Chapter 11 Alimentary System
The alimentary system (digestive system) is the digestive tract from the mouth to the anus with all its associated glands and organs. The primordial gut forms during the fourth week as the head, caudal eminence (tail), and lateral folds incorporate the dorsal part of the umbilical vesicle (yolk sac) into the embryo (see Fig. 5-1, Chapter 5). The primordial gut is initially closed at its cranial end by the oropharyngeal membrane (see Fig. 9-1E), and at its caudal end by the cloacal membrane (Fig. 11-1B). The endoderm of the primordial gut gives rise to most of the gut, epithelium, and glands. The epithelium at the cranial and caudal ends of the alimentary tract is derived from ectoderm of the stomodeum and anal pit (proctodeum), respectively (Fig. 11-1A and B).
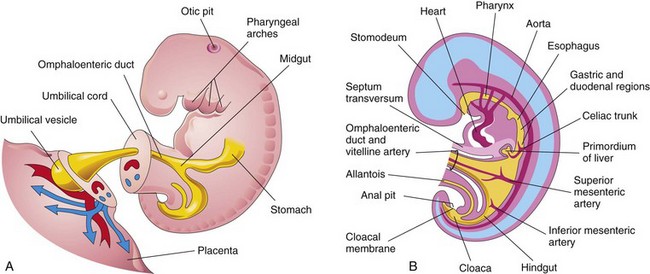
FIGURE 11–1 A, Lateral view of a 4-week embryo showing the relationship of the primordial gut to the omphaloenteric duct (yolk sac). B, Drawing of median section of the embryo showing early alimentary system and its blood supply.
Fibroblast growth factors (FGFs) are involved in early anterior-posterior axial patterning, and it appears that FGF-4 signals from the adjacent ectoderm and mesoderm induce the endoderm. Other secreted factors, such as activins, members of the transforming growth factor β superfamily, contribute to the formation of the endoderm. The endoderm specifies temporal and positional information, which is essential for the development of the gut. The muscular, connective tissue and other layers of the wall of the alimentary tract are derived from the splanchnic mesenchyme surrounding the primordial gut (digestive tract).
Mesenchymal factors, FoxF proteins, control proliferation of the endodermal epithelium that secretes sonic hedgehog (Shh). For descriptive purposes, the primordial gut is divided into three parts: foregut, midgut, and hindgut. Molecular studies indicate that Hox and ParaHox genes, as well as Shh, BMP, and Wnt signals, regulate the regional differentiation of the primordial gut to form its three parts.
Foregut
The derivatives of the foregut are:
These foregut derivatives, other than the pharynx, lower respiratory tract, and most of the esophagus, are supplied by the celiac trunk, the artery of the foregut (Fig. 11-1B).
 Development of Esophagus
Development of Esophagus
The esophagus develops from the foregut immediately caudal to the pharynx (Fig. 11-1B). The partitioning of the trachea from the esophagus by the tracheoesophageal septum is described in Chapter 10 (Fig. 10-2). Initially, the esophagus is short, but it elongates rapidly, mainly because of the growth and relocation of the heart and lungs.
The esophagus reaches its final relative length by the seventh week. Its epithelium and glands are derived from endoderm. The epithelium proliferates and, partly or completely, obliterates the lumen of the esophagus; however, recanalization of the esophagus normally occurs by the end of the eighth week. The striated muscle forming the muscularis externa of the superior third of the esophagus is derived from mesenchyme in the fourth and sixth pharyngeal arches. The smooth muscle, mainly in the inferior third of the esophagus, develops from the surrounding splanchnic mesenchyme. Recent studies indicate transdifferentiation of smooth muscle cells in the superior part of the esophagus to striated muscle, which is dependent on myogenic regulatory factors. Both types of muscle are innervated by branches of the vagus nerves (cranial nerve X), which supply the caudal pharyngeal arches (see Chapter 9, Table 9-1).
Esophageal Atresia
Blockage (atresia) of the esophageal lumen occurs with an incidence of 1 in 3000 to 4500 live births. Approximately one third of affected infants are born prematurely. Esophageal atresia is associated with tracheoesophageal fistula in more than 90% of cases (see Fig. 10-5). Esophageal atresia results from deviation of the tracheoesophageal septum in a posterior direction (see Fig. 10-2), and incomplete separation of the esophagus from the laryngotracheal tube. Isolated esophageal atresia (5%–7% of cases) results from failure of recanalization of the esophagus during the eighth week of development.
A fetus with esophageal atresia is unable to swallow amniotic fluid; consequently, this fluid cannot pass to the intestine for absorption and transfer through the placenta to the maternal blood for disposal. This results in polyhydramnios, the accumulation of an excessive amount of amniotic fluid. Neonates with esophageal atresia usually appear healthy initially. Excessive drooling may be noted soon after birth and the diagnosis of esophageal atresia should be considered if the infant rejects oral feeding with immediate regurgitation and coughing.
Inability to pass a catheter through the esophagus into the stomach strongly suggests esophageal atresia. A radiographic examination demonstrates the anomaly by imaging the nasogastric tube arrested in the proximal esophageal pouch. Surgical repair of esophageal atresia now results in survival rates of more than 85%.
Esophageal Stenosis
Narrowing of the lumen of the esophagus (stenosis) can occur anywhere along the esophagus, but it usually occurs in its distal third, either as a web or a long segment with a thread-like lumen. Stenosis results from incomplete recanalization of the esophagus during the eighth week or from a failure of esophageal blood vessels to develop in the affected area.
Development of Stomach
Initially the distal part of the foregut is a tubular structure (Fig. 11-1B). During the fourth week, a slight dilation indicates the site of the primordium of the stomach. The dilation first appears as a fusiform enlargement of the caudal (distal part) of the foregut and is initially oriented in the median plane (Figs. 11-1 and 11-2B). The primordial stomach soon enlarges and broadens ventrodorsally. During the next 2 weeks, the dorsal border of the stomach grows faster than its ventral border; this demarcates the developing greater curvature of the stomach (Fig. 11-2D).
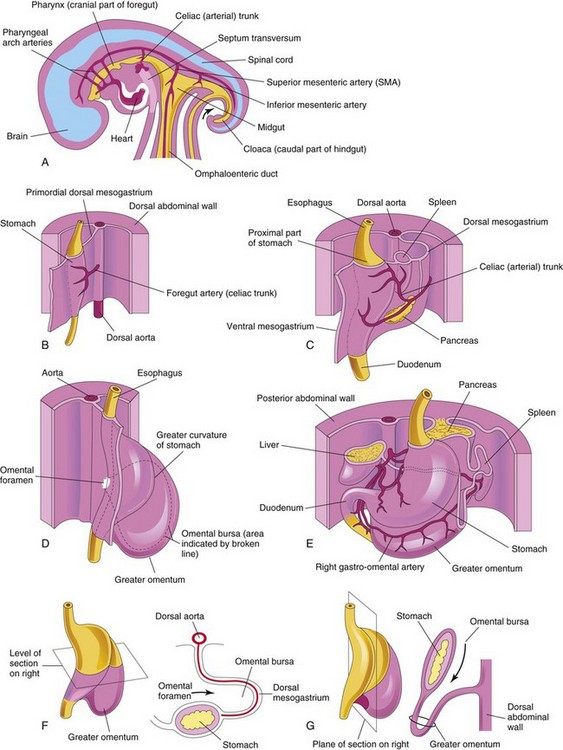
FIGURE 11–2 Development of the stomach and formation of the omental bursa and greater omentum. A, Median section of the abdomen of a 28-day embryo. B, Anterolateral view of the above embryo. C, Embryo of approximately 35 days. D, Embryo of approximately 40 days. E, Embryo of approximately 48 days. F, Lateral view of the stomach and greater omentum of an embryo of approximately 52 days. G, Sagittal section showing the omental bursa and greater omentum. The arrow in F and G indicates the site of the omental foramen.
Rotation of Stomach
Enlargement of the mesentery and adjacent organs, as well as growth of the stomach walls, contribute to the rotation of the stomach. As the stomach enlarges and acquires its final shape, it slowly rotates 90 degrees in a clockwise direction (viewed from the cranial end) around its longitudinal axis. The effects of rotation on the stomach are (Figs. 11-2 and 11-3):

FIGURE 11–3 Development of stomach and mesenteries and formation of omental bursa. A, Embryo of 5 weeks. B, Transverse section showing clefts in the dorsal mesogastrium. C, Later stage after coalescence of the clefts to form the omental bursa. D, Transverse section showing the initial appearance of the omental bursa. E, The dorsal mesentery has elongated and the omental bursa has enlarged. F and G, Transverse and sagittal sections, respectively, showing elongation of the dorsal mesogastrium and expansion of the omental bursa. H, Embryo of 6 weeks, showing the greater omentum and expansion of the omental bursa. I and J, Transverse and sagittal sections, respectively, showing the inferior recess of the omental bursa and the omental foramen. The arrows in E, F, and I indicate the site of the omental foramen. In J, the arrow indicates the inferior recess of the omental bursa.
Mesenteries of Stomach
The stomach is suspended from the dorsal wall of the abdominal cavity by a dorsal mesentery—the primordial dorsal mesogastrium (Figs. 11-2B and C and 11-3A). This mesentery is originally in the median plane, but it is carried to the left during rotation of the stomach and formation of the omental bursa or lesser sac of peritoneum (Fig. 11-3A to E). The dorsal mesentery also contains the spleen and celiac artery. The primordial ventral mesogastrium attaches to the stomach, and it also attaches the duodenum to the liver and ventral abdominal wall (Fig. 11-2C).
Omental Bursa
Isolated clefts develop in the mesenchyme forming the thick dorsal mesogastrium (Fig. 11-3A and B). The clefts soon coalesce to form a single cavity, the omental bursa or lesser peritoneal sac (Fig. 11-3C and D). Rotation of the stomach pulls the dorsal mesogastrium to the left, thereby enlarging the bursa, a large recess of the peritoneal cavity. The omental bursa expands transversely and cranially and soon lies between the stomach and the posterior abdominal wall. This pouch-like bursa facilitates movements of the stomach.
The superior part of the omental bursa is cut off as the diaphragm develops, forming a closed space—the infracardiac bursa. If it persists, it usually lies medial to the base of the right lung. The inferior region of the superior part of the omental bursa persists as the superior recess of the omental bursa (Fig. 11-3C).
As the stomach enlarges, the omental bursa expands and acquires an inferior recess of the omental bursa between the layers of the elongated dorsal mesogastrium—the greater omentum. This membrane overhangs the developing intestines (Fig. 11-3J). The inferior recess disappears as the layers of the greater omentum fuse (see Fig. 11-15F). The omental bursa communicates with the peritoneal cavity through an opening—the omental foramen (Figs. 11-2D and F and 11-3C and F).
Hypertrophic Pyloric Stenosis
Anomalies of the stomach are uncommon except for hypertrophic pyloric stenosis. This anomaly affects one in every 150 males and one in every 750 females. In infants there is a marked muscular thickening of the pylorus, the distal sphincteric region of the stomach (Fig. 11-4). The circular and, to a lesser degree, the longitudinal muscles in the pyloric region are hypertrophied. This results in severe stenosis of the pyloric canal and obstruction of the passage of food. As a result, the stomach becomes markedly distended (Fig. 11-4C ), and the infant expels the stomach’s contents with considerable force (projectile vomiting).
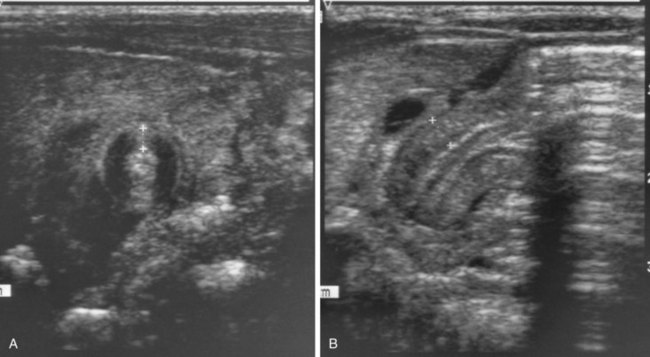
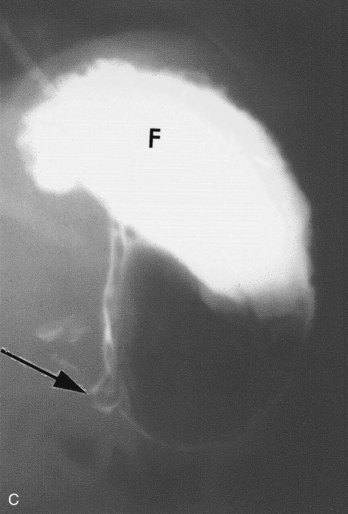
FIGURE 11–4 A, Transverse abdominal sonogram demonstrating a pyloric muscle wall thickness of greater than 4 mm (distance between crosses). B, Horizontal image demonstrating a pyloric channel length greater than 14 mm in an infant with hypertrophic pyloric stenosis. C, Contrast radiograph of the stomach in a 1-month-old male infant with pyloric stenosis. Note the narrowed pyloric end (arrow) and the distended fundus (F) of the stomach, filled with contrast material.
(A and B, From Wyllie R: Pyloric stenosis and other congenital anomalies of the stomach. In Behrman RE, Kliegman RM, Arvin AM [eds]: Nelson Textbook of Pediatrics, 15th ed. Philadelphia, WB Saunders, 1996; C, Courtesy of Dr. Prem S. Sahni, formerly of the Department of Radiology, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Surgical relief of the pyloric obstruction (pyloromyotomy) is the usual treatment. The cause of congenital pyloric stenosis is unknown, but the high rate of concordance in monozygotic twins suggests genetic factors may be involved.
Development of Duodenum
Early in the fourth week, the duodenum begins to develop from the caudal part of the foregut, the cranial part of the midgut, and the splanchnic mesenchyme associated with these parts of the primordial gut (Fig. 11-5A). The junction of the two parts of the duodenum is just distal to the origin of the bile duct (Fig. 11-5D). The developing duodenum grows rapidly, forming a C-shaped loop that projects ventrally (Fig. 11-5B to D).
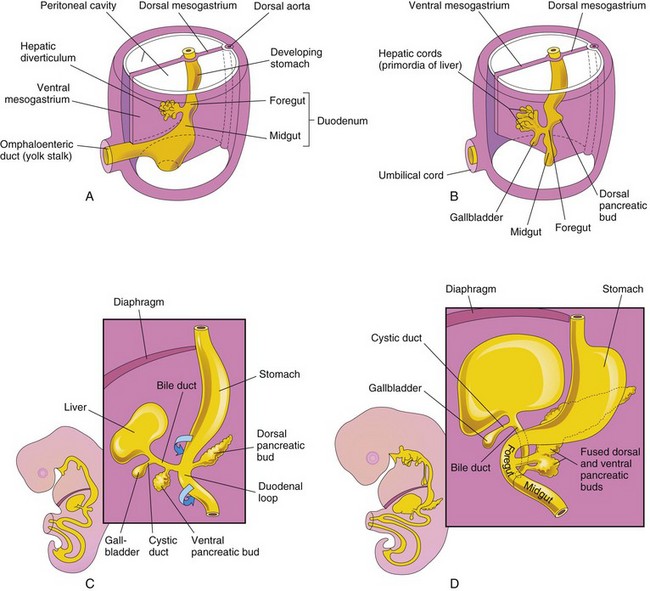
FIGURE 11–5 Progressive stages in the development of the duodenum, liver, pancreas, and extrahepatic biliary apparatus. A, Embryo of 4 weeks. B and C, Embryo of 5 weeks. D, Embryo of 6 weeks. The pancreas develops from the dorsal and ventral pancreatic buds that fuse to form the pancreas. Note that the entrance of the bile duct into the duodenum gradually shifts from its initial position to a posterior one. This explains why the bile duct in the adult passes posterior to the duodenum and the head of the pancreas.
As the stomach rotates, the duodenal loop rotates to the right and becomes pressed against the posterior wall of the abdominal cavity—retroperitoneal (external to peritoneum). Because of its derivation from the foregut and midgut, the duodenum is supplied by branches of the celiac and superior mesenteric arteries that supply these parts of the primordial gut (Fig. 11-1).
During the fifth and sixth weeks, the lumen of the duodenum becomes progressively smaller and is temporarily obliterated because of the proliferation of its epithelial cells. Normally vacuolation occurs as the epithelial cells degenerate; as a result, the duodenum normally becomes recanalized by the end of the embryonic period (Fig. 11-6C and D). By this time, most of the ventral mesentery of the duodenum has disappeared.
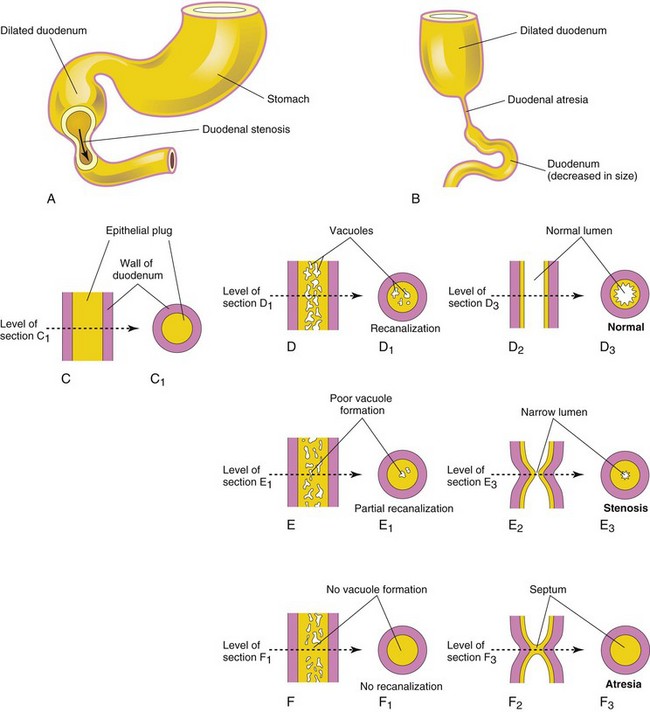
FIGURE 11–6 Drawings showing the embryologic basis of the common types of congenital intestinal obstruction. A, Duodenal stenosis. B, Duodenal atresia. C to F, Diagrammatic longitudinal and transverse sections of the duodenum showing (1) normal recanalization (D to D3), (2) stenosis (E to E3), and atresia (F to F3).
Duodenal Stenosis
Partial occlusion of the duodenal lumen—duodenal stenosis (Fig. 11-6A)—usually results from incomplete recanalization of the duodenum resulting from defective vacuolization (Fig. 11-6E3). Most stenoses involve the horizontal (third) and/or ascending (fourth) parts of the duodenum. Because of the stenosis, the stomach’s contents (usually containing bile) are often vomited.
Duodenal Atresia
Complete occlusion of the duodenal lumen—duodenal atresia (Fig. 11-6B)—is not common. During early duodenal development, the lumen is completely occluded by epithelial cells. If complete recanalization of the lumen fails to occur (Fig. 11-6D3), a short segment of the duodenum is occluded (Fig. 11-6F3). The blockage occurs nearly always at the junction of the bile and pancreatic ducts (hepatopancreatic ampulla); occasionally the blockage involves the horizontal (third) part of the duodenum. Investigation of families with familial duodenal atresia suggests an autosomal recessive inheritance.
In infants with duodenal atresia, vomiting begins a few hours after birth. The vomitus almost always contains bile; often there is distention of the epigastrium—the upper central area of the abdomen—resulting from an overfilled stomach and superior part of the duodenum. Duodenal atresia is associated with bilious emesis (vomiting of bile) because the blockage occurs distal to the opening of the bile duct. Duodenal atresia may occur as an isolated birth defect, but other defects are often associated with it, such as annular pancreas (see Fig. 11-11C ), cardiovascular defects, anorectal anomalies, and malrotation of the gut (see Fig. 11-20). Importantly, approximately one third of affected infants have Down syndrome and an additional 20% are premature.
Polyhydramnios also occurs because duodenal atresia prevents normal intestinal absorption of swallowed amniotic fluid. The diagnosis of duodenal atresia is suggested by the presence of a “double-bubble” sign on plain radiographs or ultrasound scans (Fig. 11-7). This appearance is caused by a distended, gas-filled stomach and proximal duodenum.
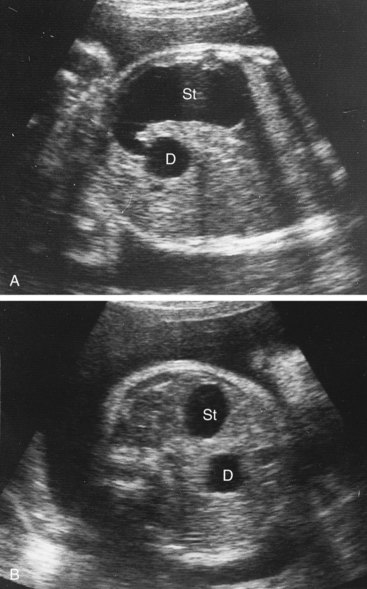
FIGURE 11–7 Ultrasound scans of a fetus of 33 weeks showing duodenal atresia. A, An oblique scan showing the dilated, fluid-filled stomach (St) entering the proximal duodenum (D), which is also enlarged because of the atresia (blockage) distal to it. B, Transverse scan illustrating the characteristic “double-bubble” appearance of the stomach and duodenum when there is duodenal atresia.
(Courtesy of Dr. Lyndon M. Hill, Magee-Women’s Hospital, Pittsburgh, PA.)
Development of Liver and Biliary Apparatus
The liver, gallbladder, and biliary duct system arise as a ventral outgrowth—hepatic diverticulum—from the distal part of the foregut early in the fourth week (Figs. 11-5A and 11-8A). Wnt/β-catenin signaling pathway plays a key role in this process. It has been suggested that both the hepatic diverticulum and the ventral bud of the pancreas develop from two cell populations in the embryonic endoderm. At sufficient levels, FGFs (fibroblast growth factors) secreted by the developing heart, interact with the bipotential cells and induce formation of the hepatic diverticulum.
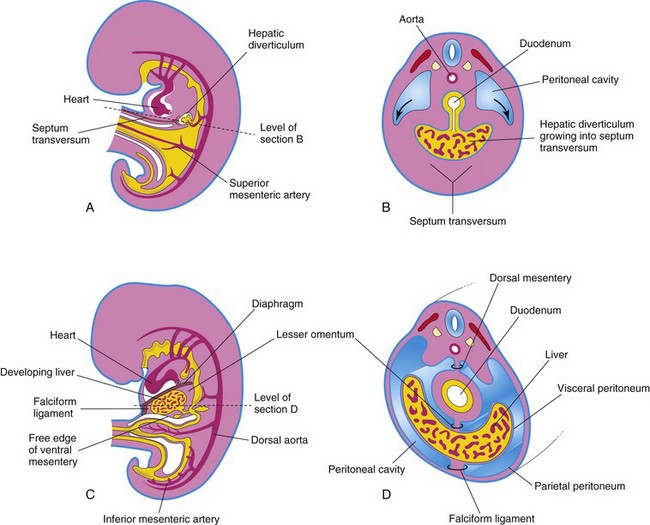
FIGURE 11–8 A, Median section of a 4-week embryo. B, Transverse section of the embryo showing expansion of the peritoneal cavity (arrows). C, Sagittal section of a 5-week embryo. D, Transverse section of the embryo after formation of the dorsal and ventral mesenteries.
The diverticulum extends into the septum transversum, a mass of splanchnic mesoderm between the developing heart and midgut. The septum transversum forms the ventral mesogastrium in this region. The hepatic diverticulum enlarges rapidly and divides into two parts as it grows between the layers of the ventral mesogastrium (Fig. 11-5A).
The larger cranial part of the hepatic diverticulum is the primordium of the liver; the smaller caudal part becomes the gallbladder. The proliferating endodermal cells give rise to interlacing cords of hepatocytes and to the epithelial lining of the intrahepatic part of the biliary apparatus. The hepatic cords anastomose around endothelium-lined spaces, the primordia of the hepatic sinusoids. Vascular endothelial growth factor Flk-1 (VEGF-Flk-1) signaling appears to be important for the early morphogenesis of the hepatic sinusoids (primitive vascular system). The fibrous and hematopoietic tissue and Kupffer cells of the liver are derived from mesenchyme in the septum transversum.
The liver grows rapidly and, from the 5th to 10th weeks, fills a large part of the upper abdominal cavity (Fig. 11-8C and D). The quantity of oxygenated blood flowing from the umbilical vein into the liver determines the development and functional segmentation of the liver. Initially, the right and left lobes are approximately the same size, but the right lobe soon becomes larger.
Hematopoiesis (formation and development of various types of blood cells) begins during the sixth week, giving the liver a bright reddish appearance. By the ninth week, the liver accounts for approximately 10% of the total weight of the fetus. Bile formation by hepatic cells begins during the 12th week.
The small caudal part of the hepatic diverticulum becomes the gallbladder, and the stalk of the diverticulum forms the cystic duct (Fig. 11-5C). Initially, the extrahepatic biliary apparatus is occluded with epithelial cells, but it is later canalized because of vacuolation resulting from degeneration of these cells. The stalk connecting the hepatic and cystic ducts to the duodenum becomes the bile duct. Initially, this duct attaches to the ventral aspect of the duodenal loop; however, as the duodenum grows and rotates, the entrance of the bile duct is carried to the dorsal aspect of the duodenum (Fig. 11-5C and D). The bile entering the duodenum through the bile duct after the 13th week gives the meconium (intestinal contents) a dark green color.
Ventral Mesentery
This thin, double-layered membrane (Fig. 11-8C and D) gives rise to:
The umbilical vein passes in the free border of the falciform ligament on its way from the umbilical cord to the liver. The ventral mesentery, derived from the mesogastrum, also forms the visceral peritoneum of the liver. The liver is covered by peritoneum except for the bare area that is in direct contact with the diaphragm (Fig. 11-9).
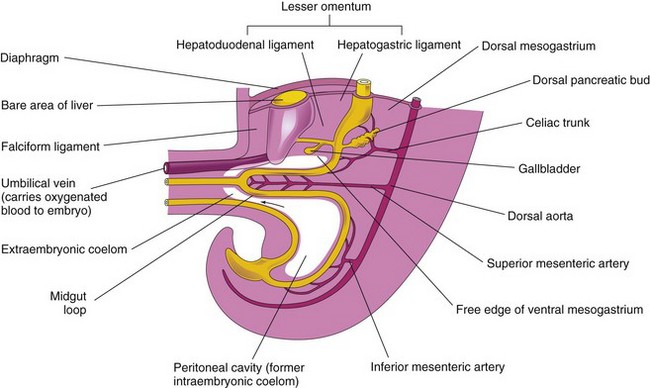
FIGURE 11–9 Median section of caudal half of an embryo at the end of the fifth week showing the liver and its associated ligaments. The arrow indicates the communication of the peritoneal cavity with the extraembryonic coelom.
Anomalies of Liver
Minor variations of liver lobulation are common, but congenital anomalies of the liver are rare. Variations of the hepatic ducts, bile duct, and cystic duct are common and clinically significant. Accessory hepatic ducts are present in approximately 5% of the population and awareness of their possible presence is of importance in surgery including liver transplantation. The accessory ducts are narrow channels running from the right lobe of the liver into the anterior surface of the body of the gallbladder. In some cases, the cystic duct opens into an accessory hepatic duct rather than into the common hepatic duct.
Extrahepatic Biliary Atresia
This is the most serious anomaly of the extrahepatic biliary system and occurs in one in 5000 to 20,000 live births. The most common form of extrahepatic biliary atresia (present in 85% of cases) is obliteration of the bile ducts at or superior to the porta hepatis—a deep transverse fissure on the visceral surface of the liver. Previous speculations that there is a failure of the bile ducts to canalize may not be true.
Biliary atresia could result from a failure of the remodeling process at the hepatic hilum or from infections or immunologic reactions during late fetal development. Jaundice occurs soon after birth, the stools are acholic (clay colored), and the urine appears dark colored. Biliary atresia can be palliated surgically in most patients, but in greater than 70% of those treated, the disease continues to progress.
Development of Pancreas
The pancreas develops between the layers of the mesentery from dorsal and ventral pancreatic buds of endodermal cells, which arise from the caudal of the foregut (Figs. 11-9 and 11-10A and B). Most of the pancreas is derived from the larger dorsal pancreatic bud, which appears first and develops a slight distance cranial to the ventral bud.
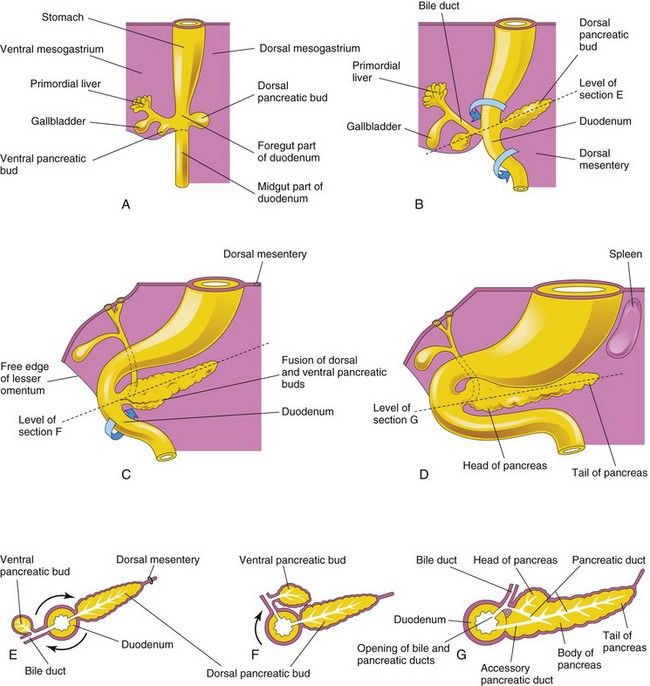
FIGURE 11–10 A to D, Successive stages in the development of the pancreas from the fifth to the eighth weeks. E to G, Diagrammatic transverse sections through the duodenum and developing pancreas. Growth and rotation (arrows) of the duodenum bring the ventral pancreatic bud toward the dorsal bud, where they subsequently fuse.
The smaller ventral pancreatic bud develops near the entry of the bile duct into the duodenum and grows between the layers of the ventral mesentery. As the duodenum rotates to the right and becomes C shaped, the ventral pancreatic bud is carried dorsally with the bile duct (Fig. 11-10C to G). It soon lies posterior to the dorsal pancreatic bud and later fuses with it.
The ventral pancreatic bud forms the uncinate process and part of the head of the pancreas. As the stomach, duodenum, and ventral mesentery rotate, the pancreas comes to lie along the dorsal abdominal wall. As the pancreatic buds fuse, their ducts anastomose. The pancreatic duct forms from the duct of the ventral bud and the distal part of the duct of the dorsal bud (Fig. 11-10G). The proximal part of the duct of the dorsal bud often persists as an accessory pancreatic duct that opens into the minor duodenal papilla, located approximately 2 cm cranial to the main duct. The two ducts often communicate with each other. In approximately 9% of people, the pancreatic ducts fail to fuse, resulting in two ducts.
Molecular studies show that the ventral pancreas develops from a bipotential cell population in the ventral region of the duodenum where the transcription factor PDX1 is expressed. A default mechanism involving FGF-2, which is secreted by the developing heart, appears to play a role. Formation of the dorsal pancreatic bud depends on the notochord secreting activin and FGF-2, which block the expression of Shh in the associated endoderm.
Histogenesis of Pancreas
The parenchyma (basic cellular tissue) of the pancreas is derived from the endoderm of the pancreatic buds, which forms a network of tubules. Early in the fetal period, pancreatic acini begin to develop from cell clusters around the ends of these tubules (primordial pancreatic ducts). The pancreatic islets develop from groups of cells that separate from the tubules and lie between the acini.
Recent studies show that chemokine SDF-1 (stromal cell derived factor), expressed in the mesechyme, controls formation and branching of the tubules. Expression of transcription factor Ngn-3 (neurogenin-3) is required for differentiation of pancreatic islet endocrine cells.
Insulin secretion begins during the early fetal period (10 weeks). The glucagon- and somatostatin-containing cells develop before differentiation of the insulin-secreting beta-cells. Glucagon has been detected in fetal plasma at 15 weeks.
The connective tissue sheath and interlobular septa of the pancreas develop from the surrounding splanchnic mesenchyme. When there is maternal diabetes mellitus, the insulin-secreting beta cells in the fetal pancreas are chronically exposed to high levels of glucose. As a result, these cells undergo hypertrophy to increase the rate of insulin secretion.
Ectopic Pancreas
Ectopic pancreas is most often located in the wall of the stomach, duodenum, or jejunum. It can present with obstruction, bleeding, or even as cancer.
Annular Pancreas
Although an annular pancreas is rare, the anomaly warrants description because it may cause duodenal obstruction (Fig. 11-11C). The ring-like or annular part of the pancreas consists of a thin, flat band of pancreatic tissue surrounding the descending or second part of the duodenum. An annular pancreas may cause obstruction of the duodenum. Infants present with symptoms of complete or partial bowel obstruction.
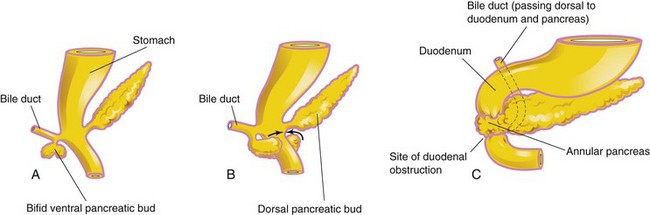
FIGURE 11–11 A and B show the probable basis of an annular pancreas. C, An annular pancreas encircling the duodenum. This birth defect produces complete obstruction (atresia) or partial obstruction (stenosis) of the duodenum.
Blockage of the duodenum develops if inflammation (pancreatitis) develops in the annular pancreas. An annular pancreas may be associated with Down syndrome, intestinal malrotation, and cardiac defects. Females are affected more frequently than males. An annular pancreas probably results from the growth of a bifid ventral pancreatic bud around the duodenum (Fig. 11-11A to C ). The parts of the bifid ventral bud then fuse with the dorsal bud, forming a pancreatic ring. Surgical intervention may be required for management of this condition.
Development of Spleen
Development of the spleen is described with the alimentary system because this organ is derived from a mass of mesenchymal cells located between the layers of the dorsal mesogastrium (Fig. 11-12). The spleen, a vascular lymphatic organ, begins to develop during the fifth week, but does not acquire its characteristic shape until early in the fetal period.
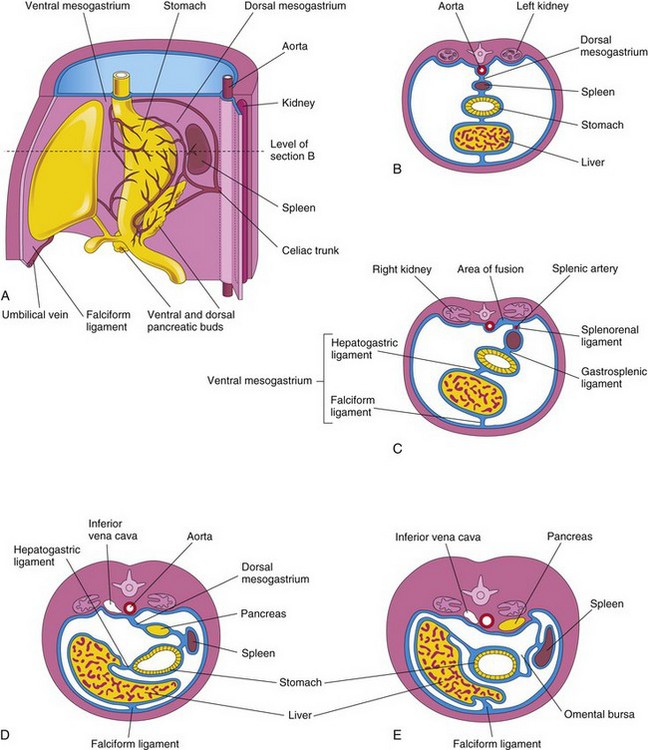
FIGURE 11–12 A, Left side of the stomach and associated structures at the end of the fifth week. Note that the pancreas, spleen, and celiac trunk are between the layers of the dorsal mesogastrium. B, Transverse section of the liver, stomach, and spleen at the level shown in A, illustrating their relationship to the dorsal and ventral mesenteries. C, Transverse section of a fetus showing fusion of the dorsal mesogastrium with the peritoneum on the posterior abdominal wall. D and E, Similar sections showing movement of the liver to the right and rotation of the stomach. Observe the fusion of the dorsal mesogastrium to the dorsal abdominal wall. As a result, the pancreas becomes retroperitoneal.
Gene-targeting experiments show that capsulin, a basic helix-loop transcription factor, and homeobox genes NKx2-5, Hox11, and Bapx1 regulate the development of the spleen.
The spleen is lobulated in the fetus, but the lobules normally disappear before birth. The notches in the superior border of the adult spleen are remnants of the grooves that separated the fetal lobules. As the stomach rotates, the left surface of the mesogastrium fuses with the peritoneum over the left kidney. This fusion explains the dorsal attachment of the splenorenal ligament and why the adult splenic artery, the largest branch of the celiac trunk, follows a tortuous course posterior to the omental bursa and anterior to the left kidney (Fig. 11-12C).
The mesenchymal cells in the splenic primordium differentiate to form the capsule, connective tissue framework, and parenchyma of the spleen. The spleen functions as a hematopoietic center until late fetal life; however, it retains its potential for blood cell formation even in adult life.
Accessory Spleens (Polysplenia)
One or more small splenic masses (about 1 cm in diameter) of fully functional splenic tissue may exist in one of the peritoneal folds, commonly near the hilum of the spleen, in the tail of the pancreas, or within the gastrosplenic ligament. These accessory spleens (polysplenia) are usually isolated but may be attached to the spleen by thin bands. An accessory spleen occurs in approximately 10% of people.
Midgut
The derivatives of the midgut are:
Herniation of Midgut Loop
As the midgut elongates, it forms a ventral, U-shaped loop of intestine—the midgut loop—that projects into the remains of the extraembryonic coelom in the proximal part of the umbilical cord. The midgut loop of the intestine is a physiologic umbilical herniation, which occurs at the beginning of the sixth week (Figs. 11-13 and 11-14). The loop communicates with the umbilical vesicle through the narrow omphaloenteric duct (yolk stalk) until the 10th week.
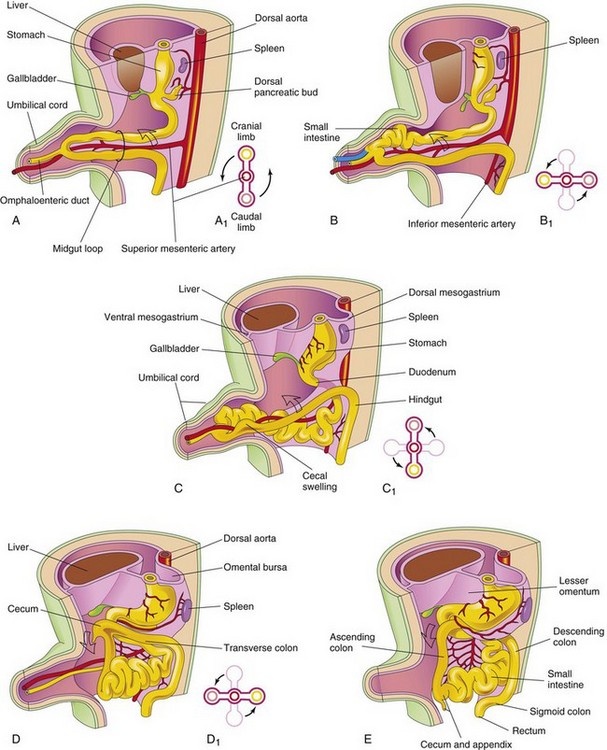
FIGURE 11–13 Drawings illustrating herniation, rotation, and retraction of the midgut from the beginning of the 6th week to the 12th week. A, Transverse section through the midgut loop, illustrating the initial relationship of the limbs of the loop to the artery. Note that the midgut loop is in the proximal part of the umbilical cord. B, Later stage showing the beginning of midgut rotation. B1, Illustration of the 90-degree counterclockwise rotation that carries the cranial limb of the midgut to the right. C, At approximately 10 weeks, showing the intestines returning to the abdomen. C1, Illustration of a further rotation of 90 degrees. D, At approximately 11 weeks showing the location of the viscera after retraction of intestines. D1, Illustration of a further 90-degree rotation of the viscera, for a total of 270 degrees. E, Later in the fetal period, showing the cecum rotating to its normal position in the lower right quadrant of the abdomen.
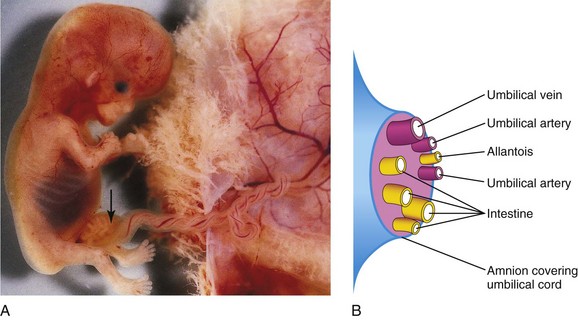
FIGURE 11–14 A, Physiologic hernia in a fetus of approximately 58 days attached to its placenta. Note the herniated intestine (arrow) in the proximal part of the umbilical cord. B, Schematic drawing showing the structures in the distal part of the umbilical cord.
(A, Courtesy of Dr. D.K. Kalousek, Department of Pathology, University of British Columbia, Children’s Hospital, Vancouver, British Columbia, Canada.)
The physiologic umbilical herniation occurs because there is not enough room in the abdominal cavity for the rapidly growing midgut. The shortage of space is caused mainly by the relatively massive liver and the kidneys that exist during this period of development.
The midgut loop has a cranial (proximal) limb and a caudal (distal) limb and is suspended from the dorsal abdominal wall by an elongated mesentery—the dorsal mesogastrium (Fig. 11-13A). The omphaloenteric duct is attached to the apex of the midgut loop where the two limbs join (Fig. 11-13A). The cranial limb grows rapidly and forms small intestinal loops, but the caudal limb undergoes very little change except for development of the cecal swelling (diverticulum), the primordium of the cecum, and appendix (Fig. 11-13B and C).
Rotation of Midgut Loop
While it is in the umbilical cord, the midgut loop rotates 90 degrees counterclockwise around the axis of the superior mesenteric artery (Fig. 11-13B). This brings the cranial limb (small intestine) of the midgut loop to the right and the caudal limb (large intestine) to the left. During rotation, the cranial limb elongates and forms intestinal loops (e.g., primordia of jejunum and ileum).
Retraction of Intestinal Loops
During the 10th week, the intestines return to the abdomen (reduction of the midgut hernia) (Fig. 11-13C and D). It is not known what causes the intestine to return; however, the enlargement of the abdominal cavity and the relative decrease in the size of the liver and kidneys are important factors. The small intestine (formed from the cranial limb) returns first, passing posterior to the superior mesenteric artery and occupies the central part of the abdomen. As the large intestine returns, it undergoes a further 180-degree counterclockwise rotation (see Fig. 11-13C1 and D1). The descending and sigmoid colon move to the right side of the abdomen. The ascending colon becomes recognizable with the elongation of the posterior abdominal wall (Fig. 11-13E).
Fixation of Intestines
Rotation of the stomach and duodenum causes the duodenum and pancreas to fall to the right. The enlarged colon presses the duodenum and pancreas against the posterior abdominal wall; as a result, most of the duodenal mesentery is absorbed (Fig. 11-15C, D, and F). Consequently, the duodenum, except for the first part (derived from foregut), has no mesentery and lies retroperitoneally. Similarly, the head of the pancreas becomes retroperitoneal (posterior to peritoneum).
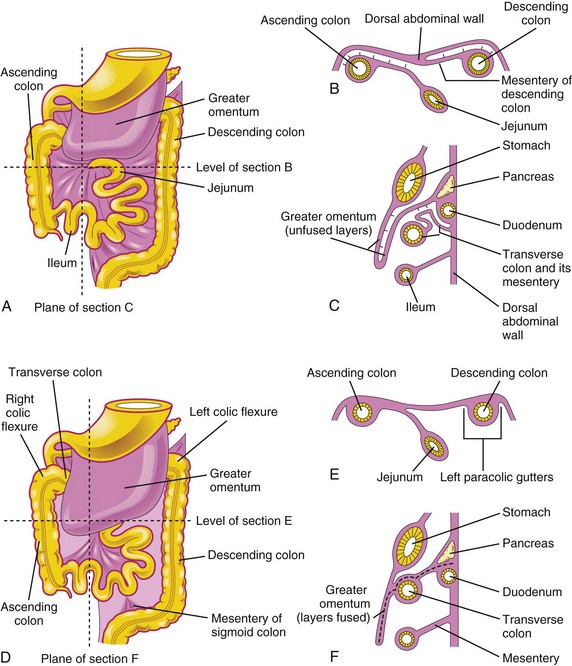
FIGURE 11–15 Illustrations showing the mesenteries and fixation of the intestines. A, Ventral view of the intestines before their fixation. B, Transverse section at the level shown in A. The arrows indicate areas of subsequent fusion, C, Sagittal section at the plane shown in A, illustrating the greater omentum overhanging the transverse colon. The arrows indicate areas of subsequent fusion. D, Ventral view of the intestines after their fixation. E, Transverse section at the level shown in D after disappearance of the mesentery of the ascending and descending colon. F, Sagittal section at the plane shown in D, illustrating fusion of the greater omentum with the mesentery of the transverse colon and fusion of the layers of the greater omentum.
The attachment of the dorsal mesentery to the posterior abdominal wall is greatly modified after the intestines return to the abdominal cavity. At first, the dorsal mesentery is in the median plane. As the intestines enlarge, lengthen, and assume their final positions, their mesenteries are pressed against the posterior abdominal wall. The mesentery of the ascending colon fuses with the parietal peritoneum on this wall and disappears; consequently, the ascending colon also becomes retroperitoneal (Fig. 11-15B and E).
Other derivatives of the midgut loop (e.g., the jejunum and ileum) retain their mesenteries. The mesentery is at first attached to the median plane of the posterior abdominal wall (Fig. 11-13B and C). After the mesentery of the ascending colon disappears, the fan-shaped mesentery of the small intestines acquires a new line of attachment that passes from the duodenojejunal junction inferolaterally to the ileocecal junction.
Cecum and Appendix
The primordium of the cecum and appendix—the cecal swelling (diverticulum)—appears in the sixth week as an elevation on the antimesenteric border of the caudal limb of the midgut loop (Fig. 11-13C and 11-16A). The apex of the cecal swelling does not grow as rapidly as the rest of it; thus, the appendix is initially a small diverticulum of the cecum (Fig. 11-16B). The appendix increases rapidly in length so that at birth it is a relatively long tube arising from the distal end of the cecum (Fig. 11-16D). After birth, the wall of the cecum grows unequally, with the result that the appendix comes to enter its medial side.
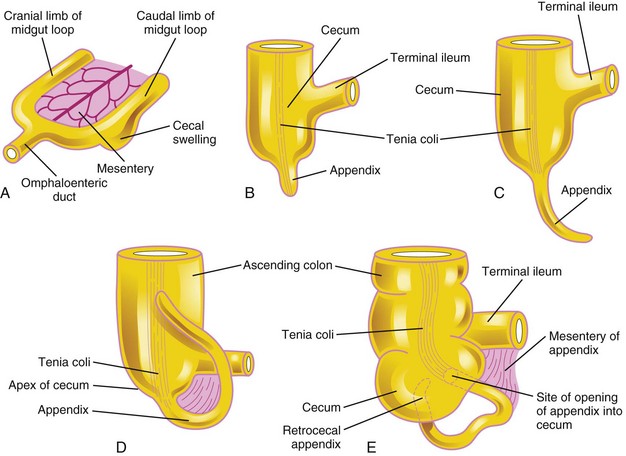
FIGURE 11–16 Successive stages in the development of the cecum and appendix. A, Embryo of 6 weeks. B, Embryo of 8 weeks. C, Fetus of 12 weeks. D, Fetus at birth. Note that the appendix is relatively long and is continuous with the apex of the cecum. E, Child. Note that the opening of the appendix lies on the medial side of the cecum. In approximately 64% of people, the appendix is located posterior to the cecum (retrocecal). The tenia coli is a thickened band of longitudinal muscle in the wall of the colon.
The appendix is subject to considerable variation in position. As the ascending colon elongates, the appendix may pass posterior to the cecum (retrocecal appendix) or colon (retrocolic appendix). It may also descend over the brim of the pelvis (pelvic appendix). In approximately 64% of people, the appendix is located retrocecally (Fig. 11-16E).
Congenital Omphalocele
This anomaly is a persistence of the herniation of abdominal contents into the proximal part of the umbilical cord (Figs. 11-17 and 11-18). Herniation of intestines into the cord occurs in approximately 1 in 5000 births, and herniation of liver and intestines in 1 in approximately 10,000 births. Up to 50% of cases are associated with chromosomal abnormalities. The abdominal cavity is proportionately small when there is an omphalocele because the impetus for it to grow is absent. Surgical repair is required. Minor omphaloceles may be treated with primary closure. A staged reduction is often planned if the viscera-abdominal disproportion is large. Infants with very large omphaloceles can also suffer from pulmonary and thoracic hypoplasia (underdevelopment).
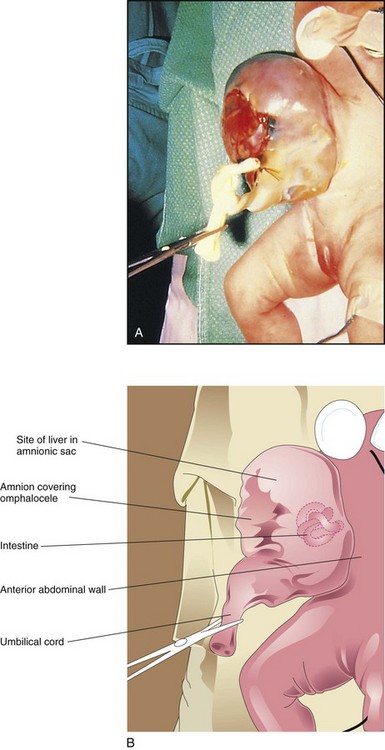
FIGURE 11–17 A, An infant with a large omphalocele. B, Drawing of the infant with an omphalocele resulting from a median defect of the abdominal muscles, fascia, and skin near the umbilicus. This defect resulted in the herniation of intra-abdominal structures (liver and intestine) into the proximal end of the umbilical cord. It is covered by a membrane composed of peritoneum and amnion.
(A, Courtesy of Dr. N.E. Wiseman, pediatric surgeon, Children’s Hospital, Winnipeg, Manitoba, Canada.)
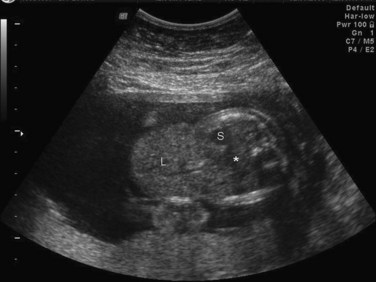
FIGURE 11–18 Sonogram of the abdomen of a fetus showing a large omphalocele, with the liver (L) protruding (herniating) from the abdomen (*). Also observe the stomach (S).
(Courtesy of Dr. G.J. Reid, Department of Obstetrics, Gynecology and Reproductive Sciences, University of Manitoba, Women’s Hospital, Winnipeg, Manitoba, Canada.)
Omphalocele results from impaired growth of the mesodermal (muscle) and ectodermal (skin) components of the abdominal wall. Because the formation of the abdominal compartment occurs during gastrulation, a critical failure of growth at this time is often associated with other birth defects involving the cardiovascular and urogenital systems. The covering of the hernial sac is the epithelium of the umbilical cord, a derivative of the amnion.
Umbilical Hernia
When the intestines return to the abdominal cavity during the 10th week and then herniate again through an imperfectly closed umbilicus, an umbilical hernia forms. This common type of hernia is different from an omphalocele. In an umbilical hernia, the protruding mass (usually the greater omentum and part of the small intestine) is covered by subcutaneous tissue and skin.
The hernia usually does not reach its maximum size until the end of the first month after birth. It usually ranges in diameter from 1 to 5 cm. The defect through which the hernia occurs is in the linea alba (fibrous band in the median line of the anterior abdominal wall between the rectus muscles). The hernia protrudes during crying, straining, or coughing, and can be easily reduced through the fibrous ring at the umbilicus. Surgery is not usually performed unless the hernia persists to the age of 3 to 5 years.
Gastroschisis
This birth defect is a relatively uncommon congenital abdominal wall anomaly (Fig. 11-19). Gastroschisis results from a defect lateral to the median plane of the anterior abdominal wall. The linear defect permits extrusion of the abdominal viscera without involving the umbilical cord. The viscera protrude into the amniotic cavity and are bathed by amniotic fluid. The term gastroschisis, which literally means a “split or open stomach,” is a misnomer because it is the anterior abdominal wall that is split, not the stomach.
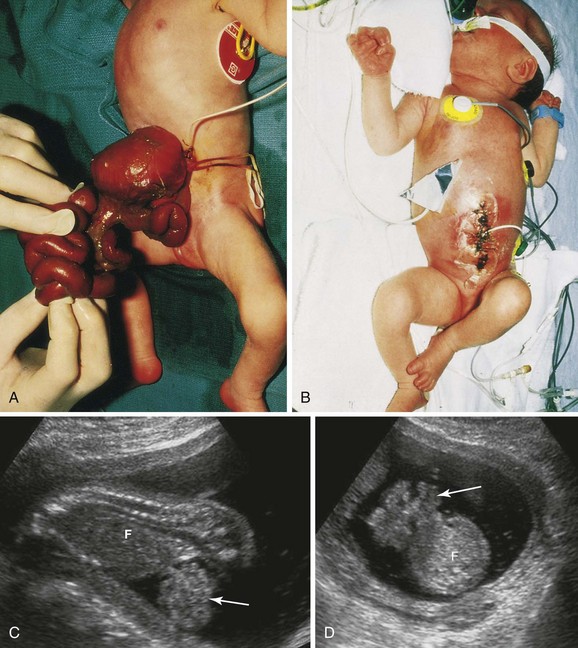
FIGURE 11–19 A, Photograph of a neonate with viscera protruding from an anterior abdominal wall birth defect—gastroschisis. The defect was 2 to 4 cm long and involved all layers of the abdominal wall. B, Photograph of the infant after the viscera were returned to the abdomen and the defect was surgically closed. C (sagittal) and D (axial) sonograms of a fetus at 18 weeks with gastroschisis. Loops of intestine (arrow) can be seen in the amniotic fluid anterior to the fetus (F).
(A and B, Courtesy of A.E. Chudley, MD, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada. C and D, Courtesy of Dr. E.A. Lyons, Departments of Radiology, Obstetrics and Gynecology, and Anatomy, Health Sciences Centre and University of Manitoba, Winnipeg, Manitoba.)
The birth defect usually occurs on the right side lateral to the umbilicus and is more common in males than females. The exact cause of gastroschisis is uncertain, but various causes have been proposed including ischemic injury to the anterior abdominal wall (absence of the right omphalomesenteric artery), rupture of the anterior abdominal wall, weakness of the wall caused by abnormal involution of the right umbilical vein, and perhaps rupture of an omphalocele before the anterior abdominal wall has folded.
Anomalies of Midgut
Birth defects of the intestines are common; most of them are anomalies of gut rotation—malrotation of the gut—that result from incomplete rotation and/or fixation of the intestines. Nonrotation of the gut occurs when the intestine does not rotate as it reenters the abdomen. As a result, the caudal limb of the midgut loop returns to the abdomen first and the small intestines lie on the right side of the abdomen and the entire large intestine is on the left. The usual 270-degree counterclockwise rotation is not completed and the cecum lies just inferior to the pylorus of the stomach. The cecum is fixed to the posterolateral abdominal wall by peritoneal bands that pass over the duodenum (Fig. 11-20B). These peritoneal bands and the volvulus (twisting) of the intestines cause duodenal obstruction. This type of malrotation results from failure of the midgut loop to complete the final 90 degrees of rotation (Fig. 11-13D). Only two parts of the intestine are attached to the posterior abdominal wall: the duodenum and proximal colon. This improperly positioned and incompletely fixed intestine may lead to a catastrophic twisting of the midgut—midgut volvulus (Fig. 11-20F ). The small intestine hangs by a narrow stalk that contains the superior mesenteric artery and vein.
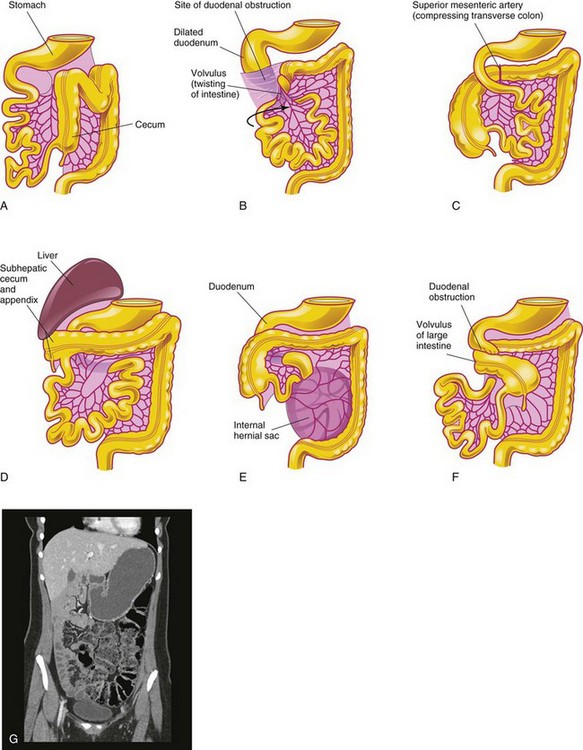
FIGURE 11–20 Birth defects of midgut rotation. A, Nonrotation. B, Mixed rotation and volvulus. C, Reversed rotation. D, Subhepatic cecum and appendix. E, Internal hernia. F, Midgut volvulus. G, CT enterographic image of nonrotation in an adolescent patient with chronic abdominal pain. The large intestine is completely on the left side of the abdomen (stool-filled). The small intestine (fluid-filled) is seen on the right.
(G, Courtesy of Dr. S. Morrison, Children’s Hospital, The Cleveland Clinic, Cleveland, Ohio.)
When midgut volvulus occurs, the superior mesenteric artery may be obstructed, resulting in infarction and gangrene of the intestine supplied by it (Fig. 11-20A and B). Infants with intestinal malrotation are prone to volvulus and present with bilious emesis (vomiting bile). A contrast x-ray study can determine the presence of rotational abnormalities.
Reversed Rotation
In rare cases, the midgut loop rotates in a clockwise rather than a counterclockwise direction (Fig. 11-20C ). As a result, the duodenum lies anterior to the superior mesenteric artery rather than posterior to it, and the transverse colon lies posterior instead of anterior to it. In these infants, the transverse colon may be obstructed by pressure from the superior mesenteric artery. In more unusual cases, the small intestine lies on the left side of the abdomen and the large intestine lies on the right side, with the cecum in the center. This unusual situation results from malrotation of the midgut followed by failure of fixation of the intestines.
Subhepatic Cecum and Appendix
If the cecum adheres to the inferior surface of the liver when it returns to the abdomen, it will be drawn superiorly as the liver diminishes in size; as a result, the cecum remains in its fetal position (Fig. 11-20D). A subhepatic cecum and appendix are more common in males and occur in approximately 6% of fetuses. A subhepatic cecum and “high-riding” appendix may be seen in adults; however, when it occurs, it may create a problem in the diagnosis of appendicitis and during surgical removal of the appendix (appendectomy).
Mobile Cecum
In approximately 10% of people, the cecum has an abnormal amount of freedom. In very unusual cases, it may herniate into the right inguinal canal. A mobile cecum results from incomplete fixation of the ascending colon. This condition is clinically significant because of the possible variations in the position of the appendix and because twisting (volvulus) of the cecum may occur.
Internal Hernia
In this rare birth defect, the small intestine passes into the mesentery of the midgut loop during the return of the intestines to the abdomen (Fig. 11-20E ). As a result, a hernia-like sac forms. This condition usually does not produce symptoms and is often detected at autopsy or during an anatomic dissection.
Stenosis and Atresia of Intestine
Partial occlusion (stenosis) and complete occlusion (atresia) of the intestinal lumen account for approximately one third of cases of intestinal obstruction (Fig. 11-6). The obstructive lesion occurs most often in the duodenum (25%) and ileum (50%). The length of the area affected varies. These birth defects result from failure of an adequate number of vacuoles to form during recanalization of the intestine. In some cases, a transverse septum or web forms, producing the blockage or atresia (Fig. 11-6F2).
Another possible cause of stenoses and atresias is interruption of the blood supply to a loop of fetal intestine resulting from a fetal vascular accident caused by impaired microcirculation that is associated with fetal distress, drug exposure, or a volvulus. The loss of blood supply leads to necrosis of the intestines and development of a fibrous cord connecting the proximal and distal ends of normal intestine. Malfixation of the gut most likely occurs during the 10th week and predisposes it to volvulus, strangulation, and impairment of its blood supply.
Ileal Diverticulum and Omphaloenteric Remnants
Outpouching of part of the ileum is a common anomaly of the alimentary tract (Figs. 11-21 and 11-22A). A congenital ileal diverticulum (Meckel diverticulum) occurs in 2% to 4% of people and is three to five times more prevalent in males than females. An ileal diverticulum is of clinical significance because it may become inflamed and cause symptoms that mimic appendicitis.
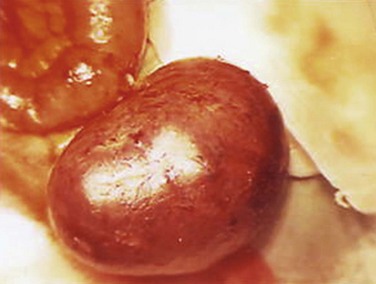
FIGURE 11–21 Photograph of a large ileal diverticulum (Meckel diverticulum). Only a small percentage of these diverticula produce symptoms. Ileal diverticula are one of the most common birth defects of the alimentary tract.
(Courtesy of Dr. M.N. Golarz De Bourne, St. George’s University Medical School, Grenada.)
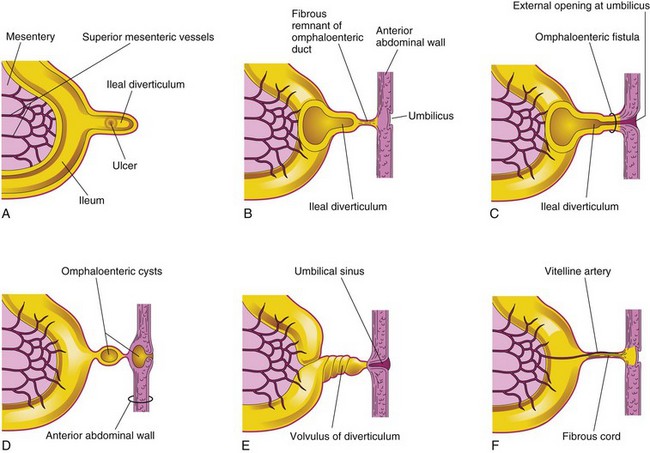
FIGURE 11–22 Ileal diverticula and remnants of the omphaloenteric duct. A, Section of the ileum and a diverticulum with an ulcer. B, A diverticulum connected to the umbilicus by a fibrous remnant of the omphaloenteric duct. C, Omphaloenteric fistula resulting from persistence of the intra-abdominal part of the omphaloenteric duct. D, Omphaloenteric cyst at the umbilicus and in the fibrous remnant of the omphaloenteric duct. E, Volvulus (twisted) ileal diverticulum and an umbilical sinus resulting from the persistence of the omphaloenteric duct in the umbilicus. F, The omphaloenteric duct has persisted as a fibrous cord connecting the ileum with the umbilicus. A persistent vitelline artery extends along the fibrous cord to the umbilicus. This artery carried blood to the umbilical vesicle from the embryo.
The wall of the diverticulum contains all layers of the ileum and may contain small patches of gastric and pancreatic tissues. This ectopic gastric mucosa often secretes acid, producing ulceration and bleeding (Fig. 11-22A). An ileal diverticulum is the remnant of the proximal part of the omphaloenteric duct. It typically appears as a finger-like pouch approximately 3 to 6 cm long that arises from the antimesenteric border of the ileum (Fig. 11-21), 40 to 50 cm from the ileocecal junction. An ileal diverticulum may be connected to the umbilicus by a fibrous cord (which may predispose to intestinal obstruction as the intestine can wrap around this cord) or an omphaloenteric fistula (Figs. 11-22B and C, and 11-23B); other possible remnants of the omphaloenteric duct are illustrated in Figure 11-22D to F.
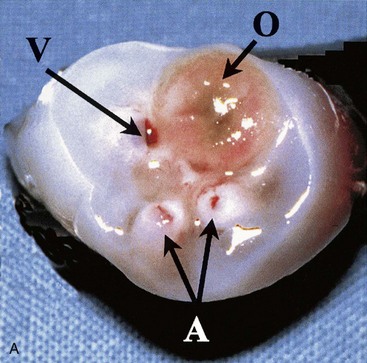
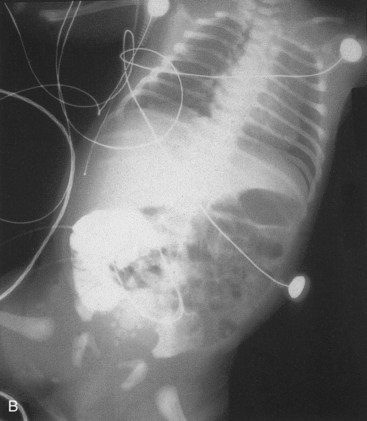
FIGURE 11–23 Male neonate with a patent omphaloenteric duct. A, The transected umbilical cord shows two umbilical arteries (A), an umbilical vein (V), and a larger lumen (O) of the omphaloenteric duct. B, An abdominal radiograph identifies contrast material injected through the omphaloenteric duct into the ileum.
(From Hinson RM, Biswas A, Mizelelle KM, Tunnessen WW Jr: Picture of the month (persistent omphalomesenteric duct). Arch Pediatr Adolesc Med 151:1161, 1997. Copyright 1997, American Medical Association. All rights reserved.)
Duplication of Intestine
Most intestinal duplications are cystic or tubular. Cystic duplications are more common (Fig. 11-24A and B). Tubular duplications usually communicate with the intestinal lumen (Fig. 11-24C ). Almost all duplications are caused by failure of normal recanalization of the small intestine; as a result, two lumina form (Fig. 11-24H and I ). The duplicated segment lies on the mesenteric side of the intestine. The duplication often contains ectopic gastric mucosa, which may result in local peptic ulceration and gastrointestinal bleeding.
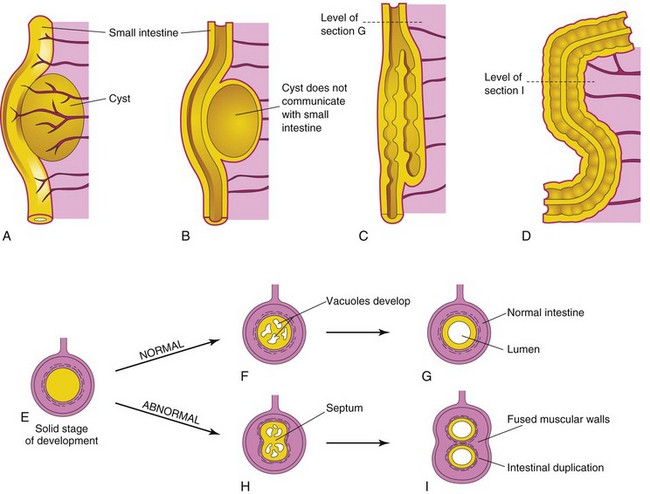
FIGURE 11–24 A, Cystic duplication of the small intestine on the mesenteric side of the intestine; it receives branches from the arteries supplying the intestine. B, Longitudinal section of the duplication shown in A; its musculature is continuous with the intestinal wall. C, A short tubular duplication. D, A long duplication showing a partition consisting of the fused muscular walls. E, Transverse section of the intestine during the solid stage. F, Normal vacuole formation. G, Coalescence of the vacuoles and reformation of the lumen. H, Two groups of vacuoles have formed. I, Coalescence of the vacuoles illustrated in H results in intestinal duplication.
Hindgut
The derivatives of the hindgut are:
All hindgut derivatives are supplied by the inferior mesenteric artery. The junction between the segment of transverse colon derived from the midgut and that originating from the hindgut is indicated by the change in blood supply from a branch of the superior mesenteric artery to a branch of the inferior mesenteric artery.
The descending colon becomes retroperitoneal as its mesentery fuses with the parietal peritoneum on the left posterior abdominal wall and then disappears (Fig. 11-15B and E). The mesentery of the fetal sigmoid colon is retained, but it is smaller than in the embryo (Fig. 11-15D).
Cloaca
The expanded terminal part of the hindgut, the cloaca, is an endoderm-lined chamber that is in contact with the surface ectoderm at the cloacal membrane (Fig. 11-25A and B). This membrane is composed of endoderm of the cloaca and ectoderm of the anal pit (Fig. 11-25D). The cloaca receives the allantois ventrally, which is a finger-like diverticulum (Fig. 11-25A).
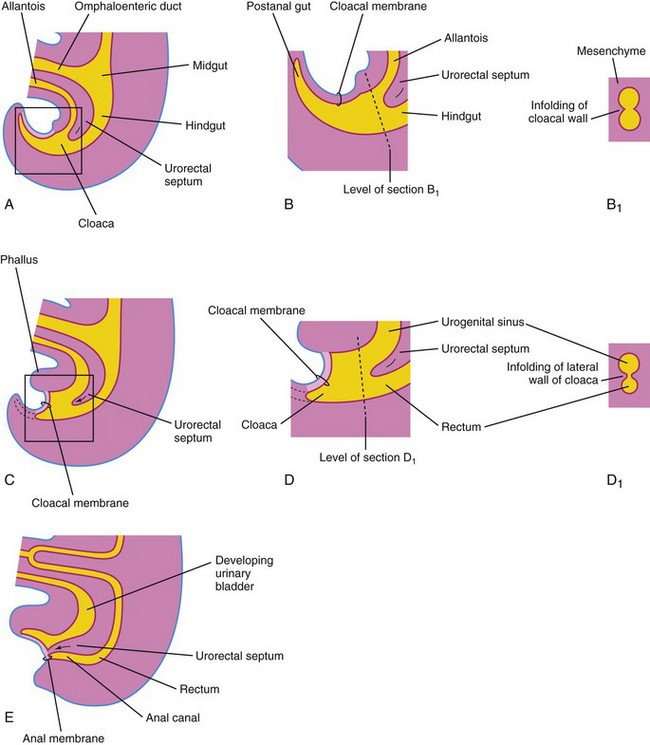
FIGURE 11–25 Successive stages in the partitioning of the cloaca into the rectum and urogenital sinus by the urorectal septum. A, C, and E, Views from the left side at 4, 6, and 7 weeks, respectively. B, D, and F, Enlargements of the cloacal region. B1 and D1, Transverse sections of the cloaca at the levels shown in B and D. Note that the postanal portion (shown in B) degenerates and disappears as the rectum forms.
Partitioning of Cloaca
The cloaca is divided into dorsal and ventral parts by a wedge of mesenchyme—the urorectal septum—that develops in the angle between the allantois and hindgut. As the septum grows toward the cloacal membrane, it develops fork-like extensions that produce infoldings of the lateral walls of the cloaca (Fig. 11-25B). These folds grow toward each other and fuse, forming a partition that divides the cloaca into two parts: the rectum, the cranial part of the anal canal, and the urogenital sinus (Fig. 11-25D and E).
The cloaca plays a crucial role in anorectal development. New information indicates that the urorectal septum does not fuse with the cloacal membrane; therefore, an anal membrane does not exist. After the cloacal membrane ruptures by apoptotic cell death, the anorectal lumen is temporarily closed by an epithelial plug, which may have been interpreted as the anal membrane (Fig. 11-25E).
Mesenchymal proliferations produce elevations of the surface ectoderm around the epithelial anal plug. Recanalization of the anorectal canal occurs by apoptotic cell death of the epithelial anal plug, which forms the anal pit (Fig. 11-25E).
Anal Canal
The superior two thirds of the adult anal canal are derived from the hindgut; the inferior one third develops from the anal pit (Fig. 11-26). The junction of the epithelium derived from the ectoderm of the anal pit and the endoderm of the hindgut is roughly indicated by the irregular pectinate line, located at the inferior limit of the anal valves. Approximately 2 cm superior to the anus is the anocutaneous line (“white line”). This is approximately where the composition of the anal epithelium changes from columnar to stratified squamous cells. At the anus, the epithelium is keratinized and continuous with the skin around the anus. The other layers of the wall of the anal canal are derived from splanchnic mesenchyme. The formation of the anal sphincter appears to be under Hox D genetic control.
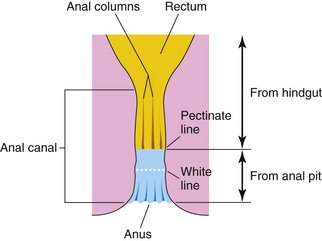
FIGURE 11–26 Sketch of the rectum and anal canal showing their developmental origins. Note that the superior two thirds of the anal canal are derived from the hindgut, whereas the inferior one third of the canal is derived from the anal pit. Because of their different embryologic origins, the superior and inferior parts of the anal canal are supplied by different arteries and nerves and have different venous and lymphatic drainages.
Because of its hindgut origin, the superior two thirds of the anal canal are mainly supplied by the superior rectal artery, the continuation of the inferior mesenteric artery (hindgut artery). The venous drainage of this superior part is mainly via the superior rectal vein, a tributary of the inferior mesenteric vein. The lymphatic drainage of the superior part is eventually to the inferior mesenteric lymph nodes. Its nerves are from the autonomic nervous system (ANS).
Because of its origin from the anal pit, the inferior one third of the anal canal is supplied mainly by the inferior rectal arteries, branches of the internal pudendal artery. The venous drainage is through the inferior rectal vein, a tributary of the internal pudendal vein that drains into the internal iliac vein. The lymphatic drainage of the inferior part of the anal canal is to the superficial inguinal lymph nodes. Its nerve supply is from the inferior rectal nerve; hence, it is sensitive to pain, temperature, touch, and pressure.
The differences in blood supply, nerve supply, and venous and lymphatic drainage of the anal canal are important clinically, such as when considering the metastasis (spread) of cancer cells. The characteristics of carcinomas in the two parts also differ. Tumors in the superior part are painless and arise from columnar epithelium, whereas tumors in the inferior part are painful and arise from stratified squamous epithelium.
Congenital Megacolon or Hirschsprung Disease
This disease is a dominantly inherited multigenic disorder with incomplete penetrance and variable expressivity. Of the genes so far identified, the RET proto-oncogene is the major susceptibility gene and accounts for most cases. This disease affects one in 5000 newborns and is defined as an absence of ganglion cells (aganglionosis) in a variable length of distal bowel.
Infants with congenital megacolon, or Hirschsprung disease (Fig. 11-27), lack autonomic ganglion cells in the myenteric plexus distal to the dilated segment of colon. The enlarged colon—megacolon—has the normal number of ganglion cells. The dilation results from failure of relaxation of the aganglionic segment, which prevents movement of the intestinal contents, resulting in dilation. In most cases, only the rectum and sigmoid colon are involved; occasionally, ganglia are also absent from more proximal parts of the colon.

FIGURE 11–27 Radiograph of the colon after a barium enema in a 1-month-old infant with congenital megacolon (Hirschsprung disease). The aganglionic distal segment (rectum and distal sigmoid colon) is narrow, with distended normal ganglionic bowel, full of fecal material, proximal to it. Note the transition zone (arrow).
(Courtesy of Dr. Martin H. Reed, Department of Radiology, University of Manitoba and Children’s Hospital, Winnipeg, Manitoba, Canada.)
Megacolon is the most common cause of neonatal obstruction of the colon and accounts for 33% of all neonatal obstructions; males are affected more often than females (4 : 1). Megacolon results from failure of neural crest cells to migrate into the wall of the colon during the fifth to seventh weeks. This results in failure of parasympathetic ganglion cells to develop in the Auerbach and Meissner plexuses.
Anorectal Anomalies
Most anorectal anomalies result from abnormal development of the urorectal septum, resulting in incomplete separation of the cloaca into urogenital and anorectal parts (see Fig. 11-29A ). Sonic hedgehog (Shh) and fibroblast growth factor (Fgf-10) have been implicated in birth defects of the hindgut. There is normally a temporary communication between the rectum and anal canal dorsally from the bladder and urethra ventrally (Fig. 11-25C ). Lesions are classified as low or high depending on whether the rectum ends superior or inferior to the puborectalis muscle, which maintains fecal continence and relaxes to allow defecation.
Low Birth Defects of Anorectal Region
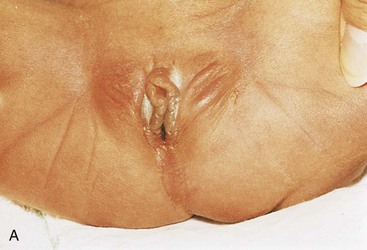
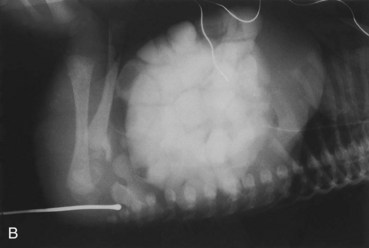
FIGURE 11–28 Imperforate anus. A, Female neonate with anal atresia (imperforate anus). In most cases, a thin layer of tissue separates the anal canal from the exterior. Some form of imperforate anus occurs approximately once in every 5000 neonates; it is more common in males. B, Radiograph of an infant with an imperforate anus. The dilated end of the radiopaque probe is at the bottom of the blindly ending anal pit. The large intestine is distended with feces and contrast material.
(A, Courtesy of A.E. Chudley, MD, Section of Genetics and Metabolism, Department of Pediatrics and Child Health, Children’s Hospital, Winnipeg, Manitoba, Canada. B, Courtesy of Dr. Prem S. Sahni, formerly of the Department of Radiology, Children’s Hospital, Winnipeg, Manitoba, Canada.)
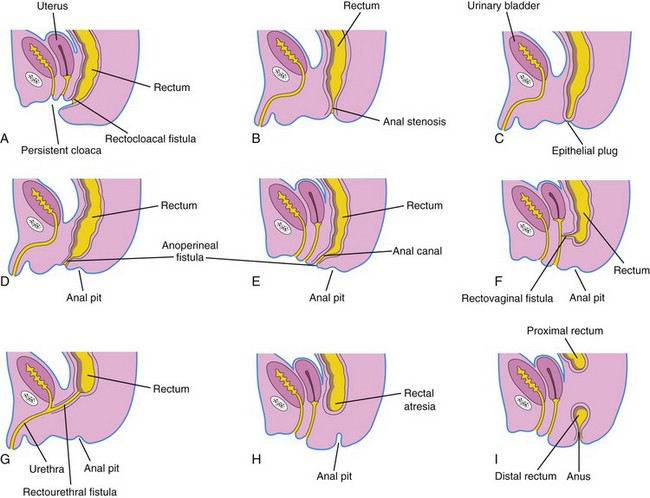
FIGURE 11–29 Various types of anorectal birth defects. A, Persistent cloaca. Note the common outlet for the intestinal, urinary, and reproductive tracts. B, Anal stenosis. C, Anal atresia (imperforate anus). D and E, Anal agenesis with a perineal fistula. F, Anorectal agenesis with a rectovaginal fistula. G, Anorectal agenesis with a rectourethral fistula. H and I, Rectal atresia.
High Birth Defects of Anorectal Region
In high anomalies of the anorectal region, the rectum ends superior to the puborectalis muscle when there is anorectal agenesis. This is the most common type of anorectal birth defect. Although the rectum ends blindly, there is usually a fistula to the bladder (rectovesical fistula) or urethra (rectourethral fistula) in males or to the vagina (rectovaginal fistula) or the vestibule of the vagina (rectovestibular fistula) in females (Fig. 11-29F and G ).
Anorectal agenesis with a fistula is the result of incomplete separation of the cloaca from the urogenital sinus by the urorectal septum (Fig. 11-25C to E ). In newborn males with this condition, meconium may be observed in the urine, whereas fistulas in females result in the presence of meconium in the vestibule of the vagina.
In rectal atresia, the anal canal and rectum are present but are separated (Fig. 11-29H and I ). Sometimes the two segments of intestine are connected by a fibrous cord, the remnant of an atretic portion of the rectum. The cause of rectal atresia may be abnormal recanalization of the colon or, more likely, defective blood supply.
Summary of Alimentary System
Clinically Oriented Problems
Case 11–1
A female infant was born prematurely at 32 weeks gestation to a 39-year-old woman whose pregnancy was complicated by polyhydramnios. Amniocentesis at 16 weeks showed that the infant had trisomy 21. The baby began to vomit within a few hours after birth. Marked dilation of the epigastrium was noted. Radiographs of the abdomen showed gas in the stomach and the superior part of the duodenum, but no other intestinal gas was observed. A diagnosis of duodenal atresia was made.
Case 11–2
The umbilicus of a newborn infant failed to heal normally. It was swollen and there was a persistent discharge from the umbilical stump. A sinus tract was outlined with contrast media during fluoroscopy. The tract was resected on the ninth day after birth and its distal end was found to terminate in a diverticulum of the ileum.
Case 11–3
A female infant was born with a small dimple where the anus should have been. Examination of the infant’s vagina revealed meconium and an opening of a sinus tract in the posterior wall of the vagina. Radiographic examination using a contrast medium injected through a tiny catheter inserted into the opening revealed a fistulous connection.
Case 11–4
A newborn infant was born with a light gray, shiny mass measuring the size of an orange that protruded from the umbilical region. The mass was covered by a thin transparent membrane.
Case 11–5
A newborn infant appeared normal at birth; however, excessive vomiting and abdominal distention developed after a few hours. The vomitus contained bile and only a little meconium was passed. Radiographic examination showed a gas-filled stomach and dilated, gas-filled loops of small bowel, but no air was present in the large intestine. This indicated a congenital obstruction of the small bowel.
Discussion of these problems appears at the back of the book.
References and Suggested Reading
Bates MD, Balistreri WF. Development and function of the liver and biliary system. In Behrman RE, Kliegman RM, Jenson HB, editors: Nelson Textbook of Pediatrics, ed 17, Philadelphia: WB Saunders, 2004.
Bronshtein M, Blazer S, Zimmer EZ. The fetal gastrointestinal tract and abdominal wall. In Callen PW, editor: Ultrasonography in Obstetrics and Gynecology, ed 5, Philadelphia: WB Saunders, 2008.
Burn SF, Hill RE. Left–right asymmetry in gut development: What happens next. BioEssays. 2009;31:1026.
Heath JK. Transcriptional networks and signaling pathway that govern vertebrate intestinal development. Curr Top Dev Biol. 2010;90:159.
Kapur RP. Practical pathology and genetics of Hirschsprung’s disease. Semin Pediatr Surg. 2009;18:212.
Kluth D, Fiegel HC, Metzger. Embryology of the hindgut. Semin Pediatr Sur. 2011;20:152.
Lade AG, Monga SPS. Beta-catenin signaling in hepatic development and progenitors: which way does WNT blow. Dev Dyn. 2011;240:486.
Lau ST, Caty MG. Hindgut abnormalities. Surg Clin North Am. 2006;86:285.
Ledbetter DJ. Gastroschisis and omphalocele. Surg Clin North Am. 2006;86:249.
Levitt MA, Pena A. Cloacal malformations: Lessons learned from 490 cases. Semin Pediatr Surg. 2010;9:118.
Magnuson DK, Parry RL, Chwals WJ. Selected abdominal gastrointestinal anomalies. In Martin RJ, Fanaroff AA, Walsh MC, editors: Fanaroff and Martin’s Neonatal–Perinatal Medicine: Diseases of the Fetus and Infant, ed 8, Philadelphia: Mosby, 2006.
Metzger R, Metzger U, Fiegel HC, et al. Embryology of the midgut. Semin Pediatr Sur. 2011;20:145.
Metzger R, Wachowiak R, Kluth Dl. Embryology of the early foregut. Semin Pediatr Sur. 2011;20:136.
Mundt E, Bates MD. Genetics of Hirschsprung disease and anorectal malformations. Semin Pediatr Surg. 2010;19:107. Wilkins, 2010
Okamoto T, Takamizawa S, Arai H, et al. Esophageal atresia: Prognostic classification revisited. Surgery. 2009;145:675.
Naik-Mathuria B, Olutoye OO. Foregut abnormalities. Surg Clin North Am. 2006;86:261.
Vakili K, Pomfret EA. Biliary anatomy and embryology. Surg Clin North Am. 2008;88:1159.
Van der Putte SCJ. The development of the human anorectum. Anat Rec. 2009;292:952.