Central Nervous System Neoplasms
INTRODUCTION
Several categories of neoplasia affect the central nervous system (CNS). Primary tumors, which may be either benign or malignant, may develop in the brain, spinal cord, or surrounding structures. Secondary, or metastatic, tumors may spread to the CNS from another site, such as the lung or breast. Paraneoplastic syndromes may occur due to remote or indirect effects on the CNS from cancer elsewhere in the body. Additionally and less commonly, leptomeningeal carcinomatosis may occur, in which carcinoma metastasizes throughout the CNS with multiple lesions to the meninges and CSF pathways of the brain and/or spinal cord.
The presence of any CNS tumor or paraneoplastic syndrome is cause for concern due to the vital functions of the brain and spinal cord. The critical areas and confined spaces in the CNS make it vulnerable to a space-occupying lesion. Most primary malignant CNS tumors are locally invasive and cause significant morbidity and mortality.77
The early effects of a CNS tumor are related to mechanical displacement of brain or spinal cord tissue, or a mild block in cerebrospinal fluid (CSF) circulation, causing increased intracranial pressure (ICP). As a tumor grows, compression or destruction of local brain or nerve tissue may occur, resulting in specific neurologic deficits. Symptoms of brain tumors may range from minimal, such as mild lethargy, to marked, such as seizures, blindness, and paralysis, as the tumor progresses. Likewise, symptoms of spinal cord tumors may range from mild to severe and include pain, sensory impairments, weakness, and paralysis. Although primary CNS tumors typically do not metastasize outside the CNS because of the lack of a CNS lymphatic system to transport cancer cells, these cells may infrequently travel through the CSF to the spinal cord as “drop metastasis” and cause spinal cord complications.
The diagnosis of a CNS tumor with its threat of significant loss of neurologic and cognitive function is devastating to the client and family. A CNS tumor robs a person of independence and dignity, and is viewed as a humiliating and inextricably fatal process.69 Difficult decisions about treatment options and quality-of-life issues add stress for the client and family. In children with brain tumors, the diagnosis creates parental fear and emotional upheaval, and requires adjusting and decision making for the different needs at varying stages of the illness.40 Caregiving and financial struggles frequently are encountered with both brain and spinal cord tumors.
Despite the inescapable realities of these difficult issues, the situation is improving, with dramatic new advances in radiologic imaging, neurosurgery, and adjuvant therapy. At present, including those with both benign and malignant tumors, approximately 50% of patients with CNS tumors can be successfully treated and have an excellent long-term prognosis.126 A knowledge and awareness of current treatment advances provide the health professional with the information and skills to care for the client and family in a sensitive, compassionate, and hopeful but realistic manner.
Classification
The major purpose of tumor classification is to facilitate communication about tumor behavior and treatment, and to design studies to learn more about the tumors.143 Primary brain tumors are classified by light microscopy according to their predominant cell type.135 The World Health Organization (WHO) classification system, which incorporates the Ringerz system for astrocytomas, is becoming the most commonly accepted system, making it easier for clinicians to accurately compare the effects of treatment.15,30,87,135 It is a three-tiered system, based on neuroembryonal origin, that is, naming a tumor by the most likely cell of origin, and adding qualifying phrases to describe its behavior.143
See Table 30-1 for the WHO classification of primary tumors. The grading, from I to IV, indicates the aggressiveness of the tumor, with grade IV being the most aggressive. The St. Anne–Mayo (Daumas-Duport)100 system is another classification system in use. It is four tiered, based on the presence or absence of four major criteria (nuclear atypia, mitoses [cells in a state of division], endothelial proliferation, and necrosis), with grade I having none of these features, grade II having one, and so on. Various other systems exist, based on a number of distinguishing criteria: neuroembryonal origin, primary versus secondary, benign versus malignant, histologic grade, anatomic location, and childhood versus adult tumors. The multiplicity of grading systems has been confusing, making it difficult for clinicians to accurately compare the effects of treatment,100 so the acceptance of one system will be beneficial.
Table 30-1
World Health Organization (WHO) Classification of Primary Brain Tumors According to Histology
| Most Common Tumors | Grade† (WHO) |
| Astrocytic tumors | |
| Pilocytic | 1 |
| Astrocytoma (diffuse, infiltrative, fibrillary) | 2 |
| Anaplastic | 3 |
| Glioblastoma multiforme | 4 |
| Oligodendroglial tumors and mixed gliomas | |
| Oligodendroglioma, well differentiated | 2 |
| Anaplastic oligodendroglioma | 3 |
| Mixed oligodendroglioma/astrocytoma* | 2 |
| Mixed anaplastic oligodendroglioma/anaplastic astrocytoma* | 3 |
| Ependymal tumors | |
| Myxopapillary ependymoma | 1 |
| Ependymoma | 2 |
| Anaplastic | 3 |
| Choroid plexus tumors | |
| Choroid plexus papilloma | 1 |
| Choroid plexus carcinoma | 3 |
| Neuronal and mixed neuronal-glial tumors | |
| Ganglioglioma | 1-2 |
| Central neurocytoma | 2 |
| Filum terminale paraganglioma | 1 |
| Dysembryoplastic neuroepithelial tumor (DNET) | 1 |
| Pineal parenchymal tumors | |
| Pineocytoma | 2 |
| Pineoblastoma | 4 |
| Embryonal tumors | |
| Medulloblastoma | 4 |
| Supratentorial primitive neuroectodermal tumor (PNET) | 4 |
| Atypical teratoid/rhabdoid tumor | 4 |
| Meningeal tumors | |
| Meningioma | 1 |
| Atypical, clear cell, chordoid | 2 |
| Rhabdoid, papillary, or anaplastic (malignant) | 3 |
| Pituitary tumors | |
| Adenomas | 1 |
| Carcinomas | 2 |
| Tumors of cranial and spinal nerves | |
| Neurinomas (schwannoma; acoustic neuroma) | 1-2 |
*Mixed tumors that consist of oligodentroglioma/anaplastic astrocytoma or anaplastic oliogodendroglioma/astrocytoma are usually graded according to the highest-grade component, although there is no consensus from the WHO on this issue.
†Arabic numbers correspond with roman numerals in text.
Adapted from Schiff D, Batchelor T: Classification of brain tumors. UpToDate website. Available on-line at http://uptodateonline.com. Accessed September 25, 2006; and data from Kleihues P, Cavenee WK, eds: Pathology and genetics—tumors of the nervous system, Lyon, 2000, International Agency for Research on Cancer.
Primary brain tumors originate from the various cells and structures normally found within the brain. Secondary or metastatic brain tumors originate from structures outside the brain, most often from primary tumors of the lungs, breast, gastrointestinal tract, or genitourinary tract,52 or from melanoma.136
Primary CNS tumors also may be subdivided into malignant tumors, such as astrocytomas, and so-called benign tumors, such as meningiomas, neurinomas, and hemangioblastomas. A histologically benign tumor has a slow growth rate and is relatively noninvasive. However, because of space-occupying properties in vital tissue with a resultant high threat of functional limitation, the use of the term benign is somewhat misleading. Some authors insist that because of location even a very slow-growing CNS tumor should be considered basically malignant.39,135 The histologically benign tumor may be surgically inaccessible or located in a vital area, such as the pons or medulla, and will continue to grow, thereby causing an increase in ICP, neurologic deficits, herniation syndromes, and, finally, death.
Malignant CNS tumors typically have a high growth rate and are invasive and infiltrative. They are capable of modulating the surrounding extracellular matrix by secretion of substances that allow for invasion of surrounding tissue by the tumor cells.100 Tumors also have the ability to create new blood vessels to sustain the tumor, a process called angiogenesis.
Anatomic brain tumor location refers to the location of the lesion in reference to the tentorium or cerebral tissue. Knowing the anatomic location helps to predict probable deficits based on the function of that particular area in the brain. Box 30-1 lists the anatomic location of the most common CNS tumors.
There are other typically recognized subdivisions. In the brain are two main groups of primary tumors: gliomas, the most common type, which includes astrocytomas and glioblastomas; and tumors arising from supporting structures, such as meningiomas, neurinomas, and pituitary adenomas. A third group arising from embryonal undifferentiated nerve cells has been termed primitive neuroectodermal tumors or PNETs47 and arise more frequently in children. Examples of primary tumors in the spinal cord include the more common neurinomas (schwannomas or neurilemomas) and the less frequent gliomas and meningiomas.
Further subgroups of gliomas have been established based on cellular atypism, the presence of mitotic figures, the incidence of endothelial hyperplasia, and the presence of necrotic areas. It is hoped that newer techniques of molecular biology, such as the ability to identify growth factors and inhibitors necessary for cell growth and differentiation, may lead to a more sophisticated subclassification. Molecular and genetic signatures may predict brain tumor behavior and may soon guide not only tumor classification and diagnosis but also tumor-specific treatment strategies.37
Because the clinical presentation, treatment, and prognosis are heavily dependent on the location of involvement and whether the tumor is primary or metastatic, this discussion is divided into four parts: (1) primary brain tumors, (2) primary intraspinal tumors, (3) metastatic tumors, and (4) childhood brain tumors.
PRIMARY BRAIN TUMORS
Tumors of the CNS are not uncommon. The National Cancer Institute projected that 18,820 new malignant primary tumors of the brain and nervous system would be diagnosed in 2006 in the United States: 10,730 in men and 8090 in women.3,24,25 This corresponds to 7.6 and 5.4 per 100,000 men and women, respectively, in the U.S. population. The National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) data indicate that in 2003 there were approximately 111,212 people alive who had had a history of CNS cancer. The mean age at diagnosis of brain and nervous system cancer is 55 years of age. The overall 5-year relative survival rate for 1996 to 2002 was reported to be 33.5%. Estimates were that 12,820 men and women would die of brain and other nervous system cancers in 2006.2,132
Benign primary brain tumors add to the total incidence. The American Brain Tumor Association reported a combined estimate of 40,900 new primary malignant and benign brain tumors in 2004, the most recent estimate available,3,4 or 14 per 100,000 U.S. population. The number of people living with either a benign or malignant brain tumor (prevalence) in the United States in 2003 was estimated to be approximately 350,000 to 360,000.3,104 Of brain tumor survivors, about 75% have a diagnosis of benign tumors, about 23% have malignant tumors, and 2% have tumors of uncertain behavior.
Although malignant brain tumors accounted for a small percentage of the approximately 1.3 million new cases of all types of cancer projected to occur in 2006,4 brain tumors kill more Americans each year than multiple sclerosis and Hodgkin’s disease.117 For all the intracranial diseases, death from intracranial neoplasms is second only to stroke.47 Approximately 12,820 deaths each year in the United States are due to primary brain and nervous system tumors, and many more are caused by metastasis.4
The incidence of primary brain and nervous system tumors peaks in the pediatric population, then increases by about 1.2% per year until it plateaus in the population over 70 years of age.100 Primary brain tumors are the second most common form of cancer in children,9,101 and primary CNS tumors are the second leading cause of death from cancer in children. Gliomas account for approximately 50% of CNS tumors. The average age of onset for all primary brain tumors is 53 years.101 Table 30-2 summarizes the frequency of primary CNS tumors.
Table 30-2
Frequency of Primary Central Nervous System Tumors
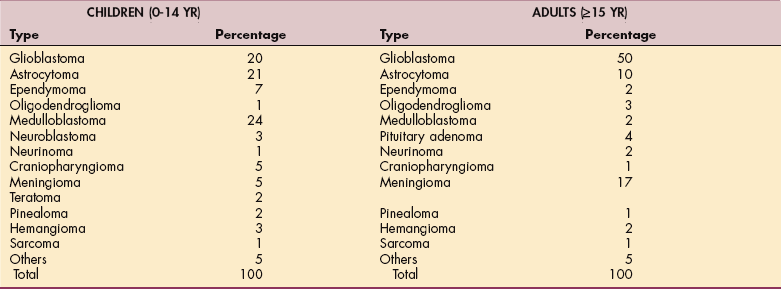
Adapted from Janus TJ, Yung WKA: Primary neurological tumors. In Goetz CG, ed: Textbook of clinical neurology, ed 2, Philadelphia, 2003, Saunders.
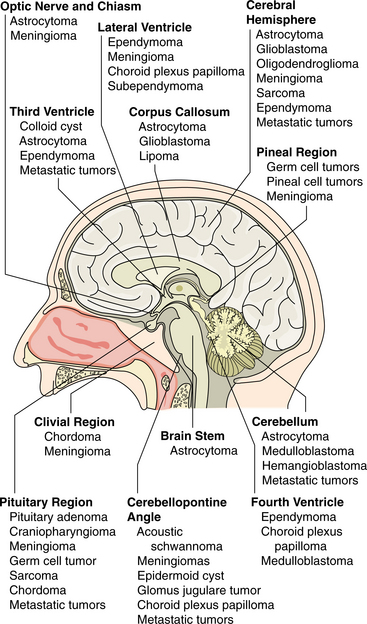
More than 60% of tumors in adults are supratentorial, or located in the cerebral hemispheres, above the tentorium. The tentorium is a flap of meninges separating the cerebral hemispheres from the posterior fossa structures. The majority of pediatric tumors are infratentorial, involving primarily the cerebellum and brainstem.26 Certain tumor types have a predilection for specific areas of the brain, although they may arise elsewhere in the brain. Topologic distribution and preferred sites of primary CNS tumors are illustrated in Fig. 30-1.
Pathogenesis
Brain tumors affect the brain through compression of cerebral tissue, including brain substance and cranial nerves; invasion or infiltration of cerebral tissue; and sometimes erosion of bone.52 These mechanisms precipitate pathophysiologic changes such as cerebral edema and increased ICP.
In most brain tumors, vasogenic edema develops in the surrounding tissue of the tumor because of compression and obstruction of CSF pathways, moving CSF across ventricular walls.47 Substances released from tumor cells altering the blood-brain barrier also may cause rapid cerebral edema. Seepage of plasma into the extracellular space and between the layers of the myelin sheath results from the increased permeability of the capillary endothelial cells of the white matter. This impairs cellular activity and causes electrochemical instability, resulting in seizures. As the edema continues to develop, signs and symptoms of increased ICP become more apparent.
Initially the brain may have a surprising tolerance to the compressive and infiltrative effects of brain tumors, particularly with slow growing tumors, and early symptoms may be few. Compensatory mechanisms to accommodate the edema and maintain normal ICP are limited but include decreasing (1) the volume of brain tissue, (2) CSF, and (3) cerebral blood volume. When the brain can no longer compensate, the resultant increase in ICP leads to more evident signs and symptoms. Intracranial herniation and herniation through the foramen magnum are potential results of serious ICP elevation. Fig. 30-2 illustrates intracranial herniation syndromes evoked by supratentorial masses.
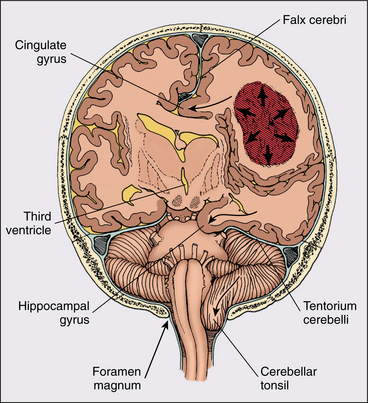
Figure 30-2 Intracranial herniation syndromes evoked by supratentorial masses. The tumor and its edema (arrows) have produced the following (curved arrows): cingulated gyrus herniation under the falx cerebri; diencephalic herniation across the midline compressing the ipsilateral ventricle and producing hydrocephalus in the contralateral ventricle; hippocampal gyrus herniation through the tentorial notch compressing the posterior cerebral artery and brainstem; and herniation of the cerebellar tonsils through the foramen magnum. (From Abeloff MD, Armitage JO, Niederhuber JE, et al: Clinical oncology, ed 3, Philadelphia, 2004, Churchill Livingstone. Adapted from Plum F, Posner JB: The diagnosis of stupor and coma, ed 2, Philadelphia, 1980, FA Davis.) FA Davis
Clinical Manifestations
The particular clinical presentation of a brain tumor depends on the compression or infiltration of specific cerebral tissue, the related cerebral edema, and the development of increased ICP.52 Cerebral edema surrounding the tumor results from the inflammatory response of tissues to the tumor and contributes to the increase in ICP. Box 30-2 lists common signs and symptoms of brain tumors.
The initial clinical signs of an intracranial tumor are related to the generalized effect of an increase in ICP. Headache is commonly present (in one third to one half of cases), is typically generalized or retro-orbital, and is typically worse in the morning and better later in the day. The headache is intensified or precipitated by any activity that tends to raise the ICP, such as stooping, straining, coughing, or exercising. Irritation, compression, or traction of pain-sensitive structures such as the dura mater and blood vessels causes the headache.105 Although tension-type headache is more common, migraine-type and other types may be exhibited.137 The sixth cranial nerve (abducens) is highly susceptible to elevated ICP because of its local anatomic relationships as the basis pontis slips caudally during transtentorial herniation, not, as previously believed, because of its long intracranial path.49 This causes weakness in the lateral rectus muscle and diplopia. Nausea and vomiting are common, often due to increased ICP. In glioblastoma multiforme (GBM), about one third of patients suffer nausea and vomiting. Box 30-3 lists signs and symptoms of intracranial hypertension.
Other common initial signs are mental clouding, lethargy, alterations in consciousness and cognition, syncope (fainting), and easy fatigability. Behavioral changes may include irritability, flat affect, emotional lability, and lack of initiative and spontaneity. Increasing intracranial CSF pressure may precipitate an increase in perioptic pressure, which in turn impedes venous drainage from the optic head area and retina, causing papilledema, or edema of the optic disc. Papilledema, present in about 70% to 75% of patients with brain tumors, is associated with visual changes, such as decreased visual acuity, an enlarged blind spot, diplopia, and deficits in the visual fields. Often deterioration in vision may be the precipitating factor in the patient’s seeking an appointment with an optometrist or ophthalmologist. A dilated ophthalmologic examination showing papilledema is fairly crucial to the diagnosis when it is not straightforward.
About 20% to 50% of adults with brain tumors develop seizure activity. The cerebral edema causes hyperactive cells, which produce abnormal, paroxysmal discharges or seizure activity.106 Seizures may be the first presenting sign of a tumor. In patients presenting with seizures, detection of low-grade gliomas is becoming increasingly frequent with magnetic resonance imaging (MRI). See Fig. 30-3 for an MRI scan of a low-grade glioma presenting with a seizure. In the later stages of illness, seizure activity is present in 70% of patients.93 A common feature of a tumor-related seizure is its repetitive nature, with seizures being very stereotypical in a given patient.137
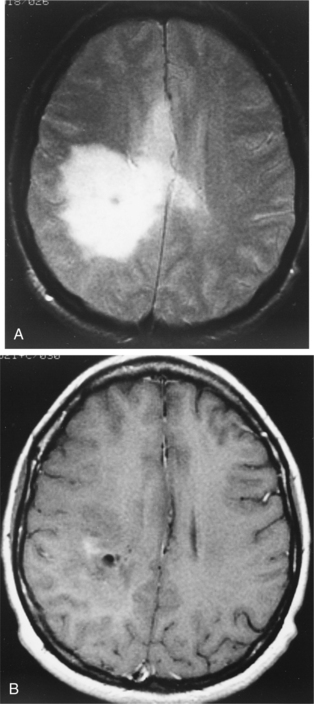
Figure 30-3 Magnetic resonance imaging (MRI) of a low-grade glioma. A, T2-weighted image. B, T1-weighted image, gadolinium contrast with minimum enhancement. The images are typical of this tumor, which is being detected with increasing frequency by MRI in seizure patients. Many are invisible on computed tomographic scans. (From Goldman LM, Ausiello D, eds: Cecil textbook of medicine, ed 22, Philadelphia, 2004, Saunders.)
As the tumor grows, causing progressive destruction or dysfunction of tissue, locally referable signs may occur (hemiparesis, specific cranial nerve dysfunction, aphasia, visual symptoms, ataxia), which may help to localize the tumor site. Table 30-3 provides a list of signs associated with localized brain lesions.
Table 30-3
Signs Associated with Localized Brain Lesions
| Location of Lesion | Associated Signs |
| Prefrontal area | Loss of judgment, failure of memory, inappropriate behavior, apathy, poor attention span, easy distractibility, release phenomena |
| Frontal eye fields | Failure to sustain gaze to opposite side, saccadic eye movements, impersistence, seizures with forced deviation of the eyes to the opposite side |
| Precentral gyrus | Partial motor seizures, jacksonian seizures, generalized seizures, hemiparesis |
| Superficial parietal lobe | Partial sensory seizures; loss of cortical sensation including two-point discrimination, tactile localization, stereognosis, and graphism |
| Angular gyrus | Agraphia, acalculia, finger agnosia, allochiria (right-left confusion) (Gerstmann’s syndrome) |
| Broca’s area | Motor dysphasia |
| Superior temporal gyrus | Receptive dysphasia |
| Midbrain | Early hydrocephalus; loss of upward gaze; pupillary abnormalities; third nerve involvement—ptosis, external strabismus, diplopia; ipsilateral cerebellar signs; contralateral hemiparesis; parkinsonism; akinetic mutism |
| Cerebellar hemisphere | Ipsilateral cerebellar ataxia with hypotonia, dysmetria, intention tremor, nystagmus to side of lesion |
| Pons | Sixth nerve involvement—diplopia, internal strabismus; seventh nerve involvement—ipsilateral facial paralysis; contralateral hemiparesis; contralateral hemisensory loss; ipsilateral cerebellar ataxia; locked-in syndrome |
| Medial surface of frontal lobe | Apraxia of gait, urinary incontinence |
| Corpus callosum | Left-hand apraxia and agraphia, generalized tonic-clonic seizures |
| Thalamus | Contralateral thalamic pain, contralateral hemisensory loss |
| Temporal lobe | Partial complex seizures, contralateral homonymous upper quadrantanopsia |
| Paracentral lobule | Progressive spastic paraparesis, urgency of micturition, incontinence |
| Deep parietal lobe | Autotopagnosia, anosognosia, contralateral homonymous lower quadrantanopsia |
| Third ventricle | Paroxysmal headache, hydrocephalus |
| Fourth ventricle | Hydrocephalus, progressive cerebellar ataxia, progressive spastic hemiparesis or quadriparesis |
| Cerebellopontine angle | Hearing loss, tinnitus, cerebellar ataxia, facial pain, facial weakness, dysphagia, dysarthria |
| Olfactory groove | Ipsilateral anosmia, ipsilateral optic atrophy, contralateral papilledema (Foster-Kennedy syndrome) |
| Optic chiasm | Incongruous bitemporal field defects, bitemporal hemianopsia, optic atrophy |
| Orbital surface frontal lobe | Partial complex seizures, paroxysmal atrial tachycardia |
| Optic nerve | Visual failure of one eye, optic atrophy |
| Uncus | Partial complex seizures with olfactory hallucinations (uncinate fits) |
| Basal ganglia | Contralateral choreoathetosis, contralateral dystonia |
| Internal capsule | Contralateral hemiplegia, hemisensory loss, homonymous hemianopsia |
| Pineal gland | Loss of upward gaze (Parinaud’s syndrome), early hydrocephalus, lid retraction, pupillary abnormalities |
| Occipital lobe | Partial seizures with elementary visual phenomena, homonymous hemianopsia with macular sparing |
| Hypothalamus, pituitary | Precocious puberty (children), impotence, amenorrhea, galactorrhea, hypothyroidism, hypopituitarism, diabetes insipidus, cachexia, diencephalic autonomic seizures |
From Gilroy J: Basic neurology, ed 2, Elmsford, NY, 1990, Pergamon Press, pp 228-229.
SPECIFIC PRIMARY BRAIN TUMORS
A wide variety of specific types of primary brain tumor exist, with similarities in medical management and implications for physical therapy. Therefore, the specific tumors are first presented individually, followed by a discussion of diagnosis, medical management, and therapy implications for all primary brain tumors.
Gliomas
Overview and Incidence.: Gliomas are the most common of the primary brain tumors, accounting for 40% to 45% of all brain tumors, with men more frequently affected than women in a 3: 2 ratio. Gliomas are divided into benign or low-grade gliomas, such as the low-grade astrocytomas, and malignant gliomas, such as anaplastic astrocytomas and GBMs. Other gliomas are oligodendrogliomas, ependymomas, and medulloblastomas. Terms such as brainstem glioma and optic nerve glioma refer to the location of these tumors, not the type of glial cell that gave rise to them. Only a tissue sample gives the specific diagnosis.
Low-grade astrocytomas account for 10% to 12% of primary brain tumors1,139 and are the most common type of intracranial tumor in children. Malignant astrocytomas (anaplastic astrocytoma and GBM) are much more common in adults than low-grade astrocytomas, making up 20% to 30% of primary brain tumors. Oligodendrogliomas and ependymomas make up another 5% to 7%. Medulloblastomas, sometimes termed embryonal tumors or PNETs, make up about 2% of primary brain tumors. Brainstem gliomas often affect children between 5 and 10 years of age but can also be found in adults between 30 and 40 years of age. Most optic gliomas occur in children under the age of 10. Table 30-4 lists the types of primary brain tumors, the cell of origin, and the distribution of primary CNS tumors by histologic type. The age of peak incidence is 45 to 55 years in adults. In children, the tumor occurs mainly between the ages of 2 and 10 years.139
Table 30-4
Cell of Origin and Distribution of Primary Central Nervous System Tumors by Histologic Type
From Abeloff MD, Armitage JO, Niederhuber JE, et al: Clinical oncology, ed 3, Philadelphia, 2004, Churchill Livingstone. Data from Central Brain Tumor Registry of the United States, 2002-2003 statistical report, Chicago, 2002, The Registry; analysis of data collected from 1995 to 1999 (n = 37,788).
Gliomas are tumors of the glial cells, the group of cells that support, insulate, and metabolically assist the neurons. Glial cells are derived from glioblasts. It is of interest to note that neurons, despite their prevalence in the CNS (100 billion in the adult brain, according to some authors), are rarely the cellular basis of neoplastic transformation.
Glial cells, which numerically exceed the number of neurons, are subdivided into astrocytes (star-shaped cells, sometimes termed long arms), which provide nutrition for neurons; oligodendrocytes (glial cells with few processes, sometimes termed short arms), which produce the myelin sheath of the axonal projections of neurons; and ependymal cells, which line the ventricles and produce cerebral spinal fluid.52 Gliomas are subdivided into astrocytomas, oligodendrogliomas, and ependymomas, named for the cell of origin of the tumor. A combination glial cell tumor may occur as well, such as an oligoastrocytoma. Medulloblastomas are tumors of the vermis of the cerebellum and are classified by some authors as gliomas and by some as PNETs or embryonal tumors. Medulloblastoma is grouped with the PNETs because of common features, but some pathologists and clinicians prefer to distinguish these two; currently the debate continues.101
Astrocytomas are given histologic grades of I through IV to indicate the rate of cell division (mitosis), nuclear atypia, endothelial proliferation, and necrosis. Grade I and II astrocytomas are the slowest-growing, and grades III and IV astrocytomas are progressively faster growing with higher rates of mitosis.96 Astrocytomas are capable at any time of converting to a higher grade.89 Refer to Table 30-1. (See the section on Grading of Tumors in Chapter 9.)
Etiologic and Risk Factors.: Relatively little is known about the cause of gliomas. They are characterized by a significant genetic heterogeneity, which makes the basic biology of glial neoplasms difficult to understand. A relationship may exist with chromosome abnormalities. Advances in the fields of molecular biology have allowed identification of mutated genes that increase the cell’s susceptibility to the development of certain cancers.89 These mutated genes that lead to the development of cancer are known as oncogenes.100 (See Chapter 9.) Another type of chromosome abnormality leads to deletion of the cell’s defense mechanism or its normal tumor-suppressing activity. This tumor suppressor gene, when altered, is unable to inhibit or limited in its normal ability to inhibit cellular proliferation.100 The presence of an oncogene and/or the absence of a tumor suppressor gene may be only one step toward tumor formation. Tumorigenesis is thought to be a multistep process, with other contributing factors in addition to chromosome abnormalities.21
Certain specific chromosome abnormalities have been linked to specific brain tumor types.117 The oncogene c-sis has been identified with GBM. The oncogene C-erbB has been identified in 30% of malignant gliomas and is associated with the transforming growth factor receptor. Chromosome 17 abnormalities have been demonstrated to be present in all grades of astrocytomas.117 Oncogenes may have some bearing on other genetic disorders associated with brain tumors. Neurofibromatosis, or von Recklinghausen’s disease (a familial condition involving the nervous system, muscles, bones, and skin and characterized by multiple soft tumors over the entire body associated with areas of pigmentation), is associated with spinal neuromas, acoustic neuromas, meningiomas, and gliomas. Tuberous sclerosis is associated with astrocytomas. Von Hippel-Lindau disease, a hereditary condition characterized by angiomatosis of the retina and cerebellum, is associated with hemangioblastomas.139 The best-described tumor suppressor genes are Rb and p53, associated with retinoblastoma and Li-Fraumeni syndrome, a familial breast cancer associated with soft tissue sarcomas and other tumors.
No risk factors have been identified for the development of brain tumors, other than exposure to ionizing radiation.103,109,143 The effects of carcinogenic viruses or agents are unclear. Associations have been made between certain viruses and brain tumors, such as the Epstein-Barr virus and primary CNS lymphoma, but they are insufficient to constitute direct cause-and-effect relationships. Sustained exposure to certain pesticides, vinyl chloride, nitrosoureas, and polycyclic hydrocarbons has been implicated in astrocytic tumors, but epidemiologic surveys of workers in the farming, petrochemical, and rubber industries have produced conflicting results.126 Certain industries such as synthetic rubber processing, vinyl chloride production, and petrochemical and oil refining do show increased risk.101,120 Infection, trauma, and immunosuppression are other suspected triggers. Radiation treatment for scalp ringworm in children is associated with an increased rate of developing brain tumors late in life.45 A history of frequent exposure to full-mouth dental x-rays, particularly at an early age, also is associated with certain brain tumors.21 Most of the extensive research in the area of nonionizing radiation exposure such as that from cellular phones, household appliances, and high-voltage electrical lines does not support an association with cancer.24 Although some of the studies may show an association with exposure to electromagnetic fields, either many other confounding variables, such as exposure to other carcinogens, may account for the association21 or a direct causal relationship cannot be proven.101 (See Chapter 4.) Increased risk of childhood tumors also has been associated with maternal diet, including consumption of cured meats containing nitrites during pregnancy.21,101
Low-Grade Astrocytoma—Grades I and II
Pathogenesis.: Low-grade astrocytomas include grades I and II. Grade I includes pilocytic astrocytoma (composed of fiber-shaped cells), sometimes termed juvenile astrocytoma, and is considered benign by some and malignant by others.73,133 Grade I astrocytoma grows slowly and often becomes cystic. It is composed of astrocytes with densely staining nuclei and scanty cytoplasm and is usually relatively acellular. The cells are uniform and closely resemble mature resting or reactive nonanaplastic astrocytes (well differentiated). Mitoses are absent or very rare.59,73 Although these are slow-growing tumors, they may become large.4 See Fig. 30-4 for a photograph of a well-differentiated astrocytoma. Grade II astrocytomas may be diffuse, infiltrative, and/or fibrillary, and have more anaplastic features. Fibrillary refers to the neuroglial fibrils. Other types are protoplasmic (cells that consist largely of protoplasm) and gemistocytic (large, densely packed cells with a globoid appearance).133 There is moderate cell density. Fig. 30-5 shows the appearance of computed tomographic (CT) and MRI astrocytoma scans with and without the use of contrast. The contrast agent, such as gadolinium, distinguishes the edema from the actual tumor. The larger the extent of the edema after administration of an intravenous contrast agent, the more malignant the lesion is likely to be.83 Cerebral astrocytoma presents as a solid, grey mass with indistinct boundaries. Differentiation falls somewhere within a spectrum from well-differentiated (grade I) tumors to more anaplastic (grade II) tumors.45 Astrocytomas in the cerebellum are often cystic and well circumscribed.
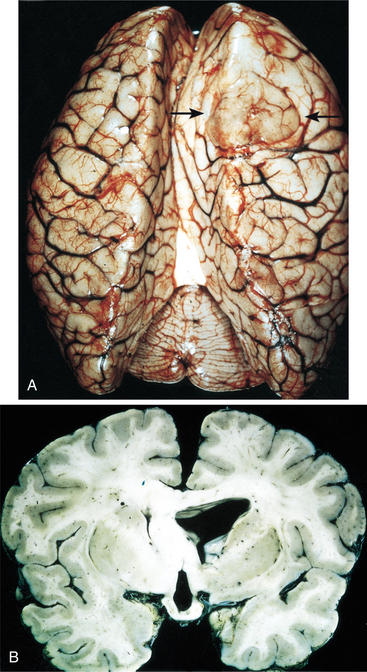
Figure 30-4 Well-differentiated astrocytoma. A, The right frontal tumor has expanded gyri, which led to flattening (arrows). B, Expanded white matter of the left cerebral hemisphere and thickened corpus callosum and fornices. (From Kumar V, Abbas, AK, Fausto N, eds: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
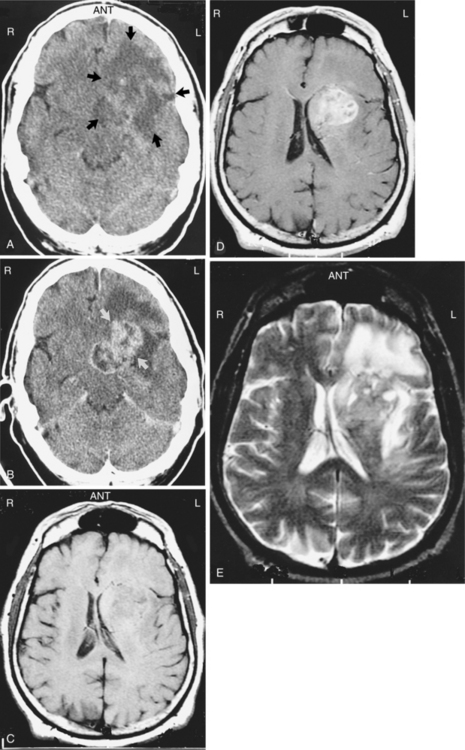
Figure 30-5 Astrocytoma. These contrasted and noncontrasted computed tomographic (CT) and magnetic resonance imaging (MRI) scans were obtained in the same patient and demonstrate a left astrocytoma with a large amount of surrounding edema. A, The noncontrasted CT scan shows only a large area of low density that represents the tumor and edema (arrows). B, A contrasted CT scan shows enhancement of the tumor (arrows) surrounded by the dark or low-density area of edema. C, A noncontrasted T1-weighted MRI scan clearly shows a mass effect due to impression of the tumor on the left lateral ventricle and some midline shift. D, A gadolinium-enhanced T1-weighted MRI scan clearly outlines the tumor, but the edema is difficult to see. E, A T2-weighted MRI scan shows the tumor rather poorly, but the surrounding edema is easily seen as an area of increased signal (white). (From Mettler FA Jr: Essentials of radiology, ed 2, Philadelphia, 2005, Saunders.)
Clinical Manifestations.: In adults, astrocytomas typically occur in the third and fourth decades of life and are usually located in the cerebrum, most commonly in the frontal lobes, but also may be found in the temporal lobes, parietal lobes, basal ganglia, and occipital lobes. Astrocytomas usually appear in the cerebellum in children.
In adults typical initial symptoms are unilateral or focal headaches that become generalized as ICP increases. Frontal lobe tumors may produce personality disorders with changes in behavior and emotional state. Parietal and temporal lobe tumors may cause seizures on one side of the body. Occipital lobe tumors produce visual changes. Involvement of the optic apparatus or optic pathways also may produce visual changes. Refer to Table 30-3 for more details of signs associated with tumor location. In time, astrocytomas, like other gliomas, tend to become more malignant.
In children, cerebellar astrocytomas lead to symptoms of unilateral cerebellar ataxia involving the limbs and trunk followed by signs of increased ICP.
Prognosis.: Individuals with low-grade astrocytomas treated optimally have 5-to 10-year survival rates of 100% for completely excised lesions, and a 60% 5-year survival and 35% 10-year survival for partially excised lesions with radiation therapy. For many there will be a period of relative clinical stability that averages 5 to 7 years.98,133 Untreated low-grade astrocytomas have a 5-year survival rate of 32% and a 10-year survival rate of 11%.126 Despite the benign categorization, it must be understood that astrocytomas are nearly always infiltrative lesions and generally progressive.
High-Grade Astrocytoma—Grades III and IV
Incidence.: High-grade malignant astrocytomas, grades III and IV, are much more common in adults than low-grade astrocytomas. Grade III is often termed anaplastic astrocytoma and grade IV is termed glioblastoma multiforme (GBM), although both are highly anaplastic. Grade III and IV astrocytomas make up 20% to 30% of primary brain tumors.
Pathogenesis.: Anaplastic astrocytomas, grades III and IV, are diffusely infiltrative tumors that invade into the cerebral parenchyma. They typically involve the white matter of the cerebral hemispheres but may occur primarily in grey matter as well as in other areas of the CNS.130 They often contains a mix of cells and cell grades but are graded by the highest-grade cell seen in the tumor. GBM is a particularly rapidly growing, aggressive, infiltrative tumor that tends to invade both cerebral hemispheres via the corpus callosum. See Fig. 30-6 for a GBM MRI and intraoperative pictures. A GBM is a pinkish grey or multicolored, well-demarcated mass with scattered areas of grossly visible hemorrhage. The blood vessels show endothelial proliferation: it is a highly vascular tumor, with vascular endothelial growth factor (VEGF) implicated, suggesting that the malignant progression from low-grade astrocytoma to GBM includes an “angiogenic switch.”130 There may be areas of cystic degeneration and a central area of creamy necrosis. The histologic distinction of an anaplastic astrocytoma from a glioblastoma is based largely on the absence or presence of tumor necrosis139 and microvascular proliferation.130 Microscopically, the tumor is pleomorphic (having various distinct forms) and hypercellular, with the cells showing hyperchromatic nuclei. There are many mitoses, giant cells, and young glial forms.
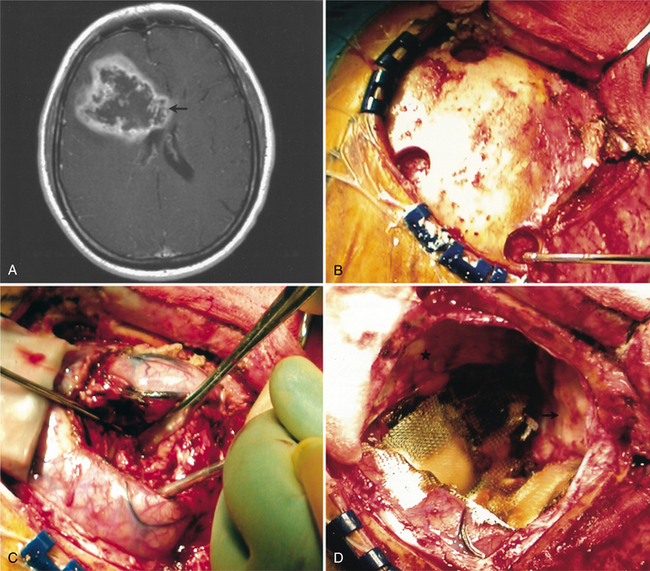
Figure 30-6 Magnetic resonance imaging (MRI) and intraoperative pictures of a patient with a right frontal glioblastoma multiforme. A, An axial T1-weighted MRI scan. The enhancing lesion demonstrates central necrosis and is causing mass effect. Infiltration along the corpus callosum is also shown (arrow). B, A frontal craniotomy is being performed. Burr holes have been placed and will be connected for bony removal. C, The brain has been incised and the tumor is being removed using a combination of suction and blunt dissection. D, The tumor and frontal lobe have been resected. The cut edge of the brain is seen at the lower left. The resection cavity has been lined with carmustine polymer (Gliadel) wafers and covered with a layer of Surgicel for hemostasis. (From Townsend CM Jr: Sabiston textbook of surgery, ed 17, Philadelphia, 2004, Saunders.)
Of interest is the advance in molecular genetics in astrocytoma. Two moderately common genetic alterations are found to occur: inactivation of the TP53 tumor suppressor gene and loss of chromosome 22q.71 Further inactivation of tumor suppressor genes on chromosomes 9p, 13q, and 19q leads to anaplastic astrocytomas.71 Many further mutations occur, and an understanding of the complexity of these mutations is beginning to suggest methods to intervene therapeutically. Also, understanding tumor stem cells that are responsible for populating and repopulating the tumors may also have therapeutic implications, as therapies that do not ablate the tumor stem cells will be ineffective in eradicating the tumor.41,74,75,130
Clinical Manifestations.: Anaplastic astrocytoma and GBM most frequently arise in the frontal and temporal lobes, with the cerebellum, brainstem, and spinal cord being rare sites for adults. They most frequently occur in the fifth and sixth decades of life. Signs and symptoms progress rapidly, with grade IV GBM being particularly aggressive. The presentation may be of unilateral headache that is followed by generalized headache, indicating an increase in ICP. The development of seizures is not unusual. Lethargy, memory loss, motor weakness, and personality changes may occur.
Prognosis.: All malignant astrocytomas will eventually recur. With optimal treatment (excision, radiation therapy) clients with anaplastic astrocytoma (grade III) have a 70% 1-year survival rate, a 40% 2-year survival rate, and a 10% to 20% 5-year survival rate. GBM (grade IV) has a grimmer prognosis with a 50% 1-year survival rate, a less than 15% 2-year survival rate, and rare long-term survival.126 The relationship between genetic alterations and prognosis is complex and may be age dependent.54,130 For patients under 50, the most significant prognostic factor is histology, with median survival for anaplastic astrocytoma 49.4 months and for GBM 13.7 months. For patients over 50, the most significant prognostic factor is the performance status. Patients with an anaplastic astrocytoma or a GBM with a high performance status live a median of 10.3 months, compared with 5.3 months for those with a lower performance status.88,101
Oligodendroglioma
Incidence.: Oligodendrogliomas make up 2% to 3% of gliomas. It is not uncommon to have a combination of cell types, such as astrocytes, creating a mixed oligodendroglioma/astrocytoma, or oligoastrocytoma. Oligodendrogliomas occur most frequently in young and middle-aged adults but can also be found in children.
Pathogenesis.: Oligodendroglioma is a slow-growing, solid, calcified tumor arising from oligodendrocytes, the myelin-producing cells of the CNS. It stains for myelin basic protein. It can be either low grade (II) or high grade (III). It is a grey-pink to red cystic area in the brain and has a honeycomb appearance at low microscopic power due to the presence of a fibrovascular stroma. On higher power the cells have a uniform appearance, with a central nucleus surrounded by a clear cytoplasm, or a fried egg appearance. Mitotic figures are infrequent. Approximately 70% of these tumors show some evidence of calcification.
Clinical Manifestations.: Oligodendrogliomas are located predominantly in the cerebral hemispheres, often in the frontal lobes. They expand toward the cortex and may spread through it and eventually attach to the dura.45 A history of partial or generalized seizures, usually of long duration and sometimes with chronic headache, is the typical presentation pattern of oligodendrogliomas. They tend to bleed spontaneously and may present with a strokelike syndrome.117 The hallmark of this tumor radiologically is calcification, which can be identified in the vast majority of people by CT. It is usually nonenhancing with gadolinium, meaning that the surrounding edema is limited.133 If an oligodendroglioma contains astrocytoma cells, it is graded at the highest level of anaplasia present.
Prognosis.: With optimal treatment, 5-and 10-year survival rates are 80% to 100% and 45% to 55%, respectively. The median overall survival is 17 years.92 Although after treatment a long interval of quiescence may occur, oligodendrogliomas eventually recur, often as a more aggressive tumor with progressing symptoms.126
Ependymoma
Incidence.: Ependymomas have a low incidence, comprising only about 2% of gliomas. Ependymoma is much more prevalent in children than adults and is the third most frequent posterior fossa neoplasm of children.
Pathogenesis.: An ependymoma is a neoplasm derived from the ependymal cell lining of the ventricular system and the central canal of the spinal cord. It is graded I to IV, depending on the degree of anaplasia. It is usually reddish, lobulated, and well circumscribed, resembling a cauliflower in shape. Pseudorosette formation, in which the cells are arranged about a clear space or a blood vessel, may occur, and blepharoplasts (small round or rod-shaped intracytoplasmic bodies) may be seen.
Clinical Manifestations.: Ependymoma is more common in the fourth ventricle and is likely to be detected early because of the signs and symptoms of increased ICP in the posterior fossa (e.g., headache, nausea, vomiting, and papilledema). However, supratentorial ependymomas often grow large before detection. Fig. 30-7 depicts an ependymoma of the fourth ventricle.
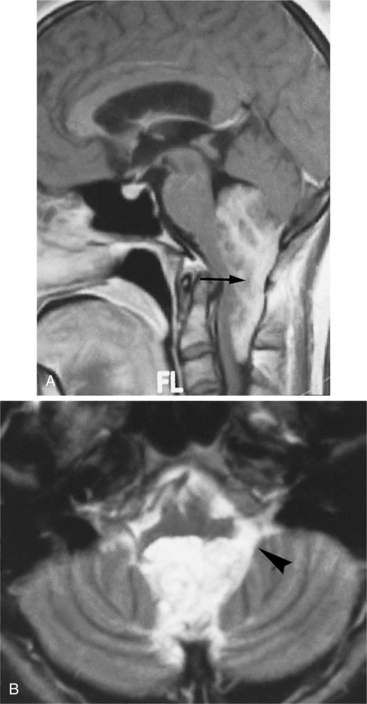
Figure 30-7 Ependymoma of the fourth ventricle. Sagittal gadolinium-enhanced T1-weighted (A) and axial T2-weighted (B) magnetic resonance images. A heterogeneously enhanced mass (arrow) fills the lower half of the fourth ventricle and extends through the foramina of Luschka (arrowhead) and Magendie to lie posterior to the medulla oblongata and upper cervical spinal cord, which are compressed from behind. There is obstructive hydrocephalus. (From Grainger and Allison’s diagnostic radiology: a textbook of medical imaging, ed 4, Philadelphia, 2001, Churchill Livingstone.)
Prognosis.: The prognosis for ependymomas is improving: 5-year survival rates exceed 80% and 10-year survival rates are 40% to 60%.126
Medulloblastoma
Incidence.: Medulloblastomas make up 3% to 5% of primary brain tumors. The age of peak incidence is 45 to 55 years in adults. In children, the tumor occurs mainly between the age of 2 and 10 years. Medulloblastoma is the most common malignant primary CNS tumor in children and the second most common posterior fossa tumor in children.
Pathogenesis.: Medulloblastoma is a rapidly growing malignant tumor. The cell of origin is unknown, but it is presumed to arise from the embryonal external granular layer of the cerebellum. It is considered to belong to a group of tumors known as primitive neuroectodermal tumors (PNETs). It characteristically metastasizes to the surface of the remaining CNS via the subarachnoid spaces. Grossly it is red and soft and is composed of many closely packed cells, with oval nuclei and many mitoses. Pseudorosette formations are common. It is highly vascular, containing numerous small blood vessels.126
Clinical Manifestations.: Medulloblastoma often develops in the cerebellar vermis and is very aggressive in younger children. Because of its proximity to the fourth ventricle, early development of hydrocephalus is common, along with other signs of cerebellar dysfunction, such as ataxia. Medulloblastomas tend to metastasize through CSF pathways, more predominantly into the spine but also into the supratentorial compartment.
Prognosis.: Early in the century medulloblastomas were uniformly fatal tumors. Improvement in therapeutic strategies during the past 30 years has dramatically improved the prognosis.124 Favorable prognostic factors include age greater than 2 years, undisseminated local disease, and greater than 75% tumor resection. In these clients, the 5-year disease-free survival rate exceeds 60% to 70% in most studies.51,126 In poorer-risk cases, the 5-year disease-free survival rate is about 45%.126
Tumors Arising from Supporting Structures in the Brain
Overview.: Meningiomas are slow-growing, usually benign lesions that occur most commonly along the dural folds and cerebral convexities, although they may occur in the spinal cord as well. The WHO classification recognizes three groups, grade I or benign, grade II or atypical, and grade III or malignant (anaplastic).139
Incidence.: Meningiomas represent up to 27% of all intracranial neoplasms and are the second most common primary intracranial tumor in adults and the most common of benign brain neoplasms. Ninety percent are considered benign and about 5% are grade III. Most are single lesions, but multiple meningiomas also occur. They are most common between the ages of 40 and 70, and are two to three times more prevalent in females than in males. They are increased in neurofibromatosis, in women who use postmenopausal hormone replacement therapy, and in patients who have had breast cancer.31 Prognostication and treatment rely on differentiation between a benign meningioma and a metastatic brain lesion originating from a breast cancer.139
Pathogenesis.: Meningiomas originate in the arachnoid layer of the meninges and are believed to be derived from the cells and vascular elements of the meninges. Cytogenetic analysis has demonstrated multiple deletions on chromosome 22 in most people with meningioma. They are most often located between or over the cerebral hemispheres, at the skull base, or in the posterior fossa. Meningiomas are typically well-circumscribed globular masses. They may infiltrate the dura, the dural sinuses, or bone, but generally do not invade the underlying brain parenchyma. See Figs. 30-8 and 30-9 for CT scans of meningiomas. Most meningiomas grow as well-encapsulated tumors, but others develop in relatively thin sheets along the dura.
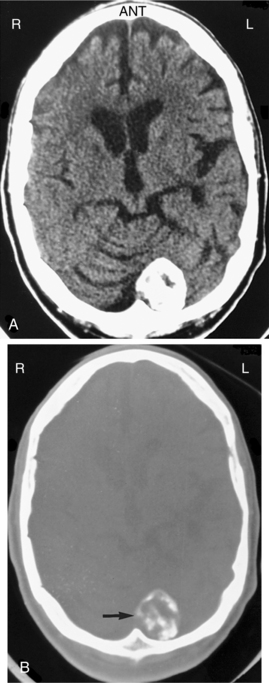
Figure 30-8 Meningioma. A, A noncontrasted computed tomographic scan shows a very dense, peripherally based lesion in the left cerebellar area. B, A bone window image obtained at the same level shows that the density is due to calcification within this lesion. (From Mettler FA Jr: Essentials of radiology, ed 2, Philadelphia, 2005, Saunders.)
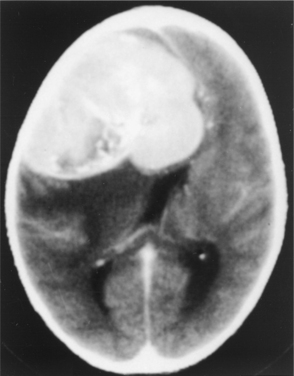
Figure 30-9 Computed tomographic scan with contrast of a meningioma in a patient who presented with mild cognitive deficits, illustrative of the size a slow-growing tumor can attain in the brain. The tumor was completely resected. (From Goldman LM, Ausiello D, eds: Cecil textbook of medicine, ed 22, Philadelphia, 2004, Saunders.
Meningiomas, because of their proximity to or invasion of the bone, are known to provoke a local osteoblastic response termed hyperostosis. This may cause a profuse local thickening of the skull. Fig. 30-10 shows diffuse reactive hyperostosis as well as facial distortion from the growing meningioma.
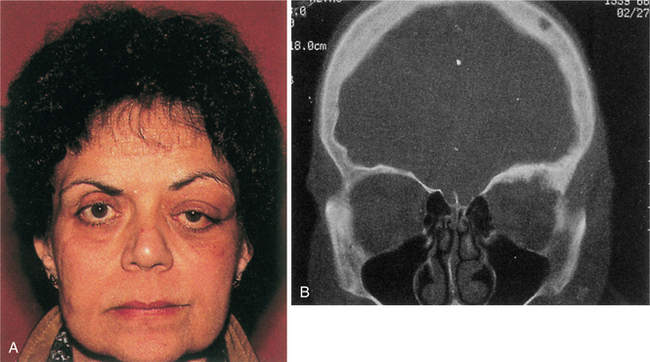
Figure 30-10 A, Upper eyelid edema, mild proptosis, and downward displacement of the eye due to en plaque sphenoid wing meningioma. B, Computed tomographic scan of the same patient demonstrating lytic bone lesions and diffuse reactive hyperostosis due to bone infiltration by meningioma. (From Abeloff MD, Armitage JO, Niederhuber JE, et al: Clinical oncology, ed 3, Philadelphia, 2004, Churchill Livingstone.)
Clinical Manifestations.: Meningiomas are more common in the later years of life and are more frequent in women. Because they are slow growing, abnormal signs and symptoms may evolve over a period of many years. When located in silent brain areas, some meningiomas can become very large before causing clinical symptoms. Also, they can be discovered incidentally as masses that show little or no growth over time. Neurologic abnormalities depend on the location of the tumor; seizures are a common finding with skull-based lesions.
Prognosis.: Meningiomas, when completely resected (surgical accessibility determines excision capabilities), have excellent prospects of long-term cure. Patients with completely excised lesions experience a 10-year survival rate of 80% to 90%. Partially resected meningiomas have a 50% to 70% 10-year progression-free survival. Malignant meningiomas, about 1% to 10% of meningiomas, have a shorter disease-free interval126 and a tendency to recur.
Pituitary Adenoma
Overview.: Pituitary adenomas are benign tumors derived from cells of the anterior portion of the pituitary gland. The pituitary gland, located at the base of the brain, sits in the sella turcica, the saddle-shaped transverse depression on the superior surface of the body of the sphenoid bone. Fig. 30-11 gives the anatomic relations of the pituitary gland, optic chiasm, and surrounding parasellar structures. Although pituitary adenomas are the most common of the pituitary tumors, infrequently other types of pituitary tumors may occur in the location of the pituitary gland and may be primary or metastatic. See also the section Pituitary Gland in Chapter 11.
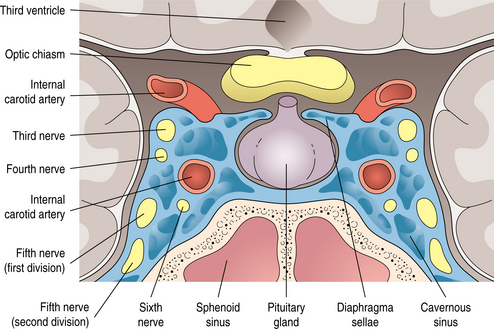
Figure 30-11 Anatomic relations of pituitary gland and surrounding parasellar structures. (From Thapar K, Laws ER: Tumors of the pituitary gland. In Murphy GP, Lawrence W, Lenhard RE, eds: American Cancer Society textbook of clinical oncology, ed 2, Atlanta, 1995, American Cancer Society.) American Cancer Society
Incidence.: Pituitary adenomas are common lesions, accounting for about 5% to 15% of all intracranial tumors, making them the third most common primary brain tumor in adults after meningiomas and the gliomas. They are usually found in middle-aged or older people. Women are more affected than men, particularly during childbearing years. Almost 70% are functional, or secreting, tumors, and these tend to occur in younger adults. Nonfunctioning tumors (nonsecreting), also called nonfunctioning adenomas or NFAs, tend to occur in older adults.
Pathogenesis.: With recent advances in molecular techniques, genetic abnormalities associated with pitu- itary tumors are becoming clearer. The great majority of pituitary adenomas are monoclonal in origin, suggesting that most arise from a single somatic cell. Additional molecular abnormalities present in aggressive pituitary adenomas and include mutations of the RAS oncogene and overexpression of the c-MYC oncogene, which suggests that these genetic events are linked to disease progression.67 Small lesions of the pituitary gland called microadenomas are less than 10 mm in diameter, and may be asymptomatic. Most grow in the front two thirds of the pituitary gland. Larger tumors, or macroadenomas, may compress the adjacent normal pituitary gland. Fig. 30-12 shows a pituitary tumor extension down into the sphenoid sinus. Extension of the tumor above the sella turcica compresses the optic chiasm.
Clinical Manifestations.: In the majority of pituitary tumors, the release of excess pituitary hormones or pituitary insufficiency results in dramatic and unique clinical syndromes. Galactorrhea and amenorrhea, gigantism and acromegaly, and the symptoms of Cushing’s disease (hypertension, facial and truncal obesity, osteoporosis, muscle weakness, menstrual abnormalities, and female hirsutism) are among the hormonal symptoms. Pituitary insufficiency, or hypopituitarism, can lead to symptoms such as fatigue, weakness, and hypogonadism. A second pattern of presentation consists of regression of secondary sexual characteristics and hypothyroidism. The third pattern of presentation is one of neurologic findings, including headache, bitemporal visual loss, and ocular palsy. Fig. 30-13 localizes masses such as a pituitary tumor by the pattern of visual field loss. Fig. 30-14 illus- trates the local effects of an expanding pituitary tumor causing visual field defects.
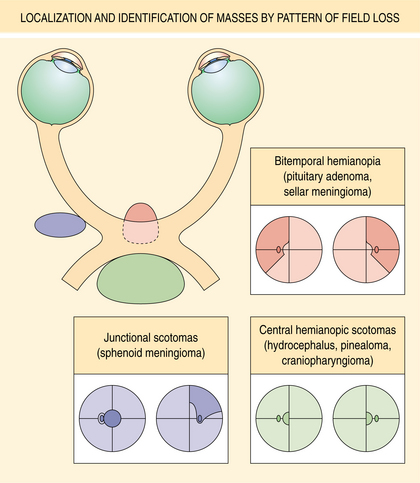
Figure 30-13 Localization and probable identification of masses by pattern of field loss. Junctional scotomas occur with compression of the anterior angle of the chiasm (sphenoid meningioma). Bitemporal hemianopia results from compression of the body of the chiasm from below (e.g., because of pituitary adenoma, sellar meningioma). Compression of the posterior chiasm and its decussating nasal fibers may cause central bitemporal hemianopic scotomas (e.g., because of hydrocephalus, pinealoma, craniopharyngioma). (From Yanoff M, Duker JS, Augsburger JJ, et al, eds: Ophthalmology, ed 2, St Louis, 2004, Mosby.)
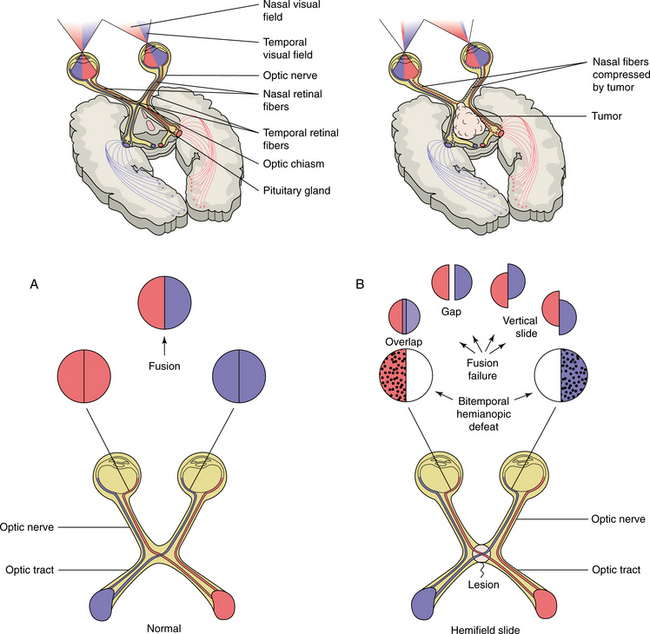
Figure 30-14 Local effects of an expanding pituitary tumor causing visual field defects: normal vision (A); bitemporal hemianopsia (B). The nasal and temporal fields lose their linkage, resulting in overlap of the preserved visual field. (From Larsen PR et al, eds: Williams textbook of endocrinology, ed 10, Philadelphia, 2003, Saunders.)
MEDICAL MANAGEMENT
Nonfunctioning tumors usually require no treatment Functional tumors may respond to hormonal therapy. Malignant tumor treatment is by surgery, transsphenoidal whenever possible, and conventional and stereotactic radiotherapy.63
PROGNOSIS.
Tumors of the pituitary have become very treatable, with the majority of people enjoying long-term survival or cure. Because visual compromise is a complicating feature of many pituitary tumors, serial recording of visual field deficits can document disease progression in addition to responses to treatment.
Neurinoma, Neuroma
Overview and Incidence.: Neurinomas are slow-growing, benign tumors originating from Schwann cells. In the brain they most commonly develop on the vestibular component of the eighth cranial nerve and are also called acoustic neurinomas, acoustic neuromas, or schwannomas. Acoustic neurinomas account for 3% to 10% of all brain tumors. They occur mainly in the fourth to sixth decades of life, with a 2: 1 female to male occurrence ratio. About 5% occur in the context of neurofibromatosis. Bilateral lesions are most likely to occur in neurofibromatosis.
Pathogenesis.: Acoustic neurinomas typically originate in the internal auditory canal in the transition zone of the oligodendroglial cells and peripheral nervous system Schwann cells. Neurinomas also may be found attached to other cranial nerves, such as the trigeminal nerve. The tumor grows into the cerebellopontine angle, eventually compressing the facial nerve, and encroaches on the brainstem. Some lesions may remain relatively quiescent for long periods of time, but the majority are slow-growing, progressive lesions. The tumor is thickly encapsulated, often highly vascular, and microscopically consists of spindle-shaped cells with rod-shaped nuclei often lying in parallel rows.
Clinical Manifestations.: Acoustic neurinomas typically present with progressive unilateral sensorineural hearing loss. Other symptoms include tinnitus, vertigo, and unsteadiness. Facial numbness, difficulty swallowing, impaired eye movement, and taste disturbances may occur. Weakness of the facial muscles is generally a late feature. Deformity and obstruction of the fourth ventricle leads to hydrocephalus with headache, vomiting, and other symptoms of increased ICP. See Fig. 30-15 for a surgical view of a large acoustic neuroma.
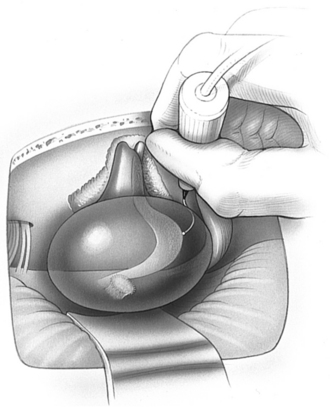
Figure 30-15 Surgical view of a large acoustic neuroma (retrosigmoid approach) showing use of a flexible-tipped probe to locate the facial nerve on the medial surface of the tumor out of direct view. Early identification of the facial nerve “around the corner” on the ventral surface of the tumor helps speed the procedure by allowing rapid removal of the remaining capsule. Tumor is drawn as if transparent to show details of anatomy on the hidden surface. (From Yingling CD, Gardi JG: Otolaryngol Clin North Am 24:413, 1992.)
Prognosis.: In the majority of cases cure is achieved with surgical resection. Stereotactic radiotherapy may be possible, reducing surgical side effects.5 As acoustic neurinomas are slow growing, and surgery often accelerates hearing loss, the decision to delay surgery until necessary may be made. However, because the likelihood of hearing retention is greatest when the tumor is small, surgery may be done as soon as possible.
Choroid Plexus Papilloma
Choroid plexus papilloma is a low-grade neoplasm of the choroid plexus, the vascular coat along the ventricles car- rying blood vessels within the pia mater to each ventricle. It is relatively rare and usually is found in children. It often is associated with overproduction of CSF and hydrocephalus. Complete removal of the tumor usually results in an excellent prognosis and resolution of the hydrocephalus. The prognosis for choroid plexus carcinoma, another variation of a choroid plexus tumor, is dismal.
Pinealoma
Overview and Incidence.: Pineal region (posterior to the third ventricle) tumors are rare (1% of all intracranial tumors), more common in children, and more common in males than females. They tend to occur in adults between 20 and 40 years of age. These are a heterogeneous group of tumors. Germ cell pinealomas have an embryonal basis, and although some are very radiosensitive, others are aggressive, highly malignant, and generally incurable. Pineal parenchymal tumors have a tendency to craniospinal dissemination.
Clinical Manifestations and Prognosis.: Pineal region tumors typically result in obstructive hydrocephalus because of the proximity of the pineal gland to the ventricular system. Symptoms include headache, nausea, vomiting, and ocular abnormalities. Management is by shunting the hydrocephalus, if present; radiation therapy; and/or surgical excision. Individuals with responsive tumors have a 5-year survival rate of 70%.126 Those with nonresponsive tumors have a 1-year survival rate of only 33%.
Craniopharyngiomas
Overview.: Craniopharyngiomas are histologically benign congenital tumors and occur most commonly in the suprasellar region in the pituitary stalk adjacent to the optic chiasm.
Incidence.: Craniopharyngiomas are rare and account for 1% to 3% of all intracranial tumors.13,22 They are the third most common intracranial tumor in children, accounting for 10% of all intracranial tumors in this age group.
Pathogenesis.: Craniopharyngiomas presumably arise from embryonic remnants of Rathke’s pouch and grow slowly from birth. They vary in size from small, solid, well-circumscribed masses to huge multilocular cysts that invade the sella turcica, reaching a large size before they are diagnosed. They often involve the pituitary gland, optic nerve, and third ventricle. Three basic histologic subtypes have been described: mucoid epithelial cysts, squamous epitheliomas, and adamantinomas.134
Clinical Manifestations.: Based on the location, craniopharyngiomas can compromise a number of important intracranial structures and produce multiple signs and symptoms. The most common presentations are pituitary hypofunction, visual difficulties, and severe headaches. Other signs are increased ICP, neuroendocrine disorders, hypothalamus involvement, cranial nerve palsies, hydrocephalus, and progressive dementia. Sexual dysfunction is the most common endocrine problem in adults, with 90% of men complaining of erectile dysfunction and most women having amenorrhea. Depression may occur, presumably because of extension of the tumor into the frontal lobes, striocapsulothalamic areas, or limbic system.134
Prognosis.: Optimal treatment is controversial, but radiation and/or surgical resection are used. Intracavitary radiation is used in select tumors. With complete resections or resections followed by radiation therapy, 10-year survival rates of 78% have been reported. The tumors do have a tendency to recur, and even though histologically they are benign, they may be better thought of as low-grade malignancies.
Epidermoid and Dermoid Tumors (Cysts)
Incidence.: Epidermoid and dermoid tumors are rare benign tumors that arise from imperfect embryogenesis of the CNS and account for 2% of intracranial tumors. The most common cysts in the brain are epidermoid, arachnoid, colloid, and dermoid.
Pathogenesis.: Cysts are fluid-filled spheres composed of desquamated epidermal cellular debris, keratin, and cholesterol. During embryologic development, groups of cells are diverted from the areas of the face or skin to the neural tube. They grow in basal regions of the brain and tend to enlarge along CSF pathways. Most cysts are benign and grow slowly, and may not cause symptoms for many years.
The epidermoid cyst often contains remnants of skin cells or tiny pieces of cartilage and occurs near the cerebellopontine angle or the pituitary gland. The arachnoid cyst is found in the subarachnoid space, often in the Sylvian fissure, the cerebellopontine angle, the cisterna magna, or the suprasellar region of the brain, and may cause increased ICP. The colloid cyst is most frequently found in the third ventricle and may block CSF, causing headache, seizures and increased ICP. The dermoid tumor has epidermal cellular debris, but it is mixed with additional dermal elements such as hair, hair follicles, sweat glands, and sebaceous glands. Dermoid cysts are usually located in the posterior fossa or the adjacent meninges, or in the lower spine.
Hemangioblastoma
Incidence.: Hemangiomas make up 2% of all intracranial tumors, are the most common adult intraaxial tumor of the posterior fossa, and occur more frequently in males. They most commonly occur in people about 40 years old.
Pathogenesis.: Hemangiomas are benign slow-growing tumors typically arising in the posterior fossa, primarily in the cerebellar vermis or pons, as solitary lesions with clearly indicated borders. The origin is thought to be cells in the blood vessel lining. Hemangioblastomas are a vascular conglomerate of endothelial cells, pericytes (peculiar elongated cells with the power of contraction, found wrapped about precapillary arterioles), and stromal cells. These highly vascular tumors attached to the wall of a surrounding cyst are often associated with von Hippel-Lindau syndrome.
Chordoma
Chordomas rarely arise in the brain and represent less than 1% of all intracranial neoplasms. They are much more typical in the axial skeleton, preferring the clivus (in the posterior cranial fossa), sacrum, and nonsacral spine. They are tumors of bone, presumed to arise from the embryonal notochord remnants. They are considered histologically benign but have a locally destructive nature, progressive course, and metastatic behavior.126 Cranial chordomas typically involve the skull base with a destructive process that invades rostrally into the optic chiasm, into the brainstem, or ventrally into the sinuses. Because of surgical inaccessibility, curative resections are difficult, if not impossible. Median survival ranges from 4.2 to 5.2 years, with recurrences likely.126
Primary Central Nervous System Lymphoma
Overview and Incidence.: Primary CNS lymphoma (PCNSL) is a non-Hodgkin’s lymphoma and occurs in the absence of systemic lymphoma. It is also called an extranodal lymphoma. This tumor was formerly quite rare, but from 1973 to 1985 tripled in frequency in immunocompetent patients and also increased in the immunosuppressed population—that is, clients with acquired immunodeficiency syndrome (AIDS) and collagen vascular disorders, organ transplant recipients, and the congenitally immunodeficient.4,139 There was a decrease in incidence in young men and patients with AIDS from 1995 to 1998, explained by the introduction of highly active antiretroviral therapy for patients with human immunodeficiency virus (HIV) infection.142 It currently accounts for 4% to 7% of all primary brain tumors.1,26,28,58,91
Pathogenesis.: The pathophysiologic basis for development of these tumors is unclear, particularly in immunocompetent patients. PCNSL most commonly originates from B lymphocytes and is associated with cytokines. In immunosuppressed patients it is almost always associated with latent infection of neoplastic B cells by Epstein-Barr virus. B cells infected with Epstein-Barr virus are immortalized and able to replicate spontaneously.58 The lymphoma cells typically assume a periventricular pattern, involving the deep white matter, basal ganglia, corpus callosum, and thalamus. PCNSL may also involve the CSF, the eyes, or the spinal cord. A large percentage of PCNSLs begin as solitary cerebral lesions but eventually develop into multiple lesions. Lesions in immunocompetent patients more often may be a single brain lesion, in a supratentorial location, and with frontoparietal lobe involvement. The diagnostic procedure of choice is a stereotactic (x-ray guided) biopsy, because patients derive no clinical benefit from surgical resection.58
Clinical Manifestations.: Symptoms and signs generally evolve over several months, including personality and behavioral changes, confusion, generalized seizures, and symptoms associated with increased ICP (headaches, nausea and vomiting). The most frequent presenting symptom in 30% to 40% of patients is impaired cognition.32 Focal neurologic signs such as hemiparesis or blurred or double vision may occur. The appearance on MRI or CT of multiple deep cerebral and periventricular lesions, along with an immunodeficient state, contributes to the diagnosis.65 Differential diagnosis includes infections, other tumors, and inflammatory disorders.
Other Miscellaneous Brain Tumor Types
Other infrequent brain tumors bear mention. They are as follows:
• Chondromas tend to arise at the base of the skull, are slow growing, and are composed of cartilage-like cells often attached to the dura mater.
• Chondrosarcomas are the malignant variant of chondromas.
• Atypical teratoid rhabdoid tumors (ATRTs) are high-grade tumors occurring most commonly in the cerebellum in children and are aggressive with frequent metastasis through the CNS.
• Dysembryoplastic neuroepithelial tumors (DNETs) are slow-growing, benign, grade I tumors, often containing a mix of neurons and glial cells, and typically found in the temporal or frontal lobe.
• Gangliocytomas and gangliogliomas arise from ganglia-type cells (groups of neurons), and are most commonly located in the temporal lobe and third ventricle.
• Germ cell tumors include the germinoma, teratoma, embryonal carcinoma and yolk sac tumor, and choriocarcinoma. These tend to arise in the pineal or suprasellar regions and occur primarily in children and young adults. Teratomas are composed of various tissue types within the tumor, often containing calcium, cysts, fat, and other soft tissues.
More details on these CNS tumors, as well as further information on the numerous other infrequent CNS tumors, are available in various references.*
Diagnosis of Primary Brain Tumors
When a brain tumor is suspected on clinical evaluation, a thorough neurologic examination as well as brain imaging studies are done to confirm its presence and exact location.
MRI has evolved as the most informative brain imaging study because of its superior imaging capabilities and lack of artifact from the temporal bones. With the addition of gadolinium contrast enhancement, which distinguishes tumor from surrounding edema, MRI detects tumors even a few millimeters in size. MRI also defines critical anatomic relationships between the tumor and surrounding neurovascular structures. The multiplanar capability of MRI allows optimal visualization of the anatomy. MRI is particularly useful in visualizing the brainstem and other posterior fossa structures.139 New MRI techniques are being developed to investigate the biochemical basis of tumors, such as the proton magnetic resonance spectroscopy (MRS), which measures the signals from nuclei other than water.101
Although MRI has many advantages over CT, CT scanning is widely accessible, convenient, and effective in revealing most brain tumors if they are large enough. The increased vessel formation or neovascularization accounts for the enhancement of these tumors and allows them to be visualized. Although its brain imaging capabilities are inferior to those of MRI, CT can identify cerebral edema, midline shift, and ventricular compression of obstructive hydrocephalus. In intraventricular masses, CT is highly sensitive in detecting calcification. CT also is better than MRI for demonstrating bone destruction. CT imaging may be needed when a patient has precautions for a magnetic study (e.g., pacemaker or other metallic implants). Intravenous contrast greatly increases the sensitivity of CT scan for brain tumors.
Once a tumor has been detected with MRI or CT, other particular parameters may help to characterize it further. For example, establishing the location of an intracranial neoplasm in either the extraaxial or intraaxial compartment is valuable in differential diagnosis.139 For example, astrocytomas are intraaxial, and meningiomas are extraaxial. The MRI or CT may detect a cleft between the brain parenchyma and the tumor, which indicates a possible extraaxial mass such as a meningioma.
There are numerous new techniques to image tumors. Single-photon emission computed tomography (SPECT) imaging uses preoperative thallium 201 emission CT in which the maximum uptake area of the brain tumor distinguishes benign from malignant tumors and localizes the area for biopsy. Iodine-123-α-methyl-L-tyrosine single-photon emission tomography (IMT-SPET) imaging uses a radioisotope to distinguish glioma recurrence from benign posttherapeutic change. The positron emission tomography (PET) scan is able to localize the areas of maximum glucose utilization within a tumor, guiding the neurosurgeon to perform biopsy of locations with the most aggressive biologic behavior and differentiating viable tumor from necrosis.26,47,101 The PET scan also maps functional areas of the brain prior to surgery or radiation in order to minimize injury to eloquent areas.139 Presurgery motor and somatosensory cortex mapping with functional MRI and PET is possible. Fluorodeoxyglucose PET (FDG-PET) measures glucose utilization and helps to differentiate recurrent tumor from radiation necrosis. It also is not influenced by corticosteroid therapy. Echo planar MRI is a new technique of functional MRI imaging that provides maps of tumor blood flow and may allow better resolution of tumor versus surrounding edema at the tumor borders. MRS may show pathologic spectra outside the area of contrast enhancement, suggesting infiltrative lesions.110,129
Additional tests may be indicated to further delineate the tumor and identify possible surgical hazards. Cerebral angiography delineates the vascularity within the brain and can help determine the best surgical approach. Visual field and funduscopic examination identifies visual defects that are specific to a particular area. Audiometric studies determine hearing loss. Chest films help to rule out lung cancer with metastatic lesions to the brain, and other studies are used to rule out a primary lesion outside the brain when a metastatic lesion is suspected. Endocrine studies are done when a pituitary adenoma or craniopharyngioma is suspected.52
A needle biopsy using CT-guided stereotactic (x-ray guided) technique through a burr hole in the cranium may be performed to identify the specific tumor type and grade. A needle biopsy may not be possible, however, with vascular tumors or tumors near vital centers for fear of precipitating bleeding or respiratory distress. As tumors may have variation in grading throughout the tumor, a needle biopsy may potentially miss the higher-graded area, limiting the accuracy of the diagnosis.
MEDICAL MANAGEMENT
Surgery, radiation therapy, chemotherapy, and immunotherapy are the treatment options for brain tumors. Management of symptoms and side effects is a major component of medical management.
TREATMENT
Surgical excision is the most important form of initial therapy, because it provides histologic confirmation of the tumor and a basis for determining the treatment and prognosis. The new stereotactic neurosurgical techniques have had a profound impact on neurosurgery efficacy and safety. Intraoperative magnification and the operating microscope have allowed stereoscopic visualization of otherwise inaccessible tissues and have reduced the morbidity and mortality of brain surgery.101 MRI scanning combined with computer-aided navigation tools helps the neurosurgeon map the exact tumor location and track its removal during the procedure. Surgery reduces tumor load and quickly relieves the ICP and mass effect, thereby reducing symptoms and improving neurologic function. The surgical cytoreduction also enhances the effectiveness of adjuvant therapy (e.g., radiation therapy).
A traditional operative technique is the craniotomy, a resection of the skull overlying the tumor, removal of the tumor, and replacement of the bone flap (Fig. 30-16). Stereotactic biopsy of the lesion without craniotomy is used when deep mass lesions are surgically unresectable or when the risk of craniotomy outweighs the benefits. Stereotactic procedures involve creating a burr hole in the brain at an exact location using a computer, radiologic equipment, and a special head-fixation device.
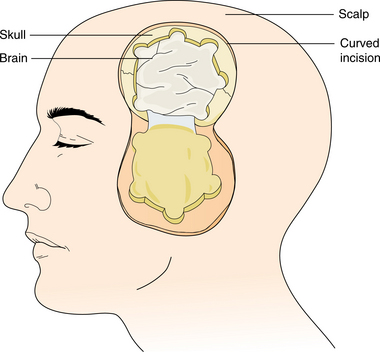
Figure 30-16 Craniotomy with osteoplastic bone flap. (From Schnell SS: Nursing care of clients with cerebral disorders. In Black JM, Matassarin-Jacobs E, eds: Luckmann and Sorensen’s medical-surgical nursing, ed 4, Philadelphia, 1993, Saunders, p 734.)
The technologic and conceptual advances in neurosurgery (e.g., intraoperative magnification, ultrasonic aspirators, microinstrumentation, computer-based stereotactic resection procedures) have allowed safer and more precise approaches to previously inaccessible tumors.118,126 Awake cortical mapping before and during surgery identifies critical areas of brain functioning to avoid and/or reduce damage to these areas.18,38 Endoscopic surgery for pituitary adenomas, tumors of the orbit, vestibular (acoustic) neuromas, meningiomas, and other skull-based tumors utilizes endoscopes attached to an endocamera and a video monitor system.119 Transsphenoidal resections are possible through the nose (transnasal), which avoid an external craniotomy. See Fig. 30-17. Actual short videos of endoscopic brain surgeries are available for viewing on the Internet.62 Facial craniotomy or endoscopy utilizes incisions positioned between facial cosmetic subunits as shown in Fig. 30-18.
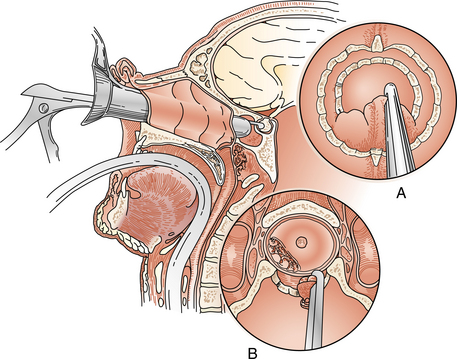
Figure 30-17 Endonasal transsphenoidal resection of the pituitary tumor. A, Removal of the sella floor with small rongeurs. B, Exposed inferior aspect of a pituitary adenoma. (From Tindall GT, Barrow DL: Disorders of the pituitary, St Louis, 1986, Mosby.)
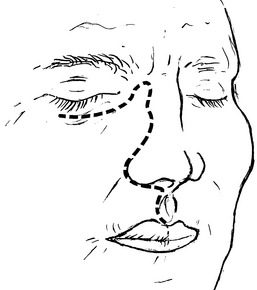
Figure 30-18 Illustration of standard location for facial incisions, with craniofacial resection completed using traditional methods. These incisions are positioned between facial cosmetic subunits (dashed lines). (From Cummings CW Jr, Haughey BH, Thomas JR, et al, eds: Cummings otolaryngology—head and neck surgery, ed 4, Philadelphia, 2005, Mosby.)
The goal of surgery is total excision, while minimizing trauma to vital neural structures. The survival rates of patients undergoing total resections for brain tumors are significantly higher than those of patients undergoing partial resections.88 In infiltrative intraaxial lesions, in which total excision is not possible, the goal is to provide a measure of temporary control by reducing mass effect and ICP. If the preoperative neurologic deficit is due to destruction of brain tissue by tumor, surgical resection will not improve the situation. However, if the deficit is related to compression from the tumor, excision may relieve the compression and allow the deficit to improve. In the case of many benign extraaxial tumors (e.g., meningiomas, schwannomas, pituitary adenomas), cure can be achieved.
Operative complications include hemorrhage, infection, seizures, hydrocephalus resulting from an impairment of CSF absorption, and neuroendocrine disturbances, especially if surgery is in the region of the pituitary. Brain edema, usually present before surgery, may be severely aggravated during surgery. Corticosteroids usually are given for several days before craniotomy to reduce preoperative edema. Improved surgical techniques have reduced the complications of hemorrhage, infection, and permanent neurologic injury to less than 10% of cases.126
Radiation Therapy.
Radiation therapy following surgical resection is of proven effectiveness for most malignant brain tumors.33 Various brain tumors have different susceptibilities to radiation therapy, but the survival advantage is unquestionable. A greater degree of tumor anaplasia and a younger age may result in a better response to radiation.11 Unresectable or incompletely resected tumors in particular are candidates for radiation therapy. Established radiation doses that avoid exceeding thresholds of CNS tolerance are in the range of 40 to 60 Gy (4000 to 6000 rad). Radiation using a linear accelerator is typically given in fractionated doses five times a week over 34 to 36 weeks.
Radiation is delivered to a localized area of the brain to minimize the volume of tissue irradiated. Acute reactions to radiation are a result of acute brain swelling, occur during or immediately after radiation, and manifest as an increase in neurologic deficit or increased ICP. Steroid therapy is given to reduce this effect during radiation therapy. A similar delayed postirradiation syndrome 1 to 3 months after radiation also can be controlled by steroids. A third brain reaction known as radiation necrosis may occur months to years after irradiation and is severe and irreversible.64,123 It is presumed to be the result of direct toxic effects on the brain and its microvasculature (Fig. 30-19). There is progressive deterioration, dementia, and focal neurologic signs.
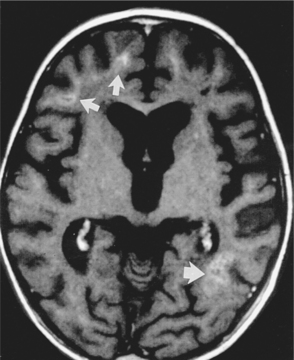
Figure 30-19 Generalized brain changes after radiotherapy. T1-weighted magnetic resonance image of a child shows both central and cortical atrophy as well as high-signal areas (arrows), owing to mineralizing microangiopathy. (From Behrman RE, Kliegman M, Jenson HB, eds: Nelson textbook of pediatrics, ed 17, Philadelphia, 2004, Saunders.)
As survival time increases, other long-term complications of irradiation are of concern. Hypopituitarism, radiation-induced occlusive disease of cerebral vessels, radiation-induced oncogenesis, leukoencephalopathy, and myelopathies from spinal axis irradiation are included in these complications (Fig. 30-20). White matter injuries have been shown to correlate significantly with radiation dose in long-term survivors (greater than 18 months). These changes correlate with functional neurologic status,30 including altered mental status, speech impairments, motor deficit, cranial nerve deficit, personality changes, altered memory, and other neurologic signs.6
Figure 30-20 Postradiotherapy encephalopathy. Axial fluid-attenuated inversion recovery magnetic resonance image. Extensive high signal in the white matter of both cerebral hemispheres is due to radiation-induced leukoencephalopathy. There are also areas of cystic necrosis in this case (arrowheads). (From Grainger and Allison’s diagnostic radiology: a textbook of medical imaging, ed 4, Philadelphia, 2001, Churchill Livingstone.)
Advances in radiation therapy have led to newer methods of radiation delivery. Interstitial radiation therapy, or brachytherapy, involves the placing of the radiation source, such as radium seeds, within the tumor for a period of several days. Brachytherapy has shown promise in treating GBM and other primary brain tumors.
Stereotactic radiosurgery is a technique to deliver a large single fraction of highly focal radiation to a brain tumor.72 It originally was used to treat functional disorders (e.g., pain and movement disorders) but is now being used in the treatment of primary and metastatic brain tumors.116 There are several methods: The linear accelerator–based systems, which are the most widely available, deliver high-energy photon beams using converging arcs, which intersect at the target site. Various modifications in the linear accelerator, or LINAC, are available. Threedimensional conformal radiation therapy (3D-CRT) allows shaping the radiation beams to match the tumor’s contours. Intensity-modulated radiation therapy (IMRT) is a refinement of 3D-CRT which ensures that maximum intensity is directed at a specific site, reducing the dose to the surrounding tissues. Gamma knife radiotherapy uses high-energy photon beams from cobalt 201 sources, each directed at a specific isocenter. A halo device is attached to the skull, and the patient’s head is positioned into a collimator that delivers focused gamma beams to the targeted tumor.
Synchrocyclotron proton beam therapy delivers heavy charged particle beams through a small number of portals in the skull.126 The CyberKnife is a frameless robotic radiosurgical device with increased fractionation flexibility and the ability to treat extracranial lesions. It is capable of changing the target of the beam delivery instantaneously.107,116
In some specific primary and metastatic brain cancers, radiosurgery may be the first line of treatment, instead of surgery. The advantages of radiosurgery compared to surgery are avoiding the risk of hemorrhage, infection, and tumor seeding; linking treatment directly to three-dimensional visualization, which reduces the chances of a marginal miss; and requiring minimal hospitalization.56,116
Although gamma knife, proton beam, and CyberKnife equipment is expensive, and treatment requires collaboration between radiation oncologists and neurosurgeons, the use of these modalities is rapidly increasing, and data on their effectiveness and neurotoxicity are becoming available. Radiotherapy may prove increasingly to be a beneficial modality of client care114 as it becomes more available and easier to deliver. Brain metastases are ideal lesions to be treated with stereotactic radiation because they can be optimally covered by the radiation distribution, which can be easily designed by radiosurgical treatment planning.28,29,128 In time, technologic modifications will allow treatment at other sites, such as the spine.
Chemotherapy.
Chemotherapy has been extensively studied in brain tumors and may have an impact on both survival and quality of life in those who have primary brain tumors, particularly for certain pediatric neoplasms such as medulloblastoma.126 The early studies in the 1960s involved the nitrosoureas (carmustine or BCNU, and lomustine or CCNU) and hydroxyurea, because of their in vitro sensitivity and lipophilic characteristics allowing them to cross the blood-brain barrier. BCNU has been the most effective cytotoxic agent against malignant glioma.139
Newer agents with antitumor activity include diaziquone, procarbazine, imidazole carboxamide (DTIC), vincristine, cisplatin, carboplatin, tamoxifen, CPT-11 (irinotecan or Camptosar), and temozolomide (Temodar or TMZ).101,139 TMZ is emerging as the chemotherapy drug of choice for high-grade gliomas in the adjuvant setting, associated with significant improvements in median progression-free survival.8,57,121,122,125 It is thought that TMZ may be especially useful for elderly patients with glioblastomas as an alternative to radiation therapy to maintain a reasonable performance status.20 A three-drug regime of procarbazine, lomustine, and vincristine (PCV) also has been shown to benefit patients with anaplastic glioma but offered no survival benefit for those with GBM.101 Current clinical trials are investigating paclitaxel (Taxol), phenylacetic acid, and other novel techniques such as the thymidine-kinase gene, continuous infusion chemotherapy, and the use of antiangiogenesis drugs such as thalidomide. Trials of intracavitary placement of carmustine polymer wafers (Gliadel) are demonstrating prolonged survival without the systemic side effects of chemotherapy.139
To bypass the blood-brain barrier, intrathecal delivery (through an Ommaya reservoir surgically placed in the scalp with its tube inserted into the lateral ventricle) can be done. Another technique is an intraarterial (intracarotid) delivery allowing much higher concentration of drugs such as methotrexate, vincristine, or cisplatin than intravenous injection, which overcomes molecular resistance. Intrathecal chemotherapy may be given when leptomeningeal involvement occurs with tumor or to increase the CSF concentration. Addition of chemotherapy to surgery and irradiation for malignant gliomas does provide some increases in 24-month survival, up to 23.4% from 15.9%.126
Hormonal Therapy.
Hormonal therapy is often used to treat functioning pituitary tumors. Dopamine agonists are used to control the production of prolactin. Somatostatin analogues are used to reduce growth hormone levels and relieve the associated symptoms. If satisfactory results are achieved, surgery and/or radiation may not be necessary.
Immunotherapy.
Immunotherapy or biotherapy is the most infrequently used and least proven therapy for brain tumors. Immunotherapy, originally the use of donor serum containing preformed antibodies, now includes the use of interferons and interleukin-2 (see the section on Interferons in Chapter 7 and Immunotherapy in Chapter 9) to boost immune function.52 The depressed immunocompetence of clients with malignant glioma gives at least a theoretical basis for the potential roles of biologic response modifiers in the treatment of these tumors. Preliminary studies have shown some promise.126
SYMPTOM MANAGEMENT.
Brain tumors lead to edema of tissue surrounding the tumor, and brain swelling can be massive and extend the neurologic deficits caused by the tumor alone. Antiinflammatory drugs (corticosteroids, such as dexamethasone [Decadron], prednisone, hydrocortisone) are used to provide prompt and effective reduction in peritumoral edema. Improvement in symptoms of ICP and in focal neurologic signs begins within 24 to 48 hours after steroid initiation, and by the fourth and fifth day the maximum degree of improvement is obtained.139
Corticosteroids generally are used perioperatively and are tapered gradually after tumor resection, because long-term high-dose corticosteroids precipitate undesirable side effects. Corticosteroids also may be used periirradiation because radiation also precipitates edema. Upon tumor recurrence or progression, corticosteroids may again be instituted to temporarily maximize residual neurologic function.
It is possible for a brain tumor to cause an acute increase in ICP that may be life-threatening because of imminent cerebral herniation. Emergency treatment is required if ICP reaches 20 mm Hg or more. Quick-acting agents are needed to lower the pressure. Mannitol is used as a temporary agent to quickly reduce brain water and relieve the pressure. Steroids are used in conjunction with mannitol to decrease edema. Table 30-5 lists emergency treatments for elevated ICP in acutely decompensating patients.
Table 30-5
Emergency Treatment of Elevated Intracranial Pressure in Acutely Decompensating Patients

PaCO2, Arterial partial pressure of carbon dioxide; IV, intravenous.
From DeAngelis, LM: Tumors of the central nervous system. In Goldman LM, Ausiello D, eds: Cecil textbook of medicine, ed 22, Philadelphia, 2004, Saunders.
Anticonvulsants also may be needed to prevent or control seizure activity. These drugs also are used before and after surgery to control symptoms and are continued as long as they are indicated.26 One of the most common anticonvulsant therapies used today in the brain tumor population is phenytoin (Dilantin).93
Brain tumors also cause a variety of motor, speech, hearing, visual, and other neurologic signs and symptoms. While control of the tumor and edema through medical management is the first priority, residual neurologic problems can significantly lower the quality of life. Timely referral to rehabilitation specialists for management of these functional deficits can improve performance status and quality of life. Strengthening, motor and balance evaluation and training, splinting, bracing, fatigue and pain management, incontinence training, home adaptation, activities of daily living (ADL) retraining, speech therapy, hearing adaptations, auditory retraining, and vision programs are available to improve and alleviate these functional deficits.
Other specialists are also part of the rehabilitation team. Enterostomal therapists provide help with ostomies; pharmacists provide assistance with pain management; the oncology nurse provides symptom management, psychosocial support, and education; the nutritionist provides diet and nutrition counseling; the social worker assists with community resources and placement in settings for necessary further care; and clergy provide assistance with personal and spiritual issues.
The psychosocial implications of brain tumors are enormous. Referral to psycho-oncologic specialists for alleviation of psychologic distress and family disruption, and promotion of role reorganization and adaptation are often of great value. Brain tumor support groups, psychotropic medications, oncology educational classes for the client and family, and enrollment in one-on-one support programs can be very helpful.
30-1 SPECIAL IMPLICATIONS FOR THE THERAPIST
Impaired Motor Function and Sensory Integrity Associated with Nonprogressive Disorders of the Central Nervous System—Acquired in Adolescence or Adulthood
Impaired Motor Function and Sensory Integrity Associated with Progressive Disorders of the Central Nervous System
Therapists will undoubtedly encounter clients with brain tumors in any practice arena because of the significant neuromuscular and cardiopulmonary impairments. Neurologic or orthopedic practices may see patients with brain tumors presenting with gait and balance instability or cervical pain. When the signs and symptoms of a yet undiagnosed brain tumor bring the person to therapy (e.g., unsteady gait and poor balance, weakness), differential diagnosis skills are needed by the therapist to determine a cluster of signs and symptoms indicating a possible tumor, such as headache, nausea and vomiting, lethargy, and so on (refer to Boxes 30-2 and 30-3 and Table 30-3) or a progression of symptoms despite physical therapy intervention requiring referral to the physician.
Knowledge Needed for Rehabilitation
Brain tumor studies are beginning to demonstrate rehabilitation effectiveness.14,46,86 As the survival of those with brain tumors increases, rehabilitation needs become more prominent.14 A general knowledge of primary brain tumor and medical treatment is needed to provide the therapist with skills for differential diagnosis, examination and evaluation, treatment planning, and goal setting. Knowledge of malignant versus benign status, disease progression expectations, complications, prognosis, and precautions such as seizures and deep vein thromboses (DVTs) are needed to plan intervention and establish goals. The therapist should also be aware of expected focal symptoms in relation to tumor location to anticipate functional changes that may require treatment modifications, as well as possible paraneoplastic syndromes that may complicate rehabilitation. In geriatric populations, managing the patient with brain cancer may require comprehensive geriatric assessment (CGA) to identify comorbidities.9,10
It is important to be knowledgeable about the more prevalent brain tumor types and be able to differentiate, for example, between a meningioma (a benign, potentially curable tumor) and a malignant glioma (a rapidly growing fatal tumor), in order to set goals, to interact with the family, and to provide appropriate intervention. The advancements in brain tumor medical management (e.g., the promising effectiveness of TMZ for malignant gliomas) and the developing preciseness of radiation therapy, sparing brain damage, brings longer survival and opportunities for better quality of life.121 The therapist must be aware, prepared, and hopeful that, as the demand for rehabilitation grows, the rehabilitation outcomes will improve.
In addition, knowledge of medical treatment complications from surgery, radiation, chemotherapy, and other interventions will give the therapist the ability to adjust the rehabilitation program as needed, to accommodate, for example, myelosuppression from chemotherapy or fatigue from radiation therapy.
Acute Postoperative Management
When a referral is received for acute postoperative rehabilitation therapy, awareness of general postoperative complications, including atelectasis, pneumonia, cardiac arrhythmias, fluid and electrolyte imbalances, infection, meningitis, intracranial hemorrhage, and renal and gastrointestinal disorders, is important. Potential symptoms after brain surgery include confusion, pain, weakness, and headache. Observing the client closely during therapy intervention for any significant signs and symptoms, and making appropriate adaptations during therapy, are part of the role of the therapist.113
Postoperative Complications of Intracranial Surgery
Potential complications of intracranial surgery may be very serious, even fatal, because of the significant functions performed by the structures involved. Some postoperative complications may improve; others may be permanent.
Increased ICP (resulting from cerebral edema or bleeding) is the major complication of intracranial surgery. Findings may include decreased level of consciousness with headaches, visual and speech disturbances, muscle weakness or paralysis, pupil changes, seizures, vomiting, and respiratory changes (see Box 30-3).
In at-risk patients, there are several methods that can be used to continuously measure ICP, including intraventricular catheters, subarachnoid or subdural screws or bolts, epidural sensors, and intraparenchymal catheters. The ICP is often displayed in the bedside monitor. Other monitored parameters include mixed venous oxygen saturation (SvO2) and jugular oxygen saturation (SjO2), which reflects oxygen saturation of the blood returning from the brain. Normal ICP ranges from 0 to 15 mm Hg, with a midrange of 8 mm Hg and fluctuations occurring with active movement of the extremities and trunk, coughing, suctioning, noxious touch, and other physical stress maneuvers. Use of pleasant sensations such as music and therapeutic touch to reduce ICP are being investigated.70 Sustained ICP above 20 mm Hg requires emergency treatment. A therapist noting a rise in ICP above 15 mm Hg should contact the nurse or physician.
Because of increased ICP, further surgical intervention may be needed to release excess fluid. A catheter may be inserted to drain excess fluid from a ventricle or other fluid-filled space, called a shunt, or a Jackson-Pratt suction drain may be needed. CSF postoperative leaks are evidenced by saturation of the surgical head dressing or leaking of a clear, thin fluid from the ear or nose that dries in concentric circles.
Management of the postoperative client with increased ICP typically includes, among other things, elevation of the head. A position with the client’s head elevated about 20 to 30 degrees is often prescribed. The client should be protected from any position that allows stasis of the CSF drainage and should be taught to observe the drainage and to be aware of signs of infection. The client also should be instructed against coughing, sneezing, or blowing the nose.
An erratic body temperature may occur after intracranial surgery. Either hypothermia or hyperthermia may be present. The therapist should check with the nursing staff before beginning therapy if concern exists regarding abnormal temperature.
DVT occurs in one third of patients who have surgery. Seizures,53 CSF leakage, and wound infection are also risks. Periocular edema is common, as are temporary visual field deficits. Patients may have other temporary deficits resulting from cerebral edema, such as communication, motor, and sensory deficits; diminished gag and swallowing reflexes; diplopia; loss of corneal reflex; and personality changes.52 Pneumocystis carinii pneumonia (PCP) is a life-threatening opportunistic infection that occurs in immunocompromised hosts, such as with corticosteroid used. Signs of PCP are fever and dyspnea with or without a prominent dry cough, though the onset may be subtle. The risk of PCP is increased while steroids are being tapered.93,112
Meningitis also may occur, caused by irritation of the meninges by infection or blood in the subarachnoid space. If it develops, meningitis typically appears 2 or 3 days after surgery. Chills, fever, nuchal rigidity, headache, irritability, increased sensitivity to light, and decreased level of consciousness are signs of meningitis. It is essential that all care providers practice infection prevention measures such as thorough handwashing when caring for clients who have had intracranial surgery.
Other signs to be aware of include ecchymosis, stress ulcer, swallowing difficulties and aspiration, and impaired airway. Respiratory changes should be monitored carefully. An abnormal respiratory rate and depth may indicate rising ICP. By carefully observing the client during therapy, protecting the client from harm, and alerting medical staff of seizure activity or other adverse signs and symptoms, the therapist can provide a valuable adjunct to postoperative care.
Positioning in the Acute Postoperative Phase
If there is any question about positioning of a client during therapy, the therapist should communicate with the nursing staff. Incorrect positioning may have serious, possibly fatal, consequences. In the acute phase after surgery above the tentorium, orders may be given to avoid lowering the head, to avoid extreme flexion of the legs, and to keep the neck in a neutral position. After surgery below the tentorium, the client may be kept flat and turned every 2 hours or have orders for elevation of the head of the bed. It is recommended that the neck not be angulated anteriorly or laterally, but there are usually no restrictions placed on turning. For infratentorial tumors that may cause dizziness on arising, elevating the head of bed gradually while concurrently monitoring vital signs is recommended. The dizziness is caused by transient edema in the area of the eighth cranial nerve. For posterior fossa surgery, the client is typically positioned on the side with a pillow under the head. This protects the operative site from pressure and minimizes tension on the suture line.52
If a bone flap was removed for decompression, the orders may be to place the client only on the nonoperated side or the back. This facilitates brain expansion. If a large tumor has been removed from a cerebral hemisphere, there may be an order to avoid positioning on the operative site to prevent shifting of the cranial contents due to gravity.
If a client is neurologically unstable, and with ICP in a critical range (more than 20 mm Hg), therapy procedures that require a flat position (e.g., lowering the head of the bed for range-of-motion [ROM] exercises) should be avoided. Placing a pillow under the head facilitates good venous outflow. When the client is side lying, protecting the hips from sharp flexion avoids an increase in intrathoracic pressure, which can in turn increase cerebral ICP.
Before physical therapy intervention, examination should include a review of the medical chart; the surgical, radiologic, and pathologic reports; laboratory values; and nursing and other reports. If indicated, physicians should be contacted for pertinent information and guidelines. Hematologic values may be of critical importance for exercise training plans. Anemia causes fatigue, leukopenia increases infection susceptibility, and thrombocytopenia increases bleeding susceptibility and is of particular concern because it may lead to intracranial hemorrhage.90,93
Familiarity with cancer terminology and the behavior of the particular tumor aids communication with others on the team. For example, knowledge of tumor staging and grading, the prognosis, the medical management such as the surgical procedure, presence of an Ommaya reservoir, and the various types of central lines and monitoring devices facilitates interdisciplinary care planning and intervention. The ICP levels, blood pressure, and other vital signs are typically available on the bedside monitor. Knowledge of the Karnofsky scale (Table 30-6), a functional performance scale used by many oncologists to indicate the client’s activity level in the hospital, home, or community, is helpful. It can facilitate communication among the team on issues such as a client’s candidacy for home discharge with home health assistance or need for more supervised care. The ECOG (Eastern Cooperative Oncology Group) 1-4 Scale is another tool that may be used to identify functional performance.
Table 30-6
Karnofsky Performance Status Scale
From Baird SB, McCorkle R, Grant M: Cancer nursing: a comprehensive textbook, Philadelphia, 1991, Saunders.
Initial outcomes expected for clients with brain surgery include full ROM, active where possible, of all extremities and optimal functioning of respiratory, cardiovascular, and other systems within the precautions indicated. When the client’s condition is stable, functional outcomes include independent bed mobility and transfers, ambulation, and self-care skills (ADLs).
Rehabilitation Examination for Postoperative Care
During the acute postoperative phase, a thorough examination within the constraints of the precautions, including strength, joint ROM, sensory and perceptual status, neurologic signs, pain patterns, presence of fatigue, and mobility, helps to identify treatable impairments that affect function.79 During examination and intervention, the therapist should avoid any Valsalva maneuver that would increase intrathoracic pressure, thus increasing ICP. The therapist also should avoid jarring the client’s bed or causing sudden movements that would increase pain.
After intracranial surgery bed rest precautions may initiated, usually for 24 hours, and should be observed by the therapist. If the client is stable, passive ROM exercises may begin.
Position changes are important, and use of a draw sheet and adequate help ensure that the patient will not strain with position change and increase ICP. When movement to the bedside chair is safe but ICP precautions preclude active supine to sitting movements, lifting the client to a reclining chair by a draw sheet with the help of several caregivers and then gradually raising the client’s back and head in the chair protects him or her from straining. Bedside sit-and-dangle exercises also may be requested. Blood pressure checks for postural hypotension, close assessment for dizziness and faintness, and monitoring of respiratory rate, heart rate, and ICP during activities are essential. General conditioning activities early in the recovery are valuable to address the fatigue cycle typical of cancer.
Subacute and Ambulatory Rehabilitation
As the patient becomes more stable and is moved into lower levels of acuity, including inpatient and ambulatory care, continued monitoring is imperative. The therapist must monitor vital signs and observe for any neurologic change; any adverse indications such as seizures, bleeding at the operative site, or signs of a DVT; or sudden changes in mental status.
Reviewing diagnostic information, laboratory reports, and medical treatment continues to be important as the patient moves into new phases of care. Medical treatment modalities and side effects have a pronounced effect on the client’s participation in therapy. Chemotherapeutic and radiation side effects, such as myelosuppression, nausea, and fatigue, may temporarily lower energy levels, requiring adjustment of the therapy program. Irradiated areas should be protected against skin injury. No heat or cold or topical agents should be used in the irradiated area during the treatment series or for several weeks after the treatment series until the skin damage has cleared. The radiation oncologist will determine the topical agent(s) to be used. If persistent trophic change occurs to the skin with obvious circulatory impairment, heat or cold should not be applied over the site because of poor dissipation effects.
Examination and intervention are based on the impairments and disabilities identified. Safety in mobility, gait and balance training, protection from falls, strengthening, equipment decisions, functional training and aerobic capacity training are important rehabilitation interventions.138 Written educational materials are increasingly requested.
The efficacy of increasing the activity level has been demonstrated, and functional training, gait training, and exercise are well-accepted postoperative interventions.102 Long-term management may include family training and education and more advanced treatment aimed at self-care and safety, return to family roles, and work and leisure activity.
More attention is being paid at the present time to cancer-related fatigue (CRF), identifying possible causes such as myelosuppression, anorexia, pain, sleep deprivation, and somnolence. Treatment modalities including surgery, radiation therapy, and chemotherapy are associated with fatigue. Understanding fatigue and the ameliorating factors is becoming the subject of more studies. In a recent glioblastoma study, fatigue was associated with decreases in almost all aspects of quality of life.76 Addressing fatigue with a structured progressive exercise program has been shown to have some efficacy.35,36,84,115 Although there is no consensus on the ideal type of exercise, frequency, intensity, duration, or mode, there is good cardiopulmonary response to interval training at 50% to 70% of heart rate reserve or working at an exertion level of 11 to 14 on the 6 to 20 rate of perceived exertion (RPE) scale. While treatment is being given, most studies recommend decreasing the intensity to the lower end of the heart rate range.27 Careful monitoring to avoid overstressing the immune system has been suggested by some studies.95 Throughout the rehabilitation process, activities that increase ICP, such as vigorous resistive exercises or isometrics, should be avoided.
Recent studies support the benefits of comprehensive and interdisciplinary rehabilitation for patients with primary and metastatic brain tumors.55,85,93 Outcomes of physical therapy intervention may be measured by standardized tools, such as the Karnofsky scale, the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire 30 (EORTCQ-30), the Psychosocial Adjustment to Illness Scale, the Functional Living Index—Cancer (FLIC) Scale, the Functional Assessment of Cancer Therapy—Brain (FACT) tool,43,78 and the Functional Assessment of Chronic Illness Therapy (FACIT) Scale. Fatigue measures include the 1 to 10 analogue scale, the Brief Fatigue Inventory (BFI),19 the Piper Fatigue Scale, and numerous other new fatigue measures. Some studies have used the Functional Independence Measure (FIM) to evaluate changes in function with inpatient rehabilitation55,78,85,90 and have demonstrated improvements. Considerations of quality of life are increasingly part of medical oncologic studies.
Corticosteroids are prescribed during surgery and again during radiation to reduce cerebral edema. Long-term effects include many adverse problems, including proximal weakness, behavioral changes, osteoporosis, increased appetite, bloating, hypertension, opportunistic infections, night sweats, hyperglycemia, yeast stomatitis, and many more. Long-term steroid use is avoided, and the drugs are typically discontinued after the acute surgical or radiation management is completed. However, with steroid tapering and discontinuation, there may be a possible increase in cerebral edema and a recurrence of symptoms previously present before surgery or radiation. For example, a person receiving brain radiation taking dexamethasone during the radiation series may demonstrate improved hemiparesis as a result of the steroid. However, when radiation therapy is completed and steroid therapy is tapered and discontinued, the hemiparesis may worsen. The therapist needs to be aware of decreasing function related to steroid tapering during this time and report it to the physician. The decision may be made to continue the steroid at a low dose. If corticosteroids are tapered too rapidly after surgery or radiation therapy, causing peritumoral edema, a bolus dose of dexamethasone followed by a more gradual weaning schedule may alleviate the symptoms.44 Edema fluctuations from the tumor effect itself may cause a puzzling improvement and regression in neurologic status. Mobility issues and goals should be planned with the client and family, keeping in mind this potential of variable symptoms from edema fluctuations. The therapist must avoid creating either false hope or pessimism when patient performance varies because of edema fluctuations.
The impact of the diagnosis may be difficult for the client to comprehend. The client who does comprehend may demonstrate extreme behavioral responses or a profound sense of hopelessness. The client should be encouraged to ask questions and express his or her feelings about the situation. Depression is difficult to distinguish from apathy caused by the brain tumor,60 but a differential diagnosis, if possible, through depression testing, is important to help the therapist to advocate for improved management of depression. Managing cognitive effects of radiation therapy is difficult for the client and family.23 The caregiver’s support, realistic reassurance, and inclusion of the client and family in the decision-making process will have a positive impact on the quality of life.52
Because the diagnosis of a brain tumor is so devastating to the client and family, causing fear and uncertainty, the therapist must have sufficient maturity and psychosocial skills to be supportive and understanding. The challenge is not necessarily to provide solutions to these psychosocial problems but to provide support and validation and to facilitate referrals to appropriate professionals while addressing the physical problem for which the person was referred.
The therapist involved in rehabilitation of the person with a brain tumor is part of a rehabilitation team of professionals that also may be involved in the care. The team may include representatives from nursing, nutrition, respiratory therapy, speech therapy, social work, psychology, the chaplain’s office, durable medical equipment (DME) suppliers, hospice staff, and the physician office staff. An interdisciplinary approach allows access to needed resources.
The therapist must understand that the term cancer rehabilitation is used by many specialists and community programs that provide services for the person with cancer and may mean different things in different contexts. The American Cancer Society has a rehabilitation program that includes support groups and services such as rides to medical appointments and supplies like wigs. Oncology nurses have become increasingly supportive of cancer rehabilitation and an interdisciplinary approach80 and include symptom control and psychosocial issues in their definition of rehabilitation. Many local groups, such as church and synagogue support groups, provide another aspect of rehabilitation. The National Cancer Institute has identified four objectives for cancer rehabilitation: psychologic support, optimal physical functioning, vocational counseling, and optimal social functioning.82 Financial issues, nutrition, spousal relationships, sexual counseling, vocational rehabilitation, employment opportunities, physician-patient communication, patient education, and coping skills are broader aspects of rehabilitation.
In some acute care and outpatient settings, the therapist may be fortunate enough to be part of a more formalized cancer rehabilitation program that includes these broader aspects of rehabilitation. The advantages for the client with access to such a program include early and appropriate referrals to skilled professionals and resources, coordination of care and information, and a smooth transition across the continuum of care. Other benefits include enhancement of program development resulting from collaboration of professionals, outcome studies that will improve the quality of care, and the potential for rehabilitation research.
PRIMARY INTRASPINAL TUMORS
Primary spinal cord tumors are about one sixth as common as primary brain tumors. The histologic types of tumor cells in the spinal cord are the same as those found in the brain, although the prevalence of certain types may differ. The most common tumor in the spinal cord is the schwannoma or neurinoma, followed by the meningioma and glioma.83
A convenient anatomic classification system of spinal cord tumors is based on the relationship of the tumor to the spinal cord and dura. Fig. 30-21 diagrams the location and relative incidence of spinal tumors. Intradural-intramedullary tumors arise within the spinal cord substance. Intradural-extramedullary tumors arise outside the spinal cord but within the dura. Extradural spinal cord tumors arise outside the spinal cord and the dura (Fig. 30-22).
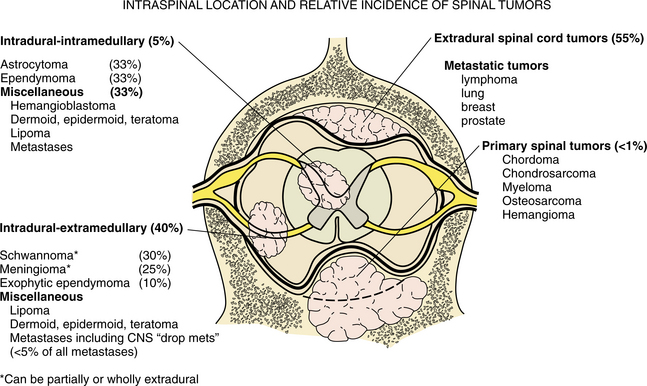
Figure 30-21 Primary and metastatic tumors of the spine and spinal cord. (Adapted from Poirier J, Gray F, Escourelle R: Manual of basic neuropathology, ed 2, Philadelphia, 1990, Saunders.)
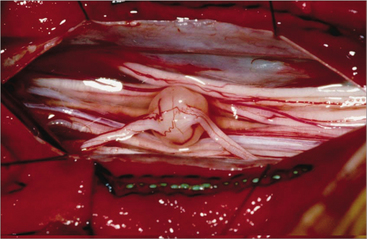
Figure 30-22 Spinal-cord neoplasms are extradural or intradural tumors according to their relation to the thecal sac. (From Lancet Oncol 8[1], 2007.)
The specific spinal cord tumor types and their incidence are discussed first, followed by the clinical presentation and medical management.
Intradural-Intramedullary Tumors
Intradural-intramedullary tumors are the least common type of primary intraspinal tumors in adults but the most common type in children. These tumors account for 5% to 10% of intraspinal tumors. The dominant tumor types are astrocytomas and ependymomas.
Pathogenesis
Because they are located within the cord itself, intradural-intramedullary tumors generally are derived from the cellular substrate of the spinal cord, such as the astrocytes and ependymal cells, or from the primitive embryonal cells. Astrocytomas may occur anywhere along the spinal cord and may span several cord segments longitudinally. In children they may run the entire length of the cord.
Although all astrocytomas are infiltrative, most are low grade and slow growing in the spinal cord. Ependymomas are generally slow growing as well and less infiltrative, and therefore more amenable to surgical excision. Other less frequent types of intradural-intramedullary tumors are hemangiomas, epidermoid and dermoid cysts, teratomas, lipomas, and neuroenteric cysts. Of interest to therapists is chemical meningitis with its significant chronic pain that can occur when epidermoid or dermoid cysts leak debris into the CSF. Very infrequently, a primary intramedullary spinal lymphoma (PCNSL) may occur.13,127
Intradural-Extramedullary Tumors
Intradural-extramedullary tumors are the most common type of primary intraspinal tumor in adults, and they account for about 45% of all spinal tumors. Neurinomas (schwannomas) and meningiomas are the dominant tumors in this group. Meningiomas are 10 times more common in women than in men and occur in middle age.
Pathogenesis
Intradural-extramedullary tumors are primarily derived from the supporting elements of the CNS, including the meninges and nerve sheath. Occasionally tumors in this compartment are carried down as drop metastases by the CSF from malignant brain tumors (medulloblastomas, ependymomas, PNETs). See Fig. 30-23 for a PNET in the brain and spinal cord surfaces. Intradural-extramedullary tumors cause compression of the spinal cord, rather than invasion of the cord. Neurinomas are soft, globular masses that arise at the sensory or dorsal nerve root. Occasionally they may straddle the intervertebral foramen and extend outside the foramen, forming the so-called dumbbell configuration. Spinal meningiomas are benign, slow-growing globular tumors that often grow in the thoracic, cervical, and foramen magnum regions. They may be present for many years before symptoms occur.
Figure 30-23 Primitive neuroectodermal tumor (PNET). A, T1-weighted coronal magnetic resonance imaging (MRI) scan after injection of contrast medium in a 5-year-old boy. There is a large multifocal tumor in the posterior fossa causing hydrocephalus. There are multiple smaller, contrast-enhancing, tumors along the surface of cerebellum and in the cerebrum. B, A T1-weighted sagittal postcontrast MRI scan of the spinal canal shows a large mass (arrow) in the junction of the cervical and thoracic spine with a syrinx and multiple small enhancing nodules (arrowheads) over the surface of the spinal cord. (From Adam A, Dixon AK, Grainger RG, et al, eds: Grainger and Allison’s diagnostic radiology: a textbook of medical imaging, ed 4, Philadelphia, 2001, Churchill Livingstone.)
Extradural-Extramedullary Tumors
Extradural-extramedullary tumors are most often metastatic tumors and are addressed in the section Metastatic Tumors. Extradural-extramedullary tumors represent about 45% of all spinal cord tumors. An occasional meningioma, neurinoma (schwannoma), or spinal chordoma arises extradurally. Spinal chordomas represent less than 1% of spinal cord tumors.
Pathogenesis
Spinal chordomas are primary tumors that arise from the vertebral bodies, usually in the cervical or sacral regions of the axial skeleton. They are prone to metastasize outside the spinal column. Lesions are characterized by expansive destruction of the bone, for example, the sacrum, or by varying degrees of vertebral collapse. Box 30-4 summarizes the type and location of spinal tumors.
Clinical Manifestations of Primary Intraspinal Tumors
Spontaneous pain caused by nerve root irritation is a common clinical feature of primary spinal tumors and is usually worse at night. Intramedullary tumors give rise to a poorly localized, deep, burning type of pain in the spinal region. Extramedullary tumors produce a knifelike radicular type of pain typically radiating to the periphery of the nerve, often aggravated by coughing, sneezing, or straining. The association of asymmetry of reflexes with nerve root pain and an insidious onset is strongly suggestive of a spinal cord tumor.
Nerve root pain may be followed by motor weakness and wasting of muscle supplied by the nerve. The motor changes of intramedullary tumors include lower motor neuron changes at the level of the lesion and may also include upper motor neuron changes at lower levels. The motor changes of extramedullary lesions begin with segmental weakness at the lesion site and progress to damage to half of the spinal cord (Brown-Séquard syndrome) and later to a transverse cord syndrome.126 The weakness is characterized by upper motor neuron signs, including spasticity. Sphincter weakness, increasing urinary frequency, and urgency may develop. In men, the development of sphincter disturbances frequently is followed by impotence.
Sensory changes in extramedullary tumors are usually along the distribution of the involved nerve roots. Intramedullary tumors result in dissociated sensory disturbances in the limbs below the level of the lesion because of their growth pattern of crossing fibers of the spinothalamic tract. People often report a feeling of temperature change, particularly a feeling of cold below the level of the lesion. Pain and temperature sensation are compromised, but proprioception and light touch are preserved.
Syringomyelia-like symptoms of loss of pain and temperature sensation below the level of the lesion on one or both sides of the body may occur from damage to the decussating lateral spinothalamic fibers. This often is accompanied by progressive spastic paraparesis caused by pressure on the descending corticospinal tracts. Anterior growth of the tumor produces anterior horn signs, such as muscle weakness, wasting, and fasciculations in the muscles supplied by the anterior horn cells.
Other symptoms of intramedullary cord tumors are papilledema, hydrocephalus, and elevations in ICP. The reason for development of papilledema is unclear, but it is more common with tumors of the thoracic and lumbosacral regions. Box 30-5 lists signs and symptoms of spinal cord tumors.
MEDICAL MANAGEMENT
After a history and neurologic examination, MRI is the method of choice for the identification of spinal cord tumors. Gadolinium-enhanced MRI is helpful to differentiate between edema and tumor. Occasionally plain films may be helpful in treatment planning. Lumbar puncture and CSF examination are no longer used as diagnostic tools.
TREATMENT AND PROGNOSIS.
Surgery is the principal treatment for all primary intraspinal tumors. Complete and curative resection is the objective. Extramedullary tumors can be cured in most cases. Radiation therapy is not required when lesions are completely excised. Intraspinal astrocytomas have a lower rate of cure, although surgery and radiation may prolong the disease-free survival. Childhood astrocytomas, however, have a more favorable prognosis, with a 5-year survival rate of 90%.
METASTATIC TUMORS
The extended survival in all types of cancer has allowed time for metastasis to occur from primary tumors elsewhere in the body. Metastatic complications are an escalating problem. Metastases to the brain and spinal cord are among the most serious complications of metastatic cancer.15,126
Incidence
The incidence of metastatic CNS tumors is estimated to be 150,000 to 180,000 per year, although exact figures are difficult to ascertain.3,104 The number of metastatic brain tumors is estimated to be 100,000 per year, and the number of metastatic spinal cord tumors is estimated to be about 80,000 per year.3 Metastatic tumors are the most common intracranial tumor in adults.135 The incidence of metastatic CNS tumors is on the rise as a result of improved life expectancies from advances in cancer treatment, allowing micrometastases time to develop in the spinal cord and brain. Here they find a safe haven behind the blood-brain barrier through which many chemotherapeutic agents for the primary cancer cannot pass. The blood-brain barrier restricts passage of high-molecular-weight compounds through its tight capillary endothelial junctions.100 The brain is a metastatic site in about 20% of people with primary cancer elsewhere, and the spinal cord is a metastatic site for 10% of primary cancers.126
Pathogenesis8
Metastatic tumors reach the brain generally through the arterial blood system. A smaller number arise by direct extension from extracranial sites such as the neck or paranasal sinuses. The cascade of events for formation of a metastasis includes tumor cells at the primary site reaching a critical volume in proximity to a blood vessel, dislodging from the primary tumor and entering blood vessels, embolizing, traveling, extravasating from the blood vessel, and growing in parenchyma of another organs.39,15 Most metastatic tumors arise in the distribution area of the middle cerebral artery to the cerebral hemispheres, with most lesions located in the parietal or frontal lobes. About 20% are found in the posterior fossa, primarily the cerebellum. About half of cases include multiple metastatic lesions in the brain (Fig. 30-24).
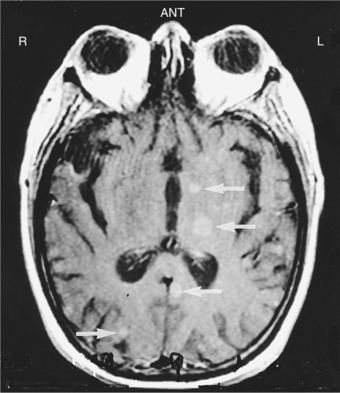
Figure 30-24 Metastatic disease to the brain. A gadolinium-enhanced T1-weighted magnetic resonance image shows multiple metastases as areas of increased signal (arrows). (From Mettler FA Jr: Essentials of radiology, ed 2, Philadelphia, 2005, Saunders.)
Metastatic tumors reach the spine and spinal cord through direct arterial dissemination to the vertebral body, by retrograde spread via the vertebral venous plexus as it perforates into the epidural space and vertebral bodies, or by direct invasion from a paravertebral tumor to the epidural space via the intervertebral foramen. The majority of spinal metastases are extradural-extramedullary, although in less than 5% of cases the location is intramedullary. The thoracic spine is the most frequent site of metastasis (70%), followed by the lumbosacral spine (20%) and the cervical area (10%).126
The most common cancers resulting in brain metastases are lung cancers, especially small cell carcinoma; cancers of the breast, kidney, and gastrointestinal tract; and melanoma. Fig. 30-25 shows dural metastasis from breast cancer. The most common cancers metastasizing to the spinal column are lung, breast, prostate, and kidney cancers and lymphomas. In about 10% of cases, the primary tumor is never found.
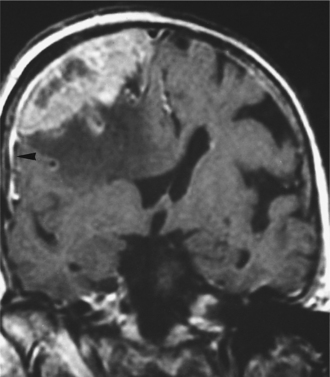
Figure 30-25 Dural metastasis from breast carcinoma. Coronal T1-weighted postcontrast magnetic resonance image. There is a heterogeneously enhancing mass with an irregular surface that arises from the dura over the right cerebral convexity. It displaces the underlying brain and causes considerable low-signal edema within it. There is a dural “tail” extending away from the tumor (arrowhead). (From Grainger and Allison’s diagnostic radiology: a textbook of medical imaging, ed 4, Philadelphia, 2001, Churchill Livingstone.)
Clinical Manifestations of Brain Metastasis
Metastatic brain tumors present with headache, seizures, elevated ICP, and similar signs of primary tumors. See Table 30-7 for a more complete listing. However, symptoms may progress much more rapidly, often in days to weeks, as a result of the significant edema that accompanies a metastasis. Cerebellar metastases may cause obstructive hydrocephalus and abrupt deterioration.
Table 30-7
Presenting Symptoms and Signs of Brain Metastasis
| Symptom | Common Signs |
| Headache | Focal weakness or unexplained falls |
| Mental status change | Focal sensory deficits |
| Altered level of consciousness | Speech difficulty |
| Seizures occurring when older than 35 yr of age | Aphasia, focal weakness |
| Papilledema or visual obscurations | Ataxia |
| Visual complaints or unexplained motor vehicle accidents | Visual field defect |
From Goetz CG: Textbook of clinical neurology, ed 2, Philadelphia, 2003, Saunders, chap 47.
MEDICAL MANAGEMENT
DIAGNOSIS, TREATMENT, AND PROGNOSIS.
MRI is the diagnostic procedure of choice because it is the most sensitive in revealing multiple small lesions. A review of the chest film is often enough to give a presumptive diagnosis of lung cancer. With a history of cancer elsewhere in the body, a solitary brain lesion has about a 90% certainty of being a metastatic deposit.126 Because meningiomas have a high prevalence in people with breast cancer, metastatic breast lesions in the brain must be differentiated pathologically from meningiomas for optimal treatment.
Medical management includes corticosteroids, surgical excision when solitary or small numbers of metastases are accessible, and irradiation in almost all cases. Of solitary brain metastases associated with non–small cell lung cancer, up to one third may be cured with surgery followed by radiation therapy. Steroids have a dramatic effect in relieving symptoms caused by the significant peritumoral swelling. Radiotherapy provides adequate palliation for many people, because death occurs from the primary cancer, not the brain metastasis.61
In general, the prognosis for people with brain metastasis is poor, because the metastasis indicates that the primary cancer has already escaped control.
Clinical Manifestations of Spinal Metastasis
Back pain is the most common and prominent symptom of metastasis to the spinal column and cord, and is present in 95% of cases. Anyone with a known cancer history who presents with new-onset back pain of unknown etiology should be considered to have spinal metastasis until proved otherwise.126 Pain is due to stretching of the periosteum, tension or traction on the spinal nerve roots and cord, or compression of the cord and meninges. It is usually a dull ache, worse at night in the recumbent position, and may be local in the spine or may be a radicular pain.
Without treatment, pain progresses in weeks or months (sometimes days) to weakness, sensory loss, and bowel and bladder sphincter disturbance. These tumors characteristically progress quickly after onset of weakness to cause paraplegia and permanent loss of sphincter control. Diagnosing and treating the metastasis early is important, because people treated while still ambulatory are likely to remain so, but those who have reached the stage of paraplegia and sphincter loss do not typically regain function.
MEDICAL MANAGEMENT
A careful neurologic examination, followed by plain films of the spine, is an important first approach and results in a diagnosis in the majority of cases. The most common findings are pedicular erosion, vertebral collapse, pathologic fracture-dislocation, and a soft tissue shadow suggestive of a paraspinal mass.126 Bone scans are the next test of choice. If results of any of these tests are positive, MRI or CT is then done for more definitive imaging of the lesion. Fig. 30-26 is an MRI showing focal spine metastases.
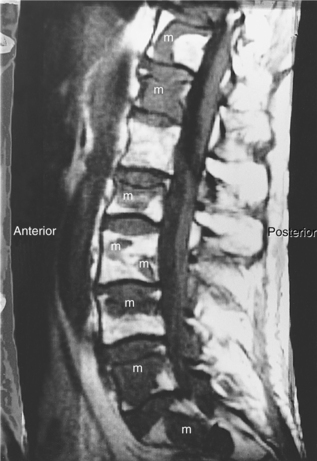
Figure 30-26 Focal spine metastases. A sagittal or lateral T1-weighted magnetic resonance image of the lumbar spine shows the normal white or high signal in fat within the bone marrow. In many of the vertebral bodies, the high signal of normal marrow has been replaced by dark areas of metastatic deposits (m). (From Mettler FA Jr: Essentials of radiology, ed 2, Philadelphia, 2005, Saunders.)
TREATMENT.
Radiotherapy is typically the treatment of choice for spinal metastasis to reduce pain, reduce tumor compression, and restore neurologic function. Radiotherapy and/or chemotherapy is also helpful in preserving spinal stability. For those tumors that are chemotherapy sensitive, urgent management with chemotherapy is indicated to preserve spinal integrity.
Surgery is reserved for people with a worsening neurologic deficit during radiation therapy, for those with a spinal instability causing cord compression, for tumors known to be radioresistant, and for individuals who already have received the maximum radiation.
It should be noted that past attempts at surgical decompression with laminectomy have proved to be disappointing, with neurologic improvement occurring in only 30% of cases.126 Surgical access via laminectomy to the typically anterior tumors compressing the cord from a ventral direction is technically difficult. Evidence exists that very high doses of corticosteroids relieve local spinal edema. Current practice is to begin very large doses of corticosteroids as soon as a spinal cord compression from metastatic tumor is diagnosed. This dose is continued for several days, then reduced, allowing time for decisions to be made for radiation or surgery.139
PROGNOSIS.
Prognosis for return of neurologic function is based on the degree of loss before radiotherapy. With radiotherapy, 80% of clients who are ambulatory at the time of treatment remain so, and 30% who are nonambulatory regain gait.126 Pain and neurologic function improve in a large percentage of people. Because the metastasis indicates loss of containment of the primary tumor, cure is beyond expectation. However, early diagnosis and treatment lead to the optimal result, the prevention of paraplegia.
Radiation to the spinal cord may cause complications of myelopathy. Although radiation has no acute effects on the cord, an early delayed radiation myelopathy after irradiation of the neck is common. Lhermitte’s sign (a sudden electric shock sensation brought on by neck flexion), the hallmark of this radiation myelopathy, is present for several months and then abates. It is not a predictor of late delayed radiation spinal cord injury.34 A late effect of radiation to the cord, occurring between 6 and 36 months after radiation, is a chronic progressive myelopathy that begins as a Brown-Séquard syndrome and progresses over weeks or months to a spastic paresis. No effective medical treatment exists, but the therapist can provide mobility management, skin precautions, and safety education for these clients.
PARANEOPLASTIC SYNDROMES
Cancer may cause effects on the nervous system that are not directly related to the primary tumor mass or a metastasis. These so-called remote effects, or paraneoplastic syndromes (see Chapter 9), include such problems as paraneoplastic cerebellar degeneration, brainstem encephalitis, myelitis of the spinal cord, and motor neuron disease.42 Paraneoplastic syndromes are also termed paraneoplastic neurologic disorders or PNDs. The cause of most paraneoplastic syndromes is unknown, although an immune mechanism is the most likely hypothesis. The response of the immune system to the antigen may be misdirected and cause neurologic dysfunction. Paraneoplastic syndromes may be the first sign of the presence of cancer.12,99,108
Although paraneoplastic syndromes involving the CNS are rare, they are often severe, often associated with an inflammatory CSF, and leave the person with severe neurologic disability. Treatment effectiveness has been limited. Some syndromes are associated with particular tumors, such as paraneoplastic cerebellar degeneration with lung cancer. In this syndrome, the early symptoms are a slight incoordination in walking, with progressive gait ataxia; incoordination of arms, legs, and trunk; dysarthria; and often nystagmus. After a few months the illness reaches its peak and stabilizes. By this time, most clients must have assistance to walk, handwriting is impossible, many cannot sit unsupported, and speech is with great effort.
It is not within the scope of this chapter to elaborate on CNS paraneoplastic syndromes. However, it is helpful for the therapist to have an acquaintance with these syndromes, because they appear in the practice of caring for people with CNS neoplasms.15
LEPTOMENINGEAL CARCINOMATOSIS
Infiltration of the meninges and CSF pathways of the CNS by neoplastic cells is a less common complication of cancer and is known as leptomeningeal carcinomatosis or neoplastic meningitis. This metastatic seeding of the meninges is widespread and multifocal. Neurologic signs depend on location. Brain symptoms may include headache, change in mental status, seizures, double vision, abducens palsy, and hemiparesis; spinal symptoms include radicular pain, numbness, and weakness.101 Meningeal carcinomatosis occurs in approximately 5% of patients with cancer but is being diagnosed with increasing frequency as patients live longer and as neuroimaging studies improve.48,99 Cancers of the breast and lung, non-Hodgkin’s lymphoma, melanomas, and adult acute leukemias are the most common primary tumors responsible for carcinomatosis. Diagnosis is by CSF studies, which show malignant cells in most cases. MRI is also done to assess bulky disease in the brain or spine. Current therapy includes radiotherapy to symptomatic sites, with concurrent intrathecal chemotherapy.126 Survival is measured in months from treatment.
PEDIATRIC TUMORS
Incidence and Pathogenesis
Approximately 2200 primary brain tumors are diagnosed in children and adolescents each year.68 The incidence of primary brain and nervous system tumors peaks in the pediatric population from age 0 to 6, drops at age 7 to 10, remains steady until age 18, then drops. In infants and young children, intracranial tumors are the second most common form of cancer, after leukemia.126 The peak incidence of these tumors occurs between birth and age 6 years. Brain tumors are the second leading cause of cancer-related deaths in children below the age of 15. Refer back to Table 30-2 for a comparison of the frequency of childhood tumors compared with adult tumors.
The etiology of pediatric brain tumors is little understood. Cranial exposure to radiation and possible evidence of a heritable syndrome are causes. Other factors being studied include maternal diet and intake of vitamins during pregnancy.
The most frequently encountered types of intracranial tumors in children are the astrocytoma, medulloblastoma, ependymoma, and brainstem glioma.45 Brain tumors in children are typically located infratentorially, primarily in the cerebellum and brainstem, although they may occur at any location. See Fig. 30-27 for relative frequency and location.
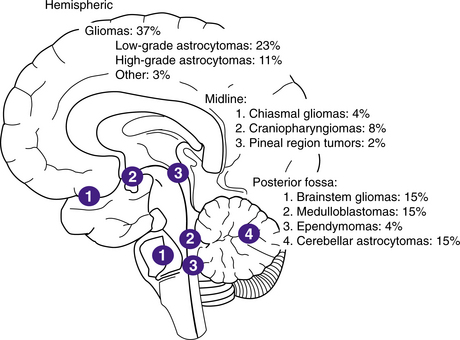
Figure 30-27 Childhood brain tumors occur at any location within the central nervous system. The relative frequency of brain tumor histologic types and the anatomic distribution are shown. (From Kliegman RM, Behrmann RE: Nelson textbook of pediatrics, ed 18, Philadelphia, 2007, Saunders.)
Astrocytomas are the most common type of pediatric intracranial tumor, accounting for about 47% of all brain tumors in children. They are usually well-differentiated grade I tumors. About half of them occur supratentorially, most commonly in the frontal lobes, but also in the temporal and parietal lobes. The cerebellum is the most common infratentorial site of the astrocytoma in children. Cerebellar astrocytoma is more common in males and usually occurs in the first two decades of life, with the median incidence at age 18. Children with grade I astrocytomas have a 10-year postoperative survival rate of 85%.45 Infrequently high-grade astrocytomas occur, with a poorer prognosis.
Medulloblastomas account for 20% to 25% of childhood brain tumors and are the most common malignant tumor in children. These aggressive tumors are most common in males and have a peak incidence at age 5 years in children. Evolution of therapeutic strategies including multimodal chemotherapy followed by surgery has dramatically improved the outlook for children with medulloblastomas.126 Medulloblastomas belong to the group of tumors known as primitive neuroectodermal tumors (PNETs) and arise in the fourth ventricle. Medulloblastomas have a predilection for meningeal seeding. The 5-year survival rate is about 50%.
Ependymomas usually arise in children from the floor of the fourth ventricle and make up 9% to 20% of childhood tumors. The 5-year survival is 85% when complete resection is possible and about 45% overall.
Brainstem gliomas may be of several tumor types; the most common is astrocytoma, but they also may be glioblastomas or ependymomas. The overall prognosis for brainstem tumors is relatively poor, but occasionally gratifying treatment results are obtained.126
Clinical Manifestations
Clinical manifestations of CNS neoplasms in children are more difficult to evaluate because children are less able to relate and report symptoms. Parents, teachers, and caretakers may notice problems before the child is aware of a change. Most supratentorial astrocytomas present initially with seizures. Cerebellar astrocytomas produce typical cerebellar findings such as ataxia, and many produce symptoms of increased ICP. Tumors of the cerebral aqueduct or the fourth ventricle, such as an ependymoma or medulloblastoma, typically present at an early stage with headache, nausea, and cranial nerve palsies but also may produce long-tract signs such as hemiparesis. Hydrocephalus is a complication in patients with ependymoma and medulloblastoma, and a late manifestation of brainstem gliomas.45
Diagnosis and Treatment
Diagnosis is by MRI or CT scan, although the CT scan may not be adequate to detect the early stages of brainstem or fourth ventricle tumors. Early treatment includes high-dose dexamethasone, emergency ventricular drainage in the case of hydrocephalus, and surgical resection. Radiation is the principal form of treatment for brainstem gliomas. Postoperative radiation is indicated for medulloblastomas and may be helpful for ependymomas and gliomas. Chemotherapy, although generally not beneficial, has been helpful for medulloblastomas.
The risks of brain radiation therapy in children are of great concern. These may include learning disabilities, hypopituitarism, occlusive disease of cerebral vessels, and radiation-induced secondary tumors. Because myelinization of the CNS is not generally complete until 2 to 3 years of age, radiation therapy performed before this age can be especially devastating and is not usually done.126 Long-term effects from childhood tumors and treatment have been reported by R.J. Packer.94
Spinal cord tumors rarely occur in children. Spinal ependymoma is the most prominent primary intraspinal tumor and has a predilection for the lumbosacral spine. It is treated with resection and radiation therapy. The risks of radiation therapy in children include myelopathy and spinal deformities.
Metastatic spinal cord tumors in children arise most commonly from sarcomas and less often from neuroblastomas, lymphomas, and leukemias. Metastatic tumors in children occur principally by direct extension from an adjacent primary cancer126 and can be the presenting feature of the primary cancer. This is in contrast to adult spinal tumors, in which access is by a vascular route and usually occurs in the setting of an advanced malignancy.
Because pediatric metastatic spinal cord tumors usually are not associated with extensive vertebral column destruction, they generally can be removed through a simple laminectomy. An aggressive approach to metastatic spinal tumors in children results in a more favorable outcome in children than in adults.126 Approximately 96% of children have improvement or stabilization of neurologic deficits, and 60% of nonambulatory children regain the ability to walk after treatment.
Prognosis
Seventy percent of children with brain tumors will be long-term survivors, according to data from the National Cancer Institute’s SEER program.68 At least half of these survivors will experience chronic problems such as focal motor and sensory abnormalities, seizures, cognitive deficits, and neuroendocrine deficiencies like hypothyroidism. Another study showed long-term sequelae in adult survivors of childhood brain tumor such as hearing losses, blindness, and coordination and motor control problems.68 Rehabilitation can be of help with functional outcomes.97
References
1. Abeloff, MD, Armitage, JO, Niederhuber, JE, et al, Chap 69. Clinical oncology. ed 3. Philadelphia: Churchill Livingstone; 2004.
2. American Brain Tumor Association Diagnosis and follow-up. Available on-line at http://www.abta.org Accessed January 30, 2007
3. American Brain Tumor Association Facts and statistics. Available on-line at http://www.abta.org Accessed January 30, 2007
4. American Brain Tumor Association Types of brain and spinal cord tumors. Available on-line at http://www.abta.org Accessed January 30, 2007
5. Andrews, DW. Stereotactic radiosurgery and fractionated stereotactic radiotherapy for the treatment of acoustic schwannomas: comparative observations of 125 patients treated at one institution. Int J Radiat Oncol Biol Phys. 2001;50(5):1265–1278.
6. Armstrong, CL. Memory performance used to detect radiation effects on cognitive functioning. Appl Neuropsychol. 2001;8(3):129–139.
7. Armstrong, TS, Gilbert, MR. Metastatic brain tumors: diagnosis, treatment, and nursing interventions. Clin J Oncol Nurs. 2000;4(5):217–225.
8. Athanassiou, H, Synodinou, M, Maragoudakis, E, et al. Randomized phase II study of temozolomide and radiotherapy compared with radiotherapy alone in newly diagnosed glioblastoma multiforme. J Clin Oncol. 2005;23(10):2372–2377.
9. Balducci, L, Extermann, M. Management of cancer in the older person: a practical approach. Oncologist. 2000;5(3):224–237.
10. Balducci, L, Stanta, G. Cancer in the frail patient: a coming epidemic. Hematol Oncol Clin North Am. 2000;14(1):235–250.
11. Barker, F, Chang, S, Larson, D, et al. Age and radiation response in glioblastoma multiforme. Neurosurgery. 2001;49(6):1288–1298.
12. Bataller, L, Dalmau, JO. Paraneoplastic disorders of the central nervous system: update on diagnostic criteria and treatment. Semin Neurol. 2004;24(4):461–471.
13. Bekar, A, Cordan, T, Evrensel, T, et al. A case of primary spinal intramedullary lymphoma. Surg Neurol. 2001;55(5):261–264.
14. Bell, KR. Rehabilitation of the patient with brain tumor. Arch Phys Med Rehabil. 1998;79(3 suppl 1):S37–S48.
15. Benjamin, RK, Das, A, Hochberg, FH. Metastatic neoplasms and paraneoplastic syndromes. In Goetz CG, ed.: Textbook of clinical neurology, ed 2, Philadelphia: Saunders, 2003. [chap 47].
16. Bertino, JR, Hait, W. Principles of cancer therapy. In Goldman LM, Ausiello D, eds.: Cecil textbook of medicine, ed 22, Philadelphia: Saunders, 2004.
17. Billings, JA. Care of the dying patients and their families. In Goldman, Ausiello D, eds.: Cecil textbook of Medicine, ed 22, Philadelphia: Saunders, 2004.
18. Bittar, R, Oliview, A, Sadikot, A, et al. Presurgical motor and somatosensory cortex mapping with functional magnetic resonance imaging and positron emission tomography. J Neurosurg. 1999;91(6):915–921.
19. Brada, M, Yung, WKA. Clinical trial end points in malignant glioma: need for effective trial design strategy. Semin Oncol. 2000;27(3 suppl 6):11–19.
20. Brandes, AA, Vastola, F, Basso, U, et al. A prospective study on glioblastoma in the elderly. Cancer. 2003;97(3):657–662.
21. Brem, S, Rozental, JM, Moskal, JR. What is the etiology of human brain tumors? A report on the first Lebow conference. Cancer. 1995;76(4):709–713.
22. Bunin, G, Surawicz, T, Witman, P, et al. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89(4):547–551.
23. Butier, JM, Rapp, SR, Shaw, EG. Managing the cognitive effects of brain tumor radiation therapy. Curr Treat Options Oncol. 2006;7(6):517–523.
24. Cancer facts and figures 2006. American Cancer Society: Atlanta, 2001:4.
25. Cancer Information Service. SEER incidence and U.S. mortality rates-brain and other nervous system. In: SEER cancer statistic review 1994-2003. Bethesda, MD: National Institutes of Health; 2004:112, 117. [NIH publication no. 00-2789].
26. Chase, M. Cancers of the central nervous system. In: Baird SB, ed. A cancer source book for nurses. ed 6. Atlanta: American Cancer Society; 1991:253–258.
27. Cheville, A, Packel, LB. Cancer. In Frontera, ed.: Essentials of physical medicine and rehabilitation, ed 1, Philadelphia: Hanley & Belfus, 2002. [chap 98].
28. Corn, B, Marcus, S, Topham, A, et al. Will central nervous system lymphoma be the most frequent brain tumor diagnosed in the year 2000? Cancer. 1997;79(12):2409–2413.
29. Corn, BW, Mehta, MP, Buatti, JM, et al. Stereotactic irradiation. Am J Clin Oncol. 1999;22(2):143–146.
30. Corn, BW, Yousem, DM, Scott, CB, et al. White matter changes are correlated significantly with radiation dose. Cancer. 1994;74:2828–2835.
31. Custer, BS, Koepsell, TD, Mueller, B. The association between breast carcinoma and meningioma in women. Cancer. 2002;94(6):1626–1635.
32. DeAngelis, I. Primary central nervous system lymphoma. In: Gilman S, Goldstein G, Waxman S, eds. Neurobase. San Diego: Arbor Publishing, 1996.
33. DeAngelis, LM. Tumors of the central nervous system and intracranial hypertension and hypotension. In Goldman LM, Ausiello D, eds.: Cecil textbook of medicine, ed 22, Saunders, 2004.
34. Delattre, JY, Posner, JB. Neurological complications of chemotherapy and radiation therapy. In: Aminoff MJ, ed. Neurology and general medicine. ed 2. New York: Churchill Livingstone; 1995:437.
35. Dimeo, FC. Strategies in managing cancer fatigue. Rehabil Oncol. 1999;17(3):27–28.
36. Dimeo, FC, Stieglitz, RD, Novelli-Fischer, U, et al. Effects of physical activity on the fatigue and psychologic status of cancer patients during chemotherapy. Cancer. 1999;85(10):2273–2277.
37. Doolittle, ND. State of the science in brain tumor classification. Sem Oncol Nurs. 2004;29(4):224–230.
38. Duffau, H. Lessons from brain mapping in surgery for low-grade glioma: insights into associations between tumour and brain plasticity. Lancet Neurol. 2005;4(8):476–489.
39. Fidler, IJ. Biology of cancer metastasis. In Abeloff MD, Armitage JO, Niederhuber JE, et al, eds.: Clinical oncology, ed 3, Philadelphia: Churchill Livingstone, 2004. [chap 4].
40. Freeman, K, O’Dell, C, Meola, C. Issues in families of children with brain tumors. Oncol Nurs Forum. 2000;27(5):843–848.
41. Galli, R, Binda, E, Orfanelli, U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–7021.
42. Gilbert, MR. Neurologic complications. In Abeloff MD, Armitage JO, Niederhuber JE, et al, eds.: Clinical oncology, ed 3, Philadelphia: Churchill Livingstone, 2004. [chap 61].
43. Gilbert, M, Armstrong, T, Meyers, C. Issues in assessing and interpreting quality of life in patients with malignant glioma. Semin Oncol. 2000;27(3 suppl 6):20–26.
44. Gillis, TA, Cheville, AL, Worsowicz, GM. Cardiopulmonary rehabilitation and cancer rehabilitation: 4. Oncologic rehabilitation. Arch Phys Med Rehabil. 2001;82(3 suppl 1):S63–S068.
45. Gilroy, J. Basic neurology, ed 2, Elmsford, NY: Pergamon Press; 1990:223–250.
46. Greenberg, E, Treger, I, Ring, H. Rehabilitation outcomes in patients with brain tumors and acute stroke: comparative study of inpatient rehabilitation. Am J Phys Med Rehabil. 2006;85(7):568–573.
47. Greenberg, JO, Polachini, I. Intracranial neoplasms. In: Greenberg JO, ed. Neuroimaging. New York: McGraw-Hill; 1995:323–383.
48. Grossman, SA, Spence, A. NCCN clinical practice guidelines for carcinomatous/lymphomatous meningitis. Oncology. 1999;13(11A):144–152.
49. Hanson, RA, Ghosh, S, Gonzalez-Gomez, I, et al. Abducens length and vulnerability. Neurology. 2004;62(1):33–36.
50. Hart, M, Grant, R, Walker, M, et al. Surgical resection and whole brain radiation therapy versus whole brain radiation therapy alone for single brain metastases. Cochrane Database Syst Review. 2005;1:CD003292.
51. Helseth, E, Due-Tonnessen, B, Wesenberg, F, et al. Posterior fossa medulloblastoma in children and young adults (0-19 years): survival and performance. Childs Nerv Syst. 1999;15(9):451–455.
52. Hickey, JV, Armstrong, T. Brain tumors. In: Hickey JV, ed. Neurological and neurosurgical nursing. ed 4. Philadelphia: Lippincott; 1997:501–539.
53. Hildebrand, J. Epileptic seizures during follow-up of patients treated for primary brain tumors. Neurology. 2005;65(2):2212–2215.
54. Houillier, C, Lejeune, J, Benouaich-Amiel, A, et al. Prognostic impact of molecular markers in a series of 220 primary glioblastomas. Cancer. 2006;106(10):2218–2223.
55. Huang, ME, Cifu, DX, Keyser-Marcus, L. Functional outcome after brain tumor and acute stroke, a comparative analysis. Arch Phys Med Rehabil. 1998;79:1386–1390.
56. Hughes, MA. Primary brain tumors treated with steroids and radiotherapy: low CD4 counts and risk of infection. Int J Radiat Oncol Biol Phys. 2005;62(5):1423–1426.
57. Huncharek, M, Kupelnick, B, Wheeler, L. Dietary cured meat and the risk of adult glioma: a meta-analysis of nine observational studies. J Environ Pathol Toxicol Oncol. 2003;22(2):129–137.
58. Iwamoto, FM, DeAngelis, LM. An update on primary central nervous system lymphoma. Hematol Oncol Clin North Am. 2006;20(6):1267–1285.
59. Janus, TJ, Yung, WKA. Primary neurological tumors. In Goetz CG, ed.: Textbook of clinical neurology, ed 2, Philadelphia: Saunders, 2003. [chap 46].
60. Johnson, AJ. Depression and apathy in patients with brain tumors: the importance of a differential diagnosis. Rehabil Oncol. 2001;19(3):8–10.
61. Kaal, EC. Therapeutic management of brain metastasis. Lancet Neurol. 2005;4(5):289–298.
62. Kabil M, Eby J, Hrayr K, Fully endoscopic transnasal vs. transseptal transnasal surgery, in Research at the Skull Base Institute, CA. Available on-line at http://www.skullbaseinstitute.com. Accessed February 20, 2007
63. Kaltsas, GA, Nomikos, P, Kontogeorgos, G, et al. Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. 2005;90(5):3089–3099.
64. Klein, M, Heimans, J, Aaronson, H, et al. Effect of radiotherapy and other treatment-related factors on mid-term to long-term cognitive sequelae in low-grade gliomas: a comparative study. Lancet. 2002;360(9343):1361–1368.
65. Küker, W, Nagele, T, Korfel, A, et al. Primary central nervous system lymphomas (PCNSL): MRI features at presentation in 100 patients. J Neurooncol. 2005;72(2):169–177.
66. Kumar, V, The central nervous system. Robbins and Cotran pathologic basis of disease. ed 7. Philadelphia: Saunders; 2005.
67. Kumar, V, Pituitary adenomas and hyperpituitarism. Robbins and Cotran pathologic basis of disease. ed 7. Philadelphia: Saunders; 2005. [chap 24].
68. Kuttesch, JF, Ater, JL. Brain tumors in childhood. In Behrman RE, Kliegman M, Jenson HB, eds.: Nelson textbook of pediatrics, ed 17, Philadelphia: Saunders, 2004. [chap 489].
69. Lenhard, RE, Jr., Osteen, RT, Gansler, T. Clinical oncology. Atlanta: American Cancer Society, 2001;655.
70. Lindamood, MO. Commentary on making headway with intracranial hypertension [original article by Voss HR:. Am J Nurs. 1993;93(2):28–35. [37-39]. AACN Nurs Scan Crit Care. 1993;3(5):10–11.
71. Pathology and genetics of tumours of the nervous system. In: Lleihues P, Cavenee WK, eds. World Health Organization Classification of Tumours of the Nervous System, Editorial and Consensus Conference Working Group. Lyon, France: IARC Press, 2000.
72. Loeffler, JS, Shrieve, DC, Wen, PY, et al. Radiosurgery for intracranial malignancies. Semin Radiat Oncol. 1995;5(3):225.
73. Lopes, MBS, Laws, ER. Low-grade central nervous system tumors. Neurosurg Focus. 2002;12(2):E1.
74. Louis, D, Cavenee, W. Molecular biology of the central nervous system tumors. In DeVita V, Jr., Hellman S, Rosenberg S, eds.: Cancer: principles and practice of oncology, ed 7, Philadelphia: Lippincott-Raven, 2004.
75. Louis, DN. Molecular pathology of malignant glioma. Annu Rev Pathol. 2006;1:97–117.
76. Lovely, MP, Miaskowski, C, Didd, M. Relationship between fatigue and quality of life in patients with glioblastoma multiformae. Oncol Nurs Forum. 1999;26:921–925.
77. Maity, A, Pruitt, AA, Judy, KD, et al. Cancer of the central nervous system. In Abeloff MD, Armitage JO, Niederhuber JE, et al, eds.: Clinical oncology, ed 3, Philadelphia: Churchill Livingstone, 2004. [chap 69, part III].
78. Marciniak, CM, Sliwa, DO, Heinemann, AW, et al. Functional outcomes of persons with brain tumors after inpatient rehabilitation. Arch Phys Med Rehabil. 2000;82(4):457–463.
79. Max, M. Pain. In Goldman LM, Ausiello D, eds.: Cecil textbook of medicine, ed 22, Philadelphia: Saunders, 2004. [chap 29].
80. Mayer, D, O’Conner, L. Rehabilitation of persons with cancer: an ONS position statement. Oncol Nurs Forum. 1989;16:433.
81. McGarvey, CL, Walton, JF. Oncology: principles and management. In: McGarvey CL, ed. Physical therapy for the cancer patient. New York: Churchill Livingstone; 1990:11.
82. McKenna, RJ, Wellisch, D, Fawzy, FI. Rehabilitation and supportive care of the cancer patient. In: Murphy GP, Lawrence W, Lenhard RE, eds. American Cancer Society textbook of clinical oncology. Atlanta: American Cancer Society; 1995:635–654.
83. Mettler, FA, Jr. Spinal neoplasms. In Mettler FA, Jr., eds.: Essentials of radiology, ed 2, Philadelphia: Saunders, 2005. [chap 8].
84. Mock, V, Ropka, ME, Rhodes, VA, et al. Forum focus: establishing mechanisms to conduct multi-institutional research-fatigue in patients with cancer: an exercise intervention. Oncol Nurs Forum. 1998;25(8):1391–1397.
85. Mukand, JA, Blackinton, DD, Crincoli, MG, et al. Incidence of neurologic deficits and rehabilitation of patients with brain tumors. Am J Phys Med Rehabil. 2001;80(5):346–350.
86. Mukand, JA, et al. Incidence of neurologic deficits and rehabilitation of patients with brain tumors. Am J Phys Med Rehabil. 2001;80(5):346–350.
87. National Cancer Institute Surveillance Epidemiology and End Results Cancer of the brain and other nervous system. Available on-line at http://www.seer.cancer.gov/statfacts/html/brain.html Accessed November 4, 2006
88. Nitta, T, Sato, K. Prognostic implications of the extent of surgical resection in patients with intracranial malignant gliomas. Cancer. 1995;75(11):2727–2731.
89. Oba-Shinjo, SM. Identification of novel differentially expressed genes in human astrocytomas by cDNA representational difference analysis. Brain Res Mol Brain Res. 2005;140(1-2):25–33.
90. O’Dell, MW, Barr, K, Spanier, D, et al. Functional outcome of inpatient rehabilitation in persons with brain tumors. Arch Phys Med Rehabil. 1998;79:1530–1534.
91. Olson, JE, Janney, CA, Rao, RD, et al. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer. 2002;95(7):1504–1510.
92. Olson, JE, Riedel, E, DeAngelis, L. Long-term outcome of low-grade oligodendroglioma and mixed glioma. Neurology. 2000;54(7):1442–1448.
93. Oncology Nursing Society 2000 conference symposia. Care: brain tumors-recent advances in the treatment of malignant brain tumors, Classification and nursing interventions. Oncology Nurse Symposium. 2000;5:31–32.
94. Packer, RJ. Long-term neurologic and neurosensory sequelae in adult survivors of childhood brain tumor: Childhood Cancer Survivor Study. J Clin Oncol. 2003;21(17):3255–3261.
95. Pederson, BK, Hoffman-Goetz, L. Exercise and the immune system: regulation, integration, and adaptation. Phys Rev. 2000;80(3):1055–1081.
96. Pfeifer, JD, Wick, MR. The pathologic evaluation of neoplastic diseases. In: Murphy GP, Lawrence W, Lenhard RE, eds. American Cancer Society textbook of clinical oncology. ed 2. Atlanta: American Cancer Society; 1995:79–80.
97. Phillip, PA. Rehabilitation outcome in children after treatment of primary brain tumor. Arch Phys Med Rehabil. 1994;75(1):36–39.
98. Pignatti, F, van den Bent, M, Curran, D, et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002;20(8):2076–2084.
99. Posner, JB. Paraneoplastic syndromes involving the nervous system. In: Aminoff MJ, ed. Neurology and general medicine. ed 2. New York: Churchill Livingstone; 1995:401–406.
100. Prados, MD, Levin, V. Biology and treatment of malignant glioma. Semin Oncol. 2000;27(3 suppl 6):1–10.
101. Prados, MD. Neoplasms of the central nervous system. In: Bast RC, Kupe DW, Pollock RE, et al, eds. Cancer medicine. ed 5. Hamilton, Ontario: BC Decker; 2000:1055–1082.
102. Pratt, M. Physical activity. In Goldman LM, Ausiello D, eds.: Cecil textbook of medicine, ed 22, Philadelphia: Saunders, 2004. [chap 13].
103. Preston, DL, Ron, E, Yonehara, S, et al. Tumors of the nervous system and pituitary gland associated with atomic bomb radiation exposure. J Natl Cancer Inst. 2002;94(20):1555–1563.
104. , Primer of brain tumors. Facts and statistics, updates. ed 7. Des Plaines, IL: American Brain Tumor Association; 2001. Available on-line at www.abta.org/primer/facts.htm
105. Purdy, RA, Kirby, S. Headaches and brain tumors. Neurol Clin. 2004;22(1):39–53.
106. Ransohoff, J, Kislow, M, Cooper, PR. Cancer of the central nervous system and pituitary. In: Holleb AI, Fink DJ, Murphy GP, eds. American Cancer Society textbook of clinical oncology. Atlanta: American Cancer Society; 1991:329.
107. Rockhill, JK, Laramore, GE. Biophysiology and clinical considerations in radiotherapy. In Cummings CW, Jr., Haughey BH, Thomas JR, et al, eds.: Cummings otolaryngology-head and neck surgery, ed 4, Philadelphia: Mosby, 2005.
108. Rugo, HS. Paraneoplastic syndromes and other non neoplastic effects of cancer. In Goldman LM, Ausiello D, eds.: Cecil textbook of medicine, ed 22, Philadelphia: Saunders, 2004. [chap 188].
109. Sadetzki, S, Flint-Richter, P, Ben-Tal, T, et al. Radiation-induced meningioma: a descriptive study of 253 cases. J Neurosurg. 2002;97(5):1078–1082.
110. Sadun, AA. Imaging in neuro-ophthalmology. In Yanoff M, Duker JS, Augsburger JJ, et al, eds.: Ophthalmology, ed 2, St Louis: Mosby, 2004. [chap 185, sect 1].
111. Sawaya, R, Bindal, RJ. Metastatic brain tumors. In: Kaye AH, Laws ER, eds. Brain tumors. New York: Churchill Livingstone; 1997:923–926.
112. Schiff, D. Pneumocystis pneumonia in brain tumor patients: risk factors and clinical features. J Neurooncol. 1996;27(2):235–240.
113. Schnell, S. Nursing care of clients with cerebral disorders. In: Black JM, Matassarin-Jacobs E, eds. Luckmann and Sorensen’s medical-surgical nursing. ed 4. Philadelphia: Saunders; 1993:705–772.
114. Schwade, JG, Wolf, AL. Future trends in radiosurgery. Semin Radiat Oncol. 1995;5:246–249.
115. Schwartz, AL. Patterns of exercise and fatigue in physically active cancer survivors. Oncol Nurs Forum. 1998;25(3):485–491.
116. Shaw, EG, Coffey, RJ, Dinapoli, RP. Neurotoxicity of radiosurgery. Semin Radiat Oncol. 1995;5(3):235.
117. Snodgrass, SM. Neurologic aspects of cancer. In: Weiner WJ, Goetz CG, eds. Neurology for the non-neurologist. ed 3. Philadelphia: JB Lippincott; 1994:259–267.
118. Snyderman, CH, Kassam, AB. Endoscopic techniques for pathology of the anterior cranial fossa and ventral skull base. J Am Coll Surg. 2006;202(3):563.
119. Stamm, AC, Pignatari, SSN. Transnasal endoscopic-assisted surgery of the skull base. In Cummings CW, Jr., Haughey BH, Thomas JR, et al, eds.: Cummings otolaryngology-head and neck surgery, ed 4, Philadelphia: Mosby, 2006.
120. Straif, K, Weiland, SK, Bungers, M, et al. Exposure to high concentrations of nitrosamines and cancer mortality among a cohort of rubber workers. Occup Environ Med. 2000;57(3):180–187.
121. Stupp, R, Dietrich, PY, Ostermann Kraljevic, S, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20(5):1375–1382.
122. Stupp, R, Mason, WP, Bent, MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996.
123. Surma-aho, O, Niemela, M, Vilkki, J, et al. Adverse long-term effects of brain radiotherapy in adult low-grade glioma patients. Neurology. 2001;56(10):1285–1290.
124. Tabori, U, Sung, L, Hukin, J, et al. Medulloblastoma in the second decade of life: a specific group with respect to toxicity and management. Cancer. 2005;103(12):1874–1880.
125. Taphoorn, MJ, Stupp, R, Coens, C, et al. Health-related quality of life in patients with glioblastoma: a randomized controlled trial. Lancet Oncol. 2005;6(12):937–944.
126. Thapar, K, Laws, ER. Tumors of the central nervous system. In: Murphy GP, Lawrence W, Lenhard RE, eds. American Cancer Society textbook of clinical oncology. ed 2. Atlanta: American Cancer Society; 1995:378–413.
127. Traul, DE, Shaffrey, ME, Schiff, D. Spinal-cord neoplasms-intradural neoplasms. Lancet Oncol. 2007;8(1):35–45.
128. Tsao, MN. Radiotherapeutic management of brain metastasis: a systemic review and met analysis. Cancer Treat Rev. 2005;31(4):256–273.
129. UpToDate Batchelor T: Clinical manifestations and diagnosis of high grade malignant astrocytoma. Available on-line at http://uptodateonline.com. Accessed July 17, 2006
130. UpToDate Batchelor T, Louis D: Pathogenesis and biology of high grade malignant astrocytoma. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
131. UpToDate Michaud D, Batchelor T: Risk factors for brain tumors. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
132. UpToDate Michaud D, Schiff D, Batchelor T: Incidence of primary brain tumors. Available on-line at http://uptodateonline.com Accessed September 25, 2006
133. UpToDate Recht L: Classification, diagnosis, and natural history of low-grade glioma. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
134. UpToDate Recht LD, Marcus KJ: Craniopharyngiomas. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
135. UpToDate Schiff D, Batchelor T: Classification of brain tumors. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
136. UpToDate Wen P, Loeffler J: Clinical manifestations and diagnosis of brain metastases. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
137. UpToDate Wong E, Wu JK: Clinical presentation and diagnosis of brain tumors. Available on-line at http://uptodateonline.com. Accessed September 25, 2006
138. Varricchio, CG, Aziz, N. Rehabilitation and survivorship. In: Lenhard RE, Osteen RT, Gansler T, eds. Clinical oncology. Atlanta: American Cancer Society; 2001:823–836.
139. Weiss, HD. Neoplasms. In: Samuels MA, ed. Manual of neurologic therapeutics. ed 6. Boston: Lippincott Williams & Wilkins; 1999:255–283.
140. Whittle, I, Smith, C, Navoo, P, et al. Meningiomas. Lancet. 2004;363(9420):1535–1543.
141. Wilkes, GM, Ingwersen, K, Barton-Burke, M. 200l oncology nursing drug handbook. Sudbury, MA: Jones & Bartlett, 2001.
142. Wolf, T, Brodt, HR, Fischtischerer, S, et al. Changing incidence and prognostic factors of survival in AIDS-related non-Hodgkin’s lymphoma in the era of highly active antiretroviral therapy (HAART). Leuk Lymphoma. 2005;46:207–215.
143. Yates, AJ. An overview of principles for classifying brain tumors. Mol Chem Neuropathol. 1992;17:106–110.
144. Yonehara, S, Brenner, AV, Kishikawa, M, et al. Clinical and epidemiologic characteristics of first primary tumors of the central nervous system and related organs among atomic bomb survivors in Hiroshima and Nagasaki, 1958-1995. Cancer. 2004;101(7):1644–1654.