
Barry W. Connors
After meticulous study of spinal reflexes, Charles Sherrington deduced that neurons somehow communicate information, one to the next, by a mechanism that is fundamentally different from the way that they conduct signals along their axons. Sherrington had merged his physiological conclusions with the anatomical observations (Fig. 13-1) of his contemporary, the preeminent neuroanatomist Santiago Ramón y Cajal. Cajal had proposed that neurons are distinct entities, fundamental units of the nervous system, that are discontinuous with each other. Discontinuous neurons must nevertheless communicate, and Sherrington in 1897 proposed that the synapse, a specialized apposition between cells, mediates the signals. The word synapse implies “contiguity, not continuity” between neurons, as Cajal himself explained it. When the fine structure of synapses was finally revealed with the electron microscope in the 1950s, the vision of Cajal and Sherrington was amply sustained. Neurons come very close together at chemical synapses, but their membranes and cytoplasm remain distinct. At electrical synapses, which are less common than chemical synapses, the membranes remain distinct, but ions and other small solutes can diffuse through the gap junctions, a form of continuity (see Chapter 8). (See Note: Sir Charles Scott Sherrington; Santiago Ramón y Cajal)
Figure 13-1 Synapses of the mammalian brain. Drawings by Ramón y Cajal taken from Golgi-stained cortex of the cerebellum. A, Basket cell (B) from the mouse, making synaptic contacts onto the somata of numerous postsynaptic Purkinje cells (A). The axon (c) of the basket cell branches several times to make baskets (a and b) that synapse onto the somata of Purkinje cells (axosomatic synapse). Ramón y Cajal used the osmic method to stain the Purkinje cell and the Golgi method to stain the basket cell. B, A single climbing fiber (a) from a human, making numerous synaptic contacts onto the dendrites of a single Purkinje cell (b). This is an example of axodendritic synapses. Ramón y Cajal used the Golgi method to stain the climbing fiber. (From Ramón y Cajal S: Histology of the Nervous System of Man and Vertebrates. Swanson N, Swanson LW, trans. New York: Oxford University Press, 1995.)
Chemical synapses use diffusible transmitter molecules to communicate messages between two cells. The first chemical synapse to be understood in detail was the neuromuscular junction (the nerve-muscle synapse) in vertebrate skeletal muscle, which is described in Chapter 8. In this chapter, we are concerned with the properties of the synapses that occur between neurons. We now know that all synapses share certain basic biochemical and physiological mechanisms, and thus many basic insights gained from the neuromuscular junction are also applicable to synapses in the brain. However, neuronal synapses differ from neuromuscular junctions in many important ways; they also differ widely among themselves, and it is the diverse properties of synapses that help make each part of the brain unique.
It is useful to begin by reviewing some of the mechanisms that are common to all chemical synapses (see Figs. 8-2 and 8-3). Synaptic transmission at chemical synapses occurs in seven steps: (See Note: Steps in Synaptic Transmission)
Step 1: Neurotransmitter molecules are packaged into membranous vesicles, and the vesicles are concentrated and docked at the presynaptic terminal.
Step 2: The presynaptic membrane depolarizes, usually as the result of an action potential.
Step 3: The depolarization causes voltage-gated Ca2+ channels to open and allows Ca2+ ions to flow into the terminal.
Step 4: The resulting increase in intracellular [Ca2+] triggers fusion of vesicles with the presynaptic membrane (see Chapter 8). The Ca2+ dependence of fusion may be conferred by a neuron-specific protein component of the fusion apparatus called synaptotagmin. The actual fusion events are incredibly fast; each individual exocytosis requires only a fraction of a millisecond to be completed.
Step 5: The transmitter is released into the extracellular space in quantized amounts and diffuses passively across the synaptic cleft.
Step 6: Some of the transmitter molecules bind to receptors in the postsynaptic membrane, and the activated receptors trigger some postsynaptic event, usually the opening of an ion channel or the activation of a G protein–coupled signal cascade.
Step 7: Transmitter molecules diffuse away from postsynaptic receptors and are eventually cleared away by continued diffusion, enzymatic degradation, or active uptake into cells. In addition, the presynaptic machinery retrieves the membrane of the exocytosed synaptic vesicle, perhaps by endocytosis from the cell surface.
The molecular machinery of synapses is closely related to components that are universal in eukaryotic cells (see Chapter 2). A large set of proteins is involved in the docking and fusion of vesicles, and the proteins present in nerve terminals are amazingly similar to the ones mediating fusion and secretion in yeast. Docking and fusion of synaptic vesicles are discussed in Chapter 8. Ligand-gated ion channels and G protein–coupled receptors (GPCRs), the receptors on the postsynaptic membrane, are also present in all eukaryotic cells and mediate processes as disparate as the recognition of nutrients and poisons as well as the identification of other members of the species. Even most of the neurotransmitters themselves are simple molecules, identical or very similar to those used in general cellular metabolism. Clearly, the evolutionary roots of synaptic transmission are much older than nervous systems themselves.
Within nervous systems, however, myriad variations on the basic molecular building blocks yield synapses with wide-ranging properties. Neuronal synapses vary widely in the size of the synaptic contact, the identity of the neurotransmitter, the nature of the postsynaptic receptors, the efficiency of synaptic transmission, the mechanism used for terminating transmitter action, and the degree and modes of synaptic plasticity. Thus, the properties of neuronal synapses can be tuned to achieve the diverse functions of the brain.
A major difference between the neuromuscular junction and most neuronal synapses is the type of neurotransmitter used. All skeletal neuromuscular junctions use acetylcholine (ACh). In contrast, neuronal synapses use many transmitters. The most ubiquitous are amino acids: glutamate and aspartate excite, whereas γ-aminobutyric acid (GABA) and glycine inhibit. Other transmitters include simple amines, such as ACh, norepinephrine, serotonin, and histamine, as well as a wide array of peptides.
Even more varied than the neuronal transmitters are their receptors. Whereas skeletal muscle manufactures a few modest variants of its ACh receptors, the nervous system typically has several major receptor variants for each neurotransmitter. Knowledge about the wide range of transmitters and receptors is essential to understanding of the chemical activity of the brain as well as the drugs that influence brain activity. For one thing, the many transmitter systems in the brain generate responses with widely varying durations that range from a few milliseconds to days (Fig. 13-2).
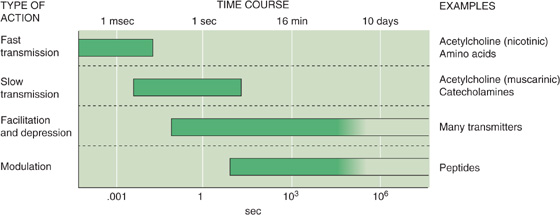
Figure 13-2 Time courses of synaptic events in the nervous system. Different transmitter systems in the brain generate responses that vary widely in how long they last in the postsynaptic cell. Note that the time axis is logarithmic. (Data from Shepherd GM: Neurobiology, 3rd ed. New York: Oxford University Press, 1994.)
Chemical synapses between neurons are generally small, often less than 1 μm in diameter, which means that their detailed structure can be seen only with an electron microscope (Fig. 13-3); under the light microscope, brain synapses are usually visible only as swellings along or at the termination of the axons (Fig. 13-1). These swellings are actually the silhouettes of the bouton terminals—the presynaptic terminals. Most presynaptic terminals arise from axons, and they can form synapses on virtually any part of a neuron. The contact site and direction of communication determine the way in which a synapse is named: axodendritic, axosomatic, and axoaxonic synapses (Fig. 13-4). These synapses are the most common types in the nervous system. In many cases, synapses occur on small outpockets of the dendritic membrane called spines and are termed axospinous synapses. However, not all synapses arise from axons, and dendrodendritic, somatosomatic, and even somatodendritic synapses may be found in the mammalian brain.
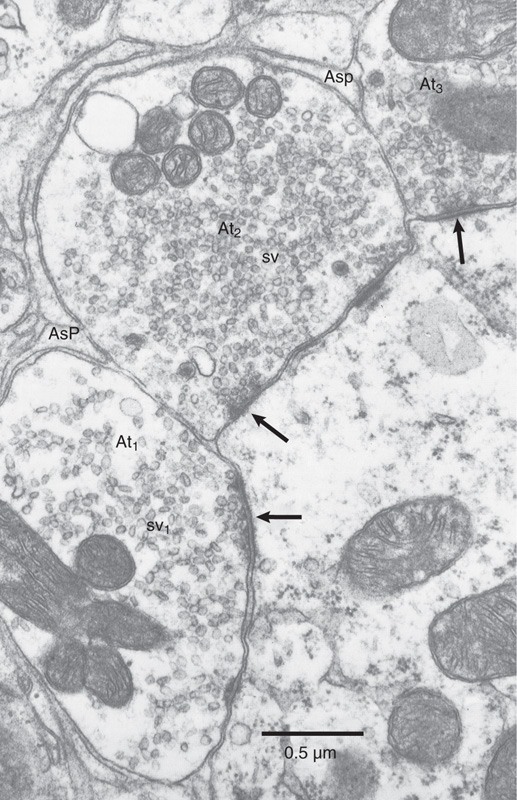
Figure 13-3 Electron micrograph of synapses in the cochlear nucleus. Three presynaptic terminals are filled with vesicles and make contact with the same postsynaptic dendrite. Postsynaptic densities (marking active zones) are indicated by arrows. (From Peters A, Palay SL, Webster HD: The Fine Structure of the Nervous System: The Neurons and Supporting Cells. Philadelphia: WB Saunders, 1976.)
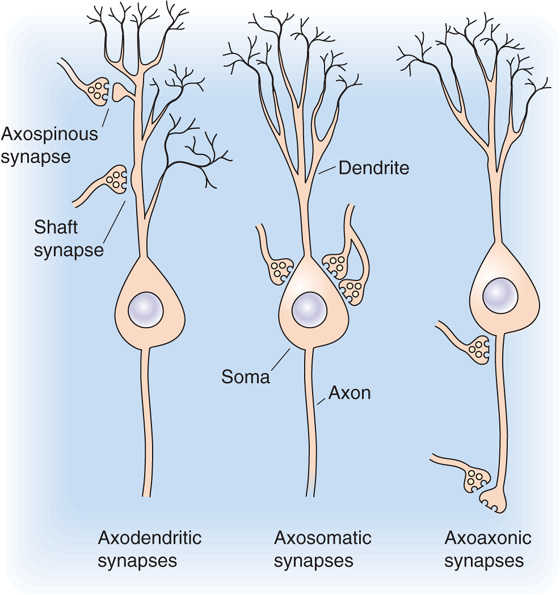
Figure 13-4 The most common synaptic arrangements in the CNS.
Despite their differences in size, site, and shape, all synapses share one basic function: they deliver a small amount of chemical transmitter onto a circumscribed patch of postsynaptic membrane. To accomplish this task, they use certain common anatomical features, most of them familiar from discussions of the neuromuscular junction (see Chapter 8).
Synapses are polarized, which means that their two apposed sides have different structures. This polarity reflects the fact that most synapses transmit information in one direction but not in the other (we will see that some rare exceptions do exist). The presynaptic side contains numerous clear vesicles, 40 to 50 nm in diameter, that appear empty when viewed by transmission electron microscopy. Synaptic termini may also contain large (100 to 200 nm in diameter), dense-core secretory granules that are morphologically quite similar to the secretory granules of endocrine cells. These granules contain neuropeptides, that is, peptides or small proteins that act as neurotransmitters and for which receptors exist in the postsynaptic membranes. Many of these neuropeptides are identical to substances secreted by “traditional” endocrine cells. Endocrine hormones such as adrenocorticotropic hormone, vasoactive intestinal polypeptide, and cholecystokinin are found in dense-core secretory granules present in the terminals of certain central and peripheral neurons.
The clear synaptic vesicles (i.e., not the dense-core granules) are anchored and shifted about by a dense network of cytoskeletal proteins. Some vesicles are clustered close to the part of the presynaptic membrane that apposes the synaptic contact; these vesicle attachment sites are called active zones. Synaptic vesicles are lined up several deep along the active zones, which are probably the regions of actual exocytosis. The number of active zones per synapse varies greatly (active zones are marked with arrows in the synapses of Fig. 13-3). Most synapses in the central nervous system (CNS) have relatively few active zones, often only 1 but occasionally as many as 10 or 20 (versus the hundreds in the neuromuscular junction). If we could view the presynaptic face of an active zone from the perspective of a synaptic vesicle, we would see filaments and particles projecting from the presynaptic membrane, often forming a regular hexagonal arrangement called a presynaptic grid. Specific points along the grid are thought to be the vesicle release sites.
Unlike the clear synaptic vesicles containing non-peptide transmitters, dense-core secretory granules are distributed randomly throughout the cytoplasm of the synaptic terminus. They are not concentrated at the presynaptic density, and they do not appear to release their contents at the active zone. Although the molecular pathways that control exocytosis of the neuronal dense-core granules are still being elucidated, it appears that a rise in [Ca2+]i is a primary stimulus.
The postsynaptic membrane lies parallel to the presynaptic membrane, and they are separated by a narrow synaptic cleft (~30 nm thick) that is filled with extracellular fluid. Transmitter molecules released from the presynaptic terminal must diffuse across the cleft to reach postsynaptic receptors. The most characteristic anatomical feature of the postsynaptic side is the postsynaptic density, a strip of granular material visible under the electron microscope on the cytoplasmic face of the membrane (Fig. 13-3). The most important molecular feature of the postsynaptic side is the cluster of transmitter receptors embedded within the postsynaptic membrane. The positions of the receptors are revealed by staining methods that use specific antibodies, toxins, or ligands coupled to some visible tag molecule.
In more than 90% of all excitatory synapses in the CNS, the postsynaptic site is a dendritic spine. The ubiquity of spines implies that they serve prominent functions, but their small size (usually less than 1 μm long) makes their function extremely difficult to study. Spines come in a variety of shapes, and their density varies from one dendrite to another (Fig. 13-5); indeed, some central neurons have no spines. The postsynaptic density of spines (as for all central synapses) contains more than 30 proteins in high concentration, including transmitter receptors, protein kinases, a host of structural proteins, and proteins that are involved in endocytosis and glycolysis.

Figure 13-5 Dendritic spines. A, Drawings of various dendrites in the neocortex, taken from Golgi-stained material. The numerous protrusions are “spines.” B, Electron micrograph of an axospinous synapse in the neocortex. Note that the dendritic spine protrudes from the dendritic shaft, making contact with a presynaptic terminal. (From Feldman ML: In Peters A, Jones EG [eds]: Cerebral Cortex: Cellular Components of the Cerebral Cortex, vol 1, pp 123-200. New York: Plenum, 1984.)
Numerous functions for spines have been proposed. It may be that spines increase the opportunity for a dendrite to form synapses with nearby axons. Many hypotheses have focused on the possibility that spines isolate individual synapses from the rest of a cell. This isolation may be electrical or chemical; the narrow spine neck may reduce current flow or the diffusion of chemicals from the spine head into the dendritic shaft. It is unlikely that the electrical resistance of the spine neck is important because given the small conductance generated by a single synapse, neck diameter would have a minimal effect on current flow. However, activation of some excitatory synapses allows substantial amounts of Ca2+ to enter the postsynaptic cell. Spines may compartmentalize this Ca2+, thus allowing it to rise to higher levels or preventing it from influencing other synapses on the cell. Because increases in postsynaptic [Ca2+]i are an essential trigger for many forms of long-term synaptic plasticity, an attractive but unproven possibility is that dendritic spines play an important role in learning mechanisms.
The brain carries out many sensory, motor, and cognitive functions that require fast, specific, spatially organized neural connections and operations. Consider the detailed neural mapping that allows you to read this sentence or the precise timing required to play the piano. These functions require spatially focused networks (Fig. 13-6A).

Figure 13-6 Synaptic connections.
Other functions, such as falling asleep, waking up, becoming attentive, or changing mood, involve more general alterations of the brain. Several systems of neurons regulate the general excitability of the CNS. Each of these modulatory systems uses a different neurotransmitter, and the axons of each make widely dispersed, diffuse, almost meandering synaptic connections to carry a simple message to vast regions of the brain. This arrangement can be achieved by a widely divergent network (Fig. 13-6B). The functions of the different systems are not well understood, but each appears to be essential for certain aspects of arousal, motor control, memory, mood, motivation, and metabolic state. The modulatory systems are of central importance to clinical medicine. Both the activity of psychoactive drugs and the pathological processes of most psychiatric disorders seem to involve alterations in one or more of the modulatory systems.
The brain has several modulatory systems with diffuse central connections. Although they differ in structure and function, they have certain similarities:
1. Typically, a small set of neurons (several thousand) forms the center of the system.
2. Neurons of the diffuse systems arise from the central core of the brain, most of them from the brainstem.
3. Each neuron can influence many others because each one has an axon that may contact more than 100,000 postsynaptic neurons spread widely across the brain.
4. The synapses made by some of these systems seem designed to release transmitter molecules into the extracellular fluid so that they can diffuse to many neurons rather than be confined to the vicinity of a single synaptic cleft.
The main modulatory systems of the brain are distinct anatomically and biochemically. Separate systems use norepinephrine, serotonin (5-hydroxytryptamine [5-HT]), dopamine, ACh, or histamine as their neurotransmitter. They all tend to involve numerous metabotropic transmitter receptors. Unlike ionotropic receptors, which are themselves channels, metabotropic receptors are coupled to enzymes such as adenylyl cyclase or phospholipase C through G proteins (see Chapter 8). For example, the brain has 10 to 100 times more metabotropic (i.e., muscarinic) ACh receptors than ionotropic (i.e., nicotinic) ACh receptors. We briefly describe the anatomy and possible functions of each major system (Fig. 13-7).
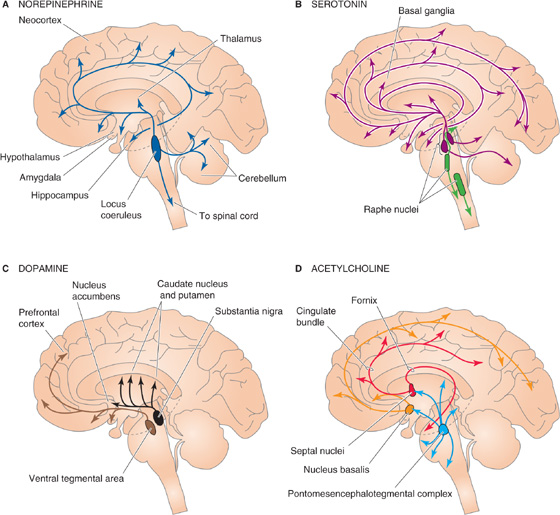
Figure 13-7 Four diffusely connected systems of central neurons using modulatory transmitters. A, Neurons containing norepinephrine are located in the locus coeruleus and innervate nearly every part of the CNS. B, Neurons containing serotonin are located in two groups of raphe nuclei and project to most of the brain. C, Neurons containing dopamine are located in the substantia nigra (and these project to the striatum) and the ventral tegmental area of the midbrain (and these project to the prefrontal cortex and parts of the limbic system). D, Neurons containing ACh are located in the basal forebrain complex, which includes the septal nuclei and nucleus basalis; the neurons project to the hippocampus and the neocortex. Other ACh-containing neurons originate in the pontomesencephalotegmental cholinergic complex and project to the dorsal thalamus and part of the forebrain.
Norepinephrine is used by neurons of the tiny locus coeruleus (from the Latin for “blue spot” because of the pigment in its cells), located bilaterally in the brainstem (Fig. 13-7A). Each human locus coeruleus has ~12,000 neurons. Axons from the locus coeruleus innervate just about every part of the brain: the entire cerebral cortex, the thalamus and hypothalamus, the olfactory bulb, the cerebellum, the midbrain, and the spinal cord. Just one of its neurons can make more than 250,000 synapses, and that cell can have one axon branch in the cerebral cortex and another in the cerebellar cortex! Locus coeruleus cells seem to be involved in the regulation of attention, arousal, and sleep-wake cycles as well as in learning and memory, anxiety and pain, mood, and brain metabolism. Recordings from awake rats and monkeys in behavioral studies show that locus coeruleus neurons are best activated by new, unexpected, nonpainful sensory stimuli in the animal’s environment. They are least active when the animals are not vigilant, just sitting around quietly digesting a meal. The locus coeruleus may participate in general arousal of the brain during interesting events in the outside world.
Serotonin-containing neurons are mostly clustered within the nine raphe nuclei (Fig. 13-7B). Raphe means “ridge” or “seam” in Greek, and indeed the raphe nuclei lie to either side of the midline of the brainstem. Each nucleus projects to different regions of the brain, and together they innervate most of the CNS in the same diffuse way as the locus coeruleus neurons. Similar to neurons of the locus coeruleus, cells of the raphe nuclei fire most rapidly during wakefulness, when an animal is aroused and active. Raphe neurons are quietest during certain stages of sleep. The locus coeruleus and the raphe nuclei are part of a venerable concept called the ascending reticular activating system, which implicates the reticular “core” of the brainstem in processes that arouse and awaken the forebrain. Raphe neurons seem to be intimately involved in the control of sleep-wake cycles as well as the different stages of sleep. Serotonergic raphe neurons have also been implicated in the control of mood and certain types of emotional behavior. Many hallucinogenic drugs, such as LSD, apparently exert their effects through interaction with serotonergic systems. Serotonin may also be involved in clinical depression; some of the most effective drugs now used to treat depression (e.g., fluoxetine [Prozac]) are potent blockers of serotonin re-uptake and thus prolong its action in the brain.
Although dopamine-containing neurons are scattered throughout the CNS, two closely related groups of dopaminergic cells have characteristics of the diffuse modulatory systems (Fig. 13-7C). One of these groups is the substantia nigra in the midbrain. Its cells project axons to the striatum, a part of the basal ganglia, and they somehow facilitate the initiation of voluntary movement. Degeneration of the dopamine-containing cells in the substantia nigra produces the progressively worsening motor disorders of Parkinson disease. Another set of dopaminergic neurons lies in the ventral tegmental area of the midbrain; these neurons innervate the part of the forebrain that includes the prefrontal cortex and parts of the limbic system. They have been implicated in neural systems that mediate reinforcement or reward as well as in aspects of drug addiction and psychiatric disorders, most notably schizophrenia. Members of the class of antipsychotic drugs called neuroleptics are antagonists of certain dopamine receptors.
Acetylcholine is the familiar transmitter of the neuromuscular junction and the autonomic nervous system. Within the brain are two major diffuse modulatory cholinergic systems: the basal forebrain complex (which innervates the hippocampus and all of the neocortex) and the pontomesencephalotegmental cholinergic complex (which innervates the dorsal thalamus and parts of the forebrain) (Fig. 13-7D). The functions of these systems are poorly understood, but interest has been fueled by evidence that they are involved in the regulation of general brain excitability during arousal and sleep-wake cycles as well as perhaps in learning and memory formation.
Collectively, the diffuse modulatory systems may be viewed as general regulators of brain function, much like the autonomic nervous system (see Chapter 14) regulates the organ systems of the body. Because their axons spread so widely within the CNS, the few modulatory neurons can have an inordinately strong influence on behavior.
Many cells are coupled to one another through gap junctions. The large and relatively unselective gap junction channels (see Chapter 6) allow ion currents to flow in both directions (in most types of gap junctions) or unidirectionally (in rare types). It follows from Ohm’s law that if two cells are coupled by gap junctions and they have different membrane voltages, current will flow from one cell into the other (see Fig. 6-18C). If the first cell generates an action potential, current will flow through the gap junction channels and depolarize the second cell; this type of current flow, for example, is the basis for conduction of excitation across cardiac muscle. Such an arrangement has all the earmarks of a synapse, and indeed, when gap junctions interconnect neurons, we describe them as electrical synapses.
Electrical synapses would seem to have many advantages over chemical synapses: they are extremely fast and limited only by the time constants of the systems involved, they use relatively little metabolic energy or molecular machinery, they are highly reliable, and they can be bidirectional. Indeed, electrical synapses have now been observed in nearly every part of the mammalian CNS. They interconnect inhibitory neurons of the cerebral cortex and thalamus, excitatory neurons of the brainstem and retina, and a variety of other neurons in the hypothalamus, basal ganglia, and spinal cord. At all of these sites, Cx36—which is expressed exclusively in CNS neurons—forms the electrical synapse (see Fig. 6-18). Glial cells in the brain express several other types of connexins. However, in all of the aforementioned sites, electrical synapses tend to be outnumbered by chemical synapses. Gap junctions universally interconnect the photoreceptors of the retina, the astrocytic glia (see Chapter 11) of all parts of the CNS, and most cells early in development.
Why are chemical synapses, as complex and relatively slow as they are, more prevalent than electrical synapses in the mature brain? Comparative studies suggest several reasons for the predominance of chemical synapses among mammalian neurons. The first is amplification. Electrical synapses do not amplify the signal passed from one cell to the next; they can only diminish it. Therefore, if a presynaptic cell is small relative to its coupled postsynaptic cell, the current that it can generate through an electrical synapse will also be small, and thus “synaptic strength” will be low. By contrast, a small bolus of neurotransmitter from a chemical synapse can trigger an amplifying cascade of molecular events that can cause a relatively large postsynaptic change.
A second advantage of chemical synapses is their ability to generate inhibition. Inhibition is difficult (although not impossible in specialized cases) to achieve in electrical synapses. Chemical synapses perform this function with ease, by simply opening channels that are selective for ions with relatively negative equilibrium potential.
A third advantage of chemical synapses is that they can transmit information over a broad time domain. By using different transmitters, receptors, second messengers, and effectors, chemical synapses can produce a wide array of postsynaptic effects with time courses ranging from a few milliseconds to minutes and even hours. The effects of electrical synapses are generally limited to the time course of the presynaptic event.
A fourth advantage of chemical synapses is that they are champions of plasticity; their strength can be a strong function of recent neural activity, and they can therefore play a role in learning and memory, which are essential to the success of vertebrate species. Electrical synapses may also be plastic, but this has not been well studied yet.
It might also be noted that the few perceived advantages of electrical synapses may be more apparent than real. Bidirectionality is clearly not useful in many neural circuits, and the difference in speed of transmission may be too small to matter in most cases. Electrical synapses serve important but specialized functions in the nervous system. They seem to be most prevalent in neural circuits in which speed or high degrees of synchrony are at a premium: quick-escape systems, the fine coordination of rapid eye movements, or the synchronization of neurons generating rhythmic activity. Gap junctions are also effective in diffusely spreading current through large networks of cells, which appears to be their function in photoreceptors and glia.
The mammalian nervous system uses dozens of different neurotransmitters that act on more than 100 types of receptors; these receptors stimulate numerous second-messenger systems, which in turn regulate several dozen ion channels and enzymes. We call these pathways of synaptic signaling the transmitter systems. It is not enough to know the identity of a transmitter to predict its effect—you also need to know the nature of the components that it interacts with, and these components may vary from one part of the brain to another and even between parts of a single neuron. The components of the transmitter systems are extremely complex. This subchapter introduces the intricate and vital web of neurotransmitters. The clinical importance of the subject is difficult to overstate. It is likely that most drugs that alter mental function do so by interacting with neurotransmitter systems in the brain. Disorders of neurotransmitter systems are also implicated in many devastating brain disorders, such as schizophrenia, depression, epilepsy, Parkinson disease, the damage of stroke, and drug addiction.
Most neurotransmitters are similar or identical to the standard chemicals of life, the same substances that all cells use for metabolism. Transmitter molecules can be large or small. The small ones, such as the amino acids glutamate, aspartate, GABA, and glycine, are also simple foods (Fig. 13-8A). Cells use amino acids as an energy source and to build essential proteins, but they have co-opted these common molecules for essential and widespread messenger functions in the brain. Another important class of small neurotransmitters is the amines, including the monoamines (e.g., ACh, serotonin, and histamine) listed in Figure 13-8B and the catecholamines (e.g., dopamine, norepinephrine, and epinephrine) listed in Figure 13-8C. Neurons synthesize these small transmitters by adding only a few chemical steps to the glucose and amino acid pathways that are present in every cell. Purine derivatives can also be important transmitters. For example, a key molecule of cell metabolism that has recently achieved neurotransmitter status is adenosine triphosphate (ATP), which is the major chemical intermediate of energy metabolism and is present in many synaptic vesicles. It is also released from various synapses in the central and peripheral nervous systems. ATP appears to be the transmitter responsible for sympathetic vasoconstriction in small arteries and arterioles, for example. ATP acts on a variety of nucleotide receptors, both ionotropic and metabotropic. Adenosine is also a transmitter in the CNS.
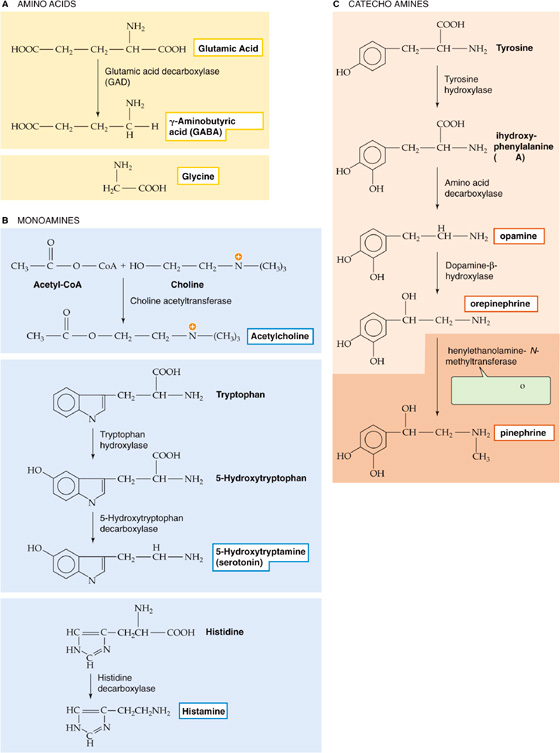
Figure 13-8 Biosynthesis of some common small transmitter molecules.
The large-molecule transmitters, which constitute a much more numerous group, are proteins or small bits of protein called neuroactive peptides. A few of the better studied neuropeptides are shown in Figure 13-9. Many were originally identified in non-neural tissues, such as the gut or endocrine glands, and were only later found in nerve terminals of the brain or peripheral nervous system. They vary in size from two–amino acid peptides to large polypeptides. Among the neuroactive peptides are the endorphins (endogenous substances with morphine-like actions), which include small peptides called enkephalins. The term opioids includes all substances with a morphine-like pharmacology—the endorphins (endogenous) as well as morphine and heroin (exogenous).

Figure 13-9 Structure of some neuroactive peptides. All peptides are presented with their NH2 termini (i.e., the first to be synthesized) to the left, as is now customary for proteins in general. However, note that for many of the peptide hormones, the amino acid residues were numbered before this convention was established. The p on the amino-terminal glutamate on some of these peptides stands for pyroglutamate. (See Note: Pyroglutamate and C-Terminal Amides)
The synthesis of most neuropeptides begins like that of any other secretory protein (see Chapter 2), with the ribosome-directed assembly of a large prehormone. The prehormone is then cleaved to form a smaller prohormone in the Golgi apparatus and further reduced into small active neuropeptides that are packaged into vesicles. Thus, the synthesis of neuropeptides differs significantly from that of the small transmitters.
In summary, then, the neurotransmitters consist of a dozen or so small molecules plus 50 to 100 peptides of various size. The small transmitters are, as a rule, each stored and released by separate sets of neurons. The peptides, however, are usually stored and released from the same neurons as one of the small transmitters (Table 13-1), an arrangement called co-localization of neurotransmitters. Thus, GABA may be paired with somatostatin in some synapses, serotonin and enkephalin in others, and so on. The co-localized transmitters may be released together, but of course each acts on its own receptors.
Table 13-1 Examples of Neuroactive Peptides That Co-localize with Small-Molecule Neurotransmitters
Small Molecule |
Peptide* |
Acetylcholine |
Enkephalin |
|
Vasoactive intestinal polypeptide |
|
Calcitonin gene–related peptide |
|
Substance P |
|
Somatostatin and enkephalin |
|
Gonadotropin-releasing hormone |
|
Neurotensin |
|
Galanin |
Dopamine |
Cholecystokinin |
|
Enkephalin |
|
Neurotensin |
Epinephrine |
Enkephalin |
|
Neuropeptide Y |
|
Neurotensin |
|
Substance P |
GABA |
Cholecystokinin |
|
Enkephalin |
|
Somatostatin |
|
Neuropeptide Y |
|
Substance P |
|
Vasoactive intestinal polypeptide |
Glutamate |
Substance P |
Glycine |
Neurotensin |
Norepinephrine |
Enkephalin |
|
Neuropeptide Y |
|
Neurotensin |
|
Somatostatin |
|
Vasopressin |
Serotonin |
Cholecystokinin |
|
Enkephalin |
|
Substance P and thyrotropin-releasing hormone |
|
Thyrotropin-releasing hormone |
*Each row gives a peptide or combination of peptides that co-localize with the small molecule on the left.
Data from Hall ZW: An Introduction to Molecular Neurobiology. Sunderland, MA: Sinauer, 1992.
One of the unique substances proposed as a transmitter is a gaseous molecule, the labile free radical nitric oxide (NO). Carbon monoxide has also been suggested to be a transmitter, although evidence thus far is meager. NO is synthesized from L-arginine by many cells of the body (see Chapter 3), and it has powerful biological effects. As a neurotransmitter, NO may have unique functions. It seems to be released from both presynaptic and what we normally think of as post synaptic neurons. Because NO is not packaged into vesicles, its release does not require an increase in [Ca2+]i, although its synthesis does. NO may sometimes act as a retrograde messenger, that is, from postsynaptic to presynaptic structures. Because NO is small and membrane permeable, it can diffuse about much more freely than other transmitter molecules, even penetrating through one cell to affect another beyond it. On the other hand, NO is evanescent, and it breaks down rapidly. The functions of gaseous transmitters are now being vigorously studied and hotly debated. (See Note: NO as a Neurotransmitter in the CNS)
The endocannabinoids are another unusual group of putative neurotransmitters. They include the endogenous lipophilic molecules anandamide (from ananda, the Sanskrit word for “internal bliss”) and arachidonyl glycerol (2-AG). These substances are called endocannabinoids because they mimic Δ9-tetrahydrocannabinol (THC), the active ingredient in marijuana, by binding to and activating specific G protein–coupled “cannabinoid” receptors. Remarkably, the brain has more cannabinoid receptors than any other GPCR type. Certain activated neurons synthesize and release endocannabinoids, which diffuse readily to presynaptic terminals, and modulate the further release of conventional transmitters such as GABA and glutamate. Their normal role in the brain is currently unknown. However, activation of cannabinoid receptors with low doses of THC leads to euphoria, relaxed sensations, decreased pain, and increased hunger; it can also impair problem-solving ability, short-term memory, and motor skills. High doses can alter personality and sometimes trigger hallucinations. THC and related drugs have shown some promise for treatment of the nausea and vomiting of cancer patients undergoing chemotherapy, suppression of chronic pain, and stimulation of appetite in some AIDS patients.
Most of the chemicals we call neurotransmitters also exist in non-neural parts of the body. Each chemical may serve dual purposes in that it can mediate communication in the nervous system but do something similar or even entirely different elsewhere. Amino acids, of course, are used to make protein everywhere. NO is a local hormone that relaxes the smooth muscle in blood vessels (see Chapter 20). Surprisingly, the cells with the highest ACh levels are in the cornea of the eye, although corneal cells lack specific receptors for ACh. It is not clear what ACh does for corneal cells, but it almost certainly is not acting as a transmitter. One of the most interesting nonmessenger functions of transmitter molecules is their role in the development of the brain, even before synapses have appeared. At these early stages of development, transmitters may regulate cell proliferation, migration, and differentiation, somehow helping to form the brain before they help operate it.
Each neuromuscular junction has a simple and stereotyped job: when an action potential fires in the motor neuron, the junction must reliably excite its muscle cell to fire an action potential and contract. Decisions about muscle contractions (where, when, and how much) are made within the CNS, and the neuromuscular junction exists simply to communicate that decision to the muscle unambiguously. To perform this function, neuromuscular transmission has evolved to be very strong so that it is fail-safe under even the most extreme of physiological conditions.
Synapses between neurons usually have a more subtle role in communication, and they use a variety of mechanisms to accomplish their more complex tasks. Like neuromuscular junctions, some neuron-neuron synapses (excitatory) can rapidly excite. However, other synapses (inhibitory) can cause profound inhibition by decreasing postsynaptic excitability directly (postsynaptic inhibition). In a third broad class of synapse (modulatory), the synapse often has little or no direct effect of its own but instead regulates or modifies the effect of other excitatory or inhibitory synapses by acting on either presynaptic or postsynaptic membranes. These three basic types of neural synapses are exemplified by their input to the pyramidal neuron of the cerebral cortex. In the example shown in Figure 13-10, a pyramidal neuron in the visual cortex receives an excitatory synaptic input from the thalamus (with use of glutamate as the neurotransmitter), an inhibitory synaptic input from an interneuron (with use of GABA as the neurotransmitter), and a modulatory input from the locus coeruleus (with use of norepinephrine as the neurotransmitter).
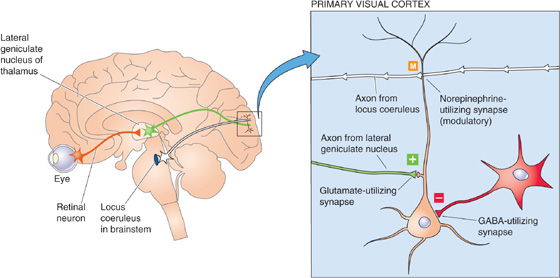
Figure 13-10 Synaptic circuitry of the visual cortex. Visual pathways that originate in the retina activate neurons in the lateral geniculate nucleus of the thalamus. These glutamate-containing neurons in turn synapse on cortical pyramidal neurons and produce some excitation. Also within the primary visual cortex, a GABA-containing neuron mediates localized inhibition. Small cells in the locus coeruleus, a brainstem nucleus, make widely divergent connections onto cortical neurons and release norepinephrine and thus produce modulation.
Pyramidal cells receive excitatory synapses from many sources, including the axons of the thalamus. Most fast excitatory synapses in the brain use glutamate or aspartate as their transmitter, and the thalamus-to-cortex synapses are no exception (Fig. 13-10). Both amino acids have similar effects on the postsynaptic excitatory amino acid receptors, and it has proved difficult to determine which excitatory amino acid is used in which synapse. For convenience, these types of synapses are often presumptuously referred to as glutamatergic. These excitatory amino acids bind to a group of fast, ligand-gated cation channels. When they are activated by synaptic glutamate, glutamate-gated channels generate an excitatory postsynaptic potential (EPSP) that is very similar to the one produced by ACh at the neuromuscular junction (see Chapter 8), except that it is much smaller than the EPSP in muscle. In the example shown in Figure 13-11 (left side), glutamate produces the EPSP by activating a nonselective cation channel that has about the same conductance for Na+ and K+. Thus, the reversal potential (see Chapter 6) of the EPSP is 0 mV, about midway between the equilibrium potential for Na+ (ENa) and that for K+ (EK). An EPSP from the activation of a single glutamatergic synapse peaks at 0.01 to a few millivolts (depending on many factors, including the size of the postsynaptic cell and the size of the synapse), whereas one neuromuscular EPSP reaches a peak of ~40 mV—a difference of 40- to 4000-fold. Obviously, most glutamatergic synapses are not designed to be fail-safe. It takes the summation of EPSPs from many such synapses to depolarize a post-synaptic neuron to the threshold for triggering of an action potential.
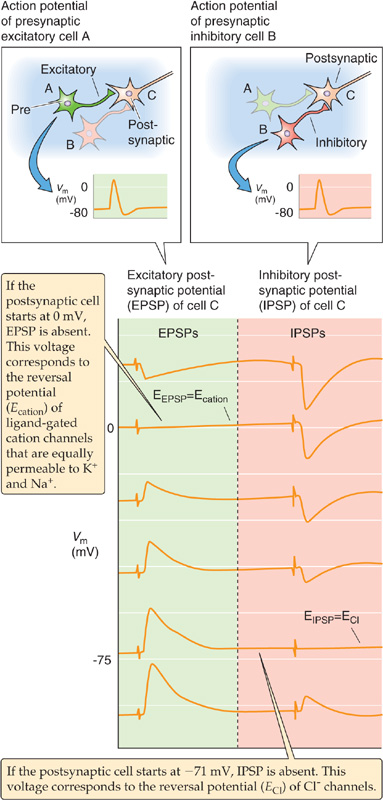
Figure 13-11 Voltage dependence of EPSPs and IPSPs of the nervous system. An excitatory presynaptic neuron (cell A) and an inhibitory presynaptic neuron (cell B) both synapse on a third neuron (cell C). In this experiment, the investigators injected enough constant current into cell C to initially set the Vm to each of the six values shown in the figure. For each record, the experimenter first stimulated the stimulatory presynaptic neuron to produce an EPSP in the postsynaptic neuron and then stimulated the inhibitory presynaptic neuron to produce an IPSP. These EPSPs and IPSPs reflect the activities of multiple synapses onto cell C. The reversal potential for the EPSP is ~0 mV (i.e., stimulating the stimulatory presynaptic neuron has no effect) because Na+ and K+ conduct through the channel equally well. The reversal potential for the IPSP is at about −71 mV (i.e., stimulating the inhibitory presynaptic neuron has no effect). This value is ECl, indicating that the IPSP is mediated by a Cl− channel.
Skeletal muscle cells in vertebrates have only excitatory synapses. On the other hand, virtually all central neurons have numerous excitatory and inhibitory synapses. Thus, the excitability of most neurons is governed by the dynamic balance of excitation and inhibition at any moment. The inhibitory transmitters GABA and glycine are the transmitters at the large majority of inhibitory synapses. Indeed, the inhibitory synapse between the interneuron and the pyramidal cell in Figure 13-10 uses GABA. Both GABA and glycine bind to receptors that gate Cl−-selective channels (see Chapter 8). Cl− conductance usually has an inhibitory influence because the equilibrium potential for Cl− (ECl) in neurons is near or slightly negative to the resting potential of the neuron. Thus, the reversal potential for the Cl−-mediated inhibitory postsynaptic potential (IPSP) is the same as the ECl. If Cl− conductance increases, the membrane potential (Vm) has a tendency to move toward ECl (Fig. 13-11, right side). The effect is inhibitory because it tends to oppose other factors (mainly EPSPs) that might otherwise move the Vm to the threshold for an action potential.
The nervous system is influenced by many forms of synaptic modulation, and their mechanisms are covered in more detail later. As an example, consider the axons arising from the locus coeruleus, which synapse widely on pyramidal cells in the cerebral cortex (Figs. 13-7A and 13-10). These axons release the transmitter norepinephrine, a classic modulator with multiple effects. Norepinephrine acts on β-adrenergic receptors in the pyramidal cell membrane (it may also act on α receptors at the same time). Unlike the actions of the fast amino acid forms of synaptic transmission, this effect of norepinephrine by itself has little or no obvious influence on the activity of a resting neuron. However, a cell exposed to norepinephrine will react more powerfully when it is stimulated by a strong excitatory input (usually by glutamatergic synapses), as shown in Figure 13-12. Thus, norepinephrine modulates the cell’s response to other inputs.
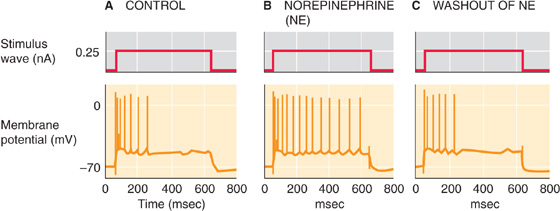
Figure 13-12 Modulatory effect of norepinephrine. A, Injecting a neuron from the hippocampus with a sustained depolarizing current pulse leads to a “phasic” action potential response: frequent spiking at the beginning but adaptation as the depolarizing current pulse is maintained. B, The application of norepinephrine causes the spiking that is elicited by the depolarizing current pulse to be sustained longer (“tonic”). C, The cell returns to its control state as in A. (Data from Madison DV, Nicoll RA: Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol 1986; 372:221-244.)
The molecular mechanisms of neuromodulators are complex and diverse, but all begin with a GPCR that activates an intracellular signal cascade (see Chapter 3). Binding of norepinephrine to the β-adrenergic receptor stimulates the intracellular enzyme adenylyl cyclase, which increases intracellular levels of cyclic adenosine monophosphate (cAMP, the second messenger), which in turn stimulates other enzymes to increase their rates of phosphorylation. Within the cortical neuron, phosphorylation of one or more types of K+ channel decreases the probability of the channels being open (see Chapter 6). Fewer open K+ channels mean higher membrane resistance, greater excitability, and less adaptation of spike firing rates during prolonged stimuli. This K+ channel pathway is but one of the many mechanisms by which norepinephrine can affect cells. Other effects are generated when norepinephrine activates other subtypes of adrenergic receptors and thus different second-messenger systems coupled to different channels or enzymes. Modulatory transmitters allow the nervous system tremendous potential and flexibility to vary its state of excitability.
GPCRs exist in every cell (see Chapter 3). In the preceding section we described one example, the receptor for norepinephrine and its second messenger–mediated effect on certain K+ channels. However, norepinephrine alone has at least five major receptor types—two α receptors and three β receptors—that act on numerous effectors. In fact, each transmitter has multiple GPCRs, and their effects are complex and interactive and engage almost all aspects of cell function through several intracellular messenger systems. The various GPCRs can recognize a wide range of transmitter types, from small molecules to peptides.
Activated G proteins can trigger a wide array of responses at synapses by either of the two general pathways introduced in Chapter 3: (1) the G protein may modulate the gating of an ion channel directly or by a very short second-messenger pathway, and (2) the G protein may activate one of several enzyme systems that involve second messengers and subsequent signal cascades.
The first—and simplest—G protein cascade involves a direct receptor–G protein–channel linkage and is sometimes called the membrane-delimited pathway. In this case, the G protein may be the only messenger between the receptor and the effector. A variety of neurotransmitter–G protein–channel systems use this pathway. For example, in heart muscle, ACh binds to a certain type of muscarinic ACh receptor (M2) that activates a G protein, the βγ subunits of which in turn cause a K+ channel to open. (We discuss this example later in Fig. 13-13B.) Other receptors in various cells can modulate other K+ and Ca2+ channels in a similar way. (See Note: The Membrane-Delimited Pathway for the Activation of Ion Channels by G Proteins)
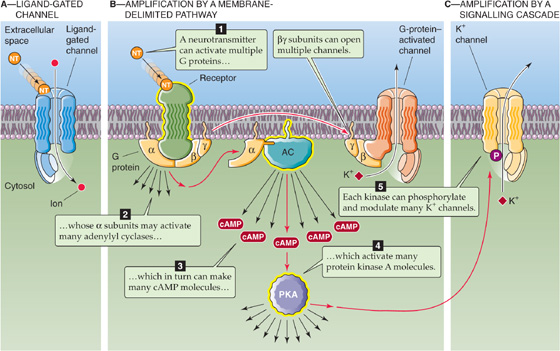
Figure 13-13 Amplification. A, The neurotransmitter (NT) binds directly to a channel, thereby activating it. B, The neurotransmitter binds to a receptor that in turn activates 10 to 20 G proteins. In this example, the βγ subunits directly activate K+ channels. In addition, each activated α subunit activates an adenylyl cyclase molecule, each of which produces many cAMP molecules that activate protein kinase A (PKA). C, Each activated PKA can phosphorylate and thereby modulate many channels. AC, adenylyl cyclase.
One advantage of the membrane-delimited pathway is that it is relatively fast, beginning within 30 to 100 ms—not quite as fast as a ligand-gated channel, which uses no intermediary between receptor and channel, but faster than the many-messenger cascades described next. The membrane-delimited pathway is also localized in comparison to the other cascades. Because the G protein cannot diffuse very far within the membrane, only channels nearby can be affected. This type of coupling also allows flexibility because many types of receptors can be coupled to a variety of channels by use of the appropriate G protein intermediate.
The other general type of G protein signaling involves enzyme systems and second messengers, often diffusing through the cytoplasm, to influence an ion channel. The terminology deserves some clarification. Traditionally, the small, diffusible intracellular chemicals (e.g., cAMP, inositol 1, 4, 5-trisphosphate [IP3]) that help carry the message between a transmitter receptor and a channel are called second messengers. The transmitter itself is counted as the first messenger, but notice that by this logic the receptor is not a messenger at all, even though it transfers a signal from the neurotransmitter to a G protein. The G protein is also not counted as a messenger, nor are the various enzymes that may come before and after the traditional “second messenger” in any signal cascade. Different cascades involve different numbers of messengers, but obviously, most have many more than two! Alas, the terminology is entrenched, although when we speak of second messengers, one should remember that a multiple-messenger cascade is almost always involved. As an added complication, two or more cascades, each with different types of messengers, may sometimes be activated by one type of receptor (an example of divergence, see later).
In Chapter 3, we discussed three of these longer, and slower, G protein signal cascades: (1) the adenylyl cyclase pathway, (2) the phospholipase C pathway, and (3) the phospholipase A2 pathway. Each is activated by a different set of receptors, each uses a different G protein, and each generates different intracellular messengers. Some of these messengers dissolve in the watery cytoplasm, whereas others diffuse within the fatty lipid bilayer. The final link in most of the messenger cascades is a kinase.
In a well-known example, cAMP binds to cAMP-dependent protein kinase (protein kinase A), which then phosphorylates amino acids on K+ or Ca2+ channels in the membrane. The addition of phosphate groups to the channel protein changes its conformation slightly, which may strongly influence its probability of being open (see Chapter 6). In Chapter 21, we discuss the stimulation of the β-adrenergic receptor by norepinephrine, which ultimately results in a stronger heartbeat through phosphorylation and opening of myocardial voltage-gated Ca2+ channels. By contrast, in the pyramidal cell of the hippocampus, stimulation of β-adrenergic receptors also results in an increase in cAMP, but it has no influence on the cell’s Ca2+ channels. Instead, it inhibits some of its K+ channels. As a result, the cell can fire more action potentials during prolonged stimuli (Fig. 13-12).
If transmitter-stimulated kinases were allowed to madly phosphorylate without some method of reversing the process, all proteins would quickly become saturated with phosphates and further regulation would become impossible. Protein phosphatases save the day. They act rapidly to remove phosphate groups (see Chapter 3), and thus the degree of channel phosphorylation at any moment depends on the dynamic balance of phosphorylation and dephosphorylation.
At this point you may be wondering about the perversity of such complex, interconnected, indirect messenger cascades. Do these long chains of command have any benefit? Why not use simple, fast ligand-gated channels (Fig. 13-13A) for all transmitter purposes? In fact, complex messenger cascades seem to have advantages.
One important advantage is amplification. When it is activated, one ligand-gated channel is just that: one ion channel in one place. However, activate one GPCR and you potentially influence many channels. Signal amplification can occur at several places in the cascade (see Chapter 3), and a few transmitter molecules can generate a sizable cellular effect. One stimulated receptor can activate perhaps 10 to 20 G proteins, each of which can activate a channel by a membrane-delimited pathway such as the βγ pathway (Fig. 13-13B). Alternatively, the α subunit of one G protein can activate an adenylyl cyclase, which can make many cAMP molecules, and the cAMP molecules can spread to activate many kinases; each kinase can then phosphorylate many channels (Fig. 13-13C). If all cascade components were tied together in a clump, signaling would be severely limited. The use of small messengers that can diffuse quickly also allows signaling at a distance, over a wide stretch of cell membrane. Signaling cascades also provide many sites for further regulation as well as interaction between cascades. Finally, signal cascades can generate long-lasting chemical changes in cells, which may form the basis for, among other things, a lifetime of memories. (See Note: Compartmentalization of Second Messenger Effects)
Two of the most common neurotransmitters in the brain are glutamate and GABA. Either molecule can bind to any of several kinds of receptors, and each of these receptors can mediate a different effect. This ability of one transmitter to activate more than one type of receptor is sometimes called divergence.
Divergence is a rule among neurotransmitters. Every well-studied transmitter can activate multiple receptor subtypes, and recent experience indicates that the number of receptors will continue to escalate as the powerful methods of molecular biology continue to be applied to each system. Divergence means that one transmitter can affect different neurons (or even different parts of the same neuron) in very different ways. It also means that if a transmitter affects different neurons in different ways, it could be because each neuron has a different type of receptor. However, among transmitter systems that use second messengers, divergence may also occur at points beyond the level of the receptor. For example, norepinephrine can turn on or turn off a variety of ion channels in different cells (Fig. 13-14A). Some of these effects occur because norepinephrine activates different receptors, but some of these receptors may each activate more than one second messenger, or a single second messenger (e.g., cAMP) may activate a kinase that influences numerous different channels. Divergence may occur at any stage in the cascade of transmitter effects.
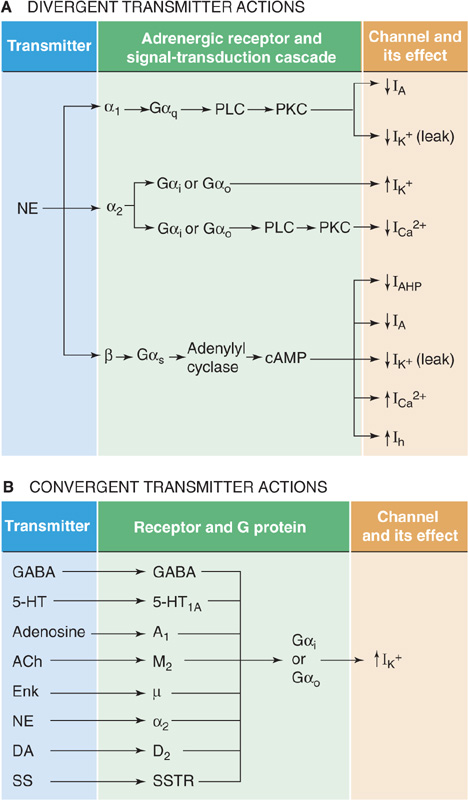
Figure 13-14 Divergence and convergence of transmitter effects on channels. A, One transmitter, norepinephrine in this case, can activate multiple receptors, which stimulate different G protein/second messengers, which in turn either stimulate or depress the gating of many types of ion channels. IAHP stands for afterhyperpolarization current, which is mediated by a Ca2+-activated K+ channel. Ih stands for hyperpolarization-activated cation current. B, Multiple transmitters bind to their specific receptors and, by the same or different second-messenger systems, influence the same set of ion channels. ACh, acetylcholine; DA, dopamine; Enk, enkephalin; 5-HT, 5-hydroxytryptamine (serotonin); NE, norepinephrine; SS, somatostatin; SSTR, somatostatin receptor.
Neurotransmitters can also exhibit convergence of effects. This property means that multiple transmitters, each activating its own receptor type, converge on a single type of ion channel in a single cell. For example, some pyramidal cells of the hippocampus have GABAB, 5-HT1A, A1 (specific for adenosine), and SS (specific for somatostatin) receptors, all of which activate the same K+ channel (Fig. 13-14B). Furthermore, in the same cells, norepinephrine, ACh, 5-HT, corticotropin-releasing hormone, and histamine all converge on and depress the slow Ca2+-activated K+ channels. Analogous to divergence, the molecular site of convergence may occur at a common second-messenger system, or different second messengers may converge on the same ion channel.
Divergence and convergence can occur simultaneously within neurotransmitter systems. Many of them have chemical feedback regulation built in as well.
Fast, amino acid–mediated synapses account for most of the neural activity that we associate with specific information processing in the brain: events directly responsible for sensory perception, motor control, and cognition, for example. Glutamate-mediated excitation and GABA-mediated inhibition have been intensively studied. In physiological terms, these are also the best understood of the brain’s synapses, and this section describes their function.
As a rule, postsynaptic events are more easily measured than are presynaptic events; thus, we know more about them. Of course, by measuring postsynaptic events, we also have a window onto the functions of the presynaptic terminal, and in fact this is usually the best view we can get of presynaptic functions. For this reason, we begin our description with the downstream, postsynaptic side of the synapse and then work backward to the presynaptic side.
Most glutamate-mediated synapses generate an EPSP with two distinct components, one much faster than the other. Both are triggered by the same presynaptic terminal releasing a single bolus of transmitter, but the two EPSP components are generated by different types of ion channels that are gated by distinct postsynaptic receptors—a case of transmitter divergence. The behavior of these channels helps understand the characteristics of the EPSP.
Glutamate can act on four major classes of receptors: one is a GPCR or metabotropic receptor, and three of them are ion channels or ionotropic receptors. As noted earlier, metabotropic receptors (mGluRs—the m stands for metabotropic) have seven membrane-spanning segments and are linked to heterotrimeric G proteins (Fig. 13-15A). At least eight metabotropic receptors have been identified, and comparisons of their primary structure have been used to infer the evolutionary relationships among receptor subunits (Fig. 13-15B). The mGluRs form three groups that differ in their sequence similarity, pharmacology, and associated signal transduction systems.
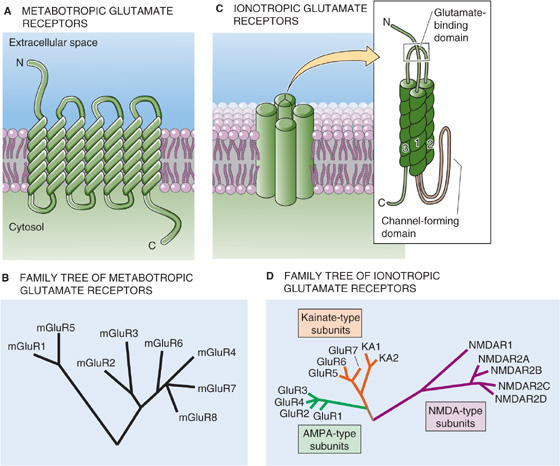
Figure 13-15 Comparison of ionotropic and metabotropic glutamate receptors. In C, the inset shows a prototypical subunit, with a large extracellular N-terminal region, a membrane-spanning segment, a short loop that partially re-enters the membrane from the cytosolic side, and two more membrane-spanning segments. The glutamate-binding domain consists of parts of both the N-terminal region and the loop connecting membrane-spanning segments 2 and 3. Four of these subunits appear to come together to form a single channel/receptor with a central pore. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; NMDA, N-methyl-D-aspartate.
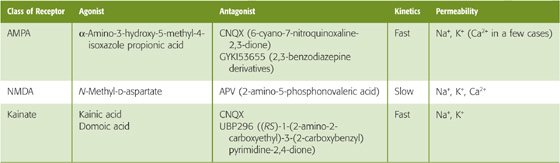
The three classes of ionotropic glutamate receptors are the AMPA, NMDA, and kainate receptors (Table 13-2). By definition, each is activated by binding glutamate, but their pharmacology differs. The receptor names are derived from their relatively specific agonists: AMPA stands for α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid. The AMPA receptor is sometimes called the quisqualate receptor, after another of its agonists, or even the quisqualate/AMPA receptor. NMDA stands for N-methyl-D-aspartate. The kainate receptor is named for one of its agonists, kainic acid, and it can also be activated by domoic acid. The three ionotropic glutamate receptors can also be distinguished by their selective antagonists. AMPA and kainate receptors, but not NMDA receptors, are blocked by drugs such as CNQX (6-cyano-7-nitroquinoxaline-2, 3-dione). Moreover, AMPA receptors can be specifically antagonized by 2, 3-benzodiazepine derivatives, such as GYKI53655. NMDA receptors, but not AMPA and kainate receptors, are blocked by APV (2-amino-5-phosphonovaleric acid). Selective antagonists of kainate receptors have also been discovered.
Table 13-2 Ionotropic Glutamate Receptors
At least 22 ionotropic glutamate receptor cDNAs—plus a variety of splice variants—have been cloned. Each of these cDNAs represents a monomer (see Fig 6-21M on p. 177) with a large extracellular region, followed by a transmembrane segment, a loop that partially enters the membrane from the cytosolic side, and then two more transmembrane segments (Fig. 13-15C). The loop appears to line the channel pore. On the basis of kinetic and structural studies, the channel appears to consist of a tetramer of four subunits. Comparisons of primary structures can be used to infer evolutionary relationships among receptor monomeric subunits. Figure 13-15D shows a hypothesized phylogenetic tree for the three classes of ionotropic glutamate receptors, with the major subtypes clustered together. Note that the various NMDA receptor subunits (e.g., NMDAR1, NMDAR2A to NMDAR2D) that combine to make the NMDA receptors are more closely related to each other than to the subunits (e.g., KA1, KA2, GluR5 to GluR7) that combine to make the kainate receptors or to the subunits (e.g., GluR1 to GluR4) that combine to make the AMPA receptors. The metabotropic and ionotropic glutamate receptors have separate family trees because, although both receptor types bind glutamate, they are so different in structure that it is highly probable they evolved from different ancestral protein lines.
As noted earlier, most glutamate-mediated synapses generate an EPSP with two temporal components (Fig. 13-16A, C). The two phases of the glutamate-mediated EPSP have different pharmacological profiles, kinetics, voltage dependencies, ion dependencies, and permeabilities, and most important, they serve distinct functions in the brain. Pharmacological analysis reveals that the faster phase is mediated by an AMPA-type glutamate receptor and the slower phase by an NMDA-type glutamate receptor. These glutamate-gated channels have been extensively studied with single-channel recording methods. Both AMPA and NMDA receptors have nearly equal permeability to Na+ and K+, but they differ in several ways.
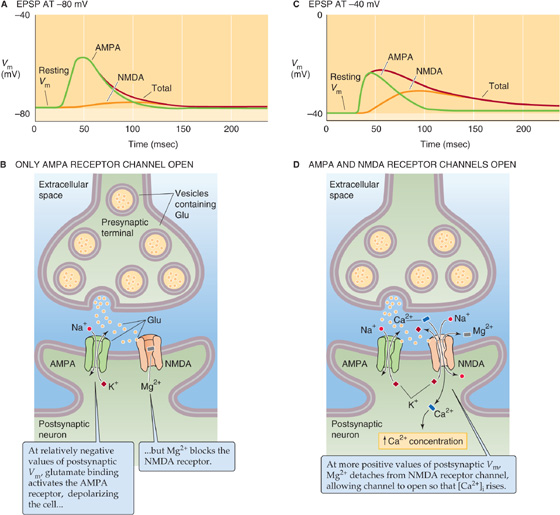
Figure 13-16 Glutamate-gated channels. A, At most glutamate-mediated synapses, the EPSP (red curve) is the sum of two components: (1) a rapid component that is mediated by an AMPA receptor channel (green curve) and (2) a slow component that is mediated by an NMDA receptor channel (orange curve). In this example, in which the postsynaptic Vm is relatively negative, the contribution of the NMDA receptor channel is very small. B, At a relatively negative initial Vm in the postsynaptic cell, as in A, the NMDA receptor channel does not open. The AMPA receptor channel, which is independent of postsynaptic Vm, opens. The result is a fast depolarization. C, In this example, in which the postsynaptic Vm is relatively positive, the contribution of the NMDA receptor channel is fairly large. D, At a relatively positive initial Vm in the postsynaptic cell, as in C, glutamate activates both the AMPA and the NMDA receptor channels. The recruitment of the NMDA receptor channels is important because unlike most AMPA receptor channels, they allow the entry of Ca2+ and have slower kinetics.
AMPA-gated channels are found in most excitatory synapses in the brain, and they mediate fast excitation, with most receptor channels normally letting very little Ca2+ into cells. Their single-channel conductance is relatively low, ~15 pS, and they show little voltage dependence.
NMDA-gated channels have more complex behavior. They have higher conductance, ~50 pS, and much slower kinetics. The ion selectivity of NMDA channels is the key to their functions: permeability to Na+ and K+ causes depolarization and thus excitation of a cell, but their high permeability to Ca2+ allows them to influence [Ca2+]i significantly. It is difficult to overstate the importance of intracellular [Ca2+]. Ca2+ can activate many enzymes, regulate the opening of a variety of channels, and affect the expression of genes. Excess Ca2+ can even precipitate the death of a cell.
The gating of NMDA channels is unusual: at normal resting voltage (about −70 mV), the channel is clogged by Mg2+, and few ions pass through it; the Mg2+ pops out only when the membrane is depolarized above about −60 mV. Thus, the NMDA channel is voltage dependent in addition to being ligand gated; both glutamate and a relatively positive Vm are necessary for the channel to open. How do the NMDA-gated channels open? NMDA-gated channels coexist with AMPA-gated channels in many synapses of the brain. When the postsynaptic cell is at a relatively negative resting potential (Fig. 13-16A, B), the glutamate released from a synaptic terminal can open the AMPA-gated channel, which is voltage independent, but not the NMDA-gated channel. However, when the postsynaptic cell is more depolarized because of the action of other synapses (Fig. 13-16C, D), the larger depolarization of the postsynaptic membrane now allows the NMDA-gated channel to open by relieving its Mg2+ block. Indeed, under natural conditions, the slower NMDA channels open only after the membrane has been sufficiently depolarized by the action of the faster AMPA channels from many simultaneously active synapses. (See Note: Differential Ca2+ Permeabilities of AMPA-and NMDA-Type Glutamate Receptors)
The physiological function of kainate-gated channels is still a mystery, although recent evidence suggests that they may contribute in a small way to some glutamate-mediated EPSPs. The kainate receptor channels also exist on presynaptic GABAergic terminals, where they regulate release of the inhibitory transmitter (i.e., GABA).
GABA mediates the large majority of synaptic inhibition in the CNS, and glycine mediates most of the rest. Both the GABAA receptor and the glycine receptor are ionotropic receptors that are, in fact, Cl−-selective channels. Note that GABA can also activate the relatively common GABAB receptor, which is a GPCR or metabotropic receptor that is linked to either the opening of K+ channels or the suppression of Ca2+ channels. Finally, GABA can activate the ionotropic GABAC receptor, which is rare in the brain.
Synaptic inhibition must be tightly regulated in the brain. Too much inhibition causes loss of consciousness and coma, whereas too little leads to a seizure. The need to control inhibition may explain why the GABAA receptor channel has, in addition to its GABA binding site, several other sites where chemicals can bind and thus dramatically modulate the function of the GABAA receptor channel. For example, two classes of drugs, benzodiazepines (one of which is the tranquilizer diazepam [Valium]) and barbiturates (e.g., the sedative phenobarbital), each binds to its own specific site on the outside face of the GABAA receptor channel. By themselves, these drugs do very little to the channel’s activity. However, when GABA is around, benzodiazepines increase the frequency of channel opening, whereas barbiturates increase the duration of channel opening. In addition, the benzodiazepines can increase Cl− conductance of the GABAA receptor channel. Figure 13-17A to D shows the effects of barbiturates on both the IPSP and the single-channel currents. The result, for both the benzodiazepines and the barbiturates, is more inhibitory Cl− current, stronger IPSPs, and the behavioral consequences of enhanced inhibition.
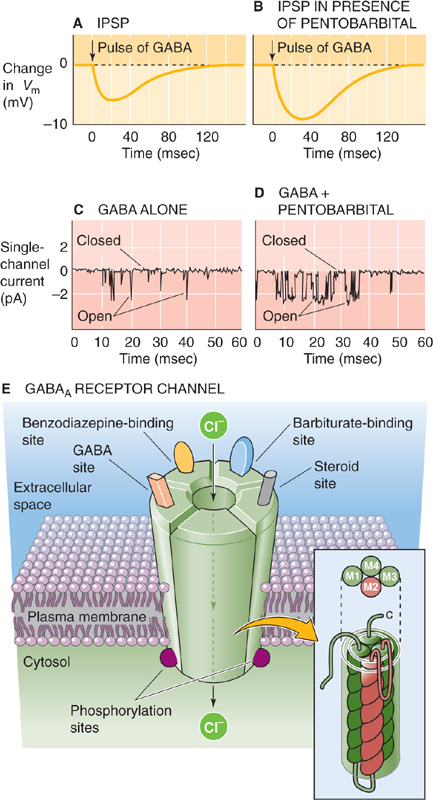
Figure 13-17 Physiology and structure of the GABAA receptor channel. A, When a pulse of GABA is released from a synapse, it elicits a small IPSP. B, In the presence of a low dose of pentobarbital, the pulse of synaptic GABA elicits a much larger IPSP. Thus, the barbiturate enhances inhibition. C, At the single-channel level, GABA by itself elicits brief channel openings. D, A barbiturate (50 μM pentobarbital, in this case) does not by itself activate the GABAA receptor channel but increases the channel open time when GABA is present. E, The channel receptor is a heteropentamer. It not only has a pore for Cl− but also separate binding sites for GABA and several classes of channel modulators. The inset shows the presumed structure of one of the five monomers. The M2 domain of each of the five subunits presumably lines the central channel pore. (Data in C and D from Puia G, Santi MR, Vicini S, et al: Neurosteroids act on recombinant human GABAA receptors. Neuron 1990; 4:759-765.)
Surely, however, the GABAA receptor did not evolve specialized binding sites just for the benefit of our modern drugs. This sort of logic has motivated research to find endogenous ligands, or natural chemicals that bind to the benzodiazepine and barbiturate sites and serve as regulators of inhibition. Figure 13-17E shows some of the binding sites on the GABAA receptor. Among the potential natural modulators of the GABAA receptor may be various metabolites of the steroid hormones progesterone, corticosterone, and testosterone. Some of these hormones increase the lifetime or opening frequency of GABA-activated single-channel currents and may thus enhance inhibition. The steroid effect is unlike the usual genomic mechanisms of steroid hormones (see Chapter 4). Instead, steroids modulate the GABAA receptor in a manner similar to barbiturates—directly, through binding sites that are distinct from the other drug binding sites on the GABAA receptor. Thus, these steroids are not the natural agonists of the benzodiazepine and barbiturate binding sites. The GABAA receptor is also subject to modulation by the effects of phosphorylation triggered by second-messenger signaling pathways within neurons.
We now know the amino acid sequences of many ligand-gated ion channels in the brain, including the receptors for ACh, serotonin, GABA, and glycine. Even though they are gated by such different ligands and have such different permeabilities, virtually all have the same overall structure: five protein subunits, with each subunit being made up of four membrane-spanning segments (as shown for the GABAA receptor channel in Fig. 13-17E and inset). For example, inhibitory GABAA and glycine receptors have structures very similar to those of excitatory nicotinic ACh receptors, even though the first two are selective for anions and the last is selective for cations. For both the glycine and nicotinic ACh receptors, the transmitters bind only to the α subunits; for the GABAA receptor, the transmitter can bind to either the α or the β subunit.
The primary structures of the many subunit types are remarkably similar, particularly within the amino acid sequences of the hydrophobic membrane-spanning segments. One such stretch, called the M2 domain, tends to have repeating sequences of the polar amino acids threonine and serine. Each of the five subunits that constitute a channel contributes one such polar M2 domain, and the set of five combine to form the water-lined pore through which ions can flow (Fig. 13-17E). For the channels gated by GABA and glycine, selectivity for Cl− may be determined by positively charged arginines and lysines near the mouth of the pore.
Not quite all ligand-gated channels belong to the same superfamily. We have already seen that the family of ionotropic glutamate receptors is distinct from the family of ligand-gated/pentameric channels. Extensive evidence also indicates that ATP is a synaptic transmitter between certain neurons and at neuron–smooth muscle cell synapses, with rapid actions similar to those of glutamate and ACh. One of the “purinoceptors,” called P2X, is an ATP-gated cation channel with relatively high Ca2+ permeability. The sequence of this receptor bears little resemblance to either the ionotropic glutamate receptor family or the ligand-gated/pentameric channel superfamily. Instead, each subunit appears to have only two membrane-spanning segments. Functionally, the ATP-gated channel closely resembles the nicotinic ACh receptor; structurally, it is much more akin to the channel family that includes voltage-gated Na+ and K+ channels (see Chapter 7) and to mechanosensitive channels. This similarity appears to be a case of convergent evolution among ion channels.
A single neuromuscular junction has ~1000 active zones (see Chapter 8). A single presynaptic impulse releases 100 to 200 quanta of transmitter molecules (i.e., ACh), which generates an EPSP of more than 40 mV in the muscle cell. This is excitation with a vengeance because the total number of quanta is far more than necessary to cause the muscle cell to fire an action potential and generate a brief contraction. Evolution has designed a neuromuscular junction that works every time, with a large margin of excess for safety. Synapses in the brain are quite different. A typical glutamatergic synapse, which has as few as one active zone, generates EPSPs of only 10 to 1000 μV. In most neurons, one EPSP is rarely enough to cause a postsynaptic cell to fire an action potential.
The basis for the small effect of central synapses has been explored by quantal analysis, with refinements of methods originally applied to the neuromuscular junction. In this approach, a single presynaptic axon is stimulated repeatedly while the postsynaptic response is recorded under voltage-clamp conditions. The frequency distribution of amplitudes of excitatory postsynaptic currents (EPSCs) is analyzed, as described for the neuromuscular junction. Recall that according to standard quantal theory:

Here, m is the total number of quanta released, n is the maximal number of releasable quanta (perhaps equivalent to the number of active zones), and p is the average probability of release. Measurement of these parameters is very difficult. Only in rare cases in central neurons is it possible to find amplitude distributions of EPSCs with clearly separate peaks that may correspond to quantal increments of transmitter. In most cases, EPSC distributions are smooth and broad, which makes quantal analysis difficult to interpret. The analysis is hampered because EPSCs are small, it is extremely difficult to identify each small synapse, the dendrites electrically filter the recorded synaptic signals, noise arises from numerous sources (including the ion channels themselves), and synapses exhibit considerable variability. Nevertheless, what is clear is that most synaptic terminals in the CNS release only a small number of transmitter molecules per impulse, often just those contained in a single quantum (i.e., 1000 to 5000 glutamate molecules). Furthermore, the probability of release of that single quantum is often substantially less than 1; in other words, a presynaptic action potential often results in the release of no transmitter at all. When a quantum of transmitter molecules is released, only a limited number of postsynaptic receptors is available for the transmitter to bind to, usually not more than 100. In addition, because not all the receptors open their channels during each response, only 10 to 40 channels contribute current to each postsynaptic response, compared with the thousands of channels opening in concert during each neuromuscular EPSP.
Because most glutamatergic synapses in the brain contribute such a weak excitatory effect, it may require the nearly simultaneous action of many synapses (and the summation of their EPSPs) to bring the postsynaptic membrane potential above the threshold for an action potential. The threshold number of synapses varies greatly among neurons, but it is roughly in the range of 10 to 100.
Some exceptions to the rule of small synaptic strengths in the CNS may be noted. One of the strongest connections in the CNS is the one between the climbing fibers and Purkinje cells of the cerebellum (Fig. 13-1B). Climbing fibers are glutamatergic axons arising from cells in the inferior olivary nucleus, and they are a critical input to the cerebellum. Climbing fibers and Purkinje cells have a dedicated, one-to-one relationship. The climbing fiber branches extensively and winds intimately around each Purkinje cell, making numerous synaptic contacts. When the climbing fiber fires, it generates a massive EPSP (~40 mV, similar to the neuromuscular EPSP) that evokes a burst of spikes in the Purkinje cell. Like the neuromuscular junction, the climbing fiber–Purkinje cell relationship seems to be designed to deliver a suprathreshold response every time it is activated. It achieves this strength in the standard way: each climbing fiber makes ~200 synaptic contacts with each Purkinje cell.
As we mentioned previously, some presynaptic terminals have two or more transmitters co-localized within them. In these cases, the small transmitters are packaged into relatively small vesicles (~40 nm in diameter), whereas neuropeptides are in larger dense-core vesicles (100 to 200 nm in diameter), as noted earlier. This dual-packaging scheme allows the neuron some control over the relative release of its two types of transmitters (Fig. 13-18A). In general, low-frequency stimulation of the presynaptic terminal triggers the release of only the small transmitter (Fig. 13-18B); co-release of both transmitters requires bursts of high-frequency stimulation (Fig. 13-18C). This frequency sensitivity may result from the size and spatial profile of presynaptic [Ca2+]i levels achieved by the different patterns of stimulation. Presynaptic Ca2+ channels are located close to the vesicle fusion sites. Low frequencies of activation yield only localized elevations of [Ca2+]i, an amount sufficient to trigger the exocytosis of small vesicles near active zones. Larger peptide-filled vesicles are farther from active zones, and high-frequency stimulation may be necessary to achieve higher, more distributed elevations of [Ca2+]i. With this arrangement, it is obvious that the synaptic effect (resulting from the mixture of transmitters released) depends strongly on the way that the synapse is activated.
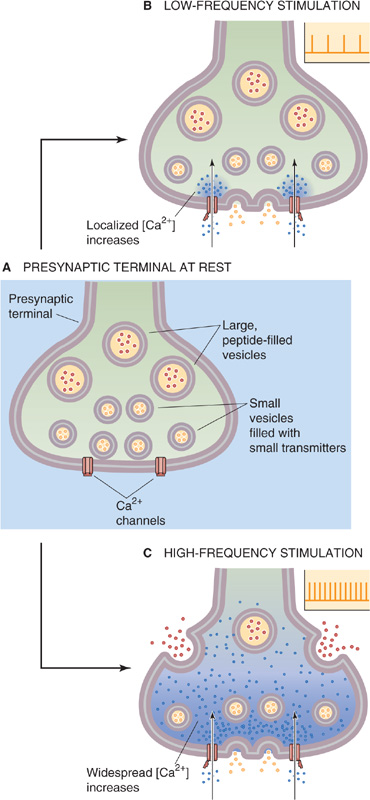
Figure 13-18 Selective release of co-localized small transmitters and neuroactive peptides. A, The presynaptic terminal at rest is filled with small vesicles (containing small transmitter molecules) and large, dense-core vesicles (containing neuroactive peptides). B, Fusion of small vesicles containing small transmitters. C, Fusion of large, dense-core vesicles.
Arguably the greatest achievement of a brain is its ability to learn and to store the experience and events of the past so that it is better adapted to deal with the future. Memory is the ability to store and to recall learned changes, and nervous systems without memory are extremely handicapped. Studies of the biological bases for learning and memory have been intense. Although they are far from understood, certain principles have become clear. First, no single mechanism can explain all forms of memory. Even within a single organism, a variety of types of memory exist—and undoubtedly a variety of mechanisms underlie them. Second, evidence is strong that synapses are the physical site of many if not all forms of memory storage in the brain. As the major points of interaction between neurons, synapses are well placed to alter the processing capabilities of a neural circuit in interesting and useful ways. Third, the synaptic strength (i.e., the mean amplitude of the postsynaptic response) of many synapses may depend on their previous activity. The sensitivity of a synapse to its past activity can lead to a long-term change in its future effectiveness, which is all we need to build memory into a neural circuit.
Some forms of memory last just a few seconds or minutes, only to be lost or replaced by new memories. Working memory is an example. It is the continual series of fleeting memories that we use during the course of a day to remember facts and events, what was just spoken to us, where we put the phone down, whether we are coming or going—things that are useful for the moment but need not be stored longer. Other forms of memory may last for hours to decades and strongly resist disruption and replacement. Such long-term memory allows the accumulation of knowledge over a lifetime. Some memories may be formed after only a single trial (recall a particularly dramatic but unique event in your life), whereas others form only with repeated practice (examples include speaking a language or playing the piano). Detailed descriptions of the many types of memory are beyond the scope of this chapter, but it seems obvious that no single synaptic mechanism would suffice to generate all of them. Neurophysiologists have identified many types of synaptic plasticity, the term for activity-dependent changes in the effectiveness of synapses, and some of their mechanisms are well understood. However, it has been very difficult to demonstrate that specific forms of memory use particular types of synaptic plasticity, and correlation of memory with synaptic plasticity remains a coveted goal of current research.
Repetitive stimulation of neuronal synapses often yields brief periods of increased or decreased synaptic strength (Fig. 13-19). The usual nomenclature for the short-term increases in strength is facilitation (which lasts tens to hundreds of milliseconds), augmentation (lasting several seconds), and potentiation (lasting tens of seconds to several minutes and outlasting the period of high-frequency stimulation). Not all of them are expressed at every type of synapse. In general, the longer lasting modifications require longer periods of conditioning stimuli. Short-term decreases in synaptic strength include depression, which can occur during high-frequency stimulation, and habituation, which is a slowly progressing decrease that occurs during relatively low-frequency activation.
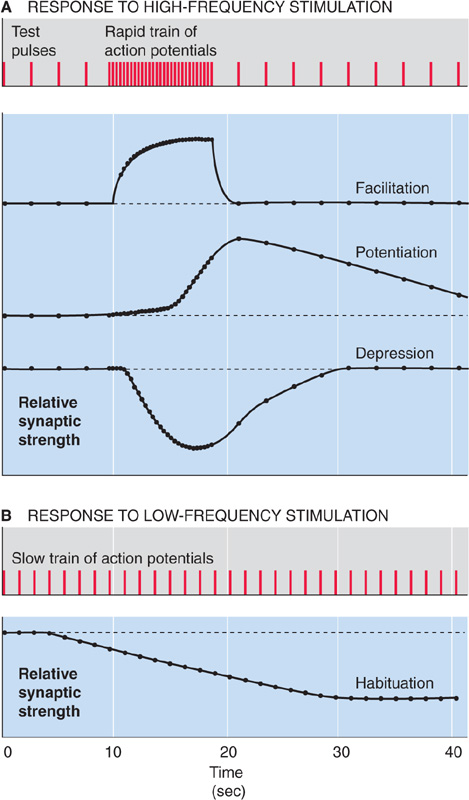
Figure 13-19 Facilitation, potentiation, depression, and habituation. (Data from Levitan IB, Kaczmarek LK: The Neuron: Cell and Molecular Biology, 2nd ed. New York: Oxford University Press, 1997.)
Three potential explanations may be offered for short-term increases in synaptic strength. First, the presynaptic terminal may release more transmitter for each action potential. Second, the postsynaptic receptors may be more responsive to transmitter because of a change in their number or sensitivity. Third, both of these explanations may occur simultaneously. Studies involving a variety of synapses suggest that the first explanation is most often true. In these cases, quantal analysis usually shows that synapses become stronger because more neurotransmitter is released during each presynaptic action potential; postsynaptic mechanisms generally do not play a role. This form of plasticity seems to depend on the influx of presynaptic Ca2+ during the conditioning tetanus. Katz and Miledi first proposed that synaptic strength is increased because of residual Ca2+ left in the terminal after a conditioning train of stimuli at the neuromuscular junction. Recent work supports this hypothesis. The idea is that (1) a tetanic stimulus leads to a substantial increase in presynaptic [Ca2+]i; (2) it takes a relatively long time for presynaptic [Ca2+]i to decline to baseline, and prolonged stimulation requires prolonged recovery times; and (3) presynaptic action potentials arriving after the conditioning tetanus generate a Ca2+ influx that sums with the residual [Ca2+]i from the preceding tetanus to yield a larger than normal peak [Ca2+]i. Because the dependence of transmitter release on presynaptic [Ca2+]i is highly nonlinear, the increase in release after conditioning stimuli can be large. It also seems likely that several types of Ca2+-sensitive proteins are present in the presynaptic terminal; the Ca2+ binding site that triggers exocytosis is distinct from Ca2+ binding sites that regulate short-term increases in transmitter release. (See Note: Short-Term Synaptic Plasticity)
The physiology of a short-term decrease in synaptic strength, namely, habituation, has been studied in the marine invertebrate Aplysia. The animal reflexively withdraws its gill in response to a stimulus to its skin. Vincent Castellucci and Eric Kandel found that withdrawal becomes less vigorous—that is, the animal habituates—when the stimulus is presented repeatedly. The basis for this behavioral habituation is, at least in part, a decrease in the strength of synapses made by skin sensory neurons onto gill withdrawal motor neurons. Using quantal analysis, Castellucci and Kandel showed that synaptic habituation is due to fewer transmitter quanta being released per action potential. Thus, as with the short-term enhancements of synaptic strength, this example of habituation is due to presynaptic modifications. (See Note: Eric R. Kandel)
In 1973, Timothy Bliss and Terje Lømo described a form of synaptic enhancement that lasted for days or even weeks. This phenomenon, now called long-term potentiation (LTP), occurred in excitatory synapses of the mammalian cerebral cortex. LTP was generated by trains of high-frequency stimulation applied to the presynaptic axons, and it was expressed as an increase in the size of EPSPs. Several properties of LTP, including its longevity and its location in cortical synapses, made it immediately attractive as a candidate for the cellular basis of certain forms of vertebrate learning. Years of intensive research have revealed many details of the molecular mechanisms of LTP. They have also provided some evidence, albeit still indirect, that LTP is involved in learning. Numerous mechanistically distinct types of LTP exist; here we discuss only the best-studied example.
LTP is easily demonstrated in several synaptic relays within the hippocampus, a part of the cerebral cortex that has often been considered essential for the formation of certain long-term memories. The best studied of these synapses is between the Schaffer collateral axons of CA3 pyramidal neurons (forming the presynaptic terminals) and CA1 pyramidal neurons (the postsynaptic neurons). In a typical experiment, the strength of synapses to the CA1 neuron is tested by giving a single shock about once every 10 seconds. Stimuli are applied separately to two sets of Schaffer collateral fibers that form two different sets of synapses (Fig. 13-20A). If we stimulate a “control” Schaffer collateral once every 10 seconds, the amplitude of the EPSPs recorded in the postsynaptic CA1 neuron remains rather constant during many tens of minutes (Fig. 13-20B). However, if we pair the presynaptic test shocks occurring once every 10 seconds to the “test” pathway with simultaneous postsynaptic depolarization of the CA1 neuron, the amplitude of the EPSP gradually increases several-fold (Fig. 13-20C), indicative of LTP. In this case, the trigger for LTP was the pairing of a low-frequency presynaptic input and a strong postsynaptic depolarization. We already saw that Bliss and Lømo originally induced LTP by pairing the low-frequency presynaptic input with brief bursts of tetanic stimulation (50 to 100 stimuli at a frequency of ~100 Hz).
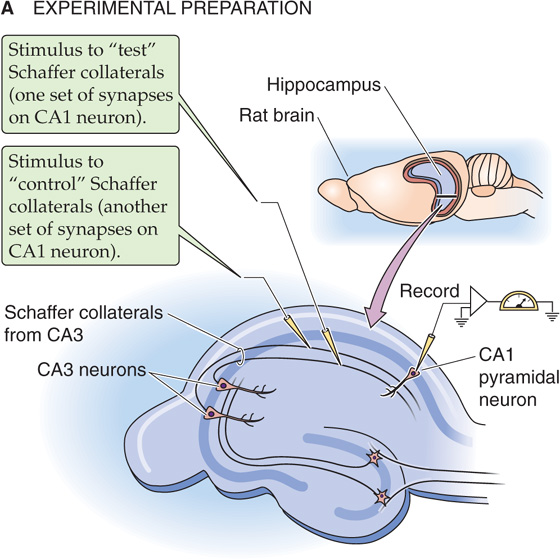
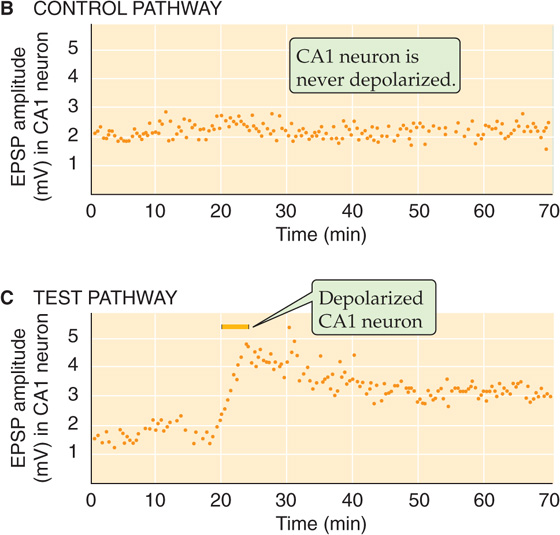
Figure 13-20 Causing long-term potentiation by pairing presynaptic and postsynaptic stimuli. A, Pyramidal CA3 neurons in the hippocampus send axons (Schaffer collaterals) to synapse on pyramidal CA1 neurons. In the case of the “control” stimulus, the stimulating electrode stimulates collaterals that activate one set of synapses on the postsynaptic CA1 neuron. In this case, the CA1 neuron receives only presynaptic stimuli. In the case of the “test” stimulus, a second electrode stimulates a different set of collaterals that activate a different set of synapses on that same CA1 neuron. However, in this case, the presynaptic stimuli will be paired with a postsynaptic depolarization that is delivered by a third microelectrode. Aside from pairing or not pairing the presynaptic stimuli with a postsynaptic stimulus, the test and control pathways are equivalent. The third microelectrode records the EPSPs from the CA1 neuron in both the test and control experiments. B, In this case, the control Schaffer collaterals are stimulated. Each test pulse is represented by a point on the graph. However, because the CA1 neuron is not depolarized, the amplitude of the EPSPs remains constant (i.e., there is no LTP). C, In this case, the test Schaffer collaterals are stimulated. When the CA1 neuron is also depolarized, the amplitude of the EPSPs greatly increases (i.e., LTP has been induced). (Data from Gustafsson B, Wigstrom H, Abraham WC, Huang YY: Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single-volley synaptic potentials. J Neurosci 1987; 7:774-780.)
The induction of LTP has several interesting features that enhance its candidacy as a memory mechanism. First, it is input specific, which means that only the activated set of synapses onto a particular cell will be potentiated, whereas unactivated synapses to that same neuron remain unpotentiated. Second, induction of LTP requires coincident activity of the presynaptic terminals plus significant depolarization of the postsynaptic membrane. We saw this effect in Figure 13-20C, which showed that inducing LTP required coincident synaptic input and depolarization. Because single hippocampal synapses are quite weak, the requirement for substantial postsynaptic activation means that LTP is best induced in an in vivo situation by cooperativity—enough presynaptic axons must cooperate, or fire coincidentally, to strongly activate the postsynaptic cell. The cooperative property of LTP can be used to form associations between synaptic inputs. Imagine that two sets of weak inputs onto one cell are, by themselves, too weak to induce LTP. Perhaps each encodes some sensory feature of an object: the sight (input 1) and sound (input 2) of your pet cat. If the two firing together are strong enough to induce LTP, both sets of synaptic input will tend to strengthen, and the features that they encode (the sight and sound of the cat) will become associated in their enhanced ability to fire the postsynaptic cell. In contrast, for example, the sight of your cat and the sound of your telephone will rarely occur together, and their neural equivalents will not become associated.
The molecular mechanisms of one form of LTP in the CA1 region of hippocampus have been partially elucidated. The synapse uses glutamate as its transmitter, and both AMPA and NMDA receptors are activated to generate an EPSP. Induction of this type of LTP depends absolutely on an increase in post synaptic [Ca2+]i levels beyond a critical level and lasting for about 1 to 2 seconds. (Recall that the short-term forms of enhancement required a pre synaptic increase in [Ca2+]i; see the preceding section.) Under most conditions, postsynaptic [Ca2+]i levels rise during a tetanic stimulus because of the activation of NMDA receptors, the only type of glutamate-activated channel that is usually permeable to Ca2+ (Fig. 13-16). Recall also that the NMDA-type glutamate receptor channel is voltage dependent; to open, it requires Vm to be relatively positive. The cooperativity requirement of LTP is really a requirement for the activation of NMDA receptors so that Ca2+ can enter—if the postsynaptic Vm is too negative (as it is when synaptic activation is weak), NMDA channels remain mostly closed. If activation is strong (as it is when multiple inputs cooperate or when tetanus occurs), Vm becomes positive enough to allow NMDA channels to open. The stimulus-induced rise in postsynaptic [Ca2+]i is thought to activate at least one essential kinase: calcium-calmodulin–dependent protein kinase II. Blocking of this kinase with drugs prevents induction of LTP. Several other types of kinases have been implicated, but the evidence that these mediate—as opposed to modulate—LTP is equivocal.
The molecular pathways leading to the expression and maintenance of LTP are more obscure after the Ca2+-induced activation of kinases. Evidence has been presented both for and against postsynaptic and presynaptic changes explaining the increase in synaptic strength. For the most commonly studied, NMDA receptor–dependent form of LTP described here, there is compelling evidence for a postsynaptic mechanism involving an increased number, and perhaps effectiveness, of AMPA receptors. There is also evidence for a presynaptic mechanism whereby the release of transmitter might be enhanced, although the molecular details of this possibility are obscure. Sometimes, postsynaptic AMPA receptors appear to be functionally “silent” until LTP mechanisms activate them or insert them into the membrane. Note that the presynaptic hypothesis requires the presence of some rapidly diffusing retrograde messenger that can carry a signal from the postsynaptic side (where rising [Ca2+]i is clearly a trigger for LTP) back to the presynaptic terminal. There are currently no convincing chemical candidates for the identity of such a retrograde messenger.
It is theoretically reasonable to suspect that memory systems have mechanisms not only to increase synaptic strength but to decrease it as well. In fact, long-term depression (LTD) can be induced in the same synapses within the hippocampus that generate the [Ca2+]i-dependent LTP described in the preceding section. The critical feature that determines whether the synapses will strengthen or weaken is simply the frequency of stimulation that they receive. For example, several hundred stimuli delivered at 50 Hz produce LTP; the same number delivered at 10 Hz has little effect, and at 1 Hz they produce LTD. One set of synapses can be strengthened or weakened repeatedly, which suggests that each process (LTP and LTD) acts on the same molecular component of the synapses. LTD induced in this way shows the same input specificity as LTP—only the stimulated synapses onto a cell are depressed.
There are multiple forms of LTD, each with distinct molecular mechanisms. One type depends on activation of mGluRs; another apparently requires activation of cannabinoid receptors. We describe the type of LTD that has been studied most extensively. The induction requirements of this form of hippocampal LTD are paradoxically similar to those of LTP: LTD induced by low-frequency stimulation depends on the activation of NMDA receptors, and it requires an increase in postsynaptic [Ca2+]i. The key determinant for whether a tetanic stimulus induces LTP or LTD may be the level to which postsynaptic [Ca2+]i rises. A simple model that illustrates the induction mechanisms of LTD and LTP is shown in Figure 13-21. Synaptic activation releases glutamate, which activates NMDA receptors, which in turn allow Ca2+ to enter the postsynaptic cell. In the case of high-frequency stimulation, postsynaptic [Ca2+]i rises to very high levels; if stimulation is of low frequency, the rise in postsynaptic [Ca2+]i is more modest. High levels of [Ca2+]i lead to a net activation of protein kinases, whereas modest levels of [Ca2+]i preferentially activate protein phosphatases. The kinases and phosphatases in turn act on a synaptic protein or phosphoprotein that somehow regulates synaptic strength. There is good evidence that LTP-inducing stimuli phosphorylate specific residues on AMPA receptors and conversely that LTD-inducing stimuli dephosphorylate other residues on AMPA receptors. These post-translational changes are, however, only part of the story of long-term synaptic plasticity. LTP increases—and LTD decreases—the numbers of AMPA receptors in the postsynaptic membrane by modulating receptor trafficking into and out of the surface membrane. Longer lasting LTP-and LTD-induced changes seem to involve mechanisms that depend on protein synthesis, including structural changes in synapses and spines.
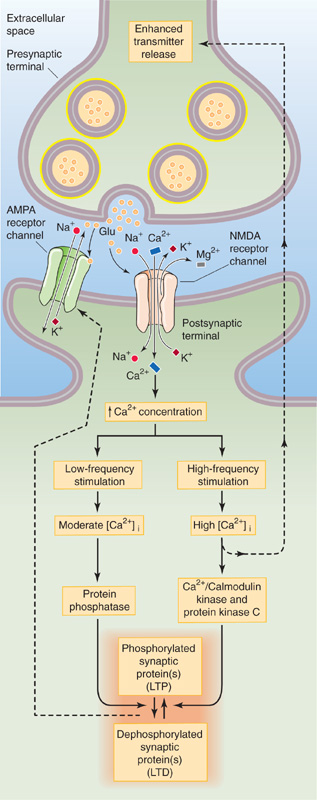
Figure 13-21 Proposed molecular mechanism for long-term potentiation and long-term depression. Glutamate release from the presynaptic terminal activates NMDA receptor channels, which allow Ca2+ to enter the postsynaptic cell. The glutamate also activates AMPA receptor channels. Whether the ultimate effect is LTP or LTD appears to depend on the extent to which [Ca2+]i rises. High levels of [Ca2+]i—produced by high-frequency stimulation—lead to a net activation of protein kinases and thus phosphorylation of one or more synaptic proteins that regulate synaptic strength. One hypothetical pathway has the phosphorylated/dephosphorylated synaptic proteins modulating the AMPA receptor channel. The result is LTP. The postsynaptic neuron also seems to be able to stimulate the presynaptic terminal. A moderate increase in [Ca2+]i—produced by low-frequency stimulation—preferentially activates protein phosphatases, which presumably dephosphorylate the same synaptic proteins as in the previous example. The result is LTD.
Note that the simple scheme shown in Figure 13-21 leaves unidentified many of the steps between the rise in postsynaptic [Ca2+]i and the change in synaptic strength; most of the molecular details remain to be determined. However, if the model is correct, it means that synaptic strength (and, by implication, some memory) is under the dynamic control of cellular processes that determine postsynaptic [Ca2+]i. Once again in physiology, [Ca2+]i has been assigned a pivotal role in a vital process.
A variety of other types of LTP and LTD have been described in other synapses and even within the same synapses of the CA1 region in the hippocampus. Clearly, multiple means and mechanisms may be used to strengthen and to weaken synapses in the brain over a range of time courses. We briefly describe one other well-studied type of synaptic modification in the mammalian brain, LTD in the cerebellum.
The cerebellum is a large brain structure that is important in motor control and strongly implicated in motor learning. The cortex of the cerebellum is a thin, multiply folded sheet of cells with an intricate but highly repetitious neural structure. The principal cell of the cerebellum is the Purkinje cell, a large neuron that uses GABA as its transmitter and whose axon forms the sole output of the cerebellar cortex. Purkinje cells receive two types of excitatory synaptic input: (1) each Purkinje cell receives powerful synaptic contact from just a single climbing fiber, which comes from a cell in the inferior olivary nucleus (Fig. 13-1B); and (2) each Purkinje cell also receives a synapse from ~150,000 parallel fibers, which originate from the tiny granule cells of the cerebellum itself. This remarkable conjunction of synaptic inputs is the basis for a theory of motor learning that was proposed by David Marr and James Albus in 1970. They predicted that the parallel fiber synapses should change their strength only if they are active at the same time as the climbing fiber onto the same cell. This idea received important experimental support from the laboratory of Masao Ito.
Ito and colleagues monitored EPSPs in a Purkinje cell while stimulating some of its inputs from parallel fibers and its one climbing fiber input. They found that the EPSPs generated by the parallel fibers became smaller when both the parallel fibers and the cell’s climbing fiber were coactivated at low frequencies. Stimulating either input alone did not cause any change. Cerebellar LTD can last at least several hours. As with the hippocampal LTP and LTD described before, cerebellar LTD showed input specificity: only those parallel fibers coactivated with the climbing fiber were depressed, whereas other synapses onto the cell were unchanged.
The mechanism of cerebellar LTD has some similarities to that of LTD in the hippocampus, but it is also distinctly different. Parallel fiber synapses weaken because of reduced effectiveness—a reduction in either number or sensitivity—of postsynaptic AMPA-type glutamate receptors. As in the hippocampus, induction of cerebellar LTD requires an increase in postsynaptic [Ca2+]i. However, unlike in the hippocampus, no NMDA-type glutamate receptors are present in the mature cerebellum to mediate Ca2+ flux. Instead, Ca2+ can enter Purkinje cells through voltage-gated Ca2+ channels that are opened during the exceptionally powerful EPSP that the climbing fiber generates. In addition to a rise in postsynaptic [Ca2+]i, cerebellar LTD induction seems to require the activation of metabotropic glutamate receptors and protein kinase C by the parallel fibers. Increases in postsynaptic [Na+]i and NO have also been implicated in LTD induction. At present, the relationships between these putative induction factors and the molecular pathways leading to the expression of cerebellar LTD are obscure.
As we pointed out for the hippocampus, most efficient memory systems need mechanisms for both weakening and strengthening of their synapses. It turns out that parallel fiber synapses of the cerebellum can be induced to generate LTP as well as LTD by stimulating them at relatively low (2 to 8 Hz) frequencies. Cerebellar LTP, unlike hippocampal LTP, requires presynaptic but not postsynaptic increases in [Ca2+]i. Potentiation seems to be a result of increased transmitter release from the presynaptic terminal.
Books and Reviews
Boehning D, Snyder SH: Novel neural modulators. Annu Rev Neurosci 2003; 26:105-131.
Carlisle HJ, Kennedy MB: Spine architecture and synaptic plasticity. Trends Neurosci 2005; 28:182-187.
Collingridge GL, Isaac JT, Wang YT: Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 2004; 5:952-962.
Connors BW, Long MA: Electrical synapses in the mammalian brain. Annu Rev Neurosci 2004; 27:393-418.
Cooper JR, Bloom FE, Roth RH: The Biochemical Basis of Neuropharmacology 8th ed. New York: Oxford University Press, 2002.
Cowan WM, Südhof TC, Stevens CF: Synapses Baltimore: Johns Hopkins University Press, 2001.
MacDonald RL, Olsen RW: GABAA receptor channels. Annu Rev Neurosci. 1994; 17:569-602.
Malenka RC, Bear MF: LTP and LTD: An embarrassment of riches. Neuron 2004; 44:5-21.
Mayer ML, Armstrong N: Structure and function of glutamate receptor ion channels. Annu Rev Physiol 2004; 66:161-181.
Mody I, Pearce RA: Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci 2004; 27:569-575.
Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 2003; 4:873-884.
Südhof TC: The synaptic vesicle cycle. Annu Rev Neurosci 2004; 27:509-547.
Zucker RS, Regehr WG: Short-term synaptic plasticity. Annu Rev Physiol 2002; 64:355-405.
Bliss TV, Lomo T: Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 1973; 232:331-356.
Dudek SM, Bear MF: Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci 1993; 13:2910-2918.
Heuser J, Reese T: Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 1973; 57:315-344.
Kamiya H, Zucker RS: Residual Ca2+ and short-term synaptic plasticity. Nature 1994; 371:603-606.
Katz B, Miledi R: The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968; 195:481-492.
Rosenmund C, Stern-Bach Y, Stevens CF: The tetrameric structure of a glutamate receptor channel. Science 1998; 280:1596-1599.
Sabatini BL, Regehr WG: Timing of neurotransmission at fast synapses in the mammalian brain. Nature 1996; 384:170-172.
Shi SH, Hayashi Y, Petralia RS, et al: Rapid spine delivery and redistribution of AMPA receptors after synaptic nmDA receptor activation. Science 1999; 284:1811-1816.
Vignes M, Collingridge GL: The synaptic activation of kainate receptors. Nature 1997; 388:179-182.
Wilson RI, Nicoll RA: Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001; 410:588-592.