Chapter 9 Treatment of Fungal Infections
Currently, only nine antifungal drugs, representing five classes, are approved for use in the United States.
Fungal Physiology
The pathogenic fungi affecting humans and animals are eukaryotes, generally existing as either filamentous molds (hyphal forms) or intracellular yeasts (Table 9-1).1 Dimorphic fungi grow in the host as a yeastlike form but as molds in vitro at room temperature. Some fungi (e.g., Coccidioides immitis, Histoplasma, and Rhinosporidium species) grow inside host cells, dividing into spores until released from the cell as it ruptures. The fungal cell wall is a target for several of the antifungal drugs (Figure 9-1). It is rigid and contains chitin, a structural component, and polysaccharides. These generally preclude Gram staining and serve as a barrier to drug penetration. The cell membrane is complex and, unlike bacteria but as with higher eukaryotes, contains sterols.2 Ergosterol is the primary sterol component of fungal cell membranes, regulating both permeability and membrane-bound enzymes. Because it also is a major component of organelle membranes, it also influences mitochondrial respiration and oxidative phosphorylation.3 The rate of chitin synthesis is influenced by ergosterol content, being inhibited at high concentrations and stimulated at low concentrations. Fungal content of ergosterol influences drug efficacy and the potential risk of resistance for some drugs. Microtubular structures found in all eukaryotic cells support the cellular cytoskeleton and mitotic spindle; they support not only cell division but also cellular integrity. Organelle position and movement are supported by microtubules. The tubules in turn are comprised of tubulin, a heterodimer containing α and β subunits. Because microtubules are constantly being assembled and dissembled, impaired microtubular synthesis alters integral cellular function.
Table 9-1 Classification of Selected Fungi of Medical Importance
| Level | Name | Disease or Infecting Organism |
|---|---|---|
| Kingdom | Fungi | |
| Subkingdom | Amastigomycotera | |
| Phylum | Zygomycota | |
| Class | Zygomycetes | |
| Order | Mucorales | |
| Family | Mucoraceae | |
| Genus | Absidia | Absidia (Zygomycosis, mucormycosis) |
| Mucor | Mucor (Zygomycosis, mucormycosis) | |
| Rhizopus | Rhizopus (Zygomycosis, mucormycosis) | |
| Family | Several others | |
| Order | Entomophthorales | |
| Family | Basidiobolaceae | |
| Genus | Basidiobolus | Basidiobolus sp. |
| Subkingdom | Eumycotera | |
| Phylum | Ascomyta | |
| Subphylum | Saccharomycotina | |
| Order | Saccharomycetales | |
| Family | Saccharomycetaceae | Budding yeasts |
| Genus | Saccharomyces | Budding yeasts |
| Genus | Debaryomyces | Candida∗ |
| Khyveromyces | Candida∗ | |
| Lodderomyces | Candida∗ | |
| Pichia | Candida∗ | |
| Subphylum | Pneumocystiodiomycetes | |
| Order | Pneumocystidiales | |
| Family | Pneumocystideaceae | |
| Genus | Pneumocystis jirovecii | Pneumocystis pneumonia |
| Subphylum | Euascomycotina | |
| Order | Eurotiales | |
| Family | Eurotiaceae | |
| Genus | Eurotium, Emericella | Aspergillus∗ |
| Genus | Talaromyces | Penicillium∗ |
| Order | Onygenales | |
| Family | Gymnoascaceae/Ajellomycetaceae | |
| Genus | Ajellomyces | Blastomyces dermatitidis∗ |
| Histoplasma capsulatum∗ | ||
| Paracoccidioides braziliensis∗ | ||
| Family | Gymnoascaceae/Arthrodermataceae | |
| Genus | Arthroderma | Microsporum |
| Trichophyton | ||
| Epidermophyton | ||
| Class | Pyrenomycetes/Sordariomycetes | |
| Order | Ophiostomatales | |
| Family | Ophiostomataceae | |
| Genus | Ophiostoma | Sporothrix schenckii∗ |
| Order | Hypocreales | |
| Family | Many | Fusarium |
| Order | Clavicipitales | |
| Family | Clavicipitaceae | |
| Genus | Ergot alkaloids | St. Anthony’s fire |
| Phylum | Basidiomycota | |
| Subphylum | Holobasidiomycont Basidiomycot | |
| Class | Phragmobasidiomycetes/Tremellomycetes | |
| Order | Trichosporonales | Trichosporon asahii∗ |
| Order | Filobasidiales | |
| Family | Filobasidiaceae | |
| Genus | Filobasidiella | Cryptococcus neoformans∗ |
| Phylum | Fungi Imperfecti/Deuteromycota | |
| Form–Class | Blastomycetes | |
| Form–Order | Cryptococcales | |
| Form–Family | Cryptococcaceae | |
| Genus | Candida | Candida |
| Genus | Cryptococcus | Cryptococcus |
| Genus | Malassezia | Malassezia |
| Genus | Pityrosporum | Pityrosporum |
| Genus | Rhodotorula | Rhodotorula |
| Genus | Trichosporon | Trichosporon |
| Form–Class | Hyphomycetes | |
| Form–Order | Moniliales | |
| Form–Family | Moniliaceae | |
| Genus | Aspergillus | Aspergillus |
| Genus | Blastomyces | Blastomyces |
| Genus | Coccidioides | Coccidioides |
| Genus | Epidermophyton | Epidermophyton |
| Genus | Geotrichum | Geotrichum |
| Genus | Hostoplasma | Hostoplasma |
| Genus | Microsporum | Microsporum |
| Genus | Paracoccidioides | Paracoccidioides |
| Genus | Penicillium | Penicillium |
| Genus | Sporothrix | Sporothrix |
| Genus | Trichophyton | Trichophyton |
| Genus | Others | |
| Form–Family | Dematiaceae | Alternaria |
| Genus | Alternaria | Bipolaris |
| Genus | Bipolaris | Cladophialophora |
| Genus | Cladophialophora | Curvularia |
| Genus | Curvularia | Helminthosporium |
∗ Teleomorphic form or sexually producing state.
(From Reference Guide to the Classification of Fungi and Fungal-like Protists, with Emphasis on the Genera with Medical Importance (circa 2007), accessed December 17, 2009, at http://www.sbs.utexas.edu/mycology/bio329/pdf_files/sp2007/refguidefungal_sp2007.pdf.)
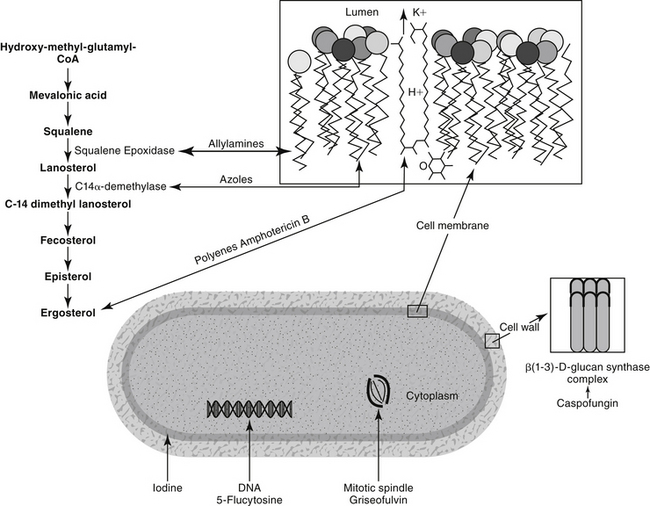
Figure 9-1 Mechanism of action of antifungal drugs. Most clinically relevant antifungals target cell wall ergosterol through different mechanisms. Amphotericin B binds to it, creating channels in the cell membrane. Azoles impair ergosterol synthesis by targeting C-14 α-demethylase while allylamines (e.g., terbinafine) inhibit squalene epoxidase. Drugs or drug classes that do not target ergosterol include 5-flucytosine, which impairs DNA synthesis upon its intracellular conversion to 5-fluouracil; griseofulvin, which impairs formation of the mitotic spindle; and caspofungin, an echinocandin that targets β-D-glucan synthesis in the fungal cell wall. Iodine may precipitate cellular proteins.
Pathophysiology of Infection
Fungal infections differ from bacterial infections in several respects, and pathogenic fungi have developed several characteristics that complicate antimicrobial therapy.4 Some of the differences in the eukaryotic structure of fungal organisms compared with the prokaryotic structures of bacteria were delineated in the preceding section. Other differences exist. For example, Cryptococcus and occasionally Sporothrix schenkii produce an external coating or slime layer that encapsulates the cells and causes them to adhere and clump together.2 Whereas many fungal organisms produce exotoxins in vivo, there is no conclusive evidence that fungi produce endotoxins. Fungal organisms are characterized by a low invasiveness and virulence. In fact, most animals will overcome a fungal infection. Factors that predispose the patient to infection include necrotic tissue, a moist environment, and immunosuppression. Innate immunity generally rapidly identifies and destroys fungal organisms that present pathogen-associated molecular patterns recognized by specific receptors on host cells. Pattern recognition receptors include the lectinlike receptor dectin-1, which binds beta-glucan in the fungal cell wall, and the Toll-like receptors. Binding by neutrophils and macrophages results in a cascade of intracellular events that cause rapid clearance in tissues of exposure. Organisms that are able to penetrate these tissues initiate an immune response. Immunity to fungal organisms appears to be T-cell mediated, with tumor necrosis factor-alpha (TNF-α) being a major signaling cytokine, although all but dermatophytes also stimulate antibody production.5 In response, fungal organisms impair an effective immune response through a number of mechanisms. A number of molecules scavenge oxygen radicals generated by inflammatory cells (melanin, mannitol, catalase). The cell wall of Histoplasma capsulatum contains β-glucan, which covers and hides α-glucan, preventing recognition by phagocytic cells. Further, it is able to survive intracellular locations in phagocytic cells by a number of mechanisms. Phenotype switching prevents receptor recognition by host cells.5 Gliotoxin is an immunosuppressive mycotoxin, produced particularly well by Aspergillus fumigatus.5
>KEY POINT 9-1
The slow-growing nature of fungal infections requires long-term therapy. However, antifungal drugs are inherently more toxic than antibacterial drugs because fungal targets are often similar to mammalian targets.
Although an adequate immune response and recruitment of appropriate inflammatory cells to the site of infection is critical to resolution of fungal infections, progression to uncontrolled, pyogranulomatous inflammation is often deleterious. Proper early cellular recruitment may determine the difference between success and failure. The positive effects of some antifungals may include their immunomodulatory capabilities. These beneficial effects, which are largely based on in vitro studies, with an occasional murine mouse model verification, have been reviewed by Ben-Ami and colleagues.5 Amphotericin B, the azoles, and the echinocandins have been studied most. Among the effects is downregulation of inflammatory cytokine genes. The use of antiinflammatories should be considered in critical situation. The use of glucocorticoids is addressed in the section on the treatment of fungal infections, but other antiinflammatories that minimally affect lymphocytic-mediated immunity (e.g., nonsteroidal antiinflammatories, leukotriene receptor antagonists) should be considered as long as attention is paid to potential negative drug interactions involving imidazoles in particular. Manipulation of the immune response in combination with antifungal drugs is a more recent area of focus that is likely to affect treatment of fungal disorders.
Fungal infections can be primarily superficial and irritating (e.g., dermatophytosis) or systemic and life threatening (e.g., dimorphic fungal infections including blastomycosis, cryptococcosis, histoplasmosis, and coccidioidomycosis). Fungal organisms may exhibit an affinity for certain tissues, such as the dermatophytes for keratin and H. capsulatum for macrophages. Animals may develop a hypersensitivity to the infecting organism (as is often seen in dermatophyte infections), which can result in a pathologic response to the infection as well as facilitate dissemination. The role of proteolytic enzymes in infections caused by dermatophytes is being investigated. On the other hand, the lack of hypersensitivity may also indicate a poorer prognosis for recovery.2
Information regarding antifungal drug use in animals is limited. Human literature often focuses on candidiasis, an infection that is much less common in dogs and cats; as such, relevance of information must be considered. The risk factors for fungal infection have been recently reviewed.7 Using candidiasis as an example, broad-spectrum antimicrobials or immunosuppressive drugs (glucocorticoids, chemotherapy) play a major role. Malnutrition, malignancy, age extremes, and neutropenia also predispose to candidiasis. Similar risk factors occur for aspergillosis, although increased organ transplantation and its accompanying immunosuppression probably has contributed to not only the increased incidence of infection but also the emergence of newer species. Likewise, the increased invasiveness of life-extending procedures coupled with increased survival is contributing to an increased incidence of infection. Improved diagnostic techniques are allowing identification of previously unknown organisms; for example, other filamentous organisms that are emerging in human medicine include the Zygometes (Mucor, Rhizopus, Rhizomucor, Absidia, and Cunninghamella), Fusarium, Paecilomyces, and Scedosporium.
Several factors can lead to therapeutic failure or relapse after antifungal therapy.4 Most antifungals are fungistatic in action, with clearance of infection largely dependent on host response.6 In humans relapsing infections are not uncommon for selected Trichophyton species and for invasive mycoses in immunocompromised patients. In the latter group, aspergillosis infections are particularly problematic. As with bacteria, the pattern of fungal disease is constantly changing. The advent of acquired immunodeficiency syndrome in human patients has been important in the development of new strains of resistant organisms, and there remains a continuing need for development of new antifungal agents. However, this represents only one of many factors that increase the risk of fungal infection. In some instances, therapeutic failure reflects poor penetration of drug into infected tissues (particularly the central nervous system and bone) or into those organisms that are encapsulated.
Several organisms, particularly the superficial pathogens and systemic opportunistic organisms, have a primary resistance to antifungal drugs, contributing to therapeutic failure. Like antibacterial resistance, antifungal resistance can be intrinsic (primary) or acquired (secondary). A third type of resistance, referred to as clinical resistance, involves the progression or relapse that occurs despite laboratory-documented susceptibility to the treatment drug.8 Primary resistance occurs with amphotericin B to filamentous fungi and dermatophytes. The risk factors for acquired resistance have not been well identified. Because resistance is an increasingly emerging problem, dosing regimens should maximize antifungal plasma concentrations. However, much more so than antibacterial drugs, antifungal drugs present a risk of toxicity causing the design of dosing regimens to be much more restrictive compared to antibacterial therapy.
Toxicity of antifungals is a common cause of therapeutic failure. Because both the antifungal target organism and the host cells are eukaryotic, the cellular targets of fungal organisms are substantially different from those of bacterial organisms. As a result, antibacterials generally are ineffective against fungal organisms, and, in contrast to most antibacterials, antifungals are often toxic or associated with undesirable side effects in the host. The incidence of side effects has limited the number of effective yet safe antifungal drugs available. Some strategies for reducing toxicity have allowed dose escalation, increasing the likelihood of efficacy and decreasing the risk of resistance. Their use (e.g., liposomal amphotericin B products) are particularly important in the immunosuppressed patient. A potentially important strategy for avoidance of resistance is combination therapy.8,9 Combination therapy, if correctly designed, should also enhance efficacy and reduce toxicity through more rapid response to therapy. Dose reduction may be possible. Finally, a common reason for therapeutic failure is discontinuing therapy after resolution of clinical signs but before eradication of infection. Therefore antifungal therapy should extend well beyond clinical cure.
As with antibacterials, efficacy is influenced by the relationship between plasma or tissue concentrations, minimum inhibitory concentration (MIC) of the infecting microbe and antifungal. Studies for antifungal organisms generally begin with killing curves followed by animal models; however, as with antibacterials, their relevance also must be supported by pharmacokinetic studies that determine drug concentrations at the site of infection coupled with pharmacodynamic studies that determine susceptibility in terms of end points of efficacy.10
Compared with bacterial testing, antifungal culture and susceptibility testing have not been well developed as a tool for the treatment of fungal infections. As with antibacterial testing, antifungal testing is principally identification of resistant microbes rather than a description of the level of susceptibility.8 In vitro susceptibility testing of antifungal agents is highly dependent on test conditions, and interlaboratory results vary markedly. Interpretation of culture and susceptibility data may be limited by a lack of standardized testing methods for some drugs and organisms. As with bacteria, the MIC for a fungal organism is the concentration of the antifungal drug that inhibits the growth of the fungus under standardized conditions (Table 9-2). The minimum lethal concentration is the concentration that kills the organisms.1 Correlation between MIC and clinical response is poor, and assessment of antifungal agents appears to be best accomplished through efficacy studies in animal models. Fortunately, the need for fungal culture and susceptibility testing may not be as critical for fungal organisms as it is for bacterial organisms because, with the exception of 5–flucytosine, fungal development of resistance to antimicrobial therapy is not common.11 Resistance is more likely with a rapidly growing organism exposed to high concentrations of an antifungal for a long period of time.1 However, resistance does occur, and as with antibacterials, use of previous antifungal drugs appears to increase the risk of resistance. This has been demonstrated for Candida, the MICs of which tend to be higher in human patients previously treated with antifungals than in drug-naïve patients. Mechanisms of resistance of fungal organisms are similar to those of bacterial organisms (e.g., failure to accumulate in cells, altered target structures, formation of alternative pathways).12 The advent of newer antifungal agents and resistance among fungal organisms is, however, likely to cause in vitro testing of antifungals to become more important to therapeutic success.
>KEY POINT 9-2
Resistance by fungal organisms is not as common as in bacterial organisms, but it is more likely in those that are rapidly growing.
One author notes that never before have so many new antifungals been under development, ranging from entirely new compounds with new targets to modifications of existing drugs (e.g., cyclodextrin–itraconazole and polyethylene glycol–amphotericin B).13 Newer concepts being explored include combinations of antifungals with one another or with nonantibiotic compounds. Newer therapies may focus on immunomodulation14 with a balance between recruitment (cytokines, chemokines, lymphokines, and growth factors) and antiinflammatory effects. The need for new therapies is timely, as the epidemiologic behavior of fungal organisms, at least as they occur in human medicine, increasingly is shifting toward opportunistic organisms for which traditional antifungals often are characterized by limited efficacy.7
Antifungal Drugs
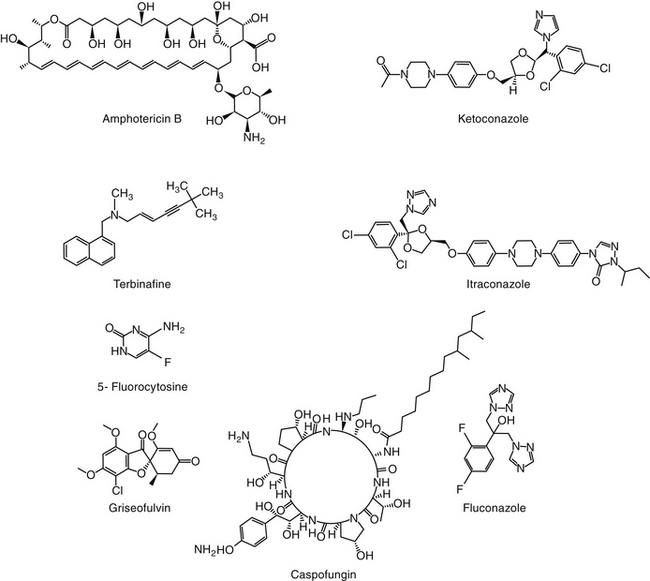
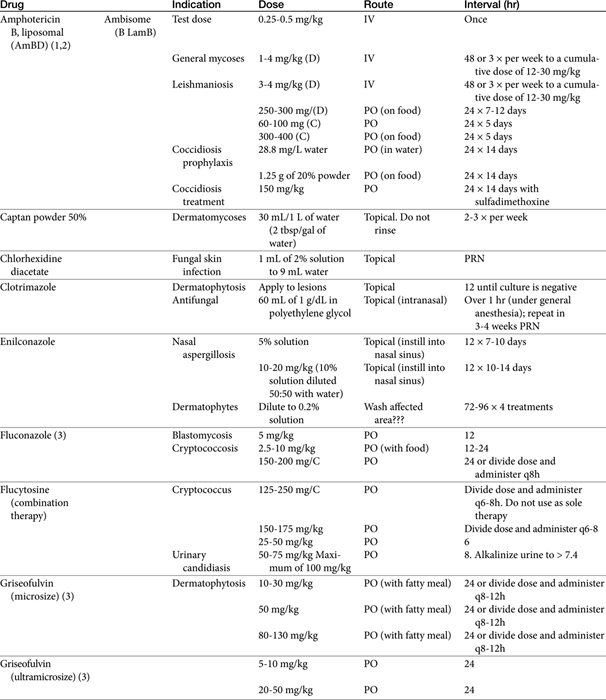
The primary agents used to treat fungal infections are the natural antibiotics; the polyene macrolides (amphotericin B as the prototype); the synthetic agents, including the azoles (ketoconazole as the prototype); and the newer allylamine antifungals (Figure 9-2). Flucytosine has a less important role in the treatment of dimorphic fungal diseases, particularly in animals. The natural antibiotic griseofulvin belongs to no group but has an important place in the armamentarium against dermatophytosis. As recently as 1988, the treatment of systemic fungal infections in humans emphasized the use of amphotericin B, ketoconazole, and flucytosine. In the decade that followed, further development of the azole derivatives has led to a new age in the treatment of systemic fungal diseases. Currently, antifungal therapy is most effective when based on an understanding of the therapeutic ratio of the drug in the infection being treated. For amphotericin B, this ratio tends to be small because of its toxicity. The newer azole derivatives have proved to provide much of the efficacy of amphotericin B without its toxicity. Doses for selected antifungal drugs are found in Table 9-3.
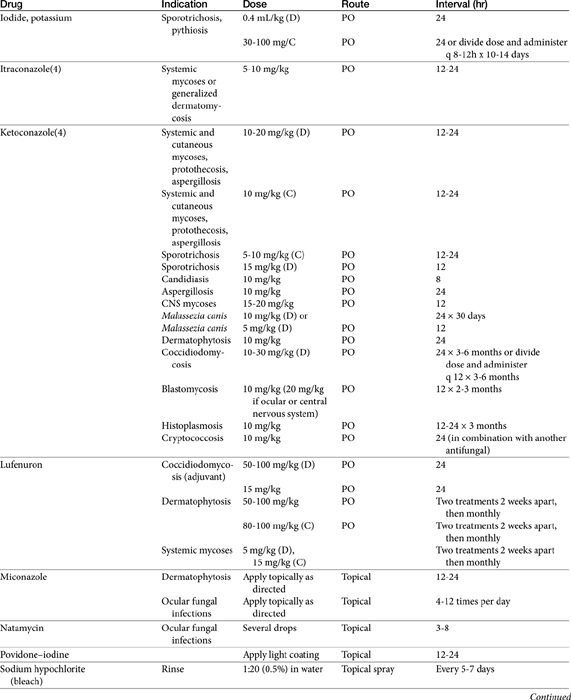
Polyene Macrolide Antibiotics: Amphotericin B
Structure–Activity Relationship
Examples of polyene (i.e., multiple double-bond; see Figure 9-1) antifungal drugs include amphotericin B, nystatin, and pimaricin. Each antibiotic is produced by a different species of Streptomyces (family Actinomyces). Amphotericin B was developed in the 1960s and was so successful in fulfilling the need for a broad-spectrum antifungal that further advancement of antifungal therapy was largely ignored. These drugs are very large molecules, consisting of a macrolide containing a large lactone ring. The polyene contains three to eight double bonds, which represent the lipophilic portion of the molecule. The number of double bonds categorizes the polyenes into trienes, tetraenes (natamycin [paramycin]), and pentanes; amphotericin B and candicidin are heptanes, whereas nystatin is classified as a pseudo–heptane/tetraene.3 A hydroxylated hydrocarbon backbone represents the hydrophilic portion of the molecule. These compounds are insoluble in water and are unstable, and they will rapidly decompose if exposed to sunlight.
Mechanism of Action
Polyene macrolides bind with the sterol portion of the phospholipids that make up the fungal cell membrane. Amphotericin has a much higher affinity for ergosterol, the major sterol component of fungal cell membranes, than for cholesterol, the major sterol in mammalian cell membranes (Figure 9-3).1 The interaction of the drug and the sterol results in the formation of channels or pores in the cell membrane (Figure 9-4). The result is an increase in cell permeability and disruption in proton gradient flow; loss of membrane fluidity may alter H+-ATPase activity.3 Altered K+/H+ exchange results in the loss of K+ and Mg2+ from the cell. Cellular metabolism is disrupted; internal acidification of the fungal cell and the loss of important organic molecules from the cell result in irreversible cell damage. The efficacy of some of the drugs can be related to their ability to bind to ergosterol. However, the polyenes also are associated with the formation of lethal reactive oxygen molecules. Polyenes also are defined according to the concentration of drug necessary to cause erythrocyte hemolysis, presumably because of cholesterol binding. Pimaricin causes lysis at low concentrations, limiting use to topical application, whereas amphotericin B and nystatin cause lysis at much higher concentrations.3
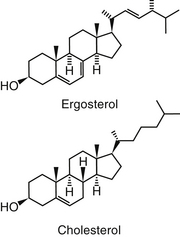
Figure 9-3 Cholesterol, the major sterol of mammalian cells, is structurally similar to ergosterol, the major sterol of fungal cell walls. Binding of amphotericin B to cholesterol results in complicated pharmacokinetics as well as host toxicity.
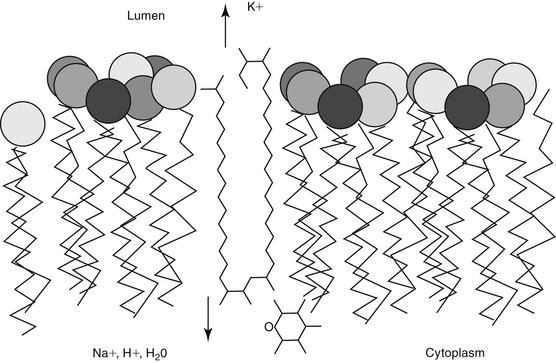
Figure 9-4 The mechanism of antifungal action and nephrotoxicity of amphotericin B reflect binding of the drug to ergosterol, the major sterol in fungal cell walls. Binding results in cell membrane permeability and the loss of critical micronutrients and electrolytes.
Amphotericin is fungistatic but can be fungicidal at high concentrations. At high concentrations the drug directly disrupts the fungal cell membrane. As with some other select antifungal drugs, amphotericin appears to have some immunomodulating characteristics. Both humoral and cell-mediated immunity may be enhanced, thus increasing the host’s ability to overcome infection. Amphotericin B activates macrophages and stimulates TNF-α and interleukin-1 (IL-1), both of which facilitate macrophage killing by way of nitric oxide–dependent pathways.5,14
Spectrum of Activity and Pharmacodynamics
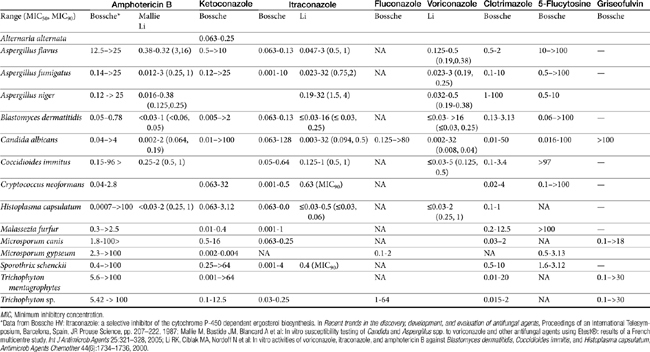
Despite the advent of the azole antifungal drugs, amphotericin B remains the most effective agent against most of the major fungal pathogens. The indications for amphotericin therapy include most systemic fungal diseases, including those caused by the dimorphic fungi (histoplasmosis, blastomycosis, cryptococcosis, and coccidioidomycosis) and disseminated sporotrichosis, phycomycosis, aspergillosis, and candidiasis. Amphotericin B has greater activity against some organisms (e.g., Candida and Aspergillus species and coccidioidal meningitis) than the newer azoles and particularly fluconazole. Amphotericin B is not effective against dermatophytes. Amphotericin MICs appear to correlate well with efficacy in animal models and human patients.10 The MICs of amphotericin B toward selected fungal microbes are listed in Table 9-4.15,16 Although these are based on humans, they represent a reasonable target for isolates infecting animals as well. In addition to the MIC, the minimum fungicidal concentration (MFC) has been reported for selected organisms infecting humans (or animals).15 The MFC50 and MFC90; μg/mL) are, respectively, Blastomyces dermatitidis (0.125, 0.5); H. capsulatum (0.5, 2), and C. immitis (4, 16). In killing-curve studies, amphotericin B consistently exhibits concentration-dependent killing. However, killing is variable among organisms, being fungicidal toward Candida sp. but not toward many yeasts and other organisms. As a concentration-dependent drug, amphotericin B may exhibit a long post-antifungal effect (PAFE), which, like the postantibiotic effect of concentration antimicrobials, varies in duration with the organism and the relationship of the MIC to the plasma drug concentration (PDC).10 However, it can prolong the dosing interval of the drug. A PAFE of only 0.5 hours occurs at approximately 2 hours exposure at drug concentrations that reflect 0.5 times the MIC, but this duration is prolonged to approximately 10 hours at 32 times the MIC for Candida and Cryptococcus neoformans; the PAFE for the latter may exceed 12 hours.10
Table 9-4 Pharmacokinetic Data for Liposomal Amphotericin B Preparation in the Dog22
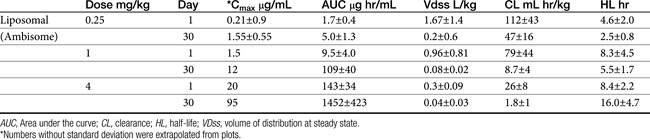
Sufficient data exist to integrate Ambisome pharmocokinetics and pharmacodynamics toward selected microbes that infect the dog (Table 9-4; see also Table 9-2). Two targets are considered: the MFC and a 12-hour PAFE (targeting PDCs that equal the MIC90 of the infecting microbe by 32). Based on the Cmax of dogs for Ambisome measured at day 1 and day 30 of dosing, the MFC of B. dermatitidis and H. capsulatum will be achieved on the first day of dosing at 1 mg/kg. Assuming the PAFE as described in animal models would be exhibited by dimorphic fungal organisms infecting dogs, based on MIC90 for B. dermatitidis of 0.05 μg/mL (see Table 9-2), a target of 1.6 μg/mL (32 times the MIC) is indicated to achieve a 10 to 12-hour PAFE. Again, the target will be reached in dogs on day 1 with a 1 mg/kg dose of Ambisome; the 12-hour PAFE could be added to the time the PDCs are above the MIC (which in turn, is based on half-life). For H. capsulatum, the MIC90 is 1 μg/mL; accordingly, a target of 32 μg/mL (for at least 2 hours) might be indicated to achieve a 10 to 12-hour PAFE. Assuming linear kinetics, reaching the target on day 1 will require a dose of approximately 4 mg/kg, which is likely not to be tolerated (an exception may occur for lipid-based products). However, the target is likely to be approached with 30 days of dosing at 2.5 mg/kg. C. immitis is less susceptible to the killing effects of amphotericin B; the MFC90 (11 μg/mL) is likely to be reached on day 1 of dosing at 2.5 mg/kg, but 30 days of dosing will be required to reach the MFC90 or the 12-hour PAFE. These calculations were based on the MIC90 of the respective organisms; if the MIC of the infecting microbe is known, then a lower dose might be reasonable.
Resistance
The incidence of resistance to amphotericin B is low and has been documented primarily for Candida. The development of resistance may be related to changes in the number of sterol components in the fungal cell wall, with low ergosterol content associated with resistance.3 Pythium insidiosum, classified as a pseudofungus, (the cause of what was previously and inappropriate referred to as fungal phycomycosis), as with other oomycetes, is largely devoid of cell membrane sterol. As such, they are inherently resistant to polyenes.1 However, other mechanisms of resistance also exist, probably reflecting multiple mechanisms. Some organisms are able to resist concentrations that exceed 2 μg/mL. The azole derivatives may contribute to amphotericin B resistance by preventing the formation of ergosterol, the target of amphotericin B.1
Amphotericin appears to act synergistically with the following: 5-flucytosine against cryptococcosis, tetracyclines against coccidioidomycosis, and imidazoles (discussed later) for a variety of fungal disorders. The use of synergistic combinations may enhance efficacy while reducing the potential of toxicity.
Preparations
Because amphotericin is unstable, it is prepared with desoxycholate as a lyophilized cake form (amphotericin B; previously Fungizone®; AmBD). Supplies were limited at the time of publication,8 perhaps reflecting declining use in human medicine in favor of newer lipid-based formulations.
Desoxycholate—also referred to as deoxycholate, a bile salt—is added to aid solubilization of the lyophilized cake form of the drug, which, when reconstituted, is in a colloidal suspension. Reconstitution should be with sterile water only; the solution will remain stable for 1 week if refrigerated. Storage should include protection from light, although protection during therapy is probably not necessary; potency is unaffected after light exposure of up to 24 hours. Labeled directions indicate that dilution should occur only with 5% dextrose to prevent drug precipitation and inactivation; Trissel’s Handbook on Injectable Drugs (http://online.statref.com/titles/titleinfopage.aspx?titleid=141) indicates that normal saline is incompatible with amphotericin B, leading to a 43% drop in drug concentrations within 2 hours. Although the drug has been mixed by veterinarians with 0.45% NaCl and 2.5% dextrose with no apparent change, the basis for this observation is unclear, and the prudent clinician would avoid the combination and provide sodium loading (see the section on therapeutic use) through a different catheter. In general, amphotericin B should not be mixed with other drugs; although known to be compatible with some, it is incompatible with many others. The drug should be administered only intravenously, with exceptions for localized treatment in selected body tissues or fluids (e.g., aqueous humor, cerebrospinal fluid [CSF]). For small animals, which do not require a complete vial, the reconstituted drug can be divided into smaller aliquots and frozen.
Amphotericin also has been complexed to lipid mixtures in an attempt to reduce nephrotoxicity. Reticuloendothelial cells phagocytize the lipid component, facilitating directed delivery to the site of fungal infection. Drug uptake by the hepatic and splenic macrophages, bone marrow, and inflammatory tissues is thus enhanced. For liposomal products, some studies have shown that amphotericin B is selectively transferred from liposomes to fungal but not host cell membranes. Prolonged antifungal activity (compared with nonliposomal preparations) has been documented for these preparations. Each product is injectable and differs in the lipid makeup as well as the ratio of lipid to amphotericin B. Lipid-based products include a lipid complex (ABLC; Abelcet) suspension that forms a tightly packed ribbonlike structure and is the largest of the lipid-based products (250 nm); a colloidal dispersion of cholesterol sulfate, which forms 122 nm disks (Amphotec or Amphocil [ABCD]); and a single-layered liposomal product, Ambisome (LAmB; see Table 9-4), which is the simplest and smallest (60 to 70 nm) of the molecules.17,18 The differences among these products have recently been reviewed.19 Amphotericin B also has been administered with a lipid vehicle (intralipid; AMB-IL) rather than 5% dextrose. Although nephrotoxocity appears to be reduced in some patients, it does not appear to be decreased in the patients at most risk for toxicity, and therefore liposomal lipid-based products should be considered.18a Administration of AMB-IL also is not recommended because of mixed product is unstable. The differences among the lipid-based or lipid-containing products and their clinical use in humans have recently been reviewed.19
Amphotericin is also available as a topical preparation (Fungizone), including cream, lotion, and ointment forms.
Pharmacokinetics
Amphotericin is not water soluble and thus is not bioavailable after oral administration. It is more than 90% bound to circulating serum lipoproteins (including cholesterol). Penetration into the pleura, peritoneum, inflamed tissues, CSF, and aqueous humor may result in a drug concentration two thirds of that in the plasma. The metabolism and excretion of amphotericin is not well characterized and is complex, being complicated by binding to cholesterol, which is structurally similar to ergosterol (see Figure 9-3). Biliary elimination may be the primary method of excretion (in humans only 3% of the drug is eliminated in the urine); however, concentrations are detectable in both bile and urine for up to 7 weeks in human patients.
The pharmacokinetics of the various amphotericin B preparations, including those complexed to lipids, vary markedly in humans; interpretation of kinetics is complicated by the lack of discrimination between free amphotericin B and that complexed to the various carriers. Because of its small size, Ambisome is characterized by the slowest uptake, and the highest PDCs. In humans, peak PDCs (μg/mL) vary with the dose (indicated in mg/kg), reflecting differences in volume of distribution that vary on account of tissue uptake: AmBD, 2.9 μg/mL at 1 mg/kg); Albecet (ABLC), 1.7 μg/mL at 5 mg/kg); Amphotec (ABCD), 3.1 μg/mL at 5 mg/kg; and Ambisome (LAmB), 83 μg/mL at 5 mg/kg. For LAmB, concentrations appear to be both dose and time dependent in dogs (Table 9-4). An advantage of the liposomal product is that it accumulates at sites of infection and targets the fungal cell wall, with release of amphotericin into the cell.19 For ABLC (and LAmB), tissue or fungal phospholipases appear to release amphotericin B at the site of infection. However, the colloidal form of amphotericin B (Amphocil), which is taken up by reticuloendothelial cells, was demonstrated in humans to accumulate in lung tissues to a greater concentration than Ambisome, suggesting that it might be preferable for respiratory infections.20 This finding requires further support by animal model or clinical studies. That LAmB may be preferred for central nervous system (CNS) infections is supported by the study and review of Ibrahim and coworkers,21 who address the potential inability of ABLC to penetrate the brain in mice or rabbit models. Comparing dosing ranges, LAmB was effective without nephrotoxicity at 5 to 20 mg/kg in animal models of disease, with highest tolerated doses being 30 to 50 mg/kg (as reviewed by Adler).19 Although ABLC was efficacious at doses ranging from 5 to 15 mg/kg, nephrotoxicity and decreased efficacy occurred at 15 mg/kg compared to 5 mg/kg. (as reviewed by Adler).19
The pharmacokinetics of Ambisone (LAmB) have been described in Beagles (n = 10 per group) as part of a toxicity study in humans and compared historically to AmBD (see Table 9-4).22 Dogs received 0.25 to 16 mg/kg/day as an intravenous infusion over 5 minutes, with PDCs determined on days 1, 14, and 30. Dogs receiving 8 and 16 mg/kg daily did not finish the treatment period because of severe weight loss (n = 17/20); this same pattern of weight loss was also described in dogs receiving AmBD at 0.75 mg/kg every other day (as reviewed by Bekersky and coworkers22). However, dogs tolerated 4 mg/kg Ambisone daily with peak concentrations at this dose were markedly variable, ranging from 18 µg/mL at day 1 to 94 μg/mL at day 30. Concentrations of Ambisone were a hundredfold higher than other formulations: A dose of 0.6 to 0.75 of AmBD resulted in peak amphotericin concentrations of 1.25 to 4.4 μg/mL after multiple dosing. This compares to a peak concentration of 54 ± 16 μg/mL at 4 mg/kg daily. Ambisome kinetics were nonlinear, with clearance and distribution decreasing as dose increased, potentially reflecting saturation of distribution and clearance processes. For example, although dose only increased 64-fold in the study, area under the curve (AUC) on day 1 at 0.25 mg/kg was 2.6 μg∗hr/mL (compared) to 2592 μg hr/mL at 16 mg/kg, representing a 2500-fold increase. Multiple dosing also resulted in accumulation at each dose, with 30-day to 1 day AUC ratios being 2.5 and 10 at low (0.25 mg/kg) and intermediate (4 and 8 mg/kg) doses, respectively. (see Table 9-4). Kinetics are likely to be equally complex in other dogs and cats, and lack of predictability may limit use.
Toxicities and Side Effects
Nephrotoxicity is the major side effect associated with the use of amphotericin B. Renal function becomes impaired in more than 80% of patients receiving amphotericin,4,23,24 with serum creatinine at least doubling in 40% to 60% in human.23 Although largely reversible, renal toxicity depends on total cumulative dose and duration of therapy. In human patients chronic renal disease occurs in close to 50% of patients receiving more than approximately 60 mg/kg total dose compared with only 17% who receive less than 57 mg/kg. The proportion decreases to 8% in those receiving less than 14 mg/kg total dose.23 Although renal function usually returns to normal before completion of therapy, some residual damage often persists after discontinuation of the drug. Two mechanisms are important in renal toxicity. Intense arterial vasoconstriction occurs within 15 minutes of administration and lasts 4 to 6 hours. The mechanism is unknown but can lead to acute tubular nephrosis secondary to ischemia. Nerve degeneration, angiotensin II receptor blockage, adrenergic blockade, and potent vasodilators do not prevent renal vasoconstrictive effects. Amphotericin B may activate formation of vasoconstrictive (thromboxane) arachidonic acid metabolites, which suggests that direct vasoconstriction may be responsible. Amphotericin B can increase calcium fluxes. Histologic lesions are most profound in regions vulnerable to hypoxia or areas rich in oxygen.23
>KEY POINT 9-5
Nephrotoxicity caused by amphotericin B might be minimized by several approaches, including pretreatment with sodium-containing fluids.
Distal renal tubular toxic effects result from binding of membrane cholesterol in the tubular cell membrane (see Figure 9-4). Altered electrolyte fluxes result in acidification abnormalities (metabolic acidosis), hypokalemia, and concentrating defects (polyuria, polydipsia). Renal tubular acidosis is common, dose related, and generally reversible (although resolution may take several months) and generally precedes significant decreases in glomerular filtration rate (GFR). Potassium and magnesium wasting in humans can lead to substantial deficits.23 Magnesium deficits appear at 2 weeks and are maximal at 4 weeks. Both potassium and magnesium should be monitored; electrolyte abnormalities may persist for several weeks after therapy is discontinued. Rarely, hyperkalemia has been reported in association with rapid infusion as potassium shifts from the intracellular compartments. The risk is greater in patients with renal failure. Concentration defects occur in essentially all human patients, are not related to azotemia, and may persist for months. Urine specific gravity may not be an effective measure by which to assess renal damage.
Ampohtericin B toxicity has been documented in dogs. Ceylan and colleagues24 described the nephrotoxicity of AmBD in dogs (n = 18; cross-bred) receiving 0.5 mg/kg in 25 mL 5% dextrose in water as a 4- to 5-minute intravenous bolus; 1 mg/kg in 50 mL as a 4- to 5-minute bolus, or 2 mg/kg in 1000 mL over 4 to 5 hours. Dogs were treated for 12 days. Side effects (vomiting, diarrhea, anorexia, fever, tachycardia, and phlebitis) were evident in all dogs by day 3 but were worse in dogs receiving 1 mg/kg and least in dogs receiving 2 mg/kg. Indicators of renal damage (blood urea nitrogen, creatinine) were increased in all groups by day 5 but were highest in the 1 mg/kg group. Serum calcium was significantly decreased compared with baseline in the 1 mg/kg and 2 mg/kg groups. Hematologic indicators of anemia were evident in the 1 mg/kg group. Among the indicators of nephrotoxicity studied was urine γ-GGT, which was significantly higher than baseline in all groups by day 5 of therapy but did not differ among groups or times during treatment. On the basis of this study, the authors suggested that infusion over a 4- to 5-hour period (or fluid support) decreases the risk of aminoglycoside toxicity. However, no study has determined the long-term effects of any method of administration of amphotericin B on renal failure in dogs or cats.
In their pharmacokinetic study of LAmB (Ambisome) in Beagles at doses ranging from 0.25 to 16 mg/kg daily for 30 days, Bekersky and coworkers22 reported that no dogs died as a result of the drug; however, dose-dependent tubular nephrosis was present at 1 mg/kg, and 70% and 100% of dogs in the 8 and 16 mg/kg treatment group, respectively, were euthanized before the end of the study because of 25% or more body weight loss. Vomiting and diarrhea were evident in all groups, including a liposomal (i.e., without amphotericin B) control, but were worse in the 4, 8, and 16 mg/kg treatment groups. Azotemia was not present in the 1 mg/kg group. Azotemia was described as moderate and clinically significant toward the end of the 30-day treatment period in the 4 mg/kg group, with creatinine increasing from a baseline of 0.7.1 ± to 2.3 ± 0.4 mg/dl by study end. Serum potassium was not clinically affected. The authors concluded that dogs tolerated up to 4 mg/kg well, despite peak amphotericin B concentrations ranging from 18 to 94 μg/mL.
Acute anaphylactic-type reactions such as vomiting, fever, and chills can occur with the use of amphotericin B and have been reported with lipid complex preparations as well.25 Up to 30% of dogs receiving AmBD develop fever.26 The frequent incidence of these reactions often leads to pretreatment for anaphylaxis (one-time use of a short-acting glucocorticoid does not enhance the toxicity of amphotericin B; see the discussion of “cocktail” in the following section).
Other side effects associated with amphotericin B include nausea and anorexia, thrombophlebitis, cardiac arrhythmias and related toxicities, hepatic dysfunction, and CNS signs (if given intrathecally) (see discussion on therapeutic use). Several side effects can be prevented by proper treatment (see following discussion of therapeutic use).
Drug Interactions
Amphotericin B interacts in an additive to synergistic fashion with a number of antifungal agents (see the discussion of combination therapy), although this effect seems to be most consistent with flucytosine. Although synergistic effects should be expected with azoles, this combination may be antagonistic if azole therapy is begun first.
In general, drugs that are renally active are not recommended for patients also receiving amphotericin B. In humans cyclosporine and tacrolimus (calcineurin inhibitors) will enhance nephrotoxicity. However, these immunomodulators tend to be safer, as far as nephrotoxicity is concerned, in both dogs and cats, and the combination may not be as dangerous. The nephrotoxicity of amphotericin also may be enhanced with catabolic drugs such as glucocorticosteroids, antineoplastic drugs, antiprostaglandins (glucocorticoids and nonsteroidal antiinflammatory drugs), and other nephrotoxic antibiotics. One study demonstrated that diuretic therapy during, but not just before, amphotericin B therapy increased the risk of nephrotoxicity more than twelvefold (as reviewed by Bagnis and Deray23). Although diuretics have been used for a “protective” effect, the potential benefits are limited to mannitol, and even it may be associated with electrolyte disturbances. Combination with digoxin may enhance nephrotoxicity and hypokalemia. Declining renal function in association with amphotericin B therapy may affect clearance of co-administered drugs. Because it is renally excreted, 5-flucytosine toxicity may be enhanced if renal function declines with amphotericin B.
Therapeutic Use
Most of the advice and studies addressing the recommended method of administration of amphotericin B are based on the non-lipid preparation (AmBD).
Multiple strategies may help reduce toxicities or side effects to amphotericin B. The first focuses on prevention of an anaphylactoid reaction by pretreatment (to prevent vomiting, fever, chills, and anaphylaxis) with antihistamines (diphenhydramine 0.5 mg/kg, administered intravenously) and short-acting glucocorticosteroids (e.g., hydrocortisone sodium succinate, 0.5 mg/kg, administered intravenously). Because the reaction appears to be associated with direct mast cell degranulation (because of the cationic nature of amphotericin B), pretreatment with a small test dose (0.1 mg/kg for cats or 0.25 mg/kg for dogs diluted in 10 mL infused over 15 minutes) may help identify animals that are likely to have an adverse reaction during infusion. Whereas lipid products may be characterized by less nephrotoxicity, they are more likely than nonlipid products to cause vomiting, nausea, and phlebitis.38 As such, pretesting is indicated with their use as well.
In addition to dilution protocols (see discussion under Toxicity), strategies are intended to minimize amphotericin-B induced nephrotoxicity. Although what constitutes amphotericin B nephrotoxicity may vary, an increase in creatinine of as little as 25% may be considered significant in some patients.18 Azotemia precedes tubular dysfunction in humans, allowing for early detection (i.e., before irreversible damage). Urine sediment initially should be monitored for evidence of nephrotoxicity. Serum chemistries tend to be less sensitive indicators of nephrotoxicity. Urine γ-gt has been used to assess lipid-based amphotericin B toxicity. The drug should be temporarily discontinued (24 to 48 hours) if the blood urea nitrogen level becomes abnormal.27
First and foremost, pretreatment with sodium-containing fluids is particularly important for preventing renal toxicity, including that associated with renal arterial vasoconstriction; posttherapy treatment might also be prudent. Secondly, amphotericin B can be administered with a “cocktail” intended to protect the kidney. Administration of the dose diluted in 5% dextrose is accompanied by mannitol (0.5 mg/kg) to maintain glomerular filtration rate and sodium bicarbonate (1 to 2 mg/kg) to prevent cellular acidification defects. The mannitol and sodium bicarbonate should not be added directly to the amphotericin B solution but given through another catheter in order to avoid precipitation of amphotericin B. The ability of cocktails to prevent nephrotoxicity is controversial. The treatment is based on one small study in dogs31 for which mannitol was demonstrated to protect the kidney. However, a subsequent study (using isolated perfused rat kidneys) found mannitol to impart a protective effect, presumably by reducing renal tubular edema by virtues of its osmotic effect.32 Binding of cellular cholesterol prevented toxicity, supporting the presumed mechanism of direct cytotoxicty. This study also demonstrated a synergistic toxic effect of amphotericin B in the presence of hypoxia. Only one clinical trial, in humans, has addressed renoprotection by mannitol, and it failed to identify an effect. However, this clinical trial, although randomized, involved only 11 patients and is probably not conclusive (as reviewed by Bagnis and Deray18,23) Although such cocktails generally are not harmful as long as the solutions are not mixed with amphotericin B, mannitol has been associated with electrolyte abnormalities in the human-medicine literature.23 Deray18 indicates that mannitol decreases renal medullary blood flow and PO2. As such, the use of mannitol remains controversial. A relatively recent study describes the nephroprotective effects of N-acetylcysteine (10 mg/kg daily) in a rat model of amphotericin-induced nephrotoxicity;33 administration began 1 day before amphotericin was started. As previously described, amphotericin should not be mixed with solutions containing electrolytes, acidic solutions, or preservatives because these materials may cause drug precipitation. The rate at which amphotericin B is administered may also reduce the incidence of nephrotoxicity.
The third strategy by which amphotericin B toxicity might be reduced is administration with lipids or as a specialized delivery system (in lipids). Conventional amphotericin B is not recommended in humans in the presence of renal insufficiency, hypokalemia, hypomagnesemia, tubular acidoses, or polyuria.18 Lipid preparations are designed to deliver more drug selectively to the site of infection,34 thus allowing higher doses (see the section on preparations). The liposomal products tend to be very expensive. In contrast to liposomes, fat emulsions are easy to prepare and administer and tend to be more cost effective. These products appear to be equal in efficacy to nonliposomal products but safer with regard to nephrotoxicity. However, the use of intralipid as the vehicle (AmB-IL) is associated with other difficulties that limit its use (see previous discussion).
The safety of liposomal or lipid-based products may allow delivery of higher doses without an increased risk of nephrotoxicity, with the incidence of nephrotoxicity reduced by 8% to 28%, depending on the preparation.18,23 Although several studies are available that address the safety of these products individually, and LAmB (Ambisome) in particular,35 none appears to have compared all three products. However, in general, the liposomal product LAmB (Ambisome) is recognized to be the safest with regard to nephrotoxicity. Ambisome achieves the highest concentrations in humans compared with other amphotericin preparations but is well tolerated renally.17,19 In general, efficacy of LAmB is equal to or exceeds that of AmBD and is consistently better tolerated. Survival rates in humans for the product ranges from 99% to 100% (visceral leishmaniasis; three studies, n ≅ 400), 37% to 78% (aspergillosis; five studies, n ≅ 400), 84% to 93% for cryptococcosis (two studies, n ≅ 280), 98% for histoplasmosis (one study, n = 55), and 20% to 69% for zygomycosis (three studies, n ≅ 130).35 That LAmB may be preferred for CNS infections is supported by the study and review of Ibrahim and coworkers,21 who found LAmB generally superior to ABLC in treating CNS infections, as indicated by response and decrease in fungal load. Abelcet (ABLC) and LamB were generally characterized as having the lowest incidence of all side effects (fever, chills, nausea, dyspnea, hypertension, hypotension, tachycardia, and nephrotoxicity) with the exception of hypokalemia (42% with LamB). Ambisome was associated with abnormal hepatic function tests in approximately 20% of human patients.17 Abelcet causes fewer infusion-related side effects in humans, and therefore a test dose is not necessarily indicated, whereas Amphotec is more likely to cause infusion side effects and should be given as a short infusion only.
One study reports the efficacy and safety of a liposomal product when used at higher than recommended cumulative doses for treatment of canine blastomycosis.36 In another study, although the products were equally efficacious, the degree of nephrotoxicity between a fat emulsion and standard amphotericin B was no different.37 Amphotericin B also has been administered with a lipid vehicle (intralipid) rather than 5% dextrose. Although nephrotoxocity appears to be reduced in some patients, it does not appear to be decreased in the patients at most risk for toxicity, and therefore liposomal lipid-based products should be considered.18 Further studies documenting the efficacy and safety of liposomal or fat emulsion products containing amphotericin B are needed.
A fourth strategy that can be used to minimize adverse events associated with amphotericin B is administration using alternative routes. Localized mycotic infections have been treated with localized administration of amphotericin, which may reduce the incidence of nephrotoxicity. Subconjunctival, intravitreal, intrathecal, intranasal (human aspergillosis: 5 mg/mL in water administered as aerosol), and intraperitoneal routes have been reported. Oral administration has been used for treatment of gastrointestinal candidiasis and presumably might be used for other gastrointestinal fungal disorders.29 Both LamB and ABLC have been demonstrated to have enhanced efficacy when administered as an aerosol for pulmonary infections (as reviewed by Adler).19 Amphotericin B (AmBD) can be mixed in sterile water to 200 mg/kg and infused into the bladder for fungal cystitis. For fungal infections of the CNS, the drug can be given (0.2 to 0.5 mg in either 5 mL of CSF or 10% dextrose) intrathecally (under general anesthesia) two to three times per week.27
Combination antifungal therapy is a fifth stategy that is strongly encouraged to enhance efficacy and thus decrease the duration of antifungal exposure to the host. For example, as reviewed by Adler,19 efficacy of LamB is enhanced when combined with micafungin (zygomycosis, aspergillosis) and voriconazole (aspergillosis).
Although a sixth strategy whereby amphotericin B toxicity can reduced includes variation in doses, frequency of administration, concomitant therapy, and duration of therapy, no protocol has proven to be superior to others. Some literature supports rapid intravenous administration (bolus) in less debilitated dogs; slow intravenous administration might be more prudent in cats. A dose (0.25 to 0.5 mg/kg) can be diluted in 300 to 1000 mL of 5% dextrose and administered in an indwelling catheter over 2 to 6 hours, or it can be diluted in as little as 10 to 60 mL (30 recommended) and given over 2 to 10 minutes through a butterfly catheter27 (flush catheter after infusion). The slow infusion method has the added advantage of additional fluids, which may reduce the incidence of nephrotoxicity,28 especially in debilitated animals. The advantages of dilution and slow infusion is preferred and was previously discussed (see Toxicity). It is recommended that the small bolus be preceded or followed up with supplemental fluid, preferably normal saline. One report of an uncontrolled clinical trial describes the successful and apparently safer administration of amphotericin B after twice- or thrice-weekly subcutaneous administration (0.5 to 0.8 mg/kg diluted in 400 to 500 mL of fluid such that amphotericin B is less than 20 mg/L) for several months for treatment of cryptococcosis in dogs and cats.30 However, only five successful cases were described, and further confirmation should be expected before this route is routinely embraced.
The sequence of repetitive treatments is also controversial. Some authors recommend alternate-day therapy, whereas others recommend daily therapy at a smaller dose. Doses also vary. Daily doses range from 0.15 to 0.5 mg/kg every other day (e.g., on Monday, Wednesday, Friday) until a cumulative dose of 4 to 12 mg/kg (depending on the organism or if therapy is combined with another antifungal) AmBD has been reached. Starting at a low dose (0.15 to 0.25 mg/kg) and gradually increasing the dose until the desired daily dose has been reached may reduce the severity of side effects. For particularly resistant infections, a dose of 1 mg/kg has been used on an alternate-day basis.
Other Polyenes
The spectrums of piramicin (natamycin) and nystatin are similar to that of amphotericin B. Pirimacin is used primarily to treat fungal keratitis, although efficacy toward Aspergillus may be questionable.3 Aerosolization of piramycin for treatment of susceptible fungal disorders also should be considered. The toxicity of nystatin precludes parenteral administration, although a liposomal product is being formulated. Currently, use of nystatin is limited to superficial and mucosal mycoses.
Azole Derivatives
Structure–Activity Relationship
The azole derivatives (imidazoles and triazoles) include a large number of predominantly synthetic drugs. These drugs consist of a five-member ring with other aromatic rings attached by a carbon nitrogen bond. Imidazoles contain two nitrogen atoms and include clotrimazole, econazole, enilconazole, miconazole, and ketoconazole. Triazoles contain three nitrogen atoms and include fluconazole, itraconazole, and voriconazole (see Figure 9-1).4,39-41 Among the newer azoles are posaconazole and voriconazole. Voriconazole, a synthetic deriviative of fluconazole, is the first of the second-generation triazole compounds to be approved by the Food and Drug Administration. Voriconazole contains a fluorine molecule and a methyl group, which greatly enhances its spectrum compared with that of fluconazole.42 Other triazoles have been patented. Most compounds generally are not available as solutions because they tend to be insoluble in water (an exception is fluconazole). They are, however, soluble in organic solvents such as propylene glycol.
Mechanism of Action
The azoles target fungal sterol ergosterol. However, in contrast to amphotericin B, the imidazole derivatives do not bind to ergosterol but rather block its synthesis. The azoles inhibit fungal cytochrome P450 enzymes with sterol14α-demethylase as the primary fungal target; however, other synthesizing enzymes also are targeted.3 Decreased cell membrane ergosterol alters cell membrane function but also is accompanied by an increase in 14-methylsterols, compounds that are potentially toxic. Cell membrane function fluidity decreases as cell permeability increases, resulting in a fungistatic effect (fluconazole and ketoconazole). At higher concentrations, selected drugs (miconazole, econazole, clotrimazole) also interfere with cell membrane fluidity and physiochemical intracellular processes (e.g., secretory vesicles, mitochondrial respiration), resulting in fungicidal effects. Efficacy of selected agents against gram-positive organisms may reflect this latter effect.3 Chitin synthesis increases in concert with decreased ergosterol synthesis, but its irregular distribution contributes to altered cell wall function. Because azoles also generate and detoxify intracellular hydrogen peroxide, selected drugs also express antibacterial, antiprotozoal, and anthelmintic activities. The imidazoles are also characterized by immunomodulatory effects, which may facilitate effective therapy.5 Because their mechanism of action depends on cell wall synthesis, the onset of action of the imidazoles may result in a lag time to therapeutic efficacy. In addition, a long elimination half-life of some members of this class (e.g., itraconazole) results in a lag time as steady-state concentrations are achieved.
Spectrum of Activity and Pharmacodynamics
Although the imidazole derivatives are more selective in their cellular activity than amphotericin B (i.e., impairing the synthesis of rather than binding to ergosterol), their spectrum of activity is broad and includes the dermatophytes (“ringworm”: Microsporum and Trichophyton species), yeasts, dimorphic fungi (blastomycosis, histoplasmosis, cryptococcosis, coccidioidomycosis), Eumycetes, Actinomyces, and some Phycomycetes.1,4,11,27,40,43 The efficacy against these organisms varies. Studies comparing the efficacies of the azoles in animals are limited at the time of this publication, although several are pending in the human-medicine literature.
>KEY POINT 9-7
Although not as efficacious as amphotericin B, the spectrum of the imidazoles is the broadest of the clinically used antifungal drugs.
In vitro MIC data regarding the relative susceptibility of selected organisms to itraconazole emphasize the variable susceptibility of fungal organisms to these drugs. Table 9-2 provides MIC90 data for selected organisms and itraconazole. For dermatophytes, the MIC90 for Microsporum species is 250 μg/mL versus for Trichophyton species 4 μg/mL. For other Eumycetes, MIC90 concentrations range from 0.130 μg/mL to more than 128 μg/mL.11,44 Fluconazole generally is considered to be fungistatic in action. This has been demonstrated for Candida sp. and C. neoformans.10 However, more recent data suggest that fungicidal activity may occur toward Candida. Like fluconazole, itraconazole more consistently exerts a time-dependent, fungistatic effect toward most organisms. The MFC50 and MFC90 (μg/mL) have been reported for B. dermatitidis (0.125, 3, respectively), H. capsulatum (2, 16), and C. immitis (>16 for both).15 However, it may exert fungicidal effects towards Aspergillus. The long half-lives of the imidazoles will facilitate convenience of dosing despite their time-dependent killing effects. Efficacy of itraconazole will be enhanced by the formation of its active metabolite, hydroxyl-itraconazole, which may surpass the parent.45 As with other azoles, the spectrum of the third-generation imidazoles, posaconazole (similar in structure to itraconazole) and voriconazole (similar in structure to fluconazole), includes a variety of infecting fungal organisms. Their spectrums are very similar.10 Posaconazole has been demonstrated to be more potent than fluconazole toward Candida and should be effective against the dimorphic fungal organisms,46 although little information is available regarding its use. As with other azoles, voriconazole is fungistatic toward yeasts, exhibiting time-dependent killing, but for some filamentous organisms may be -cidal. Its spectrum includes Candida sp., with MIC generally being 1 to 2 log lower than fluconazole; however, MICs are higher for fluconazole-resistant strains than for nonresistant strains. Voriconazole also is effective against C. neoformans, Trichosporon beigelii, and Saccharomyces cerevisiae. Additionally, it is very effective against Aspergillus sp., including some strains resistant to amphotericin B; indeed, it is approved for the treatment of Aspergillosis in humans. Time-killing studies with Aspergillus demonstrate that, in general, amphotericin B is more efficacious but itraconazole less efficacious than voriconazole. Activity against B. dermatitidis, C. immitis, and H. capsulatum appears to be “reasonable” but “less” toward S. schenckii. Li15 compared the MICs of amphotericin B, itraconazole, and voriconazole and found voriconazole to be more active in vitro than amphotericin B for the mold forms of H. capsulatum, B. dermatitidis, and C. immitis, with activity being fungicidal (MFC50, MFC90, respectively) toward B. dermatitidis (0.125, 4) and some H. capsulatum (8, ≥ 32) (see Table 9-2). Fungicidal activity toward C. immitis is difficult to achieve (>32 for the MFC50). Activity Aspergillus also is fungicidal (MFC not provided) toward several dematiaceous molds.15 Many dematiaceous and hyaline molds resistant to amphotericin B (e.g., Scedosporium, Fusarium, Paecilomyces, Alternaria) are susceptible to voriconazole. However, zygomycetes are not susceptible.42 Cutaneous infections by Leishmania species are clinically susceptible to ketoconazole.47 Clotrimazole and miconazole are common drugs used topically for treatment of dermatophytosis (e.g., Conofite) or yeast (e.g., otic preparations such as Otomax). Both drugs, as well as other imidazoles (e.g,) appear to exhibit efficacy against gram-positive organisms including staphylococci and anaerobes.
Resistance
Resistance to the azoles has been sporadic, generally occurring in immunocompromised patients receiving long-term therapy. Failed drug accumulation in the cell is a major mechanism of resistance (e.g., Candida sp., C. neoformans, Aspergillus flavus, and A. fumigatus), caused by either decreased influx or formation of efflux proteins. Two major efflux transport proteins have been identified, particularly for Candida sp. toward fluconazole: the multidrug-resistant protein, major facilitator superfamily (MFS; a proton-motive-force–based mechanism; e.g., CaMDR1) and the ATP-binding cassette (e.g., CDR1 and 2). Resistance to azoles also may reflect altered interaction between the drug and targeted fungal CYP 450 enzyme; fluconazole appears more susceptible than itraconazole to mutations that alter drug-receptor fit. Gene amplification leading to enhanced synthesis of the target protein also has been identified as a mechanism of resistance to azole derivatives. Finally, fungal organisms may circumvent the synthetic pathway inhibited by azoles or compensate for altered enzyme activity; for example, inhibition of ergosterol may result in the accumulation of 14-methylated sterols that are less toxic than other compounds.3 Resistance to fluconazole appears to be increasing. Widespread use of fluconazole for treatment of Candida has been associated with increased resistance of the drug for selected species. Whereas many molds (e.g., A. fumigatus) are inherently resistant to fluconazole,3 resistance of Aspergillus isolates to itraconazole is rare. Combinations with nonantagonistic antifungal agents might be considered for treatment of infections by organisms associated with antifungal resistance.
Pharmacokinetics
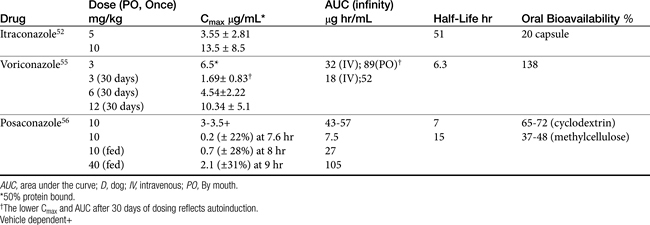
Only selected pharmacokinetic information is available for the imidazoles in dogs or cats (Table 9-5).Oral absorption of the imidazole derivatives varies with the drug and among animals. For many drugs oral preparations are not available. For example, enilconazole (imazalil) is not orally bioavailable. For other imidazoles the rate of absorption varies from 1 to 4 hours. Oral absorption often depends on gastric pH, product preparation, and the presence of other drugs.48 Absorption of itraconazole and ketoconazole is enhanced by gastric acidity; these drugs should be administered with food.49 Alkalinizing drugs administered orally will decrease their absorption. Cyclodextrins are used to form complexes with the drugs, thus rendering the liphophilic drug soluble in solution, making oral solutions available for some drugs (e.g., Sporonix solution). In general, oral bioavailability should be anticipated to be better for these solutions than for capsules. Peak PDCs of itraconazole occur between 1 to 5 hours in cats and dogs. Bioavailability of capsules is approximately 20% in dogs and may be as little as 10% in cats, compared with close to 50% for the solution in cats and dogs.50,51 Decreased bioavailability may be responsible for therapeutic failure associated with low PDCs in some animals (cats and dogs). Unpredictable oral administration of itraconazole prepared as a capsule limits its usefulness in humans; the solution, on the other hand, generates more predictable concentrations.17 Care should be taken with compounded preparations; oral absorption generally is not verified for these products, and adequate oral bioavailability should not be assumed. Fluconazole is characterized by the best oral bioavailability among the imidazoles, being completely absorbed in cats;52 however, its efficacy, compared with that of itraconazole, is limited.
>KEY POINT 9-8
Absorption of itraconazole and ketoconazole is enhanced by gastric acidity; these drugs should be administered with food.
Distribution to tissues also varies among the imidazoles. Ketoconazole is up to 99% protein bound; the highest tissue levels occur in the liver, lung, and kidney (and cerumen). Itraconazole is also very highly protein bound in humans.4 There is minimal penetration of the CSF by ketoconazole, although fluconazole penetrates the CSF well, with serum to CSF PDCs ranging from 0.58 to 0.89 μg/mL. The volume of distribution of ketoconazole is only 0.87 L/kg in dogs compared with 17 L/kg 5 L/kg for itraconazole in dogs51 and cats, respectively.50 The volume of distribution of fluconazole is 1.14 L/kg in cats, with high concentrations occurring in the CSF and aqueous humor.52 The difference in distribution volume reflects, in part, distribution and accumulation to fat.51 Drug concentrations of itraconazole in the skin may exceed that in plasma by threefold to tenfold, with drug detectable 2 to 4 weeks after therapy is discontinued.51 Although distribution of itraconazole to the CSF appears to be limited, therapeutic concentrations appear to be achieved in patients suffering from cryptococcal meningitis. Among the azole derivatives, fluconazole has the best tissue distribution pattern and can achieve effective concentrations in CSF.
>KEY POINT 9-9
Among the imidazoles, fluconazole is characterized by the best oral bioavailability and distribution into the central nervous system.
With the exception of fluconazole, the azole derivatives are eliminated by extensive oxidative (cytochrome P450) metabolism with excretion as inactive metabolites into the bile and urine. However, metabolism of itraconazole generates an active hydroxylated metabolite, whose AUC may exceed that of the parent compound.53 Metabolism may be dose dependent; elimination rate constants are lower and half-lives are longer at higher doses and with longer therapy. In contrast to the other imidazoles, fluconazole is eliminated principally (70%) in the urine. The half-life of the imidazoles varies, with that of ketoconazole being relatively short (1.4 hours in dogs). Fluconazole and itraconazole have longer half-lives, ranging from 22 to 32 hours in humans. The half-life of fluconazole in cats is 25 hours.52 The half-life of itraconazole in dogs is 51 hours (itraconazole)52 versus 40 to 70 hours in cats (itraconazole);50 The longer drug elimination half-life must be taken into account because it results in a longer time to steady-state concentration and maximum therapeutic effect. However, it also allows the flexibility of once-daily (10 mg/kg) rather than twice-daily (5 mg/kg) dosing.
The disposition of voriconazole is complicated. Its disposition has been reported in dogs as part of a preclinical study (see Table 9-5).55 Beagles (n = 4) received multiple doses (8) of 6 mg/kg orally or 3 mg/kg intravenously using a crossover design; disposition was studied on day 1 and day 30 for each route. The AUC after single and multiple dosing (μg∗h/mL) after the intravenous dose was 32 and 18, respectively, and after the oral dose 89 and 52, respectively, suggesting autoinduction. Other relevant parameters after single oral dosing were as follows: Cmax, 6.5 μg/mL at Tmax of 3 hours and apparent bioavailability of 138%. The drug was 51% protein bound, indicating a concentration of approximately 3.25 μg/mL of active drug. For a safety study, Beagles (n = 6) also received oral doses of 3, 6, or 12 mg/kg for 1 month. The apparent volume of distribution was 1.3 L/kg and clearance was 24 mL/min/kg, resulting in a calculated half-life of 6.3 hours. The Cmax increased in a slightly disproportionate dose-dependent manner, being 1.69 ± 0.83 μg/mL at 3 mg/kg and 10.3 ± 5.1 μg/mL at 12 mg/kg 30 days after oral dosing. However, cytochrome P450 increased in a dose-related manner, resulting in a 1.7-fold increase in relative liver weight at the highest compared with the lowest dose. Autoinduction in dogs, which does not occur in humans, resulted in an increased clearance after multiple dosing, with these effects dissipating approximately 1 month after the drug was discontinued. For example, the mean Cmax after a single oral dose of 3 mg/kg in dogs was 6.5 μg/mL but only 1.7 after 30 days dosing at the same dose. Although 55% to 87% of the drug ultimately was eliminated in the urine in dogs, only 5% was as the parent drug, indicating extensive hepatic metabolism. A major pathway of metabolism was generation of the N-oxide and hydroxylation metabolites as well as glucuronidation. The dose of voriconazole recommended for dogs can be based on pharmacokinetic–pharmacodynamic integration. The MIC90 of most infecting microbes for which data are available will be achieved after 30 days at 3 mg/kg, even accounting for 50% protein binding in dogs. An exception occurs, however, for H. capsulatum, for which a higher dose is indicated. With a half-life of 6 hours, based on time-dependent killing, at least twice the MIC of the infecting microbe should be targeted to allow for a 12-hour dosing interval. In humans dosing regimens are designed to reach 3 to 6 μg/mL in the plasma; accordingly, a dose of 3 to 6 mg/kg twice daily is recommended. However, the half-life in humans is long (6 to 24 hours, depending on the dose), and steady-state concentration requires 5 to 6 days of dosing. As such, a loading dose consisting of a double daily dose is recommended for the first day of therapy. This is not necessary in dogs: the shorter half-life precludes accumulation to a steady state. However, the shorter half-life will necessitate twice-daily dosing. Because of autoinduction in dogs, dosing regimens should not be extrapolated for cats from dogs without the support of pharmacokinetic studies.
Voriconazole is cleared primarily by hepatic metabolism to inactive metabolites by CYP 2C19 (the primary isoenzyme), 2C9, and 3A4 being involved. Selected humans are considered “poor metabolizers” of the drug because of variation in CYP 2C19. In humans the dose is halved in the presence of mild to moderate liver disease. The relevance of this to dogs, which autoinduce, is not known.
Very limited pharmacokinetic information is available for dogs receiving posaconazole (see Table 9-5).56 It was studied in two different vehicles (cyclodextrin or methylcellulose) during preclinical investigations. Absorption from the cyclodextrin vehicle was better, resulting in a higher Cmax, greater AUC, and better oral bioavailability. Although the half-life following intravenous administration was approximately 8 hours, the effective half-life is 15 hours after oral administration, probably reflecting slow absorption, as is indicated by a Tmax of 8 to 9 hours. Food enhanced absorption, increasing Cmax and AUC fourfold. Although Cmax increased more than two fold with multiple dosing, AUC was the same, indicating accumulation is not likely to be a clinical concern.
Preparations
Ketoconazole, itraconazole, and fluconazole are available for oral administration. Solutions are available for some products. Although their safety has not been documented for animals, fluconaozle (Diflucan) is available as an intravenous preparation that appears to be safe in cats when administered as a slow intravenous drip at 5 mg/kg. The solubility of imidazoles is poor and potentially toxic; solubilizing agents may cause adverse reactions. Cats also appear to tolerate a slow intravenous drip of itraconazole (5 mg/kg) with no adverse effects, although neither product is commercially available. Ketoconazole also is available in a topical preparation and a shampoo. Clotrimazole and miconazole are recommended only for localized dermatophyte or yeast infections susceptible to topical treatment. Clotrimazole has been used topically to treat nasal aspergillosis. Enilconazole is a topically effective azole that has been used to treat nasal aspergillosis but is available in the United States only as a 13.8% poultry dip. It is available in Canada as a 10% solution approved for use in dogs and horses. The poultry dip has been used topically in the United States at a dilution of 1:50 in water in dogs and cats with no apparent adverse effects. Terconazole is a new, topically active triazole that apparently has not yet been used for animals. However, an otic preparation containing posaconazole (with orbifloxacin and an anti-inflammatory) has recently been approved for dogs.
Drug Interactions
The azoles may interact synergistically with a number of antifungal agents. Synergism with polymyxin B is benefited in otic preprations.3 Ketoconazole and, presumably, other azole antifungals have synergistic antifungal activities with 5–flucytosine against Candida and Cryptococcus and with amphotericin against a variety of organisms. However, timing of amphotericin B and azole therapy is important. Azoles impair ergosterol synthesis, and therefore their use before amphotericin B may decrease its efficacy which is dependent on active cell wall synthesis. Amphotericin B should begin either simultaneously with (azoles are characterized by a lag time to effect) or before azole administration. Enhanced efficacy has also been demonstrated for terbenifine and topical therapy (see Therapeutic Use).
Because the efficacy of the azoles depends on interaction with P450 (an oxidative enzyme responsible for drug metabolism), drug interactions at the level of drug or steroid metabolism should be anticipated in the patient. The azoles are both inhibitors and inducers of CYP isoenzymes; the extent to which an azole inhibits metabolism often depends on its relative affinity for host compared to fungal CYP 450.3,57 All of the clinically relevant imidazoles inhibit some CYP. These include: CYP3A4 (ketoconazole, itraconazole, fluconazole, voriconazole; (ketoconzole has a higher affinity than itraconazole); 2C19 and 2C9 (ketoconazole, fluconazole, voriconazole); 2D6 (ketoconazole); 2C8 (ketoconazole and voriconazole); and 1A2 and 2E1 (ketoconazole). However, as has been demonstrated in dogs, voriconazole is an autoinducer and species differences in CYP interactions may be profound. When present, inhibition can be clinically relevant. It can be beneficial (e.g., the combination of ketoconazole with cyclosporine in an attempt to prolong cyclosporine clearance in order to decrease the daily cost of this drug (see Chapter 31).58 In the author’s laboratory, the effect of ketoconaozle on cyclosporine concentrations appears to be quite variable, ranging from no effect to a dramatic increase. Kukanich and Borum59 found that ketoconazole (approximately 13 mg/kg for 5 days) did not appear to affect the intravenous disposition of morphine in Greyhounds.
However, more commonly, inhibition is associated with adverse reactions. The author is aware of two cases of marked increases in phenobarbital concentrations (to over 85 µg/mL) in epileptic dogs receiving fluconazole or itraconazole, respectively, for treatment of Malassezia; one of the dogs was euthanized resulting from (assumed) liver disease. More recently, a dog receiving deracoxib and also developed a perforated duodenal ulcer receiving ketaconazole for a skin yeast infection. Itraconazole appears to cause autoinhibition; changes in the elimination half-life of intractonaozle have been documented in cats receiving long-term therapy (>6 weeks).50 In humans, the risk of drug interactions involving voriconazole is high, perhaps more so than with the other azoles. Their magnitude and impact in dogs and cats needs to be clarified. Clinically relevant drug interactions have been documented for a number of drugs administered in concert with voriconazole; avoidance of other drugs administered by the liver would be prudent.42
>KEY POINT 9-10
All imidazoles appear to impact drug-metabolizing enzymes, with inhibition being most common and clinically relevant.
Not all drug interactions involve inhibition of CYP. Clotrimazole is a potent inducer of CYP3A and miconazole of CYP 1A and 2E; even ketoconazole is an inducer of CYP 2, although it is less potent than clotrimazole (see Chapter 2 for drugs metabolized by CYP isozymes). As previously discussed, voriconazole is an inducer of CYP in dogs. Ketoconazole interferes with sex hormones and corticosteroids by displacing them from globulins and perhaps by interfering with their synthesis. Ketoconazole inhibits lanosterol14-demethylase (cholesterol synthesis; CYP51) and two hydroxylase enzymes responsible for steroid metabolism as well as a key enzyme involved in testosterone synthesis. As a result of its effects on steroid synthesis, ketoconazole has been used to treat hyperadrenocorticism and to impair testosterone synthesis in patients with prostatic hypertrophy or prostatic cancer. Ketoconazole has caused lightening of the hair coat of some dogs.60 Willard and coworkers60 reported depressed basal cortisol and testosterone concentrations and ACTH response by cortisol at 30 mg/kg/day. A rebound response was seen after ketoconazole was discontinued. Serum progesterone concentrations were also decreased. Aldosterone was not decreased. Cats receiving 30 mg/kg/day for 30 days developed dry hair coat and weight loss but no changes in testosterone or progesterone concentrations.61
Finally, not all clinically relevant drug interactions involving imidazoles reflect effects on cytochrome P450. The imidazoles in general are substrates for P-glycoprotein, and ketoconazole is a known inhibitor.62 Accordingly, other drugs that serve as substrates for P–glycoprotein are likely to be absorbed to a greater extent. Thus ketoconazole can affect (increase) cyclosporine A concentrations following oral administration without affecting cyclosporine elimination half-life as has been documented in the author’s laboratory.
In general, the imidazoles are not characterized by the complex toxicities that are associated with amphotericin B. Because the azoles interfere with synthesis of ergosterol rather than binding the sterol, the host toxicities typical of those induced by amphotericin do not occur.63 Gastrointestinal toxicities are the most common and are not severe.4,40,27,54,64 Nausea and vomiting can usually be prevented by administration of the drug with food. Hepatotoxicity with ketoconazole has been reported in humans. Mayer and colleagues65 retrospectively reported adverse effects of ketoconazole in dogs. Medical records in Australia (n = 296), Germany (n = 35), and the United States (n = 301) were reviewed, and adverse events were reported in 92 (14.6%). Doses ranged from 2.6 to 33.4 mg/kg, with an average daily dose of 11.2 mg/kg. The frequency of adverse events was as follows: vomiting (7.1%), anorexia (4.9%), diarrhea (1.1%), and lethargy (1.9%). Uncommon side effects included pruritis (0.6%) and ataxia, polyuria, and polydipsia; causal relationships were difficult to establish for the uncommon reactions.
Side effects to itraconazole are limited to gastrointestinal symptoms (nausea and vomiting), which may be related in part to the vehicle if associated with the oral solution (a cyclodextrin carrier).3 One case of cutaneous drug eruption typical of erythema multiforme caused by itraconazole has been reported in a dog;67 idiopathic vasculitis has also been reported.54 Fluconazole is associated with very few side effects; hematologic disorders may occur particularly in profoundly ill patients; otherwise, side effects appear to be limited to gastroinestinal and cutaneous reactions.
Isolated cases of hepatotoxicity have been reported with itraconazole and fluconazole. A retrospective study of dogs with blastomycosis found 5% to 10% of dogs treated with itraconaxole (5 to 10 mg/kg bid) developed hepatotoxicity with the effect dose dependent.119 A dose dependency has been documented in one case with fluconazole. Patients with impaired liver function may be predisposed to worsening hepatic function induced by the azole antifungal drugs. The occurrence of liver disease in animals treated with itraconazole is controversial. Cats receiving 5 and 10 mg/kg twice daily showed no adverse effects (including weight loss) after receiving itraconazole for 6 weeks.50 However, one study of enilconazole applied as a 0.2% solution once every 3 days to Persian cats in a cattery indicated potential gastrointestinal signs in the cats, including salivation, anorexia, increased liver enzymes, and emesis and muscle weakness.66 Nausea, vomiting, skin rash, thrombocytopenia, and hypokalemia have also been reported with fluconazole therapy. The author dealt with acute hepatopathy in a cat treated topically with a commercial over the counter preparation of clotrimazole; the owner treated a large cutaneous wound topically several times a day. The hepatopathy resolved within several days of discontinuing therapy.
Voriconazole is associated with a number of side effects. Interestingly, voriconazole (but no other azole) causes vision disturbances in humans. Disturbances are characterized by loss of color discrimination, blurred vision, bright spots, wavy lines, and photophobia; up to 30% of human patients are afflicted, although the drug rarely is discontinued because of this effect. Visual hallucinations are reported in 5% of human patients. Experimentally, voriconazole produced dose-dependent changes in the electroretinogram of dogs at plasma levels comparable to those in humans.68 Skin rashes also commonly occur; although most are mild, occasional severe reactions (e.g., Stevens–Johnson syndrome) occur. Clinical signs resolve when the drug is discontinued. As with other azoles, voriconazole is associated with increased hepatic leakage enzymes as well as serum alanine phosphatase; increased liver weight occurred experimentally in dogs after 1 month of dosing at 3 mg/kg.68 Hepatotoxicity should be assumed to be dose and duration dependent. Although most changes are asymptomatic, severe hepatopathy has been reported in humans receiving voriconazole, with effects apparently dose dependent. Hepatic function might be measured before and 2 weeks into therapy and then every 2 to 4 weeks. The use of N-acetylcysteine, S–adenosylmethionine, or other hepatoprotectants might be considered. Again, because dogs appear to autoinduce, the impact of voriconazole on the liver is not clear. Other gastrointestinal side effects include nausea, vomiting, diarrhea, and abdominal pain.42 Although voriconazole is not nephrotoxic, nor is it eliminated in the urine, the carrier of the intravenous preparation may accumulate in patients with impaired renal function, and therefore the intravenous preparation should not be used in these patients.42
Therapeutic Use
In general, itraconazole and fluconazole are more efficacious against many organisms than ketoconazole; however, the spectrum of fluconazole is limited compared with that of itraconazole. Ketoconazole has been used effectively for dermatophyte infections; mucocutaneous candidiasis; and many systemic mycoses in both dogs and cats. Ketoconazole has been reported to be effective in the treatment of dermatophytosis,70,71 blastomycosis,72 histoplasmosis,73 coccidioidomycosis,74 and cryptococcosis.75 Ketoconazole probably should not be used alone for treatment of canine blastomycosis; recommendations are to use amphotericin in addition to ketoconazole. Higher doses also are indicated for systemic cryptococcosis and coccidioidomycosis. Ketoconazole also has proved effective for treatment of Malassezia dermatitis, and candidiasis. It is available as a topical shampoo that can be useful for treatment of dermatophytosis or Malassezia. Ketoconazole has little efficacy (43%) against Aspergillosis species, fluconazole more efficacy (although some isolates, including A. fumigatus, are inherently resistant), and itraconazole most efficacy (60% to 70%). Fluconazole has been used successfully to treat ketoconazole-resistant strains of Candida. Equal efficacies of itraconazole and fluconazole have been shown for cryptococcal meningitis, despite relatively poor penetration of the CSF by itraconazole. Both are equally effective in Candida-induced pyelonephritis. Comparison of ketoconazole and fluconazole reveals fluconazole to be more active against coccidioidal meningitis.
Among the imidazoles, itraconazole and fluconazole are being used more consistently than the others for systemic treatment of susceptible fungal infections. For itraconazole76 conditions successfully treated include blastomycosis54 (including ocular77), histoplasmosis,78 cryptococcosis (including meningitis),79–81 sporotrichosis,82 aspergillosis,83 dermatophytosis,64 dermatophytic pseudomycetomas,84,85 phaeohyphomycosis,86 and cutaneous Alternaria.87 Efficacy against Aspergillosis is better than that clinically recognized for any other agent (not including newer drugs), and although resistance is rare, treatment failure rates of up to 50% have been reported for itraconazole. In animals administration of itraconazole at a rate of 5 mg/kg twice daily is efficacious in the treatment of blastomycosis and histoplasmosis. After administration of 10 mg/kg, the Cmax for itraconazole in dogs was 13.5 ± 8.5 μg/mL versus 3.55 ± 2.81 μg/mL at 5 mg/kg. Although the MIC90 of most infecting fungal organisms will be achieved at 5 mg/kg, at the higher 10 mg/kg dose, the MFC for itraconazole will be achieved for B. dermatitidis, almost reached for H. capsulatum, but not reached for C. immitus. However, the incidence of adverse effects may be greater at this higher dose.54 Concentrations will be higher at steady state. The efficacy of itraconazole against coccidioidomycosis is equivocal, requiring long-term therapy. Relapse of disease appears to be common. Despite the larger MIC for dermatophytes compared to other susceptible fungal organisms, itraconazole at 1.5 to 3 mg/kg every 24 hours was effective in 8 of 15 cats in one uncontrolled clinical trial for treatment of dermatophytosis.64 Fluconazole is only modestly effective toward sporotrichosis; itraconazole should be considered first-line therapy.69
>KEY POINT 9-11
In general, itraconazole and fluconazole are more efficacious against many organisms than ketoconazole; however, the spectrum of fluconazole is limited compared with that of itraconazole.
Itraconazole has been used to treat canine blastomycosis (5 mg/kg/day); sporotrichosis (7.5 mg/kg/day); and, in conjunction with surgery, nasal aspergillosis (10 mg/kg/day). In cats it has proved effective for treatment of cryptococcosis and histoplasmosis. The dermatologic pharmacokinetics of itraconazole support pulse therapy, which has been used in human medicine for treatment of selective dermatologic fungal disorders. Treatment occurs for 2 consecutive weeks of daily administration each month for 3 consecutive months.88 One report of itraconazole used to treat dermatophytosis noted similar success with this technique.64
There are few reports regarding the efficacy of fluconazole for treatment of fungal infections in animals; infections that have been treated include blastomycosis,89 cryptococcosis,74 and nasal aspergillosis.90 Efficacy has, however, been demonstrated toward a variety of fungal disorders in humans. Pulse dosing (once weekly) of fluconazole also has been described for treatment of skin infections in people.
Enilconazole has excellent in vitro activity against a number of organisms, but its topical use is limited to dermatophytes and nasal aspergillosis. Miconazole also is limited to topical use; combination with polymyxin B yields synergistic activity.
Newer azoles are likely to prove even more efficacious than fluconazole and itraconazole for the treatment of aspergillosis and coccidioidomycosis. Voriconazole is approved for treatment of selected infections, but in particular aspergillosis. The maintenance dose in humans is approximately 2 to 5 mg/kg twice daily. In human patients who have not responded adequately to traditional therapy for treatment of aspergillosis, 38% had a partial to complete response to voriconazole used as salvage therapy. In a study of invasive aspergillosis in humans, 53% of patients receiving voriconaozle responded (partial to complete) within 12 weeks compared with only 32% of patients receiving amphotericin B; survival rates were 71% and 58%, for each drug, respectively.42 Because of superior clinical response, safety, and survival, treatment of immuncompromised human patients with voriconazole was demonstrated to be economically superior to treatment with amphotericin B.91 For Pseudallescheria/Scedosporium, major pathogens in human immunocompromised hosts normally resistant to amphotericin B and Fusarium sp., response rate was 30% to 63% and 50%, respectively, for the two species. Despite excellent in vitro efficacy against C. neoformans and good CNS penetration, voriconazole is not recommended for treatment of Cryptococcosis, in part because of its failure in human patients. The same may be true for blastomycosis, histoplasmosis, and coccidiodomycosis. Although effective in animal models, successful therapy with voriconazole for these organisms has not been demonstrated in humans. A salvage approach might be considered for these organisms.42
A single case report describes the successful treatment with posoconazole of a fungal disorder caused by Mucor (Zygomycetes class; Mucorales order) species on the nose of a 15-year-old cat.91a After poor response to fluconazole, the cat was treated at 5 mg/kg daily for 3 months, with initial response evident in 2 weeks and continued response for the remainder of the 3-month treatment period.
Terbinafine was shown to enhance efficacy when combined with itraconaozle or fluconazole when treating candidiasis characterized by low susceptibility to the azoles.92 Indeed, combination with benzoyl peroxide topically enhanced treatment of candidiasis associated with Pseudomonas and Staphylococcus aureus infections in humans.93
Benzimidazoles
Benzimidazoles (e.g., thiabendazole) may be better known for their anthelmintic activity, but many also are characterized by a broad range of antifungal activity at relatively low doses. Benzimidazoles bind to β-tubulin of the microtubule. Not only is mitosis blocked, but selected organelles also are displaced, such as mitochondria in hyphal tips, which alters linear growth. Unfortunately, resistance generally caused by point mutations in the β-tubulin genes, limits the antifungal activity of the drugs. The spectrum of thiabendazole is limited to the dermatophytes (toward which activity can be -cidal) and, to a lesser degree, A. fumigatus, penicillinosis, and some Fusarium species. Thiabendazole also is effective against Pneumocystis carinii. Its use as an antifungal is largely limited to topical therapy (e.g., otitis externa).
Echinocandins
The echinocandins are the first new antifungal drugs to be developed in the last 15 years.94 Originally elucidated from different fungal organisms, including Aspergillus, they are synthetically modified lipoproteins derived from fermentation broth of a number of organisms.95 Included in the chemicals identified thus far are aculeacin A, echinocandin B, pneumocandin B, enfumafungin, and papulacandins. These chemicals inhibit the synthesis of β-D-glucan (the major glucan in the cell wall of Aspergillus), disrupting the fungal cell wall (see Figure 9-1). Their novel mechanism of action results in rapid fungicidal effects for some organisms with minimal side effects.96 Caspofungin (see Figure 9-2) is the first of the drugs to undergo approval in the United States. Its spectrum of activity is limited but does include Candida sp. and Aspergillus sp.; the drug is approved to treat the latter. Other drugs include micafungin and anidulafungin, both approved to treat candidiasis.
The large molecular weight of the echinocandins and poor oral absorption limit use to intravenous administration. Drug does not distribute well into the urine or CNS. Concentrations in the lungs approximate those in the plasma. The compounds undergo phase I metabolism to inactive metabolites. Despite its metabolism by the liver,97 capsofungin does not serve as a substrate for major CYP 450. However, selected drugs will alter capsofungin, disposition, including cyclosporine (which increases it). In humans caspofungin acetate is characterized by an elimination half-life of 9 to 10 hours but is administered once daily. Capsofungin is involved in few drug interactions. Side effects are unusual but include phlebitis, fever, nausea, skin rash, and abnormal liver function. Despite low CNS concentrations, human cases of cerebral aspergillosis have responded to caspofungin treatment. The ability to kill Aspergillus is controversial; generation of cell wall–deficient colonies in vitro may be associated with organism viability. The optimal dose has not yet been established in humans; generally a loading dose of 70 mg is followed up by doses of 50 to 70 mg daily. A ceiling dose of 1 mg/kg has been suggested, but doses of micafungin as high as 300 mg daily (up to 8 mg/kg daily) was not associated with toxicity in humans.94 Efficacy has been demonstrated against fluconazole-resistant strains of Candida. However, Cryptococcus is resistant. Although Fusarium is resistant, Scedosporium is moderately susceptible and Saccharomyces is susceptible.
Flucytosine
Structure–Activity Relationship
5-Flucytosine (FLU; 5-fluorocytosine) was originally developed as an anticancer drug much the same as its sister anticancer drug, 5-fluorouracil. It is a water-soluble powder.
Mechanism of Action
As an antimetabolite, FLU interferes with DNA synthesis after its conversion to 5–fluorouracil, a substitution compound that prevents synthesis in the fungal cell.4,40 The compound enters the cell by way of cytosine permease, which also takes up adenine, guanine, hypoxanthine, and cytosine.3 The enzyme responsible for conversion of FLU to 5–fluorouracil is a cytosine deaminase, an enzyme whose absence in mammalian cells renders FLU relatively specific for fungal cells. The effect of FLU depends on subsequent metabolism: if converted by pyrimidine processing enzymes to a uridine monophosphate derivative, inhibition of thymidylate synthase and DNA synthesis yields -cidal effects. Alternatively, it can be converted by way of a pyrimidine salvage pathway resulting in metabolism to a uridine-5’-triphosphate derivative. Subsequent incorporation into RNA results in impaired protein synthesis and fungistatic effects (Candida, Cryptococcosis). Many organisms are intrinsically resistant to FLU because they either lack the permease enzyme or have a defective deaminase enzyme. Resistance also develops relatively rapidly, particularly for aspergillosis followed by cryptococcosis and candidiasis (especially Candida krusei).3 Secondary resistance usually reflects a decrease in an enzyme responsible for formation of uridine monophosphate. Use in combination with another antifungal agent reduces the development of resistance.
Spectrum of Activity
The spectrum of activity of FLU is limited and includes cryptococcosis, candidiasis and some cladosporiosis, aspergillosis, chromomycosis, and sporotrichosis. It has been the treatment of choice for cryptococcosis in humans.4,40 Combination therapy is usually indicated (e.g., amphotericin, ketoconazole). When FLU is used alone, resistance develops rapidly. Synergism occurs with amphotericin B and probably with ketoconazole (or other imidazoles).
Pharmacokinetics
Oral absorption of FLU is rapid and close to complete. Peak plasma concentrations occur in 1 to 2 hours. Distribution is large, to total body water. Protein binding is minimal, and CSF concentrations reach up to 90% of plasma concentrations. Penetration of aqueous humor and joints is good. The half-life of FLU is 3 to 6 hours. Most of the drug is excreted into the urine unchanged. Renal clearance is similar to that of creatinine and thus may be significantly decreased if renal dysfunction is present. Doses will probably need to be modified for patients with renal disease.
Side Effects
Because FLU interferes with DNA synthesis, body systems composed of rapidly dividing cells are adversely affected. Bone marrow depression is manifested as anemia, leukopenia, and thrombocytopenia (pancytopenia). This toxicity may be serious and is more common in patients with renal disease. Gastrointestinal toxicity is manifested as nausea, vomiting, and diarrhea, but it is not usually serious. Reversible, erythemic, alopecic dermatitis has been reported in dogs.
Griseofulvin
Structure–Activity Relationship
Griseofulvin (see Figure 9-1) is produced from a Penicillium species bacterium. The drug is insoluble in water.
Mechanism of Action
Griseofulvin enters fungi through an energy-dependent transport system. Griseofulvin inhibits fungal mitosis by binding to the microtubules that form the mitotic spindle. The formation of microtubules from tubulin is inhibited. Formation of cytoplasmic microtubules responsible for transport of endogenous compounds also is inhibited. Other drugs, such as colchicine and vincristine, which also bind to and inhibit the microtubule, do so at a site that is different from that of griseofulvin. Griseofulvin also probably inhibits nucleic acid and fungal wall synthesis. It is not certain if griseofulvin is fungistatic or fungicidal. Resistance probably reflects decreased drug uptake.
Spectrum of Activity
The spectrum of activity of griseofulvin reflects the presence of an energy-dependent transport system in the fungal organism. Those with prolonged energy-dependent transport systems are susceptible, whereas those with independent systems of short duration are not. Efficacy is limited to dermatophytes: Microsporum, Trichophyton, and Epidermophyton. Because of its distribution into keratin, griseofulvin remains the drug of choice for fungal infections of the nails.
Pharmacokinetics
Oral absorption varies because of water insolubility and depends on particle size and preparation. Absorption is increased in the presence of fat. The rates of dissolution and disaggregation alter the bioavailability of different products. Bioavailability of the ultramicrosize is at least 50% greater than that of the microsize. Although griseofulvin penetrates the stratum corneum, it does not achieve effective concentrations topically. Griseofulvin is widely distributed to most tissues, but it is deposited and concentrated in keratin precursor cells. Thus it is incorporated in new keratin of skin, nails, and hair and (in humans) is secreted in perspiration. Although new keratin formed during treatment with griseofulvin is resistant to fungus, griseofulvin does not destroy fungi that infect the outer layers of the skin. New hair, skin, or nail growth accompanied by shedding of older growth is necessary before the fungus is affected; new growth is the first to be free of disease. Thus skin infections require 4 to 6 weeks of therapy, whereas toenails may require up to a year of therapy. Long hair breeds probably should be treated for a longer period of time compared to short hair breeds.
>KEY POINT 9-12
Treatment with griseofulvin must be sufficiently long for new growth to replace infected growth.
Hepatic metabolism of griseofulvin by dealkylation is significant; metabolites are not active. The half-life reportedly is 24 hours in the dog. Half of the drug is excreted as metabolites in the urine. The rest is excreted through the bile unchanged in the feces.
Preparations
Griseofulvin is available for oral use as either a microsize (particle size 10 μm) or ultramicrosize (particle size 2.7 μm; e.g., Fulvicin, Gris-PEG) tablets. The drug should be administered with a fatty meal, particularly if the microsize preparation is used. Duration of therapy is at least 4 to 6 weeks (new hair growth must occur) and possibly longer. The drug should be administered at least once a day despite initial reports that recommend one weekly administration.
Side Effects and Drug Interactions
Side effects to griseofulvin are not uncommon. Nausea, vomiting, and diarrhea can be minimized by administration of the dose in divided increments with a meal. Hepatotoxicity may occur, and use in liver disease should be avoided. Idiosyncratic toxicity has been reported in the cat, manifested as gastrointestinal upset, neurologic disease, and bone marrow suppression.98 The reaction appears to be both dose and duration independent. Signs may not be reversible, depending on the severity. Cats with feline immunodeficiency disorders may be more likely to develop neutropenia.99 Certain feline breeds (e.g., Persian, Siamese, Abyssinian) may be more commonly affected.98 At very high doses, the drug is teratogenic and carcinogenic in animals. The drug should not be given during the first two trimesters of pregnancy. Use with a shampoo (miconazole or chlorhexidine) enhances efficacy.
Griseofulvin is a potent inducer of microsomal enzymes. The clinical sequelae of this drug interaction are not well known, although increased metabolism of other drugs should be anticipated.
Allylamines and Thiocarbamates
The allylamines (e.g., terbinafine, naftifine) and the much older thiocarbamates (e.g., tolnaftate) competitively inhibit squalene epoxidase, blocking conversion of squalene to lanosterol, leading to squaline accumulation and ergosterol depletion in the cell membrane. Terbinafine has a much higher affinity for fungal compared with mammalian squaline epoxidase. Aviod uptake of terbinafine into body fat and epidermis enhances and potentially limits its efficacy to dermatophytes and superficial pathogens of the skin. Antifungal effects are -cidal in these organisms; it has proved more efficacious than griseofulvin for both acute and chronic dermatophyte infections in humans. Efficacy has also been demonstrated against S. schenckii and Aspergillus (A. flavus more so than A. fumigatus). Although it is not clear if effective tissue concentrations are acheived, in vitro activity toward B. dermatitidis, C. immitis, and H. capsulatum has also been described as excellent. Some strains of Crypotococus sp. also are susceptible. In vitro activity also has been demonstrated toward Rhizopus, Alternaria, Phialophora, Chrysosporium, and Exophiala spp.3,100 Fungistatic efficacy has been demonstrated against yeasts,101 with activity being poor toward Candida.3 Increasingly, terbinafine has enhanced the efficacy of other antifungal drugs when used in combination for treatment of a variety of fungal disorders and pythiosis.100b Terbinafine expresses some antibacterial activity; for example, when combined with benzoyl peroxide, its topical spectrum (in human) is expanded to include Pseudomonas and Staphylococcus.100b In contrast to terbinafine, tolnaftate is limited to treatment of dermatophytes. Resistance to the allylamines is rare, but the drugs potentially can be affected by multidrug resistance efflux mechanisms.3
Terbinafine, available in oral and topical preparations, is well absorbed (80% in humans) after oral administration, although fat facilitates absorption. High concentrations occur in the stratum corneum, sebum, and hair. The drug is metabolized by the liver in humans; the elimination half-life is sufficiently long to allow once-daily administration, with steady state not occurring for 10 to 14 days in humans.3,102 An abstract reporting pharmacokinetics in cats suggested that a dose of 20 to 40 mg/kg once daily provided sufficient concentration of drug in the skin. The drug was well tolerated at this dose.103 Side effects of terbinafine after oral administration are limited to gastrointestinal and skin symptoms; hepatobillary dysfunction is a rare adverse event.3 Because inhibition of ergosterol synthesis occurs at a step before cytochrome P450 involvement, the allylamines do not affect steroid synthesis as do the imidazoles.
Iodides
The mechanism of antifungal action of the iodides is not known. Iodide is rapidly and completely absorbed orally. Distribution is to the extracellular fluid. Thyroid uptake will concentrate the drug up to 50 times that in plasma. Iodide is available as a 20% Na and K+ salt oral or intravenous preparation. Both salts have been used successfully to treat canine and feline cutaneous or lymphocutaneous forms of sporotrichosis, and, as such, it remains the drug of choice.104,105 Oral Na+ preparations are usually used. Iodide toxicity is more common in cats and is manifested as sweating; tachycardia; dry, scaly coat; diarrhea; and polyuria/polydipsia. Cardiomyopathy has been reported in cats. Treatment causing clinical signs of iodinism should be discontinued for 1 week and then reinstituted at a lower dose. Iodine has also been reported to be effective for various other fungal diseases, particularly as a topical ointment for localized skin infections. Topical iodine preparations continue to be available and might be used to treat fungal rhinitis (discussed later).
Lufenuron
Lufenuron is a chitin synthetase inhibitor used for the control of fleas in dogs and cats. Its use for the treatment of dermatophytes is controversial. A retrospective study of dogs and cats found that dogs treated with once-daily administration of 50 to 60 mg/kg responded (based on skin scraping) within 21 days. Cats received doses that ranged from 50 to 266 mg/kg, with response in 8 to 12 days. However, clinical trials have not been able to accomplish what the retrospective study implied. A study describing efficacy at a single dose of about 80 mg/kg every 2 weeks until culture cure was followed by clinical trials that failed to eradicate or prevent infection at 140 mg/kg. When combined with weekly enilconazole shampoos, 60 mg/kg every 30 days caused clinical response in most cats in a cattery after several weeks of therapy; however, not all animals became culture negative, and relapse occurred.3 Several abstract reports have failed to demonstrate efficacy, despite differences in duration or dose. This includes studies using established animal models for which itraconazole is effective.106 Moriello and colleagues107 could not demonstrate a protective effect of lufenuron (30 or 122 mg/kg monthly for 2 months) when used before experimental infection of juvenile cats with Microsporum canis. Studies based on combination rather than sole therapy may provide more information.
Combination Therapy
Combination antifungal therapy has been reviewed in human medicine,108 including the molecular basis.9 Indications include patients at risk (immunocompromised). Few clinical studies provide conclusive evidence for or against combination therapy.108 In vitro studies examining combination therapy often used different methods, limiting the ability to determine a consensus. Among the difficulties with clinical studies is the more common use of combinations in patients with greater severity of disease, thus biasing results. Not surprisingly, sample size is often too small to demonstrate significant differences. In vitro data for C. neoformans exposed to ampohtericin B, when combined with imidazoles, indicates, in order of minimal to most synergistic effect, fluconazole = itraconazole < posaconazole.9 The combination of flucytosine with itraconazole indicated the most synergistic activity toward C. neoformans. In contrast, for candidiasis, the order of least to most synergistic activity when imidazoles are combined with terbinafine was fluconazole < posaconazole < itraconazole < voriconazole.9 In animal (mice) models, the addition of fluconazole offered no benefit to amphotericin B for treatment of cryptococcosis. Flucytosine potentiated the effect of fluconazole but not posaconazole for treatment of C. neoformans. The most effective combination was amphotericin with flucytosine; the addition of fluconazole to this combination reduced the fungal burden even more. The efficacy of voriconazole toward aspergillosis was enhanced by caspofungin.
The combination of antifungals with other drugs not traditionally considered antifungal may offer enhanced clinical response. For example, combination therapy with antibacterials that target DNA may enhance efficacy. Examples include the fluroquinolones and the rifamycins.9 Rifampin has shown some efficacy against fungal microorganisms (H. capsulatum, Aspergillus sp., and B. dermatitidis) when combined with amphotericin B. Amphotericin B apparently facilitates movement of rifampin through the fungal cell wall into the organism, where RNA polymerase then can be accessed. A beneficial effect has been demonstrated in vivo (but not in vitro) for trovafloxacin combined with fluconazole or amphotericin G. In vitro efficacy has been demonstrated for the latter. The combination of traditional antifungal agents with terbinafine or chitin synthesis inhibitors (e.g., caspofungin) may also prove to be effective combinations for treatment of selected infections (e.g., coccidioidomycosis, aspergillosis, and others).
Among the drug combinations currently being actively researched are antifungal agents with immunomodulators. Included are cytokines such as granulocyte or macrophage colony-stimulating factors or interferon γ-1b. In contrast, the impact of calcineurin antagonists (e.g., tacrolimus and cyclosporine) is one of exacerbation, at least for cryptococcal meningitis.
Therapeutic use of Antifungal Agents
Dermatologic Fungal Infections: Dermatophytosis
A number of fungal organisms inhabit the hair coats of dogs and cats. Alternaria, Cladosporium, and yeasts may be associated with dermatitis. Dermatophytes can be isolated from normal animals or can be a cause of infection. Dermatophyte infections generally are self-limiting, with ability to mount an inflammatory response being an important determinant of infection control. Accordingly, drugs such as glucocorticoids, which mute the inflammatory response, predispose a patient to dermatophyte infection; dermatophytosis infection is 3 times more prevalent in cats infected with feline immunodeficiency virus than in noninfected cats.66 The route of drug administration (topical versus systemic therapy) depends on the extent of infection, with the exception of Trichophyton infections, which should be treated systemically.
Topical therapy is indicated for all patients with dermatophytosis and may be the sole therapy for local, nondiffuse lesions. Hair coat preparation before medication should include clipping and bathing to remove hair and crusts. Several medicaments are available as shampoo, ointment, or cream formulations for topical therapy (see Table 9-5). Active ingredients include povidone–iodine, chlorhexidine, and imidazole. Other active ingredients that can be applied topically include captan, lime sulfur, and sodium hypochlorite. Short-term topical glucocorticoid therapy might be considered to control acute inflammation when present. Topical administration of enilconazole emulsion has been useful for treatment of feline dermatophytosis.
Moriello66,107 retrospectively evaluated in vitro and in vivo studies of treatments for dermatophytosis in dogs and cats. Topical treatments consistently found to be effective when administered once or twice weekly were lime sulfur (1:16), 0.2% enilconazole rinse (twice weekly; response in as early as 5 weeks), and a combined 2% miconazole–chlorhexidine shampoo. Captan, chlorhexidine (as sole agent), and povidine–iodine were generally ineffective.
Response to systemic therapy is likely to be more rapid if combined with topical therapy.66 Systemic therapy should probably be preceded with a total body clip. Griseofulvin is the treatment of choice for long-term systemic antifungal therapy of dermatophytosis,109 although expense mandates that a diagnosis of dermatophytosis be confirmed. Care should be taken that the proper dose and duration of therapy are followed; treatment probably should be longer for longer-haired animals. Cure was reported in 63 to 70 days in one study with a mean of 41 days at 50 mg/kg in another study (as reviewed by Moriello).66 For infections that do not respond to griseofulvin, an imidazole can be used. Ketoconazole has been used successfully, particularly when dosed at 10 mg/kg daily for 20 days. Itraconazole has, however, proved more efficacious for treatment of dermatophytes in human patients and has proved efficacious experimentally in cats infected with M. canis.110 Itraconazole was effective at 10 mg/kg once daily for 56 days or for 28 days followed by a week–on week–off pulse dosing for 56 to 70 days.111 Interestingly, low-dose itraconazole (1.5 to 3 mg/kg) in 15-day cycles required a shorter time period at 1 to 3 cycles or 15 to 45 days. However, only 8 of 15 cats were successfully treated in this study, and two of these cats required a second cycle. Using an open clinical trial, a protocol using itraconazole (10 mg/kg once daily) and lime sulphur rinses (cat coat saturated with rinse at 8 ounces per gallon of water) every 7 days for 21 days was associated with successful treatment of dermatophytosis in shelter cats.111a Cure was longer with higher fungal load (18.4+ 9.5 days) compared to smaller loads (14.5+5.7 days). Cats tolerated the rinses well. Efficacy of lufenuron was not substantiated in controlled studies. Terbinafine has demonstrated efficacy toward dermatophytosis in dogs and cats, although higher doses (>30 to 40 mg/kg) generally are required to achieve a mycologic cure. Time to cure in dogs varied from 21 to more than 126 days and from 28 to 84 days in dogs. A lower dose (10 to 30 mg/kg) may also be effective in both dogs and cats, although treatment should be expected to be approximately 60 to 90 days.107 Use of terbinafine at 30 mg/kg for 2 weeks resulted in a cure of 11 of 12 cats with M. canis, and at 8.25 mg/kg daily, it eradicated spores from asymptomatic carrier cats (as reviewed by Bossche3). The use of systemic terbenafine at either a low (10 to 20 mg/kg) or high (30 to 40 mg/kg) dose once daily was compared with placebo treatment in cats (n = 9 per group) ranging between 1.5 and 4.5 months of age that were experimentally infected with M. canis. Cats were treated for 120 days. Response in the low-dose treatment group did not differ from that of the control group but did in the high-dose group; the number of cats cured per group was not provided. PDCs did not differ between the two dose groups, but concentrations in the hair were significantly higher with the higher dose.112 Drug accumulated with multiple dosing, with concentrations at 3 months higher than at 2 months, suggesting peak effects may take up to 3 months (or more). Median concentrations in plasma at 9 and 120 days were 1.4 mg/L and 4.1 mg/L respectively, in the low-dose cats compared with 1.7 and 5.5 mg/L respectively, in the high-dose cats. Median concentrations in the hair at 9 and 120 days were 1 mg/L and 1.2 mg/L, respectively, in the low-dose cats compared with 1.9 and 3.6 mg/L respectively, in the high-dose cats. The highest concentration achieved in the hair in any one cat was 7.92 μg/mL; this compares with humans receiving only 6 mg/kg, for which concentrations reach 2.40 to 55 μg/g.
Additional topical therapies for treatment of dermatophytosis or other dermatologic fungal disorders are also addressed in Chapter 22. Environmental cleansing may be important to treatment of dermatophytosis. This might be accomplished with either 2% chlorhexidine or 0.5% sodium hypochlorite.
Yeast or Yeastlike Infections
Malassezia
Malassezia (Pityrosporum) is a commensal organism that inhabits the skin, ear canal, anal sacs, vagina, and rectum of dogs. It is now recognized to be the causative agent of either localized or generalized pruritic inflammatory skin disease in dogs. The pathogenesis of the infection is controversial and appears to involve hypersensitivity to the organism. Postulated predisposing factors include allergic disease such as atopic dermatitis, diseases of cornification, chronic inflammatory skin disease, and previous therapy with antibiotics or glucocorticoids.
Therapy for Malassezia is directed toward removing predisposing factors and killing the causative agent.113 Antimicrobial therapy ideally should include both systemic and topical drugs. Ketoconazole and itraconazole are the systemic drugs of choice and should be given for at least 30 days. Topical therapy may be sufficient in some cases. Antifungal shampoos containing chlorhexidine, miconazole, or ketoconazole should be given at least twice weekly for a minimum of 6 weeks. Shampoos that resolve any exudate (e.g., benzoyl peroxide) may facilitate topical penetration of the antifungal drug. An acetic acid rinse (white vinegar and water at a ratio of 1:1) used twice weekly as a degreasing agent after shampooing may also prove beneficial as well as inexpensive. Application of eniloconazole emulsion may also be beneficial. The emulsion can be applied with a sponge or by whole-body immersion; the diluted product appears to remain stable for 4 to 6 weeks when protected against light, although use of a fresh dilution is recommended for each treatment.
Cole and coworkers114 studied in vitro the addition of 0.1% ketaconaozle to an ear rinse containing EDTA tromethamine and benzyl alcohol (T8 Solution) for treatment of canine otitis associated with Malassezia. The low concentration was sufficient to inhibit fungal growth. By itself, EDTA and thromethamine had no effect. Ahman and coworkers115 studied pulse dosing of itraconazole (5 mg/kg orally qd, 7 days on, 7 days off, 7 days on) for treatment of Malassezia pachydermatitis associated with greasy seborrheic dermatitis in Devon Rex cats (n = 6). Assessment was not blinding; control cats were not studied. Clinical signs associated with seborrhea resolved in all cats, with dramatic improvement recorded in the second week, supporting the role of Malassezia in the disease.
Negre and coworkers116 systematically reviewed the literature (before 2007) with regard to the treatment of skin disorders associated with Malassezia. Clinical trials in peer-reviewed veterinary literature were considered if they focused on treatment of Malessezia associated with dermatitis and involved more than five dogs. Studies were assessed for quality based on methods intended to select subjects that adequately represented the target population and minimized bias. Trials were classified by size of the study groups. Outcome measure assessment had to include both clinical evidence of response as well as the extent of reduction in fungal colony counts. Only 8 of 35 studies met the full criteria and another 6 met all but the mycologic requirement. Eleven studies were evaluated for evidence of efficacy for azole derivatives. Based on their review, 2% miconazole with 2% chlorhexidene was the only topical product for which good evidence existed. Fair evidence existed for either ketoconazole at 10 mg/kg daily or itraconazole at 5 mg/kg daily for 3 weeks.
Candidiasis
In the yeast phase, candidiasis normally occurs in the gastrointestinal, respiratory, or urogenital mucosa. The organism is acquired at birth and occurs at mucocutaneous junctions in the skin and in several organs inside the body. Factors that alter normal microflora (e.g., prolonged, high-dose, broad-spectrum antimicrobial therapy) predispose to the development of candidiasis. Cell-mediated immunity is important in the control of disease, and prolonged immunosuppression increases the risk of further spread. Generally, microcirculation of the organs filters organisms, leading to embolization.
Topical infection can be treated with topical antifungal products, including polyene macrolides, imidazoles, and gentian violet (1:10,000). Systemic therapy can be treated with amphotericin B, 5-flucytosine (combined with another antifungal drug), or the imidazoles.
Systemic Fungal Diseases
Therapeutic success with antifungal drugs can be enhanced by long-term therapy, generally one to several months beyond the resolution of clinical signs; avoidance of immunosuppressive drugs; and use of combination therapy, particularly for infections that are difficult to penetrate or are life or organ threatening.
Blastomycosis
Blastomyces organisms become established in the lungs and then disseminate throughout the body. The presence of clinical signs in dogs is indicative of disseminated disease and the need for aggressive therapy. Preferred sites of infection in dogs are the skin, eyes, bones, lymph nodes, subcutaneous tissues, nasal passages, and brain. These tissues are difficult to penetrate with most antifungal drugs, thus increasing the likelihood of therapeutic failure. Immunosuppression is common in dogs with blastomycosis, further hindering therapeutic success. The use of radioimmunoassay tests based on the major surface protein W1-1 is predictive of active infection, confirming absence of infection 100% of the time.117 The authors concluded that the test is more accurate than those based on the A antigen and thus may be more relevant to assessing response to therapy. When the titer is used, concentrations are high initially but decline during therapy, persisting for months.117
Amphotericin B has been the treatment of choice for blastomycosis.118 Although high doses are more effective, the risk of nephrotoxicity may necessitate a less aggressive approach; lipid-based products should be considered particularly in patients at risk. The total cumulative dose for amphotericin B (AmBD) generally is 4 to 6 mg/kg for dogs and 4 mg/kg in cats; a higher dose should be anticipated for lipid-based products. A maintenance dose of 0.15 to 0.25 mg/kg intravenously once monthly after the cumulative dose was reached was recommended in the early 1980s, but the scientific basis of this approach is questionable. Doses range from 0.15 to 0.5 mg/kg thrice weekly in dogs and cats, depending on renal function. Severe cases or intolerance to amphotericin B might be treated with a lipid complexed drug (see Table 9-3). Renal-sparing measures should be taken for patients with preexisting renal disease. Despite aggressive therapy, a relapse rate of 17% has been reported in dogs.26 Combination therapy of amphotericin B with an imidazole should be considered whenever possible and is particularly important for infections in tissues that are difficult to penetrate, such as the brain and eye. However, because of mechanisms of action, it is important to begin the two drugs simultaneously. Of the imidazoles, ketoconazole has been used alone to treat blastomycosis in humans, but it is less successful for animals as a sole agent. Ketoconazole can be used at 10 to 10 mg/kg/day (up to 30 mg/kg/day for difficult-to-penetrate tissues [e.g., eye, CNS]) when combined with amphotericin B. Because imidazoles are characterized by variation in drug disposition among animals, efficacy might be enhanced by increasing the dose. Although sequential use of amphotericin B followed by ketoconazole has been recommended, the two apparently can be used in combination immediately with little to no increased risk of toxicity. The rapid effects of amphotericin B are critical for life-threatening or organ-threatening infections. Itraconazole or possibly fluconazole are more likely to be effective than ketoconazole for the treatment of blastomycosis. Although itraconazole is more likely to be effective as sole therapy,119 combination therapy with amphotericin B is still recommended. Therapy should continue for at least 60 days or 1 month beyond resolution of disease indicators, whichever is longer. In their retrospective study of dogs with pulmonary blastomycosis, Crews119a found 79 dogs survived, 38 died, and 8 were euthanized. Most dogs were treated with itraconazole alone (n = 89) with does in 14 of these dogs less than recommended. No information was provided regarding survival rates among the different treatments, including 20 dogs treated with a combination of amphotericin B and an imidazole. No significant effect could be demonstrated in dogs loaded for 5 days (dose not given) with itraconazole.
With proper therapy up to 80% of dogs with blastomycosis can be effectively treated.54 The severity of pulmonary involvement appears to be a prognostic factor for both initial survival and the likelihood of relapse. Therapy may result in an initial worsening of respiratory disease, presumably because of an inflammatory response to dying organisms. A short course of short-acting glucocorticoids might be considered concurrently as therapy is initiated.120 Of the remaining 20% of animals that survive initial therapy, some may die within the first 2 weeks of therapy. Relapse can occur in up to 20% of infected animals within the first 6 months after therapy, but relapse after 1 year is rare.
Histoplasmosis
Host macrophages phagocytize the yeast phase of Histoplasma, and the organism then undergoes replication. The intracellular location is a mitigating factor in the hematogenous and lymphatic dissemination of the organisms from the lungs to other tissues. In most patients cell-mediated immunity brings the infection under control. The gastrointestinal tract may also be a primary site of infection, although dissemination from the lungs appears to be more likely.121 Although pulmonary infection may be self-limiting, therapy is indicated to prevent dissemination of infection.
Ketoconazole has been the drug of choice for mild pulmonary histoplasmosis.121 One study, however, reported that only itraconazole (5 mg/kg orally twice daily for 60 to 130 days; because the half-life is sufficiently long in cats, 10 mg/kg once a day for the same duration might be used) was effective against histoplasmosis in cats after ketoconazole had failed,78 and consequently, it is the preferred treatment. Fluconazole might be considered if the CNS is involved. For more severe pulmonary infection or gastrointestinal infection, therapy should be more aggressive and include itraconazole combination therapy with amphotericin B (0.15 to 0.5 mg/kg intravenously 3 times a week, increasing to up to 1 mg/kg every other day to a total cumulative dose of 7 to 8.5 mg/kg). Both of the latter drugs are much more effective (up to a hundredfold) than ketoconazole against histoplasmosis. The prognosis for patients with pulmonary histoplasmosis is fair to good but guarded when the disease has disseminated. Acute respiratory distress might warrant a course of antiinflammatories. In a retrospective study of airway obstruction associated with chronic histoplasmosis in dogs,122 resolution of respiratory clinical signs occurred in less than 1 week in those dogs treated with glucocorticoids alone compared with a mean of 2.6 weeks if combined with an antifungal and 9 weeks if receiving an antifungal only. The use of glucocorticoids was not associated with development of active or disseminated histoplasmosis. Treatment should continue for at least 60 days or 1 month beyond resolution of disease indicators, whichever is longer.
Cryptococcosis
Cryptococcus organisms infect the upper respiratory tract or the alveoli, potentially causing granulomas at both sites. Once established in the respiratory system, they can disseminate to other tissues. Infection of the CNS by either dissemination or direct extension is common. Cutaneous and ocular lesions are common in cats.
Cell-mediated immunity is critical to the host’s ability to overcome a cryptococcal infection Cryptococcal organisms have several features that affect their virulence. The capsule inhibits plasma cell function, phagocytosis, and leukocyte activity. Fever is uncommon (25% of dogs), particularly in cats. Immunosuppression is essentially necessary for cryptococcosis to develop in humans. Underlying diseases are, however, not often identified in cats or dogs with cryptococcosis. A risk factor for therapeutic failure in cats, which are generally more commonly infected with Cryptococcus than are dogs, is co-infection with immunosuppressive viruses.123 The impact of the use of glucocorticoids in patients with severe Cryptococcus infection is not clear. Using a murine model of CNS and pulmonary cryptococcosis treated with fluconazole (5 or 15 mg/kg every 8 hours), investigators found that whereas dexamethasone (0.15 mg/kg every 8 hours intraperitoneally) for 3 days, 30 minutes before fluconazole) administration did not have a deleterious effect on successful therapy with fluconazole, response was much better with early (1 day) as opposed to later (8 days) into infection. Because pharmacokinetics confirmed that fluconazole concentrations in the plasma and tissue remained above the MIC at both time points and did not change across time, the authors concluded that the number of fungal organisms influenced outcome. Although dexamethasone was not associated with beneficial or deleterious effects, the authors did not address whether sufficient animals had been studied to detect a difference;124 however, their study does support the early rather than later use of glucocorticoids.
Amphotericin B has been the treatment of choice for cryptococcosis.79,80,85 Doses should start at 0.5 to 0.8 mg/kg 2 to 3 times a week (0.25 to 0.5 mg/kg in cats); the drug can be given subcutaneously once diluted as long as the concentration does not exceed 20 mg/mL. A total cumulative dose at the higher end is recommended for cryptococcosis (4 to 8 mg/kg in one source, but up to 9 to 12 mg/kg in another for cats), reflecting longer duration of therapy. Initial therapy with a low dose and a subsequent increase in dose has been suggested for cats. Combination with 5–flucytosine or the imidazoles is likely to improve therapeutic success and is indicated for CNS infections because amphotericin B cannot sufficiently penetrate the blood–brain barrier. Ketoconazole has been used successfully to treat cryptococcosis. Itraconazole (10 mg/kg orally once daily), however, and fluconazole are more effective than ketoconazole against cryptococcosis. Cats appear to tolerate itraconazole better than ketoconazole.79 Both drugs, but especially fluconazole, are characterized by better tissue penetrability and can be used to treat CNS infection. One study with cats reported that 16 of 28 were cured of cryptococcosis after treatment with itraconazole (100 mg orally once daily) for a mean of 4 to 16 months,81 with duration dependent on resolution of disease indicators. Combination with either flucytosine (50 mg/kg every 6 hours) or terbinafine should also be considered.100 Response to therapy can be correlated to a decline in antigen titer.125 The prognosis for recovery is favorable if the CNS is not involved.
Coccidioidomycosis
Coccidioidomycosis begins as an alveolar infection that spreads to peribronchiolar tissues and the lung surface. Cell-mediated immunity is important in overcoming the infection. In immunodepressed animals or in animals with massive exposure, pulmonary infection becomes extensive, and the infection disseminates first to mediastinal and tracheobronchial lymph nodes and then to other tissues. Organs that are subsequently infected include, in order of likelihood, bone, joints, visceral organs, the heart and pericardium, testicles, eyes, and brain.
Coccidiodomycosis is the most difficult of the dimorphic fungal infections to treat, and antifungal therapy is particularly long.126 The duration varies with both the site of infection and the drug and can be for the life of the animal in some cases of disseminated disease. Whereas respiratory infections may spontaneously resolve, untreated disseminated infections will result in death. Ketoconazole has been the drug of choice for treatment in the past and is associated with a 60% cure, with CNS or orthopedic involvement being associated with worse prognosis. Ketoconazole therapy should extend at least 1 year beyond resolution of clinical signs. However, itraconazole is more effective and should be considered; the duration is likely to be the same. Amphotericin B is also effective for the treatment of coccidioidomycosis; encapsulated or otherwise modified formulations should be considered such that therapy can continue for a longer period of time. A lower maintenance dose of either ketoconazole or amphotericin B has been recommended once clinical signs are in remission, although this approach (i.e., using lower doses) should be used very cautiously. Therapy with itraconazole may occur for a shorter time, although this has not been well established in animals. Fluconazole is indicated for CNS infections. Combination therapy (i.e., amphotericin B with an imidazole) should be strongly considered for treatment of coccidioidomycosis. Deterioration of clinical signs and a rising complement fixation test are both indications for combination therapy with amphotericin B. In general, duration of therapy should be at least 8 to 12 months; relapse is common, particularly in cats.127
Paracoccidioidomycosis
Paracoccidiodomycosis, caused by Paracoccidioides brasiliensis, is a severe infection of the lungs and other body systems associated with granulamatous response in humans. Although rare in animals, one case was reported in South America in a Doberman Pinscher that presented with cervical lymphadenopathy.128
Aspergillosis
Aspergillosis can occur as either the localized form, involving cavities of the ears, nose, or sinuses; or the disseminated form, occurring primarily in the lung of immunocompromised animals. Both systemic and topical therapy should be implemented with either form of the disease. Systemic therapy should consist of an imidazole; among the drugs itraconazole is the most efficacious against aspergillosis. Itraconazole was effective in treatment of aspergillosis or penicillinosis in two cats, although hepatotoxicity developed in a third; topical treatment with clotrimazole resulted in cure for this animal.129 Itraconazole therapy for 10 weeks cured one dog infected with systemic and subsequent respiratory infection with Aspergillus niger.130 Topical therapy has included amphotericin B; thiabendazole; and enilconazole, a topical imidazole available in Europe but not the United States. Enilconazole might be the most effective treatment when directly infused into the nasal passages through fenestrated tubes.131 The 10% solution is diluted 1:1 with water and administered through surgically placed nasal tubes within 2 to 3 minutes. The solution emulsifies within several minutes of mixing; nasal tubes must be flushed after treatment. The total daily dose of 20 mg/kg is administered in two divided doses daily. Topical clotrimazole might be considered instead of enilconazole. One study reported response of refractory fungal rhinitis to surgical débridement combined with a povodine–iodine (see Chapter 11 for definition) wound dressing changed every 2 to 3 days; treatment continued until granulation tissue was present.132
Steinbach and coworkers133 retrospectively examined the impact of combination or sequential antifungal therapy on invasive aspergillosis in humans or in experimental animals and found interactions ranging from synergy to antagonism. Amphotericin B combined with 5-fluorocytosine was the most commonly used combination (49%), with others including amphotericin B with itraconazole (16%) or rifampin (11%). Combination therapy was associated with improvement in 63% of patients, generally with amphotericin B combined with either 5-fluorocytosine or rifampin. However, combination of rifampin with the azoles was discouraged because induction by the former was apt to decrease concentrations of the latter, potentially below that considered effective. Combinations of amphotericin B with itraconazole were considered indifferent. In retrospective studies of animal model reports, amphotericin B plus 5-fluorocytosine, rifampin, or itraconazole was described as indifferent, whereas amphotericin B with micafungin was described as a positive interaction. Sequential therapy also was associated with benefits: Improvement was noted with amphotericin B or itraconazole followed by voriconazole but not with itraconazole followed by amphotericin B.133 Indeed, azoles followed by amphotericin B (rather than opposite sequence) were found to be antagonistic, as might be expected based on the mechanism of action of the two drug classes. The use of capsofungin alone or with other antifungal drugs may provide alternative therapies for treatment of aspergillosis; terbinafine might also be considered in combination.
The use of voriconazole in animals for treatment of aspergillosis is very appealing but has not yet been described. The potential side effects of voriconazole and its complex pharmacokinetics suggest that use should be based on scientific studies that establish dosing regimens necessary to achieve MICs demonstrated for targeted fungal organisms, as well as studies that establish safety after several weeks of therapy at that dose.
Subcutaneous Mycoses
Sporotrichosis
Sporotrichosis occurs in three clinical forms: cutaneous, cutaneolymphatic, and disseminated. Disseminated disease generally involves most internal organs. The treatment of choice for both dogs and cats is supersaturated potassium iodide (SSKI).134 Itraconazole at 5 to 7.5 mg/kg every 12 to 24 hours has been recommended.126 Cats are more sensitive than dogs to the side effects of SSKI. Treatment should continue for at least 30 days beyond clinical remission. Immunosuppressive drugs should be avoided, if possible, for the duration of the animal’s life; recurrence in clinically cured animals has been reported after immunosuppressive doses of glucocorticoids. Imidazoles (e.g., ketaconazole or itraconazole) should be used to treat animals that cannot tolerate or do not respond to SSKI. Other antifungals (e.g., terbinafine) might be combined with iodine.
Rhinosporidiosis
Rhinosporidium rarely causes disseminated disease in animals and is essentially limited to the nasal tissues. Surgical excision is the treatment of choice.135 For recurrences dapsone may be useful. Alternatively, ketoconazole or itraconazole may be successful.
Pythiosis
Originally referred to as phycomycosis, pythiosis, a granulomatous disease, is caused by a number of taxonomically diverse nonseptated hyphal Oomycetes. The cell wall of Oomycetes differs from that of true fungi. Most notably is the limited amount of sterols in the fungal cell wall; hence antifungal agents are often ineffective against infections caused by these organisms. No antifungal agent has proved efficacious against this organism. However, two cases of gastrointestinal pythiosis were treated postoperatively (and apparently successfully) with a combination of itraconazole (10 mg/kg daily) or combined with terbinafine (7.5 mg/kg qd).136 Up to 20 mg/kg itraconazole daily has been recommended.126 Brown137 described the MIC of Pythium insidiosum and Lagenidium sp toward a number of antifungal agents and found that the relative efficacy to be azoles (limited) < terbinafine and caspofungin (minimal to moderate inhibition) < mefenoaxam (profound efficacy). The latter drug is a fungicide used in agriculture and is approved by the Environmental Protection Agency. Its mechanism is inhibition of RNA polymerase.
1. McGinnis M.R., Rinaldi M.G. Antifungal drugs: mechanisms of action, drug resistance, susceptibility testing, and assays of activity in biological fluids. In: Lorian V., editor. Antibiotics in laboratory medicine. ed 4. Baltimore: Williams & Wilkins; 1996:176-211.
2. Carter G.R., Chengappa M.M. Essentials of veterinary bacteriology and mycology, ed 4. Philadelphia: Lea & Febiger; 1991.
3. Bossche V.H., Engelen M., Rochette F. Antifungal agents of use in animal health—chemical, biochemical and pharmacological aspects. J Vet Pharmacol Therap. 2003;26:5-29.
4. Bennett J.E. Antimicrobial agents: antifungal agents. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, 2006.
5. Ben-Ami R., Lewis R.E., Kontoyiannis D.P. Immunocompromised hosts: immunopharmacology of antifungal drugs. Clin Infect Dis. 2008;47:226-235.
6. Marichal P. Mechanisms of resistance to azole antifungal compounds. Current Opinion in Anti-Infective Investigational Drugs. 1999;1:318-333.
7. Richardson M., Lass-Flörl C. Changing epidemiology of systemic fungal infections. Clin Microbiol Infect. 2008;14(Suppl 4):5-24.
8. Kontoyiannis D.P., Lewis R.E. Antifungal drug resistance of pathogenic fungi. Lancet. 2002;359:1135-1144.
9. Lupetti A., Nibbering P.H., Campa M., et al. Molecular targeted treatments for fungal infections: the role of drug combinations. Trends Mol Med. 2003;9(6):269-276.
10. Groll A.H., Piscitelli S.C., Walsh T.J. Antifungal pharmacodynamics: concentration-effect relationships in vitro and in vivo. Pharmacotherapy. 2001;21(8s):133s-148s.
11. Grant S.M., Clissold S.P. Itraconazole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in superficial and systemic mycoses. Drugs. 1989;3:310-344.
12. Ghannoum M.A., Rice L.B. Antifungal agents: Mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin Microbiol Rev. 1999;12:501-517.
13. Steinbach W.J., Stevens D.A., Denning D.W., et al. Advances against Aspergillosis. Clin Infect Dis. 2003;37(Suppl 3):S155-S156.
14. Committee on New Directions in the Study of Antimicrobial Therapeutics: New Classes of Antimicrobials, Committee on New Directions in the Study of Antimicrobial Therapeutics: Immunomodulation, National Research Council. Promising Approaches to the Development of Immunomodulation for the Treatment of Infectious Diseases: Report of a Workshop. In: Treating Infectious Diseases in a Microbial World: Report of Two Workshops on Novel Antimicrobial Therapeutics. National Academies Press. Accessed July 10, 2010 at http://books.nap.edu/openbook.php?record_id=11471&page=37.
15. Li R.K., Ciblak M.A., Nordoff N., et al. In vitro activities of voriconazole, itraconazole, and amphotericin B against Blastomyces dermatitidis, Coccidioides immitis, and Histoplasma capsulatum. Antimicrob Agents Chemother. 2000;44(6):1734-1736.
16. Mallie M., Bastide J.M., Blancard A., et al. In vitro susceptibility testing of Candida and Aspergillus spp. to voriconazole and other antifungal agents using Etest®: results of a French multicentre study. Int J Antimicrob Agents. 2005;25:321-328.
17. Quilitz R: The use of lipid formulations of amphotericin B in cancer patients, Cancer Control: Journal of the Moffitt Cancer Center, September/October 1998. http://www.moffitt.org/moffittapps/ccj/v5n5/toc. Accessed on November 18, 2009.
18. Deray G. Amphotericin B nephrotoxicity. J Antimicrobol Chemother. 2002;49(Suppl S1):37-41.
18a. Adler-Moore J.P., Proffit R.T. Amphotericin B lipid preparations: what are the differences? Clin Microbiol Infect. 2008;14(Suppl 4):25-36.
19. Ranchère J.Y., Latour J.F., Fuhrmann C., et al. Amphotericin B intralipid formulation: stability and particle size. J Antimicrob Chemother. 1996;37(6):1165-1169.
20. Vogelsinger H., Weller S., Djanani A., et al. Amphotericin B tissue distribution in autopsy material after treatment with liposomal amphotericin B and amphotericin B colloidal dispersion. J Antimicrob Chemother. 2006;57:1153-1160.
21. Ibrahim A.S., Gebremariam T., Husseiny M.I., et al. Comparison of lipid amphotericin B preparations in treating murine zygomycosis. Antimicrob Agents Chemother. 2008;52(4):1573-1576.
22. Bekersky I., Boswell G.W., Hiles R. Safety and toxicokinetics of intravenous liposomal amphotericin B (AmBisome®) in beagle dogs. Pharm Res. 1999;16(11):1694-1701.
23. Bagnis C.I., Deray G. Amphotericin B nephrotoxicity. Saudi J Kidney Dis Transplant. 2002;13(4):481-491.
24. Ceylan E., Akkan H.A., Tutuncu M., et al. Nephrotoxic effect of amphotericin B administered in different doses and infusion model in dogs. Acta Vet Brno. 2003;72:229-234.
25. Schneider P., Klein R.M., Dietze L., et al. Anaphylactic reaction to liposomal amphotericin (AmBisome®). Br J Haematol. 1998;102:1108.
26. Legendre A.M., Selcer B.A., Edwards D.F., et al. Treatment of canine blastomycosis with amphotericin B and ketoconazole. J Am Vet Med Assoc. 1984;184(10):1249-1254.
27. Greene C.E. Antifungal chemotherapy. In Green C.E., editor: Infectious disease of the dog and cat, ed 3, St Louis: Saunders, 2006.
28. Rubin S.I., Krawiec D.R., Gelberg H., et al. Nephrotoxicity of amphotericin B in dogs: a comparison of two methods of administration. Can J Vet Res. 1989;53:23-28.
29. Stevens D.A. Oral amphotericin B as an antifungal agent. J Mycol Med. 1996;6(S II):1-2.
30. Malik R., Craig A.J., Wigney D.I., et al. Combination chemotherapy of canine and feline cryptococcosis using subcutaneously administered amphotericin B. Aust Vet J. 1996;73:124-128.
31. Olivero J.J., Loazno-Mendez J., Ghafary E.M., et al. Mitigation of amphotericin B nephrotoxocity by mannitol. Br J Med. 1975;1:550-551.
32. Zager R.A., Bredl C.R., Schimpf B.A. Direct amphotericin B-mediated tubular toxicity: assessments of selected cytoprotective agents. Kidney Int. 1992;41:1588-1594.
33. Feldman L., Efrati S., Daishy V., et al. N-acetylcysteine ameliorates amphotericin-induced nephropathy in rats. Nephron Physiol. 2005;99:23-27.
34. Leenders A.C.A.P., de Marie S. The use of lipid formulations of amphotericin B for systemic fungal infections. Leukemia. 1996;10:1570-1575.
35. Lanternier F., Lortholary O. Liposomal amphotericin B: what is its role in 2008? Clin Microbiol Infect. 2008;14(Suppl 4):71-83.
36. Krawiec D.R., McKiernan B.C., Twardock R.A., et al. Use of an amphotericin B lipid complex for treatment of blastomycosis in dogs. J Am Vet Med Assoc. 1996;209:2073-2075.
37. Randal S.R., Adams L.G., Whate M.R., et al. Nephrotoxicity of amphotericin B administered to dogs in a fat emulsion versus five percent dextrose solution. Am J Vet Res. 1996;57:1054-1058.
38. Greene C.E. Infectious disease of the dog and cat, ed 3. St Louis: Saunders; 2006.
39. Cleary J.D., Taylor J.W., Chapman S.W. New drug developments. Itraconazole in antifungal therapy. Ann Pharmacother. 26. 1992. 502-509.
40. Benson J.M., Nahata M.C. Clinical use of systemic antifungal agents. Clin Pharm. 1988;7:424-438.
41. Pasko M.T., Piscitelli S.C., Slooten V., et al. Fluconazole: a new triazole antifungal agent. DICP Ann Pharmacother. 1990;24:860-867.
42. Johnson L.B., Kauffman C.A. Voriconazole: A new triazole antifungal agent. Clin Infect Dis. 2003;36:630-637.
43. van Custum J., van Gerven F., Janssen P.A.J. The in vitro and in vivo antifungal activity of itraconazole. In Recent trends in the discovery, development, and evaluation of antifungal agents, Proceedings of an International Telesymposium. Barcelona, Spain: JR Prouse Science; 1987. pp 177–192
44. Borgers M. Changes in fungal ultrastructure after itraconazole treatment. In Recent trends in the discovery, development, and evaluation of antifungal agents, Proceedings of an International Telesymposium. Barcelona, Spain: JR Prouse Science; 1987. pp 193–206
45. Abdel-Rahman S.M., Jacobs R.F., Massarella J., et al. Single-dose pharmacokinetics of intravenous itraconazole and hydroxypropyl-beta-cyclodextrin in infants, children, and adolescents. Antimicrob Agents Chemother. 2007;51(8):2668-2673.
46. Carrillo-Munoz A.J., Quindos G., Rusega M., et al. Antifungal activity of posaconazole compared with fluconazole and amphotericin B against yeasts from oropharyngeal candidiasis and other infections. J Antimicrob Chemother. 2005;55:317-319.
47. Hart D.T., Lauwers W.J., Willemens G., et al. Perturbation of sterol biosynthesis by itraconazole and ketoconazole in Leishmania mexicana infected macrophages. Mol Biochem Parasitol. 1989;33:123-134.
48. Hardin T.C., Graybill J.R., Techick R., et al. Pharmacokinetics of itraconazole following oral administration to normal volunteers. Antimicrob Agents Chemother. 1988;9:1310-1313.
49. Van Peer A., Woestenbourghs R., Heykants J., et al. The effects of food and dose on the oral systemic availability of itraconazole in healthy subjects. Eur J Clin Pharmacol. 1989;36:423-426.
50. Boothe D.M., Herring I., Calvin J., et al. Itraconazole disposition following single oral and intravenous and multiple oral dosing in healthy cats. Am J Vet Res. 1997;58:872-877.
51. Heykants J., Michiels M., Meuldermans W., et al. The pharmacokinetics of itraconazole in animals and man. An overview. In Recent trends in the discovery, development, and evaluation of antifungal agents, Proceedings of an International Telesymposium. Barcelona, Spain: JR Prouse Science; 1987. pp 223–250
52. Vaden S.L., Heit M.C., Hawkins E.C., et al. Fluconazole in cats: pharmacokinetics following intravenous and oral administration and penetration into cerebrospinal fluid, aqueous humor and pulmonary epithelial lining fluid. J Vet Pharmacol Ther. 1997;20:181-186.
53. Groll A.H., Piscitelli S.C., Walsh T.J. Antifungal pharmacodynamics: concentration-effect relationships in vitro and in vivo. Pharmacotherapy. 2001;21(8 Pt 2):133S-148S.
54. Legendre A.M., Rohrback R., Toal R.L., et al. Treatment of blastomycosis with itraconazole in 112 dogs. J Vet Intern Med. 1996;10:365-371.
55. Roffey S.J., Cole S., Comby P., et al. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog and human. Drug Metab Dispos. 2003;31(6):731-741.
56. Nomeir A.A., Kumari P., Hilbert M.J., et al. Pharmacokinetics of SCH 56592, a new azole broad-spectrum antifungal agent, in mice, rats, rabbits, dogs, and cynomolgus monkeys. Antimicrob Agents Chemother. 2000;44(3):727-731.
57. De Coster R., Beerens D., Haelterman C., et al. Effects of itraconazole on the pituitary-testicular-adrenal axis: an overview of preclinical and clinical studies. In Recent trends in the discovery, development, and evaluation of antifungal agents, Proceedings of an International Telesymposium. Barcelona, Spain: JR Prouse Science; 1987. pp 251-262
58. Bossche H.V. Itraconazole: a selective inhibitor of the cytochrome P-450 dependent ergosterol biosynthesis. In Recent trends in the discovery, development, and evaluation of antifungal agents, Proceedings of an International Telesymposium. Barcelona, Spain: JR Prouse Science; 1987. pp 207–222
59. Kukanich B., Borum S.L. Effects of ketoconazole on the pharmacokinetics and pharmacodynamics of morphine in healthy greyhounds. Am J Vet Res. 2008;69:664-669.
60. Willard M.D., Nachreiner R., MacDonald R., et al. Ketoconazole-induced changes in selected canine hormone concentrations. Am J Vet Res. 1986;47:2504-2509.
61. Willard M.D., Nachreiner R.F., Howard V.C., et al. Effect of long term administration of ketoconazole in cats. Am J Vet Res. 1986;47:2510-2513.
62. Kim R.B. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34(1&2):47-54.
63. van Cauteren H., Coussement W., Vandenberghe J., et al. The toxicological properties of itraconazole. In Recent trends in the discovery, development, and evaluation of antifungal agents, Proceedings of an International Telesymposium. Barcelona, Spain: JR Prouse Science; 1987. pp 262-271
64. Manciati F., Pedonses F., Zulline C. Efficacy of oral administration of itraconazole to cats with dermatophytosis caused by. Microsporum canis, J Am Vet Med Assoc. 1998;213:993-995.
65. Mayer U.K., Glos K., Schmid M., et al. Adverse effects of ketoconazole in dogs—a retrospective study. Vet Dermatol. 2008;9:199-208.
66. Moriello K.A. Treatment of dermatophytosis in dogs and cats: review of published studies. Vet Dermatol. 2004;15:99-107.
67. Plotnick A.N., Boshoven E.W., Prsychuk R.A. Primary cutaneous coccidioidomycosis and subsequent drug eruption to intraconazole in a dog. J Am Anim Hosp Assoc. 1997;33:129-143.
68. Background document for the Antiviral Drug Products Advisory Committee meeting. Voriconazole, Food and Drug Administration Center for Drug Evaluation and Research, Division of Special Pathogen and Immunologic Drug Products. Pfizer Global Research & Development NDAs; 2001. pp 21–266 and 21-267
69. Castro L.G.M., Belda W.Jr., Cuce L.C., et al. Successful treatment of sporotrichosis with oral fluconazole: a report of three cases. Br J Dermatol. 1993;128(3):352-356.
70. Medleau L., Chalmers S.A. Ketoconazole for treatment of dermatophytosis in cats. J Am Vet Med Assoc. 1992;100:77-78.
71. Mundell A.C. New therapeutic agents in veterinary dermatology. Vet Clin North Am Small Anim Pract. 1990;20:1541-1556.
72. Dunbar M., Pyle L.R., Boring J.G., et al. Treatment of canine blastomycosis with ketoconazole. J Am Vet Med Assoc. 1983;182:156-157.
73. Noxon J.O., Diglio K., Schmidt D.A. Disseminated histoplasmosis in a cat: successful treatment with ketoconazole. J Am Vet Med Assoc. 1982;181:817-819.
74. Malik R., Wigney D.I., Muir D.B., et al. Cryptococcosis in cats: clinical and mycological assessment of 29 cases and evaluation of treatment using orally administered fluconazole. J Med Vet Mycol. 1992;30:133-144.
75. Noxon J.O., Monroe We, Chinn D.R. Ketoconazole therapy in canine and feline cryptococcosis. J Am Anim Hosp Assoc. 1986;22:179-183.
76. Tucker R.M., Williams P.L., Arathoon E.G., et al. Treatment of mycoses with itraconazole. Ann NY Acad Sci. 1988;544:451-470.
77. Finn M.J., Stiles J., Krohne S.G. Visual outcome in a group of dogs with ocular blastomycosis treated with systemic antifungals and systemic corticosteroids. Vet Ophthalmol. 2007;10(5):299-303.
78. Hodges R.D., Legendre A.M., Adams L.G., et al. Itraconazole for the treatment of histoplasmosis in cats. J Vet Intern Med. 1994;8:409-413.
79. Medleau L., Green C.E., Rakich P.M. Evaluation of ketoconazole and itraconazole for treatment of disseminated cryptococcosis in cats. Am J Vet Res. 1990;51:1454-1548.
80. Malik R., Krockenberger M., O’Brien C.R., et al. Cryptococcosis. In Greene C.E., editor: Infectious diseases of the dog and cat, ed 3, St Louis: Saunders, 2006.
81. Medleau L., Jacobs G.J., Marks M.A. Itraconazole for the treatment of cryptococcosis in cats. J Vet Intern Med. 1995;9:39-42.
82. Peaston A., Sporotrichosis. J Vet Intern Med. 1993;7:44-45.
83. Legendre A.M. Antimycotic drug therapy. In: Bonagura J.D., Kirk R.W., editors. Current veterinary therapy XII. Philadelphia: Saunders; 1995:327-331.
84. DeBoer D.J., Moriello K.M., Cairns R. Clinical update on feline dermatophytosis—part II. Compend Contin Educ Pract Vet. 1995;17:1471-1480.
85. Medleau L., Rakich P.M. Microsporum canis pseudomycetomas in a cat. J Am Anim Hosp Assoc. 1994;30:573-576.
86. Michaud A.J. Phaeohyphomycotic rhinitis due to Exophiala jeanselmei in a domestic cat. Feline Pract. 1993;21:19-21.
87. Simons E.G. Phaeohyphomycosis in a cat caused by Alternaria infectoria. Mycoses. 1993;36:451-454.
88. Guptal A.K., Sibbald G.R., Lynde C.W., et al. Onychomycosis in children: prevalence and treatment strategies. J Am Acad Dermatol. 1997;36:395-402.
89. Hill B., Moriello K.A., Shaw S.E. A review of systemic antifungal agents. Vet Dermatol. 1995;6:59-66.
90. Sharp N.H., Harvey C.E., Obrien J.A. Treatment of canine nasal aspergillosis/penicillinosis with fluconazole. J Small Anim Pract. 1991;32:513-516.
91. Wenzel R., Del Favero A., Killber C., et al. Economic evaluation of voriconazole compared with conventional amphotericin B for the primary treatment of aspergillosis in immunocompromised patients. J Antimicrob Chemother. 2005;55:352-361.
91a. Wray J.D., Sparkes A.H., Johnson E.M. Infection of the subcutis of the nose in a cat caused by Mucor species: successful treatment using posaconazole. J Fel Med Surg. 2008;10:523-527.
92. Barchiesi F., de Francesco L.F., Scalise G. In vitro activities of terbinafine in combination with fluconazole and itraconazole against isolates of Candida albicans with reduced susceptibility to azoles. Antimicrob Agents Chemother. 1997;41(8):1812-1814.
93. Burkhart C.G., Burkhart C.N., Isham N. Synergistic antimicrobial activity by combining an allylamine with benzoyl peroxide with expanded coverage against yeast and bacterial species. Br J Dermatol. 2006;154:341-344.
94. Denning D.W. Echinocandins: a new class of antifungal. J Antimicrob Chemother. 2002;49:889-991.
95. Renslo A.R. The echinocandins: total and semi-synthetic approaches in antifungal drug discovery. Anti-Infective Agents in Medicinal Chemistry. 2007;6:201-212.
96. Denning D.W. Echinocandin antifungal drugs. Lancet. 2003;362(9390):1142-1151.
97. Anhthu H. Caspofungin acetate: an antifungal agent. Am J Health-Syst Pharm. 2001;58(13):1206-1214.
98. Helton K.A., Nesbitt G.H., Caciolo O.L. Griseofulvin toxicities in cats: literature review and report of seven cases. J Am Anim Hosp Assoc. 1986;22:453-458.
99. Sheltin G.H., Grant C.K., Linenberg M.L., et al. Severe neutropenia associated with griseofulvin therapy in cats with feline immunodeficiency virus infection. J Vet Intern Med. 1990;4:317-319.
100. Ryder N.S. Activity of terbinafine against serious fungal infections. Mycoses. 1999;42(Suppl 2):115-119.
100a. Burkhart C.G., Burkhart C.N., Isham N. Synergistic antimicrobial activity by combining an allylamine with benzoyl peroxide with expanded coverage against yeast and bacterial species. Br J Dermatol. 2006;154(2):341-344.
100b. Cavalheiro A.S., Maboni G., de Azevedo M.I., et al. In vitro activity of terbinafine combined with caspofungin and azoles against. Pythium insidiosum, Antimicrob Agents Chemother. 2009;53(5):2136-2138.
101. Balfour J.A., Faulds D. Terbinafine: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in superficial mycosis. Drugs. 1992;43:259-284.
102. Feargemann J., Zehender H., Jones T., et al. Terbinafine levels in serum, stratum corneum, dermis-epidermis (without stratum corneum), hair, sebum, and eccrine sweat during and after 250 mg terbinafine orally once per day in man. J Invest Dermatol. 1990;24:523-528.
103. Sparks A.H. Terbinafine in cats: a pharmacokinetic study. 1996. Proc Third World Cong Vet Dermatol. Edinburgh, Scotland.
104. Werner A.H., Werner B.E. Feline sporotrichosis. Compend Contin Educ Pract Vet. 1993;15:1189-1198.
105. Moriello K.A., Granks P., Delany-Lewis D., et al. Cutaneous-lymphatic and nasal sporotrichosis in a dog. J Am Anim Hosp. 1988;24:621-626.
106. Ben-Ziony A. Use of lufenuron for treating fungal infections of dogs and cats: 297 cases (1997-1999). J Am Vet Assoc. 2000;217:1510-1513.
107. Moriello K.A., Deboer D.J., Schenker R., et al. Efficacy of pre-treatment with lufenuron for the prevention of Microsporum canin infection in a feline direct topical challenge model. Vet Dermatol. 2004;15:357-362.
108. Ostrosky-Zeichner L. Combination antifungal therapy: a critical review of the evidence. Clin Microbiol Infect. 2008;14(Suppl 4):65-70.
109. DeBoer D.J., Moriello K.A. Cutaneous fungal infections: dermatophytosis. In Greene C.E., editor: Infectious diseases of the dog and cat, ed 3, St Louis: Saunders, 2006.
110. Moriello K.A., DeBoer D.J. Efficacy of griseofulvin and itraconazole in the treatment of experimentally induced dermatophytosis in cats. J Am Vet Med Assoc. 1995;207(4):439-443.
111. Colombo S., Cornegliani L., Vercelli A. Efficacy of Itraconazole as a combined continuous/pulse therapy in feline dermatophytosis: preliminary results in nine cases. Vet Dermatol. 2001;12(6):347-350.
111a. Newbury S., Moriello K., Verbrugge M., et al. Use of lime sulphur and itraconazole to treat shelter cats naturally infected with Microsporum canis in an annex facility: an open field trial. Vet Derm. 2007;18:324-331.
112. Kotnik T., Kozuh Erzen N., Kuzner J. Drobnic-Kosorok M: Terbinafine hydrochloride treatment of Microsporim canis experimentally induced ringworm in cats. Vet Microbiol. 2001;83:161-168.
113. Ihrke P.J. Malassezia dermatitis and hypersensitivity. In Antimicrobial therapy: applications in dermatology, Proceedings from an International Symposium. Orlando, Florida: the North American Veterinary Conference; January, 1996. pp 45–48
114. Cole L.K., Luu D.H., Rajala-Schultz P.J., et al. In vitro activity of an ear rinse containing tromethamine, EDTA, benzyl alcohol and 0.1% ketoconazole on Malassezia organisms from dogs with otitis externa. Vet Dermatol. 2007;18(2):115-119.
115. Åhman S., Perrins N., Bond R. Treatment of Malassezia pachydermatis–associated seborrhoeic dermatitis in Devon Rex cats with itraconazole—a pilot study. Vet Dermatol. 2007;8:171-174.
116. Negre A., Bensignor E., Guillot J. Evidence-based veterinary dermatology: a systematic review of interventions for Malassezia dermatitis in dogs. Vet Dermatol 0. 2008:1-12.
117. Klein B.S., Squires R.A., Lloyd J.K., et al. Canine antibody response to Blastomyces dermatitidis WI-1 antigen. Am J Vet Res. 2000;61(5):554-558.
118. Legendre A.M., Selcer B.A., Edwards D.F., Stevens R. Treatment of canine blastomycosis with amphotericin B and ketoconazole. J Am Vet Med Assoc. 1984;184(10):1249-1254.
119. Areceneaux K.A., Taboada J., Hosgood G. Blastomycosis in dogs: 115 cases (1980–1995). J Am Vet Med Assoc. 1998;213:658-664.
119a. Crews L.J., Feeney D.A., Jesse D.R., et al. Utility of diagnostic tests for and medical treatment of pulmonary blastomycosis in dogs: 125 cases (1989-2006). J Am Vet Med Assoc. 2008;232:222-227.
120. Legendre A.M. Antimycotic Drug therapy. In: Bonagura J., editor. Kirk’s current veterinary therapy small animal pract XIII. Philadelphia: Saunders; 2000:327-334.
121. Greene C.E. Histoplasmosis. In Greene C.E., editor: Infectious diseases of the dog and cat, ed 3, St Louis: Saunders, 2006.
122. Schulman R.L., McKiernan R.C., Schaeffer D.J., et al. Use of corticosteroids for treating dogs with airway obstruction secondary to hilar lymphadenopathy caused by chronic histoplasmosis: 16 cases (1979-1997). J Am Vet Med Assoc. 1999;214:1345-1348.
123. Jacobs G.J., Medleau L., Calvert C., et al. Cryptococcal infection in cats: factors influencing treatment outcome, and results of sequential serum antigen titers in 35 cats. J Vet Intern Med. 1997;11(1):1-4.
124. Lortholary O., Nicolas M., Soreda S., et al. Fluconazole, with or without dexamethasone for experimental cryptococcosis: impact of treatment timing. J Antimicrob Chemother. 1999;43:817-824.
125. Flatland B., Greene R.T., Lappin M.R. Clinical and serological evaluation of cats with cryptococcosis. J Am Vet Med Assoc. 1996;209:1110.
126. Greene R. Coccidioidomycosis and paracoccidioidomycosis. In Greene C.E., editor: Infectious diseases of the dog and cat, ed 3, St Louis: Saunders, 2006.
127. Greene R.T., Troy G.C. Coccidioidomycosis in 48 cats: a retrospective study (1984–1993). J Vet Intern Med. 1995;9:86-91.
128. Ricci G., Mota F.T., Wakamatsu A., et al. Canine paracoccidioidomycosis. Med Mycol. 2004;42(4):379-383.
129. Tomsa K., Gluas T.M. Zimmer fungal rhinitis and sinusitis in three cats. J Am Vet Med Assoc. 2003;222(10):1380-1384.
130. Kim S.H., Yong H.C., Yoon J.H., et al. Aspergillus niger pulmonary infection in a dog. J Vet Med Sci. 2003;65:1139-1140.
131. Kuehn N.F. Nasal aspergillosis. In: Bonagura J.D., Twedt D.C., editors. Current veterinary therapy XIV. St Louis: Saunders, 2009.
132. Moore A.H. Use of topical povidone-iodine dressings in the management of mycotic rhinitis in three dogs. J Small Anim Pract. 2003;44(7):326-329.
133. Steinbach W.J., Stevens D.A., Denning D.W., et al. Combination and sequential antifungal therapy for invasive Aspergillosis: review of published in vitro and in vivo interactions and 6281 clinical cases from 1966 to 2001. Clin Infect Dis. 2003;37(Suppl 3):S188-S224.
134. Rosser E.J., Dunstan R.W. Sporotrichosis. In Greene C.E., editor: Infectious diseases of the dog and cat, ed 3, St. Louis: Saunders, 2006.
135. Castellano M.C., Breitschwerdt E.B. Rhinosporidiosis. In Greene C.E., editor: Infectious diseases of the dog and cat, ed 3, St Louis: Saunders, 2006.
136. Rakich P.M., Grooter A.M., Tang K.-N. Gastrointestinal pythiosis in two cats. J Vet Diagn Invest. 2005;17:262-269.
137. Brown T.A., Grooters A.M., Hosgood G.L. In vitro susceptibility of Pythium insidiosum and a Lagenidium sp to itraconazole, posaconazole, voriconazole, terbinafine, caspofungin, and mefenoxam. Am J Vet Res. 2008;69(11):1463-1468.