2 Factors Affecting Drug Disposition
Ideally, fixed dosing regimens are based on scientific studies performed with the drug of interest in the target species. Often, however, the dose for a drug used in dogs or cats is extrapolated from other species, especially humans to dogs and humans or dogs to cats, rather than based on scientific studies. For those drugs backed by scientific evidence, sample numbers are often too small, resulting in marked variability in the pharmacokinetic parameters. Further, as in human medicine, animals in which pharmacokinetic studies are performed are generally healthy and may not represent the state of disease in the animals treated with the drug. A number of factors can alter plasma drug concentrations (PDCs) in the patient due to changes in drug disposition, thereby increasing the risk of therapeutic failure. Many of these can be anticipated, allowing for adjustments in the dosing regimen. In addition to the disposition of a drug, these factors might also alter patient response to the drug. These factors might be categorized as physiologic factors such as species and age; pathologic factors, particularly cardiac, renal, or hepatic disease; and pharmacologic factors that occur when one drug alters the kinetics or response to another drug (Box 2-1).
Physiologic Factors
Age-Induced Differences
Drug Disposition in the Geriatric Animal
This discussion focuses on some of the clinically important changes that are likely to alter response to drug therapy in geriatric patients (Table 2-1) and on actions that can be implemented to compensate or reduce the sequelae of these changes. Age is associated with changes both in pharmacokinetics (gerontokinetics) and pharmacodynamics, with changes generally compared with those of the young adult.1-3 Data in animals are limited, but in general, disposition pattern changes described for humans appear to extrapolate adequately to dogs and cats.4 Geriatric animals are at a greater risk for therapeutic failure because of normal aging changes in physiology, an increased incidence of disease, and the likelihood of polypharmacy in response to disease; moreover, a decrease in normal organ protective mechanisms may increase the risk of adverse events. The age at which body functions shift from a period of growth to a period of decay (16 to 18 years in humans) has not been established in dogs and cats. The age probably differs among canine breeds. Aging is accompanied by permanent loss of up to 30% of body cells, with a parallel loss in oxygen consumption. Body composition changes, as do regional blood flow rates. Physiologic functions generally decline steadily with increasing age. In humans basal metabolic rate decreases by 0.4% per year2; according to the National Research Council,5 energy requirements of dogs decrease as they age. Changes among the body systems can influence all four drug movements.
KEY POINT 2-1
As the geriatric animal ages, organ mass reduces in size and function by approximately 25%, resulting in corresponding changes in drug disposition.
Cardiovascular
As animals age, cardiac output decreases and circulation transit time increases. In humans cardiac output decreases by about 1% per year for a total decline of 30% to 40% in the aged. Regional and organ blood flow rates similarly decrease.2 The net effect of these changes depends on the state of disease but can influence each drug movement. Absorption, metabolism, and excretion are likely to decrease, whereas distribution may increase or decrease depending on the state of vascular responses or fluid retention.2,6,7 As cardiac function decreases, secondary compensatory responses can lead to further risks of adverse reaction.7 Blood flow is preferably redistributed to the brain and heart, increasing the risk of toxicity of drugs toxic to these tissues.
Central and peripheral nervous systems
As the geriatric patient ages, brain weight and peripheral fiber numbers decrease. Connective tissue infiltrates peripherally.2 Oxygen consumption and cerebral blood flow decrease. In addition, decreased amounts of selected neurotransmitters have been documented. Morita and coworkers8 reported alterations in the blood–brain barrier in elderly dogs.
Respiratory
In human geriatric patients, residual lung volume decreases by 50% with accompanying decreases in vital capacity, arterial oxygen pressure (PO2), and maximum oxygen uptake. In addition, the central response to hypoxia and hypercapnia, such as that induced by opioid analgesics, decreases.2 Anesthetic or other sedating agents must be used more cautiously.
Gastrointestinal
As animals age, deglutition decreases as a result of decreased salivation and pharyngeal and esophageal motility. Gastric function is characterized by atrophy of the mucosa with a reduction of hydrochloric acid secretion and a subsequent increase in gastric pH. Gastrointestinal motility is generally reduced. The intestinal macrovilli and microvilli also atrophy, increasing the risk of bacterial overgrowth. These sequelae tend to reduce the absorption and thus the PDC of orally administered drugs. Changes in gastrointestinal function (including reduction of gastroprotective effects) may also predispose the geriatric patient to adverse effects induced by toxic drugs such as chemotherapeutic agents and nonsteroidal antiinflammatory drug (NSAID) analgesics. The latter should be used with caution; the clinician should anticipate and be prepared to treat toxicities. Digestibility of key nutrients, such as fat, tends to decrease in elderly cats.9 As with humans, the intestinal microbiota population shifts in elderly dogs and cats, with lactobacilli decreasing and clostridia increasing in dogs10 and bifidobacteria decreasing in cats.11 In addition to response to antimicrobials, these changes might influence enterohepatic circulation of bile acids and thus, potentially, some drugs.12
Hepatic
Changes in hepatic function are important to the geriatric animal because of the liver’s role in the metabolism of drugs.13 Hepatocyte number and function decrease, as do hepatic and splanchnic blood flow, hepatic oxidation, and cytochrome P450 content (the primary drug-metabolizing enzyme). Both flow-limited and capacity-limited drugs are affected. For example, hepatic clearance of both opioid analgesics (which are characterized by first-pass metabolism; i.e., flow limited) and nonsteroidal analgesics (eliminated principally by hepatic metabolism; i.e., capacity limited) is decreased in geriatric patients. Increased response of human geriatric patients to opioid analgesics—they require 60% to 75% less drug than younger patients do—has been attributed to changes in drug elimination.14,15 Changes in hepatic function, oxygenation, and nutrition may also predispose the liver to drug-induced hepatotoxicity. Because of reduced hepatic function, the geriatric patient may be less able to generate endogenous hepatoprotectant agents, increasing the risk of drug-induced hepatotoxicity (Chapter 4).
Urinary
As renal blood flow decreases, the glomerular filtration rate and active secretory capacity of the nephron unit progressively decrease with age. Both result in a similar decline in renal clearance. Renal excretion is the major route of elimination of many drugs. Changes in renal clearance tend to prolong the elimination and thus increase PDCs in the geriatric patient. Changes in renal function also render the geriatric patient more susceptible to adverse drug reactions such as those induced by aminoglycosides, angiotensin-converting enzyme inhibitors, and NSAID analgesics.
Body weight and composition
Changes in body composition may be among the most complex in the geriatric animal. Like humans, dogs tend to lose lean body mass and accumulate body fat as they age (Figure 2-1).16 In male humans, fat increases from approximately 18% in young adults up to 50% in the aged.2 Increased proportion of body fat is accompanied by a decrease in total body water and cell mass. Although extracellular fluid does not change in total amount, the relative proportion of total body water that it makes up increases. Thus the proportion of intracellular to extracellular fluid decreases. The sequelae of these changes depend on the drug. As distribution of water-soluble drugs decreases with total body water, PDCs tend to increase. The distribution of lipid-soluble drugs increases as the proportion of body fat increases, however, which tends to decrease PDCs unless the patient is dosed on a mg/kg basis. The impact of aging on body composition in dogs varies with breeds. For example, in one study body fat increased with age in Great Danes but not Labrador Retrievers or Papillons,17 although this did occur within 1 year of death in elderly Labrador Retrievers in another study.16 Although middle-aged cats tend to be overweight, geriatric cats often become thin.9 Thus PDCs of water-soluble compounds might be expected to be higher in older animals compared with those of young adults, even if dosing on an mg/kg body weight basis, whereas for lipid soluble drugs, dosing on an mg/kg body weight basis might compensate for potential changes in plasma concentrations.
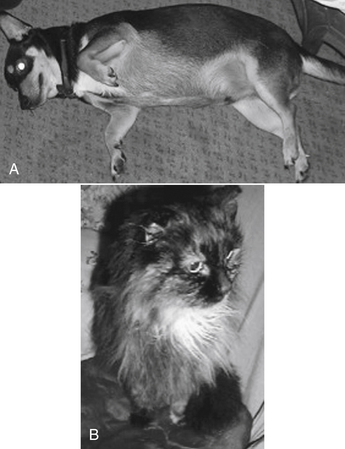
Figure 2-1 The geriatric dog and cats may represent two extremes in regard to the impact of aging on disposition. Body mass frequently increases in dogs (A) but decreases in geriatric cats (B). Mass loss in cat reflects lean tissue and fat. In contrast to the geriatric animal, adult cats, like dogs, may be characterized by marked differences in body composition, as is exemplified by the obese cat or dog. Water-soluble drugs generally do not distribute to fat, and dosing of water-soluble drug on the basis of total body weight may result in overdose. Dosing on lean body weight is more prudent. Distribution of a lipid-soluble drug to the fat compartment, on the other hand, may increase the volume of distribution, decreasing drug concentrations. For such animals dosing on total body weight (per kg) is more reasonable.
Serum albumin
Although total serum plasma protein content probably remains the same in the geriatric animal, the proportion represented by albumin decreases and that by gamma globulins increases. Changes in serum albumin can be clinically important to patients receiving highly protein-bound drugs, such as NSAIDs. Decreased albumin can result in a greater proportion of free drug: Most NSAIDs are close to 99% protein bound. A decrease of only 1% (i.e., 99% to 98% binding) doubles the concentration of a pharmacologically active drug. The sequelae of increased PDC may be offset by a compensatory increased clearance, because only unbound drugs are generally conducive to hepatic or renal clearance. However, this balance might be minimized if organs of clearance are negatively affected.
Receptor sensitivity and pharmacodynamics
Geriatric patients respond differently to some drugs, which suggests that tissue receptor sensitivity to the drugs is altered. Changes in receptor number or responsiveness have been implicated but not documented.2,3 Physiologic changes such as altered neurotransmission or intracellular constituents have also been suggested. For example, geriatric patients are less likely to perceive, appreciate, or express pain. Thus the need for analgesic therapy is often not detected. In addition, geriatric patients are less able to respond to many analgesic drugs.
Disease
Aged animals are more likely to be suffering from diseases that affect not only drug disposition but also tissue receptivity to drugs and organ protection.15 The immune system of the geriatric patient is not as effective as that of the adult,18 leading to the use of bactericidal antimicrobials and minimizing the use of immunosuppressive drugs. In addition, the geriatric patient is more likely to be receiving multiple drugs, which increases the likelihood of drug interactions. Finally, diseases of selected organs may predispose these organs to drug-induced toxicity.
Drug Disposition in the Pediatric Animal
With regard to dogs and cats, pediatric generally refers to the first 12 weeks of life.19 Important developmental changes occurring within this time spectrum, however, justify further staging into neonatal (0 to 2 weeks), infant (>2 to 6 weeks), and pediatric (>6 to 12 weeks) periods of growth. Changes associated with each of these periods cause accompanying changes in drug disposition, thus rendering the pediatric patient more susceptible to drug-induced adverse reactions. All four determinants of drug disposition (i.e., absorption, distribution, metabolism, and excretion) undergo dramatic changes as the neonate matures (see Table 2-1).20,21 However, the clinical significance of these sequelae varies.
KEY POINT 2-2
Drug clearance generally does not reach adult capacity until approximately 3 months of age.
Absorption
Young kittens have decreased energy, carbohydrate, and organic matter digestibility compared with kittens older than 19 weeks of age.22 However, because the surface area of the small intestine is large, even in neonates, the extent of drug absorption probably does not clinically differ between normal pediatric and adult animals. The rate of absorption tends to be slower in pediatric animals, however, probably because of decreased gastric emptying and irregular intestinal peristalsis. As a result, peak PDCs may be lower in pediatric patients. The decreased rate of absorption might actually protect against toxic drug concentrations.23,24 However, protection may not be present before absorption of colostrum. During this period the permeability of the intestinal mucosa is increased, leading to increased rate and extent of drug absorption. Occasionally, drugs that normally are not absorbed from the gastrointestinal tract (e.g., aminoglycosides, carbenicillin, and other acid-sensitive beta-lactams and enteric sulfonamides) can reach systemic circulation. Intestinal permeability decreases rapidly after the ingestion of colostrum,24,25 possibly because of the endogenous release of hydrocortisone or adrenocorticotropic hormone. Exogenous supplementation of either of these hormones by the 24-hour prepartum mother prevents increased permeability and colostrum absorption in the neonate.
A number of other factors may alter small intestinal drug absorption in pediatric patients. Gastric pH is neutral in the newborn; adult levels are not reached until sometime after birth, depending on the species.23,25 Increased gastric pH (achlorhydria) may decrease the absorption of many drugs that require disintegration and dissolution or are ionized in a less acidic environment (e.g., weak acids such as penicillins). Milk diets can reduce drug absorption by either decreasing gastric emptying or directly interacting with drugs (e.g., milk can impair absorption of tetracyclines). The “unstirred water layer” adjacent to the surface area of the mucosal cells is thicker in the neonate than in the older pediatric patient and may limit the rate of absorption of some drugs. As biliary function matures, the absorption of fat-soluble drugs (e.g., griseofulvin and fat-soluble vitamins) increases. Microbial colonization of the gastrointestinal tract may alter response to antimicrobial drugs, extrahepatic metabolism, or enterohepatic circulation.26,27
Absorption from the rectal mucosa is rapid. Rectal administration of drugs or fluids can be used for pediatric patients when venous catheterization is difficult, to reduce complications associated with intravenous administration (e.g., sedation, anesthesia), or when oral administration is undesirable (e.g., antiemetics). Several pediatric drugs intended for systemic effects are available as rectal suppositories. Limited data from studies of human infants indicate that peak plasma concentrations after rectal administration may be higher than those obtained by other routes.26
Absorption of drugs administered parenterally to pediatric animals also varies from that of adults. The rate of absorption after intramuscular administration changes with age as muscle mass and its accompanying blood flow increase and as vasomotor responses mature.26 Because muscle mass is small, subcutaneous administration is frequently preferred for pediatric patients. Again, variability in subcutaneous absorption rates can be anticipated with age. Less fat but greater water may result in faster absorption compared with that in adults.28 Environmental temperature probably influences subcutaneous absorption, particularly in newborns whose thermoregulatory mechanism functions poorly. Cold environments are likely to reduce subcutaneous drug absorption if the neonate is not kept warm. The same is true for patients in a state of hypothermia. Intraperitoneal administration can be a lifesaving route of blood and fluid administration, particularly for the newborn with inaccessible central veins. Isotonic fluids are rapidly absorbed, and up to 70% of red blood cells are absorbed in 48 to 72 hours.29 Blood and fluids can also be administered into the medullary cavity of large bones.30,31
Absorption of volatile anesthetics from the pediatric respiratory tract is rapid because minute ventilation is greater.19 Thus young animals are more sensitive to the effects of gas anesthetics. Although not a common route of drug administration, percutaneous absorption of drugs is likely to be greater in pediatric patients. Percutaneous absorption is directly related to skin hydration, which is greatest in neonates. Topical administration of potentially toxic lipid-soluble drugs (e.g., hexachlorophene and organophosphates) is not recommended.
Distribution
The most important factors contributing to differences in drug distribution in pediatric patients are differences in body fluid compartments and drug binding to serum proteins. Body fluid compartments undergo profound changes with the growth of the neonate. Both the percentage of total body water and the ratio of compartmental volumes change with maturation. The percentage of total body water decreases with age, but the decrease is more substantial in the extracellular versus the intracellular compartment (see Table 2-1; Figure 2-2).32 Daily fluid requirements are greater in neonatal and pediatric patients, in part because a larger proportion of their body weight is represented by body water (see Figure 2-2). The sequelae of these body compartment differences depend on the normal distribution of the drug. Most water-soluble drugs are distributed to extracellular fluids. In pediatric patients the volume to which these drugs is distributed is therefore higher than in adults; PDCs correspondingly decrease. Thus it may be necessary to increase doses to prevent therapeutic failure. A different pattern might be expected for lipid-soluble drugs because they tend to be distributed to total body water. Such drugs should be dosed according to body weight (e.g., mg/kg). Although decreased PDCs resulting from increased distribution may protect the pediatric patient from potentially toxic drug concentrations,33 a poor therapeutic response may result from failure to generate therapeutic drug concentrations of water-soluble drugs. The disposition of several antimicrobials in neonatal animals has been reviewed by Baggott,34 with differences in disposition generally found to be similar to those described for humans. Ampicillin is characterized by volumes that are larger in puppies and kittens (threefold to fourfold higher) compared with adults, resulting in clinically significant lower drug concentrations.35,36 Distribution of enrofloxacin, a lipid-soluble drug, is somewhat unpredictable and age dependent in kittens, being smaller at 2 to 4 weeks but greater at 4 to 6 weeks.37 Changes in the half-life of each drug parallel changes in distribution. Because many drugs are distributed to a larger volume in pediatric patients, a longer half-life should be anticipated, and it may be necessary to prolong the dosing interval. However, for enrofloxacin in kittens this effect was balanced by increased clearance at 6 weeks.37

Figure 2-2 Differences in body composition can be appreciated between the two age extremes by following changes as the animal ages. The extracellular compartment and total body water of dogs and cats is greatest in the neonate and gradually decreases toward adult proportions as the animal ages. Increased extracellular fluid and increased total body water result in larger volumes of distribution of most drugs in neonates or pediatric animals, which may decrease plasma drug concentrations and prolong half-life. As the adult ages, fluid content declines, especially in the geriatric cat. The impact of these changes might be greater on water-soluble drugs because the extracellular component represents a greater proportion of total body water in the young animal.
Because the proportion of body fat is smaller in pediatric patients, the distribution of lipid-soluble drugs that accumulate in fat (e.g., organophosphates, chlorinated hydrocarbons, ultrashort thiobarbiturates) may be proportionately decreased. Although drug half-life would decrease, PDCs may become toxic. Many lipid-soluble drugs have a high affinity for and are bound by plasma proteins, thus facilitating their movement through the body. Binding, however, limits their distribution to tissues. Predicting the distribution of highly protein-bound drugs is complicated in the pediatric patient. Serum concentrations of both serum albumin, the protein to which most drugs are bound, and α1-glycoproteins (to which basic drugs preferentially bind) are decreased in pediatric patients.38 Protein binding of drugs may also be reduced because of differences in albumin structure or because drugs compete with endogenous substrates (e.g., bilirubin) for binding sites.24,39 As drugs are displaced, the concentration of free, pharmacologically active drugs and the risk of adverse reactions increases. These changes are significant, however, only if the drug is highly (i.e., >80%) protein bound and characterized by a small therapeutic index. Although the concentration of free drug increases, that of total drug in the plasma tends to decrease because unbound drug is free to distribute into tissue.39 Consequently, drug half-life may increase, and longer dosing intervals may be indicated for potentially toxic drugs. Increased clearance of unbound drug may ultimately normalize a half-life that has been lengthened by an increased volume of distribution.
Differences in regional organ blood flow might cause clinically important changes in drug disposition in pediatric animals. Differences in renal blood flow have been documented40,41 and result in clinically important differences in drug excretion. Blood flow to vessel-rich tissues of the body (i.e., heart and brain) is greater and faster;19 the pediatric patient is thus more susceptible to drug-induced cardiac and central nervous system (CNS) toxicity. The potential for CNS toxicity is further increased because the blood–brain barrier is poorly developed immediately after birth. Increased permeability protects the neonatal brain from a deficiency of nutritional fuels in stressful states (e.g., hypoglycemia, hypoxia, acidosis) by allowing the movement of oxidizable substrates such as lactate into brain cells.42 The status of efflux proteins is not yet established. Drugs normally incapable of reaching the adult brain are, however, also able to reach brain cells, which are very susceptible to their effects, thus increasing the risk of CNS toxicity.43,44
Metabolism
Drug elimination, including both hepatic metabolism and renal excretion, is limited in neonatal and pediatric patients. Thus many drugs administered to the young animal are characterized by decreased clearance.20,24 In contrast to human infants, hepatic metabolism of drugs is incompetent in the near-term and neonatal puppy.45-47 Both phase I (e.g., oxidative) and phase II (e.g., glucuronidation) reactions are reduced. The various pathways of metabolism mature at different rates. Phase I activity may not occur in the neonatal puppy and may not be evident until day 9 if it does. Activity appears to progressively increase after day 25, not reaching adult levels until 135 days postpartum.46 Drug metabolism in pediatric patients, however, is quite complex. For example, Ecobichon and coworkers48 found that in Beagle puppies, phase I metabolites decreased with age, and for phase II metabolism, glucuronidation was the most predominant, with sulfonation decreasing as puppies got older.
Generally, decreased hepatic drug metabolism is reflected as decreased plasma clearance, increased plasma half-life, and potentially toxic PDCs. Dose reduction, dose prolongation of intervals, or both may be indicated for some drugs. Oral bioavailability of drugs characterized by significant first-pass metabolism in adults (e.g., propranolol) is probably greater in puppies and kittens. Response to prodrugs (e.g., primidone; prednisone; enalapril; and, potentially, methylprednisolone) may be reduced because of decreased formation of active drug products. Pediatric hepatic drug-metabolizing enzymes do appear to be inducible by phenobarbital and other drugs. Nonhepatic drug-metabolizing enzymes also appear to be decreased in pediatric patients. For example, pediatric lower plasma cholinesterase can result in increased sensitivity to organophosphates, succinylcholine, and procaine.
Excretion
Reduced renal excretion, characteristic of the pediatric puppy, results in decreased clearance of renally excreted parent drugs and products of phase II drug metabolism. Although the number of glomeruli remains constant throughout pediatric development, both glomerular filtration and renal tubular function progressively increase.41,49 Adult values may not be reached until approximately 2.5 months of age. In contrast to glomerular filtration and secretion, renal tubular reabsorption in puppies appears to be similar to that in adults as long as body fluids and electrolytes are maintained.50,51 The sequelae of developmental changes in pediatric renal function include decreased clearance and prolonged half-life of drugs (primarily water soluble) excreted by the kidneys. Such a pattern has been shown for several drugs. Compared with current recommendations for adults, pediatric patients may require a higher dose (owing to increased volume of distribution) and longer intervals (owing to increased distribution and decreased clearance) for gentamicin administration. More important, modifications should be anticipated in the gentamicin dosing regimen of unhealthy puppies because they are likely to be affected by conditions that increase the potential for gentamicin-induced nephrotoxicity (e.g., dehydration). However, underdeveloped glomeruli may actually protect the pediatric patient from aminoglycoside-induced nephrotoxicity.21 Further investigations are needed to establish safe yet effective doses of gentamicin for the neonatal puppy or kitten.
Specific Drug Therapy for the Pediatric Patient
Fluid therapy
Pediatric patients are predisposed to dehydration because extracellular fluid is increased, renal capacity to conserve water is decreased, the ratio of surface area to body weight is large, and fluid loss through immature skin is greater.52 Fluids can be administered by several routes. Crystalloids administered rectally should be isotonic; rapid rectal absorption of hyperosmolar solutions can lead to life-threatening hyperosmolarity. Subcutaneous administration may be an acceptable route if small volumes of isotonic fluids are administered in patients with normal hydration. Intraosseous fluid administration is an acceptable route of administration if a central vein is not accessible.53 Oral rehydration is recommended as the preferred therapy for dehydration caused by diarrhea in human pediatric patients.54
Antimicrobial therapy
As for adults, an appreciation of the chemotherapeutic triangle (i.e., relationship among host, drug, and microorganism) is necessary for the appropriate use of antimicrobials with pediatric patients. Several antimicrobials are not recommended for pediatric patients. These include chloramphenicol, tetracyclines, doxycycline, and other drugs that undergo enterohepatic circulation (e.g., clindamycin) and thus are more likely to disrupt the normal colonization of the alimentary tract in pediatric patients.
Beta-lactam antibiotics are generally the drugs of choice for pediatric patients whenever possible. Although drug half-lives are likely to be prolonged, they tend to be safe because they are characterized by a wide therapeutic index. Higher doses may be necessary to achieve desired peak PDCs because their distribution is greater. The time interval of administration can be prolonged to compensate for the longer half-life. Therapeutic drug monitoring should be used to improve the safety and efficacy of aminoglycosides whenever possible. Higher doses and longer intervals may be necessary to achieve recommended peak and trough concentrations. Amikacin, which is potentially less nephrotoxic (and more effective against Pseudomonas spp.) than gentamicin, might be preferred. Quinolones are very effective and, for most patients, safe antimicrobials. They are characterized by excellent tissue distribution. However, these drugs are not appropriate for large-breed pediatric animals because they can cause destructive lesions in the cartilage of long bones. Thus the author does not recommend these drugs as first choice for any pediatric patient. The use of disease-modifying agents containing glucosamine should be encouraged in any animal in which cartilage is growing (or repairing). The combination of a sulfonamide with trimethoprim or ormetroprim tends to be safe and effective for kittens and puppies. Oral tetracyclines should be avoided in nursing animals and before tooth extraction. Therapeutic indications for lincosamides and macrolides are limited for pediatric patients. Because both groups of drugs undergo extensive biliary secretion and enterohepatic circulation, they should not be used as first-choice antimicrobials. An exception should be made for Mycoplasma infections for which tylosin is the drug of choice. Metronidazole is the drug of choice for Giardia infections in dogs and cats, and it is often used for the treatment of anaerobic infections. Decreased clearance and prolonged half-life should be anticipated in kittens and puppies; lower doses and longer intervals may be necessary to prevent CNS toxicity. Enrofloxacin is one of the few drugs that has been studied well in neonatal to pediatric kittens.37 After oral administration, bioavailability of enrofloxacin (5 mg/kg) was at least 33% at 2 weeks of age, increasing to 50% at 4 and 70% at 6 and 8 weeks of age. Following intravenous and subcutaneous administration, the area under the curve compared to adult cats varied with the age of the kittens and route of administration. Neonates (younger than 2 weeks of age) presented as most “different,” with area under the curve at least twofold higher than that of any other age (4, 6, or 8 weeks), although elimination half-life was similar among the groups studied. In general, volume of distribution (Vd) was greater (up to twofold) at 4 and 6 weeks compared with the younger age groups and adults, indicating the need for a higher dose; because clearance was faster as well, elimination half-life was actually shorter than with adults.
Sedation, anesthesia, and analgesia
Opioid agonists are the preferred sedative, premedicant, or analgesic of some veterinary clinicians for pediatric patients.19 Although associated with marked cardiac and respiratory depression, the effects of opioid agonists are largely reversible with opioid antagonists. Bradycardia can be prevented in older pediatric patients by premedication with atropine or glycopyrrolate. Whereas the duration of fentanyl analgesia (nontransdermal patch) is generally too short to justify its use for adult patients, some clinicians prefer it for short-term intraoperative analgesia for pediatric patients because it minimally affects the cardiovascular system. Ketamine can be administered subcutaneously, intramuscularly, or intravenously for the restraint and immobilization of young cats. Response to ultrashort barbiturates such as thiopental and methohexital or similar agents (e.g., propofol) should be exaggerated in young animals because of decreased body fat and hepatic clearance. Dilution to a 1% to 2% solution is indicated to prevent over-administration.
Complications associated with intravenous administration can be reduced by rectal administration of either thiopental or methohexital in human pediatric patients. As a class the benzodiazepines can probably be used safely in pediatric patients. Elimination occurs primarily by hepatic metabolism and is likely to be slower in pediatric patients. Benzodiazepines are, however, characterized by a wide therapeutic index. Midazolam, the newest member of this group, is more potent, has a faster onset of action, and is more rapidly eliminated than diazepam. Although not approved for use in human pediatric patients, it has been used in this age group successfully to induce sedation.55 Inhalant anesthetics are preferred for maintenance anesthesia in veterinary pediatric patients. Halothane, methoxyflurane, enflurane, and isoflurane, and to a lesser degree, sevoflurane have been used. Hypotension is a complication of all gas anesthetics, however, and variable patient response necessitates close monitoring.
Pregnancy and Lacation
Maternal–Fetal–Placental Unit
Even the most simple pharmacokinetic representation of the maternal–fetal system is complex, being composed of at least three compartments: maternal, placental, and fetal. The pharmacokinetics of each compartment is determined, in turn, by its own rate of absorption, distribution, metabolism, and elimination.56-58 Pregnancy is further complicated by its dynamic nature, with dramatic changes in placental and fetal growth and in the physiology of the pregnant animal. All pharmacokinetic processes change in concert with the progression of pregnancy.
KEY POINT 2-3
Assuming that most drugs administered to the pregnant or lactating bitch or queen will reach the offspring is prudent. Water-soluble drugs should be chosen when possible if therapy is necessary.
The placenta transfers nutrients and oxygen from the mother to the fetus and facilitates waste removal from the fetus. However, the placenta also has a number of metabolic functions, among them the synthesis of hormones, peptides, and steroids that are vital for a successful pregnancy. The placenta also presents a barrier to drug distribution from maternal blood into the fetus. Transporter proteins (e.g., P-glycoprotein [P-gp]) on both the maternal and fetal side influence drug movement; placental phase I and II drug-metabolizing enzymes also have been identified throughout gestation in the human placenta. Thus far, CYP1A1, 2E1, 3A4, 3A5, 3A7, and 4B1 and uridine diphosphate glucuronosyltransferases have been detected in the term human placenta. However, these barriers to drug movement from the mother to fetus presented by the placenta are not impenetrable. Extrapolation of data among species regarding maternal to fetal transfer of compounds is complicated by differences in placentation.59 Humans and rats have the least number of layers. However, the endotheliochorial placentation of carnivores differs from hemochorial placentation of humans only by the presence of maternal endothelium, and it is likely that this single fenestrated layer does not present a significantly greater barrier.60 The idea of absolute placental selectivity has been replaced with the realization that any drug administered to a pregnant animal might prudently be anticipated to cross the placenta, regardless of the degree of intimacy between fetal and placental membranes.56,57,61 The route of administration is also likely to determine the amount of placental transfer: Routes that result in higher plasma peak concentrations (i.e., intravenously, as an intravenous infusion, and in multiple doses) are likely to expose the fetus to higher drug concentrations. Further, a “depot phenomenon” described in the human placenta reflects the accumulation of selected lipid-soluble xenobiotics in the placenta, with the placenta possibly serving as a storage site.61
The primary mechanism of drug movement is passive diffusion, with the amount of transfer dependent on physiochemical properties; less commonly, active transporters and facilitated diffusion contribute to drug movement.61 Although many factors determine the rate and extent of drug transfer across the placenta, the lipid solubility of the drug and a steep maternal–fetal drug concentration gradient are probably the most important.57 In general, nonionized compounds with high lipid solubility cross rapidly, whereas drugs with little lipid solubility cross slowly. Impermeability of the placenta to polar compounds, which generally do not penetrate cell membranes, has been described as relative rather than absolute.57 A number of drugs that are polar at physiologic pH can cross the placenta rapidly.57 Molecular weight influences drug movement, with those below 500 Dalton more likely and those above 1000 less likely to transfer. Protein binding precludes drug movement, whereas differences in fetal and maternal blood pH increase the accumulation of weak bases in fetal blood (humans).
Fetus and Neonate
Unique differences in drug disposition predispose the near-term fetus and neonate to adverse drug reactions. The pharmacologic principles that address risk of fetal exposure to potential teratogens have been reviewed.61,62 The drug approval process generally precludes availability of those compounds with a demonstrated risk, but premarket assessment does not necessarily occur in dogs or cats. The responses of the fetus and newborn to individual drugs, however, vary. Differences in responses reflect, in part, differences in placental kinetics of drugs. The fetus is most sensitive to adverse effects particularly in the first trimester. The risk is positively correlated with the duration of pregnancy, with the risk greater in shorter pregnancies (e.g., dogs and cats) because this period represents a longer proportion of the pregnancy.63
Current efforts in human neonatology are concerned with the characterization of the pharmacokinetic differences between drugs in the near-term fetus. Differences in drug disposition compared with that of both pediatric animals and adults can lead to adverse reactions in the near-term fetus receiving drugs through the placenta. The amount of fetal protein is generally less in the neonate, which is, in turn, less than that in the adult.56,57 Thus higher concentrations of unbound and pharmacologically active drugs can be anticipated. It is not clear if increased concentrations of unbound drug will be compensated for by increased clearance, as will occur in the adult animal. Perhaps more important are anatomic peculiarities of fetal circulation. Because the fetal liver and lungs are largely bypassed, blood reaching the heart and brain contain essentially the same concentration of drugs as present in the umbilical vein. Although fetal metabolism of drugs can contribute to the ultimate elimination of drugs in the human neonate, the amount of drug-metabolizing enzymes present in near-term animals is negligible.57
Although drugs administered to pregnant animals may be detectable in the fetus, they may not produce clinically important effects. Examples of drugs that have been shown to reach detectable and potentially clinically important concentrations in the fetus include salicylates and other NSAIDs, anticonvulsants (phenytoin and diazepam), local anesthetics such as lidocaine, gentamicin (in some species), and narcotic analgesics. In human infants the ratio of maternal to fetal concentration of beta-lactams approximates 1.64 Because predicting the effects of a drug crossing the placenta is difficult, drug selection for the mother should be based, in part, on anticipated safety to the near-term fetus.
Maternal
The effects of pregnancy can alter all phases of disposition in the mother. Gastrointestinal motility and gastric acid secretion decrease and may lead to decreased drug absorption. Distribution may be influenced by decreased serum albumin, and increased Vd of drugs, resulting in lower PDCs. However, drug clearance may be more rapid as cardiac output, renal blood flow, and glomerular filtration rate increase. High progesterone concentrations may induce hepatic microsomal enzymes and increase drug metabolism.65
Lactation
As is the fetus, the nursing animal is an inadvertent recipient of drugs administered to the mother. Most of the pertinent information in the veterinary literature is concerned with excretion of drugs in the milk of food animals; there appears to be no information regarding small animals. Studies of humans indicate that drugs diffuse into the milk from maternal circulation. Low-molecular-weight (<200), un-ionized, highly lipid-soluble drugs that are minimally protein bound diffuse into the lactating mammary gland rapidly, whereas water-soluble drugs diffuse more slowly.66 The pKa of a drug largely determines its concentration in milk. Animal milk tends to be acidic compared with plasma pH. Consequently, although a drug may be nonionized in the plasma and thus more likely to diffuse into milk, it may become ionized and nondiffusible once in the milk. Such “ion trapping” can concentrate drugs in milk. The ratio of drugs in milk to plasma is predictable, being greater for weak bases and weak acids whose pKas differ from the pH of milk by 2 pH units (+2 for acids and −2 for bases).67 Generally, the amount of drugs excreted in milk is less than 2% of the maternal dose.66 Greater concentrations can be expected, however, if a drug is administered to the mother intravenously, as an intravenous infusion, or in multiple doses.
Not all drugs ingested with milk during nursing will be absorbed from the gastrointestinal tract of the nursing animal. For example, milk may decrease the absorption of some drugs, whereas the pharmacokinetic properties of other drugs (i.e., aminoglycosides) preclude their absorption except in the very young. Not all drugs, however, must be absorbed to cause clinically important adverse effects. For example, antimicrobials can sometimes alter the developing flora of the pediatric alimentary tract.27,68 Thus it is prudent to refrain from administering potentially toxic drugs to the lactating bitch or queen.
Sex
Pregnancy and lactation are obvious differences between the sexes that affect the disposition of and response to drugs. However, sex differences are relevant without the influence of active reproduction. The impact of sex on drug disposition is not well characterized in human or other animal medicine. In general, female humans, compared with male humans, are characterized by reduced smooth muscle motility, greater body fat, greater content and fluctuation in plasma volume and increased organ blood flow, xenobiotic metabolism (increased CYP3A4, others not clear), and transporter protein activity.69,70 It should be anticipated that sex differences are likely to be complex, as has been demonstrated for CYP enzymes in cats.71 Female cats were characterized by greater CYP2D and lower CYP3A compared with males.
Roles of Species and Breed Differences in Drug Disposition
Differences among species in the kinetics and response to drugs are profound; Riviere72 has offered a review in animals. Few studies have compared disposition among species, and when reported, data are generally oriented toward human medicine, with the intent to identify the (laboratory) animal most predictive of human drug disposition. Presumably, species that are physiologically similar tend to have similar drug disposition patterns, and the same dosing regimen can often be used for a particular drug.73 However, pharmacokinetic data (Vd, clearance, and mean residence time) after intravenous administration have been compared among humans, monkeys, dogs, and rats for over 100 molecules (including many drugs).74 Correlations between chemical structures of the compounds with pharmacokinetics allowed two categories of compounds to be identified: those characterized by high clearance, for which mathematical models would allow general extrapolation of data from one species to another, and those whose characteristics yielded incorrect extrapolations. Although the authors concluded that such an approach might be useful for extrapolation, more to the point is the reality that drugs may not be extrapolatable even with the most sophisticated of mathematical models. The combination of computation analysis of the molecular characteristics of each compound with in vivo pharmacokinetic data might ultimately be helpful in identifying the most appropriate species for predicting pharmacokinetic behavior of a particular molecule in another species. Thus caution needs to continue when extrapolating information from humans to the dog, and dog to cat. Likewise, pharmacodynamic responses may be profound.
Absorption
Gastric pH profiles have been described for the dog in anticipation of extrapolation to humans.75 Dogs are described as poor gastric acid secretors, compared with humans as good secretors, with gastric pH fluctuating in dogs from 2.7 to 8.3 (mean 6.8). The shorter gastrointestinal tract of dogs compared with that of humans, results in a transit time (111 minutes) that is 50% of that of humans, although this may be offset by taller villi and greater bile salt concentrations. Accordingly, differences in absorption might be expected between dogs and humans in the oral absorption of drugs, particularly for enteric-coated or altered-release products.75 Different affinities for P-gps have been demonstrated among humans, nonhuman primates, and Beagle dogs.76 In dogs absorption of some hydrophilic compounds is more similar to that in rats, whereas absorption of others is more similar to that found in humans.77 Another retrospective comparison between dogs and humans in the oral bioavailability of 43 drugs found close to 50% to be completely absorbed in both species. For the remaining drugs, 12 were absorbed more rapidly and to a greater (15% to 200%) extent.78 However, the extent of absorption for drugs not well absorbed correlated poorly between the two species. In general, if the drug was well absorbed in humans, it tended to be well absorbed in dogs. Both lipid- and water-soluble drugs appear to be better absorbed in dogs, the latter suggesting more paracellular transport. The rate and magnitude of drug absorption for many drugs may be similar between dogs and cats, regardless of the route of administration. However, extrapolations must be done with caution, as is exemplified with ciprofloxacin, whose oral bioavailability in the dog is 40% (compared to 80% or better in humans) but 0% to 20% (the latter more likely with multiple dosing) in cats.79,80 Another example is prednisone. An exception also may need to be made for slow-release preparations; rates and extent of absorption do vary among species. Dye81,82 has demonstrated not only differences in bioavailability in cats versus dogs but also differences in pharmacokinetics related to morning versus evening dosing. Because slow-release preparations used in human patients are designed to maintain therapeutic concentrations in humans, absorption kinetics of these products can be profoundly different in the dog and cat. Use of such drugs should be based on clinical studies of these preparations in dogs and cats.
Distribution
Because most determinants of xenobiotic distribution are largely affected by cardiac output, regional blood flow, and xenobiotic chemistry, distribution differences among species might be predictable through allometric scaling.83 However, they must also take into account differences in transport proteins. Differences in drug distribution can result in important differences in drug response. Blood volume of the cat (70 mL/kg) is less than that of the dog (90 mL/kg); PDCs of drugs whose distribution is confined to the plasma compartment may therefore differ between the species. The same amount of drug (on a per-kilogram basis) is diluted less in cats because the plasma volume is smaller. Thus drug concentrations after administration of a mg/kg dose might initially be higher in cats than in dogs. Organs that are well perfused (i.e., heart, brain) may be more susceptible to toxicity. Cats are approximately the same size as the smaller dog breeds. Thus doses determined for medium-size to large-size dogs may not be appropriate for the cat because the smaller animals have a greater body surface area. In larger animals body water makes up a larger proportion of body weight, which tends to dilute the drug. A higher dose may be needed for larger animals. Because the drug half-life may be longer (owing to increased distribution), however, the dosing interval may need to be longer.
Differences in plasma protein-binding characteristics (particularly albumin) may alter the distribution of drugs that are highly protein bound. The degree to which various drugs are protein bound varies dramatically among the species, although the clinical implication is not clear. Although the elimination characteristics of many drugs have been established for cats, few studies have determined the extent of protein binding.
The interaction between disease and species should be considered when considering species differences in drug distribution. For example, the unhealthy cat does not maintain hydration as well as the dog; fluid imbalances resulting from dehydration or edema alter drug distribution. The obese cat can represent a “sink” for drugs that are lipid soluble, thus lowering PDCs potentially to submaximal levels if the dose is not appropriately increased. Weight loss in a hyperthyroid cat can have the opposite effect.
Sight hounds (e.g., Salukis, Greyhounds; Figure 2-3) offer an example of potential breed differences in drug distribution. Their lean body weight provides little fat tissue for drug distribution. As a result, they are more susceptible to overdosing with drugs that redistribute, such as thiobarbiturates.
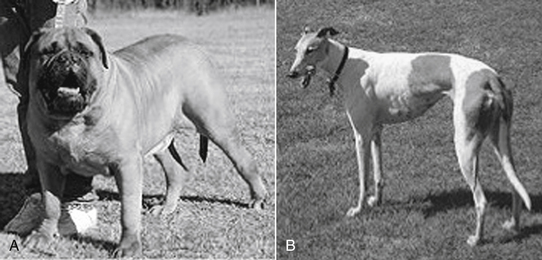
Figure 2-3 Differences in breeds may affect body composition (A) as well as other aspects of drug disposition. The sight hounds, represented by the Greyhound (B) is at risk for selected adverse drug events, in part because of a larger lean body mass compared with fat. Drugs that normally accumulate in fat may achieve higher drug concentrations in such breeds.
Distribution is influenced by the multidrug transport protein P-gp. Polymorphism has been demonstrated in the Collie, yielding differences in P-gp content. Two P-gp transporting proteins are encoded by ABCB1 (previously MDR1 or PGY1) and MDR3 (also named MDR2 and PGY3); only the MDR1 gene product is thought to significantly influence drug metabolism. P-gp acts as an efflux pump by translocating drugs from the intracellular to extracellular compartments (Table 2-2) (see Chapter 3). The protein transports a large number of drugs that are chemically divergent; further, these drugs are associated with a specific CYP450 responsible for metabolism of the drugs that have been transported. Polymorphism of the MDR1 gene and P-gp have been reported in humans and are associated with altered drug disposition and thus susceptibility to adverse drug events. Interestingly, polymorphism also has been associated with an increased risk of certain illnesses (e.g., refractory seizures, Parkinson’s disease, inflammatory bowel disease biliary mucoceole in dogs∗). Polymorphism reflecting a mutation deletion of MDR1 that causes nonfunctional P-gp has been documented in Collie and related working-breed dogs. The incidence of the deletion is impressively high: in the U.S., in one study, 35% of Collies were homozygous and another 42% heterozygous for the mutation deletion.84 A similarly high incidence was found in dogs in France: 20% of Collies and related breeds were found to be homozygous for the normal allele, 32% heterozygous for the deletion (carrier), and 48% homozygous for the mutant allele (affected dogs).85 The impact of the mutation on drug safety in afflicted animals can be profound (see Chapter 3). Substrate specificity for P-pg appears to be similar among species, suggesting that human data can be used to predict which drugs might be more likely to cause adverse effects in these breeds.86 However, protein type and amount may vary among species and breed.
Table 2-2 Substrates for P-glycoprotein Transport Pump and Known Inhibitors and Inducers of Drug Transport
| Substrates | Inhibitors | Inducers |
|---|---|---|
| Antimicrobial Drugs | ||
| Erythromycin | Bromocriptine | Clotrimazole |
| Tetracycline | Carvedilol | Dexamethasone |
| Itraconazole | Cyclosporine | Morphine |
| Fluorinated quinolones (selected) | Erythromycin | Rifampin |
| Anticancer Drugs | Fluoxetine | Phenothiazine |
| Doxorubicin | Intraconazole | St. John’s Wort |
| Vinblastine | Ketaconazole | |
| Vincristine | Meperidine | |
| Mitoxantrone | Pentazocine | |
| Anthelmintics | Progesterone | |
| Ivermectin | Quinidine | |
| Cardioactive Drugs | Tacrolimus | |
| Digoxin | Verapamil | |
| Quinidine | ||
| Diltiazem | ||
| Verapamil | ||
| CNS-Active Drugs | ||
| Phenothiazines | ||
| Amitryptyline | Inhibitor of CYP3a | |
| Morphine | Substrate CYP3a | |
| Endogenous Substrates | ||
| Bilirubin | ||
| Steroidal Hormones | ||
| Cortisol | ||
| Aldosterone | ||
| Gastrointestinal Drugs | ||
| Cimetidine | ||
| Loperamide | ||
| Ondansetron | ||
| Tacrolimus | ||
| Immunomodulators | ||
| Dexamethasone | ||
| Methylprednisolone | ||
| Cyclosporine | ||
| Tacrolimus | ||
| Colchicine | ||
CNS, Central nervous system.
Metabolism
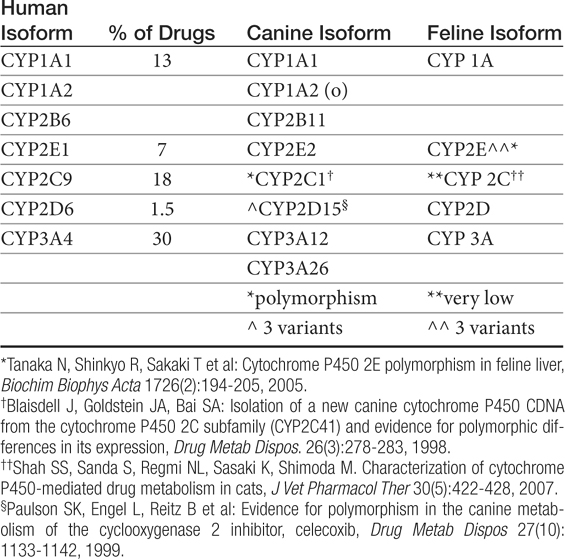
Human polymorphisms in CYP metabolic enzymes have been associated with therapeutic failure resulting from extremely rapid metabolism of a drug and toxic effects caused by decreased metabolism.87 Species extrapolation among the CYP450 appears to be predictable in order of most to least: 2E1 (reasonably predictable) greater than 1A1, 1A2, and 4A (cautiously predictable) greater than 2D and 3A greater than 2A, 2B, and 2C (major caution with extrapolation).88 Studies attempting to identify similarities between dog and human CYP activity suggest general extrapolation between the species is not prudent.87 Although significant differences among species were not detected among rodents, rabbits, and dogs for CYP2E1, 3A2, and 4A1, comparison of animal CYP activity with human found only 2D6 to be most similar to dog.89 Other enzymes present in dogs are CYP1A, 2C, and 2D families and subfamilies (Table 2-3).90
Table 2-3 Cytochrome P450 Families, Their Contribution to Drug Metabolism in Humans, and Their Orthologs in Other Species
KEY POINT 2-4
The genetic basis for differences in drug transport and metabolism limits extrapolation of dosing regimens among species, and potentially, breeds. However, understanding of these polymorphisms is increasing our ability to predict increased risks associated with drug administration in some breeds.
Species differences have been documented in the handling of racemic isomers of selected drugs. Inversion (from one isomer to the other) patterns differ and are likely to result in different pharmacologic or toxic effects of the drugs. Induction (or presumably inhibition) of drug-metabolizing enzymes also will differentially affect the isomers.91
As in humans, polymorphism in drug-metabolizing enzymes has been reported in dogs92 but is not as well described. Differences in response to anesthesia recognized in sight hounds reflect both differences in drug distribution (to lean versus fat compartments; see Chapter 1) as well as differences in metabolism. Cytochrome-mediated clearance of several anesthetic agents is less in Greyhounds compared with other (nonsight hound) dogs; documented drugs include thiopental, thiamylal, and methohexital. Clearance of propofol by Greyhounds is three times less than that by Beagles. Ketoconazole plasma concentrations were twofold higher than expected in Greyhounds in one study.93 Further, Greyhound disposition of celecoxib, a cyclooxygenase-1 protective NSAID, indicates that breed differences may predispose this breed to adverse drug reactions. Polymorphism also has been described for CYP2C isoenzymes, again in Beagles94 and possibly Greyhounds.95 Polymorphism in celecoxib metabolism was attributed to CYP2D15, for which three canine variants were found.96 In a study of 242 Beagles receiving celecoxib, approximately 50% were considered efficient metabolizers and 50% poor metabolizers, with bioavailability and maximum PDC in the latter group almost twofold higher. The impact of species differences in pharmacokinetic and pharmacodynamic considerations of enantiomers has been described.91,97 For example, for many NSAIDs the S-isomer has a much greater affinity for cyclooxygenase-2, but the proportion of the S isomer varies among species. Further, species differ in their ability to interconvert S and R isomers.97 For example, chiral inversion has been described for ketoprofen in cats.98Other polymorphisms that have been described include thiopurine methyltransferase (TPMT), which is one of several enzymes responsible for the metabolism of the active metabolite of the prodrug azathioprine; polymorphisms resulting in deficiencies in humans have been associated with an increase in the toxic bone marrow effects of the drug. Differences in dogs have been demonstrated as well: Kidd99 and coworkers have demonstrated that Giant Schnauzers have significantly less and Alaskan Malamutes significantly more TPMT compared with other canine breeds.
The most significant and best-characterized differences in drug disposition between the dog and cat probably result from differences in drug metabolism. Identification of phase I enzymes and their specific drug substrates is difficult, and few species differences have been described in the cat or dog. However, a recent review of CYP450 activity–based substrate metabolism in cats suggests that cats have very low activity of CYP2C, but activity of CYP2D and CYP3A approximates that of dogs or humans, depending on sex.77 Deficiencies in demethylation and hydroxylation have been described in the cat and may be responsible for different patterns of prodrug activation (e.g., primidone; see Chapter 27) or adverse reactions to selected drugs (i.e., chloramphenicol).100 Deficiencies in phase I demethylation and hydroxylation as well as phase II glucuronidation lead to much slower elimination of phenols and aromatic acids and amines in the cat compared with other species (Figure 2-4).101,102 The described reaction of cats to diazepam may represent differences in the metabolites produced, as may the susceptibility of the feline liver to metabolite-induced damage.103 Polymorphisms in drug-metabolizing enzymes have also been described in cats, which have at least three variants of CYP2E,104, although breeds were not cited.104
Figure 2-4 Examples of drugs (many containing phenols) whose metabolism in cats is slower owing to deficiencies in drug phase I and phase II drug-metabolizing enzymes.
Deficiencies in phase II metabolism have long been recognized in cats. The deficiency reflects extremely low concentrations of some glucuronyl transferases. Thus many drugs excreted as glucuronide conjugates in other species are characterized by a prolonged clearance rate and half-life in the cat. Toxic levels may accumulate much more quickly in the cat, and exaggerated pharmacologic responses or toxicities occur more easily (Figure 2-4). Dosing regimens must be modified for such drugs by either decreasing the dose (especially for drugs whose dosing interval is shorter than the elimination half-life) or prolonging the dosing interval. The prototypic example is aspirin, whose half-life approximates 36 hours in cats compared with 8 hours in dogs. To prevent toxicity in the cat, aspirin is dosed every 48 to 72 hours, compared with twice daily in dogs.
Not all drugs that are conjugated with glucuronide are predisposed to toxicity in the cat. This is true for several reasons. First, the cat is deficient only in certain families of glucuronyl transferase. Cats can conjugate and excrete endogenous substrates such as bilirubin, thyroxine, and steroid hormones as well as other species. Metabolism of a variety of exogenous drugs, however, particularly phenols and aromatic acids and amines, occurs at a much slower rate in the cat than in other species.101,102 The degrees of deficiency and potential toxicity depend on the drug substrate. For example, some phenolic compounds are sufficiently conjugated, whereas others are not. Second, glucuronide-conjugated drugs characterized by a wide safety margin are associated with few adverse reactions even if accumulation occurs. Finally, in the absence of glucuronide, drugs may be sufficiently metabolized by an alternative pathway. Some sulfates may be particularly well developed in the cat, and many drugs that are excreted as glucuronide-conjugates by the dog may be excreted as sulfated compounds by the cat. Other sulfate-conjugating systems, however, appear to be easily saturated in the cat. Unfortunately, alternate pathways of drug metabolism may also contribute to the toxicity of some drugs because they may involve phase I enzymes that catalyze the formation of toxic metabolites. Thus drugs shunted to another pathway in the cat may be very toxic to the cat but minimally toxic in other species. Deficiencies in both glucuronide and (potentially) glutathione transferase may also predispose the cat to poor scavenging systems in erythrocytes and hepatocytes, limiting the otherwise protective effects that might be realized by these systems and further contributing to toxicity. Acetaminophen is an excellent example of the potential sequelae of phase II deficiences in the cat (See Chapter 4). Because glucuronide is deficient, excessive acetaminophen is shunted to phase I enzymes, which produce toxic oxygen radicals. More metabolites are produced than can be handled, and the glutathione-scavenging system of feline erythrocytes and hepatocytes is overwhelmed, resulting in life-threatening methemoglobinemia and (potentially) hepatic necrosis. The rationale for cimetidine treatment can be understood in the context of the role of phase I metabolism, as well as that of N-acetylcysteine, a glutathione precursor.
Not all deficiences in metabolism occur in the cat. Although acetylation is not a common route of elimination for xenobiotics, it is an enzyme system whose deficiency in the dog is clinically relevant (e.g., procainamide). For example, the antiarrhythmic procainamide is acetylated in humans to an active metabolite. Procainamide is less potent than its acetylated metabolite and the canine dose for procainamide is considerably higher than that in humans on a mg/kg basis in order to achieve an equivalent pharmacologic response (see Chapter 14, Cardiac). A second example might be sulfonamide elimination. Sulfonamides are detoxified by N-acetylation in humans. In the face of deficient acetylation, shunting of the xenobiotic to an alternative pathway in dogs, with the production of the cytotoxic metabolite hydroxylamine, may be one mechanism of sulfonamide toxicity in dogs, although alternative mechanisms are likely to be responsible.105
Renal Excretion
In contrast to hepatic metabolism, differences in renal excretion between the dog and cat do not appear to be profoundly important to drug disposition. Glomerular filtration and active tubular secretion parallel cardiac output and thus should be predictable among species based on allometric scaling.83 The glomerular filtration rate of cats (2.5 to 3.5 mL/min/kg) is less than that of dogs (3 to 5 mL/min/kg), suggesting that renal clearance of drugs may be faster in dogs. Although this is true of inulin, differences have not been established for most drugs. Renal disease profoundly alters the rate of drug excretion in all species. In general, serum creatinine concentrations can be used to modify the dose (decrease in proportion) or interval (prolong in proportion to abnormality). The modification should be applied only to that portion of the drug eliminated by the kidney. Note that fluid imbalances in renal disease can also alter drug distribution. Finally, differences in active transport and passive resorption—the latter influenced by differences in urinary pH—may result in differential excretion among species. However, because urine tends to be acidic in both dogs and cats, differences in the latter may not be profound.
Role of Species Differences in Target Tissues
It is difficult to predict differences in drug reaction that can be ascribed to differences in target tissues because very little is known about cats. Differences in response to selected drugs (e.g., opioids, insulin, chlorinated hydrocarbons) are known to be or are thought to be reflections of differences in tissues; however, often these differences turn out to be pharmacokinetic differences reflecting decreased or increased PDCs of active (including toxic) compounds rather than differences at the receptor level.
Feline erythrocytes (hemoglobin) appear to be more susceptible to oxidation and thus to methemoglobinemia. Drugs reported to cause methemoglobinemia in the cat include urinary antiseptics containing methylene blue106 or azodyes, acetaminophen101,107,108 and related compounds, benzocaine,109 and propylthiouracil.110 Several mechanisms have been postulated to explain the potential increased sensitivity of cats to methemoglobin formation. Lower concentrations or activities of the intracellular repair enzyme methemoglobin reductase have been postulated but not confirmed.111,112 Faster metabolism of specific drugs to toxic metabolites that overwhelm scavenging systems has already been discussed as a likely cause for some drugs, particularly those whose elimination is shunted to alternate (toxic) pathways (i.e., acetaminophen).107 Differences in the structure of feline hemoglobin have also been postulated. Feline hemoglobin contains up to 20 sulfhydryl groups compared with a maximum of four in other species. Sulfhydryl groups tend to be reactive and thus are susceptible to interaction with reactive parent drugs or metabolites. Thus more sulfhydryl groups would need to be maintained in a reduced state in cats.113 Other unique considerations for the cat might include its propensity for drug-induced retinal damage; hepatic lipidosis associated with anorexia; and, potentially, an increased risk of nephrotoxicity.
Canine breed differences in response to drugs may also reflect pharmacodynamic responses. For example, brachycephalic breeds (e.g., Boxers) are more susceptible to cardiac arrhythmias (sinoatrial block) caused by acepromazine.
Miscellaneous
Differences in circadian rhythm (i.e., diurnal versus nocturnal) play a role in some differences between the dog and cat. Aminoglycosides are less likely to cause toxicity if administered during active periods. This also has been established for theophylline, for which clearance occurs more rapidly at night in the dog compared with early morning in cats. Dosing of glucocorticoids at night has been recommended for cats in order to mimic endogenous release patterns. The clinical significance of these differences has not been determined.
Recommendations Regarding Extrapolation of Dosing Regimens
Drug Information Sources
Extrapolation of doses of human drugs to dogs and cats and from dogs to cats should be based on a knowledge of the clinical pharmacology of the drug to be administered and on the physiologic differences of the target species. The safer the drug, the safer the extrapolation. Numerous resources are available for human drug information (e.g., Facts and Comparisons, USP Pharmacopeia, the package insert from the product, Physicians’ Desk Reference)114 to determine the safety and the determinants of disposition of a new drug. The veterinary literature and clinicians with expertise in the field, including diplomates of the American College of Veterinary Clinical Pharmacology, are additional sources.
Note that every drug that has been approved for use in humans has been studied in dogs. The studies generally have focused on safety, however, not efficacy. The information regarding safety (often including pertinent pharmacokinetic data, such as volume of distribution, bioavailability, and drug elimination half-life) may be obtainable through a Freedom of Information Act request, which would be processed by the Food and Drug Administration.115
Extrapolation of dosing regimens should be limited to relatively healthy animals, if possible, to avoid the effects of disease on drug disposition. Likewise, extrapolation to geriatric and pediatric patients is discouraged. Administration by the oral route is generally safer (although gastric irritation may be more likely). Oral administration is less preferred if the drug undergoes first-pass metabolism, however, because this can vary dramatically among animals. A 50% change in first-pass metabolism may double the pharmacologically active dose in a patient. Intravenous administration is not recommended; when it is absolutely necessary, the drug should be administered slowly (over 5 to 10 minutes or more). Drugs with long half-lives (>12 hours) should generally be avoided. If a drug is administered at an interval that is less than the drug half-life, accumulation should be anticipated and accounted for in the dosing extrapolation. Note that maximal adverse effects may not appear until accumulation is complete at steady state. Also, a drug half-life can change (as a result of disease or drug interactions). Thus a drug that initially did not accumulate (and whose dose is based primarily on volume of distribution) may begin to accumulate as disease worsens. Unless the drug can be monitored, a change in drug half-life will be missed. In such instances a dosage reduction is again indicated. On the other hand, as a patient improves, response to therapy may again change disposition, perhaps leading to therapeutic failure. The veterinarian should be prepared to treat adverse effects if they occur. If the drug half-life is long, the time necessary for abatement of the adverse reaction will also be long. In general, extrapolation of lipid-soluble drugs is discouraged because of the risk of too many species differences.
Water-Soluble Drugs
As a general rule, extrapolation of doses for drugs that are water soluble is more appropriate because these drugs are distributed to extracellular fluids (normalizing Vd) (Box 2-2); protein binding is likely to be negligible; and hepatic metabolism is minimized. Drug Vd and renal elimination may be similar among species, and the interval used for such drugs can often be extrapolated among species. The dose administered, however, probably should be reduced to compensate for differences in blood volume among animals. Increased doses are indicated for pediatric patients and for patients with edema; decreased doses are indicated for geriatric and dehydrated patients.
Lipid-Soluble Drugs
Lipid-soluble drugs tend to be distributed to total body water and beyond, leading to a greater risk of differences among species. They are more likely to be highly protein bound, leading to a risk of differences in tissue distribution and in the proportion of pharmacologically active drug. In contrast to water-soluble drugs, lipid-soluble drugs are more likely to require hepatic metabolism (Box 2-3). In general, the clinician should anticipate a longer half-life in cats for drugs that undergo phase I metabolism in other species. Note that species differences in phase I metabolism can be very profound. If acetylation is a major phase II route of elimination, it is likely that the drug may be metabolized faster in cats than in dogs. If phase I metabolism and glucuronidation is the major route of elimination, a longer half-life should be anticipated in cats. Although glucuronidation does not necessarily indicate that elimination of the drug will be slower in cats, until an appropriate study has established the kinetics of the drug in cats, its use is discouraged. An exception might be made if the drug can be monitored or the drug is characterized by a wide therapeutic window. Altered-release preparations are not recommended because rates of absorption among the species can be dramatic. Finally, it might be necessary to refrain from administering preparations containing propylene glycol and other unknown carriers because of adverse reactions in cats.
All Drugs
Drugs with large (>2 L/kg) Vds are not recommended because accumulation or tissue binding of the drug may occur. These factors are likely to vary among species. Prodrugs, drugs for which active metabolites contribute significant activity and slow-release drugs are not appropriate because the amount of active drug is not predictable among the species. Body surface area should be used whenever possible to determine doses of toxic drugs. Drug disposition may change as the animal improves, particularly if a disease that affects drug disposition (i.e., cardiac, renal, hepatic) is being treated. Changes in dosing regimen may again be indicated. Finally, the veterinarian should be aware of the laws regulating the use of drugs labeled for use in humans.
Pathologic Factors
The diseased patient is more likely to react adversely to a drug. Although such reactions occasionally reflect disease-induced differences in receptor number or sensitivity, most often they reflect differences in drug disposition. The dosage regimens recommended for a pharmaceutical preparation generally are based on controlled studies in the normal, healthy animal. However, drugs are most frequently administered to the diseased patient. Pathophysiologic changes in most body systems can alter all phases of drug disposition, predisposing the patient to adverse drug reactions (Table 2-4). The sequelae of disease on drug disposition often but not always leads to increased PDCs and thus to a greater potential for adverse drug reactions. Occasionally, however, PDCs are lower than anticipated (e.g., increased Vd, decreased oral bioavailability), and therapeutic failure may occur. Diseases most likely to contribute to adverse drug reactions are those affecting the kidneys, liver, and heart. Less significant effects accompany gastrointestinal, pulmonary, endocrine, and metabolic disorders. Pathologic responses that generate clinical signs of disease also may be associated with changes in drug disposition. For example, following oral administration, amoxicillin area under the curve increased in dogs rendered febrile by administration of endotoxin.116 The cause of the increase was not clear but may have reflected increased absorption caused by lipopolysaccharide reduction of gastrointestinal transit time or fever-induced increased absorption or endotoxin-induced decreased renal clearance. The sequelae of hypovolemia affects multiple aspects of drug disposition. Because blood flow to the brain and heart are maintained, drug that might have been distributed to peripheral organs is distributed to these two organs, increasing their risk of toxicity while simultaneously limiting clearance by organs of excretion. Drug distribution to peripheral organs will then increase with successful management.
Table 2-4 Impact of Disease on Drug Disposition
| Pharmacokinetic Changes | Impact | Sequelae |
|---|---|---|
| Liver disease | Decreased hepatic blood flow | Increased oral bioavailability, decreased clearance of highly extracted compounds |
| Decreased phase I enzymes | Decreased hepatic clearance (longer half-life) | |
| Decreased phase II enzymes | Decreased hepatic clearance (longer half-life) | |
| Decreased hepatoprotection (increased risk of hepatoxicity) | ||
| Decreased albumin | Increased concentration of free drug (may not be cleared more rapidly in presence of liver disease) | |
| Ascites | Decreased distribution of water-soluble drugs, higher drug concentrations, shorter half-life | |
| Decreased production of phase II enzymes | Decreased cytoprotection | |
| Renal disease | Decreased cytoprotection | Increased risk of xenobiotic-induced nephrotoxicity |
| Decreased autoregulation | Increased risk of xenobiotic-induced nephrotoxicity | |
| Decreased body mass (dehydration) | Decreased volume of distribution (higher xenobiotic concentration, shorter half-life) | |
| Increased fluid retention | Increased distribution of drugs, decreased concentration, longer half-life | |
| Decreased renal blood flow | Decreased renal clearance (longer half-life) | |
| Tubular disease | Decreased concentration of drug, decreased efficacy of urinary antibiotics, decreased clearance | |
| Glomerular disease | Increased risk of xenobiotic-induced nephrotoxicity | |
| Deceased protein-binding (increased fraction of unbound xenobiotic) | ||
| Increased clearance of unbound fraction of highly protein-bound xenobiotics | ||
| Cardiac disease | Decreased renal blood flow | See renal disease |
| Decreased hepatic blood flow | See hepatic disease | |
| Decreased regional blood flow | Decreased organ delivery (higher concentrations delivered to brain and heart) | |
| Gastrointestinal disease | Decreased cytoprotection | Increased risk of gastrointestinal toxicity |
| Altered permeability | Altered absorption | |
| Altered gastrointestinal motility | Decreased rate of absorption | |
| Altered P-glycoprotein | Altered absorption |
Renal Disease
Drug toxicity in renal failure may result either from increased sensitivity to the drug owing to uremia-induced alterations in tissue receptors or from decreased or increased PDCs caused by disease-induced changes in pharmacokinetics. Changes induced by renal disease have been best characterized (see Table 2-4).117-120
Renal blood flow is often profoundly decreased in patients with renal disease. Changes in glomerular filtration and tubular secretion tend to parallel changes in renal blood flow. The effects of changes in renal blood flow (usually decreased) on drug excretion are most profound if renal extraction of the drug is high (e.g., penicillins, sulfates, glucuronide conjugates) but are less significant for drugs that are slowly extracted (e.g., aminoglycosides, diuretics, digoxin).
Glomerular filtration of drugs and other compounds is also adversely affected in renal disease independent of changes in renal blood flow. The determinants of glomerular filtration include protein binding, glomerular integrity, and the number of functional (filtering) nephrons. The molecular size of the drug is also important because drugs with a molecular weight greater than approximately 70,000 usually cannot be filtered. Drugs that are tightly protein bound (such as NSAIDs) are not filtered until they are displaced from the protein. Factors that tend to displace such drugs from protein-binding sites may increase the rate of drug excretion in renal disease and include hypoalbuminemia, competition for protein-binding sites owing to accumulation of uremic toxins, or changes in the conformation and thus binding affinity of the protein (e.g., albumin). Changes in protein binding that have been measured in renal disease include decreased binding of acidic drugs (e.g., furosemide, NSAIDs, selected penicillins and anticonvulsants) and normal or increased binding of basic drugs owing to increased concentrations of inflammatory proteins (e.g., propranolol, diazepam, prazosin). The impact of increased clearance of otherwise protein-bound drug may be offset by the increase in plasma concentration of the free drug displaced from the protein, thus increasing elimination of unbound drug.
Although changes in active tubular reabsorption that may accompany renal disease probably do not profoundly influence the rate of drug excretion, changes in active tubular secretion can be significant. Active tubular secretion occurs in the pars recta (straight segment) of the proximal tubule. A transport system exists for a variety of organic acids (e.g., penicillins, cephalosporins, NSAIDs, sulfonamides, several diuretics) as well as bases (e.g., cimetidine, procainamide, some morphine derivatives). Distal nephron active transport may also be important for some drugs (e.g., digoxin). Excretion of these drugs is most likely to be decreased in the presence of renal disease because of decreased nephron mass, decreased renal blood flow, and decreased tubular function.
In addition to these changes, renal disease can also alter drug disposition because of changes in electrolyte, acid–base, and fluid balance. Changes in electrolytes and acid–base balance may also be important in altering receptor sensitivity to drugs, such as those affecting the cardiovascular system (e.g., hyperkalemia and its effects on responses to digitalis, quinidine, and procainamide).
KEY POINT 2-6
Serum creatinine is a reasonable indicator of the impact of renal disease on renal drug clearance.
For drugs whose elimination depends on renal function and whose clearance is known to be decreased in renal disease (Box 2-4), dosing regimens can be appropriately altered to reduce the incidence of adverse reactions.119 Either serum creatinine or creatinine clearance is generally used to estimate glomerular filtration rate; however, because of its ease of measurement, serum creatinine is most commonly used. Decreases in the renal elimination of a renally excreted drug tend to parallel increases in serum creatinine, and dosing regimens can be altered by either lengthening the dosing interval or decreasing the dose by a proportional decrease in creatinine clearance or the increase in serum creatinine (Box 2-5).
Box 2-4 Examples of Drugs Characterized by Changes in Drug Disposition in Patients with Renal Disease
Box 2-5 Dosing Regimens for Drugs Dependent on Renal Function
New dose = old dose × pt CrCl/normal CrCl or × normal Cr/pt Cr
New interval = old interval × normal CrCl/pt CrCl or × pt Cr/normal Cr
pt = patient, Cr =creatinine, and CrCl = creatinine clearance
For example, a patient has a serum creatinine level of 2.5 mg/dL (normal is 1.2), a dosing interval for a drug given every 12 hours would be prolonged to every 24 hours, or the dose of 2 mg/kg could be reduced to 1 mg/kg.
The parameter of the dosing regimen that should be altered depends on the drug. Lengthening the interval results in wider swings in PDCs during a single dosing interval and thus may not be desirable for time-dependent antimicrobial therapy but would be acceptable for concentration-dependent drugs or lengthening the interval also should be avoided for anticonvulsant or cardioactive drugs that depend on maintenance of a minimum drug concentration within a specified therapeutic range. Thus decreasing the dose may be more appropriate for these drugs. For drugs with a long elimination half-life or drugs whose effects persist in the absence of detectable drug (e.g., selected antimicrobials [e.g., concentration-dependent], glucocorticoids, nonsteroidal agents), however, it may be more appropriate to prolong the interval. Aminoglycosides previously were dosed at 12-hour intervals. Because their efficacy depends on a high PDC, yet safety is based on allowing PDCs to fall below a recommended trough concentration, prolonging the interval (i.e., from 12 to 18 or 24 hours) was more appropriate than lowering the dose in patients with renal disease sufficient to alter creatinine clearance. However, aminoglycosides are currently administered at 24-hour dosing intervals, precluding the need to prolong intervals in some renal disease patients. For other drugs whose toxicity is related to peak drug concentrations, the dose might be decreased by 50%, or the interval prolonged one half-life. Few clinical studies have addressed the impact of renal disease on the clearance of drugs in dogs or cats. Experimentally induced renal disease sufficient to cause serum creatinine to increase to 145 ± 122 μmole/L in dogs caused a slight decrease in renal clearance but no clinically significant change in elimination half-life of marbofloxacin.121
Hepatic Disease
The efficiency of hepatic elimination is determined by hepatic clearance and the hepatic extraction ratio of the drug.122-125 Both, in turn, depend on hepatic blood flow; the extent of drug protein binding; and intrinsic hepatic clearance, which itself consists of hepatic uptake (the rate-limiting step of hepatic clearance), intracellular transport, metabolism, and (if applicable) biliary elimination. Drugs that are eliminated by the liver can be categorized according to their rate of extraction.126 Flow-limited drugs (e.g., lidocaine, propranolol, verapamil) are so rapidly extracted by the liver that their rate of elimination depends only on the rate at which it is delivered to the liver (e.g., hepatic blood flow). Such drugs are insensitive to changes in hepatic metabolism but are very sensitive to changes in hepatic blood flow. Capacity-limited drugs (e.g., diazepam, prednisolone, phenylbutazone, phenytoin, theophylline, cimetidine, and antipyrine) are extracted slowly by the liver, and their elimination depends on hepatic uptake and metabolism but is independent of hepatic blood flow. The elimination of such drugs is affected by changes in hepatic metabolism but not by changes in hepatic blood flow. Some drugs are intermediate, being partially dependent on hepatic blood flow and hepatic metabolism.123,126
Protein binding can affect the elimination of some capacity-limited drugs because only unbound drug can be extracted by the liver. Flow-limited drugs tend to be binding insensitive in that hepatic extraction is so fast that binding to proteins does not alter their rate of elimination. Some capacity-limited drugs are not significantly protein bound and thus are also binding insensitive (e.g., antipyrine). In contrast, some capacity-limited drugs are binding sensitive (e.g., theophylline, phenytoin) because their slow rate of extraction can be increased by decreasing or increasing, respectively, protein binding.127
The effects of hepatic disease on drug disposition are very complex, particularly for drugs that are affected by changes in hepatic blood flow, hepatic metabolism, and protein binding (Box 2-6).123,126 Each of these parameters may be altered in various ways in patients with liver disease. Hepatic blood flow is generally reduced in chronic liver disease because of formation of portosystemic shunts and intrahepatic shunting. Drug delivery bypasses many functional hepatocytes that would otherwise clear the drug. Plasma and tissue drug concentrations are markedly higher when dosing regimens are not appropriately altered. This is particularly important for highly extracted drugs when they are administered orally. The dose of such drugs (e.g., propranolol, verapamil, prazosin, morphine derivatives) in the presence of normal hepatic blood flow is based on decreased bioavailability owing to first-pass extraction: a large percentage of the drug does not reach systemic circulation because it is removed from portal blood by the liver the first time it passes through the liver. Decreased hepatic blood flow and intrahepatic shunting of blood can markedly increase systemic bioavailability of such drugs (Figure 2-5).128 Studies in human patients with liver disease suggest that the intrinsic metabolism of highly extracted drugs also is reduced in patients with liver disease. In human patients with liver disease, the dose of many highly extracted drugs is reduced by 50%; such an approach is probably reasonable for the veterinary patient.
Box 2-6 Drugs Characterized by Changes in Drug Disposition in Patients with Hepatic Disease
(From Williams RL: Drugs and the liver: clinical applications. In Benet LZ, Massoud N, Gambertoglio JG, editors: Pharmacokinetic basis for drug treatment, New York, 1984, Raven Press, pp 53-75.)
Figure 2-5 Portosystemic shunting affects drug disposition at multiple sites. Clearance of flow-limited drugs will be decreased in proportion to the fraction of blood shunted around the liver. Capacity-limited drugs may also be decreased if hepatic mass is sufficiently reduced. Accumulation of fluid in response to sodium retention may be profound, particularly in the abdomen. Albumin may be decreased, increasing the fraction of unbound drug, which may not be cleared as rapidly in the presence of disease.
The effect of liver disease on poorly extracted (capacity-limited) drugs is more difficult to predict, particularly for drugs that are highly protein bound. Generally, hepatic metabolism is reduced in patients with liver disease. Disease is generally quite profound, however, by the time changes in drug disposition become evident. The severity of disease is manifested as a decrease in serum albumin and blood urea nitrogen levels.This has been shown by studies that have measured the clearance of antipyrine and caffeine, which are capacity-limited, binding-sensitive and binding-insensitive drugs, respectively, in dogs with experimentally induced liver disease.129,130
KEY POINT 2-7
If liver disease has impacted serum albumin or blood urea nitrogen, ensuring that drug metabolism is also impacted is prudent.
Because elimination of these drugs depends entirely on hepatic metabolism, their clearance might be used as a hepatic function test. Because their elimination does not necessarily correlate with the elimination of other drugs that also depend on hepatic clearance, however, they are not used to predict changes in dosing regimens for the patient with liver disease. Recommendations regarding dosing regimens for drugs not highly extracted by the liver thus are difficult to make for the patient with liver disease, in part because there is no simple test that will assess or quantitate hepatic function.125,129-133 In general, clearance of capacity-limited drugs is probably not impaired in patients whose serum albumin and blood urea nitrogen levels are within normal limits. Common sense dictates, however, that discretion be used when administering potentially toxic drugs that depend on hepatic clearance for elimination.
Diseases of the biliary tract alter the disposition of drugs eliminated through the bile. This route of elimination is complex, however, with drugs being eliminated in feces and undergoing enterohepatic circulation. Characterizing these changes is difficult without catheterization of the biliary duct, and recommendations are very difficult to offer. Cholestasis decreases the content or activity of cytochrome P450 drug-metabolizing enzymes and thus can affect the elimination of drugs that are not secreted in bile.134,135 In general, doses of drugs eliminated principally in the bile should be reduced, particularly if the drug is characterized by a narrow therapeutic window. Examples include selected antimicrobials (doxycycline and clindamycin), digitoxin, and naproxen.
The effects that changes in protein binding may have on hepatic drug clearance contribute to the unpredictable nature of liver disease–induced changes in drug disposition.127,136,137 Decreased protein binding of drugs that may accompany liver disease (i.e., caused by decreased synthesis of albumin, competition for binding sites by endogenous compounds, or changes in conformation of the binding site) may increase hepatic clearance and thus compensate for reduced hepatic metabolism, although the ability of the diseased liver to compensate is not clear. Increased rather than decreased binding may occur particularly for basic drugs (e.g., lidocaine) as a result of increased production of acute-phase proteins, which may have the effect of decreasing clearance. Changes in protein binding will also affect drug distribution. Protein binding decreases the amount of drug that distributes to peripheral tissues. Drug distribution in the patient with liver disease will be further complicated by the effects of fluid, electrolyte, and acid–base imbalances, which are also likely to occur. For example, an ascitic compartment may lead to overdosing if a patient is not dosed on the basis of lean body weight and the drug does not distribute to the ascitic compartment, which may constitute up to 30% of body weight (Figure 2-6).
Figure 2-6 Disease can affect body composition. The ascitic compartment in an animal with liver or other disease can represent up to 30% or more of body weight. If this compartment is not one to which a drug distributes and the animal is dosed on the basis of total body weight, plasma drug concentrations may be more than 30% higher than expected. If the drug is distributed to the ascitic compartment, prolonged distribution from the compartment back to plasma may result in a longer elimination half-life.
Cardiac Disease
Cardiac disease profoundly affects drug disposition (see Table 2-4). Primary or compensatory disturbances that lead to altered drug disposition include renal sodium and water retention, increased pulmonary and systemic venous pressures, and increased sympathetic nervous system output.6 Sodium and water retention can cause profound changes in drug distribution owing to changes in the sizes of body compartments. In addition, increased sympathetic outflow results in redistribution of blood flow such that the heart and brain receive a higher proportion of blood and thus are exposed to more drug. Because other tissues, particularly skeletal muscle, represent a large volume to which drug is normally distributed, reduced distribution of drug to these tissues results in even higher PDCs in blood going to the heart and brain. Thus the heart and brain are more susceptible to toxicity. CNS and cardiac toxicities to lidocaine and cardiac toxicity to digoxin have been described in some patients with cardiac disease; these toxicities have been attributed to blood redistribution, which accompanies cardiac failure.
KEY POINT 2-8
Cardiac disease can affect every aspect of drug disposition. Variability may be greater as the patient responds or fails to respond to therapy.
As cardiac output decreases, reduced blood flow to the kidney and liver will have profound effects on clearance of drugs through either of these organs. Reduced blood flow reflects, in part, redistribution mediated by sympathetic output. As cardiac disease progresses, however, decreases in blood flow to both the liver and kidneys parallel decreases in cardiac output. The effects of decreased hepatic blood flow on drug elimination depend on the drug; as with liver disease, hepatic clearance of flow-limited drugs may be profoundly decreased. In the kidney, sympathetically mediated intrarenal redistribution of blood from cortical to juxtaglomerular tubules increases the likelihood of tubular reabsorption, which prolongs drug half-life.
Tissue hypoxia and decreased delivery of nutrients to the kidney and liver also contribute to decreased clearance by these organs. The metabolic capacity of the liver is reduced; thus clearance of capacity-limited drugs is impaired. Similarly, renal tubular function is impaired.
Drug absorption may be impaired in the patient with cardiac disease. This is particularly true for parenterally administered drugs. The rate of absorption is more likely to be affected than the extent of absorption; hence peak concentrations may be less, although the extent of drug absorption may not be affected. Redistribution of blood away from skeletal muscle and skin decreases the rate of drug absorption after intramuscular and subcutaneous injections. Autonomic disturbances, consisting of increased sympathetic activity and decreased autonomic tone, tissue hypoxia, and mucosal edema may decrease both the rate and the extent of gastrointestinal absorption. Decreased blood flow to the intestinal villus may decrease absorption of drugs that are normally very rapidly absorbed from the gastrointestinal tract. Finally, the effects of cardiac disease on hepatic clearance of flow-limited drugs and thus on systemic bioavailability of orally administered drugs must be considered.
Several recommendations can be made for administering drugs to the patient with cardiac disease (Box 2-7):
Box 2-7 Examples of Drugs Characterized by Changes in Drug Disposition in Patients with Cardiac Disease
(From Benowitz NL: Effects of cardiac disease on pharmacokinetics: pathophysiologic considerations. In Benet LZ, Massoud N, Gambertoglio JG, editors: Pharmacokinetic basis for drug treatment, New York, 1984, Raven Press, pp 89-104.)
∗ Clearance of digoxin is affected by other drugs used to treat cardiac disease.
Thyroid and Other Diseases
Both hyperthyroidism and hypothyroidism can profoundly affect drug disposition, although the manner is unpredictable.138 The effects thus far involve metabolism. In human patients with hyperthyroidism, the activities of some cytochrome P450 enzymes (e.g., hydroxylation) are increased, whereas those of others (e.g., N-demethylation) are decreased. In rats in which hyperthyroid disease had been induced, enzymes that act as cofactors were increased. Thyroidectomized animals have a general decrease in drug metabolism, although the sequelae are not always predictable. The effects of thyroid disease on drug disposition also depend on sex, with male rats having a general decrease in cytochrome P450 enzymes. The clinical sequelae of changes in drug disposition induced by thyroid disease are not well described in scientific studies, although several examples are provided in the human literature. Digoxin doses necessary to induce a clinical response are in general increased for patients with hyperthyroidism, whereas smaller doses than normal are needed for patients with hypothyroidism. Interestingly, propranolol clearance is decreased in cats with hyperthyroidism.139
Diseases of other body systems can also dramatically alter drug disposition. For example, gastrointestinal disease (e.g., chronic inflammatory bowel disease) alters absorption of orally administered drugs.140 Diseases of any system may alter the response of that system to a drug. Nutrition also can alter drug-metabolizing enzymes. The effects of disease, regardless of the system, on drug absorption, distribution, metabolism, and excretion are very complex, often subtle, and very difficult to predict. Finally, as therapy becomes successful and the clinical signs of disease resolve, the sequelae of disease on drug disposition also resolve. Therapy once again may need to be adjusted as the animal responds.
Pharmacologic Factors
Drug interactions occur whenever the action of one drug is modified by the presence of another, concurrently administered drug. The incidence of interactions increases with the number of drugs included in the preparation and with the duration of treatment.141 As such, the critical care patient may be particularly at risk; indeed, 81% of patients in a small animal intensive care unit were at risk for clinically relevant drug interactions in one study. The risk was greater in dogs with disease of the abdominal cavity and cats with cardiovascular disease.142 Drug interactions should be expected to have a potentially profound impact on animals. In human medicine more than 50% of recent drug withdrawals in the United States have occurred because of serious drug–drug interactions, which underscores the increasing recognition of the role of interactions in adversity.143 The author is aware of several instances of euthanasia in patients resulting from drug interactions that were not recognized until significant pathology had occurred.
The route of administration can influence the type of drug interaction. Drug interactions are not limited to interactions with active drug ingredients but may also involve interactions with additives or excipients. Interactions may occur among drugs, herbs, botanicals, and endogenous nutraceutical preparations and with foods (i.e., drug–diet interactions (Table 2-5). Interactions may also occur with containers in which the drug is stored or through which it is administered and the environment in which the drug is stored. Drug interactions can be categorized according to the phase of drug administration in which they occur: pharmaceutical, pharmacokinetic, or pharmacodynamic.
Table 2-5 Drug-Supplement Interactions
| Drug | Supplement | Side Effect |
|---|---|---|
| Alprazolam | Kava | Enhanced effect |
| Anesthetics, sedatives | St. John’s wort, valerian, kava | Increased CNS effects |
| Antiplatelet/anticoagulants | Garlic, ginger, ginkgo, chamomile, feverfew, bromelain, dang gui | Prolonged bleeding time |
| Antiviral | Garlic (allium) | Decreased effect because of CYP3A4 and P-glycoprotein induction |
| Cardiac stimulants | Ephedra, ginseng | Increased risk of cardiac arrhythmias |
| Chlorpropamide | Garlic (allium) | Hypoglycemia |
| CNS stimulants | Ephedra, yohimbine, guarana, ginseng, caffeine | Increased CNS stimulation |
| Cyclosporine | St. John’s wort | Decreased effect because of CYP3A4 and P-glycoprotein induction |
| Digoxin | St. John’s wort | Decreased effect because of P-glycoprotein induction |
| Diuretics | Senna, licorice | Electrolyte disturbances |
| Hypoglycemics | Ginseng, bilberry, dandelion, garlic, bitter melon | Hypoglycemia |
| Immunosuppressants | Echinacea, astragalus | Antagonizes immunosuppressive effects |
| Loperamide | St. John’s wort | Inhibition of monoamine oxidase (enhanced effect) |
| Oral drugs | Senna | Decreased absorption |
| Propranolol | Piperine | Enhanced effect (CYP inhibition) |
| Selective serotonin reuptake inhibitors | St. John’s wort, SAMe, silamaryn | Increased risk of side effects, serotonergic crisis |
| Theophylline | St. John’s wort | Decreased effect because of CYP3A4 and P-glycoprotein induction |
| Theophylline | Piperine | Enhanced effect (CYP inhibition) |
| Thiazide diuretics | Ginkgo | Decreased clearance (enhanced effect) |
CNS, Central nervous system; SAMe, S-adenosylmethionine.
KEY POINT 2-9
The risk of drug interactions increases with the number of drugs the patient is simultaneously receiving. The negative sequealae of the interactions is likely to be greater when complicated by the presence of physiologic and pathologic factors.
Pharmaceutical Drug Interactions
Pharmaceutical drug interactions occur before the drug is absorbed and may occur before administration (Figure 2-7). Interactions can occur between two drugs or between a drug and a carrier (solvent), a receptacle (including intravenous tubing), or the environment in which it is administered (e.g., gastric environment).144 In human medicine pharmaceutical interactions most frequently result from the addition of drugs to intravenous fluid preparations. In veterinary medicine they are most likely to occur in the critical care environment, where intravenous administration and multiple drug administration are common, or after inappropriate compounding of drugs. Drug incompatibilities can change the chemical or physical nature of a drug (Figure 2-8). Incompatible reactions can reflect degradation caused by changes in pH, binding by drugs with different charges, or other molecular interactions; changes in temperature; or exposure to ultraviolet radiation.145 Incompatibilities may occur both with (approved) finished dosing forms or compounded preparations. A number of sources can be used to minimize the risk of drug interactions involving admixtures of drugs.146-150
Figure 2-7 An example of a drug interaction is exemplified by drug efflux from calcium hydroxyapatite (plaster of Paris) beads. Individually, amikacin or vancomycin is characterized by an initial rapid release, followed by slow release from the beads over several weeks. However, when both drugs are mixed in the beads, efflux of both amikacin and vancomycin is much more rapid, being nearly complete in 1 to several days.
Intravenous Preparations
A number of interactions involve drugs intended for injection (Table 2-6). Drugs that are unstable generally have a short shelf life when in solution. Reconstituted parenteral solutions should always be labeled with the new expiration date and used with strict adherence to the product label instructions after reconstitution. If directed by the label, refrigeration or freezing can prolong the shelf life. It is risky, however, to assume that cold storage will prolong the shelf life of the drug unless efficacy has been documented at the intended conditions. Freezing can increase the degradation (e.g., ampicillin), crystallization (e.g., heparin, dobutamine, furosemide), or precipitation (e.g., insulin) of drugs. Refreezing of a previously frozen and defrosted solution increases the risk of efficacy loss.Often, if specifically queried, the manufacturer of the product can provide specifit information regarding drug stability in conditions beyond those stated on the label. The proper reconstituting fluid should be used to prevent inactivation of drugs. For example, amphotericin B should be diluted only with 5% dextrose because precipitates will otherwise form; whole blood or packed red blood cells should be diluted only with 0.9% saline to prevent damage to infused cells.
Table 2-6 Examples of Drug Interactions in Solution
| Drug or Drug Class | Incompatible Drugs | Other Risks |
|---|---|---|
| Amino acid solutions | Many drugs | |
| Aminoglycosides | Semisynthetic beta-lactams, heparin, many others; check manufacturer’s label | Adsorbs to glass; use plastic for monitoring |
| Aminophylline | Should not be mixed with other drugs | |
| Amphotericin B | Use only 5% dextrose (or manufacturer suggests sterile water). | Light exposure |
| Ampicillin sodium | Selected diluents and drugs; check manufacturer’s label | |
| Atropine sulfate | Bicarbonate, methicillin, promazine, warfarin, others | |
| Beta-lactams | ||
| Cephalosporins | Many drugs, depending on specific antimicrobial; check manufacturer’s label | Check manufacturer’s recommendations regarding stability upon reconstruction. |
| Penicillins | As for cephalosporins; aminoglycosides | |
| Bicarbonate | Many drugs | |
| Buprenorphine | Should not be mixed with dimenhydrinate, pentobarbital | |
| Butorphanol | Should not be mixed with diazepam | |
| Blood, red blood cells | Any intravenous solution except 0.9% saline | |
| Calcium-containing solutions | Many drugs | |
| Calcium disodium EDTA | Should not be mixed with many drugs, including dextrose, metal salts | |
| Chloramphenicol | Many drugs | |
| Carbenicillin disodium | Should not be mixed with many drugs | |
| Cefazolin | Should not be mixed with any other drug | |
| Cephalothin | Should not be mixed with any other drug | |
| Diazepam | Cloudiness when mixed with many other drugs indicates precipitation, which will include drug; potency may be reduced. | Adsorbs to intravenous tubing and plastic containers |
| Protect from light. | ||
| Digitoxin (not digoxin) | Calcium, epinephrine, vitamin B complex | |
| Diphenhydramine | Furosemide, methylprednisolone, pentobarbital | |
| Dobutamine | Alkaline solutions | Check manufacturer’s label regarding discoloration. |
| Doxorubicin | Bicarbonate, heparin, insulin, others | Avoid prolonged contact with aluminum. |
| Doxycycline | Selected drugs, including lidocaine, heparin, isoproterenol, vitamin B complex | |
| Epinephrine | Calcium-containing solutions, ampicillin, other penicillins, pentobarbital, prochlorperazine, others | |
| Erythromycin | Several drugs, including selected cephalosporins, chloramphenicol, heparin, tetracyclines, and vitamin B complex | |
| Flunixin meglumine | Most solutions | |
| Furosemide | Acidic solutions cause hydrolysis; precipitates when combined with many drugs | Yellow discoloration; protect from light |
| Gentamicin | Many drugs, including dopamine, furosemide, heparin (see also beta-lactams, amphotericin B) | |
| Glycopyrrolate | Alkaline solutions | Strongly acidic solution |
| Should not be diluted with saline or bicarbonate for intravenous infusion | ||
| Other drugs | ||
| Heparin | Many drugs | Strongly acidic solution |
| Slightly yellow discoloration is acceptable. | ||
| Hydrocortisone sodium esters | Acid pH causes hydrolysis. | Use proper dilution volume to prevent precipitation. |
| Incompatible with many selected drugs | ||
| Imipenem | Do not freeze. | |
| Insulin | Check package label regarding diluents and refrigeration need and mixing lente insulin kinetics; incompatible with many drugs | Binds to intravenous tubing, selected types of glass, and plastics |
| Iron dextran | Oxytetracycyline, sulfonamides | |
| Kanamycin | See gentamicin, aminoglycosides | |
| Ketamine | Barbiturates, diazepam | |
| Lidocaine | Alkaline solutions | Loss of drug when stored in polyvinyl chloride bags (adsorption to polyvinyl chloride) |
| Magnesium sulfate | Many drugs, including calcium-containing drugs, sodium bicarbonate, tetracyclines, others | |
| Mannitol | Blood, strongly acidic, or alkaline solutions | Crystallization of high (25%) concentrations in glass containers generally can be redistributed by warming; crystallization in plastic solutions is difficult to resolve |
| Methylprednisolone sodium succinate | Normosol-R, Normosol-M, selected drugs | Do not dilute with a volume that is too small; precipitation may occur otherwise. |
| Metronidazole | Reconstituted lyophilized product is very acidic and must be buffered with bicarbonate. Ready-to-use product requires no additional handling. It is light sensitive; however, discoloration induced by light is not accompanied by loss of potency. Do not freeze. |
|
| Metoclopramide | Beta-lactams, erythromycin, sodium bicarbonate | Protect from light. |
| Morphine | Many drugs | |
| Multiple vitamin | Bicarbonate, selected cephalosporins, aminophylline, others | Decreased potency as been documented; complexes 8 hours after dilution. |
| Nitrofurantoin | Many drugs | |
| Oxyglobin | Vitamin K (see phytonadione) | Removal from foil wrap exposes hemoglobin to oxidation, with subsequent formation of methemoglobinemia (indicated by brown discoloration). |
| Oxytetracycline | See tetracycline | |
| Oxytocin | Do not mix with any other drug. | Refrigerate at <25° C; do not freeze. |
| Penicillin G | See also beta-lactams | Rapidly inactivates in pH <6-7 or >8 |
| Polyethylene glycol | ||
| Prochlorperazine, pentobarbital, sulfadiazine | ||
| Others | ||
| Pentobarbital | Acid pH | Prepared as extremely alkaline solution |
| Many drugs | ||
| Phenobarbital | Many drugs, especially acidic solutions | |
| Phenylephrine | Penicillin, pentobarbital, phenobarbital, phenytoin, sodium bicarbonate | |
| Phenytoin | Do not mix with any other drug or intravenous solution. | |
| Phytonadione | Do not mix with ascorbic acid, barbiturates, phenytoin. | |
| Potassium chloride | Do not mix with amphotericin B. | |
| Pralidoxime chloride (2PAM) | Reconstitute with sterile water only; do not mix with any other drug. | |
| Procaine | Many solutions, especially alkaline | |
| Procainamide | Dextrose | Light yellow (but not amber) discoloration acceptable |
| Prochlorperazine | Many drugs; do not mix in same syringe | |
| Promazine | Many drugs | |
| Promethazine | Selected drugs | |
| Protein hydrolysate | Many drugs | |
| Propofol | Do not mix with any other drugs. | |
| Propranolol | Rapidly decomposes in alkaline solution | |
| Ringer’s lactate | Alcohol in 5% dextrose, epinephrine, oxytetracycline, sodium bicarbonate, sulfadiazine | |
| Sodium bicarbonate | Many drugs; check package insert | |
| Sodium iodide | Several drugs | |
| Sulfonamides, sodium salts | Many drugs | |
| Tetracycline | Highly acidic solution may render this incompatible with many drugs. | Dark solution indicates decomposition; discoloration in multiple electrolyte solution does not indicate potency loss. |
| Thiopental | Many drugs | |
| Vancomycin | Selected drugs | |
| Vitamin B complex | Magnesium sulfate, erythromycin, selected others | |
| Warfarin | Many drugs |
Changing the pH of a solution by improperly diluting it or mixing it with another drug can be risky. The release of some insulins is pH dependent. Diluting insulin with a solution other than that provided by the manufacturer may change the pH and thus the rate of insulin release. The pH of a solution may be needed to keep the active drug dissolved or stable; changing the pH may result in precipitation or loss of stability. For example, acid-labile drugs (e.g., penicillins) can be destroyed in a low-pH solution. Drugs prepared as an acid salt (e.g., lidocaine hydrogen chloride) or in acidic solutions (i.e., sodium heparin) should not be combined with alkaline solutions (i.e., sodium bicarbonate).
Drugs can bind to and inactivate one another, often because of ionic attractions. Calcium in solutions causes precipitation when combined with solutions containing carbonates (e.g., sodium bicarbonate). Heparin is incompatible with several drugs, such as aminoglycoside and beta-lactam antibiotics. Thus saline rather than heparin should be used whenever possible to maintain patency of catheters through which drugs are administered. When present in sufficiently high concentrations, penicillins inactivate aminoglycosides. In fact, ticarcillin can be used therapeutically to bind gentamicin in cases of overdose. Although plasma concentrations after therapeutic dosing of either drug probably do not achieve concentrations necessary to inactivate aminoglycosides, in the critical care situation, and as a once-daily dosing regimen of aminoglycosides becomes more generally acceptable, the risk of aminoglycoside (or a fluorinated quinolone) antimicrobial inactivation by a penicillin may become greater.
Often, a pharmaceutical drug interaction involving intravenous solutions can be detected by a visual change in the appearance of the drugs. Discoloration, cloudiness, and formation of precipitate generally are indications of an interaction, and with some exceptions use of the drug should be reconsidered. Not all interactions will result in a physical change of the appearance, however. Likewise, the change in physical appearance of a drug combination does not necessarily indicate that the activity of the drug has been changed. For example, diazepam has been mixed with other preanesthetics with no observable change in drug efficacy, despite a cloudy discoloration. Whereas a pink discoloration of dopamine indicates inactivation, discoloration of dobutamine does not preclude efficacy if the drug is used within 24 hours. Slight yellow discoloration of procainamide is acceptable; dark discoloration indicates a loss of efficacy.
Several drugs can bind to receptacles. For example, lipid-soluble drugs (e.g., diazepam) can bind to plastic containers; insulin binds to selected glasses and to many plastics, including polyethylene and polyvinyl; aminoglycosides bind to glass. Binding to catheters and intravenous lines can be minimized by flushing each new system with a sufficient volume of solution (50 mL) before drug administration. Drugs packaged in brown bottles (e.g., diazepam, furosemide) are somewhat protected as such from ultraviolet light, and protection should be continued if they are transferred to another vial.
Oral Preparations
Oral preparations may involve drug–diet or drug–drug interactions (see Box 2-7 and Tables 2-5 and 2-6). Many drugs bind luminal contents (drug–diet interactions; Table 2-7), and oral absorption is impaired.151 Food can alter splanchnic blood flow, gastric motility (and thus mixing and drug dissolution as well as gastric emptying), and gastric secretions. Changes in gastric secretions can alter gastric pH, which can change the percentage of ionized and thus diffusible drug. The net effect of food on drug absorption depends on the pKa of the drug, whether the drug is labile to the effects of pH and enzymes, and the site of absorption of the drug (i.e., stomach versus intestine).151 The sequelae may affect either the rate or extent of drug absorption. The effect of food on the rate of drug absorption is most important for drugs with a narrow therapeutic window and for drugs with a steep dose–response curve, for which a small change in PDC can cause profound differences in response to the drug. Predicting drug–diet interactions on the basis of chemical structure should be done cautiously. Whereas food will impair the oral absorption of most tetracyclines, doxycycline is minimally affected. Food reduces the absorption of ampicillin but not amoxicillin;152 food prolongs the time to peak plasma concentrations of cefadroxil (and increases time above the minimum inhibitory concentration) but has no impact on the absorption of cephalexin.153 Oral absorption of selected drugs is enhanced rather than decreased in the presence of food (see Table 2-2). Interactions between drugs and foods were recently well reviewed using a body-systems approach.154
Table 2-7 Examples of Sites of Drug–Drug Interactions
| Interaction | Example Drugs |
|---|---|
| Binding to luminal contents | Tetracyclines (except doxycycline), fluorinated quinolones, antacids, sucralfate, |
| Absorption | |
| Gastric motility | Metoclopramide, anticholinergics, erythromycin |
| Gastric pH | Antisecretory drugs, antacids |
| Competition for transport proteins | See Table 2-2 |
| Inhibition or induction of drug metabolizing enzymes | See Table 2-7 |
| Increased splanchnic blood flow (nutrients) | |
| Distribution | |
| Competition for transport proteins in circulation | See Table 2-2 |
| Competition for transport proteins in tissues | See Table 2-2 |
| Altered regional blood flow | Fluid therapy, ACE inhibitors, other cardioactive drugs, phenothiazines, |
| Metabolism | |
| Competition for transport proteins | See Table 2-2 |
| Induction of phase I enzymes (CYP 450) | See Table 2-8 |
| Inhibition of phase I enzymes (CYP 450) | See Table 2-8 |
| Increased phase II enzymes (glutathione) | See Table 2-8 |
| Excretion | |
| Competition for transport proteins | See Box 2-8 |
| Altered renal blood flow | Cardioactive drugs, NSAIDs, amphotericin B, aminoglycosides |
| Altered urinary pH | See Box 2-9 |
NSAIDs, Nonsteroidal antiinflammatory drugs.
Drug–drug interactions (see Table 2-7) in the lumen include, but are not limited to, changes in the diffusibility, dissolution rate, and particle size of orally administered drugs. Drug–drug interactions in the gastrointestinal tract can inactivate or prevent absorption of drugs. Sucralfate, cimetidine, aluminum hydroxide, and attapulgite (previously Kaopectate®) are examples of drugs that bind to and prevent the absorption of many drugs. Other drugs alter the rate of absorption by altering gastric motility (see Table 2-4; see also the discussion of pharmacokinetic interactions later in this chapter). To minimize these effects, none of these drugs should be given simultaneously with another orally administered drug.
Topical Preparations
Pharmaceutical interactions in topical preparations may occur between drugs or between drugs and the vehicle in which they are carried.155,156 Both the rate and extent of drug absorption can be affected adversely. For example, macromolecular additives may bind chemically with the active drug. Methyl, ethyl, hydroxyethyl, and carboxymethyl cellulose frequently form complexes with drugs that can lead to drug precipitation. Vehicles are often selected because of their effect on drug absorption. For example, retardant vehicles (e.g., polyethylene glycol 300) interact with the drug, decreasing its absorption, whereas dimethyl sulfoxide (DMSO) is well recognized for its ability to enhance absorption of many topically applied drugs. The pluronic-lecithin vehicle of PLO gels (PLO) designed for transdermal drug delivery is thermoreversible, meaning they are semisolid at room temperature but become liquefied when refrigerated (see Figure 2-8).
The concentration of drug available for skin penetration depends on its dissolution in the vehicle. Drugs must generally dissolve in an aqueous layer of fluids that collects under the ointment base before percutaneous absorption can occur. Few drugs (and particularly water-soluble drugs) are sufficiently soluble to be dissolved in petrolatum bases; most drugs mixed in such bases are present as particles. Because few particles are located at the vehicle–skin interface, and dissolution is very slow for the particles, drugs in such preparations are likely to be ineffective. As such, lipid-soluble drugs tend to be better absorbed, particularly when prepared in lipid vehicles. Because only dissolved drug can move, the solubility of a drug in the vehicle is an important determinant of drug movement into the skin. However, a drug that has too great an affinity for the vehicle also may not be well absorbed simply because it will not leave the vehicle.156
Compounded Preparations
Many U.S. pharmacies now cater specifically to veterinarians and thus are prepared to address special problems in drug therapy through compounding. As important as compounding can be to the safe and effective administration of drugs to animals, compounded products present more risks of therapeutic failure than encountered with approved drugs.157,158 Among the risks are the potential failure of the drug at the pharmaceutical phase because of interactions among the active or inactive ingredients. These risks are addressed in more depth in Chapter 4.
Pharmacokinetic Drug Interactions
Drug interactions that occur inside the body may lead to life-threatening adversities because of either therapeutic failure or increased side effects. Pharmacokinetic interactions occur when one drug alters the disposition of another drug.159 Each stage of disposition of a drug—absorption, distribution, metabolism, or elimination—can be altered by another drug. The majority of drug interactions leading to serious adverse events in humans reflect pharmacokinetic interactions, involving either transport proteins or drug-metabolizing enzymes.143 Identifying the role of drug interactions in causing adverse effects is difficult; large numbers of patients generally must be studied to identify even common interactions. Predictions of drug–diet or drug–drug interactions might be facilitated by evaluation of chemistry, but caution is necessary.154 Models that predict drug interactions are complicated by complex interactions (e.g., species, disease, and multiple drug interactions).160 Identification of drug interactions involving dietary supplements is handicapped by the lack of a mandated surveillance mechanism; information in human medicine generally is obtained through reviews, case reports, or case series or based on open, uncontrolled studies.161 Evidence-based information in veterinary medicine is even further limited. Several human-medicine reviews have described interactions between drugs and botanicals or herbs161 and foods.154 Mechanisms are often difficult to identify, in part because of broad substrate specificity. Some examples of drug–drug or drug–supplement interactions follow.
Absorption
Absorption of one drug may be hindered as a result of changes in the drug’s passage through biological phases and changes in local pH; the integrity of biological membranes; regional blood flow; and, in the case of orally administered drugs, gastrointestinal motility. Each of these changes can be induced by a concurrently administered drug. Examples include the impact of an antisecretory drug on the ratio of un-ionized (diffusible) to ionized (nondiffusible) drugs. For weakly acidic drugs, a decrease in the ratio may decrease oral absorption; for weakly basic drugs, the opposite may occur. Sucralfate and cimetidine are examples of drugs that bind to and prevent the absorption of other drugs. Likewise, tetracycline and enrofloxacin are bound by divalent or trivalent cations that might be found in antacids (see Table 2-4). Finally, drugs that alter gastric motility might alter the rate of oral drug absorption. Most drugs are absorbed from the small intestine. Administration of anticholinergics decreases gastric emptying, allowing a longer time to elapse before a drug moves to the small intestine. Although extent of absorption may not be affected, peak PDCs may be lower. Metoclopramide probably has an opposite effect. In contrast to gastric motility, increasing motility of the small intestine is unlikely to alter the oral absorption of drugs because the surface area is so large that it is difficult to manipulate. However, a few drugs alter drug absorption by causing malabsorption or changing gastric blood flow (see Table 2-4). Phenytoin and phenobarbital have been associated with decreased oral absorption of vitamins.
KEY POINT 2-10
The role of drug induction and inhibition on drug transporter proteins such as P-glycoprotein is only now being delineated, but can be particularly profound for drug absorption and distribution.
Drug interactions that have an impact on absorption may also reflect altered metabolism and/or uptake in the enterocyte or liver. Drugs entering enterocytes are subject to metabolism by CYP3A, efflux mediated by P-gp, or passage into portal system and exposure to metabolic activities of the liver. Cytochrome P450 3A is the most predominant drug-metabolizing enzyme in the intestinal cell and mediates biotransformation of more than half of all drugs currently available (for humans).162 Transporters such as the multidrug export pump P-gp, facilitate either drug absorption or efflux from the enterocyte, and drugs may act as substrates or inhibit or compete at these proteins (see Table 2-2). Both proteins (CYP 3A and P-gp) share drug substrates, and interplay between metabolic enzymes and transporters appears to confound the disposition of many orally administered drugs (Table 2-8; see also Table 2-2). Poor oral bioavailability may reflect a coordinated action of intestinal drug-metabolizing enzymes and efflux transporters. Experimentally, drug efflux by intestinal P-gp is known to prevent absorption and thus decrease the bioavailability of many CYP3A4 substrates. The interaction between P-gp and CYP3A4 at the apical intestinal membrane increases drug metabolism of those drugs that are absorbed.163 In contrast, selected drug interactions also involve inhibition of the P-gp, resulting in increased oral bioavailability. Food–drug interactions most commonly occur in the gastrointestinal tract154 and reflect therapeutic failure owing to reduced bioavailability of the drug. Less commonly, increased drug concentrations reflect altered appetite or gastrointestinal acidity. Those most commonly identified drug–diet (nutrient or supplement) interactions involve altered drug pharmacokinetics caused by interference with either P-gp or xenobiotic metabolizing enzymes (see the section on metabolism). Of the potential drug–nutrient interactions affecting absorption, competition among substrates for transport proteins have probably been the best described.143,164 For example, flavenoids (found in grapefruits) are inhibitors of several P-gp substrates, which increases the risk of diet–diet or diet–drug interactions during both absorption and distribution.164
Table 2-8 Members of Cytochrome P450 Superfamilies and Example Substrates, Inducers, and Inhibitors
| Substrate | Inducer | Inhibitor |
|---|---|---|
| CYP1A2 | ||
| Acetaminophen | Insulin | Amiodarone |
| Amitriptyline | Omeprazole | Cimetidine |
| Clomipramine | Tobacco | Fluoroquinolones |
| Clozapine | Interferon | |
| Imipramine | Ticlopidine | |
| Naproxen | ||
| Ondansetron | ||
| Propranolol | ||
| Theophylline | ||
| Verapamine | ||
| Warfarin | ||
| Zileuton | ||
| CYP2B6 | ||
| Cyclophosphamide | Phenobarbital | Ticlopidine |
| Rifampin | ||
| CYP2C19 | ||
| Cyclophosphamide | Carbamazepine | Cimetidine |
| Lansoprazole | Prednisone? | Felbamate |
| Omeprazole | Rifampin? | Fluoxetine |
| Diazepam | Indomethacin | |
| Phenytoin | Ketoconazole | |
| Phenobarbital | Lansoprazole | |
| Amitriptyline | Omeprazole | |
| Clomipramine | Paroxetine | |
| Cyclophosphamide | Probenecid | |
| Imipramine | Ticlopidine | |
| Indomethacin | Topiramate | |
| Primidone | ||
| Progesterone | ||
| Propranolol | ||
| Warfarin | ||
| CYP2C9 | ||
| NSAIDS | Rifampin | Amiodarone |
| Ibuprofen | Fluconazole | |
| Diclofenac | Isoniazid | |
| Meloxicam | Lovastatin | |
| Naproxen | Paroxetine | |
| Piroxicam | Phenylbutazone | |
| Celecoxib | ||
| Tolbutamine | Probenecid | |
| Glipizide | Sulfamethoxazole | |
| Losartan | Trimethoprim | |
| Amitriptyline | Zafirlukast | |
| Fluoxetine | ||
| Phenytoin | ||
| Warfarin | ||
| CYP2D6 | ||
| Carvedilol | Dexamethasone | Amiodarone |
| Metaprolol | Rifampin | Celecoxib |
| Timolol | Chlorpromazine | |
| Amitriptyline | Chlorpheniramine | |
| Clomipramine | Cimetidine | |
| Paroxetine | Clomipramine | |
| Chlorpheniramine | Cocaine | |
| Chlorpromazine | Doxorubicin | |
| Codeine | Fluoxetine | |
| Encainide | Metoclopramide | |
| Fluoxetine | Proxetin | |
| Flecaine | Quinidine | |
| Lidocaine | Ranitidine | |
| Metoclopramide | Terbinafine | |
| Nortriptyline | ||
| Ondansetron | ||
| Propranolol | ||
| Tramadol | ||
| CYP2E1 | ||
| Enflurane | Ethanol | |
| Halothane | Isoniazid | |
| Isoflurane | ||
| Methoxyflurane | ||
| Sevoflurane | ||
| Ethanol | ||
| Theophylline | ||
| CYP3A4,5,7 | ||
| Clarithromycin | Barbiturates | Amiodarone |
| Erythromycin | Carbamazepine | Cimetidine |
| (not azithromycin) | Glucocorticoids | Ciprofloxacin |
| Quinidine | Phenobarbital | Clarithromycin |
| Alprazolam | Phenytoin | Diltiazem |
| Diazepam | Rifampin | Erythromycin |
| Midazolam | St. John’s wort | Fluconazole |
| Cyclosporine | Troglitazone | Itraconazole |
| Tacrilium | Pioglitazone | Ketoconazole |
| Cisapride | Norfloxacin | |
| Chlorpheniramine | Verapamil | |
| Amlodipine | ||
| Diltiazem | ||
| Felodipine | ||
| Nifedipine | ||
| Verapamil | ||
| Lovastatin | ||
| Hydrocortisone | ||
| Progesterone | ||
| Testosterone | ||
| Buspirone | ||
| Cocaine | ||
| Dapsone | ||
| Codeine | ||
| Dextromethorphan | ||
| Finasteride | ||
| Fentanyl | ||
| Ondansetron | ||
| Lidocaine | ||
| Propranolol | ||
| Quinine | ||
| Terfenadine | ||
| Vincristine | ||
NSAIDs, Nonsteroidal antiinflammatory drugs.
Distribution
Pharmacokinetic drug interactions that alter drug distribution from the central compartment to peripheral tissues usually result from competition for protein-binding sites between two or more concurrently administered drugs. Because protein binding is reversible, the drug with the highest affinity for protein (usually albumin) displaces the drug with less affinity (Box 2-8). If a highly (>80%) protein-bound drug is displaced by only a small fraction, the amount of unbound, pharmacologically active drug markedly increases, and the risk of toxicity is initially increased. Because NSAIDs are generally more than 90% protein bound, even slight displacement of the drug from its binding sites can initially result in concentrations that might increase the risk of adverse effects. Increased hepatic or renal clearance of the unbound drug tend to balance displacement of bound to unbound drug such that PDCs of free unbound drug only minimally increase (see Chapter 1).165 However, according to its package insert, co-administration of cefovecin with other highly protein bound drugs increases the concentration of the latter. It is not clear whether increased clearance of unbound drug will be sufficient in the presence of hepatic (or renal) disease. Most drug interactions involving protein binding reflect competition for albumin-binding sites because albumin is the most common binding protein, particularly for weak acids. Lipoproteins; globulins (increased with acute-phase protein increase); and, to a lesser extent, albumin bind weak bases (e.g., bupivacaine, lidocaine) (see Box 2-8).
Use of drugs that alter drug distribution to peripheral organs can alter drug delivery to the organs. For example, the use of afterload reducers may increase renal blood flow; renal clearance of drugs may also increase. Use of glucocorticoids before volume replacement in a hypovolemic patient may enhance peripheral vasoconstriction, reducing tissue distribution of other drugs. Rarely, drug interactions occur at the tissue site. For example, drugs can compete with each other at tissue-binding sites. Quinidine increases digoxin toxicity because it displaces digoxin from cardiac tissues; in contrast, hypokalemia facilitates binding of digoxin to cardiac tissue, thus enhancing digoxin cardiotoxicity.
Because P-gp is located on the cell membrane of many organs, this transporting protein can influence tissue distribution of drug. The most notable example is its presence in the blood–brain barrier, where it serves to efflux many drugs that are able to penetrate the barrier. Genetic deficiency of this protein has been associated with an increased risk of CNS toxicity in certain breeds its role in drug interactions should be anticipated (see Chapter 4 and later discussion). Drug–nutrient interactions resulting in competition for transport proteins may also affect distribution of drugs.143,164
Metabolism
Pharmacokinetic drug interactions frequently alter the metabolism of a concurrently administered drug (see Table 2-4).166-170 When administering a drug metabolized by the liver, it is wise to anticipate a drug interaction if a second drug also metabolized by the liver is added to therapy. Most of the interactions result from modulation of hepatic (phase I) drug-metabolizing enzymes (see Table 2-8).
KEY POINT 2-11
Interactions at the level of drug-metabolizing enzymes are among the most dangerous, yet their predictability is complicated by the large number of enzymes, the lack of substrate specificities, and polymorphisms among animals.
Induction of drug-metabolizing enzymes should be considered as a protective mechanism that facilitates excretion of potentially toxic compounds. However, induction is a double-edged sword: although the elimination of a potentially toxic drug increases, so does potential formation of toxic or carcinogenic metabolites. Most CYP enzymes are inducible, with response to inducers varying within and among species, age, and gender. Human CYP known to be influenced by inducers include CYP 1A1/2, 2A6, 2C9, 2C19, 2E1, and 3A4 (see Table 2-8). Inducers generally act as substrates for the induced enzymes, and induction generally is dose dependent.171 Inducers often induce more than one CYP and may significantly increase the activity of an enzyme that otherwise (constitutively) is either absent or present only in very low concentrations. Induction is accompanied by an increase in the transcription and thus intracellular concentration of the induced enzyme. Maximal induction of transcription generally requires 10 to 12 hours of exposure to a drug. Although transcription may return to baseline 18 to 24 hours (depending on dose) after the inducer is discontinued, the impact of the inducer may persist. The duration of the effect varies with the rate of enzyme degradation, which normally results in half-lives that range from 8 to 30 hours. Although not common, induction also may reflect a decrease in the rate of CYP degradation—that is, a prolongation of enzyme half-life.172
Barbiturates are recognized inducers of CYP; indeed, the observation that “tolerance” to hypnotics developed in dogs chronically exposed to barbiturates led to the recognition of the phenomenon of induction.172 Phenobarbital is one of the most potent microsomal enzyme inducers known and can enhance the hepatotoxicity of other hepatotoxic drugs. Likewise, it increases the formation of and response to prodrugs and decreases the effects of itself and other drugs metabolized by the liver as clearance of these drugs is increased.73,173,141 The CYP2B9 family, responsible for metabolism of a large number of drugs in rodents, is induced by phenobarbital. Therapeutic doses of phenobarbital have been associated with induction of CYPIA activity, as well as alpha-glycoproteins in dogs.174 The clinical impact of this effect is demonstrated in epileptic dogs treated with phenobarbital: initial concentrations may decrease within several months of therapy despite no dose change. Interestingly, oral pentobarbital also increases the amount of CYP2C present in the intestinal tract of dogs when administered at doses consistent with that consumed by dogs fed commercial dog foods prepared from animals euthanized by pentobarbital (10 to 60 μg/gm). The impact on the metabolism of selected subtrates varies and is greatest for higher doses, but changes are also present at low doses, suggesting a potential clinical impact on therapeutic drugs.171
Drug interactions that reflect inhibition of drug-metabolizing enzymes may be at greater risk in causing serious adverse events. A number of inhibitors of CYP enzymes have been identified. As with inducers, the extent of inhibition is dose dependent,172,175 with inhibitors often acting as substrates at the inhibited enzyme, and either characterized by broad enzyme interaction, being inhibitory for a number of different CYPs, or acting selectively for a single enzyme (see Table 2-8).172 Currently, three types of drug-metabolizing enzyme inhibitors have been described: reversible, quasi-reversible, and irreversible, with reversible inhibitors being the most commonly involved in drug–drug interactions. Reversible inhibition is transient, resolving when therapy with the inhibitor is discontinued. Like induction, reversible inhibition appears to be dose dependent.175 Reversible inhibition most commonly reflects interactions that are competitive or noncompetitive. Competitive inhibition is exemplified by a drug that blocks access to the catalytic site of the enzyme of another structurally similar drug. The drug may or may not be a substrate for the enzyme. Noncompetitive inhibition occurs when substrate binding occurs at a different site but nonetheless changes the catalytic activity of the enzyme such that it is inactivated.172
Generally, clearance of a concurrently administered drug metabolized by the liver is prolonged in the presence of inhibitors, increasing the potential for toxicity or for an exaggerated pharmacologic response. Additionally, prodrugs (e.g., enalapril, primidone) are less likely to be activated. Chloramphenicol, cimetidine, and imidazole antifungal drugs are examples of potent microsomal enzyme inhibitors.73,173,141 Co-administration with potentially toxic drugs that are also metabolized by the liver should be done cautiously. Fluorinated quinolones such as enrofloxacin and marbofloxacin can increase theophylline plasma concentrations to toxic levels, presumably because of impaired hepatic clearance of theophylline.176 Ketoconazole inhibits cytochrome P450 enzymes, including CYP3A4 in the dog.177 However, it does not necessarily clinically affect all drugs metabolized by the liver. For example, morphine clearance was not changed in Greyhounds treated with an average of 12.7 mg/kg ketaconazole per day.93 Although area under the curve was greater in the presence of morphine, the change was neither statistically nor clinically significant. This may reflect, however, the flow-limited nature of morphine clearance or, potentially, breed differences in drug metabolism. Other imidazoles (fluconazole and, to a lesser degree, itraconazole) also have broad enzyme inhibitory activity (see chapter 9).
Drug-induced inhibition of drug metabolism can be used for therapeutic benefit. Both cyclosporine and ketaconazole are substrates and inhibitors of both P-gp and CYP3A. As such, the combined use of the drugs may result in marked prolongation of the half-life of either drug. Ketoconazole has been used to increase blood drug concentrations of cyclosporine (owing to both inhibition of drug-metabolizing enzymes and competition with P-gp) in dogs.178 A twofold increase in expected PDCs may occur, as indicated by a 50% dose reduction in Beagles179 or a twofold increase in Cmax in Greyhounds.93 Indeed, the impact may be greater in some dogs: our laboratory has documented an elimination half-life of over 150 hours in dogs simultaneously receiving ketoconazole, yielding concentrations that exceed 4500 ng/mL. In cats, cyclosporine (4 mg/kg orally) concentrations can be expected to increase approximately twofold when administered with ketoconazole (10 mg/kg).180 The macrolides also have broad inhibitory effects. The inhibitory effect of erythromycin may reflect inhibition of CYP3A macrolides, which appear to inhibit cyclosporine clearance. Again, in our laboratory azithromycin co-administration was associated with marked prolongation of cyclosporine half-life in a cat, leading to cyclosporine concentrations that exceeded 4500 ng/mL (therapeutic range 800-1400 ng/mL). The effect of cimetidine, another drug-metabolizing inhibitor with broad substrate specificity, is variable, ranging from no significant effect in Beagles (n=10; 15 mg/kg cimetidine every 8 hours for 8 days and 5 mg/kg once daily for cyclosporine) to 50% decrease in clearance in humans.181 Cimetidine-induced enzyme inhibition, however, has been used to prevent metabolism of acetaminophen in humans and cats into potentially lethal toxic metabolites.182,183 Cilastatin inhibits renal tubular drug metabolism of imipenem; the net effect may prolong the half-life of imipenem, but hepatotoxicity or renal toxicity resulting from metabolites might also be reduced (see Chapter 4). Nutrition, sex, age, and other factors can influence the way drug-metabolizing enzymes respond to drugs. Alcohol and 4-methylpyrazole competitively inhibit alcohol dehydrogenase, the drug-metabolizing enzyme that converts ethylene glycol to its lethal metabolite.
Although less common, drug clearance may also be affected by drugs that change hepatic blood flow, solely due to substrate delivery, not a limitation on metabolic rate. This interaction is significant, however, only for drugs that are characterized by extensive and rapid hepatic clearance (e.g., propanol, lidocaine) and probably is not clinically relevant.
Among the most commonly identified drug–diet (nutrient or supplement) interactions that alter drug pharmacokinetics are those reflecting drug metabolism, particularly CYP3A4.154 Several dietary supplements are known to interact with drugs.161 These include, but are not limited to, St. John’s wort (induction of CYP3A4, particularly intestinal), echinacea (induction and inhibition of intestinal CYP), ginkgo biloba (induction of CYP219A), and grapefruit (inhibition of CYP3A4 and inhibition of P-gp).
Excretion
Pharmacokinetic drug interactions may alter urinary excretion because of changes in glomerular filtration, competition between the drug for active tubular secretion, or both (Box 2-9). Competition for carrier proteins responsible for active tubular secretion usually involves acidic drugs. Probenecid is still occasionally used to prolong the elimination of an expensive penicillin because it competes with the penicillin for a carrier protein. Renal excretion may also be affected by drugs that alter urinary pH and tubular reabsorption. Changes in urinary pH conducive to formation of a greater proportion of un-ionized drug (e.g., an acidic urinary pH and an acidic drug) encourage tubular reabsorption of a drug, thus decreasing its clearance and prolonging its elimination half-life (Box 2-10).73,173,141 For example, overdosing of some drugs (e.g., aminoglycoside or strychnine poisoning) can be treated by hastening elimination with urinary acidifiers.
Pharmacodynamic Drug Interactions
Pharmacodynamic drug interactions occur when one drug alters the chemical or physiologic response to another drug (see Table 2-6). Pharmacodynamic interactions may increase response to a drug in an additive or synergistic fashion at the same receptor (e.g., the permissive effect of glucocorticoids on alpha-adrenergic receptors; phenobarbital and clorazepate at gamma-aminobutyric receptors), at an intracellular site (e.g., epinephrine and theophylline in bronchial smooth muscle), or at different sites but with the same physiologic reaction (e.g., hypokalemia induced by cardiac glycosides and diuretics; many interactions of antimicrobials). Pharmacodynamic interactions may also decrease the response of some drugs owing to competitive antagonism at the same receptor site (e.g., atropine and anticholinesterases or atropine and metoclopramide) or antagonistic responses mediated at distant but physiologically related sites. Antagonistic pharmacodynamic interactions have been used therapeutically: oxymorphone or other mu agonistic effects are reversed with naloxone, xylazine and other chemical sedatives are reversed with tolazoline or yohimbine, and medatomidine is reversed with antipamazole.
The most familiar pharmacodynamic interactions are probably those that act in an additive or synergistic manner to augment response to a drug. Augmentation can occur through different mechanisms of action (i.e., controlling vomiting by combining a drug active at the chemoreceptor triggering zone with a drug that acts peripherally, controlling seizures by combining phenobarbital with bromide, controlling tachycardia by combining diltiazem with digoxin or atenolol). Less commonly, augmentation may occur through similar actions at a receptor site; more often, drugs will compete with one another at the same receptor, thus resulting in antagonism. Unfortunately, often forgotten is the fact that augmentation of the desired pharmacologic response may be accompanied by augmentation of an undesirable adverse drug event (ADE). For example, drugs that impair renal prostaglandin synthesis (e.g., NSAIDs, angiotensin-converting enzyme inhibitors, aminoglycosides) should be used in combination cautiously because their combination increases the risk of renal failure. Likewise, ulcerogenic drugs (NSAIDs, glucocorticoids) enhance the risk of gastrointestinal ulceration when used in combination.
Pharmacodynamic interactions may decrease the response to a drug because of competitive antagonism at the same receptor site. Most commonly, these actions are desirable and are frequently the target of combined drug therapy: atropine to treat organophosphate toxicity, reversal agents for opioids and anesthetic agents. Antagonistic pharmacodynamic responses can also occur through different receptor sites or different mechanisms. The combination of a bacteriostatic antimicrobial (one that slows the growth of an organism) with a bactericidal antimicrobial (one whose efficacy depends on rapid growth) might be considered as an example. Also, the prokinetic effects of cisapride and metoclopramide are prevented by anticholinergics; calcium-containing solutions should not be combined with blood or blood components because the loss of anticoagulant effects increases the risk of microthrombi formation in the transfused blood.
Hu161 has reviewed pharmacodynamic interactions between drugs and herbs or botanicals. These can be clinically significant, as is exemplified for selected organosulfur components of garlic, which act as anticoagulants. Increased bleeding times have been documented in patients taking garlic supplements and subsequently treated with warfarin compared with increases with warfarin alone.
1. Massoud N. Pharmacokinetic considerations in geriatric patients. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:269-282.
2. Ritschel W.A. Gerontokinetics: the pharmacokinetics of drugs in the elderly. Caldwell, NJ: Telford Press; 1988. pp 1-16
3. Feely J., Coakley D. Altered pharmacodynamics in the elderly. Clin Pharm. 1990;6:269-283.
4. Dowling P. Geriatric pharmacology. Vet Clin North Am Small Anim Pract. 2005;35(3):557-569.
5. National Research Council. Nutrient requirements of dogs and cats. Washington, DC: National Academies Press; 2006.
6. Benowitz N.L. Effects of cardiac disease on pharmacokinetics: pathophysiologic considerations. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:89-104.
7. Aucoin D.P. Drug therapy in the geriatric animal: the effect of aging on drug disposition. Vet Clin North Am Small Anim Pract. 1989;19:41-48.
8. Morita T.Y., Sawada M.M., A., Shimada A. Immunohistochemical and ultrastructural findings related to the blood–brain barrier in the blood vessels of the cerebral white matter in aged dogs. J Comp Path. 2005;133:14-22.
9. Perez-Camargo G. Cat nutrition: what is new in the old? Compend Cont Educ Pract Vet. 2004;26(2A):5-14.
10. Benno Y., Nakao H., Uchida K., et al. Impact of the advances in age on the gastrointestinal microflora of beagle dogs. J Vet Med Sci. 1992;54:703-706.
11. Patil A.R., Czarnecki-Maulden G.L., Dowling K.E. Effect of advances in age on fecal microflora of cats. FASEB J. 2000;14(4):A488.
12. Hickman M.A., Bruss M.L., Morris J.G., et al. Dietary-protein source (soybean vs casein) and taurine status affect kinetics of the enterohepatic circulation of taurocholic acid in cats. J Nutr. 1992;122:1019-1028.
13. Sheaker S., Bay M. Drug disposition and hepatotoxicity in the elderly. J Clin Gastroenterol. 1994;18:232-237.
14. Enck R.E. Pain control in the ambulatory elderly. Geriatrics. 1991;46:49-60.
15. Workaman B.S., Ciccone V., Christophidis N. Pain management for the elderly. Aust Fam Phys. 1989;18:1515-1527.
16. Lawler D.E., Evans R.H., Larson B.T., et al. Influence of lifetime food restriction on causes, time, and predictors of death in dogs. J Am Vet Med Assoc. 2005;226:225-231.
17. Speakman J.R., van Acker A., Harper E.J. Age-related changes in the metabolism and body composition of three dog breeds and their relationship to life expectancy. Aging Cell. 2003;2:265-275.
18. Schultz R.D. The effects of aging on the immune system. Compend Contin Educ Pract Vet. 1984;6(12):1096-1105.
19. Robinson E.P. Anesthesia of pediatric patients. Compend Contin Educ Pract Vet. 1983;5(12):1004-1011.
20. Green T.P., Mirkin B.L. Clinical pharmacokinetics: pediatric considerations. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:269-282.
21. Boothe D.M., Tannert K. Special considerations for drug and fluid therapy in the pediatric patient. Compend Contin Educ Pract Vet. 1991;14:313-329.
22. Harper E.J., Turner C.L. Age-related changes in apparent digestibility in growing kittens. Reprod Nutr Dev. 2000;40:249-260.
23. Heimann G. Enteral absorption and bioavailability in children in relation to age. Eur J Clin Pharmacol. 1980;18:43-50.
24. Rane A., Wilson J.T. Clinical pharmacokinetics in infants and children. In: Gibaldi M., Prescott L., editors. Handbook of clinical pharmacokinetics. New York: ADIS Health Science Press; 1983:142-168.
25. Gillette D.D., Filkins M. Factors affecting antibody transfer in the newborn puppy. Am J Physiol. 1966;210(2):419-422.
26. Morselli P.L., Morselli R.F., Bossi L. Clinical pharmacokinetics in newborns and infants: age-related differences and therapeutic implications. In: Gibaldi M., Prescott L., editors. Handbook of clinical pharmacokinetics. New York: ADIS Health Science Press; 1983:99-141.
27. Jones R.L. Special considerations for appropriate antimicrobial therapy in neonates. Vet Clin North Am Small Anim Pract. 1987;17(3):577-601.
28. Shifrine M., Munn S.L., Rosenblatt L.S., et al. Hematologic changes to 60 days of age in clinically normal beagles. Lab Anim. 1973;23(6):894-898.
29. Authement J.M., Wolfsheimer K.J., Catchings S., et al. Canine blood component therapy: product preparation, storage, and administration. J Am Anim Hosp Assoc. 1987;23:483-493.
30. Fiser D.H. Intraosseous infusion. N Engl J Med. 1990;322(22):1579-1581.
31. Hodge D. Intraosseous flow rates in hypovolemic “pediatric” dogs. Ann Emerg Med. 1987;16:305-307.
32. Sheng H.-P., Huggins R.A. Growth of the beagle: changes in the body fluid compartments. Proc Soc Exp Biol Med. 1972;139:330-335.
33. Davis L.E., Westfall B.A., Short C.R. Biotransformation and pharmacokinetics of salicylate in newborn animals. Am J Vet Res. 1973;34(8):1105-1108.
34. Baggott J.D. Principles of antimicrobial bioavailability and disposition. In: Prescott J.F., Baggot J.D., Walker R.D., editors. Antimicrobial therapy in veterinary medicine. ed 3. Ames, Iowa: Blackwell Publishing; 2000:50-87.
35. Goldstein R., Lavy E., Shem-Tov M., et al. Pharmacokinetics of ampicillin administered intravenously and intraosseously to kittens. Res Vet Sci. 1995;59:186-187.
36. Lavy E., Goldstein R., Shem-Tov M., et al. Disposition kinetics of ampicillin administered intravenously and intraosseously to canine puppies. J Vet Pharmacol Ther. 1995;18:379-381.
37. Seguin M.A., Papich M.G., Sigle K. Pharmacokinetics of enrofloxacin in neonatal kittens. Am J Vet Res. 2004;65:350-356.
38. Poffenbarger E.M., Ralston S.L., Chandler M.L., et al. Canine neonatology, Part I. Physiological differences between puppies and adults. Compend Contin Educ Pract Vet. 1990;12(11):1601-1609.
39. Ehrnebo M., Agurell S., Jalling B., et al. Age differences in drug binding by plasma proteins: studies on human fetuses, neonates and adults. Eur J Clin Pharmacol. 1973;3:189-193.
40. Horster M., Kemler B.J., Valtin H. Intracortical distribution of number and volume of glomeruli during postnatal maturation in the dog. J Clin Invest. 1971;50:796-800.
41. Horster M., Valtin H. Postnatal development of renal function: micropuncture and clearance studies in the dog. J Clin Invest. 1971;50:779-795.
42. Hellmann J., Vannucci R.C., Nardis E.E. Blood-brain barrier permeability to lactic acid in the newborn dog: lactate as a cerebral metabolic fuel. Pediatr Res. 1982;16:40-44.
43. Ginsberg G., Hattis D., Russ A., et al. Pharmacokinetic and pharmacodynamic factors that can affect sensitivity to neurotoxic sequelae in elderly individuals. Environ Health Perspect. 2005;113:1243-1249.
44. Hurria A., Lichtman S.M. Clinical pharmacology of cancer therapies in older adults. Br J Cancer. 2008;98:517-522.
45. Reiche R. Drug disposition in the newborn. In: Ruckesbusch P., Toutain P., Koritz D., editors. Veterinary pharmacology and toxicology. Westport, Conn: AVI Publishing; 1983:49-55.
46. Peters E.L., Farber T.M., Heider A., et al. The development of drug-metabolizing enzymes in the young dog. Fed Proc Am Soc Biol. 1971;30:560.
47. Inman R.C., Yeary R.A. Sulfadimethoxine pharmacokinetics in neonatal and young dogs. Fed Proc Am Soc Biol. 1971;30:560.
48. Ecobichon D.J., D’Ver A.S., Ehrhart W. Drug disposition and biotransformation in the developing beagle dog. Fundam Appl Toxicol. 1988;11:29-37.
49. Cowan R.H., Jukkola A.F., Arant B.S. Pathophysiologic evidence of gentamicin nephrotoxicity in neonatal puppies. Pediatr Res. 1980;14:1204-1211.
50. Kleinman L.I. Renal bicarbonate reabsorption in the newborn dog. J Physiol. 1978;281:487-498.
51. Bovee K.C., Jezyk P.F., Segal S.C. Postnatal development of renal tubular amino acid reabsorption in canine pups. Am J Vet Res. 1984;45(4):830-832.
52. Kerner J.A., Sunshine P. Parenteral alimentation. Semin Perinatol. 1979;3(4):417-434.
53. Otto C.M., Kaufman G.M., Crowe D.T. Intraosseous infusion of fluids and therapeutics. Compend Contin Educ Pract Vet. 1989;11(4):421-431.
54. Hirschhorn N. Oral rehydration therapy for diarrhea in children: a basic primer. Nutr Rev. 1982;40(4):97-104.
55. Nahata M.C. Sedation in pediatric patients undergoing diagnostic procedures. Drug Intell Clin Pharm. 1988;22(Sept):711-715.
56. Levy G. Pharmacokinetics of fetal and neonatal exposure to drugs. Obstet Gynecol. 1981;58(Suppl):9S-16S.
57. Welsch F. Placental transfer and fetal uptake of drugs. J Vet Pharmacol Ther. 1982;5:91-104.
58. Krauer B., Krauer F. Drug kinetics in pregnancy. In: Gibaldi M., Prescott L., editors. Handbook of clinical pharmacokinetics. New York: ADIS Health Science Press; 1991:1-17.
59. Carter A.M. Animal models of human placentation. Plactenta. 2007;28(SA):S41-S47.
60. Renfree M.B. Implantation and placentation. In: Austin C.R., Short R.V., editors. Reproduction in mammals 2. Embryonic and fetal development. ed 2. Cambridge: Cambridge University Press; 1982:26-69.
61. Syme M.R., Paxton J.W., Keelan J.A. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet. 2004;43(8):487-514.
62. Farrar H.C., Blumer J.L. Fetal effects of maternal drug exposure. Annu Rev Pharmacol Toxicol. 1991;31:525-547.
63. Autio K., Rassnick K.M., Bedford-Guaus S.J. Chemotherapy during pregnancy: a review of the literature. Vet Comp Oncol. 2007;5:61-75.
64. Nau H. Clinical pharmacokinetics in pregnancy and perinatology. II. Penicillins. Dev Pharmacol Ther. 1987;10(3):174-198.
65. Papich M.G., Davis L.E. Drug therapy during pregnancy and in the neonate. Vet Clin North Am Small Anim Pract. 1986;16(3):525-538.
66. Berlin C.M. Pharmacologic considerations of drug use in the lactating mother. Obstet Gynecol. 1981;58(5):17S-23S.
67. Rasmussen F. Excretion of drugs by milk. In: Brodie B.B., Gilette J.R., editors. Handbook of experimental pharmacology, vol 28, Concepts in biochemical pharmacology, part I. New York: Springer-Verlag; 1979:390.
68. Smith H.W. The development of the flora of the alimentary tract in young animals. J Pathol Bacteriol. 1965;90:495-513.
69. Tanaka T. Gender-related differences in pharmacokinetics and their clinical significance. J Clin Pharm Ther. 1999;24:339-346.
70. Gandhi M., Aweeka F., Greenblatt R.M., et al. Sex differences in pharmacokinetics and pharmacodynamics. Annu Rev Pharmacol Toxicol. 2004;44:499-523.
71. Shah S.S., Sanda S., Regmi N.L., et al. Characterization of cytochorme P450-mediated drug metabolism in cats. J Vet Pharmacol Ther. 2007;30:422-428.
72. Riviere J.E. Comparative pharmacokinetics: principles, techniques and applications. Ames, Iowa: Blackwell Publishing; 2003.
73. Vessey D.A. Hepatic metabolism of drugs and toxins. In: Zakim D., Boyer T., editors. A textbook of liver disease. Philadelphia: Saunders; 1982:197-230.
74. Jolivette L.J., Ward K.W. Extrapolation of human pharmacokinetic parameters from rat, dog, and monkey data: molecular properties associated with extrapolative success or failure. J Pharm Sci. 2005;94:1467-1483.
75. Akimoto M., Nagahata N., Furuya A., et al. Gastric pH profiles of beagle dogs and their use as an alternative to human testing. Eur J Pharm Biopharm. 2000;49:99-102.
76. Xia C.Q., Xiao G., Liu N., et al. Comparison of species differences of P-gps in beagle dog, rhesus monkey, and human using AtPase activity assays. Mol Pharm. 2005;3:78-86.
77. He Y.-L., Murby S., Warhurst G., et al. Species differences in size discrimination in the paracellular pathway reflected by oral bioavailability of poly(ethylene glycol) and D-peptides. J Pharm Sci. 1998;87:626-633.
78. Chiou W.L., Jeong H.Y., Chung S.M., et al. Evaluation of using dog as an animal model to study the fraction of oral dose absorbed of 43 drugs in humans. Pharm Res. 2000;17(2):135-140.
79. Abadia A.R., Aramayona J.J., Munoz M.J. Ciprofloxacin pharmacokinetics in dogs following oral administration. J Vet Med Assoc. 1995;42:505-511.
80. Albarellos G.A., Kreil V.E., Landoni M.F. Pharmacokinetics of ciprofloxacin after single intravenous and repeat oral administration to cats. J Vet Pharmacol Ther. 2004;27:155-162.
81. Dye J.A., McKiernan B.C., Neft Davis C.A., et al. Chronopharmacokinetics of theophylline in cats. J Vet Pharmacol Ther. 1989;12:133-140.
82. Dye J.A., McKiernan B.C., Jones S.D. Sustained-release theophylline pharmacokinetics in cats. J Vet Pharmacol Ther. 1990;13:278-286.
83. Riviere J.E., Martin-Jimenez T., Sundlof S.F., et al. Interspecies allometric analysis of the comparative pharmacokinetics of 44 drugs across veterinary and laboratory animal species. J Vet Pharmacol Therap. 1997;20:453-463.
84. Mealey K.L., Bentjen S.A., Waiting D.K. Frequency of the mutant MDR1 allele associated with ivermectin sensitivity in a sample population of collies from the northwestern United States. Am J Vet Res. 2002;63:479-481.
85. Hugnet C., Bentjen S.A., Mealey K.L. Frequency of the mutant MDR1 allele associated with multidrug sensitivity in a sample of collies from France. J Vet Pharmacol Ther. 2004;27:227-229.
86. Martinez M., Modric S., Sharkey M., et al. The pharmacogenomics of P-glycoprotein and its role in veterinary medicine. J Vet Pharmacol Ther. 2008;31(4):285-300.
87. Ingelman-Sundberg M. Polymorphism of cytochrome P450 and xenobiotic toxicity. Toxicology. 2002;181-182:447-452.
88. Guengerich F.P. Comparisons of catalytic selectivity of cytochrome P450 subfamily enzymes from different species. Chem Biol Interact. 1997;106:161-182.
89. Bogaards J.J.P., Bertrandoe M., Jacksonoe P., et al. Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica. 2000;30:1131-1152.
90. Chauret N., Gatuhter A., Martin J., et al. In vitro comparison of cytochrome p450 mediated metabolic activities in human, dog, cat and horse. Drug Metab Dispos. 1997;25:1120-1126.
91. Landoli M., Soraci A.L., Delatour P., et al. Enantioselective behavior of drugs used in domestic animals: a review. J Vet Pharmacol Ther. 1997;20:1-16.
92. Court M.H., Hay-Kraus B.L., Hill D.W., et al. Propofol hydroxylation by dog live microsomes: assay development and dog breed differences. Drug Metab Dispos. 1999;27(11):1293-1299.
93. Kukanich B., Borum S.L. Effects of ketoconazole on the pharmacokinetics and pharmacodynamics of morphine in healthy greyhounds. Am J Vet Res. 2008;69:664-669.
94. Blaisdell J., Goldstein J.A., Bai S.A. Isolation of a new canine cytochrome P450 CDNA from the cytochrome P4502C subfamily (CYP2C41) and evidence for polymorphic differences in its expression. Drug Metab Dispos. 1998;26:278-283.
95. Mealey K. Pharmacogenetics. Vet Clin North Am Small Anim Pract Pharm. 2006;36:961-973.
96. Paulson S.K., Engel L., Reitz B., et al. Evidence for polymorphism in the canine metabolism of the cyclooxygenase 2 inhibitor, celecoxib. Drug Metab Dispos. 1999;27:1133-1142.
97. Lees P., Landoni M.F., Giraudel J., et al. Pharmacodynamics and pharmacokinetics of nonsteroidal anti-inflammatory drugs in species of veterinary interest. J Vet Pharmacol Ther. 2004;27:479-490.
98. Castro E., Soraci A., Fogel F., et al. Chiral inversion of R(-) fenoprofen and ketoprofen enantiomers in cats. J Vet Pharmacol Ther. 2000;23:265-271.
99. Kidd L.B., Salavaggione O.E., Szumlanski C.L., et al. Thiopurine methyltransferase activity in red blood cells of dogs. J Vet Intern Med. 2004;18(2):214-218.
100. Watson A.D.J. Chloramphenicol toxicosis in cats. Am J Vet Res. 1978;89:1199-1203.
101. Welch R.M., Conney A.H., Burns J.J. The metabolism of acetophenetidin and N-acetyl-p-aminophenol in the cat. Biochem Pharmacol. 1966;15:521-532.
102. Baggott J.D. Principles of drug disposition in domestic animals: the basis of veterinary clinical pharmacology. Philadelphia: Saunders; 1977. pp 73-112
103. Elston TH, Rosen O, Rodar I et al: Seven cases of acute diazepam toxicity, Proc Am Anim Hosp Assoc Oct: 343–349, 1993.
104. Tanaka N., Shinkyo R., Sakaki T., et al. Cytochrome P450 2E polymorphism in feline liver. Biochim Biophys Acta: General Subjects. 2005;1726:194-205.
105. Trepanier L.A. Cytochrome P450 and its role in veterinary drug interactions. Vet Clin North Am Small Anim Pract. 2006;36:975-985.
106. Shecter R.D., Schalm C.W., Kanek J.J. Heinz-body hemolytic anemia associated with the use of urinary antiseptics in the cat. J Am Vet Med Assoc. 1983;162:37-44.
107. Cullison R.F. Acetaminophen toxicosis in small animals: clinical signs, mode of action and treatment. Compend Contin Educ Pract Vet. 1984;6:315-323.
108. Savides M.C., Oehme F.W., Leipold H.W. Effect of various antidotal treatments on acetaminophen toxicosis and biotransformation in cats. Am J Vet Res. 1985;46:1485-1489.
109. Wilkie D.A., Kirby R. Methemoglobinemia associated with dermal application of benzocaine cream in a cat. J Am Vet Med Assoc. 1988;192:85-86.
110. Peterson M.E., Horvitz A.I., Leib M.S. Propylthiouracil-associated hemolytic anemia, thrombocytopenia and antinuclear antibodies in cats with hyperthyroidism. J Am Vet Med Assoc. 1984;184:806-808.
111. Stolk J.M., Smith R.P. Species differences in methemoglobin reductase activity. Biochem Pharmacol. 1966;15:343-351.
112. Boothe D.M. Drug therapy in cats. Mechanisms and avoidance of adverse drug reactions. J Vet Med Assoc. 1990;196:1297-1306.
113. Harvey J.W., Kaneko J.J. Oxidation of human and animal hemoglobins with ascorbate, acetylphenylhydrazine, nitrite, and hydrogen peroxide. Br J Haematol. 1976;32:193-203.
114. Physician’s desk reference, ed 64. New York: Thomas Reuters; 2010.
115. FCC Freedom of Information Act (FOIA). Accessed October 14, 2009: http://www.fcc.gov/foia/.
116. Marier J.F., Beaudry F., Ducharme M.P. A pharmacokinetic study of amoxycillin in febrile beagle dogs following repeated administrations of endotoxin. J Vet Pharmacol Ther. 2001;24:379-383.
117. Brater D.C., Chennavasin P. Effects of renal disease. Altered pharmacokinetics. In: Benetz L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1985:149-172.
118. Stern A. Drug metabolism in renal failure. Compend Cont Educ Pract Vet. 1983;5:913-919.
119. Riviere J.E. Calculation of dosage regimens of antimicrobial drugs in animals with renal and hepatic dysfunction. J Am Vet Med Assoc. 1984;185(10):1094-1097.
120. Carlson G.P., Kaneko J.J. Sulfanilate clearance in clinical renal disease in the dog. J Am Vet Med Assoc. 1971;158(7):1235-1239.
121. Lefebvre H.P., Schneider M., Dupouy M. Effect of experimental renal impairment on disposition of marbofloxacin and its metabolites in the dog. J Vet Pharmacol Ther. 1998;1:453-461.
122. Wilkinson G.R., Shand D.G. A physiological approach to hepatic drug clearance. Clin Pharmacol Ther. 1975;18:377-390.
123. Wilkinson G.R., Branch R.A. Effects of hepatic disease on clinical pharmacokinetics. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:49-70.
124. Ahmad A.B., Bennet P.N., Rowland M. Models of hepatic drug clearance: discrimination between the “well stirred” and “parallel-tube” models. J Pharm Pharmacol. 1983;35:219-224.
125. Boothe D.M. Effects of hepatic disease on drug disposition. In: Bonagura J., editor. Current veterinary therapy (XII), small animal practice. Philadelphia: Saunders; 1995:758-763.
126. Williams R.L. Drugs and the liver: clinical applications. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:53-75.
127. Blaschke T.F., Rubin P.C. Hepatic first-pass metabolism in liver disease. In: Gibaldi M., Prescott L., editors. Handbook of clinical pharmacokinetics. New York: ADIS Health Science Press; 1989:140-149.
128. Blaschke T.F. Protein binding and kinetics of drugs in liver diseases. In: Gibaldi M., Prescott L., editors. Handbook of clinical pharmacokinetics. New York: ADIS Health Science Press; 1989:126-139.
129. Boothe D.M., Brown S.A., Jenkins W.L., et al. Disposition of indocyanine green in dogs with experimentally induced dimethylnitrosamine hepatotoxicity. Am J Vet Res. 1992;53:382-388.
130. Boothe D.M., Jenkins W.L., Brown S.A., et al. Antipyrine and caffeine disposition kinetics in dogs with progressive dimethylnitrosamine-induced hepatotoxicity. Am J Vet Res. 1994;55:254-261.
131. Morgan D.J., Smallwood R.A. Hepatic drug clearance in chronic liver disease: can we expect to find a universal, quantitative marker of hepatic function? Hepatology. 1989;10:893-895.
132. Poulsen H.E., Loft S. Antipyrine as a model drug to study hepatic drug-metabolizing capacity. J Hepatol. 1988;6(3):374-382.
133. Van Thiel D.H., Hassanein T. Assessment of liver function: the current situation. J Okla State Med Assoc. 1995;88:11-16.
134. Rollins D.E. Pharmacokinetics and drug excretion in bile. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:249-268.
135. Kawata S., Imai Y., Inada M., et al. Selective reduction of hepatic cytochrome P450 content in patients with intrahepatic cholestasis. Gastroenterology. 1987;92:299-303.
136. Evans G.H., Nics A.S., Shand D.G. The disposition of propranolol. III. Decreased half-life and volume of distribution as a result of plasma binding in man, monkey, dog and rat. J Pharmacol Exp Ther. 1973;186:114-122.
137. Belpaire F.M., DeRick A., Dello C., et al. Alpha 1-acid glycoprotein and serum binding of drugs in healthy and diseased dogs. J Vet Pharmacol Ther. 1987;10(1):43-48.
138. Eichelbaum M. Drug metabolism in thyroid disease. Clin Pharmacokinet. 1976;1(5):339-350.
139. Jacobs G., Whittem T., Sams R., et al. Pharmacokinetics of propranolol in healthy cats during euthyroid and hyperthyroid states. Am J Vet Res. 1997;58(4):398-403.
140. Nimmo W.S. Drugs, diseases and altered gastric emptying. Clin Pharmacokinet. 1976;1(3):189-203.
141. Griffin J.P., D’Arcy P.F. A manual of adverse drug interactions, ed 2. Chicago: Billing & Sons; 1979. 3–51
142. Larson K, Gay JM, Sellon RK et al: Potential occurrence of drug interactions in small animal intensive care unit (ICU) patients, Proceedings of the American College of Veterinary Internal Medicine, no. 117, 2003.
143. Huang S.-M., Lesko L.J. Drug-drug, drug-dietary supplement, and drug-citrus fruit and other food interactions: what have we learned? J Clin Pharmacol. 2004;44:559-569.
144. Ansel H.C. Ointment, creams, lotions and other dermatological preparations. In: Howard C., editor. Introduction to pharmaceutical dosage forms. ed 2. Philadelphia: Lea & Febiger; 1976:301-332.
145. Papich M.G. Incompatible critical care drug combinations. In: Bonagura J.D., editor. Current veterinary therapy XII, small animal practice. Philadelphia: Saunders; 1995:194-199.
146. University of the Sciences. Remington: the science and practice of pharmacy, ed 21. Philadelphia: Lippincott Williams & Wilkins; 2005.
147. Trissel L.A. Stability of compounded formulations. Washington DC: American Pharmaceutical Association; 1996.
148. Trissel L.A. Handbook on injectable drugs, ed 8. Bethesda, Md: American Society of Hospital Pharmacists; 2000.
149. King’s Guide to Parenteral Admixtures. Napa, Calif: King Guide Publications; 1998.
150. American Society of Hospital Pharmacists. Committee on extemporaneous formulations, Handbook on extemporaneous formulations. Bethesda, Md: The Society; 1997. pp 1–54
151. Toothaker R.D., Welling P.G. The effect of food on drug bioavailability. Annu Rev Pharmacol Toxicol. 1980;20:173-199.
152. Watson A.D., Emslie D.R., Martin I.C., et al. Effect of ingesta on systemic availability of penicillins administered orally in dogs. J Vet Pharmacol Ther. 1986;9(2):140-149. Jun
153. Campbell B.G., Rosin E. Effect of food on absorption on cephalexin and cefadroxil in dogs. J Vet Pharmacol Ther. 1998;21:418-420.
154. Schmidt L.E., Dalhoff K. Food-drug interactions. Drugs. 2002;62(10):1481-1502.
155. Idson B. Vehicle effects in percutaneous absorption. Drug Metab Rev. 1983;14(2):207-222.
156. Boothe D.M. Topical drugs: component interactions, vehicles and the consequences of alterations. Dermatol Rep. 1987;6:1-8.
157. Boothe D.M. Veterinary compounding in small animals: a clinical pharmacologist’s perspective. Vet Clin North Am Small Anim Pract. 2006;36(5):1129-1173.
158. Nordenberg T. Pharmacy compounding: customizing prescription drugs. FDA Consumer Magazine. July 2000.
159. Pond S.M. Pharmacokinetic drug interactions. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1984:49-62.
160. Chien J.Y., Mohutsky M.A., Wrighton S.A. Physiological approaches to the prediction of drug-drug interactions in study populations. Curr Drug Metab. 2003;4:347-356.
161. Hu Z., Yang X., Ho P.C.L., et al. Herb-drug interactions. Drugs. 2005;65:1239-1282.
162. Sagir A., Schmitt M., Dilger, et al. Inhibition of cytochrome P450 3A: relevant drug interactions in gastroenterology. Digestion. 2003;68(1):41-48.
163. Benet L.Z., Cummins C.L., Wu C.Y. Transporter-enzyme interactions: implications for predicting drug-drug interactions from in vitro data. Curr Drug Metab. 2003;4:393-398.
164. Zhang S., Morris M.E. Effects of the flavonoids biochanin A, morin, phloretin, and silymarinon P-glycoprotein-mediated transport. J Pharmacol Exp Ther. 2003;304:1258-1267.
165. Toutain P.L., Bousquet-Melou A. Free drug fraction vs. free drug concentration: a matter of frequent confusion. J Vet Pharmacol Ther. 2002;25:460-463.
166. Hostetler K.A., Wrighton S.A., Molowa D.T., et al. Coinduction of multiple hepatic cytochrome P-450 proteins and their mRNAs in rats treated with imidazole antimycotic agents. Mol Pharmacol. 1988;35:279-285.
167. Ohnhaus E.E., Taras G.-E., Park B.K. Enzyme-inducing drug combinations and their effects on liver microsomal enzyme activity in man. Eur J Clin Pharmacol. 1983;24:247-250.
168. Bresnick E. The molecular biology of the induction of the hepatic mixed function oxidases. In: Schenkman J.B., Kupfer D., editors. Hepatic cytochrome P-450 monooxygenase system. New York: Pergamon Press; 1982:99-156.
169. Snyder R., Remmer H. Class of hepatic microsomal mixed function oxidase inducers. Pharmacol Ther. 1979;7(2):203-244.
170. Schenkman J.P., Kupfer D. Hepatic cytochrome P-450 monooxygenase system. New York: Pergamon Press; 1982. pp 99-156
171. Kawalek J.C., Howard K.D., Farrell D.E., et al. Effect of oral administration of low doses of pentobarbital on the induction of cytochrome P450 insoforms and cytochrome P450-mediated reactions in immature beagles. Am J Vet Res. 2003;64:1167-1175.
172. Hollenberg P.F. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34(1-2):17-35.
173. Pond S.M. Pharmacokinetic drug interaction. In: Benet L.Z., Massoud N., Gambertoglio J.G., editors. Pharmacokinetic basis for drug treatment. New York: Raven Press; 1985:195-220.
174. Hojo T., Ohno R., Shimoda M. Enzyme and plasma protein induction by multiple oral administrations of phenobarbital at a therapeutic dosage regimen in dogs. J Vet Pharmacol Therap. 2002;25:121-127.
175. Levy R.H., Hachad H., Yao C., et al. Relationship between extent of inhibition and inhibitor dose: literature evaluation based on the metabolism and transport drug interaction database. Curr Drug Metab. 2003;4(5):371-380.
176. Hirt R.A., Teinfalt M., Dederichs D. The effect of orally administered marbofloxacin on the pharmacokinetics of theophylline. J Am Vet Med Assoc. 2003;50:246-250.
177. Kuhora M., Kuze Y. In vitro characterization of the inhibitory effect of ketaconazole on metabolic activities of CYP 450 in canine hepatic microsomes. Am J Vet Res. 2002;63:900-905.
178. Mouatt J.G. Cyclosporin and ketoconazole interaction for treatment of perianal fistulas in the dog. Aust Vet J. 2002;80(4):207-211.
179. Dahlinger J., Gregory C., Bea J. Effect of ketoconazole on cyclosporine dose in healthy dogs. Vet Surg. 1998;27(1):64-68.
180. McAnulty J.F., Lensmeyer G.L. The effects of ketoconazole on the pharmacokinetics of cyclosporine A in cats. Vet Surg. 1999;28:448-455.
181. D’Souza M.J., Pollock S.H., Solomon H.M. Cyclosporine-cimetidine interaction. Drug Metab Dispos. 1988;16(1):57-59.
182. Jackson J.E. Cimetidine protects against acetaminophen toxicity. Life Sci. 1982;31:31-35.
183. Ruffalo R.L., Thompson J.F. Cimetidine and acetylcysteine as antidote for acetaminophen overdose. South Med J. 1982;75:954-958.