Chapter 5 Pathogenesis of microbial disease
If a microorganism is capable of causing disease, it is called a pathogen. Fortunately, only a minority of the vast multitude of microorganisms in nature are pathogenic. Whereas some organisms are highly virulent and cause disease in healthy individuals, even with a small inoculum, others cause disease only in compromised individuals when their defences are weak. The latter are called opportunistic organisms, as they take the opportunity offered by reduced host defences to cause disease. These opportunists are frequently members of the body’s normal flora.
General aspects of infection
Virulence
Virulence is a quantitative measure of pathogenicity and is related to an organism’s toxigenic potential and invasiveness. Virulence can be measured by the number of organisms required to cause disease and is designated as LD50 or ID50: the LD50 (50% lethal dose) is the number of organisms needed to kill half the hosts, and ID50 (50% infectious dose) is the number needed to cause infection in half the hosts. These values are determined by inoculation of laboratory animals.
Communicable diseases
Infections are called ‘communicable diseases’ if they are spread from host to host. Many, but not all, infections are communicable; for example, tuberculosis is communicable, as it is spread by airborne droplets produced by coughing, but staphylococcal food poisoning is not, as the exotoxin produced by the organism and present in the contaminated food affects only those eating that food. If a disease is highly communicable, it is called a ‘contagious disease’ (e.g. chickenpox).
Depending on the degree of incidence and prevalence of an infectious disease in a community, it may be called an endemic, an epidemic or a pandemic infection:
Natural history of infectious disease
An acute infection generally progresses through four stages:
A number of organisms may elicit an inapparent or subclinical infection, without overt symptoms, where the individual remains asymptomatic although infected with the organism. On the other hand, once infected, the body may not completely eliminate the pathogen after recovery and some individuals may become chronic carriers of the organism (e.g. Salmonella typhi, hepatitis B virus); they may shed the organism while remaining healthy. Some infections result in a latent state, after which reactivation of the growth of the organism and recurrence of symptoms may occur at a later stage (e.g. after primary herpes infection, the virus may reside in a latent state in the trigeminal ganglion, causing recurrent herpes labialis from time to time). All the above groups may unknowingly shed pathogenic organisms and spread disease.
Pathogenesis of bacterial disease
Determinants of bacterial pathogenicity
Bacterial pathogenicity is a vast subject. The following is a brief outline of the ways and means by which bacteria cause disease. The major steps are transmission, adherence to host surfaces, invasiveness and toxigenicity.
Transmission
Most infections are acquired by transmission from external sources; i.e. they are exogenous in origin. Others are caused by members of the normal flora behaving as opportunist pathogens; i.e. they are endogenous in origin. Transmission can be by:
There are four important portals (or gates) of entry of pathogens (Table 5.1):
Table 5.1 Portals of entry of some common pathogens
| Portal of entry | Pathogen | Disease |
|---|---|---|
| Skin | Clostridium tetani | Tetanus |
| Hepatitis B virus | Hepatitis B | |
| Respiratory tract | Streptococcus pneumoniae | Pneumonia |
| Neisseria meningitidis | Meningitis | |
| Haemophilus influenzae | Meningitis | |
| Mycobacterium tuberculosis | Tuberculosis | |
| Influenza virus | Influenza | |
| Rhinovirus | Common cold | |
| Epstein–Barr virus | Infectious mononucleosis | |
| Gastrointestinal tract | Shigella dysenteriae | Dysentery |
| Salmonella typhi | Typhoid fever | |
| Vibrio cholerae | Cholera | |
| Hepatitis A virus | Infectious hepatitis | |
| Poliovirus | Poliomyelitis | |
| Genital tract | Neisseria gonorrhoeae | Gonorrhoea |
| Treponema pallidum | Syphilis | |
| Human immunodeficiency virus (HIV) | Acquired immune deficiency syndrome (AIDS) | |
| Candida albicans (fungus) | Vaginitis |
Adherence to host surfaces
Adherence is the first step in infection. Unless organisms have the ability to stick or adhere to host surfaces, they will be unable to cause infection. Some bacteria and fungi have specialized structures or produce substances that facilitate their attachment to the surface of human cells or prostheses (e.g. dentures, artificial heart valves), thereby enhancing their ability to colonize and cause disease. These adherence mechanisms are critical for organisms that attach to mucous membranes; mutants that lack these mechanisms are often non-pathogenic (e.g. the hair-like pili of Neisseria gonorrhoeae and Escherichia coli mediate their attachment to the urinary tract epithelium; the extracellular polysaccharides of Streptococcus mutans help it adhere to enamel surfaces).
Biofilm formation
Once the organisms adhere to a host surface they usually tend to aggregate and form intelligent communities of cells called biofilms. A biofilm is defined as an aggregate of interactive bacteria attached to a solid surface (such as a denture prosthesis or an intravenous catheter) or to each other, encased in an extracellular polysaccharide matrix. Up to 65% of human infections are thought to be associated with microbial biofilms. Dental plaque on solid enamel surfaces is a classic example of a biofilm. As biofilms are ubiquitous in nature and form on hulls of ships, warm water pipes, dental unit water systems and so on, their study has rapidly evolved during the past few decades, leading to many discoveries on communal behaviour of microbes.
As mentioned, biofilms are intelligent communities. Structurally, they are not flat and compressed but comprise a complex architecture with towers and mushroom or dome-shaped structures with water channels that permit transport of metabolites and nutrients (Figs 5.1-5.3). Bacteria in biofilms maintain the population level by constantly secreting low levels of chemicals called quorum-sensing molecules (e.g. homoserine lactone), which tend to repulse incoming bacteria or activate the communal bacteria to seek new abodes. Further, specific gene activation may lead to production of virulence factors or reduction in metabolic activity (especially those living deep within the matrix).
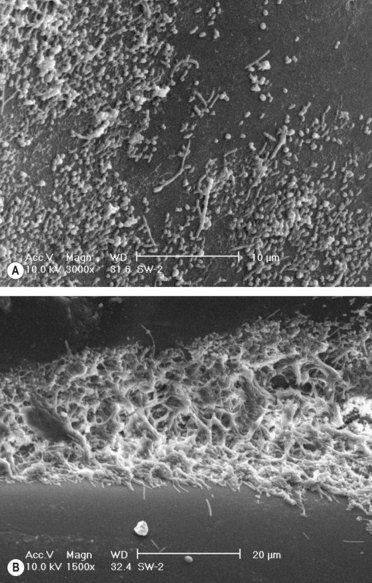
Fig. 5.1 The ultrastructure of (A) an early biofilm on a dental appliance showing the deposition of coccal and bacillary forms; (B) a mature dental plaque biofilm on a dental appliance showing the advancing edge and the complex architecture.
(Courtesy of Dr Bernard Low.)
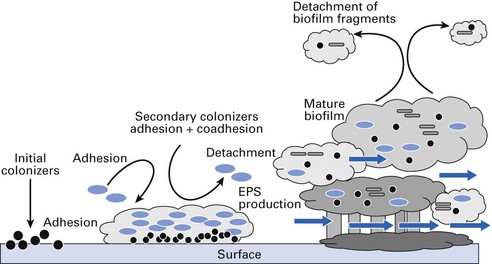
Fig. 5.2 A schematic diagram depicting the various developmental stages of a biofilm from the initial adherent phase (left) of the organisms to gradual maturation and subsequent fully developed polymicrobial biofilm (extreme right). EPS, extracellular polysaccharide.

Fig. 5.3 A mature Candida albicans biofilm showing water channels (arrows) that mediate metabolite and nutrition transfer to and from the biofilm (inset: magnified channel architecture).
It is now known that infections associated with biofilms are difficult to eradicate as sessile organisms in biofilms exhibit higher resistance to antimicrobials than their free-living or planktonic counterparts. The reasons for this appear to be (Fig. 5.4):
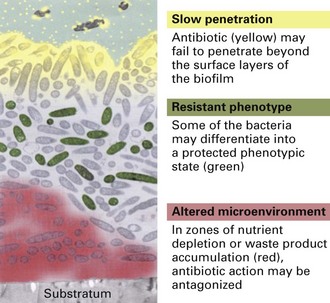
Fig. 5.4 Postulated mechanisms of antibiotic resistance in biofilms: the attachment surface is shown at the bottom and the aqueous phase containing the antibiotic at the top.
(Modified from Stewart and Costerton Lancet 2001; 358; 135–138 with permission.)
Some examples of important recalcitrant human infections mediated by biofilms, difficult to manage by antimicrobials alone, include Pseudomonas aeruginosa infections of the respiratory tract in cystic fibrosis patients, Staphylococcus aureus infections in central venous catheters, chronic candidal infections of HIV-infected individuals and chronic periodontal infections due to dental plaque.
Invasiveness
Invasiveness of bacteria plays a critical role in pathogenesis; this property is dependent upon secreted bacterial enzymes. A few examples are:
Other factors also contribute to invasiveness by interfering with the host defence mechanisms, especially phagocytosis:
Table 5.2 Examples of surface virulence factors that interfere with host defences
| Organism | Virulence factor | Used in vaccine |
|---|---|---|
| Bacteria | ||
| Streptococcus pneumoniae | Polysaccharide capsule | Yes |
| Streptococcus pyogenes | M protein | No |
| Staphylococcus aureus | Protein A | No |
| Neisseria meningitidis | Polysaccharide capsule | Yes |
| Haemophilus influenzae | Polysaccharide capsule | Yes |
| Klebsiella pneumoniae | Polysaccharide capsule | No |
| Escherichia coli | Protein pili | No |
| Salmonella typhi | Polysaccharide capsule | No |
| Mycobacterium tuberculosis | Mycolic acid cell wall | No |
| Fungi | ||
| Cryptococcus neoformans | Capsule | No |
Bacterial infection may lead to two categories of inflammation: pyogenic (pus-producing) and granulomatous (granuloma-forming).
Pyogenic inflammation
The neutrophils are the predominant cells in this type of inflammation. Streptococcus pyogenes, Staphylococcus aureus and Streptococcus pneumoniae are the common pyogenic bacteria.
Granulomatous inflammation
Macrophages and T cells predominate in this type of inflammation. The most notable organism in this category is Mycobacterium tuberculosis. Here, the bacterial antigens stimulate the cell-mediated immune system, resulting in sensitized T-lymphocyte and macrophage activity. Although the phagocytic activity of macrophages kills most of the tubercle bacilli, some survive and grow within these cells, leading to granuloma formation. The organisms reside within phagosomes, which are unable to fuse with lysosomes, resulting in protection from degradative enzymes therein. Many fungal diseases are also characterized by granulomatous lesions.
Toxigenicity
Toxin production or toxigenicity is another major mediator of bacterial disease. Toxins are of two categories: endotoxins and exotoxins. Their main features are shown in Table 5.3.
Table 5.3 Comparison of the main features of exotoxins and endotoxins
| Property | Exotoxin | Endotoxin |
|---|---|---|
| Source | Some species of some Gram-positive and Gram-negative bacteria | Cell walls of Gram-negative bacteria |
| Origin | Secreted from cell | Cell wall constituent |
| Chemistry | Polypeptide | Lipopolysaccharide |
| Toxicity | High (fatal dose of the order of 1 µg) | Low (fatal dose in the order of hundreds of micrograms) |
| Clinical effects | Variable | Fever, shock |
| Antigenicity | Induces high-titre antibodies called antitoxins | Poorly antigenic |
| Vaccines | Toxoids used as vaccines | No toxoids formed and no vaccine available |
| Heat stability | Most are thermolabile (destroyed rapidly at 60°C) | Thermostable at 100°C for 1 h |
| Typical diseases | Cholera, tetanus, diphtheria | Sepsis by Gram-negative rods, endotoxic shock |
Toxin production
Endotoxins
Endotoxins are the cell wall lipopolysaccharides of Gram-negative bacteria (both cocci and bacilli) and are not actively released from the cell. (Note: thus, by definition, Gram-positive organisms do not possess endotoxins.) Endotoxins cause fever, shock and other generalized symptoms.
A number of biological effects of endotoxin are described below. These are mainly due to the production of host factors such as interleukin-1 (IL-1) and tumour necrosis factor (TNF) from macrophages.
Endotoxin-like effects may also occur in Gram-positive bacteraemic infections. However, as endotoxin is absent in Gram-positive bacteria, other cell wall components, such as teichoic acid or peptidoglycan, are thought to trigger the release of TNF and IL-1 from macrophages.
Exotoxins
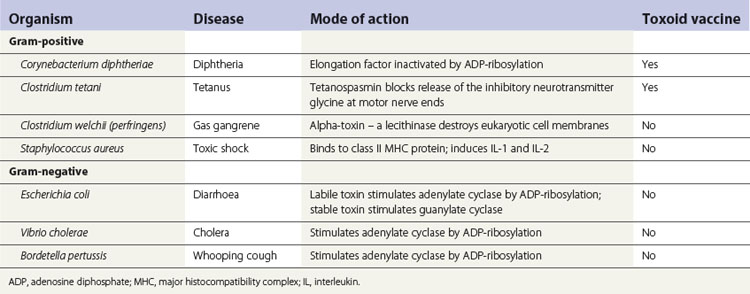
Both Gram-positive and Gram-negative bacteria (Table 5.4) secrete exotoxins, whereas endotoxin is an integral component of the cell wall of Gram-negative organisms. Exotoxins in particular can cause disease in distant parts of the body as a result of diffusion or carriage of the toxin via systemic routes (e.g. tetanus bacillus infecting a lesion in the foot produces an exotoxin, which causes ‘lockjaw’ or spasm of masseter muscles on the face).
Exotoxins are polypeptides whose genes are frequently located on plasmids or lysogenic bacterial viruses. Essentially, these polypeptides consist of two domains or subunits: one for binding to the cell membrane and entry into the cell, and the other possessing the toxic activity.
Exotoxins are highly toxic (e.g. the fatal dose of tetanus toxin for a human can be less than 1 µg). Fortunately, exotoxin polypeptides are good antigens and induce the synthesis of protective antibodies called antitoxins, useful in the prevention or treatment of diseases such as tetanus. The toxicity of the polypeptides can be neutralized when treated with formaldehyde (or acid or heat), and these toxoids are used in protective vaccines because they retain their antigenicity.
Bacterial exotoxins can be broadly categorized as:
Neurotoxins
Tetanus toxin, diphtheria toxin and botulinum toxin are all neurotoxins and their action is mediated via neuronal pathways.
Tetanus toxin, produced by Clostridium tetani, is a neurotoxin that prevents the release of the inhibitory neurotransmitter glycine, thus causing muscle spasms (see Fig. 13.4). Tetanus toxin (tetanospasmin) comprises two polypeptide subunits: a heavy chain and a light chain. The former binds to the gangliosides in the membrane of the neuron, while the latter is the toxic component. The toxin is liberated at the peripheral wound site but is transmitted to the neurons of the spinal cord either by retrograde axonal transport or in the blood stream. There it blocks the release of the inhibitory transmitter, which leads to sustained and convulsive contractions of the voluntary muscles (e.g. risus sardonicus, contraction of the facial muscles; lockjaw, contraction of the masseter muscles).
Diphtheria toxin, produced by Corynebacterium diphtheriae, is synthesized as a single polypeptide with two functional domains. Once secreted, one domain mediates the binding of the toxin to cell membrane receptors; the other domain possesses enzymatic activity and inhibits protein synthesis in all eukaryotic cells. The enzyme activity is highly potent: a single molecule can kill a cell within a few hours. E. coli, Vibrio cholerae and Bordetella pertussis also possess exotoxins that act in a similar manner.
Botulinum toxin, produced by Clostridium botulinum, is one of the most toxic compounds known (1 µg will kill a human). The toxin blocks the release of acetylcholine at the synapse, producing paralysis of both voluntary and involuntary muscles. The toxin, encoded by the genes of a bacteriophage, comprises two polypeptide subunits.
Enterotoxins
These toxins act on the gut mucosa and cause gastrointestinal disturbances.
E. coli enterotoxin is of two types: one heat labile and one heat stable. The heat-labile toxin (inactivated at 65°C in 30 min) is composed of two domains: one binds to a ganglioside in the cell membrane, while the other is the active component and mediates synthesis of cyclic adenosine monophosphate (cAMP) in the mucosal cells of the small intestine. This leads to an increase in the concentration of cAMP, which promotes cellular chloride ion excretion and inhibition of sodium ion absorption. The net result is fluid and electrolyte loss into the lumen of the gut (diarrhoea).
The heat-stable toxin of E. coli (not inactivated by boiling for 30 min) stimulates guanylate cyclase and thus increases the concentration of cyclic guanosine monophosphate (cGMP), which inhibits the reabsorption of sodium ions and causes diarrhoea (compare with heat-labile toxin). The genes for both toxins are carried on a plasmid.
The enterotoxins produced by the diarrhoea-causing organisms V. cholerae and Bacillus cereus act in a manner similar to that of the heat-labile toxin of E. coli.
Miscellaneous exotoxins
An array of exotoxins are produced by C. welchii and other species of clostridia that cause gas gangrene. These include the α-toxin (a phospholipase that hydrolyses lecithin, present in all eukaryotic cell membranes), collagenase, protease, hyaluronidase and deoxyribonuclease (DNAase). As the names imply, they destroy the cells and the connective tissue by a multiplicity of actions. In addition, a heterogeneous group of toxins with a haemolytic and necrotizing activity have been identified in clostridia.
Pathogenesis of viral disease
Viral pathogenesis can be defined as the methods by which viruses produce disease in the host. The vast majority of viral infections are subclinical (symptomless) and go almost unrecognized. One individual may succumb to disease with an infection by a virus, while another may be entirely asymptomatic when infected by the identical strain of virus; genetic factors, immunity, nutrition and other factors influence the results of infection. The study of viral pathogenesis can be considered at two levels: first, at the level of the virus (parasite) and, second, at the level of the host.
Entry of viral infections
As in bacterial infections, viruses gain entry into the host by:
See Figure 5.5. (Note: although in this section viruses are considered separately, very similar host defence mechanisms operate to prevent the entry of all other pathogens through these portals.)
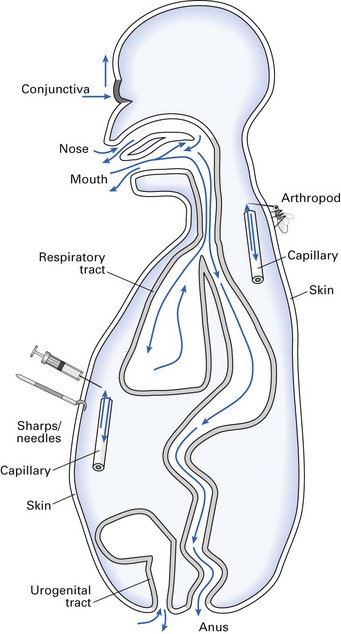
Skin and mucosa
The skin is an effective barrier against viral infection as the dead cells of the stratum corneum cannot support viral replication. Breach of skin integrity occurs:
Oropharynx and intestinal tract
Natural defence mechanisms of the mouth and the gastrointestinal tract that prevent viral entry are:
Respiratory tract
A number of defence mechanisms operate to prevent viral entry through the respiratory tract. These include:
To gain access to the respiratory tract, viruses need to be primarily in the form of aerosol particles or droplets. Other factors that affect viral respiratory infection include the humidity and air temperature (e.g. influenza is more common in the winter months) and the physical and chemical properties and structure of the virus particle.
Genitourinary tract
The vagina and urethra can be portals for entry of viral infection. The host factors that can influence viral entry via these routes include:
Sexual activity may cause tears or abrasions of the vaginal epithelium or trauma to the urethra, allowing viral ingress. Sexually transmitted viruses in humans include HIV, herpesviruses, human papillomaviruses and most hepatitis viruses.
Mechanisms of viral spread in the body
Viruses, unlike some bacteria, are completely devoid of organelles of transport, and they spread throughout the body by a number of routes. These include:
Local spread on body surfaces
A number of viruses cause disease on epithelial surfaces without systemic spread. Such infections are characterized by:
Once an invading virus overcomes the epithelial barrier, it is exposed to the second line of body defences in the form of phagocytic cells, predominantly histiocytes of the macrophage series. When the virus is phagocytosed, it will be destroyed not only by the low pH conditions in the phagocytic vesicle but also by enzymes in the phagolysosome. Some viruses have developed mechanisms to evade this type of defence and, indeed, replicate within the macrophages.
Lymphatic spread
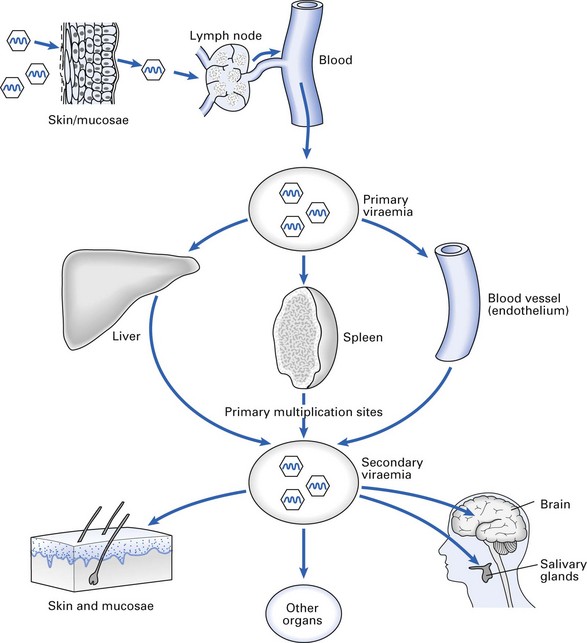
The phagocytosed and free viral particles lurking beneath the epithelium rapidly enter the subepithelial/mucosal network of lymphatic capillaries and are carried to regional lymph nodes (Fig. 5.6). Lymph nodes serve two main functions:
Soon after entering the lymph node, viruses are exposed to the macrophages lining the marginal sinus. If the virus is phagocytosed, antigens are presented to the underlying lymphoid cells to evoke an immune response, on the success of which depends the outcome of the infection. If the virus is inactivated, the infection resolves. However, the organism may infect macrophages and lymphocytes if the immune response at this stage is inadequate (e.g. herpesviruses, measles). The virus particles that escape the ‘nodal filter’ can then enter the blood stream via the efferent lymphatics and the thoracic duct (Fig. 5.6).
There is a constant bidirectional movement of macrophages and lymphocytes from the blood into lymph nodes and vice versa. Thus, if a virus infects cells in lymph nodes without damaging them, these cells can act as vehicles of virus dissemination. Sometimes, the virus infects and multiplies in lymphatic endothelium, further increasing the virus load reaching the node and hence the lymphatic system. Viruses do not appear to enter the local blood vessels directly, except perhaps when these are damaged mechanically by trauma (e.g. needlestick injury, bites).
These events are closely followed by a local inflammatory response that alters the eventual outcome of the viral infection, as described below.
Viraemia and spread to organs
The entry of virus into the blood and its subsequent spread is called viraemia. Once a virus reaches the blood stream, it is effectively disseminated within minutes. The first episode of viral entry into the blood is called a primary viraemia (Fig. 5.6). The virus may then be seeded in various distant organs, after which there is further replication at these sites and a second wave of viral entry into the blood stream – a secondary viraemia. This is usually larger than the primary viraemia and the virus is more easily detected in blood samples. The secondary viraemia often leads to infection of other organs.
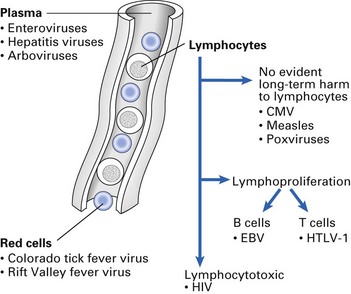
Viruses may be free in the plasma, in blood cells, or in both (Fig. 5.7). Those in the plasma can be relatively easily cleared, but viruses in leukocytes are not easily destroyed. If the infected leukocyte remains healthy, it may disseminate infection to distant body sites. Once a virus reaches an organ, its localization depends on its ability to attach to and grow in vascular endothelial cells, and on phagocytosis by reticuloendothelial cells.
Central nervous system and peripheral nerve spread
During a viraemia, circulating viruses invade the central nervous system by localizing in the blood vessels of:
The process of localization is enhanced when there is an associated inflammatory focus. Peripheral nerves act as an effective path of transmission for some viruses, such as herpes simplex virus. Viral passage can be either centripetal (from body surface inwards), as in rabies, or centrifugal, as in reactivation of herpes simplex (herpes labialis) or varicella-zoster. This mode of transport is a slow process (mm/h) compared with viraemic spread. Four possible routes of viral transmission in peripheral nerves are known:
Virus and host cell interactions
Once the virus enters the host cell, it can interact with the host cell in two main ways:
Permissive infection
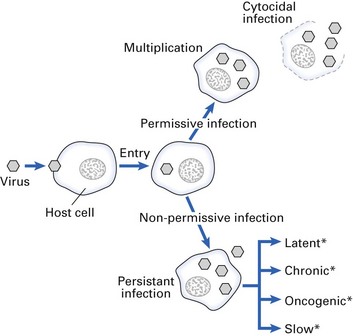
The infection of a cell by a virus may have one or more sequelae (Fig. 5.8). The most common sequela is for the virus to replicate in a lytic or cytocidal infection, causing the cells to die and producing an acute illness. A virus-infected cell may die as a result of:
The adverse cellular consequence of viral infection, particularly that observed in virus-infected cells in tissue culture, is termed the cytopathic effect (see Chapter 7). During the early phase of infection, before cell death, characteristic alterations in the infected cell membrane may occur. Haemadsorption and giant cell formation are two examples.
Haemadsorption
In viruses that leave the cell by budding through the plasma membrane, viral glycoproteins (destined for the envelope) are first inserted into the membrane. A common envelope protein is haemagglutinin; this protein enables an infected cell to attract red cells at its surface, a phenomenon called haemadsorption. Haemadsorption can be used in the laboratory to detect cells infected with certain viruses (e.g. orthoviruses and paramyxoviruses).
Non-permissive infection
Cell death is not an inevitable accompaniment of viral replication. Sometimes a persistent infection may ensue in which there may be viral replication within the cell but the cell remains alive. Many viruses can produce persistent infections. Some relevant examples are hepatitis B virus, papillomaviruses, herpesviruses and retroviruses. Factors that favour persistence include:
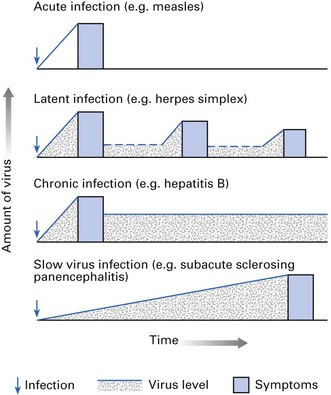
There are four categories of persistent infection: latent, chronic, oncogenic and slow (Fig. 5.9).
Latent viral infections
These occur when viral nucleic acids persist in the cell, usually integrated into the host DNA as a provirus (e.g. HIV, herpes simplex, varicella-zoster). As herpes simplex infection can be considered as the classic example of latent infection, the mechanism of its persistence is described. After an acute infection, the herpes simplex virus travels along sensory nerve fibres (intra-axonal transport) to the appropriate dorsal root ganglion (e.g. in oral herpes, the virus travels to the trigeminal ganglion). During latency, infectious virus is undetectable, but virus may be recovered by growing ganglion fragments in tissue culture. The re-emergence of virus is prevented, possibly due to host cell-mediated immunity, but when this wanes, there may be recrudescence and shedding of the virus in secretions from the area. Similarly, varicella-zoster virus remains latent for many years and may spontaneously recur as zoster (shingles) on dermatomes supplied by the specific sensory ganglion in which the virus is latent. Latent viral infections are reactivated particularly in immunocompromised patients, who subsequently suffer from infection and excrete the virus (see Chapter 30).
Chronic infections
These occur when viruses persist in quantity in the body over a prolonged period, with or without a history of disease. These chronically infected individuals, who are often asymptomatic, are called ‘carriers’ and are an important potential source of infection for others. Carriers make up a significant but unknown proportion of patients treated by dental health care workers (see Chapter 36). The main difference between chronic and latent infections is that the virus is continuously detectable in the former but not in the latter.
Host determinants of viral infection
The outcome of viral infection in a host depends not only on the type and virulence of the virus but also on host factors, including:
Koch’s postulates
A wide spectrum of microbes inhabit the human body. Some are permanent residents living as commensals, others are transient organisms and still others are commensals that behave as pathogens under suitable conditions (opportunistic pathogens). Hence when infection supervenes, it is important to differentiate a commensal from a pathogen in order to identify and eliminate the latter. This problem was encountered by Robert Koch, a German general practitioner, in 1877 when he tried to determine the cause of an infection called anthrax in cattle and tuberculosis in humans. Koch defined the criteria for attributing an organism as the cause of specific disease. These criteria, called Koch’s postulates, are as follows:
Currently, these four postulates are complemented by another:
Clearly, these are ideal criteria and are not always attainable in practice (e.g. Mycobacterium leprae, the leprosy bacillus, cannot be cultured in vitro), but they provide a framework for establishing an aetiological role of organisms in infectious diseases.
Key facts
Costerton J.W., Stewart P.S., Greenberg E.P. Bacterial biofilms: A common cause of persistent infections. Science. 1999;284:1318-1322.
Filoche S., Wong L., Sissons C.H. Oral biofilms: Emerging concepts in microbial ecology. Journal of Dental Research. 2010;89:8-18.
Inglewski B.H., Clark L.V., editors. The bacteria. Molecular basis of bacterial pathogenesis, Vol. 11. London: Academic Press, 1990.
Mims C., Dimmock N., Nash A., Stephen J. Mims’ pathogenesis of infectious disease, 4th ed. London: Academic Press; 1995.
Scully C., Samaranayake L.P. Clinical virology in oral medicine and dentistry, Ch. 3. Cambridge: Cambridge University Press. 1992.
Stewart P.S., Costerton J.W. Antibiotic resistance of bacterial biofilms. Lancet. 2001;358:135-138.
Review questions (answers on p. 351)
Please indicate which answers are true, and which are false.