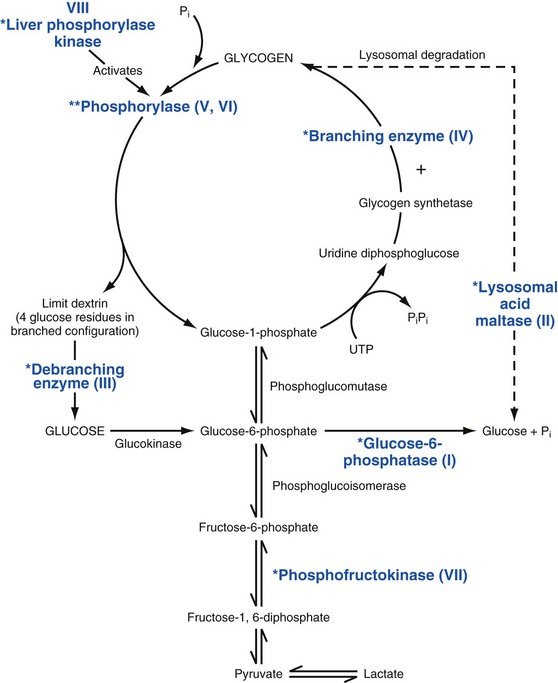
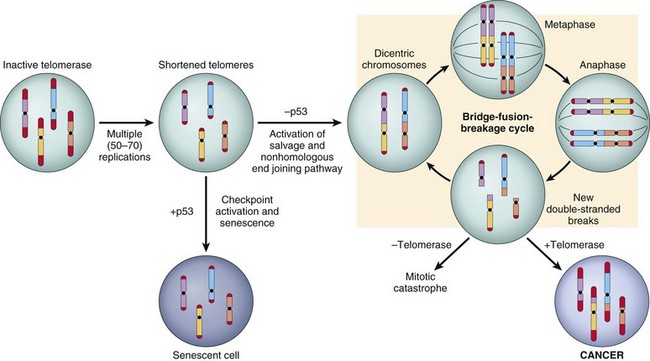
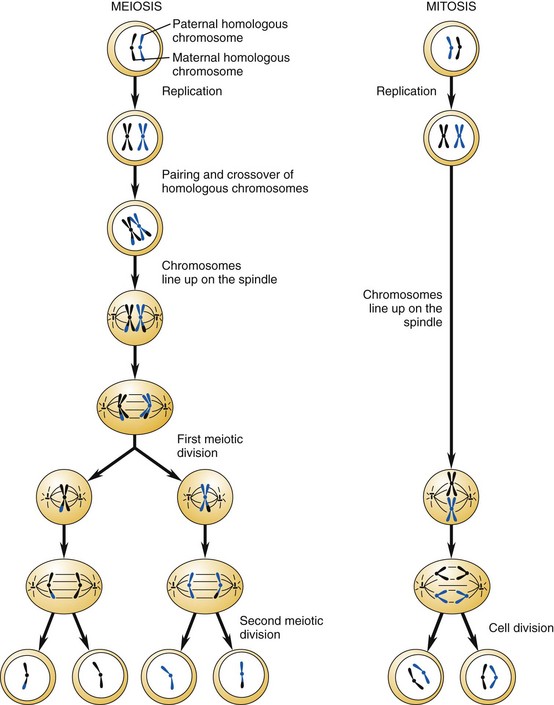
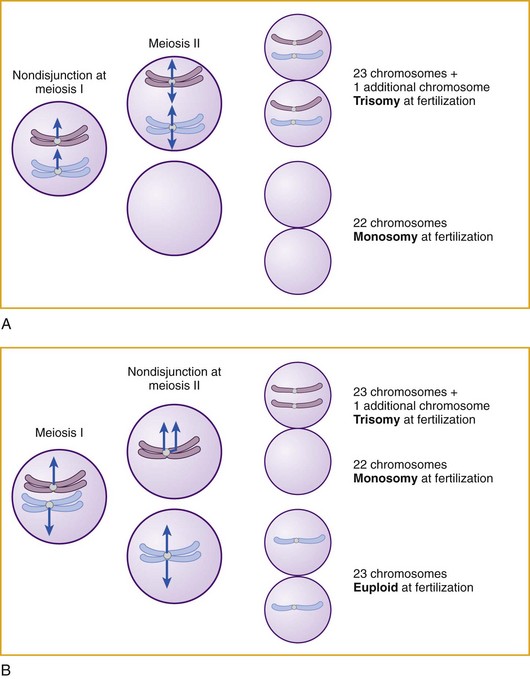
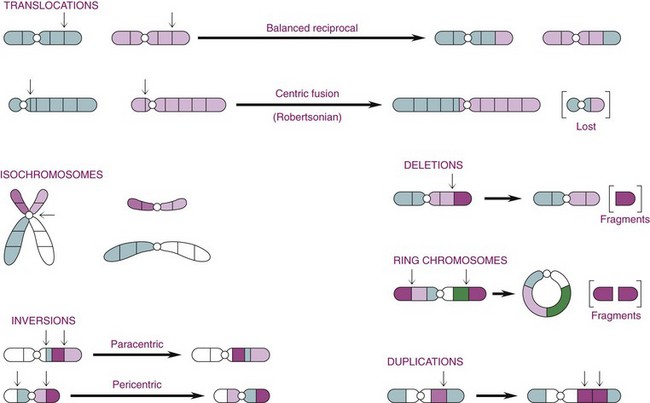
Other Extracellular Accumulations
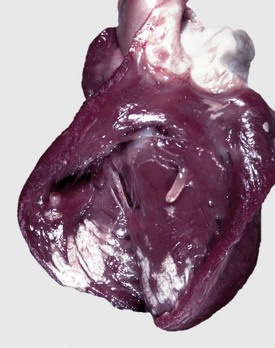
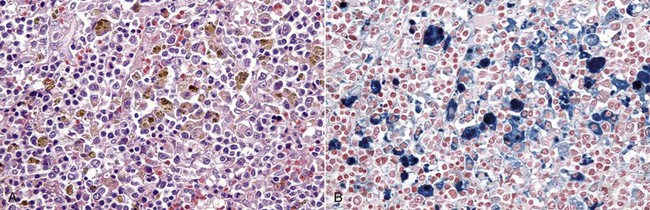
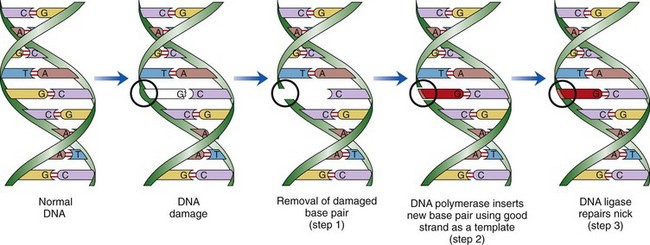
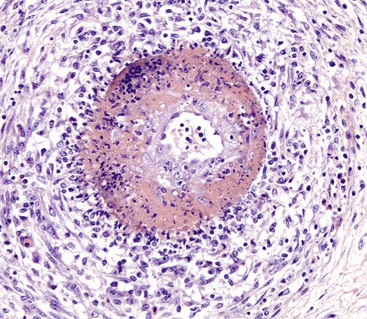
Fibrinoid Change: Fibrinoid change, also known as fibrinoid necrosis and fibrinoid degeneration, is a term applied to a pattern of lesions most often observed in the vascular system. The terms fibrinoid degeneration and fibrinoid necrosis are inappropriate because the process is not a true regressive alteration of cells. Rather, fibrinoid change is the result of the deposition of immunoglobulin, complement, and/or plasma proteins, including fibrin in the wall of a vessel. This lesion is caused by injury to the intima and media such as occurs in the immune-mediated vasculitides.
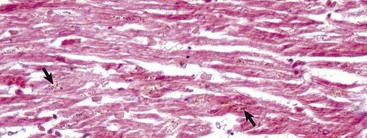
Grossly, fibrinoid change cannot be observed; however, it is often accompanied by thrombosis and hemorrhage, and when these two lesions are present in a vascular pattern of distribution, fibrinoid change of the vasculature should be considered. Microscopically, direct injury to endothelial cells, basement membrane, or myocytes, such as caused by viruses or toxins, or indirect injury such as caused by activation of complement proteins, can lead to activation of the acute inflammatory cascade and the deposition of plasma proteins in the vessel walls. These proteins, especially fibrin, stain intensely red (eosinophilic) with H&E stains and involve the vessel wall circumferentially to varying depths of the tunica intima and tunica media (Fig. 1-52). This lesion is also often accompanied by cellular and nuclear debris from injured vascular cells and inflammatory cells. These proteins contribute to the vascular “eosinophilia,” which has been described somewhat differently by different pathologists. There is general agreement that the material is eosinophilic, which is sometimes described as “smudgy” or “deeply eosinophilic.” Some pathologists add “homogeneous” and others “amorphous” to the descriptive terminology of fibrinoid change.
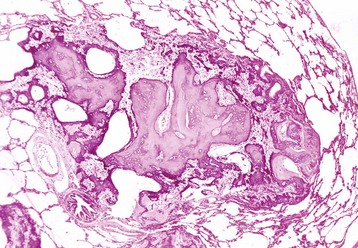
Gout: Gout is the deposition of sodium urate crystals or urates in tissue. It occurs in humans, birds, and reptiles but has not been reported in domestic mammals.
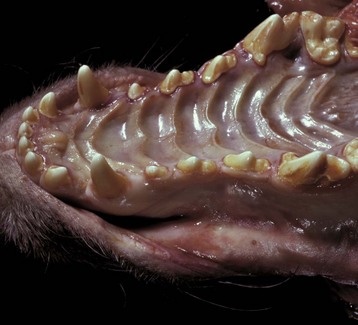
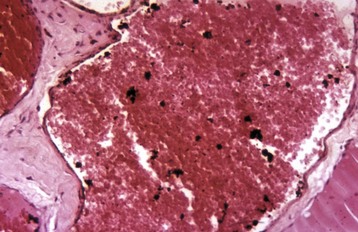
In the most common form in humans, urate crystals are deposited in the articular and periarticular tissues and elicit an acute inflammatory response characterized by the presence of neutrophils and macrophages and large aggregations of urate crystals called tophi. These tophi may be visible grossly and are pathognomonic of gout. Later in the course of the disease, the inflammation becomes chronic and a foreign body reaction to the tophi develops. Microscopically, urate crystals are acicular and birefringent and they, or the spaces left after they have been dissolved during the preparation of paraffin-embedded histologic sections, are visibly surrounded by numerous neutrophils, macrophages, and giant cells.
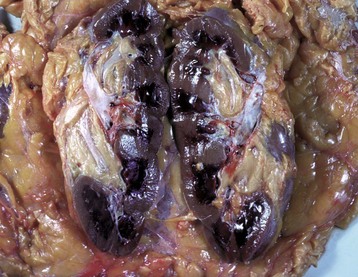
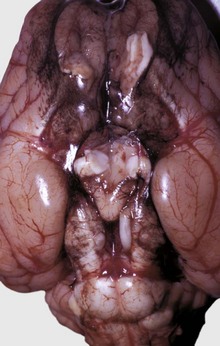
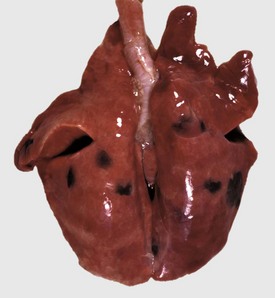
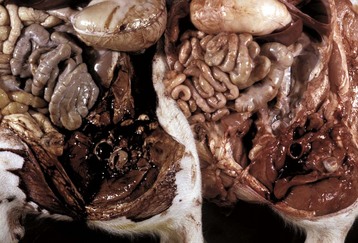
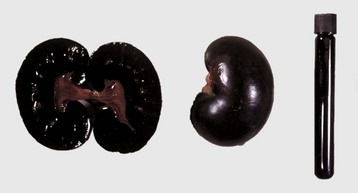
In birds and reptiles, there are two forms, namely (1) the articular type, which is rare, and (2) a visceral type. The latter characteristically affects the visceral serosae, particularly the parietal pericardium, and the kidneys. The serosa is covered with a thin layer of gray granules, and the gross appearance is diagnostic. In the renal form, urate deposits are visible in renal tubules and ureters. Uric acid and urates are the end-products of purine metabolism, and in birds and reptiles, these products are eliminated as semisolid urates. Visceral gout is usually diagnosed only at necropsy and is seen sporadically as the result of vitamin A deficiency, high-protein diets, and renal injury.
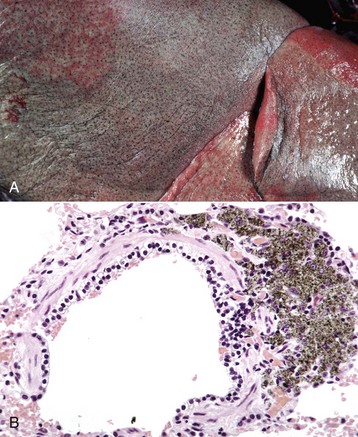
Pseudogout: Pseudogout is characterized by deposits of calcium pyrophosphate crystals. It is well recognized in humans but has been reported in the dog, in which it is rare. The pathogenesis of the canine disease is unknown, but in humans, one form is inherited as an autosomal dominant trait. Grossly, there are chalky white deposits in the joints, which histologically show a chronic reaction with aggregates of crystalline material, macrophages, and fibrosis. The disease may be differentiated from gout by the chemical analysis of the crystalline deposits.
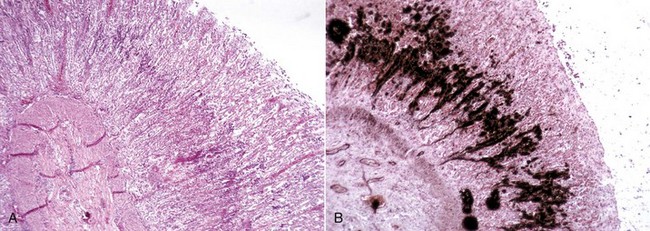
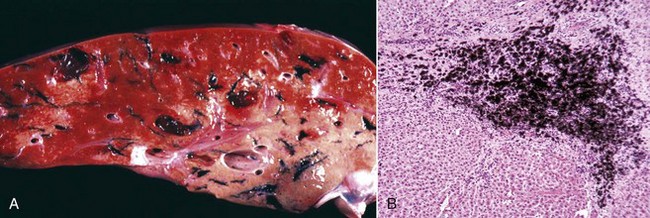
Cholesterol: Cholesterol crystals are the by-products of hemorrhage and necrosis. They are dissolved out of the tissue specimen during the preparation of paraffin-embedded sections, leaving characteristic clefts that, in tissue sections, resemble shards of glass. Actually, the crystals are thin rhomboidal plates with one corner notched out, their outline resembling that of the state of Utah. Cholesterol crystals in tissue have no significance except that they indicate the site of an old hemorrhage or tissue necrosis, and they may be present in atheromas (i.e., mass of degenerated, thickened arterial intima occurring in atherosclerosis). However, in the choroid plexus of the lateral ventricles of old horses, cholesterol crystals can induce a granulomatous response, and the resultant cholesterol granuloma or cholesteatoma can become so large as to obstruct the outflow of cerebrospinal fluid through the interventricular foramen (foramen of Munro), resulting in obstructive hydrocephalus. It is thought that these granulomas are secondary to cholesterol crystals from hemorrhages into the choroid plexus. Grossly, the cholesterol appears as firm, crumbly gray nodules in the cholesteatomas.