Peter Igarashi
In this chapter, we discuss general principles of gene structure and expression as well as mechanisms underlying the regulation of tissue-specific and inducible gene expression. We will see that proteins (transcription factors) control gene transcription by interacting with regulatory elements in DNA (e.g., promoters and enhancers). Because many transcription factors are effector molecules in signal transduction pathways, these transcription factors can coordinately regulate gene expression in response to physiological stimuli. Finally, we describe the important roles of chromatin structure and post-transcriptional regulation of gene expression. Because many of the proteins and DNA sequences are known by abbreviations, the Glossary at the end of the chapter identifies these entities.
The haploid human genome contains 30,000 to 40,000 distinct genes, but only a fraction of that number—10,000 or so—is actively translated into proteins in any individual cell. Cells from different tissues have distinct morphological appearances and functions and respond differently to external stimuli, even though their DNA content is identical. For example, although all cells of the body contain an albumin gene, only liver cells (hepatocytes) can synthesize and secrete albumin into the bloodstream. Conversely, hepatocytes cannot synthesize myosin and some other contractile proteins that skeletal muscle cells produce. The explanation for these observations is that expression of genes is regulated so that some genes are active in hepatocytes and others are silent. In skeletal muscle cells, a different set of genes is active; others, such as those expressed only in the liver, are silent. How is one cell type programmed to express liver-specific genes, whereas another cell type expresses a set of genes that are appropriate for skeletal muscle? This phenomenon is called tissue-specific gene expression.
A second issue is that genes in individual cells are generally not expressed at constant, unchanging levels (constitutive expression). Rather, their expression levels often vary widely in response to environmental stimuli. For example, when blood glucose levels decrease, α cells in the pancreas secrete the hormone glucagon. Glucagon circulates in the blood until it reaches the liver, where it causes a 15-fold increase in expression of the gene that encodes phosphoenolpyruvate carboxykinase (PEPCK), an enzyme that catalyzes the rate-limiting step in gluconeogenesis (see Chapter 51). Increased gluconeogenesis then contributes to restoration of blood glucose levels toward normal. This simple regulatory loop, which necessitates that the liver cells perceive the presence of glucagon and stimulate PEPCK gene expression, illustrates the phenomenon of inducible gene expression.
The “central dogma of molecular biology” states that genetic information flows unidirectionally from DNA to proteins. Deoxyribonucleic acid (DNA) is a polymer of nucleotides, each containing a nitrogenous base (adenine, T; guanine, G; cytosine, C; or thymine, T) attached to deoxyribose 5′-phosphate. The polymerized nucleotides form a polynucleotide strand in which the sequence of the nitrogenous bases constitutes the genetic information. With few exceptions, all cells in the body share the same genetic information. Hydrogen bond formation between bases (A and T, or G and C) on the two complementary strands of DNA produces a double-helical structure. DNA has two functions. The first is to serve as a self-renewing data repository that maintains a constant source of genetic information for the cell. This role is achieved by DNA replication, which ensures that when cells divide, the progeny cells receive exact copies of the DNA. The second purpose of DNA is to serve as a template for the translation of genetic information into proteins, which are the functional units of the cell. This second purpose is broadly defined as gene expression.
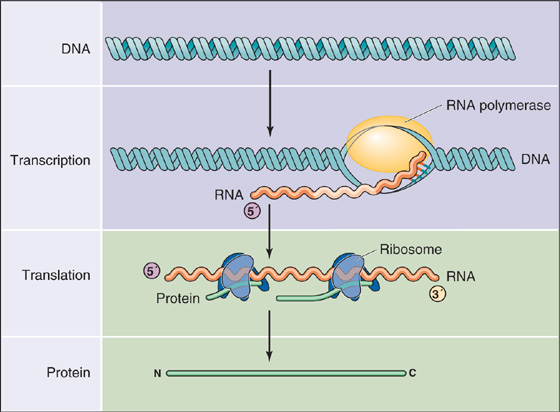
Gene expression involves two major processes (Fig. 4-1). The first process—transcription—is the synthesis of RNA from a DNA template, mediated by an enzyme called RNA polymerase II. The resultant RNA molecule is identical in sequence to one of the strands of the DNA template except that the base uracil (U) replaces thymine (T). The second process—translation—is the synthesis of protein from RNA. During translation, the genetic code in the sequence of RNA is “read” by transfer RNA (tRNA), and then amino acids carried by the tRNA are covalently linked together to form a polypeptide chain. In eukaryotic cells, transcription occurs in the nucleus, whereas translation occurs on ribosomes located in the cytoplasm. Therefore, an intermediary RNA, called messenger RNA (mRNA), is required to transport the genetic information from the nucleus to the cytoplasm (see Chapter 2). The complete process, proceeding from DNA in the nucleus to protein in the cytoplasm, constitutes gene expression.
Figure 4-1 Pathway from genes to proteins. Gene expression involves two major processes. First, the DNA is transcribed into RNA by RNA polymerase. Second, the RNA is translated into protein on the ribosomes.
Figure 4-2 depicts the structure of a typical eukaryotic gene. The gene consists of a segment of DNA that is transcribed into RNA. It extends from the site of transcription initiation to the site of transcription termination. The region of DNA that is immediately adjacent to and upstream (i.e., in the 5′ direction) from the transcription initiation site is called the 5′ flanking region. The corresponding domain that is downstream (3′) from the transcription termination site is called the 3′ flanking region. (Recall that DNA strands have directionality because of the 5′ to 3′ orientation of the phosphodiester bonds in the sugar-phosphate backbone of DNA. By convention, the DNA strand that has the same sequence as the RNA is called the coding strand, and the complementary strand is called the noncoding strand. The 5′ to 3′ orientation refers to the coding strand.) Although the 5′ and 3′ flanking regions are not transcribed into RNA, they frequently contain DNA sequences, called regulatory elements, that control gene transcription. The site where transcription of the gene begins, sometimes called the cap site, may have a variant of the nucleotide sequence 5′-ACTT(T/C)TG-3′ (called the cap sequence), where T/C means T or C. The A is the transcription initiation site. Transcription proceeds to the transcription termination site, which has a less defined sequence and location in eukaryotic genes. Slightly upstream from the termination site is another sequence called the polyadenylation signal, which often has the sequence 5′-AATAAA-3′.
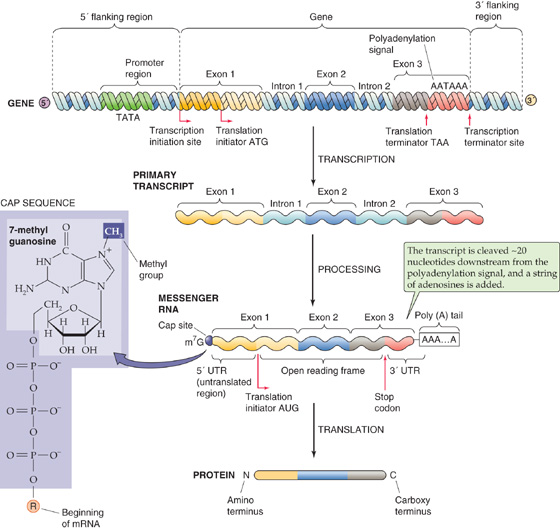
Figure 4-2 Structure of a eukaryotic gene and its products. The figure depicts a gene, a primary RNA transcript, the mature mRNA, and the resulting protein. The 5′ and 3′ numbering of the gene refers to the coding strand. m7G, 7-methyl guanosine; ATG, AATAAA, and the like are nucleotide sequences.
The RNA that is initially transcribed from a gene is called the primary transcript (Fig. 4-2) or heterogeneous nuclear RNA (hnRNA). Before it can be translated into protein, the primary transcript must be processed into a mature mRNA in the nucleus. Most eukaryotic genes contain exons, DNA sequences that are present in the mature mRNA, alternating with introns, which are not present in the mRNA. The primary transcript is colinear with the coding strand of the gene and contains the sequences of both the exons and the introns. To produce a mature mRNA that can be translated into protein, the cell must process the primary transcript in four steps.
First, the cell removes the sequences of the introns from the primary transcript by a process called pre-mRNA splicing. Splicing involves the joining of the sequences of the exons in the RNA transcript and the removal of the intervening introns. As a result, mature mRNA (Fig. 4-2) is shorter and not colinear with the coding strand of the DNA template.
Second, the cell adds an unusual guanosine base, which is methylated at the 7 position, through a 5′-5′ phosphodiester bond to the 5′ end of the transcript. The result is a 5′ methyl cap. The presence of the 5′ methyl cap is required for export of the mRNA from the nucleus to the cytoplasm as well as for translation of the mRNA.
The third processing step is cleavage of the RNA transcript about 20 nucleotides downstream from the polyadenylation signal, near the 3′ end of the transcript.
The fourth step is the addition of a string of 100 to 200 adenine bases at the site of the cleavage to form a poly(A) tail. This tail contributes to mRNA stability.
The mRNA produced by RNA processing contains a coding region that is translated into protein as well as sequences at the 5′ and 3′ ends that are not translated into protein (the 5′ and 3′ untranslated regions, respectively). Translation of the mRNA on ribosomes always begins at the codon AUG, which encodes methionine, and proceeds until the ribosome encounters one of the three stop codons (UAG, UAA, or UGA). Thus, the 5′ end of the mRNA is the first to be translated and provides the N terminus of the protein; the 3′ end is the last to be translated and contributes the C terminus.
Although DNA is commonly depicted as linear, chromosomal DNA in the nucleus is actually organized into a higher order structure called chromatin. This packaging is required to fit DNA with a total length of ~1 m into a nucleus with a diameter of 10−5 m. Chromatin consists of DNA associated with histones and other nuclear proteins. The basic building block of chromatin is the nucleosome (Fig. 4-3), each of which consists of a protein core and 147 bp of associated DNA. The protein core is an octamer of the histones H2A, H2B, H3, and H4. DNA wraps twice around the core histones to form a solenoid-like structure. A linker histone, H1, associates with segments of DNA between nucleosomes. Regular arrays of nucleosomes have a beads-on-a-string appearance and constitute the so-called 11-nm fiber of chromatin, which can condense to form the 30-nm fiber.
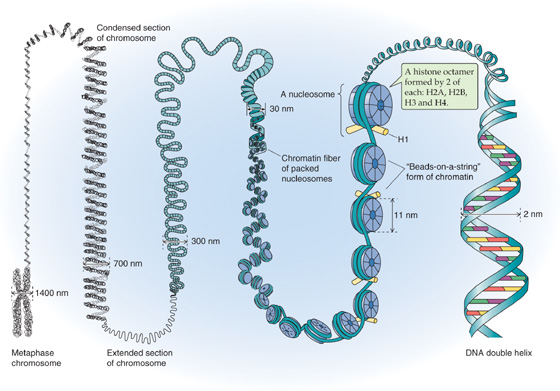
Figure 4-3 Chromatin structure.
Transcription from DNA in chromatin requires partial disruption of the regular nucleosome structure and some unwinding of the DNA. The alteration in the interaction between DNA and histones is called chromatin remodeling. One mechanism of chromatin remodeling involves histone acetylation (Fig. 4-4). The N termini of core histone proteins contain many lysine residues that impart a highly positive charge. These positively charged domains can bind tightly to the negatively charged DNA through electrostatic interactions. Tight binding between DNA and histones is associated with gene inactivity. However, if the 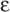 -amino groups of the lysine side chains are chemically modified by acetylation, the positive charge is neutralized and the interaction with DNA is weakened. This modification is believed to result in a loosening of chromatin structure, which permits transcriptional regulatory proteins to gain access to the DNA. Certain enzymes can acetylate histones (histone acetyltransferases) or deacetylate them (histone deacetylases). Histone acetyltransferases (HATs) acetylate histones and thus produce local alterations in chromatin structure that are more favorable for transcription. Conversely, histone deacetylases (HDACs) remove the acetyl groups, leading to tighter binding between DNA and histones and inhibition of transcription.
-amino groups of the lysine side chains are chemically modified by acetylation, the positive charge is neutralized and the interaction with DNA is weakened. This modification is believed to result in a loosening of chromatin structure, which permits transcriptional regulatory proteins to gain access to the DNA. Certain enzymes can acetylate histones (histone acetyltransferases) or deacetylate them (histone deacetylases). Histone acetyltransferases (HATs) acetylate histones and thus produce local alterations in chromatin structure that are more favorable for transcription. Conversely, histone deacetylases (HDACs) remove the acetyl groups, leading to tighter binding between DNA and histones and inhibition of transcription.
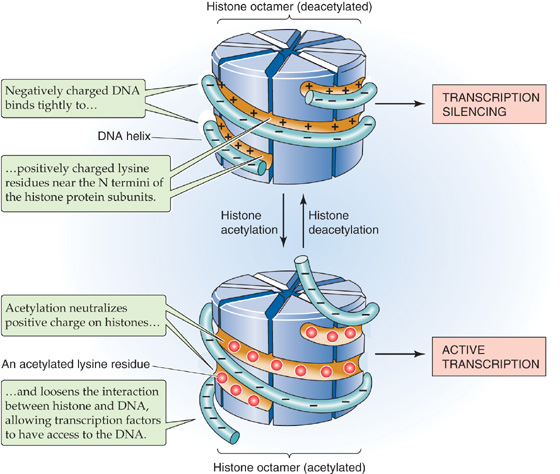
Figure 4-4 Effect of histone acetylation on the interaction between histone proteins and DNA. When the histone octamer is deacetylated (top), positively charged lysine groups on the histone strongly attract a strand of DNA. When the histone octamer becomes acetylated (bottom), the acetyl groups neutralize the positive charge on the histone and allow the DNA strand to loosen.
In addition to histone acetylation and deacetylation, another mechanism of chromatin remodeling involves the SWI/SNF family of proteins. SWI/SNF (switching mating type/sucrose non-fermenting) are large multiprotein complexes, initially identified in yeast but evolutionarily conserved in all animals. SWI/SNF chromatin-remodeling complexes can inhibit the association between DNA and histones by using the energy of ATP to peel the DNA away from the histones, thereby making this DNA more accessible to transcription factors.
Gene expression involves eight steps (Fig. 4-5):
Step 1: Chromatin remodeling. Before a gene can be transcribed, some local alteration in chromatin structure must occur so that the enzymes that mediate transcription can gain access to the DNA. Chromatin remodeling may involve histone acetylation or SWI/SNF chromatin remodeling proteins.
Step 2: Initiation of transcription. In this step, RNA polymerase is recruited to the gene promoter and begins to synthesize RNA that is complementary in sequence to one of the strands of the template DNA. For most eukaryotic genes, initiation of transcription is the critical, rate-limiting step in gene expression.
Step 3: Transcript elongation. During transcript elongation, RNA polymerase proceeds down the DNA strand and sequentially adds ribonucleotides to the elongating strand of RNA. (See Note: Role of Tat in Transcript Elongation)
Step 4: Termination of transcription. After producing a full-length RNA, the enzyme halts elongation.
Step 5: RNA processing. As noted before, RNA processing involves (1) pre-mRNA splicing, (2) addition of a 5′ methylguanosine cap, (3) cleavage of the RNA strand, and (4) polyadenylation.
Step 6: Nucleocytoplasmic transport. The next step in gene expression is the export of the mature mRNA through pores in the nuclear envelope (see Chapter 2) into the cytoplasm. Nucleocytoplasmic transport is a regulated process that is important for mRNA quality control.
Step 7: Translation. The mRNA is translated into proteins on ribosomes. During translation, the genetic code on the mRNA is read by tRNA, and then amino acids carried by the tRNA are added to the nascent polypeptide chain.
Step 8: mRNA degradation. Finally, the mRNA is degraded in the cytoplasm by a combination of endonucleases and exonucleases.
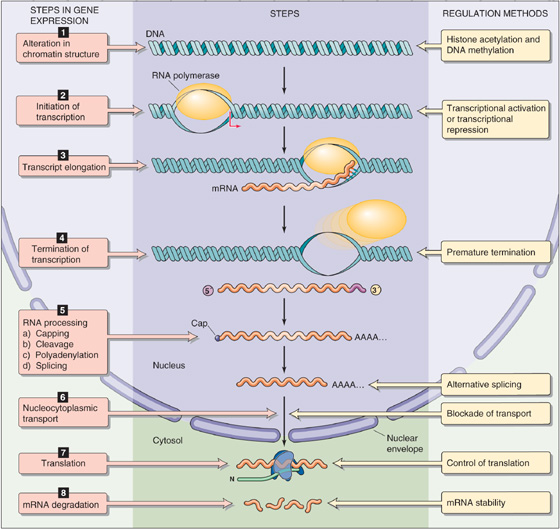
Figure 4-5 Steps in gene expression. Nearly all of the eight steps in gene expression are potential targets for regulation.
Each of these steps is potentially a target for regulation (Fig. 4-5):
1. Gene expression may be regulated by global as well as by local alterations in chromatin structure.
2. An important, related alteration in chromatin structure is the state of methylation of the DNA.
3. Initiation of transcription can be regulated by transcriptional activators and transcriptional repressors.
4. Transcript elongation may be regulated by premature termination in which the polymerase falls off (or is displaced from) the template DNA strand; such termination results in the synthesis of truncated transcripts.
5. Pre-mRNA splicing may be regulated by alternative splicing, which generates different mRNA species from the same primary transcript.
6. At the step of nucleocytoplasmic transport, the cell prevents expression of aberrant transcripts, such as those with defects in mRNA processing. In addition, aberrant transcripts containing premature stop codons may be degraded in the nucleus through a process called nonsense-mediated decay.
7. Control of translation of mRNA is a regulated step in the expression of certain genes, such as the transferrin receptor.
8. Control of mRNA stability contributes to steady-state levels of mRNA in the cytoplasm and is important for the overall expression of many genes.
Although any of these steps may be critical for regulating a particular gene, transcription initiation is the most frequently regulated (step 2) and is the focus of this chapter. At the end of the chapter, we describe examples of control of gene expression at steps that are subsequent to the initiation of transcription—post-transcriptional regulation.
A general principle is that gene transcription is regulated by interactions of specific proteins with specific DNA sequences. The proteins that regulate gene transcription are called transcription factors. These proteins are sometimes referred to as trans-acting factors because they are encoded by genes that reside elsewhere in the genome from the genes that they regulate. Many transcription factors recognize and bind to specific sequences in DNA. The binding sites for these transcription factors are called regulatory elements. Because they are located on the same piece of DNA as the genes that they regulate, these regulatory elements are sometimes referred to as cis-acting factors.
Figure 4-6 illustrates the overall scheme for the regulation of gene expression. Transcription requires proteins (transcription factors) that bind to specific DNA sequences (regulatory elements) located near the genes they regulate (target genes). Once the proteins bind to DNA, they stimulate (or inhibit) transcription of the target gene. A particular transcription factor can regulate the transcription of multiple target genes. In general, regulation of gene expression can occur at the level of either transcription factors or regulatory elements. Examples of regulation at the transcription factor level include variation in the abundance of the proteins, their DNA-binding activities, and their ability to stimulate (or to inhibit) transcription. Examples of regulation at the regulatory element level include alterations in chromatin structure (which influences accessibility to transcription factors) and covalent modifications of DNA, especially methylation.
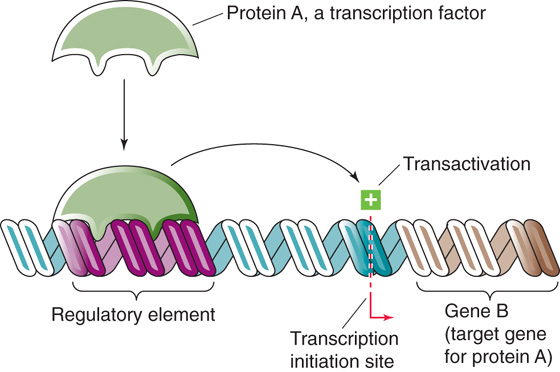
Figure 4-6 Regulation of transcription. Protein A, a transcription factor that is encoded by gene A (not shown), regulates another gene, gene B. Protein A binds to a DNA sequence (a regulatory element) that is upstream from gene B; this DNA sequence is a cis-acting element because it is located on the same DNA as gene B. In this example, protein A stimulates (transactivates) the transcription of gene B. Transcription factors also can inhibit transcription.
Genes are transcribed by an enzyme called RNA polymerase, which catalyzes the synthesis of RNA that is complementary in sequence to a DNA template. Eukaryotes have three distinct RNA polymerases: RNA polymerase I (Pol I) transcribes genes encoding ribosomal RNA. RNA polymerase II (Pol II or RNAPII) transcribes genes into mRNA, which is later translated into protein. Finally, RNA polymerase III (Pol III) transcribes genes that encode tRNA and small nuclear RNA. This discussion is confined to the protein-encoding genes transcribed by Pol II (so-called class II genes).
Pol II is a large protein (molecular mass of 600 kDa) comprising 10 to 12 subunits (the largest of which is structurally related to bacterial RNA polymerase) and is capable of transcribing RNA from synthetic DNA templates in vitro. Although Pol II catalyzes mRNA synthesis, by itself it is incapable of binding to DNA and initiating transcription at specific sites. The recruitment of Pol II and initiation of transcription requires an assembly of proteins called general transcription factors. Six general transcription factors are known, TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH, each of which contains multiple subunits. These general transcription factors are essential for the transcription of all class II genes, which distinguishes them from the transcription factors discussed later that are involved in the transcription of specific genes. Together with Pol II, the general transcription factors constitute the basal transcriptional machinery, which is also known as the RNA polymerase holoenzyme or preinitiation complex because its assembly is required before transcription can begin. The basal transcriptional machinery assembles at a region of DNA that is immediately upstream from the gene and includes the transcription initiation site. This region is called the gene promoter (Fig. 4-7).
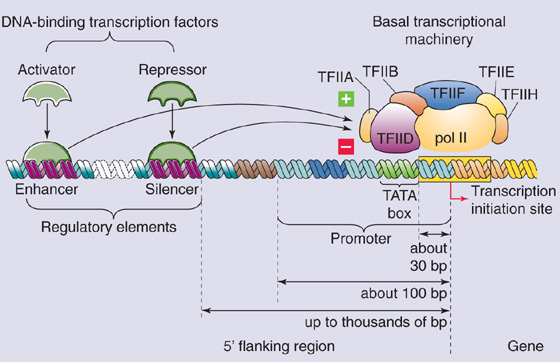
Figure 4-7 Promoter and DNA regulatory elements. The basal transcriptional machinery assembles on the promoter. Transcriptional activators bind to enhancers, and repressors bind to negative regulatory elements.
In vitro, the general transcription factors and Pol II assemble in a stepwise, ordered fashion on DNA. The first protein that binds to DNA is TFIID, which induces a bend in the DNA and forms a platform for the assembly of the remaining factors. Once TFIID binds to DNA, the other components of the basal transcriptional machinery assemble spontaneously by protein-protein interactions. The next general transcription factor that binds is TFIIA, which stabilizes the interaction of TFIID with DNA. Assembly of TFIIA is followed by assembly of TFIIB, which interacts with TFIID and also binds DNA. TFIIB then recruits a preassembled complex of Pol II and TFIIF. Entry of the Pol II–TFIIF complex into the basal transcriptional machinery is followed by binding of TFIIE and TFIIH. TFIIF and TFIIH may assist in the transition from basal transcriptional machinery to an elongation complex, which may involve unwinding of the DNA that is mediated by the helicase activity of TFIIH. Although this stepwise assembly of Pol II and general transcription factors occurs in vitro, the situation in vivo may be different. In vivo, Pol II has been observed in a multiprotein complex containing general transcription factors and other proteins. This preformed complex may be recruited to DNA to initiate transcription. (See Note: Sequential Assembly of General Transcription Factors)
The promoter is a cis-acting regulatory element that is required for expression of the gene. In addition to locating the site for initiation of transcription, the promoter also determines the direction of transcription. Perhaps somewhat surprisingly, no unique sequence defines the gene promoter. Instead, the promoter consists of modules of simple sequences (elements). The most important element in many promoters is the Goldberg-Hogness TATA box. Examination of the sequences of a large number of promoters reveals that the TATA box has the consensus sequence 5′-GNGTATA(A/T)A(A/T)-3′, where N is any nucleotide. The TATA box is usually located ~30 bp upstream (5′) from the site of transcription initiation. The general transcription factor TFIID—the first component of the basal transcriptional machinery—recognizes the TATA box, which is thus believed to determine the site of transcription initiation. TFIID itself is composed of TATA-binding protein (TBP) and at least 10 TBP-associated factors (TAFs). The TBP subunit is a sequence-specific DNA-binding protein that binds to the TATA box. Reconstitution studies indicate that recombinant TBP can replace TFIID in basal transcription, but it fails to support elevated levels of transcription in the presence of transcriptional activators. Thus, TBP-associated factors are involved in the activation of gene transcription (more on this later). (See Note: Binding of Specific Transcription Factors to Promoter Elements on DNA)
Many eukaryotic genes, especially the ubiquitously expressed “housekeeping” genes, do not contain a TATA box in their promoters. What determines the site of transcription initiation in TATA-less promoters? At least part of the answer appears to be a series of small DNA sequence elements, collectively called the initiator (Inr). Inr functions analogously to the TATA box to position the basal transcriptional machinery in these genes. Interestingly, it appears that TFIID can also bind to the Inr element, so it may function to establish assembly of the basal transcriptional machinery on both TATA-containing and TATA-less gene promoters. However, in TATA-less promoters, the site of transcription initiation appears to be less precisely defined, and often several transcripts that originate at several distinct but neighboring sites are produced.
In addition to the TATA box and Inr, gene promoters contain additional DNA elements that are necessary for initiating transcription. These elements consist of short DNA sequences and are sometimes called promoter-proximal sequences because they are located within ~100 bp upstream from the transcription initiation site. Promoter-proximal sequences are a type of regulatory element that is required for the transcription of specific genes. Well-characterized examples include the GC box (5′-GGGCGG-3′) and the CCAAT box (5′-CCAAT-3′) as well as the CACCC box and octamer motif (5′-ATGCAAAT-3′). These DNA elements function as binding sites for additional proteins (transcription factors) that are necessary for initiating transcription of particular genes. The proteins that bind to these sites are believed to help recruit the basal transcriptional machinery to the promoter. Examples include the transcription factor NF-Y, which recognizes the CCAAT box, and Sp1, which recognizes the GC box. The CCAAT box is often located ~50 bp upstream from the TATA box, whereas multiple GC boxes are frequently found in TATA-less gene promoters. Some promoter-proximal sequences are present in genes that are active only in certain cell types. For example, the CACCC box found in gene promoters of β-globin is recognized by the erythroid-specific transcription factor EKLF (erythroid Kruppel-like factor). (See Note: Typical Eukaryotic Gene Promoters)
Although the promoter is the site where the basal transcriptional machinery binds and initiates transcription, the promoter alone is not generally sufficient to initiate transcription at a physiologically significant rate. High-level gene expression generally requires activation of the basal transcriptional machinery by specific transcription factors, which bind to additional regulatory elements located near the target gene. Two general types of regulatory elements are recognized. First, positive regulatory elements or enhancers represent DNA-binding sites for proteins that activate transcription; the proteins that bind to these DNA elements are called activators. Second, negative regulatory elements (NREs) or silencers are DNA-binding sites for proteins that inhibit transcription; the proteins that bind to these DNA elements are called repressors (Fig. 4-7).
A general property of enhancers and silencers is that they consist of modules of relatively short sequences of DNA, generally 6 to 12 bp. Sometimes they contain distinct sequences, such as direct or inverted repeats, but often they do not. Regulatory elements are generally located in the vicinity of the genes that they regulate. Typically, regulatory elements do not reside within the portion of the gene that encodes protein but rather are located in noncoding regions, most frequently in the 5′ flanking region that is upstream from the promoter. However, some enhancers and silencers are located downstream from the transcription initiation site and are embedded either in introns or in the 3′ flanking region of the gene. In fact, some enhancers and silencers can function at great physical distances from the gene promoter, many hundreds of base pairs away. Moreover, the distance between the enhancer or silencer and the promoter can often be varied experimentally without substantially affecting transcriptional activity. In addition, many regulatory elements work equally well if their orientation is inverted. Thus, in contrast to the gene promoter, enhancers and silencers exhibit position independence and orientation independence. Another property of regulatory elements is that they are active on heterologous promoters; that is, if enhancers and silencers from one gene are placed near a promoter for a different gene, they can stimulate or inhibit transcription of the second gene.
After transcription factors (activators or repressors) bind to regulatory elements (enhancers or silencers), they may interact with the basal transcriptional machinery to alter gene transcription. How do transcription factors that bind to regulatory elements physically distant from the promoter interact with components of the basal transcriptional machinery? Regulatory elements may be located hundreds of base pairs from the promoter. This distance is much too great to permit proteins that are bound at the regulatory element and promoter to come into contact along a two-dimensional linear strand of DNA. One model that has been proposed to explain the long-range effects of transcription factors is the DNA looping model. According to this model, the transcription factor binds to the regulatory element, and the basal transcriptional machinery assembles on the gene promoter. Looping out of the intervening DNA permits physical interaction between the transcription factor and the basal transcriptional machinery, which subsequently leads to alterations in gene transcription.
In addition to enhancers and silencers, which regulate the expression of individual genes, some cis-acting regulatory elements are involved in the regulation of chromosomal domains containing multiple genes.
The first of this type of element to be discovered was the locus control region (LCR), also called the locus-activating region or dominant control region. The LCR is a dominant, positive-acting cis-element that regulates the expression of several genes within a chromosomal domain. LCRs were first identified at the β-globin gene locus, which encodes the β-type subunits of hemoglobin. Together with α-type subunits, these β-globin–like subunits form embryonic, fetal, and adult hemoglobin (see the box on this topic in Chapter 29). The β-globin gene locus consists of a cluster of five genes (, γG, γA, δ, β) that are distributed over 90 kb on chromosome 11. During ontogeny, the genes exhibit highly regulated patterns of expression in which they are transcribed only in certain tissues and only at precise developmental stages. Thus, embryonic globin () is expressed in the yolk sac, fetal globins (γG, γA) are expressed in fetal liver, and adult globins (δ, β) are expressed in adult bone marrow. This tightly regulated expression pattern requires a regulatory region that is located far from the structural genes. This region, designated the LCR, extends from 6 to 18 kb upstream from the -globin gene. The LCR is essential for high-level expression of the β-globin–like genes within red blood cell precursors because the promoters and enhancers near the individual genes permit only low-level expression. (See Note: Locus Control Region for the β-Globin Gene Family)
The β-globin LCR contains five sites, each with an enhancer-like structure that consists of modules of simple sequence elements that are binding sites for the erythrocyte-specific transcription factors GATA-1 and NF-E2. It is believed that the LCRs perform two functions: one is to alter the chromatin structure of the β-globin gene locus so that it is more accessible to transcription factors, and the second is to serve as a powerful enhancer of transcription of the individual genes. In one model, temporal-dependent expression of β-type globin genes is achieved by sequential interactions involving activator proteins that bind to the LCR and promoters of individual genes (Fig. 4-8).
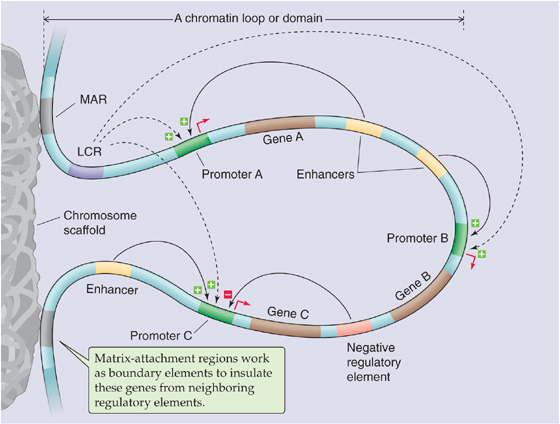
Figure 4-8 cis-Acting elements that regulate gene transcription. This model shows a loop of chromatin that contains genes A, B, and C. The matrix-attachment region (MAR) is a boundary element on the DNA. Matrix-attachment regions attach to the chromosome scaffold and thus isolate this loop of chromatin from other chromosomal domains. Contained within this loop are several cis-acting elements (i.e., DNA sequences that regulate genes on the same piece of DNA), including promoters, enhancers, negative regulatory elements, and the LCR.
A potential problem associated with the existence of LCRs that can exert transcriptional effects over long distances is that the LCRs may interfere with the expression of nearby genes. One solution to this problem is provided by boundary elements, which function to insulate genes from neighboring regulatory elements. Boundary elements (or matrix-attachment regions) are believed to represent sites of attachment of DNA to the chromosome scaffold, and loops of physically separated DNA are generated that may correspond to discrete functional domains.
Figure 4-8 summarizes our understanding of the arrangement of cis-acting regulatory elements and their functions. Each gene has its own promoter where transcription is initiated. Enhancers are positive-acting regulatory elements that may be located either near or distant from the transcription initiation site; silencers are regulatory elements that inhibit gene expression. A cluster of genes within a chromosomal domain may be under the control of an LCR. Finally, boundary elements (or matrix-attachment regions) functionally insulate one chromosomal domain from another.
The preceding discussion has emphasized the structure of the gene and the cis-acting elements that regulate gene expression. We now turn to the proteins that interact with these DNA elements and thus regulate gene transcription. Because the basal transcriptional machinery—Pol II and the general transcription factors—is incapable of efficient gene transcription alone, additional proteins are required to stimulate the activity of the enzyme complex. The additional proteins include transcription factors that recognize and bind to specific DNA sequences (enhancers) located near their target genes as well as others that do not bind to DNA.
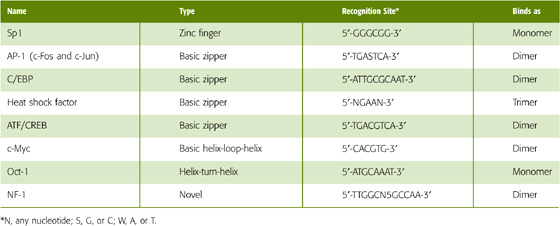
Examples of DNA-binding transcription factors are shown in (Table 4-1). The general mechanism of action of a specific transcription factor is depicted in Figure 4-7. After the basal transcriptional machinery assembles on the gene promoter, it can interact with a transcription factor that binds to a specific DNA element, the enhancer. Looping out of the intervening DNA permits physical interaction between the transcription factor and the basal transcriptional machinery, which subsequently leads to stimulation of gene transcription. The specificity with which transcription factors bind to DNA depends on the interactions between the amino acid side chains of the transcription factor and the purine and pyrimidine bases in DNA. Most of these interactions consist of noncovalent hydrogen bonds between amino acids and DNA bases. A peptide capable of a specific pattern of hydrogen bonding can recognize and bind to the reciprocal pattern in the major (and to a lesser extent the minor) groove of DNA. Interaction with the DNA backbone may also occur and involves electrostatic interactions (salt bridge formation) with anionic phosphate groups. The site that a transcription factor recognizes (Table 4-1) is generally short, usually less than a dozen or so base pairs.
Table 4-1 DNA-Binding Transcription Factors and the DNA Sequences They Recognize
Abnormalities of Regulatory Elements in β-Thalassemias
The best characterized mutations affecting DNA regulatory elements occur at the gene cluster encoding the β-globin–like chains of hemoglobin. Some of these mutations result in thalassemia, whereas others cause hereditary persistence of fetal hemoglobin. The β-thalassemias are a heterogeneous group of disorders characterized by anemia caused by a deficiency in production of the β chain of hemoglobin. The anemia can be mild and inconsequential or severe and life-threatening. The thalassemias were among the first diseases to be characterized at the molecular level. As described in the text, the β-globin gene locus consists of five β-globin–like genes that are exclusively expressed in hematopoietic cells and exhibit temporal colinearity. As expected, many patients with β-thalassemia have mutations or deletions that affect the coding region of the β-globin gene. These patients presumably have thalassemia because the β-globin gene product is functionally abnormal or absent. In addition, some patients have a deficiency in β-globin as a result of inadequate levels of expression of the gene. Of particular interest are patients with the Hispanic and Dutch forms of β-thalassemia. These patients have deletions of portions of chromosome 11. However, the deletions do not extend to include the β-globin gene itself. Why, then, do these patients have β-globin deficiency? It turns out that the deletions involve the region 50 to 65 kb upstream from the β-globin gene, which contains the LCR. In these cases, deletion of the LCR results in failure of expression of the β-globin gene, even though the structural gene and its promoter are completely normal. These results underscore the essential role that the LCR plays in β-globin gene expression.
DNA-binding transcription factors do not recognize single, unique DNA sequences; rather, they recognize a family of closely related sequences. For example, the transcription factor AP-1 (activator protein 1) recognizes the sequences
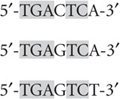
and so on, as well as each of the complementary sequences. That is, some redundancy is usually built into the recognition sequence for a transcription factor. An important consequence of these properties is that the recognition site for a transcription factor may occur many times in the genome. For example, if a transcription factor recognizes a 6-bp sequence, the sequence would be expected to occur once every 46 (or 4096) base pairs, that is, 7 × 105 times in the human genome. If redundancy is permitted, recognition sites will occur even more frequently. Of course, most of these sites will not be relevant to gene regulation but will instead have occurred simply by chance. This high frequency of recognition sites leads to an important concept: transcription factors act in combination. Thus, high-level expression of a gene requires that a combination of multiple transcription factors binds to multiple regulatory elements. Although it is complicated, this system ensures that transcription activation occurs only at appropriate locations. Moreover, this system permits greater fine-tuning of the system, inasmuch as the activity of individual transcription factors can be altered to modulate the overall level of transcription of a gene.
An important general feature of DNA-binding transcription factors is their modular construction (Fig. 4-9A). Transcription factors may be divided into discrete domains that bind DNA (DNA-binding domains) and domains that activate transcription (transactivation domains). This property was first directly demonstrated for a yeast transcription factor known as GAL4, which activates certain genes when yeast grows in galactose-containing media. GAL4 has two domains. One is a so-called zinc finger domain (discussed later) that mediates sequence-specific binding to DNA. The other domain is enriched in acidic amino acids (i.e., glutamate and aspartate) and is necessary for transcriptional activation. This “acidic blob” domain of GAL4 can be removed and replaced with the transactivation domain from a different transcription factor, VP16 (Fig. 4-9B). The resulting GAL4-VP16 chimera binds to the same DNA sequence as normal GAL4 but mediates transcriptional activation through the VP16 transactivation domain. This type of “domain swapping” experiment indicates that transcription factors have a modular construction in which physically distinct domains mediate binding to DNA and transcriptional activation (or repression). (See Note: Grouping of Transcription Factors According to Transactivation Domain)
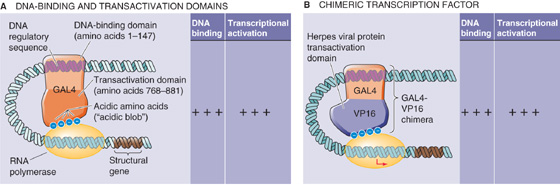
Figure 4-9 Modular design of specific transcription factors. A, DNA-binding transcription factors have independent domains for binding to DNA regulatory sequences and for activating transcription. In this example, amino acids 1 through 147 of the GAL4 transcription factor bind to DNA, whereas amino acids 768 through 881 activate transcription. B, Replacement of the transactivation domain of GAL4 with that of VP16 results in a chimera that is a functional transcription factor.
On the basis of sequence conservation as well as structural determinations from x-ray crystallography and nuclear magnetic resonance spectroscopy, DNA-binding transcription factors have been grouped into families. Members of the same family use common structural motifs for binding DNA (Table 4-1). These structures include the zinc finger, basic zipper (bZIP), basic helix-loop-helix (bHLH), helix-turn-helix (HTH), and β sheet. Each of these motifs consists of a particular tertiary protein structure in which a component, usually an α helix, interacts with DNA, especially the major groove of the DNA.
Zinc Finger The term zinc finger describes a loop of protein held together at its base by a zinc ion that tetrahedrally coordinates to either two histidine residues and two cysteine residues or four cysteine residues. Sometimes two zinc ions coordinate to six cysteine groups. Figure 4-10A shows a zinc finger in which Zn2+ coordinates to two residues on an α helix and two residues on a β sheet of the protein. The loop (or finger) of protein can protrude into the major groove of DNA, where amino acid side chains can interact with the base pairs and thereby confer the capacity for sequence-specific DNA binding. Zinc fingers consist of 30 amino acids with the consensus sequences Cys-X2-4-Cys-X12-His-X3-5-His, where X can be any amino acid. Transcription factors of this family contain at least two zinc fingers and may contain dozens. Three amino acid residues at the tips of each zinc finger contact a DNA subsite that consists of three bases in the major groove of DNA; these residues are responsible for site recognition and binding (Table 4-1). Zinc fingers are found in many mammalian transcription factors, including several that we discuss in this chapter—Egr-1, Wilms tumor protein (WT-1), and stimulating protein 1 (Sp1; Table 4-1)—as well as the steroid hormone receptors.
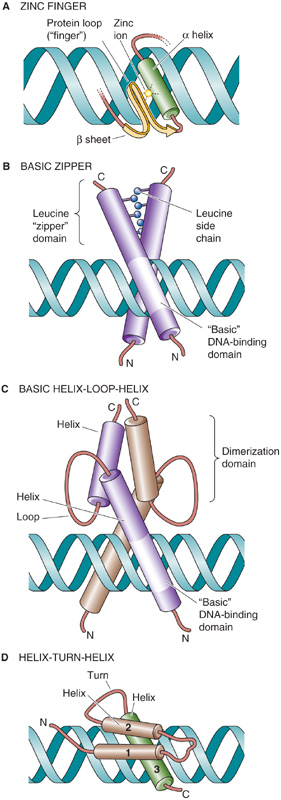
Figure 4-10 Families of transcription factors.
Basic Zipper This bZIP family (also known as the leucine zipper family) consists of transcription factors that bind to DNA as dimers (Fig. 4-10B). Members include C/EBPβ (CCAAT/enhancer binding protein β), c-Fos, c-Jun, and CREB (cAMP response element binding protein). Each monomer consist of two domains, a basic region that contacts DNA and a leucine zipper region that mediates dimerization. The basic region contains about 30 amino acids and is enriched in arginine and lysine residues. This region is responsible for sequence-specific binding to DNA through an α helix that inserts into the major groove of DNA. The leucine zipper consists of a region of about 30 amino acids in which every seventh residue is a leucine. Because of this spacing, the leucine residues align on a common face every second turn of an α helix. Two protein subunits that both contain leucine zippers can associate because of hydrophobic interactions between the leucine side chains; they form a tertiary structure called a coiled coil. Proteins of this family interact with DNA as homodimers or as structurally related heterodimers. Dimerization is essential for transcriptional activity because mutations of the leucine residues abolish both dimer formation and the ability to bind DNA and activate transcription. The crystal structure reveals that these transcription factors resemble scissors in which the blades represent the leucine zipper domains and the handles represent the DNA-binding domains (Fig. 4-10B).
Basic Helix-Loop-Helix Similar to the bZIP family, members of the bHLH family of transcription factors also bind to DNA as dimers. Each monomer has an extended α-helical segment containing the basic region that contacts DNA, linked by a loop to a second α helix that mediates dimer formation (Fig. 4-10C). Thus, the bHLH transcription factor forms by association of four amphipathic α helices (two from each monomer) into a bundle. The basic domains of each monomer protrude into the major grooves on opposite sides of the DNA. bHLH proteins include the MyoD family, which is involved in muscle differentiation, and E proteins (E12 and E47). MyoD and an E protein generally bind to DNA as heterodimers. (See Note: Dimerization of Basic Helix-Loop-Helix Transcription Factors)
Helix-Turn-Helix In prokaryotes such as Escherichia coli, the HTH family consists of two α helices that are separated by a β turn. In eukaryotes, a modified HTH structure is represented by the so-called homeodomain (Fig. 4-10D), which is present in some transcription factors that regulate development. The homeodomain consists of a 60–amino acid sequence that forms three α helices. Helices 1 and 2 lie adjacent to one another, and helix 3 is perpendicular and forms the DNA recognition helix. Particular amino acids protrude from the recognition helix and contact bases in the major groove of the DNA. Examples of homeodomain proteins include the Hox proteins, which are involved in mammalian pattern formation; engrailed homologues that are important in nervous system development; and the POU family members Pit-1, Oct-1, and unc-86. (See Note: Novel Families of Transcription Factors)
Some transcription factors that are required for the activation of gene transcription do not directly bind to DNA. These proteins are called coactivators. Coactivators work in concert with DNA-binding transcriptional activators to stimulate gene transcription. They function as adapters or protein intermediaries that form protein-protein interactions between activators bound to enhancers and the basal transcriptional machinery assembled on the gene promoter (Fig. 4-7). Coactivators often contain distinct domains, one that interacts with the transactivation domain of an activator and a second that interacts with components of the basal transcriptional machinery. Transcription factors that interact with repressors and play an analogous role in transcriptional repression are called corepressors.
One of the first coactivators found in eukaryotes was the VP16 herpesvirus protein discussed earlier (Fig. 4-9B). VP16 has two domains. The first is a transactivation domain that contains a region of acidic amino acids that in turn interacts with two components of the basal transcriptional machinery, general transcription factors TFIIB and TFIID. The other domain of VP16 interacts with the ubiquitous activator Oct-1, which recognizes a DNA sequence called the octamer motif (Table 4-1). Thus, VP16 activates transcription by bridging an activator and the basal transcriptional machinery.
Coactivators are of two types. The first plays an essential role in the transcriptional activation of many, perhaps all, eukaryotic genes. These coactivators include the TBP-associated factors and Mediator. As discussed previously, TAFs were first identified as subunits of the general transcription factor TFIID. Although TAFs are not required for basal transcription, they are essential for transcriptional activation by an activator protein, with which they interact directly. For example, the transcriptional activator Sp1 binds to a 250-kDa TAF called TAF250. TAF250 binds to a smaller TAF110, which in turn binds to TBP. This sequence establishes an uninterrupted linkage between Sp1 and the TBP component of TFIID that binds to the TATA box in the gene promoter. Mediator, a multiprotein complex consisting of 28 to 30 subunits, also appears to be required for activated gene transcription but not basal transcription. Consistent with their essential roles, TAFs and Mediator are present in the basal transcriptional machinery or preinitiation complex.
A second type of coactivator is involved in the transcriptional activation of specific genes. This type of coactivator is not a component of the basal transcriptional machinery. Rather, these coactivators are recruited by a DNA-binding transcriptional activator through protein-protein interactions. An example is the coactivator CBP (CREB-binding protein), which interacts with a DNA-binding transcription factor called CREB (Table 4-1).
Once transcriptional activators bind to enhancers (i.e., positive regulatory elements on the DNA) and recruit coactivators, how do they stimulate gene transcription? We discuss three mechanisms by which transactivation might be achieved. These mechanisms are not mutually exclusive, and more than one mechanism may be involved in the transcription of a particular gene.
Recruitment of the Basal Transcriptional Machinery We have already introduced the concept by which looping out of DNA permits proteins that are bound to distant DNA enhancer elements to become physically juxtaposed to proteins that are bound to the gene promoter (Fig. 4-11, pathway 1). The interaction between the DNA-binding transcription factor and general transcription factors, perhaps with coactivators as protein intermediaries, enhances the recruitment of the basal transcriptional machinery to the promoter. Two general transcription factors, TFIID and TFIIB, are targets for recruitment by transcriptional activators. For example, the acidic transactivation domain of VP16 binds to TFIIB, and mutations that prevent the interaction between VP16 and TFIIB also abolish transcriptional activation. Conversely, mutations of TFIIB that eliminate the interaction with an acidic activator also abolish transactivation but have little effect on basal transcription.
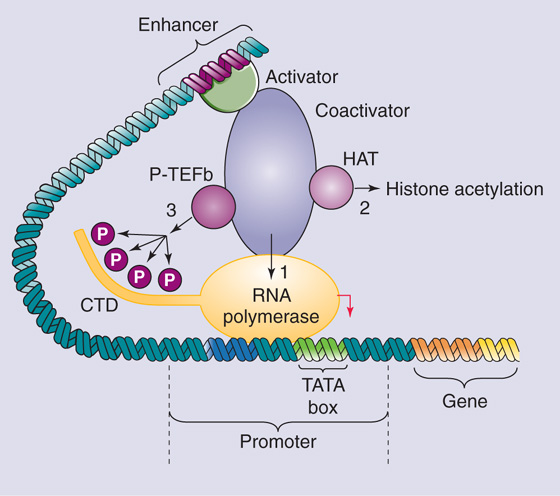
Figure 4-11 Mechanisms of transcriptional activation. The transcriptional activator binds to the enhancer and directly or indirectly (through coactivators) activates transcription by recruiting RNA polymerase to the promoter 1, recruiting histone acetyltransferases that remodel chromatin 2, or stimulating the phosphorylation of the C-terminal domain (CTD) of RNA polymerase 3.
Chromatin Remodeling A second mechanism by which transcriptional activators may function is alteration of chromatin structure. Transcription factors can bind to HATs either directly or indirectly through coactivators (Fig. 4-11, pathway 2). As discussed previously, HATs play an important role in chromatin remodeling before the initiation of gene transcription. By acetylating lysine residues on histones, they inhibit the electrostatic interaction between histones and DNA, which facilitates the binding of additional transcriptional activators and the basal transcriptional machinery. Interestingly, several coactivator proteins that mediate transcriptional activation, such as CBP, possess intrinsic histone acetylase activity. These observations suggest that transcriptional activation is mediated by coactivator proteins that not only bind to components of the basal transcriptional machinery but also promote histone acetylation and thus produce local alterations in chromatin structure that are more favorable for transcription.
Stimulation of RNA Polymerase II A third mechanism by which transcriptional activators function is by stimulating RNA Pol II (Fig. 4-11, pathway 3). The C-terminal domain (CTD) of the largest subunit of Pol II contains 52 repeats of the sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser, which can be phosphorylated at multiple serine and threonine residues. A cyclin-dependent kinase called positive transcription elongation factor b (P-TEFb) phosphorylates the CTD. Phosphorylation of the CTD occurs coincident with initiation of transcription and is required for chain elongation. Thus, transcriptional activators that interact with P-TEFb may stimulate the conversion of the Pol II holoenzyme from an initiation complex into an elongation complex. (See Note: Phosphorylation of CTD)
Taken together, these three mechanisms of interaction lead to an attractive model for activation of transcription. The transcriptional activator that is bound to an enhancer presents a functional domain (e.g., an acidic domain) that either directly or through coactivators interacts with histone acetylases and components of the basal transcriptional machinery. These interactions result in chromatin remodeling and facilitate the assembly of the basal transcriptional machinery on the gene promoter. Subsequent interactions— with the CTD of Pol II, for example—may stimulate transcriptional elongation.
Two or more activators may increase the rate of transcription by an amount that is greater than the sum of each of the activators alone. Almost all naturally occurring promoters contain more than one site for binding of transcriptional activators. A promoter region that contains only a single copy of an enhancer element shows only weak stimulation, whereas a promoter containing multiple copies of an enhancer exhibits substantial activation (Fig. 4-12). Two mechanisms for synergy have been proposed.

Figure 4-12 Synergism of transcriptional activators. The promoter contains three DNA enhancer elements A, B, and C. Binding of a transcription factor to only one of the enhancer elements (A, B, or C) causes a modest activation of transcription. Simultaneous binding of different transcription factors to each of the three enhancer elements can produce a supra-additive increase in transcription (i.e., synergy).
In the first, synergy may reflect cooperative binding to DNA; that is, binding of one transcription factor to its recognition site enhances binding of a second transcription factor to a different site. This phenomenon occurs with the glucocorticoid receptor (GR), which binds to a site on DNA known as the glucocorticoid response element (GRE). Binding of GRs to the multiple GREs is cooperative in that binding of the first receptor promotes binding of additional receptors. Thus, the presence of multiple copies of the GRE greatly stimulates gene expression in comparison to a single copy of the GRE.
In the second case, synergy reflects cooperative protein-protein interactions between transcription factors and multiple sites on the basal transcriptional machinery. For example, a transcriptional activator that recruits TFIID could synergize with another activator that recruits TFIIB. Similarly, a transcriptional activator that interacts with a HAT could synergize with another activator that interacts with components of the basal transcriptional machinery. Here, the effect on transcription depends on the cumulative effects of multiple transcription factors, each bound to its cognate recognition site and interacting with chromatin remodeling proteins and the basal transcriptional machinery.
Transcription factors may act in combination by binding to DNA as homodimers or heterodimers. This synergism is particularly true for members of the bZIP and bHLH families but also for steroid and thyroid hormone receptors. Often, different combinations of monomers have different DNA binding affinities. For example, the thyroid hormone receptor (TR) can bind to DNA as a homodimer, but the heterodimer formed from the thyroid receptor and the 9-cis-retinoic acid receptor (TR/RXR) has much higher binding affinity. As we saw earlier, the transcription factor MyoD, which is involved in muscle differentiation, requires heterodimerization with the ubiquitous proteins E12 and E47 for maximal DNA binding. Different combinations of monomers may also have different DNA binding specificities and thus be targeted to different sites on the DNA. Finally, different combinations of monomers may have different transactivational properties. For example, the c-Myc protein can bind to DNA as a homodimer or as a heterodimer with Max, but c-Myc/Max heterodimers have greater transcriptional activity.
Cells can regulate transcription not only positively through transcriptional activators but also negatively through transcriptional repressors. Repression of transcription is important for tissue specificity in that it allows cells to silence certain genes where they should not be expressed. Repression is also important for regulating inducible gene expression by rapidly turning off transcription after removal of the inducing stimulus. (See Note: Modes of Action of Transcriptional Repression)
Transcriptional repressors may act by three mechanisms. First, some repressors inhibit the binding of transcriptional activators because they compete for DNA-binding sites that are identical to, or overlap with, those for activators. An example is the CCAAT displacement protein (CDP), which binds to the CAAT box in the promoter of the γ-globin gene and thereby prevents binding of the transcriptional activator CP1. This action helps prevent inappropriate expression of the fetal globin gene in adults.
Second, some repressors inhibit the activity of transcriptional activators not by interfering with DNA binding but by a direct protein-protein interaction with activators. This form of repression is termed quenching. A classic example in yeast is the GAL80 repressor, which inhibits transcriptional activation by GAL4. By binding to the transactivation domain of GAL4, GAL80 blocks transcriptional activation. Dissociation of GAL80 (which occurs in the presence of galactose) relieves the inhibition of GAL4, which can then induce expression of galactose-metabolizing genes. Transcriptional repression can also be mediated by proteins that prevent transcriptional activators from entering the nucleus. For example, the heat shock protein hsp90 binds to GR and prevents this transcriptional activator from entering the nucleus.
A third class of repressors binds to a silencer (or NRE) and then directly inhibits transcription. This mechanism is referred to as active repression. The opposite of transcriptional activators, these proteins contain domains that mediate repression. Repression domains may directly interact with and inhibit the assembly or activity of the basal transcriptional machinery. Alternatively, transcriptional repressors may inhibit transcription through protein-protein interactions with corepressors. Some transcriptional corepressors, such as the N-CoR adapter protein that mediates repression by steroid hormone receptors, can interact with HDAC. By removing the acetyl groups from lysine residues in histones, HDACs promote tighter binding between DNA and histones and inhibit transcription initiation.
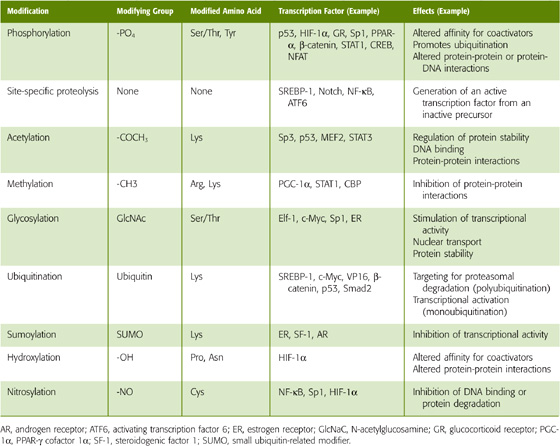
Cells can regulate the activity of transcription factors by controlling the amount of transcription factor they synthesize. In addition, cells can modulate the activity of preformed transcription factors by three general mechanisms of post-translational modification (Table 4-2).
Table 4-2 Post-translational Modifications of Transcription Factors
Phosphorylation The best studied post-translational modification affecting transcription factor activity is phosphorylation, which increases or decreases (1) transport of the transcription factor from the cytoplasm into the nucleus, (2) the affinity with which the transcription factor binds to DNA, and (3) transcriptional activation.
For transcription factors that reside in the cytoplasm under basal conditions, migration from the cytoplasm into the nucleus is a necessary step. Many proteins that are transported into the nucleus contain a sequence that is relatively enriched in basic amino acid residues (i.e., arginine and lysine). This sequence, the nuclear localization signal, is required for transport of the protein into the nucleus. Phosphorylation at sites within or near the nuclear localization signal can dramatically change the rate of nuclear translocation. Phosphorylation can also modulate import into the nucleus by regulating the binding of transcription factors to cytoplasmic anchors. In the case of the transcription factor NF-κB (Fig. 4-13A), binding to the cytoplasmic anchor IκB conceals the nuclear localization signal on the p50 and p65 subunits of NF-κB from the nuclear translocation machinery. Only after these two other subunits dissociate from the phosphorylated IκB is the transcription factor dimer free to enter the nucleus.
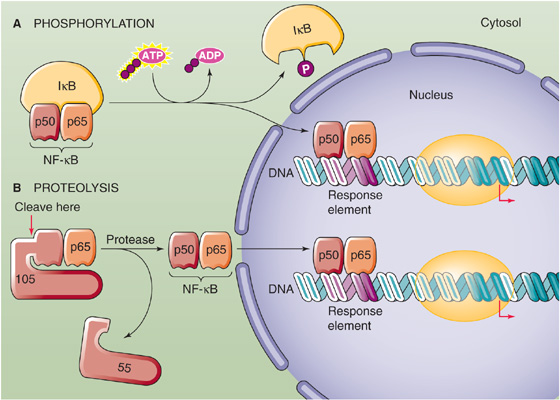
Figure 4-13 Regulation of transcription factors by post-translational modification. A, The phosphorylation of the cytoplasmic anchor IκB releases the p50 and p65 subunits of NF-κB, allowing them to translocate into the nucleus. B, Proteolytic cleavage of a 105-kDa precursor releases the p50 subunit of NF-κB. Together with the p65 subunit, the p50 subunit can now translocate to the nucleus.
Phosphorylation can also regulate transcription factor activity by altering the affinity of the transcription factor for its target recognition sequences on DNA. As a result, phosphorylation increases or decreases DNA-binding activity. For example, phosphorylation of SRF (serum response factor), a transcription factor that activates the c-fos gene in response to growth factors, enhances DNA binding. In contrast, phosphorylation of the transcription factor c-Jun by casein kinase II inhibits binding to DNA.
Phosphorylation can greatly influence the transactivation properties of transcription factors. c-Jun is an example in which transcriptional activity is increased by the phosphorylation of serine residues located within the transactivation domain near the N terminus of the protein. Phosphorylation of a transcriptional activator may stimulate its activity by increasing its binding affinity for a coactivator. Phosphorylation can also inhibit transcriptional activation by reducing transcriptional activation or stimulating active transcriptional repression.
Effects of phosphorylation on nuclear translocation, DNA binding, and transactivation are not mutually exclusive. Moreover, in addition to phosphorylation by protein kinases, dephosphorylation by protein phosphatases may also regulate transcriptional activity.
Site-Specific ProteolysisMany transcription factors undergo proteolytic cleavage at specific amino acid residues, particularly in response to exogenous signals. Site-specific proteolysis often converts an inactive precursor protein into an active transcriptional regulator. One example is NF-κB. Although phosphorylation can regulate NF-κB by controlling its binding to IκB (Fig. 4-13A), proteolysis can also regulate NF-κB (Fig. 4-13B). A 105-kDa precursor of the 50-kDa subunit of NF-κB (p50), which we mentioned earlier, binds to and thereby retains the 65-kDa subunit of NF-κB (p65) in the cytoplasm. Proteolysis of this larger precursor yields the 50-kDa subunit that together with the 65-kDa subunit constitutes the active NF-κB transcription factor.
Another form of site-specific proteolysis, which creates active transcription factors from inactive membrane-tethered precursors, is called regulated intramembranous proteolysis (RIP). The best characterized example is the sterol regulatory element binding protein (SREBP), a membrane protein that normally resides in the endoplasmic reticulum. In response to depletion of cellular cholesterol, SREBP undergoes RIP, which releases an N-terminal fragment containing a bHLH motif. The proteolytic fragment translocates to the nucleus, where it binds to DNA and activates transcription of genes that encode enzymes involved in cholesterol biosynthesis and the LDL receptor (see Chapter 2). (See Note: Examples of RIP)
Other Post-translational Modifications In addition to phosphate groups, a variety of other covalent attachments can affect the activity of transcription factors (Table 4-3). These small molecules—such as acetyl groups, methyl groups, sugars or peptides, hydroxyl groups, or nitro groups—attach to specific amino acid residues in the transcription factor. Post-translational modifications of transcription factors can affect their stability, intracellular localization, dimerization, DNA-binding properties, or interactions with coactivators. For example, acetylation of lysine residues in the p53 transcription factor increases binding to DNA and inhibits degradation. Methylation of an arginine residue in the coactivator CBP inhibits its interaction with the transcription factor CREB. The O-glycosylation, a covalent modification in which sugar groups attach to serine or threonine residues, stimulates NF-κB. Ubiquitin is a small peptide that is covalently attached to lysine groups in proteins. Addition of multiple ubiquitin groups (polyubiquitination) frequently results in degradation of the protein by the proteosome (see Chapter 2). However, addition of a single ubiquitin group (monoubiquitination) may stimulate the activity of a transcription factor, perhaps by increasing its affinity for transcriptional elongation factors. Conversely, sumoylation, covalent modification of lysine residues with small ubiquitin-like modifiers (SUMO), may inhibit activity by altering the localization of a transcription factor within the nucleus. As we will see in the next section, extracellular signals often trigger post-translational modifications to regulate the activity of transcription factors.
Table 4-3 Examples of Transcription Factors That Regulate Gene Expression in Response to Physiological Stimuli
Physiological Stimulus |
Transcription Factor |
Target Genes (Example) |
Hypoxia |
HIF-1α |
Vascular endothelial growth factor, erythropoietin, glycolytic enzymes |
DNA damage |
p53 |
CIP1/WAF1, GADD45, PCNA, MDM2 |
Cholesterol depletion |
SREBP-1 |
HMG-CoA reductase, fatty acid synthase, LDL receptor |
Viruses, oxidants |
NF-κB |
Tumor necrosis factor α, interleukin 1β, interleukin 2, granulocyte colony-stimulating factor, inducible nitric oxide synthase, intercellular cell adhesion molecule |
Heat stress |
HSF1 |
Heat shock proteins, αB-crystallin |
Fatty acids |
PPAR-α |
Lipoprotein lipase, fatty acid transport protein, acyl-CoA synthetase, carnitine palmitoyltransferase I |
Some transcription factors are ubiquitous, either because these transcription factors regulate the transcription of genes that are expressed in many different tissues or because they are required for the transcription of many different genes. Examples of ubiquitous transcription factors are the DNA-binding transcription factors Sp1 and NF-Y, which bind to regulatory elements (i.e., GC boxes and CCAAT boxes, respectively) that are present in many gene promoters. Other transcription factors are present only in certain tissues or cell types; these transcription factors are involved in the regulation of tissue-specific gene expression.
Tissue-specific activators bind to enhancers present in the promoters and regulatory regions of genes that are expressed in a tissue-specific manner. Conversely, tissue-specific repressors bind to silencers that prevent transcription of a gene in nonexpressing tissues. Each tissue-specific transcription factor could regulate the expression of multiple genes. Because the short sequences of enhancers and silencers may occur by chance, the combined effect of multiple transcription factors—each binding to distinct regulatory elements near the gene—prevents illegitimate transcription in nonexpressing tissues. In addition to activation by transcriptional activators, tissue-specific gene expression may also be regulated by transcriptional repression. In this case, transcriptional repressors prevent transcription of a gene in nonexpressing tissues. Tissue-specific expression probably also involves permanent silencing of nonexpressed genes through epigenetic modifications, such as DNA methylation, that we discuss later.
Pit-1 is an HTH-type tissue-specific transcription factor that regulates the pituitary-specific expression of genes encoding growth hormone, thyroid-stimulating hormone, and prolactin. MyoD and myogenin are bHLH-type transcription factors that bind to the E box sequence CANNTG of promoters and enhancers of many genes expressed in skeletal muscle, such as myosin heavy chain and muscle creatine kinase. EKLF as well as GATA-1 and NF-E2 mediate the erythroid-specific expression of β-globin genes. The combined effects of HNF-1, HNF-3, HNF-4, C/EBP, and other transcription factors—each of which may individually be present in several tissues—mediate the liver-specific expression of genes such as albumin and α1-antitrypsin. Many tissue-specific transcription factors play important roles in embryonic development. For example, myogenin is required for skeletal muscle differentiation, and GATA-1 is required for the development of erythroid cells.
What is responsible for the tissue-specific expression of the transcription factor itself? Although the answer is not known, many tissue-specific transcription factors are themselves under the control of other tissue-specific factors. Thus, a transcriptional cascade involving multiple tissue-specific proteins may regulate tissue-specific gene expression. Ultimately, however, tissue specificity is likely to arise from external signals that direct gene expression down a particular pathway.
How do cells activate previously quiescent genes in response to environmental cues? How are such external signals transduced to the cell nucleus to stimulate the transcription of specific genes? Transcription factors may be thought of as effector molecules in signal transduction pathways (see Chapter 3) that modulate gene expression. Several such signaling pathways have been defined. Lipid-soluble steroid and thyroid hormones can enter the cell and interact with specific receptors that are themselves transcription factors. However, most cytokines, hormones, and mitogens cannot diffuse into the cell interior and instead bind to specific receptors that are located on the cell surface. First, we consider three pathways for transducing signals from cell surface receptors into the nucleus: a cAMP-dependent pathway, a Ras-dependent pathway, and the JAK-STAT pathway. Next, we examine the mechanisms by which steroid or thyroid hormones act through nuclear receptors. Finally, we discuss how transcription factors coordinate gene expression in response to physiological stimuli.
cAMP is an important second messenger in the response to agonists binding to specific cell surface receptors. Increases in [cAMP]i stimulate the transcription of certain genes, including those that encode a variety of hormones, such as somatostatin (see Chapter 48), the enkephalins (see Chapter 13), glucagon (see Chapter 51), and vasoactive intestinal polypeptide (see Chapter 41). Many genes that are activated in response to cAMP contain within their regulatory regions a common DNA element called CRE (cAMP response element) that has the consensus sequence 5′-TGACGTCA-3′. Several different transcription factors bind to CRE, among them CREB, a 43-kDa member of the bZIP family. As shown in Figure 4-14, increases in [cAMP]i stimulate protein kinase A (PKA) by causing dissociation of the PKA regulatory subunit. The catalytic subunit of PKA then translocates into the nucleus, where it phosphorylates CREB and other proteins. Activation of CREB is rapid (30 minutes) and declines gradually during a 24-hour period. This phosphorylation greatly increases the affinity of CREB for the coactivator CBP. CBP is a 245-kDa protein that contains two domains, one that binds to phosphorylated CREB and another that activates components of the basal transcriptional machinery. Thus, CBP serves as a “bridge” protein that communicates the transcriptional activation signal from CREB to the basal transcriptional machinery. In addition, because CBP has intrinsic HAT activity, its recruitment by CREB also results in chromatin remodeling that facilitates gene transcription. The result of phosphorylating CREB is a 10-to 20-fold stimulation of CREB’s ability to induce the transcription of genes containing a CRE.
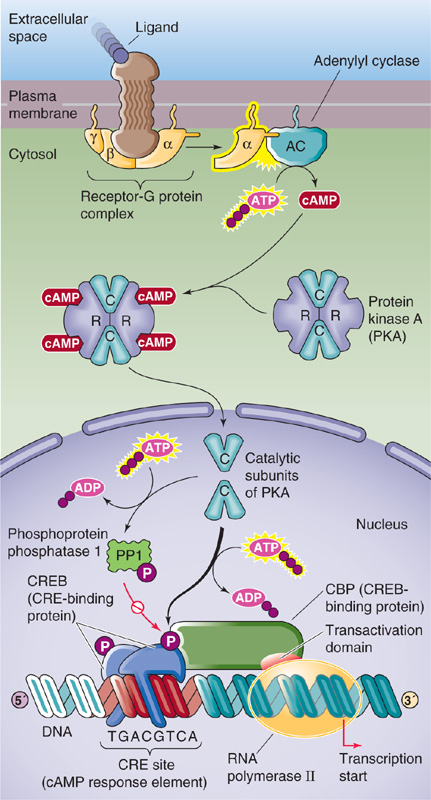
Figure 4-14 Regulation of gene transcription by cAMP. Phosphorylated CREB binds CBP, which has a transactivation domain that stimulates the basal transcriptional machinery. In parallel, phosphorylation activates PP1, which dephosphorylates CREB, terminating the activation of transcription.
How is the transcriptional signal terminated? When [cAMP]i is high, PKA phosphorylates and activates phosphoprotein phosphatase 1 in the nucleus. When cAMP levels fall, the still-active phosphatase dephosphorylates CREB.
As discussed in Chapter 3, many growth factors bind to cell surface receptors that when activated by the ligand have tyrosine kinase activity. Examples of growth factors that act through such receptor tyrosine kinases (RTKs) are epidermal growth factor (EGF), platelet-derived growth factor (PDGF), insulin, insulin-like growth factor type 1 (IGF-1), fibroblast growth factor (FGF), and nerve growth factor (NGF). The common pathway by which activation of RTKs is transduced into the nucleus is a cascade of events that increase the activity of the small guanosine triphosphate (GTP)–binding protein Ras. This Ras-dependent signaling pathway culminates in the activation of MAP kinase (MAPK), which translocates to the nucleus, where it phosphorylates a number of nuclear proteins that are transcription factors. Phosphorylation of a transcription factor by MAPK can enhance or inhibit binding to DNA and can stimulate either transactivation or transrepression. Transcription factors that are regulated by the Ras-dependent pathway include c-Myc, c-Jun, c-Fos, and Elk-1. Many of these transcription factors regulate the expression of genes that promote cell proliferation. (See Note: Transcription Factors Phosphorylated by MAP Kinase)
A group of cell surface receptors termed tyrosine kinase–associated receptors lack intrinsic tyrosine kinase activity. The ligands that bind to these receptors include several cytokines, growth hormone, prolactin, and interferons (IFN-α, IFN-β, and IFN-γ). Although the receptors themselves lack catalytic activity, their cytoplasmic domains are associated with the JAK family of protein tyrosine kinases.
Binding of ligand to certain tyrosine kinase–associated receptors activates a member of the JAK family, which results in phosphorylation of cytoplasmic proteins, among which are believed to be latent cytoplasmic transcription factors called STATs (signal transducers and activators of transcription). When phosphorylated on tyrosine residues, the STAT proteins dimerize and thereby become competent to enter the nucleus and induce transcription.
A well-characterized example of the JAK-STAT pathway is the activation of interferon-responsive genes by IFN-α and IFN-γ. IFN-α activates the JAK1 and Tyk2 kinases that are associated with its receptor (Fig. 4-15A). Subsequent phosphorylation of two different STAT monomers causes the monomers to dimerize. This STAT heterodimer enters the nucleus, where it combines with a third 48-kDa protein to form a transcription factor that binds to a DNA sequence called the IFN-α–stimulated response element (ISRE). In the case of IFN-γ (Fig. 4-15B), the receptor associates with the JAK1 and JAK2 (rather than Tyk2) kinases, and subsequent phosphorylation of a single kind of STAT monomer causes these monomers to dimerize. These STAT homodimers also enter the nucleus, where they bind to the DNA at IFN-γ response elements called γ activation sites (GAS), without requiring the 48-kDa protein.
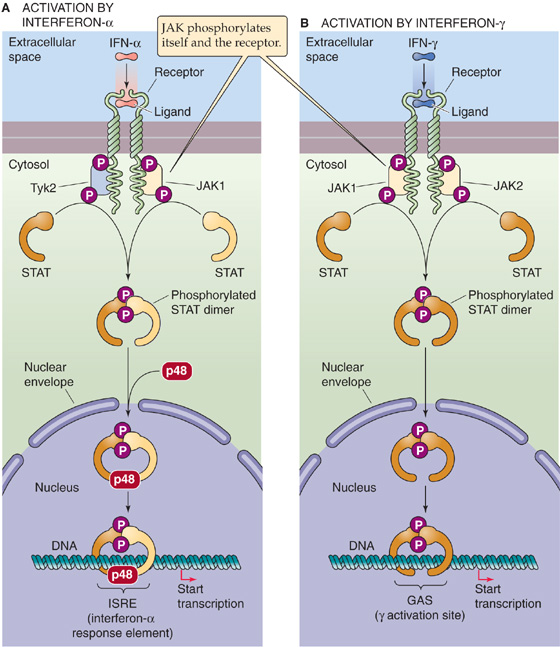
Figure 4-15 The JAK-STAT pathway. A, Binding of a ligand such as IFN-α to a tyrosine kinase–associated receptor causes JAK1 and Tyk2 to phosphorylate themselves, the receptor, and two different STAT monomers. The phosphorylation of the STAT monomers leads to the formation of a heterodimer, which translocates to the nucleus and combines with a third protein (p48). The complex binds to the ISRE and activates gene transcription. B, Binding of a ligand such as IFN-γ to a tyrosine kinase–associated receptor causes JAK1 and JAK2 to phosphorylate themselves, the receptor, and two identical STAT monomers. The phosphorylation of the STAT monomers leads to the formation of a homodimer, which translocates to the nucleus. The complex binds to the GAS response element and activates gene transcription.
Steroid and thyroid hormones are examples of ligands that activate gene expression by binding to cellular receptors that are themselves transcription factors. Members of the steroid and thyroid hormone receptor superfamily, also called the nuclear receptor superfamily, are grouped together because they are structurally similar and have similar mechanisms of action. After these hormones enter the cell, they bind to receptors in the cytoplasm or nucleus. Ligand binding converts the receptors into active transcription factors. The transcription factors bind to specific regulatory elements on the DNA, called hormone response elements, and activate the transcription of cis-linked genes. The family of nuclear receptors includes receptors that bind glucocorticoids (GR), mineralocorticoids (MR), estrogens (ER), progesterone (PR), androgens (AR), thyroid hormone (TR), vitamin D (VDR), retinoic acid (RAR), lipids (peroxisome proliferator–activated receptor, PPAR), and 9-cis-retinoic acid (retinoid X receptor, RXR) as well as bile acids (bile acid receptor, FXR) and xenobiotics (steroid and xenobiotic receptor, SXR; constitutive androstane receptor, CAR) (see Chapter 46).
With the exception of the thyroid hormones, the hormones that bind to these receptors are lipophilic molecules that enter cells by diffusion and do not require interaction with cell surface receptors. The thyroid hormones differ in that they are electrically charged and may cross the cell membrane through transporters (see Chapter 49).
Modular Construction The nuclear receptors have a modular construction consisting of an N-terminal transactivation domain, a DNA-binding domain, and a C-terminal ligand-binding domain. These receptors bind to specific DNA sequences by two zinc fingers, each of which contains four cysteine residues rather than the two histidines and two cysteines that are typical of many other zinc finger proteins (Fig. 4-10A). Particularly important for DNA recognition is the P box motif in the hormone receptor, a sequence of six amino acids at the C-terminal end of each finger. These P boxes make base pair contacts in the major groove of DNA and determine the DNA-binding specificities of the zinc finger.
Dimerization GR, MR, PR, ER, and AR bind to DNA as homodimers (Table 4-4). The recognition sites for these receptors (except for ER) consist of two 6-bp DNA sequences that are separated by three other base pairs. The 6-bp DNA sequences, commonly called half-sites, represent binding sites for each zinc finger monomer.
Table 4-4 Nuclear Receptors
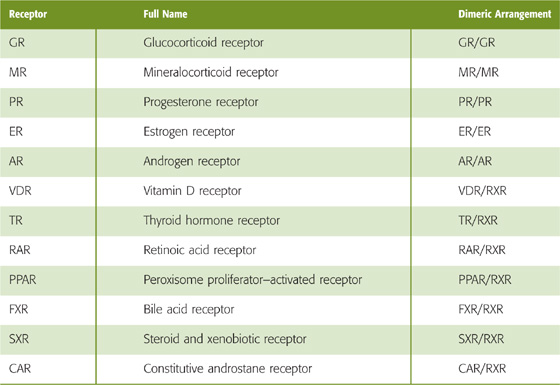
In contrast, VDR, TR, RAR, and PPAR preferentially bind to DNA as heterodimers formed with RXR, the receptor for 9-cis-retinoic acid. Thus, the dimers are VDR/RXR, TR/RXR, RAR/RXR, and PPAR/RXR. Interestingly, these heterodimers work even in the absence of the ligand of RXR (i.e., 9-cis-retinoic acid). Only the VDR, TR, RAR, or PPAR part of the dimer needs to be occupied by its hormone ligand. These heterodimers recognize a family of DNA sites containing a DNA sequence such as 5′-AGGTCA-3′, followed by a DNA spacer and then by a direct repeat of the previous 6-bp DNA sequence. Moreover, because VDR/RXR, TR/RXR, and RAR/RXR may each recognize the same 6-bp sequences, binding specificity also depends on the length of the spacer between the direct repeats. The VDR/RXR, TR/RXR, and RAR/RXR heterodimers preferentially recognize separations of 3 bp, 4 bp, and 5 bp, respectively, between the repeats of 5′-AGGTCA-3′. This relationship forms the basis for the so-called 3-4-5 rule.
Activation of Transcription Ligand binding activates nuclear receptors through two main mechanisms: regulation of subcellular localization and interactions with coactivators. Some nuclear receptors, such as GR, are normally located in the cytoplasm and are maintained in an inactive state by association with a cytoplasmic anchoring protein (Fig. 4-16A). The protein that retains GR in the cytoplasm is a molecular chaperone, the 90-kDa heat shock protein hsp90. GR must bind to hsp90 to have a high affinity for a glucocorticoid hormone. When glucocorticoids bind to the GR, hsp90 dissociates from the GR and exposes a nuclear localization signal that permits the transport of GR into the nucleus. The receptor must remain hormone bound for receptor dimerization, which is a prerequisite for binding to the GRE on the DNA. Other receptors, such as TR, are normally already present in the nucleus before binding the hormone (Fig. 4-16B). For these receptors, binding of hormone is evidently not essential for dimerization or binding to DNA. However, ligand binding is necessary at a subsequent step for transactivation.
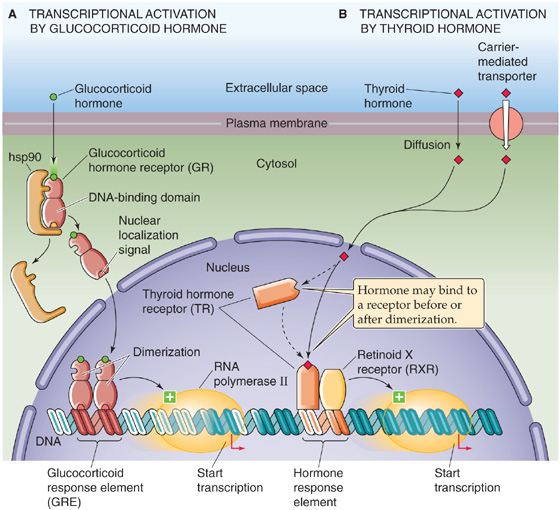
Figure 4-16 Transcriptional activation by glucocorticoid and thyroid hormones. A, The binding of a glucocorticoid hormone to a cytoplasmic receptor causes the receptor to dissociate from the chaperone hsp90 (90-kDa heat shock protein). The free hormone-receptor complex can then translocate to the nucleus, where dimerization leads to transactivation. B, The binding of thyroid hormone to a receptor in the nucleus leads to transactivation. The active transcription factor is a heterodimer of the thyroid hormone receptor and the retinoid X receptor.
Although nuclear receptors may stimulate gene expression by interacting directly with components of the basal transcriptional machinery, full transcriptional activation requires coactivators that interact with the receptor in a ligand-dependent manner. More than 200 coactivators may interact directly or indirectly with nuclear receptors through mechanisms that include the following:
1. Recruitment of basal transcriptional machinery. Coactivators that belong to the SRC (steroid receptor coactivator)/p160 family bind only to the ligand-bound form of the receptor. On binding to the nuclear receptor, SRC/p160 coactivators recruit a second coactivator, CBP, which then promotes recruitment of the basal transcriptional machinery. Nuclear receptors also bind in a ligand-dependent manner to the coactivator TRAP220, a component of Mediator, which is part of the basal transcriptional machinery.
2. Binding to a chromatin remodeling complex. Nuclear receptors also interact with Brg1 (Brahma-related gene 1), the central motor component of the chromatin remodeling complex SWI/SNF.
3. Histone acetylation. Several coactivators have enzymatic activities that mediate chromatin remodeling. Both SRC-1 and CBP have intrinsic HAT activity.
4. Histone methylation. The coactivator CARM1 is a methyltransferase that methylates specific arginine residues in histones, thereby enhancing transcriptional activation.
5. Ubiquitination. Nuclear receptors recruit components of the ubiquitin-proteosome pathway (see Chapter 2) to the promoter region of nuclear receptor target genes. Ubiquitination appears to promote transcript elongation.
Repression of Transcription Nuclear receptors sometimes function as active repressors, perhaps acting by several alternative mechanisms. First, a receptor may form inactive heterodimers with other members of the nuclear receptor family. Second, a receptor may compete with other transcription factors for DNA-binding sites. For example, when the TR—without bound thyroid hormone—interacts with its own DNA response element, the TR acts as a repressor. In addition, the receptor TRa can dimerize with one of the retinoic acid receptors (RXRb) to interfere with binding of ER to its response element. This competition may be one of the mechanisms that retinoids use to inhibit estrogen-induced alterations in gene expression and growth in mammary tissue. Finally, nuclear receptors can also inhibit gene transcription by interacting with corepressors, such as N-CoR, Sin3A, and Sin3B. These corepressors can recruit HDACs that enhance nucleosome assembly, resulting in transcriptional repression.
In response to physiological stimuli, some transcription factors regulate the expression of several genes (Table 4-3). As an example, we discuss how oxygen concentration ([O2]) controls gene expression.
When chronically exposed to low [O2] (hypoxia), many cells undergo dramatic changes in gene expression. For example, cells switch from oxidative metabolism to glycolysis, which requires the induction of genes encoding glycolytic enzymes. Many tissues activate the gene encoding the vascular endothelial growth factor (VEGF), which stimulates angiogenesis and improves the blood supply to chronically hypoxic tissues. The kidney activates the gene encoding erythropoietin, a hormone that stimulates red cell production in the bone marrow. These changes in gene expression promote survival of the cell or organism in a hypoxic environment. A key mediator in the response to hypoxia is a transcription factor called hypoxia-inducible factor 1α (HIF-1α).
Role of a Chimeric Transcription Factor in Acute Promyelocytic Leukemia
Correct regulation of gene expression involves both transcription factors and the DNA regulatory elements to which they bind. Abnormalities of either could and do result in abnormal regulation of gene expression, which is often manifested as disease. An example of a transcription factor abnormality is acute promyelocytic leukemia (APL), a hematologic malignant disease in which cells of the granulocyte lineage (promyelocytes) fail to differentiate. Normally, retinoic acid (RA) binds to retinoic acid receptor α (RARα), a member of the steroid-thyroid hormone receptor superfamily. RARα forms heterodimers with retinoid X receptor (RXR) and binds to retinoic acid response elements (RAREs) that are present in genes involved in cell differentiation. In the absence of RA, RARα/RXR heterodimers bind to RAREs and recruit the corepressor N-CoR, which in turn recruits HDACs that inhibit gene transcription. Binding of RA to RARα leads to dissociation of N-CoR, which permits binding of the CBP coactivator and activation of RARα-responsive genes that promote cell differentiation. Ninety percent of patients with APL have a translocation affecting chromosomes 15 and 17, t(15;17), that produces a chimeric transcription factor containing the DNA-and hormone-binding domains of RARα, fused to the nuclear protein PML. The PML/RARα chimeric protein also binds to RA and forms heterodimers with RXR but has an abnormally high affinity for N-CoR. At physiological levels of RA, N-CoR remains bound to PML/RARα, blocking promyelocytic differentiation. However, high concentrations of RA induce dissociation of N-CoR and permit differentiation. This mechanism explains why high concentrations of exogenous RA can be used to induce clinical remissions in patients with APL.
HIF-1α (Fig. 4-17A) belongs to the bHLH family of transcription factors. In addition, it contains a PAS domain that mediates dimerization. HIF-1α binds to DNA as a heterodimer with HIF-1β. HIF-1β is expressed at constant levels in cells, but the abundance of HIF-1α changes markedly in response to changes in [O2]. At a normal [O2] (normoxia), HIF-1α levels are low. Under hypoxic conditions, the abundance of HIF-1α increases. HIF-1α together with HIF-1β binds to an enhancer, called a hypoxia response element, that is present in many genes activated during hypoxia, including genes encoding glycolytic enzymes, VEGF, and erythropoietin. (See Note: Dimerization of Basic Helix-Loop-Helix Transcription Factors)
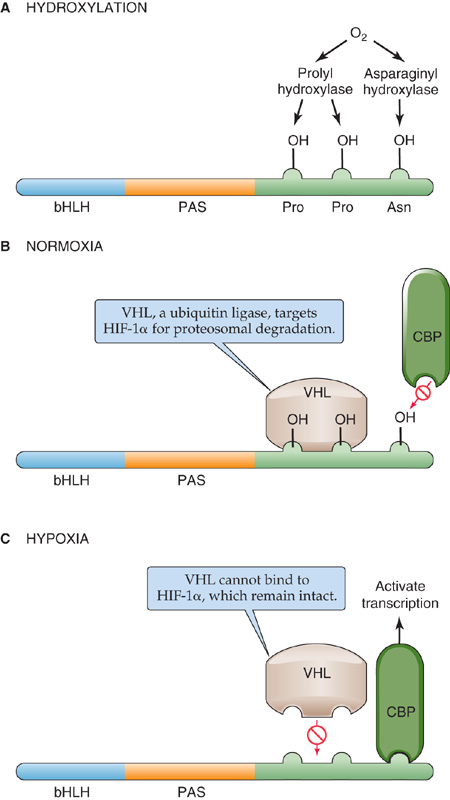
Figure 4-17 Regulation of HIF-1α by oxygen. A, In the presence of oxygen, HIF-1α is hydroxylated on proline and asparagine by hydroxylases. B, Hydroxylation of HIF-1α promotes its degradation and inhibits its interaction with coactivators. C, In hypoxic conditions, dehydroxylation of HIF-1α promotes its stabilization and transcriptional activity.
The cell regulates the abundance of HIF-1α by hydroxylation—a post-translational modification—at specific proline and asparagine residues. Oxygen activates the prolyl and asparaginyl hydroxylases (Fig. 4-17A). Proline hydroxylation stimulates the interaction of HIF-1α with VHL, a protein that targets HIF-1α for proteosomal degradation (Fig. 4-17B). Asparagine hydroxylation inhibits the interaction of HIF-1α with the transcriptional coactivator CBP. Because both of these hydroxylations reduce transcriptional activity, normoxic conditions lower the expression of HIF-1α target genes.
In contrast, under hypoxic conditions, the hydroxylases are inactive, and HIF-1α is not hydroxylated on proline and asparagine residues. HIF-1α accumulates in the nucleus and interacts with CBP, which activates the transcription of downstream target genes, including VEGF and erythropoietin (Fig. 4-17C). The net result is a system in which the expression of multiple hypoxia-inducible genes is coordinately and tightly regulated through post-translational modification of a common transcriptional activator.
You might recall that at the outset of this chapter we noted that gene expression entails several steps, beginning with alteration of chromatin structure (Fig. 4-1, step 1). Having already discussed the control of transcription (Fig. 4-1, step 2) in the previous three sections, we return in this penultimate section to the first step and consider how changes in chromatin structure and the methylation of both histones and DNA have long-term effects on gene expression. In the last section, we examine several of the later steps in gene expression.
As discussed earlier, DNA in the nucleus is organized into a higher order structure called chromatin, which consists of DNA and associated histones. Chromatin exists in two general forms that can be distinguished cytologically by their different degrees of condensation. Heterochromatin is a highly condensed form of chromatin that is transcriptionally inactive. In general, highly organized chromatin structure is associated with repression of gene transcription. Heterochromatin contains mostly repetitive DNA sequences and relatively few genes. Euchromatin has a more open structure and contains genes that are actively transcribed. Even in the transcriptionally active “open” euchromatin, local chromatin structure may influence the activity of individual genes. We have already seen that transcriptional activators recruit HATs that remodel chromatin and promote binding of additional transcription factors and the basal transcriptional machinery. Conversely, transcriptional repressors recruit HDACs that promote nucleosome assembly and inhibit gene transcription.
Chemical modification of the histones regulates the establishment and maintenance of euchromatin and heterochromatin. Especially important is methylation, in which a methyltransferase covalently attaches methyl groups to arginine residues and, most significantly, to specific lysine residues in the core histones. In euchromatin, methylation of histone H3 at Lys-4, Lys-36, and Lys-79 correlates with transcriptional activation. In heterochromatin, demethylation of these residues but methylation of H3 at Lys-9 and Lys-27 and of H4 at Lys-20 correlates with transcriptional repression. This pattern of differential methylation in transcriptionally active and inactive chromatin is referred to as a histone code.
Methylation of H3 at Lys-9 recruits heterochromatin protein 1 (HP1), which then self-dimerizes to produce higher order structures (Fig. 4-18A). In addition, HP1 recruits HDAC, which promotes nucleosome assembly. Together, these modifications produce a closed chromatin conformation.
Figure 4-18 Gene silencing by chromatin modification. A, HP1 binds to methylated Lys-9 in histone H3. Because the HP1 self-dimerizes, the result is chromatin condensation. B, HP1 recruits a histone methyltransferase (HMT) that promotes further lysine methylation, leading to recruitment of additional HP1, propagating histone methylation. C, MBD1 binds to methylated cytosine groups in DNA and can also recruit HMT.
The structure of chromatin can have a long-term influence on gene expression. The following are three examples of long-term regulation of gene expression.
X-inactivation. Females carry two X chromosomes (see Chapter 53), whereas males carry only one copy. To express X-linked genes at the same levels as males, females during development permanently inactivate one of the X chromosomes by globally converting one X chromosome from euchromatin to heterochromatin.
Imprinting. Cells contain two copies of every gene, one inherited from each parent, and usually express each copy identically. In a few cases, however, genes are differentially expressed, depending on whether they are inherited from the mother or the father. This phenomenon is called genomic imprinting. For example, the insulin-like growth factor 2 gene (IGF2; see Chapter 48) is paternally imprinted—only the copy inherited from the father is expressed; the maternal copy is not expressed.
Tissue-specific gene silencing. Many tissue-specific genes are globally inactivated during embryonic development, later to be reactivated only in particular tissues. For example, globin genes are silenced except in erythroid cells. The silencing of genes in nonexpressing tissues is associated with chromatin modifications that are similar to those found in heterochromatin.
X-inactivation, imprinting, and tissue-specific silencing require long-term inactivation of gene expression and the maintenance of this inactivation during DNA replication and cell division. For example, the inactivated X chromosome remains inactivated in the two progeny cells after mitosis. Similarly, genes silenced by imprinting or by tissue-specific silencing remain inactive in progeny cells. The maintenance of gene silencing is an example of epigenetic regulation of gene expression— “epigenetic” because the heritable changes do not depend on DNA sequences.
Chromatin modification mediates the epigenetic regulation of gene expression. As is the case for transcriptional repression in heterochromatin, the methylation of histone H3 at Lys-9 (H3-K9) is characteristic of silencing by X-inactivation, imprinting, and tissue-specific gene silencing. Cells maintain this H3-K9 methylation during division, possibly by using the HP1 discussed earlier (Fig. 4-18A). After it binds to methylated histones, HP1 recruits a histone methyltransferase (HMT) that methylates other H3-K9 residues (Fig. 4-18B), providing a mechanism for propagating histone methylation. During DNA replication, the HMT recruited to a silenced gene on a parental strand of chromatin adds methyl groups to histones on the daughter strands, which maintains gene silencing in the progeny.
Methylation of cytosine residues at the N5 position is the only well-documented postsynthetic modification of DNA in higher eukaryotes. Approximately 5% of cytosine residues are methylated in mammalian DNA. Methylation usually occurs on cytosine residues that are immediately upstream from guanosines (i.e., CpG dinucleotides).
Several lines of evidence implicate DNA methylation in the control of gene expression.
1. Although CpG dinucleotides are relatively underrepresented in mammalian genomes, they are frequently clustered near the 5′ ends of genes (forming so-called CpG islands). Moreover, methylation of cytosines in these locations is associated with inhibition of gene expression. For example, the inactivated X chromosome in females contains heavily methylated genes.
2. Methylation/demethylation may explain tissue-specific and stage-dependent gene expression. For example, globin genes are methylated in nonexpressing tissues but hypomethylated in erythroid cells. During fetal development, fetal globin genes are demethylated and then become methylated in the adult.
3. Foreign genes that are introduced into cells are transcriptionally inactive if they are methylated but active if demethylated at the 5′ end.
4. Chemical demethylating agents, such as 5-azacytidine, can activate previously inactive genes.
How does DNA methylation cause gene inactivation? One simple mechanism is that methylation inhibits the binding of an essential transcriptional activator. For example, methylation of CpG dinucleotides within the GFAP (glial fibrillary acidic protein) promoter prevents STAT3 binding. A more common mechanism is that methylation produces binding sites for proteins that promote gene inactivation. Cells contain a protein called MeCP2 that binds specifically to methylated CpG dinucleotides as well as to the HDAC. Thus, DNA methylation may silence genes by promoting histone deacetylation. In addition, methylated DNA binds to methyl-CpG binding protein 1 (MBD1), a protein that complexes with HMT (Fig. 4-18C). These last two interactions provide mechanisms coupling DNA methylation to histone modifications that promote heterochromatin formation and gene silencing.
Although initiation of transcription (Fig. 4-5, step 2) is the most frequently regulated step in gene expression, for certain genes subsequent steps are more important for determining the overall level of expression. These processes are generally classified as post-transcriptional regulation. The mechanisms for regulating these steps are less well understood than are those for regulating transcription initiation, but some information comes from the study of model genes. Post-transcriptional processes that we review here are pre-mRNA splicing (step 5) and transcript degradation (step 8).
Eukaryotic genes contain introns that must be removed from the primary transcript to create mature mRNA; this process is called pre-mRNA splicing. Splicing involves the joining of two sites on the RNA transcript, the 5′ splice-donor site and the 3′ splice-acceptor site, and removal of the intervening intron (Fig. 4-19). The first step involves cleavage of the pre-mRNA at the 5′ splice-donor site. Second, joining of the 5′ end of the intron to an adenosine residue located within the intron forms a “lariat” structure. Third, ligation of the 5′ and 3′ splice sites releases the lariat intron. The splicing reaction occurs in the nucleus, mediated by ribonucleoprotein particles (snRNPs) that are composed of proteins and small nuclear RNA (snRNA). Together, the assembly of pre-mRNA and snRNPs forms a large complex called the spliceosome.

Figure 4-19 Mechanism of pre-mRNA splicing. This example illustrates how a 5′ splice-donor site at one end of exon 1 can link to the 3′ splice-acceptor site at the end of exon 2, thereby splicing out the intervening intron. The process can be divided into three steps: (1) cleavage of the pre-mRNA at the 5′ splice-donor site; (2) joining of the 5′ end of the intron to an adenosine residue that is located within the intron, forming a lariat structure; and (3) ligation of the 5′ and 3′ splice sites and release of the lariat intron.
The location of the 5′ and 3′ splice sites is based, at least in part, on the sequences at the ends of the introns. The 5′ splice-donor site has the consensus sequence 5′-(C/A)AG ↓ GU(G/A)AGU-3′; the vertical arrow represents the boundary between the exon and the intron. The 3′ splice-acceptor site has the consensus sequence 5′-YnNCAG ↓ G-3′; Yn represents a polypyrimidine tract (i.e., a long sequence of only C and U), and N represents any nucleotide. An intronic site located more than 17 nucleotides upstream from the 3′ acceptor site (5′-YNCUG AC-3′), called the branch point, is also present and contains the adenosine (red background in Fig. 4-19) that contributes to formation of the lariat structure.
Many genes undergo alternative splicing, which refers to differential splicing of the same primary transcript to produce mature transcripts that contain different combinations of exons. If the coding region is affected, the resulting splicing variants will encode proteins with distinct primary structures that may have different physiological functions. Thus, alternative splicing is a mechanism for increasing the diversity of proteins that a single gene can produce. Figure 4-20 summarizes seven patterns of alternative splicing.
Figure 4-20 Types of alternative splicing. CGRP, calcitonin gene–related peptide; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; Poly-A, polyadenylic acid.
Retained Intron In some cases, the cell may choose whether to splice out a segment of RNA. For example, the γA isoform of rat γ-fibrinogen lacks the seventh intron, whereas the γB isoform retains the intron (Fig. 4-20A). The retained intron encodes a unique 12–amino acid C terminus in γB-fibrinogen.
Alternative 3′ Splice Sites In this case, the length of an intron is variable because the downstream boundary of the intron can be at either of two or more different 3′ splice-acceptor sites (Fig. 4-20B). For example, in rat fibronectin, a single donor site may be spliced to any of three acceptor sites. The presence or absence of the amino acids encoded by the sequence between the different splice-acceptor sites results in fibronectin isoforms with different cell adhesion properties.
Alternative 5′ Splice Sites Here also, the length of the intron is variable. However, in this case, it is the upstream boundary of the intron that can be at either of two or more different 5′ splice-donor sites (Fig. 4-20C). For example, cells can generate mRNA encoding 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase (see Chapter 46) with different 5′ untranslated regions by splicing from multiple donor sites for the first intron to a single acceptor site.
Cassette Exons In some cases, the cell may choose either to splice in an exon or group of exons (cassette exons) or to not splice them in (Fig. 4-20D). An example is the α-tropomyosin gene, which contains 12 exons. All α-tropomyosin transcripts contain the invariant exons 1, 4 to 6, 8, and 9. All muscle-like cells splice in exon 7, but hepatoma (i.e., liver tumor) cells do not splice in exon 7; they directly link exon 6 to exon 8.
Mutually Exclusive Exons In yet other cases, the cell may splice in mutually exclusive exons (Fig. 4-20E). One of the Na/K/Cl cotransporter genes (NKCC2) is an example. Isoforms containing distinct 96-bp exons are differentially expressed in the kidney cortex and medulla. Because the encoded amino acid sequence is predicted to reside in the membrane, the isoforms may have different kinetic properties. The α-tropomyosin gene again is another example. Smooth muscle cells splice in exon 2 but not exon 3. Striated muscle cells and myoblasts splice in exon 3 but not exon 2. Fibroblasts and hepatoma cells do not splice in either of these two exons.
Alternative 5′ Ends Cells may differentially splice the 5′ end of the gene (Fig. 4-20F) and thereby select different promoters. In the case of the myosin light chain gene (see Chapter 9), which consists of nine exons, one transcript is initiated from a promoter that is located upstream from exon 1, skips exons 2 and 3, and includes exons 4 to 9. The other transcript is initiated instead at a promoter located in the first intron and consists of exons 2, 3, and 5 to 9. Because the coding region is affected, the two transcripts encode proteins that differ at their N-terminal ends. These splice variants are found in different cells or different developmental stages. α-Amylase (see Chapter 45) is another example. Transcription can begin from two different sites and produce mRNA that contains different first exons. Because the two mRNAs have different promoters, this alternative splicing permits differential regulation of gene expression in liver and salivary glands.
Alternative 3′ Ends Finally, cells may differentially splice the transcript near the 3′ end of the gene (Fig. 4-20G) and thereby alter the site of cleavage and polyadenylation. Such splicing may also affect the coding region. Again, α-tropomyosin is an example. Striated muscle cells splice in exon 11, which contains one alternative 3′ untranslated region. Smooth muscle cells splice in exon 12 instead of exon 11. Another example is the calcitonin gene, which encodes both the hormone calcitonin (see Chapter 52) and calcitonin gene–related peptide α (CGRPα). Thyroid C cells produce one splice variant that includes exons 1 to 4 and encodes calcitonin. Sensory neurons, on the other hand, produce another splice variant that excludes exon 4 but includes exons 5 and 6. It encodes a different protein, CGRPα.
These examples illustrate that some splicing variants are expressed only in certain cell types but not in others. Clearly, control of alternative splicing must involve steps other than initiation of transcription because many splice variants have identical 5′ ends. In some genes, the control elements that are required for alternative splicing have been identified, largely on the basis of deletion mutations that result in aberrant splicing. These control elements can reside in either introns or exons and are located within or near the splice sites. The proteins that interact with such elements remain largely unknown, although some RNA-binding proteins that may be involved in regulation of splicing have been identified.
The stability of mRNA in cytoplasm varies widely for different transcripts. Transcripts that encode cytokines and immediate-early genes are frequently short-lived, with half-lives measured in minutes. Other transcripts are much more stable, with half-lives that exceed 24 hours. Moreover, cells can modulate the stability of individual transcripts and thus use this mechanism to affect the overall level of expression of the gene.
Degradation of mRNA is mediated by enzymes called ribonucleases. These enzymes include 3′-5′ exonucleases, which digest RNA from the 3′ end; 5′-3′ exonucleases, which digest from the 5′ end; and endonucleases, which digest at internal sites. A structural feature of typical mRNA that contributes to its stability in cytoplasm is the 5′ methyl cap, in which the presence of the 5′-5′ phosphodiester bond makes it resistant to digestion by 5′-3′ exonucleases. Similarly, the poly(A) tail at the 3′ end of the transcript often protects messages from degradation. Deadenylation (i.e., removal of the tail) is often a prerequisite for mRNA degradation. Accordingly, transcripts with long poly(A) tails may be more stable in cytoplasm than are transcripts with short poly(A) tails.
Regulatory elements that stabilize mRNA, as well as elements that accelerate its degradation, are frequently located in the 3′ untranslated region of the transcripts. A well-characterized example of a gene that is primarily regulated by transcript stability is the transferrin receptor (Fig. 4-21). The transferrin receptor is required for uptake of iron into most of the cells of the body (see Chapter 2). During states of iron deprivation, transferrin receptor mRNA levels increase, whereas transcript levels decrease when iron is plentiful. Regulation of transferrin receptor gene expression is primarily post-transcriptional; these alterations in the level of transferrin receptor mRNA are achieved through changes in the half-life of the message.
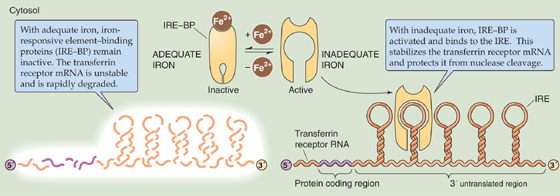
Figure 4-21 The role of iron in regulating the stability of the mRNA for the transferrin receptor. The mRNA that encodes the transferrin receptor has a series of IREs in its 3′ untranslated region.
Regulation of transferrin receptor mRNA stability depends on elements that are located in the 3′ untranslated region called iron response elements (IREs). An IRE is a stem-loop structure that is created by intramolecular hydrogen bond formation. The human transferrin receptor transcript contains five IREs in the 3′ untranslated region. The IRE binds a cellular protein called IRE-binding protein (IRE-BP), which stabilizes transferrin receptor mRNA in the cytoplasm. When IRE-BP dissociates, the transcript is rapidly degraded. IRE-BP can also bind to iron, and the presence of iron decreases its affinity for the IRE. During states of iron deficiency, less iron binds to IRE-BP, and thus more IRE-BP binds to the IRE on the mRNA. The increased stability of the transcript allows the cell to produce more transferrin receptors. Conversely, when iron is plentiful and binds to IRE-BP, IRE-BP dissociates from the IRE, and the transferrin receptor transcript is rapidly degraded. This design prevents cellular iron overload.
Small regulatory RNAs, called small interfering RNAs (siRNA), may modulate gene expression both at the post-transcriptional level and at the level of chromatin structure. These siRNAs are short (~22 bp) double-stranded RNA molecules, one strand of which is complementary in sequence to a target mRNA. The process in which siRNAs silence the expression of specific genes is called RNA interference (RNAi).
Recall that RNA is usually single stranded. However, certain non–protein-coding sequences in the genome may yield RNA transcripts that contain inverted repeats, allowing double-stranded hairpins to form through intramolecular hydrogen bonds (Fig. 4-22). Cleavage of the hairpin structure by an endonuclease called Dicer produces the mature siRNA.
Figure 4-22 Regulation of gene expression by RNA interference. The siRNA is produced from hairpin RNA by Dicer. 1, Assembly of siRNA in the RISC complex results in cleavage of the target mRNA. 2, The siRNA can also inhibit mRNA translation. 3, Assembly of siRNA in the RITS complex promotes DNA methylation and gene silencing.
Mature siRNA can assemble into a ribonucleoprotein complex called RNA-induced silencing complex (RISC), which specifically cleaves a target mRNA that is complementary in sequence to one of the strands of the siRNA (Fig. 4-22A). In addition, the binding of an siRNA to a complementary mRNA can inhibit translation of the mRNA into protein (Fig. 4-22B). Finally, siRNAs can assemble into another ribonucleoprotein complex called RNA-induced transcriptional silencing (RITS), which promotes DNA and histone methylation and thus the formation of heterochromatin (Fig. 4-22C).
Hundreds of genes that are potentially regulated by RNAi have been identified, and it is likely that this number will continue to grow. Because the expression of siRNAs is often tissue specific and developmentally regulated, RNAi may be an important mechanism for silencing gene expression during cell differentiation.
Books and Reviews
Conaway RC, Conaway JW: General initiation factors for RNA polymerase II. Annu Rev Biochem 1993; 63:161-190.
Karin M: Signal transduction from the cell surface to the nucleus through the phosphorylation of transcription factors. Curr Opin Cell Biol 1994; 6:415-424.
Maniatis T, Goodbourn S, Fischer JA: Regulation of inducible and tissue-specific gene expression. Science 1987; 236:1237-1245.
McKeown M: Alternative mRNA splicing. Annu Rev Cell Biol 1992; 8:133-155.
Pabo CO, Sauer RT: Transcription factors: Structural families and principles of DNA recognition. Annu Rev Biochem 1992; 61:1053-1095.
Ptashne M, Gann A: Genes & Signals. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2002.
Turner BM: Chromatin and Gene Regulation: Molecular Mechanisms in Epigenetics. Oxford: Blackwell Science, 2001.
Journal Articles
Casey JL, Koeller DM, Ramin VC, et al: Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′ untranslated region of the mRNA. EMBO J 1989; 8:3693-3699.
Gillies SD, Morrison SL, Oi VT, Tonegawa S: A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 1983; 33:717-728.
Koleske AJ, Young RA: An RNA polymerase II holoenzyme responsive to activators. Nature 1994; 368:466-469.
Schindler C, Shuai K, Prezioso VR, Darnell JE Jr: Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 1992; 257:809-813.
van der Ploeg LH, Flavell RA: DNA methylation in the human γδβ-globin locus in erythroid and nonerythroid tissues. Cell 1980; 19:947-958.
AP-1 activator protein 1, a Jun/Fos heterodimer that is a transcription factor (Table 4-1).
AR androgen receptor.
ATF-2 activating transcription factor 2, a transcription factor (Table 4-1).
bHLH basic helix-loop-helix family of transcription factors.
Brg1 Brahma-related gene 1, central motor component of SWI/SNF.
bZIP basic zipper family of transcription factors.
CAR constitutive androstane receptor.
CARM1 a coactivator and methyltransferase that methylates histones.
cAMP cyclic adenosine monophosphate.
CBP CREB-binding protein, 245 kDa, a coactivator.
C/EBPβ CCAAT/enhancer binding protein β, a transcription factor (Table 4-1).
c-Fos a transcription factor.
c-Jun a transcription factor.
c-Myc a transcription factor (Table 4-1).
CRE cAMP response element, a DNA sequence.
CREB CRE binding protein, 43 kDa.
CREM cAMP response element modifier, a transcriptional repressor.
CTD C-terminal domain of the largest subunit of Pol II.
DNA deoxyribonucleic acid.
E box sequence of six nucleotides (CANNTG, where N is any nucleotide) recognized by transcription factors MyoD and myogenin.
EGF epidermal growth factor.
Egr-1 a transcription factor (activator) that binds through zinc fingers to the same DNA site as WT-1 (repressor).
EKLF erythroid Kruppel-like factor (a transcription factor).
Elk-1 a transcription factor.
ER estrogen receptor.
FGF fibroblast growth factor.
FXR bile acid receptor.
GAL4 a yeast transcription factor that activates certain genes when yeast grows in galactose-containing media.
GAS interferon γ activation site.
GATA-1 a transcription factor.
GFAP glial fibrillary acidic protein.
GR glucocorticoid receptor.
GRB2 growth factor receptor-bound protein 2, a protein that contains SH2 domains that bind to phosphotyrosine residues on an activated receptor tyrosine kinase. GRB2 also contains SH3 domains that bind to proline-rich regions of SOS.
GRE glucocorticoid response element.
HAT histone acetyltransferase.
HDAC histone deacetylase.
HIF-1α and HIF-1β hypoxia-inducible factors.
HMT histone methyltransferase.
HNF-1, HNF-3, HNF-4 transcription factors.
hnRNA heterogeneous nuclear RNA, primary transcript of DNA, unprocessed.
HP1 heterochromatin protein 1.
hsp90 90-kDa heat shock protein, a molecular chaperone.
HTH helix-turn-helix family of transcription factors.
IFN-α, β, γ interferons α, β, and γ.
IGF-1 and IGF-2 insulin-related growth factors.
Inr “initiator,” a promoter sequence in both TATA-containing and TATA-less genes.
IRE iron response element.
ISRE interferon-stimulated response element.
JAK1, JAK2 Janus kinase, a protein tyrosine kinase.
LCR locus control region, a site distant from the structural genes.
MAPK or MAP kinase mitogen-activated protein kinase; also known as ERK-1, ERK-2 for extracellular signal-regulated kinase.
MBD1 methyl-CpG binding protein 1.
MeCP2 binds to methylated CpG dinucleotides on DNA.
MEK MAP kinase kinase (MAPKK). In the Ras cascade, it is phosphorylated by Raf-1 (MAPKKK) and phosphorylates MAP kinase (MAPK). It is also activated by JAK, part of the receptor-associated tyrosine kinase pathway.
mRNA messenger RNA.
MR mineralocorticoid receptor.
MyoD a bHLH-type transcription factor.
N-CoR a corepressor of transcription; also known as SMRT.
NF-1 nuclear factor 1, a transcription factor (Table 4-1).
NF-E2 nuclear factor E2, a heterodimeric protein complex composed of p45 and small Maf family proteins considered crucial for the proper differentiation of erythrocytes and megakaryocytes in vivo.
NF-κB a transcription factor and protein complex responsible for regulating the immune response to infection.
NF-Y ubiquitous multisubunit CCAAT binding protein composed of three subunits: NF-YA, NF-YB, NF-YC.
NGF nerve growth factor.
NRE negative regulatory element.
Oct-1 ubiquitous DNA-binding protein that recognizes a DNA sequence called the octamer motif (Table 4-1).
P box sequence of six amino acids at the C terminus of a zinc finger.
p62TCF ternary complex factor, a transcription factor.
PDGF platelet-derived growth factor.
PEPCK phosphoenolpyruvate carboxykinase, the enzyme that catalyzes the rate-limiting step in gluconeogenesis.
Pit-1 an HTH-type pituitary-specific transcription factor.
Pol II RNA polymerase II, the polymerase that transcribes DNA to mRNA.
PPAR peroxisome proliferator–activated (i.e., lipid) receptor.
PR progesterone receptor.
P-TEFb positive transcription elongation factor b, a kinase that phosphorylates the CTD of Pol II.
Raf-1 a serine/threonine kinase, also known as MAP kinase kinase kinase (MAPKKK).
RAR retinoic acid receptor.
RARE retinoic acid response element.
Ras a low-molecular-weight GTP-binding protein.
RIP regulated intramembraneous proteolysis.
RISC RNA-induced silencing complex.
RITS RNA-induced transcriptional silencing.
RNA ribonucleic acid.
RNAi RNA interference.
RNAPII an alternative designation for RNA polymerase II (Pol II).
RTK receptor tyrosine kinase.
RXR retinoid X receptor.
SH2 Src homology domain 2, a domain on a protein that binds to phosphotyrosine-containing sites.
SH3 Src homology domain 3, a domain on a protein that binds to proline-rich sequences.
Sin3A, Sin3B corepressors.
siRNA small interfering RNA.
snRNA small nuclear RNA.
snRNP a complex of proteins and snRNA.
SOS son of “sevenless” protein, a guanine nucleotide exchange protein that is part of the Ras signaling cascade. It becomes active when it binds to GRB2. It promotes the conversion of inactive GDP-Ras to active GTP-Ras.
Sp1 stimulating protein 1, a transcription factor.
SRC steroid receptor coactivator.
SREBP sterol regulatory element binding protein.
SRF serum response factor, a transcription factor.
STAT signal transducer and activator of transcription.
SUMO small ubiquitin-like modifiers.
SWI/SNF multiprotein complexes initially identified in yeast as “switching mating type/sucrose non-fermenting.”
SXR steroid and xenobiotic receptor.
TAFs TBP-associated factors.
TATA box common gene promoter sequence.
TBP TATA-binding protein.
TFIIA transcription factor IIA.
TFIIB transcription factor IIB.
TFIID transcription factor IID.
TFIIE transcription factor IIE.
TFIIF transcription factor IIF.
TFIIH transcription factor IIH.
TR thyroid hormone receptor.
TRAP220 a component of Mediator.
tRNA transfer RNA.
Tyk a protein tyrosine kinase related to JAK.
UTR untranslated region of mRNA.
VDR vitamin D receptor.
VEGF vascular endothelial growth factor.
VHL a protein that targets HIF-1α for proteosomal degradation.
VP16 viral protein from herpes simplex virus, a transcription factor.
WT-1 Wilms tumor protein, a transcriptional repressor that binds through zinc fingers to the same DNA site as Egr-1 (activator).