Soft Tissue Tumors
Inflammatory Papillary Hyperplasia
Peripheral Giant Cell Granuloma
Palisaded Encapsulated Neuroma
Multiple Endocrine Neoplasia Type 2B
Melanotic Neuroectodermal Tumor of Infancy
Hemangioma and Vascular Malformations
Hemangiopericytoma–Solitary Fibrous Tumor
Osseous and Cartilaginous Choristomas
Malignant Fibrous Histiocytoma
FIBROMA (IRRITATION FIBROMA; TRAUMATIC FIBROMA; FOCAL FIBROUS HYPERPLASIA; FIBROUS NODULE)
The fibroma is the most common “tumor” of the oral cavity. However, it is doubtful that it represents a true neoplasm in most instances; rather, it is a reactive hyperplasia of fibrous connective tissue in response to local irritation or trauma.
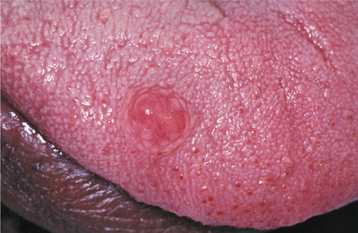
CLINICAL FEATURES: Although the irritation fibroma can occur anywhere in the mouth, the most common location is the buccal mucosa along the bite line. Presumably, this is a consequence of trauma from biting the cheek (Figs. 12-1 and 12-2). The labial mucosa, tongue, and gingiva also are common sites (Figs. 12-3 and 12-4). It is likely that many gingival fibromas represent fibrous maturation of a preexisting pyogenic granuloma. The lesion typically appears as a smooth-surfaced pink nodule that is similar in color to the surrounding mucosa. In black patients, the mass may demonstrate gray-brown pigmentation. In some cases the surface may appear white as a result of hyperkeratosis from continued irritation. Most fibromas are sessile, although some are pedunculated. They range in size from tiny lesions that are only a couple of millimeters in diameter to large masses that are several centimeters across; however, most fibromas are 1.5 cm or less in diameter. The lesion usually produces no symptoms, unless secondary traumatic ulceration of the surface has occurred. Irritation fibromas are most common in the fourth to sixth decades of life, and the male-to-female ratio is almost 1:2 for cases submitted for biopsy.
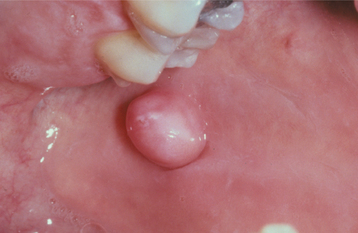
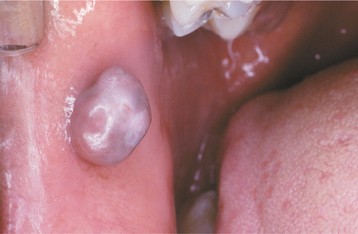
Fig. 12-2 Fibroma. Black patient with a smooth-surfaced pigmented nodule on the buccal mucosa near the commissure.
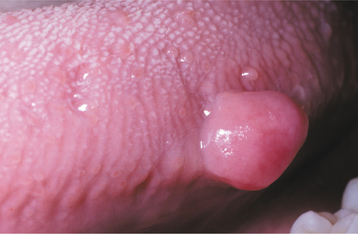
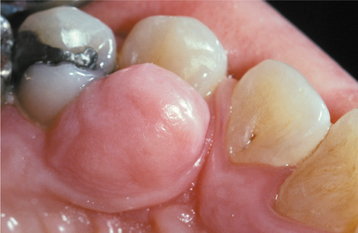
Fig. 12-4 Fibroma. Smooth-surfaced, pink nodular mass of the palatal gingiva between the cuspid and first bicuspid.
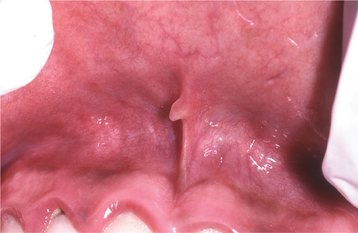
The frenal tag is a commonly observed type of fibrous hyperplasia, which most frequently occurs on the maxillary labial frenum. Such lesions present as small, asymptomatic, exophytic growths attached to the thin frenum surface (Fig. 12-5).
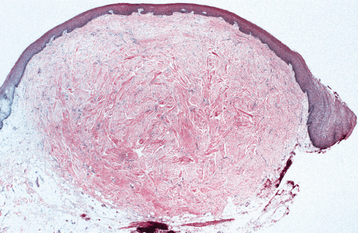
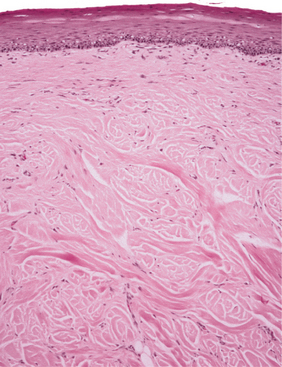
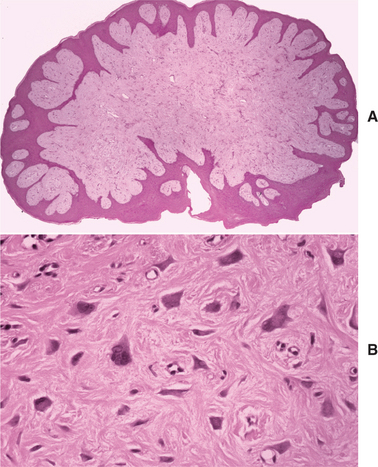
HISTOPATHOLOGIC FEATURES: Microscopic examination of the irritation fibroma shows a nodular mass of fibrous connective tissue covered by stratified squamous epithelium (Figs. 12-6 and 12-7). This connective tissue is usually dense and collagenized, although in some cases it is looser in nature. The lesion is not encapsulated; the fibrous tissue instead blends gradually into the surrounding connective tissues. The collagen bundles may be arranged in a radiating, circular, or haphazard fashion. The covering epithelium often demonstrates atrophy of the rete ridges because of the underlying fibrous mass. However, the surface may exhibit hyperkeratosis from secondary trauma. Scattered inflammation may be seen, most often beneath the epithelial surface. Usually this inflammation is chronic and consists mostly of lymphocytes and plasma cells.
TREATMENT AND PROGNOSIS: The irritation fibroma is treated by conservative surgical excision; recurrence is extremely rare. However, it is important to submit the excised tissue for microscopic examination because other benign or malignant tumors may mimic the clinical appearance of a fibroma.
Because frenal tags are small, innocuous growths that are easily diagnosed clinically, no treatment is usually necessary.
GIANT CELL FIBROMA
The giant cell fibroma is a fibrous tumor with distinctive clinicopathologic features. Unlike the traumatic fibroma, it does not appear to be associated with chronic irritation. The giant cell fibroma represents approximately 2% to 5% of all oral fibrous proliferations submitted for biopsy.
CLINICAL FEATURES: The giant cell fibroma is typically an asymptomatic sessile or pedunculated nodule, usually less than 1 cm in size (Fig. 12-8). The surface of the mass often appears papillary; therefore, the lesion may be clinically mistaken for a papilloma. Compared with the common irritation fibroma, the lesion usually occurs at a younger age. In about 60% of cases, the lesion is diagnosed during the first 3 decades of life. Some studies have suggested a slight female predilection. Approximately 50% of all cases occur on the gingiva. The mandibular gingiva is affected twice as often as the maxillary gingiva. The tongue and palate also are common sites.
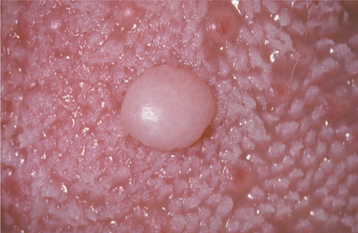
The retrocuspid papilla is a microscopically similar developmental lesion that occurs on the gingiva lingual to the mandibular cuspid. It is frequently bilateral and typically appears as a small, pink papule that measures less than 5 mm in diameter (Fig. 12-9). Retrocuspid papillae are quite common, having been reported in 25% to 99% of children and young adults. The prevalence in older adults drops to 6% to 19%, suggesting that the retrocuspid papilla represents a normal anatomic variation that disappears with age.
HISTOPATHOLOGIC FEATURES: Microscopic examination of the giant cell fibroma reveals a mass of vascular fibrous connective tissue, which is usually loosely arranged (Fig. 12-10). The hallmark is the presence of numerous large, stellate fibroblasts within the superficial connective tissue. These cells may contain several nuclei. Frequently, the surface of the lesion is pebbly. The covering epithelium often is thin and atrophic, although the rete ridges may appear narrow and elongated.
EPULIS FISSURATUM (INFLAMMATORY FIBROUS HYPERPLASIA; DENTURE INJURY TUMOR; DENTURE EPULIS)
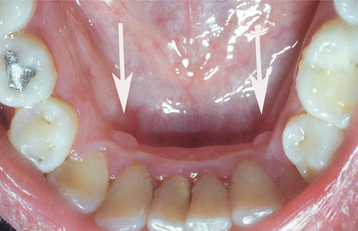
The epulis fissuratum is a tumorlike hyperplasia of fibrous connective tissue that develops in association with the flange of an ill-fitting complete or partial denture. Although the simple term epulis sometimes is used synonymously for epulis fissuratum, epulis is actually a generic term that can be applied to any tumor of the gingiva or alveolar mucosa. Therefore, some authors have advocated not using this term, preferring to call these lesions inflammatory fibrous hyperplasia or other descriptive names. However, the term epulis fissuratum is still widely used today and is well understood by virtually all clinicians. Other examples of epulides include the giant cell epulis (peripheral giant cell granuloma) (see page 520), ossifying fibroid epulis (peripheral ossifying fibroma) (see page 521), and congenital epulis (see page 537).
CLINICAL FEATURES: The epulis fissuratum typically appears as a single or multiple fold or folds of hyperplastic tissue in the alveolar vestibule (Figs. 12-11 and 12-12). Most often, there are two folds of tissue, and the flange of the associated denture fits conveniently into the fissure between the folds. The redundant tissue is usually firm and fibrous, although some lesions appear erythematous and ulcerated, similar to the appearance of a pyogenic granuloma. Occasional examples of epulis fissuratum demonstrate surface areas of inflammatory papillary hyperplasia (see page 512). The size of the lesion can vary from localized hyperplasias less than 1 cm in size to massive lesions that involve most of the length of the vestibule. The epulis fissuratum usually develops on the facial aspect of the alveolar ridge, although occasional lesions are seen lingual to the mandibular alveolar ridge (Fig. 12-13).
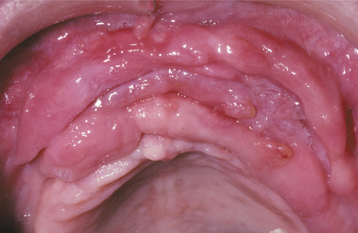
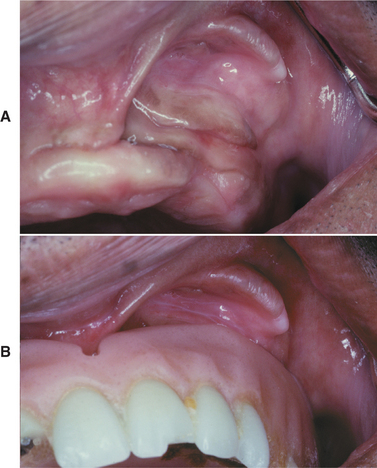
Fig. 12-12 Epulis fissuratum. A, Several folds of hyperplastic tissue in the maxillary vestibule. B, An ill-fitting denture fits into the fissure between two of the folds. (Courtesy of Dr. William Bruce.)
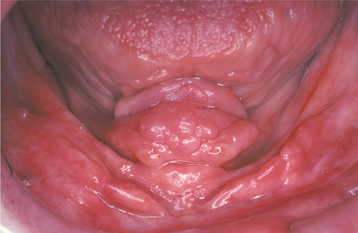
Fig. 12-13 Epulis fissuratum. Redundant folds of tissue arising in the floor of the mouth in association with a mandibular denture.
The epulis fissuratum most often occurs in middle-aged and older adults, as would be expected with a denture-related lesion. It may occur on either the maxilla or mandible. The anterior portion of the jaws is affected much more often than the posterior areas. There is a pronounced female predilection; most studies show that two thirds to three fourths of all cases submitted for biopsy occur in women.
Another similar but less common fibrous hyperplasia, often called a fibroepithelial polyp or leaflike denture fibroma, occurs on the hard palate beneath a maxillary denture. This characteristic lesion is a flattened pink mass that is attached to the palate by a narrow stalk (Fig. 12-14). Usually, the flattened mass is closely applied to the palate and sits in a slightly cupped-out depression. However, it is easily lifted up with a probe, which demonstrates its pedunculated nature. The edge of the lesion often is serrated and resembles a leaf.
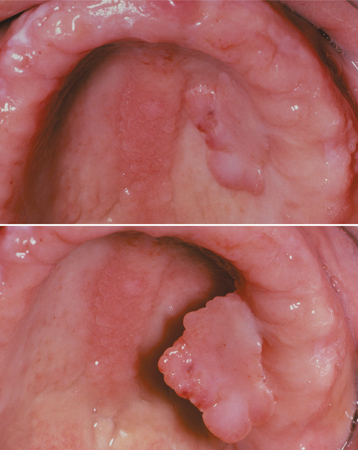
Fig. 12-14 Fibroepithelial polyp. Flattened mass of tissue arising on the hard palate beneath a maxillary denture; note its pedunculated nature. Because of its serrated edge, this lesion also is known as a leaflike denture fibroma. Associated inflammatory papillary hyperplasia is visible in the palatal midline.
HISTOPATHOLOGIC FEATURES: Microscopic examination of the epulis fissuratum reveals hyperplasia of the fibrous connective tissue. Often multiple folds and grooves occur where the denture impinges on the tissue (Fig. 12-15). The overlying epithelium is frequently hyperparakeratotic and demonstrates irregular hyperplasia of the rete ridges. In some instances, the epithelium shows inflammatory papillary hyperplasia (see page 513) or pseudoepitheliomatous (pseudocarcinomatous) hyperplasia. Focal areas of ulceration are not unusual, especially at the base of the grooves between the folds. A variable chronic inflammatory infiltrate is present; sometimes, it may include eosinophils or show lymphoid follicles. If minor salivary glands are included in the specimen, then they usually show chronic sialadenitis.
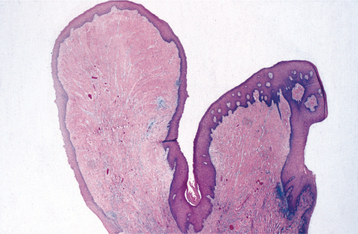
Fig. 12-15 Epulis fissuratum. Low-power photomicrograph demonstrating folds of hyperplastic fibrovascular connective tissue covered by stratified squamous epithelium.
In rare instances, the formation of osteoid or chondroid is observed. This unusual-appearing product, known as osseous and chondromatous metaplasia, is a reactive phenomenon caused by chronic irritation by the ill-fitting denture (see page 318). The irregular nature of this bone or cartilage can be microscopically disturbing, and the pathologist should not mistake it for a sarcoma.
The denture-related fibroepithelial polyp has a narrow core of dense fibrous connective tissue covered by stratified squamous epithelium. Like the epulis fissuratum, the overlying epithelium may be hyperplastic.
INFLAMMATORY PAPILLARY HYPERPLASIA (DENTURE PAPILLOMATOSIS)
Inflammatory papillary hyperplasia is a reactive tissue growth that usually, although not always, develops beneath a denture. Some investigators classify this lesion as part of the spectrum of denture stomatitis (see page 216). Although the exact pathogenesis is unknown, the condition most often appears to be related to the following:
Approximately 20% of patients who wear their dentures 24 hours a day have inflammatory papillary hyperplasia. Candida organisms also have been suggested as a cause, but any possible role appears uncertain.
CLINICAL FEATURES: Inflammatory papillary hyperplasia usually occurs on the hard palate beneath a denture base (Figs. 12-16 and 12-17). Early lesions may involve only the palatal vault, although advanced cases cover most of the palate. Less frequently, this hyperplasia develops on the edentulous mandibular alveolar ridge or on the surface of an epulis fissuratum. On rare occasions, the condition occurs on the palate of a patient without a denture, especially in people who habitually breathe through their mouth or have a high palatal vault. Candida-associated palatal papillary hyperplasia also has been reported in dentate patients with human immunodeficiency virus (HIV) infection.
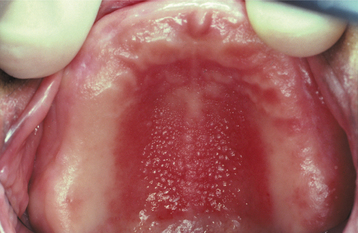
Fig. 12-16 Inflammatory papillary hyperplasia. Erythematous, pebbly appearance of the palatal vault.
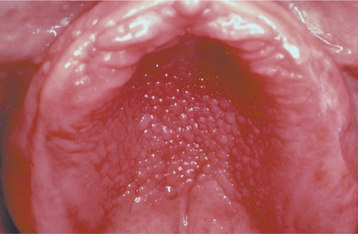
Fig. 12-17 Inflammatory papillary hyperplasia. An advanced case exhibiting more pronounced papular lesions of the hard palate.
Inflammatory papillary hyperplasia is usually asymptomatic. The mucosa is erythematous and has a pebbly or papillary surface. Many cases are associated with denture stomatitis.
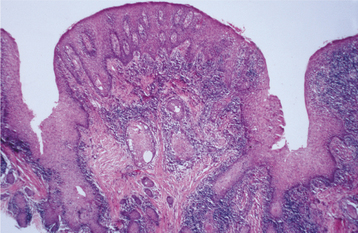
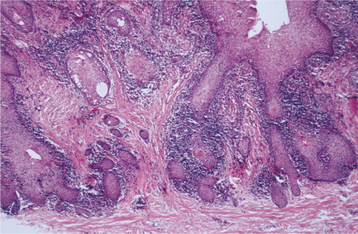
HISTOPATHOLOGIC FEATURES: The mucosa in inflammatory papillary hyperplasia exhibits numerous papillary growths on the surface that are covered by hyperplastic, stratified squamous epithelium (Fig. 12-18). In advanced cases, this hyperplasia is pseudoepitheliomatous in appearance, and the pathologist should not mistake it for carcinoma (Fig. 12-19). The connective tissue can vary from loose and edematous to densely collagenized. A chronic inflammatory cell infiltrate is usually seen, which consists of lymphocytes and plasma cells. Less frequently, polymorphonuclear leukocytes are also present. If underlying salivary glands are present, then they often show sclerosing sialadenitis.
TREATMENT AND PROGNOSIS: For very early lesions of inflammatory papillary hyperplasia, removal of the denture may allow the erythema and edema to subside, and the tissues may resume a more normal appearance. The condition also may show improvement after topical or systemic antifungal therapy. For more advanced and collagenized lesions, many clinicians prefer to excise the hyperplastic tissue before fabricating a new denture. Various surgical methods have been used, including the following:
After surgery, the existing denture can be lined with a temporary tissue conditioner that acts as a palatal dressing and promotes greater comfort. After healing, the patient should be encouraged to leave the new denture out at night and to keep it clean.
FIBROUS HISTIOCYTOMA
Fibrous histiocytomas are a diverse group of tumors that exhibit fibroblastic and histiocytic differentia-tion. Although the cell of origin is still uncertain, it may arise from the tissue histiocyte, which then assumes fibroblastic properties. Because of the variable nature of these lesions, an array of terms has been used for them, including dermatofibroma, sclerosing hemangioma, fibroxanthoma, and nodular subepidermal fibrosis. Unlike other fibrous growths discussed previously in this chapter, the fibrous histiocytoma is generally considered to represent a true neoplasm.
CLINICAL FEATURES: The fibrous histiocytoma can develop almost anywhere in the body. The most common site is the skin of the extremities, where the lesion is called a dermatofibroma. Tumors of the oral and perioral region are uncommon. Although oral tumors can occur at any site, the most frequent location is the buccal mucosa and vestibule. Rare intrabony lesions of the jaws have also been reported. Oral fibrous histiocytomas tend to occur in middle-aged and older adults; cutaneous examples are most frequent in young adults. The tumor is usually a painless nodular mass and can vary in size from a few millimeters to several centimeters in diameter (Fig. 12-20). Deeper tumors tend to be larger.
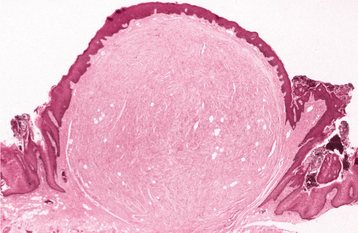
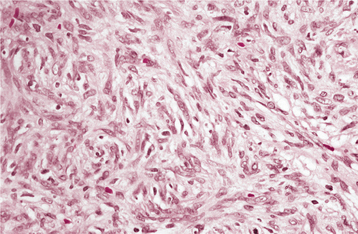
HISTOPATHOLOGIC FEATURES: Microscopically, the fibrous histiocytoma is characterized by a cellular proliferation of spindle-shaped fibroblastic cells with vesicular nuclei (Figs. 12-21 and 12-22). The margins of the tumor often are not sharply defined. The tumor cells are arranged in short, intersecting fascicles, known as a storiform pattern because of its resemblance to the irregular, whorled appearance of a straw mat. Rounded histiocyte-like cells, lipid-containing xanthoma cells, or multinucleated giant cells can be seen occasionally, as may scattered lymphocytes. The stroma may demonstrate areas of myxoid change or focal hyalinization.
FIBROMATOSIS
The fibromatoses are a broad group of fibrous proliferations that have a biologic behavior and histopathologic pattern that is intermediate between those of benign fibrous lesions and fibrosarcoma. A number of different forms of fibromatosis are recognized throughout the body, and they often are named based on their particular clinicopathologic features. In the soft tissues of the head and neck, these lesions are frequently called juvenile aggressive fibromatoses or extraabdominal desmoids. Similar lesions within the bone have been called desmoplastic fibromas (see page 658). Individuals with familial adenomatous polyposis and Gardner syndrome (see page 651) have a greatly increased risk for developing aggressive fibromatosis.
CLINICAL AND RADIOGRAPHIC FEATURES: Soft tissue fibromatosis of the head and neck is a firm, painless mass, which may exhibit rapid or insidious growth (Fig. 12-23). The lesion most frequently occurs in children or young adults; hence, the term juvenile fibromatosis. However, cases also have been seen in middle-aged adults. The most common oral site is the paramandibular soft tissue region, although the lesion can occur almost anywhere. The tumor can grow to considerable size, resulting in significant facial disfigurement. Destruction of adjacent bone may be observed on radiographs and other imaging studies.
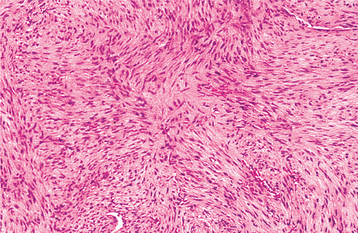
HISTOPATHOLOGIC FEATURES: Soft tissue fibromatosis is characterized by a cellular proliferation of spindle-shaped cells that are arranged in streaming fascicles and are associated with a variable amount of collagen (Fig. 12-24). The lesion is usually poorly circumscribed and infiltrates the adjacent tissues. Hyperchromatism and pleomorphism of the cells should not be observed.
TREATMENT AND PROGNOSIS: Because of its locally aggressive nature, the preferred treatment for soft tissue fibromatosis is wide excision that includes a generous margin of adjacent normal tissues. Adjuvant chemotherapy or radiation therapy sometimes has been used for incompletely resected or recurrent tumors. A 23% recurrence rate has been reported for oral and paraoral fibromatosis, but a higher recurrence rate has been noted for other head and neck sites. Metastasis does not occur.
MYOFIBROMA (MYOFIBROMATOSIS)
Myofibroma is a rare spindle cell neoplasm that consists of myofibroblasts (i.e., cells with both smooth muscle and fibroblastic features). Such cells are not specific for this lesion, however, because they also can be identified in other fibrous proliferations. Most myofibromas occur as solitary lesions, but some patients develop a multicentric tumor process known as myofibromatosis.
CLINICAL AND RADIOGRAPHIC FEATURES: Although myofibromas are rare neoplasms, they demonstrate a predilection for the head and neck region. Solitary tumors develop most frequently in the first 4 decades of life, with a mean age of 22 years. The most common oral location is the mandible, followed by the tongue and buccal mucosa. The tumor is typically a painless mass that sometimes exhibits rapid enlargement. Intrabony tumors create radiolucent defects that usually tend to be poorly defined, although some may be well defined or multilocular (Fig. 12-25). Multicentric myofibromatosis primarily affects neonates and infants who may have tumors of the skin, subcutaneous tissue, muscle, bone, and viscera. The number of tumors can vary from several to more than 100.
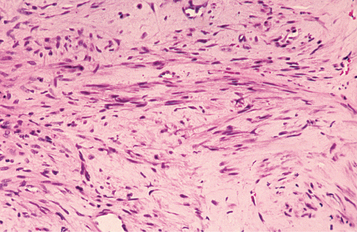
HISTOPATHOLOGIC FEATURES: Myofibromas are composed of interlacing bundles of spindle cells with tapered or blunt-ended nuclei and eosinophilic cytoplasm (Fig. 12-26). Nodular fascicles may alternate with more cellular zones, imparting a biphasic appearance to the tumor. Scattered mitoses are not uncommon. Centrally, the lesion is often more vascular with a hemangiopericytoma-like appearance. The tumor cells are positive for smooth muscle actin and muscle-specific actin with immunohistochemistry, but they are negative for desmin.
TREATMENT AND PROGNOSIS: Solitary myofibromas are usually treated by surgical excision. A small percentage of tumors will recur after treatment, but typically, these can be controlled with reexcision. Multifocal tumors arising in soft tissues and bone rarely recur after surgical excision. Spontaneous regression may occur in some cases. However, myofibromatosis involving the viscera or vital organs in infants can act more aggressively and sometimes proves to be fatal within a few days after birth.
ORAL FOCAL MUCINOSIS
Oral focal mucinosis is an uncommon tumorlike mass that is believed to represent the oral counterpart of cutaneous focal mucinosis or a cutaneous myxoid cyst. The cause is unknown, although the lesion may result from overproduction of hyaluronic acid by fibroblasts.
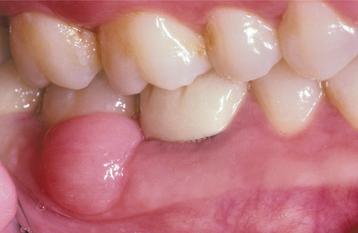
CLINICAL FEATURES: Oral focal mucinosis is most common in young adults and shows a 2:1 female-to-male predilection. The gingiva is the most common site; two thirds to three fourths of all cases are found there. The hard palate is the second most common location. The mass rarely appears at other oral sites. The lesion usually presents as a sessile or pedunculated, painless nodular mass that is the same color as the surrounding mucosa (Fig. 12-27). The surface is typically smooth and nonulcerated, although occasional cases exhibit a lobulated appearance. The size varies from a few millimeters up to 2 cm in diameter. The patient often has been aware of the mass for many months or years before the diagnosis is made.
HISTOPATHOLOGIC FEATURES: Microscopic examination of oral focal mucinosis shows a well-localized but nonencapsulated area of loose, myxomatous connective tissue surrounded by denser, normal collagenous connective tissue (Figs. 12-28 and 12-29). The lesion is usually found just beneath the surface epithelium and often causes flattening of the rete ridges. The fibroblasts within the mucinous area can be ovoid, fusiform, or stellate, and they may demonstrate delicate, fibrillar processes. Few capillaries are seen within the lesion, especially compared with the surrounding denser collagen. Similarly, no significant inflammation is observed, although a perivascular lymphocytic infiltrate often is noted within the surrounding collagenous connective tissue. No appreciable reticulin is evident within the lesion, and special stains suggest that the mucinous product is hyaluronic acid.
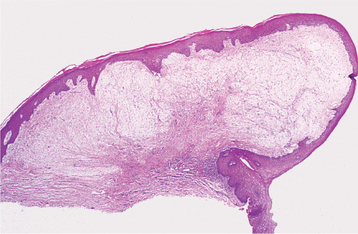
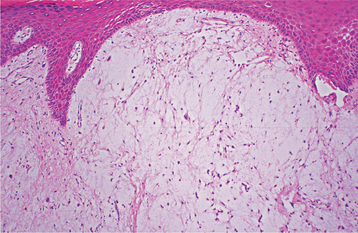
PYOGENIC GRANULOMA
The pyogenic granuloma is a common tumorlike growth of the oral cavity that traditionally has been considered to be nonneoplastic in nature.* Although it was originally thought to be caused by pyogenic organisms, it is now believed to be unrelated to infection. Instead, the pyogenic granuloma is thought to represent an exuberant tissue response to local irritation or trauma. In spite of its name, it is not a true granuloma.
CLINICAL FEATURES: The pyogenic granuloma is a smooth or lobulated mass that is usually pedunculated, although some lesions are sessile (Figs. 12-30 to 12-32). The surface is characteristically ulcerated and ranges from pink to red to purple, depending on the age of the lesion. Young pyogenic granulomas are highly vascular in appearance; older lesions tend to become more collagenized and pink. They vary from small growths only a few millimeters in size to larger lesions that may measure several centimeters in diameter. Typically, the mass is painless, although it often bleeds easily because of its extreme vascularity. Pyogenic granulomas may exhibit rapid growth, which may create alarm for both the patient and the clinician, who may fear that the lesion might be malignant.
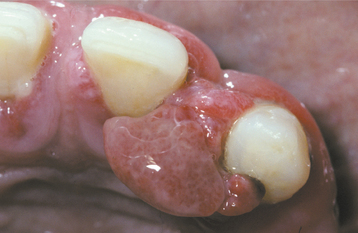
Fig. 12-30 Pyogenic granuloma. Erythematous, hemorrhagic mass arising from the maxillary anterior gingiva.
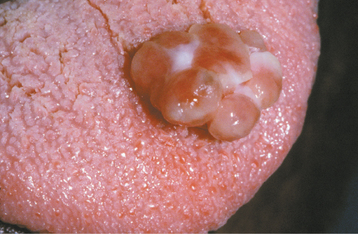
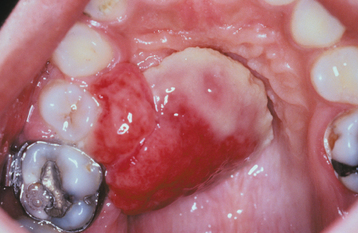
Fig. 12-32 Pyogenic granuloma. Unusually large lesion arising from the palatal gingiva in association with an orthodontic band. The patient was pregnant.
Oral pyogenic granulomas show a striking predilection for the gingiva, which accounts for 75% of all cases. Gingival irritation and inflammation that result from poor oral hygiene may be a precipitating factor in many patients. The lips, tongue, and buccal mucosa are the next most common sites. A history of trauma before the development of the lesion is not unusual, especially for extragingival pyogenic granulomas. Lesions are slightly more common on the maxillary gingiva than the mandibular gingiva; anterior areas are more frequently affected than posterior areas. These lesions are much more common on the facial aspect of the gingiva than the lingual aspect; some extend between the teeth and involve both the facial and the lingual gingiva.
Although the pyogenic granuloma can develop at any age, it is most common in children and young adults. Most studies also demonstrate a definite female predilection, possibly because of the vascular effects of female hormones. Pyogenic granulomas of the gingiva frequently develop in pregnant women, so much so that the terms pregnancy tumor or granuloma gravidarum often are used. Such lesions may begin to develop during the first trimester, and their incidence increases up through the seventh month of pregnancy. The gradual rise in development of these lesions throughout pregnancy may be related to the increasing levels of estrogen and progesterone as the pregnancy progresses. After pregnancy and the return of normal hormone levels, some of these pyogenic granulomas resolve without treatment or undergo fibrous maturation and resemble a fibroma (Fig. 12-33).
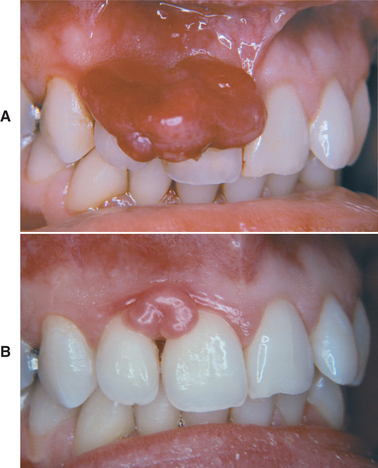
Fig. 12-33 Pyogenic granuloma. A, Large gingival mass in a pregnant woman just before childbirth. B, The mass has decreased in size and undergone fibrous maturation 3 months after childbirth. (Courtesy of Dr. George Blozis.)
Epulis granulomatosa is a term used to describe hyperplastic growths of granulation tissue that sometimes arise in healing extraction sockets (Fig. 12-34). These lesions resemble pyogenic granulomas and usually represent a granulation tissue reaction to bony sequestra in the socket.
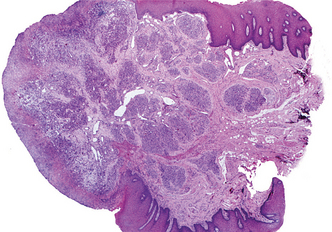
HISTOPATHOLOGIC FEATURES: Microscopic examination of pyogenic granulomas shows a highly vascular proliferation that resembles granulation tissue (Figs. 12-35 and 12-36). Numerous small and larger endothelium-lined channels are formed that are engorged with red blood cells. These vessels sometimes are organized in lobular aggregates, and some pathologists require this lobular arrangement for the diagnosis (lobular capillary hemangioma). The surface is usually ulcerated and replaced by a thick fibrinopurulent membrane. A mixed inflammatory cell infiltrate of neutrophils, plasma cells, and lymphocytes is evident. Neutrophils are most prevalent near the ulcerated surface; chronic inflammatory cells are found deeper in the specimen. Older lesions may have areas with a more fibrous appearance. In fact, many gingival fibromas probably represent pyogenic granulomas that have undergone fibrous maturation.
TREATMENT AND PROGNOSIS: The treatment of patients with pyogenic granuloma consists of conservative surgical excision, which is usually curative. The specimen should be submitted for microscopic examination to rule out other more serious diagnoses. For gingival lesions, the excision should extend down to periosteum and the adjacent teeth should be thoroughly scaled to remove any source of continuing irritation. Occasionally, the lesion recurs and reexcision is necessary. In rare instances, multiple recurrences have been noted.
For lesions that develop during pregnancy, usually treatment should be deferred unless significant functional or aesthetic problems develop. The recurrence rate is higher for pyogenic granulomas removed during pregnancy, and some lesions will resolve spontaneously after parturition.
PERIPHERAL GIANT CELL GRANULOMA (GIANT CELL EPULIS)
The peripheral giant cell granuloma is a relatively common tumorlike growth of the oral cavity. It probably does not represent a true neoplasm but rather is a reactive lesion caused by local irritation or trauma. In the past, it often was called a peripheral giant cell reparative granuloma, but any reparative nature appears doubtful. Some investigators believe that the giant cells show immunohistochemical features of osteoclasts, whereas other authors have suggested that the lesion is formed by cells from the mononuclear phagocyte system. The peripheral giant cell granuloma bears a close microscopic resemblance to the central giant cell granuloma (see page 626), and some pathologists believe that it may represent a soft tissue counterpart of this central bony lesion.
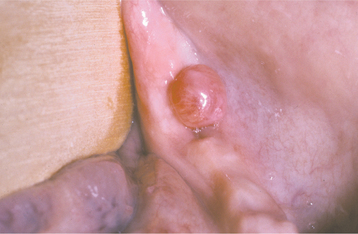
CLINICAL AND RADIOGRAPHIC FEATURES: The peripheral giant cell granuloma occurs exclusively on the gingiva or edentulous alveolar ridge, presenting as a red or red-blue nodular mass (Figs. 12-37 and 12-38). Most lesions are smaller than 2 cm in diameter, although larger ones are seen occasionally. The lesion can be sessile or pedunculated and may or may not be ulcerated. The clinical appearance is similar to the more common pyogenic granuloma of the gingiva (see page 517), although the peripheral giant cell granuloma often is more blue-purple compared with the bright red of a typical pyogenic granuloma.
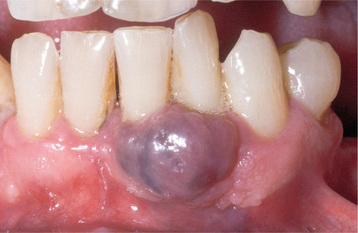
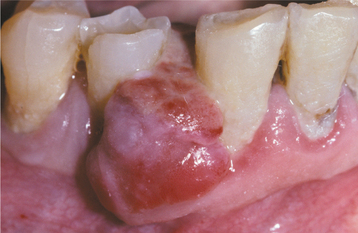
Peripheral giant cell granulomas can develop at almost any age, especially during the first through sixth decades of life. The mean age in several large series ranges from 31 to 41 years. Approximately 60% of cases occur in females. It may develop in either the anterior or posterior regions of the gingiva or alveolar mucosa, and the mandible is affected slightly more often than the maxilla. Although the peripheral giant cell granuloma develops within soft tissue, “cupping” resorption of the underlying alveolar bone sometimes is seen. On occasion, it may be difficult to determine whether the mass arose as a peripheral lesion or as a central giant cell granuloma that eroded through the cortical plate into the gingival soft tissues.
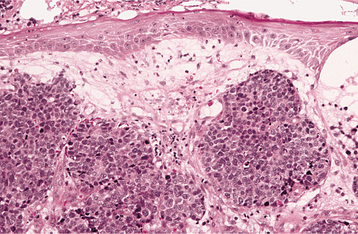
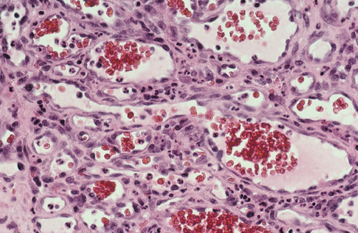
HISTOPATHOLOGIC FEATURES: Microscopic examination of a peripheral giant cell granuloma shows a proliferation of multinucleated giant cells within a background of plump ovoid and spindle-shaped mesenchymal cells (Figs. 12-39 and 12-40). The giant cells may contain only a few nuclei or up to several dozen. Some of these cells may have large, vesicular nuclei; others demonstrate small, pyknotic nuclei. Mitotic figures are fairly common in the background mesenchymal cells. Abundant hemorrhage is characteristically found throughout the mass, which often results in deposits of hemosiderin pigment, especially at the periphery of the lesion.
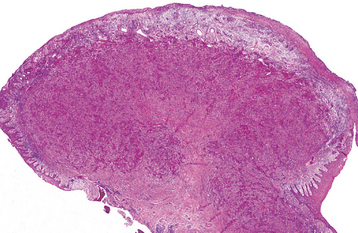
Fig. 12-39 Peripheral giant cell granuloma. Low-power view showing a nodular proliferation of multinucleated giant cells within the gingiva.
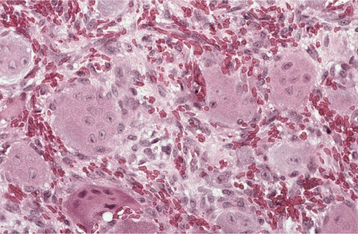
Fig. 12-40 Peripheral giant cell granuloma. High-power view showing scattered multinucleated giant cells within a hemorrhagic background of ovoid and spindle-shaped mesenchymal cells.
The overlying mucosal surface is ulcerated in about 50% of cases. A zone of dense fibrous connective tissue usually separates the giant cell proliferation from the mucosal surface. Adjacent acute and chronic inflammatory cells are frequently present. Areas of reactive bone formation or dystrophic calcifications are not unusual.
TREATMENT AND PROGNOSIS: The treatment of the peripheral giant cell granuloma consists of local surgical excision down to the underlying bone. The adjacent teeth should be carefully scaled to remove any source of irritation and to minimize the risk of recurrence. Approximately 10% of lesions are reported to recur, and reexcision must be performed. On rare occasions, lesions indistinguishable from peripheral giant cell granulomas have been seen in patients with hyperparathyroidism (see page 838). They apparently represent the so-called osteoclastic brown tumors associated with this endocrine disorder. However, the brown tumors of hyperparathyroidism are much more likely to be intraosseous in location and mimic a central giant cell granuloma.
PERIPHERAL OSSIFYING FIBROMA (OSSIFYING FIBROID EPULIS; PERIPHERAL FIBROMA WITH CALCIFICATION; CALCIFYING FIBROBLASTIC GRANULOMA)
The peripheral ossifying fibroma is a relatively common gingival growth that is considered to be reactive rather than neoplastic in nature. The pathogenesis of this lesion is uncertain. Because of their clinical and histopathologic similarities, researchers believe that some peripheral ossifying fibromas develop initially as pyogenic granulomas that undergo fibrous maturation and subsequent calcification. However, not all peripheral ossifying fibromas may develop in this manner. The mineralized product probably has its origin from cells of the periosteum or periodontal ligament.
Considerable confusion has existed over the nomenclature of this lesion, and several terms have been used to describe its variable histopathologic features. In the past, the terms peripheral odontogenic fibroma (see page 727) and peripheral ossifying fibroma often were used synonymously, but the peripheral odontogenic fibroma is now considered to be a distinct and separate entity. In addition, in spite of the similarity in names, the peripheral ossifying fibroma does not represent the soft tissue counterpart of the central ossifying fibroma (see page 646).
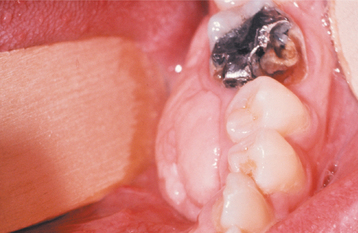
CLINICAL FEATURES: The peripheral ossifying fibroma occurs exclusively on the gingiva. It appears as a nodular mass, either pedunculated or sessile, that usually emanates from the interdental papilla (Figs. 12-41 and 12-42). The color ranges from red to pink, and the surface is frequently, but not always, ulcerated. The growth probably begins as an ulcerated lesion; older ones are more likely to demonstrate healing of the ulcer and an intact surface. Red, ulcerated lesions often are mistaken for pyogenic granulomas; the pink, nonulcerated ones are clinically similar to irritation fibromas. Most lesions are less than 2 cm in size, although larger ones occasionally occur. The lesion often has been present for many weeks or months before the diagnosis is made.
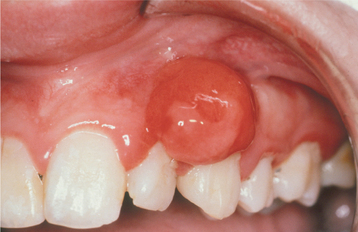
Fig. 12-41 Peripheral ossifying fibroma. Red, ulcerated mass of the maxillary gingiva. Such ulcerated lesions are easily mistaken for a pyogenic granuloma.
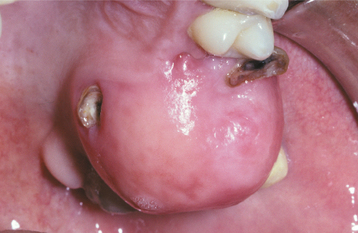
Fig. 12-42 Peripheral ossifying fibroma. Pink, nonulcerated mass arising from the maxillary gingiva. The remaining roots of the first molar are present.
The peripheral ossifying fibroma is predominantly a lesion of teenagers and young adults, with peak prevalence between the ages of 10 and 19. Almost two thirds of all cases occur in females. There is a slight predilection for the maxillary arch, and more than 50% of all cases occur in the incisor-cuspid region. Usually, the teeth are unaffected; rarely, there can be migration and loosening of adjacent teeth.
HISTOPATHOLOGIC FEATURES: The basic microscopic pattern of the peripheral ossifying fibroma is one of a fibrous proliferation associated with the formation of a mineralized product (Figs. 12-43 and 12-44). If the epithelium is ulcerated, then the surface is covered by a fibrinopurulent membrane with a subjacent zone of granulation tissue. The deeper fibroblastic component often is cellular, especially in areas of mineralization. In some cases, the fibroblastic proliferation and associated mineralization is only a small component of a larger mass that resembles a fibroma or pyogenic granuloma.
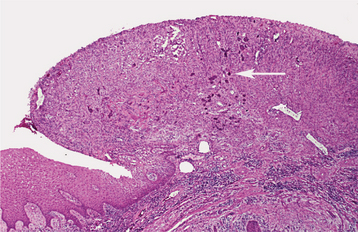
Fig. 12-43 Peripheral ossifying fibroma. Ulcerated gingival mass demonstrating focal early mineralization (arrow).
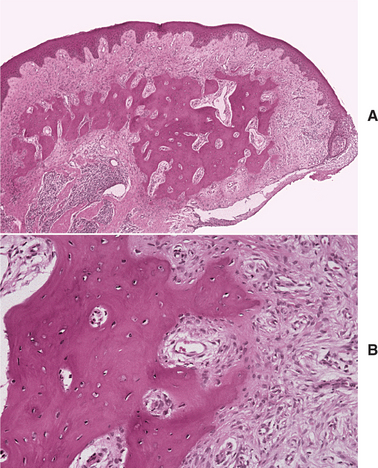
Fig. 12-44 Peripheral ossifying fibroma. A, Nonulcerated fibrous mass of the gingiva showing central bone formation. B, Higher-power view showing trabeculae of bone with adjacent fibrous connective tissue.
The type of mineralized component is variable and may consist of bone, cementum-like material, or dystrophic calcifications. Frequently, a combination of products is formed. Usually, the bone is woven and trabecular in type, although older lesions may demonstrate mature lamellar bone. Trabeculae of unmineralized osteoid are not unusual. Less frequently, ovoid droplets of basophilic cementum-like material are formed. Dystrophic calcifications are characterized by multiple granules, tiny globules, or large, irregular masses of basophilic mineralized material. Such dystrophic calcifications are more common in early, ulcerated lesions; older, nonulcerated examples are more likely to demonstrate well-formed bone or cementum. In some cases, multinucleated giant cells may be found, usually in association with the mineralized product.
TREATMENT AND PROGNOSIS: The treatment of choice for the peripheral ossifying fibroma is local surgical excision with submission of the specimen for histopathologic examination. The mass should be excised down to periosteum because recurrence is more likely if the base of the lesion is allowed to remain. In addition, the adjacent teeth should be thoroughly scaled to eliminate any possible irritants. Periodontal surgical techniques, such as repositioned flaps or connective tissue grafts, may be necessary to repair the gingival defect in an aesthetic manner. Although excision is usually curative, a recurrence rate of 8% to 16% has been reported.
LIPOMA
The lipoma is a benign tumor of fat. Although it represents by far the most common mesenchymal neoplasm, most examples occur on the trunk and proximal portions of the extremities. Lipomas of the oral and maxillofacial region are much less frequent. The pathogenesis of lipomas is uncertain, but they appear to be more common in obese people. However, the metabolism of lipomas is completely independent of the normal body fat. If the caloric intake is reduced, then lipomas do not decrease in size, although normal body fat may be lost.
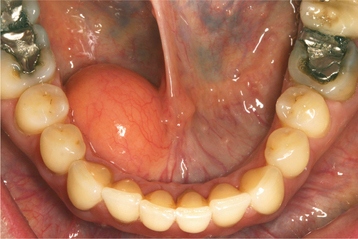
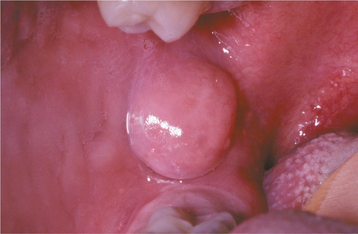
CLINICAL FEATURES: Oral lipomas are usually soft, smooth-surfaced nodular masses that can be sessile or pedunculated (Figs. 12-45 and 12-46). Typically, the tumor is asymptomatic and often has been noted for many months or years before diagnosis. Most are less than 3 cm in size, but occasional lesions can become much larger. Although a subtle or more obvious yellow hue often is detected clinically, deeper examples may appear pink. The buccal mucosa and buccal vestibule are the most common intraoral sites and account for 50% of all cases. Some buccal cases may not represent true tumors, but rather herniation of the buccal fat pad through the buccinator muscle, which may occur after local trauma in young children or subsequent to surgical removal of third molars in older patients. Less common sites include the tongue, floor of the mouth, and lips. Most patients are 40 years of age or older; lipomas are uncommon in children. Lipomas of the oral and maxillofacial region have shown a fairly balanced sex distribution in some studies, although one recent large series demonstrated a marked male predilection.
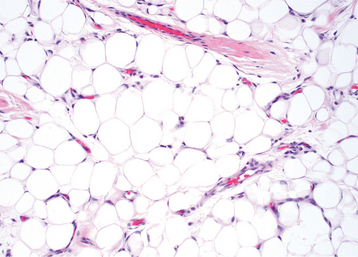
HISTOPATHOLOGIC FEATURES: Most oral lipomas are composed of mature fat cells that differ little in microscopic appearance from the surrounding normal fat (Figs. 12-47 and 12-48). The tumor is usually well circumscribed and may demonstrate a thin fibrous capsule. A distinct lobular arrangement of the cells often is seen. On rare occasions, central cartilaginous or osseous metaplasia may occur within an otherwise typical lipoma.
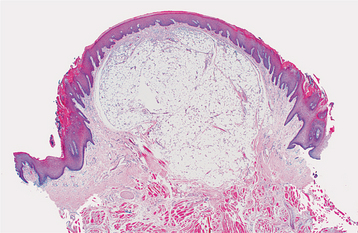
Fig. 12-47 Lipoma. Low-power view of a tumor of the tongue demonstrating a mass of mature adipose tissue.
A number of microscopic variants have been described. The most common of these is the fibrolipoma, which is characterized by a significant fibrous component intermixed with the lobules of fat cells. The remaining variants are rare.
The angiolipoma consists of an admixture of mature fat and numerous small blood vessels. The spindle cell lipoma demonstrates variable amounts of uni-form-appearing spindle cells in conjunction with a more typical lipomatous component. Some spindle cell lipomas exhibit a mucoid background (myxoid lipoma) and may be confused with myxoid liposarcomas. Pleomorphic lipomas are characterized by the presence of spindle cells plus bizarre, hyperchromatic giant cells; they can be difficult to distinguish from a pleomorphic liposarcoma. Intramuscular (infiltrating) lipomas often are more deeply situated and have an infiltrative growth pattern that extends between skeletal muscle bundles.
TREATMENT AND PROGNOSIS: Lipomas are treated by conservative local excision, and recurrence is rare. Most microscopic variants do not affect the prognosis. Intramuscular lipomas have a higher recurrence rate because of their infiltrative growth pattern, but this variant is rare in the oral and maxillofacial region.
TRAUMATIC NEUROMA (AMPUTATION NEUROMA)
The traumatic neuroma is not a true neoplasm but a reactive proliferation of neural tissue after transection or other damage of a nerve bundle. After a nerve has been damaged or severed, the proximal portion attempts to regenerate and reestablish innervation of the distal segment by the growth of axons through tubes of proliferating Schwann cells. If these regenerating elements encounter scar tissue or otherwise cannot reestablish innervation, then a tumorlike mass may develop at the site of injury.
CLINICAL AND RADIOGRAPHIC FEATURES: Traumatic neuromas of the oral mucosa are typically smooth-surfaced, nonulcerated nodules. They can develop at any location but are most common in the mental foramen area, tongue, and lower lip (Figs. 12-49 and 12-50). A history of trauma often can be elicited; some lesions arise subsequent to tooth extraction or other surgical procedures. Intraosseous traumatic neuromas may demonstrate a radiolucent defect on oral radiographs. Examples also may occur at other head and neck sites; it has been estimated that traumatic neuromas of the greater auricular nerve develop in 5% to 10% of patients undergoing surgery for pleomorphic adenomas of the parotid gland.
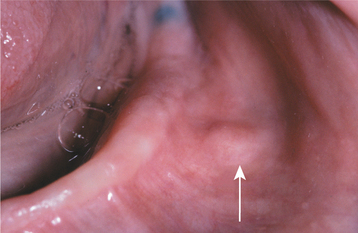
Fig. 12-49 Traumatic neuroma. Painful nodule of the mental nerve as it exits the mental foramen (arrow).
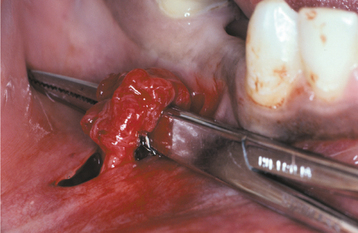
Fig. 12-50 Traumatic neuroma. Note the irregular nodular proliferation along the mental nerve that is being exposed at the time of surgery.
Traumatic neuromas can occur at any age, but they are diagnosed most often in middle-aged adults. They appear to be slightly more common in women. Many traumatic neuromas are associated with altered nerve sensations that can range from anesthesia to dysesthesia to overt pain. Although pain has been traditionally considered a hallmark of this lesion, studies indicate that only one fourth to one third of oral traumatic neuromas are painful. This pain can be intermittent or constant and ranges from mild tenderness or burning to severe radiating pain. Neuromas of the mental nerve are frequently painful, especially when impinged on by a denture or palpated.
HISTOPATHOLOGIC FEATURES: Microscopic examination of traumatic neuromas shows a haphazard proliferation of mature, myelinated and unmyelinated nerve bundles within a fibrous connective tissue stroma that ranges from densely collagenized to myxomatous in nature (Figs. 12-51 and 12-52). An associated mild chronic inflammatory cell infiltrate may be present. Traumatic neuromas with inflammation are more likely to be painful than those without significant inflammation.
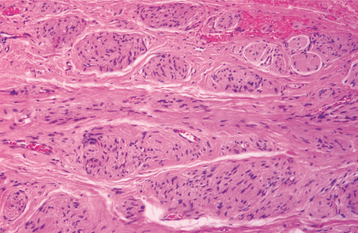
PALISADED ENCAPSULATED NEUROMA (SOLITARY CIRCUMSCRIBED NEUROMA)
The palisaded encapsulated neuroma is a benign neural tumor with distinctive clinical and histopathologic features. Although it was first recognized only as recently as 1972, it represents one of the more common superficial nerve tumors, especially in the head and neck region. The cause is uncertain, but some authors have speculated that trauma may play an etiologic role; the tumor is generally considered to represent a reactive lesion rather than a true neoplasm.
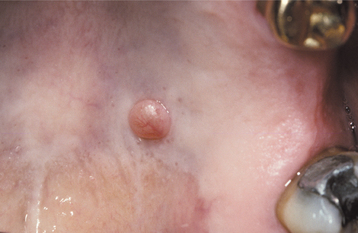
CLINICAL FEATURES: The palisaded encapsulated neuroma shows a striking predilection for the face, which accounts for approximately 90% of reported cases. The nose and cheek are the most common specific sites. The lesion is most frequently diagnosed between the fifth and seventh decades of life, although the tumor often has been present for many months or years. It is a smooth-surfaced, painless, dome-shaped papule or nodule that is usually less than 1 cm in diameter. There is no sex predilection.
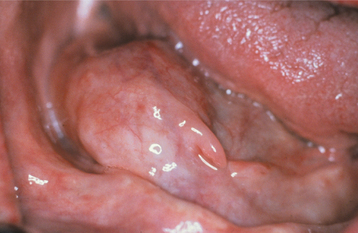
Oral palisaded encapsulated neuromas are not uncommon, although many are probably diagnosed microscopically as neurofibromas or neurilemomas. The lesion appears most frequently on the hard palate (Fig. 12-53) and maxillary labial mucosa, although it also may occur in other oral locations.
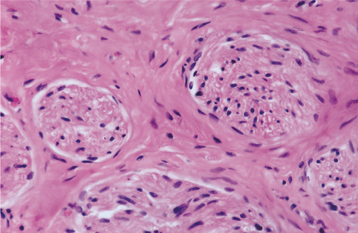
HISTOPATHOLOGIC FEATURES: Palisaded encapsulated neuromas appear well circumscribed and often encapsulated (Fig. 12-54), although this capsule may be incomplete, especially along the superficial aspect of the tumor. Some lesions have a lobulated appearance. The tumor consists of moderately cellular interlacing fascicles of spindle cells that are consistent with Schwann cells. The nuclei are characteristically wavy and pointed, with no significant pleomorphism or mitotic activity. Although the nuclei show a similar parallel orientation within the fascicles, the more definite palisading and Verocay bodies typical of the Antoni A tissue of a neurilemoma are usually not seen. Special stains reveal the presence of numerous axons within the tumor and the cells show a positive immunohistochemical reaction for S-100 protein (Fig. 12-55). Because the tumor is not always encapsulated and the cells are usually not truly palisaded, some pathologists prefer solitary circumscribed neuroma as a better descriptive term for this lesion.
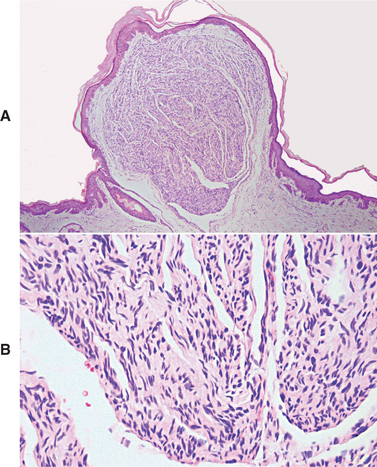
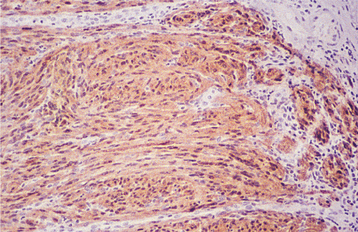
TREATMENT AND PROGNOSIS: The treatment for the palisaded encapsulated neuroma consists of conservative local surgical excision. Recurrence is rare. However, specific recognition of this lesion is important because it is not associated with neurofibromatosis or multiple endocrine neoplasia (MEN) type 2B.
NEURILEMOMA (SCHWANNOMA)
The neurilemoma is a benign neural neoplasm of Schwann cell origin. It is relatively uncommon, although 25% to 48% of all cases occur in the head and neck region. Bilateral neurilemomas of the auditory-vestibular nerve are a characteristic feature of the hereditary condition, neurofibromatosis type II (NF2).
CLINICAL AND RADIOGRAPHIC FEATURES: The solitary neurilemoma is a slow-growing, encapsulated tumor that typically arises in association with a nerve trunk. As it grows, it pushes the nerve aside. Usually, the mass is asymptomatic, although tenderness or pain may occur in some instances. The lesion is most common in young and middle-aged adults and can range from a few millimeters to several centimeters in size.
The tongue is the most common location for oral neurilemomas, although the tumor can occur almost anywhere in the mouth (Fig. 12-56). On occasion, the tumor arises centrally within bone and may produce bony expansion. Intraosseous examples are most common in the posterior mandible and usually appear as either unilocular or multilocular radiolucencies on radiographs. Pain and paresthesia are not unusual for intrabony tumors.
NF2 is an autosomal dominant condition caused by a mutation of a tumor suppressor gene on chromosome 22, which codes for a protein known as merlin. In addition to bilateral neurilemomas (“acoustic neuromas”) of the vestibular nerve, patients also develop neurilemomas of peripheral nerves, plus meningiomas and ependymomas of the central nervous system (CNS). Characteristic symptoms include progressive sensorineural deafness, dizziness, and tinnitus.
HISTOPATHOLOGIC FEATURES: The neurilemoma is usually an encapsulated tumor that demonstrates two microscopic patterns in varying amounts: (1) Antoni A and (2) Antoni B. Streaming fascicles of spindle-shaped Schwann cells characterize Antoni A tissue. These cells often form a palisaded arrangement around central acellular, eosinophilic areas known as Verocay bodies (Fig. 12-57). These Verocay bodies consist of reduplicated basement membrane and cytoplasmic processes. Antoni B tissue is less cellular and less organized; the spindle cells are randomly arranged within a loose, myxomatous stroma. Typically, neurites cannot be demonstrated within the tumor mass. The tumor cells will show a diffuse, positive immunohistochemical reaction for S-100 protein.
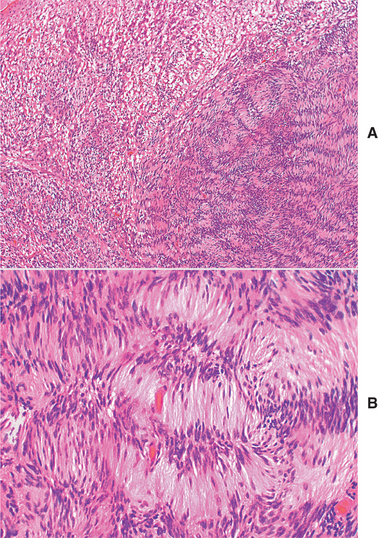
Fig. 12-57 Neurilemoma. A, Low-power view showing well-organized Antoni A tissue (right) with adjacent myxoid and less organized Antoni B tissue (left). B, The Schwann cells of the Antoni A tissue form a palisaded arrangement around acellular zones known as Verocay bodies.
Degenerative changes can be seen in some older tumors (ancient neurilemomas). These changes consist of hemorrhage, hemosiderin deposits, inflammation, fibrosis, and nuclear atypia. However, these tumors are still benign, and the pathologist must be careful not to mistake these alterations for evidence of a sarcoma.
TREATMENT AND PROGNOSIS: The solitary neurilemoma is treated by surgical excision, and the lesion should not recur. Malignant transformation does not occur or is extremely rare.
Vestibular schwannomas in patients with NF2 are difficult to manage. Surgical removal is indicated for large symptomatic tumors, but this almost always results in total deafness and risks facial nerve damage. Stereotactic radiosurgery may be considered for older adult or frail patients, as well as for individuals who decline traditional surgery.
NEUROFIBROMA
The neurofibroma is the most common type of peripheral nerve neoplasm. It arises from a mixture of cell types, including Schwann cells and perineural fibroblasts.
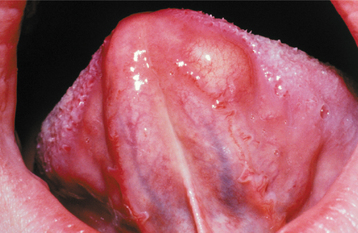
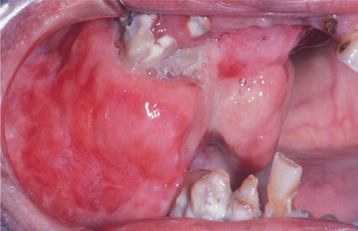
CLINICAL AND RADIOGRAPHIC FEATURES: Neurofibromas can arise as solitary tumors or be a component of neurofibromatosis (see page 529). Solitary tumors are most common in young adults and present as slow-growing, soft, painless lesions that vary in size from small nodules to larger masses. The skin is the most frequent location for neurofibromas, but lesions of the oral cavity are not uncommon (Figs. 12-58 and 12-59). The tongue and buccal mucosa are the most common intraoral sites. On rare occasions, the tumor can arise centrally within bone, where it may produce a well-demarcated or poorly defined unilocular or multilocular radiolucency (Fig. 12-60).
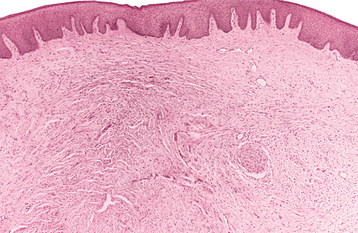
HISTOPATHOLOGIC FEATURES: The solitary neurofibroma often is well circumscribed, especially when the proliferation occurs within the perineurium of the involved nerve. Tumors that proliferate outside the perineurium may not appear well demarcated and tend to blend with the adjacent connective tissues.
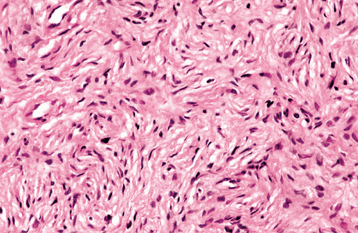
The tumor is composed of interlacing bundles of spindle-shaped cells that often exhibit wavy nuclei (Figs. 12-61 and 12-62). These cells are associated with delicate collagen bundles and variable amounts of myxoid matrix. Mast cells tend to be numerous and can be a helpful diagnostic feature. Sparsely distributed small axons usually can be demonstrated within the tumor tissue by using silver stains. Immunohistochemically, the tumor cells show a scattered, positive reaction for S-100 protein.
TREATMENT AND PROGNOSIS: The treatment for solitary neurofibromas is local surgical excision, and recurrence is rare. Any patient with a lesion that is diagnosed as a neurofibroma should be evaluated clinically for the possibility of neurofibromatosis (see next topic). Malignant transformation of solitary neurofibromas can occur, although the risk appears to be remote, especially compared with that in patients with neurofibromatosis.
NEUROFIBROMATOSIS TYPE I (VON RECKLINGHAUSEN’S DISEASE OF THE SKIN)
Neurofibromatosis is a relatively common hereditary condition that is estimated to occur in one of every 3000 births. At least eight forms of neurofibromatosis have been recognized, but the most common form is neurofibromatosis type I (NF1), which is discussed here. This form of the disease, also known as von Recklinghausen’s disease of the skin, accounts for 85% to 97% of cases and is inherited as an autosomal dominant trait (although 50% of all patients have no family history and apparently represent new mutations). It is caused by a variety of mutations of the NF1 gene, which is located on chromosome region 17q11.2 and is responsible for a tumor suppressor protein product known as neurofibromin.
CLINICAL AND RADIOGRAPHIC FEATURES: The diagnostic criteria for NF1 are summarized in Box 12-1. Patients have multiple neurofibromas that can occur anywhere in the body but are most common on the skin. The clinical appearance can vary from small papules to larger soft nodules to massive baggy, pendulous masses (elephantiasis neuromatosa) on the skin (Figs. 12-63 and 12-64). The plexiform variant of neurofibroma, which feels like a “bag of worms,” is considered pathognomonic for NF1. The tumors may be present at birth, but they often begin to appear during puberty and may continue to develop slowly throughout adulthood. Accelerated growth may be seen during pregnancy. There is a wide variability in the expression of the disease. Some patients have only a few neurofibromas; others have literally hundreds or thousands of tumors. However, two thirds of patients have relatively mild disease.
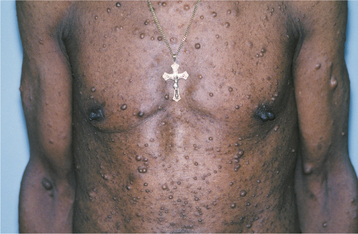
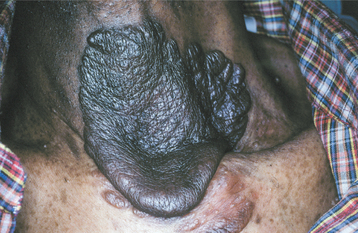
Another highly characteristic feature is the presence of café au lait (coffee with milk) pigmentation on the skin (Fig. 12-65). These spots are smooth-edged, yellow-tan to dark-brown macules that vary in diameter from 1 to 2 mm to several centimeters. They are usually present at birth or may develop during the first year of life. Axillary freckling (Crowe’s sign) is also a highly suggestive sign.
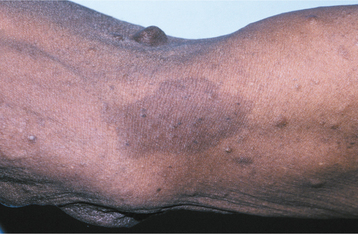
Fig. 12-65 Neurofibromatosis type I. Same patient as depicted in Fig. 12-63. Note the café au lait pigmentation on the arm.
Lisch nodules, translucent brown-pigmented spots on the iris, are found in nearly all affected individuals. The most common general medical problem is hypertension, which may develop secondary to coarctation of the aorta, pheochromocytoma, or renal artery stenosis. Other possible abnormalities include CNS tumors, macrocephaly, mental deficiency, seizures, short stature, and scoliosis.
In the past, oral lesions were estimated to occur in 4% to 7% of cases (Fig. 12-66). However, two studies suggest that oral manifestations may occur in as many as 72% to 92% of cases, especially if a detailed clinical and radiographic examination is performed. The most common reported finding is enlargement of the fungiform papillae (in about 50% of all affected patients); however, the specificity of this finding for neurofibromatosis is unknown. Only about 25% of patients examined in these two studies exhibited actual intraoral neurofibromas. Radiographic findings may include enlargement of the mandibular foramen, enlargement or branching of the mandibular canal, increased bone density, concavity of the medial surface of the ramus, and increase in dimension of the coronoid notch.
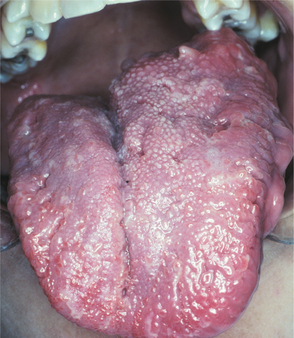
Fig. 12-66 Neurofibromatosis type I. Intraoral involvement characterized by unilateral enlargement of the tongue.
Several unusual clinical variants of NF1 have been described. On occasion, the condition can include unilateral enlargement that mimics hemifacial hyperplasia (see page 38). In addition, several patients with NF1 have been described with associated Noonan syndrome or with central giant cell granulomas of the jaw.
TREATMENT AND PROGNOSIS: There is no specific therapy for NF1, and treatment often is directed toward prevention or management of complications. Facial neurofibromas can be removed for cosmetic purposes. Carbon dioxide (CO2) laser and dermabrasion have been used successfully for extensive lesions.
One of the most feared complications is the development of cancer, most often a malignant peripheral nerve sheath tumor (neurofibrosarcoma; malignant schwannoma), which has been reported to occur in about 5% of cases. These tumors are most common on the trunk and extremities, although head and neck involvement is occasionally seen (Figs. 12-67 to 12-69). The prognosis for malignant peripheral nerve sheath tumors associated with neurofibromatosis is poor, with a 5-year survival rate of only 15%. Other malignancies also have been associated with neurofibromatosis, including CNS tumors, pheochromocytoma, leukemia, rhabdomyosarcoma, and Wilms’ tumor. The average lifespan of individuals with NF1 is 15 years less than the general population, mostly related to vascular disease and malignant neoplasms.
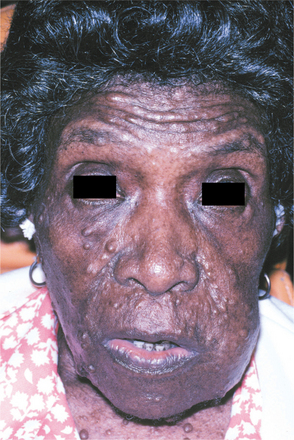
Fig. 12-67 Neurofibromatosis type I. Malignant peripheral nerve sheath tumor of the left cheek in a patient with type I neurofibromatosis. (From Neville BW, Hann J, Narang R et al: Oral neurofibrosarcoma associated with neurofibromatosis type I, Oral Surg Oral Med Oral Pathol 72:456-461, 1991.)
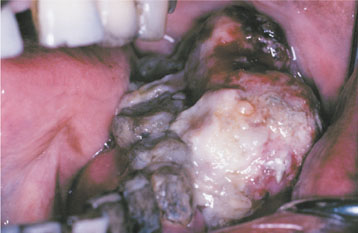
Fig. 12-68 Neurofibromatosis type I. Same patient as depicted in Fig. 12-67. Note the intraoral appearance of malignant peripheral nerve sheath tumor of the mandibular buccal vestibule. The patient eventually died of this tumor. (From Neville BW, Hann J, Narang R et al: Oral neurofibrosarcoma associated with neurofibromatosis type I, Oral Surg Oral Med Oral Pathol 72:456-461, 1991.)
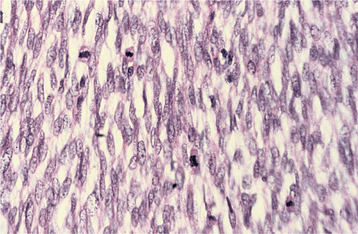
Fig. 12-69 Malignant peripheral nerve sheath tumor. High-power view of an intraoral tumor that developed in a patient with neurofibromatosis type I. There is a cellular spindle cell proliferation with numerous mitotic figures.
In recent years, there has been considerable interest in Joseph (not John) Merrick, the so-called Elephant Man. Although Merrick once was mistakenly considered to have neurofibromatosis, it is now generally accepted that his horribly disfigured appearance was not because of neurofibromatosis, but that he most likely had a rare condition known as Proteus syndrome. Because patients with neurofibromatosis may fear acquiring a similar clinical appearance, they should be reassured that they have a different condition. The phrase “Elephant Man disease” is incorrect and misleading, and it should be avoided. Genetic counseling is extremely important for all patients with neurofibromatosis.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2B (MULTIPLE ENDOCRINE NEOPLASIA TYPE 3; MULTIPLE MUCOSAL NEUROMA SYNDROME)
The multiple endocrine neoplasia (MEN) syndromes are a group of rare conditions characterized by tumors or hyperplasias of the neuroendocrine tissues. For example, patients with MEN type 1 have benign tumors of the pancreatic islets, adrenal cortex, parathyroid glands, and pituitary gland. MEN type 2A, also known as Sipple syndrome. includes the development of adrenal pheochromocytomas and medullary thyroid carcinoma. In addition to pheochromocytomas and medullary thyroid carcinoma, patients with MEN type 2B have mucosal neuromas that especially involve the oral mucous membranes. Because oral manifestations are most prominent in MEN type 2B, the remainder of the discussion is limited to this condition.
Similar to the other MEN syndromes, MEN type 2B is inherited as an autosomal dominant trait. However, researchers believe that 50% of cases represent spontaneous mutations. The condition is caused by a mutation of the RET protooncogene on chromosome 10, which has been detected in 95% of affected individuals.
CLINICAL FEATURES: Patients with MEN type 2B usually have a marfanoid body build characterized by thin, elongated limbs with muscle wasting. The face is narrow, but the lips are characteristically thick and protuberant because of the diffuse proliferation of nerve bundles. The upper eyelid sometimes is everted because of thickening of the tarsal plate (Fig. 12-70). Small, pedunculated neuromas may be observable on the conjunctiva, eyelid margin, or cornea.
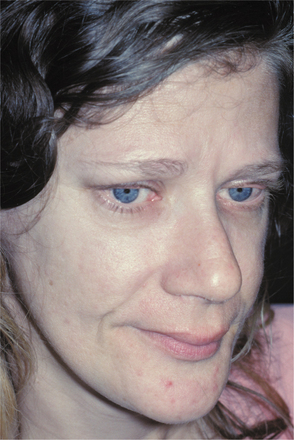
Fig. 12-70 Multiple endocrine neoplasia (MEN) type 2B. Note the narrow face and eversion of the upper eyelids.
Oral mucosal neuromas are usually the first sign of the condition. These neuromas appear as soft, painless papules or nodules that principally affect the lips and anterior tongue but also may be seen on the buccal mucosa, gingiva, and palate (Fig. 12-71). Bilateral neuromas of the commissural mucosa are highly characteristic.
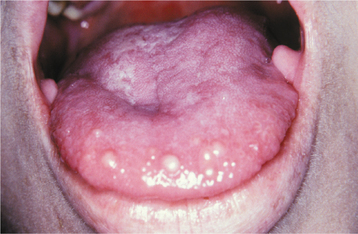
Fig. 12-71 Multiple endocrine neoplasia (MEN) type 2B. Multiple neuromas along the anterior margin of the tongue and bilaterally at the commissures. (Courtesy of Dr. Emmitt Costich.)
Pheochromocytomas of the adrenal glands develop in at least 50% of all patients and become more prevalent with increasing age. These neuroendocrine tumors are frequently bilateral or multifocal. The tumor cells secrete catecholamines, which result in symptoms such as profuse sweating, intractable diarrhea, headaches, flushing, heart palpitations, and severe hypertension.
The most significant aspect of this condition is the development of medullary carcinoma of the thyroid gland, which occurs in more than 90% of cases. This aggressive tumor arises from the parafollicular cells (C cells), which are responsible for calcitonin production. Medullary carcinoma most often is diagnosed in patients between the ages of 18 and 25, and it shows a marked propensity for metastasis. The average age at death from this neoplasm is 21 years.
LABORATORY VALUES: If medullary carcinoma of the thyroid gland is present, then serum or urinary levels of calcitonin are elevated. An increase in calcitonin levels may herald the onset of the tumor, and calcitonin also can be monitored to detect local recurrences or metastases after treatment. Pheochromocytomas may result in increased levels of urinary vanillylmandelic acid (VMA) and increased epinephrine-to-norepinephrine ratios.
HISTOPATHOLOGIC FEATURES: The mucosal neuromas are characterized by marked hyperplasia of nerve bundles in an otherwise normal or loose connective tissue background (Figs. 12-72 and 12-73). Prominent thickening of the perineurium is typically seen.
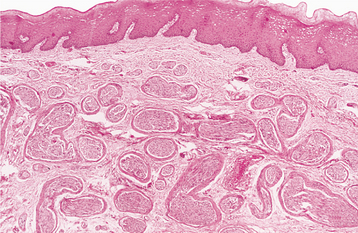
Fig. 12-72 Multiple endocrine neoplasia (MEN) type 2B. Low-power view of an oral mucosal neuroma showing marked hyperplasia of nerve bundles.
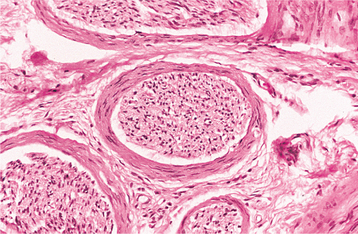
Fig. 12-73 Multiple endocrine neoplasia (MEN) type 2B. High-power view of the same neuroma as depicted in Fig. 12-72. Note the prominent thickening of the perineurium.
TREATMENT AND PROGNOSIS: The prognosis for patients with MEN type 2B centers on early recognition of the oral features, given the serious nature of the medullary thyroid carcinoma. Some investigators advocate prophylactic removal of the thyroid gland at an early age because medullary carcinoma is almost certain to occur. Once it has developed, this tumor often exhibits an aggressive behavior with a poor prognosis. The patient also should be observed for the development of pheochromocytomas because they may result in a life-threatening hypertensive crisis, especially if surgery with general anesthesia is performed.
MELANOTIC NEUROECTODERMAL TUMOR OF INFANCY
The melanotic neuroectodermal tumor of infancy is a rare pigmented neoplasm that usually occurs during the first year of life. It is generally accepted that this lesion is of neural crest origin. In the past, however, a number of tissues were suggested as possible sources of this tumor. These included odontogenic epithelium and retina, which resulted in various older terms for this entity, such as pigmented ameloblastoma, retinal anlage tumor, and melanotic progonoma. Because these names are inaccurate, however, they should no longer be used. Melanotic (pigmented) neuroectodermal tumor of infancy is the preferred term.
CLINICAL AND RADIOGRAPHIC FEATURES: Melanotic neuroectodermal tumor of infancy almost always develops in young children during the first year of life; only 9% of cases are diagnosed after the age of 12 months. There is a striking predilection for the maxilla, which accounts for 61% of reported cases. Less frequently reported sites include the skull (16%), epididymis and testis (9%), mandible (6%), and brain (6%). A slight male predilection has been noted.
The lesion is most common in the anterior region of the maxilla, where it classically appears as a rapidly expanding mass that is frequently blue or black (Fig. 12-74). The tumor often destroys the underlying bone and may be associated with displacement of the developing teeth (Fig. 12-75). In some instances, there may be an associated osteogenic reaction, which exhibits a “sun ray” radiographic pattern that can be mistaken for osteosarcoma.
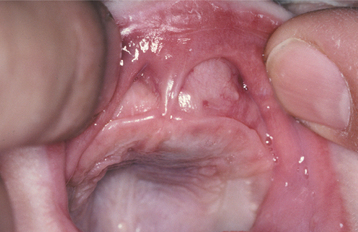
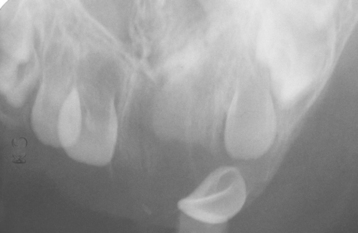
LABORATORY VALUES: High urinary levels of vanillylmandelic acid (VMA) often are found in patients with melanotic neuroectodermal tumor of infancy. These levels may return to normal once the tumor has been resected. This finding supports the hypothesis of neural crest origin because other tumors from this tissue (e.g., pheochromocytoma, neuroblastoma) often secrete norepinephrine-like hormones that are metabolized to VMA and excreted in the urine.
HISTOPATHOLOGIC FEATURES: The tumor consists of a biphasic population of cells that form nests, tubules, or alveolar structures within a dense, collagenous stroma (Figs. 12-76 and 12-77). The alveolar and tubular structures are lined by cuboidal epithelioid cells that demonstrate vesicular nuclei and granules of dark-brown melanin pigment. The second cell type is neuroblastic in appearance and consists of small, round cells with hyperchro-matic nuclei and little cytoplasm. These cells grow in loose nests and are frequently surrounded by the larger pigment-producing cells. Mitotic figures are rare.
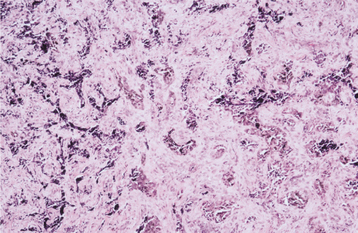
Fig. 12-76 Melanotic neuroectodermal tumor of infancy. Low-power view showing nests of epithelioid cells within a fibrous stroma.
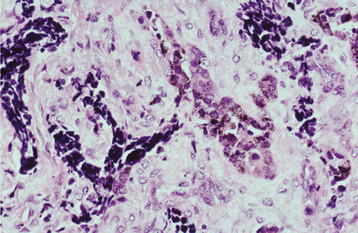
Fig. 12-77 Melanotic neuroectodermal tumor of infancy. High-power view of a tumor nest demonstrating two cell types: (1) small, hyperchromatic round cells and (2) larger epithelioid cells with vesicular nuclei. Some stippled melanin pigment is also present.
Because of the tumor’s characteristic microscopic features, immunohistochemistry usually is not essential to establish the diagnosis. However, the larger epithelioid cells typically are positive for cytokeratin and also may express neuron-specific enolase. In addition, the smaller cells usually are positive for neuron-specific enolase and CD56, and sometimes they will express other neuroendocrine markers such as glial fibrillary acidic protein and synaptophysin.
TREATMENT AND PROGNOSIS: Despite their rapid growth and potential to destroy bone, most melanotic neuroectodermal tumors of infancy are benign. The lesion is best treated by surgical removal. Some clinicians prefer simple curettage, although others advocate that a 5-mm margin of normal tissue be included with the specimen. Recurrence of the tumor has been reported in about 20% of cases. In addition, about 6% of reported cases, mostly from the brain or skull, have acted in a malignant fashion, resulting in metastasis and death. Although this estimation of 6% is probably high (because unusual malignant cases are more likely to be reported), it underscores the potentially serious nature of this tumor and the need for careful clinical evaluation and follow-up of affected patients.
PARAGANGLIOMA (CAROTID BODY TUMOR; CHEMODECTOMA; GLOMUS JUGULARE TUMOR; GLOMUS TYMPANICUM TUMOR)
The paraganglia are specialized tissues of neural crest origin that are associated with the autonomic nerves and ganglia throughout the body. Some of these cells act as chemoreceptors, such as the carotid body (located at the carotid bifurcation), which can detect changes in blood pH or oxygen tension and subsequently cause changes in respiration and heart rate. Tumors that arise from these structures are collectively known as paragangliomas, with the term preferably preceded by the anatomic site at which they are located. Therefore, tumors of the carotid body are appropriately known as carotid body paragangliomas (carotid body tumors); those that develop in the temporal bone and middle ear are called jugulotympanic paragangliomas. Jugulotympanic paragangliomas also are commonly known as glomus jugulare tumors, although some authors prefer to reserve this term only for those examples that arise from the jugular bulb and to use the term glomus tympanicum tumors for those that arise in the middle ear.
CLINICAL AND RADIOGRAPHIC FEATURES: Although paragangliomas are rare, the head and neck area is the most common site for these lesions. The most common paraganglioma is the carotid body tumor, which develops at the bifurcation of the internal and external carotid arteries. This tumor usually occurs in middle-aged adults. Most often it is a slowly enlarging, painless mass of the upper lateral neck below the angle of the jaw. It is seen more frequently in patients who live at high altitudes, indicating that some cases may arise from chronic hyperplasia of the carotid body in response to lower oxygen levels. Angiography can help to localize the tumor and demonstrate its characteristic vascular nature.
Jugulotympanic paragangliomas are the second most common type of these tumors. They also are most frequent in middle-aged individuals but show a 2:1 female predilection. The most common symptoms include dizziness, tinnitus (a ringing or other noise in the ear), hearing loss, and cranial nerve palsies. Imaging studies, especially three-dimensional (3D) time-of-flight magnetic resonance angiography, can help to detect and characterize such lesions. Other less common paragangliomas of the head and neck include vagal, nasopharyngeal, laryngeal, and orbital paragangliomas.
Approximately 10% to 20% of affected patients have multifocal tumors. In 10% of all cases, there is a family history of such tumors, with an autosomal dominant pattern of inheritance that is modified by genomic imprinting. The gene responsible for familial paragangliomas has been mapped to chromosome 11q23. In genomic imprinting, the gene is transmitted in a mendelian manner, but expression of that gene is determined by the sex of the transmitting parent. Paternal transmission results in development of tumors in the offspring, even if the father is clinically unaffected. Maternal transmission does not result in development of tumors in the offspring, although these children will carry the gene and have the ability to pass it down to subsequent generations. Hereditary cases have an even greater chance of being multicentric; about one third of affected patients have more than one tumor.
HISTOPATHOLOGIC FEATURES: The paraganglioma is characterized by round or polygonal epithelioid cells that are organized into nests or zellballen (Fig. 12-78). The overall architecture is similar to that of the normal paraganglia, except the zellballen are usually larger and more irregular in shape. These nests consist primarily of chief cells, which demonstrate centrally located, vesicular nuclei and somewhat granular, eosinophilic cytoplasm. The tumor is typically vascular and may be surrounded by a thin fibrous capsule.
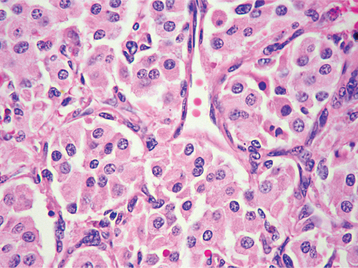
TREATMENT AND PROGNOSIS: The treatment of paragangliomas may include surgery, radiation therapy, or both, depending on the extent and location of the tumor. Localized carotid body paragangliomas often can be treated by surgical excision with maintenance of the vascular tree. If the carotid artery is encased by tumor, it also may need to be resected, followed by vascular grafting. Radiation therapy may be used as adjunctive treatment or for unresectable carotid body tumors.
Although most carotid body paragangliomas are benign and can be controlled with surgery and radiation therapy, vascular complications can lead to considerable surgical morbidity or mortality. In addition, 6% to 9% of carotid body paragangliomas metastasize, either to regional lymph nodes or distant sites. Unfortunately, it is usually difficult to predict which tumors will act in a malignant fashion based on their microscopic features. Because such metastases may develop many years after the original diagnosis is made, long-term follow-up is important.
Because of their location near the base of the brain, jugulotympanic paragangliomas are more difficult to manage. Recent advances in both diagnostic radiology and neurosurgery have greatly improved the potential for resection of these tumors. Radiation therapy may be used in conjunction with surgery or as a primary treatment for unresectable tumors. Stereotactic radiosurgery (gamma knife treatment) has shown promise in the management of primary or recurrent glomus jugulare tumors in patients who are poor surgical candidates. This technique allows the delivery of a focused, large, single dose of radiation under stereotactic guidance. Malignant behavior has been documented in approximately 4% of jugulotympanic paragangliomas.
GRANULAR CELL TUMOR
The granular cell tumor is an uncommon benign soft tissue neoplasm that shows a predilection for the oral cavity. The histogenesis of this lesion has long been debated. Originally, it was believed to be of skeletal muscle origin and was called the granular cell myoblastoma. However, more recent investigations do not support a muscle origin but point to a derivation from Schwann cells (granular cell schwannoma) or neuroendocrine cells. At present, it seems best to use the noncommittal term granular cell tumor for this lesion.
CLINICAL FEATURES: Granular cell tumors are most common in the oral cavity and on the skin. The single most common site is the tongue, which accounts for one third to half of all reported cases. Tongue lesions most often occur on the dorsal surface. The buccal mucosa is the second most common intraoral location. The tumor most frequently occurs in the fourth to sixth decades of life and is rare in children. There is a 2:1 female predilection.
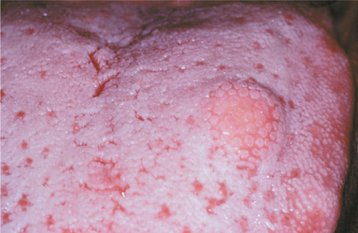
The granular cell tumor is typically an asymptomatic sessile nodule that is usually 2 cm or less in size (Figs. 12-79 and 12-80). The lesion often has been noted for many months or years, although sometimes the patient is unaware of its presence. The mass is typically pink, but occasional granular cell tumors appear yellow. The granular cell tumor is usually solitary, although multiple, separate tumors sometimes occur, especially in black patients.
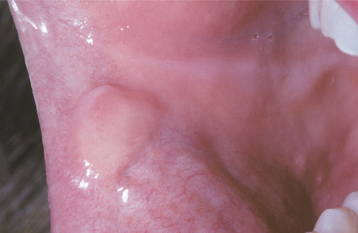
HISTOPATHOLOGIC FEATURES: The granular cell tumor is composed of large, polygonal cells with abundant pale eosinophilic, granular cytoplasm and small, vesicular nuclei (Fig. 12-81). The cells are usually arranged in sheets, but they also may be found in cords and nests. The cell borders often are indistinct, which results in a syncytial appearance. The lesion is not encapsulated and sometimes appears to infiltrate the adjacent connective tissues. Often, there appears to be a transition from normal adjacent skeletal muscle fibers to granular tumor cells; this finding led earlier investigators to suggest a muscle origin for this tumor. Less frequently, one may see groups of granular cells that envelop small nerve bundles. Immunohistochemical analysis reveals positivity for S-100 protein within the cells—a finding that is supportive, but not diagnostic, of neural origin.
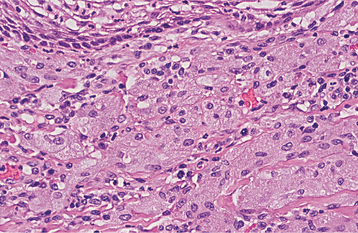
Fig. 12-81 Granular cell tumor. Medium-high–power view showing polygonal cells with abundant granular cytoplasm.
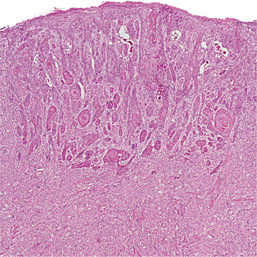
An unusual and significant microscopic finding is the presence of acanthosis or pseudoepitheliomatous (pseudocarcinomatous) hyperplasia of the overlying epithelium, which has been reported in up to 50% of all cases (Fig. 12-82). Although this hyperplasia is usually minor in degree, in some cases it may be so striking that it results in a mistaken diagnosis of squamous cell carcinoma and subsequent unnecessary cancer surgery. The pathologist must be aware of this possibility, especially when dealing with a superficial biopsy sample or a specimen from the dorsum of the tongue—an unusual location for oral cancer.
TREATMENT AND PROGNOSIS: The granular cell tumor is best treated by conservative local excision, and recurrence is uncommon. Extremely rare examples of malignant granular cell tumor have been reported.
CONGENITAL EPULIS (CONGENITAL EPULIS OF THE NEWBORN; CONGENITAL GRANULAR CELL LESION): The congenital epulis is an uncommon soft tissue tumor that occurs almost exclusively on the alveolar ridges of newborns. It is often known by the redundant term, congenital epulis of the newborn. Rare examples also have been described on the tongue; therefore, some authors prefer using the term congenital granular cell lesion, because not all cases present as an epulis on the alveolar ridge. It also has been called gingival granular cell tumor of the newborn, but this term should be avoided. Although it bears a light microscopic resemblance to the granular cell tumor (discussed previously), it exhibits ultrastructural and immunohistochemical differences that warrant its classification as a distinct and separate entity. However, the histogenesis of this tumor is still uncertain.
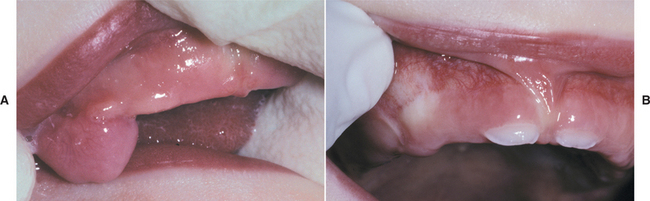
CLINICAL FEATURES: The congenital epulis typically appears as a pink-to-red, smooth-surfaced, polypoid mass on the alveolar ridge of a newborn (Fig. 12-83). Most examples are 2 cm or less in size, although lesions as large as 7.5 cm have been reported. On occasion, the tumor has been detected in utero via ultrasound examination. Multiple tumors develop in 10% of cases. A few rare examples on the tongue have been described in infants who also had alveolar tumors.
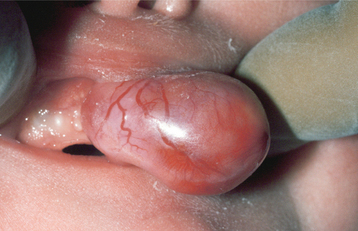
The tumor is two to three times more common on the maxillary ridge than on the mandibular ridge. It most frequently occurs lateral to the midline in the area of the developing lateral incisor and canine teeth. The congenital epulis shows a striking predilection for females, which suggests a hormonal influence in its development, although estrogen and progesterone receptors have not been detected. Nearly 90% of cases occur in females.
HISTOPATHOLOGIC FEATURES: The congenital epulis is characterized by large, rounded cells with abundant granular, eosinophilic cytoplasm and round to oval, lightly basophilic nuclei (Figs. 12-84 and 12-85). In older tumors, these cells may become elongated and separated by fibrous connective tissue. In contrast to the granular cell tumor, the overlying epithelium never shows pseudoepitheliomatous hyperplasia but typically demonstrates atrophy of the rete ridges. In addition, in contradistinction to the granular cell tumor, immunohistochemical analysis shows the tumor cells to be negative for S-100 protein.
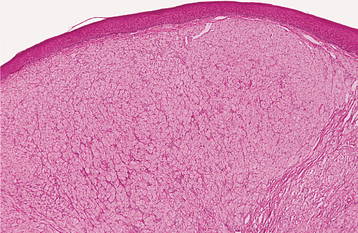
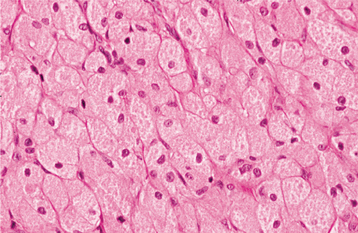
TREATMENT AND PROGNOSIS: The congenital epulis is usually treated by surgical excision. The lesion never has been reported to recur, even with incomplete removal.
After birth, the tumor appears to stop growing and may even diminish in size. Eventual complete regression has been reported in a few patients, even without treatment (Fig. 12-86).
HEMANGIOMA AND VASCULAR MALFORMATIONS
In recent years, great progress has been made in the classification and understanding of tumors and tumorlike proliferations of vascular origin. A modified classification scheme for these vascular anomalies is presented in Box 12-2.
The term hemangioma has traditionally been used to describe a variety of developmental vascular anomalies. Currently, hemangiomas are considered to be benign tumors of infancy that display a rapid growth phase with endothelial cell proliferation, followed by gradual involution. Most hemangiomas cannot be recognized at birth, but arise subsequently during the first 8 weeks of life. On the other hand, vascular malformations are structural anomalies of blood vessels without endothelial proliferation. By definition, vascular malformations are present at birth and persist throughout life. They can be categorized according to the type of vessel involved (capillary, venous, arteriovenous) and according to hemodynamic features (low flow or high flow).
CLINICAL AND RADIOGRAPHIC FEATURES:
HEMANGIOMA OF INFANCY: Hemangiomas are the most common tumors of infancy, occurring in 5% to 10% of 1-year-old children. They are much more common in females than in males (ratio: 3:1 to 5:1), and they occur more frequently in whites than in other racial groups. The most common location is the head and neck, which accounts for 60% of all cases. Eighty percent of hemangiomas occur as single lesions, but 20% of affected patients will have multiple tumors.
Fully developed hemangiomas are rarely present at birth, although a pale macule with threadlike telangiectasias may be noted on the skin. During the first few weeks of life, the tumor will demonstrate rapid development that occurs at a faster pace than the infant’s overall growth. Superficial tumors of the skin appear raised and bosselated with a bright-red color (“strawberry” hemangioma) (Fig. 12-87). They are firm and rubbery to palpation, and the blood cannot be evacuated by applying pressure. Deeper tumors may appear only slightly raised with a bluish hue.
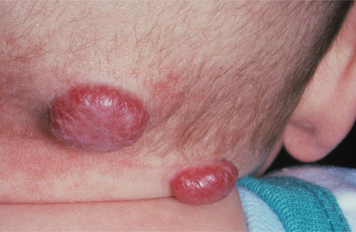
Fig. 12-87 Hemangioma. Infant with two red, nodular masses on the posterior scalp and neck (“strawberry” hemangioma).
The proliferative phase usually lasts for 6 to 10 months, after which the tumor slows in growth and begins to involute. The color gradually changes to a dull-purple hue and the lesion feels less firm to palpation. By age 5, most of the red color is usually gone. About half of all hemangiomas will show complete resolution by 5 years of age, with 90% resolving by age 9. After tumor regression is complete, normal skin will be restored in about 50% of patients; however, up to 40% of affected individuals will show perma-nent changes such as atrophy, scarring, wrinkling, or telangiectasias.
Complications occur in about 20% of hemangiomas. The most common problem is ulceration, which may occur with or without secondary infection. Although hemorrhage may be noted, significant blood loss does not usually occur. Hemangiomas that occur in crucial areas can be associated with significant morbidity. Periocular tumors often result in amblyopia (dimness of vision), strabismus, or astigmatism. Patients with multiple cutaneous hemangiomas or large facial hemangiomas are at increased risk for concomitant visceral hemangiomas. Tumors in the neck and laryngeal region can lead to airway obstruction.
Large, segmental cervicofacial hemangioma can be a component of a well-recognized hemangioma syndrome—PHACE(S) syndrome. This acronym stands for the following:
• Posterior fossa brain anomalies (usually Dandy-Walker malformation)
• Hemangioma (usually cervical segmental hemangioma)
Kasabach-Merritt phenomenon is a serious coagulopathy that has been associated with two rare vascular tumors known as tufted hemangioma and kaposiform hemangioendothelioma. This disorder is characterized by severe thrombocytopenia and hemorrhage because of platelet trapping within the tumor. The mortality rate is as high as 20% to 30%.
VASCULAR MALFORMATIONS: In contrast to hemangiomas, vascular malformations are present at birth and persist throughout life. Port wine stains are relatively common capillary malformations that occur in 0.3% to 1.0% of newborns. They are most common on the face, particularly along the distribution of the trigeminal nerve. In Sturge-Weber angiomatosis, associated intracranial lesions are present (see page 543). Port wine stains are typically pink or purple macular lesions that grow commensurately with the patient. As the patient gets older, the lesion often darkens and becomes nodular because of vascular ectasia.
Low-flow venous malformations encompass a wide spectrum of lesions, from small isolated ectasias to complex growths that involve multiple tissues and organs. They are present at birth, although they may not always be immediately apparent. Typically, venous malformations are blue and are easily compressible (Fig. 12-88). They often grow proportionately with the patient, but they may swell when dependent or with increased venous pressure. Secondary thrombosis and phlebolith formation can occur.
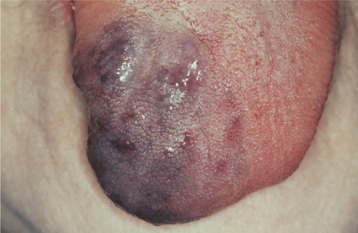
Arteriovenous malformations are high-flow lesions that result from persistent direct arterial and venous communication. Although they are present from birth, they may not become noticeable until later in childhood or adulthood. Because of the fast vascular flow through these lesions, a palpable thrill or bruit often is noticeable. The overlying skin typically feels warmer to touch. Presenting symptoms may include pain, bleeding, and skin ulceration.
INTRABONY VASCULAR MALFORMATIONS: Intrabony “hemangiomas” also may occur and probably represent either venous or arteriovenous malformations. In the jaws, such lesions are detected most often during the first 3 decades of life. They are slightly more common in females than in males and occur three times more often in the mandible than the maxilla. The lesion may be completely asymptomatic, although some examples are associated with pain and swelling. Mobility of teeth or bleeding from the gingival sulcus may occur. A bruit or pulsation may be apparent on auscultation and palpation.
The radiographic appearance of intrabony vascular malformations is variable. Most commonly, the lesion shows a multilocular radiolucent defect. The individual loculations may be small (honeycomb appearance) or large (soap bubble appearance). In other cases the lesion may present as an ill-defined radiolucent area or a well-defined, cystlike radiolucency (Fig. 12-89). Large malformations may cause cortical expansion, and occasionally a “sunburst” radiographic pattern is produced (Fig. 12-90). Angiography can be helpful in demonstrating the vascular nature of the lesion (Fig. 12-91).
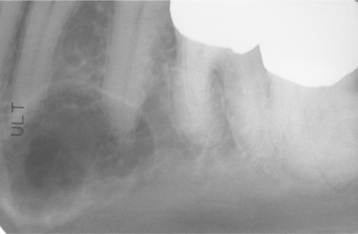
Fig. 12-89 Intrabony venous malformation. Well-circumscribed radiolucency that contains fine trabeculations.
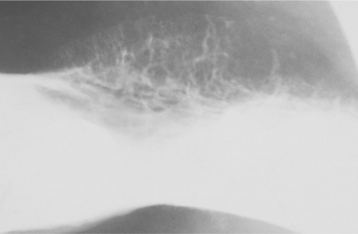
Fig. 12-90 Intrabony venous malformation. Occlusal radiograph demonstrating cortical destruction and a “sunburst” periosteal reaction resembling osteosarcoma.
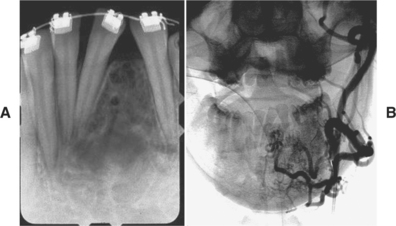
Fig. 12-91 Intrabony arteriovenous malformation. A, Periapical radiograph showing an expansile, mottled radiolucency in the mandibular incisor region. Pulsatile hemorrhage was encountered when a biopsy of this lesion was attempted. B, Angiogram demonstrating a vascular proliferation between the mandibular incisors. (Courtesy of Dr. Larry Cunningham and Dr. Jason Ford.)
HISTOPATHOLOGIC FEATURES: Early hemangiomas of infancy are characterized by numerous plump endothelial cells and often-indistinct vascular lumina (Figs. 12-92 and 12-93). At this stage, such lesions often are known microscopically as juvenile or cellular hemangiomas. Because of their cellular nature, these lesions also have been called juvenile hemangioendothelioma, although this term should be avoided because hemangioendothelioma also is used to designate other vascular tumors of intermediate malignant potential. As the lesion matures, the endothelial cells become flattened, and the small, capillary-sized vascular spaces become more evident (Fig. 12-94). As the hemangioma undergoes involution, the vascular spaces become less prominent and are replaced by fibrous connective tissue.
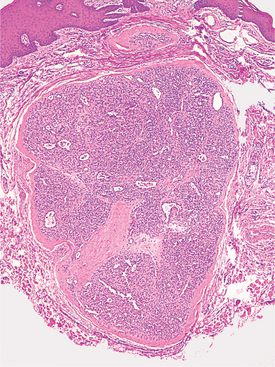
Fig. 12-92 Juvenile (cellular) hemangioma. Low-power photomicrograph showing a circumscribed cellular mass of vascular endothelial cells arranged in lobular aggregates.
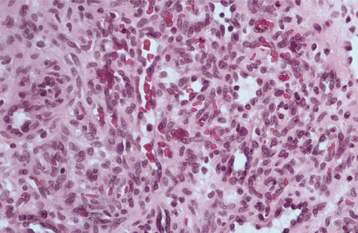
Fig. 12-93 Juvenile (cellular) hemangioma. High-power view showing a highly cellular endothelial proliferation forming occasional indistinct vascular lumina.
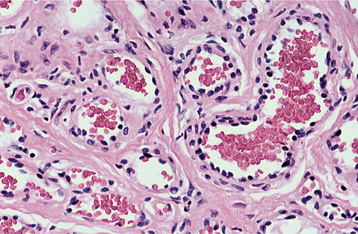
Fig. 12-94 Capillary hemangioma. High-power photomicrograph demonstrating well-formed capillary-sized vessels.
Vascular malformations do not show active endothelial cell proliferation, and the channels resemble the vessels of origin. Therefore, capillary malformations may be similar to the capillary stage of hemangioma (Fig. 12-95), whereas venous malformations may show more dilated vessels (Fig. 12-96). Because of their similar features, many vascular malformations are incorrectly categorized as hemangiomas. Arterio-venous malformations demonstrate a mixture of thick-walled arteries and veins, along with capillary vessels.
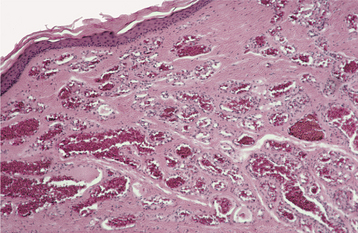
Fig. 12-95 Capillary malformation. Low-power view of a vascular proliferation forming multiple capillary blood vessels.
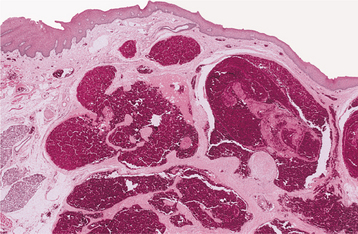
Fig. 12-96 Venous malformation. Low-power photomicrograph showing multiple large, dilated blood vessels.
GLUT1 is an immunohistochemical marker that is consistently positive in the hemangioma of infancy. In contrast, this marker is negative in other developmental vascular tumors and anomalies listed in Box 12-2 (rapidly involuting congenital hemangioma [RICH], noninvoluting congenital hemangioma [NICH], tufted angioma, kaposiform hemangioendothelioma, pyogenic granuloma, and vascular malformations).
TREATMENT AND PROGNOSIS: Because most hemangiomas of infancy undergo involution, management often consists of “watchful neg-lect.” It is important to educate parents that although rapid growth may be seen, regression will occur. Surgical resection is rarely warranted during infancy. For problematic or life-threatening hemangiomas, pharmacologic therapy may be indicated. Systemic corticosteroids may help to reduce the size of the lesion and are associated with a 70% to 90% response rate. Intra-lesional and topical corticosteroids also have been used for smaller localized, problematic lesions. Intravenous (IV) vincristine is currently the drug of choice for complicated tumors that are unresponsive to systemic corticosteroid therapy. Interferon a-2a is no longer widely used because of the reported risk of permanent spastic diplegia.
Flashlamp pulsed dye lasers can be effective in the treatment of port wine stains. The management of venous malformations depends on the size, location, and associated complications of the lesion. Small, stable malformations may not require treatment. Larger, problematic lesions may be treated with a combination of sclerotherapy and surgical excision. Sclerotherapy involves the injection of sclerosing agents, such as 95% ethanol, directly into the lesion to induce fibrosis. Sclerotherapy alone may be sufficient for smaller lesions; for larger lesions, subsequent surgical resection can be accomplished with less risk of bleeding after sclerotherapy.
The treatment of arteriovenous malformations is more challenging and also depends on the size of the lesion and degree of involvement of vital structures. For cases that require resection, radiographic embolization often is performed 24 to 48 hours before surgery to minimize blood loss.
Vascular malformations of the jaws are potentially dangerous lesions because of the risk of severe bleeding, which may occur spontaneously or during surgical manipulation. Needle aspiration of any undiagnosed intrabony lesion before biopsy is a wise precaution to rule out the possibility of a vascular malformation. Severe and even fatal hemorrhages have occurred after incisional biopsy or extraction of teeth in the area of such lesions.
STURGE-WEBER ANGIOMATOSIS (ENCEPHALOTRIGEMINAL ANGIOMATOSIS; STURGE-WEBER SYNDROME)
Sturge-Weber angiomatosis is a rare, nonhereditary developmental condition that is characterized by a hamartomatous vascular proliferation involving the tissues of the brain and face. It is believed to be caused by the persistence of a vascular plexus around the cephalic portion of the neural tube. This plexus develops during the sixth week of intrauterine development but normally undergoes regression during the ninth week.
CLINICAL AND RADIOGRAPHIC FEATURES: Patients with Sturge-Weber angiomatosis are born with a dermal capillary vascular malformation of the face known as a port wine stain or nevus flammeus because of its deep-purple color. This port wine stain usually has a unilateral distribution along one or more segments of the trigeminal nerve. Occasionally, patients have bilateral involvement or additional port wine lesions elsewhere on the body. Not all patients with facial port wine nevi have Sturge-Weber angiomatosis. In one study of patients with facial port wine nevi, only slightly more than 10% had Sturge-Weber angiomatosis. Only patients with involvement along the distribution of the ophthalmic branch of the trigeminal nerve were at risk for the full condition (Figs. 12-97 and 12-98).
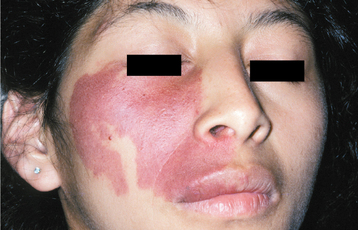
Fig. 12-97 Port wine stain. Nevus flammeus of the malar area in a patient without Sturge-Weber angiomatosis. Unless the vascular lesion includes the region innervated by the ophthalmic branch of the trigeminal nerve, usually the patient does not have central nervous system (CNS) involvement.
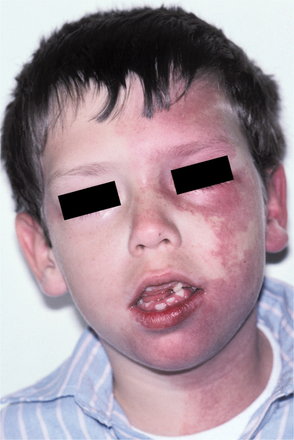
Fig. 12-98 Sturge-Weber angiomatosis. Port wine stain of the left face, including involvement along the ophthalmic branch of the trigeminal nerve. The patient also was mentally retarded and had a seizure disorder.
In addition to the facial port wine nevus, affected individuals also have leptomeningeal angiomas that overlie the ipsilateral cerebral cortex. This meningeal angiomatosis is usually associated with a convulsive disorder and often results in mental retardation or contralateral hemiplegia. Imaging studies of the brain may reveal gyriform “tramline” calcifications on the affected side (Fig. 12-99). Ocular involvement may be manifested by glaucoma and vascular malformations of the conjunctiva, episclera, choroid, and retina.
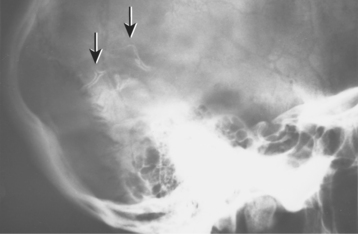
Fig. 12-99 Sturge-Weber angiomatosis. Skull film showing “tramline” calcifications (arrows). (Courtesy of Dr. Reg Munden.)
Intraoral involvement in Sturge-Weber angiomatosis is common, resulting in hypervascular changes to the ipsilateral mucosa (Fig. 12-100). The gingiva may exhibit slight vascular hyperplasia or a more massive hemangiomatous proliferation that can resemble a pyogenic granuloma. Such gingival hyperplasia may be attributable to the increased vascular component, phenytoin therapy used to control the epileptic seizures, or both. Destruction of the underlying alveolar bone has been reported in rare instances.
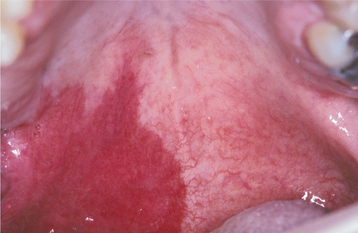
HISTOPATHOLOGIC FEATURES: The port wine nevus is characterized by excessive numbers of dilated blood vessels in the middle and deep dermis. The intraoral lesions show a similar vascular dilatation. Proliferative gingival lesions may resemble a pyogenic granuloma.
TREATMENT AND PROGNOSIS: The treatment and prognosis of Sturge-Weber angiomatosis depend on the nature and severity of the possible clinical features. Usually, facial port wine nevi can be improved by using the newer flashlamp pulsed dye lasers. Cortical excision of angiomatous meningeal lesions may be necessary in some cases. Patients with intractable epilepsy and progressive mental retardation eventually may require more extensive neurosurgical treatment, including lobectomy or hemispherectomy.
Port wine nevi that affect the gingiva can make flossing and dental prophylaxis difficult. Great care must be taken when performing surgical procedures in affected areas of the mouth because severe hemorrhage may be encountered. Lasers also may be helpful in the removal of hyperplastic oral lesions.
NASOPHARYNGEAL ANGIOFIBROMA
The nasopharyngeal angiofibroma is a rare vascular and fibrous tumorlike lesion that occurs only in the nasopharynx. Although microscopically benign, it frequently exhibits locally destructive and aggressive behavior. It may represent a vascular malformation rather than a true neoplasm.
CLINICAL AND RADIOGRAPHIC FEATURES: Nasopharyngeal angiofibromas occur almost exclusively in males. The tumor is exceedingly rare in females—so much so that the diagnosis in a female patient should be viewed with skepticism and closely scrutinized. The lesion also shows a striking predilection for adolescents between the ages of 10 and 17 and often has been called the juvenile nasopharyngeal angiofibroma. However, rare examples also have been reported in slightly younger and older patients. Because of its almost exclusive occurrence in adolescent boys, a hormonal influence seems likely, although no endocrine abnormalities have been detected.
Nasal obstruction and epistaxis are common early symptoms. The lesion is currently presumed to arise in the pterygopalatine fossa and expands medially into the nasal cavity via the sphenopalatine foramen. Some cases will show extension into the paranasal sinuses, orbits, or middle cranial fossa. Invasion into the oral cavity or cheek rarely has been reported. Computed tomography (CT) scans and magnetic resonance imaging (MRI) studies are helpful adjuncts in visualizing the extent of the lesion and degree of adjacent tissue destruction. Anterior bowing of the posterior wall of the maxillary sinus is a characteristic feature (Fig. 12-101). Angiograms can be used to confirm the vascular nature of the lesion (Fig. 12-102).
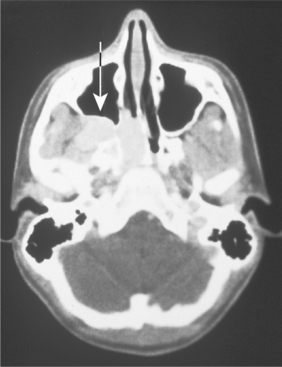
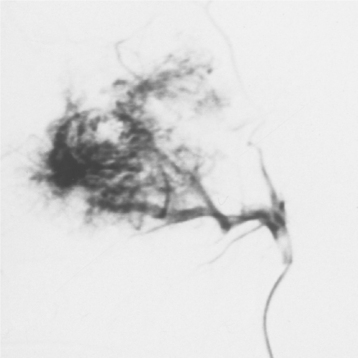
HISTOPATHOLOGIC FEATURES: The nasopharyngeal angiofibroma consists of dense fibrous connective tissue that contains numerous dilated, thin-walled blood vessels of variable size (Fig. 12-103). Typically, the vascular component is more prominent at the periphery of the tumor, especially in lesions from younger patients.
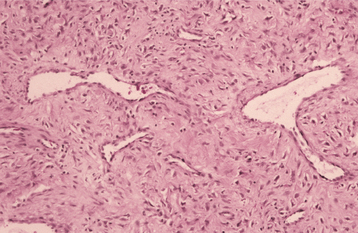
TREATMENT AND PROGNOSIS: The primary treatment of nasopharyngeal angiofibroma usually consists of surgical excision. Depending on the extent of the lesion, this may be accomplished via endoscopic surgery, lateral rhinotomy, midfacial degloving procedure, infratemporal fossa approach, or combined craniofacial resection. Preoperative embolization of the tumor is helpful in controlling blood loss. Radiation therapy is usually reserved for recurrent lesions and extensive tumors with unusual vascular supplies or intracranial extension.
The recurrence rate varies from 20% to 40% in most recent studies. Such recurrences are usually retreated with further surgery or radiation therapy. Malignant transformation into fibrosarcoma has rarely been reported and is probably associated with prior radiation therapy.
HEMANGIOPERICYTOMA–SOLITARY FIBROUS TUMOR
As originally described, the hemangiopericytoma (HPC) was a rare neoplasm that presumably was derived from pericytes (i.e., cells with processes that encircled endothelial cells of capillaries). However, in many examples it was difficult to confirm pericytic differentiation, making diagnostic reproducibility among pathologists difficult. The solitary fibrous tumor (SFT) was initially described as a pleural neoplasm that was believed to be derived from either mesothelial cells or submesothelial fibroblasts. However, examples of this tumor were later identified in a number of other anatomic sites, including the head and neck region. Many of these lesions exhibit microscopic features that overlap those of HPC. In recent years, experts have questioned the concept of the HPC, suggesting that many purported examples should be reclassified as SFTs or designated by the combined name, heman-giopericytoma–solitary fibrous tumor (HPC-SFT). As more knowledge is gained, it appears likely that the classification and nomenclature of this spectrum of tumors will undergo further revision.
In addition, a microscopically similar neoplasm (hemangiopericytoma-like tumor) of the sinonasal region is recognized, which is felt to represent a distinct, separate entity.
CLINICAL FEATURES: HPC-SFTs have been reported primarily in adults and are rare in children. The tumor is usually described as a slow-growing, painless, submucosal, or deep soft tissue mass that is easily removed from the surrounding tissues. One of the most common oral locations is the buccal mucosa, which accounts for 75% of cases reported under the designation of SFT.
Hemangiopericytoma-like tumors of the nasal cavity and paranasal sinuses primarily occur in middle-aged and older adults. Common presenting symptoms include nasal obstruction and epistaxis.
HISTOPATHOLOGIC FEATURES: HPC-SFTs are usually well-circumscribed lesions that exhibit a variable microscopic appearance. At the traditional HPC end of the spectrum, the tumor exhibits tightly packed cells that surround endothelium-lined vascular channels. The cells are haphazardly arranged and demonstrate round to ovoid nuclei and indistinct cytoplasmic borders. The blood vessels often show irregular branching, which results in a characteristic “staghorn” and “antlerlike” appearance (Fig. 12-104, A).
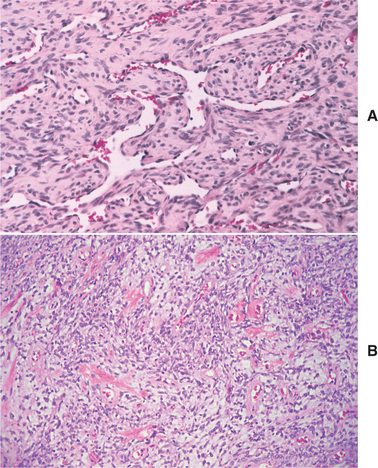
Fig. 12-104 Hemangiopericytoma–solitary fibrous tumor. A, “Staghorn” blood vessels with surrounding pericytes. B, Moderately cellular fibrous proliferation (“patternless pattern”) with prominent vascularity, slightly myxoid areas, and scattered dense collagen bundles.
At the SFT end of the spectrum, the cells are more spindled and arranged in either short fascicles or in a disorganized fashion (“patternless pattern”) (Fig. 12-104, B). The tumor often demonstrates alternat-ing hypercellular and hypocellular zones with a variable degree or myxoid background change. Prominent hyalinized collagen bundles are characteristically observed in the hypocellular areas. Immunohistochemical studies show the lesional cells to be positive for CD34.
The identification of four or more mitoses per 10 high-power fields suggests a rapidly growing tumor that is capable of metastasis. The presence of necrosis also suggests malignancy. However, it is difficult to predict microscopically whether a particular tumor will act in a benign or malignant fashion.
Hemangiopericytoma-like tumors of the sinonasal region have a more prominent spindle cell pattern, with the cells arranged in a more orderly fashion. Mitotic figures are rare or absent. The vascular component is less intricate, and less interstitial collagen is found among the tumor cells.
TREATMENT AND PROGNOSIS: For HPC-SFTs with a benign histopathologic appearance, local excision is the treatment of choice. More extensive surgery is required for tumors with malignant characteristics. Oral examples usually behave in a benign fashion, although up to 10% of extraplural SFTs have been reported to show malignant behavior. One recent paper reported a 5-year survival rate of 86% for HPC. Therefore, long-term follow-up of all patients with this tumor spectrum is recommended.
Studies show that patients with hemangiopericytoma-like tumors of the sinonasal region have a better prognosis than do those with tumors at other sites.
LYMPHANGIOMA
Lymphangiomas are benign, hamartomatous tumors of lymphatic vessels. It is doubtful that they are true neoplasms; instead, they most likely represent developmental malformations that arise from sequestrations of lymphatic tissue that do not communicate normally with the rest of the lymphatic system.
There are three types of lymphangioma:
1. Lymphangioma simplex (capillary lymphangioma), which consists of small, capillary-sized vessels
2. Cavernous lymphangioma, which is composed of larger, dilated lymphatic vessels
3. Cystic lymphangioma (cystic hygroma), which exhibits large, macroscopic cystic spaces
However, this classification system is rather arbitrary because all three sizes of vessels often can be found within the same lesion.
The subtypes are probably variants of the same pathologic process, and the size of the vessels may depend on the nature of the surrounding tissues. Cystic lymphangiomas most often occur in the neck and axilla, where the loose adjacent connective tissues allow for more expansion of the vessels. Cavernous lymphangiomas are more frequent in the mouth, where the denser surrounding connective tissue and skeletal muscle limit vessel expansion.
CLINICAL FEATURES: Lymphangiomas have a marked predilection for the head and neck, which accounts for 50% to 75% of all cases (Fig. 12-105). About half of all lesions are noted at birth, and around 90% develop by 2 years of age.
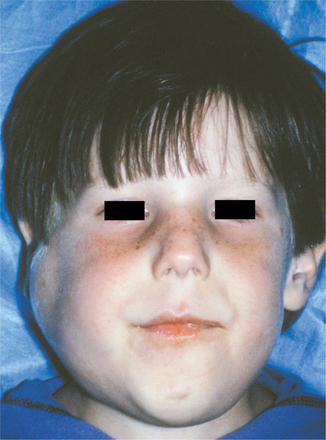
Fig. 12-105 Lymphangioma. Young boy with a cystic hygroma primarily involving the right side of the face. (Courtesy of Dr. Frank Kendrick.)
Cervical lymphangiomas are more common in the posterior triangle and are typically soft, fluctuant masses. They occur less frequently in the anterior triangle, although lesions in this location are more likely to result in respiratory difficulties or dysphagia if they grow large. Occasionally, cervical lymphangiomas extend into the mediastinum or upward into the oral cavity. Such tumors can become massive and can measure 15 cm or greater in size. Rapid tumor enlargement may occur secondary to an upper respiratory tract infection, presumably because of increased lymph production, blocked lymphatic drainage, or secondary infection of the tumor.
Oral lymphangiomas may occur at various sites but are most frequent on the anterior two thirds of the tongue, where they often result in macroglossia (Figs. 12-106 and 12-107). Usually, the tumor is superficial in location and demonstrates a pebbly surface that resembles a cluster of translucent vesicles. The surface has been likened to the appearance of frog eggs or tapioca pudding. Secondary hemorrhage into the lymphatic spaces may cause some of these “vesicles” to become purple. Deeper tumors present as soft, ill-defined masses.
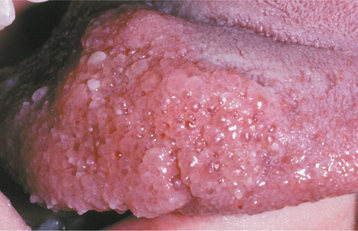
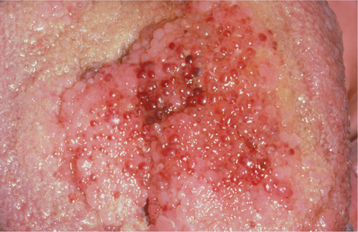
Fig. 12-107 Lymphangioma. Dorsal tongue lesion demonstrating a purple color, which can be caused by secondary hemorrhage or an associated hemangiomatous component.
Small lymphangiomas less than 1 cm in size occur on the alveolar ridge in around 4% of black neonates. These lesions often occur bilaterally on the mandibular ridge and show a 2:1 male-to-female distribution. Most of these alveolar lymphangiomas apparently resolve spontaneously because they are not observed in older people.
HISTOPATHOLOGIC FEATURES: Lymphangiomas are composed of lymphatic vessels that may show marked dilatation (cavernous lymphangioma) (Figs. 12-108 and 12-109) or macroscopic cystlike structures (cystic hygroma) (Fig. 12-110). The vessels often diffusely infiltrate the adjacent soft tissues and may demonstrate lymphoid aggregates in their walls. The lining endothelium is typically thin, and the spaces contain proteinaceous fluid and occasional lymphocytes. Some channels also may contain red blood cells, which creates uncertainty as to whether they are lymphatic or blood vessels. Although many of these likely represent secondary hemorrhage into a lymphatic vessel, some actually may be examples of mixed lymphangioma and hemangioma.
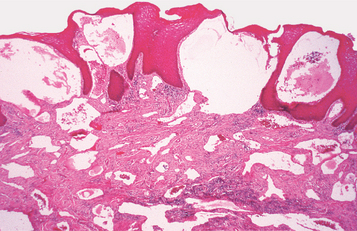
Fig. 12-108 Cavernous lymphangioma. Lesion of the tongue showing dilated lymphatic vessels beneath the epithelium and in the deeper connective tissues.
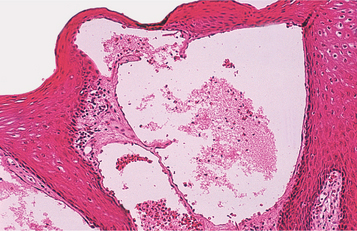
Fig. 12-109 Cavernous lymphangioma. High-power photomicrograph showing dilated, lymph-filled vessels immediately below the atrophic surface epithelium.
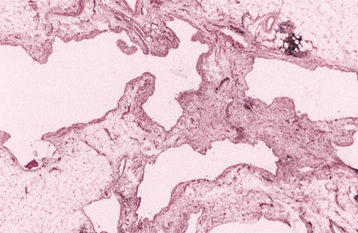
In intraoral tumors, the lymphatic vessels are characteristically located just beneath the epithelial surface and often replace the connective tissue papillae. This superficial location results in the translucent, vesicle-like clinical appearance. However, extension of these vessels into the deeper connective tissue and skeletal muscle also may be seen.
TREATMENT AND PROGNOSIS: The treatment of lymphangiomas usually consists of surgical excision, although total removal may not be possible in all cases because of large size or involvement of vital structures. Recurrence is common, especially for cavernous lymphangiomas of the oral cavity, because of their infiltrative nature. Some clinicians do not recommend treatment for nonenlarging lymphangiomas of the tongue because of the difficulty in removal and high recurrence rate. Cystic lymphangiomas of the cervical region are often well circumscribed and have a lower rate of recurrence. Spontaneous regression of lymphangiomas is rare.
Unfortunately, lymphangiomas do not respond to sclerosing agents as do hemangiomas. However, some success with sclerosant therapy for unresectable lymphangiomas has been reported using OK-432, a lyophilized incubation mixture of a low-virulent strain of Streptococcus pyogenes with penicillin G potassium, which has lost its streptolysin S–producing ability.
The prognosis is good for most patients, although large tumors of the neck or tongue may result in airway obstruction and death. The mortality rate for cystic hygromas ranges from 2% to 5% in most series.
LEIOMYOMA
Leiomyomas are benign tumors of smooth muscle that most commonly occur in the uterus, gastrointestinal tract, and skin. Leiomyomas of the oral cavity are rare. Most of these probably have their origin from vascular smooth muscle.
The three types are as follows:
Almost all oral leiomyomas are either solid or vascular in type; angiomyomas account for nearly 75% of all oral cases.
CLINICAL AND RADIOGRAPHIC FEATURES: The oral leiomyoma can occur at any age and is usually a slow-growing, firm, mucosal nodule (Fig. 12-111). Most lesions are asymptomatic, although occasional tumors can be painful. Solid leiomyomas are typically normal in color, although angiomyomas may exhibit a bluish hue. The most common sites are the lips, tongue, palate, and cheek, which together account for 80% of cases. Extremely rare intraosseous examples may present as unilocular radiolucencies of the jaws.
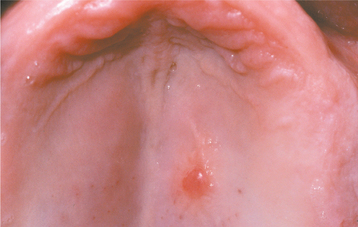
HISTOPATHOLOGIC FEATURES: Solid leiomyomas are well-circumscribed tumors that consist of interlacing bundles of spindle-shaped smooth muscle cells (Figs. 12-112 and 12-113). The nuclei are elongated, pale staining, and blunt ended. Mitotic figures are uncommon. Angiomyomas also are well-circumscribed lesions that demonstrate multiple tortuous blood vessels with thickened walls caused by hyperplasia of their smooth muscle coats (Fig. 12-114). Intertwining bundles of smooth muscle may be found between the vessels, sometimes with intermixed adipose tissue. As its name implies, the epithelioid leiomyoma is composed primarily of epithelioid cells rather than spindle cells.
Fig. 12-112 Leiomyoma. Low-power view showing a well-circumscribed cellular mass of spindle-shaped smooth muscle cells.
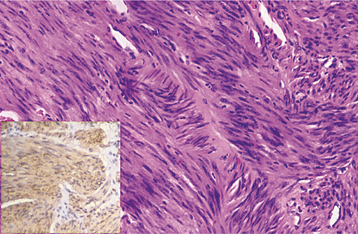
Fig. 12-113 Leiomyoma. High-power view showing spindle-shaped cells with blunt-ended nuclei. Immuno-histochemical analysis shows strong positivity for smooth muscle actin (inset).
Fig. 12-114 Angiomyoma. Well-circumscribed tumor exhibiting prominent blood vessels surrounded by smooth muscle.
Special stains and immunohistochemistry may be helpful to confirm the smooth muscle origin if the diagnosis is in doubt. The smooth muscle stains bright red with the Masson trichrome stain (Fig. 12-115). Immunohistochemical analysis usually reveals the tumor cells to be positive for vimentin, smooth muscle actin, and muscle-specific actin; desmin positivity also may be seen.
RHABDOMYOMA
Benign neoplasms of skeletal muscle are called rhabdomyomas. The term rhabdomyoma also is used to describe a hamartomatous lesion of the heart that often is associated with tuberous sclerosis (see page 757). Despite the great amount of skeletal muscle throughout the body, benign skeletal muscle tumors are extremely rare. However, these extracardiac rhabdomyomas show a striking predilection for the head and neck. Rhabdomyomas of the head and neck can be subclassified into two major categories: (1) adult rhabdomyomas and (2) fetal rhabdomyomas.
ADULT RHABDOMYOMAS: Adult rhabdomyomas of the head and neck occur primarily in middle-aged and older patients, with about 70% of cases found in men. The most frequent sites are the pharynx, oral cavity, and larynx; intraoral lesions are most common in the floor of the mouth, soft palate, and base of tongue. The tumor appears as a nodule or mass that can grow to many centimeters before discovery (Figs. 12-116 and 12-117). Laryngeal and pharyngeal lesions often lead to airway obstruction. Sometimes, the tumor is multinodular in nature, with two or more discrete nodules found in the same anatomic location. Occasional cases are multicentric, with separate, distinct tumors at different sites.
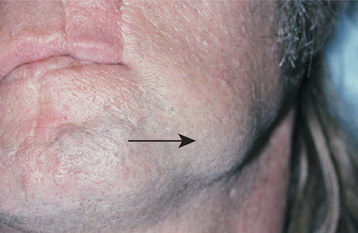
Fig. 12-116 Adult rhabdomyoma. Nodular mass (arrow) in the left cheek. (Courtesy of Dr. Craig Little.)
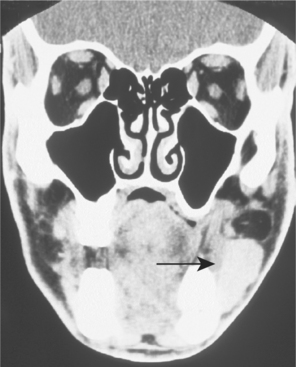
Fig. 12-117 Adult rhabdomyoma. Computed tomography (CT) scan of the same tumor depicted in Fig. 12-116. Note the mass (arrow) lateral to the left body of the mandible. (Courtesy of Dr. Craig Little.)
ADULT RHABDOMYOMAS: The adult rhabdomyoma is composed of well-circumscribed lobules of large, polygonal cells, which exhibit abundant granular, eosinophilic cytoplasm (Fig. 12-118). These cells often demonstrate peripheral vacuolization that results in a “spider web” appearance of the cytoplasm. Focal cells with cross striations can be identified in most cases (Fig. 12-119). Although rarely necessary for the diagnosis, immunohistochemical examination will show the tumor cells to be positive for myoglobin, desmin, and muscle-specific actin.
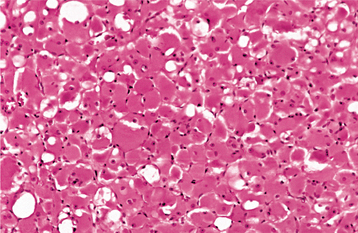
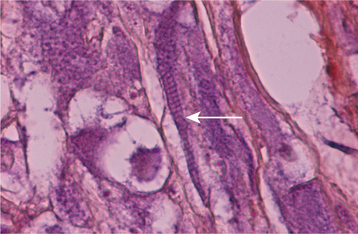
Fetal Rhabdomyomas: The fetal rhabdomyoma has a less mature appearance and consists of a haphazard arrangement of spindle-shaped muscle cells that sometimes are found within a myxoid stroma. Some tumors may show considerable cellularity and mild pleomorphism, which makes them easily mistaken for rhabdomyosarcomas.
OSSEOUS AND CARTILAGINOUS CHORISTOMAS
A choristoma is a tumorlike growth of microscopically normal tissue in an abnormal location. Several different tissue types may occur in the mouth as choristomas. These include gastric mucosa, glial tissue, and tumorlike masses of sebaceous glands. However, the most frequently observed choristomas of the oral cavity are those that consist of bone, cartilage, or both. These lesions sometimes have been called soft tissue osteomas or soft tissue chondromas, but choristoma is a better term because they do not appear to be true neoplasms.
CLINICAL FEATURES: Osseous and cartilaginous choristomas show a striking predilection for the tongue, which accounts for 85% of cases. The most common location is the posterior tongue near the foramen cecum, although rare examples also have been reported elsewhere on the tongue and at other oral locations. The lesion is usually a firm, smooth-surfaced, sessile or pedunculated nodule between 0.5 and 2.0 cm in diameter (Fig. 12-120). Many patients are unaware of the lesion, although some complain of gagging or dysphagia. More than 70% of osseous choristomas have been reported in women.
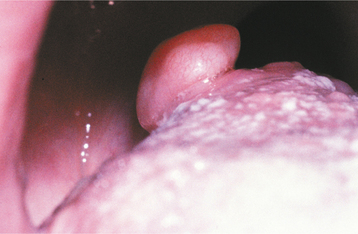
HISTOPATHOLOGIC FEATURES: Microscopic examination of choristomas shows a well-circumscribed mass of dense lamellar bone or mature cartilage that is surrounded by dense fibrous connective tissue (Fig. 12-121). Sometimes a combination of bone and cartilage is formed. The bone has a well-developed haversian canal system and occasionally demonstrates central fatty or hematopoietic marrow.
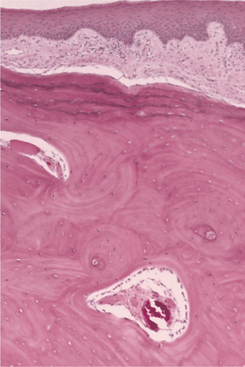
Soft Tissue Sarcomas
Fortunately, soft tissue sarcomas are rare in the oral and maxillofacial region and account for less than 1% of the cancers in this area. Because of their relative rarity, it is beyond the scope of this book to give a complete, detailed discussion of each of these tumors. However, a review of these entities is included in the following section.
Over the past two decades, clinicians have seen a radical shift in the classification and nomenclature of many soft tissue sarcomas on the basis of detailed histochemical, immunohistochemical, and ultrastructural studies. Because of this, a number of previous tumor concepts have changed or are being critically reevaluated.
FIBROSARCOMA
The fibrosarcoma is a malignant tumor of fibroblasts. At one time, it was considered one of the most common soft tissue sarcomas. However, the diagnosis of fibrosarcoma is made much less frequently today because of the recognition and separate classification of other spindle cell lesions that have similar microscopic features. The tumor is most common in the extremities; only 10% occur in the head and neck region.
CLINICAL FEATURES: Fibrosarcomas most often present as slow-growing masses that may reach considerable size before they produce pain (Fig. 12-122). They can occur anywhere in the head and neck region. A number of cases have been reported in the nose and paranasal sinuses, where they often result in obstructive symptoms. They can occur at any age but are most common in young adults and children.
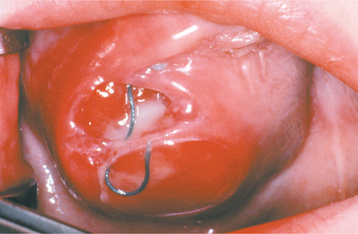
HISTOPATHOLOGIC FEATURES: Well-differentiated fibrosarcomas consist of fascicles of spindle-shaped cells that classically form a “herringbone” pattern (Fig. 12-123). The cells often show little variation in size and shape, although variable numbers of mitotic figures can usually be identified. In poorly differentiated tumors, the cells are less organized and may appear rounder or ovoid. Mild pleomorphism along with more frequent mitotic activity may be seen. Poorly differentiated tumors tend to produce less collagen than do well-differentiated tumors.
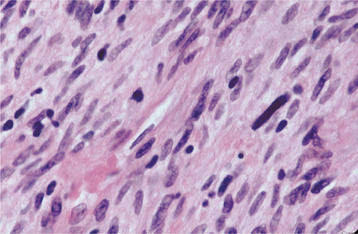
MALIGNANT FIBROUS HISTIOCYTOMA
The malignant fibrous histiocytoma is considered to be a sarcoma with both fibroblastic and histiocytic features. After the introduction of this term in 1963, this tumor concept rapidly gained acceptance and became the most common soft tissue sarcoma diagnosed in adults. However, some experts today question the concept of malignant fibrous histiocytoma and now favor reclassification of the various tumors in this family into other categories, including undifferentiated pleomorphic sarcoma, liposarcoma, myxofibrosarcoma, melanoma, and anaplastic carcinoma. On the other hand, other authors still support the concept of this neoplasm. The following brief discussion reviews the traditional features of malignant fibrous histiocytoma, although it is possible that use of this term may fall further out of favor in the future.
CLINICAL FEATURES: The malignant fibrous histiocytoma is primarily considered to be a tumor of older age groups. The most common complaint is an expanding mass that may or may not be painful or ulcerated. Tumors of the nasal cavity and paranasal sinuses produce obstructive symptoms.
HISTOPATHOLOGIC FEATURES: Several histopathologic subtypes have been described. The storiform-pleomorphic type is the most common. This pattern is characterized by short fascicles of plump spindle cells arranged in a storiform pattern, admixed with areas of pleomorphic giant cells (Fig. 12-124). Myxoid, giant cell, inflammatory, and angiomatoid subtypes also have been recognized.
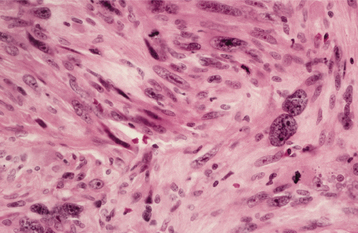
TREATMENT AND PROGNOSIS: The malignant fibrous histiocytoma is considered to be an aggressive tumor that is usually treated by radical surgical resection. Approximately 40% of patients have had local recurrences. A similar percentage has developed metastases, usually within 2 years of the initial diagnosis. The survival rate for patients with oral tumors appears to be worse than for those with tumors at other body sites.
LIPOSARCOMA
The liposarcoma is a malignant neoplasm of fatty origin. It currently is considered to be the most common soft tissue sarcoma and accounts for 20% of all soft tissue malignancies in adults. The most common sites are the thigh, retroperitoneum, and inguinal region. Liposarcomas of the head and neck are rare.
CLINICAL FEATURES: Liposarcomas are primarily seen in adults, with peak prevalence between the ages of 40 and 60. The tumor is typically a soft, slow-growing, ill-defined mass that may appear normal in color or yellow. Pain or tenderness is uncommon; when present, it is usually a late feature. The neck is the most common site for liposarcomas of the head and neck region. The most frequent oral locations are the tongue and cheek.
Histopathologic Features: Most liposarcomas can be divided into three major categories:
The most common of these variants in the oral cavity is the well-differentiated liposarcoma, which ac-counts for 55% to 90% of all cases. These tumors resemble benign lipomas but demonstrate scattered lipoblasts and atypical, hyperchromatic stromal cells (Fig. 12-125).
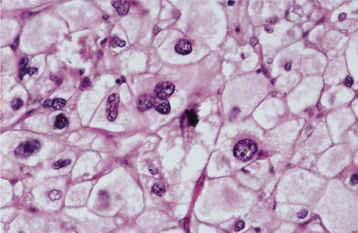
Myxoid liposarcomas demonstrate proliferating lipoblasts within a myxoid stroma that contains a rich capillary network. The round cell liposarcoma is a more aggressive form of myxoid liposarcoma with less differentiated, rounded cells.
Pleomorphic liposarcomas exhibit extreme cellular pleomorphism and bizarre giant cells. Dedifferentiated liposarcomas are characterized by the combination of well-differentiated liposarcoma with poorly differentiated, nonlipogenic sarcomatous changes. These features may coexist in the same neoplasm, or the dedifferentiated changes may develop in a recurrent tumor or metastasis.
TREATMENT AND PROGNOSIS: Radical excision is the treatment of choice for most liposarcomas throughout the body. In spite of this, around 50% of all tumors recur. The overall 5-year survival rate ranges from 59% to 70%. There is a 10-year survival rate of approximately 50%. The histopathologic subtype is extremely important in predicting the prognosis; the outlook for pleomorphic liposarcomas is much worse than for myxoid and well-differentiated tumors.
In contrast, the prognosis for oral liposarcoma is more favorable because of the predominance of well-differentiated subtypes and because most tumors are small when diagnosed. Local recurrence has been reported in 15% to 20% of cases, but metastasis and death as a result of tumor is rare.
MALIGNANT PERIPHERAL NERVE SHEATH TUMOR (MALIGNANT SCHWANNOMA; NEUROFIBROSARCOMA; NEUROGENIC SARCOMA)
The principal malignancy of peripheral nerve origin is preferably called a malignant peripheral nerve sheath tumor. These tumors account for 5% of all soft tissue sarcomas, with about half of such cases occurring in patients with neurofibromatosis type I (see page 529). The lesion is most common on the proximal portions of the extremities and the trunk; only 10% to 15% of cases occur in the head and neck.
CLINICAL AND RADIOGRAPHIC FEATURES: Malignant peripheral nerve sheath tumors are most common in young adults. The mean age in patients with neurofibromatosis (29 to 36 years) is about one decade younger than in those without this condition (40 to 46 years). The tumor is an enlarging mass that sometimes exhibits rapid growth. Associated pain or a nerve deficit is common.
Oral tumors may occur anywhere, but the most common sites are the mandible, lips, and buccal mucosa (see Figs. 12-67 and 12-68, page 531). Radiographic examination of intraosseous tumors of the mandible may reveal widening of the mandibular canal or the mental foramen, with or without irregular destruction of the surrounding bone.
HISTOPATHOLOGIC FEATURES: The malignant peripheral nerve sheath tumor shows fascicles of atypical spindle-shaped cells, which often resemble the cells of fibrosarcoma (see Fig. 12-69, page 531). However, these cells are frequently more irregular in shape with wavy or comma-shaped nuclei. In addition to streaming fascicles, less cellular myxoid areas also may be present. With some tumors, there can be heterologous elements, which include skeletal muscle differentiation (malignant Triton tumor), cartilage, bone, or glandular structures.
A definitive diagnosis of neural origin is often difficult, especially in the absence of neurofibromatosis. Positive immunostaining for S-100 protein is a helpful clue, but this is found in only about 50% of all cases.
TREATMENT AND PROGNOSIS: The treatment of malignant peripheral nerve sheath tumors consists primarily of radical surgical excision, possibly along with adjuvant radiation therapy and chemotherapy. The prognosis is poor, especially in patients with neurofibromatosis. One study showed the 5-year survival rate in individuals with neurofibromatosis type I to be only 16%. For other patients, the 5-year survival rate was 53%; this rate dropped to 38% at 10 years. However, another study showed an overall 5-year survival rate of 44%, which was nearly equal between both groups.
OLFACTORY NEUROBLASTOMA (ESTHESIONEUROBLASTOMA)
The olfactory neuroblastoma is a rare neuroectodermal neoplasm of the upper nasal vault that shows some similarities to neuroblastomas seen elsewhere in the body. Traditionally, it is believed to arise from the olfactory epithelium.
CLINICAL AND RADIOGRAPHIC FEATURES: Unlike the usual neuroblastoma, the olfactory neuroblastoma is rare in patients younger than the age of 10 years. Instead, it is more common in adults and occurs over a wide age range. The tumor arises high in the nasal cavity close to the cribriform plate. From there it may extend into the adjacent paranasal sinuses (especially the ethmoid sinus), the orbit, and the anterior cranial fossa (Fig. 12-126). The most common symptoms are nasal obstruction, anosmia, epistaxis, and pain.
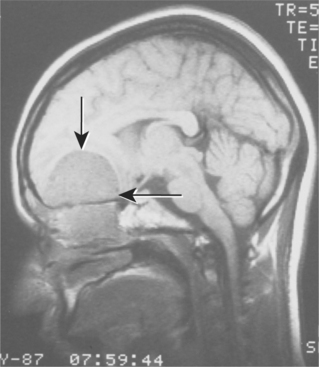
HISTOPATHOLOGIC FEATURES: Olfactory neuroblastomas consist of small, round to ovoid basophilic cells that are arranged in sheets and lobules (Fig. 12-127). Rosette and pseudorosette formation and areas of delicate neurofibrillary material may be seen.
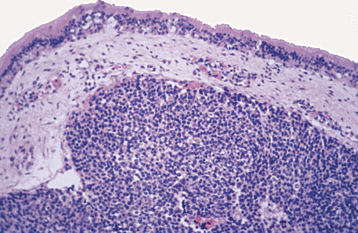
TREATMENT AND PROGNOSIS: The treatment of olfactory neuroblastoma consists of surgical excision, often with adjuvant radiation therapy. A combined craniofacial surgical approach frequently is used. Chemotherapy also has been administered, especially in advanced cases.
The prognosis depends on the stage of the disease. For patients with stage A lesions (tumor confined to the nasal cavity), the 5-year survival rate ranges from 72% to nearly 90%. The 5-year survival rate drops to 59% to 70% for stage B disease (tumor extending into the paranasal sinuses). For stage C disease (tumor extending beyond the nasal cavity and sinuses), the 5-year survival rate has improved to nearly 50% or even greater with newer treatment regimens. Death is usually a result of local recurrence; metastasis occurs in approximately 20% to 37% of cases.
ANGIOSARCOMA
Angiosarcoma is a rare malignancy of vascular endothelium, which may arise from either blood or lymphatic vessels. More than 50% of all cases occur in the head and neck region, with the scalp and forehead being the most common sites. Oral lesions are quite rare.
The term hemangioendothelioma is used to describe vascular tumors with microscopic features intermediate between those of hemangiomas and angiosarcomas. Such tumors also are rare and are considered to be of intermediate malignancy.
CLINICAL FEATURES: Cutaneous angiosarcomas of the head and neck are most common in older adult patients. Early lesions often resemble a simple bruise, which may lead to a delay in diagnosis. However, the lesion continues to enlarge, which results in an elevated, nodular, or ulcerated surface. Many examples appear multifocal in nature. Oral angiosarcomas have been reported in various locations; the tongue and mandible are two of the more common sites (Fig. 12-128).
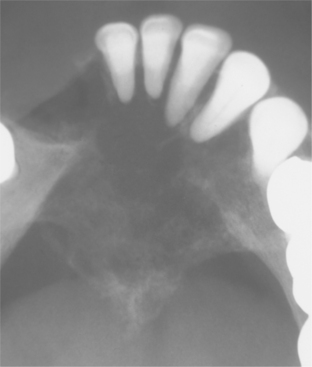
HISTOPATHOLOGIC FEATURES: Angiosarcoma is characterized by an infiltrative proliferation of endothelium-lined blood vessels that form an anastomosing network (Fig. 12-129). The endothelial cells appear hyperchromatic and atypical; they often tend to pile up within the vascular lumina. Increased mitotic activity may be seen. Immunohistochemical studies show the tumor cells to be positive for CD31 and factor VIII–related antigen in most cases, whereas CD34 positivity is observed less consistently.
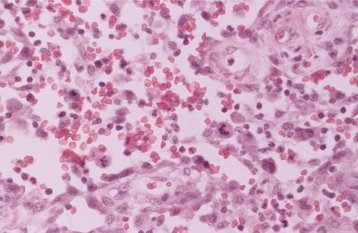
TREATMENT AND PROGNOSIS: Treatment usually consists of radical surgical excision, radiation therapy, or both. The prognosis for angiosarcoma of the face and scalp is poor, with a reported 10-year survival rate of only 21%. However, angiosarcomas of the oral cavity and salivary glands appear to have a better outcome. One recent study showed 11 of 14 patients with oral and salivary angiosarcoma to be free of tumor on follow-up (mean follow-up period: 8.6 years).
KAPOSI’S SARCOMA
Kaposi’s sarcoma is an unusual vascular neoplasm that was first described in 1872 by Moritz Kaposi, a Hungarian dermatologist. Before the advent of the acquired immunodeficiency syndrome (AIDS) epidemic, it was a rare tumor; however, beginning in the early 1980s, Kaposi’s sarcoma became quite common because of its propensity to develop in individuals infected by the human immunodeficiency virus (HIV). Since the introduction of highly active antiretroviral therapy (HAART) in the mid- to late 1990s, the prevalence of AIDS-related Kaposi’s sarcoma in the Western world has declined. Unfortunately, however, the frequency of this tumor still appears to be increasing in certain developing African nations.
Current evidence suggests that Kaposi’s sarcoma is caused by human herpesvirus 8 (HHV-8; Kaposi’s sarcoma–associated herpesvirus [KSHV]). The lesion most likely arises from endothelial cells, with some evidence of lymphatic origin. Four clinical presentations are recognized:
The first three forms are discussed here; AIDS-related Kaposi’s sarcoma is covered in the section on HIV disease (see page 270).
CLASSIC TYPE: Classic (chronic) Kaposi’s sarcoma is primarily a disease of late adult life, and 70% to 90% of cases occur in men. It mostly affects individuals of Italian, Jewish, or Slavic ancestry. Multiple blue-purple macules and plaques are present on the skin of the lower extremities (Fig. 12-130). These lesions grow slowly over many years and develop into painless tumor nodules. Oral lesions are rare and most frequently involve the palate. Some earlier reports suggested that patients with classic Kaposi’s sarcoma had an increased prevalence of lymphoreticular malignancies, but more recent analysis has questioned any significant association.
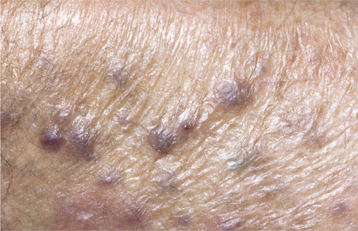
ENDEMIC TYPE: Endemic Kaposi’s sarcoma in Africa has been divided into four subtypes:
1. A benign nodular type, similar to classic Kaposi’s sarcoma
2. An aggressive or infiltrative type, characterized by progressive development of locally invasive lesions that involve the underlying soft tissues and bone
3. A florid form, characterized by rapidly progressive and widely disseminated, aggressive lesions with frequent visceral involvement
4. A unique lymphadenopathic type, occurring primarily in young black children and exhibiting generalized, rapidly growing tumors of the lymph nodes, occasional visceral organ lesions, and sparse skin involvement
IATROGENIC TYPE: Iatrogenic immunosuppression-associated Kaposi’s sarcoma most often occurs in recipients of organ transplants. It affects 0.5% of renal transplant patients, usually several months to a few years after the transplant. It is probably related to the loss of cellular immunity, which occurs as a result of immunosuppressive drugs. Like classic Kaposi’s sarcoma, iatrogenic immunosuppression-associated cases are most common in individuals of Italian, Jewish, and Slavic ancestry; however, the disease may run a more aggressive course.
HISTOPATHOLOGIC FEATURES: Kaposi’s sarcoma typically evolves through three stages:
A proliferation of miniature vessels characterizes the patch stage. This results in an irregular, jagged vascular network that surrounds preexisting vessels. Sometimes normal structures, such as hair follicles or preexisting blood vessels, may appear to protrude into these new vessels (promontory sign). The lesional endothelial cells have a bland appearance and may be associated with scattered lymphocytes and plasma cells.
The plaque stage demonstrates further proliferation of these vascular channels along with the development of a significant spindle cell component.
In the nodular stage, the spindle cells increase to form a nodular tumorlike mass that may resemble a fibrosarcoma or other spindle cell sarcomas (Figs. 12-131 and 12-132). However, numerous extravasated erythrocytes are present, and slitlike vascular spaces may be discerned.
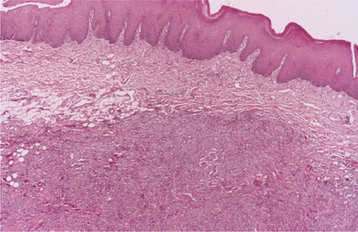
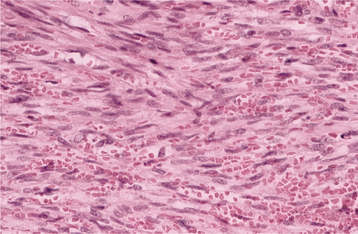
TREATMENT AND PROGNOSIS: The treatment of Kaposi’s sarcoma depends on the clinical subtype and stage of the disease. For skin lesions in the classic form of the disease, radiation therapy (especially electron beam) often is used. Radiation therapy for oral lesions must be approached with caution, because an unusually severe mucositis can develop. Surgical excision can be performed for the control of individual lesions of the skin or mucosa. Systemic chemotherapy, especially vinblastine, also may be helpful. Intralesional injection of chemotherapeutic agents is used to control individual lesions.
The prognosis is variable, depending on the form of the disease and the patient’s immune status. The classic form of the disease is slowly progressive; only 10% to 20% of patients develop disseminated lesions. The mean survival time is 10 to 15 years, and patients often die from unrelated causes. The benign nodular, endemic African form of the disease is similar in behavior to classic non-African Kaposi’s sarcoma. However, the other endemic African forms are more aggressive and the prognosis is poorer. The lymphadenopathic form runs a particularly fulminant course, usually resulting in the death of the patient within 2 to 3 years. In transplant patients, the disease also may be somewhat more aggressive, although the tumors may regress if immunosuppressive therapy is discontinued or reduced.
LEIOMYOSARCOMA
The leiomyosarcoma is a malignant neoplasm of smooth muscle differentiation, which accounts for 5% to 10% of all soft tissue sarcomas. The most common sites are the uterine wall and gastrointestinal tract. Leiomyosarcomas of the oral cavity are rare.
CLINICAL FEATURES: In general, leiomyosarcomas are most common in middle-aged and older adults. However, tumors in the oral and maxillofacial region occur over a wide age range without a predilection for any age group. They have been reported at various sites, but half of all oral cases occur in the jawbones. The clinical appearance is nonspecific; there is usually an enlarging mass that may or may not be painful (Fig. 12-133). Secondary ulceration of the mucosal surface may occur.
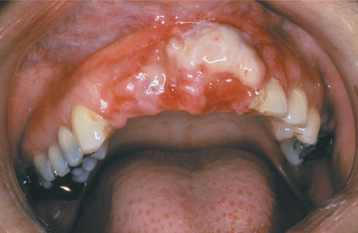
HISTOPATHOLOGIC FEATURES: The microscopic examination of a leiomyosarcoma shows fascicles of spindle-shaped cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei (Fig. 12-134). Some tumors may be composed primarily of rounded epithelioid cells that have either eosinophilic or clear cytoplasm (epithelioid leiomyosarcoma). The degree of pleomorphism varies from one tumor to the next, but smooth muscle tumors with the presence of five or more mitoses per 10 high-power fields should be considered malignant. Glycogen can usually be demonstrated within the cells with a periodic acid-Schiff (PAS) stain, and the cell cytoplasm appears bright red with a Masson trichrome stain. Immunohistochemical analysis usually reveals the presence of one or more of the following myogenic markers: desmin, muscle-specific actin (HHF 35), smooth muscle myosin (SMMS), and smooth muscle actin.
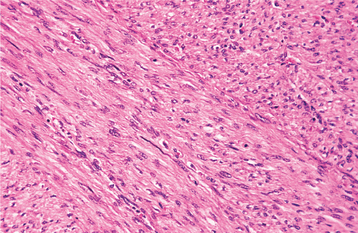
TREATMENT AND PROGNOSIS: The treatment of leiomyosarcoma primarily consists of radical surgical excision, sometimes with adjunctive chemotherapy or radiation therapy. The prognosis for oral tumors is guarded, with the potential for local recurrence and distant metastasis. Although few cases are available for analysis, a 5-year survival rate of 62% has been estimated.
RHABDOMYOSARCOMA
Rhabdomyosarcoma is a malignant neoplasm that is characterized by skeletal muscle differentiation. These tumors are much more common in young children, accounting for 60% of soft tissue sarcomas in childhood. In contrast, rhabdomyosarcoma comprises only 2% to 5% of soft tissue sarcomas in adults. The most frequent site is the head and neck, which accounts for 35% of all cases. The genitourinary tract is the second most common location. Several microscopic patterns of pediatric rhabdomyosarcoma are recognized (Table 12-1), although discussion here will be limited primarily to the embryonal and alveolar subtypes.
Table 12-1
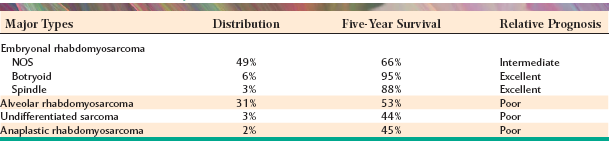
Adapted from Hicks J, Flaitz C: Rhabdomyosarcoma of the head and neck in children, Oral Oncol 38:450-459, 2002. NOS, Not otherwise specified.
CLINICAL FEATURES: Rhabdomyosarcoma primarily occurs during the first decade of life but also may occur in teenagers and young adults. It is rare in people older than 45 years, and approximately 60% of all cases occur in males. Embryonal rhabdomyosarcomas are most common in the first 10 years of life and account for about 60% of all cases. Alveolar rhabdomyosarcomas occur most often in persons between 10 and 25 years of age; they account for 20% to 30% of all tumors. Pleomorphic rhabdomyosarcomas represent less than 5% of all cases and show a peak prevalence in patients older than 40 years of age. Most head and neck lesions are embryonal or alveolar types; pleomorphic rhabdomyosarcomas primarily occur on the extremities.
The tumor is most often a painless, infiltrative mass that may grow rapidly (Figs. 12-135 and 12-136). In the head and neck region, the face and orbit are the most frequent locations, followed by the nasal cavity. The palate is the most frequent intraoral site, and some lesions may appear to arise in the maxillary sinus and break through into the oral cavity. Some embryonal rhabdomyosarcomas that arise within a cavity, such as the vagina or oropharynx, demonstrate an exophytic, polypoid growth pattern that resembles a cluster of grapes. The term botryoid (grapelike) rhabdomyosarcoma has been used for these lesions.
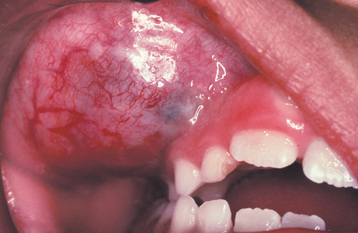
Fig. 12-135 Embryonal rhabdomyosarcoma. Young child with a mass of the right maxilla. (Courtesy of Dr. Robert Achterberg.)
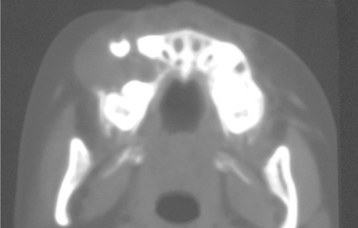
Fig. 12-136 Embryonal rhabdomyosarcoma. Computed tomography (CT) scan of patient from Fig. 12-135 showing expansile lytic lesion of the maxilla. (Courtesy of Dr. Robert Achterberg.)
EMBRYONAL TYPE: The embryonal rhabdomyosarcoma resembles various stages in the embryogenesis of skeletal muscle. Poorly differentiated examples may be difficult to diagnose and consist of small round or oval cells with hyperchromatic nuclei and indistinct cytoplasm (Fig. 12-137). Alternating hypercellular and myxoid zones may be seen. Better-differentiated lesions show round to ovoid rhabdomyoblasts with distinctly eosinophilic cytoplasm and fibrillar material around the nucleus. Cross striations are rarely found. Some tumors show better-differentiated, elongated, strap-shaped rhabdomyoblasts.
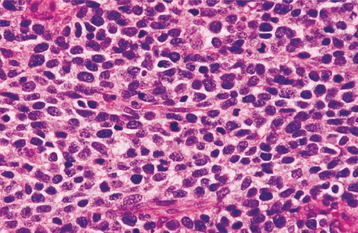
Fig. 12-137 Embryonal rhabdomyosarcoma. Medium-power view showing a sheet of small, round cells with hyperchromatic nuclei.
The botryoid subtype of embryonal rhabdomyosarcoma is sparsely cellular and has a pronounced myxoid stroma. Increased cellularity, or a so-called cambium layer, is usually seen just beneath the mucosal surface.
Immunohistochemical analysis for the presence of desmin, myogenin, and muscle-specific actin can be helpful in supporting the muscular nature of the tumor. However, the intensity of the immunostaining can vary depending on the degree of rhabdomyoblastic differentiation.
ALVEOLAR TYPE: Both classic and solid variants of alveolar rhabdomyosarcoma are recognized. The classic pattern is characterized by aggregates of poorly differentiated round to oval cells separated by fibrous septa. These cells demonstrate a central loss of cohesiveness, which results in an alveolar pattern. The peripheral cells of these aggregates adhere to the septal walls in a single layer. The central cells appear to float freely within the alveolar spaces. Mitoses are common, and multinucleated giant cells also may be seen. In contrast, solid alveolar rhabdomyosarcoma demonstrates cellular fields of small round basophilic cells without fibrovascular septa.
Cytogenetic and molecular studies play an important role in the diagnosis of rhabdomyosarcoma. Two distinct translocations have been identified in alveolar rhabdomyosarcoma (PAX3-FKHR and PAX7-FKHR). Embryonal rhabdomyosarcoma is characterized by a consistent loss of heterozygosity or loss of imprinting at chromosome 11p15.
TREATMENT AND PROGNOSIS: Before 1960 the prognosis for a patient with rhabdomyosarcoma was extremely poor, with more than 90% of patients dying. With the advent of multimodal therapy during the past several decades, the prognosis has improved dramatically.
Treatment typically consists of local surgical excision followed by multiagent chemotherapy (vincristine, actinomycin D, and cyclophosphamide). Postoperative radiation therapy also is used, except for localized tumors that have been completely resected at initial surgery. The 5-year survival rate for embryonal rhabdomyosarcoma (not otherwise specified [NOS]) is around 66%, although the figures for botryoid (95%) and spindle cell variants (88%) are much better. The 5-year survival rate for alveolar rhabdomyosarcoma is only 53%, and survival drops to slightly less than 50% for anaplastic rhabdomyosarcoma and undifferentiated sarcomas.
SYNOVIAL SARCOMA
Synovial sarcoma is an uncommon malignancy that represents 5% to 10% of all soft tissue sarcomas. The tumor occurs primarily near large joints and bursae, especially in the extremities, but most authorities now agree that this lesion probably does not arise from the synovium. Although it is often para-articular in location, the tumor rarely occurs within the joint capsule. In some instances, it arises in areas without any obvious relationship to synovial structures. Synovial sarcomas of the head and neck are rare (only 4% to 9% of all cases), and many of these apparently are unrelated to joint areas.
Over 90% of synovial sarcomas exhibit a specific balanced reciprocal translocation between the X chromosome and chromosome 18: t(X;18)(p11.2;q11.2). Detection of this translocation can be helpful in making the diagnosis, evaluating tumor margins, and confirming the presence of metastatic disease.
CLINICAL FEATURES: Synovial sarcomas most frequently occur in teenagers and young adults, and there is a slight male predilection. The most common presentation is a gradually enlarging mass that often is associated with pain or tenderness. Tumors in the head and neck region are most common in the paravertebral and parapharyngeal areas. Often, they produce symptoms of dysphagia, dyspnea, or hoarseness. Oral tumors most often have been reported in the tongue and cheek.
HISTOPATHOLOGIC FEATURES: Classic synovial sarcoma is a biphasic tumor that consists of a combination of spindle cells and epithelial cells (Fig. 12-138). The spindle cells usually predominate and produce a pattern that is similar to fibrosarcoma. Within this spindle cell background are groups of cuboidal to columnar epithelial cells that surround glandlike spaces or form nests, cords, or whorls. Calcifications are seen in around 30% of cases.
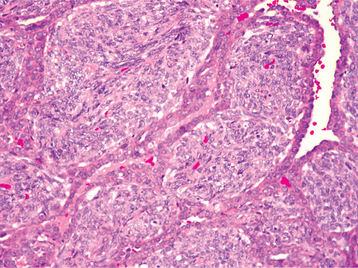
Fig. 12-138 Synovial sarcoma. Biphasic tumor consisting of spindle cells intermixed with cuboidal to columnar epithelial cells that line glandlike spaces.
Less frequently, the tumor is monophasic and consists primarily or entirely of spindle cells. The diagnosis of these tumors is difficult, but most lesions demonstrate at least focal positive immunostaining of spindle cells for cytokeratin or epithelial membrane antigen. Rare examples of monophasic epithelial synovial sarcomas also have been reported.
TREATMENT AND PROGNOSIS: Treatment of synovial sarcoma usually consists of radical surgical excision, possibly with adjunctive radiation therapy or chemotherapy. The prognosis is poor because the tumor has a high rate of recurrence and metastasis. The reported 5-year survival rate ranges from 36% to 64%. However, the 10-year survival rate drops to 20% to 38% because of the high rate of late metastases.
ALVEOLAR SOFT-PART SARCOMA
The alveolar soft-part sarcoma is a rare neoplasm of uncertain histogenesis. About 25% of all cases occur in the head and neck.
CLINICAL FEATURES: The alveolar soft-part sarcoma is usually a slow-growing, painless mass. The tumor is most common in young adults and children. In adults, the lower extremity is the most frequent location; in younger patients, the head and neck region is the most common site. The orbit and tongue are the most common head and neck locations, and the median age for lingual tumors is only 5 to 8 years. During the first 2 decades of life, the tumor shows nearly a 2:1 female predilection. However, cases that develop after the age of 30 are more common in men.
HISTOPATHOLOGIC FEATURES: Alveolar soft-part sarcomas are composed of groups of large, polygonal cells that are arranged around central alveolar spaces (Fig. 12-139). These cells have abundant granular, eosinophilic cytoplasm and one to several vesicular nuclei. Mitoses are rare. Special stains will reveal PAS-positive, diastase-resistant crystals that are highly characteristic for this tumor. Under the electron microscope, these crystals appear as rhomboid, polygonal, or rod-shaped structures with a regular latticework pattern.
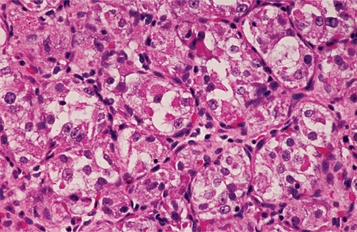
Fig. 12-139 Alveolar soft-part sarcoma. Alveolar collections of large, polygonal cells containing abundant granular cytoplasm.
Immunohistochemistry has played a limited role in the specific diagnosis of alveolar soft-part sarcoma, except to rule out other tumors with more characteristic staining patterns. However, molecular analysis of fresh tissue may be helpful in difficult cases because the tumor shows a characteristic genetic translocation, der(17)t(X;17)(p11.2;q25), which results in an ASPL-TFE3 fusion gene. Recently, an antibody against the C-terminus of the ASPL-TFE3 fusion protein has been created, allowing the development of a sensitive immunohistochemical marker for this tumor.
TREATMENT AND PROGNOSIS: Most patients with alveolar soft-part sarcomas are treated by radical surgical excision, possibly in conjunction with radiation therapy and chemotherapy. The prognosis is poor, often as a result of late metastasis. One study reported a 5-year survival rate of 60%, but the 20-year survival rate dropped to only 15%. Another series showed a 5-year disease-free survival of 71% for patients with localized disease, compared with only 20% for patients who presented with metastatic disease. However, the prognosis for children appears to be better than for adults. Lingual and orbital tumors have very high survival rates, possibly because of smaller tumor size at diagnosis and younger patient age.
METASTASES TO THE ORAL SOFT TISSUES
Metastatic tumors to the oral cavity are uncommon and represent approximately 1% of all oral malignancies. Such metastases can occur to bone (see page 669) or to the oral soft tissues. The mechanism by which tumors can spread to the oral cavity is poorly understood. Primary malignancies from immediately adjacent tissues might be able to spread by a lymphatic route; however, such a mechanism cannot explain metastases from tumors from lower parts of the body, which are almost certainly blood-borne and should be filtered out by the lungs. One possible explanation for blood-borne metastases to the head and neck, especially in the absence of pulmonary metastases, is Batson’s plexus, a valveless vertebral venous plexus that might allow retrograde spread of tumor cells, bypassing filtration through the lungs.
CLINICAL FEATURES: The most common site for oral soft tissue metastases is the gingiva, which accounts for slightly more than 50% of all cases. The next most common site is the tongue, which accounts for 25% of cases. The lesion usually appears as a nodular mass that often resembles a hyperplastic or reactive growth, such as a pyogenic granuloma (Figs. 12-140 to 12-142). Occasionally, the lesion appears as a surface ulceration. Adjacent teeth may become loosened by an underlying destruction of the alveolar bone. The presence of teeth may play an important role in the preference of metastases for the gingiva. Once malignant cells reach the oral cavity, the rich vascular network of inflamed gingival tissues may serve as a fertile site for further growth.
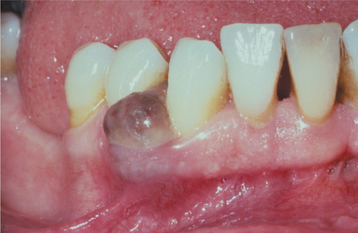
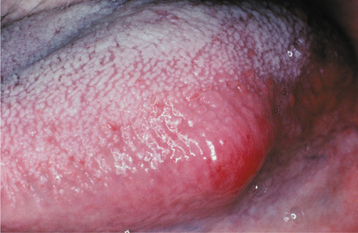
Fig. 12-141 Metastatic renal carcinoma. Nodular mass of the left lateral border of the tongue. (Courtesy of Dr. Mark Bowden.)
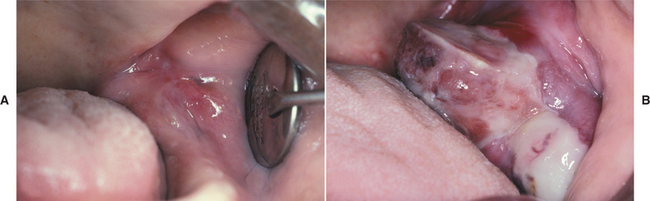
Fig. 12-142 Metastatic adenocarcinoma of the colon. A, Focal swelling of the left retromolar pad area. B, Same patient 4 weeks later. Note the marked enlargement of the lesion. (Courtesy of Dr. George Blozis.)
Oral soft tissue metastases are more common in males and are seen most frequently in middle-aged and older adults. Almost any malignancy from any body site is capable of metastasis to the oral cavity, and a wide variety of tumors have been reported to spread to the mouth. (However, there is probably a bias in the literature toward reporting more unusual cases.) In the cases reported, lung cancer is responsible for more than one third of all oral soft tissue metastases in men, followed by renal carcinoma and melanoma. Although prostate cancer is common in men, metastases from these tumors have an affinity for bone and rarely occur in soft tissues. For women, breast cancer accounts for 25% of all cases, followed by malignancies of the genital organs, lung, bone, and kidney. It is probable that in the future we will see an increased number of metastatic lung cancers in women (today this is the most common cancer killer of women in the United States).
In most cases the primary tumor already is known when the metastatic lesion is discovered. In some cases, however, the oral lesion is the first sign of the malignant disease.
HISTOPATHOLOGIC FEATURES: The microscopic appearance of the metastatic neoplasm should resemble the tumor of origin (Fig. 12-143). Most cases represent carcinomas; metastatic sarcomas to the oral region are rare.