48 Antiviral Agents and Infection Control
Unlike bacteria, viruses are obligate intracellular parasites that use the host cell’s biosynthetic machinery and enzymes for replication (see Chapter 44). Hence it is more difficult to inhibit viral replication without also being toxic to the host. Most antiviral drugs are targeted toward viral-encoded enzymes or structures of the virus that are important for replication. Most of these compounds are classic biochemical inhibitors of viral-encoded enzymes. Some antiviral drugs are actually stimulators of host innate immune protective responses.
Unlike antibacterial drugs, the activity of most antiviral drugs is limited to a specific virus. Antiviral drugs are available for viruses that cause significant morbidity and mortality, which also provide reasonable targets for drug action (Box 48-1). As has occurred with antibacterial drugs, however, resistance to antiviral drugs is becoming more of a problem because of the high rate of mutation for viruses and the long-term treatment of some patients, especially those who are immunocompromised (e.g., patients with the acquired immunodeficiency syndrome [AIDS]).
Targets for Antiviral Drugs
The different targets for antiviral drugs (e.g., structures, enzymes, or processes important or essential for virus production) are discussed with respect to the steps of the viral replication cycle they inhibit. These targets and their respective antiviral agents are listed in Table 48-1 (see also Chapter 44, Figure 44-9).
Table 48-1 Examples of Targets for Antiviral Drugs
| Replication Step or Target | Agent | Targeted Virus* |
|---|---|---|
| Attachment | Peptide analogues of attachment protein | HIV (CCR5 co-receptor antagonist) |
| Neutralizing antibodies | Most viruses | |
| Heparan and dextran sulfate | HIV, HSV | |
| Penetration and uncoating | Amantadine, rimantadine | Influenza A virus |
| Tromantadine | HSV | |
| Arildone, disoxaril, pleconaril | Picornaviruses | |
| Transcription | Interferon | HCVs, papillomavirus |
| Antisense oligonucleotides | – | |
| Protein synthesis | Interferon | HCV, papillomavirus |
| DNA replication (polymerase) | Nucleoside analogues | Herpesviruses; HIV; hepatitis B virus, poxviruses, etc. |
| Phosphonoformate, phosphonoacetic acid | Herpesviruses | |
| Nucleoside biosynthesis | Ribavirin | Respiratory syncytial virus, Lassa fever virus, HCV |
| Nucleoside scavenging (thymidine kinase) | Nucleoside analogues | HSV, varicella-zoster virus |
| Glycoprotein processing | — | HIV |
| Assembly (protease) | Hydrophobic substrate analogues | HIV, HCV |
| Assembly (neuraminidase) | Oseltamivir, zanamivir | Influenza A, B virus |
| Virion integrity | Nonoxynol-9 | HIV, HSV |
CCR5, C-C chemokine receptor 5; DNA, deoxyribonucleic acid; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HSV, herpes simplex virus.
* Therapies may not have received approval for human use.
Virion Disruption
Enveloped viruses are susceptible to certain lipid and detergent-like molecules that disperse or disrupt the envelope membrane, thereby preventing acquisition of the virus. Nonoxynol-9, a detergent-like component in birth control jellies, can inactivate herpes simplex virus (HSV) and human immunodeficiency virus (HIV) and prevent sexual acquisition of the viruses. Rhinoviruses are susceptible to acid, and citric acid can be incorporated into facial tissues as a means of blocking virus transmission.
Attachment
The first step in viral replication is mediated by the interaction of a viral attachment protein with its cell surface receptor. This interaction can be blocked by neutralizing antibodies, which bind to the viral attachment protein, or by receptor antagonists. The administration of specific antibodies (passive immunization) is the oldest form of antiviral therapy. Receptor antagonists include peptide or sugar analogues of the cell receptor or the viral attachment protein that competitively blocks the interaction of the virus with the cell. Compounds that bind to the C-C chemokine receptor 5 (CCR5) molecule block binding of HIV to macrophages and some CD4 T cells to prevent the initial infection. Acidic polysaccharides, such as heparan and dextran sulfate, interfere with viral binding and have been suggested for the treatment of infection with HIV, HSV, and other viruses.
Penetration and Uncoating
Penetration and uncoating of the virus are required to deliver the viral genome into the cytoplasm of the host cell. Arildone, disoxaril, pleconaril, and other methylisoxazole compounds block uncoating of picornaviruses by fitting into a cleft in the receptor-binding canyon of the capsid and preventing disassembly of the capsid. For viruses that enter through endocytic vesicles, uncoating may be triggered by conformational changes in attachment proteins that promote fusion or by membrane disruption resulting from the acidic environment of the vesicle. Amantadine, rimantadine, and other hydrophobic amines (weak organic bases) are antiviral agents that can neutralize the pH of these compartments and inhibit virion uncoating. Amantadine and rimantadine have a more specific activity against influenza A. These compounds bind to and block the hydrogen ion (H+) channel formed by the viral M2 protein. Without the influx of H+, the M1 matrix proteins do not dissociate from the nucleocapsid (uncoating), so movement of the nucleocapsid to the nucleus, transcription, and replication are prevented. Blockage of this proton pore also disrupts the proper processing of the hemagglutinin protein late in the replication cycle. In the absence of a functional M2 proton pore, the hemagglutinin inopportunely changes its conformation into its “fusion form” and is inactivated as it traverses the normally acidic Golgi environment. Tromantadine, a derivative of amantadine, also inhibits penetration of HSV. Penetration and uncoating of HIV are blocked by a 33-amino acid peptide, T20 (enfuvirtide [Fuzeon]), which inhibits the action of the viral fusion protein gp41.
RNA Synthesis
Although messenger ribonucleic acid (mRNA) synthesis is essential for the production of virus, it is not a good target for antiviral drugs. It would be difficult to inhibit viral mRNA synthesis without affecting cellular mRNA synthesis. Deoxyribonucleic acid (DNA) viruses use the host cell’s transcriptases for mRNA synthesis. The RNA polymerases encoded by RNA viruses may not be sufficiently different from host cell transcriptases to selectively inhibit this activity, and the high rate at which RNA viruses mutate results in the generation of many drug-resistant strains. Guanidine and 2-hydroxybenzylbenzimidine are two compounds that can block picornavirus RNA synthesis by binding to the 2C picornavirus protein, which is essential for RNA synthesis. Ribavirin resembles riboguanosine and inhibits nucleoside biosynthesis, mRNA capping, promotes hypermutation, and other processes (cellular and viral) important to the replication of many viruses.
The proper processing (splicing) and translation of viral mRNA can be inhibited by interferon and antisense oligonucleotides. Isatin-β-thiosemicarbazone induces mRNA degradation in poxvirus-infected cells and was used as a treatment for smallpox. Viral infection of an interferon-treated cell triggers a cascade of biochemical events that block viral replication. Specifically, the degradation of viral and cellular mRNA is enhanced, and ribosomal assembly is blocked, preventing protein synthesis and viral replication. Interferon is described further in Chapter 10. Interferon and artificial interferon inducers (Ampligen, poly rI:rC) have been approved for clinical use (papilloma, hepatitis B and C) or are in clinical trials.
Genome Replication
Most antiviral drugs are nucleoside analogues, which are nucleosides with modifications of the base, sugar, or both (Figure 48-1). The viral DNA polymerases of the herpesviruses and the reverse transcriptases of HIV and hepatitis B virus (HBV) are the prime targets for most antiviral drugs because they are essential for virus replication and are different from host enzymes. Before being used by the polymerase, the nucleoside analogues must be phosphorylated to the triphosphate form by viral enzymes (e.g., HSV thymidine kinase), cellular enzymes, or both. For example, the thymidine kinase of HSV and varicella-zoster virus (VZV) applies the first phosphate to acyclovir (ACV), and cellular enzymes apply the rest. HSV mutants lacking thymidine kinase activity are resistant to ACV. Cellular enzymes phosphorylate azidothymidine (AZT) and many other nucleoside analogues.
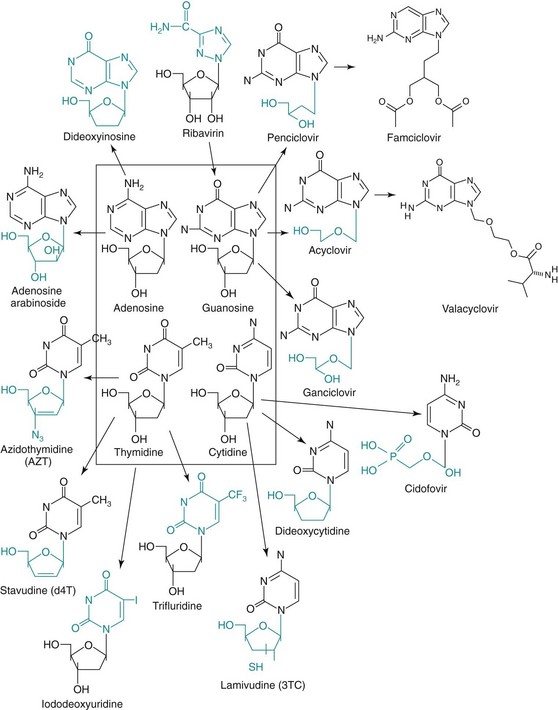
Figure 48-1 Structure of the most common nucleoside analogues that are antiviral drugs. The chemical distinctions between the natural deoxynucleoside and the antiviral drug analogues are highlighted. Arrows indicate related drugs. Valacyclovir is the L-valyl ester of acyclovir. Famciclovir is the diacetyl 6-deoxyanalogue of penciclovir. Both of these drugs are metabolized to the active drug in the liver or intestinal wall.
Nucleoside analogues selectively inhibit viral polymerases because these enzymes are less accurate than host cell enzymes. The viral enzyme binds nucleoside analogues with modifications of the base, sugar, or both several hundred times better than the host cell enzyme. These drugs either prevent chain elongation, as a result of the absence of a 3′-hydroxyl on the sugar, or alter recognition and base pairing, as a result of a base modification, and induce inactivating mutations (see Figure 48-1). Antiviral drugs that cause termination of the DNA chain by means of modified nucleoside sugar residues include ACV, ganciclovir (GCV), valacyclovir, penciclovir, famciclovir, adefovir, cidofovir, adenosine arabinoside (vidarabine, ara-A), zidovudine (AZT), lamivudine (3TC), dideoxycytidine, and dideoxyinosine. Antiviral drugs that become incorporated into the viral genome and cause errors in replication (mutation) and transcription (inactive mRNA and proteins) because of modified nucleoside bases include ribavirin, 5-iododeoxyuridine (idoxuridine), and trifluorothymidine (trifluridine). The rapid rate and large extent of nucleotide incorporation during viral replication make retrovirus and DNA virus replication especially susceptible to these drugs. A variety of other nucleoside analogues is also being developed as antiviral drugs.
Pyrophosphate analogues resembling the byproduct of the polymerase reaction, such as phosphonoformic acid (foscarnet, PFA) and phosphonoacetic acid, are classic inhibitors of the herpesvirus polymerases. Nevirapine, delavirdine, and other nonnucleoside reverse transcriptase inhibitors bind, as noncompetitive inhibitors of the enzyme, to sites on the enzyme other than the substrate site.
Deoxyribonucleotide scavenging enzymes (e.g., the thymidine kinase and ribonucleoside reductase of the herpesviruses) are also enzyme targets of antiviral drugs. Inhibition of these enzymes reduces the levels of deoxyribonucleotides necessary for the replication of the DNA virus genome, preventing virus replication.
Integration of the cDNA of HIV into the host chromosome catalyzed by the viral integrase enzyme is essential for virus replication. An inhibitor of the integrase is now approved for anti-HIV therapy.
Protein Synthesis
Although bacterial protein synthesis is the target for several antibacterial compounds, viral protein synthesis is a poor target for antiviral drugs. The virus uses host cell ribosomes and synthetic mechanisms for replication, so selective inhibition is not possible. Interferon-α (IFN-α) and interferon-β (IFN-β) stop a virus by promoting the inhibition of viral protein synthesis in the infected cell. Inhibition of the posttranslational modification of proteins, such as the proteolysis of a viral polyprotein or glycoprotein processing (castanospermine, deoxynojirimycin), can inhibit virus replication.
Virion Assembly and Release
The HIV protease is unique and essential to the assembly of virions and the production of infectious virions. Computer-assisted molecular modeling was used to design inhibitors of the HIV protease, such as saquinavir, ritonavir, and indinavir (navir, “no virus”), by modeling inhibitors that would fit into the active site of the enzyme. The enzyme structures were defined by x-ray crystallographic and molecular biologic studies. Bocepravir and telaprevir are two new protease inhnibitors for treatment of hepatitis C virus (HCV). Proteases of other viruses are also targets for antiviral drugs.
The neuraminidase of influenza has also become a target for antiviral drugs. Zanamivir (Relenza) and oseltamivir (Tamiflu) act as enzyme inhibitors and, unlike amantadine and rimantadine, can inhibit influenza A and B. Amantadine and rimantadine also inhibit release of influenza A.
Stimulators of Host Innate Immune Protective Responses
The best antiviral agents are those of the host’s innate and immune antiviral response. Stimulation or supplementation of the natural response is an effective approach to limit or treat viral infections. Innate responses of dendritic cells, macrophages, and other cells can be stimulated by imiquimod, resiquimod, and CpG oligodeoxynucleotides, which bind to Toll-like receptors to stimulate release of protective cytokines, activation of natural killer cells, and subsequent cell-mediated immune responses. Interferon and interferon inducers, including mismatched polynucleotides and double-stranded RNA (e.g., Ampligen, poly rI:rC), facilitate the treatment of chronic diseases of hepatitis C and papillomaviruses. Antibodies, acquired naturally or by passive immunization (see Chapters 10 and 11), prevent both the acquisition and the spread of the virus. For example, passive immunization is administered after exposure to rabies and hepatitis A virus (HAV) and HBV.
Nucleoside Analogues
Most of the antiviral drugs approved by the U.S. Food and Drug Administration (FDA) (Table 48-2) are nucleoside analogues that inhibit viral polymerases. Resistance to the drug is usually caused by a mutation of the polymerase.
Table 48-2 Some Antiviral Drug Therapies Approved by the U.S. Food and Drug Administration
| Virus | Antiviral Drug | Trade Name |
|---|---|---|
| Herpes simplex and varicella-zoster viruses | Acyclovir* | Zovirax |
| Valacyclovir* | Valtrex | |
| Penciclovir | Denavir | |
| Famciclovir* | Famvir | |
| Iododeoxyuridine (idoxuridine)† | Stoxil | |
| Trifluridine | Viroptic | |
| Cytomegalovirus | Ganciclovir | Cytovene |
| Valganciclovir | Valcyte | |
| Cidofovir | Vistide | |
| Phosphonoformate (foscarnet) | Foscavir | |
| Influenza A virus | Amantadine | Symmetrel |
| Rimantadine | Flumadine | |
| Influenza A and B viruses | Zanamivir | Relenza |
| Oseltamivir | Tamiflu | |
| Hepatitis B virus | Lamivudine | Epivir |
| Adefovir dipivoxil | Hepsera | |
| Hepatitis C virus | Interferon-α, ribavirin boceprevir, telaprevir | Various Victrelis, Incivek |
| Papillomavirus | Interferon-α | Various |
| Respiratory syncytial virus and Lassa virus | Ribavirin | Virazole |
| Picornaviruses | Pleconaril | Picovir |
| Human immunodeficiency virus | ||
| Nucleoside analogue reverse transcriptase inhibitors | Azidothymidine (zidovudine) | Retrovir |
| Dideoxyinosine (didanosine) | Videx | |
| Dideoxycytidine (zalcitabine) | Hivid | |
| Stavudine (d4T) | Zerit | |
| Lamivudine (3TC) | Epivir | |
| Nonnucleoside reverse transcriptase inhibitors | Nevirapine | Viramune |
| Delavirdine | Rescriptor | |
| Protease inhibitors | Saquinavir | Invirase |
| Ritonavir | Norvir | |
| Indinavir | Crixivan | |
| Nelfinavir | Viracept | |
| Integrase inhibitor | Raltegravir | Isentriss |
| CCR5 co-receptor antagonist | Maraviroc | Selzentry |
| Fusion inhibitor | Enfuvirtide | Fuzeon |
CCR5, C-C chemokine receptor 5.
* Also active against varicella-zoster virus.
Acyclovir, Valacyclovir, Penciclovir, and Famciclovir
ACV (acycloguanosine) and its valyl derivative, valacyclovir, differ in pharmacologic ways. ACV differs from the nucleoside guanosine by having an acyclic (hydroxyethoxymethyl) side chain instead of a ribose or deoxyribose sugar. ACV has selective action against HSV and VZV, the herpesviruses that encode a thymidine kinase (Figure 48-2). The viral thymidine kinase is required to activate the drug by phosphorylation, and host cell enzymes complete the progression to the diphosphate form and finally to the triphosphate form. Because there is no initial phosphorylation in uninfected cells, there is no active drug to inhibit cellular DNA synthesis or to cause toxicity. The ACV triphosphate causes termination of the growing viral DNA chain because there is no 3′-hydroxyl group on the ACV molecule to allow chain elongation. The minimal toxicity of ACV is also a result of a 100-fold or greater use by the viral DNA polymerase than by cellular DNA polymerases. Resistance to ACV develops by mutation of either the thymidine kinase, so that activation of ACV cannot occur, or the DNA polymerase, to prevent ACV binding.
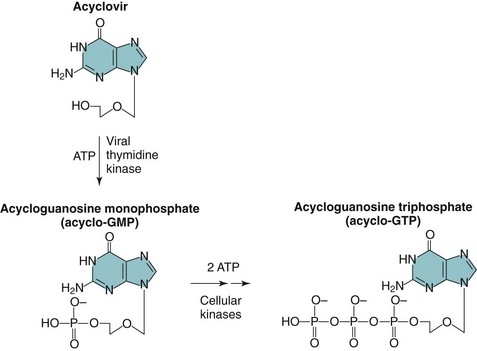
Figure 48-2 Activation of acyclovir (ACV) (acycloguanosine) in herpes simplex virus–infected cells. ACV is converted to acycloguanosine monophosphate (acyclo-GMP) by herpes-specific viral thymidine kinase, then to acycloguanosine triphosphate (acyclo-GTP) by cellular kinases. ATP, Adenosine triphosphate.
ACV is effective against all HSV infections, including encephalitis, disseminated herpes, and other serious herpes diseases. The fact that it is not toxic to uninfected cells allows use of it and its analogues as a prophylactic treatment to prevent recurrent outbreaks, especially in immunosuppressed people. A recurrent episode may be prevented if it is treated before or soon after the triggering event. ACV inhibits the replication of HSV but cannot resolve the latent HSV infection.
Valacyclovir, the valyl ester derivative of ACV, is more efficiently absorbed after oral administration and rapidly converted into ACV, increasing the bioavailability of ACV for the treatment of HSV and serious VZV. ACV and valacyclovir can also be used for the treatment of VZV infection, although higher doses are required. VZV is less sensitive to the agent because ACV is phosphorylated less efficiently by the VZV thymidine kinase.
Penciclovir inhibits HSV and VZV in the same way ACV does but is concentrated and persists in the infected cells to a greater extent than ACV. Penciclovir also has some activity against the Epstein-Barr virus and cytomegalovirus (CMV). Famciclovir is a prodrug derivative of penciclovir that is well absorbed orally and then is converted to penciclovir in the liver or intestinal lining. Resistance to penciclovir and famciclovir develops in the same manner as for ACV.
Ganciclovir
GCV (dihydroxypropoxymethyl guanine) differs from ACV in having a single hydroxymethyl group in the acyclic side chain (see Figure 48-1). The remarkable result of this addition is that it confers considerable activity against CMV. CMV does not encode a thymidine kinase, but a viral-encoded protein kinase phosphorylates GCV. Once activated by phosphorylation, GCV inhibits all herpesvirus DNA polymerases. The viral DNA polymerases have nearly 30 times greater affinity for the drug than the cellular DNA polymerase. Similar to ACV, a valyl ester of GCV (valganciclovir) was developed to improve the pharmacologic properties of GCV.
GCV is effective in the treatment of CMV retinitis and shows some efficacy in the treatment of CMV esophagitis, colitis, and pneumonia in patients with AIDS. The potential for bone marrow and other toxicity limits its use. Of interest, this potential toxicity has been used as the basis for the development of an antitumor therapy. In one application, an HSV thymidine kinase gene was incorporated into the cells of a brain tumor with the use of a retrovirus vector. The retrovirus replicated only in the growing cells of the tumor, and the thymidine kinase was expressed only in the tumor cells, making the tumor cells susceptible to GCV.
Cidofovir and Adefovir
Cidofovir and adefovir are both nucleotide analogues and contain a phosphate attached to the sugar analogue. This obviates the need for the initial phosphorylation by a viral enzyme. Compounds with this type of sugar analogue are substrates for DNA polymerases or reverse transcriptases and have an expanded spectrum of susceptible viruses. Cidofovir, a cytidine analogue, is approved for CMV infections in AIDS patients but can also inhibit replication of polyomavirus, papillomaviruses, and inhibit the polymerases of herpesviruses, adenoviruses, and poxvirus. Adefovir and adefovir dipivoxil (a diester prodrug) are analogues of adenosine and are approved for treatment of HBV.
Azidothymidine
Originally developed as an anticancer drug, AZT was the first useful therapy for HIV infection. AZT (Retrovir), a nucleoside analogue of thymidine, inhibits the reverse transcriptase of HIV (see Figure 48-1). Similar to other nucleosides, AZT must be phosphorylated by host cell enzymes. It lacks the 3′-hydroxyl necessary for DNA chain elongation and prevents complementary DNA synthesis. The selective therapeutic effect of AZT stems from the 100-fold lower sensitivity of the host cell DNA polymerase in comparison with the HIV reverse transcriptase.
Continuous oral AZT treatment is administered to HIV-infected people with depleted CD4 T-cell counts to prevent progression of disease. AZT treatment of pregnant HIV-infected women can reduce the likelihood of or prevent transmission of the virus to the baby. Side effects of AZT range from nausea to life-threatening bone marrow toxicity.
The high error rate of the HIV polymerase creates extensive mutations and promotes the development of antiviral drug-resistant strains. This problem is being addressed by the administration of multidrug therapy as initial therapy (highly active antiretroviral therapy [HAART]). It is more difficult for the HIV to develop resistance to multiple drugs with multiple target enzymes. Multidrug-resistant HIV strains are likely to be much weaker than the parent strains.
Dideoxyinosine, Dideoxycytidine, Stavudine, and Lamivudine
Several other nucleoside analogues have been approved as anti-HIV agents. Dideoxyinosine (didanosine) is a nucleoside analogue that is converted to dideoxyadenosine triphosphate (see Figure 48-1). Similar to AZT, dideoxyinosine, dideoxycytidine, and stavudine (d4T) lack a 3′-hydroxyl group. The modified sugar attached to lamivudine (2′-deoxy-3′-thiacytidine [3TC]) also inhibits the HIV reverse transcriptase by preventing DNA chain elongation and HIV replication. These drugs are available for the treatment of AIDS that is unresponsive to AZT therapy, or they can be given in combination with AZT. Lamivudine is also active on the reverse transcriptase polymerase of HBV. Most of the anti-HIV agents have potential toxic side effects.
Ribavirin
Ribavirin is an analogue of the nucleoside guanosine (see Figure 48-1) but differs from guanosine in that its base ring is incomplete and open. Similar to other nucleoside analogues, ribavirin must be phosphorylated. The drug is active in vitro against a broad range of viruses.
Ribavirin monophosphate resembles guanosine monophosphate and inhibits nucleoside biosynthesis, mRNA capping, and other processes important to the replication of many viruses. Ribavirin depletes the cellular stores of guanine by inhibiting inosine monophosphate dehydrogenase, an enzyme important in the synthetic pathway of guanosine. It also prevents the synthesis of the mRNA 5′ cap by interfering with the guanylation and methylation of the nucleic acid base. In addition, ribavirin triphosphate inhibits RNA polymerases and promotes hypermutation of the viral genome. Its multiple sites of action may explain the lack of ribavirin-resistant mutants.
Ribavirin is administered in an aerosol to children with severe respiratory syncytial virus bronchopneumonia and potentially to adults with severe influenza or measles. The drug may be effective for the treatment of influenza B, as well as Lassa, Rift Valley, Crimean-Congo, Korean, and Argentine hemorrhagic fevers, for which it is administered orally or intravenously. Ribavirin is approved for use against HCV in combination with IFN-α. Treatment can have serious side effects.
Other Nucleoside Analogues
Idoxuridine, trifluorothymidine (see Figure 48-1), and fluorouracil are analogues of thymidine. These drugs either (1) inhibit the biosynthesis of thymidine, a nucleotide essential for DNA synthesis, or (2) replace thymidine and become incorporated into the viral DNA. These actions inhibit further synthesis of the virus or cause extensive misreading of the genome, leading to mutation and inactivation of the virus. These drugs target cells in which extensive DNA replication is taking place, such as those infected with HSV, and spare nongrowing cells from harm.
Idoxuridine was the first anti-HSV drug approved for human use but has been replaced by trifluridine and other more effective, less toxic agents. Fluorouracil is an antineoplastic drug that kills rapidly growing cells but has also been used for the topical treatment of warts caused by human papillomaviruses.
Adenine arabinoside was the principal anti-HSV drug until ACV was introduced but is no longer used. Ara-A is an adenosine nucleoside analogue with an arabinose substituted for deoxyribose as the sugar moiety (see Figure 48-1). This agent is phosphorylated by cellular enzymes even in uninfected cells. Ara-A has a greater potential for causing toxicity than ACV and is difficult to administer. The viral enzyme is 6 to 12 times more sensitive than the cellular enzyme. Resistance can develop as a result of a mutation of the viral DNA polymerase.
Many other nucleoside analogues that have antiviral activity are being investigated for clinical use against the herpesviruses, HBV, and HIV.
Nonnucleoside Polymerase Inhibitors
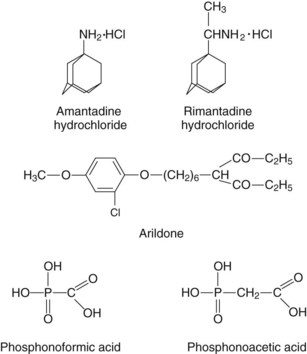
Foscarnet (PFA) and the related phosphonoacetic acid (PAA) are simple compounds that resemble pyrophosphate (Figure 48-3). These drugs inhibit viral replication by binding to the pyrophosphate-binding site of the DNA polymerase to block nucleotide binding. PFA and PAA do not inhibit cellular polymerases at pharmacologic concentrations, but they can cause renal and other problems because of their ability to chelate divalent metal ions (e.g., calcium) and become incorporated into bone. PFA inhibits the DNA polymerase of all herpesviruses and the HIV reverse transcriptase without having to be phosphorylated by nucleoside kinases (e.g., thymidine kinase). PFA has been approved for the treatment of CMV retinitis in patients with AIDS.
Nevirapine, delavirdine, efavirenz, and other nonnucleoside reverse transcriptase inhibitors bind to sites on the enzyme different from the substrate. Because these drugs’ mechanisms of action differ from those of the nucleoside analogues, the mechanism of HIV resistance to the agents is also different. As a result, these drugs are very useful in combination with nucleoside analogues for the treatment of HIV infection.
Protease Inhibitors
The unique structure of the HIV protease and its essential role in the production of a functional virion have made this enzyme a good target for antiviral drugs. Saquinavir, indinavir, ritonavir, nelfinavir, amprenavir, and other agents work by slipping into the hydrophobic active site of the enzyme to inhibit its action. As occurs with the other anti-HIV drugs, drug-resistant strains arise through mutation of the protease. The combination of a protease inhibitor with AZT and a second nucleoside analogue (HAART) can reduce blood levels of HIV to undetectable levels. Development of resistance to a “cocktail” of anti-HIV drugs is also less likely than that to a single drug. Protease inhibitors have great potential for treatment of hepatitis C and other viruses.
Antiinfluenza Drugs
Amantadine and rimantadine are amphipathic amine compounds with clinical efficacy against the influenza A but not the influenza B virus (see Figure 48-3). These drugs have several effects on influenza A replication. Both compounds are acidotrophic and concentrate in and buffer the contents of the endosomal vesicles involved in the uptake of the influenza virus. This effect can inhibit the acid-mediated change in conformation in the hemagglutinin protein that promotes the fusion of the viral envelope with cell membranes. However, the specificity for influenza A is a result of its ability to bind to and block the proton channel formed by the M2 membrane protein of the influenza A virus. Resistance is the result of an altered M2 or hemagglutinin protein.
Amantadine and rimantadine may be useful in ameliorating an influenza A infection if either agent is taken within 48 hours of exposure. They are also useful as a prophylactic treatment in lieu of vaccination. In addition, amantadine is an alternative therapy for Parkinson disease. The principal toxic effect is on the central nervous system, with patients experiencing nervousness, irritability, and insomnia.
Zanamivir (Relenza) and oseltamivir (Tamiflu) inhibit influenza A and B as enzyme inhibitors of the neuraminidase of influenza. Without the neuraminidase, the hemagglutinin of the virus binds to sialic acid on other glycoproteins, forming clumps and preventing assembly and virus release. These drugs can be taken prophylactically as an alternative to vaccination or to reduce the length of illness, if taken within the first 48 hours of infection.
Immunomodulators
Genetically engineered forms of IFN-α have been approved for human use. Interferons work by binding to cell surface receptors and initiating a cellular antiviral response. In addition, interferons stimulate the immune response and promote the immune clearance of viral infection.
IFN-α is active against many viral infections, including hepatitis A, B, and C; HSV; papillomavirus; and rhinovirus. It has been approved for the treatment of condyloma acuminatum (genital warts, a presentation of papillomavirus) and hepatitis C (especially with ribavirin). Attachment of polyethylene glycol to IFN-α (pegylated IFN-α) increases its potency. Pegylated IFN-α is used with ribavirin to treat hepatitis C infections. Natural interferon causes the influenza-like symptoms observed during many viremic and respiratory tract infections, and the synthetic agent has similar side effects during treatment. Interferon is discussed further in Chapters 10 and 45.
Imiquimod, a Toll-like receptor ligand, stimulates innate responses to attack the virus infection. This therapeutic approach can activate local protective responses against papillomas, which generally escape immune control.
Infection Control
Infection control is essential in hospital and health care settings. The spread of respiratory viruses is the most difficult to prevent. Viral spread can be controlled in the following ways:
1. Limiting personnel contact with sources of infection (e.g., wearing gloves, mask, goggles; using quarantine)
2. Improving hygiene, sanitation, and disinfection
3. Ensuring that all personnel are immunized against common diseases
4. Educating all personnel regarding points 1, 2, and 3 and in the ways to decrease high-risk behaviors
Methods for disinfection differ for each virus and depend on its structure. Most viruses are inactivated by 70% ethanol, 15% chlorine bleach, 2% glutaraldehyde, 4% formaldehyde, or autoclaving (as described in Guidelines for Prevention of Transmission of Human Immunodeficiency Virus and Hepatitis B Virus to Health-Care and Public-Safety Workers, issued in 1989 by the U.S. Centers for Disease Control and Prevention [CDC]). Most enveloped viruses do not require such rigorous treatment and are inactivated by soap and detergents. Other means of disinfection are also available.
Special “universal” precautions are required for the handling of human blood; that is, all blood should be assumed to be contaminated with HIV or HBV and should be handled with caution. In addition to these procedures, special care must be taken with syringe needles and surgical tools contaminated with blood. Specific guidelines are available from the CDC.
Control of an outbreak usually requires identification of the source or reservoir of the virus, followed by cleanup, quarantine, immunization, or a combination of these measures. The first step in controlling an outbreak of gastroenteritis or hepatitis A is identification of the food, water, or possibly the day-care center that is the source of the outbreak.
Education programs can promote compliance with immunization programs and help people change lifestyles associated with viral transmission. Such programs have had a significant impact in reducing the prevalence of vaccine-preventable diseases, such as smallpox, polio, measles, mumps, and rubella. It is hoped that educational programs will also promote changes in lifestyles and habits, to restrict the spread of the blood-borne and sexually transmitted HBV and HIV.
1. List the steps in viral replication that are poor targets for antiviral drugs. Why?
2. Which viruses can be treated with an antiviral drug? Distinguish the viruses treatable with an antiviral nucleoside analogue.
3. A mutation in the gene for which enzymes or proteins would confer resistance to the following antiviral drugs: ACV, ara-A, phosphonoformate, amantadine, AZT?
4. A patient has been exposed to influenza A virus and is in his third day of symptoms. He has heard that an antiinfluenza drug is available and requests therapy. You tell him that therapy is not appropriate. To what therapeutic agents is the patient referring, and why did you decline to use the treatment?
5. What disinfection procedures are sufficient for inactivating the following viruses: HAV, HBV, HSV, and rhinovirus?
6. What precautions should health care workers take to protect themselves from infection with the following viruses: HBV, influenza A virus, HSV (whitlow), and HIV?
1. Steps in viral replication that depend on cellular processes are generally poor antiviral drug targets. These include protein synthesis and mRNA synthesis and processing (e.g., splicing, capping).
HSV (treatable with nucleoside analogues)
VZV (treatable with nucleoside analogues)
CMV (treatable with nucleoside analogues)
Smallpox (treatable with nucleoside analogues)
Hepatitis B (treatable with nucleoside analogues)
Respiratory syncytial virus (treatable with nucleoside analogues)
HCV (treatable with nucleoside analogues)
HIV (treatable with nucleoside analogues)
3. ACV: DNA polymerase, thymidine kinase of HSV or VZV
Phosphonoformate: DNA polymerase of herpesviruses (e.g., CMV)
Amantadine: M2 protein of influenza A
AZT: RNA-dependent DNA polymerase of HIV
4. Amantadine and rimantidine inhibit influenza A virus replication by preventing uncoating of the virus in the cytoplasm. These drugs are effective as prophylactics and before inflammatory and immune responses are generated. Oseltamivir and zanamivir are neuraminidase inhibitors that inhibit both influenza A and B virus by preventing proper release of the virus.
5. HAV and rhinovirus are picornaviruses with very different requirements for disinfection. HAV is an enterovirus and resistant to detergents and acid. Disinfection requires rigorous treatment for disinfection, such as 2% glutaraldehyde (a protein cross-linking fixative), toilet bowl cleaner with 23% hydrochloric acid and quaternary ammonium compounds, or ≈10% bleach (sodium hypochlorite). Autoclaving is also appropriate. In contrast, rhinoviruses can be disinfected by mild acid, including citric acid as well as the treatments described for HAV. For enveloped viruses, such as HBV and HSV, detergent, 70% isopropanol or ethanol, acid, greater than 3% hydrogen peroxide, ≈10% bleach, and iodophors will inactivate the virus.
6. For HBV and Influenza A, the best protection is vaccination. Aerosol spread of influenza A is so contagious that vaccination is the best prevention. For HBV, as for HIV and HCV, universal precautions for preventing transmission and contact with blood-borne infections must be used. All human blood is treated as if infected with these viruses. Universal precautions include wearing protective clothes; gloves; eye shields; and not bending, recapping, or removing contaminated needles and other contaminated sharps. Contaminated items must be discarded or disinfected as described for question 5. For HSV, gloves must be worn to prevent infection. There is no vaccine for HSV.