CHAPTER 2
Michael J. Caplan
In the minds of many students, the discipline of physiology is linked inextricably to images from its past. This prejudice is not surprising because many experiments from physiology’s proud history, such as those of Pavlov and his dogs, have transcended mere scientific renown and entered the realm of popular culture. Some might believe that the science of physiology devotes itself exclusively to the study of whole animals and is therefore an antique relic in this era of molecular reductionism. Nothing could be further from the truth. Physiology is and always has been the study of the homeostatic mechanisms that allow an organism to persist despite the ever-changing pressures imposed by a hostile environment. These mechanisms can be appreciated at many different levels of resolution.
Certainly it would be difficult to understand how the body operates unless one appreciates the functions of its organs and the communication between these organs that allows them to influence one another’s behaviors. It would also be difficult to understand how an organ performs its particular tasks unless one is familiar with the properties of its constituent cells and molecules.
The modern treatment of physiology that is presented in this textbook is as much about the interactions of molecules in cells as it is about the interactions of organs in organisms. It is necessary, therefore, at the outset to discuss the structure and characteristics of the cell. Our discussion focuses first on the architectural and dynamic features of a generic cell. We then examine how this generic cell can be adapted to serve in diverse physiological capacities. Through adaptations at the cellular level, organs acquire the machinery necessary to perform their individual metabolic tasks.
The chemical composition of the cell interior is very different from that of its surroundings. This observation applies equally to unicellular paramecia that swim freely in a freshwater pond and to neurons that are densely packed in the cerebral cortex of the human brain. The biochemical processes involved in cell function require the maintenance of a precisely regulated intracellular environment. The cytoplasm is an extraordinarily complex solution, the constituents of which include myriad proteins, nucleic acids, nucleotides, and sugars that the cell synthesizes or accumulates at great metabolic cost. The cell also expends tremendous energy to regulate the intracellular concentrations of numerous ions. If there were no barrier surrounding the cell to prevent exchange between the intracellular and extracellular spaces, all of the cytoplasm’s hard-won compositional uniqueness would be lost by diffusion in a few seconds.
The requisite barrier is provided by the plasma membrane, which forms the cell’s outer skin. The plasma membrane is impermeable to large molecules such as proteins and nucleic acids, thus ensuring their retention within the cytosol. It is selectively permeable to small molecules such as ions and metabolites. However, the metabolic requirements of the cell demand a plasma membrane that is much more sophisticated than a simple passive barrier that allows various substances to leak through at different rates. Frequently, the concentration of a nutrient in the extracellular fluid is several orders of magnitude lower than that required inside the cell. If the cell wishes to use such a substance, therefore, it must be able to accumulate it against a concentration gradient. A simple pore in the membrane cannot concentrate anything; it can only modulate the rate at which a gradient dissipates. To accomplish the more sophisticated feat of creating a concentration gradient, the membrane must be endowed with special machinery that uses metabolic energy to drive the uphill movements of substances—active transport—into or out of the cell. In addition, it would be useful to rapidly modulate the permeability properties of the plasma membrane in response to various metabolic stimuli. Active transport and the ability to control passive permeabilities underlie a wide range of physiological processes, from the electrical excitability of neurons to the resorptive and secretory functions of the kidney. In Chapter 5, we will explore how cells actively transport solutes across the plasma membrane. The mechanisms through which the plasma membrane’s dynamic selectivity is achieved, modified, and regulated are discussed briefly later in this chapter and in greater detail in Chapter 7.
Our understanding of biological membrane structure is based on studies of red blood cells, or erythrocytes, that were conducted in the early part of the 20th century. The erythrocyte lacks the nucleus and other complicated intracellular structures that are characteristic of most animal cells. It consists of a plasma membrane surrounding a cytoplasm that is rich in hemoglobin. It is possible to break open erythrocytes and release their cytoplasmic contents. The plasma membranes can then be recovered by centrifugation, providing a remarkably pure preparation of cell surface membrane. Biochemical analysis reveals that this membrane is composed of two principal constituents: lipid and protein.
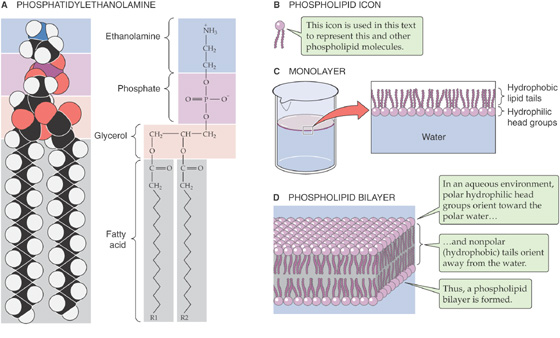
Most of the lipid associated with erythrocyte plasma membranes belongs to the molecular family of phospholipids. In general, phospholipids share a glycerol backbone, two hydroxyl groups of which are esterified to various fatty acid or acyl groups (Fig. 2-1A). These acyl groups may have different numbers of carbon atoms and also may have double bonds between carbons. For glycerol-based phospholipids, the third glycerolic hydroxyl group is esterified to a phosphate group, which is in turn esterified to a small molecule referred to as a head group. The identity of the head group determines the name as well as many of the properties of the individual phospholipids. For instance, glycerol-based phospholipids that bear an ethanolamine molecule in the head group position are categorized as phosphatidyl-ethanolamines (Fig. 2-1A).
Figure 2-1 Phospholipids.
The unique structure and physical chemistry of each phospholipid (Fig. 2-1B) underlie the formation of biological membranes and explain many of their most important properties. Fatty acids are nonpolar molecules. Their long carbon chains lack the charged groups that would facilitate interactions with water, which is polar. Consequently, fatty acids dissolve poorly in water but readily in organic solvents; thus, fatty acids are hydrophobic. On the other hand, the head groups of most phospholipids are charged or polar. These head groups interact well with water and consequently are very water soluble. Thus, the head groups are hydrophilic. Because phospholipids combine hydrophilic heads with hydrophobic tails, their interaction with water is referred to as amphipathic.
When mixed with water, phospholipids organize themselves into structures that prevent their hydrophobic tails from making contact with water while simultaneously permitting their hydrophilic head groups to be fully dissolved. When added to water at fairly low concentrations, phospholipids form a monolayer (Fig. 2-1C) on the water’s surface at the air-water interface. It is energetically less costly to the system for the hydrophobic tails to stick up in the air than to interact with the solvent.
At higher concentrations, phospholipids assemble into micelles. The hydrophilic head groups form the surfaces of these small spheres, whereas the hydrophobic tails point toward their centers. In this geometry, the tails are protected from any contact with water and instead are able to participate in energetically favorable interactions among themselves. At still higher concentrations, phospholipids spontaneously form bilayers (Fig. 2-1D). In these structures, the phospholipid molecules arrange themselves into two parallel sheets or leaflets that face each other tail to tail. The surfaces of the bilayer are composed of hydrophilic head groups; the hydrophobic tails form the center of the sandwich. The hydrophilic surfaces insulate the hydrophobic tails from contact with the solvent, leaving the tails free to associate exclusively with one another.
The physical characteristics of a lipid bilayer largely depend on the chemical composition of its constituent phospholipid molecules. For example, the width of the bilayer is determined by the length of the fatty acid side chains. Dihexadecanoic phospholipids (whose two fatty acid chains are each 16 carbons long) produce bilayers that are 2.47 nm wide; ditetradecanoic phospholipids (bearing 14-carbon fatty acids) generate 2.3-nm bilayers. Similarly, the nature of the head groups determines how densely packed adjacent phospholipid molecules are in each leaflet of the membrane.
Detergents can dissolve phospholipid membranes because like the phospholipids themselves, they are amphipathic. They possess very hydrophilic head groups and hydrophobic tails and are water soluble at much higher concentrations than are the phospholipids. When mixed together in aqueous solutions, detergent and phospholipid molecules interact through their hydrophobic tails, and the resulting complexes are water soluble, either as individual dimers or in mixed micelles. Therefore, adding sufficient concentrations of detergent to phospholipid bilayer membranes disrupts the membranes and dissolves the lipids. Detergents are extremely useful tools in research into the structure and composition of lipid membranes.
Despite its highly organized appearance, a phospholipid bilayer is a fluid structure. An individual phospholipid molecule is free to diffuse within the entire leaflet in which it resides. The rate at which this two-dimensional diffusion occurs is extremely temperature dependent. At high temperatures, the thermal energy of any given lipid molecule is greater than the interaction energy that would tend to hold adjacent lipid molecules together. Under these conditions, lateral diffusion can proceed rapidly, and the lipid is said to be in the sol state. At lower temperatures, interaction energies exceed the thermal energies of most individual molecules. Thus, phospholipids diffuse slowly because they lack the energy to free themselves from the embraces of their neighbors. This behavior is characteristic of the gel state.
The temperature at which the bilayer membrane converts from the gel to the sol phase (and vice versa) is referred to as the transition temperature. The transition temperature is another characteristic that depends on the chemical makeup of the phospholipids in the bilayer. Phospholipids with long, saturated fatty acid chains can extensively interact with one another. Consequently, a fair amount of thermal energy is required to overcome these interactions and permit diffusion. Not surprisingly, such bilayers have relatively high transition temperatures. For example, the transition temperature for dioctadecanoic phosphatidylcholine (which has two 18-carbon fatty acid chains, fully saturated) is 55.5°C. In contrast, phospholipids that have shorter fatty acid chains or double bonds (which introduce kinks) cannot line up next to each other as well and hence do not interact as well. Considerably less energy is required to induce them to participate in diffusion. For example, if we reduce the length of the carbon chain from 18 to 14, the transition temperature falls to 23°C. If we retain 18 carbons but introduce a single, double bond (making the fatty acid chains monounsaturated), the transition temperature also falls dramatically.
By mixing other types of lipid molecules into phospholipid bilayers, we can markedly alter the membrane’s fluidity properties. The glycerol-based phospholipids, the most common membrane lipids, include the phosphatidylethanolamines described earlier (Fig. 2-1A) as well as the phosphatidylinositols (Fig. 2-2A), phosphatidylserines (Fig. 2-2B), and phosphatidylcholines (Fig. 2-2C). The second major class of membrane lipids, the sphingolipids (derivatives of sphingosine), are made up of three subgroups: sphingomyelins (Fig. 2-2D), glycosphingolipids such as the galactocerebrosides (Fig. 2-2E), and gangliosides (not shown). Cholesterol (Fig. 2-2F) is another important membrane lipid. Because these other molecules are not shaped exactly like the glycerol-based phospholipids, they participate to different degrees in intermolecular interactions with phospholipid side chains. The presence of these alternative lipids changes the strength of the interactions that prevent lipid molecules from diffusing. Consequently, the membrane has a different fluidity and a different transition temperature. This behavior is especially characteristic of the cholesterol molecule, whose rigid steroid ring binds to and partially immobilizes fatty acid side chains. Therefore, at modest concentrations, cholesterol decreases fluidity. However, when it is present in high concentrations, cholesterol can substantially disrupt the ability of the phospholipids to interact among themselves, which increases fluidity and lowers the gel-sol transition temperature. This issue is significant because animal cell plasma membranes can contain substantial quantities of cholesterol. (See Note: Sphingomyelins; Diversity of Lipids in a Bilayer)
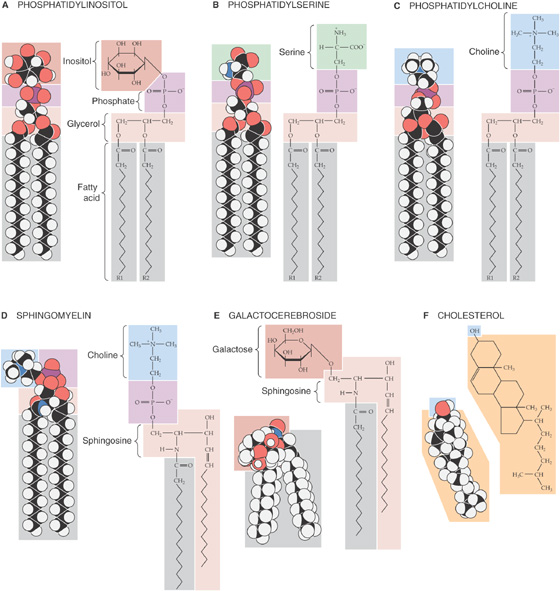
Figure 2-2 Structures of some common membrane lipids.
Bilayers composed of several different lipids do not undergo the transition from gel to sol at a single, well-defined temperature. Instead, they interconvert more gradually over a temperature range that is defined by the composition of the mixture. Within this transition range in such multi-component bilayers, the membrane can become divided into compositionally distinct zones. The phospholipids with long-chain, saturated fatty acids will adhere to one another relatively tightly, which results in the formation of regions with “gel-like” properties. Phospholipids bearing short-chain, unsaturated fatty acids will be excluded from these regions and migrate to sol-like regions. Hence, “lakes” of lipids with markedly different physical properties can exist side-by-side in the plane of a phospholipid membrane. Thus, the same thermodynamic forces that form the elegant bilayer structure can partition distinct lipid domains within the bilayer. As discussed later, the segregation of lipid lakes in the plane of the membrane may be important for sorting membrane proteins to different parts of the cell.
Although phospholipids can diffuse in the plane of a lipid bilayer membrane, they do not diffuse between adjacent leaflets (Fig. 2-3). The rate at which phospholipids spontaneously “flip-flop” from one leaflet of a bilayer to the other is extremely low. As mentioned earlier, the center of a bilayer membrane consists of the fatty acid tails of the phospholipid molecules and is an extremely hydrophobic environment. For a phospholipid molecule to jump from one leaflet to the other, its highly hydrophilic head group would have to transit this central hydrophobic core, which would have an extremely high energy cost. This caveat does not apply to cholesterol (Fig. 2-3), whose polar head is a single hydroxyl group. The energy cost of dragging this small polar hydroxyl group through the bilayer is relatively low, thus permitting relatively rapid cholesterol flip-flop.
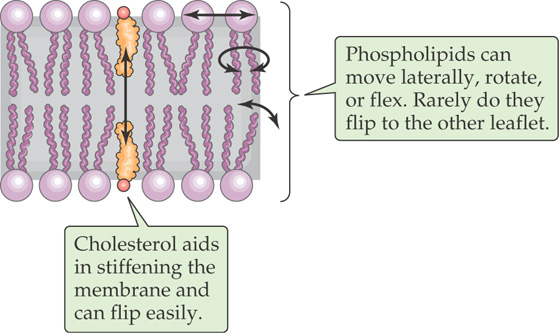
Figure 2-3 Mobility of lipids within a bilayer.
The lipid bilayer is ideally suited to separate two aqueous compartments. Its hydrophilic head groups interact well with water at both membrane surfaces, whereas the hydrophobic center ensures that the energetic cost of crossing the membrane is prohibitive for charged atoms or molecules. Pure phospholipid bilayer membranes are extremely impermeable to almost any charged water-soluble substance. Ions such as Na+, K+, Cl−, and Ca2+ are insoluble in the hydrophobic membrane core and consequently cannot travel from the aqueous environment on one side of the membrane to the aqueous environment on the opposite side. The same is true of large water-soluble molecules, such as proteins, nucleic acids, sugars, and nucleotides.
Whereas phospholipid membranes are impermeable to water-soluble molecules, small uncharged polar molecules can cross fairly freely. This is often true for O2, CO2, NH3, and, remarkably, water itself. Water molecules may, at least in part, traverse the membrane through transient cracks between the hydrophobic tails of the phospholipids, without having to surmount an enormous energetic barrier. The degree of water permeability (and perhaps that of CO2 and NH3 as well) varies extensively with lipid composition; some phospholipids (especially those with short or kinked fatty acid chains) permit a much greater rate of transbilayer water diffusion than others do.
As may be inferred from the preceding discussion, the membrane at the cell surface is, in fact, a phospholipid bilayer. The truth of this statement was established by a remarkably straightforward experiment. In 1925, Gorter and Grendel measured the surface area of the lipids they extracted from erythrocyte plasma membranes. They used a device called a Langmuir trough in which the lipids are allowed to line up at an air-water interface (Fig. 2-1C) and are then packed together into a continuous monolayer by a sliding bar that decreases the surface available to them. The area of the monolayer that was created by the erythrocyte lipids was exactly twice the surface area of the erythrocytes from which they were derived. Therefore, the plasma membrane must be a bilayer.
Confirmation of the bilayer structure of biological membranes has come from x-ray diffraction studies performed on the repetitive whorls of membrane that form the myelin sheaths surrounding neuronal axons (see Chapter 11). The membrane’s bilayer structure can be visualized directly in the high-magnification electron micrograph depicted in Figure 2-4. The osmium tetraoxide molecule (OsO4), with which the membrane is stained, binds to the head groups of phospholipids. Thus, both surfaces of a phospholipid bilayer appear black in electron micrographs, whereas the membrane’s unstained central core appears white.
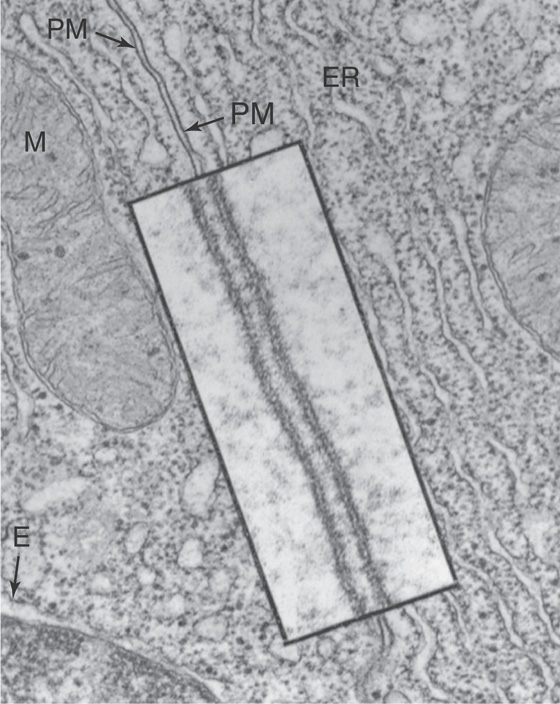
Figure 2-4 Transmission electron micrograph of a cell membrane. The photograph shows two adjacent cells of the pancreas of a frog (magnification ×43,000). The inset is a high-magnification view (×216,000) of the plasma membranes (PM) of the cells. Note that each membrane includes two dense layers with an intermediate layer of lower density. The dense layers represent the interaction of the polar head groups of the phospholipids with the OsO4 used to stain the preparation. ER, endoplasmic reticulum; M, mitochondrion. (From Porter KR, Bonneville MR: Fine Structure of Cells and Tissues, 4th ed. Philadelphia: Lea & Febiger, 1973.)
The phospholipid compositions of the two leaflets of the plasma membrane are not identical. Labeling studies performed on erythrocyte plasma membranes reveal that the surface that faces the cytoplasm contains phosphatidylethanolamine and phosphatidylserine, whereas the outward-facing leaflet is composed almost exclusively of phosphatidylcholine. As is discussed later in this chapter, this as ymmetry is created during the biosynthesis of the phospholipid molecules. It is not entirely clear what advantage this distribution provides to the cell. It appears likely that the interactions between certain proteins and the plasma membrane may require this segregation. The lipid asymmetry may be especially important for those phospholipids that are involved in second-messenger cascades (see Chapter 3). Finally, the phospholipids that are characteristic of animal cell plasma membranes generally have one saturated and one unsaturated fatty acid residue. Consequently, they are less likely to partition into sol-like or gel-like lipid domains than are phospholipids that bear identical fatty acid chains. (See Note: Membrane Microdomains)
The demonstration that the plasma membrane’s lipid components form a bilayer leaves open the question of how the membrane’s protein constituents are organized. Membrane proteins can belong to either of two broad classes, peripheral or integral. Peripherally associated membrane proteins are neither embedded within the membrane nor attached to it by covalent bonds; instead, they adhere tightly to the cytoplasmic or extracellular surfaces of the plasma membrane (Fig. 2-5A). They can be removed from the membrane, however, by mild treatments that disrupt ionic bonds (very high salt concentrations) or hydrogen bonds (very low salt concentrations).
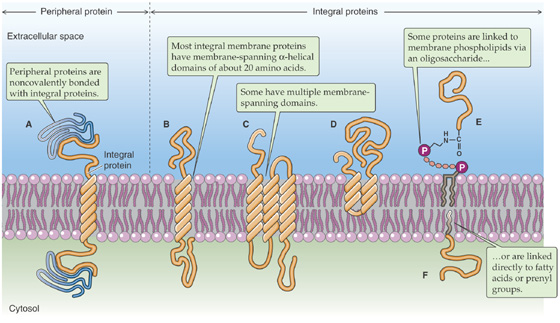
Figure 2-5 Classes of membrane proteins. In E, protein is coupled by a GPI linkage.
In contrast, integral membrane proteins are intimately associated with the lipid bilayer. They cannot be eluted from the membrane by these high-or low-salt washes. To dislodge integral membrane proteins, the membrane itself must be dissolved by adding detergents. Integral membrane proteins can be associated with the lipid bilayer in any of three ways. First, some proteins actually span the lipid bilayer once or several times (Fig. 2-5B, C) and hence are referred to as transmembrane proteins. Experiments performed on erythrocyte membranes reveal that these proteins can be labeled with protein-tagging reagents applied to either side of the bilayer.
The second group of integral membrane proteins is embedded in the bilayer without actually crossing it (Fig. 2-5D). A third group of membrane-associated proteins is not actually embedded in the bilayer at all. Instead, these lipid-anchored proteins are attached to the membrane by a covalent bond that links them either to a lipid component of the membrane or to a fatty acid derivative that intercalates into the membrane. For example, proteins can be linked to a special type of glycosylated phospholipid molecule (Fig. 2-5E), which is most often glycosylphosphatidylinositol (GPI), on the outer leaflet of the membrane. This family is referred to collectively as the glycophospholipid-linked proteins. Another example is a direct linkage to a fatty acid (e.g., a myristyl group) or a prenyl (e.g., farnesyl) group that intercalates into the inner leaflet of the membrane (Fig. 2-5F).
How can membrane-spanning proteins remain stably associated with the bilayer in a conformation that requires at least some portion of their amino acid sequence to be in continuous contact with the membrane’s hydrophobic central core? The answer to this question can be found in the special structures of those protein domains that actually span the membrane.
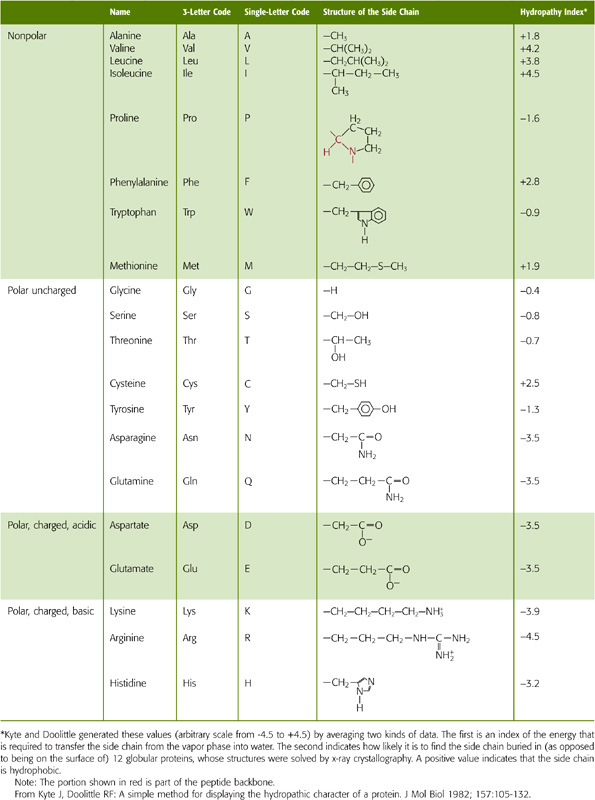
The side chains of the eight amino acids listed in the upper portion of Table 2-1 are hydrophobic. These aromatic or uncharged aliphatic groups are almost as difficult to solvate in water as are the fatty acid side chains of the membrane phospholipids themselves. Not surprisingly, therefore, these hydrophobic side chains are quite comfortable in the hydrophobic environment of the bilayer core. Most membrane-spanning segments—that is, the short stretch of amino acids that passes through the membrane once—are composed mainly of these nonpolar amino acids, in concert with polar, uncharged amino acids.
Table 2-1 Classification of the Amino Acids Based on the Chemistry of Their Side Chains
The hydrophobic, membrane-spanning segments of transmembrane proteins are specially adapted to the hydrophobic milieu in which they reside. The phospholipid molecules of the membrane bilayer actually protect these portions of transmembrane proteins from energetically unfavorable interactions with the aqueous environment. Transmembrane proteins tend to be extremely insoluble in water. If we separate the membrane-spanning segments of these proteins from the amphipathic phospholipids that surround them, these hydrophobic sequences tend to interact tightly with one another rather than with water. The resulting large protein aggregates are generally insoluble and precipitate out of solution. If, however, we disrupt the phospholipid membrane by adding detergent, the amphipathic detergent molecules can substitute for the phospholipids. The hydrophobic membrane-spanning sequences remain insulated from interactions with the aqueous solvent, and the proteins remain soluble as components of detergent micelles. This ability of detergents to remove transmembrane proteins from the lipid bilayer—while maintaining the solubility and native architectures of these proteins—has proved important for purifying individual membrane proteins.
Transmembrane proteins can have a single membrane-spanning segment (Fig. 2-5B) or several (Fig. 2-5C). Those with a single transmembrane segment can be oriented with either their amino (N) or their carboxyl (C) termini facing the extracellular space. Multispanning membrane proteins weave through the membrane like a thread through cloth. Again, the N or C termini can be exposed to either the cytoplasmic or extracellular compartments. The pattern with which the transmembrane protein weaves across the lipid bilayer defines its membrane topology.
The amino acid sequences of membrane-spanning segments tend to form α helices, with ~3.6 amino acids per turn of the helix (Fig. 2-5B). In this conformation, the polar atoms of the peptide backbone are maximally hydrogen bonded to one another—from one turn of the helix to the next—so they do not require the solvent to contribute hydrogen bond partners. Hence, this structure ensures the solubility of the membrane-spanning sequence in the hydrophobic environment of the membrane. Whereas most transmembrane proteins appear to traverse the membrane with α-helical spans, it is clear that an intriguing subset of membrane polypeptides makes use of a very different structure. The best studied member of this class is the porin protein, which serves as a channel in bacterial membranes. As discussed in Chapter 5, the membrane-spanning portions of porin are arranged as a β barrel.
In the case of multispanning membrane proteins, their transmembrane helices probably pack together tightly (Fig. 2-5C). Molecular analysis of a number of known membrane-spanning sequences has helped in the development of algorithms predicting the likelihood that a given amino acid sequence can span the membrane. These algorithms are widely used to assess the likelihood that newly identified genes encode transmembrane proteins and to predict the number and location of membrane-spanning segments.
Many membrane proteins form tight, noncovalent associations with other membrane proteins in the plane of the bilayer. These multimeric proteins can be composed of a single type of polypeptide or of mixtures of two or more different proteins. The side-to-side interactions that hold these complexes together can involve the membrane-spanning segments or regions of the proteins that protrude at either surface of the bilayer. By assembling into multimeric complexes, membrane proteins can increase their stability. They can also increase the variety and complexity of the functions that they are capable of performing.
As is true for phospholipid molecules (Fig. 2-3), some transmembrane proteins can also diffuse within the surface of the membrane. In the absence of any protein-protein attachments, transmembrane proteins are free to diffuse over the entire surface of a membrane. This fact was demonstrated by Frye and Edidin in 1970 (Fig. 2-6). They labeled the surface proteins of a population of mouse lymphocytes with a lectin (a plant protein that binds strongly to certain sugar groups attached to proteins) that was linked to the fluorescent dye fluorescein. They also tagged the surface proteins of a second population of human lymphocytes with a lectin that was conjugated to a different fluorescent dye, rhodamine. Because fluorescein glows green and rhodamine glows red when excited by the light of the appropriate wavelengths, these labeling molecules can be easily distinguished from one another in a fluorescence microscope. Frye and Edidin mixed the two lymphocyte populations and treated them with a reagent that caused the cells to fuse to each other. Immediately after fusion, the labeled surface proteins of the newly joined cells remained separate; half of the fused cell surface appeared red, whereas the other half appeared green. During a period of ~30 minutes, however, the green and red protein labels intermixed until the entire surface of the fused cell was covered with both labeling molecules. The rate at which this intermingling occurred increased with temperature, which is not surprising, given the temperature dependence of membrane fluidity.
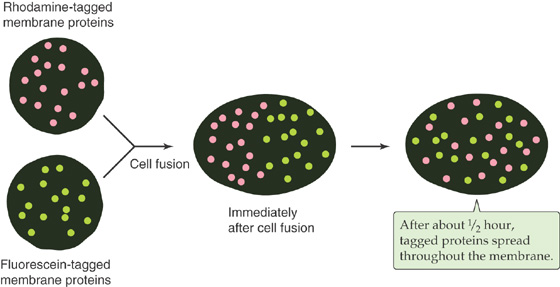
Figure 2-6 Diffusion of membrane proteins within the plane of the cell membrane. The surface proteins of a human lymphocyte are tagged with a lectin conjugated to rhodamine, a fluorescent dye; the surface proteins of a mouse lymphocyte are tagged with a lectin linked to fluorescein, another fluorescent dye. Immediately after fusion of the two cells, the labeled surface proteins remained segregated. However, the membrane proteins intermingled during a period of ~30 minutes.
Because transmembrane proteins are large molecules, their diffusion in the plane of the membrane is much slower than that of lipids. Even the fastest proteins diffuse ~1000 times more slowly than the average phospholipid. The diffusion of many transmembrane proteins appears to be further impeded by their attachments to the cytoskeleton, just below the surface of the membrane. Tight binding to this meshwork can render proteins essentially immobile. Other transmembrane proteins appear to travel in the plane of the membrane by directed processes that are much faster and less directionally random than diffusion is. Motor proteins that are associated with the cytoplasmic cytoskeleton (discussed later) appear to grab onto certain transmembrane proteins, dragging them in the plane of the membrane like toy boats on strings. Finally, like phospholipids, proteins can diffuse only in the plane of the bilayer. They cannot flip-flop across it. The energetic barrier to dragging a transmembrane protein’s hydrophilic cytoplasmic and extracellular domains across the bilayer’s hydrophobic core is very difficult to surmount. Thus, a membrane protein’s topology does not change over its life span.
All communication between a cell and its environment must involve or at least pass through the plasma membrane. For the purposes of this discussion, we define communication rather broadly as the exchange of any signal between the cell and its surroundings. Except for lipid-soluble signaling molecules such as steroid hormones, essentially all communication functions served by the plasma membrane occur through membrane proteins. From an engineering perspective, membrane proteins are perfectly situated to transmit signals because they form a single, continuous link between the two compartments that are separated by the membrane.
Ligand-binding receptors comprise the group of transmembrane proteins that perhaps most clearly illustrate the concept of transmembrane signaling (Fig. 2-7A). For water-soluble hormones such as epinephrine to influence cellular behavior, their presence in the extracellular fluid compartment must be made known to the various intracellular mechanisms whose behaviors they modulate. The interaction of a hormone with the extracellular portion of the hormone receptor, which forms a high-affinity binding site, produces conformational changes within the receptor protein that extend through the membrane-spanning domain to the intracellular domain of the receptor. As a consequence, the intracellular domain either becomes enzymatically active or can interact with cytoplasmic proteins that are involved in the generation of so-called second messengers. Either mechanism completes the transmission of the hormone signal across the membrane. The transmembrane disposition of a hormone receptor thus creates a single, continuous communication medium that is capable of conveying, through its own structural modifications, information from the environment to the cellular interior. The process of transmembrane signal transduction is discussed in Chapter 3.
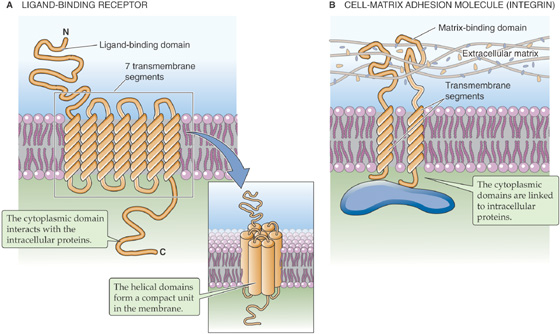
Figure 2-7 Integral membrane proteins that transmit signals from the outside to the inside of a cell. A, The ligand may be a hormone, a growth factor, a neurotransmitter, an odorant, or another local mediator. B, An integrin is an adhesion molecule that attaches the cell to the extracellular matrix.
Cells can also exploit integral membrane proteins as adhesion molecules that form physical contacts with the surrounding extracellular matrix (i.e., cell-matrix adhesion molecules) or with their cellular neighbors (i.e., cell-cell adhesion molecules). These attachments can be extremely important in regulating the shape, growth, and differentiation of cells. The nature and extent of these attachments must be communicated to the cell interior so that the cell can adapt appropriately to the physical constraints and cues that are provided by its immediate surroundings. Numerous classes of transmembrane proteins are involved in these communication processes. The integrins are examples of matrix receptors or cell matrix adhesion molecules. They comprise a large family of transmembrane proteins that link cells to components of the extracellular matrix (e.g., fibronectin, laminin) at adhesion plaques (Fig. 2-7B). These linkages produce conformational changes in the integrin molecules that are transmitted to their cytoplasmic tails. These tails, in turn, communicate the linkage events to various structural and signaling molecules that participate in formulating a cell’s response to its physical environment.
In contrast to matrix receptors, which attach cells to the extracellular matrix, several enormous superfamilies of cell-cell adhesion molecules attach cells to each other. These cell-cell adhesion molecules include the Ca2+-dependent cell adhesion molecules (cadherins) and Ca2+-independent neural cell adhesion molecules (N-CAMs). The cadherins are glycoproteins (i.e., proteins with sugars attached) with one membrane-spanning segment and a large extracellular domain that binds Ca2+. The N-CAMs, on the other hand, generally are members of the immunoglobulin superfamily. The two classes of cell-cell adhesion molecules mediate similar sorts of transmembrane signals that help organize the cytoplasm and control gene expression in response to intercellular contacts. Some cell-cell adhesion molecules belong to the GPI-linked class of membrane proteins. These polypeptides lack a transmembrane and cytoplasmic tail. It is not clear, therefore, how (or if) interactions mediated by this unique class of adhesion molecules are communicated to the cell interior.
Adhesion molecules orchestrate processes that are as diverse as the directed migration of immune cells and the guidance of axons in the developing nervous system. Loss of cell-cell and cell-matrix adhesion is a hallmark of metastatic tumor cells.
Earlier in this discussion, we noted that a pure phospholipid bilayer does not have the permeability properties that are normally associated with animal cell plasma membranes. Pure phospholipid bilayers also lack the ability to transport substances uphill. Transmembrane proteins endow biological membranes with these capabilities. Ions and other membrane-impermeable substances can cross the bilayer with the assistance of transmembrane proteins that serve as pores, channels, carriers, and pumps. Pores and channels serve as conduits that allow water, specific ions, or even very large proteins to flow passively through the bilayer. Carriers can either facilitate the transport of a specific molecule across the membrane or couple the transport of a molecule to that of other solutes. Pumps use the energy that is released through the hydrolysis of adenosine triphosphate (ATP) to drive the transport of substances into or out of cells against energy gradients. Each of these important classes of proteins is discussed in Chapter 5.
Channels, carriers, and pumps succeed in allowing hydrophilic substances to cross the membrane by creating a hydrophilic pathway in the bilayer. Previously, we asserted that membrane-spanning segments are as hydrophobic as the fatty acids that surround them. How is it possible for these hydrophobic membrane-spanning domains to produce the hydrophilic pathways that permit the passage of ions through the membrane? The solution to this puzzle appears to be that the α helices that make up these membrane-spanning segments are amphipathic. That is, they possesses both hydrophobic and hydrophilic domains.
For each α helix, the helical turns produce alignments of amino acids that are spaced at regular intervals in the sequence. Thus, it is possible to align all the hydrophilic or hydrophobic amino acids along a single edge of the helix. In amphipathic helices, hydrophobic amino acids alternate with hydrophilic residues at regular intervals of approximately three or four amino acids (recall that there are ~3.6 amino acids per turn of the helix). Thus, as the helices pack together, side-by-side, the resultant membrane protein has distinct hydrophilic and hydrophobic surfaces. The hydrophobic surfaces of each helix will face either the membrane lipid or the hydrophobic surfaces of neighboring helices. Similarly, the hydrophilic surfaces of each helix will face a common central pore through which water-soluble particles can move. Depending on how the protein regulates access to this pore, the protein could be a channel, a carrier, or a pump. The mix of hydrophilic amino acids that line the pore presumably determines, at least in part, the nature of the substances that the pore can accommodate. In some instances, the amphipathic helices that line the pore are contributed by several distinct proteins—or subunits—that assemble into a single multimeric complex. Figure 2-8 shows an example of a type of K+ channel that is discussed in Chapter 7. This channel is formed by the apposition of four identical subunits, each of which has six membrane-spanning segments. The pore of this channel is created by the amphipathic helices as well as by short, hydrophilic loops (P loops) contributed by each of the four subunits.
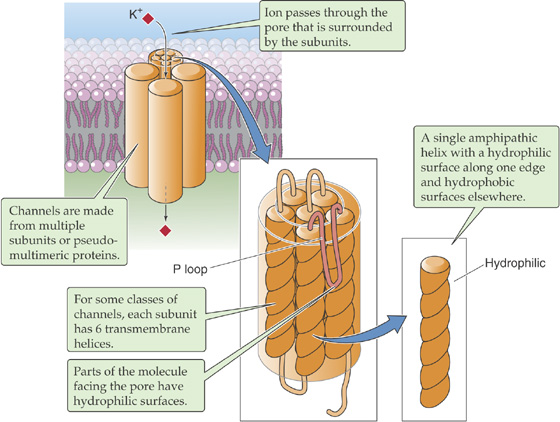
Figure 2-8 Amphipathic α helices interacting to form a channel through the cell membrane. This is an example of a potassium channel.
Ion pumps are actually enzymes. They catalyze the hydrolysis of ATP and use the energy released by that reaction to drive ion transport. Many other classes of proteins that are embedded in cell membranes function as enzymes as well. Membrane-bound enzymes are especially prevalent in the cells of the intestine, which participate in the final stages of nutrient digestion and absorption (see Chapter 45). These enzymes—located on the side of the intestinal cells that faces the lumen of the intestine—break down small polysaccharides into single sugars, or break down polypeptides into shorter polypeptides or amino acids, so that they can be imported into the cells. By embedding these enzymes in the plasma membrane, the cell can generate the final products of digestion close to the transport proteins that mediate the uptake of these nutrient molecules. This theme is repeated in numerous other cell types. Thus, the membrane can serve as an extremely efficient two-dimensional reaction center for multistep processes that involve enzymatic reactions or transport.
Many of the GPI-linked proteins are enzymes. Several of the enzymatic activities that are classically thought of as extracellular markers of the plasma membrane, such as alkaline phosphatase and 5′-nucleotidase, are anchored to the external leaflet of the bilayer by covalent attachment to a GPI. The biological utility of this arrangement has yet to be determined. However, the GPI linkage is itself a substrate for enzymatic cleavage. Phospholipase C, which is present at appreciable levels in the serum, can cleave the covalent bond between the protein and its lipid anchor, thereby releasing the protein from the membrane. The released protein subsequently behaves like a soluble polypeptide.
Some integral proteins associate with the cytoplasmic surface of the plasma membrane by covalently attaching to fatty acids or prenyl groups that in turn intercalate into the lipid bilayer (Fig. 2-5F). The fatty acids or prenyl groups act as hydrophobic tails that anchor an otherwise soluble protein to the bilayer. These proteins are all located at the intracellular leaflet of the membrane bilayer and often participate in intracellular signaling and growth regulation pathways. The family of lipid-linked proteins includes the small and heterotrimeric guanosine triphosphate (GTP)–binding proteins, kinases, and oncogene products (see Chapter 3). Many of these proteins are involved in relaying the signals that are received at the cell surface to the effector machinery within the cell interior. Their association with the membrane, therefore, brings these proteins close to the cytoplasmic sides of receptors that transmit signals from the cell exterior across the bilayer. The medical relevance of this type of membrane association is beginning to be appreciated. For example, denying certain oncogene products their lipid modifications—and hence their membrane attachment—eliminates their ability to induce tumorigenic transformation.
Peripheral membrane proteins attach loosely to the lipid bilayer but are not embedded within it. Their association with the membrane can take one of two forms. First, some proteins interact through ionic interactions with phospholipid head groups. Many of these head groups are positively or negatively charged and thus can participate in salt bridges with adherent proteins.
For a second group of peripheral membrane proteins, attachment is based on the direct binding of peripheral membrane proteins to the extracellular or cytoplasmic surfaces of integral membrane proteins (Fig. 2-5A). This form of attachment is epitomized by the cytoskeleton. For instance, the cytoplasmic surface of the erythrocyte plasma membrane is in close apposition to a dense meshwork of interlocking protein strands known as the subcortical cytoskeleton. It consists of a long, fibrillar molecule called spectrin, short polymers of the cytoskeletal protein actin, and other proteins including ankyrin and band 4.1 (Fig. 2-9).
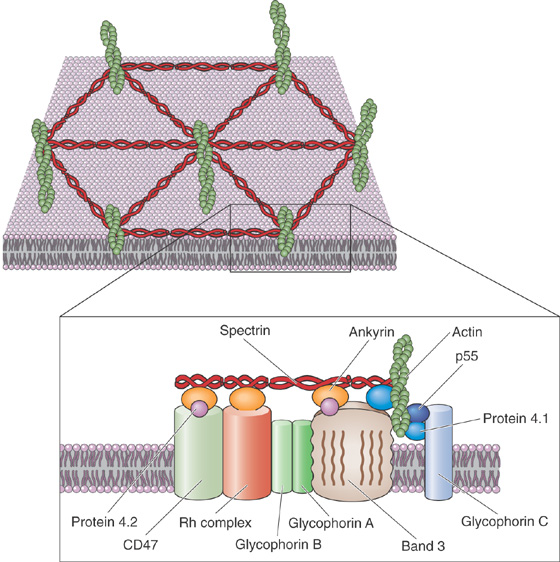
Figure 2-9 Attachments of the cell membrane to the submembranous cytoskeleton in red blood cells. Integral membrane proteins form the bridges that link the cell membrane to the interlocking system of proteins that form the subcortical cytoskeleton.
Two closely related isoforms of spectrin (α and β) form dimers, and two of these dimers assemble head-to-head with one another to form spectrin heterotetramers. The tail regions of spectrin bind the globular protein band 4.1, which in turn can bind to actin fibrils. Each actin fibril can associate with more than one molecule of band 4.1 so that, together, spectrin, actin, and band 4.1 assemble into an extensive interlocking matrix. The protein known as ankyrin binds to spectrin as well as to the cytoplasmic domain of band 3, the integral membrane protein responsible for transporting Cl− and HCO−3 ions across the erythrocyte membrane. Thus, ankyrin is a peripheral membrane protein that anchors the spectrin-actin meshwork directly to an integral membrane protein of the erythrocyte.
The subcortical cytoskeleton provides the erythrocyte plasma membrane with strength and resilience. People who carry mutations in genes encoding their components have erythrocytes that do not have the characteristic biconcave disk shape. These erythrocytes are extremely fragile and are easily torn apart by the shear stresses (see Chapter 17) associated with circulation through capillaries. It would appear, therefore, that the subcortical cytoskeleton forms a scaffolding of peripheral membrane proteins whose direct attachment to transmembrane proteins enhances the bilayer’s structural integrity.
The subcortical cytoskeleton is not unique to erythrocytes. Numerous cell types, including neurons and epithelial cells, have submembranous meshworks that consist of proteins very similar to those first described in the erythrocyte. In addition to band 3, transmembrane proteins found in a wide variety of cells (including ion pumps, ion channels, and cell adhesion molecules) bind ankyrin and can thus serve as focal points of cytoskeletal attachment. In polarized cells (e.g., neurons and epithelial cells), the subcortical cytoskeleton appears to play a critically important role in organizing the plasma membrane into morphologically and functionally distinct domains.
When a eukaryotic cell is viewed through a light microscope, a handful of recognizable intracellular structures can be discerned. The intracellular matrix, or cytoplasm, appears grainy, suggesting the presence of components that are too small to be discriminated by this technique. With the much higher magnifications available with an electron microscope, the graininess gives way to clarity that reveals the cell interior to be remarkably complex. Even the simplest nucleated animal cell possesses a wide variety of intricate structures with specific shapes and sizes. These structures are the membrane-enclosed organelles, the functional building blocks of cells.
Figure 2-10 illustrates the interior of a typical cell. The largest organelle in this picture is the nucleus, which houses the cell’s complement of genetic information. This structure, which is visible in the light microscope, is usually round or oblong, although in some cells it displays a complex, lobulated shape. Depending on the cell type, the nucleus can range in diameter from 2 to 20 μm. With some exceptions, including skeletal muscle and certain specialized cells of the immune system, each animal cell has a single nucleus.
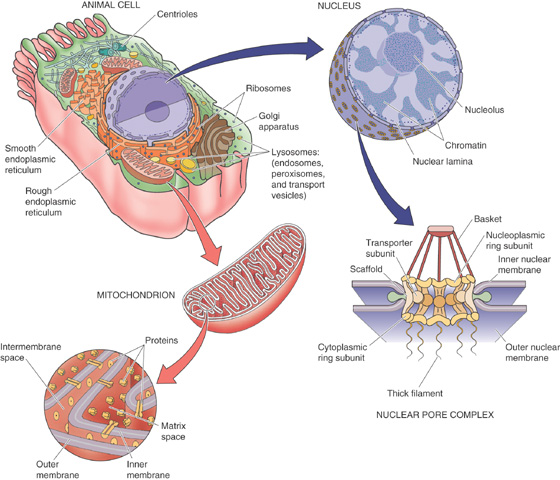
Figure 2-10 Ultrastructure of a typical animal cell.
Surrounding the nucleus is a web of tubules or saccules known as the endoplasmic reticulum (ER). This organelle can exist in either of two forms, rough or smooth. The surfaces of the rough ER tubules are studded with ribosomes, the major sites of protein synthesis. Ribosomes can also exist free in the cytosol. The surfaces of the smooth ER, which participates in lipid synthesis, are not similarly endowed. The ER also serves as a major reservoir for calcium ions. The ER membrane is endowed with a Ca2+ pump that uses the energy released through ATP hydrolysis to drive the transport of Ca2+ from the cytoplasm into the ER lumen (see Chapter 5). This Ca2+ can be rapidly released in response to messenger molecules and plays a major role in intracellular signaling (see Chapter 3).
The Golgi complex resembles a stack of pancakes. Each pancake in the stack represents a discrete, flat saccule. The number and size of the saccules in the Golgi stack vary among cell types. The Golgi complex is a processing station that participates in protein maturation and targets newly synthesized proteins to their appropriate subcellular destinations.
Perhaps the most intriguing morphological appearance belongs to the mitochondrion, which is essentially a balloon within a balloon. The outer membrane and inner membrane define two distinct internal compartments: the intermembrane space and the matrix space. The surface of the inner membrane is thrown into dramatic folds called cristae. This organelle is ~0.2 μm in diameter, placing it at the limit of resolution of the light microscope. The mitochondrion is the power plant of the cell, a critical manufacturer of ATP. Many cellular reactions are also catalyzed within the mitochondrion.
The cell’s digestive organelle is the lysosome. This large structure frequently contains several smaller round vesicles called exosomes within its internal space.
The cytoplasm contains numerous other organelles whose shapes are not quite as distinguishing, including endosomes, peroxisomes, and transport vesicles.
Despite their diversity, all cellular organelles are constructed from the same building blocks. Each is composed of a membrane that forms the entire extent of its surface. The membranes of the subcellular organelles are what can be visualized in electron micrographs. The biochemical and physical properties of an organelle’s limiting membrane dictate many of its functional properties.
The nucleus serves as a cell’s repository for its complement of chromosomal DNA. To conceive of the nucleus as simply a hermetically sealed vault for genetic information, however, is a gross oversimplification. All of the machinery necessary to maintain, to copy, and to transcribe DNA is in the nucleus, which is the focus of all of the cellular pathways that regulate gene expression and cell division. Transcriptional control is discussed in Chapter 4. The focus of this section is nuclear structure.
The nucleus is surrounded by a double membrane (Fig. 2-10). The outer membrane is studded with ribosomes and is continuous with the membranes of the rough ER. The inner membrane is smooth and faces the intranuclear space, or nucleoplasm. The space between these concentric membranes is continuous with the lumen of the rough ER. The inner and outer nuclear membranes meet at specialized structures known as nuclear pores, which penetrate the nuclear envelope and provide a transport pathway between the cytoplasm and the nuclear interior (see Chapter 5). All RNA transcripts that are produced in the nucleus must pass through nuclear pores to be translated in the cytoplasm. Similarly, all the signaling molecules that influence nuclear function as well as all proteins of the nuclear interior (which are synthesized in the cytoplasm) enter the nucleus through nuclear pores.
Nuclear pores are selective in choosing the molecules that they allow to pass. Cytoplasmic proteins destined for the nuclear interior must be endowed with a nuclear localization sequence to gain entry. Several nuclear localization sequences have been characterized, and all seem to share common structural elements. For example, they all have short stretches of four to eight basic amino acids that can be located anywhere in the protein’s sequence. Evidence implies that the ability of these signals to mediate nuclear localization can be modulated by phosphorylation, which suggests that the entry of proteins into the nucleus may be under the control of the cell’s second-messenger systems.
The selectivity of the nuclear pore is surprising, considering its size. The outer diameter of the entire nuclear pore is ~100 nm, considerably larger than the proteins whose passage it controls. The nuclear pore’s specificity is provided by the nuclear pore complex (NPC), an intricate matrix of protein that is distributed in a highly organized octagonal array. In its resting state, the NPC forms an aqueous channel that is ~9 nm in diameter, restricting the movement of any protein larger than 60 kDa. However, when it is confronted with a protein bearing a nuclear localization signal or a messenger RNA (mRNA) transcript, the pore complex can dilate to many times this size. The mechanisms by which the pore’s permeability is regulated remain unknown. The NPC has a barrier that prevents the diffusion of intrinsic membrane proteins between the outer and inner membranes of the nuclear envelope. Thus, although the inner and outer nuclear membranes are continuous with one another at nuclear pores, their protein contents remain distinct.
Between mitoses, the chromosomal DNA is present in the nucleus as densely packed heterochromatin and more loosely arrayed euchromatin. Chromatin is a complex between DNA and numerous DNA-binding proteins, which organize the chromosome into a chain of tightly folded DNA-protein assemblies called nucleosomes (see Chapter 4). Interspersed within the nucleoplasm are round, dense nucleoli, where the transcription of ribosomal RNA and the assembly of ribosomal subunits appear to occur.
The interior surface of the inner nuclear membrane is apposed to a fibrillar protein skeleton referred to as the nuclear lamina. This meshwork, composed of proteins known as lamins, is presumably involved in providing structural support to the nuclear envelope. The nuclear lamina may also play a role in orchestrating nuclear reassembly. During mitosis, the nuclear envelope breaks down into small vesicles, and the contents of the nucleoplasm mix with the cytoplasm. After mitosis, these vesicles fuse with one another to regenerate the double-walled nuclear membrane. The means by which these vesicles find one another and assemble correctly is the subject of intense study. Similarly, the mechanisms involved in maintaining the compositional discreteness of the inner and outer membranes during vesiculation and reassembly have yet to be determined. After reconstitution of the nuclear envelope, the proteins of the nucleoplasm are re-imported from the cytoplasm through the nuclear pores by virtue of their nuclear localization sequences.
In the course of normal daily living, cells accumulate waste. Organelles become damaged and dysfunctional. Proteins denature and aggregate. New materials are constantly being brought into the cells from the extracellular environment through the process of endocytosis (discussed later). In specialized cells of the immune system, such as macrophages, the collection of foreign materials (in the form of pathogens) from the extracellular milieu is the cellular raison d’être. If this material were allowed to accumulate indefinitely, it would ultimately fill the cell and essentially choke it to death. Clearly, cells must have mechanisms for disposing of this waste material.
The lysosome is the cell’s trash incinerator. It is filled with a broad assortment of degradative enzymes that can break down most forms of cellular debris. Proton pumps embedded within the lysosome’s limiting membrane ensure that this space is an extremely acidic environment, which aids in protein hydrolysis. A rare group of inherited disorders, called lysosomal storage diseases (see the box on page 43 about this topic), result from the deficiency of lysosomal enzymes that are involved in the degradation of a variety of substances.
The lysosomal membrane is specially adapted to resist digestion by the enzymes and the acid that it encapsulates, thus ensuring that the harsh conditions necessary for efficient degradation are effectively contained. Loss of lysosomal membrane integrity may underlie some clinically important inflammatory conditions, such as gout.
Material that has been internalized from the cell exterior by endocytosis is surrounded by the membrane of an endocytic vesicle. To deliver this material to the lysosome, the membranes of the endocytic vesicles fuse with the lysosomal membrane and discharge their cargo into the lysosomal milieu.
Intracellular structures that are destined for degradation, such as fragments of organelles, are engulfed by the lysosome in a process called autophagy. Autophagy results in the formation of membrane-enclosed structures within the lysosomal lumen; hence, the lysosome is often referred to as a multivesicular body.
Oxygen-dependent ATP production—or oxidative phosphorylation—occurs in the mitochondrion. Like the nucleus, the mitochondrion (Fig. 2-10) is a double-membrane structure. The inner mitochondrial membrane contains the proteins that constitute the electron transport chain, which generates pH and voltage gradients across this membrane. According to the “chemiosmotic” model (see Chapter 5), the inner membrane uses the energy in these gradients to generate ATP from adenosine diphosphate (ADP) and inorganic phosphate.
The mitochondrion maintains and replicates its own genome. This circular DNA strand encodes mitochondrial transfer RNAs (tRNAs) and (in humans) 13 mitochondrial proteins. Several copies of the mitochondrial genome are located in the inner mitochondrial matrix, which also has all of the machinery necessary to transcribe and to translate this DNA, including ribosomes. Whereas the proteins encoded in mitochondrial DNA contribute to the structure and function of the mitochondrion, they account for a relatively small fraction of total mitochondrial protein. Most mitochondrial proteins are specified by nuclear DNA and are synthesized on cytoplasmic ribosomes.
The two mitochondrial membranes enclose two distinct compartments: the intermembrane space and the inner mitochondrial matrix space. The intermembrane space lies between the two membranes; the inner mitochondrial matrix space is completely enclosed by the inner mitochondrial membrane. These compartments have completely different complements of soluble proteins, and the two membranes themselves have extremely different proteins.
In addition to its role in energy metabolism, the mitochondrion also serves as a reservoir for intracellular Ca2+. It is not clear whether—under physiological conditions—the mitochondrion releases Ca2+ from this reservoir. The mitochondrial Ca2+ stores are released as a consequence of energy starvation, which leads to cell injury and death. Finally, the mitochondrion plays a central role in the process called apoptosis, or programmed cell death (see Chapter 62). Certain external or internal signals can induce the cell to initiate a signaling cascade that leads ultimately to the activation of enzymes that bring about the cell’s demise. One of the pathways that initiates this highly ordered form of cellular suicide depends on the participation of the mitochondrion. Apoptosis plays an extremely important role during tissue development and is also involved in the body’s mechanisms for identifying and destroying cancer cells.
Our discussion thus far has focused almost exclusively on the cell’s membranous elements. We have treated the cytoplasm as if it were a homogeneous solution in which the organelles and vesicles carry out their functions while floating about unimpeded and at random. Rather, the cytoplasm is enormously complex with an intricate local structure and the capacity for locomotion.
The cytoplasmic cytoskeleton is composed of protein filaments that radiate throughout the cell, serving as the beams, struts, and stays that determine cell shape and resilience. On the basis of their appearance in the electron microscope, these filaments were initially divided into several classes (Table 2-2): thick, thin, and intermediate filaments as well as microtubules. Subsequent biochemical analysis has revealed that each of these varieties is composed of distinct polypeptides and differs with respect to its formation, stability, and biological function.
Table 2-2 Components of the Cytoskeleton
|
Subunits |
Diameter (nm) |
Intermediate filaments |
Tetramer of two coiled dimers |
8-10 |
Microtubules |
Heterodimers of α and β tubulin form long protofilaments, 5 nm in diameter |
25 |
Thin filaments |
Globular or G-actin, 5 nm in diameter, arranged in a double helix to form fibrous or F-actin |
5-8 |
Thick filaments |
Assembly of myosin molecules |
10 |
Intermediate filaments are so named because their 8-to 10-nm diameters, as measured in the electron microscope, are intermediate between those of the actin thin filaments and the myosin thick filaments. As with all of the cytoskeletal filaments that we will discuss, intermediate filaments are polymers that are assembled from individual protein subunits. There is a very large variety of biochemically distinct subunit proteins that are all structurally related to one another and that derive from a single gene family. The expression of these subunit polypeptides can be cell type specific or restricted to specific regions within a cell. Thus, vimentin is found in cells that are derived from mesenchyme, and the closely related glial fibrillary acidic protein is expressed exclusively in glial cells (see Chapter 11). Neurofilament proteins are present in neuronal processes. The keratins are present in epithelial cells as well as in certain epithelially derived structures. The nuclear lamins that form the structural scaffolding of the nuclear envelope are also members of the intermediate filament family.
Intermediate filament monomers are themselves fibrillar in structure. They assemble to form long, intercoiled dimers that in turn assemble side-to-side to form the tetrameric subunits. Finally, these tetrameric subunits pack together, end-to-end and side-to-side, to form intermediate filaments. Filament assembly can be regulated by the cell and in some cases appears to be governed by phosphorylation of the subunit polypeptides. Intermediate filaments appear to radiate from and to reinforce areas of a cell that are subject to tensile stress. They emanate from the adhesion plaques that attach cells to their substrata. In epithelial cells, they insert at the desmosomal junctions that attach neighboring cells to one another. The toughness and resilience of the meshworks formed by these filaments is perhaps best illustrated by the keratins, the primary constituents of nails, hair, and the outer layers of skin.
Microtubules are polymers formed from heterodimers of the proteins α and β tubulin (Fig. 2-11A). These heterodimers assemble head to tail, creating a circumferential wall of a microtubule, which surrounds an empty lumen. Because the tubulin heterodimers assemble with a specific orientation, microtubules are polar structures, and their ends manifest distinct biochemical properties. At one tip of the tubule, designated the plus end, tubulin heterodimers can be added to the growing polymer at three times the rate that this process occurs at the opposite minus end. The relative rates of microtubule growth and depolymerization are controlled in part by an enzymatic activity that is inherent in the tubulin dimer. Tubulin dimers bind to GTP, and in this GTP-bound state they associate more tightly with the growing ends of microtubules. Once a tubulin dimer becomes part of the microtubule, it hydrolyzes the GTP to guanosine diphosphate (GDP), which lowers the binding affinity of the dimer for the tubule and helps hasten disassembly. Consequently, the microtubules can undergo rapid rounds of growth and shrinkage, a behavior termed dynamic instability. Various cytosolic proteins can bind to the ends of microtubules and serve as caps that prevent assembly and disassembly and thus stabilize the structures of the microtubules. A large and diverse family of microtubule-associated proteins appears to modulate not only the stability of the tubules but also their capacity to interact with other intracellular components.
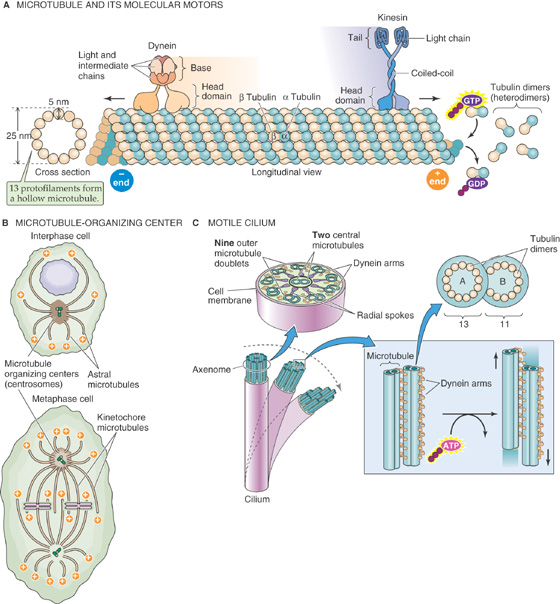
Figure 2-11 Microtubules. A, Heterodimers of α and β tubulin form long protofilaments, 13 of which surround the hollow core of a microtubule. The microtubule grows more rapidly at its plus end. The molecular motor dynein moves along the microtubule in the plus-to-minus direction, whereas the molecular motor kinesin moves in the opposite direction. ATP is the fuel for each of these motors. B, The microtubules originate from a microtubule-organizing center or centrosome, which generally consists of two centrioles (green cylinders). C, A motile cilium can actively bend as its microtubules slide past each other. The molecular motor dynein produces this motion, fueled by ATP.
In most cells, all of the microtubules originate from the microtubule-organizing center or centrosome. This structure generally consists of two centrioles, each of which is a small (~0.5 μm) assembly of nine triplet microtubules that are arranged obliquely along the wall of a cylinder (see upper portion of Fig. 2-11B). The two centrioles in a centrosome are oriented at right angles to one another. The minus ends of all of a cell’s microtubules are associated with proteins that surround the centrosome, whereas the rapidly growing plus ends radiate throughout the cytoplasm in a star-like arrangement (“astral” microtubules).
Microtubules participate in a multitude of cellular functions and structures. For example, microtubules project down the axon of neurons. Microtubules also provide the framework for the lacy membranes of the ER and Golgi complex. Disruption of microtubules causes these organelles to undergo dramatic morphological rearrangements and vesicularization. Microtubules also play a central role in cell division. Early in mitosis, the centrioles that make up the centrosomes replicate, forming two centrosomes at opposite poles of the dividing nucleus. Emanating from these centrosomes are the microtubules that form the spindle fibers, which in turn align the chromosomes (see lower portion of Fig. 2-11B). Their coordinated growth and dissolution at either side of the chromosomes may provide the force for separating the genetic material during the anaphase of mitosis. A pair of centrioles remains with each daughter cell.
The architectural and mechanical capacities of microtubules are perhaps best illustrated by their role in motility. An electron microscopic cross section of a cilium demonstrates the elegance, symmetry, and intricacy of this structure (Fig. 2-11C). Every cilium arises out of its own basal body, which is essentially a centriole that is situated at the ciliary root. Cilia are found on the surfaces of many types of epithelial cells, including those that line the larger pulmonary airways (see Chapter 26). Their oar-like beating motions help propel foreign bodies and pathogens toward their ultimate expulsion at the pharynx. At the center of a cilium is a structure called the axoneme, which is composed of a precisely defined 9 + 2 array of microtubules. Each of the 9 (which are also called outer tubules) consists of a complete microtubule with 13 tubulin monomers in cross section (the A tubule) to which is fused an incomplete microtubule with 11 tubulin monomers in cross section (the B tubule). Each of the 2, which lie at the core of the cilium, is a complete microtubule. This entire 9 + 2 structure runs the entire length of the cilium. The same array forms the core of a flagellum, the serpentine motions of which propel sperm cells (see Chapter 56).
Radial spokes connect the outer tubules to the central pair, and outer tubules attach to their neighbors by two types of linkages. One is composed of the protein dynein, which acts as a molecular motor to power ciliary and flagellar motions. Dynein is an ATPase that converts the energy released through ATP hydrolysis into a conformational change that produces a bending motion. Because dynein attached to one outer tubule interacts with a neighboring outer tubule, this bending of the dynein molecule causes the adjacent outer tubules to slide past one another. It is this sliding-filament motion that gives rise to the coordinated movements of the entire structure. To some extent, this coordination is accomplished through the action of the second linkage protein, called nexin. The nexin arms restrict the extent to which neighboring outer tubules can move with respect to each other and thus prevent the dynein motor from driving the dissolution of the entire complex.
The utility of the dynein motor protein is not restricted to its function in cilia and flagella. Cytoplasmic dynein, which is a close relative of the motor molecule found in cilia, and a second motor protein called kinesin provide the force necessary to move membrane-bound organelles through the cytoplasm along microtubular tracks (Fig. 2-11A). The ability of vesicular organelles to move rapidly along microtubules was first noted in neurons, in which vesicles carrying newly synthesized proteins must be transported over extremely long distances from the cell body to the axon tip. Rather than trust this critical process to the vagaries of slow, nondirected diffusion, the neuron makes use of the kinesin motor, which links a vesicle to a microtubule. Kinesin hydrolyzes ATP and, like dynein, converts this energy into mechanical transitions that cause it to “walk” along the microtubule. Kinesin will move only along microtubules and thereby transport vesicles in the minus-to-plus direction. Thus, in neurons, kinesin-bound vesicles move from the microtubular minus ends, originating at the centrosome in the cell body, toward the plus ends in the axons. This direction of motion is referred to as anterograde fast axonal transport. Cytoplasmic dynein moves in the opposite plus-to-minus (or retrograde) direction.
The motor-driven movement of cellular organelles along microtubular tracks is not unique to neurons. This process, involving both kinesin and cytoplasmic dynein, appears to occur in almost every cell and may control the majority of subcellular vesicular traffic.
Thin filaments, also called microfilaments, are 5 to 8 nm in diameter. They are helical polymers composed of a single polypeptide called globular actin or G-actin. Thin filaments are functionally similar to microtubules in two respects: (1) the actin polymers are polar and grow at different rates at their two ends, and (2) actin binds and then hydrolyzes a nucleotide. However, whereas tubulin binds GTP and then hydrolyzes it to GDP, actin binds ATP and then hydrolyzes it to ADP. After G-actin binds ATP, it may interact with another ATP-bound monomer to form an unstable dimer (Fig. 2-12A). Adding a third ATP-bound monomer, however, yields a stable trimer that serves as a nucleus for assembly of the polymer of fibrous actin or F-actin. Once it is part of F-actin, the actin monomer hydrolyzes its bound ATP, retaining the ADP and releasing the inorganic phosphate. The ADP-bound actin monomer is more likely to disengage itself from its neighbors, just as GDP-bound tubulin dimers are more likely to disassemble from tubulin. Even though the length of the F-actin filament may remain more or less constant, the polymer may continually grow at its plus end but disassemble at its minus end (Fig. 2-12B). This “treadmilling” requires the continuous input of energy (i.e., hydrolysis of ATP) and illustrates the unique dynamic properties of actin filament elongation and disassembly.
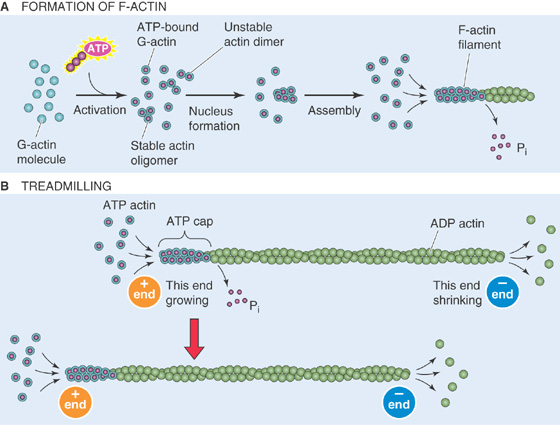
Figure 2-12 Thin filaments. A, Single molecules of G-actin form F-actin filaments. B, F-actin can grow at the plus end while shrinking at the minus end, with no change in length.
Thick filaments are composed of dimers of a remarkable force-generating protein called myosin. All myosin molecules have helical tails and globular head groups that hydrolyze ATP and act as motors to move along an actin filament. The energy liberated by ATP hydrolysis is invested in bending the myosin molecule around a pivot point called the hinge region, which marks the junction between the globular and tail regions. By means of this bending, myosin, like the dynein and kinesin that interact with microtubules, acts as a molecular motor that converts chemical into mechanical energy.
In muscle, the myosin molecules are in the myosin II subfamily and exist as dimers with their long tails intertwined (Fig. 2-13A). In muscle, each of the two myosin II heads binds two additional protein subunits that are referred to as myosin light chains. Non-muscle cells, in addition to myosin II, may have a variety of other, smaller myosin molecules. These other myosins, the most widely studied of which is myosin I, have shorter tails and, at least in some cases, act as molecular motors that move vesicles along actin filaments.
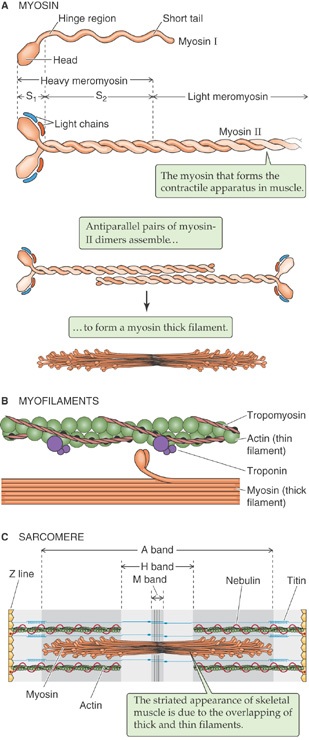
Figure 2-13 Thick filaments. A, Myosin I is one of a large number of widely distributed myosins that have short tails. Myosin II is the myosin that participates in muscle contraction. B, The pivoting action of the myosin head, fueled by ATP, moves the thick filament past the thin filament. C, In skeletal and cardiac muscle, the sarcomere is the fundamental contractile unit.
In muscle, the myosin II dimers stack as antiparallel arrays to form a bipolar structure with a bare central region that contains only tails (Fig. 2-13A). The ends of the thick filament contain the heads that bend toward the filament’s central region. The pivoting action of the myosin head groups drags the neighboring thin filament (Fig. 2-13B), which includes other molecules besides actin. This sliding-filament phenomenon underlies muscle contraction and force generation (Fig. 2-13C).
Actin as well as an ever-growing list of myosin isoforms is present in essentially every cell type. The functions of these proteins are easy to imagine in some cases and are less obvious in many others. Many cells, including all of the fibroblast-like cells, possess actin filaments that are arranged in stress fibers. These linear arrays of fibers interconnect adhesion plaques to one another and to interior structures in the cell. They orient themselves along lines of tension and can, in turn, exert contractile force on the substratum that underlies the cell. Stress fiber contractions may be involved in the macroscopic contractions that are associated with wound healing. Frequently, actin filaments in non-muscle cells are held together in bundles by cross-linking proteins. Numerous classes of cross-linking proteins have been identified, several of which can respond to physiological changes by either stabilizing or severing filaments and filament bundles.
In motile cells, such as fibroblasts and macrophages, arrays of actin-myosin filaments are responsible for cell locomotion. A Ca2+-stimulated myosin light chain kinase regulates the assembly of myosin and actin filaments and thus governs the generation of contractile force. The precise mechanism by which these fibers cooperate in causing the cell to crawl along a substrate remains poorly understood. (See Note: Cell Locomotion)
In contrast to fibroblasts and circulating cells of the immune system, cells such as neurons and epithelial cells generally do not move much after their differentiation is complete. Despite this lack of movement, however, these cells are equipped with remarkably intricate actin and myosin filament networks. In some cases, these cytoskeletal elements permit the cell to extend processes to distant locations. This is the case in neurons, in which the growth and migration of axons during development or regeneration of the nervous system bear a striking morphological resemblance to the crawling of free-living amoebae. The tip of a growing axon, known as a growth cone, is richly endowed with contractile fibers and is capable of the same types of crawling motions that characterize motile cells.
In epithelial cells, the role of the actin-myosin cytoskeleton is somewhat less obvious but still important to normal physiological function. The microvilli at the apical surfaces of many epithelial cell types (e.g., those that line the renal proximal tubule and the small intestine) are supported by an intricate scaffolding of actin filaments that form their cores (Fig. 2-14). This bundle of actin fibers is held together and anchored to the overlying plasma membrane by a variety of cross-linking proteins, including various myosin isoforms. The roots of the microvillar actin filament bundles emerge from the bases of the microvilli into a dense meshwork of cytoskeletal filaments known as the terminal web. Included among the components of the terminal web network are fodrin (the nonerythroid homologue of spectrin) and myosin. It remains unclear whether the myosin in the terminal web is present simply to interconnect the actin filaments of neighboring microvilli or if this actin-myosin complex is capable of generating contractile movements.
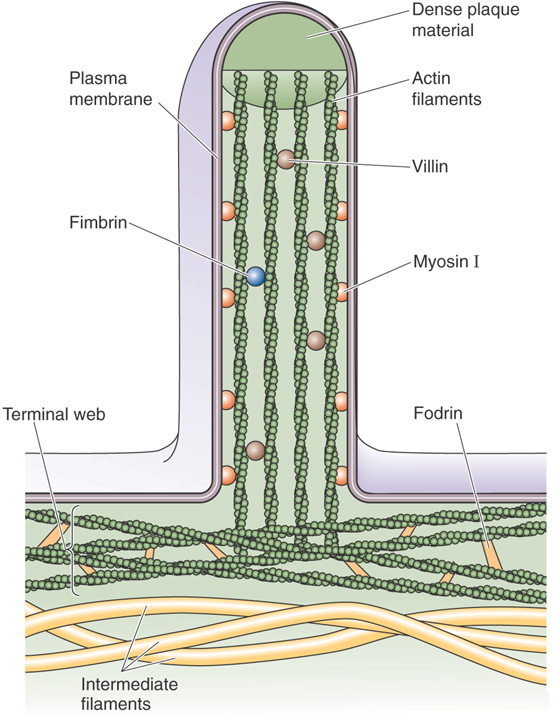
Figure 2-14 Actin filaments at the brush border of an epithelial cell.
Actin and myosin filaments also form an adhesion belt that encircles the cytoplasmic surface of the epithelial plasma membrane at the level of the tight junctions that interconnect neighboring cells. These adhesion belts are apparently capable of contraction and thus cause epithelial cells that normally form a continuous sheet to pull away from one another, temporarily loosening tight junctions and creating direct passages that connect the luminal space to the extracellular fluid compartment.
Actin and myosin also participate in processes common to most if not all cell types. The process of cytokinesis, in which the cytoplasm of a dividing cell physically separates into two daughter cells, is driven by actin and myosin filaments. Beneath the cleavage furrow that forms in the membrane of the dividing cell is a contractile ring of actin and myosin filaments. Contraction of this ring deepens the cleavage furrow; this invagination ultimately severs the cell and produces the two progeny. (See Note: Other Roles of Actin and Myosin)
Transmembrane proteins are composed of hydrophobic domains that are embedded within the phospholipid bilayer and hydrophilic domains that are exposed at the intracellular and extracellular surfaces. These proteins do not “flip” through the membrane. How, then, do intrinsic membrane proteins overcome the enormous energetic barriers that should logically prevent them from getting inserted into the membrane in the first place?
The cell has developed several schemes to address this problem. Mammalian cells have at least three different membrane insertion pathways, each associated with specific organelles. The first two are mechanisms for inserting membrane proteins into peroxisomes and mitochondria. The third mechanism inserts membrane proteins destined for delivery to the plasma membrane and to the membranes of organelles (the endomembranous system) other than the peroxisome and mitochondrion. This same mechanism is involved in the biogenesis of essentially all proteins that mammalian cells secrete and is the focus of the following discussion.
The critical work in this field centered on studies of the rough ER. The membrane of the rough ER is notable for the presence of numerous ribosomes that are bound to its cytosol-facing surface. Whereas all nucleated mammalian cells have at least some rough ER, cells that produce large quantities of secretory proteins—such as the exocrine cells of the pancreas, which function as factories for digestive enzymes (see Chapter 43)—are endowed with an abundance of rough ER. Roughly half of the cytoplasmic space in an exocrine pancreatic acinar cell is occupied by rough ER.
In early experiments exploring cell fractionation techniques, membranes that were derived from the rough ER were separated from the other membranous and cytoplasmic components of pancreatic acinar cells. The mRNAs associated with rough ER membranes were isolated and the proteins they encoded were synthesized by in vitro translation. Analysis of the resultant polypeptides revealed that they included the cell’s entire repertoire of secretory proteins. It is now appreciated that the mRNA associated with the ER also encodes the cell’s entire repertoire of membrane proteins, with the exception of those destined for either the peroxisome or the mitochondrion. When the same experiment was performed with mRNAs isolated from ribosomes that are freely distributed throughout the cytoplasm, the products were not secretory proteins but rather the soluble cytosolic proteins. Later work showed that the ribosomes bound to the ER are biochemically identical to and in equilibrium with those that are free in the cytosol. Therefore, a ribosome’s subcellular localization—that is, whether it is free in the cytosol or bound to the rough ER—is somehow dictated by the mRNA that the ribosome is currently translating. A ribosome that is involved in assembling a secretory or membrane protein will associate with the membrane of the rough ER, whereas the same ribosome will be free in the cytosol when it is producing cytosolic proteins. Clearly, some localization signal that resides in the mRNA or in the protein that is being synthesized must tell the ribosome what kind of protein is being produced and where in the cell that production should occur.
The nature of this signal was discovered in 1972 during studies of the biosynthesis of immunoglobulin light chains. Light chains synthesized in vitro, in the absence of rough ER membranes, have a 15–amino acid extension at their amino terminus that is absent from the same light chains synthesized and secreted in vivo by B lymphocytes. Similar amino-terminal extensions are present on most secretory or membrane proteins but never with the soluble proteins of the cytosol. Although they vary in length and composition, these extensions are present on most acids that are interspersed with occasional basic residues. These signal sequences, as they have come to be known, serve as the localization devices discussed earlier. As it emerges from a ribosome and is freely floating in the cytosol, the signal sequence of a nascent protein (Fig. 2-15, stage 1) targets the ribosome-mRNA complex to the surface of the rough ER where the protein’s biogenesis will be completed. Ribosome-mRNA complexes that lack a signal sequence complete the translation of the mRNA—which encodes neither secretory nor membrane proteins—without attaching to the rough ER. For his work on signal sequences, Günter Blobel received the 1999 Nobel Prize for Physiology or Medicine. (See Note: Günter Blobel)
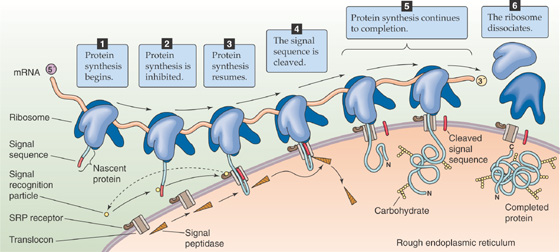
Figure 2-15 Synthesis and translocation of a secretory protein.
Why does the cell bother to segregate the synthesis of different protein populations to different cellular locales? Proteins that are destined either to reside in a membrane or to be secreted are inserted into or across the membrane of the rough ER at the same time that they are translated; this is called cotranslational translocation. As the nascent polypeptide chain emerges from the ribosome, it traverses the rough ER membrane and ultimately appears at the ER’s luminal face. There, an enzyme cleaves the amino-terminal signal sequence while the protein is still being translocated. This is why proteins that are synthesized in vitro in the absence of membranes are longer than the same proteins that are produced by intact cells.
The information embodied within a signal sequence explains how a nascent protein can direct a cell to complete that protein’s translation at the time of translocation in the rough ER. However, the signal sequence by itself is not sufficient. Two critical pieces of targeting machinery are also necessary to direct the ribosome and its attached nascent peptide to the ER. The first is a ribonucleoprotein complex called the signal recognition particle (SRP), which binds to the signal sequence on the nascent peptide (Fig. 2-15, stage 2). The SRP is composed of seven distinct polypeptides and a short strand of RNA. When the SRP binds to a nascent chain, it also binds a GTP molecule. The second vital piece of targeting machinery is a transmembrane component of the rough ER, the SRP receptor, also called the docking protein. Interaction between a signal sequence and the SRP, and subsequently between the SRP–nascent peptide–ribosome complex and the docking protein, directs the nascent chain to the rough ER’s translocation apparatus.
Because the membrane of the rough ER has a finite number of docking sites, the cell must coordinate the synthesis of secretory and membrane proteins with the availability of docking sites. If all docking sites were occupied, and if the synthesis of nascent secretory and membrane proteins were allowed to continue unabated, these nascent peptides would be synthesized entirely in the cytoplasm on free ribosomes. As a consequence, these newly synthesized proteins would never arrive at their proper destination. The SRP serves as a regulatory system that matches the rate of secretory and membrane protein syntheses to the number of unoccupied translocation sites. By associating with a nascent signal sequence, the SRP causes the ribosome to halt further protein synthesis (Fig. 2-15, stage 2). This state of translation arrest persists until the SRP–nascent peptide–ribosome complex finds an unoccupied docking protein with which to interact. Thus, SRP prevents secretory and membrane proteins from being translated until their cotranslational translocation can be ensured. Because SRP interacts only with nascent chains that bear signal sequences, ribosomes that synthesize proteins destined for release into the cytosol never associate with SRP, and their translation is never arrested. Thus, SRP serves as a highly specific spatial and temporal sorting machine, guaranteeing the accurate and efficient targeting of secretory and membrane proteins.
How does the cell terminate the translation arrest of the SRP–nascent peptide–ribosome complex? When this complex interacts with a docking protein (Fig. 2-15, stage 3), one of the SRP’s subunits hydrolyzes the previously bound GTP, thereby releasing the SRP from a successfully targeted nascent peptide–ribosome complex. In this way, the docking protein informs the SRP that its mission has been accomplished and it can return to the cytosol to find another ribosome with a signal peptide. A second GTP hydrolysis step transfers the nascent peptide from the docking protein to the actual translocation tunnel complex. GTP hydrolysis is a common event and is involved in the transmission of numerous cellular messages (see Chapter 3). In this case, the two separate instances of GTP hydrolysis serve a quality-control function because the activation of the GTPase activity depends on the delivery of the nascent peptide to the appropriate component in the translocation apparatus.
Adjacent to the docking protein in the membrane of the rough ER is a protein translocator termed a translocon (Fig. 2-15, stage 3), which contains a tunnel through which the nascent protein will pass across the rough ER membrane. It appears that delivery of a nascent chain to the translocon causes the entrance of the translocator’s tunnel, which is normally closed, to open. This opening of the translocon also allows the flow of small ions. The electrical current carried by these ions can be measured by the patch-clamp technique (see Chapter 6). By “gating” the translocon so that it opens only when it is occupied by a nascent protein, the cell keeps the tunnel’s entrance closed when it is not in use. This gating prevents the Ca2+ stored in the ER from leaking into the cytoplasm.
Because the tunnel of the translocon is an aqueous pore, the nascent secretory or membrane protein does not come into contact with the hydrophobic core of the ER membrane’s lipid bilayer during cotranslational translocation. Thus, this tunnel allows hydrophilic proteins to cross the membrane. As translation and translocation continue and the nascent protein enters the lumen of the rough ER, an enzyme called signal peptidase cleaves the signal peptide, which remains in the membrane of the rough ER (Fig. 2-15, stage 4). Meanwhile, translation and translocation of the protein continue (Fig. 2-15, stage 5). In the case of secretory proteins (i.e., not membrane proteins), the peptide translocates completely through the membrane. The ribosome releases the complete protein into the lumen of the rough ER and then dissociates from the rough ER (Fig. 2-15, stage 6).
Unlike soluble proteins, nascent membrane proteins do not translocate completely through the membrane of the rough ER (Fig. 2-16A, stage 1). The current concept is that the hydrophobic amino acid residues that will ultimately become the transmembrane segment of a membrane protein also function as a stop-transfer sequence (Fig. 2-16A, stage 2). When a stop-transfer sequence emerges from a ribosome, it causes the translocon to disassemble, releasing the hydrophobic membrane-spanning segment into the hospitable environment of the rough ER membrane’s hydrophobic core (Fig. 2-16A, stage 3). In the meantime, the ribosomal machinery continues to translate the rest of the nascent protein. If the signal peptidase cleaves the amino terminus at this time, the end result is a protein with a single transmembrane segment, with the amino terminus in the lumen of the rough ER and the carboxyl terminus in the cytoplasm (Fig. 2-16A, stage 4).
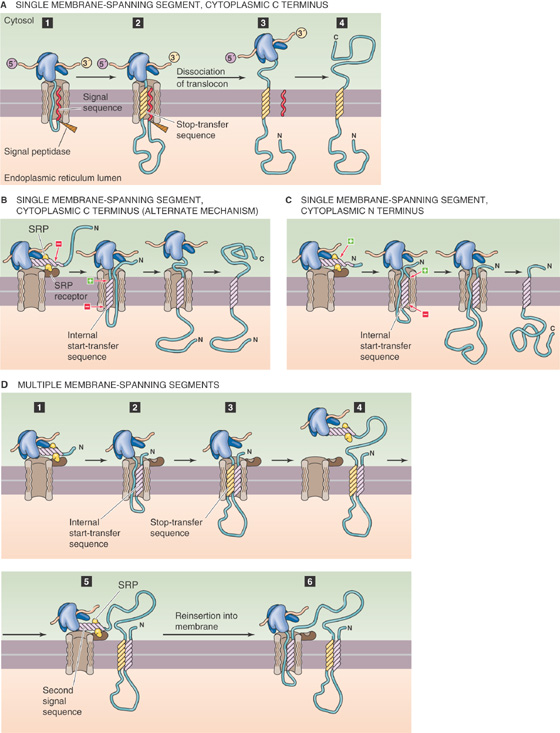
Figure 2-16 Synthesis of integral membrane proteins. A, Like a secreted protein, the membrane protein can have a cleavable signal sequence. In addition, it has a stop-transfer sequence that remains in the membrane as a membrane-spanning segment. B, The emerging protein lacks a signal sequence but instead has an internal start-transfer sequence, which is a bifunctional sequence that serves both as a signal sequence that binds signal recognition particles and as a hydrophobic membrane-spanning segment. In this example, the positively charged region flanking the internal start-transfer sequence is on the carboxyl-terminal end of the internal start-transfer sequence. Therefore, the C-terminal end is in the cytoplasm. C, The example is similar to that in B except that the positively charged region flanking the internal start-transfer sequence is on the amino-terminal end of the internal start-transfer sequence. D, The emerging peptide has alternating internal start-transfer and stop-transfer sequences.
There is another way of generating a protein with a single transmembrane segment. In this case, the protein lacks a signal sequence at the N terminus but instead has—somewhere in the middle of the nascent peptide—a bifunctional sequence that serves both as a signal sequence that binds SRP and as a hydrophobic membrane-spanning segment. This special sequence is called an internal start-transfer sequence. The SRP binds to the internal start-transfer sequence and brings the nascent protein to the rough ER, where the internal start-transfer sequence binds to the translocon in such a way that the more positively charged residues that flank the start-transfer sequence face the cytosol. Because these positively charged flanking residues can either precede or follow the hydrophobic residues of the internal start-transfer sequence, either the carboxyl (C) terminus or the N terminus can end up in the cytosol. If the more positively charged flanking residues are at the carboxyl-terminal end of the internal start-transfer sequence (Fig. 2-16B), the protein will be oriented with its carboxyl terminus in the cytosol. If the more positively charged flanking residues are at the amino-terminal end of the internal start-transfer sequence (Fig. 2-16C), the protein will be oriented with its amino terminus in the cytosol.
By alternating both stop-transfer sequences (Fig. 2-16A) and internal start-transfer sequences (Fig. 2-16B, C), the cell can fabricate membrane proteins that span the membrane more than once. Figure 2-16 shows how the cell could synthesize a multispanning protein with its N terminus in the cytosol. The process starts just as in Figure 2-16C, as the translation machinery binds to the rough ER (Fig. 2-16D, stage 1) and the protein’s first internal start-transfer sequence inserts into the translocon (Fig. 2-16D, stage 2). However, when the first stop-transfer sequence reaches the translocon (Fig. 2-16D, stage 3), the translocon disassembles, releasing the protein’s first two membrane-spanning segments into the membrane of the rough ER. Note that the first membrane-spanning segment is the internal start-transfer sequence and the second is the stop-transfer sequence. In the meantime, an SRP binds to the second internal start-transfer sequence (Fig. 2-16D, stage 4) and directs it to the rough ER (Fig. 2-16D, stage 5) so that cotranslational translocation can once again continue (Fig. 2-16D, stage 6). If there are no further stop-transfer sequences, we will end up with a protein with three membrane-spanning segments.
Several points from the preceding discussion deserve special emphasis. First, translocation through the ER membrane can occur only cotranslationally. If a secretory or membrane protein were synthesized completely on a cytoplasmic ribosome, it would be unable to interact with the translocation machinery and consequently would not be inserted across or into the bilayer. As discussed later, this is not true for the insertion of either peroxisomal or mitochondrial proteins. Second, once a signal sequence emerges from a ribosome, there is only a brief period during which it is competent to mediate the ribosome’s association with the ER and to initiate translocation. This time constraint is presumably due to the tendency of nascent polypeptide chains to begin to fold and acquire tertiary structure very soon after exiting the ribosome. This folding quickly buries hydrophobic residues of a signal sequence so that they cannot be recognized by the translocation machinery. Third, because the translocation channel appears to be fairly narrow, the nascent protein cannot begin to acquire tertiary structure until after it has exited at the ER’s luminal face. Thus, the peptide must enter the translocation tunnel as a thin thread immediately after emerging from the ribosome. These facts explain why translocation is cotranslational. In systems in which post-translational translocation occurs (e.g., peroxisomes and mitochondria), special adaptations keep the newly synthesized protein in an unfolded state until its translocation can be consummated.
Finally, because the protein cannot flip once it is in the membrane, the scheme just outlined results in proteins that are inserted into the rough ER membrane in their final or “mature” topology. The number and location of a membrane protein’s transmembrane segments, as well as its cytoplasmic and extracytoplasmic loops, are entirely determined during the course of its cotranslational insertion into the ER membrane. The order in which signal, internal start-transfer, and stop-transfer sequences appear in a membrane protein’s primary structure completely determines how that protein will be arrayed across whatever membrane it ultimately comes to occupy.
As a newly synthesized secretory or membrane protein exits the tunnel of the translocon and enters the lumen of the rough ER, it may undergo a series of post-translational modifications that will help it to acquire its mature conformation. The first alteration, as discussed earlier, is cleavage of the signal sequence (if present) and is accomplished very soon after the signal sequence has completed its translocation. Other covalent modifications that occur as translocation continues include glycosylation and formation of intramolecular disulfide bonds. Glycosylation refers here to the enzymatic, en bloc coupling of preassembled, branched oligosaccharide chains that contain 14 sugar molecules (Fig. 2-17A) to asparagine (Asn) residues that appear in the sequence Asn-X-Ser or Asn-X-Thr (X can be any amino acid except proline). These N-linked sugars (N is the single-letter amino acid code for asparagine) will go on to be extensively modified as the protein passes through other organellar compartments. The addition of sugar groups to proteins can serve numerous functions, which include increasing the protein’s stability and endowing it with specific antigenic, adhesive, or receptor properties.
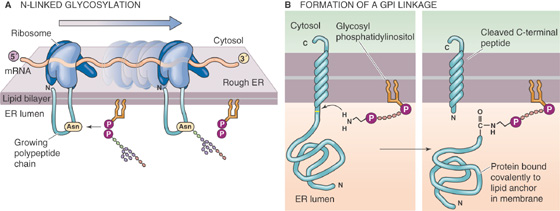
Figure 2-17 Post-translational modifications of integral membrane proteins. A, An enzyme in the ER lumen attaches a preassembled, branched, oligosaccharide chain to an asparagine (Asn or N) residue on the nascent protein. B, An enzyme in the ER lumen cleaves the protein and couples the protein’s new terminal carboxyl group to the terminal amino group on the GPI molecule.
Disulfide bond formation is catalyzed by protein disulfide isomerase, an enzyme that is retained in the ER lumen through noncovalent interactions with ER membrane proteins. Because the cytoplasm is a reducing environment, disulfide bonds can form only between proteins or protein domains that have been removed from the cytosolic compartment through translocation to the ER’s interior. Other, more specialized modifications also take place in the lumen of the rough ER. For example, the ER contains the enzymes responsible for the hydroxylation of the proline residues that are present in newly synthesized collagen chains.
The ER also catalyzes the formation of GPI linkages to membrane proteins (Fig. 2-17B). GPI-linked proteins are synthesized as transmembrane polypeptides, with a typical membrane-spanning region. Shortly after their translation, however, their lumen-facing domains are cleaved from the membrane-spanning segments and covalently transferred to the GPI phospholipid. They retain this structure and orientation throughout the remainder of their journey to the cell surface. A defect in the synthesis of GPI-linked proteins underlies the human disease paroxysmal nocturnal hematuria (see the box on this topic).
Perhaps the most important maturational process for a nascent chain emerging into the ER lumen is the acquisition of tertiary structure. The folding of a secretory or membrane protein is determined during and immediately after its cotranslational translocation. The progress of protein folding influences—and is influenced by—the addition of sugar residues and the formation of disulfide bridges. Proteins fold into conformations that minimize their overall free energies. Their extramembranous surfaces are composed of hydrophilic residues that interact easily with the aqueous solvent. Hydrophobic residues are hidden in internal globular domains where they can be effectively isolated from contact with water or charged molecules. Left to its own devices, a linear strand of denatured protein will spontaneously fold to form a structure that reflects these thermodynamic considerations. Thus, protein folding requires no catalysis and can occur without help from any cellular machinery. However, the cell is not content to allow protein folding to follow a random course and instead orchestrates the process through the actions of molecular chaperones.
The chaperones constitute a large class of ATP-hydrolyzing proteins that appear to participate in a wide variety of polypeptide-folding phenomena, including the initial folding of newly synthesized proteins as well as the refolding of proteins whose tertiary structures have been damaged by exposure to high temperature (i.e., heat shock) or other denaturing conditions. Chaperones bind to unfolded protein chains and stabilize them in an unfolded conformation, thus preventing them from spontaneously folding into what might be an energetically favorable but biologically useless arrangement. Using energy that is provided through ATP hydrolysis, the chaperones sequentially release domains of unfolded proteins and thus allow them to fold in an ordered fashion. Distinct subclasses of chaperones are present in several cell compartments, including the cytoplasm, the mitochondrion, and the lumen of the rough ER. Newly synthesized secretory and membrane proteins appear to interact with ER chaperones as they exit from the tunnel of the translocon and subsequently disengage from the chaperones to assume their mature tertiary structure.
The acquisition of tertiary structure is followed quickly by the acquisition of quaternary structure. As noted earlier in this chapter, many membrane proteins assemble into oligomeric complexes in which several identical or distinct polypeptides interact with one another to form a macromolecular structure. Assembly of these multimers occurs in the ER. It is unknown whether the oligomeric assembly process occurs entirely spontaneously or if, like folding, it is orchestrated by specialized cellular mechanisms. Cells clearly go to great trouble to ensure that proteins inserted into or across their ER membranes are appropriately folded and oligomerized before allowing them to continue with their postsynthetic processing. As discussed later, proteins destined for secretion from the cell or for residence in the cell membrane or other organellar membranes depart the ER for further processing in the membranous stacks of the Golgi complex. This departure is entirely contingent on successful completion of the protein folding and assembly operations.
Paroxysmal Nocturnal Hematuria
The list of proteins embedded in the plasma membrane through a GPI linkage is remarkably long and ever-growing. In red blood cells, the inventory of GPI-linked proteins includes a pair of polypeptides, decay-accelerating factor (DAF) and CD59, which help protect the erythrocytes from being accidentally injured by constituents of the immune system. One of the mechanisms that the immune system uses to rid the body of invading bacteria involves the activation of the complement cascade. Complement is a complex collection of proteins that circulate in the blood plasma. The complement system recognizes antibodies that are bound to the surface of a bacterium or polysaccharides in the bacterial membrane. This recognition initiates a cascade of enzymatic cleavages that results in the assembly of a subset of complement proteins to form the membrane attack complex, which inserts itself into the membrane of the target organism and forms a large pore that allows water to rush in (see Chapter 5). The target bacterium swells and undergoes osmotic lysis. Unfortunately, the complement system’s lethal efficiency is not matched by its capacity to discriminate between genuine targets and normal host cells. Consequently, almost every cell type in the body is equipped with surface proteins that guard against a misdirected complement attack.
DAF and CD59 are two such proteins that interfere with distinct steps in the complement activation pathway. Because GPI linkages couple both proteins to the membrane, any dysfunction of the enzymes that participate in the transfer of GPI-linked proteins from their transmembrane precursors to their GPI tails in the ER would interfere with the delivery of DAF and CD59 to their sites of functional residence at the cell surface. One of the proteins that participates in the synthesis of the GPI anchor is a sugar transferase encoded by the phosphatidylinositol glycan class A (PIG-A) gene. This gene is located on the X chromosome. Because every cell has only one working copy of the X chromosome (although female cells are genetically XX, one of the two X chromosomes is inactivated in every cell), if a spontaneous mutation occurs in the PIG-A gene in a particular cell, that cell and all of its progeny will lose the ability to synthesize GPI-linked proteins.
In paroxysmal nocturnal hemoglobinuria (i.e., hemoglobin appearing in the urine at night, with a sharp onset), a spontaneous mutation occurs in the PIG-A gene in just one of the many precursor cells that give rise to erythrocytes. All of the erythrocytes that arise from this particular precursor, therefore, are deficient in GPI-linked protein synthesis. Consequently, these cells lack DAF and CD59 expression and are susceptible to complement attack and lysis. For reasons that are largely unknown, the complement system is somewhat more active during sleep, so the hemolysis (lysis of erythrocytes) occurs more frequently at night in these patients. Some of the hemoglobin released by this lysis is excreted in the urine.
Because the PIG-A gene product is required for the synthesis of all GPI-linked proteins, the plasma membranes of affected red blood cells in patients with paroxysmal nocturnal hemoglobinuria are missing a number of different proteins that are found in the surface membranes of their normal counterparts. It is the lack of DAF and CD59, however, that renders the cells vulnerable to complement-mediated killing and that creates the symptoms of the disease. Paroxysmal nocturnal hemoglobinuria is an uncommon disease. Because it is the result of an acquired mutation, it is much more likely to occur in people of middle age rather than in children. Patients with paroxysmal nocturnal hemoglobinuria are likely to become anemic and can suffer life-threatening disorders of clotting and bone marrow function. It is a chronic condition, however, and more than half of patients survive at least 15 years after diagnosis.
Misfolded or unassembled proteins are retained in the ER and ultimately degraded. The ER chaperone proteins play a critical role both in identifying proteins with incorrect tertiary or quaternary structures and in actively preventing their egress to the Golgi complex. Proteins that have not folded or assembled correctly are destroyed through a process known as ERAD (endoplasmic reticulum–associated degradation). The sequential, covalent addition of ubiquitin monomers results in the formation of a branched-chain ubiquitin polymer that marks these proteins for destruction. Ubiquitin is a small protein of 76 amino acid residues. The process known as retrotranslocation removes ubiquitin-tagged proteins from the ER membrane, and a large cytoplasmic complex of proteolytic enzymes—the proteosome—degrades the ubiquitinated proteins.
The rough ER is the common point of origin for the cell’s secretory and membrane proteins. Most of these proteins are not retained in the rough ER but depart for distribution to their sites of ultimate functional residence throughout the cell. As is true for their arrival in the rough ER, the departure of these proteins is a highly organized and regimented affair. In fact, the rough ER is the first station along the secretory pathway, which is the route followed (at least in part) by all secretory and membrane proteins as they undergo their post-translational modifications (Fig. 2-18).
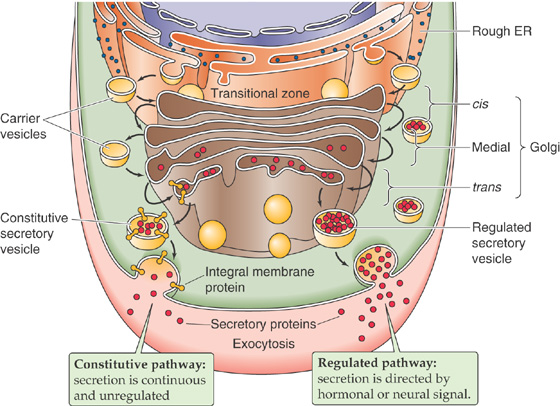
Figure 2-18 The secretory pathway. After their synthesis in the rough ER, secretory and membrane proteins destined for the plasma membrane move through the Golgi stacks and secretory vesicles. In the constitutive pathway, vesicles fuse spontaneously with the plasma membrane. In the regulated pathway, the vesicles fuse only when triggered by a signal such as a hormone.
The elucidation of the secretory pathway occurred in the 1960s, mainly in the laboratory of George Palade. For his contribution, Palade was awarded the 1975 Nobel Prize in Physiology or Medicine. This work also exploited the unique properties of pancreatic acinar cells to illuminate the central themes of secretory protein biogenesis. Because ~95% of the protein that is synthesized by pancreatic acinar cells are digestive enzymes destined for secretion (see Chapter 43), when these cells are fed radioactively labeled amino acids, the majority of these tracer molecules are incorporated into secretory polypeptides. Within a few minutes after the tracer is added, most of the label is associated with a specialized subregion of the rough ER. Known as transitional zones, these membranous saccules are ribosome studded on one surface and smooth at the opposite face (Fig. 2-18). The smooth side is directly apposed to one pole of the pancake-like membrane stacks (or cisternae) of the Golgi complex. Smooth-surfaced carrier vesicles crowd the narrow moat of cytoplasm that separates the transitional zone from the Golgi. These vesicles “pinch off” from the transitional zone and fuse with a Golgi stack. From this first or cis-Golgi stack, carrier vesicles ferry the newly synthesized proteins sequentially and vectorially through each Golgi stack, ultimately delivering them to the trans-most saccule of the Golgi. Finally, the newly synthesized secretory proteins appear in secretory vesicles (also called secretory granules in many tissues). (See Note: George Palade)
The journey from the rough ER to the secretory vesicle takes ~45 minutes in pancreatic acinar cells and requires the expenditure of metabolic energy. Each nucleated eukaryotic cell possesses a secretory pathway that shares this same general outline, although the specific features reflect the cell’s particular function. The secretory pathway of the pancreatic acinar cell, for example, is specially adapted to accommodate the controlled secretion of protein by the so-called regulated pathway. Instead of being released from the cell continuously as they are produced, newly synthesized secretory proteins are held in specialized secretory vesicles that serve as an intracellular storage depot. This type of storage occurs in several cells, including those of endocrine and exocrine secretory tissues, and neurons. When the cells receive the requisite message, the storage vesicles fuse with the plasma membrane, sometimes at a specialized structure called a porosome, in a process known as exocytosis. The vesicles then dump their contents into the extracellular space. In the case of the pancreatic acinar cells, the enzymes are secreted into the pancreatic ductules and then make their way to the site of digestion in the duodenum (see Chapter 43). (See Note: The Porosome)
Most cell types, however, deliver newly synthesized secretory and membrane proteins to the cell surface in a continuous and unregulated fashion, which is referred to as the constitutive pathway. Specialized cells that have the capacity for regulated delivery also send an important subset of their secretory and membrane protein synthetic products to the cell surface constitutively. The regulated and constitutive secretory pathways are identical except for the final station of the Golgi complex. At this point, the “regulated” proteins divert to the specialized secretory vesicles described in the previous paragraph. The “constitutive” proteins, at the trans-most cisterna of the Golgi complex, sort into other secretory vesicles, which move directly to the cell surface. There, the constitutive membrane proteins are delivered to the plasma membrane, and the constitutive secretory proteins are immediately exocytosed.
This section has provided a broad overview of the secretory pathway. In the following sections, we examine the details of how newly synthesized proteins move between organellar compartments of the secretory pathway, how the proteins are processed during this transit, and how they are sorted to their final destination.
As the preceding discussion suggests, the secretory pathway is not a single, smooth, continuous highway but rather a series of saltatory translocations from one discrete organellar compartment to the next. Each of these steps requires some orchestration to ensure that the newly synthesized proteins arrive at their next terminus.
The cell solves the problem of moving newly synthesized proteins between membranous organelles by using membrane-enclosed carrier vesicles (or vesicular carriers). Each time proteins are to be moved from one compartment to the next, they are gathered together within or beneath specialized regions of membrane that subsequently evaginate or pinch off to produce a carrier vesicle (Fig. 2-18). Secretory proteins reside within the lumen of the carrier vesicle, whereas membrane proteins span the vesicle’s own encapsulating bilayer. On arrival at the appropriate destination, the carrier vesicle fuses with the membrane of the acceptor organelle, thus delivering its contents of soluble proteins to the organelle’s lumen and its cargo of membrane proteins to the organelle’s own membrane. Carrier vesicles mediate the transport of secretory and membrane proteins across the space between the ER’s transition zone and the cis-Golgi stack and also between the rims of the Golgi stacks themselves. The movement between one vesicular compartment and the next is mediated by the cytoskeleton and molecular motors that were discussed earlier.
A few critical facts deserve emphasis. First, throughout the formation, transit, and fusion of a carrier vesicle, no mixing occurs between the vesicle lumen and cytosol. The same principle applies to the carrier vesicle’s membrane protein passengers, which were inserted into the membrane of the rough ER with a particular topology. Those domains of a membrane protein that are exposed to the cytosol in the rough ER remain exposed to the cytosol as the protein completes its journey through the secretory pathway.
Second, the flow of vesicular membranes is not unidirectional. The rate of synthesis of new membrane lipid and protein in the ER is less than the rate at which carrier vesicles bud off of the ER that is bound for the Golgi. Because the sizes of the ER and Golgi are relatively constant, the membrane that moves to the Golgi by carrier vesicles must return to the ER. This return is again accomplished by vesicular carriers. Each discrete step of the secretory pathway must maintain vesicle-mediated backflow of membrane from the acceptor to the donor compartment so that each compartment can retain a constant size.
Finally, we have already noted that each organelle along the secretory pathway is endowed with a specific set of “resident” membrane proteins that determines the properties of the organelle. Despite the rapid forward and backward flow of carrier vesicles between successive stations of the secretory pathway, the resident membrane proteins do not get swept along in the flow. They are either actively retained in their home organelles’ membranes or actively retrieved by the returning “retrograde” carrier vesicles. Thus, not only the size but also the composition of each organelle of the secretory pathway remains essentially constant despite the rapid flux of newly synthesized proteins that it constantly handles.
The formation of a vesicle through evagination appears to be geometrically indistinguishable from its fusion with a target membrane. In both cases, a cross-sectional view in the electron microscope reveals an “omega” profile, which is so named because the vesicle maintains a narrow opening to the organellar lumen that resembles the shape of the Greek letter omega (Ω). However, different problems are confronted during the formation and fusion of membrane vesicles.
Vesicle Formation in the Secretory Pathway To form a spherical vesicle from a planar membrane, the mechanism that pulls the vesicle off from the larger membrane must grab onto the membrane over the entire surface of the nascent vesicle. The mechanism that achieves this makes use of a scaffolding that is composed of coat proteins. The cell has at least two and probably more varieties of coat proteins. The best characterized of these is clathrin, which mediates the formation of secretory vesicles from the trans Golgi. Clathrin also mediates the internalization of membrane from the cell surface during the process of endocytosis, which is the reverse of exocytosis. Another major protein coat, which is involved in nonselective trafficking of vesicles between the ER and Golgi and between the stacks of the Golgi, is a protein complex known as coatamer. Both clathrin and coatamer proteins form the borders of a cage-like lattice.
In the case of clathrin, the coat proteins preassemble in the cytoplasm to form three-armed “triskelions” (Fig. 2-19A). A triskelion is not planar but resembles the three adjoining edges of a tetrahedron. As triskelions attach to one another, they produce a three-dimensional structure resembling a geodesic dome with a roughly spherical shape. A triskelion constitutes each vertex in the lattice of hexagons and pentagons that form the cage.
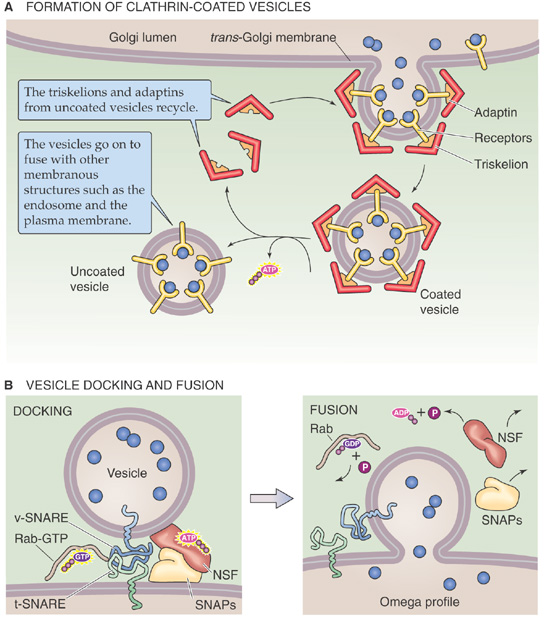
Figure 2-19 Vesicle formation and fusion. A, Clathrin mediates the formation of secretory vesicles that bud off from the trans Golgi as well as the internalization of membrane from the cell surface during the process of endocytosis. B, A complex of proteins forms a bridge between the vesicle and the target membranes. ATP provides the fuel for fusion. The Rab appears to be a molecular switch. NSF, N-ethylmaleimide–sensitive factor; SNAP, soluble NSF attachment protein; SNARE, SNAP receptor.
The triskelions of clathrin attach indirectly to the surface of the membrane that is to be excised by binding to the cytosolic tails of membrane proteins. Mediating this binding are adapter proteins, called adaptins, that link the membrane protein tails to the triskelion scaffold. The specificity for particular membrane proteins is apparently conferred by specialized adaptins. Triskelions assemble spontaneously to form a complete cage that attaches to the underlying membrane and pulls it up into a spherical configuration. Completion of the cage occurs simultaneously with the pinching off of the evaginated membrane from the planar surface, forming a closed sphere.
The pinching off, or fission, process appears to involve the action of a GTP-binding protein called dynamin, which forms a collar around the neck of the forming vesicle and may sever it. The fission process must include an intermediate that resembles the structure depicted in Figure 2-19A. According to the prevalent view, each of the lumen-facing leaflets of membrane lipids fuse, leaving only the cytoplasmic leaflets to form a continuous bridge from the vesicle to the donor membrane. This bridge then breaks, and fission is complete.
Once formed, the clathrin-coated vesicle cannot fuse with its target membrane until it loses its cage, which prevents the two membranes from achieving the close contact required to permit fusion. Because formation of the clathrin cage is spontaneous and energetically favorable, dissolution of the cage requires energy. Uncoating is accomplished by a special class of cytoplasmic enzymes that hydrolyze ATP and use the energy thus liberated to disassemble the scaffold (Fig. 2-19A).
The function of coatamers is similar to that of clathrin in that coatamer forms a cage around the budding membrane. However, coatamer coats differ from clathrin in several respects. First, coatamer coats are composed of several coatamer proteins, one of which is related to the adaptins. Second, unlike the spontaneous assembly of the clathrin triskelions, assembly of the coatamer coat around the budding vesicle requires ATP. Third, a coatamer-coated vesicle retains its coat until it docks with its target membrane.
Vesicle Fusion in the Secretory Pathway Membrane fusion occurs when the hydrophobic cores of two bilayers come into contact with one another, a process that requires the two membranes to be closely apposed. Because the cytoplasmic leaflets of most cellular membranes are predominantly composed of negatively charged phospholipids, electrostatic repulsion prevents this close apposition from occurring spontaneously. To overcome this charge barrier and perhaps to assist in targeting as well, a multicomponent protein complex forms and acts as a bridge, linking vesicular membrane proteins to membrane proteins in the target bilayer (Fig. 2-19B). Investigators have established the components of this complex by use of three approaches: studies of the membrane fusion steps involved in vesicular transport between successive Golgi stacks, genetic analysis of protein secretion in yeast, and molecular dissection of the protein constituents of the synaptic vesicles of nerve terminals. In each case, the same proteins are instrumental in attaching the donor and target membranes to one another.
The central components of the bridge are proteins known as SNAREs (so named because they act as receptors for the SNAPs discussed in the next paragraph). There are SNAREs in both the vesicular membrane (v-SNAREs) and the membrane of the target organelle (t-SNAREs). The best studied SNARE family members are those that participate in the fusion of neurotransmitter-containing synaptic vesicles with the plasma membrane of the axons in neurons (see Chapter 8). In that setting, the v-SNARE is known as synaptobrevin, and proteins known as syntaxin and SNAP-25 together act as t-SNAREs. The t-SNAREs and v-SNAREs bind to each other extremely tightly, pulling the vesicular and target membranes close together. This proximity alone may be sufficient to initiate fusion, although this point remains controversial. In cells that employ rapid and tightly regulated membrane fusion, such as neurons, increases in the cytoplasmic concentration of Ca2+, sensed by the SNARE fusion complex, trigger fusion (see Chapter 8). Although the nature of the fusion event itself remains unclear, clues have emerged about its regulation. Fusion requires the participation of a class of small GTP-binding proteins called Rabs that are important for signaling. Rabs appear to act as molecular switches that assemble with the SNARE fusion complex when they are binding GTP but dissociate from the complex after they hydrolyze the GTP to GDP. Rab-GTP must associate with the fusion complex for fusion to occur. Numerous Rab isoforms exist, each isoform associated with a different vesicular compartment and a distinct membrane-to-membrane translocation step.
Once fusion occurs, the former vesicle generally loses its spherical shape rapidly as it becomes incorporated into the target membrane. This “flattening out” is the result of surface tension, inasmuch as the narrow radius of curvature demanded by a small spherical vesicle is energetically unfavorable. After fusion, it is also necessary to disassemble the v-SNARE/t-SNARE complex so that its components can be reused in subsequent fusion events. The dissociation step involves the activity of two additional components that participate in the SNARE complex. The first is an ATP-hydrolyzing enzyme; because it is inhibited by the alkylating agent N-ethylmaleimide (NEM), it was named NEM-sensitive factor (NSF). Soluble NSF attachment proteins (the SNAPs mentioned before), which target NSF to the SNARE complex, are the second. Hydrolysis of ATP by NSF causes dissociation of the SNARE complex, thus regenerating the fusion machinery. Homologues of the neuronal t-SNARE and v-SNARE proteins are found in almost every cell type in the body and are thought to participate in most if not all membrane fusion events.
While in the rough ER, newly synthesized secretory and membrane proteins undergo the first in a series of post-translational modifications. As discussed earlier, this first group includes glycosylation, disulfide bond formation, and the acquisition of tertiary structure. On delivery to the cis stack of the Golgi complex, these proteins begin a new phase in their postsynthetic maturation. For many proteins, the most visible byproduct of this second phase is the complete remodeling of their N-linked sugar chains, originally attached in the rough ER.
Of the 14 sugar residues transferred en bloc to newly synthesized proteins during N-linked glycosylation, nine are mannose and three are glucose (Fig. 2-20A). Enzymes called glucosidases and one called a mannosidase are associated with the luminal face of the ER; these enzymes remove the three glucose residues and one mannose. As proteins arrive from the ER, mannosidases in the cis Golgi attack the N-linked sugar trees, thereby shearing off all except two N-acetylglucosamine and five mannose residues. As the proteins pass from the cis-Golgi cisterna to the medial cisterna and ultimately to the trans-Golgi cisterna, another mannosidase removes two additional mannose residues, and other enzymes add sugars to the stump of the original sugar tree in a process referred to as complex glycosylation.
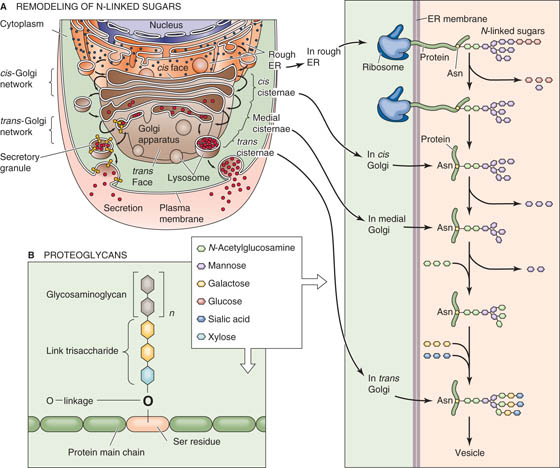
Figure 2-20 Modification and assembly of the sugar chains on proteins in the Golgi. A, Remodeling of N-linked sugars. B, Proteoglycans. A trisaccharide links glycosaminoglycan chains to the protein by the -OH group of a serine residue. The glycosaminoglycan is made up of n repeating disaccharide units, one of which is always an amino sugar.
The addition of new sugars occurs one residue at a time and is accomplished by enzymes called sugar transferases that face the lumens of the Golgi stacks. Each sugar is transported from the cytoplasm to the Golgi lumen by a carrier protein that spans the Golgi membrane. Throughout the maturation process, the N-linked sugar chains are always exposed only to the luminal face of the Golgi. (See Note: Sugar Uptake into the Golgi)
Each cisterna of the Golgi is characterized by a different set of sugar transferases and sugar transporters. Thus, each Golgi compartment catalyzes a distinct step in the maturation of the N-linked chains. Complex glycosylation, therefore, proceeds like an assembly line from one modification station to the next. Because proteins have different shapes and sizes, however, the degree to which a sugar chain of any given polypeptide has access to each transferase can vary quite extensively. Thus, each protein emerges from the assembly line with its own particular pattern of complex glycosylation. The Golgi’s trans-most cisterna houses the enzymes responsible for adding the terminal sugars, which cap the N-linked chain. The final residue of these terminal sugars is frequently N-acetylneuraminic acid, also known as sialic acid. At neutral pH, sialic acid is negatively charged. This acidic sugar residue therefore is responsible for the net negative electrostatic charge that is frequently carried by glycoproteins.
The Golgi’s function is not limited to creating N-linked sugar tree topiaries. It oversees a number of other post-translational modifications, including the assembly of O-linked sugars. Many proteins possess O-linked sugar chains, which attach not to asparagine residues but to the hydroxyl groups (hence, O) of serine and threonine residues. The O-linked sugars are not preassembled for en bloc transfer the way that the original 14-sugar tree is added in the rough ER in the case of their N-linked counterparts. Instead, the O-linked sugars are added one residue at a time by sugar transferases such as those that participate in the remodeling of complex N-linked glycosylation. O-linked chains frequently carry a great deal of negatively charged sialic acid.
Proteoglycans contain a very large number of a special class of O-linked sugar chains that are extremely long (Fig. 2-20B). Unlike other O-linked sugars that attach to the protein core by an N-acetylglucosamine, the sugar chain in a proteoglycan attaches by a xylose-containing three-sugar “linker” to a serine residue on the protein backbone. One or more glycosaminoglycan side chains are added to this linker, one sugar at a time, to form the mature proteoglycan.
As the sugar chains grow, enzymes can add sulfate groups and greatly increase the quantity of negative charge that they carry. Sulfated proteoglycans that are secreted proteins become important components of the extracellular matrix and are also constituents of mucus. Proteoglycan chains can also be attached to membrane proteins that eventually reach the plasma membrane. The negatively charged sugars that are associated with the glycosaminoglycan groups, which are present both in mucus and on cell surface glycoproteins, can help form a barrier that protects cells from harsh environmental conditions such as those inside the stomach (see Chapter 42). In the upper portion of the respiratory tract, the mucus assists in the removal of foreign bodies (see Chapter 26).
From their common point of origin at the rough ER, newly synthesized secretory and membrane proteins must be distributed to a wide variety of different subcellular destinations. How can a cell recognize an individual protein from among the multitudes that are inserted into or across the membranes of the rough ER and ensure its delivery to the site of its ultimate functional residence? Such a sorting operation has two prerequisites: (1) each protein to be sorted must carry some manner of address or “sorting signal” that communicates its destination, and (2) the cell must possess machinery capable of reading this sorting signal and acting on the information it embodies.
Little is known about the molecular correlates of sorting signals, and even less is established about the sorting machinery. However, for many proteins, it is clear that sorting occurs in the trans-Golgi network (TGN). The trans-most cisterna of the Golgi complex is morphologically and biochemically distinct from the other Golgi stacks. Viewed in cross section, it appears as a complex web of membranous tubules and vesicles (Fig. 2-21). This structure befits the TGN’s apparent function as a staging area from which carrier vesicles depart to transport their specific protein cargoes to one of many distinct subcellular locales.
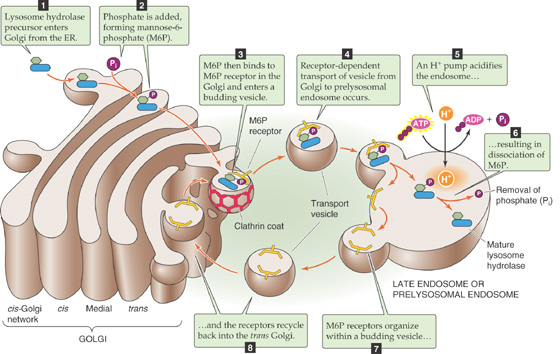
Figure 2-21 Sorting of lysosomal enzymes.
Sorting machinery within or at the TGN appears to segregate classes of proteins—each bound for a common destination—into small discrete clusters. Each cluster is subsequently incorporated into a separate carrier vesicle, which evaginates from the TGN membrane and mediates the final stage of delivery. In the case of secretory proteins, this clustering happens within the lumen of the TGN. In fact, such clusters of secretory proteins can be directly visualized in the electron microscope. Membrane proteins gather into two-dimensional clusters in the plane of the TGN membrane. Carrier vesicles incorporate these clusters into their own bilayers. Proteins bound for different destinations co-cluster in different subdomains of the TGN. Secretory and membrane proteins that are earmarked for the same destination can cluster in the same subdomain of the TGN and can be incorporated into the same carrier vesicle. Therefore, the TGN appears to function as a cellular transportation terminal that is able to direct groups of passengers who are carrying the same tickets to a common waiting area and ultimately to load them onto a common shuttle. Ticket agents herd passengers bearing different tickets into different waiting lounges.
The most thoroughly established sorting paradigm is the pathway for newly synthesized lysosomal enzymes. Like secretory proteins, lysosomal enzymes carry amino-terminal signal sequences that direct their cotranslational translocation across the membrane of the rough ER. Their N-linked glycosylation and folding proceed in the usual fashion, after which they join all of the other simultaneously synthesized proteins in the Golgi complex (Fig. 2-21, stage 1).
A special sugar transferase in the cis-Golgi cisterna recognizes newly synthesized lysosomal enzymes and adds a unique sugar. This enzyme adds N-acetylglucosamine phosphate to the mannose residues at the termini of the lysosomal enzymes’ N-linked sugar trees. This enzyme differs from the usual sugar transferases in that it adds a phospho sugar group to the mannose residue, rather than just a sugar. This enzyme is also unique in recognizing specific amino acid sequences that are exclusively in these lysosomal enzymes. A second cis-Golgi enzyme removes the additional N-acetylglucosamine sugar, leaving its phosphate group behind. As a result, the sugar trees of the lysosomal enzymes terminate in mannose 6-phosphate residues (Fig. 2-21, stage 2).
A special class of mannose 6-phosphate receptors, localized predominantly in the elements of the trans Golgi, recognize proteins that carry mannose 6-phosphate groups (Fig. 2-21, stage 3). This recognition step constitutes the first stage of the cosegregation and clustering process discussed earlier. The mannose 6-phosphate receptors are transmembrane proteins. Their luminal portions bind to the newly synthesized lysosomal enzymes, whereas their cytoplasmically facing tails possess a particular signal that allows them to interact with adaptins and hence to be incorporated into clathrin-coated vesicles. The assembly of the clathrin lattice causes the mannose 6-phosphate receptors to cluster, along with their associated lysosomal enzymes, in the plane of the TGN membrane. Completion of the clathrin cage results in the formation of a vesicle whose membrane contains the mannose 6-phosphate receptors that bind their cargo of lysosomal enzymes.
After departing the TGN, these transport vesicles lose their clathrin coats (Fig. 2-21, stage 4) and fuse with structures referred to as late endosomes or prelysosomal endosomes. Proton pumps in the membranes of these organelles ensure that their luminal pH is acidic (Fig. 2-21, stage 5). When exposed to this acidic environment, the mannose 6-phosphate receptors undergo a conformational change that releases the mannose 6-phosphate–bearing lysosomal enzymes (Fig. 2-21, stage 6). Consequently, the newly synthesized enzymes are dumped into the lumen of the prelysosomal endosome, which will go on to fuse with or mature into a lysosome. The empty mannose 6-phosphate receptors join vesicles that bud off from the lysosome (Fig. 2-21, stage 7) and return to the TGN (Fig. 2-21, stage 8). The luminal environment of the TGN allows the receptors to recover their affinity for mannose 6-phosphate, thus allowing them to participate in subsequent rounds of sorting.
Disruption of lysosomal sorting can be produced in several ways. For example, a drug called tunicamycin blocks the addition of N-linked sugars to newly synthesized proteins and thereby prevents attachment of the mannose 6-phosphate recognition marker. Compounds that elevate the luminal pH of the prelysosomal endosomes prevent newly synthesized enzymes from dissociating from the mannose 6-phosphate receptors and consequently block recycling of the receptor pool back to the TGN. The resulting shortage of unoccupied receptors allows mannose 6-phosphate–bearing proteins to pass through the TGN unrecognized (see the box titled Lysosomal Storage Diseases). Thus, instead of diverting to the lysosomes, these lysosomal enzymes continue along the secretory pathway and are ultimately released from the cell by constitutive secretion.
The same fundamental mechanisms in the secretory pathway that produce vesicles by evaginating regions of Golgi membrane can also move material in the opposite direction by inducing vesicle formation through the invagination of regions of the plasma membrane. Vesicles created in this fashion are delimited by membrane that had formerly been part of the cell surface, and their luminal contents derive from the extracellular compartment.
This internalization process, referred to as endocytosis, serves the cell in at least four ways. First, certain nutrients are too large to be imported from the extracellular fluid into the cytoplasm by transmembrane carrier proteins; they are instead carried into the cell by endocytosis. Second, endocytosis of hormone-receptor complexes can terminate the signaling processes that are initiated by numerous hormones. Third, endocytosis is the first step in remodeling or degrading of portions of the plasma membrane. Membrane that is delivered to the surface during exocytosis must be retrieved and ultimately returned to the TGN. Fourth, proteins or pathogens that need to be cleared from the extracellular compartment are brought into the cell by endocytosis and subsequently condemned to degradation in the lysosomes. Because endocytosed material can pursue a number of different destinies, there must be sorting mechanisms in the endocytic pathway, just as in the secretory pathway, that allow the cell to direct the endocytosed material to its appropriate destination.
Fluid-phase endocytosis is the uptake of the materials that are dissolved in the extracellular fluid (Fig. 2-22, stage 1) and not specifically bound to receptors on the cell surface. This process begins when a clathrin cage starts to assemble on the cytoplasmic surface of the plasma membrane. Earlier we discussed the physiology of clathrin-coated vesicles in the secretory pathway (Fig. 2-19). The clathrin attaches to the membrane through interactions with adaptin proteins, which in turn adhere to the cytoplasmic tail domains of certain transmembrane polypeptides. Construction of the cage causes its adherent underlying membrane to invaginate and to form a coated pit (Fig. 2-22, stage 2A). Completion of the cage creates a closed vesicle, which detaches from the cell surface through the process of membrane fission (Fig. 2-22, stage 3). The resultant vesicle quickly loses its clathrin coat through the action of the uncoating ATPase and fuses with an organelle called an endosome.
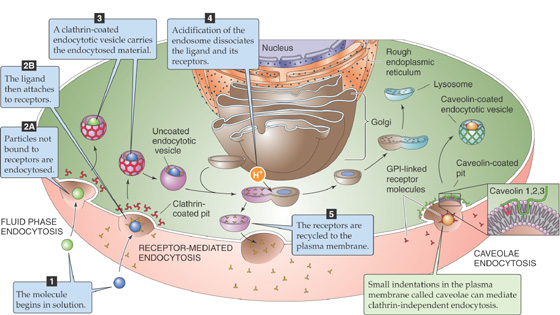
Figure 2-22 Endocytosis.
Most of the proteins that a cell seeks to import by endocytosis are present in the extracellular fluid in extremely low concentrations. Furthermore, the volume of extracellular fluid that is internalized by an individual coated vesicle is very small. Consequently, the probability that any particular target molecule will enter the cell during a given round of fluid-phase endocytosis is low. To improve the efficiency of endocytosis and to ensure that the desired extracellular components are gathered in every endocytic cycle, the cell has devised a method for concentrating specific proteins at the site of endocytosis before initiating their uptake.
This concentration is achieved in a process known as receptor-mediated endocytosis, in which molecules to be internalized (Fig. 2-22, stage 1) bind to cell surface receptors with high affinity (Fig. 2-22, stage 2B). Through this interaction, the substrates for endocytosis become physically associated with the plasma membrane, thus greatly enhancing the probability that they will be successfully internalized. Cells increase this probability even further by ensuring that the receptors themselves cluster in regions of the membrane destined to be endocytosed. The cytoplasmic tails of these receptors are endowed with recognition sequences that allow them to serve as binding sites for adaptins. Consequently, these receptors congregate in regions of the cell membrane where clathrin cages are assembling and are incorporated into coated pits as they are forming. The affinity of these receptors for the endocytic machinery ensures that their ligands are internalized with maximum efficiency.
Most endocytic receptors are constitutively associated with coated pits and are endocytosed whether or not they have bound their specific ligands. The cytoplasmic tails of certain receptors, however, interact with adaptins only when the receptor is in the bound state. For example, in the absence of epidermal growth factor (EGF), the EGF receptor is excluded from regions of the membrane in which coated pits are assembling. Modifications induced by ligand binding alter these receptors’ tails, which allows them to participate in coated vesicle formation and hence in endocytosis.
After the clathrin-coated vesicle forms (Fig. 2-22, stage 3), it quickly loses its clathrin coat, as described earlier for fluid-phase endocytosis, and fuses with an endosome. Although endosomes can be wildly pleomorphic, they frequently have a frying pan–like appearance in which a round vesicular body is attached to a long tubular “handle” (Fig. 2-22, stage 4). The cytoplasmic surfaces of the handles are often decorated with forming clathrin lattices and are the sites of vesicular budding.
Lysosomal Storage Diseases
The experimental elucidation of lysosomal enzyme sorting was achieved only because of the existence of a remarkable, naturally occurring human disease that was traced to a genetic defect in the sorting machinery. In lysosomal storage diseases, the absence of a particular hydrolase—or group of hydrolases—from the lysosome prevents the lysosomes from degrading certain substances, resulting in the formation of overstuffed lysosomes that crowd the cytoplasm and impede cell function.
In I-cell disease, most hydrolases are missing from the lysosomes of many cell types. As a result, lysosomes become engorged with massive quantities of undigested substrates. The enormously swollen lysosomes that characterize this disease were named inclusion bodies, and the cells that possess them were designated inclusion cells, or I cells for short. Whereas I cells lack most lysosomal enzymes, the genes that encode all of the hydrolases are completely normal. The mutation responsible for I-cell disease resides in the gene for the phosphosugar transferase that creates the mannose 6-phosphate recognition marker (Fig. 2-21). Without this enzyme, the cell cannot sort any of the hydrolases to the lysosomes. Instead, the hydrolases pass through the trans-Golgi network unnoticed by the mannose 6-phosphate receptors and are secreted constitutively from the affected cells. Certain cell types from I-cell individuals can sort newly synthesized hydrolases normally, suggesting that alternative, as yet unelucidated pathways for the targeting of lysosomal enzymes must also exist.
In some other lysosomal storage diseases, specific hydrolases are not missorted but rather are genetically defective. For example, children who suffer from Tay-Sachs disease carry a homozygous mutation in the gene that encodes the lysosomal enzyme hexosaminidase A (HEX A). Consequently, their lysosomes are unable to degrade substances that contain certain specific sugar linkages. Because they cannot be broken down, these substances accumulate in lysosomes. Over time, these substances fill the lysosomes, which swell and crowd the cytoplasm. The resulting derangements of cellular function are toxic to a number of cell types and ultimately underlie this disease’s uniform fatality within the first few years of life. Carriers of the Tay-Sachs trait can be detected either by HEX A enzyme testing or by DNA analysis of the HEX A gene. Among the Ashkenazi Jewish population, in which 1 in 27 individuals is a carrier, three distinct HEX A mutations account for 98% of all carrier mutations.
In many cell types, endocytosis is so rapid that each hour, the cell internalizes a quantity of membrane that is equivalent in area to the entire cell surface. To persist in the face of this tremendous flux of membrane, the cell must retrieve most of the endocytosed membrane components and return them to the plasmalemma. However, substances that a cell wishes to degrade must be routed to lysosomes and prevented from escaping back to the surface. The sophisticated sorting operation required to satisfy both of these conditions takes place in the endosome.
Proton pumps embedded in its membrane ensure that like the lysosome, the endosome maintains an acidic luminal pH (Fig. 2-22, stage 4). This acidic environment initiates the separation of material that is destined for lysosomal destruction from those proteins that are to be recycled. Most endocytic receptors bind their ligands tightly at neutral pH but release them rapidly at pH values below 6.0. Therefore, as soon as a surface-derived vesicle fuses with an endosome, proteins that are bound to receptors fall off and enter the endosomal lumen. The receptor proteins segregate in the membranes of the handles of the frying pan–shaped endosomes and are ultimately removed from the endosome in vesicles that shuttle them back to the cell surface (Fig. 2-22, stage 5). The soluble proteins of the endosome lumen, which include the receptors’ former ligands, are ultimately delivered to the lysosome. This sorting scheme allows the receptors to avoid the fate of their cargo and ensures that the receptors are used in many rounds of endocytosis.
The low-density lipoprotein (LDL) receptor follows this regimen precisely. On arrival of the LDL-laden receptor at the endosome, the acidic environment of the endosome induces the LDL to dissociate from its receptor, which then promptly recycles to the cell surface. The LDL travels on to the lysosome, where enzymes destroy the LDL and liberate its bound cholesterol. (See Note: Familial Hypercholesterolemia: A Defect in Receptor-Mediated Endocytosis)
A variation on this paradigm is responsible for the cellular uptake of iron. Iron circulates in the plasma bound to a protein called transferrin. At the mildly alkaline pH of extracellular fluid, the iron-transferrin complex binds with high affinity to a transferrin receptor in the plasma membranes of almost every cell type. Bound transferrin is internalized by endocytosis and delivered to endosomes. Instead of inducing transferrin to fall off its receptor, the acid environment of the endosome lumen causes iron to fall off transferrin. Apotransferrin (i.e., transferrin without bound iron) remains tightly bound to the transferrin receptor at an acidic pH. The released iron is transported across the endosomal membrane for use in the cytosol. The complex of apotransferrin and the transferrin receptor recycles to the cell surface, where it is again exposed to the extracellular fluid. The mildly alkaline extracellular pH causes the transferrin receptor to lose its affinity for apotransferrin and promptly releases it. Thus, the cell uses the pH-dependent sorting trick twice to ensure that both the transferrin receptor and apotransferrin recycle for subsequent rounds of iron uptake.
Clathrin-coated pits are not the only cellular structures involved in receptor-mediated internalization. Electron microscopic examination of vascular endothelial cells that line blood vessels long ago revealed the presence of clusters of small vesicles that display a characteristic appearance, in close association with the plasma membrane. These caveolae were thought to be involved in the transfer of large molecules across the endothelial cells, from the blood space to the tissue compartment. Actually, caveolae are present in most cell types. The caveolae are rich in cholesterol and sphingomyelin. Rather than having a clathrin lattice, they contain intrinsic membrane proteins called caveolins, which face the cytosol (Fig. 2-22). In addition, caveolae appear to be rich in membrane-associated polypeptides that participate in intracellular signaling, such as the Ras-like proteins as well as heterotrimeric GTP-binding proteins (see Chapter 5). They are also enriched in the receptor for folate, a vitamin required by several metabolic pathways (see Chapter 45). Unlike the receptors in the plasma membrane discussed earlier, the folate receptor has no cytoplasmic tail that might allow it to associate with coated pits. Instead, it belongs to the GPI-linked class of proteins that are anchored to the membrane through covalent attachment to phospholipid molecules. It appears that caveolae mediate the internalization of folate. In fact, a large number and variety of GPI-linked proteins are embedded in the outer leaflet of the caveolar membrane that faces its lumen.
The role of caveolae in the uptake of other substances, the significance of the large inventory of GPI-linked proteins in caveolae, and the functions served by their cache of signaling molecules remain to be determined. It is clear, however, that the caveolae represent a novel endocytic structure that participates in pathways distinct from those involving coated vesicles and endosomes.
All cells are constructed of the same basic elements and share the same basic metabolic and biosynthetic machinery. What distinguishes one cell type from another? Certainly, cells have different shapes and molecular structures. In addition, out of an extensive repertory of molecules that cells are capable of making, each cell type chooses which molecules to express, how to organize these molecules, and how to regulate them. It is this combination of choices that endows them with specific physiological functions. These specializations are the product of cell differentiation. Each of these cell types arises from a stem cell. Stem cells are mitotically active and can give rise to multiple, distinct cellular lineages; thus, they are referred to as pluripotent. Clearly, the zygote is the ultimate stem cell because through its divisions, it gives rise to every cell lineage present in the complete organism. Specific cell types arise from stem cells by activating a differentiation-specific program of gene expression. The interplay of environmental signals, temporal cues, and transcription factors that control the processes of cellular differentiation constitutes one of the great unraveling mysteries of modern biology.
How can an organism tightly regulate its internal fluid environment (i.e., internal milieu) without allowing this environment to come into direct and disastrous contact with the external world (i.e., external milieu)? The body has solved these problems by arranging a sheet of cells—an epithelium—between two disparate solutions. Because of their unique subcellular designs and intercellular relationships, epithelial cells form a dynamic barrier that can import or expel substances, sometimes against steep concentration gradients.
Two structural features of epithelia permit them to function as useful barriers between two very different solutions (Fig. 2-23). First, epithelial cells connect to one another by tight junctions, which constrain the free diffusion of solutes and fluids around the epithelial cells, between the internal and external compartments. Second, the tight junctions define a boundary between an apical and a basolateral domain of the plasma membrane. Each of these two domains is endowed with distinct protein and lipid components, and each subserves a distinct function. Thus, the surface membranes of epithelial cells are polarized. In Chapter 5, we discuss the mechanisms by which polarized epithelial cells exploit their unique geometry to transport salts and water from one solution to the other. However, it is worth touching on a few of the cellular specializations that characterize polarized epithelia and permit them to perform their critical roles.
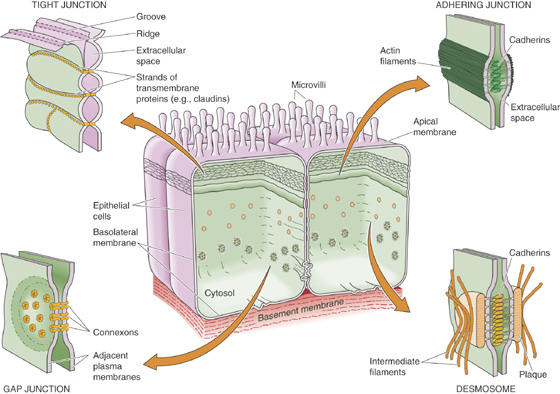
Figure 2-23 Epithelial cells. In an epithelial cell, the tight junction separates the cell membrane into apical and basolateral domains that have very different functional properties.
The apical membranes of the epithelial cells (Fig. 2-23) face the lumen of a compartment that is often topologically continuous with the outside world. For example, in the stomach and intestine, apical membranes form the inner surface of the organs that come into contact with ingested matter. The apical membranes of many epithelial cells, including those lining kidney tubules, are endowed with a single nonmotile cilium. Known as the central cilium, this structure may sense the mechanical deformation associated with fluid flow. Mutations that disrupt individual components of the central cilium are associated with cystic disease of the kidney, in which the normal architecture of the kidney is replaced by a collection of large fluid-filled cysts.
The basolateral membranes of epithelial cells face the extracellular fluid compartment—which indirectly makes contact with the blood—and rest on a basement membrane. The basement membrane is composed of extracellular matrix proteins that the epithelial cells themselves secrete and include collagens, laminin, and proteoglycans. The basement membrane provides the epithelium with structural support and, most important, serves as an organizing foundation that helps the epithelial cells to establish their remarkable architecture.
Each epithelial cell is interconnected to its neighbors by a variety of junctional complexes (Fig. 2-23). The lateral surfaces of epithelial cells participate in numerous types of cell-cell contacts, including tight junctions, adhering junctions, gap junctions, and desmosomes.
Tight Junctions A tight junction (or zonula occludens) is a complex structure that impedes the passage of molecules and ions between the cells of the epithelial monolayer. This pathway between the cells is termed the paracellular pathway. Although the complete molecular structure of the tight junction has yet to be elucidated, it is clear that its functional properties are related to its intriguing architecture (Fig. 2-23). Viewed by transmission electron microscopy, tight junctions include regions of apparent fusion between the outer leaflets of the lipid bilayer membranes of neighboring epithelial cells. Freeze-fracture electron microscopy reveals that the tight junction comprises parallel strands of closely packed particles, which presumably represent the transmembrane proteins participating in the junction’s formation. The degree of an epithelium’s impermeability—or “tightness”—is roughly proportional to the number of these parallel strands. The claudins, a large family of proteins, are the principal structural elements of the tight junction. Interactions between the claudins present in the apposing membranes of neighboring cells form the permeability barrier (see Chapter 5).
Tight junctions play several roles. First, they are barriers in that they separate one compartment from another. In some epithelial cells, such as those of the renal thick ascending limb, the tight junctions form an essentially impenetrable boundary that completely blocks the flow of ions and water between cells. In contrast, the tight junctions of the renal proximal tubule are leaky, permitting significant transepithelial movement of fluid and solutes.
Second, tight junctions can act as selective gates in that they permit certain solutes to flow more easily than others. Examples are the leaky tight junctions of tissues such as the proximal tubule. As discussed in Chapter 5, the permeability and selectivity of an epithelium’s tight junctions are critical variables for determining that epithelium’s transport characteristics. Moreover, the permeability properties of the gate function of tight junctions can be modulated in response to various physiological stimuli. The inventory of claudins expressed by an epithelium appears to determine in large measure the permeability properties of the tight junctions.
Third, tight junctions act as fences that separate the polarized surfaces of the epithelial plasma membrane into apical and basolateral domains. The presence of distinct populations of proteins and lipids in each plasma membrane domain is absolutely essential for an epithelium to mediate transepithelial fluid and solute transport (see Chapter 5).
Adhering Junction An adhering junction (or zonula adherens) is a belt that encircles an entire epithelial cell just below the level of the tight junction. Epithelial cells need two pieces of information to build themselves into a coherent epithelium. First, the cells must know which end is up. The extracellular matrix (see earlier) provides this information by defining which side will be basolateral. Second, the cells must know that there are like neighbors with which to establish cell-cell contacts. Adhering junctions provide epithelial cells with clues about the nature and proximity of their neighbors. These cell-cell contacts are mediated by the extracellular domains of members of the cadherin family, transmembrane proteins discussed earlier. Epithelial cells will organize themselves into a properly polarized epithelium—with differentiated apical and basolateral plasma membranes—only if the cadherins of neighboring cells have come into close enough apposition to form an adhering junction. Formation of these junctions initiates the assembly of a subcortical cytoskeleton, in which anchor proteins (e.g., vinculin, catenins, α-actinin) link the cytosolic domains of cadherins to a network of actin filaments that is associated with the cytosolic surfaces of the lateral membranes. Conversely, the disruption of adhering junctions can lead to a loss of epithelial organization. In epithelial tumors, for example, loss of expression of the adhering junction cadherins tends to correlate with the tumor cell’s loss of controlled growth and its ability to metastasize, that is, to leave the epithelial monolayer and form a new tumor at a distant site in the body. (See Note: Role of Cell-Cell Adhesion Molecules in Development)
Gap Junctions Gap junctions, which are discussed in Chapter 6, are channels that interconnect the cytosols of neighboring cells. They allow small molecules (less than ~1000 in molecular weight) to diffuse freely between cells. In some organs, epithelial cells are interconnected by an enormous number of gap junctions, which organize into paracrystalline hexagonal arrays. Because ions can flow through gap junctions, cells that communicate through gap junctions are electrically coupled. The permeability of gap junctions, and hence the extent to which the cytoplasmic compartments of neighboring cells are coupled, can be regulated in response to a variety of physiological stimuli.
Desmosome A desmosome (or macula adherens) holds adjacent cells together tightly at a single, round spot. Desmosomes are easily recognized in thin-section electron micrographs by the characteristic dense plaques of intermediate filaments. The extracellular domains of transmembrane proteins in the cadherin family mediate the interaction of adjacent cells. Anchor proteins link the cytosolic domains of the cadherins to intermediate filaments that radiate into the cytoplasm from the point of intercellular contact (Fig. 2-23). These filaments interact with and organize the cytoplasmic intermediate filaments, thus coupling the structurally stabilizing elements of neighboring cells to one another. Epithelial cells are often coupled to adjacent cells by numerous desmosomes, especially in regions where the epithelium is subject to physical stress.
In many epithelia, the apical surface area is amplified by the presence of a brush border that is composed of hundreds of finger-like, microvillar projections (Fig. 2-23). In the case of the small intestine and the renal proximal tubule, the membrane covering each microvillus is richly endowed with enzymes that digest sugars and proteins as well as with transporters that carry the products of these digestions into the cells. The presence of a microvillar brush border can amplify the apical surface area of a polarized epithelial cell by as much as 20-fold, thus greatly enhancing its capacity to interact with, to modify, and to transport substances present in the luminal fluid.
The basolateral surface area of certain epithelial cells is amplified by the presence of lateral interdigitations and basal infoldings (Fig. 2-23). Although they are not as elegantly constructed as microvilli, these structures can greatly increase the basolateral surface area. In epithelial cells that are involved in large volumes of transport—or in transport against steep gradients—amplifying the basolateral membrane can greatly increase the number of basolateral Na-K pumps that a single cell can place at its basolateral membrane.
Although the morphological differences between apical and basolateral membranes can be dramatic, the most important distinction between these surfaces is their protein composition. As noted earlier, the “fence” function of the tight junction separates completely different rosters of membrane proteins between the apical and basolateral membranes. For example, the Na-K pump is restricted to the basolateral membrane in almost all epithelial cells, and the membrane-bound enzymes that hydrolyze complex sugars and peptides are restricted to apical membranes in intestinal epithelial cells. The polarized distribution of transport proteins is absolutely necessary for the directed movement of solutes and water across epithelia. Furthermore, the restriction of certain enzymes to the apical domain limits their actions to the lumen of the epithelium and therefore offers the advantage of not wasting energy putting enzymes where they are not needed. The polarity of epithelial membrane proteins also plays a critical role in detecting antigens present in the external milieu and in transmitting signals between the external and internal compartments.
The maintenance of epithelial polarity involves complex intermolecular interactions that are only beginning to be understood. When tight junctions are disrupted, diffusion in the plane of the membrane leads to intermingling of apical and basolateral membrane components and thus a loss of polarity. The subcortical cytoskeleton beneath the basolateral surface may play a similar role by physically restraining a subset of membrane proteins at the basolateral surface.
However, such mechanisms for stabilizing the polarized distributions of membrane proteins do not explain how newly synthesized proteins come to be distributed at the appropriate plasma membrane domain. We give two examples of mechanisms that cells can use to direct membrane proteins to either the basolateral or apical membrane. The first example focuses on protein-protein interactions. As noted during our discussion of the secretory protein pathway, the sorting operation that separates apically from basolaterally directed proteins apparently occurs in the TGN. Some proteins destined for the basolateral membrane have special amino acid motifs that act as sorting signals. Some of these motifs are similar to those that allow membrane proteins to participate in endocytosis. Members of the adaptin family may recognize these motifs during the formation of clathrin-coated vesicles at the TGN and segregate the basolateral proteins into a vesicle destined for the basolateral membrane.
Another example of mechanisms that cells use to generate a polarized distribution of membrane proteins focuses on lipid-lipid interactions. In many epithelia, GPI-linked proteins are concentrated exclusively at the apical surface. It appears that the phospholipid components of GPI-linked proteins are unusual in that they cluster into complexes of fairly immobile gel-phase lipids during their passage through the Golgi apparatus. We saw earlier how lakes of phospholipids with different physical properties may segregate within a membrane. The “glycolipid rafts” of GPI-linked proteins incorporate into apically directed vesicles so that sorting can occur through lipid-lipid interactions in the plane of the membrane rather than through protein-protein interactions at the cytoplasmic surface of the Golgi membrane. From these two examples, it should be clear that a number of different mechanisms may contribute to protein sorting and the maintenance of epithelial polarity.
Books and Reviews
Goldstein JL, Brown MS, Anderson RGW, et al: Receptor-mediated endocytosis: Concepts emerging from the LDL receptor system. Annu Rev Cell Dev Biol 1985; 1:1-39.
Mellman I: Endocytosis and molecular sorting. Annu Rev Cell Dev Biol 1996; 12:575-625.
Palade GE: Intracellular aspects of the process of protein synthesis. Science. 1985; 189:347-358.
Rodriguez-Boulan E, Powell SK: Polarity of epithelial and neuronal cells. Annu Rev Cell Dev Biol 1992; 8:395-427.
Rothman JE: The protein machinery of vesicle budding and fusion. Protein Sci. 1995; 5:185-194.
Sheetz MP: Microtubule motor complexes moving membranous organelles. Cell Struct Funct. 1996; 21:369-373.
Journal Articles
Frye LD, Edidin M: The rapid intermixing of cell surface antigens after formation of mouse-human heterokaryons. J Cell Sci 1970; 7:319-335.
Griffiths G, Hoflack B, Simons K, et al: The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 1988; 52:329-341.
Kyte J, Doolittle RF: A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157:105-132.
Walter P, Ibrahimi I, Blobel G: Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in vitro assembled polysomes synthesizing secretory protein. J Cell Biol 1981; 91:545-550.