Infectious Disorders of the Central Nervous System
OVERVIEW
Infection of the central nervous system (CNS) is relatively rare in that many protective responses limit the access of harmful organisms to the nervous tissue. However, neurologic infections are a major cause of mortality and morbidity worldwide. Bacterial infections can be serious and life-threatening. Biologic adaptations of infectious agents and altered modes of transmission present new challenges. Drug-resistant strains create new threats, and travel has increased the exposure to both viruses and the bacteria that can cause infection of the nervous system.15,26
Once there is access to the brain, viruses produce a large range of neuropathologic conditions. They can be oncogenic, producing astrocytomas.40 CNS infections can affect the brain’s parenchyma, directly causing abscess. Remote infectious processes, such as bacterial endocarditis, resulting in infected emboli, can cause infectious intracranial aneurysms.32 Sepsis can cause diffuse, multifactorial involvement of the CNS, including invasion of the meninges with resulting meningitis.39 The cerebral spinal fluid (CSF) can be contaminated when an object penetrates the meninges. This is often a result of trauma or a neurosurgical procedure.8 Trauma to the front of the face that causes damage or fracture of nasal structures or the cribriform plate can lead to infection in the CSF. Infection of the inner ear can spread to the brain via the CSF.33
MENINGITIS
Definition
In meningitis, the meninges of the brain and spinal cord become inflamed. The three layers of the meningeal membranes (dura mater, arachnoid, and pia mater) can be involved. The relationship of the meninges to the brain tissue is shown in Fig. 29-1. The pia mater and arachnoid become congested and opaque. The inflammation extends into the first and second layers of the cortex and spinal cord and can produce thrombosis of the cortical veins. There is an increased chance of infarction, and the scar tissue can restrict the flow of CSF, especially around the base of the brain. This block of CSF can result in hydrocephalus or a subarachnoid cyst. Stretch or pressure on the meninges will cause the cardinal sign of headache.1
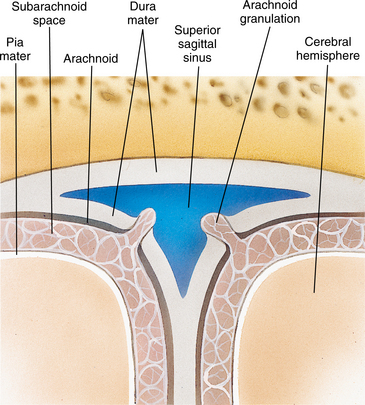
Incidence
The estimated incidence of meningitis is 2 to 6 per 100,000 adults per year in developed countries and is up to 10 times higher in less-developed countries.44 There appears to be increased susceptibility in some populations, but this may also be related to living conditions and temperate climates. There is a meningitis belt in sub-Saharan Africa, where the incidence is five to ten times higher than in the developed world. There is a genetically determined deficiency that increases susceptibility and risk, but this accounts for only a small portion of those who develop infection.22,35
Etiologic and Risk Factors
Bacterial meningitis once was predominantly a disease of young children and older adults. Vaccines developed in the past 15 years to protect against the development of meningitis, primarily against Haemophilus influenzae type B (Hib) infection, have dramatically decreased the incidence in the countries where there is access to the vaccine. There appears to be a second period of increased susceptibility during late adolescence. In adulthood, bacterial meningitis is mostly associated with conditions that affect the defense mechanisms of the host.15 Individuals with compromised immune function related to other conditions, such as human immunodeficiency virus (HIV), remain at high risk to develop meningitis.26 When there is damage or removal of the spleen, a person becomes more susceptible to pneumococcal disease. Otitis, mastoiditis, and sinusitis are common predisposing conditions that may need specialized treatment. Neoplastic meningitis is a complication that occurs infrequently but is characterized by neurologic signs and symptoms and has a poor outcome.19 Meningitis associated with cutaneous anthrax became an urgent health concern with the 2001 bioterrorism threat. Although there is meningeal involvement in only 5% of persons exposed, studies of experimental inhalation anthrax in monkeys have demonstrated meningeal involvement in 40% to 50% of cases.
Pathogenesis
The process of infection is complex, but a series of events can be established. Immune responses destroy organisms at the site of the infection and in the blood. Bacteria and viruses are removed from the blood by the reticuloendothelial system. Infection can be carried by blood products or other fluids and can cause changes in the cerebral capillary endothelium. The blood-brain barrier then fails to prevent entry of infectious organisms into the brain or CSF. The blood-brain barrier is a mechanism of the endothelium that selectively inhibits certain substances from entering the interstitial spaces of the brain or CSF. It is believed that the astrocytes and tight junctions with high electrical resistance between the endothelial cells of the brain capillaries create this barrier. The barrier opposes passage of most ions, small peptides, proteins, and high-molecular-weight compounds.39
Once there is penetration of the blood-brain barrier and infectious agents move into the CSF and parenchyma of the brain, there is less immune protection than in the rest of the body. The CSF has about 1/200 the amount of antibody as blood, and the number of white blood cells is very low compared with the blood. The brain lacks a lymphatic system to fight infection.14
During infection and inflammation, the level of leukocytes in the brain increases. Cytokines, chemokines, macrophages, and microglia respond to viral and bacterial infections. The polymorphonuclear cells recruited to the infection cause damage to the surrounding brain tissue by the release of cytotoxic free radicals and excitatory amino acids such as glutamate. Oxidative stress may be responsible for apoptosis in the hippocampus. Responses to inflammation in the brain can block CSF, resulting in hydrocephalus, edema, and increased intracranial pressure. Vasculitis can lead to infarction and decreases in cerebral blood flow, causing a drop in the glucose level of the CSF.7,35 Drugs that scavenge for free radicals and the use of N-methyl-D-aspartate (NMDA) receptor blockers can help reduce tissue injury.40
Aseptic (Viral) Meningitis.: Viral infection is the most common cause of inflammation of the CNS. Aseptic meningitis is primarily caused by contamination of the CSF by a virus or fungus. Aseptic meningitis most often is caused by enteroviruses, which are the major cause of meningitis in 40% of those 30 to 60 years old. The second most common cause of meningitis is herpes simplex virus 2, which is detected in approximately 20% of individuals with meningitis. Epstein-Barr virus (EBV) can also be responsible and is more often seen in late adolescence and early adulthood. Systemic lupus erythematosus (SLE), a disorder of connective tissue, can cause aseptic meningitis. Sarcoid tumors and other intracranial tumors or cysts can lead to aseptic meningitis through rupture.3 Often the meningitis occurs days or weeks after the exposure. Recurrent aseptic meningitis is two or more episodes with a disease-free interval between. This must be distinguished from the waxing and waning of chronic meningitis.16
Certain drugs or chemicals can cause aseptic meningitis. The drugs most commonly involved are the nonsteroidal antiinflammatory medications (NSAIDs). Chemicals can cause direct meningeal irritation and are often related to surgical procedures that expose the chemical.
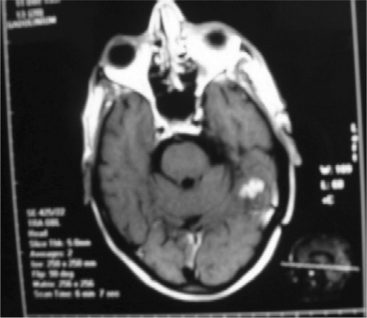
Tuberculous Meningitis.: Tuberculous meningitis is an infection by Mycobacterium tuberculosis, which enters the body by inhalation.15,42 CNS involvement includes abscess or spinal cord disease. A computed tomography (CT) image of a tuberculoma is seen in Fig. 29-2. Tuberculous brain abscesses may produce mass effect and edema. CSF may demonstrate formation of multiple cysts with lymphocytes and an elevated protein. Infected bacilli enter the subarachnoid space to cause diffuse meningitis.28
Bacterial Meningitis.: The organisms generally responsible for bacterial meningitis are those found in mucosal surfaces in the upper respiratory tract. Bacteria in the birth canal can be transferred from the mother to the infant during birth. Group B streptococcus, Escherichia coli, and Listeria monocytogenes are bacteria that can cause infection in the neonate, although antibodies are passed through the placenta. As these antibodies decline, the susceptibility to Hib, pneumococcus, and meningococcus increases, especially in the second half of the first year of life. On the other end of the spectrum, Streptococcus pneumoniae and Neisseria meningitis are the most common bacteria causing infection in the adult and geriatric populations.31,35
In bacterial meningitis, inflammation initially is confined to the subarachnoid space, then spreads to the adjacent brain parenchyma. Vasculitis starts in the small subarachnoid vessels. Thrombotic obstruction of vessels and decreased cerebral perfusion pressure can lead to focal ischemic lesions. Veins are more frequently affected than arteries, probably because of their thinner vessel walls and the slower blood flow. Damage to the cell bodies causes the production of amyloid-β precursor protein (APP) that is carried through the axon and accumulates within terminal axonal swellings, or spheroids. This axonal pathology contributes to neurologic sequelae seen after bacterial meningitis.29
Clinical Manifestations
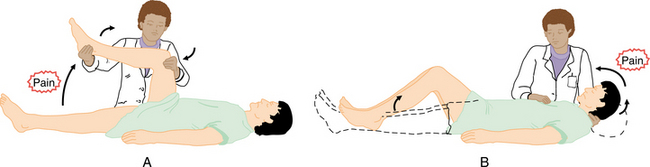
Early features of meningitis include fever and headache associated with a stiff and painful neck. There is often pain in the lumbar area and the posterior aspects of the thigh. Kernig’s sign, or pain with combined hip flexion and knee extension, is positive. As the inflammation progresses, flexion of the neck will produce flexion of the hips and knees. This is known as a positive Brudzinski’s sign.36 The positions for Kernig’s and Brudzinski’s tests are shown in Fig. 29-3. If the infection remains unde- tected or untreated, the brainstem centers may be affected. The individual may then experience seizures and coma, vomiting, and papilledema. Focal neurologic signs, including cranial nerve palsies and deafness, can also be seen when the brainstem is affected.
MEDICAL MANAGEMENT
Lumbar puncture is the only absolute means of substantiating a diagnosis of meningitis. The viruses causing viral meningitis can be isolated in CSF and include enteroviruses, lymphocytic choriomeningitis, and herpes simplex virus. Lumbar puncture reveals mononuclear cells in the hundreds, a normal glucose level, a mild increase in protein, and absence of bacterial organisms (see the section on Laboratory Values in Chapter 40). Radiographs are taken to rule out fracture, sinusitis, and mastoiditis. A CT scan or magnetic resonance imaging (MRI) will reveal evidence of brain abscess or infarction that may be responsible for the symptoms. Fig. 29-2 shows evidence of abnormal MRI with tuberculous meningitis.
Viral infection is the most common cause of inflammation of the CNS in children. Differentiation from bacterial infection of the CNS is made on the basis of signs and symptoms and CSF changes. Clinical symptoms consistent with meningeal involvement are milder but overlap with those of bacterial infection.31
Prompt diagnosis is critical in bacterial meningitis since death can occur without antibiotic treatment. Because determining the bacterial etiology can take up to 48 hours with CSF cultures, an alternative diagnostic test should be considered. Gram stain examination of CSF is recommended when meningitis is suspected. It is fast, inexpensive, and accurate up to 90% of the time. Polymerase chain reaction (PCR) is useful for excluding a diagnosis of bacterial meningitis and may eventually, with further refinement, be used for determining etiology.14
When CSF findings suggest bacterial meningitis, but Gram stain and culture results are negative, a combination of laboratory tests is necessary to distinguish bacterial from viral meningitis. In bacterial meningitis, opening pressure generally is between 200 and 500 mm H2O (lower in children), white blood cell count and protein concentration are elevated, glucose concentration may be low, and there may be a neutrophil or lymphocyte predominance.40
When there is a neoplasm or brain tumor, the infection may be the result of pleomorphic manifestations of neoplastic meningitis and co-occurrence of disease at other sites. Useful tests to establish diagnosis and guide treatment include MRI of the brain and spine, CSF cytology, and radioisotope CSF flow studies. Assessment of the extent of disease of the CNS is valuable because large-volume subarachnoid disease or CSF flow obstruction is prognostically significant.19
Anthrax meningitis produces CSF that is marked by polymorphonuclear pleocytosis and hemorrhage. CSF protein elevation is generally present, and CSF glucose concentrations are generally decreased, as is seen with causes of other bacterial meningitis.38
The time course after onset of the disease indicates the type of organism involved. Viral meningitis is hyperacute, with symptoms developing within hours. Acute pyogenic bacterial meningitis can also develop in 4 to 24 hours. Individuals with fungal meningitis or tuberculous meningitis develop symptoms over days to weeks. In tuberculous meningitis, there is a predominance of mononuclear cells, the glucose level is decreased, and protein is increased. It is difficult to identify the tuberculosis bacterium, so clinical signs are important to follow.15
TREATMENT.
Guidelines for the diagnosis and treatment of bacterial meningitis from the Infectious Diseases Society of America (IDSA) can be found at http://www.journals.uchicago.edu/CID/journal/issues/v39n9/34796/34796.text.html.40
When acute bacterial meningitis is suspected, antimicrobial therapy should begin as soon as possible. Bacterial meningitis is a neurologic emergency; progression to more severe disease reduces the likelihood of a full recovery. Targeted antimicrobial therapy can begin in adults following a positive CSF Gram stain result. Antibiotic therapy should not be delayed pending the results of Gram stain or other diagnostic tests. Antimicrobial therapy should be modified as soon as the pathogen has been isolated. Duration of therapy depends on individual responses.40
Suspected bacterial meningitis in a child or infant is considered a medical emergency. The general picture involves fever, decreased feeding, vomiting, bulging fontanel (in infants), seizures, and a high-pitched cry. In neonates with meningitis caused by gram-negative bacilli, the duration of therapy should be determined in part by repeated lumbar punctures documenting CSF sterilization. If there is no response after 48 hours of appropriate therapy, repeated CSF analysis may be necessary. Because any complications of bacterial meningitis usually occur within the first 2 or 3 days of treatment, outpatient management requires close follow-up. Criteria for outpatient therapy are inpatient antimicrobial therapy for 6 or more days; no fever for at least 24 to 48 hours; no significant neurologic dysfunction, focal findings, or seizure activity; stable or improving condition; ability to take fluids by mouth. There should be an established plan for physician and nurse visits, laboratory monitoring, and emergencies. Seizures can be controlled with antiseizure medications. As the infection is controlled, the seizures are resolved, so a short course is all that is usually necessary.40
The addition of dexamethasone can reduce the subarachnoid space inflammatory response that is related to morbidity and mortality and may therefore alleviate many of the pathologic consequences of bacterial meningitis related to cerebral edema or cerebral vasculitis. Change in cerebral blood flow, increase in intracranial pressure, and neuronal injury can be controlled by judicious steroid use.
Radiologic treatment is effective with neoplastic meningitis. Because neoplastic meningitis affects the entire neuraxis, chemotherapy treatment can include intra-CSF fluid. Neoplastic meningitis is often a part of a progressive systemic disease, and consequently treatment is palliative.19
Usual treatment for viral meningitis is symptomatic. Medication is given for the headache and nausea. The prognosis in viral meningitis is excellent, and most individuals recover within 1 to 2 weeks. Treatment of acute episodes of herpes meningitis with acyclovir has been shown to decrease the duration and severity of symptoms. It may work as well for prophylactic control of episodes. Tuberculous meningitis is managed with drugs given to treat the tuberculosus. In addition, adjunctive therapy with corticosteroids may reduce mortality and decrease neurologic sequelae in severe meningitis.
PROGNOSIS.
Mortality ranges from 5% to 25% depending on the infecting bacteria and the health and age of the person infected. At least one neurologic complication, such as impairment of consciousness, seizures, or focal neurologic abnormalities, typically develops in 75% of individuals with bacterial meningitis. Systemic complications, cardiorespiratory failure, or sepsis are also common and found about 40% of the time. Hyponatremia occurs about 30% of the time, with an average duration of 3 days, well managed by fluid restriction.
Cranial nerve palsies occur about 30% of the time, with hearing impairment during hospitalization a common complaint, but more than half have full return of hearing. The severity of hearing loss was graded as mild one-third of the time, moderate one-third, and profound in another third. When there is hearing loss, it is more likely to be bilateral than unilateral.44
In children, long-term neurologic consequences of bacterial meningitis include developmental impairment, hearing loss, blindness, hydrocephalus, hypothalamic dysfunction, hemiparesis, and tetraparesis. There is a 30% mortality rate, with increasing death with individuals over 60 years. Most death occurs within 2 weeks, as a result of both systemic and neurologic complications. Aseptic or viral meningitis is usually self-limiting, and there is not the same degree of neurologic sequelae involved. Mortality rates for tuberculous meningitis range from 20% to 50%, and survivors may be left with neurologic sequelae similar to those seen in acute bacterial meningitis.22,23 With better understanding of the role of cytokines, therapies targeting these processes are under study and show promise. These therapies may help to further control damage to the nervous system during the infectious or inflammatory process.7
ENCEPHALITIS
Definition
Encephalitis is an acute inflammatory disease of the parenchyma, or tissue of the brain, caused by direct viral invasion or hypersensitivity initiated by a virus. Encephalitis is characterized by inflammation primarily in the gray matter of the CNS. Neuronal death can result in cerebral edema. There can also be damage to the vascular system and inflammation of the arachnoid and pia mater.2 Viruses carried by mosquitoes or ticks are responsible for most of the worldwide known cases of primary CNS infection. In many cases, such as West Nile virus and herpes simplex virus, the individual can develop either encephalitis or meningitis. This is reflected in the different levels of impairment that may be experienced after exposure.
Incidence
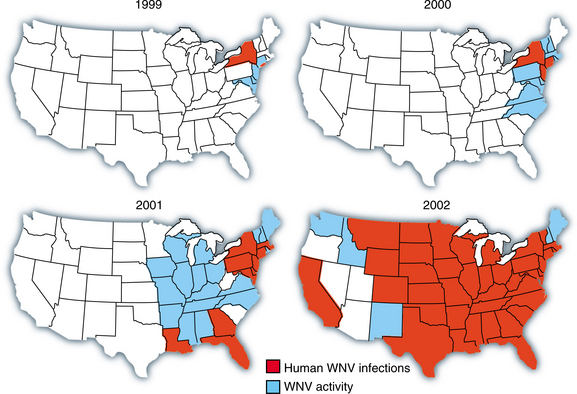
Before 1994, outbreaks of West Nile virus were sporadic and occurred primarily in the Mediterranean region, Africa, and eastern Europe. Since 1994, outbreaks have occurred with a higher incidence of severe human disease, particularly affecting the nervous system. By 2002, incidence was 4 to 14 per 100,000 population in the Midwest. Fig. 29-4 illustrates the spread of the virus through the United States. The virus has caused meningitis, encephalitis, and poliomyelitis, resulting in significant morbidity and mortality.27 Norwegian scientists have reported that an unexpectedly large number of dogs in the Arendal area of southern Norway have antibodies to tick-borne encephalitis virus caused by flavivirus passed on by forest-living, blood-sucking ticks. Human beings, dogs, sheep, cows, and other mammals become infected when fed on by a tick, although the virus also spreads through the consumption of raw milk from infected animals, and it can cross the placenta from mother to fetus. Human symptoms include meningitis and encephalitis. Up to 20% of infected people suffer permanent neurologic damage, and 2% die.6
Etiologic and Risk Factors
The cause cannot be identified in almost two-thirds of cases of viral encephalitis. Viral infection may cause encephalitis as a primary manifestation or as a secondary complication. Risk of neuroinvasive disease in an organ recipient infected with West Nile virus is estimated as 40% compared with less than 1% in the general population.11
Acute viral encephalitides, such as eastern and western equine encephalitis, St Louis encephalitis, and California virus encephalitis, and the most recent outbreak of West Nile virus depend on mosquitos for transmission and tend to occur in the mid to late summer. The eastern variety is the least common but most deadly. It occurs in outbreaks along the entire east coast of the United States. It is rapidly progressive with lesions in the basal ganglia. It carries high mortality and morbidity rates. The western version has a much lower mortality but appears to be particularly severe in infants and children.11
West Nile virus is a flavivirus that was originally isolated in 1937 from the blood of a febrile woman in the West Nile province of Uganda. The virus is widely distrib- uted in Africa, Europe, Australia, and Asia, and since 1999, it has spread rapidly throughout the western hemisphere, including the United States, Canada, Mexico, and the Caribbean and into parts of Central and South America. Table 29-1 shows the human illness associated with West Nile infections since 1999.
Table 29-1
Human Illness From West Nile Virus Infections Reported in the United States, 1999-2005
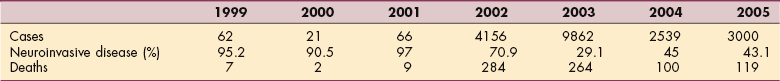
Data from the US Centers for Disease Control and Prevention as of January 10, 2006; Kramer LD: West Nile virus, Lancet Neurol 6(2):171-181, 2007.
West Nile virus is transmitted primarily between avian hosts and mosquitos. Fig. 29-5 shows the cycle of transmission. Mosquitoes of the genus Culex carry the virus, and it is maintained during the dormant period of the adult mosquito and reintroduction of the virus by migratory birds from their winter breeding grounds or from locations where the virus may be transmitted all year round.13
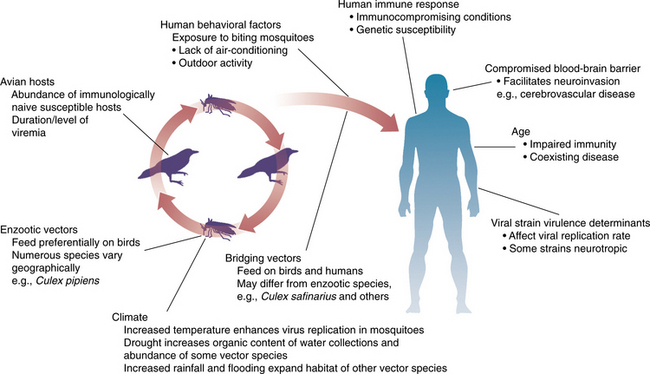
Figure 29-5 West Nile virus transmission cycle and examples of modifying climatologic, vertebrate, mosquito, and human factors on infection and illness. (From Tsai TF, Vaughn DW, Solomon T: Flaviviruses. In Mandell GL, Bennett JE, Dolin R, eds: Principles and practice of infectious diseases, ed 6, New York, 2005, Churchill Livingstone.)
Pathogenesis
Encephalitis produces an inflammatory response and pathologic changes in the brain. Ballooning of infected cells and degeneration of the cellular nuclei can lead to cell death. Plasma membranes are destroyed, and cells form multinucleated giant cells. There is perivascular cuffing causing damage to the lining of a vessel and hemorrhagic necrosis. The oligodendrocytes are affected, creating gliosis, or scarring. Widespread destruction of white matter can occur through inflammation and thrombosis of perforating vessels. Focal damage can hit discrete areas such as the optic nerve.20
West Nile virus is thought to initially replicate in dendritic cells after the host has been bitten by an infected mosquito. The infection then spreads to regional lymph nodes and into the bloodstream. The way in which the virus invades the nervous system is still unknown; retrograde transport along peripheral nerve axons has been proposed. Histologic CNS findings of West Nile virus infection are usually characterized by perivascular lymphoplasmacytic infiltration, microglia, astrocytes, necrosis, and neuronal loss with predilection to structures like the thalamus, brainstem, and cerebellar Purkinje cells. These variable anatomic involvements explain different clinical presentations.21
Herpes simplex virus is found in neonatal infants and appears to arise from maternal genital infection with the virus. It is acquired as the baby passes through the birth canal. Fifty percent of those who contract herpes simplex virus will develop CNS disease, whereas others may only develop skin, eye, and mouth disease. Herpes simplex encephalitis is found after the age of 3 months and is often a latent infection found in the gray matter of the temporal lobe and surrounding structures of the limbic system and the frontal lobe. It is the most common cause of sporadic nonepidemic encephalitis in the United States. Possible genetic factors are undergoing study, and in animal studies, there appears to be a connection to the γ1 34.5 gene.1
Encephalomyelitis can result from viral infections such as measles, mumps, rubella, or varicella. Mumps is usually benign and self-limited, but it can trigger encephalitis and other CNS complications such as acute, hydrocephalus, ataxia, transverse myelitis, and deafness.41 Vaccines that contain neuronal antigens have been known to precede these infections, particularly for rabies or smallpox. When there is an illness at the time of vaccination, the risk of developing infection increases. Neurologic problems typically occur within 3 weeks of the illness or vaccination.3
EBV and hepatitis A have been associated with CNS disorders of an infectious nature. Acute toxic encephalitis occurs during the course of a system infection with a common virus. Parasites, bacteria, and toxic drug reactions can lead to infection of the brain and cause encephalitis or encephalopathy.
Clinical Manifestations
Signs and symptoms of encephalitis depend on the etiologic agent, but in general, headache, nausea, and vomiting are followed by altered consciousness. If the person becomes comatose, the coma may persist for days or weeks. Agitation can be associated with the degree of infection and may be associated with abnormal sensory processing. Depending on the area of the brain involved, there may be focal neurologic signs, with hemiparesis, aphasia, ataxia, or disorders of limb movement. There can be symptoms of meningeal irritation with stiffness of the back and neck. With herpes simplex encephalitis, there can be repeated seizure activity, hallucinations, and disturbance of memory, reflecting involvement of the temporal lobe.33
Although many individuals infected with West Nile virus are asymptomatic, symptoms develop in 20% to 40% of people with West Nile virus infection. The incubation period is 2 to 14 days before symptom onset. Most complaints are of flulike symptoms. West Nile virus is characterized by fever, headache, malaise, myalgia, fatigue, skin rash, lymphadenopathy, vomiting, and diarrhea. Kernig’s and Brudzinski’s signs may be found on physical examination. Less than 1% of infected individuals develop severe neuroinvasive diseases. West Nile meningitis usually presents with fever and signs of meningeal irritation such as headache, stiff neck, nuchal rigidity, and photophobia. Box 29-1 lists the findings that are most critical to watch for to determine potential for high level of disability or death. In addition, West Nile virus can present as acute flaccid paralysis. Fig. 29-6 shows the pattern of weakness found in some individuals with this form of West Nile virus infection. The lesion of spinal anterior horns results in a paralysis similar to polio and reaches a plateau within hours. Deep tendon reflex can be diminished in severely paralysed limbs. Reports of substantial muscle ache in the lower back; bowel and bladder functions are common. There is minimal or no sensory disturbance.21,43
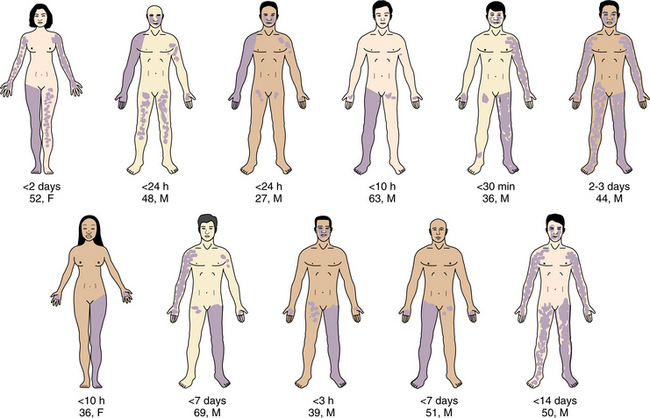
Figure 29-6 Clinical features induced by West Nile virus paralysis in 11 representative individuals. Weak limbs at the peak of paralysis are darkened. Degree of darkness corresponds to the severity of weakness. Duration of weakness, age and sex are noted. (From Kramer LD: West Nile virus, Lancet Neurol 6(2):171-181, 2007.)
Encephalitic lesions appear to alter sleep patterns as sequelae of brain-immune interactions. Responses of the immune system to invading pathogens are detected by the CNS, which responds by orchestrating complex changes in behavior and physiology. Sleep is one of the behaviors altered in response to immune challenge. Cytokines may play an active role in infectious challenge by regulating sleep.30
MEDICAL MANAGEMENT
Differential diagnosis of the types of infections of the brain has improved with the use of MRI and PCR to diagnosis herpes simplex encephalitis. The electroencephalogram (EEG) will show seizure activity in the temporal lobe in herpes simplex. In general, lumbar puncture is abnormal with increased proteins. The glucose level, however, may be normal or moderately increased. CT scans do not show much until the damage is extensive. MRI shows cerebral edema and vascular damage earlier in the process and leads to earlier detection.1 In West Nile virus, lesions can sometimes be seen in the white matter, pons, substantia nigra, and thalamus. An important MRI finding is the focal abnormal signal intensity within the anterior horns; the level of abnormal spinal MRI findings corresponds to the paralysis. Change can be seen in the spinal roots, possibly a result of axonal degeneration secondary to spinal motor neuron loss or Wallerian degeneration in the spinal roots. Fig. 29-7 shows the imaging studies of individuals with West Nile virus.
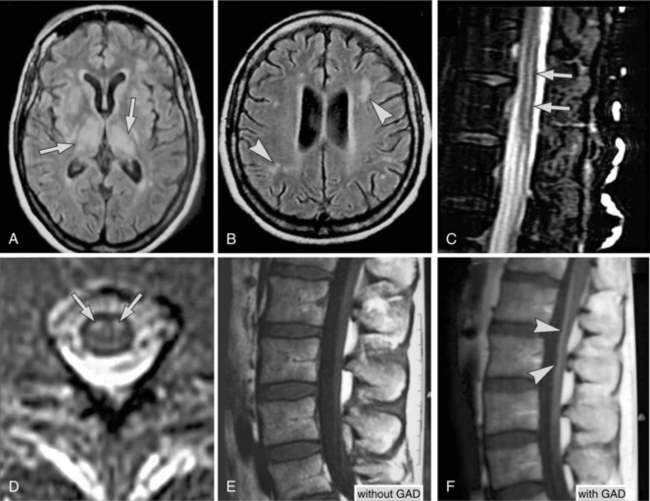
Figure 29-7 Abnormal MRI findings in patients with West Nile virus. A, Image of the brain from a 57-year-old woman with encephalitis shows abnormal signals in bilateral thalamus and weighted MRI in other areas of basal ganglion. B, Focal white matter lesions are also seen. C, Sagittal T2-weighted MRI of the lumbar spinal cord shows abnormal signal intensity (arrows) conspicuous within the cord. D, Transverse view of the cord at the midlumbar level; abnormal signal intensity (arrows) is confined to the anterior horns. E, T1-weighted lumbar spine MRI from a patient with both meningitis and acute flaccid paralysis shows no discernible abnormality; however, after giving gadolinium contrast, spinal roots are significantly enhanced (F). (From Kramer LD: West Nile virus, Lancet Neurol 6(2):171-181, 2007; C and D from Li J, Loeb JA, Shy ME, et al: Asymmetric flaccid paralysis: a neuromuscular presentation of West Nile virus infection, Ann Neurol 53:703-710, 2003.)
West Nile virus infection begins with nonspecific symptoms, making early clinical diagnosis challenging. Immunoglobulin M (IgM) antibodies against the virus (usually by enzyme-linked immunoassay [ELISA]) are usually indicative of a recent West Nile virus infection. Blood samples that are collected between the eighth and twenty-first day after onset are likely to give the best yield. IgM antibodies are only detectable 8 days after symptom onset. There may be a negative result from a blood sample obtained before the eighth day after symptom onset. After the twenty-first day, the titre of IgM could decline. The lymphocyte count, particularly the degree of relative lymphopenia, is a readily available test; the degree of relative lymphopenia (≥10%) appears to have prognostic importance in West Nile encephalitis. Clinicians should maintain a high index of suspicion for West Nile virus infection during the epidemic season, particularly when evaluating the elderly with neurologic or gastrointestinal symptoms.12,27
West Nile meningitis and encephalitis have similar degrees of pleocytosis, or multiple cystic lesions. However, West Nile encephalitis tends to create higher concentrations of total protein in the CSF and leads to a more severe outcome. Electrophysiologic studies are helpful for the diagnosis of paralysis induced by West Nile virus. Motor-nerve conduction studies may reveal severely reduced amplitudes of compound muscle action potentials in symptomatic limbs. However, if the nerve conduction study is done in the early phase of the illness, compound muscle action potentials can be normal because Wallerian degeneration can take 7 to 10 days to complete. Nerve conduction velocities are usually preserved, and sensory nerve conduction is typically normal. Needle electromyography shows severe denervation in muscles of weak limbs and its corresponding paraspinal muscles. Taken together, these abnormalities in the paralysed limbs localize the lesions to the anterior horn motor neurons or their ventral nerve roots. The localization is typically consistent with the MRI findings. Individuals presenting with an otherwise unexplained facial palsy during the summer should be tested not only for neuroborreliosis but also for West Nile virus. West Nile encephalitis may present with cranial nerve abnormalities involving the cranial nerves VI or VII.13,21
TREATMENT.
Treatment varies with the infectious agent. No antiviral treatment is available for encephalitis except for that caused by the herpes simplex virus. Acyclovir appears to improve the outcome in herpes simplex encephalitis.
There is no specific regime currently available for treatment of flavivirus infections. Close monitoring of the symptoms is critical, especially with the complication of cerebral edema, which may require surgical decompression, hyperventilation, or administration of mannitol. The use of corticosteroids is controversial because of the potential suppression of antibody protection within the CNS. Because no effective therapy is known, only supportive care is now available. However, there may be advantages to immunosuppression in postinfectious disorders with possible repair of a damaged blood-brain barrier. Human vaccines for flavivirus infections are currently available only for yellow fever, Japanese encephalitis, and tick-borne encephalitis.11
PROGNOSIS.
The prognosis depends on the infectious agent. The rate of recovery can range from 10% to 50% even in individuals who may have been very ill at the onset. Individuals with mumps meningoencephalitis and Venezuelan equine encephalitis have an excellent prognosis. Other encephalitides, such as western equine, St Louis, and California encephalitis and West Nile virus, have a moderate-to-good rate of survival. Although, with the use of medication, herpes simplex encephalitis has a moderately good outcome (20% mortality), neurologic sequelae are common in 50% of persons.2,11 Recovery for paralysis is remarkably variable. The variation may be caused by different degrees of motor neuron or motor unit loss.21
Some recovery is complete within weeks, but outcome is highly variable. The severity of the original illness does not always predict the final outcome. Prediction of post–West Nile virus poliomyelitis syndrome similar to post-polio syndrome is not yet possible. Permanent cerebral problems are more likely to occur in infants. Young children will take longer to recover than adults with similar infections. Anti–West Nile virus IgM can persist for 1 year or longer.
Development of West Nile virus vaccines have been explored, including immunization of animals with recombinant viral proteins, inactivated West Nile virus, deoxyribonucleic acid (DNA) that expresses viral antigens, or attenuated West Nile virus isolates.21
BRAIN ABSCESS
Definition
Brain abscess is an uncommon disorder accounting for only 2% of intracranial mass lesions. CNS abscesses are circumscribed, enlarging, focal infections that produce symptoms and findings similar to those of other space-occupying lesions such as brain tumors. Brain abscesses, however, often progress more rapidly than tumors and more frequently affect meningeal structures. Brain abscesses occur when microorganisms reach the brain and cause a local infection. There may be only one area of abscess, or many areas may be infected because of spread by blood-borne pathogens. Although the site and size of the abscess influence the initial symptoms, evidence of increased intracranial pressure is common.39
Risk Factors and Pathogenesis
Persons with a compromised immune system receiving steroids, immunosuppressants, or cytotoxic chemotherapy or persons with a systemic illness, such as HIV infection, have an increased risk of developing a brain abscess. Whereas viruses tend to cause diffuse brain infections as described previously, most bacteria, fungi, and other parasites cause localized brain disease. Brain abscesses may develop from other infections in the cranium such as sinusitis or mastoiditis.
Infections leading to brain abscess can come from extracerebral locations; blood-borne metastases; infection from lung or heart; or infections within the cranium such as otitis, cranial osteomyelitis, and sinusitis. Recent or remote head trauma or neurosurgical procedures may be the cause. Blood-borne infections seed the brain and spread and produce abscesses in brain regions in proportion to the blood flow; accordingly, parietal lobe abscesses predominate. Extension of infection from otitis and mastoiditis involves contiguous brain regions of the temporal lobe and cerebellum, whereas abscesses resulting from sinusitis affect the frontal and temporal lobes. Table 29-2 presents typical sources of brain abscesses.
Table 29-2
Potential Causes of Brain Abscess and Related Organisms Based on 123 Patients Treated Between 1986 and 2000
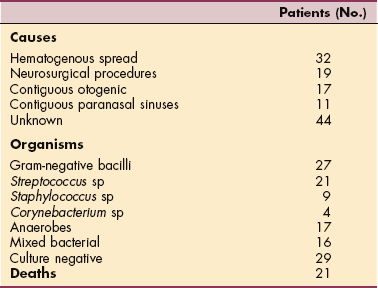
Data modified from Lu CH, Chang WN, Lin YC, et al: Bacterial brain abscess: microbiological features, epidemiological trends and therapeutic outcomes, QJM 95:501-509, 2002; Goldman L, ed: Cecil textbook of medicine, ed 22, Philadelphia, 2004, WB Saunders.
Most brain abscesses evolve over a number of stages, with involvement of the cerebrum occurring during the first 1 to 3 days. Inflammatory infiltrates of polymorphonuclear cells, lymphocytes, and plasma cells follow within 24 hours. By 3 days, the surrounding area shows an increase in perivascular inflammation. The late cerebritis phase develops approximately 4 to 9 days after infection, during which time the center becomes necrotic, containing a mixture of debris and inflammatory cells Early reactive astrocytes surround the zone of infection and proceed to early capsule formation between approximately 10 and 13 days. At this time, the necrotic center shrinks slightly, and a well-developed peripheral fibroblast layer evolves. The late capsule stage continues to evolve between 14 days and 5 weeks, with continual shrinking of the necrotic center and a relative decrease in the inflammatory cells. The capsule thickens with astrocyte scarring.39
If the infection is carried in the blood from another site in the body, the abscess will usually develop at the junction of the gray and white matter. Anaerobic bacteria are found in over one-half of brain abscesses. The infection usually begins as local encephalitis with necrosis and inflammation of the neurons and glial cells. As the process continues, a capsule wall is formed by the proliferation of fibroblasts. There is usually an area of cerebral edema around the abscess. Bacterial and fungal abscesses will continue to grow until they become lethal. Abscesses caused by other parasites are usually self-limiting.33
Clinical Manifestations
Normal body temperature is common, and white cell counts are not always high. Neck stiffness is rare in the absence of increased intracranial pressure. Otherwise, the presenting features resemble those of any expanding intracranial mass, like a slow-growing neoplasm, or it may be rapid and progress to possible herniation and brainstem compression, causing death. A headache of recent onset is the most common symptom, representing distortion or irritation of pain-sensitive structures within the cranial vault, especially those of the great venous sinuses and the dura mater about the base of the brain. If the process continues untreated, isolated headache increases in severity and becomes accompanied by focal signs, such as hemiparesis or aphasia, followed by obtundation and coma. The period of evolution may be as brief as hours or as long as many days to weeks with more indolent organisms. Seizures may occur with abscesses that involve the cortical gray matter.39 Lethargy and confusion progress with the increased intracranial pressure present with the growing mass. Focal signs reflect the area of the brain that is affected, with paresis resulting from frontal and parietal lesions and visual disturbances noted with occipital lobe dysfunction.33 Table 29-3 lists typical manifestations of brain abscess.

MEDICAL MANAGEMENT
A history of infection or immunosuppression will lead to suspicion of abscess rather than neoplasm. CT and MRI will support the diagnosis based on the different configurations of tumor and abscess. Although the early signs are similar to meningitis, the focal signs of compression in one area of the brain distinguish the abscess over time. The EEG is often abnormal.20
TREATMENT.
Appropriate and timely antibiotic protocol and surgical drainage is required to reduce the mass effect. Careful clinical observation is necessary with multiple abscesses, and CT scans should be repeated often to determine if the abscess continues to expand. Initially, corticosteroids may be used to control the cerebral edema caused by the abscess, but these are used for a short time only because of their interference with capsule formation and immunosuppressive action in the brain.
PROGNOSIS.
Mortality from brain abscess can be lowered from 65% to 30% with control of the infection with antibiotics and surgery. Nearly one-half of the clients are left with some neurologic sequelae, which may include focal signs and seizure activity.39
PRION DISEASE
Definition
Rare forms of encephalopathies include the prion diseases of Creutzfeldt-Jakob disease, kuru, and bovine spongiform encephalopathy, also known as mad cow disease. The incubation period is slow and can be up to 5 to 8 years. Most often the disease has been found in young adults ranging from 16 to 30 years.10
Classic Creutzfeldt-Jacob disease (CJD) is a rare, fatal, and degenerative neurologic disease with a long asymptomatic latent period that was first described in 1920. The causative agent of CJD is thought by most experts to be a prion protein (PrPsc), an abnormal conformation of a normal cellular protein (PrPc) that can recruit additional PrPc to PrPsc, resulting in deposition of insoluble precipitates in neural tissue. CJD is one of a variety of prion diseases of humans that occur spontaneously at a rate of approximately 1 per million throughout the world and can be transmitted vertically in familial conditions, such as Gerstmann-Sträussler-Scheinker syndrome, or through ritualistic cannibalism (kuru).
Like classic CJD, variant CJD (vCJD) is a fatal, degenerative neurologic disease, although it occurs in younger persons and has distinctive clinical, histopathologic, and biochemical features, including the presence of readily detectable prion protein in non–CNS lymphoreticular tissues such as appendix, spleen, tonsil, and lymph nodes. In contrast to classical CJD, vCJD disease is new, first reported in the United Kingdom in 1996. The causative agent of vCJD, a prion, is the same agent that causes bovine spongiform encephalopathy (BSE). A massive epidemic of BSE occurred in Great Britain in the 1980s and early 1990s as a result of the recycling and processing of material from dead sheep and cattle into food meal for cattle. Although this practice was stopped in the mid-1990s after appreciation of the BSE epidemic, an estimated 250,000 cattle had already been infected with BSE. Transmission of the BSE prions to humans occurred by oral consumption of beef and other cattle products containing reticular endothelial or neural tissue, resulting in a delayed outbreak of vCJD in the United Kingdom.18
Etiologic and Risk Factors
In Italy the incidence of genetic transmissible spongiform encephalopathy diseases is the second highest among European countries.24 Infection with BSE-derived or vCJD-derived prions depends on the host’s genetic makeup, which means that there could be a substantial number of symptom-free human carriers of these infectious agents. There is now evidence that vCJD prions can be transmitted through blood transfusion and other iatrogenic routes.4,5
Iatrogenic transmission has occurred via corneal and dural transplants and depth cerebral electrodes. Hormone therapy using human pituitary products can be a cause. Transmission of prion disease is also possible through ingesting nervous system products that contain infected material. Eating nervous system products of beef that had been fed rendered protein led to the increased incidence in the United Kingdom in the 1990s.37 Skeletal muscle tissue from CWD-infected deer has shown prion infectivity, however, the implications of these findings for human beings are unclear. It is very difficult, if not impossible, to predict how prions from one species will behave when they transit species barriers.34
Pathogenesis
Like oncogenes, mutations of the normal cellular prion protein gene cause disease. Abnormal prion protein differs from oncogene products in that the prion protein programs its own creation from normal cellular proteins and then instructs the normal protein to change so that it is functionally similar to the abnormal protein. The microscopic alterations consist of spongy change in neuropil, neuronal loss, and gliosis. The spongiform change is similar to that seen after anoxia and in some cases of Alzheimer’s disorders. The prion is a substance that contains no nucleic acid but can replicate within the nervous system. There appears to be accelerated death of Purkinje’s cells, resulting in the ataxia that is commonly seen.17
Clinical Manifestations
Movement becomes abnormal, with dementia that also worsens over time. Sleep-wake symptoms develop with severe sleep EEG abnormalities with loss of sleep spindles, very low sleep efficiency, and virtual absence of rapid eye movement (REM) sleep.25 Akinetic mutism will eventually develop with overriding myoclonic jerks.
MEDICAL MANAGEMENT
Detection of spongy change alone is not sufficient for the neuropathologic diagnosis of prion disease but must be corroborated with Western blot testing. If familial prion proteinopathy is suspected, molecular genetic analysis of DNA from lymphocytes can be performed.17
TREATMENT AND PROGNOSIS.
At this time there is no known treatment for prion diseases, although clinical trials of methods to disrupt replication are being considered. Once diagnosed, these disorders are rapidly progressive and eventually fatal. Control of the use of potentially infected animal feed has increased in the early 2000s, and it is believed that this will help to eliminate the current concerns.17
References
1. Arvin, A. Herpes simplex infections of the central nervous system. In: Peterson PK, Remington JS, eds. New concepts in the immunopathogenesis of CNS infections. Malden, MA: Blackwell Science, 2000.
2. Baringer, JR. Herpes simplex virus encephalitis. In: Davis LE, Kennedy PG, eds. Infectious diseases of the nervous system. Oxford: Butterworth-Heinemann, 2000.
3. Baringer, JR, Viral infections. Asbury, AK, McKhann, GM, McDonald, WI, eds. Diseases of the nervous system: clinical neurobiology, 2. Philadelphia: Saunders, 1992.
4. Bradbury, J. Genetic susceptibility to prion disease: new phenotypes? Lancet Neurol. 2005;4(1):17.
5. Bradbury, J. Are there healthy human carriers for vCJD? Lancet Neurol. 2006;5(8):652.
6. Burton, A. Tick-borne encephalitis emerges in Norway. Lancet Infect Dis. 2004;4(4):189.
7. Campbell, IL. Cytokines and chemokines in defense and damage in the intact CNS. In: Peterson PK, Remington JS, eds. New concepts in the immunopathogenesis of CNS infections. Malden, MA: Blackwell Science, 2000.
8. Cardona-Bonet, LL. Management of perioperative infectious complications in the neurologic patient. Neurol Clin. 2004;22(2):329–345.
9. Carew, TJ. Posture and locomotion. In Kandel ER, Schwartz JH, eds.: Principles of neural science, ed 2, New York: Elsevier, 1985.
10. Chesebro, B. Transmissible spongiform encephalopathies and prion protein. In: Peterson PK, Remington JS, eds. New concepts in the immunopathogenesis of CNS infections. Malden, MA: Blackwell Science, 2000.
11. Coyle, PK. Postinfectious encephalitis. In: Davis LE, Kennedy PG, eds. Infectious diseases of the nervous system. Oxford: Butterworth-Heinemann, 2000.
12. Cunha, BA. Prognostic importance of lymphopenia in West Nile encephalitis. Am J Med. 2004;117(9):710–711.
13. Cunha, BA. West Nile encephalitis relapse presenting with abducens and facial nerve palsies. Am J Med. 2006;119(6):2.
14. Davis, LE. Central nervous system infections. In Weiner WJ, Goetz CG, eds.: Neurology for the non-neurologist, ed 3, Philadelphia: Lippincott, 1994.
15. Davis, LE. Tuberculous meningitis. In: Davis LE, Kennedy PG, eds. Infectious diseases of the nervous system. Oxford: Butterworth-Heinemann, 2000.
16. DeBiasi, RL, Tyler, KL. Recurrent aseptic meningitis. In: Davis LE, Kennedy PG, eds. Infectious diseases of the nervous system. Oxford: Butterworth-Heinemann, 2000.
17. DeSilva, R, Will, RG. Human prion diseases. In: Davis LE, Kennedy PG, eds. Infectious diseases of the nervous system. Oxford: Butterworth-Heinemann, 2000.
18. Fiebig, EW. Emerging infections in transfusion medicine. Clin Lab Med. 2004;24(3):797–823.
19. Gleissner, B. Neoplastic meningitis. Lancet Neurol. 2006;5(5):443–452.
20. Gilroy, J, Holliday, PL. Basic neurology. New York: Macmillan, 1982.
21. Kramer, LD. West Nile virus. Lancet Neurol. 2007;6(2):171–181.
22. Kroll, JS, MacLennan, JM. Vaccines to prevent bacterial meningitis. In: Davis LE, Kennedy PG, eds. Infectious diseases of the nervous system. Oxford: Butterworth-Heinemann, 2000.
23. Kupila, L. Etiology of aseptic meningitis and encephalitis in an adult population. Neurology. 2006;66(1):75–80.
24. Ladogana, A. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. 2005;64(9):1592–1597.
25. Landolt, HP. Sleep-wake disturbances in sporadic Creutzfeldt-Jakob disease. Neurology. 2006;66(9):1418–1424.
26. Masur, H. Treatment of HIV infection and aids. In Goldman L, ed.: Cecil Textbook of Medicine, ed 22, Philadelphia: WB Saunders, 2004.
27. Mazurek, JM. The epidemiology and early clinical features of West Nile virus infection. Am J Emerg Med. 2005;23(4):536–543.
28. Myers, JN. Miliary, central nervous system, and genitourinary tuberculosis. Dis Mon. 2007;53(1):22–31.
29. Nau, R. Axonal injury, a neglected cause of CNS damage in bacterial meningitis. Neurology. 2004;62(3):509–511.
30. Opp, MR. Sleep and psychoneuroimmunology. Neurol Clin. 2006;24(3):493–506.
31. Overturf, GD. Defining bacterial meningitis and other infections of the central nervous system. Pediatr Crit Care Med. 2005;6(3 suppl):14–18.
32. Peters, PJ, Harrison, T, Lennox, JL. A dangerous dilemma: management of infectious intracranial aneurysms complicating endocarditis. Lancet Infect Dis. 2006;6(11):742–748.
33. Porter, RE. Inflammatory and infectious disorders of the brain. In Umphred DA, ed.: Neurological rehabilitation, ed 4, St Louis: Mosby, 2001.
34. Quirk, M. Infectious prions discovered in deer muscle. Lancet Infect Dis. 2006;6(3):133.
35. Scheld, WM. Bacterial meningitis: epidemiology, pathophysiology, and immunobiology. In: Peterson PK, Remington JS, eds. New concepts in the immunopathogenesis of CNS infections. Malden, MA: Blackwell Science, 2000.
36. Schnell, SS. Nursing care of clients with cerebral disorders. In Black JM, Matassarin-Jacobs E, eds.: Luckman and Sorensen’s medical-surgical nursing, ed 4, Philadelphia: WB Saunders, 1993.
37. Schonberger, LB. New variant of Creutzfeldt-Jakob disease and bovine spongiform encephalopathy. In: Hughes JM, Conte JE, eds. Infectious disease clinics of North America. Philadelphia: WB Saunders, 1998.
38. Sejvar, JJ. Management of anthrax meningitis. Lancet Infect Dis. 2005;5(5):287–295.
39. Simon, RP. Parameningeal infections. In Goldman L, ed.: Cecil textbook of medicine, ed 22, Philadelphia: WB Saunders, 2004.
40. Smith, L. Management of bacterial meningitis: new guidelines from the IDSA. Am Fam Physician. 2005;71(10):2003–2010.
41. Sonmez, FM. Brainstem encephalitis and acute disseminated encephalomyelitis following mumps. Pediatr Neurol. 2004;30(2):132–134.
42. Tsenova, L, Freedman, VH, Kaplan, G. Experimental tuberculous meningitis. In: Peterson PK, Remington JS, eds. New concepts in the immunopathogenesis of CNS infections. Malden, MA: Blackwell Science, 2000.
43. Tyler, KL. CSF findings in 250 patients with serologically confirmed West Nile virus meningitis and encephalitis. Neurology. 2006;66(3):361–365.
44. Weisfelt, M. Clinical features, complications, and outcome in adults with pneumococcal meningitis: a prospective case series. Lancet Neurol. 2006;5(2):123–129.