Chapter 9 Structure, activity and drug design
Introduction
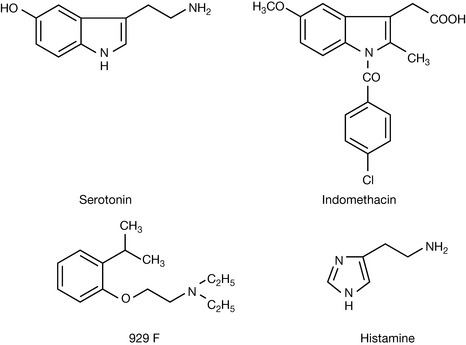
Much of modern drug discovery took place between 1930 and 1980 before there was a comprehensive understanding of how drugs can be specifically designed to work in a particular way. For example, the non-steroidal anti-inflammatory drug indomethacin (Fig. 9.1) was produced since there was a theory that serotonin was involved in inflammatory processes.1 However, although there are links between serotonin and inflammation, it acts at specific transmembrane-spanning receptors, whereas indomethacin is an inhibitor of cyclooxygenase enzymes. Alternatively, bioactive lead compounds, bearing no direct resemblance to the biomolecule of interest, have often been found by random testing. For example, compound 929 F (Fig. 9.1) was tested for its activity in blocking the action of histamine and most subsequent antihistamine compounds bear some resemblance to it although it does not closely resemble histamine itself.
The additional tranquillising effects shown by antihistamines then gave rise to antipsychotic agents such as chlorpromazine. Thus, to some extent, the formulation of the rules governing structure activity relationships of drugs has been carried out retrospectively. As outlined in Chapter 1, there are several types of intramolecular bonding involved in drug action, ranging from covalent bonds through to hydrogen bonds and van der Waals forces. All of these forces are utilised in the modification of a lead compound into a successful drug. Since the focus of this book is on pharmaceutical, rather than medicinal, chemistry, this chapter will concentrate more on explaining relationships between existing drugs rather than focusing on how to discover new ones.
Isosteres
Isosteres are structural elements that are deemed to have more or less equivalent chemical or physical properties, and they have been used extensively in modification of lead compounds in order to enhance drug action. Table 9.1 shows some classical isosteres. The definition of an isostere is an atom or a group in which the peripheral layers of electrons are identical.2 However, this definition is not adhered to and, for instance, F is often considered as a substitute for H although it has three lone pairs of electrons in its peripheral layer whereas H does not have any. Thus the strict definition of isosteres quite quickly breaks down and, for instance, benzene can be substituted for by a number of ring types and carbonyl and carboxylate substituted by groups which are not that close to them in structure. Thus isosteric substitution is not an exact science.
| Type of group | Isosteres |
|---|---|
| Univalent | H, F, Cl |
| Univalent | CH3, NH2, OH, F, SH, Cl, CF3, |
| Univalent | Br, i-Pr |
| Univalent | I, t-Bu |
| Bivalent | -CH2-, -NH-, -O-, -S-. |
| Trivalent | -CH=, -N= |
| Rings |
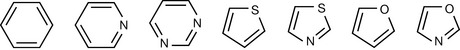 |
| Carbonyl CO |
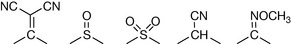 |
| Carboxyl COOH | 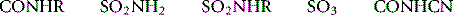 |
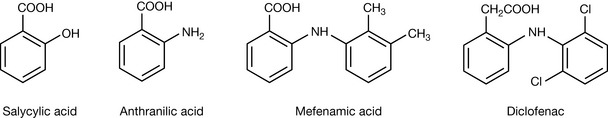
Substitution of isosteric components can alter the activity of a compound. For example, one of the first non-steroidal anti-inflammatory (NSAID) compounds produced was mefenamic acid. Its structure derives from that of salicylic acid, the first non-steroidal anti-inflammatory drug. First there is an isosteric substitution of NH2 for OH in salicylic acid (Fig. 9.2), which causes loss of anti-inflammatory activity. Then a dimethylbenzene group is added to the NH2 group as a substituent to produce the NSAID mefenamic acid (Fig 9.2). This has the effect of making the resultant NH group into a very weak base so it is hardly charged at stomach pH and not at all at plasma pH. In diclofenac (Fig. 9.2) an isosteric substitution of chlorine for the methyl groups in mefenamic acid is made and the carboxyl group is separated from the ring by a CH2 group. These small alterations cause increase in potency, with diclofenac having about five times the potency of mefenamic acid. The exact reasons for the increased potency cannot be defined although one site of metabolism is removed in substituting Cl for methyl since in mefenamic acid the 3′-methyl group is a site of metabolism being converted to a carboxylic acid (cf. chlorpropamide and tolbutamide, Ch. 4) and moving the carboxyl group away from the aromatic ring weakens the acidic group slightly.
Similar types of isosteric substitution can be seen in the evolution of antihistamines with the addition of ring to chain transformations and ring formation. The conversion between rings and chains is a common modification in the evolution of a drug structure. The introduction of a ring restricts the stereochemical conformations that a drug can take, which can potentially increase potency. Cyclizine (Fig. 9.3) was one of the first antihistamines. Its structure was evolved in a number of different directions. Isosteric substitution of nitrogen for carbon, conversion of the ring to an open chain and locking of the two benzene rings together with a sulphur atom led to promethazine which is somewhat more potent than cyclizine. Both drugs are currently used to treat nausea. Addition of an extra methylene unit into the side chain of promethazine and removal of the methyl group substituted onto the side chain led to its isomer, promazine. The small structural change led to the development of a series of antipsychotic agents based on promazine, which is still used as an antipsychotic. The simplest modifications were isosteric substitutions of hydrogen by chlorine and by trifluoromethyl, producing chlorpromazine and flupromazine which have higher partition coefficients than promazine and thus greater CNS penetration and higher potency.
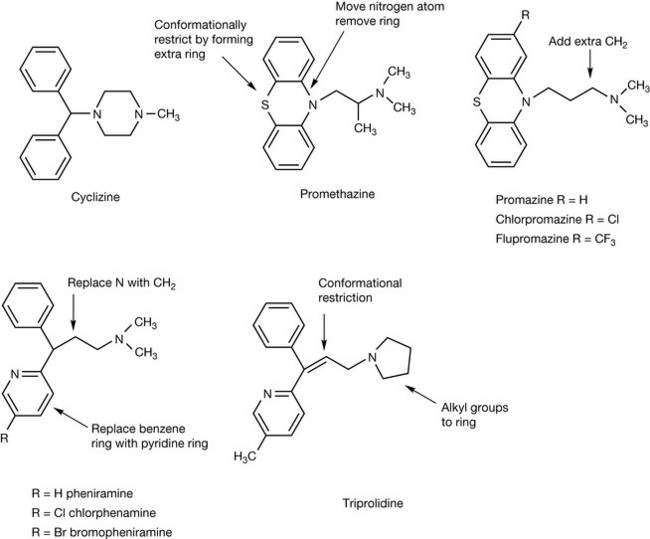
The replacement of the nitrogen atom in the side chain of cyclizine by carbon and isosteric replacement of one of the benzene rings by a pyridine ring led to pheniramine which was further elaborated by replacement of a hydrogen atom by chlorine or bromine thus increasing potency. In triprolidine conformational restriction is introduced into the side chain via a double bond and the methyl groups attached to the nitrogen in the side chain are changed into a ring, increasing potency further.
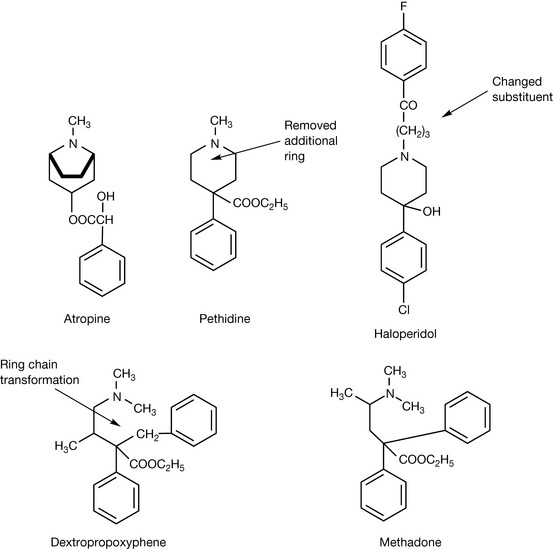
During the golden era of drug discovery between 1930 and 1980 there was often no clear awareness of isosteres in drug design, but retrospectively it is possible to see a pharmacore evolving through a series of compounds, often starting with one type of biological activity and finishing with another. For example, the biological effects of atropine (Fig. 9.4) which occurs in some members of the solanaceous plants, e.g. deadly nightshade, was known for centuries due to its effects as an antagonist of cholinergic receptors. Thus plant extracts containing atropine were used to treat colic by relaxing the smooth muscle of the gut and for pain relief in neuralgia. The structure of atropine is close to that of the synthetic analogue pethidine, which is used in analgesia, and the basic pharmacophore is also retained in haloperidol, which is used as a sedative to control hyperactive psychotic states. In moving to dextropropoxyphene, the piperidine ring was converted into a chain producing an analgesic which does not have the addictive potential of pethidine. The structure of methadone is also based on this basic pharmacophore and again has analgesic properties and is used as a morphine substitute since it is orally bioavailable.
 Self Test 9.1
Self Test 9.1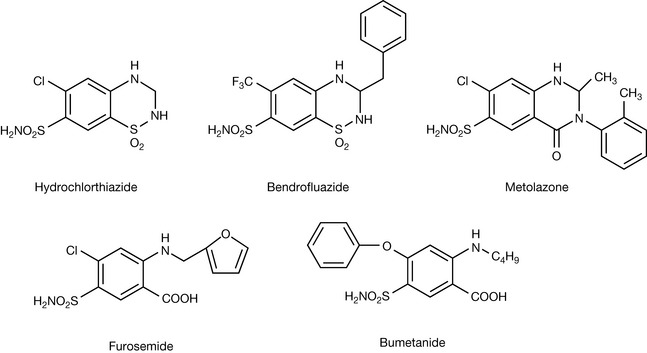
Intramolecular forces governing drug action
Lipophilicity
The importance of lipophilicity in the interaction of drugs with membranes has been discussed in Chapter 2. Lipophilicity is governed by van der Waals forces and these have been discussed in Chapter 1. It is difficult to focus on one type interaction within a drug since the overall interaction of the drugs with proteins and membranes depends on all the forces outlined in Chapter 1. Thus the overall interaction may involve covalent bonding, ionic bonding, dipole–dipole interaction, hydrogen bonding, charge transfer or van der Waals interaction. However, it is simplest to consider one factor at a time. The simplest type of structure activity relationships can be viewed when one factor is varied while everything else is held constant. A classical series for illustrating how substituent groups affect partition coefficient is provided by the barbiturates, which are rarely used because of their addictive potential. The Hansch approach to estimating partition coefficients (log P values) uses substituent coefficients (π). Table 9.2 gives some Hansch substituent coefficients. The values can be applied in the calculation of theoretical partition coefficients for the barbiturates shown in Figure 9.5. The higher the partition coefficient the greater the penetration into the CNS and thus the higher the sedative potency. The larger the lipophilic subsituent the higher the partition coefficient. Many compounds do not fall into such a convenient series for the calculation of log P.
| Group/residue | π |
|---|---|
| barbiturate ring | −1.35 |
| N-methyl barbiturate ring | −1.07 |
| -CH-/-CH2-/CH3-/-C= | 0.50 |
| C6H5-(phenyl) | 1.96 |
| Cyclohexen-1-yl | 2.21 |
| -CH2-(ring residue) | 0.40 |
The following increments are deducted if the alkyl group is either branched or contains a double bond or both:
Each branch (π) = −0.2
Each double bond (π) = −0.3
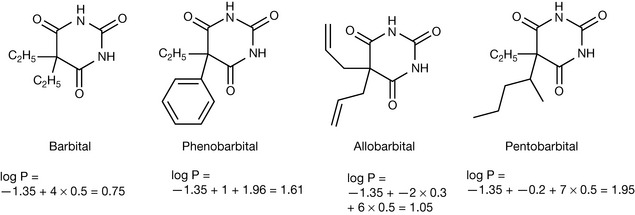
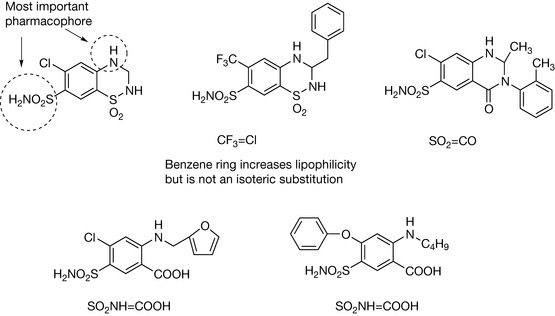
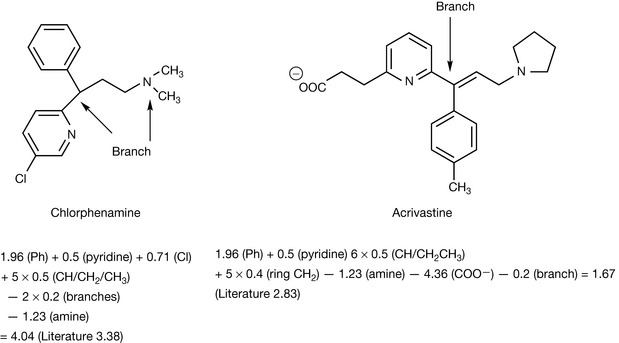
Table 9.3 shows some further substituent constants, and some further calculations are shown for some antihistamine drugs. The calculated values are not in complete agreement with the literature; however, calculated values are never perfect and the π-constants vary within the literature. Comprehensive lists of these are available.3 The calculated octanol/water partition coefficient for acrivastine does not agree well with the literature. However, the literature is reported as if it were possible for it to be a neutral species but since it is an amphoteric compound it can only be neutral at its isoelectric point where the amine group and the carboxyl group are equally ionised (see Ch. 7 for discussion of isoelectric points). Taking a naive approach and putting all the charge on the carboxyl group produces a partition coefficient which is too low. An important point is made by the examples shown in Figure 9.6, which is that partition coefficient alone does not predict activity. Chlorphenamine and acrivastine are of a similar potency as antihistamines but differ in their partition coefficients. The Hansch approach has been refined over the years and an example of the Hansch equation is shown below:
Table 9.3 Some examples of Hansch π and σ constants
| Substituent | π | ρσ |
|---|---|---|
| -COOH | −0.32 | 0.41 |
| -COO- | −4.36 | −0.1 |
| -OH | −1.12 | −0.38 |
 |
−1.23 | −0.66 |
| -CN | −0.57 | 0.5 |
| -H | 0 | 0 |
| -Cl | 0.71 | 0.23 |
| -F | 0.14 | 0.06 |
| -CF3 | 0.1 | 0.54 |
| -Br | 0.86 | 0.23 |
| -OCH3 | −0.02 | −0.27 |
| Pyridine ring | 0.5 | 0.44 |
| CONH | −1.49 | 0.36 |
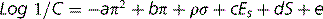
The terms in the equation can be explained as follows:
Log1/C gives an indication of the potency of the drug and is the molar concentration which gives 50% of maximal response, whether this is enzyme inhibition (IC50) or receptor agonism/antagonism (LD50). The lower the value of this term, the lower the potency of the drug.
The term −aπ2 relates to the substituent constants that we have already considered. It takes account of the fact that for any drug there is an optimum partition coefficient and thus a plot of potency against partition coefficient is a parabola. The term a is experimentally derived from the observation of how large the partition coefficient has to be before potency starts to reduce. The bπ reflects the positive contribution of partition coefficient to activity and obviously the constant b has a much larger value than a. The term ρσ gives an indication of the contribution of the electronic nature of functional groups to the activity of the molecule; in simple terms it indicates which groups can be substituted for each other in optimising structure activity. Of course, not all groups in the molecule are critical to its activity. Table 9.2 shows some σ constants; negative values indicate that the substituent donates electrons to the structure and positive values indicate electron withdrawal. The values given in the table only apply to substituents attached to benzene ring and this is a weakness of the Hansch approach. The terms Es and dS relate to the shape of the molecule. The complexity of the parameters within the Hansch equation means that complex computer models have to be set up in order to predict activity from structure. Still, the vast majority of drugs on the market have not been designed in a particularly rational way. One modern approach which will be considered throughout this book is drug optimisation based on a better understanding of the structures of the proteins which are the targets of drugs. The availability of protein crystal structures and sometimes the structures of co-crystals with a ligand bound to a protein assist in getting a better understanding of what governs the binding of a drug to an enzyme or a receptor. Where the structure of a co-crystal of the drug with its target is available, it is possible to see directly what the important interactions between the drug and amino acids in the protein are. In the absence of a co-crystal, molecular docking studies can be carried out where computer simulation of the interaction of the drug with its binding site is carried out in order to determine the likely mode of binding and how binding might be optimised in order to design more potent analogues of the drug.