Developmental Defects of the Oral and Maxillofacial Region
OROFACIAL CLEFTS
The formation of the face and oral cavity is complex in nature and involves the development of multiple tissue processes that must merge and fuse in a highly orchestrated fashion. Disturbances in the growth of these tissue processes or their fusion may result in the formation of orofacial clefts.
Development of the central face begins around the end of the fourth week of human development, with the appearance of the nasal (olfactory) placodes on either side of the inferior aspect of the frontonasal process. Proliferation of ectomesenchyme on both sides of each placode results in the formation of the medial and lateral nasal processes. Between each pair of processes is a depression, or nasal pit, that represents the primitive nostril.
During the sixth and seventh weeks of development, the upper lip forms when the medial nasal processes merge with each other and with the maxillary processes of the first branchial arches. Thus the midportion of the upper lip is derived from the medial nasal processes, and the lateral portions are derived from the maxillary processes. The lateral nasal processes are not involved in the formation of the upper lip, but they give rise to the alae of the nose.
The primary palate also is formed by the merger of the medial nasal processes to form the intermaxillary segment. This segment gives rise to the premaxilla, a triangular-shaped piece of bone that will include the four incisor teeth. The secondary palate, which makes up 90% of the hard and soft palates, is formed from the maxillary processes of the first branchial arches.
During the sixth week, bilateral projections emerge from the medial aspects of the maxillary processes to form the palatal shelves. Initially, these shelves are oriented in a vertical position on each side of the developing tongue. As the mandible grows, the tongue drops down, allowing the palatal shelves to rotate to a horizontal position and grow toward one another. By the eighth week, sufficient growth has occurred to allow the anterior aspects of these shelves to begin fusion with one another. The palatal shelves also fuse with the primary palate and the nasal septum. The fusion of the palatal shelves begins in the anterior palate and progresses posteriorly; it is completed by the twelfth week.
Defective fusion of the medial nasal process with the maxillary process leads to cleft lip (CL). Likewise, failure of the palatal shelves to fuse results in cleft palate (CP). Frequently, CL and CP occur together. Approximately 45% of cases are CL + CP, with 30% being CP only (CPO) and 25% being isolated CL. Both isolated CL and CL associated with CP are thought to be etiologically related conditions and can be considered as a group: CL, with or without CP (i.e., CL ± CP). Isolated CPO appears to represent a separate entity from CL ± CP.
The cause of CL ± CP and CPO is still being debated. First of all, distinguishing isolated clefts from cases associated with specific syndromes is important. Although many facial clefts are isolated anomalies, more than 350 developmental syndromes have been identified that may be associated with CL ± CP or CPO. Recent studies have suggested that up to 30% of patients with CL ± CP and 50% of those with CPO have associated anomalies. Some of these cases are single-gene syndromes that may follow autosomal dominant, autosomal recessive, or X-linked inheritance patterns. Other syndromes are the result of chromosome anomalies or are idiopathic.
The cause of nonsyndromic clefts does not follow any simple mendelian pattern of inheritance but appears to be heterogeneous (Box 1-1). Thus the propensity for cleft development may be related to a number of major genes, minor genes, and environmental factors that can combine to surpass a developmental threshold. A number of candidate clefting genes and loci have been identified on different chromosome regions, such as 1q, 2p, 4q, 6p, 14q, 17q, and 19q. Maternal alcohol consumption has been associated with an increased risk for both syndromic and nonsyndromic clefts. Maternal cigarette smoking at least doubles the frequency of cleft development compared with nonsmoking mothers. Multiple studies have demonstrated that a deficiency of folic acid increases the risk for CL and CP. Maternal corticosteroid use has been associated with a 3.4 times greater risk of orofacial clefting. An increased frequency also has been related to anticonvulsant therapy, especially phenytoin, which causes a nearly tenfold greater risk of cleft formation.
CL ± CP and CPO represent the vast majority of orofacial clefts. However, other rare clefts also may occur.
The lateral facial cleft is caused by lack of fusion of the maxillary and mandibular processes and represents 0.3% of all facial clefts. This cleft may be unilateral or bilateral, extending from the commissure toward the ear, resulting in macrostomia. The lateral facial cleft may occur as an isolated defect, but more often it is associated with other disorders, such as the following:
• Mandibulofacial dysostosis (see page 45)
The oblique facial cleft extends from the upper lip to the eye. It is nearly always associated with CP, and severe forms often are incompatible with life. The oblique facial cleft may involve the nostril, as in CL, or it may bypass the nose laterally as it extends to the eye. This cleft is rare, representing only 1 in 1300 facial clefts. Some of these clefts may represent failure of fusion of the lateral nasal process with the maxillary process; amniotic bands may cause others.
Median cleft of the upper lip is an extremely rare anomaly that results from failure of fusion of the medial nasal processes. It may be associated with a number of syndromes, including the oral-facial-digital syndromes and Ellis-van Creveld syndrome. Most apparent median clefts of the upper lip actually represent agenesis of the primary palate associated with holoprosencephaly.
CLINICAL AND RADIOGRAPHIC FEATURES: Clefting is one of the most common major congenital defects in humans. Considerable racial variation in prevalence is seen. In whites, CL ± CP occurs in 1 of every 700 to 1000 births. The frequency of CL ± CP in Asian populations is about 1.5 times higher than in whites. In contrast, the prevalence of CL ± CP in blacks is much lower, occurring in 0.4 of 1000 births. Native Americans appear to have the highest frequency, around 3.6 of 1000 births. CPO is less common than CL ± CP, with a frequency of 0.4 of 1000 births in whites and blacks.
CL ± CP is more common in males than in females. The more severe the defect, the greater the male predilection; the male-to-female ratio for isolated CL is 1.5:1; the ratio for CL + CP is 2:1. In contrast, CPO is more common in females. Likewise, the more severe the cleft, the greater the female predilection. Clefts of both the hard and soft palates are twice as common in females, but the ratio is nearly equal for clefts of the soft palate only.
Approximately 80% of cases of CL will be unilateral, with 20% bilateral (Fig. 1-1). Approximately 70% of unilateral CLs occur on the left side. In addition, about 70% of unilateral CLs will be associated with CP, whereas the frequency of concomitant CP increases to 85% for patients with bilateral CL. A complete CL extends upward into the nostril, but an incomplete CL does not involve the nose. Complete clefts involving the alveolus usually occur between the lateral incisor and cuspid. It is not unusual for teeth, especially the lateral incisor, to be missing in the cleft area. Conversely, supernumerary teeth may be discovered. The bony defect can be observed on radiographs.
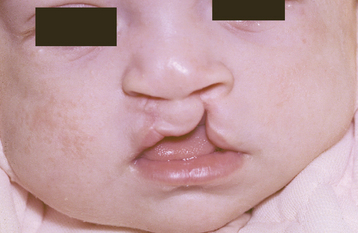
Fig. 1-1 Cleft lip (CL). Infant with bilateral cleft of the upper lip. (Courtesy of Dr. William Bruce.)
A CP shows considerable range in severity (Fig. 1-2). The defect may involve the hard and soft palates or the soft palate alone. The minimal manifestation of CP is a cleft or bifid uvula (Fig. 1-3). The prevalence of cleft uvula is much higher than that of CP, with a frequency of 1 in every 80 white individuals. The frequency in Asian and Native American populations is as high as 1 in 10. Cleft uvula is less common in blacks, occurring in 1 out of every 250 persons.
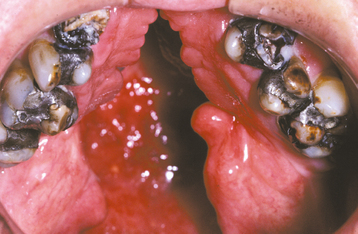
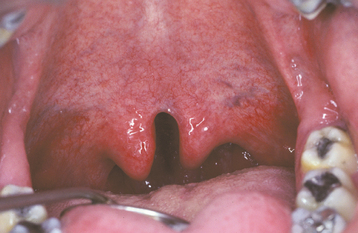
In some instances a submucous palatal cleft develops. The surface mucosa is intact, but a defect exists in the underlying musculature of the soft palate (Fig. 1-4). Frequently a notch in the bone is present along the posterior margin of the hard palate. This incomplete cleft occasionally appears as a bluish midline discoloration but is best identified by palpation with a blunt instrument. An associated cleft uvula is also usually seen.
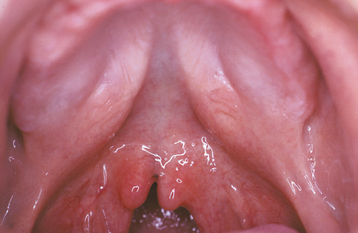
Fig. 1-4 Submucous palatal cleft. A cleft of the midline palatal bone exists, but the overlying mucosa is intact. A bifid uvula also is present.
The Pierre Robin sequence (Pierre Robin anomalad) (Fig. 1-5) is a well-recognized presentation characterized by CP, mandibular micrognathia, and glossoptosis (airway obstruction caused by lower, posterior displacement of the tongue). The Pierre Robin sequence may occur as an isolated phenomenon, or it may be associated with a wide variety of syndromes or other anomalies. Stickler syndrome and velocardiofacial syndrome are the two most frequently associated genetic disorders. Researchers have theorized that constraint of mandibular growth in utero results in failure of the tongue to descend, thus preventing fusion of the palatal shelves. The retruded mandible results in the following:
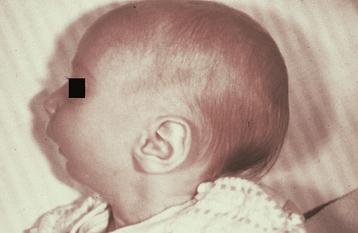
Fig. 1-5 Pierre Robin sequence. Micrognathic mandible in an infant with cleft palate (CP). (Courtesy of Dr. Robert Gorlin.)
Respiratory difficulty, especially when the child is in a supine position, is usually noted from birth and can cause asphyxiation. The palatal cleft is often U-shaped and wider than isolated CP.
The patient with a cleft is burdened with a variety of problems, some obvious and some less so. The most obvious problem is the clinical appearance, which may lead to psychosocial difficulties. Feeding and speech difficulties are inherent, especially with CP. Malocclusion is caused by collapse of the maxillary arch, possibly along with missing teeth, supernumerary teeth, or both.
TREATMENT AND PROGNOSIS: The management of the patient with an orofacial cleft is challenging. Ideally, treatment should involve a multidisciplinary approach, including (but not limited to) a pediatrician, oral and maxillofacial surgeon, otolaryngologist, plastic surgeon, pediatric dentist, orthodontist, prosthodontist, speech pathologist, and geneticist.
Surgical repair often involves multiple primary and secondary procedures throughout childhood. The specific types of surgical procedures and their timing will vary, depending on the severity of the defect and the philosophy of the treatment team. A detailed discussion of these procedures is beyond the scope of this text. However, primary lip closure is usually accomplished during the first few months of life, followed later by repair of the palate. Prosthetic and orthopedic appliances often are used to mold or expand the maxillary segments before closure of the palatal defect. Later in childhood, autogenous bone grafts can be placed in the area of the alveolar bone defect. Secondary soft tissue and orthognathic procedures may be used to improve function and cosmetic appearance. Distraction osteogenesis of the maxilla can prove useful in patients in whom palatal scarring limits the amount of advancement possible at the time of osteotomy.
Genetic counseling is important for the patient and family. In nonsyndromic cases, the risk for cleft development in a sibling or offspring of an affected person is 3% to 5% if no other first-degree relatives also are affected. The risk increases to 10% to 20% if other first-degree relatives are affected. The risk may be even higher for those with clefts that are associated with syndromes, depending on the possible inheritance pattern.
COMMISSURAL LIP PITS
Commissural lip pits are small mucosal invaginations that occur at the corners of the mouth on the vermilion border. Their location suggests that they may represent a failure of normal fusion of the embryonal maxillary and mandibular processes.
Commissural lip pits appear to be common in adults, where they have been reported in 12% to 20% of the population. Their prevalence in children is considerably lower, ranging from 0.2% to 0.7% of those examined.
Although commissural lip pits are generally considered to be congenital lesions, these figures suggest that these invaginations often develop later in life. Commissural pits are seen more often in males than in females. A family history suggestive of autosomal dominant transmission has been noted in some cases.
CLINICAL FEATURES: Commissural lip pits are usually discovered on routine examination, and the patient often is unaware of their presence. These pits may be unilateral or bilateral. They manifest as blind fistulas that may extend to a depth of 1 to 4 mm (Fig. 1-6). In some cases a small amount of fluid may be expressed from the pit when the pit is squeezed, presumably representing saliva from minor salivary glands that drain into the depth of the invagination.
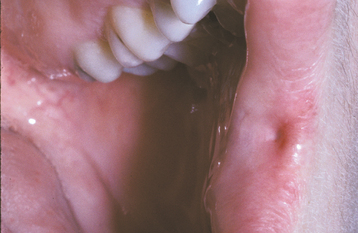
Unlike paramedian lip pits (described in the following section), commissural lip pits are not associated with facial or palatal clefts. However, there does appear to be a significantly higher prevalence of preauricular pits (aural sinuses) in these patients.
PARAMEDIAN LIP PITS (CONGENITAL FISTULAS OF THE LOWER LIP; CONGENITAL LIP PITS)
Paramedian lip pits are rare congenital invaginations of the lower lip. They are believed to arise from persistent lateral sulci on the embryonic mandibular arch. These sulci normally disappear by 6 weeks of embryonic age.
CLINICAL FEATURES: Paramedian lip pits typically appear as bilateral and symmetric fistulas on either side of the midline of the vermilion of the lower lip (Fig. 1-7). Their appearance can range from subtle depressions to prominent humps. These blind sinuses can extend down to a depth of 1.5 cm and may express salivary secretions. Occasionally, only a single pit is present that may be centrally located or lateral to the midline.
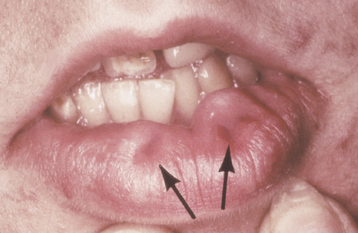
Fig. 1-7 Paramedian lip pits. Bilateral pits (arrows) on the lower lip in a patient with van der Woude syndrome.
The greatest significance of paramedian lip pits is that they are usually inherited as an autosomal dominant trait in combination with cleft lip (CL) and/or cleft palate (CP) (van der Woude syndrome). Van der Woude syndrome is the most common form of syndromic clefting and accounts for 2% of all cases of CL and CP. Associated hypodontia also may be observed. Genetic studies have shown that this condition is caused by mutations in the gene that encodes interferon regulatory factor 6, which has been mapped to chromosome locus 1q32-q41. Some people who carry the trait may not demonstrate clefts or may have a submucous CP; however, they may pass the full syndrome to their offspring.
Paramedian lip pits also may be a feature of the popliteal pterygium syndrome and Kabuki syndrome. Popliteal webbing (pterygia), CL and/or CP, genital abnormalities, and congenital bands connecting the upper and lower jaws (syngnathia) characterize popliteal pterygium syndrome, which is closely related to van der Woude syndrome. Kabuki syndrome received its name because affected patients exhibit eversion of the lower lateral eyelids, which is reminiscent of the makeup used by actors in Kabuki, the traditional form of Japanese theater. Other common findings include mental retardation, large ears, CL and/or CP, hypodontia, joint laxity, and various skeletal abnormalities.
DOUBLE LIP
Double lip is a rare oral anomaly characterized by a redundant fold of tissue on the mucosal side of the lip. It is most often congenital in nature, but it may be acquired later in life. Congenital cases are believed to arise during the second to third month of gestation as a result of the persistence of the sulcus between the pars glabrosa and pars villosa of the lip. Acquired double lip may be a component of Ascher syndrome, or it may result from trauma or oral habits, such as sucking on the lip.
CLINICAL FEATURES: In a patient with double lip, the upper lip is affected much more often than the lower lip; occasionally, both lips are involved. With the lips at rest, the condition is usually unnoticeable, but when the patient smiles or when the lips are tensed, the excess fold of tissue is visible (Fig. 1-8).
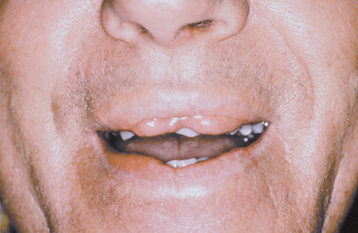
Fig. 1-8 Double lip. Redundant fold of tissue on the upper lip in a patient with Ascher syndrome. (Courtesy of Dr. R.C. Zeigler.)
Ascher syndrome is characterized by a triad of features:
In a person with blepharochalasis, recurring edema of the upper eyelid leads to sagging of the lid at the outer canthus of the eye (Fig. 1-9). This drooping may be severe enough to interfere with vision. Both the double lip and blepharochalasis usually occur abruptly and simultaneously, but in some cases they develop more gradually.
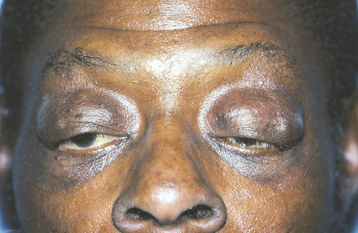
The nontoxic thyroid enlargement occurs in as many as 50% of patients with Ascher syndrome and may be mild in degree. The cause of Ascher syndrome is not certain; autosomal dominant inheritance has been suggested in some cases.
FORDYCE GRANULES
Fordyce granules are sebaceous glands that occur on the oral mucosa. Similar lesions also have been reported on the genital mucosa. Because sebaceous glands are typically considered to be dermal adnexal structures, those found in the oral cavity often have been considered to be “ectopic.” However, because Fordyce granules have been reported in more than 80% of the population, their presence must be considered a normal anatomic variation.
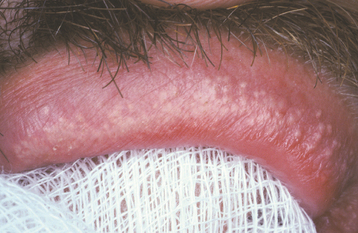
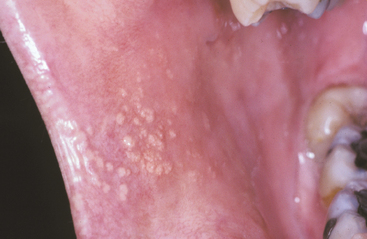
CLINICAL FEATURES: Fordyce granules appear as multiple yellow or yellow-white papular lesions that are most common on the buccal mucosa and the lateral portion of the vermilion of the upper lip (Figs. 1-10 and 1-11). Occasionally, these glands also may appear in the retromolar area and anterior tonsillar pillar. They are more common in adults than in children, probably as a result of hormonal factors; puberty appears to stimulate their development. The lesions are typically asymptomatic, although patients may be able to feel a slight roughness to the mucosa. Considerable clinical variation may exist; some patients may have only a few lesions, whereas others may have literally hundreds of these “granules.”
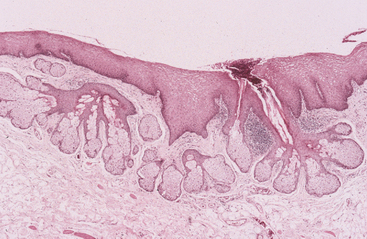
HISTOPATHOLOGIC FEATURES: Except for the absence of associated hair follicles, Fordyce granules are closely similar to normal sebaceous glands found in the skin. Acinar lobules can be seen immediately beneath the epithelial surface, often communicating with the surface through a central duct (Fig. 1-12). The sebaceous cells in these lobules are polygonal in shape, containing centrally located nuclei and abundant foamy cytoplasm.
TREATMENT AND PROGNOSIS: Because Fordyce granules represent a normal anatomic variation and are asymptomatic, no treatment is indicated. Usually, the clinical appearance is characteristic and biopsy is not necessary for diagnosis.
On occasion, Fordyce granules may become hyperplastic or may form keratin-filled pseudocysts. Tumors arising from these glands are exceedingly rare.
LEUKOEDEMA
Leukoedema is a common oral mucosal condition of unknown cause. It occurs more commonly in blacks than in whites, supporting the likelihood of an ethnic predisposition to its development. Leukoedema has been reported in 70% to 90% of black adults and in 50% of black children. The prevalence in whites is considerably less, although published reports have ranged from less than 10% to more than 90%. This variation may reflect differing population groups, examination conditions, and stringency of criteria used to make the diagnosis. At any rate, leukoedema shows a much milder presentation in whites and often is hardly noticeable. The difference in racial predilection may be explained by the presence of background mucosal pigmentation in blacks that makes the edematous changes more noticeable.
Because leukoedema is so common, it can reasonably be argued that it represents a variation of normal rather than a disease. The finding of similar edematous mucosa in the vagina and larynx further supports this argument. Although leukoedema appears to be developmental in nature, some studies have indicated that it is more common and more severe in smokers and becomes less pronounced with cessation of smoking.
CLINICAL FEATURES: Leukoedema is characterized by a diffuse, gray-white, milky, opalescent appearance of the mucosa (Fig. 1-13). The surface frequently appears folded, resulting in wrinkles or whitish streaks. The lesions do not rub off. Leukoedema typically occurs bilaterally on the buccal mucosa and may extend forward onto the labial mucosa. On rare occasions, it can also involve the floor of the mouth and palatopharyngeal tissues. Leukoedema can be easily diagnosed clinically because the white appearance greatly diminishes or disappears when the cheek is everted and stretched (Fig. 1-14).
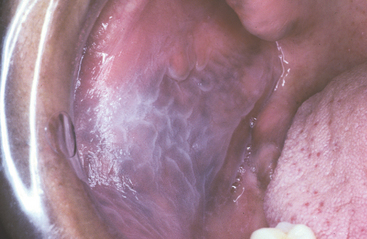
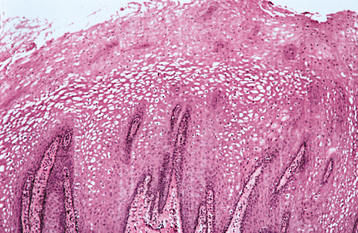
HISTOPATHOLOGIC FEATURES: Biopsy specimens of leukoedema demonstrate an increase in thickness of the epithelium, with striking intracellular edema of the spinous layer (Fig. 1-15). These vacuolated cells appear large and have pyknotic nuclei. The epithelial surface is frequently parakeratinized, and the rete ridges are broad and elongated.
TREATMENT AND PROGNOSIS: Leukoedema is a benign condition, and no treatment is required. The characteristic milky-white, opalescent lesions of the buccal mucosa that disappear when stretched help distinguish it from other common white lesions, such as leukoplakia, candidiasis, and lichen planus. The affected mucosa always should be stretched during clinical examination to rule out any underlying lesions that may be hidden by the edematous change.
MICROGLOSSIA (HYPOGLOSSIA)
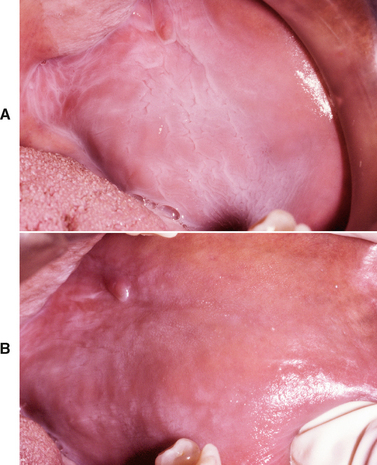
CLINICAL FEATURES: Microglossia is an uncommon developmental condition of unknown cause that is characterized by an abnormally small tongue. In rare instances, virtually the entire tongue may be missing (aglossia). Isolated microglossia is known to occur, and mild degrees of microglossia may be difficult to detect and may go unnoticed. However, most reported cases have been associated with one of a group of overlapping conditions known as oromandibular-limb hypogenesis syndromes. These syndromes feature associated limb anomalies, such as hypodactylia (i.e., absence of digits) and hypomelia (i.e., hypoplasia of part or all of a limb). Other patients have had coexisting anomalies, such as cleft palate, intraoral bands, and situs inversus. Micro-glossia frequently is associated with hypoplasia of the mandible, and the lower incisors may be missing (Fig. 1-16).
MACROGLOSSIA
Macroglossia is an uncommon condition characterized by enlargement of the tongue. The enlargement may be caused by a wide variety of conditions, including congenital malformations and acquired diseases. The most frequent causes are vascular malformations and muscular hypertrophy. Box 1-2 lists the most common and important causes of macroglossia. Many of these diseases are discussed in greater detail in subsequent chapters of this book.
CLINICAL FEATURES: Macroglossia most commonly occurs in children and can range from mild to severe (Fig. 1-17). In infants, macroglossia may be manifested first by noisy breathing, drooling, and difficulty in eating. The tongue enlargement may result in a lisping speech. The pressure of the tongue against the mandible and teeth can produce a crenated lateral border to the tongue (Fig. 1-18), open bite, and mandibular prognathism. If the tongue constantly protrudes from the mouth, it may ulcerate and become secondarily infected or may even undergo necrosis. Severe macroglossia can produce airway obstruction.
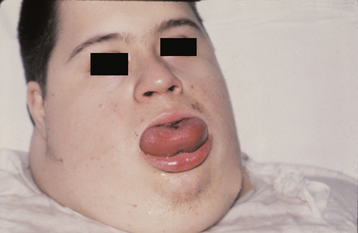
Fig. 1-17 Macroglossia. Large tongue in a patient with Down syndrome. (Courtesy of Dr. Sanford Fenton.)
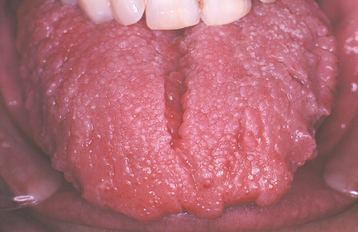
Fig. 1-18 Macroglossia. The tongue enlargement has resulted in a crenated border that corresponds to the embrasures between the teeth.
Macroglossia is a characteristic feature of Beckwith-Wiedemann syndrome, a rare hereditary condition that includes many other possible defects, such as the following:
• Omphalocele (i.e., protrusion of part of the intestine through a defect in the abdominal wall at the umbilicus)
Individuals with Beckwith-Wiedemann syndrome have an increased risk for several childhood visceral tumors, including Wilms’ tumor, adrenal carcinoma, hepatoblastoma, rhabdomyosarcoma, and neuroblastoma. Facial features may include nevus flammeus of the forehead and eyelids, linear indentations of the earlobes, and maxillary hypoplasia (resulting in relative mandibular prognathism). Most examples of Beckwith-Wiedemann syndrome are sporadic, but 10% to 15% of cases show autosomal dominant inheritance with preferential maternal transmission. The genetic basis is complex, involving a variety of alterations within two domains of imprinted growth-regulatory genes on chromosome 11p15.
In patients with hypothyroidism (see page 843) or Beckwith-Wiedemann syndrome, the tongue usually shows a diffuse, smooth, generalized enlargement. In those with other forms of macroglossia, the tongue usually has a multinodular appearance. Examples of this nodular type include amyloidosis (see page 822) and neoplastic conditions, such as neurofibromatosis (see page 529) and multiple endocrine neoplasia, type 2B (see page 532).
In patients with lymphangiomas (see page 547), the tongue surface is characteristically pebbly and exhibits multiple vesicle-like blebs that represent superficial dilated lymphatic channels. The enlarged tongue in those with Down syndrome typically demonstrates a papillary, fissured surface.
In patients with hemifacial hyperplasia (see page 38), the enlargement will be unilateral. Some patients with neurofibromatosis also can have unilateral lingual enlargement.
In edentulous patients, the tongue often appears elevated and tends to spread out laterally because of loss of the surrounding teeth; as a result, wearing a denture may become difficult.
HISTOPATHOLOGIC FEATURES: The microscopic appearance of macroglossia depends on the specific cause. In some cases, such as the tongue enlargement seen with Down syndrome or in edentulous patients, no histologic abnormality can be detected. When macroglossia is due to tumor, a neoplastic proliferation of a particular tissue can be found (e.g., lymphatic vessels, blood vessels, neural tissue). Muscular enlargement occurs in those with hemihyperplasia and Beckwith-Wiedemann syndrome. In the patient with amyloidosis, an abnormal protein material is deposited in the tongue.
ANKYLOGLOSSIA (TONGUE-TIE)
Ankyloglossia is a developmental anomaly of the tongue characterized by a short, thick lingual frenum resulting in limitation of tongue movement. It has been reported to occur in 1.7% to 4.4% of neonates and is four times more common in boys than in girls. In adults, mild forms are not unusual, but severe ankyloglossia is a relatively uncommon condition that has been estimated to occur in about 2 to 3 of every 10,000 people.
CLINICAL FEATURES: Ankyloglossia can range in severity from mild cases with little clinical significance to rare examples of complete ankyloglossia in which the tongue is actually fused to the floor of the mouth (Fig. 1-19). Sometimes the frenum extends forward and attaches to the tip of the tongue, and slight clefting of the tip may be seen.
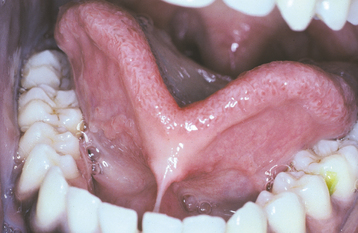
Some investigators have speculated that ankyloglossia may contribute to the development of an anterior open bite because the inability to raise the tongue to the roof of the mouth prevents development of the normal adult swallowing pattern. However, others have questioned this theory. It also is possible that a high mucogingival attachment of the lingual frenum may lead to periodontal problems.
It has been suggested that tongue-tie may result in speech defects. Usually, however, the shortened frenum results in only minor difficulties because most people can compensate for the limitation in tongue movement. Yet there are rare examples of patients who have experienced an immediate noticeable improvement in speech after surgical correction of ankyloglossia. With the increase in popularity of breast-feeding over the past several decades, some clinicians have related tongue-tie with feeding problems, such as nipple pain or difficulty in the baby attaching to the breast. Recent reports from Japan have theorized that some ankyloglossia cases can be associated with an upward and forward displacement of the epiglottis and larynx, resulting in various degrees of dyspnea.
TREATMENT AND PROGNOSIS: Because most cases of ankyloglossia result in few or no clinical problems, treatment is often unnecessary. For infants with specific breast-feeding problems, a frenotomy (“clipping” or simple release of the frenulum) can be performed. In children or adults with associated functional or periodontal difficulties, a frenuloplasty (release with plastic repair) may allow greater freedom of tongue movement. In young children it often is recommended that surgery be postponed until age 4 or 5. Because the tongue is always short at birth, assessing the degree of tongue limitation caused by ankyloglossia is difficult in the infant’s early life. As the infant grows, the tongue becomes longer and thinner at the tip, often decreasing the severity of the tongue-tie. The condition probably is self-correcting in many cases because it is less common in adults.
LINGUAL THYROID
During the third to fourth week of fetal life, the thyroid gland begins as an epithelial proliferation in the floor of the pharyngeal gut. By the seventh embryonic week, this thyroid bud normally descends into the neck to its final resting position anterior to the trachea and larynx. The site where this descending bud invaginates later becomes the foramen cecum, located at the junction of the anterior two thirds and posterior third of the tongue in the midline. If the primitive gland does not descend normally, ectopic thyroid tissue may be found between the foramen cecum and the epiglottis. Of all ectopic thyroids, 90% are found in this region.
CLINICAL FEATURES: Based on autopsy studies, small asymptomatic remnants of thyroid tissue can be discovered on the posterior dorsal tongue in about 10% of both men and women. However, clinically evident or symptomatic lingual thyroids are much less common and are four to seven times more frequent in females, presumably because of hormonal influences. Symptoms most often develop during puberty, adolescence, pregnancy, or menopause. In 70% of cases, this ectopic gland is the patient’s only thyroid tissue.
Lingual thyroids may range from small, asymptomatic, nodular lesions to large masses that can block the airway (Fig. 1-20). The most common clinical symptoms are dysphagia, dysphonia, and dyspnea. The mass often is vascular, but the physical appearance is variable and there are no reliable features to distinguish it from other masses that might develop in this area. Hypothyroidism has been reported in up to 33% of patients. Many authors say that lingual thyroid enlargement is a secondary phenomenon, compensating for thyroid hypofunction. Interestingly, as many as 75% of patients with infantile hypothyroidism have some ectopic thyroid tissue.
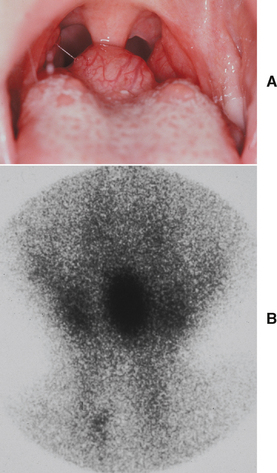
Fig. 1-20 Lingual thyroid. A, Nodular mass of the posterior dorsal midline of the tongue in a 4-year-old girl. B, Thyroid scan of the same patient. The scan shows localization (central dark zone) of iodine isotope in the tongue mass and minimal uptake in the neck.
Diagnosis is best established by thyroid scan using iodine isotopes or technetium-99m. Computed tomography (CT) and magnetic resonance imaging (MRI) can be helpful in delineating the size and extent of the lesion. Biopsy is often avoided because of the risk of hemorrhage and because the mass may represent the patient’s only functioning thyroid tissue. In some cases, incisional biopsy may be needed to confirm the diagnosis or to rule out malignant changes.
TREATMENT AND PROGNOSIS: No treatment except periodic follow-up is required for patients with asymptomatic lingual thyroids. In symptomatic patients, suppressive therapy with supplemental thyroid hormone often can reduce the size of the lesion. Some authors advise that this treatment also should be tried in asymptomatic patients to prevent possible subsequent enlargement. If hormone therapy does not eliminate symptoms, surgical removal or ablation with radioactive iodine-131 can be performed. If the mass is surgically excised, autotransplantation to another body site can be attempted to maintain functional thyroid tissue and to prevent hypothyroidism.
Rare examples of carcinomas arising in lingual thyroids have been reported; malignancy develops in about 1% of identified cases. Although lingual thyroids are decidedly more common in females, this predilection for females is less pronounced for lingual thyroid carcinomas. Because a disproportionate number of these malignancies have been documented in males, some authors have advocated prophylactic excision of lingual thyroids in men older than 30 years of age.
FISSURED TONGUE (SCROTAL TONGUE)
Fissured tongue is a relatively common condition that is characterized by the presence of numerous grooves, or fissures, on the dorsal tongue surface. The cause is uncertain, but heredity appears to play a significant role. Evidence indicates that the condition may be either a polygenic trait or an autosomal dominant trait with incomplete penetrance. Aging or local environmental factors also may contribute to its development.
CLINICAL FEATURES: Patients with fissured tongue exhibit multiple grooves, or furrows, on the surface of the tongue, ranging from 2 to 6 mm in depth (Fig. 1-21). Considerable variation can be seen. In the most severe cases, numerous fissures cover the entire dorsal surface and divide the tongue papillae into multiple separate “islands.” Some patients have fissures that are located mostly on the dorsolateral areas of the tongue. Other patients exhibit a large central fissure, with smaller fissures branching outward at right angles. The condition is usually asymptomatic, although some patients may complain of mild burning or soreness.
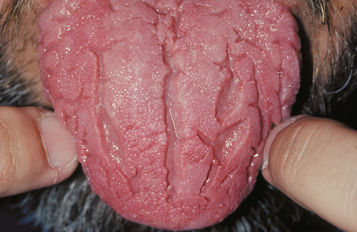
Fig. 1-21 Fissured tongue. Extensive fissuring involving the entire dorsal tongue surface. (Courtesy of Chris Neville.)
Most studies have shown that the prevalence of fissured tongue ranges from 2% to 5% of the overall population. The condition may be seen in children or adults, but the prevalence and severity appear to increase with age, with some studies noting the presence of fissured tongue in as many as 30% of older adults. In some investigations, a male predilection has been noted.
A strong association has been found between fissured tongue and geographic tongue (see page 779), with many patients having both conditions. A hereditary basis also has been suggested for geographic tongue, and the same gene or genes may possibly be linked to both conditions. In fact, it even has been suggested that geographic tongue may cause fissured tongue. Fissured tongue also may be a component of Melkersson-Rosenthal syndrome (see page 342).
HISTOPATHOLOGIC FEATURES: Microscopic examination of fissured tongue reveals hyperplasia of the rete ridges and loss of the keratin “hairs” on the surface of the filiform papillae. The papillae vary in size and often are separated by deep grooves. Polymorphonuclear leukocytes can be seen migrating into the epithelium, often forming microabscesses in the upper epithelial layers. A mixed inflammatory cell infiltrate is present in the lamina propria.
HAIRY TONGUE (BLACK HAIRY TONGUE; COATED TONGUE)
Hairy tongue is characterized by marked accumulation of keratin on the filiform papillae of the dorsal tongue, resulting in a hairlike appearance. The condition apparently represents an increase in keratin production or a decrease in normal keratin desquamation. Hairy tongue is found in about 0.5% of adults. Although the cause is uncertain, many affected people are heavy smokers. Other possible associated factors include general debilitation, poor oral hygiene, and a history of radiation therapy to the head and neck.
CLINICAL FEATURES: Hairy tongue most commonly affects the midline just anterior to the circumvallate papillae, sparing the lateral and anterior borders (Fig. 1-22). The elongated papillae are usually brown, yellow, or black as a result of growth of pigment-producing bacteria or staining from tobacco and food. Sometimes most of the dorsal tongue may be involved, resulting in a thick, matted appearance (Fig. 1-23). Multiple individual elongated filiform papillae may be elevated by using gauze or a dental instrument. The condition is typically asymptomatic, although occasionally patients complain of a gagging sensation or a bad taste in the mouth. Because the diagnosis usually can be made from the clinical appearance, biopsy is unnecessary in most instances.
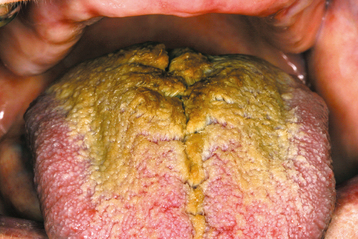
Fig. 1-22 Hairy tongue. Elongated, yellow-brown filiform papillae on the posterior dorsal surface of the tongue.
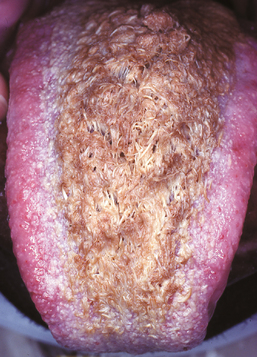
Fig. 1-23 Hairy tongue. Marked elongation and brown staining of the filiform papillae, resulting in a hairlike appearance.
Because of the similarity in names, care should be taken to avoid confusing hairy tongue with hairy leukoplakia (see page 268), which typically occurs on the lateral border of the tongue. Hairy leukoplakia is caused by the Epstein-Barr virus and is usually associated with human immunodeficiency virus (HIV) infection or other immunosuppressive conditions.
In some individuals, numerous bacteria and desquamated epithelial cells accumulate on the dorsal tongue surface, but without the hairlike filiform projections (Fig. 1-24). Such cases, which are often designated as a coated tongue, also may be the source of oral malodor.
HISTOPATHOLOGIC FEATURES: On histopathologic examination, hairy tongue is characterized by marked elongation and hyperparakeratosis of the filiform papillae (Fig. 1-25). Usually, numerous bacteria can be seen growing on the epithelial surface.
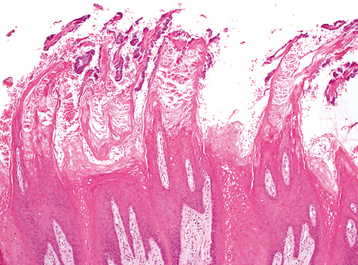
TREATMENT AND PROGNOSIS: Hairy or coated tongue is a benign condition with no serious sequelae. The major concern is often the aesthetic appearance of the tongue along with possible associated bad breath. Any predisposing factors, such as tobacco, antibiotics, or mouthwashes, should be eliminated, and excellent oral hygiene should be encouraged. Periodic scraping or brushing with a toothbrush or tongue scraper can promote desquamation of the hyperkeratotic papillae and surface debris. Keratolytic agents, such as podophyllin, also have been tried with success, but for safety reasons their use probably should not be encouraged.
VARICOSITIES (VARICES)
Varicosities, or varices, are abnormally dilated and tortuous veins. Age appears to be an important etiologic factor because varices are rare in children but common in older adults. This suggests that their development may be an age-related degeneration, in which a loss of connective tissue tone supporting the vessels occurs. Oral varices have not been associated with systemic hypertension or other cardiopulmonary diseases, although one study did find that people with varicose veins of the legs were more likely to have varicosities of the tongue.
CLINICAL FEATURES: The most common type of oral varicosity is the sublingual varix, which occurs in two thirds of people older than 60 years of age. Sublingual varicosities classically present as multiple blue-purple, elevated or papular blebs on the ventral and lateral border of the tongue (Fig. 1-26). The lesions are usually asymptomatic, except in rare instances when secondary thrombosis occurs.
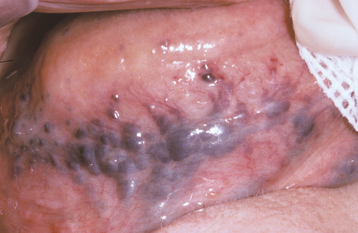
Fig. 1-26 Varicosities. Multiple purple dilated veins on the ventral and lateral surface of the tongue.
Less frequently, solitary varices occur in other areas of the mouth, especially the lips and buccal mucosa. These isolated varicosities often are first noticed after they have become thrombosed (Fig. 1-27). Clinically, a thrombosed varix presents as a firm, nontender, blue-purple nodule that may feel like a piece of buckshot beneath the mucosal surface.
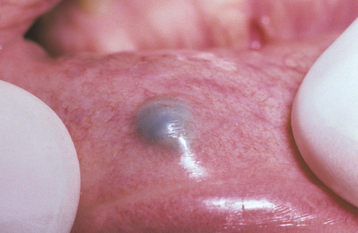
HISTOPATHOLOGIC FEATURES: Microscopic examination of a varix reveals a dilated vein, the wall of which shows little smooth muscle and poorly developed elastic tissue. If secondary thrombosis has occurred, then the lumen may contain concentrically layered zones of platelets and erythrocytes (lines of Zahn). The clot can undergo organization via granulation tissue, with subsequent recanalization. Older thrombi may exhibit dystrophic calcification, resulting in formation of a phlebolith (phlebo = vein; lith = stone).
CALIBER-PERSISTENT ARTERY
A caliber-persistent artery is a common vascular anomaly in which a main arterial branch extends up into the superficial submucosal tissues without a reduction in its diameter. Similar to oral varices, caliber-persistent arteries are seen more frequently in older adults. This suggests that their development may be an age-related degenerative phenomenon in which there is a loss of tone in the surrounding supporting connective tissue.
CLINICAL FEATURES: The caliber-persistent artery occurs almost exclusively on the lip mucosa. Either lip may be affected, and some patients have bilateral lesions or lesions on both lips. The average patient age is 58 years, and the gender ratio is nearly equal. The lesion presents as a linear, arcuate, or papular elevation that ranges from pale to normal to bluish in color (Fig. 1-28). Stretching the lip usually causes the artery to become inconspicuous. The unique feature is pulsation—not only vertically but also in a lateral direction. However, usually it is not possible to feel a pulse in a caliber-persistent artery with gloved fingers.
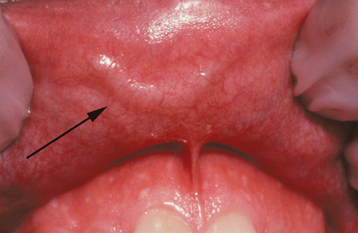
Fig. 1-28 Caliber-persistent artery. Linear, arcuate lesion on the upper labial mucosa (arrow). (Courtesy of Dr. John Lovas.)
The lesion is usually asymptomatic, being discovered as an incidental finding during an oral examination; rarely a patient may notice a pulsatile lip nodule. A few cases have been associated with ulceration of the overlying mucosa. In addition, a couple of examples have been found adjacent to labial squamous cell carcinomas, although this is probably coincidental.
HISTOPATHOLOGIC FEATURES: Microscopic examination shows a thick-walled artery situated close to the mucosal surface (Fig. 1-29).
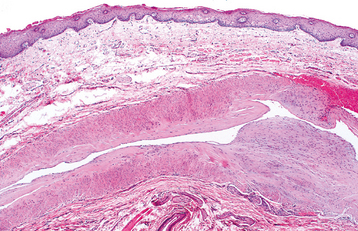
TREATMENT AND PROGNOSIS: If the true nature of the caliber-persistent artery can be recognized clinically, no treatment is necessary. Oftentimes a biopsy is performed when the lesion is mistaken for a mucocele or another vascular lesion, such as a varix or hemangioma. Brisk bleeding is typically encountered if the lesion is removed.
LATERAL SOFT PALATE FISTULAS
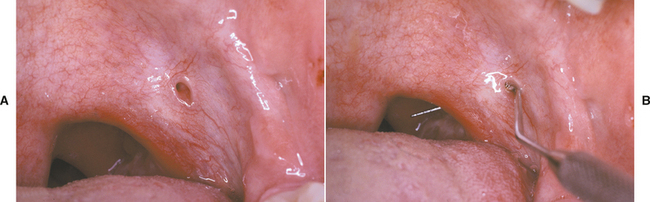
Lateral soft palate fistulas are rare anomalies of uncertain pathogenesis. Many cases appear to be congenital, possibly related to a defect in the development of the second pharyngeal pouch. Some fistulas may be the result of infection or surgery of the tonsillar region.
CLINICAL FEATURES: Lateral soft palate fistulas are usually bilateral, but they may occur only on one side. They are more common on the anterior tonsillar pillar (Fig. 1-30), but they also may involve the posterior pillar. The perforations are typically asymptomatic, ranging from a few millimeters to more than 1 cm. A few cases have been associated with other anomalies, such as absence or hypoplasia of the palatine tonsils, hearing loss, and preauricular fistulas.
CORONOID HYPERPLASIA
Hyperplasia of the coronoid process of the mandible is a rare developmental anomaly that may result in limitation of mandibular movement. The cause of coronoid hyperplasia is unknown, but the overall male-to-female ratio is 5:1. Because most cases have been seen in pubertal males, an endocrine influence has been suggested. Heredity also may play a role, because cases have been noted in siblings.
Coronoid hyperplasia may be unilateral or bilateral, although bilateral cases are nearly five times more common than unilateral examples. Unilateral enlargement of the coronoid process also can result from a true tumor, such as an osteoma or osteochondroma, and such cases should be distinguished from pure coronoid hyperplasia. However, some cases reported as tumors of the coronoid process actually may have been hyperplastic processes rather than true neoplasms.
CLINICAL AND RADIOGRAPHIC FEATURES: In a person with unilateral coronoid hyperplasia, the enlarged coronoid process impinges on the posterior surface of the zygoma, restricting mandibular opening. In addition, the mandible may deviate toward the affected side. Usually, there is no pain or associated abnormality in occlusion. Radiographs may reveal an irregular, nodular growth of the tip of the coronoid process.
In bilateral coronoid hyperplasia, the limitation of mandibular opening may progressively worsen over several years during childhood, reaching maximum severity during the late teens. The radiographic appearance is characterized by regular elongation of both processes. Because the coronoid process is often superimposed on the zygoma on conventional radiographs, tomograms or CT scans often demonstrate the hyperplasia more effectively.
TREATMENT AND PROGNOSIS: Treatment of coronoid hyperplasia consists of surgical removal of the elongated coronoid process or processes to allow freedom of mandibular motion. Coronoidectomy or coronoidotomy is usually accomplished via an intraoral approach. Although initial improvement in oral opening can be effected, the long-term results sometimes can be disappointing because of surgically induced fibrosis and the tendency for coronoid regrowth. Postoperative physiotherapy is important for reestablishing normal function.
CONDYLAR HYPERPLASIA
Condylar hyperplasia is an uncommon malformation of the mandible created by excessive growth of one of the condyles. The cause of this hyperplasia is unknown, but local circulatory problems, endocrine disturbances, and trauma have been suggested as possible etiologic factors.
Condylar hyperplasia can be difficult to distinguish from hemifacial hyperplasia (see page 38); however, in the latter condition the associated soft tissues and teeth also may be enlarged.
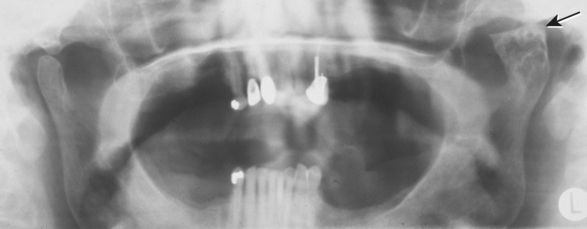
CLINICAL AND RADIOGRAPHIC FEATURES: Condylar hyperplasia may manifest itself in a variety of ways, including facial asymmetry, prognathism, crossbite, and open bite (Fig. 1-31). Sometimes compensatory maxillary growth and tilting of the occlusal plane occurs. The condition most commonly is discovered in adolescents and young adults.
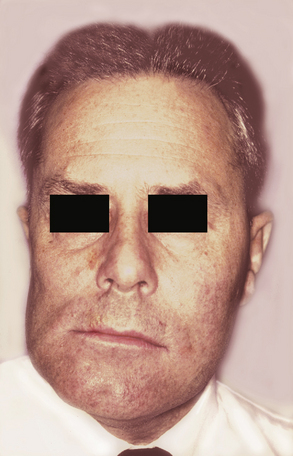
Fig. 1-31 Condylar hyperplasia. Enlargement of the patient’s left condyle has displaced the mandible to the right and resulted in facial asymmetry.
The radiographic features are quite variable. Some patients have an enlargement of the condylar head, and others show elongation of the condylar neck (Fig. 1-32). Many cases also demonstrate hyperplasia of the entire ramus, suggesting that the condition sometimes affects more than just the condyle. Scintigraphy using 99mTc-MDP has been advocated as a useful method for assessing the degree of bone activity in condylar hyperplasia.
HISTOPATHOLOGIC FEATURES: During active growth, proliferation of the condylar cartilage is noted. Once condylar growth has ceased, the condyle has a normal histologic appearance.
TREATMENT AND PROGNOSIS: Condylar hyperplasia is a self-limiting condition, and treatment is determined by the degree of functional difficulty and aesthetic change. Some patients can be treated with unilateral condylectomy, whereas others require unilateral or bilateral mandibular osteotomies. In patients with compensatory maxillary growth, a maxillary osteotomy also may be needed. Concomitant orthodontic therapy frequently is necessary.
CONDYLAR HYPOPLASIA
Condylar hypoplasia, or underdevelopment of the mandibular condyle, can be either congenital or acquired. Congenital condylar hypoplasia often is associated with head and neck syndromes, including mandibulofacial dysostosis (see page 45), oculoauriculovertebral syndrome (Goldenhar syndrome), and hemifacial microsomia. In the most severe cases, complete agenesis of the condyle or ramus (condylar aplasia) is seen.
Acquired condylar hypoplasia results from disturbances of the growth center of the developing condyle. The most frequent cause is trauma to the condylar region during infancy or childhood. Other causes include infections, radiation therapy, and rheumatoid or degenerative arthritis.
CLINICAL AND RADIOGRAPHIC FEATURES: Condylar hypoplasia can be unilateral or bilateral, producing a small mandible with a Class II malocclusion. Unilateral hypoplasia results in distortion and depression of the face on the affected side. The mandibular midline shifts to the involved side when the mouth is opened, accentuating the deformity. Ankylosis of the temporomandibular joint (TMJ) can develop in cases caused by trauma.
The deformity is observed easily on panoramic films and can range in severity. In severe cases the condyle or ramus may be totally absent. Milder types demonstrate a short condylar process, shallow sigmoid notch, and poorly formed condylar head. A prominent antegonial notch may be present. CT scans may be helpful in evaluating the condyles.
TREATMENT AND PROGNOSIS: Treatment of the patient with condylar hypoplasia depends on the cause and severity of the defect, but surgery often is required. If the condyle is missing, then a costochondral rib graft can be placed to help establish an active growth center. In addition, osteotomies sometimes provide a cosmetically acceptable result. In certain instances, distraction osteogenesis can be used to stimulate new bone formation.
BIFID CONDYLE
A bifid condyle is a rare developmental anomaly characterized by a double-headed mandibular condyle. Most bifid condyles have a medial and lateral head divided by an anteroposterior groove. Some condyles may be divided into an anterior and posterior head.
The cause of bifid condyle is uncertain. Anteroposterior bifid condyles may be of traumatic origin, such as a childhood fracture. Mediolaterally divided condyles may result from trauma, abnormal muscle attachment, teratogenic agents, or persistence of a fibrous septum within the condylar cartilage.
CLINICAL AND RADIOGRAPHIC FEATURES: A bifid condyle is usually unilateral, but occasionally both sides may be affected. The malformation is often asymptomatic and may be discovered on routine radiographs, although some patients may have a “pop” or “click” of the TMJ when opening their mouths. Panoramic radiographs and CT scans will demonstrate a bilobed appearance of the condylar head (Fig. 1-33).
EXOSTOSES
Exostoses are localized bony protuberances that arise from the cortical plate. These benign growths frequently affect the jaws. The best-known oral exostoses, the torus palatinus and the torus mandibularis, are described later in the chapter. Other types of exostoses also may affect the jaws and are considered here.
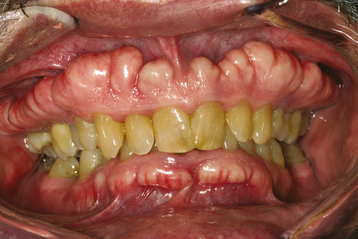
CLINICAL AND RADIOGRAPHIC FEATURES: Exostoses are discovered most often in adults. Buccal exostoses occur as a bilateral row of bony hard nodules along the facial aspect of the maxillary and/or mandibular alveolar ridge (Fig. 1-34). They are usually asymptomatic, unless the thin overlying mucosa becomes ulcerated from trauma. One study reported that buccal exostoses were found in nearly 1 of every 1000 adults (0.09%); however, a more recent survey found a much higher prevalence of nearly 19%. This variation may be due to the different populations being studied or to the clinical criteria used to make the diagnosis.
Palatal exostoses (palatal tubercles) are similar bony protuberances that develop from the lingual aspect of the maxillary tuberosities. These lesions are usually bilateral but may affect only one side (Fig. 1-35). They are more common in males and have been reported in 8% to 69% of various populations. Many patients with buccal or palatal exostoses also will have palatal or mandibular tori.
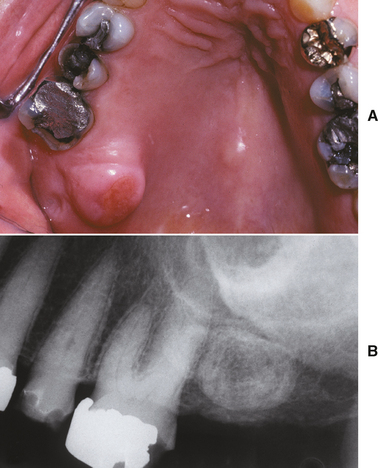
Fig. 1-35 Exostosis. A, Secondarily ulcerated palatal exostosis. B, Radiograph shows an ovoid radiopacity distal to the molar.
Less commonly, solitary exostoses may occur, possibly in response to local irritation. Such lesions may develop from the alveolar bone beneath free gingival grafts and skin grafts. Presumably placement of the graft acts as a stimulant to the periosteum to form new bone.
Another uncommon, interesting variant is the reactive subpontine exostosis (subpontic osseous proliferation, subpontic osseous hyperplasia), which may develop from the alveolar crestal bone beneath the pontic of a posterior bridge (Fig. 1-36).
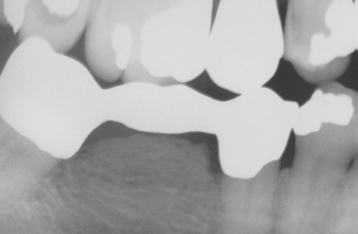
Fig. 1-36 Reactive subpontine exostosis. Nodular growth of bone beneath the pontic of a posterior mandibular bridge.
If enough excess bone is present, exostoses may exhibit a relative radiopacity on dental radiographs (see Fig. 1-35, B). In rare instances an exostosis may become so large that distinguishing it from a tumor, such as an osteoma, is difficult (see page 650).
HISTOPATHOLOGIC FEATURES: Microscopic examination reveals a mass of dense, lamellar, cortical bone with a small amount of fibrofatty marrow. In some cases an inner zone of trabecular bone also is present.
TREATMENT AND PROGNOSIS: Most exostoses are distinctive enough clinically to make biopsy unnecessary. If the diagnosis is uncertain, biopsy should be performed to rule out other bony pathosis. Sometimes the exostosis must be removed if it repeatedly has been exposed to trauma or has become ulcerated and painful. In addition, surgical removal may be required to accommodate a dental prosthesis or to allow for proper flap adaptation during periodontal surgery. Reactive subpontine exostoses may need to be removed if they interfere with oral hygiene or are associated with adjacent periodontal disease.
TORUS PALATINUS
The torus palatinus is a common exostosis that occurs in the midline of the vault of the hard palate. The pathogenesis of these tori has long been debated, with arguments centering on genetic versus environmental factors, such as masticatory stress. Some authorities have suggested that the torus palatinus is inherited as an autosomal dominant trait. However, others believe that the development of this lesion is multifactorial, including both genetic and environmental influences. In this model, patients are affected by a variety of hereditary and local environmental factors. If enough of these factors are present, then a “threshold” is surpassed and the trait (torus palatinus) will be expressed.
CLINICAL AND RADIOGRAPHIC FEATURES: The torus palatinus presents as a bony hard mass that arises along the midline suture of the hard palate (Figs. 1-37 to 1-39). Tori sometimes are classified according to their morphologic appearance:
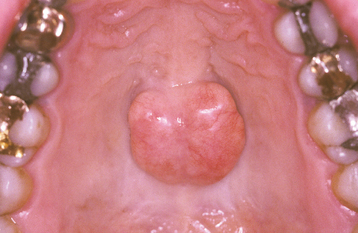
• The flat torus has a broad base and a slightly convex, smooth surface. It extends symmetrically onto both sides of the midline raphe.
• The spindle torus has a midline ridge along the palatal raphe. A median groove is sometimes present.
• The nodular torus arises as multiple protuberances, each with an individual base. These protuberances may coalesce, forming grooves between them.
• The lobular torus is also a lobulated mass, but it rises from a single base. Lobular tori can be either sessile or pedunculated.
Most palatal tori are small, measuring less than 2 cm in diameter; however, they can slowly increase in size throughout life, sometimes to the extent that they fill the entire palatal vault. Most tori cause no symptoms, but in some cases the thin overlying mucosa may become ulcerated secondary to trauma.
The torus palatinus does not usually appear on routine dental radiographs. Rarely it may be seen as a radiopacity on periapical films if the film is placed behind the torus when the radiograph is taken.
The prevalence of palatal tori has varied widely in a number of population studies, ranging from 9% to 60%. Some of this variation may be due to the criteria used to make the diagnosis and also may be based on whether the study was conducted on live patients or skulls. There appear to be significant racial differences, however, with a higher prevalence in Asian and Inuit (i.e., Eskimo) populations. In the United States, most studies have shown a prevalence of 20% to 35%, with little difference between whites and blacks. Almost all studies from around the world have shown a pronounced female-to-male ratio of 2:1. The prevalence peaks during early adult life, tapering off in later years. This finding supports the theory that tori are dynamic lesions that are related, in part, to environmental factors; in later life, some may undergo resorption remodeling in response to decreased functional stresses.
HISTOPATHOLOGIC FEATURES: Microscopic examination of the torus shows a mass of dense, lamellar, cortical bone. An inner zone of trabecular bone sometimes is seen.
TREATMENT AND PROGNOSIS: Most palatal tori can be diagnosed clinically based on their characteristic appearance; therefore biopsy rarely is necessary. In edentulous patients, the torus may need to be removed surgically to accommodate a denture base. Surgical removal may also be indicated for palatal tori that become repeatedly ulcerated or that interfere with oral function.
TORUS MANDIBULARIS
The torus mandibularis is a common exostosis that develops along the lingual aspect of the mandible. As with torus palatinus, the cause of mandibular tori is probably multifactorial, including both genetic and environmental influences.
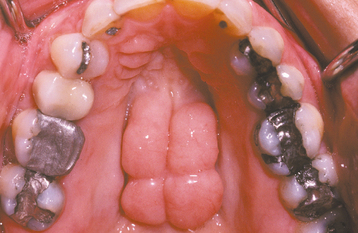
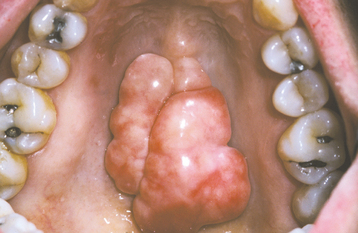
CLINICAL AND RADIOGRAPHIC FEATURES: The mandibular torus presents as a bony protuberance along the lingual aspect of the mandible above the mylohyoid line in the region of the premolars (Fig. 1-40). Bilateral involvement occurs in more than 90% of cases. Most mandibular tori occur as single nodules, although multiple lobules paralleling the teeth are not unusual. Patients often are unaware of their presence unless the overlying mucosa becomes ulcerated secondary to trauma. In rare instances, bilateral tori may become so large that they almost meet in the midline (Fig. 1-41). A large mandibular torus may appear on periapical radiographs as a radiopacity superimposed on the roots of the teeth (Fig. 1-42), especially on anterior films. Mandibular tori are easily visualized on occlusal radiographs (Fig. 1-43).
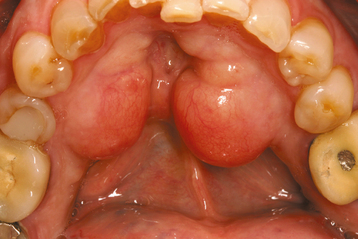
Fig. 1-40 Torus mandibularis. Bilateral lobulated bony protuberances of the mandibular lingual alveolar ridge.
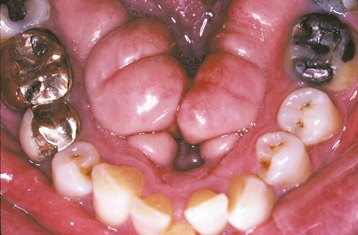
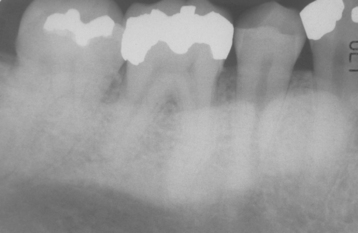
Fig. 1-42 Torus mandibularis. Torus is causing a radiopacity that is superimposed over the roots of the mandibular teeth.
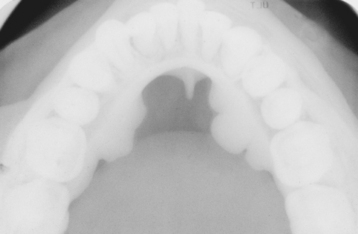
Most studies indicate that the torus mandibularis is not as common as the torus palatinus; the prevalence ranges from 5% to 40%. Like the torus palatinus, the mandibular torus appears to be more common in Asians and the Inuit. The prevalence in the United States ranges from 7% to 10%, with little difference between blacks and whites. A slight male predilection has been noted.
The prevalence of mandibular torus peaks in early adult life, tapering slightly in later years. In addition, the prevalence has been correlated with both bruxism and the number of teeth remaining present. These findings support the theory that the torus mandibularis is multifactorial in development and responds to functional stresses.
HISTOPATHOLOGIC FEATURES: The histopathologic appearance of the torus mandibularis is similar to that of other exostoses, consisting primarily of a nodular mass of dense, cortical lamellar bone (Fig. 1-44). An inner zone of trabecular bone with associated fatty marrow sometimes is visible.
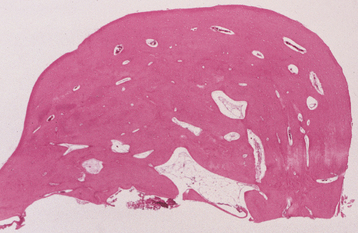
EAGLE SYNDROME (STYLOHYOID SYNDROME; CAROTID ARTERY SYNDROME; STYLALGIA)
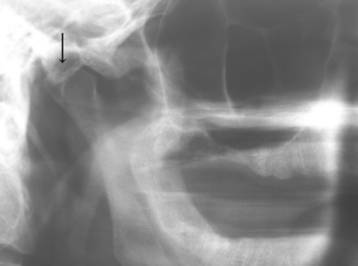
The styloid process is a slender bony projection that originates from the inferior aspect of the temporal bone, anterior and medial to the stylomastoid foramen. It is connected to the lesser cornu of the hyoid bone by the stylohyoid ligament. The external and internal carotid arteries lie on either side. Elongation of the styloid process or mineralization of the stylohyoid ligament complex is not unusual, having been reported in 18% to 40% of the population in some radiographic reviews. Such mineralization is usually bilateral, but it may affect only one side. Most cases are asymptomatic; however, a small number of such patients experience symptoms of Eagle syndrome, caused by impingement or compression of adjacent nerves or blood vessels.
CLINICAL AND RADIOGRAPHIC FEATURES: Eagle syndrome most commonly affects adults. The patient experiences vague facial pain, especially while swallowing, turning the head, or opening the mouth. Other symptoms may include dysphagia, dysphonia, otalgia, headache, dizziness, and transient syncope.
Elongation of the styloid process or mineralization of the stylohyoid ligament complex can be seen on panoramic or lateral-jaw radiographs (Fig. 1-45). The mineralized stylohyoid complex may be palpated in the tonsillar fossa area, and pain often is elicited.
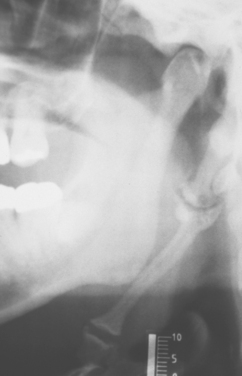
Fig. 1-45 Eagle syndrome. Mineralization of the stylohyoid ligament is visible posterior to the mandibular ramus.
Classic Eagle syndrome occurs after a tonsillectomy. Development of scar tissue in the area of a mineralized stylohyoid complex then results in cervicopharyngeal pain in the region of cranial nerves V, VII, IX, and X, especially during swallowing. Some authors reserve the term Eagle syndrome only for those cases in which the ossification of the stylohyoid chain occurs as a result of the tonsillectomy or other neck trauma.
A second form of this condition unrelated to tonsillectomy is sometimes known as carotid artery syndrome or stylohyoid syndrome. The elongated, mineralized complex is thought to impinge on the internal or external carotid arteries and associated sympathetic nerve fibers. The patient may complain of pain in the neck when turning the head, and this pain may radiate to other sites in the head or neck.
Traumatic Eagle syndrome also has been reported, in which symptoms develop after fracture of a mineralized stylohyoid ligament.
TREATMENT AND PROGNOSIS: Treatment of Eagle syndrome depends on the severity of the symptoms. For mild cases, no treatment may be necessary (except reassurance of the patient). Local injection of corticosteroids sometimes provides relief. In more severe cases, partial surgical excision of the elongated styloid process or mineralized stylohyoid ligament is required. Usually, this is accomplished via an intraoral approach, although an extraoral approach also can be used. The prognosis is good.
STAFNE DEFECT (STAFNE BONE CYST; LINGUAL MANDIBULAR SALIVARY GLAND DEPRESSION; LATENT BONE CYST; STATIC BONE CYST; STATIC BONE DEFECT; LINGUAL CORTICAL MANDIBULAR DEFECT)
In 1942, Stafne described a series of asymptomatic radiolucent lesions located near the angle of the mandible. Subsequent reports of similar lesions have shown that this condition represents a focal concavity of the cortical bone on the lingual surface of the mandible. In most cases, biopsy has revealed histologically normal salivary gland tissue, suggesting that these lesions represent developmental defects containing a portion of the submandibular gland. However, a few of these defects have been reported to be devoid of contents or to contain muscle, fibrous connective tissue, blood vessels, fat, or lymphoid tissue.
Similar lingual cortical defects also have been noted more anteriorly in the mandible, in the area of the incisor, canine, or premolar teeth. These rare defects have been related to the sublingual gland or to aberrant salivary gland tissue. In addition, one report has implicated the parotid gland as the cause of an apparent cortical defect in the upper mandibular ramus. Therefore, all of the major salivary glands appear to be capable of causing such cortical concavities.
In rare examples, the radiolucent defect has been reported to be totally surrounded by intact bone. Such cases might be explained by entrapment of embryonic salivary gland tissue within the jawbone.
CLINICAL AND RADIOGRAPHIC FEATURES: The classic Stafne defect presents as an asymptomatic radiolucency below the mandibular canal in the posterior mandible, between the molar teeth and the angle of the mandible (Fig. 1-46). The lesion is typically well circumscribed and has a sclerotic border. Sometimes the defect may interrupt the continuity of the inferior border of the mandible, with a palpable notch observed clinically in this area. Most Stafne defects are unilateral, although bilateral cases may be seen. Anterior lingual salivary defects associated with the sublingual gland present as well-defined radiolucencies that may appear superimposed over the apices of the anterior teeth (Figs. 1-47 and 1-48).
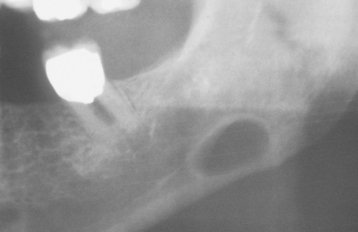
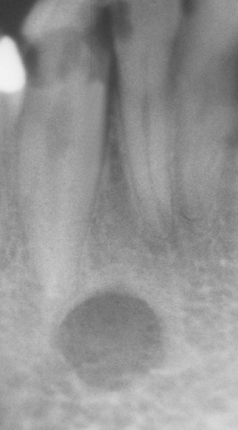
Fig. 1-47 Stafne defect. Anterior radiolucent lesion of the body of the mandible associated with the sublingual gland.
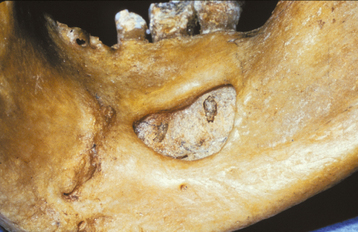
Fig. 1-48 Stafne defect. Lingual surface of the mandible showing an anterior cortical defect caused by the sublingual gland.
Posterior Stafne defects are not rare, having been reported in 0.3% of panoramic radiographs. A striking male predilection is observed, with 80% to 90% of all cases seen in men.
Although the defect is believed to be developmental in nature, it does not appear to be present from birth. Most cases have been reported in middle-aged and older adults, with children rarely affected; this implies that the lesion usually “develops” at a later age. Stafne defects typically remain stable in size; hence the name static bone cyst. In a few cases, however, the lesion has increased in size over time (Fig. 1-49). This also indicates that these lesions are not congenital.
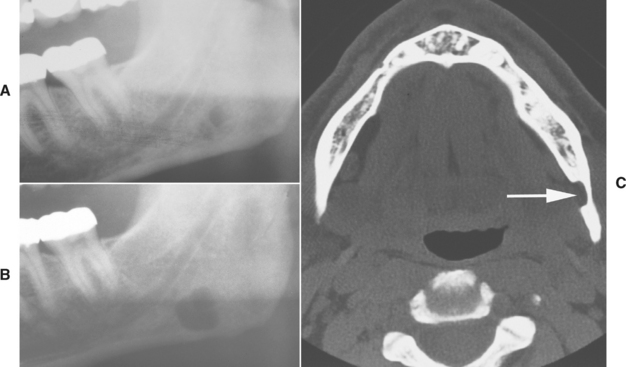
Fig. 1-49 Stafne defect. A, Ill-defined radiolucency near the angle of the mandible. B, Appearance of the same defect several years later showing enlargement of the lesion. C, Computed tomography (CT) image of the same lesion showing a left lingual cortical defect (arrow). (Courtesy of Dr. Carroll Gallagher.)
The diagnosis can usually be made on a clinical basis by the typical radiographic location and lack of symptoms. If the clinical diagnosis is in doubt, then it can be confirmed by CT scans, MRI, or sialography. CT scans and MRIs show a well-defined concavity on the lingual surface of the mandible. Sialograms may be able to demonstrate the presence of salivary gland tissue in the area of the defect.
HISTOPATHOLOGIC FEATURES: Because of the typical radiographic appearance, biopsy is usually not necessary to establish the diagnosis of Stafne defects of the posterior mandible. If biopsy is performed, normal submandibular gland tissue is usually seen. However, some defects are devoid of tissue or contain muscle, blood vessels, fat, connective tissue, or lymphoid tissue. In cases reported to be devoid of contents, it is possible that the gland was simply displaced at the time of biopsy.
Developmental Cysts
By definition, a cyst is a pathologic cavity (often fluid-filled) that is lined by epithelium. A number of different developmental cysts of the head and neck have been described. Some of these have been considered historically as “fissural” cysts because they were thought to arise from epithelium entrapped along embryonal lines of fusion. However, the concept of a fissural origin for many of these cysts has been questioned in more recent years. In many instances the exact pathogenesis of these lesions is still uncertain. Regardless of their origin, once cysts develop in the oral and maxillofacial region, they tend to slowly increase in size, possibly in response to a slightly elevated hydrostatic luminal pressure.
PALATAL CYSTS OF THE NEWBORN (EPSTEIN’S PEARLS; BOHN’S NODULES)
Small developmental cysts are a common finding on the palate of newborn infants. Researchers have theorized that these “inclusion” cysts may arise in one of two ways. First, as the palatal shelves meet and fuse in the midline during embryonic life to form the secondary palate, small islands of epithelium may become entrapped below the surface along the median palatal raphe and form cysts. Second, these cysts may arise from epithelial remnants derived from the development of the minor salivary glands of the palate.
As originally described, Epstein’s pearls occur along the median palatal raphe and presumably arise from epithelium entrapped along the line of fusion. Bohn’s nodules are scattered over the hard palate, often near the soft palate junction and are believed to be derived from the minor salivary glands. However, these two terms have been used almost interchangeably in the literature and also have often been used to describe gingival cysts of the newborn (see page 691), similar-appearing lesions of dental lamina origin. Therefore, the term palatal cysts of the newborn may be preferable to help distinguish them from gingival cysts of the newborn. In addition, because these cysts are most common near the midline at the junction of the hard and soft palates, it is usually difficult to ascertain clinically whether they are arising from epithelium entrapped by fusion of the palate or from the developing minor salivary glands.
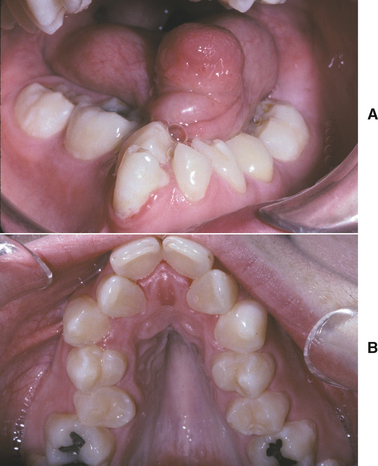
CLINICAL FEATURES: Palatal cysts of the newborn are quite common and have been reported in as many as 65% to 85% of neonates. The cysts are small, 1- to 3-mm, white or yellow-white papules that appear most often along the midline near the junction of the hard and soft palates (Fig. 1-50). Occasionally, they may occur in a more anterior location along the raphe or on the posterior palate lateral to the midline. Frequently a cluster of two to six cysts is observed, although the lesions also can occur singly.
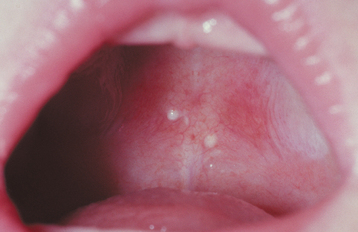
HISTOPATHOLOGIC FEATURES: Microscopic examination reveals keratin-filled cysts that are lined by stratified squamous epithelium. Sometimes these cysts demonstrate a communication with the mucosal surface.
TREATMENT AND PROGNOSIS: Palatal cysts of the newborn are innocuous lesions, and no treatment is required. They are self-healing and rarely observable several weeks after birth. Presumably the epithelium degenerates, or the cysts rupture onto the mucosal surface and eliminate their keratin contents.
NASOLABIAL CYST (NASOALVEOLAR CYST; KLESTADT CYST)
The nasolabial cyst is a rare developmental cyst that occurs in the upper lip lateral to the midline. The pathogenesis is uncertain, although there are two major theories. One theory considers the nasolabial cyst to be a “fissural” cyst arising from epithelial remnants entrapped along the line of fusion of the maxillary, medial nasal, and lateral nasal processes. A second theory suggests that these cysts develop from misplaced epithelium of the nasolacrimal duct because of their similar location and histologic appearance.
CLINICAL AND RADIOGRAPHIC FEATURES: The nasolabial cyst usually appears as a swelling of the upper lip lateral to the midline, resulting in elevation of the ala of the nose. The enlargement often elevates the mucosa of the nasal vestibule and obliterates the maxillary mucolabial fold (Fig. 1-51). On occasion, this expansion may result in nasal obstruction or may interfere with the wearing of a denture. Pain is uncommon unless the lesion is secondarily infected. The cyst may rupture spontaneously and may drain into the oral cavity or nose.
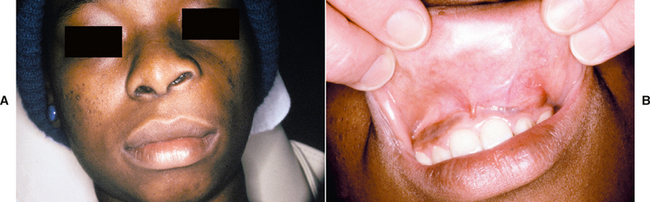
Fig. 1-51 Nasolabial cyst. A, Enlargement of the left upper lip with elevation of the ala of the nose. B, Intraoral swelling fills the maxillary labial fold. (Courtesy of Dr. Jim Weir.)
Nasolabial cysts are most commonly seen in adults, with a peak prevalence in the fourth and fifth decades of life. A significant predilection exists for women, with a female-to-male ratio of 3:1. Approximately 10% of the reported cases have been bilateral.
Because the nasolabial cyst arises in soft tissues, in most cases no radiographic changes are seen. Occasionally, pressure resorption of the underlying bone may occur.
HISTOPATHOLOGIC FEATURES: The nasolabial cyst is characteristically lined by pseudostratified columnar epithelium, often demonstrat-ing goblet cells and cilia (Fig. 1-52). Areas of cuboidal epithelium and squamous metaplasia are not unusual. Apocrine changes also have been reported. The cyst wall is composed of fibrous connective tissue with adjacent skeletal muscle. Inflammation may be seen if the lesion is secondarily infected.
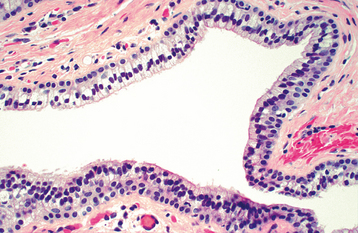
TREATMENT AND PROGNOSIS: Complete surgical excision of the cyst via an intraoral approach has been the treatment of choice. Because the lesion is often close to the floor of the nose, it is sometimes necessary to sacrifice a portion of the nasal mucosa to ensure total removal. Recurrence is rare. Recently an alternative transnasal approach has been suggested that allows endoscopic marsupialization of the cystic cavity.
“GLOBULOMAXILLARY CYST”
As originally described, the “globulomaxillary cyst” was purported to be a fissural cyst that arose from epithelium entrapped during fusion of the globular portion of the medial nasal process with the maxillary process. This concept has been questioned, however, because the globular portion of the medial nasal process is primarily united with the maxillary process and a fusion does not occur. Therefore, epithelial entrapment should not occur during embryologic development of this area.
Virtually all cysts in the globulomaxillary region (between the lateral incisor and canine teeth) can be explained on an odontogenic basis. Many are lined by inflamed stratified squamous epithelium and are consistent with periapical cysts (see page 130). Some exhibit specific histopathologic features of an odontogenic keratocyst (see page 683) or developmental lateral periodontal cyst (see page 692). Researchers have also theorized that some of these lesions may arise from inflammation of the reduced enamel epithelium at the time of eruption of the teeth.
On rare occasions, cysts in the globulomaxillary area may be lined by pseudostratified, ciliated, columnar epithelium. Such cases may lend credence to the fissural theory of origin. However, this epithelium may be explained by the close proximity of the sinus lining. In addition, respiratory epithelium also has been reported in periapical cysts, dentigerous cysts, and glandular odontogenic cysts found in other locations.
Because a fissural cyst in this region probably does not exist, the term globulomaxillary cyst should no longer be used. When a radiolucency between the maxillary lateral incisor and canine is encountered, the clinician should first consider an odontogenic origin for the lesion.
NASOPALATINE DUCT CYST (INCISIVE CANAL CYST)
The nasopalatine duct cyst is the most common nonodontogenic cyst of the oral cavity, occurring in about 1% of the population. The cyst is believed to arise from remnants of the nasopalatine duct, an embryologic structure connecting the oral and nasal cavities in the area of the incisive canal.
In the 7-week-old fetus, the developing palate consists of the primary palate, which is formed by the fusion of the medial nasal processes. Behind the primary palate, downgrowth of the nasal septum produces two communications between the oral and nasal cavities, the primitive nasal choanae. Formation of the secondary palate begins around the eighth intrauterine week, with downward growth of the medial parts of the maxillary processes (palatine processes) to a location on either side of the tongue.
As the mandible develops and the tongue drops down, these palatine processes grow horizontally, fusing with the nasal septum in the midline and with the primary palate along their anterior aspect. Two passageways persist in the midline between the primary and secondary palates (the incisive canals). Also formed by this fusion and found within the incisive canals are epithelial structures—the nasopalatine ducts. These ducts normally degenerate in humans but may leave epithelial remnants behind in the incisive canals.
The incisive canals begin on the floor of the nasal cavity on either side of the nasal septum, coursing downward and forward to exit the palatal bone via a common foramen in the area of the incisive papilla. In addition to the nasopalatine ducts, these canals contain the nasopalatine nerve plus anastomosing branches of the descending palatine and sphenopalatine arteries. Occasionally, two smaller foramina carrying the nasopalatine nerves—the canals of Scarpa—are found within the incisive foramen.
In some mammals the nasopalatine ducts remain patent and provide communication between the oral and nasal cavities. On rare occasions, patent or partially patent nasopalatine ducts may be encountered in humans. In mammals the nasopalatine ducts may communicate with the vomer-nasal organ of Jacobson, acting as an accessory olfactory organ. However, in humans, Jacobson’s organ usually recedes in uterine life to become a vestigial structure.
Researchers have suggested that the nasopalatine duct cyst may arise from the epithelium of Jacobson’s organ, but this appears highly unlikely. Trauma or infection of the duct and mucous retention of adjacent minor salivary glands also have been mentioned as possible etiologic factors, but the role of each has been questioned. Although the pathogenesis of this lesion is still uncertain, the lesion most likely represents a spontaneous cystic degeneration of remnants of the nasopalatine duct.
CLINICAL AND RADIOGRAPHIC FEATURES: The nasopalatine duct cyst may develop at almost any age but is most common in the fourth to sixth decades of life. In spite of its being a “developmental” cyst, the nasopalatine duct cyst is rarely seen during the first decade. Most studies have shown a male predilection.
The most common presenting symptoms include swelling of the anterior palate, drainage, and pain (Fig. 1-53). Patients sometimes relate a long history of these symptoms, probably because of their intermittent nature. However, many lesions are asymptomatic and are discovered on routine radiographs. Rarely a large cyst may produce a “through-and-through” fluctuant expansion involving the anterior palate and labial alveolar mucosa.
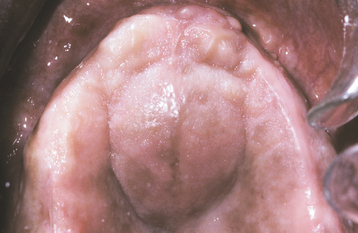
Radiographs usually demonstrate a well-circumscribed radiolucency in or near the midline of the anterior maxilla, between and apical to the central incisor teeth (Figs. 1-54 and 1-55). Root resorption is rarely noted. The lesion most often is round or oval with a sclerotic border. Some cysts may have an inverted pear shape, presumably because of resistance of adjacent tooth roots. Other examples may show a classic heart shape as a result of superimposition of the nasal spine or because they are notched by the nasal septum.
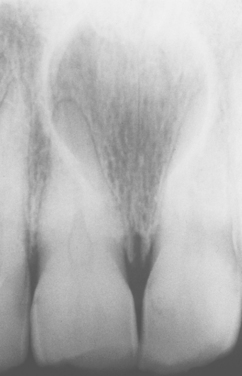
Fig. 1-54 Nasopalatine duct cyst. Well-circumscribed radiolucency between and apical to the roots of the maxillary central incisors.
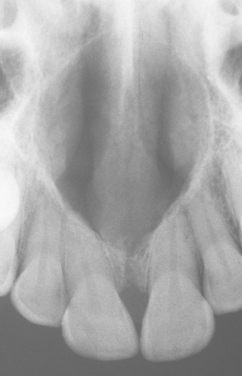
The radiographic diameter of nasopalatine duct cysts can range from small lesions, less than 6 mm, to destructive lesions as large as 6 cm. However, most cysts are in the range of 1.0 to 2.5 cm, with an average diameter of 1.5 to 1.7 cm. It may be difficult to distinguish a small nasopalatine duct cyst from a large incisive foramen. It is generally accepted that a diameter of 6 mm is the upper limit of normal size for the incisive foramen. Therefore, a radiolucency that is 6 mm or smaller in this area is usually considered a normal foramen unless other clinical signs or symptoms are present.
In rare instances, a nasopalatine duct cyst may develop in the soft tissues of the incisive papilla area without any bony involvement. Such lesions often are called cysts of the incisive papilla. These cysts frequently demonstrate bluish discoloration as a result of the fluid contents in the cyst lumen (Fig. 1-56).
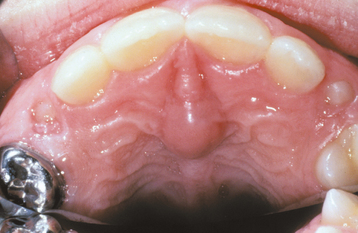
HISTOPATHOLOGIC FEATURES: The epithelial lining of nasopalatine duct cysts is highly variable (Figs. 1-57 and 1-58). It may be composed of the following:
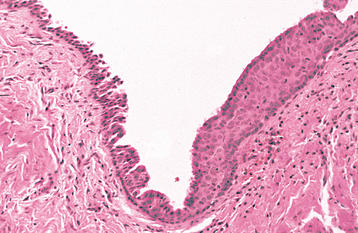
Fig. 1-57 Nasopalatine duct cyst. Cystic lining showing transition from pseudostratified columnar to stratified squamous epithelium.
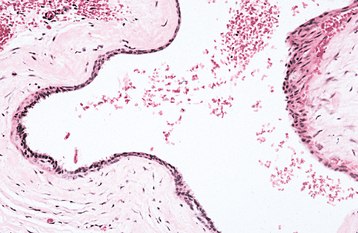
Frequently, more than one epithelial type is found in the same cyst. Stratified squamous epithelium is most common, present in at least three fourths of all cysts. Pseudostratified columnar epithelium has been reported in from one third to three fourths of all cases. Simple cuboidal and columnar epithelium are discovered less frequently.
Cilia and goblet cells may be found in association with columnar linings. The type of epithelium may be related to the vertical position of the cyst within the incisive canal. Cysts developing within the superior aspect of the canal near the nasal cavity are more likely to demonstrate respiratory epithelium; those in an inferior position near the oral cavity are more likely to exhibit squamous epithelium.
The contents of the cyst wall can be a helpful diagnostic aid. Because the nasopalatine duct cyst arises within the incisive canal, moderate-sized nerves and small muscular arteries and veins are usually found in the wall of the cyst (Fig. 1-59). Small mucous glands have been reported in as many as one third of cases. Occasionally, the clinician may see small islands of hyaline cartilage. Frequently, an inflammatory response is noted in the cyst wall and may range from mild to heavy. This inflammation is usually chronic in nature and is composed of lymphocytes, plasma cells, and histiocytes. Associated acute inflammatory cells (neutrophils) sometimes may be seen.
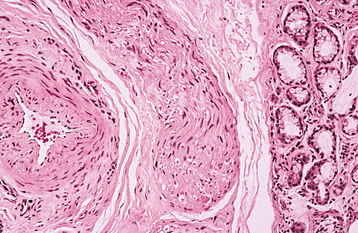
TREATMENT AND PROGNOSIS: Nasopalatine duct cysts are treated by surgical enucleation. Biopsy is recommended because the lesion is not diagnostic radiographically; other benign and malignant lesions have been known to mimic the nasopalatine duct cyst. The lesion is best approached with a palatal flap that is reflected after an incision is made along the lingual gingival margin of the anterior maxillary teeth. Recurrence is rare. Malignant transformation has been reported in a couple of cases, but this is an extremely rare complication.
MEDIAN PALATAL (PALATINE) CYST
The median palatal cyst is a rare fissural cyst that theoretically develops from epithelium entrapped along the embryonic line of fusion of the lateral palatal shelves of the maxilla. This cyst may be difficult to distinguish from a nasopalatine duct cyst. In fact, most “median palatal cysts” may represent posteriorly positioned nasopalatine duct cysts. Because the nasopalatine ducts course posteriorly and superiorly as they extend from the incisive canal to the nasal cavity, a nasopalatine duct cyst that arises from posterior remnants of this duct near the nasal cavity might be mistaken for a median palatal cyst. On the other hand, if a true median palatal cyst were to develop toward the anterior portion of the hard palate, then it could easily be mistaken for a nasopalatine duct cyst.
CLINICAL AND RADIOGRAPHIC FEATURES: The median palatal cyst presents as a firm or fluctuant swelling of the midline of the hard palate posterior to the palatine papilla. The lesion appears most frequently in young adults. Often it is asymptomatic, but some patients complain of pain or expansion. The average size of this cyst is 2 × 2 cm, but sometimes it can become quite large. Occlusal radiographs demonstrate a well-circumscribed radiolucency in the midline of the hard palate (Fig. 1-60). Occasional reported cases have been associated with divergence of the central incisors, although it may be difficult to rule out a nasopalatine duct cyst in these instances.
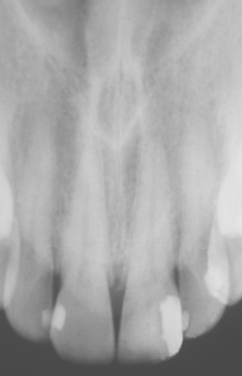
Fig. 1-60 Median palatal cyst. Well-circumscribed radiolucency apical to the maxillary incisors in the midline. At surgery the lesion was unrelated to the incisive canal. (Courtesy of Dr. Timothy Armanini.)
To differentiate the median palatal cyst from other cystic lesions of the maxilla, Gingell and associates suggested the following diagnostic criteria:
• Grossly appears symmetrical along the midline of the hard palate
• Located posterior to the palatine papilla
• Appears ovoid or circular radiographically
• Not intimately associated with a nonvital tooth
• Does not communicate with the incisive canal
• Shows no microscopic evidence of large neurovascular bundles, hyaline cartilage, or minor salivary glands in the cyst wall
It must be stressed that a true median palatal cyst should exhibit clinical enlargement of the palate. A midline radiolucency without clinical evidence of expansion is probably a nasopalatine duct cyst.
“MEDIAN MANDIBULAR CYST”
The “median mandibular cyst” is a controversial lesion of questionable existence. Theoretically, it represents a fissural cyst in the anterior midline of the mandible that develops from epithelium entrapped during fusion of the halves of the mandible during embryonic life. However, the mandible actually develops as a single bilobed proliferation of mesenchyme with a central isthmus in the midline. As the mandible grows, this isthmus is eliminated. Therefore, because no fusion of epithelium-lined processes occurs, entrapment of epithelium should not be possible.
Because respiratory prosoplasia is not uncommon in odontogenic cysts, it appears likely that most (if not all) of these midline cysts are of odontogenic origin. Many purported cases would be classified today as examples of the glandular odontogenic cyst (see page 697), which has a propensity for occurrence in the midline mandibular region. Others could be classified as periapical cysts, odontogenic keratocysts, or lateral periodontal cysts. Because a fissural cyst in this region probably does not exist, the term median mandibular cyst should no longer be used.
FOLLICULAR CYSTS OF THE SKIN
Follicular cysts of the skin are common keratin-filled lesions that arise from one or more portions of the hair follicle. The most common type, which is derived from the follicular infundibulum, is known as an epidermoid or infundibular cyst. These cysts often arise after localized inflammation of the hair follicle and probably represent a nonneoplastic proliferation of the infundibular epithelium resulting from the healing process. The term sebaceous cyst sometimes is used mistakenly as a synonym for both the epidermoid cyst and another cyst of the scalp known as a pilar, tricholemmal, or isthmus-catagen cyst. However, because both the epidermoid cyst and pilar cyst are derived from the hair follicle rather than the sebaceous gland, the term sebaceous cyst should be avoided.
Keratin-filled cysts of the skin may occasionally arise after traumatic implantation of epithelium, although such lesions may be difficult to distinguish from an infundibular cyst. Rarely, such epidermal inclusion (implantation) cysts also can develop in the mouth. These small inclusion cysts should be distinguished from oral epidermoid cysts that occur in the midline floor of mouth region and represent the minimal manifestation of the teratoma-dermoid cyst-epidermoid cyst spectrum (see page 33).
CLINICAL FEATURES: Epidermoid (infundibular) cysts account for approximately 80% of follicular cysts of the skin and are most common in the acne-prone areas of the head, neck, and back. They are unusual before puberty unless they are associated with Gardner syndrome (see page 651). Young adults are more likely to have cysts on the face, whereas older adults are more likely to have cysts on the back. Males are affected more frequently than females.
Epidermoid cysts present as nodular, fluctuant subcutaneous lesions that may or may not be associated with inflammation (Figs. 1-61 and 1-62). If a noninflamed lesion presents in an area of thin skin, such as the earlobe, then it may be white or yellow.
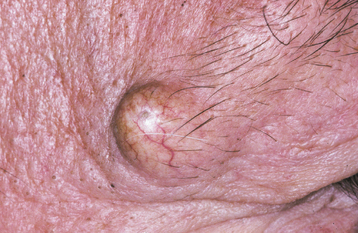
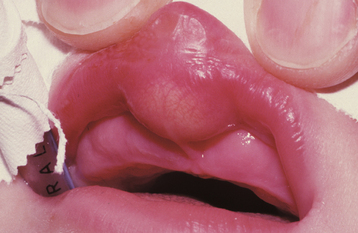
Pilar (tricholemmal) cysts comprise approximately 10% to 15% of skin cysts, occurring most frequently on the scalp (Fig. 1-63). They are twice as common in women as in men. The lesion is usually movable and shells out easily.
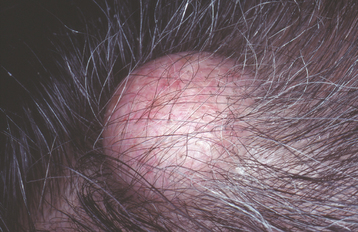
HISTOPATHOLOGIC FEATURES: Microscopic examination of an epidermoid cyst reveals a cavity that is lined by stratified squamous epithelium resembling epidermis (Fig. 1-64). A well-developed granular cell layer is seen, and the lumen is filled with degenerating orthokeratin. Not infrequently, the epithelial lining will be disrupted. When this occurs, a prominent granulomatous inflammatory reaction, including multinucleated giant cells, can be present in the cyst wall because the exposed keratin is recognized as a foreign material.
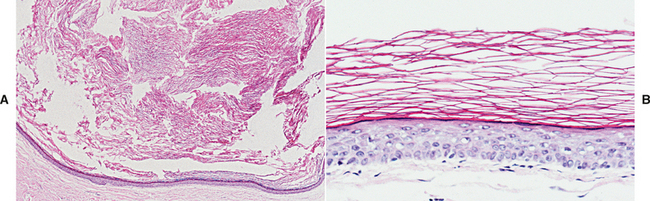
Fig. 1-64 Epidermoid cyst. A, Low-power view showing a keratin-filled cystic cavity. B, High-power view showing stratified squamous epithelial lining with orthokeratin production.
The pilar cyst is also lined by stratified squamous epithelium, although a granular cell layer is usually absent or greatly diminished (Fig. 1-65). The keratinocytes remain large in the upper epithelial layers with an abrupt transition to dense, compact keratin that fills the cyst lumen.
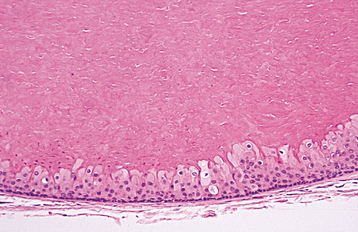
DERMOID CYST
The dermoid cyst is an uncommon developmental cystic malformation. The cyst is lined by epidermis-like epithelium and contains dermal adnexal structures in the cyst wall. It is generally classified as a benign cystic form of teratoma.
By definition, a true teratoma is a developmental tumor composed of tissue from all three germ layers: (1) ectoderm, (2) mesoderm, and (3) endoderm. Such tumors are believed to arise from germ cells or entrapped totipotent blastomeres, which can produce derivatives of all three germ layers.
Teratomatous malformations have a spectrum of complexity. In their most complex form, these lesions produce multiple types of tissue that are arranged in a disorganized fashion. These “complex” teratomas are most common in the ovaries or testes and can be benign or malignant. Occasionally, ovarian teratomas (or “dermoids”) produce well-formed teeth, or even partially complete jaws. Complex teratomas of the oral cavity are rare and are usually congenital in nature. When they occur, they usually extend through a cleft palate from the pituitary area via Rathke’s pouch. Cervical teratomas also have been reported.
The term teratoid cyst has been used to describe a cystic form of teratoma that contains a variety of germ layer derivatives:
1. Skin appendages, including hair follicles, sebaceous glands, and sweat glands
2. Connective tissue elements, such as muscle, blood vessels, and bone
Rarely, oral cysts may be lined entirely by gastrointestinal epithelium. These heterotopic oral gastrointestinal cysts (enterocystomas, enteric duplication cysts) are usually considered to be choristomas, or histologically normal tissue found in an abnormal location. However, these lesions probably can be included under the broad umbrella of teratomatous lesions, especially because they are occasionally found in combination with dermoid cysts.
Dermoid cysts are simpler in structure than complex teratomas or teratoid cysts. Although they do not contain tissue from all three germ layers, they probably represent a forme fruste of a teratoma. Similar cysts of the oral cavity can be seen that are lined by epidermis-like epithelium, but they contain no dermal appendages in the cyst wall. These lesions have been called epidermoid cysts and represent the simplest expression of the teratoma spectrum. These intraoral epidermoid cysts should not be confused with the more common epidermoid cyst of the skin (see page 32), a nonteratomatous lesion that arises from the hair follicle.
CLINICAL AND RADIOGRAPHIC FEATURES: Dermoid cysts most commonly occur in the midline of the floor of the mouth (Fig. 1-66), although occasionally they are displaced laterally or develop in other locations. If the cyst develops above the geniohyoid muscle, then a sublingual swelling may displace the tongue toward the roof of the mouth and create difficulty in eating, speaking, or even breathing. Cysts that occur below the geniohyoid muscle often produce a submental swelling, with a “double-chin” appearance.
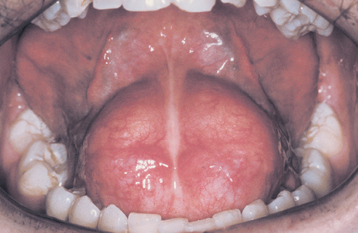
Fig. 1-66 Dermoid cyst. Fluctuant midline swelling in the floor of the mouth. (From Budnick SD: Handbook of pediatric oral pathology, Chicago, 1981, Year Book Medical.) Year Book Medical
Oral dermoid cysts can vary in size from a few millimeters to 12 cm in diameter. They are most common in children and young adults; 15% of reported cases have been congenital. The lesion is usually slow growing and painless, presenting as a doughy or rubbery mass that frequently retains pitting after application of pres sure. Secondary infection can occur, and the lesion may drain intraorally or onto the skin. MRIs, CT scans, or contrast medium radiographs may be helpful in delineating the extent of the lesion.
HISTOPATHOLOGIC FEATURES: Dermoid cysts are lined by orthokeratinized stratified squamous epithelium, with a prominent granular cell layer. Abundant keratin often is found within the cyst lumen. On rare occasions, areas of respiratory epithelium can be seen. The cyst wall is composed of fibrous connective tissue that contains one or more skin appendages, such as sebaceous glands, hair follicles, or sweat glands (Fig. 1-67).
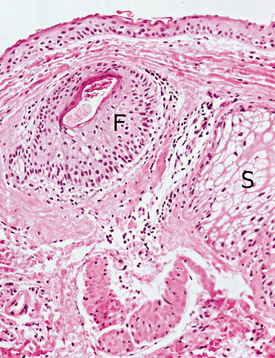
TREATMENT AND PROGNOSIS: Dermoid cysts are treated by surgical removal. Those located above the geniohyoid muscle can be removed by an intraoral incision, and those below the geniohyoid muscle may require an extraoral approach. Recurrence is uncommon. Malignant transformation into squamous cell carcinoma has been reported only rarely.
THYROGLOSSAL DUCT CYST (THYROGLOSSAL TRACT CYST)
The thyroid gland begins its development at the end of the third week of embryonic life as a proliferation of endodermal cells from the ventral floor of the pharynx, between the tuberculum impar and copula of the developing tongue—a point that later becomes the foramen cecum. This thyroid anlage descends into the neck as a bilobed diverticulum anterior to the developing hyoid bone and reaches its definitive level below the thyroid cartilage by the seventh embryonic week. Along this path of descent, an epithelial tract or duct is formed, maintaining an attachment to the base of the tongue. This thyroglossal duct becomes intimately associated with the developing hyoid bone. As the hyoid matures and rotates to its adult position, the thyroglossal duct passes in front and beneath the hyoid, looping upward and behind it before curving downward again into the lower neck. The caudal segment of this duct often persists, forming the pyramidal lobe of the thyroid gland.
The thyroglossal duct epithelium normally undergoes atrophy and is obliterated. However, remnants of this epithelium may persist and give rise to cysts along this tract known as thyroglossal duct cysts. The impetus for cystic degeneration is uncertain. Inflammation is the most frequently suggested stimulus, especially from adjacent lymphoid tissue that may react to draining infections of the head and neck. Retention of secretions within the duct is another possible factor. In addition, there are several reports of familial occurrence of such cysts.
CLINICAL FEATURES: Thyroglossal duct cysts classically develop in the midline and may occur anywhere from the foramen cecum area of the tongue to the suprasternal notch. Suprahyoid cysts may be submental in location. In 60% to 80% of cases, the cyst develops below the hyoid bone. Intralingual cysts are rare. Cysts that develop in the area of the thyroid cartilage often are deflected lateral to the midline because of the sharp anterior margin of the thyroid cartilage.
Thyroglossal duct cysts may develop at any age, but they are most commonly diagnosed in the first two decades of life; about 50% of cases occur before the age of 20. There is no sex predilection. The cyst usually presents as a painless, fluctuant, movable swelling unless it is complicated by secondary infection (Fig. 1-68). Lesions that develop at the base of the tongue may cause laryngeal obstruction. Most thyroglossal duct cysts are smaller than 3 cm in diameter, but occasional cysts may reach 10 cm in size. If the cyst maintains an attachment to the hyoid bone or tongue, it will move vertically during swallowing or protru-sion of the tongue. Fistulous tracts to the skin or mucosa develop in as many as one third of cases, usually from rupture of an infected cyst or as a sequela of surgery.
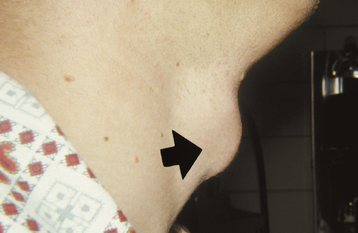
HISTOPATHOLOGIC FEATURES: Thyroglossal duct cysts are usually lined by columnar or stratified squamous epithelium, although occasionally, cuboidal or even small intestine epithelium may be documented (Fig. 1-69). Sometimes a mixture of epithelial types is present. Thyroid tissue may occur in the cyst wall, but this is not a constant finding.
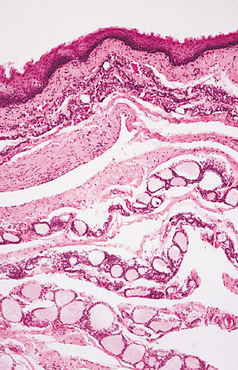
TREATMENT AND PROGNOSIS: Thyroglossal duct cysts are best treated by a Sistrunk procedure. In this operation the cyst is removed in addition to the midline segment of the hyoid bone and a generous portion of muscular tissue along the entire thyroglossal tract. The recurrence rate associated with this procedure is less than 10%. A much higher recurrence rate can be expected with less aggressive surgery.
Carcinoma arising in a thyroglossal duct cyst is a rare complication that occurs in approximately 1% of cases. Most of these have been papillary thyroid adenocarcinomas. Fortunately, metastases from thyroglossal carcinoma are rare, and the prognosis for people with these tumors is good.
BRANCHIAL CLEFT CYST (CERVICAL LYMPHOEPITHELIAL CYST)
The branchial cleft cyst, a developmental cyst of the lateral neck, has a disputed pathogenesis. The classic theory holds that the cyst develops from remnants of the branchial clefts because it occurs in the area of the embryonic gill arch apparatus. A second theory considers that it arises from cystic changes in parotid gland epithelium that becomes entrapped in the upper cervical lymph nodes during embryonic life. However, immunohistochemical analysis supports the classic branchial cleft theory of pathogenesis for this lesion. About 95% of these cysts are believed to arise from the second branchial arch, with the remaining 5% originating from the first, third, and fourth branchial arches.
CLINICAL FEATURES: The branchial cleft cyst most commonly occurs in the upper lateral neck along the anterior border of the sternocleidomastoid muscle (Figs. 1-70 and 1-71). It most frequently affects young adults between the ages of 20 and 40. Clinically, the cyst appears as a soft, fluctuant mass that can range from 1 to 10 cm in diameter. Associated tenderness or pain sometimes may occur with secondary infection. Occasionally, the lesion becomes evident after an upper respiratory tract infection or trauma. Some lesions appear as sinuses or fistulae that may produce a mucoid discharge onto the skin. Two thirds of branchial cleft cysts occur on the left side of the neck, and one third are found on the right side. In rare instances, bilateral cysts may develop.
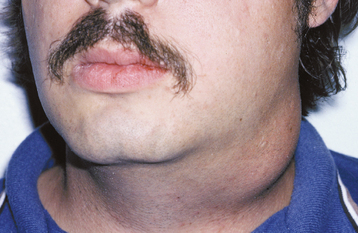
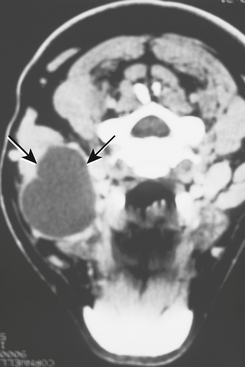
Fig. 1-71 Cervical lymphoepithelial cyst. Imaging study of the same cyst depicted in Figure 1-70, showing a well-circumscribed lesion of the lateral neck (arrows).
Although one theory suggests that these cysts are derived from parotid epithelium that becomes entrapped within lymph node tissue, lymphoepithelial cysts are uncommon within the parotid gland itself. In recent years, however, increased numbers of parotid lymphoepithelial cysts have been reported in patients with HIV infection. These are probably related to intraparotid lymphadenopathy associated with HIV infection.
HISTOPATHOLOGIC FEATURES: More than 90% of branchial cleft cysts are lined by stratified squamous epithelium that may or may not be keratinized (Fig. 1-72). Some cysts demonstrate respiratory epithelium. The wall of the cyst typically contains lymphoid tissue, often demonstrating germinal center formation. However, occasional cysts have been reported without lymphoid tissue.
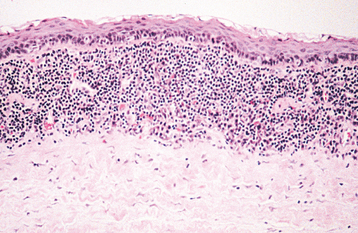
TREATMENT AND PROGNOSIS: The branchial cleft cyst is treated by surgical removal. The lesion almost never recurs.
Rare examples of malignant transformation in these cysts have been reported. Although such an occurrence is theoretically possible, most suspected cases actually represent cystic metastases from previously undetected carcinomas of the head and neck region, especially from the base of tongue, lingual tonsil, or palatine tonsil. When evaluating patients with cystic neck masses, fine-needle aspiration biopsy can be helpful to rule out the possibility of malignancy before surgery.
ORAL LYMPHOEPITHELIAL CYST
The oral lymphoepithelial cyst is an uncommon lesion of the mouth that develops within oral lymphoid tissue. It is microscopically similar to the branchial cleft cyst (cervical lymphoepithelial cyst) but much smaller in size.
Lymphoid tissue is normally found in the oral cavity and pharynx, principally consisting of Waldeyer’s ring, which includes the palatine tonsils, lingual tonsils, and pharyngeal adenoids. In addition, accessory oral tonsils or lymphoid aggregates may occur in the floor of the mouth, ventral surface of the tongue, and soft palate.
Oral lymphoid tissue has a close relationship with the overlying mucosal epithelium. This epithelium demonstrates invaginations into the tonsillar tissue, resulting in blind pouches or tonsillar crypts that may fill up with keratin debris. The tonsillar crypt may become obstructed or pinched off from the surface, producing a keratin-filled cyst within the lymphoid tissue just below the mucosal surface. It also is possible that oral lymphoepithelial cysts may develop from salivary or surface mucosal epithelium that becomes enclaved in lymphoid tissue during embryogenesis. It even has been suggested that these cysts may arise from the excretory ducts of the sublingual gland or minor salivary glands, and that the associated lymphoid tissue represents a secondary immune response.
CLINICAL FEATURES: The oral lymphoepithelial cyst presents as a small submucosal mass that is usually less than 1 cm in diameter; rarely will the lesion be greater than 1.5 cm (Figs. 1-73 and 1-74). The cyst may feel firm or soft to palpation, and the overlying mucosa is smooth and nonulcerated. The lesion is typically white or yellow and often contains creamy or cheesy keratinous material in the lumen. The cyst is usually asymptomatic, although occasionally, patients may complain of swelling or drainage. Pain is rare but may occur secondary to trauma.
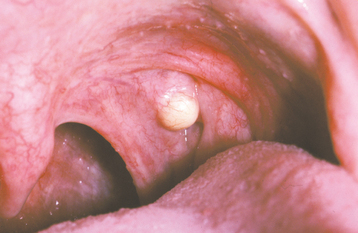
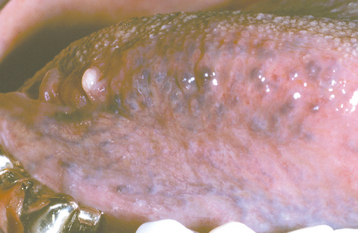
Fig. 1-74 Oral lymphoepithelial cyst. Small white nodule of the posterior lateral border of the tongue.
Oral lymphoepithelial cysts may develop in people of almost any age, but they are most common in young adults. The most frequent location is the floor of the mouth, with at least half of all cases found there. The ventral surface and posterior lateral border of the tongue are the next most common sites. These cysts also may develop in the area of the palatine tonsil or soft palate. All of these locations represent sites of normal or accessory oral lymphoid tissue.
HISTOPATHOLOGIC FEATURES: Microscopic examination of the oral lymphoepithelial cyst demonstrates a cystic cavity that is lined by stratified squamous epithelium without rete ridges (Fig. 1-75). This epithelium is typically parakeratinized, with desquamated epithelial cells seen filling the cyst lumen. In rare instances the epithelial lining also may contain mucous cells. Occasional cysts may communicate with the overlying mucosal surface.
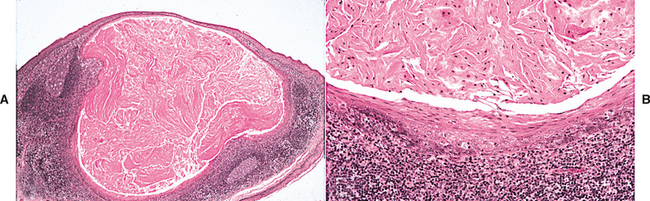
Fig. 1-75 Oral lymphoepithelial cyst. A, Low-power view showing a keratin-filled cyst below the mucosal surface. Lymphoid tissue is present in the cyst wall. B, High-power view showing lymphoid tissue adjacent to the cystic lining.
The most striking feature is the presence of lymphoid tissue in the cyst wall. In most instances, this lymphoid tissue encircles the cyst, but sometimes it involves only a portion of the cyst wall. Germinal centers are usually, but not always, present.
Other Rare Developmental Anomalies
HEMIHYPERPLASIA (HEMIHYPERTROPHY)
Hemihyperplasia is a rare developmental anomaly characterized by asymmetric overgrowth of one or more body parts. Although the condition is known more commonly as hemihypertrophy, it actually represents a hyperplasia of the tissues rather than a hypertrophy. Hemihyperplasia can be an isolated finding, but it also may be associated with a variety of malformation syndromes (Box 1-3).
Almost all cases of isolated hemihyperplasia are sporadic. A number of possible etiologic factors have been suggested, but the cause remains obscure. Various theories include vascular or lymphatic abnormalities, central nervous system disturbances, endocrine dysfunctions, and aberrant twinning mechanisms. Occasionally, chromosomal anomalies have been documented.
CLINICAL AND RADIOGRAPHIC FEATURES: In a person with hemihyperplasia, one entire side of the body (complex hemihyperplasia) may be affected or the enlargement may be limited to a single limb (simple hemihyperplasia). If the enlargement is confined to one side of the face, the term hemifacial hyperplasia (or hemifacial hypertrophy) may apply. The condition can occasionally be crossed, involving different areas on both sides of the body. Hemihyperplasia shows a 2:1 female-to-male predilection, and it occurs more often on the right side of the body.
Asymmetry often is noted at birth, although in some cases the condition may not become evident until later in childhood (Fig. 1-76). The enlargement becomes more accentuated with age, especially at puberty. This disproportionate growth continues until the patient’s overall growth ceases, resulting in permanent asymmetry.
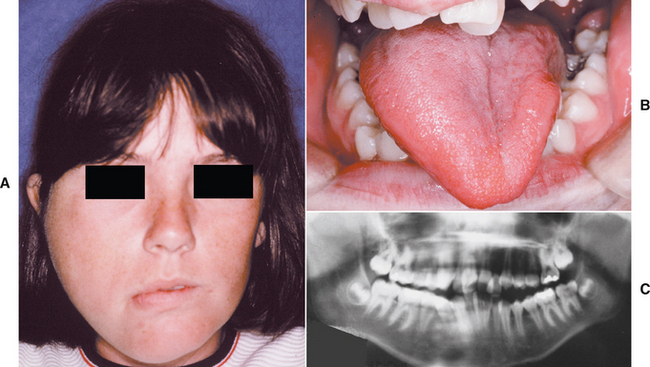
Fig. 1-76 Hemihyperplasia. A, Enlargement of the right side of the face. B, Same patient with associated enlargement of the right half of the tongue. C, Panoramic radiograph of the same patient showing enlargement of the mandible and teeth on the right side. (Courtesy of Dr. George Blozis.)
The changes may involve all the tissues on the affected side, including the underlying bone. Often the skin is thickened and may demonstrate increased pigmentation, hypertrichosis, telangiectasias, or nevus flammeus. About 20% of those affected are mentally retarded. One of the most significant features is an increased prevalence of abdominal tumors, especially Wilms’ tumor, adrenal cortical carcinoma, and hepatoblastoma. These tumors have been reported in 5.9% of patients with isolated hemihyperplasia, and they do not necessarily occur on the same side as the somatic enlargement.
Unilateral macroglossia, featuring prominent tongue papillae, is common. Enlargement of other oral soft tissues and bone can occur. The mandibular canal may be increased in size on radiographs. The crowns of the teeth on the affected side, especially the permanent cuspids, premolars, and first molars, can be larger. Premature development of these teeth, along with precocious eruption, may be obvious. The roots also may be larger, but some reports have described root resorption. Malocclusion with open bite is not unusual.
HISTOPATHOLOGIC FEATURES: Microscopic examination shows an increase in thickness of the epithelium, with hyperplasia of the underlying connective tissues.
TREATMENT AND PROGNOSIS: A complete workup should be undertaken to rule out other possible causes of unilateral growth, such as Beckwith-Wiedemann syndrome, Proteus syndrome, and neurofibromatosis type I (see page 529), which can exhibit hemihyperplasia. During childhood, periodic ultrasound examination should be performed to rule out development of abdominal tumors. After the patient’s growth has ceased, cosmetic surgery can be performed, including soft tissue debulking, face lifts, and orthognathic surgery. Orthodontic therapy is also frequently needed.
PROGRESSIVE HEMIFACIAL ATROPHY (PROGRESSIVE FACIAL HEMIATROPHY; ROMBERG SYNDROME; PARRY-ROMBERG SYNDROME)
Progressive hemifacial atrophy is an uncommon and poorly understood degenerative condition characterized by atrophic changes affecting one side of the face. The cause of these changes remains obscure. Speculation has considered trophic malfunction of the cervical sympathetic nervous system. A history of trauma has been documented in some cases, although a number of recent reports have considered Borrelia spp. infection (Lyme disease) in the cause. Usually, the condition is sporadic, but a few familial cases have been reported, suggesting a possible hereditary influence. Progressive hemifacial atrophy exhibits many features similar to a localized form of scleroderma (see page 798), indicating a close relationship between these two disorders.
CLINICAL AND RADIOGRAPHIC FEATURES: The onset of the syndrome is usually during the first two decades of life. The condition begins as atrophy of the skin and subcutaneous structures in a localized area of the face (Fig. 1-77). This atrophy progresses at a variable rate and affects the dermatome of one or more branches of the trigeminal nerve. Hypoplasia of the underlying bone also may occur. Osseous hypoplasia is more common when the condition begins during the first decade. Occasionally, bilateral facial atrophy may occur, or the condition may affect one side of the entire body. Females are affected more often than males.
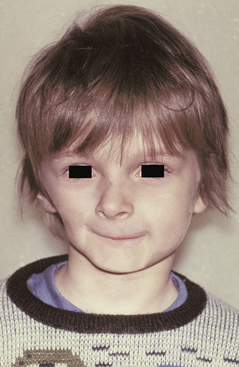
The overlying skin often exhibits dark pigmentation. Some patients have a sharp line of demarcation, resembling a large linear scar, between normal and abnormal skin near the midline of the forehead, known as linear scleroderma “en coup de sabre” (i.e., “strike of the sword”). Ocular involvement is common, and the most frequent manifestation is enophthalmos because of loss of periorbital fat. Local alopecia may occur. Occasionally, trigeminal neuralgia, facial paresthe-sia, migraine, or epilepsy may develop. MRI studies may reveal a variety of central nervous system abnormalities.
The mouth and nose are deviated toward the affected side. Atrophy of the upper lip may expose the maxillary teeth. Unilateral atrophy of the tongue also can occur. Unilateral posterior open bite often develops as a result of mandibular hypoplasia and delayed eruption of the teeth. The teeth on the affected side may exhibit deficient root development or root resorption.
HISTOPATHOLOGIC FEATURES: Microscopic examination of the affected skin reveals atrophy of the epidermis and a variable perivascular infiltrate of lymphocytes and monocytes. In cases showing clinical features of linear scleroderma, dermal fibrosis can be seen. Degenerative changes in the vascular endothelium can be identified with electron microscopy.
SEGMENTAL ODONTOMAXILLARY DYSPLASIA (HEMIMAXILLOFACIAL DYSPLASIA)
Segmental odontomaxillary dysplasia is a recently recognized developmental disorder that affects the jaw and (sometimes) the overlying facial tissues. The cause is unknown. Clinically, it is frequently mistaken for craniofacial fibrous dysplasia or hemifacial hyperplasia, but it represents a distinct and separate entity.
CLINICAL AND RADIOGRAPHIC FEATURES: Segmental odontomaxillary dysplasia is usually discovered during childhood and is characterized by painless, unilateral enlargement of the maxillary bone, along with fibrous hyperplasia of the overlying gingival soft tissues (Fig. 1-78). One or both developing maxillary premolars frequently are missing, and the primary teeth in the affected area may be hypoplastic or show enamel defects. Radiographic examination reveals thickened trabeculae that often are vertically oriented, which results in a relatively radiopaque, granular appearance. The maxillary sinus may be smaller on the affected side. Several cases have been associated with hypertrichosis or rough erythema of the overlying facial skin. One patient was described with a Becker’s nevus (hypertrichosis and hyperpigmentation) of the ipsilateral face and neck.
Fig. 1-78 Segmental odontomaxillary dysplasia. A, Unilateral enlargement of the maxilla and overlying gingival soft tissues. B, Periapical radiograph showing coarse trabecular pattern with absence of the first premolar. C, Panoramic radiograph showing irregular bone pattern of the left maxilla expanding into the maxillary sinus.
HISTOPATHOLOGIC FEATURES: The gingival soft tissues may show nonspecific fibrosis. The affected maxillary bone consists of irregular trabeculae with a woven appearance. This bone shows numerous resting and reversal lines, but it lacks significant osteoblastic and osteoclastic activity. Deciduous teeth in the involved area may exhibit irregular dentinal tubules, a focally deficient odontoblastic layer, and external resorption.
TREATMENT AND PROGNOSIS: Because segmental odontomaxillary dysplasia has been recognized only recently as a distinct entity, not much is known about its natural evolution. Once diagnosed, the condition seems to remain stable and may not require surgical intervention. However, orthodontic therapy and orthognathic surgery may be considered in some cases.
CROUZON SYNDROME (CRANIOFACIAL DYSOSTOSIS)
Crouzon syndrome is one of a rare group of syndromes characterized by craniosynostosis, or premature closing of the cranial sutures. It is believed to be caused by one of a variety of mutations of the fibroblast growth factor receptor 2 (FGFR2) gene on chromosome 10q26. The condition occurs in about 1 of every 65,000 births and is inherited as an autosomal dominant trait. A significant number of cases, however, represent new mutations, often apparently related to increased paternal age.
CLINICAL AND RADIOGRAPHIC FEATURES: Crouzon syndrome exhibits a wide variability in expression. The premature sutural closing leads to cranial malformations, such as brachycephaly (short head), scaphocephaly (boat-shaped head), or trigonocephaly (triangle-shaped head). The most severely affected patients can demonstrate a “cloverleaf” skull (kleeblatt schädel deformity). The orbits are shallow, resulting in characteristic ocular proptosis (Fig. 1-79). Visual impairment or total blindness and a hearing deficit may occur. Some patients report headaches, attributable to increased intracranial pressure. Marked mental deficiency is rarely seen. Skull radiographs typically show increased digital markings (i.e., “beaten-metal” pattern).
Fig. 1-79 Crouzon syndrome. Ocular proptosis and midface hypoplasia. (Courtesy of Dr. Robert Gorlin.)
The maxilla is underdeveloped, resulting in midface hypoplasia. Often the maxillary teeth are crowded, and occlusal disharmony usually occurs. Cleft lip and cleft palate are rare, but lateral palatal swellings may produce a midline maxillary pseudocleft.
TREATMENT AND PROGNOSIS: The clinical defects of Crouzon syndrome can be treated surgically, but multiple procedures may be necessary. Early craniectomy often is needed to alleviate the raised intracranial pressure. Frontoorbital advancement can be performed to correct the ocular defects, with midfacial advancement used to correct the maxillary hypoplasia.
APERT SYNDROME (ACROCEPHALOSYNDACTYLY)
Like Crouzon syndrome, Apert syndrome is a rare condition that is characterized by craniosynostosis. It occurs in about 1 of every 65,000 births and is caused by one of two point mutations in the fibroblast growth factor receptor 2 (FGFR2) gene, which is located on chromosome 10q26. Although it is inherited as an autosomal dominant trait, most cases represent sporadic new mutations, which are thought to be exclusively of paternal origin and often associated with increased paternal age.
CLINICAL AND RADIOGRAPHIC FEATURES: Craniosynostosis typically produces acrobrachycephaly (tower skull); severe cases may demonstrate the kleeblattschädel deformity (cloverleaf skull). The occiput is flattened, and a tall appearance to the forehead is noted. Ocular proptosis is a characteristic finding, along with hypertelorism and downward-slanting lateral palpebral fissures (Fig. 1-80). Visual loss can result from the following:
Skull films may demonstrate digital impressions similar to those of Crouzon syndrome (Fig. 1-81).
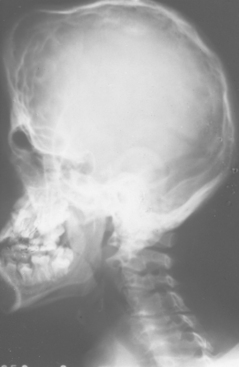
Fig. 1-81 Apert syndrome. Radiograph showing “tower skull,” midface hypoplasia, and digital markings. Similar digital impressions are apparent in people with Crouzon syndrome. (Courtesy of Dr. Robert Gorlin.)
The middle third of the face is significantly retruded and hypoplastic, resulting in a relative mandibular prognathism. The reduced size of the nasopharynx and narrowing of the posterior choanae can lead to respiratory distress in the young child. To compensate for this, most infants become mouth breathers, contributing to an “open-mouth” appearance. Sleep apnea may develop. Middle-ear infections are common, as is conductive hearing loss.
Characteristic limb defects help distinguish Apert syndrome from other craniosynostosis syndromes. Syndactyly of the second, third, and fourth digits of the hands and feet always is observed (Fig. 1-82). Associated synonychia also may occur. The first and fifth digits may be separate or joined to the middle digits. Synostosis of adjacent phalanges may be observed on radiographs. The average height of affected patients is below that of the general population.
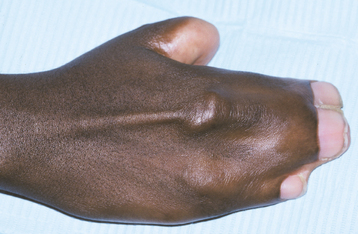
Mental retardation is common in a large proportion of patients with Apert syndrome. An unusual acnelike eruption develops in most of the patients and involves the forearms.
Specific oral manifestations include a trapezoid-shaped appearance to the lips when they are relaxed, resulting from the midface hypoplasia and mouth breathing. Three fourths of all patients exhibit either a cleft of the soft palate or a bifid uvula. The maxillary hypoplasia leads to a V-shaped arch and crowding of the teeth. Class III malocclusion typically occurs and may be associated with anterior open bite plus anterior and posterior crossbite. Swellings are observed along the lateral hard palate from the accumulation of glycosaminoglycans, especially hyaluronic acid (Fig. 1-83). These swellings often enlarge with age to produce a pseudocleft of the hard palate. Gingival thickening may be associated with delayed eruption of the teeth. Shovel-shaped incisors have been reported in one third of patients.
TREATMENT AND PROGNOSIS: The cosmetic and functional defects of Apert syndrome can be treated by an interdisciplinary approach using multiple surgical procedures. Craniectomy often is performed during the first year of life to treat the craniosynostosis. Frontofacial advancement and midface advancement can be done later to correct the proptosis and midface hypoplasia. Coordinated orthodontic therapy often is necessary to bring unerupted teeth into place and to improve occlusion. Surgery also can be used to separate the fused fingers.
MANDIBULOFACIAL DYSOSTOSIS (TREACHER COLLINS SYNDROME; FRANCESCHETTI-ZWAHLEN-KLEIN SYNDROME)
Mandibulofacial dysostosis is a rare syndrome that is characterized primarily by defects of structures derived from the first and second branchial arches. It is inherited as an autosomal dominant trait and occurs in around 1 of every 25,000 to 50,000 live births. The condition has variable expressivity, and the severity of the clinical features often tends to be greater in subsequent generations of the same family. Approximately 60% of cases represent new mutations, and these often are associated with increased paternal age. The gene for mandibulofacial dysostosis (treacle or TCOF1) has been mapped to chromosome 5q32-q33.1.
CLINICAL AND RADIOGRAPHIC FEATURES: Individuals with mandibulofacial dysostosis exhibit a characteristic facies (Fig. 1-84). The zygomas are hypoplastic, resulting in a narrow face with depressed cheeks and downward-slanting palpebral fissures. In 75% of patients, a coloboma, or notch, occurs on the outer portion of the lower eyelid. Approximately half of the patients have no eyelashes medial to the coloboma. Often the sideburns show a tongue-shaped extension toward the cheek.
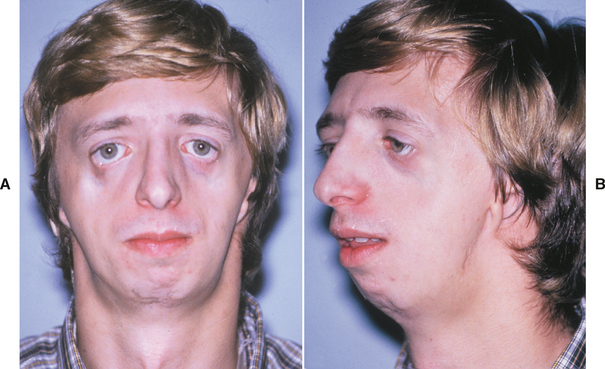
Fig. 1-84 Mandibulofacial dysostosis. Patient exhibits a hypoplastic mandible, downward-slanting palpebral fissures, and ear deformities. (Courtesy of Dr. Tom Brock.)
The ears may demonstrate a number of anomalies. The pinnae often are deformed or misplaced, and extra ear tags may be seen. Ossicle defects or absence of the external auditory canal can cause conductive hearing loss.
The mandible is underdeveloped, resulting in a markedly retruded chin. Radiographs often demonstrate hypoplasia of the condylar and coronoid processes, with prominent antegonial notching. The mouth is downturned, and about 15% of patients have lateral facial clefting (see page 2) that produces macrostomia. Cleft palate is seen in about one third of cases. The parotid glands may be hypoplastic or may be totally absent (see page 453).
A number of infants may experience respiratory and feeding difficulties because of hypoplasia of the nasopharynx, oropharynx, and hypopharynx. Choanal atresia is a common finding, and the larynx and trachea are often narrow. Combined with the mandibular hypoplasia and resultant improper tongue position, these defects can lead to the infant’s death from respiratory complications.
TREATMENT AND PROGNOSIS: Patients with mild forms of mandibulofacial dysostosis may not require treatment. In more severe cases the clinical appearance can be improved with cosmetic surgery. Because of the extent of facial reconstruction required, multiple surgical procedures are usually necessary. Individual operations may be needed for the eyes, zygomas, jaws, ears, and nose. Combined orthodontic therapy is needed along with the orthognathic surgery.
BIBLIOGRAPHY
Avery, JK, Chiego, DJ, Jr. Development of the face and palate. In Essentials of oral histology and embryology: a clinical approach, ed 3, St Louis: Mosby; 2006:51–61.
Berkovitz, BK, Holland, GR, Moxham, BJ. Development of the face and development of the palate. In Oral anatomy, histology and embryology, ed 3, St Louis: Mosby; 2002:269–283.
Carinci, F, Pezzetti, F, Scapoli, L, et al. Recent developments in orofacial cleft genetics. J Craniofac Surg. 2003;14:130–143.
Derijcke, A, Eerens, A, Carels, C. The incidence of oral clefts: a review. Br J Oral Maxillofac Surg. 1996;34:488–494.
Eppley, BL, van Aalst, JA, Robey, A, et al. The spectrum of orofacial clefting. Plast Reconstr Surg. 2005;115:101e–114e.
Evans, CA. Orthodontic treatment for patients with clefts. Clin Plast Surg. 2004;31:271–290.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Orofacial clefting syndromes: general aspects. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:850–876.
Gosain, AK, Conley, SF, Marks, S, et al. Submucous cleft palate: diagnostic methods and outcomes of surgical treatment. Plast Reconstr Surg. 1996;97:1497–1509.
Harada, K, Sato, M, Omura, K. Long-term maxillomandibular skeletal and dental changes in children with cleft lip and palate after maxillary distraction. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:292–299.
Krapels, IP, Vermeij-Keers, C, Müller, M, et al. Nutrition and genes in the development of orofacial clefting. Nutr Rev. 2006;64:280–288.
Marazita, ML, Mooney, MP. Current concepts in the embryology and genetics of cleft lip and cleft palate. Clin Plast Surg. 2004;31:125–140.
Millard, DR, Jr., Latham, RA. Improved primary surgical and dental treatment of clefts. Plast Reconstr Surg. 1990;86:856–871.
Murray, JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256.
Rintala, A, Leisti, J, Liesmaa, M, et al. Oblique facial clefts. Scand J Plast Reconstr Surg. 1980;14:291–297.
Shprintzen, RJ. Pierre Robin, micrognathia, and airway obstruction: the dependency of treatment on accurate diagnosis. Int Anesthesiol Clin. 1988;26:64–71.
St-Hilaire, H, Buchbinder, D. Maxillofacial pathology and management of Pierre Robin sequence. Otolaryngol Clin North Am. 2000;33:1241–1256.
Stoll, C, Alembik, Y, Dott, B, et al. Associated malformations in cases with oral clefts. Cleft Palate Craniofac J. 2000;37:41–47.
Ten Cate, AR, Nanci, A. Embryology of the head, face, and oral cavity. In: Nanci A, ed. Ten Cate’s oral histology: development, structure, and function. ed 6. St Louis: Mosby; 2003:30–53.
Thornton, JB, Nimer, S, Howard, PS. The incidence, classification, etiology, and embryology of oral clefts. Semin Orthod. 1996;2:162–168.
van den Elzen, AP, Semmekrot, BA, Bongers, EM, et al. Diagnosis and treatment of the Pierre Robin sequence: results of a retrospective clinical study and review of the literature. Eur J Pediatr. 2001;160:47–53.
Vanderas, AP. Incidence of cleft lip, cleft palate, and cleft lip and palate among races: a review. Cleft Palate J. 1987;24:216–225.
Weinberg, SM, Neiswanger, K, Martin, RA, et al. The Pittsburgh oral-facial cleft study: expanding the cleft phenotype. Background and justification. Cleft Palate Craniofac J. 2006;43:7–20.
Baker, BR. Pits of the lip commissures in caucasoid males. Oral Surg Oral Med Oral Pathol. 1966;21:56–60.
Everett, FG, Wescott, WB. Commissural lip pits. Oral Surg Oral Med Oral Pathol. 1961;14:202–209.
Gorsky, M, Buchner, A, Cohen, C. Commissural lip pits in Israeli Jews of different ethnic origin. Community Dent Oral Epidemiol. 1985;13:195–196.
Burdick, AB, Bixler, D, Puckett, CL. Genetic analysis in families with van der Woude syndrome. J Craniofac Genet Dev Biol. 1985;5:181–208.
Cervenka, J, Gorlin, RJ, Anderson, VE. The syndrome of pits of the lower lip and cleft lip and/or palate: genetic considerations. Am J Hum Genet. 1967;19:416–432.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Popliteal pterygium syndrome (facio-genito-popliteal syndrome). In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:775–778.
Kondo, S, Schutte, BC, Richardson, RJ, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289.
Matsumoto, N, Niikawa, N. Kabuki make-up syndrome: a review. Am J Med Genet C Semin Med Genet. 2003;117:57–65.
Onofre, MA, Brosco, HB, Brosco, JU, et al. Congenital fistulae of the lower lip in van der Woude syndrome: a histomorphological study. Cleft Palate Craniofac J. 1999;36:79–85.
Onofre, MA, Brosco, HB, Taga, R. Relationship between lower-lip fistulae and cleft lip and/or palate in van der Woude syndrome. Cleft Palate Craniofac J. 1997;34:261–265.
Rizos, M, Spyropoulos, MN. Van der Woude syndrome: a review. Cardinal signs, epidemiology, associated features, differential diagnosis, expressivity, genetic counseling and treatment. Eur J Orthod. 2004;26:17–24.
Schutte, BC, Basart, AM, Watanabe, Y, et al. Microdeletions at chromosome bands 1q32-q41 as a cause of van der Woude syndrome. Am J Med Genet. 1999;84:145–150.
Shotelersuk, V, Punyashthiti, R, Srivuthana, S, et al. Kabuki syndrome: report of six Thai children and further phenotypic and genetic delineation. Am J Med Genet. 2002;110:384–390.
Barnett, ML, Bosshardt, LL, Morgan, AF. Double lip and double lip with blepharochalasis (Ascher’s syndrome). Oral Surg Oral Med Oral Pathol. 1972;34:727–733.
Eski, M, Nisanci, M, Atkas, A, et al. Congenital double lip: review of 5 cases. Br J Oral Maxillofac Surg. 2007;45:68–70.
Gomez-Duaso, AJ, Seoane, J, Vazquez-Garcia, J, et al. Ascher syndrome: report of two cases. J Oral Maxillofac Surg. 1997;55:88–90.
Kenny, KF, Hreha, JP, Dent, CD. Bilateral redundant mucosal tissue of the upper lip. J Am Dent Assoc. 1990;120:193–194.
Daley, TD. Pathology of intraoral sebaceous glands. J Oral Pathol Med. 1993;22:241–245.
Fordyce, JA. A peculiar affection of the mucous membrane of the lips and oral cavity. J Cutan Genito-Urin Dis. 1896;14:413–419.
Halperin, V, Kolas, S, Jefferis, KR, et al. The occurrence of Fordyce spots, benign migratory glossitis, median rhomboid glossitis, and fissured tongue in 2,478 dental patients. Oral Surg Oral Med Oral Pathol. 1953;6:1072–1077.
Miles, AEW. Sebaceous glands in the lip and cheek mucosa of man. Br Dent J. 1958;105:235–248.
Sewerin, I. The sebaceous glands in the vermilion border of the lips and in the oral mucosa of man. Acta Odontol Scand. 1975;33(suppl 68):13–226.
Sewerin, I, Prætorius, F. Keratin-filled pseudocysts of ducts of sebaceous glands in the vermilion border of the lip. J Oral Pathol. 1974;3:279–283.
Archard, HO, Carlson, KP, Stanley, HR. Leukoedema of the human oral mucosa. Oral Surg Oral Med Oral Pathol. 1968;25:717–728.
Axéll, T, Henricsson, V. Leukoedema—an epidemiologic study with special reference to the influence of tobacco habits. Community Dent Oral Epidemiol. 1981;9:142–146.
Durocher, RT, Thalman, R, Fiore-Donno, G. Leukoedema of the oral mucosa. J Am Dent Assoc. 1972;85:1105–1109.
Martin, JL. Leukoedema: an epidemiological study in white and African Americans. J Tenn Dent Assoc. 1997;77:18–21.
Martin, JL, Crump, EP. Leukoedema of the buccal mucosa in Negro children and youth. Oral Surg Oral Med Oral Pathol. 1972;34:49–58.
Sandstead, HR, Lowe, JW. Leukoedema and keratosis in relation to leukoplakia of the buccal mucosa in man. J Natl Cancer Inst. 1953;14:423–437.
van Wyk, CW, Ambrosio, SC. Leukoedema: ultrastructural and histochemical observations. J Oral Pathol. 1983;12:319–329.
Dunham, ME, Austin, TL. Congenital aglossia and situs inversus. Int J Pediatr Otorhinolaryngol. 1990;19:163–168.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Oromandibular-limb hypogenesis syndromes. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:822–826.
Hall, BD. Aglossla-adactylia. Birth Defects Orig Artic Ser. 1971;7:233–236.
Shah, RM. Palatomandibular and maxillo-mandibular fusion, partial aglossia and cleft palate in a human embryo: report of a case. Teratology. 1977;15:261–272.
Yasuda, Y, Kitai, N, Fujii, Y, et al. Report of a patient with hypoglossia-hypodactylia syndrome and a review of the literature. Cleft Palate Craniofac J. 2003;40:196–202.
Cohen, MM, Jr. Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatr Dev Pathol. 2005;8:287–304.
Engström, W, Lindham, S, Schofield, P. Wiedemann-Beckwith syndrome. Eur J Pediatr. 1988;147:450–457.
Maturo, SC, Mair, EA. Submucosal minimally invasive lingual excision: an effective, novel surgery for pediatric tongue base reduction. Ann Otol Rhinol Laryngol. 2006;115:624–630.
Morgan, WE, Friedman, EM, Duncan, NO, et al. Surgical management of macroglossia in children. Arch Otolaryngol Head Neck Surg. 1996;122:326–329.
Myer, CM, III., Hotaling, AJ, Reilly, JS. The diagnosis and treatment of macroglossia in children. Ear Nose Throat J. 1986;65:444–448.
Rimell, FL, Shapiro, AM, Shoemaker, DL, et al. Head and neck manifestations of Beckwith-Wiedemann syndrome. Otolaryngol Head Neck Surg. 1995;113:262–265.
Siddiqui, A, Pensler, JM. The efficacy of tongue resection in treatment of symptomatic macroglossia in the child. Ann Plast Surg. 1990;25:14–17.
Vogel, JE, Mulliken, JB, Kaban, LB. Macroglossia: a review of the condition and a new classification. Plast Reconstr Surg. 1986;78:715–723.
Wang, J, Goodger, NM, Pogrel, MA. The role of tongue reduction. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:269–273.
Weksberg, R, Shuman, C, Smith, AC. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2005;137:12–23.
Wolford, LM, Cottrell, DA. Diagnosis of macroglossia and indications for reduction glossectomy. Am J Orthod Dentofac Orthop. 1996;110:170–177.
Dollberg, S, Botzer, E, Grunis, E, et al. Immediate nipple pain relief after frenotomy in breast-fed infants with ankyloglossia: a randomized, prospective study. J Pediatr Surg. 2006;41:1598–1600.
Ewart, NP. A lingual mucogingival problem associated with ankyloglossia: a case report. N Z Dent J. 1990;86:16–17.
Flinck, A, Paludan, A, Matsson, L, et al. Oral findings in a group of newborn Swedish children. Int J Paediatr Dent. 1994;4:67–73.
Hall, DMB, Renfrew, MJ. Tongue tie. Arch Dis Child. 2005;90:1211–1215.
Lalakea, ML, Messner, AH. Ankyloglossia: does it matter? Pediatr Clin N Am. 2003;50:381–397.
Lalakea, ML, Messner, AH. Ankyloglossia: the adolescent and adult perspective. Otolaryngol Head Neck Surg. 2003;128:746–752.
Mukai, S, Mukai, C, Asaoka, K. Ankyloglossia with deviation of the epiglottis and larynx. Ann Otol Rhinol Laryngol. 1991;100:3–20.
Mukai, S, Mukai, C, Asaoka, K. Congenital ankyloglossia with deviation of the epiglottis and larynx: symptoms and respiratory function in adults. Ann Otol Rhinol Laryngol. 1993;102:620–624.
Batsakis, JG, El-Naggar, AK, Luna, MA. Thyroid gland ectopias. Ann Otol Rhinol Laryngol. 1996;105:996–1000.
Baughman, RA. Lingual thyroid and lingual thyroglossal tract remnants: a clinical and histopathologic study with review of the literature. Oral Surg Oral Med Oral Pathol. 1972;34:781–799.
Chanin, LR, Greenberg, LM. Pediatric upper airway obstruction due to ectopic thyroid: classification and case reports. Laryngoscope. 1988;98:422–427.
Diaz-Arias, AA, Bickel, JT, Loy, TS, et al. Follicular carcinoma with clear cell change arising in lingual thyroid. Oral Surg Oral Med Oral Pathol. 1992;74:206–211.
Kalan, A, Tariq, M. Lingual thyroid gland: clinical evaluation and comprehensive management. Ear Nose Throat J. 1999;78:340–341. [345-349].
Massine, RE, Durning, SJ, Koroscil, TM. Lingual thyroid carcinoma: a case report and review of the literature. Thyroid. 2001;11:1191–1196.
Montgomery, ML. Lingual thyroid: a comprehensive review. West J Surg Obstet Gynecol. 1935;43:661–669. [44:54-62, 122-128, 189-195, 237-247, 303-309, 373-379, 442-446, 1936.].
Prasad, KC, Bhat, V. Surgical management of lingual thyroid: a report of four cases. J Oral Maxillofac Surg. 2000;58:223–227.
Williams, JD, Sclafani, AP, Slupchinskij, O, et al. Evaluation and management of the lingual thyroid gland. Ann Otol Rhinol Laryngol. 1996;105:312–316.
Bouquot, JE, Gundlach, KKH. Odd tongues: the prevalence of common tongue lesions in 23,616 white Americans over 35 years of age. Quintessence Int. 1986;17:719–730.
Eidelman, E, Chosack, A, Cohen, T. Scrotal tongue and geographic tongue: polygenic and associated traits. Oral Surg Oral Med Oral Pathol. 1976;42:591–596.
Halperin, V, Kolas, S, Jefferis, KR. The occurrence of Fordyce spots, benign migratory glossitis, median rhomboid glossitis, and fissured tongue in 2,478 dental patients. Oral Surg Oral Med Oral Pathol. 1953;6:1072–1077.
Jainkittivong, A, Aneksuk, V, Langlais, RP. Oral mucosal conditions in elderly dental patients. Oral Dis. 2002;8:218–223.
Kullaa-Mikkonen, A. Familial study of fissured tongue. Scand J Dent Res. 1988;96:366–375.
Kullaa-Mikkonen, A, Sorvari, T. Lingua fissurata: a clinical, stereomicroscopic and histopathological study. Int J Oral Maxillofac Surg. 1986;15:525–533.
Yarom, N, Cantony, U, Gorsky, M. Prevalence of fissured tongue, geographic tongue and median rhomboid glossitis among Israeli adults of different ethnic origins. Dermatology. 2004;209:88–94.
Bouquot, JE, Gundlach, KKH. Odd tongues: the prevalence of common tongue lesions in 23,616 white Americans over 35 years of age. Quintessence Int. 1986;17:719–730.
Celis, A, Little, JW. Clinical study of hairy tongue in hospital patients. J Oral Med. 1966;21:139–145.
Danser, MM, Mantilla Gómez, S, Van der Weijden, GA. Tongue coating and tongue brushing: a literature review. Int J Dent Hygiene. 2003;1:151–158.
Farman, AG. Hairy tongue (lingua villosa). J Oral Med. 1977;32:85–91.
Gómez, SM, Danser, MM, Sipos, PM, et al. Tongue coating and salivary bacterial counts in healthy/gingivitis subjects and periodontitis patients. J Clin Periodontol. 2001;28:970–978.
Manabe, M, Lim, HW, Winzer, M, et al. Architectural organization of filiform papillae in normal and black hairy tongue epithelium. Arch Dermatol. 1999;135:177–181.
Sarti, GM, Haddy, RI, Schaffer, D, et al. Black hairy tongue. Am Fam Physician. 1990;41:1751–1755.
Standish, SM, Moorman, WC. Treatment of hairy tongue with podophyllin resin. J Am Dent Assoc. 1964;68:535–540.
Ettinger, RL, Manderson, RD. A clinical study of sublingual varices. Oral Surg Oral Med Oral Pathol. 1974;38:540–545.
Jainkittivong, A, Aneksuk, V, Langlais, RP. Oral mucosal conditions in elderly dental patients. Oral Dis. 2002;8:218–223.
Kleinman, HZ. Lingual varicosities. Oral Surg Oral Med Oral Pathol. 1967;23:546–548.
Southam, JC, Ettinger, RL. A histologic study of sublingual varices. Oral Surg Oral Med Oral Pathol. 1974;38:879–886.
Weathers, DR, Fine, RM. Thrombosed varix of oral cavity. Arch Dermatol. 1971;104:427–430.
Jaspers, MT. Oral caliber-persistent artery: unusual presentations of unusual lesions. Oral Surg Oral Med Oral Pathol. 1992;74:631–633.
Lovas, JG, Goodday, RH. Clinical diagnosis of caliber-persistent labial artery of the lower lip. Oral Surg Oral Med Oral Pathol. 1993;76:480–483.
Lovas, JGL, Rodu, B, Hammond, HL, et al. Caliber-persistent labial artery: a common vascular anomaly. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:308–312.
Miko, T, Adler, P, Endes, P. Simulated cancer of the lower lip attributed to a “caliber persistent” artery. J Oral Pathol. 1980;9:137–144.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Fistulas of lateral soft palate and associated anomalies. In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:1160.
Miller, AS, Brookreson, KR, Brody, BA. Lateral soft-palate fistula: report of a case. Arch Otolaryngol. 1970;91:200.
Gerbino, G, Bianchi, SD, Bernardi, M, et al. Hyperplasia of the mandibular coronoid process: long-term follow-up after coronoidotomy. J Craniomaxillofac Surg. 1997;25:169–173.
Giacomuzzi, D. Bilateral enlargement of the mandibular coronoid processes: review of the literature and report of case. J Oral Maxillofac Surg. 1986;44:728–731.
Hall, RE, Orbach, S, Landesberg, R. Bilateral hyperplasia of the mandibular coronoid processes: a report of two cases. Oral Surg Oral Med Oral Pathol. 1989;67:141–145.
Izumi, M, Isobe, M, Toyama, M, et al. Computed tomographic features of bilateral coronoid process hyperplasia with special emphasis on patients without interference between the process and the zygomatic bone. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:93–100.
McLoughlin, PM, Hopper, C, Bowley, NB. Hyperplasia of the mandibular coronoid process: an analysis of 31 cases and a review of the literature. J Oral Maxillofac Surg. 1995;53:250–255.
Tucker, MR, Guilford, WB, Howard, CW. Coronoid process hyperplasia causing restricted opening and facial asymmetry. Oral Surg Oral Med Oral Pathol. 1984;58:130–132.
Bruce, RA, Hayward, JR. Condylar hyperplasia and mandibular asymmetry: a review. J Oral Surg. 1968;26:281–290.
Eslami, B, Behnia, H, Javadi, H, et al. Histopathologic comparison of normal and hyperplastic condyles. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:711–717.
Gray, RJM, Horner, K, Testa, HJ, et al. Condylar hyperplasia: correlation of histological and scintigraphic features. Dentomaxillofac Radiol. 1994;23:103–107.
Iannetti, G, Cascone, P, Belli, E, et al. Condylar hyperplasia: cephalometric study, treatment planning, and surgical correction (our experience). Oral Surg Oral Med Oral Pathol. 1989;68:673–681.
Motamedi, MHK. Treatment of condylar hyperplasia of the mandible using unilateral ramus osteotomies. J Oral Maxillofac Surg. 1996;54:1161–1169.
Obwegeser, HL, Makek, MS. Hemimandibular hyperplasia-hemimandibular elongation. J Maxillofac Surg. 1986;14:183–208.
Slootweg, PJ, Müller, H. Condylar hyperplasia: a clinico-pathological analysis of 22 cases. J Maxillofac Surg. 1986;14:209–214.
Arun, T, Kayhan, F, Kiziltan, M. Treatment of condylar hypoplasia with distraction osteogenesis: a case report. Angle Orthod. 2002;72:371–376.
Berger, SS, Stewart, RE. Mandibular hypoplasia secondary to perinatal trauma: report of case. J Oral Surg. 1977;35:578–582.
Jerrell, RG, Fuselier, B, Mahan, P. Acquired condylar hypoplasia: report of case. ASDC J Dent Child. 1991;58:147–153.
Sapp, JP, Cherrick, HM. Pathological aspects of developmental, inflammatory, and neoplastic disease. In: Sarnat BG, Laskin DM, eds. The temporomandibular joint: a biological basis for clinical practice. ed 4. Philadelphia: WB Saunders; 1991:150–151.
Svensson, B, Larsson, Å, Adell, R. The mandibular condyle in juvenile chronic arthritis patients with mandibular hypoplasia. A clinical and histological study. Int J Oral Maxillofac Surg. 2001;30:306–312.
Antoniades, K, Hadjipetrou, L, Antoniades, V, et al. Bilateral bifid mandibular condyle. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:535–538.
Cowan, DF, Ferguson, MM. Bifid mandibular condyle. Dentomaxillofac Radiol. 1997;26:70–73.
Gundlach, KKH, Fuhrmann, A, Beckmann-Van der Ven, G. The double-headed mandibular condyle. Oral Surg Oral Med Oral Pathol. 1987;64:249–253.
Loh, FC, Yeo, JF. Bifid mandibular condyle. Oral Surg Oral Med Oral Pathol. 1990;69:24–27.
Stefanou, EP, Fanourakis, IG, Vlastos, K, et al. Bilateral bifid mandibular condyles. Report of four cases. Dentomaxillofac Radiol. 1998;27:186–188.
Szentpétery, A, Kocsis, G, Marcsik, A. The problem of the bifid mandibular condyle. J Oral Maxillofac Surg. 1990;48:1254–1257.
Antoniades, DZ, Belazi, M, Papanayiotou, P. Concurrence of torus palatinus with palatal and buccal exostoses. Case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:552–557.
Blakemore, JR, Eller, DJ, Tomaro, AJ. Maxillary exostoses: surgical management of an unusual case. Oral Surg Oral Med Oral Pathol. 1975;40:200–204.
Bouquot, JE, Gundlach, KKH. Oral exophytic lesions in 23,616 white Americans over 35 years of age. Oral Surg Oral Med Oral Pathol. 1986;62:284–291.
Frazier, KB, Baker, PS, Abdelsayed, R, et al. A case report of subpontic osseous hyperplasia in the maxillary arch. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:73–76.
Hegtvedt, AK, Terry, BC, Burkes, EJ, et al. Skin graft vestibuloplasty exostosis: a report of two cases. Oral Surg Oral Med Oral Pathol. 1990;69:149–152.
Jainkittivong, A, Langlais, RP. Buccal and palatal exostoses: prevalence and concurrence with tori. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:48–53.
Morton, TH, Jr., Natkin, E. Hyperostosis and fixed partial denture pontics: report of 16 patients and review of literature. J Prosthet Dent. 1990;64:539–547.
Pack, ARC, Gaudie, WM, Jennings, AM. Bony exostosis as a sequela to free gingival grafting: two case reports. J Periodontol. 1991;62:269–271.
Sonnier, KE, Horning, GM, Cohen, ME. Palatal tubercles, palatal tori, and mandibular tori: prevalence and anatomical features in a U.S. population. J Periodontol. 1999;70:329–336.
Wasson, DJ, Rapley, JW, Cronin, RJ. Subpontic osseous hyperplasia: a literature review. J Prosthet Dent. 1991;66:638–641.
Torus Palatinus and Torus Mandibularis
Al Quran, FA, Al-Dwairi, ZN. Torus palatinus and torus mandibularis in edentulous patients. J Contemp Dent Pract. 2006;7:112–119.
Eggen, S. Torus mandibularis: an estimation of the degree of genetic determination. Acta Odontol Scand. 1989;47:409–415.
Eggen, S, Natvig, B. Relationship between torus mandibularis and number of present teeth. Scand J Dent Res. 1986;94:233–240.
Eggen, S, Natvig, B. Variation in torus mandibularis prevalence in Norway: a statistical analysis using logistic regression. Community Dent Oral Epidemiol. 1991;19:32–35.
Gorsky, M, Bukai, A, Shohat, M. Genetic influence on the prevalence of torus palatinus. Am J Med Genet. 1998;75:138–140.
Haugen, LK. Palatine and mandibular tori: a morphologic study in the current Norwegian population. Acta Odontol Scand. 1992;50:65–77.
Kerdpon, D, Sirirungrojying, S. A clinical study of oral tori in southern Thailand: prevalence and the relation to parafunctional activity. Eur J Oral Sci. 1999;107:9–13.
Kolas, S, Halperin, V, Jefferis, K, et al. The occurrence of torus palatinus and torus mandibularis in 2,478 dental patients. Oral Surg Oral Med Oral Pathol. 1953;6:1134–1141.
Reichart, PA, Neuhaus, F, Sookasem, M. Prevalence of torus palatinus and torus mandibularis in Germans and Thai. Community Dent Oral Epidemiol. 1988;16:61–64.
Suzuki, M, Sakai, T. A familial study of torus palatinus and torus mandibularis. Am J Phys Anthropol. 1960;18:263–272.
Bafaqeeh, SA. Eagle syndrome: classic and carotid artery types. J Otolaryngol. 2000;29:88–94.
Blatchford, SJ, Coulthard, SW. Eagle’s syndrome: an atypical cause of dysphonia. Ear Nose Throat J. 1989;68:48–51.
Camarda, AJ, Deschamps, C, Forest, D. I. Stylohyoid chain ossification: a discussion of etiology. Oral Surg Oral Med Oral Pathol. 1989;67:508–514.
Camarda, AJ, Deschamps, C, Forest, D. II. Stylohyoid chain ossification: a discussion of etiology. Oral Surg Oral Med Oral Pathol. 1989;67:515–520.
Correll, RW, Jensen, JL, Taylor, JB, et al. Mineralization of the stylohyoid-stylomandibular ligament complex: a radiographic incidence study. Oral Surg Oral Med Oral Pathol. 1979;48:286–291.
Eagle, WW. Elongated styloid processes: report of two cases. Arch Otolaryngol. 1937;25:584–587.
Montalbetti, L, Ferrandi, D, Pergami, P, et al. Elongated styloid process and Eagle’s syndrome. Cephalalgia. 1995;15:80–93.
Rechtweg, JS, Wax, MK. Eagle’s syndrome: a review. Am J Otolaryngol. 1998;19:316–321.
Smith, RG, Cherry, JE. Traumatic Eagle’s syndrome: report of a case and review of the literature. J Oral Maxillofac Surg. 1988;46:606–609.
Apruzzese, D, Longoni, S. Stafne cyst in an anterior location. J Oral Maxillofac Surg. 1999;57:333–338.
Ariji, E, Fujiwara, N, Tabata, O, et al. Stafne’s bone cavity: classification based on outline and content determined by computed tomography. Oral Surg Oral Med Oral Pathol. 1993;76:375–380.
Barker, GR. A radiolucency of the ascending ramus of the mandible associated with invested parotid salivary gland material and analogous with a Stafne bone cavity. Br J Oral Maxillofac Surg. 1988;26:81–84.
Bouquot, JE, Gnepp, DR, Dardick, I, et al. Intraosseous salivary tissue: jawbone examples of choristomas, hamartomas, embryonic rests, and inflammatory entrapment: another histogenetic source for intraosseous adenocarcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:205–217.
Branstetter, BF, Weissman, JL, Kaplan, SB. Imaging of a Stafne bone cavity: what MR adds and why a new name is needed. AJNR Am J Neuroradiol. 1999;20:587.
Buchner, A, Carpenter, WM, Merrell, PW, et al. Anterior lingual mandibular salivary gland defect. Evaluation of twenty-four cases. Oral Surg Oral Med Oral Pathol. 1991;71:131–136.
Correll, RW, Jensen, JL, Rhyne, RR. Lingual cortical mandibular defects: a radiographic incidence study. Oral Surg Oral Med Oral Pathol. 1980;50:287–291.
de Courten, A, Küffer, R, Samson, J, et al. Anterior lingual mandibular salivary gland defect (Stafne defect) presenting as a residual cyst. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:460–464.
Miller, AS, Winnick, M. Salivary gland inclusion in the anterior mandible: report of a case with a review of the literature on aberrant salivary gland tissue and neoplasms. Oral Surg Oral Med Oral Pathol. 1971;31:790–797.
Oikarinen, VJ, Wolf, J, Julku, M. A stereosialographic study of developmental mandibular bone defects (Stafne’s idiopathic bone cavities). Int J Oral Surg. 1975;4:51–54.
Stafne, EC. Bone cavities situated near the angle of the mandible. J Am Dent Assoc. 1942;29:1969–1972.
Burke, GW, Jr., Feagans, WM, Elzay, RP, et al. Some aspects of the origin and fate of midpalatal cysts in human fetuses. J Dent Res. 1966;45:159–164.
Cataldo, E, Berkman, MD. Cysts of the oral mucosa in newborns. Am J Dis Child. 1968;116:44–48.
Donley, CL, Nelson, LP. Comparison of palatal and alveolar cysts of the newborn in premature and full term infants. Pediatr Dent. 2000;22:321–324.
Flinck, A, Paludan, A, Matsson, L, et al. Oral findings in a group of newborn Swedish children. Int J Paediatr Dent. 1994;4:67–73.
Fromm, A. Epstein’s pearls, Bohn’s nodules and inclusion-cysts of the oral cavity. J Dent Child. 1967;34:275–287.
Jorgenson, RJ, Shapiro, SD, Salinas, CF, et al. Intraoral findings and anomalies in neonates. Pediatrics. 1982;69:577–582.
Liu, MH, Huang, WH. Oral abnormalities in Taiwanese newborns. J Dent Child. 2004;71:118–120.
Monteleone, L, McLellan, MS. Epstein’s pearls (Bohn’s nodules) of the palate. J Oral Surg. 1964;22:301–304.
Moreillon, MC, Schroeder, HE. Numerical frequency of epithelial abnormalities, particularly microkeratocysts, in the developing human oral mucosa. Oral Surg Oral Med Oral Pathol. 1982;53:44–55.
Allard, RHB. Nasolabial cyst: review of the literature and report of 7 cases. Int J Oral Surg. 1982;11:351–359.
Choi, JH, Cho, JH, Kang, HJ, et al. Nasolabial cyst: a retrospective analysis of 18 cases. Ear Nose Throat J. 2002;81:94–96.
Kuriloff, DB. The nasolabial cyst—nasal hamartoma. Otolaryngol Head Neck Surg. 1987;96:268–272.
López-Ríos, F, Lassaletta-Atienza, L, Domingo-Carrasco, C, et al. Nasolabial cyst. Report of a case with extensive apocrine change. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:404–406.
Roed-Petersen, B. Nasolabial cysts: a presentation of five patients with a review of the literature. Br J Oral Surg. 1969;7:84–95.
Su, C-Y, Chien, C-Y, Hwang, C-F. A new transnasal approach to endoscopic marsupialization of the nasolabial cyst. Laryngoscope. 1999;109:1116–1118.
Vasconcelos, RF, Souza, PE, Mesquita, RA. Retrospective analysis of 15 cases of nasolabial cyst. Quintessence Int. 1999;30:629–632.
Christ, TF. The globulomaxillary cyst: an embryologic misconception. Oral Surg Oral Med Oral Pathol. 1970;30:515–526.
D’Silva, NJ, Anderson, L. Globulomaxillary cyst revisited. Oral Surg Oral Med Oral Pathol. 1993;76:182–184.
Ferenczy, K. The relationship of globulomaxillary cysts to the fusion of embryonal processes and to cleft palates. Oral Surg Oral Med Oral Pathol. 1958;11:1388–1393.
Little, JW, Jakobsen, J. Origin of the globulomaxillary cyst. J Oral Surg. 1973;31:188–195.
Steiner, DR. A lesion of endodontic origin misdiagnosed as a globulomaxillary cyst. J Endod. 1999;25:277–281.
Vedtofte, P, Holmstrup, P. Inflammatory paradental cysts in the globulomaxillary region. J Oral Pathol Med. 1989;18:125–127.
Wysocki, GP. The differential diagnosis of globulomaxillary radiolucencies. Oral Surg Oral Med Oral Pathol. 1981;51:281–286.
Wysocki, GP, Goldblatt, LI. The so-called “globulomaxillary cyst” is extinct. Oral Surg Oral Med Oral Pathol. 1993;76:185–186.
Abrams, AM, Howell, FV, Bullock, WK. Nasopalatine cysts. Oral Surg Oral Med Oral Pathol. 1963;16:306–332.
Allard, RHB, van der Kwast, WAM, van der Waal, I. Nasopalatine duct cyst: review of the literature and report of 22 cases. Int J Oral Surg. 1981;10:447–461.
Anneroth, G, Hall, G, Stuge, U. Nasopalatine duct cyst. Int J Oral Maxillofac Surg. 1986;15:572–580.
Brown, FH, Houston, GD, Lubow, RM, et al. Cyst of the incisive (palatine) papilla: report of a case. J Periodontol. 1987;58:274–275.
Chapple, IL, Ord, RA. Patent nasopalatine ducts: four case presentations and review of the literature. Oral Surg Oral Med Oral Pathol. 1990;69:554–558.
Hisatomi, M, Asaumi, J, Konouchi, H, et al. MR imaging of nasopalatine duct cysts. Eur J Radiol. 2001;39:73–76.
Swanson, KS, Kaugars, GE, Gunsolley, JC. Nasopalatine duct cyst: an analysis of 334 cases. J Oral Maxillofac Surg. 1991;49:268–271.
Takagi, R, Ohashi, Y, Suzuki, M. Squamous cell carcinoma in the maxilla probably originating from a nasopalatine duct cyst: report of case. J Oral Maxillofac Surg. 1996;54:112–115.
Vasconcelos, RF, de Aguiar, MF, Castro, WH, et al. Retrospective analysis of 31 cases of nasopalatine duct cyst. Oral Dis. 1999;5:325–328.
Courage, GR, North, AF, Hansen, LS. Median palatine cysts. Oral Surg Oral Med Oral Pathol. 1974;37:745–753.
Donnelly, JC, Koudelka, BM, Hartwell, GR. Median palatal cyst. J Endod. 1986;12:546–549.
Gingell, JC, Levy, BA, DePaola, LG. Median palatine cyst. J Oral Maxillofac Surg. 1985;43:47–51.
Gordon, NC, Swann, NP, Hansen, LS. Median palatine cyst and maxillary antral osteoma: report of an unusual case. J Oral Surg. 1980;38:361–365.
Gardner, DG. An evaluation of reported cases of median mandibular cysts. Oral Surg Oral Med Oral Pathol. 1988;65:208–213.
Soskolne, WA, Shteyer, A. Median mandibular cyst. Oral Surg Oral Med Oral Pathol. 1977;44:84–88.
White, DK, Lucas, RM, Miller, AS. Median mandibular cyst: review of the literature and report of two cases. J Oral Surg. 1975;33:372–375.
Boatman, BW, Headington, JT. Epidermoid and tricholemmal cysts. In: Demis DJ, ed. Clinical dermatology. Philadelphia: Lippincott-Raven, 1996. [chapter 4-57].
Golden, BA, Zide, MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613–1619.
Kligman, AM. The myth of the sebaceous cyst. Arch Dermatol. 1964;89:253–256.
López-Ríos, F, Rodríguez-Peralto, JL, Castaño, E, et al. Squamous cell carcinoma arising in a cutaneous epidermal cyst. Am J Dermatopathol. 1999;21:174–177.
Maize, JC, Burgdorf, WHC, Hurt, MA, et al. Follicular cysts. In: Cutaneous pathology. Philadelphia: Churchill Livingstone; 1998:540–541.
McGavran, MH, Binnington, B. Keratinous cysts of the skin. Arch Dermatol. 1966;94:499–508.
Rajayogeswaran, V, Eveson, JW. Epidermoid cyst of the buccal mucosa. Oral Surg Oral Med Oral Pathol. 1989;67:181–184.
Arcand, P, Granger, J, Brochu, P. Congenital dermoid cyst of the oral cavity with gastric choristoma. J Otolaryngol. 1988;17:219–222.
Crivelini, MM, Soubhia, AM, Biazolla, ÉR, et al. Heterotopic gastrointestinal cyst partially lined with dermoid cyst epithelium. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;91:686–688.
Edwards, PC, Lustrin, L, Valderrama, E. Dermoid cysts of the tongue: report of five cases and review of the literature. Pediatr Dev Pathol. 2003;6:531–535.
King, RC, Smith, BR, Burk, JL. Dermoid cyst in the floor of the mouth. Review of the literature and case reports. Oral Surg Oral Med Oral Pathol. 1994;78:567–576.
Lipsett, J, Sparnon, AL, Byard, RW. Embryogenesis of enterocystomas-enteric duplication cysts of the tongue. Oral Surg Oral Med Oral Pathol. 1993;75:626–630.
Meyer, I. Dermoid cysts (dermoids) of the floor of the mouth. Oral Surg Oral Med Oral Pathol. 1955;8:1149–1164.
Said-Al-Naief, N, Fantasia, JE, Sciubba, JJ, et al. Heterotopic oral gastrointestinal cyst. Report of 2 cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;88:80–86.
Shigematsu, H, Dobashi, A, Suzuki, S, et al. Delayed recurrence of teratoid cyst 17 years after enucleation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001;92:539–542.
Allard, RHB. The thyroglossal cyst. Head Neck Surg. 1982;5:134–146.
Brousseau, VJ, Solares, CA, Xu, M, et al. Thyroglossal duct cysts: presentation and management in children versus adults. Int J Pediatr Otorhinolaryngol. 2003;67:1285–1290.
Dedivitis, RA, Camargo, DL, Peixoto, GL, et al. Thyroglossal duct: a review of 55 cases. J Am Coll Surg. 2002;194:274–277.
Fernandez, JF, Ordoñez, NG, Schultz, NA, et al. Thyroglossal duct carcinoma. Surgery. 1991;110:928–935.
Katz, AD, Hachigian, M. Thyroglossal duct cysts: a thirty-year experience with emphasis on occurrence in older patients. Am J Surg. 1988;155:741–744.
Kuint, J, Horowitz, Z, Kugel, C, et al. Laryngeal obstruction caused by lingual thyroglossal duct cyst presenting at birth. Am J Perinatol. 1997;14:353–356.
Patel, SG, Escrig, M, Shaha, AR, et al. Management of well-differentiated thyroid carcinoma presenting within a thyroglossal duct cyst. J Surg Oncol. 2002;79:134–139.
Plaza, CP, López, ME, Carrasco, CE, et al. Management of well-differentiated thyroglossal remnant thyroid carcinoma: time to close the debate? Report of five new cases and proposal of a definitive algorithm for treatment. Ann Surg Oncol. 2006;13:745–752.
Schader, I, Robertson, S, Maoate, K, et al. Hereditary thyroglossal duct cysts. Pediatr Surg Int. 2005;21:593–594.
Bhaskar, SN, Bernier, JL. Histogenesis of branchial cysts: a report of 468 cases. Am J Pathol. 1959;35:407–423.
Elliott, JN, Oertel, YC. Lymphoepithelial cysts of the salivary glands. Am J Clin Pathol. 1990;93:39–43.
Foss, RD, Warnock, GR, Clark, WB, et al. Malignant cyst of the lateral aspect of the neck: branchial cleft carcinoma or metastasis? Oral Surg Oral Med Oral Pathol. 1991;71:214–217.
Goldenberg, D, Sciubba, J, Koch, WM. Cystic metastasis from head and neck squamous cell cancer: a distinct disease variant? Head Neck. 2006;28:633–638.
Kadhim, AL, Sheahan, P, Colreavy, MP, et al. Pearls and pitfalls in the management of branchial cyst. J Laryngol Otol. 2004;118:946–950.
Little, JW, Rickles, NH. The histogenesis of the branchial cyst. Am J Pathol. 1967;50:533–547.
Mandel, L, Reich, R. HIV parotid gland lymphoepithelial cysts: review and case reports. Oral Surg Oral Med Oral Pathol. 1992;74:273–278.
Regauer, S, Gogg-Kamerer, M, Braun, H, et al. Lateral neck cysts—the branchial theory revisited. APMIS. 1997;105:623–630.
Skouteris, CA, Patterson, GT, Sotereanos, GC. Benign cervical lymphoepithelial cyst: report of cases. J Oral Maxillofac Surg. 1989;47:1106–1112.
Thompson, LD, Heffner, DK. The clinical importance of cystic squamous cell carcinomas in the neck: a study of 136 cases. Cancer. 1998;82:944–956.
Bhaskar, SN. Lymphoepithelial cysts of the oral cavity: report of twenty-four cases. Oral Surg Oral Med Oral Pathol. 1966;21:120–128.
Buchner, A, Hansen, LS. Lymphoepithelial cysts of the oral cavity. Oral Surg Oral Med Oral Pathol. 1980;50:441–449.
Chaudhry, AP, Yamane, GM, Scharlock, SE, et al. A clinico-pathological study of intraoral lymphoepithelial cysts. J Oral Med. 1984;39:79–84.
Giunta, J, Cataldo, E. Lymphoepithelial cysts of the oral mucosa. Oral Surg Oral Med Oral Pathol. 1973;35:77–84.
Ballock, RT, Wiesner, GL, Myers, MT, et al. Hemihypertrophy. Concepts and controversies. J Bone Joint Surg. 1997;79:1731–1738.
Bell, RA, McTigue, DJ. Complex congenital hemihypertrophy: a case report and literature review. J Pedod. 1984;8:300–313.
Dalal, AB, Phadke, SR, Pradhan, M, et al. Hemihyperplasia syndromes. Indian J Pediatr. 2006;73:609–615.
Elliott, M, Bayly, R, Cole, T, et al. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin Genet. 1994;46:168–174.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Hemihyperplasia (hemihypertrophy). In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:405–408.
Horswell, BB, Holmes, AD, Barnett, JS, et al. Primary hemihypertrophy of the face: review and report of two cases. J Oral Maxillofac Surg. 1987;45:217–222.
Hoyme, HE, Seaver, LH, Jones, KL, et al. Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet. 1998;79:274–278.
Progressive Hemifacial Atrophy
Abele, DC, Bedingfield, RB, Chandler, FW, et al. Progressive facial hemiatrophy (Parry-Romberg syndrome) and borreliosis. J Am Acad Dermatol. 1990;22:531–533.
Blaszczyk, M, Królicki, L, Krasu, M, et al. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol. 2003;30:1997–2004.
Fayad, S, Steffensen, B. Root resorptions in a patient with hemifacial atrophy. J Endod. 1994;20:299–303.
Foster, TD. The effects of hemifacial atrophy on dental growth. Br Dent J. 1979;146:148–150.
Iñigo, F, Rojo, P, Ysunza, A. Aesthetic treatment of Romberg’s disease: experience with 35 cases. Br J Plast Surg. 1993;46:194–200.
Orozco-Covarrubias, L, Guzmán-Meza, A, Ridaura-Sanz, C, et al. Scleroderma “en coup de sabre” and progressive facial hemiatrophy. Is it possible to differentiate them? J Eur Acad Dermatol Venereol. 2002;16:361–366.
Pensler, JM, Murphy, GF, Mulliken, JB. Clinical and ultrastructural studies of Romberg’s hemifacial atrophy. Plast Reconstr Surg. 1990;85:669–674.
Roddi, R, Riggio, E, Gilbert, PM, et al. Clinical evaluation of techniques used in the surgical treatment of progressive hemifacial atrophy. J Craniomaxillofac Surg. 1994;22:23–32.
Sommer, A, Gambichler, T, Bacharach-Buhles, M, et al. Clinical and serological characteristics of progressive facial hemiatrophy: a case series of 12 patients. J Am Acad Dermatol. 2006;54:223–227.
Segmental Odontomaxillary Dysplasia
Armstrong, C, Napier, SS, Boyd, RC, et al. Histopathology of the teeth in segmental odontomaxillary dysplasia: new findings. J Oral Pathol Med. 2004;33:246–248.
Becktor, KB, Reibel, J, Vedel, B, et al. Segmental odontomaxillary dysplasia: clinical, radiological and histological aspects of four cases. Oral Dis. 2002;8:106–110.
Danforth, RA, Melrose, RJ, Abrams, AM, et al. Segmental odontomaxillary dysplasia. Report of eight cases and comparison with hemimaxillofacial dysplasia. Oral Surg Oral Med Oral Pathol. 1990;70:81–85.
Jones, AC, Ford, MJ. Simultaneous occurrence of segmental odontomaxillary dysplasia and Becker’s nevus. J Oral Maxillofac Surg. 1999;57:1251–1254.
Miles, DA, Lovas, JL, Cohen, MM, Jr. Hemimaxillofacial dysplasia: a newly recognized disorder of facial asymmetry, hypertrichosis of the facial skin, unilateral enlargement of the maxilla, and hypoplastic teeth in two patients. Oral Surg Oral Med Oral Pathol. 1987;64:445–448.
Packota, GV, Pharoah, MJ, Petrikowski, CG. Radiographic features of segmental odontomaxillary dysplasia. A study of 12 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;82:577–584.
Paticoff, K, Marion, RW, Shprintzen, RJ, et al. Hemimaxillofacial dysplasia. A report of two new cases and further delineation of the disorder. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;83:484–488.
David, DJ, Sheen, R. Surgical correction of Crouzon syndrome. Plast Reconstr Surg. 1990;85:344–354.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Crouzon syndrome (craniofacial dysostosis). In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:658–659.
Katzen, JT, McCarthy, JG. Syndromes involving craniosynostosis and midface hypoplasia. Otolaryngol Clin North Am. 2000;33:1257–1284.
Kreiborg, S. Crouzon syndrome. Scand J Plast Reconstr Surg Suppl. 1981;18:1–198.
Mulliken, JB, Steinberger, D, Kunze, S, et al. Molecular diagnosis of bilateral coronal synostosis. Plast Reconstr Surg. 1999;104:1603–1615.
Posnick, JC. The craniofacial dysostosis syndromes. Staging of reconstruction and management of secondary deformities. Clin Plast Surg. 1997;24:429–446.
Singer, SL, Walpole, I, Brogan, WF, et al. Dentofacial features of a family with Crouzon syndrome. Case reports. Aust Dent J. 1997;42:11–17.
Cohen, MM, Jr., Kreiborg, S. A clinical study of the craniofacial features in Apert syndrome. Int J Oral Maxillofac Surg. 1996;25:45–53.
Ferraro, NF. Dental, orthodontic, and oral/maxillofacial evaluation and treatment in Apert syndrome. Clin Plast Surg. 1991;18:291–307.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Apert syndrome (acrocephalosyndactyly). In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:654–658.
Ibrahimi, OA, Chiu, ES, McCarthy, JG, et al. Understanding the molecular basis of Apert syndrome. Plast Reconstr Surg. 2005;115:264–270.
Katzen, JT, McCarthy, JG. Syndromes involving craniosynostosis and midface hypoplasia. Otolaryngol Clin North Am. 2000;33:1257–1284.
Kreiborg, S, Cohen, MM, Jr. The oral manifestations of Apert syndrome. J Craniofac Genet Dev Biol. 1992;12:41–48.
Marsh, JL, Galic, M, Vannier, MW. Surgical correction of the craniofacial dysmorphology of Apert syndrome. Clin Plast Surg. 1991;18:251–275.
Mulliken, JB, Steinberger, D, Kunze, S, et al. Molecular diagnosis of bilateral coronal synostosis. Plast Reconstr Surg. 1999;104:1603–1615.
Fuente del Campo, A, Martinez Elizondo, M, Arnaud, E. Treacher Collins syndrome (mandibulofacial dysostosis). Clin Plast Surg. 1994;21:613–623.
Gorlin, RJ, Cohen, MM, Jr., Hennekam, RCM. Mandibulofacial dysostosis (Treacher Collins syndrome, Franceschetti-Zwahlen-Klein syndrome). In Syndromes of the head and neck, ed 4, New York: Oxford University Press; 2001:799–802.
Marszalek, B, Wójcicki, P, Kobus, K, et al. Clinical features, treatment and genetic background of Treacher Collins syndrome. J Appl Genet. 2002;43:223–233.
Posnick, JC. Treacher Collins syndrome: perspectives in evaluation and treatment. J Oral Maxillofac Surg. 1997;55:1120–1133.
Posnick, JC, Ruiz, RL. Treacher Collins syndrome: current evaluation, treatment, and future directions. Cleft Palate Craniofac J. 2000;37:434.