Chapter 46 Congenital abnormalities
Anticipating the arrival of a healthy baby is every prospective parent’s dream. Sadly for some this dream is shattered when the presence of an abnormality is recognized either prenatally, at birth or in the neonatal period. The incidence of major congenital abnormalities is 2–3% of all births, although this figure is subject to familial, cultural and geographic variations. It is therefore very likely that every practising midwife will at some time in their career be confronted with the challenge of providing appropriate care and support for such babies and their families.
Communicating the news
It is often the midwife who first notices an abnormality in the baby either during the process of the birth or on routine newborn examination. All abnormalities, identified or suspected, should be notified to medical staff but there is sometimes a difference of opinion as to who should communicate the news to the parents.
There is a very strong argument for suggesting that this should be done by the midwife present at the birth. The midwife–client relationship is, or ought to be, one of mutual trust and respect. Honesty is an implicit tenet of such a relationship. It is well recognized that one of the first questions a mother will ask the midwife after the birth is ‘is the baby all right?’ For the midwife to be non-committal or economical with the truth is to betray that trust. It is preferable that the midwife tells both parents sensitively but honestly that she has concerns, and shows them any obvious abnormality in the baby.
Where there is doubt in the midwife’s mind, for example in cases of suspected chromosomal abnormalities, it could be argued that the issue is less clear cut. Discretion could therefore be exercised in the precise form of words used, but the intention of inviting a second opinion should be made clear to the parents. It is advisable that both the parents and the midwife be present when an experienced paediatrician examines the baby and that the midwife be present during any dialogue between the parents and medical staff so that she is aware of exactly what has been said. She is then in a position to clarify or repeat any points that were not fully understood. Opportunities for follow up consultation with the paediatrician should be offered as and when the parents desire. Patience, tact and understanding are prerequisites for midwives caring for these families.
Some abnormalities are slight and cause no further problems for the parents or child, whereas others are profound and cause the subsequent daily care to be fraught with difficulties. Unlike most others, abnormalities involving the face cannot be disguised or hidden and therefore must rate among the most distressing for parents. This is confirmed by Wirt et al (1992) who claim that congenital defects of the head, face and neck often precipitate a major family crisis. The psychological impact on parents of being told or shown, or both, that their baby has a congenital abnormality is often not dissimilar to the grieving process discussed in Chapter 38. Great sensitivity is required on the part of the midwife when showing the baby to the parents for the first time.
Whatever the abnormality it is essential that families receive accurate, consistent and appropriate information about their baby’s condition. Since a comprehensive discussion of every abnormality is clearly not possible, selection has therefore been made of those the midwife is most likely to encounter.
Definition and causes
By definition, a congenital abnormality is any defect in form, structure or function. Identifiable defects can be categorized as follows (Fig. 46.1):
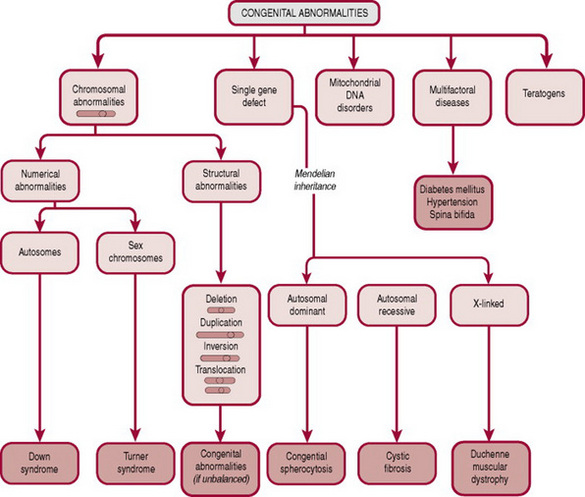
Chromosomal abnormalities
Box 46.1 provides definitions of terms used in this and subsequent sections.
| Karyotyping | The process of identifying chromosomes by size |
| Gamete | Oocyte or spermatozoon |
| Zygote | Formed by gametes combining at fertilization |
| Meiosis | The type of cell reduction division that occurs in the formation of gametes, in which one of each chromosome pair is ‘lost’ |
| Mitosis | The type of cell division that occurs in somatic cells where each new cell gets a full set of chromosomes |
| Aneuploidies | Numerical chromosomal abnormalities |
| Non-disjunction | Failure of a chromosome pair to separate during meiosis or paired chromatids during mitosis |
| Trisomy | A situation where a particular chromosome is represented three times in the nucleus |
| Deletion | Breaking off or loss of part of a chromosome |
| Duplication | Repetition of part of a chromosome |
| Inversion | Rearrangement of part of a chromosome |
| Translocation | Transfer of material from one chromosome to another of a different kind. Should this occur during mitosis, the result will be a balanced or reciprocal translocation where the total chromosomal complement is normal. This would be discovered only during karyotyping; there is no clinical manifestation. If, however, translocation happens during meiosis an unbalanced translocation will result in either an excess or deficit of chromosomal material in the gamete formed. |
Every human cell carries a blueprint for reproduction in the form of 44 chromosomes (autosomes) and two sex chromosomes. Each chromosome comprises a number of genes, which are specific sequences of DNA coding for particular proteins. The zygote should have 22 autosomes and one sex chromosome from each parent. Should a fault occur in either the formation of the gametes or following fertilization (see Ch. 10), abnormalities in chromosome number (aneuploidies) or structure (deletions, duplications, inversions, translocations) may occur. Each abnormal chromosomal pattern has a characteristic clinical presentation, the most common of which will be discussed further.
Gene defects (Mendelian inheritance)
Genes are composed of DNA and each is concerned with the transmission of one specific hereditary factor. Genetically inherited factors may be dominant or recessive.
A dominant gene will produce its effect even if present in only one chromosome of a pair. An autosomal dominant condition can usually be traced through several generations although the severity of clinical expression may vary from generation to generation. Congenital spherocytosis, achondroplasia, osteogenesis imperfecta, adult polycystic kidney disease and Huntington’s chorea are examples of dominant conditions.
A recessive gene needs to be present in both chromosomes before producing its effect. An individual who is carrying only one abnormal copy of the gene (a heterozygote) is unaffected. Examples of autosomal recessive conditions are cystic fibrosis or phenylketonuria.
Some congenital abnormalities are a consequence of single gene defects. In a dominantly inherited disorder the risk of an affected fetus is 1:2 (50%) for each and every pregnancy. In a recessive disorder, the risk is 1:4 (25%) for each and every pregnancy. In an X-linked recessive inheritance the condition affects almost exclusively males, although females can be carriers. X-linked recessive inheritance is responsible for conditions such as haemophilia A and B and Duchenne muscular dystrophy. Spontaneous mutations commonly arise in X-linked recessive disorders. When a woman is a carrier of an X-linked condition, there is a 50% chance of each of her sons being affected and an equal chance that each of her daughters will be carriers.
The recent work on the human genome is likely to clarify further some of these disorders and may offer a way into treatment in the future; for example, polycystic kidney disease (see p. 896) arises from a mutation on chromosome 6 and cystic fibrosis is due to a defect on chromosome 7.
Mitochondrial inheritance
Mitochondria are cellular structures responsible for energy production. Mitochondria are always inherited from the mother. Symptoms and signs of mitochondrial disorders can be diverse but tend to occur in tissues which have high energy requirements such as the brain and muscles. Examples are very rare but include lactic acidosis, encephalopathy and stroke- like episodes (MELAS) and myoclonic epilepsy with myopathy (MERFF).
Teratogenic causes
A teratogen is any agent that raises the incidence of congenital abnormality. The list of known and suspected teratogens is continually growing but includes: prescribed drugs (e.g. anticonvulsants, anticoagulants and preparations containing large concentrations of vitamin A such as those prescribed for the treatment of acne), drugs used in substance abuse (e.g. heroin, alcohol and nicotine), environmental factors such as radiation and chemicals (e.g. dioxins, pesticides), infective agents (e.g. rubella, cytomegalovirus) and maternal disease (e.g. diabetes). It should be borne in mind that several factors influence the effect(s) produced by any one teratogen, such as gestational age of the embryo or fetus at the time of exposure, length of exposure and toxicity of the teratogen. Direct cause and effect is sometimes difficult to establish. Accurate recording of all congenital abnormalities on a central register, such as the Scottish Anomaly Register, facilitates the early recognition of new teratogens.
The role of preconception advice
Although the midwife may advise on modulation of behaviour or diet during pregnancy, by the time the majority of women present for a booking visit the damage has been done. The burden of prevention therefore lies with dissemination of information and appropriate counselling in preconception clinics. The increasing awareness and availability of preconception advice has helped reduce the incidence of some categories of abnormality, notably those associated with poorly controlled diabetes and neural tube defects. Whyte (1995) cites a 72% decrease in neural tube defects as a result of folic acid supplements being taken prior to conception.
Prenatal screening and diagnosis
Improved prenatal screening and diagnostic techniques (see Ch. 18), have led to increased recognition of abnormality, particularly in early pregnancy. This has resulted in some families making the decision to have their pregnancy terminated, while allowing many others time to adjust and try to come to terms with the news that their baby will be born with a particular problem. One advantage of prenatal diagnosis is that, if necessary, arrangements can be made for the mother to have prenatal transfer to a unit where neonatal surgical or intensive care facilities are available. The disadvantage of transfer is that the mother may then be separated from family, friends and the support of the midwives she knows best. This makes it all the more imperative that the staff in these units are sensitive to the needs of such women.
Chromosomal abnormalities
Trisomy 21 (Down syndrome)
The classic features of what is now known as Down syndrome were first described in 1866 by physician John Down (Fig. 46.2). He recognized a commonly occurring combination of facial features among mentally subnormal individuals. His description included features such as widely set and obliquely slanted eyes, small nose and thick rough tongue. In addition to these, it is now accepted that other signs are evident: a small head with flat occiput, squat broad hands with an incurving little finger, a wide space between the thumb and index finger, a single palmar (simian) crease, Brushfield spots in the eyes, and generalized hypotonia. Not all of these manifestations need be present and any of them can occur alone without implying chromosomal aberration. Babies born with Down syndrome also have a higher incidence of cardiac anomalies, cataracts, hearing loss, leukaemia and hypothyroidism. Intelligence quotient is below average, at 40–80.

Figure 46.2 (A) Baby with Down syndrome: note slant of eyes and incurving little finger. (B) With good parental involvement and stimulus these infants can reach maximum potential.
(Photographs courtesy of Scottish Down’s Syndrome Association.)
Although there may be little doubt in the midwife’s mind that a baby has Down syndrome, she should be careful not to make any definitive statements. Family likeness alone may explain some babies’ appearance. Parents themselves may voice their suspicions. If they do not, a sensitive but honest approach should be made by either the midwife or paediatrician to alert them to the possibility and to request permission to conduct further investigations. It is inappropriate to transfer the baby to the special care nursery in order to carry out these investigations under the guise of the baby being cold or sleepy. Investigations indicated include chromosome analysis and echocardiography, because of the increased risk of congenital heart disease. Some centres offer rapid diagnosis (see Ch. 18).
Down syndrome arising sporadically as a result of a non-disjunction process occurs in 95% of cases. Unbalanced translocation occurs in 2.5% of cases, usually between chromosomes 14 and 21. Mosaic forms also occur. There is no difference between the types in clinical appearance. Parents who have a baby with Down syndrome, therefore, should be offered genetic counselling to establish the risk of recurrence. The overall incidence of Down syndrome is 1 in 700.
An individual baby’s needs will vary depending on whether there are any co-existing abnormalities. Apart from any emotional support the mother may require, the midwife may also offer help with feeding. Problems are likely to be encountered because of the baby’s generalized hypotonia. Breastfeeding should be encouraged if the mother had so planned. Providing audiovisual or reading material about Down syndrome for the parents may be helpful, or the address of the local branch of the Down Syndrome Association (see Useful addresses, below).
Trisomy 18 (Edwards syndrome)
This condition is found in about 1 in 5000 births. An extra 18th chromosome is responsible for the characteristic features. The lifespan for these children is short and the majority die during their 1st year. The head is small with a flattened forehead, a receding chin and frequently a cleft palate. The ears are low set and maldeveloped.
The sternum tends to be short, the fingers often overlap each other and the feet have a characteristic rocker-bottom appearance. Malformations of the cardiovascular and gastrointestinal systems are common.
Trisomy 13 (Patau syndrome)
An extra copy of the 13th chromosome leads to multiple abnormalities. These children have a short life. Only 5% live beyond 3 years. Affected infants are small and are microcephalic. Midline facial abnormalities such as cleft lip and palate are common and limb abnormalities are frequently seen. Brain, cardiac and renal abnormalities may coexist with this trisomy.
Turner syndrome (XO)
In this monosomal condition, only one sex chromosome exists: an X. The absent chromosome is indicated by ‘O’. The child is a girl with a short, webbed neck, widely spaced nipples and oedematous feet. The genitalia tend to be underdeveloped and the internal reproductive organs do not mature. The condition may not be diagnosed until puberty fails to occur. Congenital cardiac defects may also be found. Mental development is usually normal.
Gastrointestinal malformations
Most of the abnormalities affecting this system call for prompt surgical intervention, for example atresias, gastroschisis and exomphalos. With the increasing use of routine ultrasound screening at 18–20 weeks’ gestation many are likely to be diagnosed prenatally (Haddock et al 1996). If prenatal diagnosis has been made, the parents will be at least partially prepared. In this event, where possible, the paediatric team of neonatal paediatrician and paediatric surgeon should speak to the parents before birth of the baby to explain the probable sequence of events. Once the baby is born, prior to obtaining their consent for surgery, the paediatric surgeon should have a full discussion with the parents. If the baby’s condition allows, the parents should be encouraged to hold the baby and photographs should be taken.
Gastroschisis and exomphalos
Gastroschisis (Fig. 46.3) is a paramedian defect of the abdominal wall with extrusion of bowel that is not covered by peritoneum, thus making it very vulnerable to infection and injury. Surgical closure of the defect is usually possible; the size of the defect will determine whether primary closure is possible or whether a temporary silo made from synthetic materials (e.g. Silastic) is necessary until the abdominal cavity is able to contain all the abdominal organs.
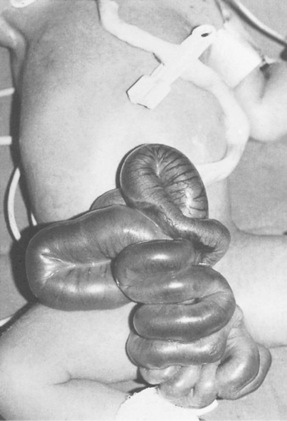
Figure 46.3 Gastroschisis showing prolapsed intestine to the right of umbilical cord.
(From Rennie & Roberton 1999, with permission of Churchill Livingstone.)
Exomphalos or omphalocele (Fig. 46.4) is a defect in which the bowel or other viscera protrude through the umbilicus. Very often these babies have other abnormalities, for example heart defects, which could be a contraindication to surgery in the immediate neonatal period. Closure of the defect may consequently be delayed as long as 1 or even 2 years.
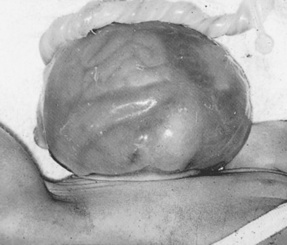
Figure 46.4 Omphalocele defect with bowel visible through sac in the lower part and abnormally lobulated liver in the sac in the upper part.
(From Rennie & Roberton 1999, with permission of Churchill Livingstone.)
The immediate management of both the above conditions is to cover the herniated abdominal contents with clean cellophane wrap (e.g. Clingfilm) or warm sterile saline swabs to reduce fluid and heat losses and to give a degree of protection. An orogastric or nasogastric tube should be passed and stomach contents aspirated. Transfer of the baby to a surgical unit is then expedited.
Atresias
Oesophageal atresia
Oesophageal atresia (Fig. 46.5) occurs when there is incomplete canalization of the oesophagus in early intrauterine development. It is commonly associated with tracheo–oesophageal fistula, which connects the trachea to the upper or lower oesophagus, or both. The commonest type of abnormality is where the upper oesophagus terminates in a blind upper pouch and the lower oesophagus connects to the trachea. This abnormality should be suspected in the presence of maternal polyhydramnios and should be screened for after birth in all such affected pregnancies. At birth the baby has copious amounts of mucus coming from the mouth. Early detection is essential. The midwife should attempt to pass a wide orogastric tube but it may travel less than 10–12 cm. Radiography will confirm the diagnosis. The baby must be given no oral fluid but a wide bore oesophageal tube should be passed into the upper pouch and connected to gentle continuous suction apparatus. Usually a double lumen 10 fg (Replogle) tube is used and the baby nursed head up. He should be transferred immediately to a paediatric surgical unit, ensuring that continuous suction is available throughout the transfer. It may be possible to anastomose the blind ends of the oesophagus. If the gap in the oesophagus is too large a series of bouginages can be carried out in an attempt to stretch the ends of the oesophagus, stimulate growth and thereby eventually facilitate repair by end-to-end anastomosis. Alternatively transplant of, for example, a section of colon may be needed at a later date. Rarely, if the repair is delayed, cervical oesophagostomy may be performed to allow drainage of secretions. Meanwhile the baby will need to be fed via a gastrostomy tube. This method of feeding obviously deprives the baby of oral stimuli. Such a baby may be given ‘sham’ feeds to allow him to taste the milk and to promote sucking, swallowing and normal development of the mandible.
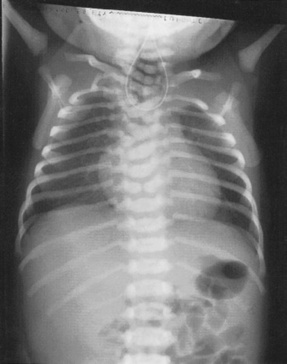
Duodenal atresia
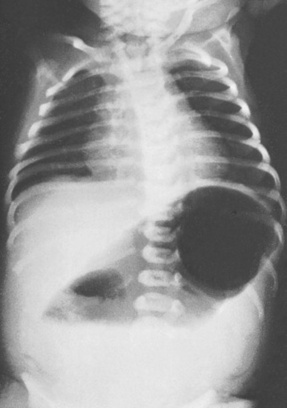
Atresia may occur at any level of the bowel but the duodenum is the most common site. If this has not already been diagnosed in the prenatal period, persistent vomiting within 24–36 hrs of birth will be the first feature encountered. The vomit will often contain bile unless the obstruction is proximal to the entrance of the common bile duct. Abdominal distension may not be present and the baby may pass meconium. A characteristic double bubble of gas may be seen on radiological examination (Fig. 46.6). Treatment is by surgical repair. Prognosis is good if the baby is otherwise healthy, but this abnormality is often associated with others; 30% of cases occur in children with Down syndrome.
Rectal atresia and imperforate anus
Careful examination of the perineum is an important aspect of any newborn examination. An imperforate anus should be obvious at birth on examination of the baby, but a rectal atresia might not become apparent until it is noted that the baby has not passed meconium. Occasionally, for the unwary, the passage of meconium through a fistulous connection to the vagina, bladder or urethra may mask an imperforate anus (see Plates 32, 33 & 34). Whatever the anatomical arrangement, all babies should be referred for surgery.
Should a baby fail to pass meconium in the first 24 hrs, three other possibilities should be considered:
Malrotation/volvulus
This is a developmental abnormality where incomplete rotation of the small bowel has taken place, giving rise to signs of obstruction. There is often bilious vomiting and abdominal distension.
Surgical assessment and correction are necessary. Because of the risks of severe bowel damage secondary to the obstruction of blood flow in the mesentery in unrecognized malrotation, any newborn infant with bile-stained vomiting requires urgent medical assessment.
Meconium ileus (cystic fibrosis)
Some 15% of children with cystic fibrosis present with meconium ileus in the neonatal period. Cystic fibrosis is an autosomal recessive condition affecting 1 in 2500 births. In the past, the majority of cases were not diagnosed until later in infancy or childhood when the child failed to thrive or had repeated chest infections. In meconium ileus the meconium is particularly viscous and causes intestinal obstruction. There is accompanying abdominal distension and bile-stained vomiting. Intravenous fluids and a Gastrografin enema may relieve the obstruction. Definitive diagnosis may be difficult initially, but the presence of a raised immunoreactive trypsin level (IRT) and of markers in the serum using the polymerase chain reaction (PCR) may demonstrate the ΔF508 deletion in both copies of the cystic fibrosis gene on chromosome 7. Histology of any resected bowel may also indicate the likelihood of cystic fibrosis, but a definitive diagnosis is not usually possible until a sweat test has been carried out at 4–6 weeks of age. Treatment of cystic fibrosis is supportive rather than curative and involves administration of pancreatic enzymes and a rigorous programme of chest physiotherapy and antibiotics as indicated by bacteriological evidence.
Hirschsprung’s disease
In this disease, which has an incidence of 1 in 5000 births, an aganglionic section of the large bowel is present. This means that peristalsis does not occur and the bowel therefore becomes obstructed. The baby develops abdominal distension and bile-stained vomiting. Definite diagnosis is made by carrying out a rectal biopsy. Resection of the aganglionic segment of bowel is indicated.
Pyloric stenosis
Pyloric stenosis arises from a genetic defect that causes hypertrophy of the muscles of the pyloric sphincter. The characteristic clinical presentation is projectile vomiting usually at around 6 weeks of age, but it may occur earlier and hence is included in this section. There is a gender-related predominance in that it is usually boys who are affected. Surgical repair is effected by pyloromyotomy (Ramstedt’s operation), which involves partial splitting of the hypertrophied pyloric muscle along its length.
The remaining abnormalities of the gastrointestinal system, while amenable to surgery, do not usually necessitate immediate action.
Cleft lip and cleft palate
The incidence of cleft lip occurring as a single deformity is 1.3 in 1000. This defect may be unilateral or bilateral. Since it is very often accompanied by cleft palate, both will be considered together.
Clefts in the palate may affect the hard palate, soft palate, or both. Some defects will include alveolar margins and some the uvula. It is recommended that, during the initial examination of the baby, the palate be examined by means of a good light source rather than by digital palpation. The greatest problem for these babies initially is feeding. If the defect is limited to unilateral cleft lip, mothers who had intended to breastfeed should be encouraged to do so. Where there is the additional problem of cleft palate, arranging for the baby to be fitted with an orthodontic plate may facilitate breastfeeding but this obviously does not afford the same stimulus as nipple-to-palate contact.
Middle ear infection is a concomitant risk for babies with cleft palate. Repeated infections of this type could impede hearing and subsequent development of speech. Danner (1992) suggests that breastfeeding be encouraged since passive immunity may protect these babies from the infections to which they are prone. Expressed breastmilk may of course be given. Cup or spoon feeding is an alternative method but for those who wish to bottle feed there is a wide variety of specially shaped teats available to accommodate the different sizes and positions of palate defects. Above all else, an unending supply of patience is required. The midwife should encourage the mother and father to find the most successful technique rather than ‘taking over’ since this may compound any feelings of guilt or inadequacy the parents may feel. Early referral to the cleft team of paediatric or plastic surgeon and orthodontists should be arranged. These teams may also include specialist nursing staff, speech and language therapists and audiologists.
Corrective surgery will be carried out at some stage but there is some debate as to the most appropriate time to carry out these procedures. Sullivan (1996) examines the arguments for both early and late repair of a cleft lip. He explains that some surgeons advocate effecting closure of the cleft lip within 2 weeks of birth in order to capitalize on the increased tissue-healing properties that are present as a short-lived legacy of intrauterine existence. They also argue that an early repair will be instrumental in encouraging healthy attachment between mother and baby. Advocates of later intervention suggest cleft lip repair at the age of 3–4 months because cleft lip often occurs as a feature of other medical conditions that may not be detected immediately. Surgery in the early neonatal period for such a baby may be too hazardous. Closure of the palate defect is suggested at around the age of 12–15 months. One of the main reasons for this apparently long delay is to allow sufficient growth to take place, which may result in reducing the size of the defect thus increasing the possibility of a more satisfactory repair. Some children have a series of cosmetic operations at some time after the initial repairs are carried out. It is often helpful for the midwife to show families ‘before and after’ photographs of babies for whom surgery has been a success (Fig. 46.7).
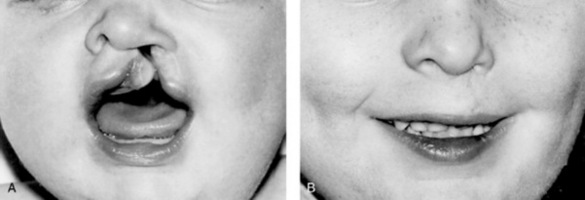
Figure 46.7 (A) Cleft lip and palate. (B) The repaired cleft.
(From Raine 1994, with permission of Churchill Livingstone.)
Clearly, although the midwife may offer valuable support in these early days, she is limited in the length of time she has available to help these families. Giving the parents the address of a support group such as the Cleft Lip And Palate Association (CLAPA) is useful (see Useful addresses, below).
Pierre Robin sequence
Pierre Robin sequence is characterized by micrognathia (hypoplasia of the lower jaw), abnormal attachment of muscles controlling the tongue, which allows it to fall backward and occlude the airway, and a central cleft palate. This triad of abnormalities presents a challenge for nursing care. Maintenance of a clear airway is paramount. In order to achieve this, the baby will need to be nursed prone and some may require the insertion of an oral airway. Nasal and nasopharyngeal constant positive airways pressure (CPAP) may be necessary for some time after birth. This is one of the few exceptions to the ‘Back to sleep’ campaign aimed at reducing cot deaths. Feeding can be problematic. There is a high risk of aspiration occurring. Suction catheter and oxygen equipment should be readily to hand. Some of these babies may be fitted with an orthodontic plate to facilitate feeding. The action of sucking will encourage development of the mandible. Parents will need considerable support during what may for some babies be a protracted period of hospitalization. Discharge will be when the lower jaw has grown sufficiently or when the parents feel comfortable about taking the baby home. Habel et al (1996) suggest that some of these babies can have an earlier transfer home if a shortened endotracheal tube is in place to ensure a patent nasopharyngeal airway. They recommend replacing the tube every 2 weeks until adequate mandibular development has taken place. Despite this, there remains a small risk of sudden death in these infants.
Abnormalities relating to respiration
Making a successful transition from fetus to neonate includes being able to establish regular respiration. Any abnormality of the respiratory tract or accessory respiratory muscles is likely to hamper this process.
Diaphragmatic hernia
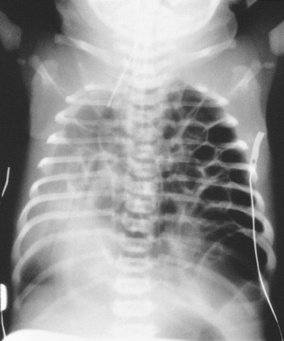
This abnormality consists of a defect in the diaphragm that allows herniation of abdominal contents into the thoracic cavity (Fig. 46.8). The extent to which lung development is compromised as a result depends on the size of the defect and the gestational age at which herniation first occurred. The condition is increasingly diagnosed antenatally by ultrasound; where there is prenatal diagnosis, delivery in a specialist unit is advisable. At birth, the condition may be suspected if the baby is cyanosed and unexpected difficulty is experienced in resuscitation. In addition, since the majority of such defects are left sided, heart sounds will be displaced to the right. The abdomen may have a flat or scaphoid appearance. Chest X-ray will confirm the diagnosis. Continuous gastric suction should be commenced. Surgical repair of the defect is necessary, but this is not urgent. It is more important to stabilize the baby’s general condition before surgery. It is especially critical to deal with the problem of persistent pulmonary hypertension and right-to-left shunting of blood within the heart. This may necessitate the use of newer ventilation techniques and pharmacological agents such as nitric oxide. Prognosis relates to the degree of pulmonary hypoplasia and reversibility of the pulmonary hypertension. There is also the possibility of co-existent abnormalities such as cardiac defects or skeletal anomalies. The incidence is one in 2000 births.
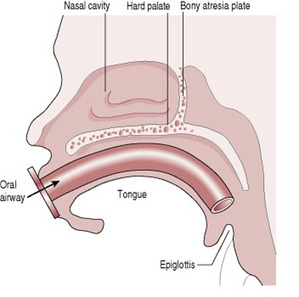
Choanal atresia
Choanal atresia describes a unilateral or bilateral narrowing of the nasal passage(s) with a web of tissue or bone occluding the nasopharynx (Fig. 46.9). Tachypnoea and dyspnoea are cardinal features, particularly when a bilateral lesion is present. The diagnosis is made relatively easily by noting that the baby mouth breathes and finds feeding impossible without cyanosis. In addition, nasal catheters cannot be passed into the pharynx and if a mirror or cold metal spoon is held under the nose no vapour will collect. A helpful diagnostic aid is that the baby’s colour will improve with crying (Bagwell 1993). Maintaining a clear airway is obviously essential and an oral airway may have to be used to affect this. A unilateral defect may not be noticed until the baby feeds for the first time. The midwife should therefore bear in mind the possibility of this problem if respiratory difficulty and cyanosis occur at this time. Surgery will be required to remove the obstructing tissue. The incidence is 1 in 8000. Occasionally choanal atresia is associated with other abnormalities such as ‘Charge’ syndrome, a syndrome in which there are defects found in the eye (coloboma), the heart, the ear, the genitalia, occasionally oesophageal atresia and usually growth retardation.
Laryngeal stridor
This is a noise made by the baby, usually on inspiration and exacerbated by crying. Most commonly the cause is laryngomalacia, which is due to laxity of the laryngeal cartilage. Although it sounds distressing, the baby generally is not at all upset. It is the parents who require comforting and reassurance (often repeatedly). It should be explained to them that the stridor may take some time to resolve, perhaps up to 2 years. If, however, the stridor is accompanied by signs of dyspnoea or feeding problems, further investigations such as bronchoscopy or laryngoscopy would become necessary to rule out a more sinister cause.
Congenital cardiac defects
Babies born with congenital heart defects comprise the second largest group of babies born with abnormalities. Approximately eight in 1000 livebirths have some degree of congenital heart disease and about one-third of these babies will be symptomatic in early infancy.
Causes
Approximately 90% of cardiac defects cannot be attributed to a single cause, chromosomal and genetic factors account for 8%, and a further 2% are thought to be caused by teratogens. The critical period of exposure to teratogens in respect of embryological development of cardiac tissue is from the 3rd to the 6th week.
Prenatal detection
An increasing number of cardiac problems are being identified by means of detailed ultrasound scanning (see Ch. 18). However, the detection of many defects is still dependent upon accurate observations and examination during the neonatal period.
Cardiac defects presenting with cyanosis
Defects included in this group are:
The persistence of central cyanosis (i.e. cyanosis of the lips and mucous membranes), tachypnoea and tachycardia may be the first signs that a cardiac defect is present. If cyanosis is present, administration of oxygen to these babies will be ineffective in improving their colour and oxygen saturation monitoring will show no improvement. Indeed, giving 100% oxygen may encourage closure of the ductus arteriosus, the patency of which, as Paul (1995) remarks, is literally a lifeline for some of these babies. If 100% oxygen therapy does not lead to improvement within 10 min of starting, the midwife should not persist at that concentration without seeking medical advice. Chest X-ray should be carried out to exclude abnormalities of the respiratory tract, respiratory disease and diaphragmatic hernia. The precise nature of the cardiac anomaly will need to be further explored by electrocardiography and echocardiography.
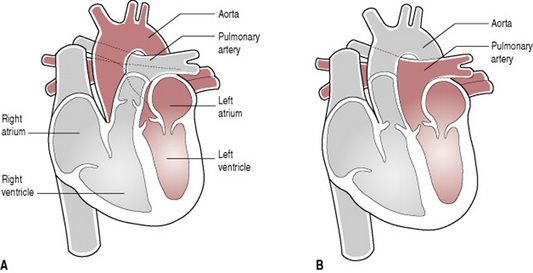
The baby with transposition of the great arteries is worthy of special mention since early detection allows intervention, which will be life saving. This is a condition wherein the aorta arises from the right ventricle and the pulmonary artery from the left ventricle (Fig. 46.10). Consequently, oxygenated blood is circulated back through the lungs and deoxygenated blood back into the systemic circuit. It is apparent therefore that unless there is an opportunity for oxygenated blood to access the systemic circulation, either by means of a patent ductus arteriosus or through accompanying septal defects, such a baby will die. Maintaining the patency of the ductus arteriosus is essential.
Prostaglandin infusion should be commenced to achieve this, but some babies develop apnoea with this treatment and may require ventilatory support. Arrangements should be made to transfer the baby to a unit where an atrial septostomy (Rashkind) can be performed. This procedure involves enlarging the foramen ovale with a balloon catheter to allow oxygenated blood to cross at atrial level and hence into the systemic circulation. Corrective surgery (arterial switch operation) is then carried out, usually within a few weeks of birth.
Pulmonary atresia usually produces early central cyanosis and responds well to prostaglandin therapy to keep the duct open while surgery (usually a palliative Blalock shunt) is planned.
Tricuspid atresia usually occurs with a ventricular septal defect or an atrial septal defect, or both, allowing mixing of the circulation.
Potentially more distressing for the parents are the defects that do not initially present with marked cyanosis. These babies may for a time be considered to be healthy.
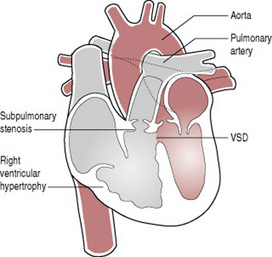
Tetralogy of Fallot (Fig. 46.11) is a good example of this. In this condition there is pulmonary outflow tract obstruction, a ventricular septal defect, right ventricular hypertrophy and an overriding aorta. It seldom presents with cyanosis in the immediate newborn period, but this a may become apparent within a few weeks of birth. Corrective surgery is available.
‘Acyanotic’ cardiac defects
Anomalies subsumed under this heading include:
Astute midwives may detect in these babies the first subtle signs of cardiac failure, that is tachypnoea, tachycardia and incipient cyanosis, especially following the exertion of crying or feeding. These signs will become more evident, sometimes dramatically so, with the closure of the ductus arteriosus if coarctation of the aorta, hypoplastic left heart syndrome or critical aortic stenosis is present. Detailed examination may disclose heart murmurs and diminution or absence of peripheral pulses. In this event resuscitation with prostaglandin is required to stabilize the baby and allow time for further assessment. While coarctation of the aorta and aortic stenosis are usually amenable to surgical correction, hypoplastic left heart syndrome is still a major surgical challenge with a poor long term outcome. There is a substantial psychological impact on the parents following confirmation of such a diagnosis, which calls for particularly supportive management.
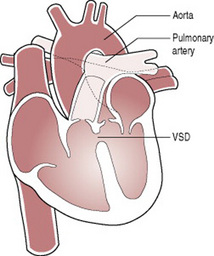
Persistent ductus arteriosus, ventricular septal defects (Fig. 46.12) and atrial septal defects seldom require medical or surgical intervention in early neonatal life, but do require careful follow-up for signs of developing heart failure and may require surgery or interventional cardiology at a later stage.
Changing patterns of postnatal care often mean early transfer home. Ideally each baby should be examined by a competent practitioner before going home. It is important to realize, however, that not all heart murmurs heard at this time are significant. Equally the absence of a murmur at discharge from hospital does not exclude significant heart disease. There is therefore increased responsibility on community midwives to be observant and to communicate effectively with parents. Parents who report any changes in the baby’s behaviour such as breathlessness or cyanosis should never be ignored, but rather encouraged to seek medical advice promptly.
Central nervous system abnormalities
Ingestion of folic acid supplements prior to conception and during the early stages of pregnancy has helped prevent such abnormalities. In addition, the ability to recognize these anomalies prenatally (see Ch. 18) has resulted in some parents choosing selective termination of pregnancies where severe neural tube defects are found. All of these measures have combined to reduce the number of babies born with abnormalities of the central nervous system.
Anencephaly
This major abnormality describes the absence of the forebrain and vault of the skull. It is a condition that is incompatible with sustained life but occasionally such a baby is born alive. The midwife should wrap the baby carefully before showing him to the mother. It is recognized that seeing and holding the baby will facilitate the grieving process. It may be beneficial for the parents then to see the full extent of the abnormality, unpleasant though it is. Seeing the whole baby will help them to accept the reality of the situation and prevent imagination of an even more gruesome picture.
Spina bifida
Spina bifida results from failure of fusion of the vertebral column. There is no skin covering the defect, which allows protrusion of the meninges, hence the term meningocele (Fig. 46.13). The meningeal membrane may be flat or appear as a membranous sac, with or without cerebrospinal fluid, but it does not contain neural tissue. Meningomyelocele, on the other hand, does involve the spinal cord (Figs 46.13, 46.14). This lesion may be enclosed, or the meningocele may rupture and expose the neural tissue. Meningomyelocele usually gives rise to neural damage, producing paralysis distal to the defect, and impaired function of urinary bladder and bowel. The lumbosacral area is the most common site for these to present, but they may appear at any point in the vertebral column. When the defect is at base of skull level it is known as an encephalocele. The added complication here is that the sac may contain varying amounts of brain tissue. Normal progression of labour may be impeded by a large lesion of this type.
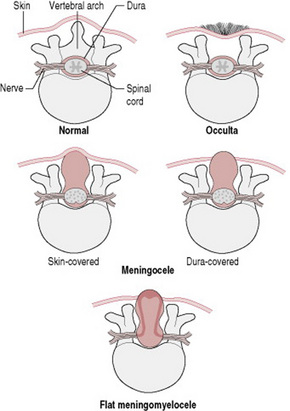
Figure 46.13 Various forms of spina bifida.
(After Wallis & Harvey 1979, with permission of Nursing Times.)
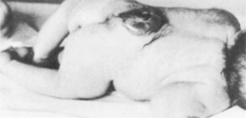
Immediate management involves covering open lesions with a non-adherent dressing. Babies with enclosed lesions should be handled with the utmost care in an attempt to preserve the integrity of the sac. This will limit the risk of meningitis occurring. A paediatric surgeon or neurosurgeon should be contacted. Surgical intervention for myelomeningocele carries a high rate of success of skin closure, but has no impact on any damage already present in the cord or more distally. There is usually associated hydrocephalus (see below), which requires surgical shunting to prevent a rapid increase in the intracranial pressure. It is seldom necessary to close the back within 24 hrs of birth. Following examination of the baby, discussion with the parents will allow them to make an informed choice about whether or not they wish their baby to have surgery.
Spina bifida occulta
Spina bifida occulta (see Fig. 46.13) is the most minor type of defect where the vertebra is bifid. There is usually no spinal cord involvement. A tuft of hair or sinus at the base of the spine may be noted on first examination of the baby. Ultrasound investigation will confirm the diagnosis and rule out any associated spinal cord involvement.
Parents who have a baby with a neural tube defect should be offered genetic counselling since the risk of recurrence is one in 25.
Hydrocephalus
This condition arises from a blockage in the circulation and absorption of cerebrospinal fluid, which is produced from the choroid plexuses within the lateral ventricles of the brain. The large lateral ventricles increase in size and eventually compress the surrounding brain tissue. It is a not infrequent accompaniment to the more severe spina bifida lesions because of a structural defect around the area of the foramen magnum known as the Arnold–Chiari malformation. Consequently, hydrocephalus may either be present at birth or develop following surgical closure of a myelomeningocele. In the absence of myelomeningocele, aqueduct stenosis is the commonest cause of hydrocephalus. The risk of cerebral impairment may be minimized by the insertion of a ventriculoperitoneal shunt. As the baby grows, this will need to be replaced. Attendant risks with these devices are that the line blocks and that the shunt is a portal for infection leading to meningitis. The midwife must be alert for the signs of increased intracranial pressure:
Microcephaly
This is where the occipitofrontal circumference is more than two standard deviations below normal for gestational age. The disproportionately small head may be the result of intrauterine infection (e.g. rubella), a feature of fetal alcohol syndrome, or part of a number of defects in some trisomic disorders. Most babies will have learning difficulties with evidence of cerebral palsy and often seizures.
Musculoskeletal deformities
These range from relatively minor anomalies, for example an extra digit, to major deficits such as absence of a limb.
Polydactyly and syndactyly
Careful examination including separation and counting of the baby’s fingers and toes during the initial examination is important, otherwise anomalies such as syndactyly (webbing) and polydactyly (extra digits) may go unnoticed.
Syndactyly more commonly affects the hands. It can appear as an independent anomaly or as a feature of a syndrome such as Apert’s syndrome; this is a genetically inherited condition in which there is premature fusion of the sutures of the vault of the skull, cleft palate and complete syndactyly of both hands and feet. Whether or not any surgical division needs to be carried out depends on the degree of webbing or fusion.
In polydactyly the extra digit(s) may be fully formed or simply extra tissue attached by a pedicle. Even where there is a rudimentary digit without bone involvement, better cosmetic results are obtained if the offending digit is surgically excised and this is mandatory in more complex cases.
Where there is a family history of either of these defects and this is common, the mother will be anxious to examine the baby for herself.
Limb reduction anomalies
Over the years various suggestions have been postulated with regard to the cause(s) of limb reduction anomalies. Amniotic band syndrome was the reason most often given for a baby being born with a limb deficit. It was thought that the amnion ruptured, then wrapped itself around a developing limb causing strangulation and necrosis. Although this may account for some instances, it cannot explain all of them. Clustering of cases in certain geographical areas, for example in close proximity to chemical waste plants, has provoked research in an attempt to identify environmental teratogens, as yet to no avail. One possibility being mooted is an iatrogenic cause, namely damage inflicted at the time of chorionic villus sampling (Carr & Lui 1994). They can also occur as part of a syndrome such as the VACTERL spectrum (vertebral anomalies, anal anomalies, cardiac, tracheoesophageal, radial aplasia, renal and limb anomalies).
Limb reduction defects, which may be due to failure of formation (arrest of development), comprise a wide range of possibilities. In some either a hand or a foot will be completely missing, whereas in others a normal hand or foot will be present on the end of a shortened limb (longitudinal arrest). Thalidomide has been proven to be teratogenic in this context.
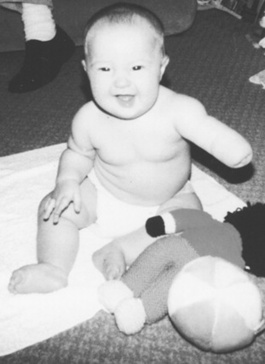
Although, as with any deviation from normal, the parents of a child with a limb defect will grieve for the loss of their perfect child, the child is not ill and will not be upset by the defect. Children usually prove themselves to be most adaptable and able to cope (Fig. 46.15). Specific management plans are often only reached after detailed assessment by an orthopaedic surgeon with a special interest in limb anomalies. For those who require them, different types of prostheses are available and can be fitted as early as 3 months of age. Innovative surgical techniques such as limb lengthening or the transferring of toe(s) to hand to serve as substitute finger(s) are proving successful for some children. Once again one of the most helpful things the midwife can do in these early days of parental adjustment is to offer the address of a support group such as Reach (see Useful addresses, below). This appropriately named support group for parents of children with upper limb deformities has branches throughout the UK.
Talipes
Talipes equinovarus (TEV, club foot) (Plate 17) is the descriptive term for a deformity of the foot where the ankle is bent downwards (plantarflexed) and the front part of the foot is turned inwards (inverted). Talipes calcaneovalgus describes the opposite position where the foot is dorsiflexed and everted. It is thought that these deformities are more likely to occur when intrauterine space has been at a premium, for example in multiple pregnancy, macrosomic fetus or oligohydramnios. TEV is also more likely to occur in conjunction with spina bifida deformities. They may be unilateral or bilateral. There is, in some instances, a family history of the defect. Statistically more boys than girls are born with talipes. In the mildest form the foot may easily be turned to the correct position. The midwife should encourage the mother to exercise the baby’s foot in this way several times a day. More severe forms will require one or more of manipulation, splinting, or surgical correction. The advice of an orthopaedic surgeon should be sought as soon as possible after birth as early treatment with manipulation or splinting may enhance results. Care should be taken to ensure that, for babies who have splints applied, the strapping is not too tight and that the baby’s toes are well perfused.
Developmental hip dysplasia
Congenital hip dysplasia is an abnormality more commonly found where there has been a breech presentation at term, oligohydramnios, a foot deformity or a family history in a first-degree relative. It most often occurs in primigravida pregnancies and there is a higher percentage of girls than boys born with this defect. The left hip is more often affected than the right. The dysplastic hip may present in one of three ways: dislocated, dislocatable or with subluxation of the joint. Prenatal diagnosis by ultrasound is possible; most, however, are diagnosed in the neonatal period. Examination of the hip should be carried out by an experienced practitioner with the baby lying relaxed on a firm surface. It will depend on individual unit policies whether this is an appropriately trained midwife, paediatrician or GP. Repeated examinations by inexperienced people may compound any pre-existing damage to the joint. Either Ortolani’s test or Barlow’s test is employed (see Ch. 40). Any abnormal findings should be reported and the baby referred for an orthopaedic opinion or ultrasound scan of the hips, or both. Where the diagnosis is confirmed it is usual for the baby to have a splint or harness such as the Pavlik harness (Fig. 46.16) applied, which will keep the hips in a flexed and abducted position of about 60%. The splint should not be removed for napkin changing or bathing. Parents will require additional support in learning how to handle and care for their baby. Particular attention should be paid to skin care and checking for signs of chafing or excoriation.
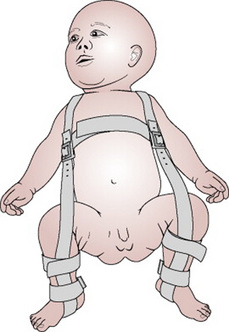
Figure 46.16 Pavlik harness for congenital dislocation of hip.
(From Barr 1992, with permission of Churchill Livingstone.)
Achondroplasia
Achondroplasia is an autosomal dominant condition where the baby is generally small with a disproportionately large head and short limbs. Some 80% of cases are new mutations and hence these families may have no anticipation of the disorder unless an antenatal diagnosis has been made. These babies often develop a marked lordosis. Cognitive development is not usually impaired.
Osteogenesis imperfecta
This autosomal dominant disorder of collagen production has at least four forms and leads to unduly brittle bones in the affected fetus and infant. In some types (II–IV) it can cause either lethal multiple fractures of the skull, ribs and long bones in utero or neonatal long bone fractures. Recognition and genetic counselling are important for future pregnancies.
Abnormalities of the skin
Vascular naevi
These defects in the development of the skin can be divided into two main types, which commonly overlap.
Capillary malformations
These are due to defects in the dermal capillaries. The most commonly observed are ‘stork marks’. These are usually found on the nape of the neck. They are generally small and will fade. No treatment is necessary.
Port wine stain
This is a purple-blue capillary malformation affecting the face. It occurs in approximately 1 in 3000 births. It is generally fully formed at birth and does not regress with time. However, laser treatment and the skillful use of cosmetics will help to disguise the problem. The parents, and later the child, may need substantial psychological support.
Should the malformation mimic the distribution of the ophthalmic branch of the trigeminal nerve, further malformations in the eyes (glaucoma), meninges or brain (epilepsy) may be suspected. This is known as the Sturge–Weber syndrome.
Capillary haemangiomata (‘strawberry marks’)
Capillary haemangiomata are not usually noticeable at birth but appear as red, raised lesions in the first few weeks of life (Plate 35). These common lesions affect up to 10% of the population by the age of 1 year. They are five times more common in girls than boys and are also commoner in preterm infants. They can appear anywhere in the body but cause particular distress to the parents when they appear on the face. However, parents may be reassured that, although the lesion will grow bigger for the first few months it will then regress with associated central pallor (Plate 36) and usually disappears completely by the age of 5–6 years. No treatment is normally required unless the haemangioma is situated in an awkward area where it is likely to be subject to abrasion, such as on the lip, or around the eye where it may interfere with vision. Treatment with steroids or pulsed laser therapy is possible.
Pigmented (melanocytic) naevi
These are brown, sometimes hairy, marks on the skin that vary in size and may be flat or raised. A percentage of this type of birthmark may become malignant. Surgical excision may be recommended to pre-empt this.
It is unlikely that treatment for any of these birthmarks will be carried out in the immediate neonatal period except in the case of larger pigmented naevi. The midwife’s responsibilities are therefore to notify appropriate medical staff and offer parents general emotional support.
Genitourinary system
At birth the first indication that there is an abnormality of the renal tract may be finding a single umbilical artery in the umbilical cord, or alternatively recognizing the abnormal facies associated with Potter’s syndrome. Attention should be paid at the time of birth to see whether the baby passes urine. If no urine is passed within 24 hrs, the baby is noted to be dribbling urine constantly, or the urine stream seems poor, the paediatrician should be informed. Dribbling of urine is a sign of nerve damage such as occurs with neural tube defects, as previously discussed, whereas a poor urine stream may indicate lower urinary tract obstruction (posterior urethral valve).
Potter syndrome
This collection of features in a series of stillborn infants was first described by Edith Potter, a perinatal pathologist, in 1946. It is due to the compressive effects of longstanding oligohydramnios (from 13–14 weeks’ gestation) in renal agenesis or severe hypoplasia.
The baby’s face will have a flattened appearance, low set ears, an antimongoloid slant to the eyes, with deep epicanthic folds, and a beaked nose. These babies are usually in poor condition at birth because they have lung hypoplasia. It is a syndrome incompatible with sustained life.
Posterior urethral valve(s)
This is an abnormality affecting boys. The presence of valves in the posterior urethra prevents the normal outflow of urine. As a result the bladder distends, causing back pressure on the ureters and to the kidneys. This will ultimately cause hydronephrosis. Prenatal diagnosis and intervention by intrauterine fetal bladder catheterization is possible. Failing this, early diagnosis in the neonatal period is clearly important but severe renal damage may already have been sustained. Different treatment strategies are possible with surgical procedures featuring prominently.
Polycystic kidneys
It is likely that problems may arise in delivering a baby with polycystic kidneys because of an increase in abdominal girth. On abdominal examination the kidneys will be palpable. Radiological or ultrasound investigations will be carried out to confirm the diagnosis. Unfortunately the prognosis is poor, as renal failure is the likely outcome. The severest forms of polycystic kidney disease are usually linked to an autosomal recessive inheritance, but an autosomal dominant variety also occurs with a less gloomy prognosis.
Hypospadias
Examination of a baby boy may reveal that the urethral meatus opens on to the undersurface of the penis. The meatus can be placed at any point along the length of the penis and in some cases will open onto the perineum. This abnormality often coexists with chordee, in which the penis is short and bent and the foreskin is present only on the dorsal side of the penis. It is anticipated that some babies will require surgery in the neonatal period to ‘release’ the chordee and enlarge the urethral meatus. It is important that the parents are made aware that circumcision should be deferred until consultation with the paediatric surgeon is completed.
Cryptorchidism
Undescended testes may be unilateral or bilateral and occur in 1–2% of male infants. If on examination of the baby after birth the scrotum is empty, the undescended testes may be found in the inguinal pouch. Sometimes the testis in this position can be manipulated into the scrotal pouch. This augurs well for future normal development. Testes that are found too high in the inguinal canal to manipulate into the scrotum may be malformed. If neither testis is palpable further investigation to exclude endocrine or chromosomal causes is required. In unilateral undescended testis parents should be encouraged to have the baby examined at regular intervals. If descent of the testis has not occurred by the time the child is 2 years old, arrangements for orchidopexy may be made.
Ambiguous genitalia
Ambiguous genitalia describe a situation in which the external appearance is neither definitely male nor female. In this situation it is vital that the midwife is honest and does not assign a gender to the baby. Examination of the baby may reveal any of the following: a small hypoplastic penis, chordee, bifid scrotum, undescended testes (careful examination should be made to detect undescended testes in the inguinal canal) or enlarged clitoris, incompletely separated or poorly differentiated labia. It can be impossible to differentiate by clinical examination alone between male under-virilization and female masculinization and expert clarification is always needed.
Congenital adrenal hyperplasia
One of the commoner reasons for ambiguous genitalia is an autosomal recessive condition called congenital adrenal hyperplasia. In this condition the adrenal gland is stimulated to overproduce androgens because of a deficiency of an enzyme called 21-hydroxylase, which is necessary for normal production of steroid from cholesterol. If aldosterone production is reduced then these babies will rapidly lose salt and may present collapsed and dehydrated. Urea and electrolyte levels, blood glucose and 17-hydroxy progesterone concentrations should be measured and appropriate fluid replacement given. In the process of eliminating or confirming the cause, a 24 hrs urine collection may be requested. It is necessary to ensure that the urine bag is correctly placed to avoid faecal contamination. Placing one end of a catheter or feeding tube in the urine bag and aspirating the contents at regular intervals will help to prevent spillage and the unnecessary trauma to the baby of repeated applications of urine bags. Babies with this condition may require later cosmetic surgery. The condition is not always recognized in boys in the neonatal period.
Intersex
This is where the internal reproductive organs are at variance with the external appearance of the genitalia. Ultrasound examination will help to identify the nature of the internal reproductive organs. True hermaphroditism is extremely rare. The decision of gender attribution is made following chromosomal studies to determine genetic make-up, hormone assays and consideration of the potential for cosmetic surgery.
Clearly, this time of waiting for results of investigations is a time of great concern for parents because they cannot tell relatives and friends the gender of the baby. Delay in naming the baby is an additional pressure. Some parents in this invidious position elect to give the baby a name that would suit either a boy or a girl. It is, however, more common for a child of truly ambiguous gender to be raised as a girl.
Teratogenic causes
Fetal alcohol syndrome/spectrum
Preconception preparation for pregnancy advice on alcohol intake, if heeded, could dramatically reduce the incidence of this phenomenon. Health promotion objectives and successful outcomes are, however, rarely totally synchronous.
The midwife may be alerted to the possibility of a baby being born with this syndrome prenatally if, in addition to psychosocial markers, clinical intrauterine growth restriction is evident. Postnatally the following characteristics are recognizable: a growth-restricted infant with microcephaly, flat facies, close-set eyes, epicanthic folds, small upturned nose, thin upper lip and low set ears. Most of these features become less pronounced as the child grows. Microcephaly, small stature and learning difficulties remain. The baby may experience acute withdrawal symptoms and require appropriate supportive therapies. The midwife will need to exercise counselling skills to provide much-needed support for the mother. Collaboration with social services is usually called for to ensure that the care options decided are in the best interests of the family.
Establishing such a direct link between a teratogen and such a complex clinical pattern remains the exception rather than the rule although as mentioned earlier accurate recording of all congenital abnormalities on a central register can aid early recognition of potential new teratogens.
Support for the midwife
Caring for a mother whose baby has some major congenital abnormality places extra demands on the midwife. This stress is compounded if the abnormality was not anticipated prior to birth or if the midwife has not previously encountered the particular problem. The exercising of effective counselling and communication skills is invaluable in helping the family to adjust and in facilitating appropriate lines of support. The extra effort expended can be costly in terms not only of time but of the emotional stress the midwife may experience.
It is important that support is available for midwives in these situations (e.g. from her supervisor of midwives). Preparatory courses on grief and bereavement counselling are also of some benefit as many parents with affected babies will experience many of these emotions. Midwives who have acquired experience in this realm should not, however, automatically be targeted as the experts and always be called upon to fulfil this role. Conversely, student midwives ought not to be deliberately shielded from being involved in caring for such families. The provision of quality care for parents who have a child with a congenital abnormality is contingent upon meeting the needs of the carers.
Midwives may also find information available via the internet, however, they should be aware of the dubious quality of some of this information. They should therefore exercise caution in how they utilize it. It might also be wise to caution parents, who often search the internet for further information, of this potential risk.
Bagwell CE. Surgical lesions of pediatric airways and lungs. In: Koff PB, Eitzman D, Neu J, editors. Neonatal and pediatric respiratory care. 2nd edn. St Louis: Mosby Year Book; 1993:132. Ch 8
Barr DGD. Disorders of bone. In: Campbell AGM, McIntosh N, editors. Forfar and Arneil’s textbook of paediatrics. Edinburgh: Churchill Livingstone; 1992:1628. Ch 23
Carr AJ, Lui DTY. Chorionic villus sampling. Advantages and disadvantages for prenatal diagnosis. Midwives Chronicle. 1994;107(1279):284-287.
Danner SC. Breast-feeding the infant with a cleft defect. NAACOGs Clinical Issues in Perinatal and Women’s Health Nursing. 1992;3(4):634-639.
Habel A, Sell D, Mars M. Management of cleft lip and palate. Archives of Disease in Childhood. 1996;74(4):360-366.
Haddock G, Davis CF, Raine PAM. Gastroschisis in the decade of prenatal diagnosis. European Journal of Paediatric Surgery. 1996;6:18-24.
Paul K. Recognition, stabilization and early management of infants with critical congenital heart disease presenting in the first days of life. Neonatal Network. 1995;14(5):13-25.
Raine P. Cleft lip and palate. In: Freeman NV, Burge DM, Grisffiths M, Malone PSJ, editors. Surgery in the newborn. Edinburgh: Churchill Livingstone; 1994:375. Ch 34
Sullivan G. Parental bonding in cleft lip and palate repair. Paediatric Nursing. 1996;8(1):21-24.
Wallis S, Harvey D. Disorders in the newborn 1. Nursing Times. 1979;75:1315-1327.
Whyte A. Folic acid fortifying the pregnancy message. Health Visitor. 1995;68(10):397-398.
Wirt S, Algren CL, Arnold SL. Cleft lip and palate: a multidisciplinary approach. Plastic Surgical Nursing. 1992;12(4):140-145.
Jones KL, editor. Smith’s recognizable patterns of human malformation, 5th edn., Philadelphia: W B Saunders, 1997.
This book provides a comprehensive and systematic approach to dysmorphic syndromes
On-line Mendelian Inheritance in Man (OMIM), www.ncbi.nlm.nih.gov/omim.
This website provides detailed information about clinical features and genetics of all inherited diseases
Contact a family Contact a family, www.cafamily.org.uk.
This website provides information and support for families with disabled children
Genetic Interest Group (GIG), www.gig.org.uk/members.htm.
This is a national alliance of organizations which support children and families affected by genetic disorders
Association for Spina Bifida and Hydrocephalus (ASBAH), www.asbah.org.
Cleft Lip and Palate Association (CLAPA), www.clapa.com.
Children’s Heart Federation, www.childrens-heart-fed.org.uk.
Cystic Fibrosis Research Trust (CF), www.cftrust.org.uk.
Down’s Syndrome Association, www.downs-syndrome.org.uk.
Scottish Down’s Syndrome Association (SDSA), www.dsscotland.org.uk.
Reach: The Association for Children with Hand or Arm Deficiency, www.reach.org.uk.
Support organization for trisomy 13/18 (SOFT) Support organization for trisomy 13/18 (SOFT), www.soft.org.uk.
STEPS (National Association for Children with Lower Limb Abnormalities), www.steps-charity.org.uk.
Antenatal Results and Choices (ARC, formerly SAFTA), www.arc-uk.org.