initial therapy. This gives a modest additional therapeutic advantage over conventional treatment, but future host modulation treatment based on counteracting specific tissue destructive molecules may widen and improve the way clinicians can treat these ubiquitous diseases.
1 Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051-2054.
2 Allesen-Holm M, Barken KB, Yang L, et al. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol. 2006;59:1114-1128.
3 Andrian E, Grenier D, Rouabhia M. Porphyromonas gingivalis-epithelial cell interactions in periodontitis. J Dent Res. 2006;85:392-403.
4 Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20-38.
5 Asai Y, Ohyama Y, Gen K, Ogawa T. Bacterial fimbriae and their peptides activate human gingival epithelial cells through Toll-like receptor 2. Infect Immun. 2001;69:7387-7395.
6 Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998;160:403-409.
7 Baba A, Kadowaki T, Asao T, Yamamoto K. Roles for Arg- and Lys-gingipains in the disruption of cytokine responses and loss of viability of human endothelial cells by Porphyromonas gingivalis infection. Biol Chem. 2002;383:1223-1230.
8 Banyer JL, Hamilton NH, Ramshaw IA, Ramsay AJ. Cytokines in innate and adaptive immunity. Rev Immunogenet. 2000;2:359-373.
9 Barbour SE, Tew JG, Schenkein HA. B-cell responses in periodontitis. In: Henderson B, Curtis MA, Seymour R, Donos N, editors. Periodontal medicine and systems biology. Oxford, UK: Wiley-Blackwell; 2009:339-356.
10 Barksby HE, Lea SR, Preshaw PM, Taylor JJ. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin Exp Immunol. 2007;149:1-9.
11 Bartold PM, Walsh LJ, Narayanan AS. Molecular and cell biology of the gingiva. Periodontol 2000. 2000;24:28-55.
12 Bartold PM, Narayanan AS. Molecular and cell biology of healthy and diseased periodontal tissues. Periodontol 2000. 2006;40:29-49.
13 Ben-Sasson SZ, Hu-Li J, Quiel J, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA. 2009;106:7119-7124.
14 Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-238.
15 Bezerra MM, de Lima V, Alencar VB, et al. Selective cyclooxygenase-2 inhibition prevents alveolar bone loss in experimental periodontitis in rats. J Periodontol. 2000;71:1009-1014.
16 Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1-5.
17 Birkedal-Hansen H. Role of cytokines and inflammatory mediators in tissue destruction. J Periodontal Res. 1993;28:500-510.
18 Birkedal-Hansen H. Role of matrix metalloproteinases in human periodontal diseases. J Periodontol. 1993;64:474-484.
19 Blumberg H, Dinh H, Trueblood ES, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204:2603-2614.
20 Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A. Chemokines and chemokine receptors: an overview. Front Biosci. 2009;14:540-551.
21 Bostanci N, Ilgenli T, Emingil G, et al. Gingival crevicular fluid levels of RANKL and OPG in periodontal diseases: implications of their relative ratio. J Clin Periodontol. 2007;34:370-376.
22 Brecx M. Histophysiology and histopathology of the gingiva. J Western Soc Periodontol. 1991;39:33-38.
23 Brecx MC, Frohlicher I, Gehr P, Lang NP. Stereological observations on long-term experimental gingivitis in man. J Clin Periodontol. 1988;15:621-627.
24 Bufler P, Azam T, Gamboni-Robertson F, et al. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci USA. 2002;99:13723-13728.
25 Calkins CC, Platt K, Potempa J, Travis J. Inactivation of tumor necrosis factor-alpha by proteinases (gingipains) from the periodontal pathogen, Porphyromonas gingivalis. Implications of immune evasion. J Biol Chem. 1998;273:6611-6614.
26 Carpenter S, O’Neill LA. How important are Toll-like receptors for antimicrobial responses? Cell Microbiol. 2007;9:1891-1901.
27 Carriere V, Roussel L, Ortega N, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282-287.
28 Cecil RL, Angevine DM. Clinical and experimental observations on focal infection, with an analysis of 200 cases of rheumatoid arthritis. Ann Intern Med. 1938;12:577-584.
29 Champagne CM, Buchanan W, Reddy MS, et al. Potential for gingival crevice fluid measures as predictors of risk for periodontal diseases. Periodontol 2000. 2003;31:167-180.
30 Christersson LA, Albini B, Zambon JJ, et al. Tissue localization of Actinobacillus actinomycetemcomitans in human periodontitis. I. Light, immunofluorescence and electron microscopic studies. J Periodontol. 1987;58:529-539.
31 Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol. 2008;79:1569-1576.
32 Commins S, Steinke JW, Borish L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J Allergy Clin Immunol. 2008;121:1108-1111.
33 Costelloe C, Watson M, Murphy A, et al. IL-1F5 mediates anti-inflammatory activity in the brain through induction of IL-4 following interaction with SIGIRR/TIR8. Journal of Neurochemistry. 2008;105:1960-1969.
34 Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771-5777.
35 Crotti T, Smith MD, Hirsch R, et al. Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J Periodontal Res. 2003;38:380-387.
36 Cutler CW, Jotwani R. Dendritic cells at the oral mucosal interface. J Dent Res. 2006;85:678-689.
37 Dale BA. Periodontal epithelium: a newly recognized role in health and disease. Periodontol 2000. 2002;30:70-78.
38 Darveau RP, Pham TT, Lemley K, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041-5051.
39 Darveau RP. Innate immunity and homeostasis in the periodontium. In: Henderson B, Curtis MA, Seymour R, Donos N, editors. Periodontal medicine and systems biology. Oxford, UK: Wiley-Blackwell; 2009:263-277.
40 Delaleu N, Bickel M. Interleukin-1 beta and interleukin-18: regulation and activity in local inflammation. Periodontol 2000. 2004;35:42-52.
41 Devine D, Cosseau C. Antimicrobial host defence peptides in oral health and periodontitis. In: Henderson B, Curtis MA, Seymour R, Donos N, editors. Periodontal medicine and systems biology. Oxford, UK: Wiley-Blackwell; 2009:279-297.
42 Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16:457-499.
43 Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519-550.
44 Dinarello CA. Interleukin-1beta and the autoinflammatory diseases. New Eng J Med. 2009;360:2467-2470.
45 Dixon DR, Darveau RP. Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid A structure. J Dent Res. 2005;84:584-595.
46 Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Current Pharmaceutical Design. 2006;12:971-975.
47 Edwards AM, Grossman TJ, Rudney JD. Fusobacterium nucleatum transports noninvasive Streptococcus cristatus into human epithelial cells. Infect Immun. 2006;74:654-662.
48 Eskan MA, Hajishengallis G, Kinane DF. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun. 2007;75:892-898.
49 Fives-Taylor P, Meyer D, Mintz K. Virulence factors of the periodontopathogen Actinobacillus actinomycetemcomitans. J Periodontol. 1996;67:291-297.
50 Frank RM. Bacterial penetration in the apical pocket wall of advanced human periodontitis. J Periodontal Res. 1980;15:563-573.
51 Fransson C, Berglundh T, Lindhe J. The effect of age on the development of gingivitis. Clinical, microbiological and histological findings. J Clin Periodontol. 1996;23:379-385.
52 Fraser ID, Germain RN. Navigating the network: signaling cross-talk in hematopoietic cells. Nat Immunol. 2009;10:327-331.
53 Fredriksson MI, Gustafsson AK, Bergstrèom KG, Asman BE. Constitutionally hyperreactive neutrophils in periodontitis. J Periodontol. 2003;74:219-224.
54 Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817-828.
55 Garrison SW, Nichols FC. LPS-elicited secretory responses in monocytes: altered release of PGE2 in patients with but not IL-1 adult periodontitis. J Periodontal Res. 1989;24:88-95.
56 Gemmell E, Seymour GJ. Interleukin 1, interleukin 6 and transforming growth factor-beta production by human gingival mononuclear cells following stimulation with Porphyromonas gingivalis and Fusobacterium nucleatum. J Periodontal Res. 1993;28:122-129.
57 Gemmell E, Marshall RI, Seymour GJ. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol 2000. 1997;14:112-143.
58 Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontol 2000. 2004;35:21-41.
59 Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000. 2007;43:14-40.
60 Giannobile WV, Beikler T, Kinney JS, et al. Saliva as a diagnostic tool for periodontal disease: current state and future directions. Periodontol 2000. 2009;50:52-64.
61 Golub LM, Ciancio S, Ramamurthy NS, et al. Low-dose doxycycline therapy: effect on gingival and crevicular fluid collagenase activity in humans. J Periodontal Res. 1990;25:321-330.
62 Golub LM, Sorsa T, Lee HM, et al. Doxycycline inhibits neutrophil (PMN)-type matrix metalloproteinases in human adult periodontitis gingiva. J Clin Periodontol. 1995;22:100-109.
63 Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol. 2003;73:213-224.
64 Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74:391-401.
65 Griffiths GS. Formation, collection and significance of gingival crevice fluid. Periodontol 2000. 2003;31:32-42.
66 Hajishengallis G, Shakhatreh MA, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359-2367.
67 Hajishengallis G. Porphyromonas gingivalis–host interactions: open war or intelligent guerilla tactics? Microbes Infect. 2009;11:637-645.
68 Hamilton HL, Domâinguez NM, Schwartz KJ, et al. Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Mol Microbiol. 2005;55:1704-1721.
69 Handfield M, Baker HV, Lamont RJ. Beyond good and evil in the oral cavity: insights into host-microbe relationships derived from transcriptional profiling of gingival cells. J Dent Res. 2008;87:203-223.
70 Hannas AR, Pereira JC, Granjeiro JM, Tjaderhane L. The role of matrix metalloproteinases in the oral environment. Acta Odontol Scand. 2007;65:1-13.
71 Hart TC, Shapira L, van Dyke TE. Neutrophil defects as risk factors for periodontal diseases. J Periodontol. 1994;65:521-529.
72 Hasturk H, Kantarci A, Goguet-Surmenian E, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021-7029.
73 Heasman PA, Collins JG, Offenbacher S. Changes in crevicular, leukotriene B (fluid levels of interleukin-14), prostaglandin E2, thromboxane B2 in experimental and tumour necrosis factor gingivitis in humans. J Periodontal Res. 1993;28:241-247.
74 Heinrich PC, Behrmann I, Haan S, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1-20.
75 Hill AV. Immunogenetics. Defence by diversity. Nature. 1999;398:668-669.
76 Hillmann G, Dogan S, Geurtsen W. Histopathological investigation of gingival tissue from patients with rapidly progressive periodontitis. J Periodontol. 1998;69:195-208.
77 Ho LH, Ohno T, Oboki K, et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol. 2007;82:1481-1490.
78 Holzhausen M, Rossa CJr, Marcantonio EJr, et al. Effect of selective cyclooxygenase-2 inhibition on the development of ligature-induced periodontitis in rats. J Periodontol. 2002;73:1030-1036.
79 Honda T, Domon H, Okui T, et al. Balance of inflammatory response in gingivitis and progressive periodontitis lesions. Clin Exp Immunol. 2006;144:35-40.
80 Hong S, Gronert K, Devchand PR, et al. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. 2003;278:14677-14687.
81 Ishimi Y, Miyaura C, Jin CH, et al. IL-6 is produced by osteoblasts and induces bone resorption. J Immunol. 1990;145:3297-3303.
82 Johnson RB, Serio FG. Interleukin-18 concentrations and the pathogenesis of periodontal disease. J Periodontol. 2005;76:785-790.
83 Johnstone AM, Koh A, Goldberg MB, Glogauer M. A hyperactive neutrophil phenotype in patients with refractory periodontitis. J Periodontol. 2007;78:1788-1794.
84 Kantarci A, Hasturk H, Van Dyke TE. Host-mediated resolution of inflammation in periodontal diseases. Periodontol 2000. 2006;40:144-163.
85 Kinane DF, Darby IB, Said S, et al. Changes in gingival crevicular fluid matrix metalloproteinase-8 levels during periodontal treatment and maintenance. J Periodontal Res. 2003;38:400-404.
86 Kishimoto T. Interleukin-6: from basic science to medicine—40 years in immunology. Annu Rev Immunol. 2005;23:1-21.
87 Koide M, Suda S, Saitoh S, et al. In vivo administration of IL-1 beta accelerates silk ligature-induced alveolar bone resorption in rats. J Oral Pathol Med. 1995;24:420-434.
88 Komai-Koma M, Gracie JA, Wei XQ, et al. Chemoattraction of human T cells by IL-18. J Immunol. 2003;170:1084-1090.
89 Komai-Koma M, Xu D, Li Y, et al. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007;37:2779-2786.
90 Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14:33-53.
91 Kornman KS. Mapping the pathogenesis of periodontitis: a new look. J Periodontol. 2008;79:1560-1568.
92 Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709-760.
93 Kurihara N, Bertolini D, Suda T, et al. IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol. 1990;144:4226-4230.
94 Kurita-Ochiai T, Amano S, Fukushima K, Ochiai K. Cellular events involved in butyric acid-induced T cell apoptosis. J Immunol. 2003;171:3576-3584.
95 Kurita-Ochiai T, Seto S, Suzuki N, et al. Butyric acid induces apoptosis in inflamed fibroblasts. J Dent Res. 2008;87:51-55.
96 Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244-1263.
97 Lamont RJ, Jenkinson HF. Subgingival colonization by Porphyromonas gingivalis. Oral Microbiol Immun. 2000;15:341-349.
98 Lee HJ, Kang IK, Chung CP, Choi SM. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. J Clin Periodontol. 1995;22:885-890.
99 Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med. 2000;343:1703-1714.
100 Lerner UH. Inflammation-induced bone remodeling in periodontal disease and the influence of post-menopausal osteoporosis. J Dent Res. 2006;85:596-607.
101 Leung BP, Culshaw S, Gracie JA, et al. A role for IL-18 in neutrophil activation. J Immunol. 2001;167:2879-2886.
102 Lin H, Ho AS, Haley-Vicente D, et al. Cloning and characterization of IL-1HY2, a novel interleukin-1 family member. J Biol Chem. 2001;276:20597-20602.
103 Lin SJ, Chen YL, Kuo MY, Li CL, Lu HK. Measurement of gp130 cytokines oncostatin M and IL-6 in gingival crevicular fluid of patients with chronic periodontitis. Cytokine. 2005;30:160-167.
104 Lindhe J, Rylander H. Experimental gingivitis in young dogs. Scand J Dent Res. 1975;83:314-326.
105 Listgarten MA. Pathogenesis of periodontitis. J Clin Periodontol. 1986;13:418-430.
106 Loe H, Anerud A, Boysen H, Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan labourers 14 to 46 years of age. J Clin Periodontol. 1986;13:431-440.
107 Lourbakos A, Potempa J, Travis J, et al. Arginine-specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect Immun. 2001;69:5121-5130.
108 MacNaul KL, Chartrain N, Lark M, et al. Discoordinate expression of stromelysin, collagenase, and tissue inhibitor of metalloproteinases-1 in rheumatoid human synovial fibroblasts. Synergistic effects of interleukin-1 and tumor necrosis factor-alpha on stromelysin expression. J Biol Chem. 1990;265:17238-17245.
109 Marcheselli VL, Hong S, Lukiw WJ, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807-43817.
110 Mariotti A. The extracellular matrix of the periodontium: dynamic and interactive tissues. Periodontol 2000. 1993;3:39-63.
111 Mathur A, Michalowicz B, Castillo M, Aeppli D. Interleukin-1 alpha, interleukin-8 and interferon-alpha levels in gingival crevicular fluid. J Periodontal Res. 1996;31:489-495.
112 Matthews JB, Wright HJ, Roberts A, et al. Hyperactivity and reactivity of peripheral blood neutrophils in chronic periodontitis. Clin Exp Immunol. 2007;147:255-264.
113 Matthews JB, Wright HJ, Roberts A, et al. Neutrophil hyper-responsiveness in periodontitis. J Dent Res. 2007;86:718-722.
114 McMahon B, Godson C. Lipoxins: endogenous regulators of inflammation. Am J Physiol Renal Physiol. 2004;286:F189-F201.
115 Miller WD. The human mouth as a focus of infection. Dental Cosmos. 1891;33:689-713.
116 Moulin D, Donze O, Talabot-Ayer D, et al. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine. 2007;40:216-225.
117 Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PloS ONE. 2008;3:e3331.
118 Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491-8496.
119 Nakajima T, Ueki-Maruyama K, Oda T, et al. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. 2005;84:639-643.
120 Nanci A, Bosshardt DD. Structure of periodontal tissues in health and disease. Periodontol 2000. 2006;40:11-28.
121 Nathan C. Points of control in inflammation. Nature. 2002;420:846-852.
122 Niederman R, Zhang J, Kashket S. Short-chain carboxylic-acid-stimulated, PMN-mediated gingival inflammation. Crit Rev Oral Biol Med. 1997;8:269-290.
123 Nonnenmacher C, Dalpke A, Zimmermann S, et al. DNA from periodontopathogenic bacteria is immunostimulatory for mouse and human immune cells. Infect Immun. 2003;71:850-856.
124 O’Brien-Simpson NM, Veith PD, Dashper SG, Reynolds EC. Antigens of bacteria associated with periodontitis. Periodontol 2000. 2004;35:101-134.
125 Offenbacher S, Salvi GE. Induction of prostaglandin release from macrophages by bacterial endotoxin. Clin Infect Dis. 1999;28:505-513.
126 Offenbacher S, Barros SP, Beck JD. Rethinking periodontal inflammation. J Periodontol. 2008;79:1577-1584.
127 Ogawa T, Uchida H. A peptide, ALTTE, within the fimbrial subunit protein from Porphyromonas gingivalis, induces production of interleukin 6, gene expression and protein phosphorylation in human peripheral blood mononuclear cells. FEMS Immunol Med Microbiol. 1995;11:197-205.
128 Okada H, Murakami S. Cytokine expression in periodontal health and disease. Crit Rev Oral Biol Med. 1998;9:248-266.
129 Orozco A, Gemmell E, Bickel M, Seymour GJ. Interleukin-1 beta, interleukin-12 and interleukin-18 levels in gingival fluid and serum of patients with gingivitis and periodontitis. Oral Microbiol Immun. 2006;21:256-260.
130 Orozco A, Gemmell E, Bickel M, Seymour GJ. Interleukin 18 and periodontal disease. J Dent Res. 2007;86:586-593.
131 Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976;33:235-249.
132 Page RC, Schroeder HE. Current status of the host response in chronic marginal periodontitis. J Periodontol. 1981;52:477-491.
133 Page RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodontal Res. 1991;26:230-242.
134 Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000. 1997;14:9-11.
135 Payne WA, Page RC, Ogilvie AL, Hall WB. Histopathologic features of the initial and early stages of experimental gingivitis in man. J Periodontal Res. 1975;10:51-64.
136 Persson GR. Immune responses and vaccination against periodontal infections. J Clin Periodontol. 2005;32(Suppl 6):39-53.
137 Potempa J, Pike RN. Corruption of innate immunity by bacterial proteases. J Innate Immun. 2009;1:70-87.
138 Preshaw PM. Host response modulation in periodontics. Periodontol 2000. 2008;48:92-110.
139 Rasmussen L, Hanstrom L, Lerner UH. Characterization of bone resorbing activity in gingival crevicular fluid from patients with periodontitis. J Clin Periodontol. 2000;27:41-52.
140 Renelli M, Matias V, Lo RY, Beveridge TJ. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology. 2004;150:2161-2169.
141 Reynolds JJ, Meickle MC. Mechanisms of connective tissue matrix destruction in periodontitis. Periodontol 2000. 1997;14:144-157.
142 Roberts FA, Hockett RDJr, Bucy RP, Michalek SM. Quantitative assessment of inflammatory cytokine gene expression in chronic adult periodontitis. Oral Microbiol Immun. 1997;12:336-344.
143 Rouabhia M, Ross G, Page N, Chakir J. Interleukin-18 and gamma interferon production by oral epithelial cells in response to exposure to Candida albicans or lipopolysaccharide stimulation. Infect Immun. 2002;70:7073-7080.
144 Ryan ME, Ramamurthy NS, Golub LM. Matrix metalloproteinases and their inhibition in periodontal treatment. Curr Opin Periodontol. 1996;3:85-96.
145 Saglie FR, Newman MG, Carranza FAJr. Pattison GL: Bacterial invasion of gingiva in advanced periodontitis in humans. J Periodontol. 1982;53:217-222.
146 Saglie FR, Marfany A, Camargo P. Intragingival occurrence of Actinobacillus actinomycetemcomitans and Bacteroides gingivalis in active destructive periodontal lesions. J Periodontol. 1988;59:259-265.
147 Sasaki H, Okamatsu Y, Kawai T, et al. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J Periodontal Res. 2004;39:432-441.
148 Schenkein HA, Barbour SE, Tew JG. Cytokines and inflammatory factors regulating immunoglobulin production in aggressive periodontitis. Periodontol 2000. 2007;45:113-127.
149 Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479-490.
150 Schroeder HE. Quantitative parameters of early human gingival inflammation. Arch Oral Biol. 1970;15:383-400.
151 Schroeder HE, Munzel-Pedrazolli S, Page RC. Correlated morphometric and biochemical analysis of gingival tissue in early chronic gingivitis in man. Arch Oral Biol. 1973;18:899-923.
152 Schroeder HE, Listgarten MA. The gingival tissues: the architecture of periodontal protection. Periodontol 2000. 1997;13:91-120.
153 Serhan CN. Lipoxin biosynthesis and its impact in inflammatory and vascular events. Biochim Biophys Acta. 1994;1212:1-25.
154 Serhan CN, Hong S, Gronert K, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025-1037.
155 Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101-137.
156 Serhan CN. Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators. J Periodontol. 2008;79:1520-1526.
157 Seymour GJ, Gemmell E, Reinhardt RA, et al. Immunopathogenesis of chronic inflammatory periodontal disease: cellular and molecular mechanisms. J Periodontal Res. 1993;28:478-486.
158 Seymour GJ, Gemmell E. Cytokines in periodontal disease: where to from here? Acta Odontol Scand. 2001;59:167-173.
159 Seymour GJ, Taylor JJ. Shouts and whispers: an introduction to immunoregulation in periodontal disease. Periodontol 2000. 2004;35:9-13.
160 Sharma M. Chemokines and their receptors: orchestrating a fine balance between health and disease. Cr Rev Biotechn. 2009.
161 Sharma S, Kulk N, Nold MF, et al. The IL-1 family member 7b translocates to the nucleus and down-regulates proinflammatory cytokines. J Immunol. 2008;180:5477-5482.
162 Silva TA, Garlet GP, Fukada SY, et al. Chemokines in oral inflammatory diseases: apical periodontitis and periodontal disease. J Dent Res. 2007;86:306-319.
163 Smith A. Focal infection. JAMA. 1952;150:490-491.
164 Sorkin BC, Niederman R. Short chain carboxylic acids decrease human gingival keratinocyte proliferation and increase apoptosis and necrosis. J Clin Periodontol. 1998;25:311-315.
165 Stashenko P, Fujiyoshi P, Obernesser MS, et al. Levels of interleukin 1 beta in tissue from sites of active periodontal disease. J Clin Periodontol. 1991;18:548-554.
166 Stashenko P, Jandinski JJ, Fujiyoshi P, et al. Tissue levels of bone resorptive cytokines in periodontal disease. J Periodontol. 1991;62:504-509.
167 Steinberger RE, Holden PA. Extracellular DNA in single- and multiple-species unsaturated biofilms. Appl Environ Microb. 2005;71:5404-5410.
168 Sun YP, Oh SF, Uddin J, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. 2007;282:9323-9334.
169 Takano T, Clish CB, Gronert K, et al. Neutrophil-mediated changes in vascular permeability are inhibited by topical application of aspirin-triggered 15-epi-lipoxin A4 and novel lipoxin B4 stable analogues. J Clin Invest. 1998;101:819-826.
170 Takashiba S, Naruishi K, Murayama Y. Perspective of cytokine regulation for periodontal treatment: fibroblast biology. J Periodontol. 2003;74:103-110.
171 Takata T, Donath K. The mechanism of pocket formation. A light microscopic study on undecalcified human material. J Periodontol. 1988;59:215-221.
172 Tanabe N, Maeno M, Suzuki N, et al. IL-1 alpha stimulates the formation of osteoclast-like cells by increasing M-CSF and PGE2 production and decreasing OPG production by osteoblasts. Life Sci. 2005;77:615-626.
173 Tato CM, Cua DJ. Alternative lifestyles of T cells. Nat Immunol. 2008;9:1323-1325.
174 Taylor JJ, Preshaw PM, Donaldson PT. Cytokine gene polymorphism and immunoregulation in periodontal disease. Periodontol 2000. 2004;35:158-182.
175 Tervahartiala T, Pirila E, Ceponis A, et al. The in vivo expression of the collagenolytic matrix metalloproteinases (MMP-2, -8, -13, and -14) and matrilysin (MMP-7) in adult and localized juvenile periodontitis. J Dent Res. 2000;79:1969-1977.
176 Thomas VC, Hiromasa Y, Harms N, et al. A fratricidal mechanism is responsible for eDNA release and contributes to biofilm development of Enterococcus faecalis. Mol Microbiol. 2009;72:1022-1036.
177 Tonetti MS, Imboden MA, Gerber L, et al. Localized expression of mRNA for phagocyte-specific chemotactic cytokines in human periodontal infections. Infect Immun. 1994;62:4005-4014.
178 Tonetti MS. Molecular factors associated with compartmentalization of gingival immune responses and transepithelial neutrophil migration. J Periodontal Res. 1997;32:104-109.
179 Tonetti MS, Imboden MA, Lang NP. Neutrophil migration into the gingival sulcus is associated with transepithelial gradients of interleukin-8 and ICAM-1. J Periodontol. 1998;69:1139-1147.
180 Towne JE, Garka KE, Renshaw BR, et al. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279:13677-13688.
181 Trombelli L, Tatakis DN, Scapoli C, et al. Modulation of clinical expression of plaque-induced gingivitis. II. Identification of “high-responder” and “low-responder” subjects. J Clin Periodontol. 2004;31:239-252.
182 Uehara A, Imamura T, Potempa J, et al. Gingipains from Porphyromonas gingivalis synergistically induce the production of proinflammatory cytokines through protease-activated receptors with Toll-like receptor and NOD1/2 ligands in human monocytic cells. Cell Microbiol. 2008;10:1181-1189.
183 Van Dyke TE. Inflammation and periodontal diseases: a reappraisal. J Periodontol. 2008;79:1501-1502.
184 Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol. 2008;79:1601-1608.
185 Van Dyke TE, Kornman KS. Inflammation and factors that may regulate inflammatory response. J Periodontol. 2008;79:1503-1507.
186 Van Snick J. Interleukin-6: an overview. Annu Rev Immunol. 1990;8:253-278.
187 Vardar S, Baylas H, Huseyinov A. Effects of selective cyclooxygenase-2 inhibition on gingival tissue levels of prostaglandin E2 and prostaglandin F2 and clinical parameters of chronic periodontitis. J Periodontol. 2003;74:57-63.
188 Waerhaug J. The angular bone defect and its relationship to trauma from occlusion and downgrowth of subgingival plaque. J Clin Periodontol. 1979;6:61-82.
189 Wang BY, Chi B, Kuramitsu HK. Genetic exchange between Treponema denticola and Streptococcus gordonii in biofilms. Oral Microbiol Immun. 2002;17:108-112.
190 Weinberg A, Krisanaprakornkit S, Dale BA. Epithelial antimicrobial peptides: review and significance for oral applications. Crit Rev Oral Biol Med. 1998;9:399-414.
191 Williams RC, Jeffcoat MK, Howell TH, et al. Ibuprofen: an inhibitor of alveolar bone resorption in beagles. J Periodontal Res. 1988;23:225-229.
192 Williams RC, Jeffcoat MK, Howell TH, et al. Three-year clinical trial of flurbiprofen treatment of human periodontitis: preliminary analysis. J Dent Res. 1988;67:370. Abstr. 2062
193 Williams RC. Understanding and managing periodontal diseases: a notable past, a promising future. J Periodontol. 2008;79:1552-1559.
194 Wu J, Xi C. Evaluation of different methods for extracting extracellular DNA from the biofilm matrix. Appl Environ Microb. 2009;75:5390-5395.
195 Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210.
196 Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154:2897-2903.
197 Yoshimoto T, Takeda K, Tanaka T, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161:3400-3407.
198 Yoshimoto T, Tsutsui H, Tominaga K, et al. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc Natl Acad Sci USA. 1999;96:13962-13966.
Suggested Readings
Bartold PM, Narayanan AS. Molecular and cell biology of healthy and diseased periodontal tissues. Periodontol 2000. 2006;40:29-49.
Champagne CM, Buchanan W, Reddy MS, et al. Potential for gingival crevice fluid measures as predictors of risk for periodontal diseases. Periodontol 2000. 2003;31:167-180.
Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817-828.
Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontol 2000. 2005;35:21-41.
Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000. 2007;43:14-40.
Handfield M, Baker HV, Lamont RJ. Beyond good and evil in the oral cavity: insights into host-microbe relationships derived from transcriptional profiling of gingival cells. J Dent Res. 2008;87:203-223.
Seymour GJ, Taylor JJ. Shouts and whispers: An introduction to immunoregulation in periodontal disease. Periodontal 2000. 2004;35:9-13.