Chapter 20 The Cellular and Molecular Basis of Development
During embryonic development, undifferentiated precursor cells differentiate and organize into the complex structures found in functional adult tissues. This process requires cells to integrate many different cues, both intrinsic and extrinsic, for development to occur properly. These cues control the proliferation, differentiation, and migration of cells to determine the final size and shape of the developing organs. Disruption of these signaling pathways can result in human developmental disorders and birth defects. Interestingly, these key developmental signaling pathways may be co-opted in the adult by diseases such as cancer.
Although there are diverse changes that occur during embryogenesis, the differentiation of many different cell types is regulated through a relatively restricted set of molecular signaling pathways:
• Intercellular communication. Cells communicate with each other in several ways, including through gap junctions, ligand-receptor interactions and specific channels that permit the passage of ions, neurotransmitters, or proteins.
• Morphogens. These are diffusible molecules that specify which cell type will be generated at a specific anatomical location. Morphogens also direct the migration of cells and their processes to their final destination. These include retinoic acid, transforming growth factor β (TGF-β)/bone morphogenetic proteins (BMPs), and the hedgehog and Wnt protein families (see Table 20-1 for gene and protein nomenclature).
• Receptor tyrosine kinases (RTKs). Many growth factors signal by binding to and activating membrane-bound RTKs. These kinases are essential for the regulation of cellular proliferation, apoptosis, and migration as well, for example, growth of new blood vessels and axonal processes in the nervous system.
• Notch/Delta. This pathway often specifies the fate of precursor cells.
• Transcription factors. This set of evolutionarily conserved proteins activates or represses downstream genes that are essential for a number of cellular processes. Many transcription factors are members of the homeobox or helix-loop-helix (HLH) families. Their activity can be regulated by all of the other pathways described in this chapter.

Epigenetics relates to the ability of heritable properties of gene function that do not occur as a result of changes to the sequence of the DNA code. This can include variations in DNA packaging and DNA chemical modification.
Intercellular Communication
Cells communicate with each other in several ways. Gap junctions are channels that permit ions and small molecules (less than 1 kD) to directly pass from one cell to another, known as gap junctional intercellular communication (GJIC). However, large proteins and nucleic acids do not transfer through gap junctions. Gap junctions are made from hemi-channels present on the surface of each cell known as connexons. Each connexon is made up of six connexin molecules that form hexamers. In early development, gap junctions are usually open, permitting exchange of small molecules in relatively large regions. However, as development proceeds, GJIC is more restricted, with establishment of boundaries, such as in the rhombomeres of the developing hindbrain. Gap junctions are particularly important for electrical coupling in the heart and brain. Mutations of specific connexin molecules, for example Cx43, are associated with human diseases, such as atherosclerosis.
Morphogens
Extrinsic signaling by morphogens guide the differentiation and migration of cells during development, determining the morphology and function of developing tissues and organs (see Chapter 6). Many morphogens are found in concentration gradients in the embryo. Different morphogens can be expressed in opposing gradients in the dorsoventral, anteroposterior, and mediolateral axes. The fate of a specific cell can be determined by its location along these gradients. Cells can also be attracted or repelled by morphogens depending on the set of receptors expressed on the cell surface.
Retinoic Acid
The anteroposterior (AP) axis of the embryo is crucial for determining the correct location for structures such as limbs and for the patterning of the nervous system. For decades, it has been clinically evident that alterations in the level of vitamin A (retinol) in the diet (excessive or insufficient amounts) can lead to the development of congenital malformations (Chapter 19). The bioactive form of vitamin A is retinoic acid that is formed by enzymatic oxidation. Free levels of retinoic acid can be modulated by cellular retinoic acid–binding proteins that sequester retinoic acid. Retinoic acid can also be actively degraded into inactive metabolites by enzymes such as CYP26 (Fig. 20-1). Normally, retinoic acid acts to “posteriorize” the body plan and either excessive retinoic acid or inhibition of its degradation leads to a truncated body axis where structures have a more posterior nature. In contrast, insufficient retinoic acid or defects in the enzymes such as retinal aldehyde dehydrogenase will lead to a more anteriorized structure. At a molecular level, retinoic acid binds to its receptors (transcription factors) inside the cell and their activation will regulate the expression of downstream genes. Hox genes are crucial targets of retinoic acid receptors in development. Because of their profound influence on early development, retinoids are powerful teratogens, especially during the first trimester.
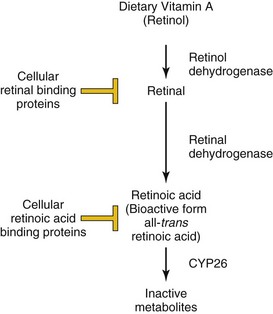
Figure 20–1 Regulation of retinoic acid metabolism and signaling. Dietary retinol (vitamin A) is converted to retinal via the action of retinol dehydrogenases. The concentration of free retinal is controlled by the action of cellular retinal-binding proteins. Similarly, retinal is converted to retinoic acid by retinal dehydrogenases, and its free level is modulated by sequestration by cellular retinoic acid–binding proteins and degradation by CYP26. The bioactive form of retinoic acid is all-trans retinoic acid.
Transforming Growth Factor β/Bone Morphogenetic Protein
Members of the TGF-β superfamily include TGF-β, BMPs, and activin. These molecules contribute to the establishment of dorsoventral patterning, cell fate decisions, and formation of specific organs and systems, including the kidneys, nervous system, the skeleton, and blood. In humans, there are three different forms of TGF-β (isoforms TGF-β1, TGF-β2, and TGF-β3).
Binding of these ligands to transmembrane kinase receptors results in phosphorylation of intracellular receptor-associated Smad proteins (R-Smads) (Fig. 20-2). The Smad proteins are a large family of intercellular proteins that are divided into three classes: receptor-activated (R-Smads), common-partner (co-Smads, Smad4), and inhibitory Smads (I-Smads). R-Smad/Smad4 complexes regulate target gene transcription by interacting with other proteins or as transcription factors by directly binding to DNA. The diversity of TGF-β ligand, receptor, and R-Smad combinations contributes to particular developmental and cell-specific processes, often in combination with other signaling pathways.
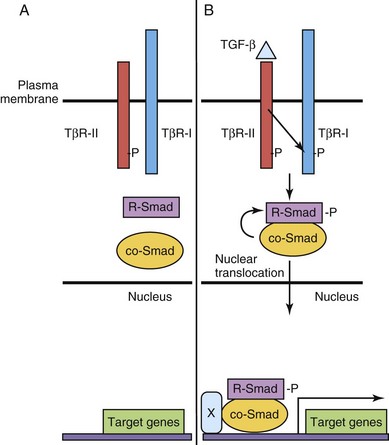
Figure 20–2 Transforming growth factor β (TGF-β)/Smad signaling pathway. A, The type II TGF-β receptor subunit (TβR-II) is constitutively active. B, Upon binding of ligand to TβR-II, a type I receptor subunit is recruited to form a heterodimeric receptor complex and the TβR-I kinase domain is transphosphorylated (-P). Signaling from the activated receptor complex phosphorylates R-Smads, which then bind to a co-Smad, translocate from the cytoplasm to the nucleus, and activate gene transcription with cofactor(s) (X).
Hedgehog
Sonic hedgehog (Shh) was the first mammalian ortholog of the Drosophila gene hedgehog to be identified. Shh and other related proteins, such as desert hedgehog and Indian hedgehog, are secreted morphogens critical to early patterning, cell migration, and differentiation of many cell types and organ systems. Cells have variable thresholds for response to the secreted Shh signal. The primary receptor for Shh is Patched (PTCH in human, PTC family in mouse), a transmembrane domain protein. In the absence of Shh, Patched inhibits transmembrane-domain, G-protein–linked protein (Smoothened [Smo]). This results in inhibitions of downstream signaling to the nucleus. However, in the presence of Shh, Ptc inhibition is blocked and downstream events follow, including transcriptional activation of target genes, such as Ptc–1, Engrailed, and others (Fig. 20-3).
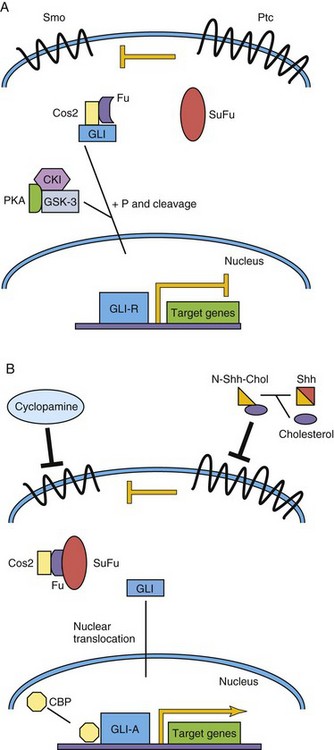
Figure 20–3 Sonic hedgehog/Patched signaling pathway. A, The Patched (Ptc) receptor inhibits signaling from the Smoothened (Smo) receptor. In a complex with Costal–2 (Cos2) and Fused (Fu), Gli is modified to become a transcriptional repressor, Gli-R. B, Sonic hedgehog (Shh) is cleaved, and cholesterol is added to its N-terminus. This modified Shh ligand inhibits the Ptc receptor, permitting Smo signaling, and ultimately activated Gli (Gli-A) translocates to the nucleus to activate target genes with CBP. CBP, cyclic AMP-binding protein; CKI, casein kinase I; GSK–3, glycogen synthase kinase–3; P, phosphate group; PKA, protein kinase A; SuFu, suppressor of Fused.
Post-translational modification of Shh protein affects its association with the cell membrane, formation of Shh multimers, and the movement of Shh, which, in turn, alters its tissue distribution and concentration gradients.
One of the best explained activities of Shh in vertebrate development is its role in patterning the ventral neural tube. Shh is secreted at high levels by the notochord, and therefore the concentration of Shh is highest in the floor plate of the neural tube and lowest in the roof plate, where members of the TGF-β family are highly expressed. The cell fates of ventral interneuron classes and motor neurons are determined by the relative Shh concentrations in the tissue and other factors.
The understanding of the requirement of Shh pathway signaling for many developmental processes has been enhanced by the discovery of human mutations of members of the Shh pathway. In addition, corresponding phenotypes of genetically modified mice, in which members of the Shh pathway are either inactivated (loss of function/knockout) or overexpressed (gain of function), have also added to this knowledge. Mutations of SHH and PTCH have been associated with holoprosencephaly in humans, a common congenital brain defect resulting in the fusion of the two cerebral hemispheres, dorsalization of forebrain structures, and anophthalmia or cyclopia (see Chapter 17). In sheep this same defect has been associated with exposure to the teratogen cyclopamine, which disrupts Shh signaling (see Fig. 20-3). Gorlin’s syndrome, often due to germline PTCH mutations, is a constellation of congenital malformations mostly affecting the epidermis, craniofacial structures, and nervous system. GLI3 mutations are associated with autosomal dominant polydactyly syndromes.
Wnt/β-Catenin Pathway
The Wnt-secreted glycoproteins are vertebrate orthologs of the Drosophila gene Wingless. Similar to the other morphogens, the nineteen Wnt family members control several processes during development, including establishment of cell polarity, proliferation, apoptosis, cell fate specification, and migration. Wnt signaling is a very complex process, and three signaling pathways have been elucidated to date; only the classic or “canonical” β-catenin-dependent pathway is discussed here (Fig. 20-4). Specific Wnts bind to 1 of 10 Frizzled (Fzd) seven-transmembrane domain, cell surface receptors, and with low-density, lipoprotein receptor–related protein (LRP5/LRP6) coreceptors, thereby activating downstream intracellular signaling events. In the absence of Wnt binding, cytoplasmic β-catenin is phosphorylated by glycogen synthase kinase (GSK–3) and targeted for degradation. In the presence of Wnts, GSK–3 is inactivated, and β-catenin is not phosphorylated and accumulates in the cytoplasm. The β-catenin translocates to the nucleus, where it activates target gene transcription, in a complex with T-cell factor (TCF) transcription factors. β-catenin/TCF target genes include vascular endothelial growth factor (VEGF) and matrix metalloproteinases.
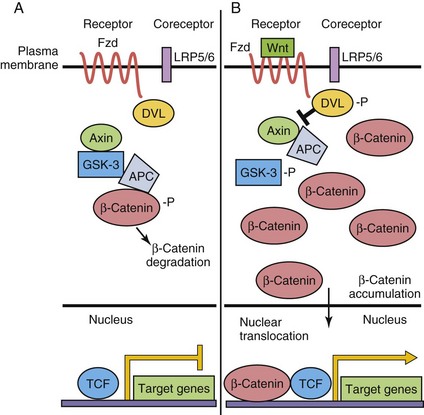
Figure 20–4 Wnt/β-catenin canonical signaling pathway. A, In the absence of Wnt ligand binding to Frizzled (Fzd) receptor, β-catenin is phosphorylated (-P) by a multiprotein complex and targeted for degradation. Target gene expression is repressed by T-cell factor (TCF). B, When Wnt binds to the Fzd receptor, LRP coreceptors are recruited, Disheveled (DVL) is phosphorylated and β-catenin then accumulates in the cytoplasm. Some β-catenin enters the nucleus to activate target gene transcription. APC, adenomatous polyposis coli; GSK–3, glycogen synthase kinase–3; LRP, lipoprotein receptor–related protein.
Dysregulated Wnt signaling is a prominent feature in many developmental disorders such as Williams-Beuren syndrome (heart, neurodevelopmental, and facial defects) and cancer. LRP5 mutations are found in the osteoporosis-pseudoglioma syndrome (congenital blindness and juvenile osteoporosis). Similar to the Shh pathway, canonical Wnt pathway mutations have been described in children with medulloblastoma.
Receptor Tyrosine Kinases
Common Features
Growth factors, such as insulin, epidermal growth factor, nerve growth factor, and other neurotrophins, and members of the platelet-derived growth factor family, bind to cell surface transmembrane receptors found on target cells. These receptors, members of the RTK superfamily, have three domains: (1) an extracellular ligand-binding domain, (2) a transmembrane domain (TMD), and (3) an intracellular kinase domain (Fig. 20-5). These receptors are found as monomers in the unbound state but become dimers upon ligand binding. This process of dimerization brings the two intracellular kinase domains into close proximity such that one kinase domain can phosphorylate and activate the other receptor. This process, transphosphorylation, is required to fully activate the receptors, which then initiate a series of intracellular signaling cascades. An inactivating mutation of one receptor subunit’s kinase domain results in abolishment of signaling; such a mutation in the kinase domain of the VEGF receptor 3 (VEGFR–3) results in the autosomal dominantly inherited lymphatic disorder called Milroy disease.
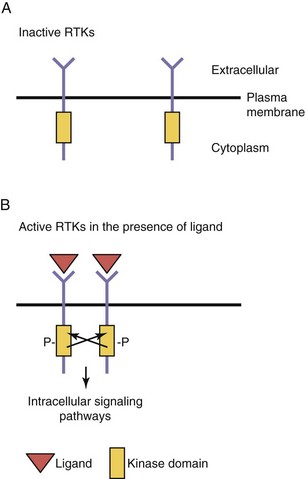
Regulation of Angiogenesis by Receptor Tyrosine Kinases
Growth factors generally promote cellular proliferation, migration, and survival (i.e., they are antiapoptotic). During embryogenesis, signaling through RTKs is crucial for normal development and affects many different processes such as the growth of new blood vessels (see Chapter 5), cellular migration, and neuronal axonal guidance.
Endothelial cells are derived from a progenitor cell (the hemangioblast) that can give rise to both the hematopoietic cell lineage and endothelial cells. The early endothelial cells proliferate and eventually coalesce to form the first primitive blood vessels. This process is termed vasculogenesis. After the first blood vessels are formed, they undergo intensive remodeling and maturation into the mature blood vessels in a process called angiogenesis. This maturation process involves the recruitment of vascular smooth muscle cells to the vessels that stabilize them. Vasculogenesis and angiogenesis are both dependent on the function of two distinct RTK classes, members of the VEGF and Tie receptor families. VEGF-A was shown to be essential for endothelial and blood cell development; VEGF-A knockout mice fail to develop blood or endothelial cells and die at early embryonic stages. A related molecule, VEGF-C, was shown to be crucial for the development of lymphatic endothelial cells. VEGF-A signals through two receptors, VEGFR–1 and VEGFR–2, that are expressed by endothelial cells but VEGFR–2 predominates in vasculogenesis in the embryo.
The process of angiogenic refinement depends on the function of the angiopoietin/Tie2 signaling pathway. Tie2 is a RTK that is specifically expressed by endothelial cells and angiopoietin 1 and angiopoietin 2 are its ligands that are expressed by the surrounding vascular smooth muscle cells. This represents a paracrine signaling system in which receptor and ligand are expressed in adjacent cells. Both the VEGF/VEGFR–2 and angiopoietin/Tie2 signaling pathways are co-opted by tumors to stimulate growth of new blood vessels, which stimulates their growth and metastasis. This demonstrates how normal signaling pathways in the embryo can be reused by disease processes, such as cancer.
Notch–Delta pathway
The Notch signaling pathway is integral for cell fate determination, including maintenance of stem-cell niches, proliferation, apoptosis, and differentiation. These processes are essential for all aspects of organ development through regulation of lateral and inductive cell-cell signaling. Notch proteins are single transmembrane receptors that interact with membrane-bound Notch ligands (e.g., Delta-like ligands and serrate-like ligands [Jagged]) on adjacent cells (Fig. 20-6). Ligand-receptor binding triggers proteolytic events leading to the release of the Notch intracellular domain (NICD). When the NICD translocates to the nucleus, a series of intranuclear events culminates in the induction of expression a transcription factor that maintains the progenitor state of the cell.
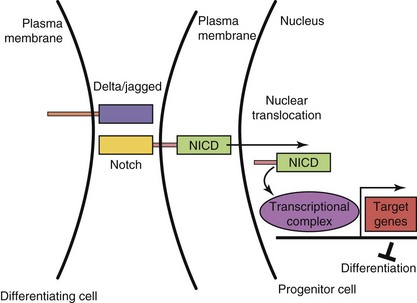
Figure 20–6 Notch–Delta signaling pathway. In progenitor cells (right), activation of Notch signaling leads to cleavage of the Notch intracellular domain (NICD). NICD translocates to the nucleus, binds to a transcriptional complex, and activates target genes, such as the bHLH gene, Hes1, that inhibit differentiation. In differentiating cells (left), Notch signaling is not active.
Lateral inhibition ensures in a population of cells with equivalent developmental potential, the correct number of two distinct cell types. In the initial cell–cell interaction, Notch receptor signaling maintains one cell as an uncommitted progenitor. The adjacent cell maintains reduced Notch signaling and undergoes differentiation. Inductive signaling with other surrounding cells expressing morphogens may overcome a cell’s commitment to a default fate to an alternative cell fate. Understanding the function of the Notch–Delta signaling pathway in mammalian development has been assisted by loss-of-function studies in the mouse. Mutations Alagille syndrome (arteriohepatic dysplasia), with liver, kidney, cardiovascular, ocular, and skeletal malformations, and NOTCH–3 gene mutations in the CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) adult vascular degenerative disease, with a tendency to early-age onset of stroke-like events, support the importance of the Notch signaling pathway in embryonic and postnatal development, respectively.
Transcription Factors
Transcription factors belong to a large class of proteins that regulate the expression of many target genes, either through activation or repression mechanisms. Typically, a transcription factor will bind to specific nucleotide sequences in the promoter/enhancer regions of target genes and regulate the rate of transcription of its target genes via interacting with accessory proteins. Transcription factors can both activate or repress target gene transcription depending on the cell in which they are expressed, the specific promoter, the chromatin context, and the developmental stage. Some transcription factors do not need to bind to DNA to regulate transcription: they may bind to other transcription factors already bound to the promoter DNA thereby regulating transcription or bind and sequester other transcription factors from their target genes, thus repressing their transcription. The transcription factor superfamily is composed of many different classes of proteins. Three examples of this diverse family of proteins are described: Hox/Homeobox, Pax, and bHLH transcription factors.
Hox/Homeobox Proteins
The Hox genes were first discovered in the fruit fly, Drosophila melanogaster. The order of the Hox genes along the AP axis is faithfully reproduced in their organization at the level of the chromosome. Mutations in these genes of the HOM-C complex lead to dramatic phenotypes (homeotic transformation) such as the Antennapedia gene in which legs instead of antennae sprout from the head of the fruit fly. In humans, the order of the Hox genes along the AP axis and chromosomal location is conserved as well. Defects in HOXA1 have been shown to impair human neural development, and mutations in HOXA13 and HOXD13 result in limb malformations.
All the Hox genes contain a 180-base-pair sequence, the homeobox, which encodes a 60-amino-acid homeodomain composed of three α helices. The third (recognition) helix binds to DNA sites that contain one or more binding motifs in the promoters of their target genes. The homeodomain is the most conserved region of the protein and is highly conserved across evolution, whereas other regions of the protein are not as well conserved. Mutations in the DNA-binding region of the homeobox gene NKX2.5 are associated with cardiac atrial-septal defects and mutations in ARX are associated with the central nervous system malformation syndrome lissencephaly.
Pax Genes
The Pax genes all contain conserved bipartite DNA-binding motifs called the Pax (or paired) domain, and most Pax family members also contain a homeodomain. PAX proteins have been shown to both activate and repress transcription of target genes. The Drosophila melanogaster ortholog of Pax6, eyeless, was shown to be essential for eye development because homozygous mutant flies had no eyes. Eyeless shares a high degree of sequence conservation with its human ortholog PAX6 and is associated with ocular malformations such as aniridia (absence of the iris) and Peter’s anomaly. In human eye diseases, the level of PAX6 expression seems to be crucial because patients with only one functional copy (haploinsufficiency) have ocular defects and patients without PAX6 function are anophthalmic. This concept of haploinsufficiency is a recurring theme for many different transcription factors and corresponding human malformations.
PAX3 and PAX7 encode both homeodomain and Pax DNA-binding domains. The human childhood cancer alveolar rhabdomyosarcoma results from a translocation that results in the formation of a chimeric protein wherein PAX3 or PAX7 (including both DNA domains) is fused to the strong activating domains of the Forkhead family transcription factor FOXO1. The autosomal dominant human disease Waardenburg’s syndrome type I has been demonstrated to result from mutations in the PAX3 gene. Patients with this syndrome have hearing deficits, ocular defects (dystopia canthorum), and pigmentation abnormalities best typified by a white forelock.
Basic Helix-Loop-Helix (bHLH) Transcription Factors
The bHLH genes are a class of transcription factors that regulate cell fate determination and differentiation in many different tissues during development. At a molecular level, bHLH proteins contain a basic (positively charged) DNA-binding region that is followed by two α helices that are separated by a loop. The α helices have a hydrophilic and a hydrophobic side (amphipathic). The hydrophobic side of the helix is a motif for protein-protein interactions between different members of the bHLH family. This domain is the most conserved region of the bHLH proteins across different species. bHLH proteins often bind other bHLHs (heterodimerize) to regulate transcription. These heterodimers are composed of tissue-specific bHLH proteins bound to ubiquitously expressed bHLH proteins. The powerful prodifferentiation effect of bHLH genes can be repressed by several different mechanisms. For example, inhibitor of differentiation (Id) proteins are HLH proteins that lack the basic DNA-binding motif. When Id proteins heterodimerize with specific bHLH proteins, they prevent binding of these bHLH proteins to their target gene promoter sequences (called E-boxes).
Growth factors, which tend to inhibit differentiation, increase the level of Id proteins that sequester bHLH proteins from their target promoters. In addition, growth factors can stimulate the phosphorylation of the DNA-binding domain of bHLH proteins, which inhibits their ability to bind to DNA. bHLH genes are crucial for the development of tissues such as muscle (MyoD/Myogenin) and neurons (NeuroD/Neurogenin) in humans. MyoD expression was shown to be sufficient to transdifferentiate several different cell lines into muscle cells, demonstrating that it is a master regulator of muscle differentiation. Studies of knockout mice confirmed that MyoD and another bHLH, Myf5, are crucial for the differentiation of precursor cells into primitive muscle cells (myoblasts). Similarly, Mash1 and Neurogenin1 are proneural genes that regulate the formation of neuroblasts from the neuroepithelium. Mouse models have shown that these genes are crucial for the specification of different subpopulations of precursors in the developing central nervous system. For example, Mash1 knockout mice have defects in forebrain development, whereas Neurogenin1 knockout mice have defects in cranial sensory ganglia and ventral spinal cord neurons. Muscle and neuronal differentiation are controlled by a cascade of bHLH genes that function at early and at late stages of cellular differentiation. In addition, both differentiation pathways are inhibited via signaling through the Notch pathway.
Epigenetics
Epigenetics refers to inherited changes that affect gene expression as a result of mechanisms other than changes in the sequence of DNA. Examples include DNA methylation and chromatin modifications, such as acetylation of histones.
DNA Methylation
DNA is methylated at cytosine residues by DNA methyltransferases at CpG sites, where cytosine and guanine nucleotides are directly paired. CpG islands are DNA regions with high concentrations of CpG sites, and are often located in proximal promoter regions of genes. DNA methylation at CpG sites, in general, leads to a reduction of gene expression or gene silencing, whereas DNA hypomethylation at CpG sites, conversely leads to gene overexpression. Silencing of tumor suppressor genes or overexpression of oncogenes may lead to cancer.
Acetylation
Histones are the positively charged nuclear proteins around which genomic DNA is coiled to tightly pack it within the nucleus. Modification of these proteins is a common pathway by which transcription factors regulate the activity of their target promoters. One such modification is acetylation. DNA is less tightly bound to acetylated histones, thus allowing for more open access of transcription factors and other proteins to the promoters of their target genes. Histone acetylation status is controlled by genes that add acetyl groups (histone transferase) or remove acetyl groups (histone deacetylases) (Fig. 20-7). Phosphorylation of histones also leads to an opening of the chromatin structure and activation of gene transcription. Disorders of chromatin remodeling include Rett, Rubinstein-Taybi, and alpha-thalassemia/X-linked mental retardation syndromes.
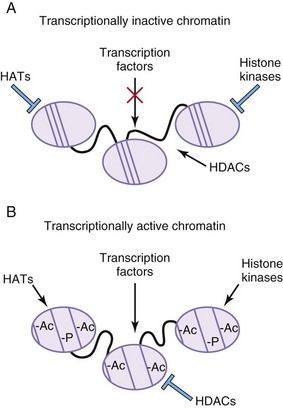
Figure 20–7 Histone modifications alter transcriptional properties of chromatin. A, In areas of transcriptionally inactive chromatin, the DNA is tightly bound to the histone cores. The histones are not acetylated or phosphorylated. Histone deacetylases (HDACs) are active, whereas histone acetyl transferases (HATs) and histone kinases are inactive. B, In areas of transcriptionally active chromatin, the DNA is not as tightly bound to the histone cores. The histone proteins are acetylated (Ac) and phosphorylated (-P). HDACs are inactive, whereas HATs and histone kinases are active.
Summary of Common Signaling Pathways Used During Development
• There are marked differences among the various signaling pathways, but they share many common features: ligands, membrane-bound receptors and coreceptors, intracellular signaling domains, adapters, and effector molecules.
• Signaling pathways are co-opted at various times during development for stem cell renewal, cell proliferation, migration, apoptosis, and differentiation.
• Pathways have “default” settings that result in generation or maintenance of one cell fate rather than another.
• Many genes and signaling pathways are highly conserved throughout evolution.
• Knowledge of gene function has been acquired by reverse genetics using model systems with loss- or gain-of-function transgenic approaches and by forward genetics beginning with the description of abnormal phenotypes arising spontaneously in mice and humans and then subsequent identification of the mutant gene.
• There is evidence of cross-talk among pathways. This communication among various signaling pathways facilitates our understanding of the far-reaching consequences of single gene mutations that result in malformation syndromes affecting the development of multiple organ systems or in cancers.