JUVENILE OSSIFYING FIBROMA (JUVENILE ACTIVE OSSIFYING FIBROMA; JUVENILE AGGRESSIVE OSSIFYING FIBROMA)
The juvenile ossifying fibroma is a controversial lesion that has been distinguished from the larger group of ossifying fibromas on the basis of the age of the patients, most common sites of involvement, and clinical behavior. Two different neoplasms have been reported under the term, and disagreement exists over the spectrum of what should be accepted as juvenile ossifying fibromas. Although the two forms demonstrate different histopathologic and clinical features, several investigators have chosen to compromise and accept two patterns of juvenile ossifying fibroma: (1) trabecular and (2) psammomatoid. Among lesions involving the craniofacial skeleton, the number of psammomatoid cases reported exceeds the number of trabecular cases reported by a ratio of approximately 4:1.
A recent study of three cases of the psammomatoid variant arising in the orbit of adolescent boys demonstrated the presence of nonrandom chromosomal breakpoints at Xq26 and 2q33 resulting in (X;2) translocation. Although no similar studies have been reported with respect to the trabecular variant, it is possible that future insights into the cytogenetic abnormalities of these two variants may aid in defining them as distinct entities.
CLINICAL AND RADIOGRAPHIC FEATURES: In most instances, the neoplasms often grow rapidly, are well-circumscribed, and lack continuity with the adjacent normal bone. The lesions are circumscribed radiolucencies that in some cases contain central radiopacities (Fig. 14-56). In some cases “ground glass” opacification may be observed. Those present within a sinus may appear radiodense and often create a clouding that may be confused with sinusitis.
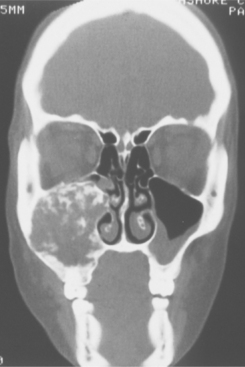
Fig. 14-56 Juvenile ossifying fibroma. Computed tomography (CT) scan showing a large tumor involving the left maxilla and maxillary sinus of a 12-year-old girl. Clinically, the tumor was growing rapidly.
The age at diagnosis varies, with reported cases occurring in patients from younger than 6 months to older than 70 years of age. Although both patterns reveal similar radiographic features and growth patterns, the trabecular form is diagnosed initially in younger patients. The mean age of trabecular juvenile ossifying fibromas is approximately 11 years, whereas the mean age of patients diagnosed with the psammomatoid variant approaches 22 years. Both forms exhibit a slight male predilection and occur in either jaw but reveal a maxillary predominance. Although many of these tumors are initially discovered on routine radiographic examination, cortical expansion may result in clinically detectable facial enlargement. The psammomatoid variant frequently appears outside the jaws, with more than 70% arising in the orbital and frontal bones and paranasal sinuses.
Complications secondary to the neoplasm are typically the result of impingement on neighboring structures. With persistent growth, lesions arising in the paranasal sinuses penetrate the orbital, nasal, and cranial cavities. Nasal obstruction, exophthalmos, or proptosis may be seen. Rarely, temporary or permanent blindness occurs.
Intracranial extension has been discovered in neoplasms arising adjacent to the cribriform plates. Because of the circumscribed growth pattern of the tumor, the frontal lobe is typically elevated without any associated neurologic signs. Rarely, intracranial extension has resulted in meningitis, with one report of a maxillary tumor leading to convulsions and death from pneumococcal meningitis.
In some cases of the psammomatoid variant, development of an aneurysmal bone cyst has been reported. Such cystic changes tend to occur in younger patients in the first and second decades of life and have been associated with large maxillary lesions exhibiting aggressive behavior.
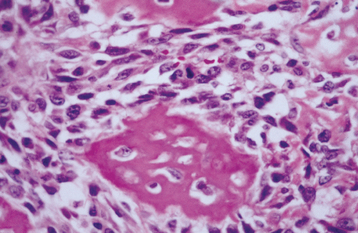
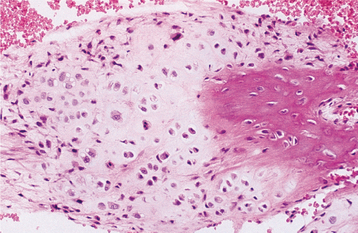
HISTOPATHOLOGIC FEATURES: Both patterns are typically nonencapsulated but well demarcated from the surrounding bone. The tumor consists of cellular fibrous connective tissue that exhibits areas that are loose and other zones that are so cellular that the cytoplasm of individual cells is hard to discern because of nuclear crowding. Myxomatous foci are not rare and often are associated with pseudocystic degeneration. Mitotic figures can be found but are not numerous. Areas of hemorrhage and small clusters of multinucleated giant cells are usually seen.
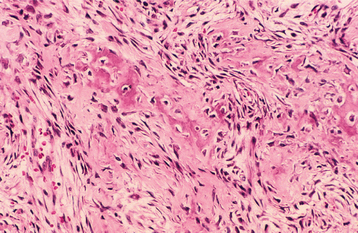
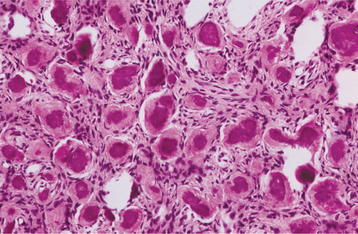
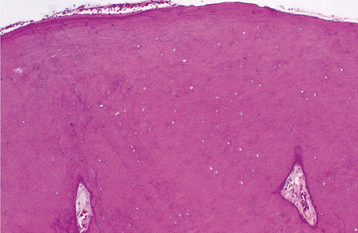
The mineralized component in the two patterns is very different. The trabecular variant shows irregular strands of highly cellular osteoid encasing plump and irregular osteocytes (Fig. 14-57). These strands often are lined by plump osteoblasts and in other areas by multinucleated osteoclasts. In contrast, the psammomatoid pattern forms concentric lamellated and spherical ossicles that vary in shape and typically have basophilic centers with peripheral eosinophilic osteoid rims (Fig. 14-58). A peripheral brush border blending into the surrounding stroma is noted in many of the ossicles. Occasionally, individual ossicles undergo remodeling and form crescentic shapes.
TREATMENT AND PROGNOSIS: The clinical management and prognosis of the juvenile ossifying fibroma are uncertain. Although many tumors demonstrate slow but progressive growth, some juvenile ossifying fibromas demonstrate rapid enlargement. The more aggressive neoplasms tend to arise in infants and young children.
For smaller lesions, complete local excision or thorough curettage appears adequate. For some rapidly growing lesions, wider resection may be required.
In contrast to the negligible recurrence rate seen in the common types of ossifying fibromas, recurrence rates of 30% to 58% have been reported for juvenile ossifying fibromas. Malignant transformation has not been documented.
OSTEOMA
Osteomas are benign tumors composed of mature compact or cancellous bone. Osteomas are essentially restricted to the craniofacial skeleton and rarely, if ever, are diagnosed in other bones. There is some question as to whether osteomas represent true neoplasms, and not all lesions designated as an osteoma may represent a single entity. Some likely repre-sent the end stage of an injury or inflammatory process or the end stage of a hamartomatous process, such as fibrous dysplasia. The common palatal and mandibular tori and buccal exostoses (see page 19) are not considered to represent osteomas, although they are histopathologically identical to osteomas. Be-cause many osteomas are small, asymptomatic lesions, there is little reliable information as to their true frequency.
CLINICAL AND RADIOGRAPHIC FEATURES: Osteomas of the jaws may arise on the surface of the bone, as a polypoid or sessile mass (periosteal, peripheral, or exophytic osteoma), or they may be located in the medullary bone (endosteal or central osteoma). Extraskeletal lesions of soft tissue, typically located within muscle or the dermis of the skin (osteoma cutis), also are possible. Most jaw osteomas are detected in young adults and are generally asymptomatic, solitary lesions. There is little valid information as to whether there is any gender predilection. The most common gnathic locations are the body of the mandible or the condyle. When located in the body, most osteomas occur posterior to the premolars on the lingual surface. Less common mandibular locations include the angle (particularly at the inferior border), coronoid process, and ramus.
Periosteal osteomas appear as slowly growing masses on the surface of the mandible or maxilla. Some types may reach a large size, resulting in facial deformity. Small endosteal osteomas are asymptomatic, but large lesions cause a slowly progressive enlargement of the affected area.
An osteoma involving the mandibular condyle may cause a slowly progressing shift in the patient’s occlusion, with deviation of the midline of the chin toward the unaffected side. Other reported signs and symptoms include facial swelling, pain, and limited mouth opening.
Condylar osteomas are considered by some to be a true neoplasm, whereas others designate them as hyperostoses. Distinguishing this process from condylar hyperplasia can be difficult; however, condylar osteomas are typically lobulated, whereas the condyle retains its original shape when hyperplastic.
Paranasal sinus lesions also are possible and are actually more common than gnathic lesions. The frontal sinus is most commonly involved, followed by the ethmoid and maxillary sinuses. Such lesions are usually asymptomatic, although pain, swelling, sinusitis, and nasal discharge are possible. In rare cases, paranasal sinus osteomas may expand into orbital structures and result in proptosis, diplopia, and decreased visual acuity. Infrequently, intracranial extension can lead to meningitis, cerebral abscesses, and intracranial mucoceles.
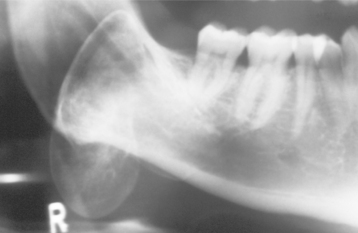
Radiographically, osteomas appear as circumscribed sclerotic masses. Periosteal osteomas may show a uniform sclerotic pattern or may demonstrate a sclerotic periphery with a central trabecular pattern (Fig. 14-59). Smaller endosteal osteomas are difficult, if not impossible, to differentiate from foci of sclerotic bone representing the end stage of an inflammatory process (condensing osteitis, focal chronic sclerosing osteomyelitis) or from noninflammatory foci of sclerotic bone (idiopathic osteosclerosis). The true nature of these osteomas can be confirmed only by documentation of continued growth.
HISTOPATHOLOGIC FEATURES: Compact osteomas are composed of normal-appearing dense bone showing minimal marrow tissue (Fig. 14-60). Cancellous osteomas are composed of trabeculae of bone and fibrofatty marrow. Osteoblastic activity may be fairly prominent.
TREATMENT AND PROGNOSIS: Larger osteomas of the mandibular body causing symptoms or cosmetic deformity are treated by conservative surgical excision. Small, asymptomatic osteomas, particularly those located endosteally, probably do not need to be treated but should be observed periodically. Because of the frequency of associated symptoms, osteomas arising in the condyle are usually removed surgically. Large lesions mandate condylectomy, whereas peripheral osteomas are treated by local resection. Paranasal sinus osteomas may not require removal unless they become large or symptomatic; small, periosteal lesions may be removed endoscopically, whereas larger lesions typically require an open surgical approach. Osteomas are completely benign, and patients do not experience malignant change. Recurrence after excision is extremely rare.
GARDNER SYNDROME
Gardner syndrome is a rare disorder that is inherited as an autosomal dominant trait with near 100% penetrance; approximately one third of cases occur spontaneously and appear to represent new gene mutations. The responsible gene has been mapped to the long arm of chromosome 5 and has been identified as the adenomatous polyposis coli (APC) tumor suppressor gene. Gardner syndrome is considered to be part of a spectrum of diseases characterized by familial colorectal polyposis. In addition to the colonic manifestations, other gastrointestinal abnormalities are seen along with a variety of findings that may involve the skin, soft tissues, retina, skeletal system, and teeth. The presence of extracolonic manifestations and the severity of gastrointestinal disease correlate with the specific position of mutations within the APC gene. However, the possibility that additional mutations in undiscovered modifier genes also may influence the severity and pattern of disease cannot be excluded. Several of the extracolonic manifestations are distinctive and have led to the discovery of the syndrome.
CLINICAL AND RADIOGRAPHIC FEATURES: The reported prevalence of Gardner syndrome varies from 1:8300 to 1:16,000 live births. The associated colonic polyps typically develop during the second decade; because these are adenomatous, they ultimately transform into adenocarcinoma. In addition, detection of extracolonic polyps is not rare in the small intestine or stomach, with a small percentage exhibiting carcinomatous transformation.
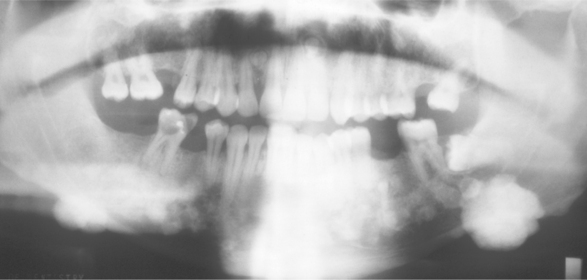
Up to 90% of patients with Gardner syndrome demonstrate skeletal abnormalities, the most common of which are osteomas. Although the osteomas may affect any part of the skeleton, the most commonly involved areas are the skull, paranasal sinuses, and the mandible. When gnathic lesions are seen, they often occur in the region of the mandibular angles and are frequently associated with prominent facial deformity (Fig. 14-61). The osteomas are usually noted during puberty and precede the development of, or any symptoms from, the bowel polyps (Fig. 14-62). Most patients demonstrate between three and six osseous lesions. The osteomas appear as areas of increased radiodensity that vary from slight thickenings to large masses. On occasion, large osteomas of the mandible or condyle will limit the mandibular opening. Dental abnormalities include an increased prevalence of odontomas, supernumerary teeth, and impacted teeth. Although up to 20% of affected patients demonstrate supernumerary teeth, the frequency of extra teeth is not nearly as high as that noted in cleidocranial dysplasia.
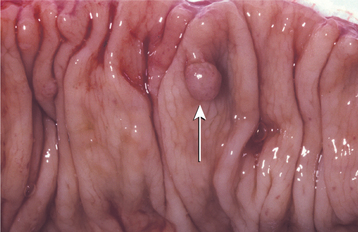
Most patients show one or several epidermoid cysts of the skin (Fig. 14-63). Desmoid tumors (locally aggressive fibrous neoplasms) of the soft tissue arise in approximately 10% of affected patients. These lesions are three times more frequent in females and often develop in the abdominal scar that forms after colectomy.
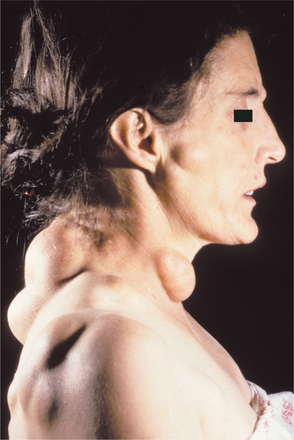
Fig. 14-63 Gardner syndrome. This patient has multiple, large epidermoid cysts. (Courtesy of Dr. William Welton.)
Although lesser known, an increased prevalence of thyroid carcinoma also is noted, with females demonstrating a 100-fold increase. In addition, pigmented lesions of the ocular fundus are evident in nearly 90% of affected patients. Identification of this ocular abnormality is useful when evaluating patients for the syndrome.
HISTOPATHOLOGIC FEATURES: Histopathologically, the osteomas are generally of the compact type. An individual lesion cannot be differentiated microscopically from a solitary osteoma.
TREATMENT AND PROGNOSIS: The major problem for patients with Gardner syndrome is the high rate of malignant transformation of bowel polyps into invasive adenocarcinoma. By age 30, about 50% of patients with Gardner syndrome will develop colorectal carcinoma. The frequency of malignant change approaches 100% in older patients.
Prophylactic colectomy is usually recommended. Removal of jaw osteomas and epidermoid cysts for cosmetic reasons sometimes may be indicated, but the long-term prognosis depends on the behavior of the bowel adenocarcinomas.
OSTEOBLASTOMA AND OSTEOID OSTEOMA
Osteoblastoma and osteoid osteoma are closely related benign bone tumors that arise from osteoblasts. There is general agreement that the histopathologic features of these two lesions are identical, but it has been shown that the tumor nidus in osteoid osteomas contains a concentration of peripheral nerves not seen in other fibro-osseous neoplasms. In addition, the tumor produces prostaglandins that result in significant pain that is relieved by prostaglandin inhibitors such as aspirin. Classically, the distinction depends on the size of the lesion, with osteoid osteoma being smaller than 2 cm and osteoblastoma being larger than 2 cm. Some authors prefer to classify both of these lesions as osteoblastomas.
The features of the cementoblastoma closely resemble those present in the osteoblastoma. A number of noted authorities in orthopedic pathology consider cementoblastoma and osteoblastoma to be identical lesions and prefer to designate both as osteoblastomas. Because of significant radiographic and histopathologic similarities, the primary difference between an osteoblastoma and a cementoblastoma depends on whether the lesion is fused to a tooth or not, and this ability is most likely because of the final histodifferentiation of the tumor cells. Because of these similarities, cementoblastoma is presented in the next section of this chapter rather than in Chapter 15.
CLINICAL AND RADIOGRAPHIC FEATURES:
OSTEOBLASTOMA: Osteoblastomas are rarely encountered and represent less than 1% of all bone tumors. The most frequently affected bones are the vertebral column, sacrum, calvarium, long bones, and the small bones of the hands and feet. For those developing within the jaws, there is a slight mandibular predilection, with most examples arising in the posterior regions. Approximately 85% of gnathic osteoblastomas occur before age 30, and there is a slight female predominance among reported cases.
Most osteoblastomas are between 2 and 4 cm, but they may be as large as 10 cm. Pain, tenderness, and swelling are common presenting features. In contrast to the osteoid osteoma, aspirin usually does not relieve the pain associated with an osteoblastoma. In many cases, pain may be misinterpreted as evidence of odontogenic infection. Lesions adjacent to teeth may lead to tooth mobility, root resorption, or tooth displacement. Radiographically, the osteoblastoma may appear as a well-defined or ill-defined radiolucent lesion often with patchy areas of mineralization (Fig. 14-64). Other lesions demonstrate considerable mineralization. Although frequently noted in osteoid osteomas, reactive sclerosis surrounding the lesion is not a constant feature. Most osteoblastomas arise within medullary bone; however, in some instances, an osteoblastoma may present as a bony outgrowth projecting from the periosteum without evidence of a more central destructive process. Such lesions are termed periosteal osteoblastomas.
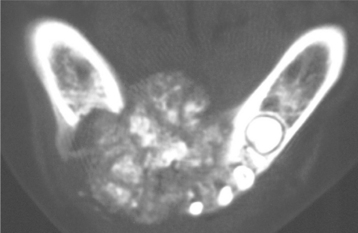
Fig. 14-64 Osteoblastoma. Computed tomography (CT) image showing a large, destructive radiolucent and radiopaque lesion of the mandible. (Courtesy of Dr. Ed Marshall.)
A small group of osteoblastomas (aggressive osteoblastomas) is characterized by more atypical histopathologic features and locally aggressive behavior. These tumors usually occur in older patients, with most being older than 30 years of age. A variety of bones, including the mandible, may be involved. Aggressive osteoblastomas tend to be larger than conventional osteoblastomas and typically measure greater than 4 cm in diameter. Pain is a common symptom and may be severe. Radiographically, these lesions show the features of conventional osteoblastomas but tend to be larger.
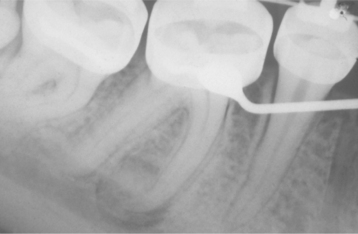
OSTEOID OSTEOMA: Osteoid osteomas occur most often in the femur, tibia, and phalanges. They are very rare in the jaws. Pain is the most common presenting symptom. It is usually nocturnal in nature and alleviated by salicylates. However, nocturnal pain relieved by aspirin has been documented more often among extragnathic lesions than jaw lesions. Radiographically, the osteoid osteoma appears as a well-circumscribed radiolucent defect, usually less than 1 cm in diameter, with a surrounding zone of reactive sclerosis of varying thickness. A small radiopaque nidus may be present, resulting in a targetlike appearance radiographically (Fig. 14-65).
HISTOPATHOLOGIC FEATURES: The lesions reveal mineralized material that demonstrates prominent reversal lines. The material may be present in large sheets or irregular trabeculae. At the periphery of the large masses and surrounding the trabeculae are scattered multinucleated osteoclast-like cells and numerous osteoblasts that have ample cytoplasm and hyperchromatic nuclei (Fig. 14-66). The supporting stroma consists of loose fibrous connective tissue that contains scattered dilated vascular channels. Focal areas of hemorrhage are not rare, and osteoblastomas occasionally exhibit a central zone of increased vascularity.
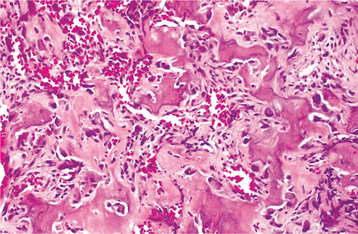
Fig. 14-66 Osteoblastoma. High-power photomicrograph showing irregular bony trabeculae with prominent osteoblastic rimming and osteoclasts.
Microscopically, aggressive osteoblastomas are characterized by the presence of large (epithelioid) osteoblasts with increased mitotic activity and nontrabecular sheets or lacelike areas of osteoid production. On occasion, osteoblastomas may demonstrate a rich cellularity that has led to erroneous diagnoses of osteosarcoma. Differentiation between some osteoblastomas and low-grade osteosarcomas may be very difficult. Some low-grade osteosarcomas may closely resemble the microscopic appearance of osteoblastomas, and some lesions may have microscopic features intermediate between osteoblastoma and osteosarcoma.
TREATMENT AND PROGNOSIS: Most cases of osteoid osteoma and osteoblastoma are treated by local excision or curettage. The prognosis is good, and some lesions will regress even after incomplete excision. A small number of lesions will recur; in rare instances, an osteoblastoma may undergo transformation into an osteosarcoma. Although about 50% of aggressive osteoblastomas will recur, metastasis or death from the tumor has not been reported.
CEMENTOBLASTOMA (TRUE CEMENTOMA)
Cementoblastoma is an odontogenic neoplasm of cementoblasts, and many authorities believe this neoplasm represents the only true neoplasm of cementum.
CLINICAL AND RADIOGRAPHIC FEATURES: Cementoblastomas are rare neoplasms, representing less than 1% of all odontogenic tumors. Greater than 75% arise in the mandible, with 90% arising in the molar and premolar region. Almost 50% involve the first permanent molar. Impacted or unerupted teeth rarely may be affected as well. Cementoblastomas rarely affect deciduous teeth. There is no significant sex predilection. The neoplasm occurs predominantly in children and young adults, with about 50% arising before the age of 20 years and 75% occurring before the age of 30 years. Pain and swelling are present in approximately two thirds of reported patients. Although most investigators consider the cementoblastoma to represent a rather innocuous neoplasm, signs of locally aggressive behavior may be observed, including bony expansion, cortical erosion, displacement of adjacent teeth, envelopment of multiple adjacent teeth, maxillary sinus involvement, and infiltration into the pulp chamber and root canals.
Radiographically, the tumor appears as a radiopaque mass that is fused to one or more tooth roots and is surrounded by a thin radiolucent rim (Fig. 14-67). The outline of the root or roots of the involved tooth is usually obscured as a result of root resorption and fusion of the tumor with the tooth.
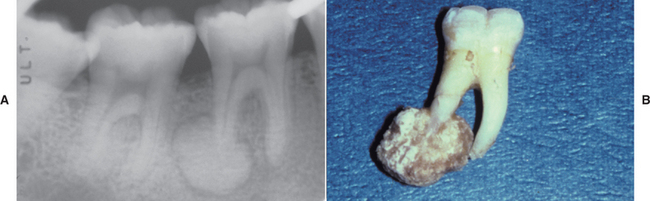
HISTOPATHOLOGIC FEATURES: The histopathologic presentation of cementoblastoma closely resembles that of osteoblastoma, with the primary distinguishing feature being tumor fusion with the involved tooth (Fig. 14-68). The majority of the tumor consists of sheets and thick trabeculae of mineralized material with irregularly placed lacunae and prominent basophilic reversal lines. Cellular fibrovascular tissue is present between the mineralized trabeculae. Multinucleated giant cells often are present, and prominent blastlike cells frequently line the mineralized trabeculae (Fig. 14-69). The periphery of the lesion, corresponding to the radiolucent zone seen on the radiograph, is composed of uncalcified matrix, which often is arranged in radiating columns. In few instances, the lesion may infiltrate the pulp chamber and root canals of the involved tooth.
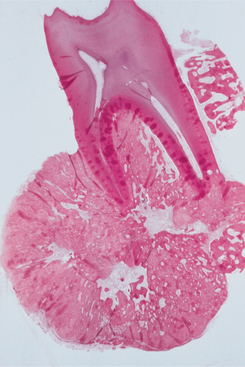
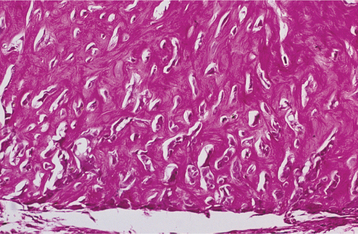
TREATMENT AND PROGNOSIS: Treatment of a cementoblastoma usually consists of surgical extraction of the tooth together with the attached calcified mass. Surgical excision of the mass with root amputation and endodontic treatment of the involved tooth may be considered. In the past, this tumor has been considered to exhibit a very low recurrence rate, although a recently reported series of a large number of cases suggests recurrences may be more common than previously thought, with an overall recurrence rate as high as 22%. Completeness of removal is most closely related to recurrence. Total removal of the mass and the associated tooth minimizes but does not completely eliminate the chance of recurrence. Progressive growth of the tumor after extraction of the involved tooth and incomplete removal of the mass has been documented.
CHONDROMA
Chondromas are benign tumors composed of mature hyaline cartilage. Chondromas are one of the more common bone tumors and are located most often in the short tubular bones of the hands and feet. However, a diagnosis of chondroma in the jaws, facial bones, and base of the skull should be viewed with great skepticism because many so-called benign chondromas of the craniofacial complex have recurred and acted in a malignant manner. No major series has reported enchondromas arising in the craniofacial bones. In spite of this, individual reports and small series of gnathic chondromas can be found, with most examples thought to arise from vestigial cartilaginous rests. Such rests are located in the anterior maxilla, symphysis, coronoid process, and condyle.
CLINICAL AND RADIOGRAPHIC FEATURES: Chondromas usually arise in the third and fourth decades without a significant sex predilection. Most gnathic examples have been found in the condyle or anterior maxilla of adult patients. When arising in the jaw, most chondromas are painless and slowly growing tumors. Tooth mobility and root resorption are noted occasionally. Radiographically, chondromas typically appear as radiolucencies with central areas of radiopacity.
In most cases, chondromas arise in a single site. Multiple and widespread involvement with a tendency to be unilateral is termed Ollier disease. In another presentation, termed Maffucci syndrome, skeletal chondromatosis is seen in association with soft tissue angiomas.
HISTOPATHOLOGIC FEATURES: Histopathologically, a chondroma appears as a circumscribed mass of mature hyaline cartilage that typically demonstrates well-formed lacunae containing small chondrocytes with pale cytoplasm and small, round nuclei. On occasion, the microscopic distinction between a benign chondroma and a low-grade chondrosarcoma of the jaws is difficult (see page 664).
CHONDROMYXOID FIBROMA
The chondromyxoid fibroma is an uncommon benign neoplasm accounting for less than 1% of all primary bone tumors. It is located most commonly in the metaphyseal region of the long bones. Chondromyxoid fibromas rarely involve the jaws. Cytogenetic analysis in several cases has demonstrated the presence of nonrandom, clonal abnormalities of chromosome 6, where a number of candidate genes important in cartilage development are located. Further characterization of these chromosomal abnormalities may aid in differentiating chondromyxoid fibroma from chondrosarcoma, a distinction that at times can be difficult.
CLINICAL AND RADIOGRAPHIC FEATURES: Chondromyxoid fibromas of the jaws have been en-countered in patients ranging in age from 10 to 67 years. The mean age of occurrence is approximately 30 years, with the majority discovered in the second and third decades. There is no sex predilection. Of the re-ported cases in the jaws, about three quarters occurred in the mandible. In about one fourth, pain was an initial symptom, and swelling was noted in approximately three fourths. Some cases have been asymptomatic, being detected only on a radiographic examination.
Radiographically, the lesion is a circumscribed radiolucent defect with sclerotic or scalloped margins. Central radiopacities sometimes are present within the lesion. On initial presentation, the size of reported chondromyxoid fibromas varied from 1.0 to 6.5 cm.
HISTOPATHOLOGIC FEATURES: The tumor consists of lobulated areas of spindle-shaped or stellate cells with abundant myxoid or chondroid intercellular substance. The lobules characteristically are separated by zones of a more cellular tissue composed of spindle-shaped or round cells with varying numbers of multinucleated giant cells (Fig. 14-70).
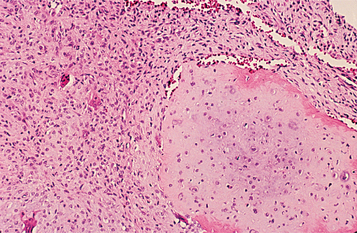
Fig. 14-70 Chondromyxoid fibroma. Myxoid connective tissue with scattered giant cells and foci of cartilaginous differentiation.
Large pleomorphic cells that may cause confusion with chondrosarcoma may be seen. Focal areas of calcification and spicules of residual bone also may be present within the tumor.
TREATMENT AND PROGNOSIS: Although the chondromyxoid fibroma is a benign tumor, approximately 25% of cases in the long bones recur after curettage. Some orthopedic surgeons recommend block excision as the initial treatment. Generally, chondromyxoid fibromas of the jaws have been small and treated by curettage; recurrence is uncommon. Because of the lobular growth and associated scalloped margins, larger gnathic lesions appear to justify resection in an attempt to prevent recurrence. Radiation therapy is contraindicated because of the risk of inducing malignant transformation or osteoradionecrosis.
Distinguishing between a chondromyxoid fibroma and myxoid chondrosarcoma histopathologically may be difficult. Examples of both underdiagnosis and overdiagnosis, with resultant improper treatment, have been described.
SYNOVIAL CHONDROMATOSIS (CHONDROMETAPLASIA)
Synovial chondromatosis is a rare, benign, nonneoplastic arthropathy characterized by the metaplastic development of cartilaginous nodules within the synovial membrane. The exact cause is unknown. In many cases an association with other joint conditions (e.g., inflammatory joint disease, noninflammatory arthropathy, joint overuse or other trauma) has been described, and thus the development of synovial chondromatosis appears to represent a secondary reactive phenomenon. Less commonly, no identifiable etiologic factors are identified, and such cases have been designated as primary synovial chondromatosis.
The process typically proceeds through three stages. In the first stage, foci of metaplastic cartilage arise in the synovial lining. With time, these foci increase in size and begin to detach, with cartilaginous material present in both the synovial membrane and the joint. In the final stage, metaplastic cartilage is found only in the joint.
CLINICAL AND RADIOGRAPHIC FEATURES: The disease most commonly affects large joints, such as the knee, elbow, hip, and shoulder. The number of reported cases involving the temporomandibular joint (TMJ) is less than 100. In recent years, there has been an increase in reported cases, possibly because of improved imaging techniques and increased awareness of the disease. The condition is usually limited to one joint.
Synovial chondromatosis of the TMJ occurs across a wide age range, but most affected patients are middle-aged. In contrast to the findings in other joints, there is a predilection for females. Periarticular swelling, pain, crepitus, and limitation of joint motion are usually present. These features are common to a number of pathoses involving the TMJ and are not diagnostic for synovial chondromatosis. In rare instances, the disease may produce no symptoms. The process is usually limited to the joint, although extraarticular extension and even intracranial extension have been reported in a few more aggressive cases.
Radiographically, the most common feature is the presence of loose bodies in the joint. These consist of rounded, irregularly shaped, and variably sized radiopaque structures in the region of the joint. Other features include irregularity of the joint space, widened joint space, and irregularity of the condylar head.
These findings, however, are not diagnostic of synovial chondromatosis and may be seen in other degenerative joint diseases. The absence of loose bodies does not preclude a diagnosis of synovial chondromatosis. Computed tomography (CT) scans and magnetic resonance imaging (MRI) have been advocated as more sensitive diagnostic imaging procedures.
HISTOPATHOLOGIC FEATURES: Nodules of cartilage are present within the synovium and lie loose in the joint space. As many as 100 nodules may be present. These cartilaginous nodules often become calcified and may ossify. The cartilage may appear atypical with hyperchromatic and binucleated chondrocytes, particularly in primary lesions (Fig. 14-71). In another clinical situation, these features would suggest a diagnosis of chondrosarcoma, but these changes are not considered significant in synovial chondromatosis.
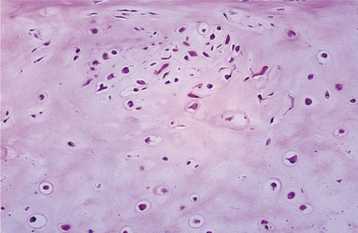
TREATMENT AND PROGNOSIS: For patients with synovial chondromatosis, the involved synovium and all loose bodies are removed surgically. Some surgeons advocate total synovectomy to prevent recurrence. Meniscectomy may be necessary if the disc cannot be repaired. A few cases of erosion of the glenoid fossa and cranial extension of the process have been reported. Surgery is performed most commonly via open arthrotomy, although arthroscopy may be used for biopsy and occasionally for treatment of limited disease with loose bodies less than 3 mm in diameter. A wider approach is typically necessary for the rare cases with extensive extraarticular involvement.
The prognosis is good, with an overall low frequency of recurrence after surgical excision. However, some investigators have noted that the less common primary lesions may exhibit more aggressive behavior with a higher recurrence rate, even after synovectomy. Thus periodic follow-up examinations would appear to be prudent. Malignant transformation of synovial chondromatosis of the TMJ has not been noted. Most patients experience improved joint function and pain relief after surgery.
DESMOPLASTIC FIBROMA
The desmoplastic fibroma of bone is an uncommon tumor of fibroblastic origin. It is thought to be the osseous counterpart of the soft tissue fibromatosis (desmoid tumor) (see page 515). In a few cases, desmoplastic fibroma-like lesions of the jaws have been reported in association with tuberous sclerosis.
CLINICAL AND RADIOGRAPHIC FEATURES: Most examples of desmoplastic fibroma of bone are discovered in patients younger than 30 years of age. The age range of reported gnathic examples is from 10 months to 59 years, with a mean of approximately 16 years. There is no significant sex predilection. The most common locations are the mandible, femur, pelvic bones, radius, and tibia. Of the reported cases involving the jaws, 84% have occurred in the mandible, most often in the molar angle-ascending ramus area.
Although some tumors are associated with limited opening, with or without malocclusion, a painless swelling of the affected area is the most common initial complaint. Tooth mobility, proptosis, concurrent infection, and dysesthesia have been reported infrequently. Radiographically, the lesion appears as a multilocular or occasionally unilocular radiolucent area. The margins may be well defined or ill defined (Fig. 14-72). The bone is expanded, and the cortex is thinned; cortical reaction mimicking the appearance of an osteosarcoma is rare. If the lesion erodes through the cortex, then an accompanying soft tissue mass will be present. When this occurs, it may be difficult to determine whether the lesion is a desmoplastic fibroma of bone with soft tissue extension or a soft tissue fibromatosis with secondary extension into bone. Adjacent teeth may exhibit displacement and root resorption.
HISTOPATHOLOGIC FINDINGS: The tumor is composed of small elongated fibroblasts and abundant collagen fibers (Fig. 14-73). The degree of cellularity may vary from area to area in a given lesion, and the cellular areas may show plumper fibroblasts and less collagen. The fibroblasts are not atypical, however, and mitoses are essentially absent. Bone spicules may be present at the interface between the tumor and adjacent bone but are never an integral part of the lesion.
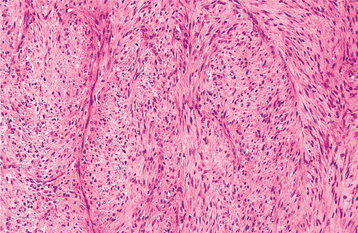
Fig. 14-73 Desmoplastic fibroma. The tumor consists of a cellular proliferation of fibroblasts arranged in interlacing fascicles.
Some authors have recommended that diagnostic biopsies be sampled generously from the center rather than the periphery of the lesion, because reactive bone at the periphery may be mistaken for osteoid production, which may lead to a mistaken diagnosis of a benign fibro-osseous lesion or osteosarcoma.
TREATMENT AND PROGNOSIS: Although the desmoplastic fibroma is considered to be a benign tumor, it often behaves in a locally aggressive fashion, with extensive bone destruction and soft tissue extension; therefore, radical surgery may be required to control the disease. Some surgeons prefer curettage, whereas others prefer wide local excision or resection with a wide margin. Curettage may be adequate for localized lesions without cortical perforation or soft tissue extension, but segmental resection is preferred for lesions exhibiting rapid growth, an ill-defined radiographic appearance, cortical perforation, or soft tissue extension. The recurrence rate for lesions treated by curettage alone is approximately 70%, compared with approximately 20% for those treated by resection. Given these high recurrence rates, patients should be monitored postoperatively for a minimum of 3 years. Although metastases do not occur and the long-term prognosis is good, the lesion may be associated with considerable morbidity.
It may be very difficult to distinguish desmoplastic fibroma of bone from well-differentiated fibrosarcoma. Some authorities suggest that all desmoplastic fibromas of bone be considered potentially malignant.
OSTEOSARCOMA (OSTEOGENIC SARCOMA)
Osteosarcoma is a malignancy of mesenchymal cells that have the ability to produce osteoid or immature bone. Excluding hematopoietic neoplasms, osteosarcoma is the most common type of malignancy to originate within bone. The majority of osteosarcomas demonstrate intramedullary origin, but a small number may be juxtacortical (discussed in the following section) or rarely, extraskeletal.
CLINICAL AND RADIOGRAPHIC FEATURES: Extragnathic osteosarcoma demonstrates a bimodal age distribution. Most arise in patients between the ages of 10 and 20 years, with a lesser number diagnosed in adults older than age 50. The initial peak occurs during the period of greatest bone growth; accordingly, most of these osteosarcomas arise in the distal femoral and proximal tibial metaphyses. In older patients, the axial skeleton and flat bones are involved most frequently; Paget’s disease and previous irradiation are associated with an increased prevalence.
Osteosarcomas of the jaws are uncommon and represent 6% to 8% of all osteosarcomas. These gnathic tumors have been diagnosed in patients ranging from young children to older adults, but they occur most often in the third and fourth decades of life. The mean age for patients with osteosarcoma of the jaw is about 33 years, which is 10 to 15 years older than the mean age for osteosarcomas of the long bones. As is seen in extragnathic locations, a slight male predominance is noted.
The maxilla and mandible are involved with about equal frequency. Mandibular tumors arise more frequently in the posterior body and horizontal ramus rather than the ascending ramus. Maxillary lesions are discovered more commonly in the inferior portion (alveolar ridge, sinus floor, palate) than the superior aspects (zygoma, orbital rim).
Swelling and pain are the most common symptoms (Fig. 14-74). Loosening of teeth, paresthesia, and nasal obstruction (in the case of maxillary tumors) also may be noted. Some patients report symptoms for relatively long periods before diagnosis, which indicates that some osteosarcomas of the jaws grow rather slowly (Fig. 14-75).
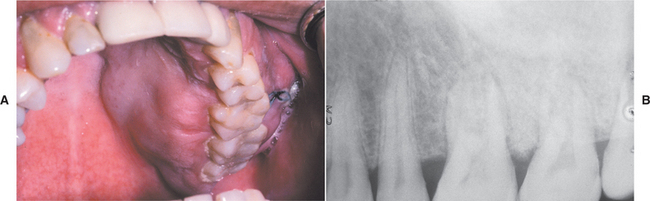
Fig. 14-74 Osteosarcoma. A, This patient shows a firm, painful swelling of the left maxilla of recent onset. B, The periapical radiograph shows a dense sclerotic change in the bone pattern. (Courtesy of Dr. Len Morrow.)
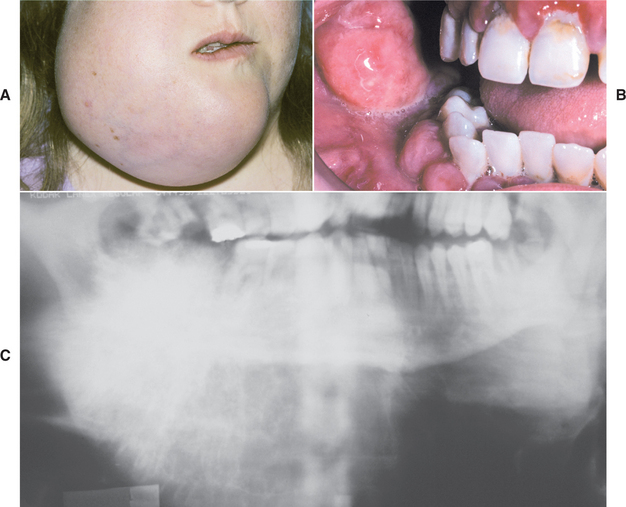
Fig. 14-75 Osteosarcoma. A, This massive tumor had been present for many months before the patient sought treatment. B, Intraoral photograph of the tumor mass. C, The panoramic radiograph shows a “sunburst” pattern of trabeculation within the tumor.
The radiographic findings vary from dense sclerosis to a mixed sclerotic and radiolucent lesion to an entirely radiolucent process. The peripheral border of the lesion is usually ill defined and indistinct, making it difficult to determine the extent of the tumor radiographically. In some cases, an extensive osteosarcoma may show only minimal and subtle radiographic change with only slight variation in the trabecular pattern. Occasionally, there is resorption of the roots of teeth involved by the tumor. This feature is often described as “spiking” resorption as a result of the tapered narrowing of the root. The “classic” sunburst or sun ray appearance caused by osteophytic bone production on the surface of the lesion is noted in about 25% of jaw osteosarcomas. Often this is appreciated best on an occlusal projection (Fig. 14-76). In few cases a triangular elevation of the periosteum, referred to as Codman’s triangle, may be observed.
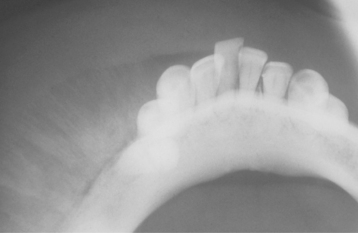
Fig. 14-76 Osteosarcoma. Occlusal radiograph demonstrating prominent exophytic tumor bone production on the buccal surface of the mandible, resulting in the “sunburst” pattern. (Courtesy of Dr. Lewis Gilbert.)
An important early radiographic change in patients with osteosarcoma consists of a symmetrical widening of the periodontal ligament space around a tooth or several teeth. This is the result of tumor infiltration along the periodontal ligament space (Fig. 14-77). Widening of the periodontal ligament space is not specific for osteosarcoma and may be seen associated with other malignancies. This radiographic finding, when accompanied by pain or discomfort and other minimal radiographic changes, may be of great importance in the early diagnosis of jaw osteosarcomas.
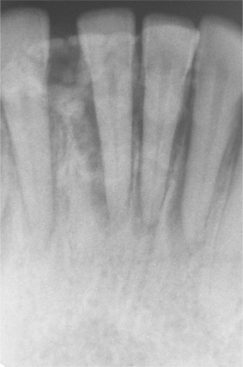
Fig. 14-77 Osteosarcoma. This 26-year-old woman had a 6-cm painful tumor of the anterior mandible. The periapical radiograph shows widening of the periodontal ligament spaces and a mottled radiopacity superimposed on the teeth. (Courtesy of Dr. Charles Ferguson.)
Although periapical, occlusal, and panoramic radiographs often lead to the initial diagnosis, CT scans are excellent for demonstrating the degree of intramedullary extension, tumor calcification, cortical involvement, and soft tissue involvement. These scans prove invaluable for determining the extent of surgery.
HISTOPATHOLOGIC FEATURES: Osteosarcomas of the jaws display considerable histopathologic variability. The essential microscopic criterion is the direct production of osteoid by malignant mesenchymal cells (Fig. 14-78). In addition to osteoid, the cells of the tumor may produce chondroid material and fibrous connective tissue. The tumor cells may vary from relatively uniform round or spindle-shaped cells to highly pleomorphic cells with bizarre nuclear and cytoplasmic shapes. The amount of matrix material produced in the tumor may vary considerably. In some instances, osteoid production may be very minimal and difficult to demonstrate. Most osteosarcomas of the jaws tend to be better differentiated than osteosarcomas of the extragnathic skeleton.
Depending on the relative amounts of osteoid, cartilage, or collagen fibers produced by the tumor, many pathologists subclassify osteosarcomas into the following types:
These histopathologic subtypes, however, do not have any great bearing on the prognosis. Other less commonly encountered histopathologic variations include malignant fibrous histiocytoma-like, small cell, epithelioid, telangiectatic, and giant cell–rich.
Chondroblastic osteosarcomas constitute a substantial proportion of all osteosarcomas of the jaws. Some examples may be composed almost entirely of malignant cartilage growing in lobules with only small foci of direct osteoid production by tumor cells being identified (Fig. 14-79). Such lesions, however, should be classified as osteosarcomas rather than chondrosarcomas.
Low-grade, well-differentiated osteosarcomas may show only minimal cellular atypia of the lesional cells and abundant bone formation. On microscopic examination, these lesions may be difficult to differentiate from benign bone lesions, such as fibrous dysplasia or ossifying fibroma.
TREATMENT AND PROGNOSIS: Many past and present investigators believe osteosarcoma of the jaws is less aggressive than those occurring in the long bones. Most gnathic osteosarcomas are low-grade, and metastases are seen less frequently. Despite these findings, many current clinicopathologic studies fail to support this contention and suggest that osteosarcomas of the jaws are aggressive neoplasms. The most important prognostic indicator is the ability to achieve initial complete surgical removal, a feat that is much more difficult to achieve in the jaws than the long bones. In particular, maxillary lesions often are more difficult to resect completely than mandibular lesions. The aggressiveness of gnathic osteosarcoma remains an area of controversy that is difficult to resolve because of the rarity of the neoplasm and the lack of consistently applied diagnostic criteria.
Multicenter investigations of different therapies to osteosarcoma of long bones have led to an improved prognosis that now appears superior to that associated with gnathic neoplasms. These protocols involve neoadjuvant (preoperative) chemotherapy followed by radical surgical excision with careful pathologic examination of the specimen to evaluate the chemotherapeutic effects on the tumor. Adjuvant (postoperative) chemotherapy is used and may be modified if poor histopathologic response to the neoadjuvant regimen is noted. Some investigators have demonstrated 4-year survival rates exceeding 80% with this approach. Limited numbers of patients with jaw osteosarcomas have been treated with these protocols, and superior results have been claimed compared with surgical treatment alone. In addition, a systematic literature review of 201 patients with craniofacial osteosarcoma demonstrated that patients treated with chemotherapy exhibited an improved long-term survival regardless of the ability to achieve complete surgical removal.
In spite of the improved prognosis in patients receiving chemotherapy, radical surgical excision remains the mainstay of therapy. Because the tumor may extend for some distance beyond the apparent clinical and radiographic margins, local recurrence after surgery is a major problem. Local uncontrolled disease is more often the cause of death for patients with jaw osteosarcoma than are the effects of distant metastases. Most deaths from uncontrolled local disease occur within 2 years of the initial treatment. Jaw osteosarcomas have less tendency to metastasize than do osteosarcomas of long bones. Although regional lymph nodes may be infrequently involved, metastases most often affect the lungs and brain. When comparing mandibular and maxillary osteosarcomas, metastasis is noted more frequently from mandibular neoplasms, whereas local recurrence is associated more frequently with maxillary tumors.
The prognosis remains serious. Various studies indicate a 30% to 70% survival rate. Survival rates of up to 80% have been reported for patients receiving initial radical surgery, the best hope for permanent cure. Additional prospective investigations of gnathic osteosarcoma treated by neoadjuvant chemotherapy followed by surgical removal and adjuvant chemotherapy are necessary in an attempt to confirm the most appropriate approach.
PERIPHERAL (JUXTACORTICAL) OSTEOSARCOMA
In contrast to the usual forms of intramedullary osteosarcoma, several varieties originate adjacent to the cortex of the bone, initially grow outward from the surface, and do not involve the underlying medullary cavity. The terminology used for these lesions by different authors is somewhat confusing. These peripheral (juxtacortical) osteosarcomas usually occur in the long bones, but examples involving the jaws have been reported.
The parosteal type of osteosarcoma is a lobulated nodule attached to the cortex by a short stalk (Fig. 14-80). There is no elevation of the periosteum and no peripheral periosteal reaction. Histopathologically, the exophytic mass demonstrates a spindle cell fibroblast-like proliferation that contains well-developed trabeculae of bone. With time, the trabeculae often coalesce and form a large mass of solid bone. Parosteal osteosarcoma is a low-grade sarcoma that has a small risk of recurrence and metastasis if treated by radical excision. With inadequate surgery, the tumor may eventually develop into a higher-grade osteosarcoma, with a resultant poor prognosis.
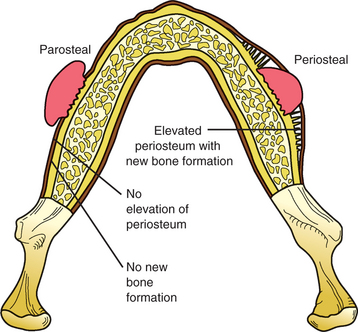
Fig. 14-80 Peripheral (juxtacortical) osteosarcoma. Illustration comparing different types of peripheral osteosarcoma. Parosteal osteosarcoma presents as a lobulated nodule without a peripheral periosteal reaction. Periosteal osteosarcoma presents as a sessile mass associated with significant periosteal new bone formation.
The periosteal form of osteosarcoma is a sessile lesion that arises within the cortex and elevates the overlying periosteum, which provokes the production of significant peripheral periosteal new bone formation (see Fig. 14-80). Often the leading edge of the tumor mass perforates the surface of the periosteum and extends into the surrounding soft tissue. Histopathologically, the tumor demonstrates primitive sarcomatous cells within a tumor that demonstrates significant chondroblastic differentiation. Close inspection will reveal foci of tumor osteoid and immature bone formation. Radical surgical excision with wide margins is the therapy of choice. Although the prognosis is better than that associated with intramedullary tumors, periosteal osteosarcoma has a poorer outcome than the parosteal variant. In reviews of patients with periosteal osteosarcomas, approximately 25% died from metastatic disease.
POSTIRRADIATION BONE SARCOMA
Sarcoma arising in a bone that has been previously subjected to radiation therapy is a well-recognized phenomenon. The jaws are situated closely to tissues that commonly receive therapeutic radiation and are a common site for postirradiation bone sarcomas. Postirradiation sarcomas may develop as early as 3 years after radiation, but the average latent period is about 14 years. The frequency of development of sarcoma is related to radiation dose. Postirradiation sarcoma develops in about 0.2% of patients receiving 7000 cGy; there is no increased prevalence of sarcoma for those receiving less than 1000 cGy.
Osteosarcoma is the most common type of postirradiation sarcoma, accounting for 50% of all cases. About 40% of postirradiation sarcomas are fibrosarcomas, with chondrosarcomas and other histopathologic types making up the rest. Postirradiation bone sarcomas have no distinctive histopathologic features that allow them to be distinguished from other bone sarcomas of the same type that arise de novo.
The prognosis for postirradiation sarcomas is about the same as for de novo tumors of the same type.
CHONDROSARCOMA
Chondrosarcoma is a malignant tumor characterized by the formation of cartilage, but not bone, by the tumor cells. Chondrosarcomas comprise about 10% of all primary tumors of the skeleton but are considered by most authorities to involve the jaws only rarely. Chondrosarcoma is about half as common as osteosarcoma and about twice as common as Ewing sarcoma. Approximately 1% to 3% of all chondrosarcomas arise in the head and neck area, and such lesions comprise only 0.1% of all head and neck malignancies. Some institutions report a somewhat greater frequency of chondrosarcomas in the jaws. This may be the result of differing criteria used by the pathologists for distinguishing chondroblastic osteosarcomas from chondrosarcomas.
CLINICAL AND RADIOGRAPHIC FINDINGS: In extragnathic bones, chondrosarcoma is primarily a neoplasm of adulthood, with peak prevalence in the sixth and seventh decades of life. Although chondrosarcoma arises across a wide age range, the majority of affected patients are older than 50 years of age; tumors arising in patients younger than age 45 are uncommon. No significant sex or race predilection is noted. The most frequently involved bones are the ileum, femur, and humerus. Involvement of the head and neck is seen infrequently.
When occurring in the head and neck, chondrosarcomas arise most frequently in the maxilla; less common sites of involvement are the mandibular body, ramus, nasal septum, and paranasal sinuses. Although chondrosarcomas most often develop in osseous locations, approximately one third of head and neck lesions are extraosseous and originate in either laryngotracheal cartilage or soft tissue. Because of the rarity of chondrosarcoma, large series of jaw and facial bone lesions are uncommon. In one of the larger series, the Mayo Clinic reviewed 56 patients with chondrosarcoma of the jaws and facial bones, and a pattern of occurrence similar to the extragnathic bones was observed. In this series the peak prevalence was noted in the seventh decade, but the age at initial diagnosis had a wide range, with approximately 20% noted in patients younger than 20 years of age. The mean patient age at the time of diagnosis was 41.6 years. No sex or race predilection was noted. Involvement of the maxilla and maxillary sinus outnumbered those in the mandible by 4 to 1.
In a recent review of 400 head and neck chondrosarcomas from the American College of Surgeons’ National Cancer Database, the age distribution for patients with conventional-type tumors was similar to that previously reported by the Mayo Clinic series. However, review of prior publications presents a conflicting picture in which the mean age is variable and often reveals a peak prevalence as early as the third decade. Some investigators have suggested that such conflicting results may be the result of difficulty in performing literature reviews that may contain chondroblastic osteosarcomas intermixed with true chondrosarcomas.
A painless mass or swelling is the most common presenting sign. This may be associated with separation or loosening of teeth. In contrast to osteosarcoma, pain is an unusual complaint. Maxillary tumors may cause nasal obstruction, congestion, epistaxis, photophobia, or visual loss.
Radiographically, the tumor usually shows features suggestive of a malignancy, consisting of a radiolucent process with poorly defined borders. The radiolucent area often contains scattered and variable amounts of radiopaque foci, which are caused by calcification or ossification of the cartilage matrix (Fig. 14-81). Some chondrosarcomas show extensive calcification and radiographically appear as a densely calcified mass with irregular peripheral margins. Penetration of the cortex can result in a sunburst pattern similar to that seen in some osteosarcomas.
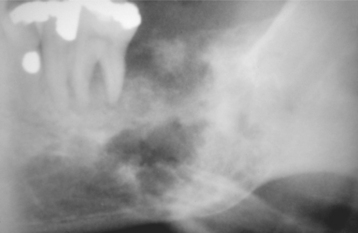
Fig. 14-81 Chondrosarcoma. Ill-defined radiolucent lesion of posterior mandible containing radiopaque foci. (Courtesy of Dr. Ben B. Henry.)
Chondrosarcomas often demonstrate extensive infiltration between the osseous trabeculae of the preexisting bone without causing appreciable resorption. In such cases the extent of the tumor is difficult to determine by radiographic examination. Root resorption or symmetrical widening of the periodontal ligament space of the teeth involved by the tumor also may be noted. Chondrosarcomas may grow in a lobular pattern with minimal or no foci of calcification. In such instances, the lesion can appear as a multilocular radiolucency and mimic a benign process.
HISTOPATHOLOGIC FEATURES: Chondrosarcomas are composed of cartilage showing varying degrees of maturation and cellularity. In most cases, typical lacunar formation within the chondroid matrix is visible, although this feature may be scarce in poorly differentiated tumors. The tumor often shows a lobular growth pattern, with tumor lobules separated by thin fibrous connective tissue septa. The central areas of the lobules demonstrate the greatest degree of maturation. The peripheral areas consist of immature cartilage and mesenchymal tissue consisting of round or spindle-shaped cells. Calcification or ossification may occur within the chondroid matrix. Neoplastic cartilage may be replaced by bone in a manner similar to normal endochondral ossification.
Chondrosarcomas may be divided into three histopathologic grades of malignancy. This grading system correlates well with the rate of tumor growth and prognosis for chondrosarcomas of the extragnathic skeleton.
GRADES: Grade I chondrosarcomas closely mimic the appearance of a chondroma, composed of chondroid matrix and chondroblasts that show only subtle variation from the appearance of normal cartilage. The distinction between benign and well-differentiated malignant cartilaginous tumors is notoriously difficult. Many believe that a tumor should be considered malignant when large, plump chondroblasts and binucleated chondrocytes are present, even in only scattered microscopic fields. Calcification or ossification of the cartilaginous matrix often is prominent, and mitoses are rare.
Grade II chondrosarcomas show a greater proportion of moderately sized nuclei and increased cellularity, particularly about the periphery of the lobules. The cartilaginous matrix tends to be more myxoid, with a less prominent hyaline matrix. The mitotic rate, however, is low (Fig. 14-82).
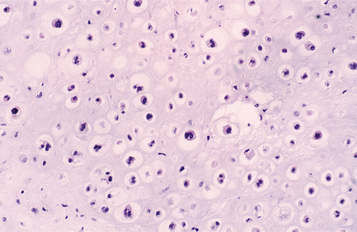
Fig. 14-82 Chondrosarcoma. This grade II chondrosarcoma shows a variation in size of chondrocyte nuclei. Occasional double nuclei are seen in the lacunae.
Grade III chondrosarcomas are highly cellular and may show a prominent spindle cell proliferation. Mitoses may be prominent. Easily recognizable cartilaginous matrix containing cells within lacunae may be scarce.
Chondrosarcomas of the jaws are predominantly of the histopathologic grades I and II. Grade III tumors are very uncommon. In the 56 cases reviewed by the Mayo Clinic, more than 75% were grade I, with the remainder being grade II; no grade III chondrosarcomas were noted in this series.
VARIANTS: Several uncommon microscopic variants of chondrosarcoma are also recognized.
The clear cell chondrosarcoma shows cells with abundant clear cytoplasm; this may lead to problems in differentiation from a metastatic clear cell carcinoma. The clear cell chondrosarcoma is considered to be a low-grade lesion.
Dedifferentiated chondrosarcoma is a high-grade malignancy that shows an admixture of well-differentiated chondrosarcoma and a malignant mesenchymal tumor resembling fibrosarcoma. If these variants occur in the jaws, then they are exceedingly rare.
Myxoid chondrosarcoma classically is described as a soft tissue tumor, although intraosseous lesions have been reported within the head and neck and elsewhere. This variant is characterized by a proliferation of cells with clear, vacuolated, or eosinophilic cytoplasm within a background of mucoid material.
Mesenchymal chondrosarcoma is discussed in the next section.
TREATMENT AND PROGNOSIS: The prognosis for chondrosarcoma is related to the size, location, and grade of the lesion. The most important factor is the location of the tumor because this has the greatest influence on the ability to achieve complete resection. The most effective treatment for chondrosarcoma is radical surgical excision. Radiation and chemotherapy are less effective when compared with osteosarcoma and are primarily used for unresectable high-grade chondrosarcomas.
Although aggressive tumors are occasionally seen, gnathic chondrosarcomas are usually slowly growing neoplasms with a lower potential for metastasis than osteosarcoma. Local recurrence leads to death by direct extension of the tumor into vital structures of the head and neck. Maxillary and antral tumors often are located centrally, obtain a larger size before diagnosis, occur adjacent to the central nervous system (CNS), and create more difficulty in surgical eradication; therefore, they are less amenable to cure. In the National Cancer Database series of 400 head and neck chondrosarcomas, only approximately 12% of patients had regional or distant metastasis at the time of diagnosis, with a tendency for metastasis in higher-grade and sinonasal lesions. The overall 5- and 10-year disease-specific survival rates in this series were 87.2% and 70.6%, respectively. Survival was greater for patients with conventional chondrosarcoma than for those with the myxoid or mesenchymal variants. In the Mayo Clinic review, no distant metastases occurred in the 56 reported patients; the 5-, 10-, and 15-year survivals were 67.6%, 53.7%, and 43.9%, respectively. As is obvious from these data, the importance of 5-year survival is minimal because recurrence often is a late sequela. Patients must be followed for their lifetime. Although these two large series suggest that the prognosis of gnathic and craniofacial chondrosarcoma is better than that associated with osteosarcoma, previous studies have suggested that the prognosis of chondrosarcoma is worse. This disagreement may be related to differences in the diagnostic categorization of many cartilage-producing tumors (chondrosarcoma versus chondroblastic osteosarcoma).
MESENCHYMAL CHONDROSARCOMA
The mesenchymal chondrosarcoma, an uncommon and distinctive tumor of bone and soft tissue, shows a biphasic histopathologic pattern. This aggressive form of chondrosarcoma represents only 3% to 9% of all chondrosarcomas.
CLINICAL AND RADIOGRAPHIC FEATURES: In contrast to other types of chondrosarcoma, the mesenchymal variant is unusual in that it most frequently affects individuals in the second or third decade of life and the jaws are among the most frequently involved bones (25% to 30%). Other commonly affected sites are the ribs, shoulder, pelvic girdle, and vertebrae. About one third to one fourth of all examples arise in the soft tissues rather than in bone.
Swelling and pain, often of fairly short duration, are the most common symptoms. Radiographically, the tumor demonstrates a radiolucency with infiltrative margins (Fig. 14-83). Stippled calcification may be present within the radiolucent area.
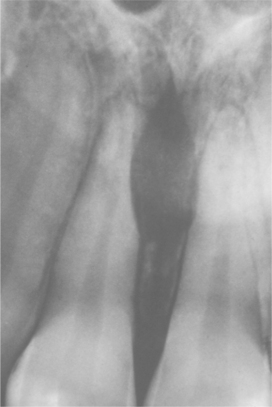
Fig. 14-83 Mesenchymal chondrosarcoma. Periapical radiograph showing a radiolucent lesion between the roots of the central incisors in a 29-year-old woman. The roots of the incisors show resorption. At surgery, the lesion was considerably larger than indicated on the radiograph. (Courtesy of Dr. Gary Baker.)
HISTOPATHOLOGIC FEATURES: Microscopically, the mesenchymal chondrosarcoma reveals sheets or patternless masses of small, undifferentiated spindle or round cells surrounding discrete nodules of cartilage (Fig. 14-84). The chondroid tissue is well differentiated, and its degree of cellularity and atypia may vary from that of a benign chondroma to a low-grade chondrosarcoma. The noncartilaginous component of the tumor is difficult to differentiate from, and may be confused with, a variety of small cell tumors of bone, such as Ewing sarcoma, lymphoma, and metastatic small cell carcinoma. In some cases a prominent, branching vascular pattern is present in the soft tissue component of a mesenchymal chondrosarcoma. If cartilaginous foci are sparse, then the tumor may be misdiagnosed as a hemangiopericytoma.
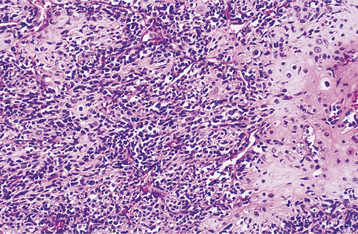
TREATMENT AND PROGNOSIS: Surgical excision with wide margins is the most appropriate therapy. Radiation and chemotherapy have not prolonged survival in a predictable manner. Local recurrence and metastasis are not rare. When metastasis occurs, hematogenous spread is seen more frequently than lymphatic involvement, with the lung being a favored site for metastatic deposits. Recurrent or metastatic disease may be discovered as long as 20 years after initial therapy. The 10-year survival rate is approximately 28%.
EWING SARCOMA
Ewing sarcoma is a distinctive primary malignant tumor of bone that is composed of small, undifferentiated round cells of uncertain histogenesis. Recent studies have provided data showing that most cases of Ewing sarcoma exhibit features consistent with neuroectodermal origin. In 85% to 90% of the cases, the tumor cells demonstrate a reciprocal translocation between chromosomes 11 and 22 [t(11;22) (q24;q12)]. Ewing sarcoma constitutes 6% to 8% of all primary malignant bone tumors and represents the third most common osseous neoplasm after osteosarcoma and chondrosarcoma. In addition, extraskeletal examples have been well documented.
CLINICAL AND RADIOGRAPHIC FEATURES: The peak prevalence of Ewing sarcoma is in the second decade of life, with approximately 80% of patients being younger than 20 years of age at the time of diagnosis and 50% of these tumors being detected in the second decade. A slight male predominance is noted. The vast majority of affected patients are white, with blacks almost never developing this tumor. The long bones, pelvis, and ribs are affected most frequently, but almost any bone can be affected. Jaw involvement is uncommon, with only 1% to 2% occurring in the gnathic or craniofacial bones.
Pain, often associated with swelling, is the most common symptom. It is usually intermittent and varies from dull to severe. Fever, leukocytosis, and an elevated erythrocyte sedimentation rate also may be present and may lead to an erroneous diagnosis of osteomyelitis. The tumor commonly penetrates the cortex, resulting in a soft tissue mass overlying the affected area of the bone (Fig. 14-85). Jaw involvement is more common in the mandible than the maxilla. Paresthesia and loosening of teeth are common findings in Ewing sarcomas of the jaws.
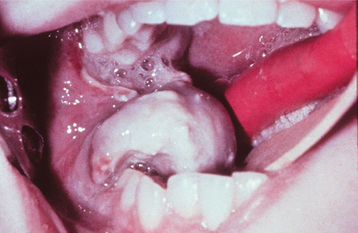
Fig. 14-85 Ewing sarcoma. A rapidly growing, ulcerated tumor of the right posterior mandible. (Courtesy of Dr. George Blozis.)
Radiographically, there is irregular lytic bone destruction with ill-defined margins. Cortical destruction or expansion may or may not be present. The characteristic “onionskin” periosteal reaction, commonly observed in Ewing sarcoma of long bones, is seldom seen in jaw lesions. Although plain radiographs often are used for initial evaluation, computed tomography (CT) is the radiographic study of choice for assessment of lesion extent.
HISTOPATHOLOGIC FEATURES: Ewing sarcoma is composed of small round cells with well-delineated nuclear outlines and ill-defined cellular borders (Fig. 14-86). The tumor cells often are arranged in broad sheets without any distinct pattern. In some cases, variable-sized nests of tumor cells are separated by fibrovascular septa, creating a lobular pattern. Large areas of necrosis and hemorrhage are commonly present. Ewing sarcomas are not as morphologically homogeneous as once was believed. Some examples contain foci or may be composed mostly of larger cells. These are designated as large cell (atypical) Ewing sarcomas.
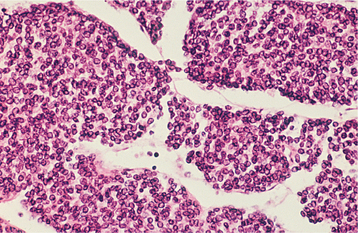
Fig. 14-86 Ewing sarcoma. Broad sheets of small round cells with well-defined nuclear outlines and ill-defined cytoplasmic borders.
About 75% of cases contain glycogen granules in the cytoplasm of the tumor cells. This is a helpful diagnostic feature, but it is not specific because glycogen also can be demonstrated in some other primitive tumors. About 25% of well-documented Ewing tumors do not show glycogen.
Diagnosis of Ewing sarcoma may be difficult. The tumor must be differentiated from other primitive small cell tumors involving bone and soft tissues in young patients. These include metastatic neuroblastoma, malignant lymphoma, small cell osteosarcoma, embryonal rhabdomyosarcoma, and the primitive neuroectodermal tumor. Metastatic small cell carcinoma also must be considered in the differential diagnosis of a suspected Ewing sarcoma in an older patient.
A battery of immunohistochemical reactions and electron microscopy may be required for confirmation of the diagnosis of Ewing sarcoma in some cases. Ewing sarcoma expresses high levels of an antigen determined by the MIC2 gene. The product of MIC2 is a glycoprotein designated CD99 that can be detected by the immunoperoxidase technique. Although useful in confirmation of Ewing sarcoma, positive reactivity to CD99 also is noted in other tumors and some normal tissues. Reverse transcription polymerase chain reaction (RT-PCR) or fluorescence in situ hybridization (FISH) may be used as more sensitive and specific methods for detecting the genetic translocation and confirming the diagnosis.
TREATMENT AND PROGNOSIS: The prognosis for patients with Ewing sarcoma has improved dramatically in recent years. Formerly, fewer than 5% of patients survived more than 5 years. Current treatment, consisting of combined surgery, radiotherapy, and multidrug chemotherapy, has led to 40% to 80% survival rates. Because of the risk for postirradiation sarcomas, some clinicians do not recommend radiation except when surgical resection of the primary site is not possible.
At the time of initial diagnosis, seemingly localized disease often is associated with occult micrometastases, making systemic chemotherapy appropriate in most cases. Ewing sarcomas frequently metastasize to the lungs, liver, lymph nodes, and other bones. The anatomic location of the tumor is a critical factor in prognosis. Pelvic lesions are associated with the poorest prognosis. Distal lesions have a better prognosis than those in a proximal location. With modern therapy, patients with Ewing sarcoma of the jaws probably have an improved prognosis; however, there is a scarcity of good information because of the small number of cases.
METASTATIC TUMORS TO THE JAWS
Metastatic carcinoma is the most common form of cancer involving bone. Autopsy studies have shown that more than two thirds of breast carcinomas, one half of all prostate carcinomas, and one third of all lung and kidney carcinomas spread to one or more bones before a patient dies. Although metastasis to a jaw bone may arise from primary carcinomas of any anatomic site, carcinomas of the breast, lung, thyroid, prostate, and kidney give rise to the majority of gnathic metastases. Metastatic spread of a carcinoma to the jaws usually occurs by the hematogenous route. Sarcomas arising in soft tissues or other bones may metastasize to the jaws, but this is very rare.
CLINICAL AND RADIOGRAPHIC FEATURES: Most patients with metastatic carcinoma are older; children are affected rarely. This finding is a reflection of the greater prevalence of carcinoma in older adults. Although metastatic lesions may be observed in any bone, the vertebrae, ribs, pelvis, and skull are the most frequent sites for metastasis.
The jaws are usually considered to be uncommon sites for metastasis but may be involved more often than generally appreciated. A study of carcinomas arising in various extraoral sites demonstrated that 10 (16%) of 62 autopsied cases of carcinoma showed histopathologic evidence of metastasis to the mandible, even though radiographic study of the mandibles removed at autopsy in these cases failed to show evidence of metastatic disease. Metastasis to the maxilla is uncommon, and more than 80% of reported metastases to the jaws have occurred in the mandible.
Metastatic involvement of the jaws exhibits a wide variety of symptoms. Often the patient experiences pain, swelling, loosening of teeth, a mass, or paresthesia. Metastasis to the mandible with involvement of the inferior alveolar nerve occasionally produces a distinctive pattern of anesthesia termed numb-chin syndrome in which there is an unexplained loss of sensation in the lower lip and chin. These symptoms, however, are not specific for metastatic disease and may be associated with primary inflammatory or neoplastic diseases of the jaws. In some instances, the patient may be completely asymptomatic, and the diagnosis of metastatic carcinoma occurs only after microscopic study of a lesion noted on radiographic examination. Not uncommonly, an osseous metastasis is discovered in a nonhealing extraction site from which the tooth was recently removed because of complaints of local pain or significant mobility.
Of particular interest are those cases in which diagnosis of a jaw metastasis is the first indication that the patient has a primary malignancy at some other anatomic site. On occasion, the oral lesion is the first indication of an undiscovered and distant malignancy. Location of the occult primary tumor may be difficult, requiring extensive evaluation.
Radiographically, metastatic deposits in the jaws usually appear as radiolucent defects. The defect may be well circumscribed, resembling a cyst, but more often it is ill defined with a “moth-eaten” appearance. Involvement of the alveolus may resemble periodontal disease clinically and radiographically (Fig. 14-87). On occasion, a metastatic tumor may cause widening of the periodontal ligament (Fig. 14-88). Some carcinomas, particularly from the prostate and breast, may stimulate new bone formation in the metastatic site, resulting in radiopaque or mixed radiolucent and radiopaque lesions.
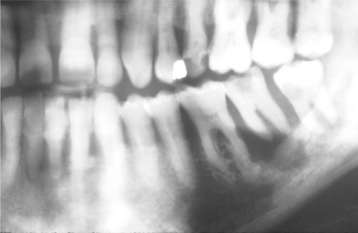
Fig. 14-87 Carcinoma metastatic to the jaws. Panoramic radiograph showing destruction of the alveolar bone surrounding the roots of the mandibular second molar. Such changes may mimic advanced periodontal disease. In this patient, the lesion originated from an occult carcinoma of the lung. (Courtesy of Dr. J.M. Sarnovsky.)
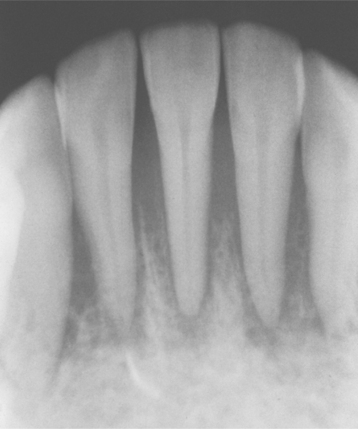
Fig. 14-88 Carcinoma metastatic to the jaws. Periapical radiograph showing widening of the periodontal ligament spaces.
Not uncommonly, patients with gnathic metastases will have symptoms at a time when conventional radiographs fail to demonstrate detectable alterations. In these instances, bone scintigraphy is occasionally used because it has a higher sensitivity and a greater ability to detect subtle osseous metastases. Some investigators recommend this technique for patients with prolonged, unexplained pain who have a history of cancer that is frequently associated with osseous metastases.
HISTOPATHOLOGIC FEATURES: The microscopic appearance of metastatic carcinoma in bone varies. In some instances, the metastatic tumor is well differentiated and closely resembles a carcinoma of a specific site, such as the kidney, colon, or thyroid. In such instances, the pathologist can say with reasonable certainty that a given metastatic tumor comes from a specific primary site (Fig. 14-89). More often, however, metastatic carcinomas are poorly differentiated and histopathologic study of the metastatic deposit gives little clue as to the primary site of the tumor. Poorly differentiated metastatic carcinoma may be difficult to differentiate from anaplastic small cell sarcomas, malignant lymphomas, and malignant melanoma. Immunohistochemical reactions are usually necessary in such cases to establish the diagnosis. Although the diagnosis of metastatic carcinoma can usually be determined by microscopic examination, the final diagnosis depends mostly on a careful medical history and complete physical examination, with appropriate laboratory studies.
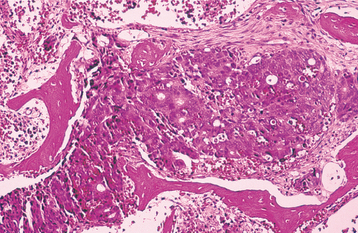
TREATMENT AND PROGNOSIS: The prognosis for metastatic carcinoma of the jaws is poor because, by definition, osseous metastasis automatically places the patient in stage IV disease. Although a solitary metastatic focus may be treated by excision or radiation therapy, jaw involvement almost always is associated with widely disseminated disease. Five-year survival after detection of metastatic carcinoma involving the jaws is exceedingly rare, and most patients do not survive more than 1 year.
BIBLIOGRAPHY
Ablin, DS. Osteogenesis imperfecta: a review. Can Assoc Radiol J. 1998;49:110–123.
Binger, T, Rücker, M, Spitzer, WJ. Dentofacial rehabilitation by osteodistraction, augmentation and implantation despite osteogenesis imperfecta. Int J Oral Maxillofac Surg. 2006;35:559–562.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Syndromes affecting bone: the osteogenesis imperfectas. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:178–191.
Lee, CYS, Ertel, SK. Bone graft augmentation and dental implant treatment in a patient with osteogenesis imperfecta: review of the literature with a case report. Implant Dent. 2003;12:291–295.
Levin, LS, Wright, JM, Byrd, DL, et al. Osteogenesis imperfecta with unusual skeletal lesions: report of three families. Am J Med Genet. 1985;21:257–269.
O’Connell, AC, Marini, JC. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:189–196.
Ormiston, IW, Tideman, H. Orthognathic surgery in osteogenesis imperfecta: a case report with management considerations. J Craniomaxillofac Surg. 1995;23:261–265.
Rauch, F, Glorieux, FH. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385.
Schwartz, S, Tsipouras, P. Oral findings in osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol. 1984;57:161–167.
Sillence, DO, Senn, A, Danks, PM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116.
Albuquerque, MA, Melo, ES, Jorge, WA, et al. Osteomyelitis of the mandible associated with autosomal dominant osteopetrosis: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:94–98.
Bakeman, RJ, Abdelsayed, RA, Sutley, SH, et al. Osteopetrosis: a review of the literature and report of a case complicated by osteomyelitis of the mandible. J Oral Maxillofac Surg. 1998;56:1209–1213.
Balemans, W, Van Wesenbeeck, L, Van Hul, W. A clinical and molecular overview of the human osteopetroses. Calcif Tissue Int. 2005;77:263–274.
Barbaglio, A, Cortelazzi, R, Martignoni, G, et al. Osteopetrosis complicated by osteomyelitis of the mandible: a case report including gross and microscopic findings. J Oral Maxillofac Surg. 1998;56:393–398.
Filho, AM, de Castro Domingos, A, de Freitas, DQ, et al. Osteopetrosis—a review and report of two cases. Oral Dis. 2005;11:46–49.
Key, LL, Jr., Ries, WL. Osteopetrosis. In: Bilezikian JP, Raisz LG, Rodan GA, eds. Principles of bone biology. ed 2. San Diego: Academic Press; 2002:1217–1227.
Satomura, K, Kon, M, Tokuyama, R, et al. Osteopetrosis complicated by osteomyelitis of the mandible: a case report including characterization of the osteopetrotic bone. Int J Oral Maxillofac Surg. 2007;36:86–93.
Tolar, J, Teitelbaum, SL, Orchard, PJ. Osteopetrosis. N Engl J Med. 2004;351:2839–2849.
Younai, F, Eisenbud, L, Sciubba, JJ. Osteopetrosis: a case report including gross and microscopic findings in the mandible at autopsy. Oral Surg Oral Med Oral Pathol. 1988;65:214–221.
Camilleri, S, McDonald, F. Runx2 and dental development. Eur J Oral Sci. 2006;114:361–373.
Counts, AL, Rohrer, MD, Prasad, H, et al. An assessment of root cementum in cleidocranial dysplasia. Angle Orthod. 2001;71:293–298.
Daskalogiannakis, J, Piedade, L, Lindholm, TC, et al. Cleidocranial dysplasia: 2 generations of management. J Can Dent Assoc. 2006;72:337–342.
Furuuchi, T, Kochi, S, Sasano, T, et al. Morphologic characteristics of masseter muscle in cleidocranial dysplasia: a report of 3 cases. Oral Med Oral Pathol Oral Radiol Endod. 2005;99:185–190.
Golan, I, Baumert, U, Hrala, BP, et al. Dentomaxillofacial variability of cleidocranial dysplasia: clinicoradiological presentation and systematic review. Dentomaxillofac Radiol. 2003;32:347–354.
Golan, I, Baumert, U, Hrala, BP, et al. Early craniofacial signs of cleidocranial dysplasia. Int J Paediatr Dent. 2004;14:49–53.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Cleidocranial dysplasia. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:306–310.
Ishii, K, Nielsen, IL, Vargervik, K. Characteristics of jaw growth in cleidocranial dysplasia. Cleft Palate Craniofac J. 1998;35:161–166.
Jansen, BL, Kreiborg, S. Dental treatment strategies in cleidocranial dysplasia. Br Dent J. 1992;172:243–247.
McNamara, CM, O’Riordan, BC, Blake, M, et al. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography. Dentomaxillofac Radiol. 1999;28:89–97.
Mundlos, S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet. 1999;36:177–182.
Visosky, AMB, Johnson, J, Bingea, B. Otolaryngological manifestations of cleidocranial dysplasia, concentrating on audiological findings. Laryngoscope. 2003;113:1508–1514.
Focal Osteoporotic Marrow Defect
Barker, BF, Jensen, JL, Howell, FV. Focal osteoporotic marrow defects of the jaws. Oral Surg Oral Med Oral Pathol. 1974;38:404–413.
Crawford, BE, Weathers, DR. Osteoporotic marrow defects of the jaws. J Oral Surg. 1970;28:600–603.
Makek, M, Lello, GE. Focal osteoporotic bone marrow defects of the jaws. J Oral Maxillofac Surg. 1986;44:268–273.
Schneider, LC, Mesa, ML, Fraenkel, D. Osteoporotic bone marrow defect: radiographic features and pathogenic factors. Oral Surg Oral Med Oral Pathol. 1988;65:127–129.
Standish, SM, Shafer, WG. Focal osteoporotic bone marrow defects of the jaws. J Oral Surg. 1962;20:123–128.
Geist, JR, Katz, JO. The frequency and distribution of idiopathic osteosclerosis. Oral Surg Oral Med Oral Pathol. 1990;69:388–393.
Halse, A, Molven, O. Idiopathic osteosclerosis of the jaws followed through a period of 20-27 years. Int Endod J. 2002;35:747–751.
Kawai, T, Hirakuma, H, Murakami, S, et al. Radiographic investigation of idiopathic osteosclerosis of the jaws in Japanese dental outpatients. Oral Surg Oral Med Oral Pathol. 1992;74:237–242.
MacDonald-Jankowski, DS. Idiopathic osteosclerosis in the jaws of Britons and of the Hong Kong Chinese: radiology and systematic review. Dentomaxillofac Radiol. 1999;28:357–363.
McDonnell, D. Dense bone island: a review of 107 patients. Oral Surg Oral Med Oral Pathol. 1993;76:124–128.
Petrikowski, CG, Peters, E. Longitudinal radiographic assessment of dense bone islands of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:627–634.
Williams, TP, Brooks, SL. A longitudinal study of idiopathic osteosclerosis and condensing osteitis. Dentomaxillofac Radiol. 1998;27:275–278.
Yonetsu, K, Yuasa, K, Kanda, S. Idiopathic osteosclerosis of the jaws: panoramic radiographic and computed tomographic findings. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:517–521.
Frederiksen, NL, Wesley, RK, Sciubba, JJ, et al. Massive osteolysis of the maxillofacial skeleton: a clinical, radiographic, histologic and ultrastructural study. Oral Surg Oral Med Oral Pathol. 1983;55:470–480.
Heffez, L, Doku, HC, Carter, BL, et al. Perspective of massive osteolysis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1983;55:331–343.
Holroyd, I, Dillon, M, Roberts, GJ. Gorham’s disease: a case (including dental presentation) of vanishing bone disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:125–129.
Kayada, Y, Yoshiga, K, Takada, K, et al. Massive osteolysis of the mandible with subsequent obstructive sleep apnea syndrome: a case report. J Oral Maxillofac Surg. 1995;53:1463–1465.
Mignogna, MD, Fedele, S, Lo Russo, L, et al. Gorham’s disease of the mandible mimicking periodontal disease on radiograph. J Clin Periodontol. 2005;32:1022–1025.
Mignogna, MD, Fedele, S, Lo Russo, L, et al. Treatment of Gorham’s disease with zoledronic acid. Oral Oncol. 2005;41:747–750.
Patel, D. Gorham’s disease or massive osteolysis. Clin Med Res. 2005;3:65–74.
Ricalde, P, Ord, RA, Sun, CCJ. Vanishing bone disease in a five year old: report of a case and review of the literature. Int J Oral Maxillofac Surg. 2003;32:222–226.
Tsang, WM, Tong, ACK, Chow, LTC, et al. Massive osteolysis (Gorham disease) of the maxillofacial skeleton: report of 2 cases. J Oral Maxillofac Surg. 2004;62:225–230.
Ankrom, MA, Shapiro, JR. Paget’s disease of bone (osteitis deformans). J Am Geriatr Soc. 1998;46:1025–1033.
Carrillo, R, Morales, A, Rodriguez-Peralto, JL, et al. Benign fibro-osseous lesions in Paget’s disease of the jaws. Oral Surg Oral Med Oral Pathol. 1991;71:588–592.
Cheng, YSL, Wright, JM, Walstad, WR, et al. Osteosarcoma arising in Paget’s disease of the mandible. Oral Oncol. 2002;38:785–792.
Daroszewska, A, Ralston, SH. Mechanisms of disease: genetics of Paget’s disease of bone and related disorders. Nat Clin Pract Rheumatol. 2006;2:270–277.
Drake, WM, Kendler, DL, Brown, JP. Consensus statement on the modern therapy of Paget’s disease of bone from a Western Osteoporosis Alliance Symposium. Clin Ther. 2001;23:620–626.
Gleich, LL, Eberle, RC, Shaha, AR, et al. Paget’s sarcoma of the mandible. Head Neck. 1995;17:425–430.
Mailander, JC. The “black beard” sign of monostotic Paget’s disease of the mandible. Clin Nucl Med. 1986;11:325–327.
Mankin, HJ, Hornicek, FJ. Paget’s sarcoma: a historical and outcome review. Clin Orthop Relat Res. 2005;438:97–102.
Selby, PL, Davie, MW, Ralston, SH, et al. Guidelines on the management of Paget’s disease of bone. Bone. 2002;31:10–19.
Siris, ES. Paget’s disease of bone. J Bone Miner Res. 1998;13:1061–1065.
Smith, J, Eveson, J. Paget’s disease of bone with particular reference to dentistry. J Oral Pathol. 1981;10:233–247.
Takata, S, Yasui, N, Nakatsuka, K, et al. Evolution of understanding of genetics of Paget’s disease of bone and related diseases. J Bone Miner Metab. 2004;22:519–523.
Tillman, H. Paget’s disease of bone: a clinical, radiographic and histopathologic study of 24 cases involving the jaws. Oral Surg Oral Med Oral Pathol. 1962;15:1225–1234.
Whyte, MP. Paget’s disease of bone. N Engl J Med. 2006;355:593–600.
Adornato, MC, Paticoff, KA. Intralesional corticosteroid injection for treatment of central giant-cell granuloma. J Am Dent Assoc. 2001;132:186–190.
Auclair, PL, Cuenin, P, Kratochvil, FJ, et al. A clinical and histomorphologic comparison of the central giant cell granuloma and the giant cell tumor. Oral Surg Oral Med Oral Pathol. 1988;66:197–208.
DeLange, J, van den Akker, HP. Clinical and radiological features of central giant-cell lesions of the jaw. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:464–470.
DeLange, J, van den Akker, HP, Klip, H. Incidence and disease-free survival after surgical therapy of central giant cell granulomas of the jaw in the Netherlands: 1990-1995. Head Neck. 2004;26:792–795.
DeLange, J, van der Akker, HP, van Zanten, GOV, et al. Calcitonin therapy in central giant cell granuloma of the jaw: a randomized double-blind placebo-controlled study. Int J Oral Maxillofac Surg. 2006;35:791–795.
DeLange, J, Rosenberg, AJWP, van den Akker, HP, et al. Treatment of central giant cell granuloma of the jaw with calcitonin. Int J Oral Maxillofac Surg. 1999;28:372–376.
Ficarra, G, Kaban, LB, Hansen, LS. Giant cell lesions of the jaws: a clinicopathologic and cytometric study. Oral Surg Oral Med Oral Pathol. 1987;64:44–49.
Goldman, KE, Marshall, MK, Alessandrini, E, et al. Complications of alpha-interferon therapy for aggressive central giant cell lesion of the maxilla. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:285–291.
Itonaga, I, Hussein, I, Kudo, O, et al. Cellular mechanisms of osteoclast formation and lacunar resorption in giant cell granuloma of the jaw. J Oral Pathol Med. 2004;32:224–231.
Kaban, LB, Mulliken, JB, Ezekowitz, RA, et al. Antiangiogenic therapy of a recurrent giant cell tumor of the mandible with interferon alfa-2a. Pediatrics. 1999;103:1145–1149.
Kaban, LB, Troulis, MJ, Ebb, D, et al. Antiangiogenic therapy with interferon alpha for giant cell lesions of the jaws. J Oral Maxillofac Surg. 2002;60:1103–1111.
Kaffe, I, Ardekian, L, Taicher, S, et al. Radiologic features of central giant cell granuloma of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:720–726.
Kaplan, I, Manor, I, Yahalom, R, et al. Central giant cell granuloma associated with central ossifying fibroma of the jaws: a clinicopathologic study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103(4):e35–e41.
Kruse-Lösler, B, Diallo, R, Gaertner, C, et al. Central giant cell granuloma of the jaws: a clinical radiologic, and histopathologic study of 26 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:346–354.
Miloro, M, Quinn, PD. Synchronous central giant cell lesions of the jaws: report of a case and review of the literature. J Oral Maxillofac Surg. 1995;53:1350–1355.
Pogrel, MA. Calcitonin treatment for central giant cell granuloma. J Oral Maxillofac Surg. 2003;61:649–653.
Pogrel, MA, Regezi, JA, Harris, ST, et al. Calcitonin treatment for central giant cell granulomas of the mandible: report of two cases. J Oral Maxillofac Surg. 1999;57:848–853.
Stavropoulos, F, Katz, J. Central giant cell granulomas: a systematic review of the radiographic characteristics with the addition of 20 new cases. Dentomaxillofac Radiol. 2002;31:213–217.
Whitaker, SB, Waldron, CA. Central giant cell lesions of the jaws: a clinical, radiologic and histopathologic study. Oral Surg Oral Med Oral Pathol. 1993;75:199–208.
Yamaguchi, T, Dorfman, HD. Giant cell reparative granuloma: a comparative clinicopathologic study of lesions in gnathic and extragnathic sites. Int J Surg Pathol. 2001;9:189–200.
Beaman, FD, Bancroft, LW, Peterson, JJ. Imaging characteristics of cherubism. AJR Am J Roentgenol. 2004;182:1051–1054.
Kaugars, GE, Niamtu, J, III., Svirsky, JA. Cherubism: diagnosis, treatment, and comparison with central giant cell granulomas and giant cell tumors. Oral Surg Oral Med Oral Pathol. 1992;73:369–374.
Mangion, J, Rahman, N, Edkins, S, et al. The gene for cherubism maps to chromosome 4p16.3. Am J Hum Genet. 1999;65:151–157.
Meng, XM, Yu, SF, Yu, GY. Clinicopathologic study of 24 cases of cherubism. Int J Oral Maxillofac Surg. 2005;34:350–356.
Peñarrocha, M, Bonet, J, Minguez, M. Cherubism: a clinical, radiographic, and histopathologic comparison of 7 cases. J Oral Maxillofac Surg. 2006;64:924–930.
Peters, WJN. Cherubism: a study of twenty cases from one family. Oral Surg Oral Med Oral Pathol. 1979;47:307–311.
Southgate, J, Sarma, U, Townend, JV, et al. Study of the cell biology and biochemistry of cherubism. J Clin Pathol. 1998;51:831–837.
Tiziani, V, Reichenberger, E, Buzzo, CL, et al. The gene for cherubism maps to chromosome 4p16. Am J Hum Genet. 1999;65:158–166.
Ueki, Y, Tiziani, V, Santanna, C, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001:125–126.
Von Wowern, N. Cherubism: a 36-year long-term follow-up of 2 generations in different families and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:765–772.
Copete, MA, Kawamata, A, Langlais, RP. Solitary bone cyst of the jaws: radiographic review of 44 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:221–225.
Kaugars, GE, Cale, AE. Traumatic bone cyst. Oral Surg Oral Med Oral Pathol. 1987;63:318–324.
Matsumura, S, Murakami, S, Kakimoto, N, et al. Histopathologic and radiographic findings of the simple bone cyst. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:619–625.
Peñarrocha-Diago, M, Sanchis-Bielsa, JM, Bonet-Marco, J, et al. Surgical treatment and follow-up of solitary bone cyst of the mandible: a report of seven cases. Br J Oral Maxillofac Surg. 2001;39:221–223.
Perdiagão, PF, Silva, EC, Sakurai, E, et al. Idiopathic bone cavity: a clinical, radiographic, and histological study. Br J Oral Maxillofac Surg. 2003;41:407–409.
Saito, Y, Hoshino, Y, Nagamine, T, et al. Simple bone cyst: a clinical and histopathologic study of fifteen cases. Oral Surg Oral Med Oral Pathol. 1992;74:487–491.
Sapp, JP, Stark, ML. Self-healing traumatic bone cysts. Oral Surg Oral Med Oral Pathol. 1990;69:597–602.
Shimoyama, T, Horie, N, Nasu, D, et al. So-called simple bone cyst of the jaw: a family of pseudocysts of diverse nature and etiology. J Oral Sci. 1999;41:93–98.
Tong, AC, Ng, IO, Yan, BS. Variations in clinical presentations of the simple bone cyst: report of cases. J Oral Maxillofac Surg. 2003;61:1487–1491.
Althof, PA, Ohmori, K, Zhou, M, et al. Cytogenetic and molecular cytogenetic findings in 43 aneurysmal bone cysts: aberrations of 17p mapped to 17p13.2 by fluorescence in situ hybridization. Mod Pathol. 2004;17:518–525.
Bataineh, AB. Aneurysmal bone cysts of the maxilla: a clinicopathologic review. J Oral Maxillofac Surg. 1997;55:1212–1216.
Kaffe, I, Naor, H, Calderon, S, et al. Radiological and clinical features of aneurysmal bone cyst of the jaws. Dentomaxillofac Radiol. 1999;28:167–172.
Kalantar Motamedi, MH. Aneurysmal bone cysts of the jaws: clinicopathological features, radiographic evaluation and treatment analysis of 17 cases. J Craniomaxillofac Surg. 1998;26:56–62.
Mankin, HJ, Hornicek, FJ, Ortiz-Cruz, E, et al. Aneurysmal bone cyst: a review of 150 patients. J Clin Oncol. 2005;23:6756–6762.
Mendenhall, WM, Zlotecki, RA, Gibbs, CP, et al. Aneurysmal bone cyst. Am J Clin Oncol. 2006;29:311–315.
Padwa, BL, Denhart, BC, Kaban, LB. Aneurysmal bone cyst—“plus”: a report of three cases. J Oral Maxillofac Surg. 1997;55:1144–1152.
Struthers, PJ, Shear, M. Aneurysmal bone cyst of the jaws. I. Clinicopathologic features. Int J Oral Surg. 1984;13:85–91.
Struthers, PJ, Shear, M. Aneurysmal bone cyst of the jaws. II. Pathogenesis. Int J Oral Surg. 1984;13:92–100.
Toljanic, JS, Lechewski, E, Huvos, AG, et al. Aneurysmal bone cyst of the jaws: a case study and review of the literature. Oral Surg Oral Med Oral Pathol. 1987;64:72–77.
Fibro-Osseous Lesions—General Aspects and Classification
Brannon, RB, Fowler, CB. Benign fibro-osseous lesions: a review of current concepts. Adv Anat Pathol. 2001;8:126–143.
Koury, ME, Regezi, JA, Perrott, DH, et al. “Atypical” fibro-osseous lesions: diagnostic challenges and treatment concepts. Int J Oral Maxillofac Surg. 1995;24:162–169.
MacDonald-Jankowski, DS. Fibro-osseous lesions of the face and jaws. Clin Radiol. 2004;59:11–25.
Slootweg, PJ. Maxillofacial fibro-osseous lesions: classification and differential diagnosis. Semin Diagn Pathol. 1996;13:104–112.
Waldron, CA. Fibro-osseous lesions of the jaws. J Oral Maxillofac Surg. 1993;51:828–835.
Wright, JM. Reactive, dysplastic and neoplastic conditions of periodontal ligament origin. Periodontol 2000. 1999;21:7–15.
Akintoye, SO, Lee, JS, Feimster, T, et al. Dental characteristics of fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:275–282.
Akintoye, SO, Otis, LL, Atkinson, JC, et al. Analyses of vari-able panoramic radiographic characteristics of maxillo-mandibular fibrous dysplasia in McCune-Albright syndrome. Oral Dis. 2004;10:36–43.
Becelli, R, Perugini, M, Cerulli, G, et al. Surgical treatment of fibrous dysplasia of the cranio-maxillo-facial area. Review of the literature and personal experience from 1984 to 1999. Minerva Stomatol. 2002;51:293–300.
Cohen, MM, Jr., Howell, RE. Etiology of fibrous dysplasia and McCune-Albright syndrome. Int J Oral Maxillofac Surg. 1999;28:366–371.
DiCaprio, MR, Enneking, WF. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87:1848–1864.
Frodel, JL, Funk, G, Boyle, J, et al. Management of aggressive midface and orbital fibrous dysplasia. Arch Facial Plast Surg. 2000;2:187–195.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. McCune-Albright syndrome. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:334–338.
MacDonald-Jankowski, D. Fibrous dysplasia in the jaws of a Hong Kong population: radiographic presentation and systematic review. Dentomaxillofac Radiol. 1999;28:195–202.
Petrikowski, CG, Pharoah, MJ, Lee, L, et al. Radiographic differentiation of osteogenic sarcoma, osteomyelitis, and fibrous dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;80:744–750.
Waldron, CA, Giansanti, JS. Benign fibro-osseous lesions of the jaws. I. Fibrous dysplasia of the jaws. Oral Surg Oral Med Oral Pathol. 1973;35:190–201.
Groot, RH, van Merkesteyn, JPR, Bras, J. Diffuse sclerosing osteomyelitis and florid osseous dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:333–342.
Higuchi, Y, Nakamura, N, Tashiro, H. Clinicopathologic study of cemento-osseous dysplasia producing cysts of the mandible. Oral Surg Oral Med Oral Pathol. 1988;65:339–342.
Kawai, T, Hiranuma, H, Kishino, M, et al. Cemento-osseous dysplasia of the jaws in 54 patients: a radiographic study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:107–114.
MacDonald-Jankowski, DS. Florid cemento-osseous dysplasia: a systematic review. Dentomaxillofac Radiol. 2003;32:141–149.
Mahomed, F, Altini, M, Meer, S, et al. Cemento-osseous dysplasia with associated simple bone cysts. J Oral Maxillofac Surg. 2005;63:1549–1554.
Melrose, RJ. The clinico-pathologic spectrum of cemento-osseous dysplasia. Oral Maxillofac Clin North Am. 1997;9:643–653.
Melrose, RJ, Abrams, AM, Mills, BG. Florid osseous dysplasia, a clinico-pathologic study of thirty-four cases. Oral Surg Oral Med Oral Pathol. 1976;41:62–82.
Schneider, LC, Dolinsky, HB, Grodjesk, JE, et al. Malignant spindle cell tumor arising in the mandible of a patient with florid osseous dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;88:69–73.
Schneider, LC, Mesa, ML. Differences between florid osseous dysplasia and diffuse sclerosing osteomyelitis. Oral Surg Oral Med Oral Pathol. 1990;70:308–312.
Su, L, Weathers, DR, Waldron, CA. Distinguishing features of focal cemento-osseous dysplasias and cemento-ossifying fibromas. I. A pathologic spectrum of 316 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:301–309.
Summerlin, DJ, Tomich, CE. Focal cemento-osseous dysplasia: a clinicopathologic study of 221 cases. Oral Surg Oral Med Oral Pathol. 1994;78:611–620.
Waldron, CA. Fibro-osseous lesions of the jaws. J Oral Maxillofac Surg. 1985;43:249–262.
Familial Gigantiform Cementoma
Abdelsayed, RA, Eversole, LR, Singh, BS, et al. Gigantiform cementoma: clinicopathologic presentation of 3 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;91:438–444.
Cannon, JS, Keller, EE, Dahlin, DC. Gigantiform cementoma: report of two cases (mother and son). J Oral Surg. 1980;38:65–70.
Coleman, H, Altini, M, Kieser, J, et al. Familial florid cemento-osseous dysplasia—a case report and review of the literature. J Dent Assoc S Afr. 1996;51:766–770.
Finical, SJ, Kane, WJ, Clay, RP, et al. Familial gigantiform cementoma. Plast Reconstr Surg. 1999;103:949–954.
Rossbach, HC, Letson, D, Lacson, A, et al. Familial gigantiform cementoma with brittle bone disease, pathologic fractures, and osteosarcoma: a possible explanation of an ancient mystery. Pediatr Blood Cancer. 2005;44:390–396.
Toffanin, A, Benetti, R, Manconi, R. Familial florid cemento-osseous dysplasia: a case report. J Oral Maxillofac Surg. 2000;58:1440–1446.
Young, SK, Markowitz, NR, Sullivan, S, et al. Familial gigantiform cementoma: classification and presentation of a large pedigree. Oral Surg Oral Med Oral Pathol. 1989;68:740–747.
Eversole, LR, Leider, AS, Nelson, K. Ossifying fibroma: a clinicopathologic study of 64 cases. Oral Surg Oral Med Oral Pathol. 1985;60:505–511.
MacDonald-Jankowski, DS. Cemento-ossifying fibromas in the jaws of Hong Kong Chinese. Dentomaxillofac Radiol. 1998;27:298–304.
Pimenta, FJ, Silveira, LFG, Tavares, GC, et al. HRPT2 gene alterations in ossifying fibroma of the jaws. Oral Oncol. 2006;42:735–739.
Slootweg, PJ. Maxillofacial fibro-osseous lesions: classification and differential diagnosis. Semin Diagn Pathol. 1996;13:104–112.
Su, L, Weathers, DR, Waldron, CA. Distinguishing features of focal cemento-osseous dysplasias and cemento-ossifying fibromas. I. A pathologic spectrum of 316 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:301–309.
Waldron, CA, Giansanti, JS. Benign fibro-osseous lesions of the jaws. II. Benign fibro-osseous lesions of periodontal ligament origin. Oral Surg Oral Med Oral Pathol. 1973;35:340–350.
El-Mofty, S. Psammomatoid and trabecular juvenile ossifying fibroma of the craniofacial skeleton: two distinct clinico-pathologic entities. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:296–304.
Johnson, LC, Yousefi, M, Vinh, TN, et al. Juvenile active ossifying fibroma: its nature, dynamics and origin. Acta Otolaryngol Suppl. 1991;488:1–40.
Makek, MS. So-called “fibroosseous lesions” of tumorous origin: biology confronts terminology. J Craniomaxillofac Surg. 1987;15:154–168.
Margo, C, Ragsdale, BD, Perman, KI, et al. Psammomatoid (juvenile) ossifying fibroma of the orbit. Ophthalmology. 1985;92:150–159.
Slootweg, PJ, Panders, AK, Koopmans, R, et al. Juvenile ossifying fibroma. An analysis of 33 cases with emphasis on histopathological aspects. J Oral Pathol Med. 1994;23:385–388.
Williams, HK, Mangham, C, Speight, PM. Juvenile ossifying fibroma. An analysis of eight cases and a comparison with other fibro-osseous lesions. J Oral Pathol Med. 2000;29:13–18.
Cutilli, BJ, Quinn, PD. Traumatically induced peripheral osteoma: report of case. Oral Surg Oral Med Oral Pathol. 1992;73:667–669.
Johann, AC, de Freitas, JB, de Aguiar, MC, et al. Peripheral osteoma of the mandible: case report and review of the literature. J Craniomaxillofac Surg. 2005;33:276–281.
Kondoh, T, Seto, K, Kobayashi, K. Osteoma of the mandibular condyle: report of a case with a review of the literature. J Oral Maxillofac Surg. 1998;56:972–979.
Ortakoglu, K, Gunaydin, Y, Aydintug, YS, et al. Osteoma of the mandibular condyle: report of a case with 5-year follow-up. Mil Med. 2005;170:117–120.
Richards, HE, Strider, JW, Jr., Short, SG, et al. Large peripheral osteoma arising from the genial tubercle area. Oral Surg Oral Med Oral Pathol. 1986;61:268–271.
Schneider, LC, Dolinski, HB, Grodjesk, JE. Solitary peripheral osteoma of the jaws: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1980;38:452–455.
Woldenberg, Y, Nash, M, Bodner, L. Peripheral osteoma of the maxillofacial region. Diagnosis and management: a study of 14 cases. Med Oral Patol Oral Cir Bucal. 2005;10(suppl 2):E139–E142.
Yassin, OM, Bataineh, AB, Mansour, MJ. An unusual osteoma of the mandible. J Clin Pediatr Dent. 1997;21:337–340.
Baykul, T, Heybeli, N, Oyar, O, et al. Multiple huge osteomas of the mandible causing disfigurement related with Gardner’s syndrome: case report. Auris Nasus Larynx. 2003;30:447–451.
Bilkay, U, Erdem, O, Ozek, C, et al. Benign osteoma with Gardner syndrome: review of the literature and report of a case. J Craniofac Surg. 2004;15:506–509.
Butler, J, Healy, C, Toner, M, et al. Gardner syndrome—review and report of a case. Oral Oncol Extra. 2005;41:89–92.
Chimenos-Küstner, E, Pascual, M, Blanco, I, et al. Hereditary familial polyposis and Gardner’s syndrome: contribution of the odontostomatology examination in its diagnosis and a case description. Med Oral Patol Oral Cir Bucal. 2005;10:402–409.
Fotiadis, C, Tsekouras, DK, Antonakis, P, et al. A case report and review of the literature. World J Gastroenterol. 2005;11:5408–5411.
Galiatsatos, P, Foulkes, WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385–398.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Gardner syndrome. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:437–444.
Ida, M, Nakamura, T, Utsunomiya, J. Osteomatous changes and tooth abnormalities found in the jaws of patients with adenomatosis coli. Oral Surg Oral Med Oral Pathol. 1981;52:2–11.
Lew, D, DeWitt, A, Hicks, RJ, et al. Osteomas of the condyle associated with Gardner’s syndrome causing limited mandibular movement. J Oral Maxillofac Surg. 1999;57:1004–1009.
Sondergaard, JO, Bulows, S, Jarvinen, H, et al. Dental anomalies in familial adenomatous polyposis. Acta Odontol Scand. 1987;45:61–63.
Thacker, NS, Evans, DGR, Horner, K, et al. Florid oral manifestations in an atypical familial adenomatous polyposis family with late presentation of colorectal polyps. J Oral Pathol Med. 1996;25:459–462.
Ahmed, MS, Nwoku, AL. Benign osteoblastoma of the mandibular ramus: review of the literature and report of a case. J Oral Maxillofac Surg. 2000;58:1310–1317.
Angiero, F, Mellone, P, Baldi, A, et al. Osteoblastoma of the jaws: report of two cases and review of the literature. In Vivo. 2006;20:665–670.
Capodiferro, S, Maiorano, E, Giardina, C, et al. Osteoblastoma of the mandible: clinicopathologic study of four cases and literature review. Head Neck. 2005;27:616–625.
Dorfman, HD, Weiss, SW. Borderline osteoblastic tumors: problems in differential diagnosis of aggressive osteoblastoma and low grade osteosarcoma. Semin Diagn Pathol. 1984;1:215–245.
Eisenbud, L, Kahn, L, Friedman, E. Benign osteoblastoma of the mandible: fifteen year follow-up showing spontaneous regression after biopsy. J Oral Maxillofac Surg. 1987;45:53–57.
Jones, AC, Prihoda, TJ, Kacher, JE, et al. Osteoblastoma of the maxilla and mandible: a report of 24 cases, review of the literature, and discussion of its relationship to osteoid osteoma of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:639–650.
O’Connell, JX, Nanthakumar, SS, Nielsen, GP, et al. Osteoid osteoma: the uniquely innervated bone tumor. Mod Pathol. 1998;11:175–180.
Peters, TED, Oliver, DR, McDonald, JS. Benign osteoblastoma of the mandible: report of a case. J Oral Maxillofac Surg. 1995;53:1347–1349.
Rawal, YB, Angiero, F, Allen, CM, et al. Gnathic osteoblastoma: clinicopathologic review of seven cases with long-term follow-up. Oral Oncol. 2006;42:123–130.
Unni, KK. Dahlin’s bone tumors: general aspects and data on 11,087 cases, ed 5, Philadelphia: Lippincott-Raven; 1996:131–142.
Biggs, JT, Benenati, FW. Surgically treating a benign cementoblastoma while retaining the involved tooth. J Am Dent Assoc. 1995;126:1288–1290.
Brannon, RB, Fowler, CB, Carpenter, WM, et al. Cementoblastoma: an innocuous neoplasm? A clinicopathologic study of 44 cases and review of the literature with special emphasis on recurrence. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:311–320.
Jelic, JS, Loftus, MJ, Miller, AS, et al. Benign cementoblastoma: report of an unusual case and analysis of 14 additional cases. J Oral Maxillofac Surg. 1993;51:1033–1037.
Ohki, K, Kumamoto, H, Nitta, Y, et al. Benign cementoblastoma involving multiple maxillary teeth: report of a case with a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:53–58.
Pacifici, L, Tallarico, M, Bartoli, A, et al. Benign cementoblastoma: a clinical case of conservative surgical treatment of the involved tooth. Minerva Stomatol. 2004;53:685–691.
Slootweg, PJ. Cementoblastoma and osteoblastoma: a comparison of histologic features. J Oral Pathol Med. 1992;21:385–389.
Ulmansky, M, Hjørting-Hansen, E, Praetorius, F, et al. Benign cementoblastoma. A review and five new cases. Oral Surg Oral Med Oral Pathol. 1994;77:48–55.
Dorfman, HD, Czerniak, B. Bone tumors. St Louis: Mosby, 1998;253–285.
Hyams, VJ, Batsakis, JG, Michaels, L. Tumors of the upper respiratory tract and ear, atlas of tumor pathology. Washington, DC: Armed Forces Institute of Pathology, 1988;163–164. [series 2, fascicle 25].
Lazow, SK, Pihlstrom, RT, Solomon, MP. Condylar chondroma: report of a case. J Oral Maxillofac Surg. 1998;56:373–378.
Unni, KK. Dahlin’s bone tumors. General aspects and data on 11,087 cases, ed 5, Philadelphia: Lippincott-Raven; 1996:25–35.
Chow, LTC, Lin, J, Yip, KMH, et al. Chondromyxoid fibroma-like osteosarcoma: a distinct variant of low-grade osteosarcoma. Histopathology. 1996;29:429–436.
Damm, DD, White, DK, Geissler, RH, et al. Chondromyxoid fibroma of the maxilla: electron microscopic findings and review of the literature. Oral Surg Oral Med Oral Pathol. 1985;59:176–183.
Hammad, HM, Hammond, HL, Kurago, ZB, et al. Chondromyxoid fibroma of the jaws: case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:293–300.
Macan, D, Cabov, T, Uglesic, V, et al. Chondromyxoid fibroma of the mandible. Br J Oral Maxillofac Surg. 2003;41:261–263.
Müller, S, Whitaker, SB, Weathers, DR. Chondromyxoid fibroma of the mandible: diagnostic image cytometry findings and review of the literature. Oral Surg Oral Med Oral Pathol. 1992;73:465–468.
Smith, CA, Magenis, RE, Himoe, E, et al. Chondromyxoid fibroma of the nasal cavity with an interstitial insertion between chromosomes 6 and 19. Cancer Genet Cytogenet. 2006;171:97–100.
Tallini, G, Dorfman, H, Brys, P, et al. Correlation between clinicopathological features and karyotype in 100 cartilaginous and chordoid tumours. A report from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. J Pathol. 2002;196:194–203.
Wu, CT, Inwards, CY, O’Laughlin, S, et al. Chondromyxoid fibroma of bone: a clinicopathologic review of 278 cases. Hum Pathol. 1998;29:438–446.
Ardekian, L, Faquin, W, Troulis, MJ, et al. Synovial chondromatosis of the temporomandibular joint: report and analysis of eleven cases. J Oral Maxillofac Surg. 2005;63:941–947.
Deahl, ST, Ruprecht, A. Asymptomatic, radiographically detected chondrometaplasia in the temporomandibular joint. Oral Surg Oral Med Oral Pathol. 1991;72:371–374.
Holmlund, AB, Eriksson, L, Reinholt, FP. Synovial chondromatosis of the temporomandibular joint: clinical, surgical and histological aspects. Int J Oral Maxillofac Surg. 2003;32:143–147.
Karlis, V, Glickman, RS, Zaslow, M. Synovial chondromatosis of the temporomandibular joint with intracranial extension. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:664–666.
Lustman, J, Zeltzer, R. Synovial chondromatosis of the temporomandibular joint: review of the literature and case report. Int J Oral Maxillofac Surg. 1989;18:90–94.
Miyamoto, H, Sakashita, H, Wilson, DF, et al. Synovial chondromatosis of the temporomandibular joint. Br J Oral Maxillofac Surg. 2000;38:205–208.
Petito, AR, Bennett, J, Assael, LA, et al. Synovial chondromatosis of the temporomandibular joint: varying presentation in 4 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:758–764.
Von Lindern, JJ, Theuerkauf, I, Niederhagen, B, et al. Synovial chondromatosis of the temporomandibular joint: clinical, diagnostic, and histomorphologic findings. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:31–38.
Yu, Q, Yang, Y, Wang, P, et al. CT features of synovial chondromatosis in the temporomandibular joint. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:524–528.
Bakaeen, G, Rajab, LD. Desmoplastic fibroma of the mandible: report of a case. Int J Paediatr Dent. 1999;9:117–121.
Bertoni, F, Present, D, Marchetti, C, et al. Desmoplastic fibroma of the jaw: the experience of the Istituto Beretta. Oral Surg Oral Med Oral Pathol. 1986;61:179–184.
Fletcher, CDM, Uni, KK, Mertens, F. WHO classification of tumors: pathology and genetics of tumors of soft tissue and bone. Lyon, France: IARC Press, 2002;288.
Freedman, PD, Cardo, VA, Kerpel, SM, et al. Desmoplastic fibroma (fibromatosis) of the jaw-bones: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1978;46:386–395.
Said-Al-Naief, N, Fernandes, R, Louis, P, et al. Desmoplastic fibroma of the jaw: a case report and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:82–94.
Templeton, K, Glass, N, Young, SK. Desmoplastic fibroma of the mandible in a child: report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:620–623.
Vargas-Gonzalez, R, San Martin-Brieke, W, Gil-Orduna, C, et al. Desmoplastic fibroma-like tumor of maxillofacial region associated with tuberous sclerosis. Pathol Oncol Res. 2004;10:237–239.
August, M, Magennis, P, Dewitt, D. Osteogenic sarcoma of the jaws: factors influencing prognosis. Int J Oral Maxillofac Surg. 1997;26:198–204.
Bennett, JH, Thomas, G, Evans, AW, et al. Osteosarcoma of the jaws: a 30-year retrospective review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:323–333.
Canadian Society of Otolaryngology-Head and Neck Surgery Oncology Study Group. Osteogenic sarcoma of the mandible and maxilla: a Canadian review (1980-2000). J Otolaryngol. 2004;33:139–144.
Fernandes, R, Nikitakis, NG, Pazoki, A, et al. Osteogenic sarcoma of the jaw: a 10-year experience. J Oral Maxillofac Surg. 2007;65:1286–1291.
Garrington, GE, Scofield, HH, Cornyn, J, et al. Osteosarcoma of the jaws: analysis of 56 cases. Cancer. 1967;20:377–391.
Givol, N, Buchner, A, Taicher, S, et al. Radiological features of osteogenic sarcoma of the jaws. A comparative study of different radiographic modalities. Dentomaxillofac Radiol. 1998;27:313–320.
Huvos, AG, Woodard, HQ, Cahan, WG, et al. Postradiation sarcoma of bone and soft tissues: a clinicopathologic study of 66 patients. Cancer. 1985;55:1244–1255.
Lewis, M, Perl, A, Som, PM, et al. Osteogenic sarcoma of the jaw. A clinicopathologic review of 12 patients. Arch Otolaryngol Head Neck Surg. 1997;123:169–174.
Nakayama, E, Sugiura, K, Ishibashi, H, et al. The clinical and diagnostic imaging findings of osteosarcoma of the jaw. Dentomaxillofac Radiol. 2005;34:182–188.
Nakayama, E, Sugiura, K, Kobayashi, I, et al. The association between the computed tomography findings, histologic features, and outcome of osteosarcoma of the jaw. J Oral Maxillofac Surg. 2005;63:311–318.
Oda, D, Bavisotto, LM, Schmidt, RA, et al. Head and neck osteosarcoma at the University of Washington. Head Neck. 1997;19:513–523.
Ogunlewe, MO, Ajayi, OF, Adeyemo, WL, et al. Osteogenic sarcoma of the jaw bones: a single institution experience over a 21-year period. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:76–81.
Patel, SG, Meyers, P, Huvos, AG, et al. Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer. 2002;95:1495–1503.
Patel, SG, See, ACH, Williamson, PA, et al. Radiation-induced sarcoma of the head and neck. Head Neck. 1999;21:346–354.
Piattelli, A, Favia, GF. Periosteal osteosarcoma of the jaws: report of 2 cases. J Periodontol. 2000;71:325–329.
Rosen, G, Caparros, B, Huvos, AG. Preoperative chemotherapy for osteogenic sarcoma: selection of postoperative chemotherapy based upon the response of the primary tumor to preoperative chemotherapy. Cancer. 1982;49:1221–1230.
Slootweg, PJ, Müller, H. Osteosarcoma of the jawbones. J Maxillofac Surg. 1985;13:158–166.
Smeele, LE, Kostense, PJ, van der Waal, I, et al. Effect of chemotherapy on survival of craniofacial osteosarcoma: a systematic review of 201 patients. J Clin Oncol. 1997;15:363–367.
Tanzawa, H, Uchiyama, S, Sato, K. Statistical observation of osteosarcoma of the maxillofacial region in Japan. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1991;2:444–448.
van Es, RJJ, Keus, RB, van der Waal, I, et al. Osteosarcoma of the jaw bones. Long-term follow up of 48 cases. Int J Oral Maxillofac Surg. 1997;26:191–197.
Aziz, SR, Miremadi, AR, McCabe, JC. Mesenchymal chondrosarcoma of the maxilla with diffuse metastasis: case report and literature review. J Oral Maxillofac Surg. 2002;60:931–935.
Garrington, GE, Collett, WK. Chondrosarcoma. I. A selected literature review. J Oral Pathol. 1988;17:1–11.
Garrington, GE, Collett, WK. Chondrosarcoma. II. Chondrosarcoma of the jaws: analysis of 37 cases. J Oral Pathol. 1988;17:12–20.
Koch, BB, Karnell, LH, Hoffman, HT, et al. National Cancer Database report on chondrosarcoma of the head and neck. Head Neck. 2000;22:408–425.
Lockhart, R, Menard, P, Martin, JP, et al. Mesenchymal chondrosarcoma of the jaws: report of four cases. Int J Oral Maxillofac Surg. 1998;27:358–362.
Nakashima, Y, Unni, KK, Shives, TC, et al. Mesenchymal chondrosarcoma of bone and soft tissue: a review of 111 cases. Cancer. 1985;57:2444–2453.
Saito, K, Unni, KK, Wollan, PC, et al. Chondrosarcoma of the jaw and facial bones. Cancer. 1995;76:1550–1558.
Vencio, EF, Reeve, CM, Unni, KK, et al. Mesenchymal chondrosarcoma of the jaw bones. Clinicopathologic study of 19 cases. Cancer. 1998;82:2350–2355.
Arafat, A, Ellis, G, Adrian, JC. Ewing’s sarcoma of the jaws. Oral Surg Oral Med Oral Pathol. 1983;55:589–596.
Berk, R, Heller, A, Heller, D, et al. Ewing’s sarcoma of the mandible: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;79:159–162.
Daw, NC, Mahmoud, HH, Meyer, WH, et al. Bone sarcomas of the head and neck in children: the St. Jude Children’s Research Hospital experience. Cancer. 2000;88:2172–2180.
de Alava, E, Gerald, WL. Molecular biology of the Ewing’s sarcoma/primitive neuroectodermal tumor family. J Clin Oncol. 2000;18:204–213.
Fiorillo, A, Tranfa, F, Canale, G, et al. Primary Ewing’s sarcoma of the maxilla, a rare and curable localization: report of two new cases, successfully treated by radiotherapy and systemic chemotherapy. Cancer Lett. 1996;103:177–182.
Infante-Cassio, P, Gutierrez-Perez, JL, Garcia-Perla, A, et al. Primary Ewing’s sarcoma of the maxilla and zygoma: report of a case. J Oral Maxillofac Surg. 2005;63:1539–1542.
Kissane, JM, Askin, FB, Foulkes, M, et al. Ewing’s sarcoma of bone: clinicopathologic aspects of 303 cases from the intergroup Ewing’s sarcoma study. Hum Pathol. 1983;14:773–779.
La, TH, Meyers, PA, Wexler, LH, et al. Radiation therapy for Ewing’s sarcoma: results from Memorial Sloan-Kettering in the modern era. Int J Radiat Oncol Biol Phys. 2006;2:544–550.
Schultze-Mosgau, S, Thorwarth, M, Wehrhan, F, et al. Ewing sarcoma of the mandible in a child: interdisciplinary treatment concepts and surgical reconstruction. J Craniofac Surg. 2005;16:1140–1146.
Vaccani, JP, Forte, V, de Jong, AL, et al. Ewing’s sarcoma of the head and neck in children. Int J Pediatr Otorhinolaryngol. 1999;48:209–216.
Wexler, LH, Kacker, A, Piro, JD, et al. Combined modality treatment of Ewing’s sarcoma of the maxilla. Head Neck. 2003;25:168–172.
Bouquot, JE, Weiland, LH, Kurland, LT. Metastasis to and from the upper aero-digestive tract in the population of Rochester, Minnesota, 1935-1984. Head Neck. 1989;11:212–218.
Clausen, F, Poulson, H. Metastatic carcinoma of the jaws. Acta Pathol Microbiol Scand. 1963;57:361–374.
D’Silva, NJ, Summerlin, DJ, Cordell, KG, et al. Metastatic tumors in the jaws: a retrospective study of 114 cases. J Am Dent Assoc. 2006;137:1667–1672.
Hashimoto, N, Kurihara, K, Yamasaki, H, et al. Pathologic characteristics of metastatic carcinoma in the human mandible. J Oral Pathol. 1987;16:362–367.
O’Carroll, MK, Krolls, SO, Mosca, NG. Metastatic carcinoma to the mandible: report of two cases. Oral Surg Oral Med Oral Pathol. 1993;76:368–374.
Zachariades, N. Neoplasms metastatic to the mouth, jaws and surrounding tissues. J Craniomaxillofac Surg. 1989;17:283–290.
*Dr. Charles A. Waldron wrote the original version of this chapter in the first edition of this book.