Hepatobiliary System and Exocrine Pancreas
Liver and Intrahepatic Biliary System
Development
Early in embryogenesis, the origins of the liver are evident. The hepatic diverticulum, also termed the liver bud, arises from embryonic endoderm as a hollow out-pouching of the primitive duodenum. Primitive hepatic epithelial cells of the hepatic diverticulum extend into the adjacent mesenchymal stroma and surround the vessels that form the vitelline venous plexus, a complex of vessels that drain the yolk sac. This relationship between the epithelial cells of the liver and the small-caliber vitelline vessels is the earliest developmental form of the hepatic sinusoids. The caudal part of the hepatic diverticulum develops into the gallbladder and the cystic duct. Hepatic connective tissue is derived from the septum transversum, a sheet of cells that incompletely separates the pericardial and peritoneal cavity, and an ingrowth of mesenchymal cells from the coelomic cavity.
The biliary epithelium also arises from the hepatic diverticulum. Intrahepatic ducts develop from a structure, termed the ductal plate, which is composed initially of a single row of hepatoblasts that surround the portal vein branches and ensheathe the mesenchyme of the primitive portal tract. A second discontinuous outer layer of primitive hepatoblasts forms subsequently, and the two-cell-thick regions remodel into tubules and become the intrahepatic biliary ductular system. Development of the ducts begins at the porta hepatis and extends to the margins of the liver until the later stages of gestation. The residual portion of the hollow outpouching of the hepatic diverticulum persists to become the extrahepatic bile ducts.
It is known that the hepatocytes and the biliary epithelial cells share a common embryonic origin, but the factors that lead to the final characteristic morphology of the primitive hepatoblasts are not well understood. Epithelial-mesenchymal interactions are believed to play a role. Primitive hepatic epithelial cells in contact with vascular endothelium are destined to become hepatocytes, and those in contact with the developing mesenchyme of the portal tracts develop into bile ducts.
Macroscopic and Microscopic Structure
The liver is the largest internal organ in the body. In adult carnivores, the liver constitutes 3% to 4% of the body weight. In adult omnivores, it is about 2% of body weight and about 1% of the body weight in herbivores. In the neonate of all species, the liver is a larger percentage of body weight than in the adult. In monogastric animals, the liver abuts the diaphragm and occupies the central area of the cranial abdomen. In ruminants the liver is displaced to the right side of the cranial abdominal cavity. A series of ligaments maintains the liver in its position. The coronary ligament attaches the liver to the diaphragm near the esophagus. The falciform ligament attaches the midline of the liver to the ventral midline of the abdomen. The round ligament, a remnant of the umbilical vein, is embedded within the falciform ligament. The liver is supplied with blood from two sources. The portal vein drains the digestive tract and provides 60% to 70% of the total afferent hepatic blood flow. The hepatic artery provides the remainder of hepatic blood flow. Blood leaves the liver via the hepatic vein, which is very short, and enters the caudal vena cava. The liver has a smooth capsular surface, and the parenchyma consists of friable red-brown tissue that is divided into lobes. Gross subdivision of the liver into lobes differs among the domestic species. At the periphery, the lobes taper to a sharp edge.
The classic functional subunit of the liver is the hepatic lobule, a hexagonal structure, 1 to 2 mm wide. At the center, the lobule has a central vein (also termed the terminal hepatic venule), which is a tributary of the hepatic vein, and at the angles of the hexagon, it has portal tracts (Fig. 8-1). The portal tracts contain bile ducts, branches of the portal vein, the hepatic artery, nerves, and lymph vessels, all supported by a collagenous stroma (Fig. 8-2). The limiting plate, a discontinuous border of hepatocytes, forms the outer boundary of the portal tract. Blood flows into the sinusoids from the terminal distributing branches of the hepatic artery and portal veins that leave the portal tracts and form an outer perimeter of the lobule (see Figs. 8-1 and 8-2). Portal blood and hepatic arterial blood mix in the sinusoids. Blood drains from the sinusoids into the central veins and to progressively larger sublobular veins and then into the hepatic veins.
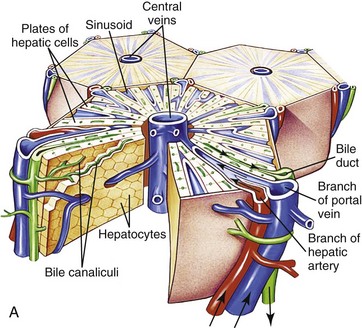
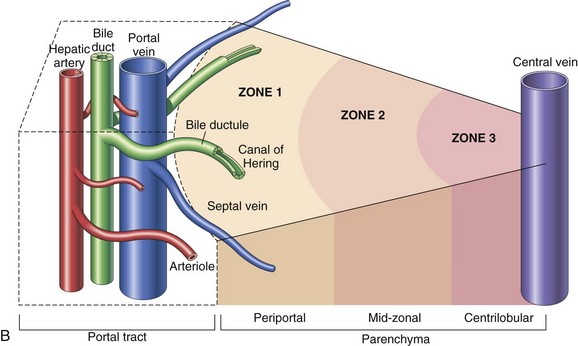
Fig. 8-1 Schematic views of the microscopic and functional organization of the liver.
A, Microscopic organization of the liver. A central vein is located in the center of the lobule with plates of hepatocytes arranged radially. Branches of the portal vein and hepatic artery are located on the periphery of the lobule, and blood from both perfuses the sinusoids. Peripherally located bile ducts drain the bile canaliculi that run between hepatocytes. B, Functional organization of the liver. Both the lobule and the acinus are represented. The lobule is a hexagonal unit with portal areas at the margin and a terminal hepatic vein (central vein) at the center. The lobule is divided into the periportal, midzonal, and centrilobular areas. The acinus is a diamond-shaped structure with the distributing branches of the vessels from the portal areas as the center of the structure. Zone 1 of the acinus is closest to the afferent blood supply, and zone 3 is at the tip of the diamond-shaped structure, close to the terminal hepatic vein. Zone 2 is between Zones 1 and 3. (A from McCance KL, Huether SE: Pathophysiology: the biologic basis for diseases in adults and children, ed 6, St Louis, 2010, Mosby. B from Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
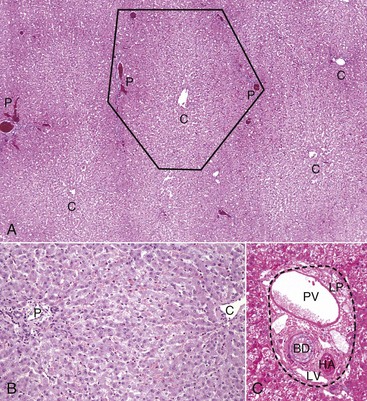
Fig. 8-2 Liver, hepatic lobules, normal dog.
A, Low magnification. A central vein (C) is located in the center of the lobule. Branches of the portal vein, hepatic artery, bile duct, and lymphatic vessels are located on the periphery of the lobule in portal tracts (P) (also Fig. 8-2, C). H&E stain. B, Higher magnification. Plates of hepatocytes arranged radially between portal tracts (P) to a central vein (C). H&E stain. C, Higher magnification, portal tract. The normal portal tract contains the hepatic artery (HA), bile duct (BD), portal vein (PV), and several lymphatic vessels (LV). These structures are surrounded by a collagenous extracellular matrix that forms an abrupt border with a circumferential row of hepatocytes, termed the limiting plate (LP—dotted line). Note that the profile of the portal vein is typically larger than those of the hepatic artery and bile duct. H&E stain. (A and C courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Alternatively, when the liver is viewed as a bile-secreting gland, the acinus is the anatomic subunit of the hepatic parenchyma. Terminal afferent branches (penetrating vessels) of the portal vein and hepatic artery project into the parenchyma, like branches from the trunk of a tree, forming the long axis of the diamond-shaped acinus. Thus terminal afferent branches of the portal vein and hepatic artery are at the center of the acinus and the terminal hepatic venule is located at the periphery. Each terminal hepatic venule (central vein) receives blood from several acini. There are three zones within the acinus. Zone 1 is closest to the afferent blood coming from the hepatic artery and the portal vein. Zone 2 is peripheral to zone 1, and zone 3 borders the terminal hepatic venule (see Fig. 8-1). In this anatomic unit, bile flow begins in the canaliculi of the hepatocytes in zone 3 and flows through zones 2 and 1 then into the interlobular bile ducts in the portal areas.
The ultrastructural appearance of hepatocytes reflects the cell’s active metabolism, bile secretion, and close contact with the plasma (Web Fig. 8-1). The surface of the hepatocyte that faces the lumen of the sinusoids contains an abundance of microvilli, which increase the hepatocytic surface area and facilitate uptake of plasma-borne substances, such as bilirubin and amino acids, and the secretion of products of hepatic metabolism, such as lipoproteins and clotting factors. Basolateral aspects of hepatocytes are characterized by the presence of canaliculi, modified portions of the cell membrane in two adjacent hepatocytes, which form a lumen for bile secretion. The cytoplasm contains glycogen and a variety of organelles, including numerous mitochondria, lysosomes, and abundant smooth and rough endoplasmic reticulum.
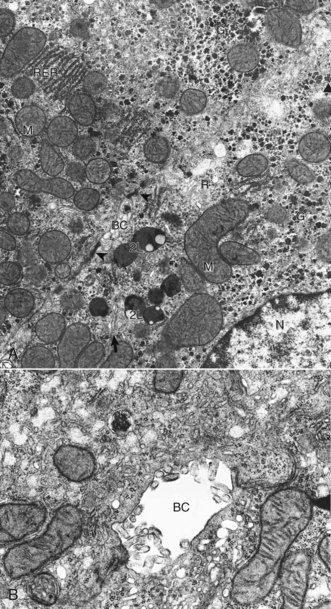
Web Fig. 8-1 Hepatocyte, ultrastructure, liver, normal dog.
A, Features to note are the nucleus (N), mitochondria (M), secondary lysosomes (2L), glycogen (G), rough endoplasmic reticulum (RER), Golgi (arrow), bile canaliculus (BC), and free ribosomes (R). Note desmosomes on both sides of the bile canaliculus (arrowheads). TEM. Uranyl citrate and lead acetate stain. B, Higher magnification of bile canaliculus (BC). Note the microvilli projecting into the lumen of the canaliculus. TCM uranyl citrate and lead acetate stain. (A and B courtesy Dr. V. Meador, Covance Inc.)
Within the liver, hepatocytes are arranged in one-cell-thick branching plates, which extend radially from the terminal hepatic venule. Hepatic plates are separated by vascular sinusoids. Blood from the terminal afferent branches of the hepatic artery and portal vein mixes in the hepatic sinusoids and flows to the terminal hepatic venule. Hepatic sinusoids differ from capillaries in that they are lined by discontinuous endothelial cells that lack a typical basement membrane (Fig. 8-3), whereas capillaries have a continuous endothelial lining and are ensheathed in the basement membrane. The sinusoids are critical for appropriate hepatic function. The architecture of the sinusoids enables efficient uptake of plasma constituents by hepatocytes and facilitates hepatocellular secretion. A fine scaffold of electron lucent basement membrane that contains collagen types III, IV, and XVIII, and other extracellular matrix (ECM) components supports the sinusoidal endothelial cells (Web Fig. 8-2; see Fig. 8-3). These elements collectively make up the “reticulin” of the liver (Fig. 8-4).
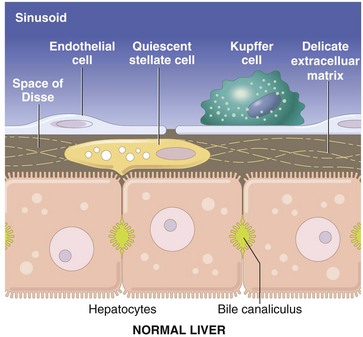
Fig. 8-3 Schematic diagram of the hepatic sinusoid.
The vascular lumen is lined by discontinuous capillaries. Kupffer cells rest on the endothelial cells and project into the sinusoid. Between the endothelial cells and the hepatocytes is a gap called the space of Disse. Microvilli extending from the luminal aspect of the hepatocytes are found in this space. Hepatic stellate cells are situated within the space of Disse and extend between hepatocytes. (Schematic based on concepts in Friedman SL: J Biol Chem 275:2247-2250, 2000; and Crawford JM: Curr Op Gastroenterol 13:175-185, 1997.)
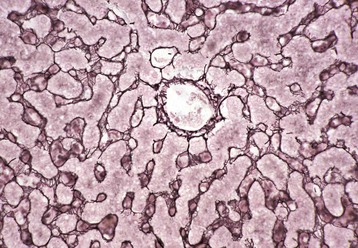
Fig. 8-4 Reticulin fibers (reticulin stain), hepatic extracellular matrix, liver, normal dog.
This stain reveals “reticulin” (black), composed of extracellular matrix found within the space of Disse that forms the scaffolding of the hepatic parenchyma. Note the radial arrangements of the hepatic plates and the single hepatocyte thickness of the plates. A central vein is evident in the center of the image. Gordon and Sweet’s reticulin stain with a nuclear fast red counterstain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
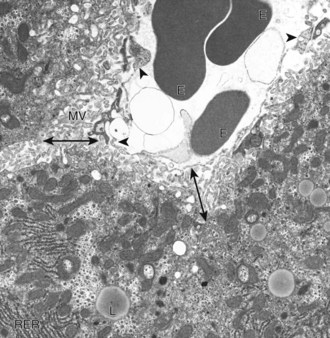
Web Fig. 8-2 Hepatic sinusoids, liver, normal dog.
A sinusoid containing erythrocytes (E) can be seen in the upper right-hand portion of the figure. The margins of the sinusoid are lined with discontinuous capillaries (arrowheads). The space of Disse (double-headed arrows) lies between the endothelial cells and the hepatocytes. Microvilli (MV) project from the hepatocytes into the space of Disse. Rough endoplasmic reticulum (RER) and lipid droplets (L) can be seen in the hepatocyte cytoplasm. TEM. Uranyl citrate and lead acetate stain. (Courtesy Dr. V. Meador, Covance Inc.)
Although blood cells are normally excluded from the space of Disse because they are too large to pass through endothelial gaps, the modified endothelial cells and basement membrane permit plasma to pass freely into a gap between the endothelial cells and the hepatocytes (see Fig. 8-3). This critical anatomic feature of the liver is termed the space of Disse. Within this space, plasma constituents come into contact with the luminal surface of the hepatocytes. This surface of the hepatocytes is characterized by the presence of numerous microvilli, which increase the surface area of the hepatocytes and facilitate uptake of a variety of plasma-borne substances, as well secretion of synthesized products. Any damage to this area has significant impact on hepatic function.
The lumen of the sinusoids contains hepatic macrophages, termed Kupffer cells (Fig. 8-5). These cells are members of the monocyte-macrophage system, and they clear infectious agents and senescent cells, such as erythrocytes, particulate material, endotoxin, and other substances, from the sinusoidal blood. They are mobile and able to migrate along the sinusoids and into areas of tissue injury and regional lymph nodes. Kupffer cells are involved in cytokine-driven interactions with hepatocytes, endothelial cells, and the stellate cells discussed later. They can express class II histocompatibility antigens and function as antigen-presenting cells, although they are not as efficient as the macrophages in other tissue. Phagocytosis and clearance of immune complexes are the primary roles of Kupffer cells. Kupffer cells are derived from in situ replication and recruitment of blood-borne monocytes.
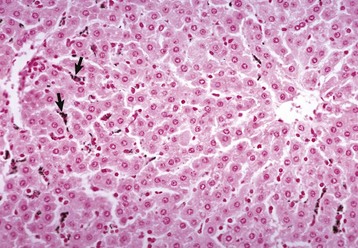
Fig. 8-5 Kupffer cells, carbon particle uptake, liver, normal calf.
Carbon particles injected into the portal vein have been phagocytosed by Kupffer cells (arrows), making them more easily detectable along the sinusoids of the liver. Nuclear fast red stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Hepatic stellate cells (also termed lipocytes or Ito cells) are found within the space of Disse and between hepatocytes at the edge of the space of Disse (Web Fig. 8-3). Normally, hepatic stellate cells are primarily responsible for storing vitamin A in their characteristic cytoplasmic vacuoles. During hepatic injury, hepatic stellate cells alter their morphology and their function. These activated hepatic stellate cells lose their vitamin A content and synthesize collagen and other ECM components that lead to hepatic fibrosis.
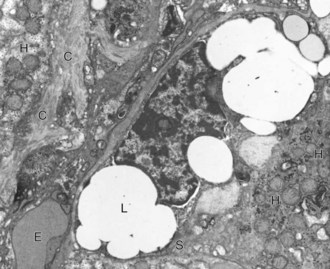
Web Fig. 8-3 Liver, normal dog.
A hepatic stellate cell (S) with its characteristic cytoplasmic lipid vacuoles (L) is found adjacent to hepatocytes (H) and within the space of Disse. Bundles of collagen (C) are found at the margins of the cells. Erythrocytes (E) are within the sinusoidal lumen. TEM. Uranyl citrate and lead acetate stain. (Courtesy Dr. V. Meador, Covance Inc.)
Bile flows within the lobule in the opposite direction to blood flow, which facilitates the concentration of bile. The biliary system commences as canaliculi within the centrilobular (periacinar) areas of the hepatic lobule. The walls of canaliculi are formed entirely by the cell membranes of adjacent hepatocytes. Just outside the limiting plate, canaliculi drain into the canals of Hering that are lined partially by hepatocytes and partially by biliary epithelium. These drain into cholangioles with low cuboidal biliary epithelium. The cholangioles converge into interlobular bile ducts that are lined with cuboidal epithelium and located in the portal areas. Bile then flows into the right and left hepatic ducts that unite to form the hepatic duct. The confluence of the common hepatic duct and the cystic duct from the gallbladder form the common bile duct by which bile is carried to the duodenum. The gallbladder is responsible for storage and concentration of bile in most species. It is absent in the horse, elephant, and rat.
Bipotential progenitor cells that have the ability to differentiate into hepatocytes or biliary epithelium are believed to reside in the area of the cholangiole, although their precise location and nature is not resolved. These cells may proliferate in circumstances in which mature hepatocytes or bile duct epithelium cannot replicate such as severe injury or nutritional deficits. When these cells proliferate, they form islands or crude tubules of small basophilic cells found initially at the margin of the limiting plate. This proliferation is termed the ductular reaction and is a hallmark of severe injury.
Both sympathetic and parasympathetic nerves running along the portal vein and the hepatic artery innervate the liver. The nerve fibers enter the liver at the hilus and ramify to the level of the portal tracts and then extend along the sinusoids. Nerve supply is believed to affect sinusoidal blood flow, the balance of hepatic blood flow from the portal vein and the hepatic artery, and metabolic functions of the liver.
Normal Function
The liver performs many critical functions, including the following:
Bilirubin Metabolism: Excretion of bile is the main exocrine function of the liver. Bile is composed of water, cholesterol, bile acids, bilirubin, inorganic ions, and other constituents. Bile formation is continuous, but the rate of secretion can vary significantly. There are three major purposes for bile synthesis. The first purpose is excretory; many of the body’s waste products, such as surplus cholesterol, bilirubin, and metabolized xenobiotics, are eliminated in bile. The second purpose is the facilitation of digestion; bile acids secreted into the intestine aid in the digestion of lipids within the intestine. The third is to provide buffers to neutralize the acid pH of the ingesta.
Bilirubin, a major component of bile, is produced from the metabolic degradation of hemoglobin, and to a lesser extent, other heme proteins including myoglobin and the hepatic hemoproteins, such as cytochromes (Fig. 8-6). The majority of bilirubin is derived from normal extrahepatic breakdown of senescent erythrocytes in cells of the monocyte-macrophage phagocytic cell series. Senescent erythrocytes normally are phagocytosed by macrophages of the spleen, bone marrow, and liver. Within the phagocyte, the globin portion is degraded and the constituents are returned to the amino acid pool. The heme iron is transferred to iron-binding proteins, such as transferrin, for recycling. The remaining portion of heme is first oxidized by heme oxygenase to biliverdin. In the next metabolic step, biliverdin reductase converts biliverdin to bilirubin. Subsequently, the bilirubin, which is poorly soluble in an aqueous medium, is then released into the blood in its unconjugated form and bound to albumin to increase its solubility in plasma.
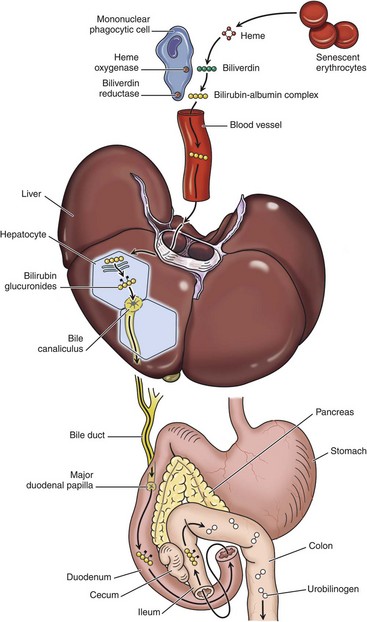
Fig. 8-6 Schematic diagram of bilirubin metabolism and elimination (as depicted in human beings).
1, Normal bilirubin production from heme (0.2 to 0.3 g per day) is derived primarily from the breakdown of senescent circulating erythrocytes, with a minor contribution from degradation of tissue heme-containing proteins. 2, Extrahepatic bilirubin is bound to serum albumin and delivered to the liver. 3, Hepatocellular uptake. 4, Glucuronidation in the endoplasmic reticulum generates bilirubin monoglucuronides and diglucuronides, which are water soluble and readily excreted into bile. 5, Gut bacteria deconjugate the bilirubin and degrade it to colorless urobilinogens. The urobilinogens and the residue of intact pigments are excreted in the feces, with some reabsorption and excretion into urine. Residual urobilinogen is metabolized by bacteria into the brown pigment stercobilin, imparting the typical color to feces.
The process of bilirubin elimination can be divided into three phases: uptake, conjugation, and secretion. Uptake refers to the process by which hepatocytes remove the bilirubin bound to albumin from the circulation. Unconjugated bilirubin is separated from albumin at the sinusoidal surface and bilirubin is taken up by hepatocytes by a carrier-mediated process. In the second phase of bilirubin metabolism, bilirubin is conjugated, principally with glucuronic acid, by bilirubin UDP-glucuronyltransferase in the endoplasmic reticulum. After conjugation, bilirubin becomes water soluble and less toxic. It is then excreted, in the third phase of bilirubin metabolism, into the bile by active transport through specialized portions of hepatocyte membranes that form the margins of the bile canaliculi. The excretion phase is the rate-limiting step in most species.
Within the gastrointestinal tract, conjugated bilirubin is converted to urobilinogen by bacteria and a fraction of this is reabsorbed into the portal blood, a process called enterohepatic circulation, and returned to the liver. The majority of urobilinogen that is absorbed from the gastrointestinal tract is resecreted into bile. Urobilinogen has a small molecular weight and is freely filtered through the glomerulus, and small amounts are normally found in the urine. Urobilinogen that is not absorbed from the intestine becomes oxidized to stercobilin, which is responsible for the color of the feces.
Bile Acid Metabolism: The three principal functions of bile acids, important constituents of bile, are maintenance of cholesterol homeostasis, stimulation of bile flow and digestion, and absorption of fats and fat-soluble vitamins. Bile acids are synthesized in the liver from cholesterol and are conjugated to glycine or taurine to facilitate their interaction with other components of bile and to prevent precipitation into calculi when they are secreted into the bile. The major bile acids are cholic acid and chenodeoxycholic acid, but there are various types and proportions of bile acids found in different species. Bile acids are actively secreted into the bile canaliculi from the hepatocyte cytoplasm by specific intramembranous molecular pumps against a concentration gradient, which creates an osmotic gradient, stimulating the inflow of water and solutes into the bile canaliculi. Conjugated bile acids are therefore the principal physiologic stimulus for bile production through a process termed bile acid–dependent flow. Bile acids are effective detergents that assist in the digestion of lipids within the intestine and increasing the solubility of lipids secreted into the bile. The quantities of bile acids required far exceed the liver’s capacity to produce them. For this reason, bile acids are avidly reabsorbed from the ileum, extracted from the portal blood, and resecreted into bile via a process known as enterohepatic circulation. This is a very efficient system. As much as 95% of secreted bile acids are recycled, and the proportion of reabsorbed bile acids in the liver greatly exceeds that of recently synthesized bile acids; bile acids may be recycled 15 times a day. Interruption of this process results in fat malabsorption and a deficiency of fat-soluble vitamins.
Carbohydrate Metabolism: The liver has an important role in the regulation of plasma glucose concentrations. After eating, the liver removes carbohydrates (i.e., glucose, fructose) from the plasma and stores them as glycogen or fatty acids. In periods of need, energy balance is maintained by glycolysis of stored glycogen or by gluconeogenesis. Production of energy by oxidative phosphorylation and β-oxidation of fatty acids in hepatic mitochondria is used to sustain the activities of the hepatocyte.
Lipid Metabolism: The liver plays a central role in lipid metabolism. It is involved in the production and degradation of plasma lipids such as cholesterol, triglycerides, phospholipids, and lipoproteins. Cholesterol is synthesized, secreted, and degraded by hepatocytes. Hepatocytes can synthesize fatty acids when energy levels are high, and they can oxidize fatty acids as an energy source when necessary.
Xenobiotic Metabolism: Foreign substances (xenobiotics), such as many therapeutic drugs, insecticides, and endogenous substances—such as steroids that are lipophilic—require conversion to water-soluble forms for elimination from the body. The cytochrome p450 enzymes of the smooth endoplasmic reticulum of the hepatocytes serve as the major site of metabolism of these substances in preparation for excretion in bile or urine. This process is discussed in detail in the section on toxic liver injury.
Protein Synthesis: Synthesis of the majority of plasma proteins, mainly within the rough endoplasmic reticulum, is a principal function of the liver. Proteins produced in the liver include plasma proteins, such as albumin; a variety of transport proteins; lipoproteins; clotting factors II, V, and VII to XIII; fibrinolysis proteins; some acute phase proteins; and components of the complement system. The liver is responsible for synthesis of approximately 15% of body proteins.
The liver is also the principal site of ammonia metabolism. Highly toxic ammonia is generated through catabolism of amino acids. Metabolic conversion of ammonia into urea, a far less toxic compound, occurs through the urea cycle, which occurs almost exclusively in the liver. Urea then enters the systemic circulation (blood urea nitrogen) and is excreted in the urine.
Immune Function: The liver has a significant immune function. It is involved in systemic, local, and mucosal immunity. Hepatocytes participate in the response to systemic inflammation through the synthesis and release of acute phase proteins. Approximately 10% of the cells in the liver belong to the adaptive immune system (T and B lymphocytes) or the innate immune system (Kupffer cells, natural killer lymphocytes, and natural killer T lymphocytes). Compared with other organs, the liver is particularly enriched with cells of the innate immune system, likely a result of the fact it is the site where foreign antigens from the gastrointestinal tract first encounter the innate immune system defenses. The liver contains the largest pool of mononuclear phagocytes and natural killer cells in the body in most species. The Kupffer cells lining the sinusoids provide the first line of defense against infectious agents, endotoxin, and foreign material absorbed from the intestines before they gain access to the systemic circulation. Most blood-borne foreign material is cleared by Kupffer cells in all domestic species, except members of the Order Artiodactyla (pigs, goats, and cattle), in which this function is performed by intravascular macrophages in the pulmonary alveolar capillaries. The liver is also involved in transport of secretory immunoglobulin A (IgA), the primary immunoglobulin of the mucosal surfaces, from plasma cells and recirculation into the biliary tree and intestine.
Response of the Liver to Injury
The epithelial cells of the liver, hepatocytes, and biliary epithelium are the principal targets of most liver diseases. Sublethal injury to hepatocytes is characterized by cell swelling (hydropic degeneration), steatosis, or atrophy. Cells that have sustained a sublethal injury often remove damaged organelles by forming autophagosomes. Material that cannot be digested further is retained as lipofuscin, which is why after sublethal injury, this pigment can often be found in affected cells and associated phagocytes.
By convention, cell death has been divided into two distinct processes. These are necrosis, which is characterized by cytoplasmic swelling, destruction of organelles, and disruption of the plasma membrane, and apoptosis, or programmed cell death, which is characterized by one of several active processes involving caspases that lead to cell shrinkage and an intact cell membrane. Necrosis is triggered by lethal injury. Necrotic cells typically exhibit karyorrhexis and fragmentation of the cell body. Coagulative necrosis results from sudden denaturation of hepatocytes and produces swollen hepatocytes with a preserved eosinophilic cytoplasmic outline and karyorrhexis or karyolysis. Lytic necrosis is characterized by a loss of hepatocytes and an influx of erythrocytes into the vacant space or condensation of the reticular connective tissue (collagen and other ECM) scaffolding of the liver that once supported the hepatocytes.
Classic apoptosis is triggered by an interaction between tumor necrosis factor-α (TNF-α) or Fas ligand and specific receptors on the cell membrane leading to caspase activation, although other pathways, including those involving mitochondrial cytochrome-c, have been identified. Apoptosis is recognized by the formation of acidophilic bodies, which are brightly eosinophilic, homogeneous, round structures that can be found between hepatocytes, within the lumen of sinusoids, or within macrophages or hepatocytes. A detailed review of cell death is beyond the scope of this section but is covered in Chapter 1. However, recent evidence reveals that there may some overlap between necrosis and apoptosis, depending on the cell type and the type and dose of injurious agent. Thus both hepatic necrosis and apoptosis can be produced by the same agent and can occur in the same liver.
Patterns of Hepatocellular Degeneration and Necrosis: Although the liver is subjected to a wide variety of different insults, the cellular degeneration and/or necrosis that results invariably occurs in one of following three morphologic patterns:
• Random hepatocellular degeneration and/or necrosis
Random Hepatocellular Degeneration: Random hepatocellular degeneration and/or necrosis is characterized by the presence either of single cell necrosis throughout the liver or multifocal areas of necrotic hepatocytes. These areas are scattered randomly throughout the liver; there is no predictable location within a lobule. This pattern is typical of many infectious agents, including viruses, bacteria, and certain protozoa. Lesions may be obvious grossly as discrete, pale, or less often, dark red foci that are sharply delineated from the adjacent parenchyma (Fig. 8-7, A). The size of such foci is variable, ranging from tiny (<1 mm) to several millimeters. Hepatocytes in affected areas are either degenerated or necrotic because of the injurious effects of the infectious agents and the stage of the process (Fig. 8-7, B).
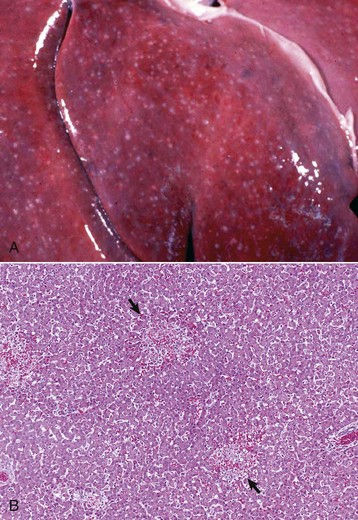
Fig. 8-7 Random hepatocellular injury, liver.
A, Equine herpes virus infection, foal. Random foci of viral-induced lytic necrosis. B, Salmonellosis, focal necrosis and inflammation, pig. The random pattern of hepatocellular necrosis and inflammation (arrows) caused by septicemic Salmonella spp. can also be seen within the hepatic lobules. H&E stain. (A courtesy Drs. J. King and L. Roth, College of Veterinary Medicine, Cornell University. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Zonal Hepatocellular Degeneration and/or Necrosis: Zonal hepatocellular degeneration and/or necrosis or as it is more simply termed, zonal change, affects hepatocytes within defined areas of the hepatic lobule. The zones are centrilobular (periacinar), midzonal (between centrilobular and periportal areas), or periportal (centroacinar) areas. Extensive zonal change within the liver, regardless of location within the lobule, typically produces a liver that is pale and modestly enlarged with rounded margins, has increased friability, and characteristically has an enhanced lobular pattern on the capsular and cut surface of the organ (Fig. 8-8). Degenerated hepatocytes swell and when the majority of hepatocytes in a zone are affected, that portion of the lobule appears pale. In contrast, once the hepatocytes in a particular zone of the lobule have become necrotic, this results in dilation and congestion of sinusoids so that the affected zone appears red. Although zonal change typically produces an enhanced lobular pattern, microscopic examination is usually required to determine the type of zonal change. Specific forms of zonal change are described next.

Fig. 8-8 Zonal hepatocellular injury, liver, horse.
Accentuation of the normal lobular pattern is evident on the capsular surface of the liver. This is not a specific change, as it may be associated with zonal hepatocellular degeneration and/or necrosis (regardless of lobular location), passive congestion, or diffuse cellular infiltration of the portal and periportal areas (often reflecting hepatic involvement of hematopoietic neoplasms, such as lymphoma and myeloproliferative disorders). (Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
Centrilobular degeneration and necrosis: Centrilobular degeneration and necrosis of hepatocytes is particularly common (Fig. 8-9), as this portion of the lobule receives the least oxygenated blood and is therefore susceptible to hypoxia, and it has the greatest enzymatic activity (mixed-function oxidases) capable of activating compounds into toxic forms. Centrilobular necrosis can result from a precipitous and severe anemia or right side heart failure. Similarly, passive congestion of the liver results in hypoxia as a result of stasis of blood and produces atrophy of centrilobular hepatocytes.
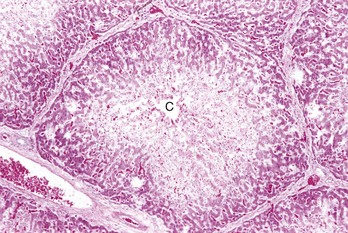
Fig. 8-9 Centrilobular necrosis, zonal hepatocellular injury, liver, pig.
Centrilobular necrosis (periacinar or zone 3) is characterized by a circumferential zone of hepatocellular necrosis surrounding the terminal hepatic venule (central vein [C]). H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Paracentral (periacinar) cellular degeneration: Paracentral (periacinar) cellular degeneration involves only a wedge around the central vein because only the periphery of one acinus is affected, typically reflecting the action of a direct-acting toxin that requires bioactivation (Fig. 8-10) or severe, acute anemia. As several acini border on a single central vein (terminal hepatic venule), changes induced by hypoxia may not be present equally in all acini, and thus hepatocytes at the periphery of one acinus can have more severe change than those in adjacent acini.
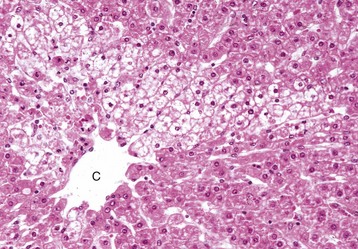
Fig. 8-10 Paracentral degeneration and necrosis, zonal hepatocellular injury, liver, cow.
Rather than a pattern of complete circumferential necrosis, a wedge-shaped area of hepatocytes is damaged. In this case, the paracentral lesion consists of necrotic hepatocytes to the left and other hepatocytes with hydropic degeneration. This wedge is the apex of the diamond-shaped liver acinus (zone 3) and reflects the partitioning of the lobule based on the inflow of blood from each of the individual portal tracts that surround the lobule. This change can be seen as an early manifestation of hepatic hypoxia in animals with anemia or right-sided heart failure and precedes centrilobular necrosis. C, Central vein. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Midzonal degeneration and necrosis: Midzonal degeneration and necrosis are unusual lesions in domestic animals but have been reported in pigs and horses with aflatoxicosis and cats exposed to hexachlorophene (Fig. 8-11).
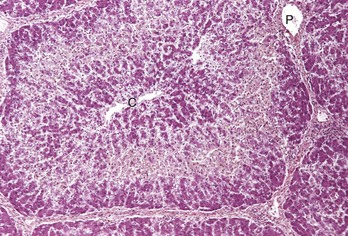
Fig. 8-11 Midzonal necrosis, zonal hepatocellular injury, liver, horse.
Midzonal necrosis is the least common pattern of hepatic injury. Hepatocytes in the middle portion of the lobule (zone 2) are affected and hepatocytes in the other regions are spared. C, Central vein; P, portal vein. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Periportal degeneration and necrosis: Periportal degeneration and necrosis are also uncommon but may occur following exposure to toxins, such as phosphorus, that do not require metabolism by mixed function oxidases (most active in the centrilobular hepatocytes) to cause injury (Fig. 8-12). Some of these compounds may be metabolized to injurious intermediates by cytoplasmic enzymes found in periportal hepatocytes. Alternatively, some of these toxins may not require metabolism and produce hepatocyte injury in the first hepatocytes that they encounter as they flow from the portal areas.
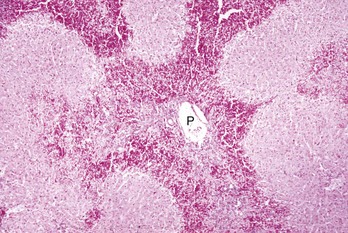
Fig. 8-12 Periportal necrosis, zonal hepatocellular injury, liver, horse.
Periportal (or zone 1) necrosis is an uncommon pattern of hepatocellular injury. Hepatocytes surrounding the portal tracts (P) are affected. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Bridging necrosis: Bridging necrosis is the result of confluence of areas of necrosis. Bridging may link centrilobular areas (central bridging) or centrilobular areas to periportal areas (Fig. 8-13).
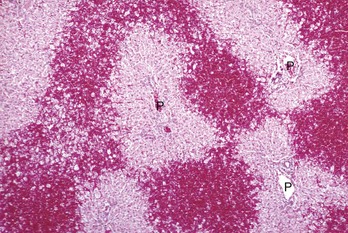
Fig. 8-13 Bridging necrosis, zonal hepatocellular injury, liver.
Bridging necrosis refers to a pattern characterized by connection of areas of necrosis between different lobules. Three patterns of bridging necrosis are recognized: central to central, as seen here; portal to portal; and central to portal. P, Portal area. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Massive Necrosis: Massive necrosis is not necessarily, as the name might be taken to imply, necrosis of the entire liver, but rather the term describes necrosis of an entire hepatic lobule or contiguous lobules (Fig. 8-14, A). All hepatocytes within affected lobules are necrotic. The gross appearance of the liver varies with the maturity of the lesion. If, in acute cases, the majority of the parenchyma is affected, the liver may initially be modestly increased in size with a smooth external surface and dark parenchyma because of extensive congestion. At first, necrotic hepatocytes lyse and the residual stroma becomes condensed. Regeneration does not occur because virtually all hepatocytes in the lobule are affected. Microscopically, affected areas consist of blood-filled spaces within a connective tissue stroma devoid of hepatocytes (Fig. 8-14, B). Later in the course of the process, stellate cells or other ECM–producing cells from the portal and centrilobular areas that may survive or migrate to the site of injury contribute new collagen (collagen I, in particular). The final result is collapse of the lobule and replacement of the lost hepatic parenchyma with a scar consisting of condensed stroma, including variable amounts and types of collagen. Grossly the liver may be smaller than normal with a wrinkled capsule. Partial involvement of the liver is characterized by depressed areas of parenchymal necrosis and vascular congestion scattered throughout the organ.
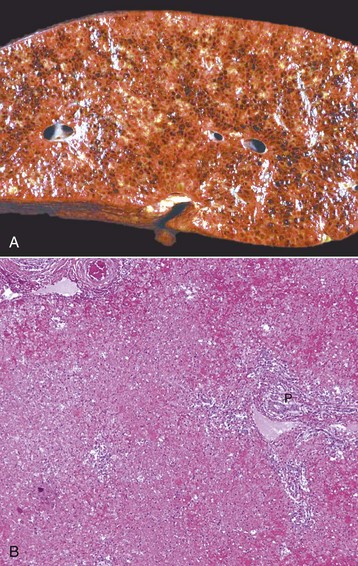
Fig. 8-14 Massive necrosis, liver.
A, Pig, cut surface. Massive necrosis refers to a pattern of necrosis that involves an entire hepatic lobule, as seen here. B, Dog. The entire population of hepatocytes within the lobule has undergone necrosis. P, Portal area. H&E stain. (A courtesy Dr. D. Cho, College of Veterinary Medicine, Louisiana State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Disturbances of Bile Flow and Icterus
Hepatic injury is frequently manifested as an increased concentration of conjugated or unconjugated bilirubin in blood called hyperbilirubinemia. High concentrations of bilirubin (>approximately 2 mg/dL) can produce jaundice (icterus), a yellow discoloration of tissue that is especially evident in tissue rich in elastin such as the aorta and sclera (Fig. 8-15). This concentration is within the reference range for horses, so horses may not be hyperbilirubinemic at this level. However, in other species, hyperbilirubinemia can occur once the concentration exceeds 0.5 mg/dL (dog) and therefore the patient is hyperbilirubinemic, but icterus will not be detected until it exceeds 2 mg/dL. Maximal accumulation of bilirubin in tissues takes about 2 days and explains why some animals with acute hepatic failure may have only slight icterus.
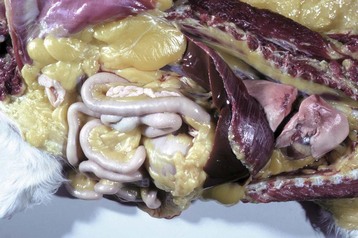
Fig. 8-15 Icterus, dog.
Icterus and jaundice are terms that refer to the yellow discoloration of tissue, by bilirubin, in this case evident in the fat and serosa. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The causes of hyperbilirubinemia include the following:
1. Overproduction of bilirubin as a consequence of hemolysis, particularly severe intravascular hemolysis, which overwhelms the liver’s capacity to remove bilirubin from the plasma and to secrete conjugated bilirubin into bile. The destruction of damaged red blood cells by extravascular hemolysis can also increase the burden of bilirubin presented to the liver. Hypoxia secondary to anemia may also play a role. Decreased uptake, conjugation, or secretion of bilirubin by hepatocytes arising as a consequence of severe, diffuse hepatic disease, whether acute or chronic.
2. Reduced outflow of bile (cholestasis). Cholestasis is defined as a defect in bile secretory mechanisms that leads to an accumulation in the blood of substances normally excreted into the bile. Cholestasis occurs as a consequence of either obstruction of the biliary ducts (extrahepatic cholestasis) or impairment of bile flow within canaliculi (intrahepatic cholestasis).
Obviously, hepatic dysfunction is not the only cause of hyperbilirubinemia and icterus. In fact, icterus in ruminants is usually a consequence of severe intravascular hemolysis and less often a sequel to hepatic damage. Horses often manifest icterus with acute hepatic dysfunction, but icterus may or may not occur in horses with chronic hepatic disease. Interestingly, “physiologic icterus” is also common in the horse, and horses deprived of feed for several days can become icteric because uptake of bilirubin from the plasma by hepatocytes is decreased. Icterus in carnivores occurs as a consequence of either hemolysis or hepatic dysfunction. Inherited metabolic abnormalities can also lead to abnormal concentrations of serum bilirubin. In Southdown sheep with certain mutations, bile is ineffectively taken up from the circulation and a persistent unconjugated hyperbilirubinemia develops, although icterus is rarely apparent because there is sufficient excretion despite the mutation. Corriedale sheep may have a mutation that leads to deficient conjugated bilirubin excretion. Affected sheep have persistently elevated plasma bilirubin concentration, but jaundice is not apparent. Other compounds that are normally excreted through conjugation also accumulate in the liver of affected sheep. The livers are dark and discolored because of accumulated polymerized catecholamine metabolites that accumulate in lysosomes. These residues resemble lipofuscin histologically.
Cholestasis can be divided into two types: intrahepatic and extrahepatic. Intrahepatic cholestasis can result from (1) a wide spectrum of liver injury affecting the ability of hepatocytes to metabolize and excrete bile; (2) hemolysis, which produces an abundance of bilirubin for excretion and diminishes the supply of oxygen for hepatocyte metabolism; or (3) inherited abnormalities of bile synthesis that inhibit the excretion of bile. Extrahepatic cholestasis is produced by obstruction of the extrahepatic bile ducts. This can occur by intraluminal obstruction (calculi or possibly parasites) or extraluminal means, including neoplasia or adjacent inflammation, often involving the pancreas. Cholestasis, if sufficiently severe, can produce a greenish brown discoloration to the liver (Fig. 8-16, A).
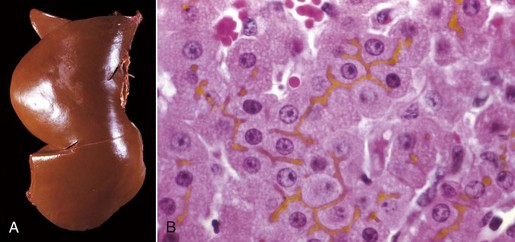
Fig. 8-16 Hepatic bilirubin retention, icterus, liver.
A, Cat. The liver is markedly yellowed by retained bilirubin. B, Canalicular bilirubin, acute hemolytic anemia, calf. Acute hemolysis caused by babesiosis has led to a dramatic increase in bilirubin production and distention of canaliculi, clearly demonstrating the location of canaliculi between hepatocytes. H&E stain. (A courtesy College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Histologically, acute intrahepatic cholestasis is characterized by formation of bile plugs within canaliculi (Fig. 8-16, B). As intrahepatic cholestasis becomes more chronic, bile that has been released from hepatocytes is taken up by Kupffer cells and can be detected within their cytoplasm.
Acute extrahepatic obstruction is characterized by edema of the portal areas, a mild neutrophilic inflammatory cell infiltrate, and a proliferative reaction by the biliary epithelium of the bile ducts. In chronic extrahepatic biliary obstruction, portal areas are enlarged by deposition of fibrosis, and there is a prominent laminar, circumferential fibrosis of bile ducts (Fig. 8-17). Biliary hyperplasia characterized by proliferation of small-caliber bile ducts is often prominent. Pigmented macrophages, containing bile, and mixed inflammatory infiltrates are also present. In severe cases, bridging fibrosis connecting portal tracts may develop.
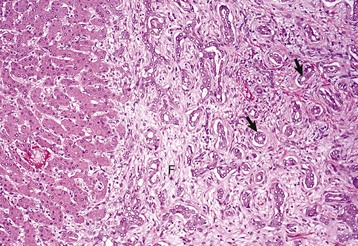
Fig. 8-17 Chronic extrahepatic cholestasis, cholelithiasis, liver, horse.
There is reduplication of bile ducts (arrows) and extensive fibrosis (F) throughout the portal tract (biliary fibrosis) as a consequence of prolonged stasis and subsequent leakage of bile. H&E stain. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Complete biliary obstruction leads to maldigestion of fats and a characteristic clay-colored stool termed acholic feces because of the lack of normal dark pigment, stercobilin, the bilirubin-derived pigment produced by bacterial metabolism (Fig. 8-18).
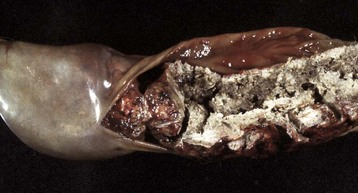
Fig. 8-18 Intrahepatic biliary obstruction, intestine, dog.
In cases of complete biliary obstruction, bile is unable to reach the intestine and as a result stool(s) lacks the characteristic dark color produced by bile pigments. (Courtesy College of Veterinary Medicine, North Carolina State University.)
Regeneration
A characteristic feature of the liver is the ability to rapidly and efficiently regenerate lost hepatic mass. Experimentally, as much as two-thirds of the liver can be excised from a healthy animal without signs of hepatic dysfunction, and the liver is rapidly regenerated. In addition to replication of hepatocytes, there is a wave of replication in bile duct epithelium, endothelium, and sinusoidal lining cells that is coordinated with hepatocyte replication.
Regeneration usually takes place by replication of mature hepatocytes. In most circumstances, this leads to an increase in the size of existing lobules; however, recent data suggest that some new lobule formation can also occur through subdivision of existing lobules. After removal of liver lobes only, the remaining liver lobes persist, and no new lobe formation occurs.
Individual cell necrosis leads to local proliferation by regeneration of adjacent hepatocytes. Scattered foci of necrotic hepatocytes are quickly replaced through cell division of adjacent hepatocytes. Necrosis in the centrilobular area of the lobule leads to a wave of hepatocyte proliferation in the hepatocytes in the remaining areas of the lobule, particularly the periportal hepatocytes. In some circumstances, such as necrosis of nearly all hepatocytes or exposure to certain chemical toxins that inhibit replication of mature hepatocytes, replacement of hepatocytes lost by necrosis occurs through proliferation of hepatocyte stem cells or oval cells. This process is most often observed in experimental manipulations of laboratory rodents but probably occurs in naturally occurring cases of hepatotoxicity also. These cells reside in the connective tissue of the portal tracts and have the ability to differentiate into hepatocyte or bile duct epithelium. Histologically, they are identified as small basophilic cells with an oval shape. They are not apparent in most circumstances of hepatic regeneration but can be abundant in experimental studies of hepatic regeneration.
The body carefully orchestrates hepatic regeneration to replace lost hepatocyte mass along with bile ducts and vessels without producing excess liver. A variety of growth factors, including transforming growth factor-α (TGF-α) and hepatocyte growth factor, stimulate hepatocyte replication. Once normal hepatic mass has been established, macrophages release TGF-β, which, in concert with other less well characterized factors, stops hepatic parenchymal cell proliferation.
A single episode of extensive hepatic necrosis is usually followed by parenchymal regeneration without scarring, as long as the normal ECM (reticulin) scaffolding of the affected portion remains intact and has not collapsed. However, repetitive injury or massive necrosis can disrupt the normal lobular architecture, and there may be parenchymal collapse after removal of the dead hepatocytes and/or stromal collapse with repair by collagen synthesis (postnecrotic scarring) (Fig. 8-19). Even when necrosis of hepatocytes is continuous, the liver attempts to regenerate its functional mass. However, prolonged regenerative effort with damage to the normal ECM scaffolding of the liver often results in nodular proliferations of parenchyma, which architecturally distort the liver. Although regenerative nodules may reconstitute a proportionately large amount of hepatic mass, adequate function is rarely attained. Blood flow into the regenerative nodules and bile flow out of the nodules is abnormal, and as a result, hepatic function cannot be reestablished. As the nodules develop, the portal tract vessels and the central veins develop communications within the fibrous septa between nodules, which leads to vascular shunts between the portal vein and the central vein that bypass hepatocytes within the nodules.
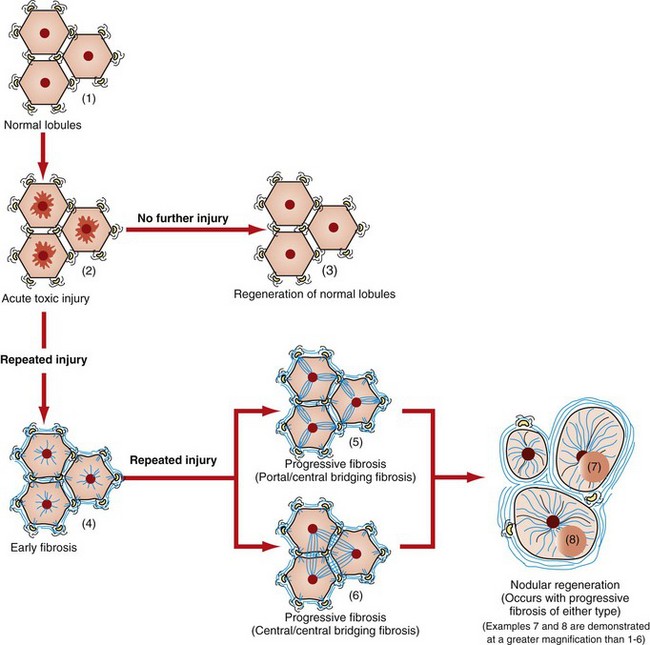
Fig. 8-19 Schematic diagram of the effects of hepatic injury on the development of fibrosis.
Acute hepatic centrilobular injury (2) that occurs only once usually resolves, and normal liver architecture returns (1, 3). Repeated bouts of injury or severe injury can initiate hepatic fibrosis (4). In the earliest stages, fibrosis may be reversible, but as fibrosis progresses it reaches a point at which repair is not likely. Fibrosis often starts as fine branches of collagen deposition between portal areas or central areas or dissecting into the hepatic parenchyma (5, 6). Over time, greater amounts of collagen and other extracellular matrix are deposited and the lobular architecture becomes progressively distorted. In the end-stage liver, nodular regeneration (7, 8) and extensive, circumferential fibrosis are typical. The regenerative nodules shown here (7, 8) are at an early stage of regeneration. As shown in Fig. 8-21, they will regenerate to form nodules that will commonly exceed the size of normal hepatic lobules. These nodules will often compress (7) the central vein(s) of hepatic lobules within and adjacent to those from which they arose.
Fibrosis
Fibrosis is one of the more common manifestations of chronic liver injury. The pattern of fibrosis is frequently a useful indicator of the type of insult that produces the lesion. The significance of fibrosis depends on its effect on hepatic function and its reversibility. Despite the considerable regenerative capacity of the liver, hepatic fibrosis, when sufficiently severe, can be lethal.
In the normal liver, fibrillar collagens I and III are confined primarily to the connective tissue of the portal tracts and immediately around the terminal hepatic venule (central vein). Collagen IV is the most abundant collagen type in the reticulin framework of the sinusoids, but it is present in only small amounts. A delicate scaffolding of collagen and other ECM components, which are produced by stellate cells, endothelial cells, and hepatocytes, make up the normal framework of the sinusoid. This stroma in the space of Disse supports the endothelial cells and maintains their relationship to the hepatocytes.
Hepatic fibrosis is an overall increase in the ECM within the liver. In a fibrotic liver, there is an increase in the amount of ECM and a change in the types of collagen and their site of deposition. A severely fibrotic liver can contain up to six times as much collagen and proteoglycan as a normal liver. Hepatic fibrosis is characterized by an increase of fibrillar collagens, type I and type III, and nonfibrillar collagen XVIII within the space of Disse, the portal areas, and the area surrounding the central veins. In addition to an increase in collagens, there is also a commensurate increase in the ECM components, proteoglycans, fibronectin, and hyaluronic acid.
The stellate cells (Ito cells and lipocytes) have a central role in hepatic fibrosis, although it should be noted that there are myofibroblastic cells with similar capabilities found within the connective tissue of the portal areas and connective tissue surrounding the central vein. In the normal liver, stellate cells occupy the space of Disse (a subendothelial position in the sinusoid) nestled between hepatocytes. They are characterized by the presence of large lipid-containing vacuoles in their cytoplasm (see Web Fig. 8-3). The vacuoles are a primary storage site for retinyl esters, including vitamin A. Stellate cells are positioned in the space of Disse around the circumference of the endothelium of the sinusoids and have been likened to pericytes in other organs, such as the mesangial cells of the renal glomerulus. Hepatic stellate cells have been shown to have a role in the control of the diameter of the sinusoids and consequently the flow of blood through the sinusoids.
When the liver is injured, these cells go through a progressive phenotypic change from the typical lipid-storing cell to a cell with a myofibroblastic appearance (Fig. 8-20). When they are activated, they express smooth muscle actin and desmin, a marker usually found in muscle cells. Once these cells have switched to the myofibroblast phenotype, they begin synthesis of collagen types I, III, and IV. They also produce other ECM components, including laminin and chondroitin sulfate proteoglycans. Hepatocytes synthesize few or no matrix proteins, and the large sum of matrix proteins is derived from the hepatic stellate cells. The type of hepatic injury does not seem to be important in the genesis of hepatic fibrosis. Chemical injury, biliary obstruction, and iron overload produce similar patterns of activation of hepatic stellate cells.
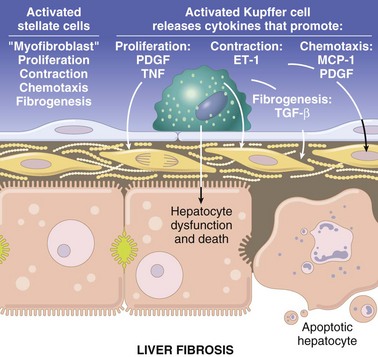
Fig. 8-20 Schematic diagram of a fibrotic hepatic sinusoid.
Sinusoidal fibrosis dramatically reduces contact between the plasma and the hepatocytes. The sinusoidal endothelial cells have lost their fenestrations. The space of Disse contains abundant collagen type I fibers that have been synthesized by activated hepatic stellate cells. The hepatic stellate cells have eliminated the lipid vacuoles and assumed a myofibroblastic morphology with cell extensions that often surround the endothelial cells (not shown here). Hepatocyte microvilli have been lost along the sinusoid, as shown on the apoptotic hepatocyte, and this loss begins with the onset of hepatocyte dysfunction and degeneration. ET-1, Endothelin-1; MCP-1, macrophage chemotactic protein-1; PDGF, platelet-derived growth factor; TGF-α, transforming growth factor-α; TNF, tumor necrosis factor. (Schematic based on concepts presented in Friedman SL: J Biol Chem 275:2247-2250, 2000; and Crawford JM: Curr Op Gastroenterol 13:175-185, 1997.)
The site in which collagen is deposited in the liver has a significant impact on liver function. Perisinusoidal fibrosis can have a severe effect on hepatic function. In addition to collagen and ECM deposits, there is a loss of gaps in the endothelial cells and a loss of microvilli on the luminal surface of the hepatocytes. These changes have been termed capillarization of the sinusoids because the alterations in the sinusoids result in a vascular structure that more closely resembles a capillary than a sinusoid. The functional effect of this microanatomic change is profound. The ability of the liver to carry out its synthetic, catabolic, and excretory roles is severely compromised by the reduced exposure of hepatocytes to plasma.
Within the hepatic lobule, the site of fibrosis can be indicative of the type of insult. Most often, chronic toxic injury produces centrilobular (periacinar) fibrosis. This region is affected because the centrilobular hepatocytes are the site of metabolism for most drugs. Long-standing right-sided heart failure can cause fibrosis in this site as well. Periportal (centroacinar) fibrosis can result from chronic inflammatory conditions or a small group of toxins that affect the periportal hepatocytes because they do not require metabolism by the cytochrome p450 enzymes to produce an injurious metabolite. Fibrosis may be limited to individual lobules, but in more severe injuries, the areas of fibrosis can be more extensive. Bridging fibrosis, which is analogous to bridging necrosis, implies fibrosis that extends from one portal tract to another or from portal tracts to central veins. Bridging fibrosis is more likely to impair hepatic function than focal forms of hepatic fibrosis; however, all forms of hepatic fibrosis, if sufficiently severe, lead to impaired hepatic function. However, because of the enormous reserve capacity of the liver, fibrosis is usually quite extensive before there are clinical signs of hepatic dysfunction.
A single event of widespread hepatocellular necrosis is sometimes followed, not by the usual regenerative response, but by fibrosis and condensation of the preexisting connective tissue stroma that results in formation of bands of dense connective tissue. This process is referred to as postnecrotic scarring.
Other patterns of hepatic fibrosis can occur, including biliary fibrosis (centered on bile ducts in the portal triads), focal or multifocal hepatic fibrosis (randomly scattered throughout the hepatic parenchyma)—which is produced, for example, by migrating nematode larvae—and diffuse hepatic fibrosis (affects all regions of the lobule and is present throughout the liver). Diffuse fibrosis with regenerative nodule formation is by definition cirrhosis. Different hepatic insults may produce different patterns of fibrosis, but when fibrosis is severe (end-stage liver disease), it frequently is impossible to determine either the cause or the initial pattern of fibrosis.
Biliary Hyperplasia
Biliary hyperplasia, which is proliferation of new biliary ducts within the portal areas and periportal regions, can be a relatively nonspecific response to a variety of insults to the liver. The mechanism responsible for this proliferation is unknown. In severe hepatic injury, proliferation of bipotential progenitor cells with the ability to differentiate into hepatocytes or biliary epithelium can form small caliber ducts and tubules in a response termed the ductular reaction. These cells can mature and replace adult biliary epithelium or hepatocytes. Biliary hyperplasia or ductular reaction can occur swiftly, particularly in young animals. Biliary hyperplasia is typically regarded as a lesion seen in long-standing hepatic injury. Biliary hyperplasia occurs particularly after diseases that obstruct normal bile drainage.
End-Stage Liver or Cirrhosis
The best accepted definition for cirrhosis was pronounced by the World Health Organization (WHO) in 1977 and is as follows: “a diffuse process characterized by fibrosis and the conversion of the normal liver architecture into structurally abnormal lobules (Fig. 8-21).” As it is the final, irreversible result of any one of several different hepatic diseases, the term end-stage liver is appropriate, particularly because the term cirrhosis is neither descriptive nor precise in meaning and originally meant “tawny yellow.” Another authority states that the hallmark is the total absence of any normal lobular architecture. The architecture of the liver is altered by loss of hepatic parenchyma, condensation of reticulin framework, and formation of tracts of fibrous connective tissue. Regeneration of hepatic tissue between fibrous bands leads to the formation of variably sized regenerative nodules (Fig. 8-22). The entire liver is thus distorted and consists of nodules of regenerating parenchyma separated by fibrous bands, which appear as depressions on the surface (see Fig. 8-21).
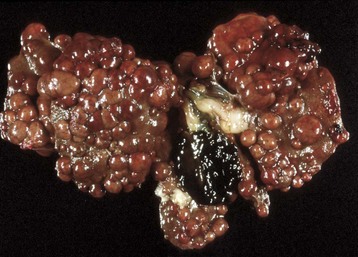
Fig. 8-21 End-stage liver (cirrhosis), dog.
End-stage liver from a dog that had received phenobarbital for many years. The liver is small, firm, and irregular with nodules of regenerative parenchyma separated by tracts of fibrous connective tissue. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
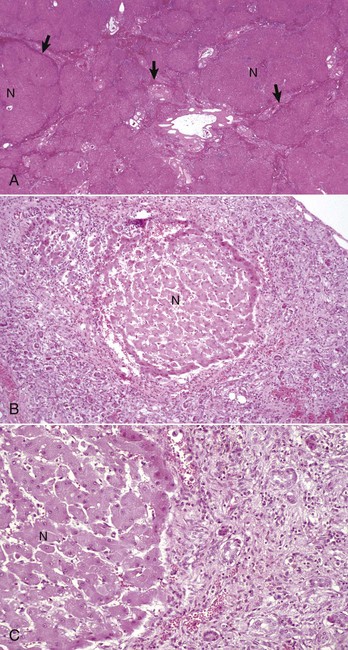
Fig. 8-22 End-stage, liver, dog.
A, Histologic appearance of end-stage liver disease. Nodules of regenerative parenchyma (N) are separated by septa of collapsed reticulin and fibrous connective tissue (arrows), which also contains numerous blood vessels and bile ducts. H&E stain. B, A single regenerative hepatic nodule (N) is surrounded by haphazardly arranged bands of fibrous connective tissue that contains numerous blood vessels and hypertrophied and hyperplastic bile ducts. H&E stain. C, Higher magnification of Fig. 8-22, B. Note the regenerative nodule (N), bands of fibrous connective tissue, hyperplastic bile ducts, and mononuclear inflammatory cells. H&E stain. (A courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University. B and C courtesy College of Veterinary Medicine, University of Illinois.)
Profound vascular abnormalities with serious consequences for the health of affected patients occur in this condition, including multiple abnormal vascular anastomoses between the portal vein and the systemic vasculature, as a consequence of the increased portal pressure. Also, venous shunts between the central veins and the portal veins and arteriovenous shunts between the hepatic arteries and the central veins can occur within the regenerative nodules.
The potential causes of an end-stage (cirrhotic) liver are numerous (Box 8-1). Chronic toxic insult results from the continued ingestion of any hepatotoxin (e.g., herbivores ingesting toxic plants, such as those that contain pyrrolizidine alkaloids, and the long-term administration of anticonvulsant drugs, such as primidone for dogs). Chronic extrahepatic biliary obstruction and cholestasis leads to extensive fibrosis, which primarily affects the portal triads, but the fibrosis can eventually extend into the adjacent hepatic parenchyma. Chronic inflammation of the liver (hepatitis) or biliary tract (cholangitis) may lead to an end-stage liver. Although infection of the liver typically is focal or multifocal, diffuse hepatitis and subsequent fibrosis occurs in disease entities such as canine chronic hepatitis. Chronic passive hepatic congestion eventually leads to fibrosis near central veins, which is sometimes termed cardiac sclerosis and which can progress to cardiac cirrhosis. The actual amount of fibrosis is usually small. Abnormal storage or metabolism of metals, particularly copper, as occurs in Bedlington terriers and several other breeds, may produce chronic inflammation and an end-stage liver. Lobular dissecting hepatitis, a specific form of end-stage liver or cirrhosis, is usually seen in young dogs and is described further under specific diseases of dogs. A variety of more poorly defined disease entities can lead to progressive hepatocellular injury and hepatic fibrosis resulting in end-stage hepatic disease.
The end-stage liver obviously cannot perform its normal functions, so the clinical manifestations of hepatic failure invariably occur in affected animals. However, the cause of the hepatic damage that leads to the end-stage liver frequently cannot be determined at the time signs of hepatic failure are observed.
Hepatic Failure
The liver has considerable functional reserve and regenerative capacity. In healthy animals, more than two-thirds of the hepatic parenchyma can be removed without significant impairment of hepatic function and normal hepatic mass can be regenerated in a matter of days. This process of tissue removal can be repeated several times, particularly in younger animals, and function is retained. In all species, clinical signs from hepatic derangement are similar, regardless of their cause. These are manifest, however, only when the liver’s considerable reserve and regenerative capacity are depleted or when biliary outflow is obstructed. Only lesions that affect the majority of the hepatic parenchyma are likely to produce the signs of hepatic failure because focal lesions rarely destroy sufficient parenchyma to deplete the liver’s reserve. The term hepatic failure implies loss of adequate hepatic function as a consequence of either acute or chronic hepatic damage; however, all hepatic functions are not usually lost at the same time. The potential consequences of hepatic dysfunction and failure include (1) hepatic encephalopathy; (2) disturbances of bile flow with a resultant hyperbilirubinemia (discussed previously); (3) a variety of metabolic disturbances; (4) vascular and hemodynamic alterations, such as shunting of portal blood into the systemic circulation bypassing the hepatocytes; (5) cutaneous manifestations, such as epidermal necrosis in dogs and photosensitization in herbivores; and (6) immunologic manifestations.
Hepatic Encephalopathy: Hepatic failure can result in a metabolic disorder of the central nervous system (CNS) termed hepatic encephalopathy (synonyms: hepatic coma or portosystemic encephalopathy). Neurologic manifestations range from depression and other behavioral changes to mania and convulsions. Affected horses may walk aimlessly. The central feature of this disorder is abnormal neurotransmission in the CNS and the neuromuscular system. Undetermined as of yet are the specific metabolites that cause the neurologic dysfunction, but increased concentrations of plasma ammonia derived from amines absorbed from the gastrointestinal tract may be responsible. Normally, amines are absorbed from the intestines into the portal blood and metabolized by the liver. If they bypass the liver and gain access to the systemic circulation, they can exert toxic effects on the brain. These toxic products can enter the systemic circulation by two mechanisms. Blood can be shunted to the systemic circulation before it reaches the liver as a result of congenital portosystemic shunts or secondary to portal vein hypertension. Shunting of more than 10% to 15% of portal flow away from the liver is considered abnormal. Alternatively the toxic products may not be fully eliminated by the liver if there is sufficient liver disease. However, abnormal ammonia concentrations are not the only possible cause of hepatic encephalopathy. An altered balance of inhibitory and excitatory amino acid neurotransmitters, γ-aminobutyric acid (GABA) and l-glutamate, respectively, and increased brain concentrations of endogenous benzodiazepines are other possible explanations. Hepatic encephalopathy is common in ruminants and horses with hepatic failure, dogs and cats with congenital portosystemic shunts, and animals with end-stage liver (hepatic fibrosis and nodular regeneration) that leads to shunting of blood within regenerative nodules.
Metabolic Disturbances of Hepatic Failure: Hepatic failure can be manifested by a variety of metabolic disturbances. The type and duration of the hepatic disorder may influence the nature of the metabolic perturbation.
Bleeding Tendencies: Bleeding tendencies (hemorrhagic diathesis) sometimes accompany hepatic failure. Impaired synthesis of clotting factors, reduced clearance of the products of the clotting process, and metabolic abnormalities affecting platelet function can affect normal clotting, individually or in combination. All clotting factors, with the possible exception of factor VIII, are synthesized in the liver. In acute liver failure, diminished synthesis of clotting factors with a short half-life, such as factors V, VII, IX, and X, impairs the ability of blood to coagulate. In chronic liver disease, factor II (prothrombin) deficiency also contributes to diminished coagulation of blood. Diminished clearance of fibrin degradation products (FDPs), activated coagulation factors, and plasminogen factors by the damaged liver also perturbs clotting. Metabolic disturbances resulting from liver failure can affect platelet function and lead to synthesis of abnormal fibrinogen, a condition termed dysfibrinogenemia. Obstruction of the biliary system prevents the release of bile into the intestinal tract. The resulting impaired fat absorption limits vitamin K uptake from the intestine, which leads to an inactivity of factors II, VII, IX, and X. Acute hepatic failure may also precipitate disseminated intravascular coagulation (DIC), which can itself cause hemorrhagic diathesis. Impaired removal of activated clotting factors by the damaged liver is one potential mechanism for this. Acute hepatic failure in the horse and perhaps other species is sometimes accompanied by severe intravascular hemolysis, the cause of which is unknown.
Hypoalbuminemia: Hypoalbuminemia, as a consequence of hepatic dysfunction, usually reflects severe and chronic liver disease rather than acute disease. This is because of the relatively long half-life of plasma albumin (which ranges from 8 days in the dog to 21 days in cattle), which masks for a period of time the diminished albumin synthesis of the diseased liver. Severe diffuse liver disease can obstruct portal vein inflow, leading to portal hypertension, which accelerates the formation of ascites in affected animals. Loss of albumin in ascitic fluid or into the intestinal tract accentuates the loss of intravascular albumin, and widespread edema can result.
Vascular and Hemodynamic Alterations of Hepatic Failure: Chronic hepatic injury typically is accompanied by extensive diffuse fibrosis of the liver, which increases resistance to blood flow through the liver. This in turn elevates pressure within the portal vein (portal hypertension). With time, collateral vascular channels open to allow blood in the portal vein to bypass the abnormal liver (acquired portosystemic vascular anastomoses, which connect the portal vein and its tributaries to the systemic venous circulation). Shunting of portal vein blood directly to the central vein can also occur within the fibrous septa formed in the liver. In addition, the increased pressure within the hepatic vasculature causes transudation of fluid (modified transudate) into the peritoneal cavity to produce ascites in several species, except in most cases horses. Transudation of fluid into the peritoneal cavity can be enhanced by hypoalbuminemia because there is a decreased colloid osmotic pressure in plasma. Hypoalbuminemia and reduced plasma colloid osmotic pressure can arise as a consequence of accelerated albumin loss into the lumen of the intestines as a result of portal hypertension or because of reduced hepatic synthesis of albumin and other plasma proteins by the diseased liver. Ascites associated with hepatic fibrosis in chronic liver disease (end-stage liver) or other causes of portal hypertension, such as right-sided heart failure, occurs most commonly in the dog and cat, occasionally in sheep, and rarely in horses and cattle.
Cutaneous Manifestations of Hepatic Failure:
Hepatocutaneous Syndrome (Necrolytic Migratory Erythema, Superficial Necrolytic Dermatitis): Hepatocutaneous syndrome is a syndrome of chronic hepatic injury and skin disease. The central diagnostic elements include crusting, erosions, and ulceration of the epidermis of the muzzle, mucocutaneous areas of the face, footpads, and pressure points of the skin in some dogs with severe hepatic disease (Web Fig. 8-4). The mechanism of cutaneous injury is not understood, but affected animals have characteristic multinodular livers, often with little fibrosis between the nodules. It seems likely that this histologically distinctive skin disorder results from abnormal hepatic metabolism, as the skin lesions of affected dogs may respond to infusions of amino acids. Typically, the cutaneous lesions are parakeratosis, edema of the superficial epithelium, and basal cell hyperplasia (see Chapter 17).
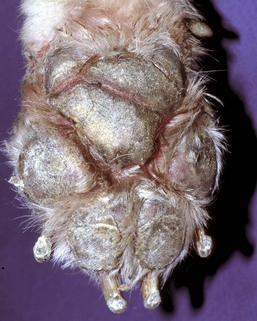
Web Fig. 8-4 Cutaneous necrosis, hepatocutaneous syndrome, foot pads, dog.
In some cases of hepatic failure, there is a syndrome of associated cutaneous necrosis termed hepatocutaneous syndrome. Footpads, as well as areas of haired skin, can be affected. (Courtesy Dr. T. Olivry, College of Veterinary Medicine, North Carolina State University.)
Photosensitization: Injury to the skin resulting from activation of photodynamic pigments by ultraviolet light in the sun’s rays is called photosensitization. Cutaneous lesions display hair loss, erythema, and necrosis (Web Fig. 8-5). Lesions are typically limited to hairless skin and to lightly or nonpigmented areas of skin. The sources of photodynamic pigments that can induce photosensitization include plants, certain drugs, and products of inherited disorders of porphyrin metabolism. The mechanism of tissue injury is not well characterized but is presumed to involve oxidative injury when photodynamic pigments that have been activated by the radiant energy of ultraviolet light subsequently release their energy to adjacent tissue. Hepatic dysfunction is only responsible for one form of photosensitization termed secondary (hepatogenous) photosensitization.
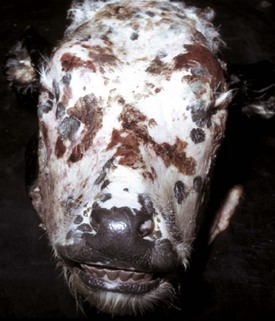
Web Fig. 8-5 Cutaneous necrosis, photosensitization, skin, cow.
When the liver of herbivores fails and can no longer remove the photodynamic pigment phylloerythrin from the portal blood, skin with little pigment or hair covering, exposed to ultraviolet light, initially becomes acutely inflamed and may be followed by necrosis, as seen on the face of this cow. (Courtesy Dr. H. Gelberg, College of Veterinary Medicine, Oregon State University.)
Primary photosensitization: Primary photosensitization occurs after a primary (preformed) photodynamic agent is ingested, absorbed into the blood, and deposited in tissue. Certain plants, such as St. John’s wort (Hypericum perforatum) and buckwheat (Fagopyrum esculentum), and pharmaceutical agents, such as tetracycline or phenothiazine, contain compounds that are photodynamic.
Secondary photosensitization: Secondary or hepatogenous photosensitization occurs in herbivores when hepatic dysfunction or biliary obstruction impairs normal excretion of phylloerythrin in bile. Phylloerythrin, a photodynamic agent, is produced by chlorophyll contained in ingested plants by gastrointestinal bacteria of herbivores. Phylloerythrin is normally absorbed from the intestines, taken up by hepatocytes, and excreted in bile, using the same pathway as bilirubin. Thus hepatocellular dysfunction or biliary obstruction prevents normal excretion and allows high concentrations of phylloerythrin to accumulate in blood and cutaneous tissue. Most often, secondary photosensitization occurs in animals with chronic liver disease and loss of 80% or more of normal hepatic function, but it can also occur in animals with acute liver disease or inflammatory or obstructive disorders of the biliary tree. Mutant Corriedale sheep have an inherited inability to excrete conjugated bilirubin and are also susceptible to secondary photosensitization because phylloerythrin concentrations are increased in these sheep.
Congenital porphyria: Congenital porphyria is a metabolic disorder involving the liver that occurs in several species, including cattle and cats. This disorder is caused by abnormal metabolism of heme, leading to abnormal excretion and an accumulation of porphyrins, which are themselves photodynamic.
Immunologic Manifestations of Hepatic Failure: Chronic liver failure leads to an impairment of normal hepatic immune function. As a consequence, the affected patient frequently develops endotoxemia and an increased risk of systemic infection. For the most part, this impairment is manifest as a reduction of blood filtration by Kupffer cells, which is primarily a result of shunting of portal blood rather than reduced phagocytic activity of the Kupffer cells.
Portals of Entry
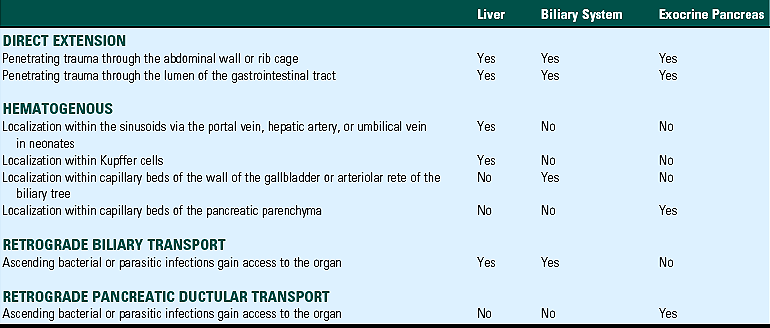
The liver and biliary systems are exposed to infectious or otherwise injurious substances via three main routes: hematogenous, biliary, and direct penetration (Table 8-1). The liver receives the entire flow of the portal vein and as a consequence is bathed in potentially injurious microbes, which inhabit and penetrate the digestive system, and toxic substances that have been ingested or produced by the intestinal flora. The distribution of blood from the portal vein to the different lobes of the liver is most likely not uniform. So-called portal streaming refers to the differential flow of portal blood from one segment of the digestive tract to particular lobes of the liver. This explains why some lobes of the liver are more severely affected by toxins that are absorbed by the small intestine rather than the large intestine. Examples of this include the preponderance of injury to the left liver lobe of sheep that ingest the mycotoxin sporidesmin. Systemic infections or intoxications can also affect the liver through the blood from the hepatic artery. Neonates and fetal animals are also at risk from infections ascending the umbilical vein. Infectious agents, such as enteric bacteria and parasites, can also gain access to the liver through the biliary tree that is in direct connection with the duodenum and enteric bacteria. Finally, direct penetration of the body cavity or from the digestive tract (i.e., traumatic reticuloperitonitis or foreign bodies in the reticulum) can deliver infectious or traumatic insults.
Defense Mechanisms
The liver is well defended from blood-borne injury by the Kupffer cells, the fixed macrophages that are distributed intermittently throughout the lumen of the sinusoids on the surface of endothelial cells (Table 8-2). They actively ingest and degrade bacteria and other organisms; senescent cells, such as erythrocytes; and particulate matter in the sinusoidal blood. They are very efficient and able to clear virtually all particulate matter in a single pass through the liver. Kupffer cells are particularly important in the removal of endotoxin from the portal blood.
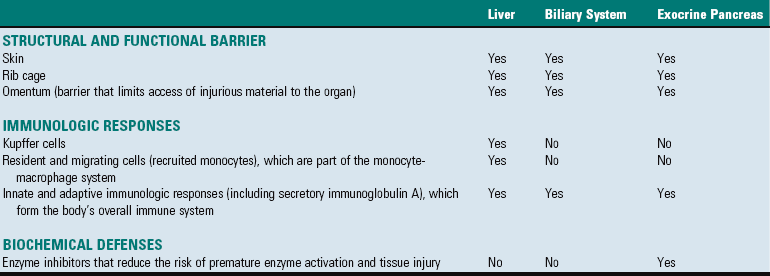
The biliary tree, like the upper gastrointestinal tract, is defended from infection by secreted IgA as part of mucosal immunity (see Table 8-2). The majority of biliary IgA is synthesized by gastrointestinal plasma cells. The majority of released IgA is taken into lymph and from there it enters the bloodstream. Hepatocytes in many species can transport IgA from blood across their cell membranes via a secretory component–mediated endocytosis. Subsequently, IgA molecules reach the bile by secretion into canaliculi. Concentrations of bile IgA are maintained by enterohepatic circulation. Within the biliary tree, IgA provides defense from infectious agents and clearance of harmful antigens as antibody-antigen complexes. In addition, the biliary tree is protected by the sphincter at the terminal end of the common bile duct, which provides a physical barrier to the ascent of enteric bacteria and the continuous flow of bile that assists in flushing bacteria out of the ducts.
The liver is protected from direct penetration by its anatomic location within the protection of the rib cage. The wall of the digestive tract also provides a certain degree of protection from penetration by ingested foreign bodies.
The high metabolic rate of hepatocytes renders them highly susceptible to metabolic disturbances that lead to cellular degeneration and necrosis (Box 8-2). This section will consider the patterns of hepatic degeneration, responses of the liver to injury, and inflammation of the liver.
Gallbladder and Extrahepatic Bile Ducts
The structure of the gallbladder and major ducts of the biliary system is similar in all species (the gallbladder is absent in the horse, the rat, and the elephant). It consists of a muscular wall (tunica muscularis) and a mucosa lined by simple columnar epithelium. The epithelium and muscularis are separated only by the lamina propria because the gallbladder lacks a muscularis mucosa. Right and left hepatic ducts carry bile from different lobes of the liver; these ducts and the cystic duct from the gallbladder unite to form the common bile duct. Location of the opening of the common bile duct into the intestine differs somewhat among the domestic species; it is as little as 2 cm from the pylorus in the pig to as much as 70 cm from the pylorus in the cow. The gallbladder stores and concentrates bile. Considerable concentration (twentyfold to thirtyfold) of bile by active transport of sodium and anions across the gallbladder epithelial cells occurs in dogs and cats, whereas little concentration occurs in pigs and ruminants. The horse lacks a gallbladder and continuously releases bile into the duodenum.
Animals that have not eaten in 24 to 48 hours or that are starving or cachectic can have notably enlarged gallbladders (Fig. 8-23). The reason is that these animals lack the paracrine stimulus (i.e., cholecystokinin), and a number of other neural and hormonal stimuli, to contract the gallbladder and relax the sphincter at the termination of the common bile duct, which regulates flow of bile into the duodenum.
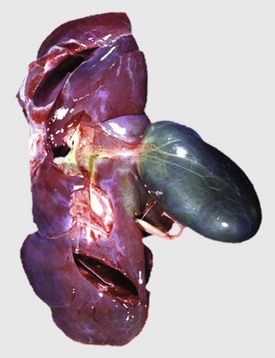
Fig. 8-23 Distended gallbladder, calf.
Note the distended gallbladder, which is common in all species after a prolonged period of fasting, because there is no stimulus for emptying the gallbladder. Thus they are frequently seen at necropsy of sick animals. (Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
Exocrine Pancreas
Development
The embryonic origin of the pancreas begins as a dorsal and a ventral bud of the duodenum. These buds fuse during embryogenesis to give rise to the entire pancreas. The major pancreatic duct arises from fusion of the ventral duct and the distal portion of the dorsal duct.
Macroscopic and Microscopic Structure
The pancreas is a lobulated, pink to gray, tubuloalveolar gland, a large portion of which is located in the mesentery immediately adjacent to the duodenum. The blood vessels, nerves, and lymph vessels that serve the pancreas are located within the delicate connective tissue septa that separate the lobules of pancreatic tissue. The pancreas contains both endocrine and exocrine elements, the endocrine portion being the islets of Langerhans. The exocrine portion constitutes the majority of the pancreas, up to 80% to 85% of the organ, and consists of acini composed of columnar to triangular secretory cells. The acinar cells have basally oriented nuclei and cytoplasm with a deeply basophilic basal margin and eosinophilic, granular zymogen granules occupying the apical portion. When the cells are appropriately signaled, the zymogen granules, containing the digestive enzymes of the pancreas, are released into the lumen of the acinus. The ductal system, in which secretions of the exocrine pancreas are conveyed to the intestinal tract, commences as fine radicals within acini and progresses to intralobular and interlobular ducts. These small ducts eventually drain into the main pancreatic duct or ducts. The arrangement of the major pancreatic ducts and how they empty into the duodenum varies among the domestic species. It is particularly variable in the dog, in which at least five different anatomic arrangements are recognized. In the cat, the major pancreatic duct enters the duodenum in close proximity to the common bile duct, and this may predispose the cat to pancreatic injury.
Normal Function
The exocrine pancreas produces secretions that contribute to digestion. The secretions contain a variety of enzymes that break down dietary lipids (lipase and phospholipase), proteins (trypsin and chymotrypsin), and carbohydrates (amylase). The secretions also contain electrolytes, which maintain the pH of the intestinal contents within a range that is optimal for enzymatic activity. Pancreatic enzymes act on the products of gastric digestion after they enter the duodenum. These enzymes often are released into secretions as inactive precursors (proenzymes), which helps prevent degradation of the pancreas by its own digestive enzymes. These are activated within the intestine. In addition, inhibitors of pancreatic enzymes are present in the pancreatic tissue. Secretion is controlled by neural stimulation regulated by the vagus nerve and by humoral factors. Secretin is one of the more important hormones involved in pancreatic secretion, and it stimulates water and bicarbonate secretion by duct cells. It is produced by neuroendocrine cells within the duodenal epithelium. The efflux of acid from the stomach and the presence of fatty acids in the duodenum stimulate its release. Cholecystokinin, another important hormone, stimulates the release of digestive enzymes from the acinar cells. It is produced by neuroendocrine cells of the duodenum in response to the presence of fatty acids, peptides, and amino acids.
Responses to Injury
The pancreas possesses modest regenerative capacity after necrosis of pancreatic exocrine epithelial cells, although more robust regeneration after partial pancreatectomy has been shown. Acinar cells can undergo replication in cases of limited injury, and precursor cells arise from cells within or adjacent to ductal epithelium in more severe injury. After acute pancreatic injury, there is usually little evidence of regeneration of the exocrine pancreas if there has been sufficient tissue destruction, and fibrosis is the principal response. Proliferated ductules and atrophic exocrine pancreatic lobules are also found after significant pancreatic injury.
Defense Mechanisms
Defense mechanisms for the exocrine pancreas are listed in Table 8-2. The pancreas is protected by the continuous flow of secretions into the duodenum, which prevents reflux of duodenal contents. The normal secretion of the pancreas contains trypsin, chymotrypsin, elastase, aminopeptidases, lipase, phospholipases, amylase, and nucleases. Trypsin is a critical enzyme because it has a role in the activation of several of the other pancreatic enzymes. Several mechanisms exist to protect the healthy pancreas from the effects of digestive enzymes it produces. Before secretion, enzymes are isolated from the acinar cell cytoplasm in membrane-bound zymogen granules. Most enzymes, except amylase and lipase, are secreted as proenzymes to prevent pancreatic injury. In particular, trypsin activation is tightly controlled because of its central role in activation of other enzymes. Consequently, the proenzyme trypsinogen is not normally activated until it enters the lumen of the duodenum through duodenal enteropeptidase. In addition, the chance of inappropriate trypsin activation in acinar cells or ducts of the pancreas is reduced by secretion of protective trypsin inhibitors. In circumstances in which trypsin or other enzymes are inappropriately activated, there are several other defenses in place. Acinar cells have an innate resistance to several of the digestive enzymes. Intrapancreatic release of active enzymes triggers the release of other enzymes that degrade the offending digestive enzymes, and lysosomal enzymes can degrade the zymogen granules when the pancreatic secretion is disturbed, reducing the burden of potentially injurious enzymes.
Disorders of the Liver and Biliary System of Domestic Animals
Developmental Anomalies and Incidental Findings
Developmental anomalies of the liver occur in domestic animals, although most are of little consequence.
Congenital Biliary Cysts
Congenital biliary cysts are found within the livers of dogs, cats, and pigs, but presumably all domestic species can be affected. The cysts are usually an incidental finding and can be found in animals of any age. Grossly, the cysts can be single or multiple and are filled with clear fluid (Fig. 8-24). Histologically, cysts have a thin wall lined by a single layer of biliary epithelium. Congenital cysts must be distinguished from parasitic cysts, particularly cysticerci, because they also have a thin wall and are fluid-filled. The presence of the larval cestode within parasitic cysts assists in distinguishing the two structures.
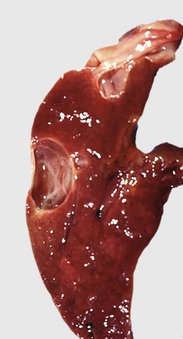
Fig. 8-24 Biliary cysts, liver, pig.
Multiple biliary cysts within the liver of a pig. Unilocular cysts replace a portion of the parenchyma in the affected portion of the liver. (Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
Multiple cysts that affect extensive areas of the liver occur in cats and occasionally dogs and are thought to represent developmental anomalies of the intrahepatic bile ducts. There are several types of these anomalies, which are collectively termed ductal plate malformations. Congenital polycystic disease, characterized by numerous epithelial-lined cysts in the liver, kidneys, and occasionally the pancreas, occurs in dogs, with Cairn terriers and West Highland white terriers predisposed; cats, with Persian cats believed to have a higher risk for the disorder; and goats and lambs. Affected animals can die of either liver or renal failure.
Hepatic Displacement
Displacement of the liver into the thoracic cavity, called a diaphragmatic hernia, can occur when there is a defect in the diaphragm. A congenital malformation that leaves an opening in the diaphragm or a traumatic event that ruptures the diaphragm can cause this condition.
Tension Lipidosis (Steatosis)
Discrete, pale areas of parenchyma at the liver margins are common in cattle and horses (Fig. 8-25). These foci typically occur adjacent to the insertion of a ligament (serosal) attachment, and it is proposed that these attachments impede blood supply to the subjacent hepatic parenchyma by exerting tension on the capsule. Affected hepatocytes most probably accumulate fat within their cytoplasm (steatosis) as a consequence of hypoxia. The lesions are of no functional significance.
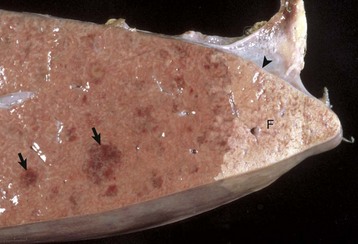
Capsular Fibrosis
Discrete fibrous tags or plaques are frequently present on the diaphragmatic surface of the liver and on the adjacent diaphragm of the horse (Fig. 8-26). Larval nematode migration tracts were originally believed to be the cause of this lesion, but this pathogenesis seems less likely in view of the widespread use of anti-parasitic treatments and the continued evidence of the lesion. Resolution of nonseptic peritonitis, possibly as a result of contact between the diaphragm and the adjacent liver capsule, has now been proposed as the cause of these regions of capsular fibrosis.
Postmortem Change
Autolysis of the liver occurs rapidly and can be advanced before it is obvious in most other tissue. At or just before death, bacteria are released from the gastrointestinal tract into the portal circulation and reach the liver where they proliferate rapidly after death. This process is especially rapid in large animals during hot weather—particularly cattle, in which fermentation in the adjacent rumen produces heat—and in pigs, which are often well insulated by fat. Pale areas appear on the capsular surface as bacterial degradation begins. In time, the organ becomes green-blue as bacteria degrade blood pigments to iron sulfide. The liver in contact with the gallbladder is quickly discolored by bile pigment, which passes through the wall of the gallbladder. The consistency of the organ becomes puttylike and gas bubbles may form beneath the capsule and in the parenchyma from bacterial fermentation. Postmortem changes are covered in detail in Chapter 1.
Morphologic Classification of Hepatobiliary Disease
The nature and distribution of inflammatory lesions in the liver are usually dictated by the route of entry, the host inflammatory response, the nature of the infectious agent (e.g., virus, bacterium, or fungus), and any predilection they have for involvement with a particular cell type in the liver. The hematogenous route of infection tends to cause a random multifocal distribution of lesions. Infections, usually bacterial, that ascend the biliary tract are typically centered in the bile ducts. Severe infections may affect the entire portal tract and extend into the adjacent parenchyma. Penetrating wounds cause discrete areas of inflammation with or without necrosis that are evident on the capsule and extend into the hepatic parenchyma. Liver injury should be characterized by the pattern of involvement (multifocal random, zonal, or massive), type of inflammatory cells involved (neutrophils, lymphocytes, plasma cells, eosinophils, or macrophages), evidence of necrosis or fibrosis, severity of these processes, evidence of regeneration, and the presence of an etiologic agent or agents. The type of inflammatory response and the duration of the injury can help identify the infectious agents.
Acute Hepatitis
Inflammation of the liver parenchyma is termed hepatitis. Acute hepatitis is characterized by inflammation, hepatocellular necrosis, and apoptosis. The proportion and type of inflammatory cells involved varies considerably, depending on the cause of inflammation, the host response, and the stage or age of the lesion. Characterization of the type of inflammation usually requires microscopic evaluation. In many forms of acute hepatitis, particularly bacterial and protozoal infections, neutrophils accumulate in response to the usual chemotactic stimuli. Random foci of neutrophilic hepatitis, as a consequence of embolic localization of bacteria, are relatively common in all species. In neonates—especially calves, lambs, and foals—bacteria, such as Escherichia coli, usually seed the liver via the umbilical veins or less often the portal venous or hepatic arterial systems. Acute hepatitis produced by viral infections, such as herpesvirus infection in many species, is more frequently characterized by a random distribution of necrosis and apoptosis with minimal inflammation or infiltrations of lymphocytes.
Chronic Hepatitis
Chronic hepatitis results when there is continued inflammation as a result of persistence of an antigenic stimulus. In the absence of such a stimulus, inflammation rapidly resolves. Chronic hepatitis is characterized by fibrosis; accumulation of mononuclear inflammatory cells, including lymphocytes, macrophages, and plasma cells, and frequently, regeneration. Neutrophils often are present in chronic unresolved hepatic inflammation such as that which characterizes some forms of canine chronic hepatitis. The proportion and distribution of each of these elements varies with the inciting cause and host response. Local variation within the liver can also occur.
Different terms are used to distinguish separate types of chronic hepatitis. Granulomatous hepatitis may be obvious grossly if it produces discrete granulomas of sufficient size. These may be focal or diffuse. Chronic suppurative hepatitis is usually manifested as discrete or multiple abscesses. Focal lesions, such as abscesses or granulomas, often are sufficiently localized so that they do not alter hepatic function. In contrast, diffuse and severe chronic hepatitis, as seen in dogs, usually leads to loss of hepatic parenchyma and architectural distortion of the liver as a consequence of fibrosis and nodular parenchymal regeneration. This process can proceed to end-stage hepatic disease with hepatic failure and its associated constellation of clinical signs. The form of chronic hepatitis that has been referred to as chronic active hepatitis is discussed in a separate section next.
Nonspecific Reactive Hepatitis
Nonspecific reactive hepatitis is a diffuse process distributed throughout the liver in response to some systemic illness, most often the gastrointestinal tract, or is the residuum of prior liver inflammation. Typically, there is a mild inflammatory infiltrate in the portal tract and possibly the parenchyma without evidence of necrosis. In acute cases, there is a minimal-to-mild infiltrate of neutrophils within the connective tissue of the portal tracts that may vary in intensity. Mononuclear cells, primarily lymphocytes and plasma cells, predominate in more chronic manifestations. Pigmented macrophages containing hemosiderin, lipofuscin, or both may be scattered throughout the parenchyma. Mononuclear inflammatory cells may also be evident within the hepatic parenchyma and at the periphery of central veins. Kupffer cells are usually reactive (i.e., they appear somewhat swollen because of abundant cytoplasm and display prominent nuclei as well).
Cholangitis
Inflammation of the biliary ducts (either intrahepatic or extrahepatic) is termed cholangitis. There are several patterns of cholangitis. The inflammatory cell population and the degree of fibrosis will vary with the type and duration of injury. Specific forms of cholangitis are discussed below.
Neutrophilic Cholangitis: Neutrophilic (suppurative) cholangitis is the most common type of cholangitis. It is characterized by the presence of neutrophils within the lumen or the epithelium of the bile ducts (Fig. 8-27). Acute and chronic forms of this process can occur. Fibrosis and the addition of mononuclear inflammatory cells are characteristic of the chronic form. Rupture of affected bile ducts can lead to hepatic abscess formation. Most neutrophilic cholangitis is believed to be caused by ascending bacterial infections from the intestine. Bacterial culture from bile most often reveals infection with Escherichia coli, Enterococcus, Bacteroides, Streptococcus, or Clostridium.
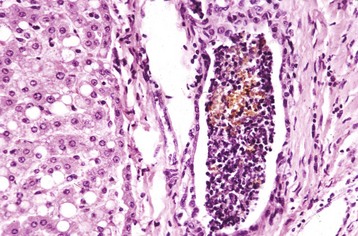
Fig. 8-27 Neutrophilic cholangitis (intrahepatic), liver, cat.
This condition is characterized by the presence of degenerate neutrophils within the lumen or the walls of bile ducts in the portal areas. Ascending bacterial infection from the intestine via the common bile duct is the most common cause. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Lymphocytic Cholangitis: Lymphocytic cholangitis occurs most often in cats and is described in detail in the section on Disorders of Cats.
Destructive Cholangitis: Destructive cholangitis is an uncommon syndrome characterized by necrosis of the epithelium of bile ducts (Fig. 8-28). Inflammation is often present around the areas of biliary destruction, but within the portal area. Certain chemicals, such as trimethoprim sulfa, have been implicated in this syndrome in dogs.
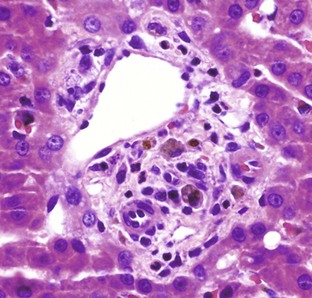
Fig. 8-28 Destructive cholangitis, liver, dog.
This condition is an uncommon disorder characterized by the destruction of bile duct epithelium, followed by regeneration in some instances. Necrotic or absent biliary epithelium, pigmented macrophages, and small numbers of mononuclear inflammatory cells in portal tracts are the typical histologic findings. H&E stain. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Cholangiohepatitis
Inflammation that affects the biliary ducts and hepatic parenchyma is termed cholangiohepatitis. In most cases of intrahepatic disease, the primary focus of inflammation can be identified as affecting either the hepatocytes or the biliary tree, but on occasion, both components of the liver are affected, usually as an extension of biliary disease, such as neutrophilic cholangitis, to involve the periportal hepatocytes; in that circumstance the term cholangiohepatitis should be used.
Circulatory Disorders
Passive Congestion (Acute and Chronic): Passive congestion of the liver can occur in any species and is almost always the consequence of cardiac dysfunction. Right-sided heart failure produces elevated pressure within the caudal vena cava that later involves the hepatic vein and its tributaries. Chronic passive congestion is particularly common in aged dogs and occurs secondary to endocardiosis (mucoid degeneration) of the right atrioventricular valve. Acute passive congestion, on the other hand, can occur as a consequence of acute right-sided heart failure, which has a wide variety of causes.
The appearance of the liver differs with the duration and severity of the congestion. Passive congestion initially causes distention of central veins and centrilobular sinusoids. Persistent centrilobular hypoxia leads to atrophy or loss of hepatocytes and eventually to centrilobular fibrosis. Fibrosis of the central vein (phlebosclerosis) may also occur.
Acute congestion of the liver produces slight enlargement of the organ, and blood flows freely from any cut surface. The intrinsic lobular pattern of the liver may be slightly more pronounced, particularly on the cut surface, because centrilobular areas are congested (dark red) in contrast to the more normal color of the remainder of the lobule.
Diffuse enlargement and rounded edges of liver lobes are the main features of chronic passive congestion (Fig. 8-29). Chronic passive congestion leads to persistent hypoxia in centrilobular areas and because of oxygen and nutrient deprivation, the centrilobular hepatocytes atrophy, degenerate, or eventually may undergo necrosis. As a result, sinusoids in these areas are dilated and congested and grossly appear red, whereas periportal hepatocytes frequently undergo steatosis (fatty degeneration) because of hypoxia, thereby causing this area of the lobule to appear yellow. The result is accentuation of the lobular pattern of the liver, referred to as an enhanced lobular or reticular pattern. It is especially evident on the cut surface of the liver, and the enhanced lobular pattern that occurs with severe chronic passive congestion has been likened to the appearance of the cut surface of a nutmeg and is termed nutmeg liver (Fig. 8-30). This pattern is not unique to passive congestion, however, and is encountered with other processes such as zonal hepatic necrosis. In addition to an enhanced lobular pattern, chronic passive congestion is characterized by focal fibrous thickening of the capsule and in severe cases, widespread hepatic fibrosis that bridges between central veins (Fig. 8-31).
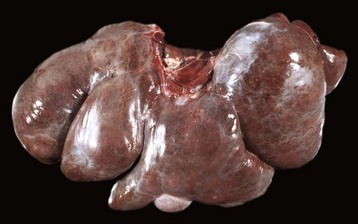
Fig. 8-29 Chronic passive congestion, liver, dog.
Chronic passive hepatic congestion in the liver of a dog with a heart base tumor that impeded venous return to the heart. The liver is enlarged with rounded margins. (Courtesy College of Veterinary Medicine, North Carolina State University.)
Fig. 8-30 Chronic passive congestion (nutmeg liver), liver, cut surface, cow.
The congestion in the centrilobular areas and the peripheral lipid accumulation give the liver a characteristic appearance that has been likened to that of the cut surface of a nutmeg, hence the term nutmeg liver. Inset, Cut surface of a nutmeg for comparison. (Figure courtesy Dr. D.A. Mosier, College of Veterinary Medicine, Kansas State University. Inset courtesy Dr. M.O. Howard, College of Veterinary Medicine, Iowa State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
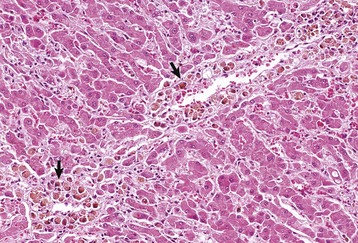
Fig. 8-31 Chronic passive congestion, liver, dog.
The liver is firm because of hepatic fibrosis that is most severe in centrilobular areas (arrows). The central vein is surrounded by a mild amount of connective tissue from which fine fibrous septa extend out into the lobule. Note the macrophages containing hemosiderin, the result of erythrocyte breakdown in this area as a result of chronic congestion. H&E stain. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Hepatic Veno-Occlusive Disease: Intimal thickening and occlusion of the central vein by fibrous connective tissue characterize the distinctive lesions of this syndrome. The consequence is passive hepatic congestion and resultant hepatic injury, which may progress to hepatic failure and its associated constellation of signs. The lesion is not etiologically specific but can follow pyrrolizidine alkaloid or aflatoxin-induced hepatic injury. An extremely high incidence is recognized in captive exotic cats, such as cheetahs, possibly because of the ingestion of large amounts of vitamin A, although the mechanism for this lesion is not known.
Disturbances of Blood Flow into the Liver
Anemia: The centrilobular (periacinar) regions of the lobule receive blood last; thus it is the least oxygenated, and the effects of hypoxia are usually manifested first in this area. Acute severe anemia, regardless of cause, can cause centrilobular or paracentral degeneration and even necrosis of hepatocytes. This typically occurs in severe anemias of precipitous onset. Chronic anemia can cause atrophy of centrilobular hepatocytes, which results in dilation and congestion of sinusoids (Fig. 8-32). Livers from animals with severe anemia, whether acute or chronic, typically have an enhanced lobular pattern that is evident on both the capsular and cut surfaces of the organ.
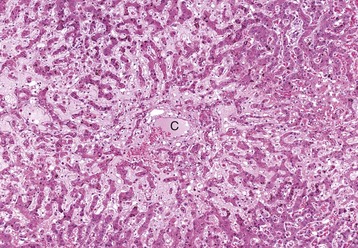
Fig. 8-32 Centrilobular hepatocyte atrophy, liver, dog.
In chronic anemia, centrilobular hepatocytes, which are the last hepatocytes in the lobule to receive oxygenated blood, become atrophic, and as a consequence, their sinusoids are more dilated. C, Central vein. H&E stain. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Congenital Portosystemic Shunts: A congenital portosystemic shunt is an abnormal vascular channel that allows blood within the portal venous system to bypass the liver and to drain into the systemic circulation. A congenital shunt can be either intrahepatic or extrahepatic in location but is usually limited to a single relatively large-caliber vessel (Fig. 8-33, A). A variety of different shunts have been described. Typically, intrahepatic portosystemic shunts involve failure of closure of the ductus venosus at birth. The ductus venosus is a normal fetal vessel that conducts blood from the portal vein to the caudal vena cava. Intrahepatic shunts, such as the patent ductus venosus, are most often located in the left side of the liver and occur most commonly in large breed dogs. Extrahepatic congenital shunts, such as portal vein to caudal vena cava anastomoses and portal vein to azygous vein anastomoses, occur more often in small breeds of dogs and cats. Shunts have been described in several species but occur most commonly in the dog and cat. Affected animals are typically stunted and frequently develop signs of hepatic encephalopathy. The liver is small and may have a characteristic histologic appearance of lobular atrophy, reduplication of arterioles, and small or absent portal veins within the portal tracts (Fig. 8-33, B). Studies have shown that the histologic appearance of the liver in animals with congenital portosystemic shunts is not a good prognostic indicator. The portal vein pressure is normal in congenital shunts and ascites does not occur. Abnormal vascular anastomoses are often difficult to identify without benefit of antemortem imaging studies. Affected dogs frequently have abnormal plasma ammonia concentrations and as a consequence, pass ammonium biurate crystals in their urine (Fig. 8-34). It should be noted that the liver has a stereotypic response to inadequate portal vein perfusion. Thus the histologic appearance of congenital portosystemic shunts and other vascular anomalies of the liver (discussed later) have considerable overlap. Clinical data, such as the presence or absence of shunt vessels and the determination of portal vein pressure, may be necessary to achieve a final diagnosis.
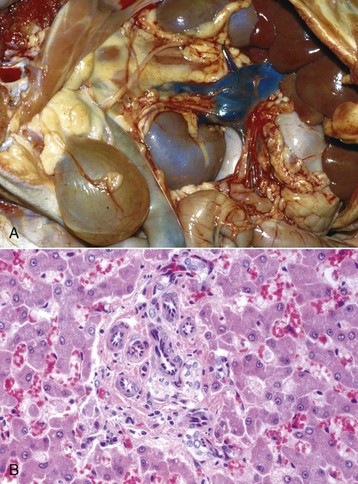
Fig. 8-33 Portosystemic shunts, liver, dog.
A, A single anomalous vessel that connects the portal circulation with the systemic circulation is the characteristic lesion of congenital portosystemic shunt. Note the small size but normal color of the liver (up under the rib cage). B, Congenital portosystemic shunt. Portal areas are abnormal because they lack a portal vein and contain numerous small-caliber arterioles. H&E stain. (A courtesy Dr. J. Sagartz, College of Veterinary Medicine, The Ohio State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
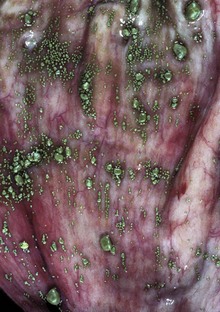
Portal Hypertension: Increased pressure within the portal vein can arise from disturbances of venous blood flow in any of the following three sites:
Prehepatic portal hypertension is relatively uncommon and occurs when blood flow through the portal vein is impaired before it enters the liver. Portal vein thrombosis can be induced by damage to the portal vein, by local inflammatory disorders, including pancreatitis, or by hypercoagulable states. Tumor emboli can also obstruct the portal vein. External compression by tumors or abscesses can restrict or obstruct portal vein flow as well. Portal vein hypoplasia (discussed later) affecting the extrahepatic segment of the portal vein is another cause.
Intrahepatic portal hypertension arises from increased resistance to blood flow to or within the sinusoids. Chronic liver disease that typically results in bridging collagenous septa, loss of normal lobular architecture, and regenerative nodule formation is the most common intrahepatic cause of portal hypertension. Sinusoidal fibrosis from diseases, such as lobular dissecting hepatitis, is another cause. Disorders, such as veno-occlusive disease, amyloidosis, and schistosomiasis, and other granulomatous disease processes can also produce intrahepatic portal hypertension. Arteriovenous fistulae (discussed later) within the hepatic parenchyma can also lead to intrahepatic portal hypertension.
Posthepatic causes of portal hypertension are uncommon and include any abnormalities that lead to increased resistance to venous outflow in the hepatic vein or adjacent vena cava. Partial or complete thrombosis of the hepatic veins (Budd-Chiari syndrome) or of the adjacent caudal vena cava are the most likely, although uncommon, causes of posthepatic portal hypertension. Congestive heart failure can also lead to portal hypertension.
Regardless of cause, persistent portal hypertension can lead to acquired portosystemic shunts with the exception of passive congestion, which rarely, if ever, results in the development of shunt vessels. These shunts are usually numerous and composed of distended thin-walled veins, which may connect the mesenteric veins and the caudal vena cava (Fig. 8-35). Ascites is common in conditions that develop acquired shunts because of the associated portal hypertension.
Fig. 8-35 Acquired portosystemic anastomoses, abdomen, dog.
Acquired portosystemic anastomoses secondary to portal hypertension (in this case as a consequence of chronic hepatitis in a dog). The numerous prominent veins that are present over the surface of the kidney allow blood within the portal venous system to bypass the liver and directly enter the systemic circulation. (Courtesy Dr. L. Hardy, College of Veterinary Medicine, North Carolina State University.)
Vascular Anomalies that May Produce Portal Hypertension:
Intrahepatic arteriovenous shunts (anastomoses): Arteriovenous shunts, either acquired or congenital, occur in the dog and cat and are direct communications between the hepatic artery and branches of the portal vein. They may occur anywhere within the liver. Affected portions of the liver contain convoluted thick-walled arteries and distended portal vein branches. Shunting of blood may lead to portal hypertension or reversal of the direction of portal blood flow, subsequent development of acquired portocaval shunts, and ascites. Clinical signs vary in intensity, most likely in relationship to the caliber of vessels that are affected, and are probably the result of the degree of portosystemic shunting of blood.
Portal vein hypoplasia (microvascular dysplasia, noncirrhotic portal hypertension): Portal vein hypoplasia is a congenital vascular anomaly that occurs in dogs and occasionally in cats. It is characterized by abnormally small extrahepatic or intrahepatic portal veins, which result in diminished hepatic perfusion by the portal vein blood flow and the potential for portal hypertension. Typically, affected animals have small livers and the typical histologic pattern of portal vein hypoperfusion: small or absent portal veins, proliferated hepatic arterioles (so-called reduplication), and hepatocyte atrophy. This disorder resembles portosystemic shunts histologically, but affected animals often have portal hypertension and resultant ascites. Portal fibrosis and biliary hyperplasia occur in about half of the cases. Because of the histologic similarities between portal vein hypoplasia and congenital portosystemic shunts, clinical data, such as imaging studies to determine the presence of a shunt vessel, are often required to make a final diagnosis from biopsy material.
Incidental Vascular Disorders
Telangiectasis: Telangiectasis is the notable dilation of sinusoids in areas where hepatocytes have been lost. Grossly, these areas appear as variably sized dark red-blue foci within the liver that vary from pinpoint to several centimeters in size (Fig. 8-36). Telangiectasis is particularly common in cattle and apparently is of no clinical significance. It also occurs in old cats, in which it can be mistaken for vascular tumors such as hemangioma or hemangiosarcoma. Histologically, there is ectasia of the sinusoidal space and a loss of hepatocytes. There is no evidence of inflammation or fibrosis associated with this lesion.
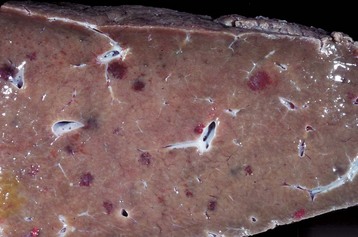
Fig. 8-36 Telangiectasia, liver, cut surface, cow.
Telangiectasia is a condition in which hepatic sinusoids become dilated and filled with blood. These lesions can be seen as dark red areas on the cut and capsular surface of the liver. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Infarction: Infarction of the liver occurs infrequently because of the organ’s dual blood supply from the hepatic artery and portal vein. Infarcts are usually sharply delineated and may be either dark red when acute or pale as they age. They tend to occur at the margins of the liver, the terminal end of parenchymal perfusion, and can affect small wedges of only a few centimeters in length or larger portions of a lobe. The cut surface of infarcted liver tends to be dry and granular. Histologically, infarcted liver is characterized by a zone of coagulative necrosis bordered by a basophilic band of inflammatory cells and an outer band of hyperemia (Fig. 8-37). Torsion of individual lobes of the liver, which occurs infrequently, can result in vascular occlusion and infarction of the affected lobe.
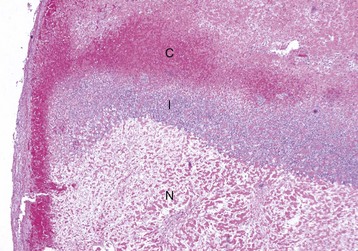
Fig. 8-37 Hepatic infarction, liver, dog.
Hepatic infarction is uncommon and usually occurs at the margins of the liver where the terminal divisions of the blood supply are found. Infarction of the liver is characterized by a zone of coagulative necrosis (N) rimmed with inflammatory cells (I) and an outer zone of congestion (C) in the viable liver. H&E stain. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Metabolic Disturbances and Hepatic Accumulations
Hepatocellular Steatosis (Lipidosis)
Lipids are normally transported to the liver from adipose tissue and the gastrointestinal tract in the form of either free fatty acids or chylomicrons, respectively. Within hepatocytes, free fatty acids are esterified to triglycerides, converted to cholesterol or phospholipids. Some oxidation of fatty acids to ketone bodies for energy production occurs within hepatocytes. Triglycerides can be complexed with apoproteins to form low-density lipoproteins, and these are released into the plasma as a readily available energy source for use by a variety of tissues. With the exception of ruminants, the liver also actively produces lipids from amino acids and glucose.
The presence of excessive lipid within the liver is termed lipidosis or steatosis (also known as fatty liver or fatty change) and occurs when the rate of triglyceride accumulation within hepatocytes exceeds either their rate of metabolic degradation or their release as lipoproteins. Hepatocellular steatosis is obviously not a specific disease entity but can occur as a sequel to a variety of perturbations of normal lipid metabolism. The potential mechanisms responsible for excessive accumulation of fat within the liver include the following:
1. Excessive entry of fatty acids into the liver, which occurs as a consequence of excessive dietary intake of fat or increased mobilization of triglycerides from adipose tissue because of increased demand (e.g., lactation, starvation, and endocrine abnormalities).
2. Excessive dietary intake of carbohydrates results in the synthesis of increased amounts of fatty acids with formation of excessive triglycerides within hepatocytes.
3. Abnormal hepatocyte function leads to accumulation of triglycerides within hepatocytes as a result of decreased energy for oxidation of fatty acids as in hypoxia or mitochondrial damage impairing oxidation of fatty acids.
4. Increased esterification of fatty acids to triglycerides in response to increased concentrations of glucose and insulin, which stimulate the rate of triglyceride synthesis from glucose or from prolonged increases in dietary chylomicrons.
5. Decreased apoprotein synthesis and subsequent decreased production and export of lipoprotein from hepatocytes.
6. Impaired secretion of lipoprotein from the liver because of secretory defects produced by hepatotoxins or drugs.
It must be stressed that these are potential mechanisms (some being more significant than others, depending on the condition of the animal) and that more than one defect might occur in any given hepatic disorder. Regardless of cause, the gross appearance of the hepatocellular steatosis is highly characteristic. With progressive accumulation of lipid, the liver enlarges and becomes yellow (Fig. 8-38, A). In mild cases, lipids may only accumulate in specific portions of each lobule, such as centrilobular regions, thereby imparting an enhanced lobular pattern to the liver. In extreme cases, the entire liver is affected, and the organ may become considerably enlarged and have an extremely greasy texture. Histologically, hepatocellular lipid appears as a clear round vacuole. The vacuoles are clear because lipid is removed in routine processing of tissue for histologic review. Macrovesicular steatosis is most common and is characterized by large vacuoles that displace the nucleus of the hepatocyte (Fig. 8-38, B). Microvesicular steatosis has multiple small, round, and clear vacuoles that do not displace the nucleus and can be associated with significant hepatocellular dysfunction. Frozen sections and special stains, such as Oil Red O, are employed to identify lipid.
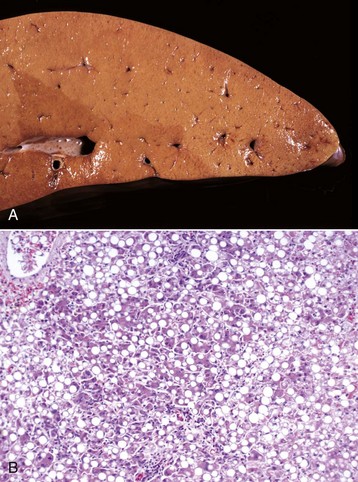
Fig. 8-38 Fatty liver syndrome (hepatic steatosis).
A, Cut surface, cow. The liver is swollen and yellow because of notable infiltration of lipid into hepatocytes. B, Steatosis or fatty degeneration, cat. Diffuse cytoplasmic accumulation of lipid is evident within the hepatocytes throughout the liver. H&E stain. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Specific causes and syndromes of hepatocellular steatosis in domestic animals include the following:
1. Dietary causes of hepatocellular steatosis include simple dietary excess in monogastric animals, such as a high-fat and/or high-cholesterol diet. Dietary deficiencies of cobalt and vitamin B12 have been implicated as causes of fatty liver in sheep and goats.
2. Toxic and anoxic causes of hepatocellular steatosis are common. Sublethal (reversible) injury to hepatocytes frequently results in accumulation of lipid within the affected cell.
3. Ketosis is a metabolic disease that results from impaired metabolism of carbohydrate and volatile fatty acids. In times of energy demand, free fatty acids are released from body fat stores, and the free fatty acids are esterified into fatty acyl CoA in the liver. Ketone bodies (acetoacetic acid and β-hydroxybutyric acid) are derived from fatty acyl CoA by oxidation in the mitochondria. In pregnant and lactating animals, there is a continuous demand for glucose and amino acids, and ketosis results when fat metabolism, which occurs in response to the increased energy demands, becomes excessive. Ketosis is characterized by increased concentrations of ketone bodies in blood (hyperketonemia), hypoglycemia, and low concentrations of hepatic glycogen. Ketosis is common in ruminants and usually occurs during peak lactation, whereas ketosis of sheep usually occurs in late gestation, particularly in ewes carrying twins; this latter disease is known as pregnancy toxemia.
4. Bovine fatty liver syndrome, also known as fatty liver disease, is mechanistically similar to ketosis and is especially common in ruminants with high energy demands. In dairy cattle, the disease is usually encountered in obese animals in late gestation, within a few days after parturition or peak lactation and is often precipitated by an event that causes anorexia such as retained placenta, metritis, mastitis, abomasal displacement, or parturient paresis. Typically, affected beef cattle are obese and the disease occurs within a few days before parturition. Accumulation of lipid within the liver is the result of both increased mobilization of lipids from adipose tissue, which results in increased influx of fatty acids to the liver, and in severe cases, defective hepatocytic function, which results in decreased export of lipoprotein from the liver.
5. Feline fatty liver syndrome is a distinct syndrome of idiopathic hepatocellular steatosis recognized in cats. Typically, affected cats are obese and anorectic and have no other diseases that could cause hepatocellular steatosis. Cats with this type of hepatocellular steatosis frequently develop hepatic failure, icterus, and subsequently hepatic encephalopathy.
6. Hepatocellular steatosis occurs in ponies, miniatures horses, and donkeys. Shetland ponies are predisposed. The condition usually occurs in overweight pregnant or lactating mares, characteristically after an event that causes stress or anorexia. In addition to notable hepatocellular steatosis, affected ponies are usually hyperlipemic and may also manifest signs of renal failure and hepatic rupture. In severe cases, hepatic encephalopathy and/or terminal DIC can occur.
7. Endocrine disorders, such as diabetes mellitus and hypothyroidism, can produce hepatocellular steatosis in a variety of species. In these cases, hepatocellular steatosis is obviously but one manifestation of abnormal metabolism. The accumulation of lipids in the liver in the diabetic animal is the result of increased fat mobilization and decreased use of lipids by injured hepatocytes.
Glycogen Accumulation
Glucose is normally stored within hepatocytes as glycogen and is often present in large amounts after feeding. Excessive hepatic accumulation of glycogen occurs with metabolic perturbations involving glucose regulation, including diabetes mellitus and the glycogen storage diseases. In these instances, hepatic involvement is just one manifestation of a systemic disease process. Excessive hepatic accumulation of glycogen is also observed in dogs secondary to excess glucocorticoids, and is explained further in the section on liver diseases in dogs.
Grossly, glycogen accumulation in the liver can lead to variable degrees of pallor and swelling. Microscopically, hepatocytes are swollen with lacy vacuolated cytoplasm. Unlike lipid vacuoles, glycogen vacuoles are poorly defined and hepatocyte nuclei are not displaced to the periphery of the cell.
Lysosomal Storage Diseases
Vacuolation of hepatocytes and Kupffer cells can be observed in several types of lysosomal storage diseases. This lesion is most often seen in addition to vacuolation of neurons, other macrophages and/or other cells. Affected animals are usually young and have an abnormal pattern of growth and development.
Amyloidosis
Hepatic amyloidosis occurs in most species of domestic animals. Amyloidosis is not a single disease entity but a term used for various diseases that lead to the deposition of proteins that are composed of β-pleated sheets of nonbranching fibrils. Affected livers are enlarged, friable, and pale (Fig. 8-39, A). Histologically, hepatic amyloid appears as bright eosinophilic amorphous deposits that are usually found in the space of Disse along the sinusoids but can be found in the portal tracts and within blood vessel walls (Fig. 8-39, B). Amyloid physical properties are responsible for its birefringence and characteristic apple green appearance in Congo red–stained sections viewed under polarized light. As many as 15 distinct amyloid proteins have been identified, but the hepatic amyloid is usually derived from one of three types. In primary amyloidosis, the amyloid fibril is designated AL (amyloid light chain) and is composed of immunoglobulin light chains derived from the amino terminal variable region of κ- and λ-light chains synthesized by plasma cell neoplasms. Secondary or reactive amyloidosis occurs as a consequence of prolonged inflammation such as chronic infection or tissue destruction. In secondary amyloidosis, by far the most common type to occur in veterinary medicine, the fibrils are composed of amyloid A (AA). The precursor protein is serum amyloid–associated (SAA) protein, an apolipoprotein that is an acute phase protein synthesized by the liver. Inherited or familiar amyloidosis is uncommon in animals but occurs in Shar-Pei dogs and Abyssinian, Siamese, and other Oriental breeds of cats.
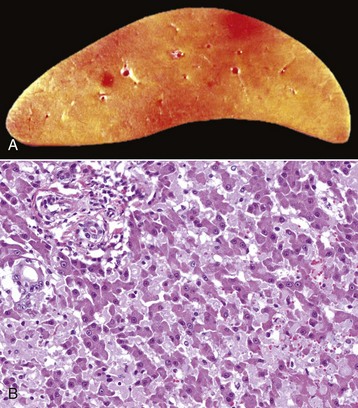
Fig. 8-39 Hepatic amyloidosis, liver.
A, Cut surface, duck. Hepatic amyloidosis has imparted a firm, waxy appearance and a pale brownish hue to the affected liver. B, Dog. The perisinusoidal spaces of Disse adjacent to the sinusoids are lined with a glassy eosinophilic (hyaline) material—amyloid. H&E stain. (A courtesy Drs. J. King and L. Roth, College of Veterinary Medicine, Cornell University. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Regardless of its cause, amyloid can accumulate in more than one pattern. It may accumulate in vessel walls in the portal area, within the connective tissue of the portal area, and in the space of Disse, where it impairs the normal access of plasma to hepatocytes. Amyloid deposits can produce varying degrees of hepatomegaly, and extensive accumulations cause the liver to appear pale. In severe cases, affected animals may have clinical signs of either hepatic dysfunction or failure because the liver is more fragile; liver rupture and exsanguination may occur, especially in the horse. Frequently, amyloid is also deposited within the kidneys, particularly the glomeruli. Renal failure often occurs before signs of hepatic dysfunction are manifested.
Copper Accumulation
Copper poisoning is included as a metabolic disorder because hepatic injury in copper poisoning of domestic animals frequently is the result of progressive accumulation of copper within the liver. This occurs in domestic animals, especially sheep, in which storage of copper is poorly regulated. Also, disorders of copper metabolism have been described in various dog breeds and rarely cats.
Copper is an essential trace element of all cells, but even a modest excess of copper can be life threatening because copper must be properly sequestered to prevent toxicosis. Normally, serum copper is bound to ceruloplasmin and the majority of hepatic copper is bound to metallothionein and stored in lysosomes. Excess copper, like excess iron, can lead to the production of reactive oxygen species that initiate destructive lipid peroxidation reactions that affect the mitochondria and other cellular membranes. In domestic animals, copper toxicosis usually occurs as a consequence of one of the following:
1. Simple dietary excess in ruminants, particularly sheep, and pigs occurring, for example, because of excessive dietary supplementation as an overcorrection for copper deficiency or from contamination of the pasture with copper from sprays or fertilizer. It also occurs in sheep that have access to copper-containing mineral blocks formulated for cattle.
2. Grazing animals on pastures with normal concentrations of copper but with inadequate concentrations of molybdenum, which antagonizes copper uptake.
3. Pasturing herbivores on fields with plants that contain hepatotoxic phytotoxins, usually pyrrolizidine alkaloids. Heliotropium, Crotalaria, and Senecio species are common examples of such plants (Web Table 8-1). Pyrrolizidine alkaloids prevent hepatocellular DNA synthesis and mitosis. This failure to replace necrotic hepatocytes leads to an ever-increasing copper load in surviving hepatocytes because these hepatocytes take up the copper released by the dying cells.
4. Metabolic disorders of copper metabolism, as occur in dogs. The disorder is best characterized in Bedlington terriers that have an autosomal recessive inheritance of a mutation in the COMMD1 gene that leads to impaired biliary excretion of copper, which results in progressive accumulation within the liver.
The consequences of excessive accumulation of copper within the liver of domestic animals are species-dependent. See specific species disease sections for more information.
Pigment Accumulation
Pigments are colored substances, some of which are normal cellular constituents, whereas others accumulate only in abnormal circumstances. They are covered in detail in Chapter 1.
Bile Pigments: Bile pigments may accumulate in excessive amounts as a consequence of either extrahepatic or intrahepatic cholestasis and typically produce icterus and green discoloration of the liver.
Hemosiderin: Hemosiderin is an iron-containing, golden-brown, granular pigment derived from ferritin, the initial iron-storage protein. As iron accumulates within the cell, aggregates of ferritin molecules form hemosiderin (Web Fig. 8-6). Most hemosiderin in Kupffer cells and other macrophages located in tissues throughout the body is derived from the breakdown of erythrocytes, whereas most hepatocellular hemosiderin is derived from iron present in transferrin and to a lesser extent hemoglobin. Hemosiderin forms in the liver when there is local or systemic excess of iron, such as when erythrocytic breakdown is excessive (e.g., hemolytic anemia), and within areas of hepatic necrosis. An excessive systemic load of iron that is characterized by abundant hemosiderin in a variety of tissues without impairment of organ function is called hemosiderosis. In contrast, hemochromatosis is an abnormally increased storage of iron within the body that can cause hepatic dysfunction. Notable accumulation of iron can produce a dark brown or even a black liver.
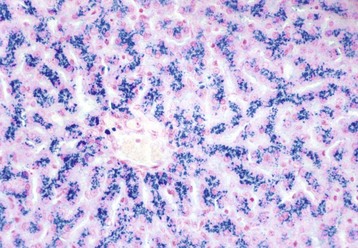
Web Fig. 8-6 Hemosiderosis, liver, dog.
When hemosiderin is identified with the Prussian blue reaction, the dark blue granules are indicative of iron stored as hemosiderin in hepatocytes. See Chapter 1 and Fig. 1-69 for more information about hemosiderin. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Lipofuscin: Lipofuscin is an insoluble pigment that is yellow-brown to dark brown and is derived from incomplete oxidation of lipids such as those in cell membranes (Web Fig. 8-7). Lipofuscin is progressively oxidized with time; thus it actually is a group of lipid pigments, all of which consist of polymers of lipid, phospholipids, protein (and minimal carbohydrate in early forms). Amounts of lipofuscin present in the liver tend to increase with age.
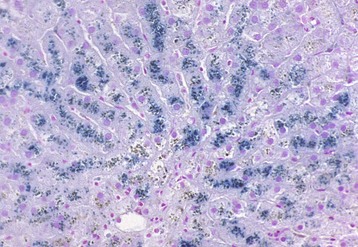
Web Fig. 8-7 Lipofuscinosis, liver, dog.
Lipofuscin can be detected by a variety of special stains. In these hepatocytes, lipofuscin granules in lysosomes are dark gray-blue to green. Schmorl’s stain. See Chapter 1 and Fig. 1-61 for more information about lipofuscin. (Courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Melanin: Melanin is an endogenous pigment that is dark brown or black. Benign disorders of melanin pigmentation are usually designated as melanosis. Congenital melanosis of the liver occurs in pigs and ruminants and produces variably sized areas of discoloration of the liver. Acquired “melanosis” of sheep has been described in Australia and is associated with the ingestion of certain plants, but the pigment has not been proved to be melanin and may be derived from a component of the ingested plants.
Parasite Hematin: Liver flukes specifically produce very dark excreta that contain a mixture of iron and porphyrin. These excreta produce the characteristic discoloration that occurs in fascioliasis (Fasciola hepatica) and is especially pronounced in the migratory tracts produced by Fascioloides magna in bovine livers (see Fig. 8-56).
Infectious Diseases of the Liver
Herpesvirus Infections: Herpesvirus infections of the liver typically occur in neonates or fetuses. A variety of abortigenic herpesviruses are described, each animal species being affected by a specific virus. Examples of these viruses include the abortigenic equine herpesvirus (equine herpesvirus 1), infectious bovine rhinotracheitis virus (bovine herpesvirus 1), caprine herpesvirus, canine herpesvirus (canine herpesvirus 1), feline viral rhinotracheitis virus (feline herpesvirus 1), and pseudorabies virus (suid herpesvirus 1).
Infection can occur via several routes, including transplacental exposure, passage through the birth canal, contact with infected littermates, and contact with oronasal secretions from the dam. In neonates, initial infection often occurs in the oronasal epithelium where virus replication first takes place. After local replication, the virus enters the bloodstream via infected mononuclear phagocytic cells. Viremia leads to dissemination of the virus to a variety of organs, and viral infection is cytolytic.
The abortigenic herpesviruses characteristically induce multifocal, randomly distributed, small (<1 mm) areas of necrosis in several fetal organs, including the liver (Fig. 8-40, A). Similar lesions occasionally are present in neonates infected with herpesviruses.
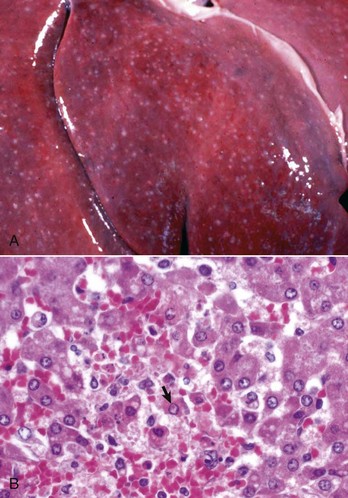
Fig. 8-40 Equine herpesvirus hepatitis, hepatic necrosis, liver, foal.
A, Note the randomly distributed gray-to-white foci of random hepatocellular necrosis caused by equine herpesvirus. B, Infection of hepatocytes with equine herpesvirus produces characteristic acidophilic intranuclear inclusions surrounded by a clear zone that separates them from the marginated chromatin (arrow). Note the individual cell necrosis. H&E stain. (A courtesy Drs. J. King and L. Roth, College of Veterinary Medicine, Cornell University. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
Histologically, herpesvirus can produce multifocal hepatic necrosis with scant inflammation in fetuses and neonates (Fig. 8-40, B). The liver is often affected, but foci of necrosis are more consistently present in the kidneys, lungs, and spleen. The virus most often affects neonates in the first 2 weeks of life and is most severe before they develop competent thermoregulation. Animals that are able to maintain a normal body temperature are less likely to be affected.
Other Viral Infections: Infectious canine hepatitis, Rift Valley fever, and Wesselsbron disease are discussed under species-specific disease sections later in the chapter.
Certain viral diseases may involve the liver, but the hepatic involvement either does not occur invariably or it may be but one manifestation of a systemic process. Such diseases include feline infectious peritonitis (FIP) that is characterized by foci of pyogranulomatous vasculitis or perivascular accumulations of lymphocytes and plasma cells within multiple organs, sometimes including the liver. Subacute and chronic forms of equine infectious anemia are characterized by cellular accumulations, particularly lymphocytes, in sinusoids and the space of Disse. Systemic adenoviral infection of lambs, calves, and goat kids may produce multifocal areas of hepatocellular necrosis, cholangitis, and necrosis of biliary epithelium. Porcine circovirus type 2 injures hepatocytes and Kupffer cells and can cause mild-to-severe necrosis.