Chapter 6 Protein structure and its relevance to drug action
Amino acids
The basic building blocks which make up proteins are the 20 amino acids. The simplest amino acid is glycine (Fig. 6.1) which has a carboxylic acid group (pKa 2.5) and a primary amine group (pKa 9.7). Like all amino acids in its free state, when not part of a protein chain, it can carry both positive and negative charges depending on the pH of the solution it is dissolved in, and there is no pH at which it is not charged. A molecule which carries both positive and negative charges in solution is known as a zwitterion, and in the case of a simple amino acid like glycine, the charges are equal and opposite at a pH half-way between the two pKa values, i.e. at pH 5.1. This point is known as the isoelectric point and its value is known as the pI of the molecule. As shown in Figure 6.1, an amino acid is completely positively charged only at low pH and completely negatively charged only at high pH. In the case of glycine, the side chain of the amino acid represented by R=H but the other 19 amino acids occurring in proteins have various R groups (Fig. 6.2).
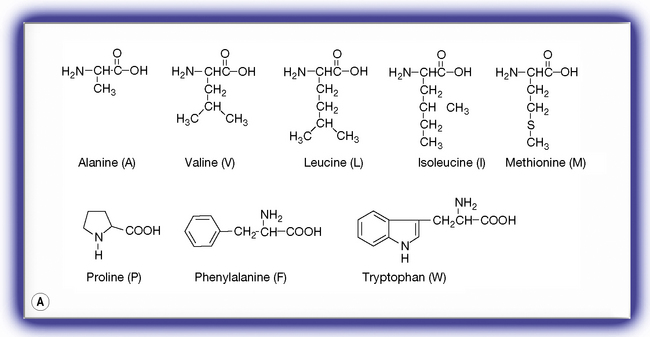
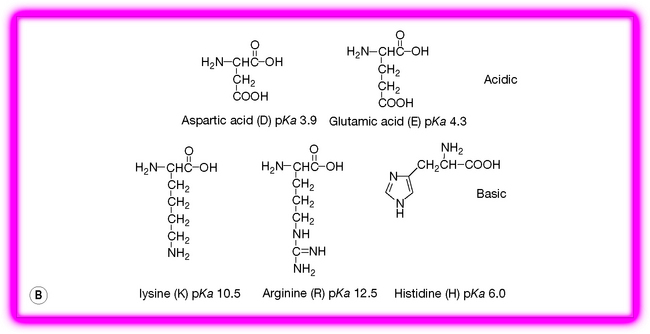
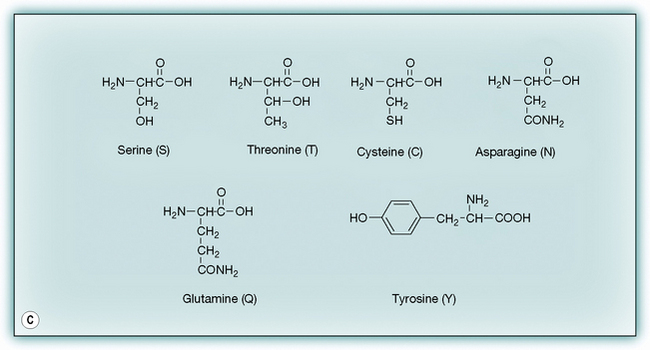
Figure 6.2 The 19 amino acids in addition to glycine occurring in proteins. (A) Hydrophobic amino acids. (B) Charged amino acids. (C) Neutral hydrophilic amino acids.
There are various ways of classifying the amino acids but perhaps the most useful in terms of protein function is classification into hydrophobic, polar and charged. Glycine can be classified on its own since it does not fall into any of these categories, although lack of a side chain gives it flexibility and it has an important role in protein structure for this reason. The importance of the different classes of amino acids will be discussed in more detail as we build up protein structures but the different categories are briefly considered below.
Hydrophobic amino acids
Hydrophobic amino acids exhibit varying degrees of hydrophobicity depending on how large the hydrophobic side chain is. Alanine is the least hydrophobic and tryptophan is the most hydrophobic even though it contains a weakly polar indole nitrogen. These amino acids are quite water soluble at extremes of pH but if the pH is adjusted to around their pI then they are effectively neutral and their hydrophobicity will cause them to come out of solution. This class of amino acid is found buried inside proteins avoiding contact with water and thus form the protein core. If the protein has a helical portion which passes through a lipophilic cell membrane this portion of the protein will be found to be rich in hydrophobic/lipophilic amino acids.
Charged amino acids
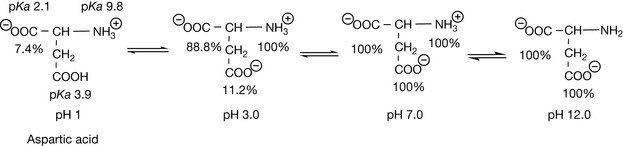
Charged amino acids are found predominantly on the surface of proteins in contact with the surrounding solution. Thus they are responsible for the solution stability of the protein and if they lose their charge the protein will become unstable and precipitate out of solution. A standard method for removing a protein from a solution is to adjust it so that it is strongly acidic so that the negatively charged side chains of the protein become uncharged, thus destabilising its structure in solution. Often, proteins cannot be re-dissolved after such treatment. A more gentle method of removing proteins from solution is to salt them out with concentrated ammonium sulphate, which effectively competes with the protein groups for the solvating water molecules required to keep the protein in solution, thus destabilising it. The charged residues in proteins are important in the action of many drug molecules, as many drugs are bases and they bind to the negatively charged aspartate or glutamate side chains in proteins. Histidine can be classified as a polar neutral amino acid residue since it is only about 4% ionised at physiological pH. However its charge state is important since its pKa value of 6 provides buffering in the pH range where many proteins exert their functions. The pI values of the charged amino acids are either higher or lower than those of neutral amino acids such as glycine. Figure 6.3 shows the ionisation states of glutamic acid, the pI is at the average of the pKa values for the two acids in the structure at pH 3.0.
Polar amino acids
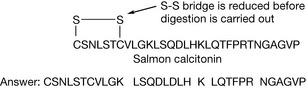
Polar amino acids have a diverse range of functions. Serine residues are important in enzyme-catalysed reactions, cysteine is important in determining the 3-D structure of proteins because of its ability to form S–S bridges with another cysteine residue, and tyrosine and serine are important because they form phosphate esters which cause an alteration of protein conformation, thus triggering other cellular events. Asparagine and glutamine are important sites for hydrogen bonding within proteins and with ligands binding to proteins. In addition, cysteine is important for its ability to bond to metal ions, which are often present at the active sites of enzymes.
The peptide bond
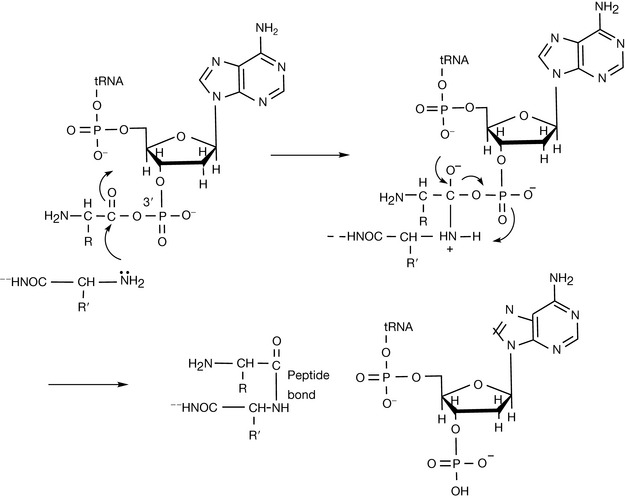
The amino group of one amino acid reacts with the carboxylic acid of another amino acid to form an amide – the peptide bond. In the process of production of the peptide the DNA sequence coding for a particular amino acid is transcribed to produce the RNA sequence corresponding to a particular amino acid. The RNA sequence then binds to a sequence in a tRNA molecule (see Ch. 7) corresponding to a particular amino acid. The tRNA is largely a single-stranded molecule but at the 5′ end of the main chain there is a short sequence of seven DNA bases. The final base at the 3′ end of this short chain is unpaired and is adenine monophosphate. The amino acid to be attached to the tRNA is first converted to its aminoacyl AMP (equation 1) and then this reactive form of the amino acid is transferred to the 3′ terminal AMP group of the acceptor sequence in the tRNA (equation 2). The aminoacyl-AMP at the 3′ then undergoes nucleophilic attack by the terminal amino group of the growing peptide (Fig. 6.4). This process occurs in the cellular ribosomes which contain a binding site which specifies the next amino acid to be added to the growing peptide and recruits the appropriate aminoacyl-tRNA to be added to the growing peptide chain which is held in another binding site within the ribosome. Thus the ribosomal RNA can be described as having ribozyme activity, indicating that it functions like an enzyme. The sequence of amino acids in a peptide constitutes its primary structure. By convention, the amino terminal (N-terminal) of a peptide is at the left-hand end of a sequence of amino acids and the carboxyl (C-terminal) of a peptide is at the right-hand end.
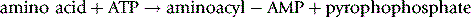
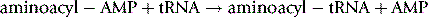
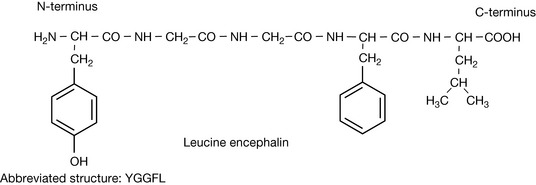
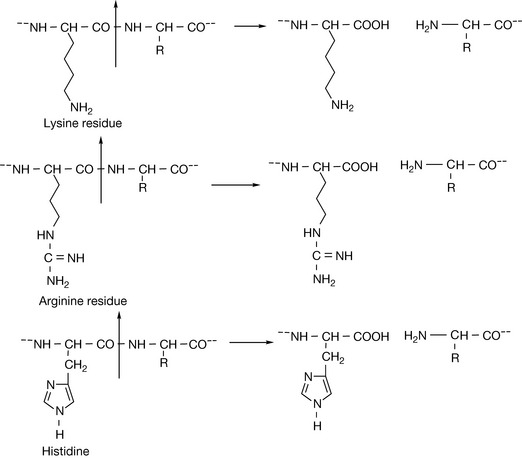
The structure of a pentapeptide (having five amino acid residues) leucine encephalin is shown in Figure 6.5. Proteins such as enzymes have many more amino acids, but there are many small peptides which have important biological activities. The encephalins are endogenous pain regulation molecules which act at opioid receptors in the body. The abbreviated version of the sequence for leucine encephalin is also shown in Figure 6.5. As instrumental methods have advanced it has become easier to determine the structure of unknown proteins. Protein molecular weights and sequences can now be rapidly determined by using mass spectrometry. The protein sequence is most often determined by carrying out a tryptic digest which cleaves proteins at the C-terminus side of one of the basic amino acid residues (Fig. 6.6) except when they are followed by a proline residue.
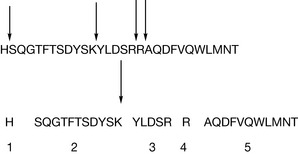
This results in a limited number of peptides which are generally in the range of 500–3000 amu and are thus amenable to analysis by mass spectrometry. Tandem mass spectrometry produces a series of fragments arising from cleavage of the peptide bonds within the molecule and the sequences for each peptide can thus be determined and the molecular structure thus determined. Considering the peptide hormone glucagon, which is involved in glucose regulation, it is possible to predict that the sequence of peptides shown in Figure 6.7 would be formed following tryptic digest. Cleavage next to the histidine, lysine and two arginine residues should, in theory, yield the five peptides and amino acid residues shown in Figure 6.7. These small peptides can then be fragmented to yield their amino acid sequence. The sequences of short peptide fragments are often sufficiently unique to yield an identity for the full peptide. Thus if the sequence of peptide 2 shown in Figure 6.7 is submitted to the Uniprot database (http://services.uniprot.org) then a series of glucogen peptides or glucogen peptide precursors is returned as containing this short sequence of amino acids. Thus the primary amino acid sequence of this undecapeptide is sufficiently unique to indicate that it originates from glucagon.
 Self Test 6.2
Self Test 6.2
Use the blast search facility at http://services.uniprot.org in order to determine which proteins the following sequences are found in. Type the sequence into the blast search form.
Protein secondary structure
The sequence of amino acids making up a protein assumes a particular three-dimensional (secondary) structure. It is possible to predict the secondary structure to some extent from the sequence of amino acids making up the primary structure. There are two fundamental structural motifs which can occur in proteins, α-helices and β-sheets, and particular protein functions tend to be associated with these motifs.
α-Helices
Alpha-helices are pharmacologically important since they are the main structural element in membrane-spanning helices which are present in many receptors, and also ion channels are formed from groups of transmembrane helices. Figure 6.8 shows a schematic diagram of an α-helix, the helix is stabilised by H-bonding between the NH and CO groups of the protein backbone and has a regular structure with 3.6 amino acids per turn.
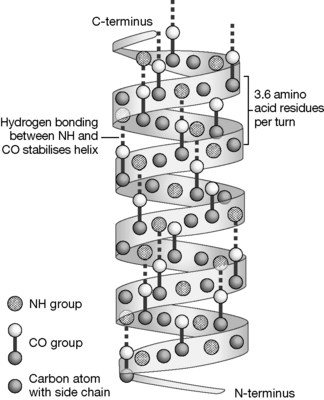
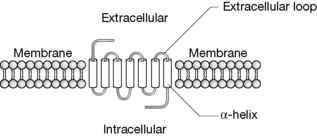
α-helices have a number of important functions and perhaps the most significant role in relation to drug action is that they form ion channels and membrane receptor binding sites. The general view of many membrane receptors is shown in Figure 6.9 where the α-helices are shown as cylinders connected by loops of peptide chain. There are usually seven membrane-spanning α-helices in receptor proteins although there may be many more in ion channel proteins. Membrane-spanning receptors are coupled to G-proteins which initiate a chain of events including binding of GTP followed by activation of adenylate cyclase, resulting in the formation of cAMP which functions as a second messenger and initiates further cellular events such as opening of ion channels in the cell membrane. Many disease states can be linked to the activity of G-proteins coupled to membrane-spanning receptors. G-protein coupled receptors include: light and olfactory receptors, receptors for adenosine, adrenaline, dopamine, bradykinin, opioids, GABA, acetylcholine, prostaglandins, glucagon, calcitonin, oxytocin, histamine, serotonin and many more. G-protein coupled receptors are also involved in drug resistance. Thus they present essentially the most important target for drugs. In addition, another mechanism of drug action is exerted via direct effects on ion channels which are also composed of membrane-spanning α-helices. There are many cases where a gene coding G-protein coupled receptors has been identified but the function of the receptor remains unknown.
The first G-protein coupled receptor to be investigated was the adrenergic receptor.1,2 Figure 6.10 shows the primary amino acid sequence of the β2-adrenergic receptor. A major goal in biological sciences is to be able to predict how primary amino acid sequences can lead to specific types of secondary protein structure, but despite some general rules this is still very difficult and where protein structures are well known this has been arrived at by crystallising them and then being able to locate the positions of the atoms in space, making up the protein using X-ray crystallography. The best way to determine the way in which a ligand binds to its receptor is to co-crystallise the ligand with the receptor protein. Again, this is technically difficult but has been achieved for a number of important receptors. In retrospect, once the protein secondary structure is known, there are often some well-established features in the primary amino acid sequence that indicate the secondary structure of the protein. The structure of the adrenoreceptor contains seven membrane-spanning helices and even if the structure of the receptor was unknown it might be possible to characterise these as areas which are rich in amino acids with lipophilic side chains which have an affinity for the hydrophobic environment of the core of the cell membrane which the helices span. The helices in the receptor are very regular with helices 1, 2, 4, 5, 6 and 7 having 24 amino acids, and helix 3 which has 22 amino acids. The helices have between 63% and 75% of lipophilic amino acid content (except for helix 7 which has 50%). In contrast, loop regions 1 and 2, which are in contact with the aqueous extra- and intra-cellular environments, have 53% and 40%, respectively. However, this is quite a crude predictive tool. The binding sites for adrenaline occur within the membrane in the helix regions. The charged amine group binds via electrostatic interaction to a glutamic acid residue in helix 3, the catechol group hydrogen bonds with the first and third serine residues in helix 5, the lipophilic portion of the benzene rings undergoes van der Waals interaction and possibly charge transfer interaction with the phenylalanine residues in helix 6, and the benzyl alcohol group hydrogen bonds with a tyrosine residue to helix 7. These interactions can be visualised as shown in Figure 6.11 and they lead to a change in the conformation of the receptor, which in turn affects the conformation of the coupled G-protein, which is bound to the long intracellular loop between residues 220 and 275, leading to the cascade of events outlined briefly above. As described in Chapter 10, only R (−) adrenaline can contact all the points of interaction within the receptor and thus the S (+) isomer is much less biologically active. The β2 agonists reinforce the reaction with the receptor. One aspect of this may be that they have higher partition coefficients than adrenaline, which encourages them to enter the lipophilic region within the transmembrane-spanning helices.

Figure 6.10 The amino acid sequence of the human β2 adrenergic receptor (ExC, extracellular; InC, intracellular). Membrane-spanning helices in bold, adrenaline binding sites in red, and serine phosphorylation sites in blue.
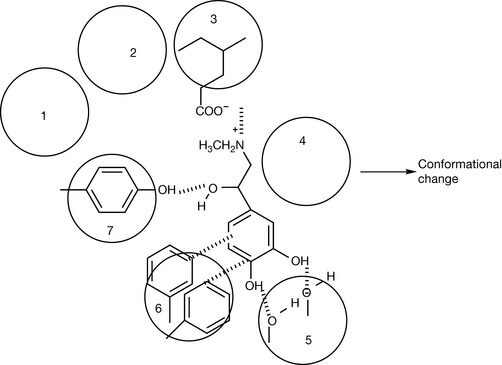
Figure 6.11 Interaction of adrenaline with the α-helices in the β2-adrenaline receptor which lead to a conformational change and activation of the coupled G-protein.
Another feature relating to the activity of the receptor can be observed in the primary structure of the final intracellular sequence at the C-terminus of the receptor. If receptors are stimulated for a long period with a ligand then they become desensitised and their action is terminated. In the adrenoreceptor, the activity of the receptor is terminated by phosphorylation of serine residues which are abundant at the C-terminus end of the receptor.
Figure 6.12 shows the primary sequence of the β1-adrenergic receptor. Like the β2-adrenergic receptor it has seven transmembrane helices which are extensively composed of lipophilic amino acids. However, there are many differences in the structure, e.g. the first extracellular loop in the β1 receptor is much longer than that in the β2 receptor. The sequences of amino acids in the helix regions of the β1 receptor are not the same as in the β2 receptor and thus the binding of noradrenaline, which is the ligand for the receptor, is not the same as adrenaline in the β2 receptor. In fact, much more is known about the β2 receptor than the β1 receptor and the binding sites in the β1 receptor are not completely elucidated, although the second and seventh helices are known to contain important binding sites.

Self Test 6.3
The primary amino acid sequence of the HT1 receptor,3 which binds the neurotransmitter serotonin, is shown below. Answer the following questions.

Another major category of α-helix rich proteins which is important in drug action is the ion channel proteins. These proteins are much larger than the receptor-coupled G-proteins and have many transmembrane-spanning helices. For example, the sodium ion channel protein associated with the action of local anaesthetics in nerve blockade has 1988 amino acid residues which form 24 transmembrane helices connected by intracellular and extracellular loops.
The β-sheet motif
The other major structural element found in proteins is the β-sheet. In this case, the primary sequence folds so that strands of the backbone are arranged parallel to each other to form a pleated sheet. Unlike the α-helix, the strands making up the sheet are not part of one continuous sequence of amino acids but have intervening loop regions. There are two ways of arranging the strands, either parallel or antiparallel, as shown in Figure 6.13. The sheets are held together by hydrogen bonding between the amide nitrogens and the carbonyls of the peptide backbone. The strands in a β-sheet can be represented by an arrow which points towards the C-terminus of the peptide. This enables parallel and antiparallel sheet motifs to be drawn, as shown in Figure 6.14. Where the ends of the strands are joined by loops of peptide, the length of the loops is greater in the case of the parallel arrangement of strands. Although the strands can be drawn in such a two-dimensional representation they have a three-dimensional structure, and a common way for a β-sheet to fold is into a barrel shape where enzymatic activity occurs within the barrel, much like the receptor binding that takes place amongst a group of α-helices. Many enzymes have a mixture of β-sheet and α-helix motifs.
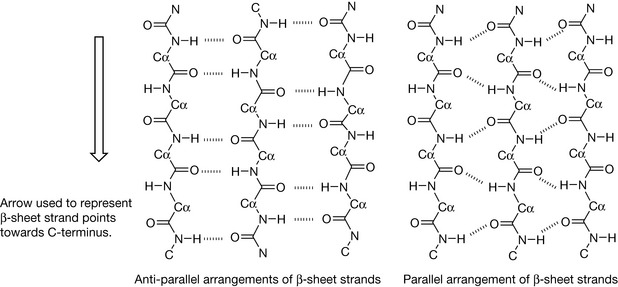
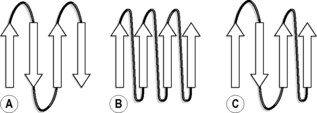
Figure 6.14 (A) Antiparallel, (B) parallel and (C) mixed parallel and antiparallel arrangement of peptide strands in β-sheets.
Retinol-binding protein provides an example of a protein that is largely composed of β-sheets. Retinol (vitamin A) is one of the fat-soluble vitamins and is important in a number of physiological functions, including vision (see Ch. 26). Since it has to be transported from the intestine where it is absorbed to its site of action it requires a carrier protein since, unlike a water-soluble vitamin such as ascorbic acid, it does not dissolve in physiological fluids. Retinol-binding protein has eight antiparallel strands which form a β-sheet which arranges itself into a barrel-like structure. Its primary sequence is shown in Figure 6.15, with the strands making up the β-sheet shown in bold. It can be viewed as having its β-sheet folded into a barrel arrangement, as shown in Figure 6.16. As can be seen from the amino acid sequences of the strands, they contain a mixture of polar and non-polar residues. The polar residues orientate themselves outwards into the hydrophilic environment while the hydrophobic residues due to amino acids such as valine, leucine and phenylalanine which occur in the residues lining the interior of the barrel and bind the hydrophobic retinol molecule (Fig. 6.17). The amino acids at the N-terminus and C-terminus ends of the molecule are partly in the form of α-helices and fold over to close the barrel so that the retinol is protected from the hydrophilic environment. These types of proteins are classed as lipocalins and they have a widespread role in binding lipophilic molecules such as essential fatty acids, pheromones and olfactory molecules. They all have the barrel-like structure shown in Figure 6.16.

Figure 6.15 Primary sequence of retinol binding protein. The eight β-sheet regions which form the barrel are shown in bold.
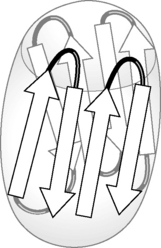
Figure 6.16 Barrel-like conformation adopted by retinol-binding protein β-sheets in order to shield retinol from a hydrophilic environment.
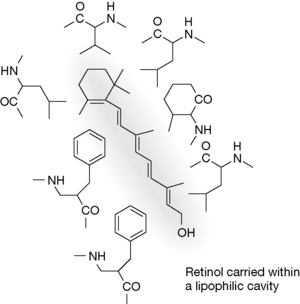
Figure 6.17 Lipophilic interaction between retinol and the groups lining the barrel formed by the β-sheet.
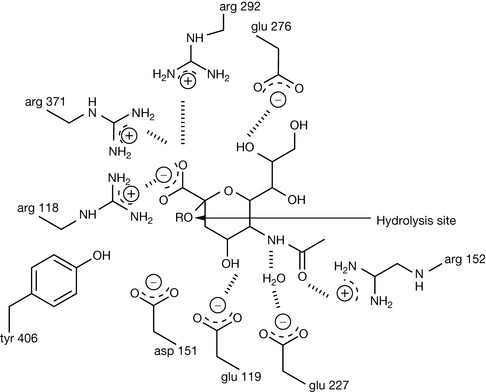
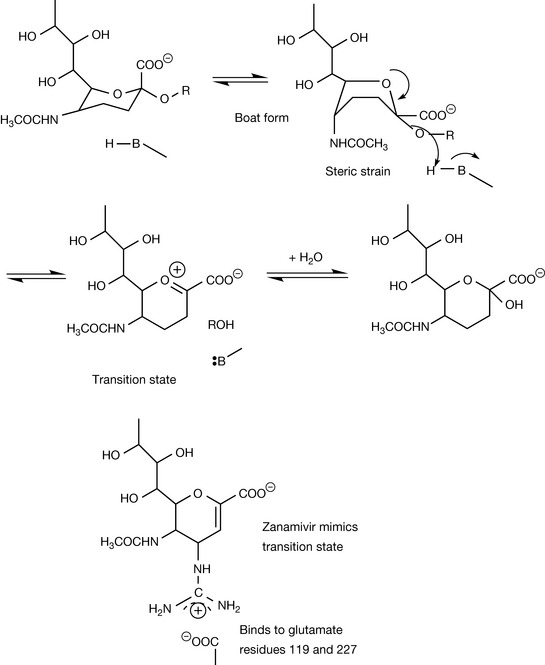
Many proteins have a mixture of α-helix and β-sheet domains. Neuraminidase is a large protein composed of β-sheet domains arranged in a propeller-like conformation surrounded by α-helices. It is a target in chemotherapy against influenza (see Ch. 23).4,5 The active site of the enzyme is located within the β-sheet domains. Once crystal structure information on the enzyme became available, it became possible to see that certain residues within the binding pocket of the enzyme were conserved between different strains of the virus and, using modelling software, that these residues would be likely to interact favourably with the substrate. Compounds with inhibitory activity were designed and the formation of crystals of the inhibitor enzyme complexes enabled refinement of the design of the inhibitor. The interactions of the substrate with the active site are shown in Figure 6.18. The substrate is strongly bound into place by the interaction of three arginine residues with its carboxyl group. The actual hydrolysis of the glycosidic bond is most probably catalysed by an aspartate or a glutamate residue (Fig. 6.19). The substrate binds very strongly to the active site and its conformation becomes distorted from a chair to a boat form. The boat form is very strained and the relief of steric strain upon hydrolysis helps to promote it. Once the rest of the sugar chain has been removed, a transition state oxonium ion is formed which instantaneously reacts with water (this is called a transition state analogue but in fact the true transition state is at the point where RO is just leaving the molecule). Zanamivir mimics the structure of the intermediate formed during hydrolysis and at the same time is strongly bound to the enzyme via interaction between its positively charged guanidine group and negatively charged aspartate residues at the active site of the enzyme.
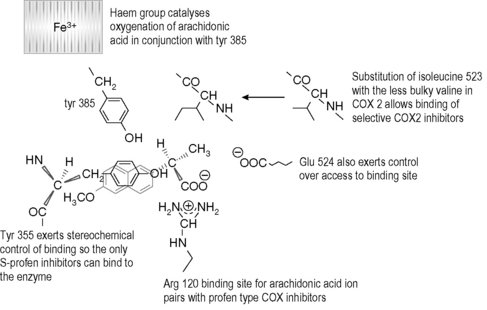
The design of selective cyclooxygenase (COX) inhibitors has attracted much interest over the last few years because of the side effects associated with the existing non-steroidal anti-inflammatory drugs (NSAIDs) which inhibit both COX-1 and COX-2.6,7 COX-1 is generally found in cells without being induced and, for instance, exerts a protective effect on the GI tract whereas COX-2 is up-regulated in response to, for instance, infection, and drives inflammatory processes and is the target of drugs used to treat inflammation. COX-1 and COX-2 have very similar primary amino acid sequences and thus drugs such as NSAIDs affect COX-1 potentially causing damage to the GI tract as well as affecting COX-2 to reduce inflammation. COX-2 is largely composed of helical regions, and the binding of substrate and the regions of catalytic activity are found largely in the loops joining the helices together. The binding site residues in COX-1 are shown in Figure 6.20. The arginine group at position 120 in the protein strongly binds to the carboxyl group of the enzyme substrate arachidonic acid or to the carboxyl group of a profen-type NSAID. The entry of the substrate to the active site is stereochemically controlled and there are a number of other key residues at the binding site, including a tyrosine at position 355 which exerts a steric hindrance effect restricting the compounds, which can enter the binding site, and a glutamate residue at position 524 which also exerts stereochemical control. The stereochemical control explains why the S-isomers of profen-type NSAIDs are active. COX-2 inhibitors were largely found by random screening and they bind specifically to COX-2 due to stereochemical effects in the region around a valine residue at position 523 which is replaced by a more bulky isoleucine group in COX-1.
1 Isogaya M., Sugimoto Y., Tanimura R., et al. Binding pockets of the β1- and β2-adrenergic receptors for subtype-selective agonists. Mol Pharmacol. 1999;56:875-885.
2 Behr B., Hoffman C., Ottolina G., Klotz K.-N. Novel mutants of the human β1-adrenergic receptor reveal amino acids relevant for receptor activation. J Biol Chem. 2006;281:18120-18125.
3 Lopez-Rodriguez M.L., Vicente B., Deupi X., et al. Design, synthesis and pharmacological evaluation of 5-hydroxytryptamine 1a receptor ligands to explore the three-dimensional structure of the receptor. Mol Pharmacol. 2002;62:15-21.
4 von Itzstein M. The war against influenza discovery and development of sialidase inhibitors. Nature Drug Discovery Rev. 2007;6:967-974.
5 Magesh S., Suzuki T., Miyagi T., Ishida H., Kiso M. Homology modeling of human sialidase enzymes NEU1, NEU3 and NEU4 based on the crystal structure of NEU2: Hints for the design of selective NEU3 inhibitors. J Mol Graph Model. 2006;25:196-207.
6 Kiefer J.R., Pawlitz J.L., Moreland K.T., et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature. 1999;405:97-101.
7 Marnett L.J., Kalgutkar A.S. Cyclooxygenase 2 inhibitors: discovery, selectivity and the future. TIPS. 1999;20:465-469.