Chapter 31 Immunomodulators or Biological Response Modifiers
Introduction and Miscellaneous Agents∗
The Immune Response
Immunomodulators, or biological response modifiers, are agents or drugs that act to regulate or modify the host’s immune response to a microbe, neoplasm, or inflammatory response. It is beyond the scope of this chapter to provide a comprehensive review of the immune system. The following is a general review only. Cytokine biology can also be reviewed elsewhere,1,2 as can interferon (IFN) biology.3
Immune Defenses
Immune defenses are composed of the innate and adaptive systems. The innate systems are nonspecific in their response and are best exemplified by barriers provided by the integument; the gastrointestinal environment (acid pH, mucosal and epithelial barriers, microbiota); the mucociliary tract of the respiratory system; and the intimate vasculature of selected organs such as the placenta, brain, and prostate. Other nonspecific defense mechanisms are exemplified fever and the antimicrobial actions of many secretions. White blood cells also represent innate immunity. These include neutrophils, eosinophils, basophils, macrophages, and dendritic cells, and unconventional T-cell subsets bearing T-cell receptors (TCR), natural killer (NK; CD1d) cells and gamma/delta (γδ) T-cells. Natural killer (NK) cells are innate (non conventional) cytotoxic T cells that work in concert with the adaptive arm of immunity, targeting compromised host cells such as tumor or virally infected cells. Released in the vicinity of a cell slated for destruction, it is stimulated by macrophage cytokines. They recognize targeted cells which containing abnormally low concentrations of major histocompatibility complex [MHC] class I antigen (“missing self”). Materials that induce apoptosis enter the target cell through the pores, thus avoiding cellular lysis, which is of benefit in the presence of virally-infected cells that might release viral materials causing host re-infection. NK cells exhibiting the FrC receptor interact with the humoral arm of adaptive immunity, causing antibody-dependent cell cytotoxicity. Gamma-delta T cells contain characteristics of both the innate and adaptive systems.
The adaptive immune system provides the host an ability to recognize and, with subsequent presentation, remember specific pathogens (B and T memory cells), and increasingly stronger responses each time the pathogen is presented. These capabilities reflect both hypermutability and irreversible genetic recombination in cells that are carried forward in subsequent progeny. The system is composed of two major arms, cell-mediated immunity and humoral immunity, each accompanied by a variable number of cells and their chemical mediators. Some of the nonspecific components are also part of the innate immune system, including phagocytic circulating leukocytes and tissue macrophages, and secretions or body fluids such as IFN, complement, and leukocyte substances such as lysozyme. The adaptive arm of the immune system provides the primary targets of pharmacologic manipulation. The adaptive response recognizes both non-self and missing-self antigens. As such, MHC plays a major role in the adaptive arm of the immune system. This large gene complex encodes proteins unique to the individual. Expression on the cell surface allows recognition of self by relevant cell types. At least two subgroups of MHC are important in adaptive immunity. All cells, save non-nucleated cells, generally express MHC class I peptides on the cell surface. They are derived from the cytosol and contain other cytosolic proteins. Abnormalities in MHC class I expression that initiate an adaptive response generally result in interactions exclusively with CD8+ cells, inducing apoptosis. The MHC class II complexes are located on antigen-presenting cells, including dendritic, macrophage, activated T cells, and B cells. These peptides generally are associated with presentation of extracellular pathogens, and presentation that results in an adaptive response generally reflects interactions only with CD4+ cells (T helper or Th cells).
KEY POINT 31-1
The primary targets of pharmacologic manipulation of the immune system are the adaptive arms of the immune system.
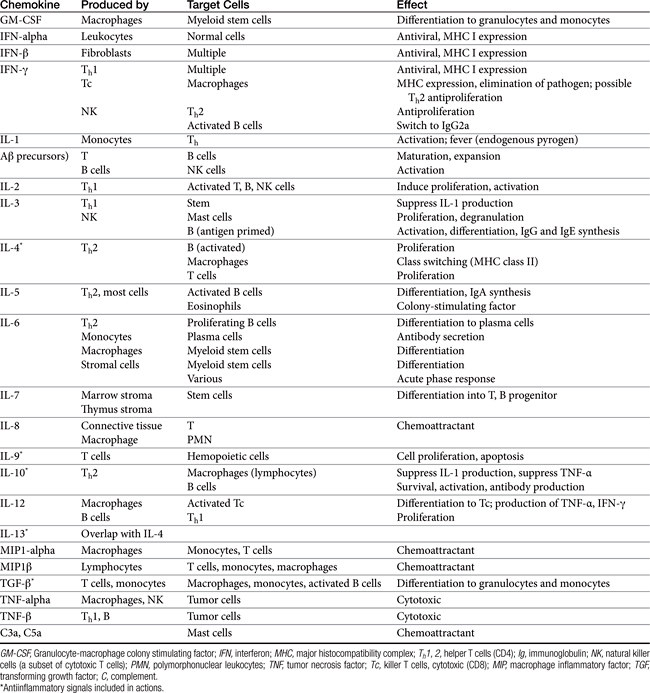
Both cell-mediated and humoral immune mechanisms are characterized by specificity toward antigenic epitopes expressed as molecular components of infectious organisms, foreign (transplanted) cells, or transformed (malignant) cells. Cytokines, including soluble growth and activation factors (Table 31-1), are a vital component of the adaptive response and are released and subsequently mediate the response of the various cell populations involved in both cell-mediated and humoral immunity.4 In addition to cytokines, a number of other molecules (e.g., adhesion or accessory molecules) are necessary as “second signals” for antigen processing, recognition, or response.4
Effectors
The following is a brief synopsis of the events following infection to implementation of the immune response (Figure 31-1). The antigen is exposed to an antigen-processing or presenting cell (APC), which includes dendritic cells (the principle APC), macrophages, and activated B cells.4 The antigen is identified by the APC as foreign and is subsequently phagocytized by the APC. The APC “processes” the antigen and “exhibits” it on the cell surface in a groove made by the MHC molecule. Peptides derived from endogenous cytosolic proteins synthesized within the cell complex (including those synthesized in response to viral stimuli) are expressed with class II MHC molecules on the cell surface, whereas exogenous intracellular proteins are expressed bound to a class I MHC groove for presentation.4
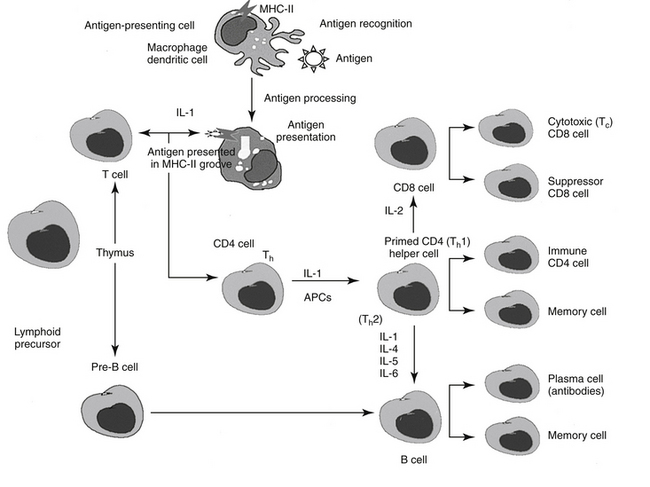
Figure 31-1 Overview of the immune response to antigen presentation. The antigen is presented to an antigen-presenting cell (APC), which processes the antigen and expresses its peptides in a groove located on the major histocompatibility complex (MHC) molecule. When presented to naïve CD4 T cells with appropriate receptors, the helper cell becomes primed to form CD8 cells, capable of directly killing cells presently containing the antigen; Th1 cells, which result in the formation of memory T cells and T cells that attract other leukocytes (type IV hypersensitivity); or Th2 cells, which stimulate B cell production of antibodies. A number of interleukins signal the activities; adhesion molecules (not shown) facilitate communication between cells as well as movement from lymph nodes through vascular endothelial cells into tissues. IL, Interleukin; Th, T helper cell.
A single APC surface may have tens of thousands of MHC molecules, each containing a different peptide; a single animal may have more than 108 APCs. The APC migrates to the T cell area of a lymph node and presents the antigen to naïve CD4 T cells. If the APC comes into contact with a CD4 T cell with a TCR that recognizes the antigen associated with the MHC, it becomes activated. The release of adhesion molecules causes the two cells to stick together, which facilitates interaction between the APC and the CD4. Cytokines move between the two cells, and production of interleukin-1 (IL-1) by the APC and interleukin-2 (IL-2) from the activated CD4 cell itself amplifies the sequelae of CD4 activation. The activated CD4 cell differentiates, proliferates, and produces a number of cytokines (see Table 31-1), which results in recruitment of other leukocytes, initiation of B cell production of immunoglobulins (Igs), and formation of other T cell colonies, including “memory” cells (see Figure 31-1).4,5
Cellular Components
Efficient pathogen elimination depends on the adaptive immune response. Two classes of lymphocytes are responsible for adaptive immunity (see Figure 31-1). B lymphocytes must be programmed to respond to antigen exposure and, after activation to plasma cells, are responsible for the production of specific Igs (humoral response). T lymphocytes provide the primary regulation of the immune response. T cell activity begins with specific antigen recognition by a receptor on the surface of the cell.4 T cells are further subdivided into several populations of cells depending on their role in immunoregulation (see Figure 31-1). Helper cell (CD4 T cells; Th cells) receptors recognize and bind to the peptide–MHC class II complex of APC cells. In response to IL-1, CD4 cells consequently proliferate and become primed as either Th1 or Th2 CD4 cells, which modulate further responses in both the cell-mediated and humoral arms (see Figure 31-1). Migration of activated lymphocytes from lymph nodes to tissues is facilitated by adhesion molecules expressed by endothelial cells in tissues. Whether a T cell becomes Th1 or Th2 reflects the stimulating cytokine: IL-12 activates signal transducer and activator of transcription 4 (STAT-4), which regulates Th1 differentiation, whereas IL-4 activates STAT-6 and Th2 differentiation.5
KEY POINT 31-2
T lymphocytes provide the primary regulation of the immune response, whereas B lymphocytes must be programmed to respond to antigen exposure.
The Th1 subsets of CD4 produce primarily proinflammatory cytokines such as IFN-γ (IFN-γ) and tumor necrosis factor-alpha (TNF-α) (cachexin; see Figure 31-1). As such, Th1 cells regulate signals that promote cell-mediated immunity and control intracellular pathogens. Th1 response is paramount to successful resistance to most microbial pathogens, including bacteria, intracellular protozoa, and fungal organisms. Additionally, Th1 cells mediate organ-specific autoimmunity and as such are crucial to the pathogenesis of autoimmune diseases. The cell-mediated response occurs when activated CD4 (Th1) cells attract other cells (polymorphonuclear leukocytes, eosinophils, and monocytes) to support cellular killing; the result is referred to as delayed hypersensitivity. Activated CD4 cells also yield, in response to IL-2, helper T cells that contain the glycoprotein CD8 and are responsible for the T cell–mediated cytotoxic response. Receptors of the CD8 T cells recognize peptide–MHC class I complexes (generally associated with endogenous peptides present on all nucleated cells) on APCs. Recognition of the CD8 receptor and subsequent interaction with CD4 cells result in the generation of cytolytic (CTL) or cytotoxic T cells (TC; killer T cells). Cytotoxic T cells are capable of directly (without further interaction with CD4 cells) causing lysis of cells expressing the targeted specific peptide–MHC complex.4 This includes normal but otherwise damaged (include virally infected or cells damaged by TNF-β or lymphotoxin) cells. CD8 cells release several different cytotoxins which cause pores in the target cell membrane. Cytotoxic T cells also undergo clonal expansion, resulting in prepared effector cells. In contrast to Th1 cells, Th2 cells secrete antiinflammatory cytokines (IL-4, IL-9, IL-10, IL-13) (in additional to pro-inflammatory cytokines) and support the humoral immune response, including host defense against intestinal helminths. Because of their antiinflammatory effects, Th2 cytokines can decrease autoimmune diseases associated with cell-mediated immunity; however, an imbalance toward Th2 cells have been implicated in the pathogenesis of asthma and allergy (see below).5 Dysregulation of the influence of STAT-4 and STAT-6—which have contrary effects—has been associated with immune-mediated diseases. For example, each STAT is reported to be involved in systemic lupus erythematosus: STAT-6 contributes to the development of glomerulosclerosis and antibody production, which is exacerbated in the absence of STAT-4.5
In contrast to immune-mediated diseases, in which Th1 cells play key roles, key effector cells in all chronic allergic diseases include eosinophils, basophils, and Th2 cells.6 Their involvement occurs through release of preformed, or formed in situ, granule proteins and cytokines.7 A focus on eosinophil regulation may offer targeted therapy for chronic allergic diseases. Cysteinyl leukotrienes appear to have an important role in the regulation of human eosinophil hematopoiesis, recruitment, and activation.7 Eosinophils are generated from CD34+ progenitors in the bone marrow upon stimulation by cytokines such as IL- 5. A single dose of IL-5 antibody profoundly inhibited circulating eosinophils and allergen-induced sputum eosinophils but not airway hyperreactivity in humans. In a follow-up study, eosinophil numbers in blood and bronchoalveolar lavage were reduced 80% or more after 3 months of anti–IL-5 therapy numbers in bone marrow and bronchial biopsies were reduced 50% to 60%. This suggests that IL-5 antibody therapy effectively reduces circulating and airway luminal eosinophils, but less so bone marrow and lung parenchymal eosinophils.8 Cys-leukotrienes (LTs) may also play a role in eosinophil regulation.Circulating eosinophil counts in humans decrease by almost 20% after treatment with cyst-LT-receptor antagonists. These effects may reflect actions at the level of the bone marrow. Eosinophil–basophil colony-forming units increase in number, adhesion, and function, including production of IL-4 when exposed in vitro in the presence of LTD4. These responses are blocked by the presence of cysLT1 antagonists.
Activation of the humoral system occurs when naïve B lymphocytes with appropriate receptors (Igs) recognize an epitope in the intact foreign antigen. The antigen binds to the Ig, is ingested, and is processed such that it is expressed on the surface of the B cell in association with the same MHC (class I or II) that presented the antigen initially.4 Binding of the Ig receptor with the peptide and subsequent interaction with a CD4 cell that recognizes the antigen in its MHC complex stimulates proliferation of B cells, their differentiation into plasma cells, and the secretion of antibodies able to bind the epitope. Ig production is stimulated by IL-1, IL-4, IL-5, and IL-6 in B lymphocytes. The complete sequence occurs over 8 to 14 days and results in an anamnestic, or secondary, response. Generation of “memory” B and T cells provides a long-term mechanism for a rapid immune response on reexposure to the epitope (antigen).4
Soluble Components
Soluble components of the immune response include the cytokines previously described, Igs described later, and the complement cascade.
Cytokines
More than 13 chemokine receptors are associated with rheumatoid arthritis, and at least 16 chemokines interact with these receptors.9 Both constitutive (responsible for physiologic trafficking and homing of the adaptive immune response) and inducible (responsible for effector white blood cell recruitment, including lymphocytes) responses are involved. A number of the chemicals also are responsible for angiogenesis. The inappropriate production of chemokines is associated with formation of an ectopic germinal center, which contributes to an uncontrolled immune response. Consequently, drugs directed toward chemokine receptors offer a therapeutic approach to control.
TNFα induces a broad spectrum of activity. Cytotoxicity of multiple cell types, including tumors, reflects both apoptosis and necrosis. A large number of proteins are targeted, along with other central mediators of the inflammatory process and immune activation. Examples include nuclear factors (e.g., NFκB), nitric oxide synthetase (NOS), cell-surface molecules, MHC classes I and II, and secreted proteins such as IL (e.g., -l, -6, -8), IFN, granulocyte-macrophage colony-stimulating factor, platelet-derived growth factor, urokinase plasminogen activator, and TNF-α itself. When administered exogenously, high concentrations of TNFα causes a toxic syndrome similar to septic shock. Bacterial lipopolysaccharide is the most potent stimulator of macrophage TNFα production. TNFα appears to mediate ischemia–reperfusion injury after transplantation of the liver, kidney, intestine, heart, lung, and pancreas and is a marker cytokine during organ rejection. Inhibitors have proved useful in human medicine for treatment of a number of autoimmune diseases, including Crohn’s disease, rheumatoid arthritis, psoriasis, and ankylosing spondylitis. Indications included corticosteroid-resistant graft-versus-host disease after bone marrow transplantation.10
KEY POINT 31-5
TNFα plays a pervasive role in the inflammatory response, interacting with a number of other cytokines and other mediators and has been identified as a major role player in the “cytokine storm” associated with some syndromes.
Not surprisingly, the pervasive role of TNFα has led to the development of drugs that inhibit its actions. Included are glucocorticoids, pentoxifylline, and monoclonal immunoglobulin G (IgG) antibodies (e.g, infliximab, etanercept and the “humanized” adalimumab) etanercept, a humanized soluble TNFα receptor (TNFR) construct, and onercept, a TNFα–binding protein.10 A variety of other drugs targeting TNFα or its receptors are currently under investigation, as well as metalloproteinase inhibitors such as TNFα-converting enzyme (TACE). The use of these drugs, not surprisingly, is associated with a number of side effects. These include immunosuppresion, acute infusion reactions, delayed-type hypersensitivity reactions, autoimmune diseases, cardiovascular and neurologic adverse events, malignancies and lymphomas, and infectious complications.
Complement
The complement cascade is a major effector mechanism of the immune response. The cascade results in highly amplified events that interact with other physiologic cascades, including the coagulation pathway, kinin formation, and fibrinolysis.11 Consequences of complement activation include opsonization, the release of biologically vasoactive peptides, and cellular lysis. Cell-mediated cytotoxicity is implemented by cytotoxic T lymphocytes, NK cells and antibody-dependent cell-mediated cytotoxicity (ADCC), mediated by a variety of cells that express surface Fc receptors. Effector cells of ADCC include monocytes, neutrophils, eosinophils, selected cytotoxic T cells, and NK cells.
Immunoglobulins
Five classes of Igs are recognized in animals. IgM is the largest, forming up to 15% of the total Ig present. IgM exists as a monomer or a large polymeric form. IgM is responsible for the primary Ig response; in animals, for some infections, it is the only defense.11 The availability of five binding sites renders IgM efficient at antigen binding and agglutination, virus neutralization, and opsonization. IgM also is a potent activator of complement. Because it is such a large molecule, unless vascular permeability is altered, most IgM stays in circulation.
IgG (with multiple subclasses) makes up the majority of total Ig. It is the most soluble of the Igs and thus is able to reach extravascular spaces. Its primary biological functions are to facilitate the removal of microorganisms; neutralize toxins; and bind to microorganisms or infected cells, initiating effector mechanisms. IgG activates complement or initiates Fc-bearing effector cells (ADCC) and promotes removal of Ig-coated cells by ADCC.
IgE has a major role in the response to parasites and in the pathogenesis of allergic diseases in part because of its unique ability to bind by way of the Fc portion of the IgE molecule to specific receptors on mast cells and basophils. Cross-linkage of two IgE molecules results in calcium-mediated mast cell degranulation and the release of a number of preformed (e.g., histamine and serotonin) and synthesized (e.g., metabolites of arachidonic acid) mediators.
IgA is produced in submucosal lymphoid tissues and regional lymph nodes. After secretion outside of the cell, it travels to epithelial cells, where the secretory component of the IgA acts as a receptor, binding to IgA and stimulating its endocytosis. The two components are eventually exocytosed and attach to the mucosal surface, where they provide a protective component, neutralize toxins, adhere to bacteria and viruses, and interact with parasites.11
Each Ig molecule monomer comprises two heavy chains and two light chains attached by covalent bonds. The number of bonds varies with the Ig; the number of dimers varies with the class. A number of fragments (generated by enzymatic cleavage) can be described, including Fab, the antigen-combining fragment, and Fc, the crystallizable fragment, which binds to Fc receptors found on cells of the innatue immune system (e.g., NK cells, macrophages, neutrophils and mast cells).11
Mucosal Immunity
The common mucosal immune system (CIMS) has evolved as a system separate from the general systemic immune system. It includes the mucosa-associated lymphoid tissue (MALT), a complex network of tissues, lymphoid- and mucous membrane–associated cells and their effector molecules. Components include gut-associated lymphoid tissue (GALT) or nasal-associated lymphoid tissue (NALT), the mucosa of the genitourinary tract, the mammary and salivary glands, and draining lymph nodes. MALT differs from systemic lymphoid tissues in its characteristic lymphoid architecture, an epithelium that is unique in its uptake of antigen, and unique APCs (e.g., dendritic cells). Most notable among the differences is the predominance of IgA and the return of immune effector cells to either the originating mucosal sites or distant mucosal sites. Innate components of the CIMS include epithelial cells and antimicrobial peptides (AMP; see later discussion), the latter being exemplified by defensins; lactoferrin; lysozyme; the lactoperoxidase, secretory phospholipase A2, and cathelin-associated peptides. Adaptive components include IgA antibody and CD4+ T cell responses, and mechanisms that impart mucosal (oral) tolerance.
Effective mucosal immunity depends on induction of effective mucosal immune responses. Oral tolerance refers to the prevention of unwanted immune reactions to food or environmental antigens; such tolerance also can be induced nasally. Integral to mucosal tolerance are Toll-like receptors (TRC). Key molecules of the innate immune system, these receptors recognize conserved microbial molecules, alerting the system to their presence when physical barriers are breeched. The term “toll” refers to effect that toll gene mutations have on the physical appearance of Drisophila flies (making them appear “toll”, the German translation meaning “wild”). Toll receptors are a type of pattern recognition receptor, able to distinquish host from microbial pathogen associated molecular patterns (PAMPS).
Induction of nasal mucosal tolerance can be manipulated, leading to mucosally induced immune therapy against selected infectious diseases (e.g, in Escherichia coli in human medicine). Mucosal tolerance has the advantage of inducing local and thus targeted Th1- or Th2- type immune responses, thus avoiding the negative sequelae of systemic cytokine injection. Mucosal adjuvants co-administered with antigens include cytokines (e.g., IL-1 and IL-12), or chemokines which promote specific CD4+ T helper cell cytokine responses (e.g., RANTES, lymphotactin, macrophage inhibitory protein–1 [MIP-1]). Therapeutic modulation of mucosal immunity generally targets unique Th cells and cytokine responses. For example, inflammatory bowel diseases (IBDs) appear to reflect failure of oral tolerance to luminal antigens, resulting in an imbalance of regulatory cytokines involving both Th1 and Th2 cell–mediated inflammation.12 Differences exist between orally and nasally induced immune responses. For example, aging more negatively influences GALT compared to NALT immunity. Nasal vaccines are more effective than oral vaccines in the promotion of protective immunity in the genitourinary tract. That these differences exist suggests that pharmacologic therapies might also be manipulated to locally target systemic effects or to limit therapy to local effects only, thus increasing benefits versus risks of treatment and that these therapies are likely to differentials, affect the different CIMS.
Antimicrobial and Host Defense Peptides
Antimicrobial peptides (AMPs, also called host defense peptides;) are evolutionarily ancient yet essential small cationic molecules found in animals, plants, and bacteria. Because they exhibit antimicrobial activity against a wide range of bacteria, fungi, and viruses, they are considered part of mucosal immunity. AMPs primarily act as cations, interacting with the anionic structure of and thus disrupting microbial membranes.13 Phosphatidylcholine in eukaryotic cell membranes is thought to be more positive compared to that in prokaryotic membranes, thus decreasing attraction to (or repelling) positively charged AMPs. However, in addition to antimicrobial actions, AMPs also serve as multifunctional mediators of immunity, inflammation, and wound repair. The importance of AMP or HDP in veterinary medicine has been reviewed by Linde and coworkers.14
KEY POINT 31-6
Antimicrobial peptides are an ancient evolutionary arm of the mucosal immune system that act to disrupt microbial membranes.
Three major classes of antimicrobial molecules have been described: defensins, cathelicidins, and the four-disulfide core proteins, secretory leukocyte proteinase inhibitor (SLPI) and elafin. Cathelicidins are produced and stored inactive yet capable of exhibiting broad-spectrum killing activity. The number of cathelicidins among species ranges from multiple (pigs and cows) to one (humans and mice). The SLPI and elafin exhibit antimicrobial activity toward a number of bacteria (e.g., E. coli, Pseudomonas aeruginosa, Staphylococcus aureus, Staphylococcus epidermis, and group A Streptococcus); fungi (e.g., Aspergillus fumigatus and Candida albicans); and selected viruses (e.g., human immunodeficiency virus). In addition to their antimicrobial properties, SLPI and elafin are potent antiproteases, neutralizing potentially harmful proteases (e.g, human neutrophil elastase) and inhibiting proinflammatory microbial products (e.g., lipopolysaccharide). Many are chemotactic for white blood cells, including T lymphocytes. Other activities include, but are not limited to stimulation of angiogenesis or inhibition of signals which activate B cells (e.g., NFκB).
The AMPs are extensively integrated into other defense systems. They are stimulated by a number of signals (e.g, macrophage cytokines such as IL-β and TNF-α). In contrast, microbes may protect themselves by downregulating AMP expression.Microorganisms have other protective mechanisms, including changes in cell wall or membrane composition, secretion of factors that block AMP action, or manipulation of host cells such that AMP activity is decreased.
It is likely that the ability of AMP to modulate inflammation, immunity, and tissue repair processes contributes to a central role in numerous essential host defenses. Pharmacologic modulation of AMP is a focus of investigation for a treatment and prevention measures for a variety of disease, including as adjuvants for tumor or infectious vaccines.
Hypersensitivity Reactions and Cytokine Storm
An imbalance in the activities of CD4 Th1 and Th2 subsets may be responsible for the onset or exacerbation of immune-mediated diseases. Increased concentrations of Th1 (and decreased Th2) are associated with response to viral and fungal infections, whereas increased concentrations of Th2 (and decreased Th1) are associated with increased production of IgE and IgA antibodies. Autoimmune diseases are associated with a predominance (reflecting an imbalance) of Th1. Imbalances also have been associated with resistance to infectious disease and malignancy.15 In contrast, allergic inflammatory diseases (eg., atopy, inflammatory bowel disease, asthma) may reflect an imbalance that favors Th2.
KEY POINT 31-7
An imbalance in the activities of CD4 Th1 and Th2 subsets may be responsible for the onset or exacerbation of immune-mediated diseases.
Multiple novel mechanisms have been identified for their underlying role in the pathogenesis of immune-mediated diseases. Most, if not all, involve transcription factors, each providing potential current of future opportunities for manipulation. Among the prominent immune-mediated diseases being intensely studied in an attempt to understand the molecular mechanisms of immune mediated diseases are diabetes type 1, rheumatoid arthritis, multiple sclerosis, IBDs, psoriasis, and systemic lupus erythematosus (SLE).16
Four types of reactions result from activation of immunologic pathways. Type I hypersensitivity results from antigen–IgE interaction (Figure 31-2). IgE that has previously interacted with the antigen binds to the surface of a basophil or mast cell. Subsequent interaction with the same antigen causes mast cell degranulation and the release of a number of mediators associated with immediate hypersensitivity. Mediator release can be instantaneous (e.g., anaphylaxis); delayed for 2 to 4 hours; or biphasic, with both an immediate and a delayed reaction. Systemic release of mediator results in systemic anaphylaxis; localized mediator release limits reaction to the site of release (eg, swelling, redness, pain). Atopy is an inherited predisposition to develop IgE antibodies to environmental antigens and is characterized by constant high levels of IgE. An anaphylactoid reaction may be similar to anaphylaxis in presentation but reflects nonimmune-mediated mast cell degranulation (e.g., cationic drug-induced) (see Chapter 4).
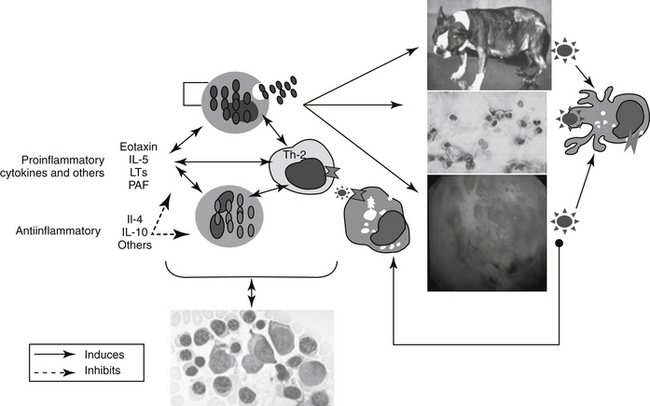
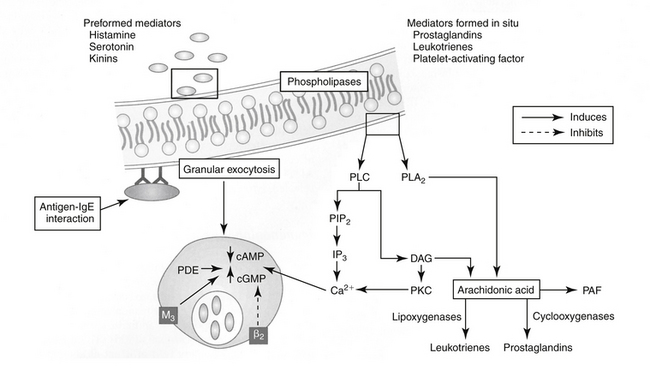
Figure 31-2 The shared pathophysiology of chronic allergic inflammatory disease begins with antigen processing at the tissue (e.g., skin, airways, gastrointestinal tract). Presentation and subsequent Th2 helper cell acitivity results in signaling to basophils and eosinophils. Their release of inflammatory mediators at the site may perpetuate the response by increasing tissue permeability, allowing access of the antigen to deeper tissues. The chemotactant cytokine eotaxin and its interaction with interleukin-5 plays a major role in stimulating local responses as well as bone marrow production of eosinophils; leukotrienes may serve as signals for these interactions. Ultimately, cells released from the bone marrow perpetuate the response at the tissue. Accordingly, therapy might target not only the affected tissue (including adjuvant therapy) but also the bone marrow itself. Inset: Diagrammatic representation of a type I hypersensitivity reaction involving antigen bound to immunoglobulin E and calcium-mediated degranulation. Degranulation results in the release of both preformed mediators and mediators formed in situ (e.g., from arachidonic acid). Stimulation of muscarinic (M3) receptors supports exocytosis, which is inhibited by stimulation of beta2-adrenergic receptors. cAMP, Cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; DAG, diacylglycerol; IP3, inositol triphosphate; PAF, platelet-aggregating factor; PDE, phosphodiesterase; PIP2, phosphatidylinositol; PKC, protein kinase C; PLA2, phospholipase A2; PLC, phospholipase C.
Type II hypersensitivity occurs when ADCC occurs after antibody binds to a cell or to an exogenous antigen associated with a cell surface or a basement membrane. Complement may be activated and contribute to the damage. Examples of type II hypersensitivity include drug hypersensitivities, autoimmune hemolytic anemia, immune-mediated thrombocytopenia, immune-mediated endocrinopathies, and immune-mediated dermatologic disorders such as bullous pemphigus.11
Type III hypersensitivity results from the formation of antigen–antibody or immune complexes that either circulate or are deposited as microprecipitates in vascular beds or basement membranes. The Arthus reaction occurs 2 to 4 hours after IgG interacts with an antigen in the vessel wall. Serum sickness occurs when circulating immune complexes develop as a result of intravascular injection of the antigen. Microprecipitates in circulation deposit in basement membranes and the vascular endothelium, resulting in immune complex diseases. The risk of serum sickness increases with the persistence of antigen. The size of the immune complex also determines the degree of damage because larger complexes are more likely to deposit and initiate inflammation. Complement both contributes to and protects against damage caused by immune complex disease.11
Type IV hypersensitivity involves sensitized T cells that initiate a cell-mediated reaction on interaction with the appropriate class II MHC antigen. Lymphocyte and macrophage influx to the site occurs over a 24- to 72-hour period. Allergic contact dermatitis is an example of a type IV hypersensitivity.11
The term cytokine storm, or hypercytokinemia, has been used to describe the inappropriate systemic reaction resulting from the release of cytokines (more than 150) from a healthy and reactive immune system. Its importance emerged in response to the impact that swine flu to have in some, but not all afflicted human patients.17 The immune system appears to lose the control normally provided through a system of checks and balances involving both inflammatory signals (e.g., TNFα, IL-1, and IL-6) and antiinflammatory signals (e.g., IL-10), as well as coagulation factors, and oxygen free radicals. The impact of these signals on body tissues and organs can be profound and is exemplified by acute respiratory distress syndrome (particularly that associated with influenza or flu), sepsis, and systemic inflammatory response syndrome. Inhibiting T cell response has been proposed as a potential mechanism of treatment or prevention. Drugs that alter production or the impact of TNFα are among those studied for possible efficacy.
Regulation of the Immune Response Transcription Factors
Regulation of the immune response reflects a balance of the integrated actions of proinflammatory and antiinflammatory cytokines that act to trigger signaling pathways. The pathways, in turn, modulate gene expression program in the cells. Transcription factors targeted by cytokines include NFκB (proinflammatory),18 activator protein 1 (AP-1), SMAD proteins (responsible for transfer of extracellular signals from transforming growth factor to intracelluar nuclear TGF-ß gene transcription), glucocorticoid receptors (antiinflammatory)19,20 (see Chapter 30), and members of the STAT protein family (proinflammatory and antiinflammatory).5
NFκB is a transcription factor that, upon induction by a number of inflammatory agents, participates in the expression of a large number of target genes, many of which regulate both innate and adaptive immunity. Because a number of the target genes activate NFκB, signal amplification may occur at very low concentrations of the inciting antigen, causing profound effects. Regulatory failure has been associated with a number of autoimmune diseases, including IBDs such as Crohn’s disease and ulcerative colitis.18 NFκB appears to play a key role in rheumatoid arthritis in humans. Metalloproteinase-1(MMP-1) and TNF-α are among the molecules it regulates.16 The NFκB system is vital to survival, but regulatory activity is very complex, and undirected inhibition may result in undesirable immune suppression. For example, pharmacologic interference with a potentially therapeutic aspect (e.g., inactivation in enterocytes such that a systemic inflammatory response is avoided) often is accompanied by potentially lethal parallel effects (e.g., severe apoptotic damage to the reperfused intestinal mucosa). Pharmacologic manipulation is most likely to succeed only when tissue- or organ-targeted inhibition is possible. Another major role player is activator protein 1 (AP-1), a transcription factor with major proinflammatory effects. It regulates gene expression stimulated by cytokines, growth factors, stress, and bacterial and viral infections, thus controls diverse cellular processes such as differentiation, proliferation, and apoptosis.
KEY POINT 31-8
Nuclear factor kappa B (NFκB), a transcription factor induced by a number of inflammatory signals, is involved in the expression of genes that regulate both innate and adaptive immunity.
Patients with glucocorticoid-sensitive versus glucocorticoid-resistant chronic inflammatory disease exhibit different cellular activation of NFκB, and AP-1 and upstream kinases; activation occurs in macrophages of the lamina propria in steroid-responsive patients but predominantly in epithelial cells of steroid resistant patients. Thus directed therapy for steroid-resistant patients might focus on inhibition of NFκB activation in epithelial cells.18
The Janus kinase (JAK)–STAT pathway is a major signaling pathway by which cytokine signals lead to the expression of genes that regulate immune cell proliferation and differentiation. The JAK–STAT signaling pathway is activated in response to cytokines (e.g., ILs and IFNs) as well as certain peptide hormones. The binding of the signaling molecules to the receptors causes activation (phosphorylation) of JAKs (e.g., JAK-1 through -3), which provide docking sites on the JAK protein for STAT proteins. The STAT proteins are then activated (phosphorylation) and translocated to the nucleus, where they bind to their response elements in the promoter of target genes. Transactivation cannot occur without co-activation by proteins such as acetyl transferase binding protein (e.g., CREB, a cyclic adenosine monophosphate [cAMP]–responsive element binding protein that increases MMP production in the synoviocytes of patients with rheumatoid arthritis).16
Cytokines that promote immune and inflammatory responses (e.g., IL-6, IFN-γ, IL-12, and IL-18) as well as those that suppress the immune response (e.g., IL-4, IL-10, IL-13) mediate cellular responses through the JAK–STAT signaling pathway. Up to seven STAT (including STAT-1 through -4, 5a and 5b, and STAT-6) proteins have been identified in mammals, each with a specific function in the immune response that modulates either proinflammatory or antiinflammatory responses. For example, STAT-3, originally discovered as an acute-phase response factor activated by IL-6, is also activated by many other cytokines and appears to be a constitutive protein that influences chronic inflammation. However, its embryonic deletion is lethal.5
Immunomodulatory Effects of Opioids
The potential of opioids to influence the immune system was first recognized close to 100 years ago when the effects of opium on phagocytic function were observed.21 Heroin addicts are more susceptible to infection. Rodent studies indicate increased mortality and morbidity rates when opioid-treated animals are exposed to infectious agents. The presence of opioid receptors on cells of the immune system has been recognized since the late 1970s.21,22 Although further studies are needed to fully elucidate the effect of opioids on immune function, thus far both the cellular and humoral immune reactions are believed to be affected.
Effects of opioids on the immune system are variable but are directed centrally, rather than peripherally, and involve primarily, but not exclusively, supraspinal mu receptors. Effects (inhibitory versus stimulatory) are dose dependent but occur at clinically relevant doses in animal models. Multiple central opioid receptors and endogenous opioids appear to be involved in complex immune responses. NK cytolytic activity and mitogen-stimulated T-cell proliferation are reduced by centrally administered morphine by way of mu receptors, whereas antibody production is both increased and decreased by met-enkephalin, which probably reflects interaction with multiple receptors. Effects can occur with single or multiple administration of exogenous opioids. Both the neuroendocrine system and the autonomic nervous system may serve as the efferent mechanisms mediating central opioid modulation of the immune system.21 Tolerance appears to develop to some of the immunomodulatory effects, but this remains controversial. The clinical implications regarding the role of opioids in modulating the immune response are not clear.
Immunomodulatory Effects of Vitamin D
Increasingly, vitamin D (1,25, dihydroxyvitamin D3) is recognized to have a noncalcemic role. Among the proposed activities is support of the immune system, particularly T cell–mediated immunity.23 Both T lymphocytes and macrophages are characterized by a high density of vitamin D receptors, particularly in immature immune cells and mature CD8 cells. Vitamin D compounds have been demonstrated in animal models to selectively immunosuppress, effectively preventing or modulating autoimmune diseases, including systemic lupus erythematosus, encephalomyelitis, rheumatoid arthritis, and IBD. Suppression or prevention of transplant rejection has been demonstrated in animal models. Mechanisms may include stimulation of the antiinflammatory mediators transforming growth factor beta 1 (TGF-β 1) and IL-4. These effects appear to occur without negatively affecting normal immune defense mechanisms. Therapeutic use may be limited by the advent of hypercalcemia, leading to investigations of noncalcemic analogs; indeed, some suggestion exists that hypercalcemia may be required for immunosuppression.
Classification of Immunomodulatory Drugs
Immunomodulators often are classified according to their source: microbial, animal, or synthetic. Immunomodulators can also be classified by either an inhibitory or a stimulatory effect on the immune system. Prohost agents augment the cellular immune response either by facilitating a normal response in the face of immunosuppression (immunorestoratives) or by stimulating the immune response (immunostimulants). Immunostimulants can be used either before antigenic challenge to protect immunocompromised patients at risk or after exposure to potentially virulent agents has occurred. Immunosuppressant agents are used to manage hypersensitivity reactions, including autoimmune diseases, and as anticancer drugs. Many immunomodulatory drugs are target specific in their effects. However, many also have nonspecific effects that affect several to many arms of the immune response. Immunostimulants may in fact inhibit components of the immune response in some instances. Care should be taken when selecting an immunomodualtory drug, particularly if all the effects of the drug are not known or anticipated. For example, selective depression of some virally induced immune reactions is beneficial if the host’s immune response to the infecting virus threatens the host’s survival. Drugs that inhibit B lymphocyte activity (e.g., cyclophosphamide) often lower the mortality risk associated with some human influenza viruses and presumably should prove beneficial in the treatment of feline viral diseases associated with poorly controlled Ig production (i.e., feline infectious peritonitis).
The major indications for pharmacologic modulation of the immune system in animals are treatment of autoimmune diseases; prevention or treatment of infections, particularly in immunocompromised hosts; and prevention and therapy of malignancies. A less common but increasingly growing use of immunomodulators in small animals is treatment of graft-versus-host reactions after organ transplantation.
Immunomodulation in Viral and Neoplastic Diseases
Biologica l response modifiers are discussed in Chapter 32. Their primary indications are for treatment of viral or neoplastic disease or (generally their antagonism) for treatment of immune-mediated disease.
Viral Diseases
Biological response modifiers potentially offer a logical and unique approach to the treatment of viral diseases because (1) viruses are capable of immunosuppression and (2) the immune response is an important determinant in the host’s ability to overcome viral infection.24,25 Resistance against and recovery from viral infections in mammals depend on three components of the immune system. The mononuclear phagocytic (reticuloendothelial) system represents the first barrier to viral infections, but it is nonspecific and not always efficient. Sensitized T lymphocytes provide specific cell-mediated immunity, which is followed later by humoral events, either narrowly specific (antibodies) or nonspecific (IFN). Multiple interactions occur among these systems. Unfortunately, several systems also contribute to the pathogenesis of viral disease. Viruses may cause immunosuppression by several mechanisms: They may directly injure or impair all classes of lymphocytes; cause the production of soluble immunosuppressing chemicals (e.g., IFN, p15[E] of feline leukemia virus [FeLV]) damage all three cell systems by infection, or cause an imbalance in immunoregulation, thus leading to overactivity of suppressor T cells.
Neoplastic Diseases
Many tumors have surface antigens toward which specific antibodies can be directed. Biological response modifiers can alter response to tumor antigen at one or several of these points, depending on the drug. Some biological response modifiers augment or restore normal host effector mechanisms by acting as either a mediator or an effector of the antitumor response. Transformation of tumor cells may be decreased or maturation of tumor cells may be increased by biological response modifiers. Host tolerance of damage caused by cytotoxic chemotherapeutic drugs also may be increased by biological response modifiers. Finally, biological response modifiers have been used to alter patient response to FeLV viremia and thus the subsequent associated neoplasms.26,27
Immunomodifying Drugs
The sequelae of immunomodulation are not always beneficial; alteration of the immune response can impair many aspects of the host’s normal defense system. The use of immunomodulators remains in its early stages despite advances; this remains particularly true for veterinary medical applications. Because of the many facets of the immune system susceptible to regulation, the dose, timing, and route of administration of these drugs are important if undesired effects are to be avoided (Table 31-2). In addition, host effects such as age and nutritional status and the nature of the disease may be important determinants in the patient’s response to these drugs.
Marked advances have been made in the use of drugs that target the adaptive immune response. For the purposes of this text, drugs used to modify the immune response will include drugs whose actions are relatively nonspecific in their immunomoldulatory effect and those products of biological (animal) origin that target specific mediators of the immune response or their receptors, the latter is the focus of Chapter 32.
The role of P-glycoprotein in drug-induced immunomodulation is an emerging area of interest.28 The protein is expressed on peripheral blood mononuclear cells. It influences the secretion of cytokines secreted from antigen-presenting cells and selected T cells, and it has been shown to influence lymphocyte survival and antigen-presenting cell differentiation. These actions appear to contribute to the immunomodulatory actions of selected drugs capable of inhibiting P-glycoprotein (e.g., verapamil, progesterone, tamoxifen) and may account for the therapeutic immunomodulatory functions of these drugs. Eventually, these effects might be redirected for therapeutic benefit in allograft rejection and cell-mediated autoimmune disorders.
Immunosuppressant Drugs
Four major classes of immunosuppressive drugs are described in human medicine: glucocorticoids (discussed in Chapter 30), calcineurin inhibitors, antiproliferative–antimetabolic drugs, and biological agents.4 The latter includes biological response modifiers (Chapter 32).
Several principles should guide use of immunosuppressant drugs.4 First, suppression of the primary immune response is more easily accomplished than is suppression of the secondary (amnestic) response. Second, successful inhibition or suppression is easier if therapy begins before exposure to the inciting immunogen (antigen). Third, immunosuppressive drugs do not cause the same effect on all aspects of the immune system. Often, opposite effects are concentration dependent. Thus failure to achieve the desired response should not necessarily lead to an increase in dose. Finally, patients often require lifelong therapy, which increases the risk of adverse effects.
Two major limitations characterize immunosuppressive therapy. Patients receiving immunosuppressive therapy are predisposed to infections of any type. In addition, the risk of lymphomas and related malignancies is increased. This latter risk is more problematic in human patients because, in part, of their longer life span, but has proven a risk in dogs or cats receiving cyclosporine. Events that immunosuppressive drugs tend to target include antigen recognition, stimulation of IL-1, synthesis and release of IL-2 or other cytokines, and lymphocyte proliferation and differentiation.4 Secondary signal molecules are also becoming increasingly important as targets of immunosuppressive therapy.
Immunosuppressive drugs are used to treat immune-mediated disease, which in this chapter include autoimmune diseases (e.g., immune-mediated anemias or thrombocytopenias) as well as chronic allergic inflammatory diseases (e.g., atopic dermatitis and asthma, IBD). A third, less common indication in veterinary medicine is prevention of organ rejection in renal transplant patients. Autoimmune diseases are characterized by sensitization to endogenous proteins that are perceived to be foreign. Both cell-mediated and humoral responses can be directed toward the protein. Many immune-mediated disorders afflicting dogs and cats respond sufficiently well to glucocorticoids (discussed in Chapter 30) and, when necessary (in severe cases), cytotoxic drugs. These include immune-mediated autoimmune hemolytic anemia or thrombocytopenia, acute glomerulopathies, and many dermatologic disorders with an immune-mediated basis. However, the approval of cyclosporine (CsA) for chronic allergic dermatitis has increased its use for autoimmune diseases, providing an alternative to cytotoxic drugs as a second tier.
Cyclosporine A
Chemistry–Structure Relationship
Cyclosporin A (CsA), now known as cyclosporine, is the most important immunosuppressive drug for human transplantation and the treatment of selective autoimmune disorders.4,30 CsA is one of nine cyclosporins (A through I), each a cyclic peptide drug (Figure 31-3) isolated from the fungi Cylindrocarpon lucidum and Trichoderma polysporum. An M designation, when present, indicates a metabolite. The drug is both very lipophilic (hydrophobic) and must be solubilized before administration.4 The oral preparation is a soft gelatin capsule (Sandimmune) or a (newer) microemulsion formulation (Neoral). The intravenous preparation is an ethanol–polyoxyethylated castor oil mixture. Historically, CsA has been formulated in peanut oil (for treatment of ophthalmic ocular disorders in dogs); however, an approved product formulated for topical use is now available.
Mechanism of Action
CsA is a unique immunosuppressant in that it specifically inhibits Th cells (both Th1 and Th2) early in their immune response to antigenic and regulatory stimuli4 without affecting suppressor cells. Suppression occurs as CsA binds to cyclophilin, a cytoplasmic receptor protein, forming a heterodimeric complex. This complex then binds to calcineurin. Binding of calcineurin inhibits calcium-stimulated phosphatase that dephosphorylates the regulatory protein. As such, CsA prevents transcription of T cell genes enhanced in response to T cell activation. Transcription mediated by IL-2, certain proto-oncogenes, and selected cytokine receptors are particularly affected.4 IL-2 production (and thus T cell proliferation and antigen-specific cytotoxic T lymphocyte generation) also is attenuated because the expression of TGF-β, a potent inhibitor of IL-2, is increased.4 B cells are not affected. The drug is most effective when administered before T cell proliferation has occurred. In addition to these immunomodulatory effects, CsA also affects other inflammatory cells. CsA inhibits skin mast cell numbers, survival, and response (secretory and histamine release) as well as secretion of IL-3, IL-4, IL-5, IL-8, and TNFα. Similarly, eosinophile response (release of granules and cytokines) and recruitment to allergic sites is impaired. CsA prevents TNFα-mediated late phase reactions, thus inhibiting IgE and mast cell–dependent cellular infiltration in the skin and bronchial mucosa. However, CsA does not inhibit IgA secretion or IgG and IgM formation in dogs and has no apparent effect on serum allergen-specific IgE levels, intradermal tests, or vaccination.
KEY POINT 31-9
Cyclosporine A specifically inhibitits T helper cells; however, little information is available regarding its impact on immune response in dogs or cats.
Surprisingly little information is available regarding immunomodulatory effects of CsA in dogs or cats. Using dermal microdialysis, Brazis and coworkers31 demonstrated that CsA at 5 mg/kg per day for 15 and 30 days decreased histamine, but not prostaglandin D2, release in sensitized Beagles challenged with Ascaris suum.
Preparations
CsA is available in a variety of preparations, two of which are approved in the United States for use in animals. CsA was first formulated for oral use in humans as vegetable oil. Oral absorption in this preparation depends on emulsification by bile acids. Poor oral bioavailability led to the formulation of a microemulsion preparation (modified CsA, ME), which, on contact with gastrointestinal fluids, disperses into a homogeneous monophasic microemulsion that mimics the mixed micellar phase of the standard formulation. In dogs the ME formulation offers a 35% bioavailability (Novartis Animal Health data on file).
Atopica is the veterinary-approved microemulstion preparation of CsA intended for systemic use in both capsules and solutions. A 0.2% ophthalmic ointment (Optimmune) is also available. A number of human-approved drugs are available, including an injectable solution, an oral solution (100 mg/mL), an ophthalmic emulsion (0.05%), and a variety of capsule sizes for the standard and the microemulsion-modified preparation. Included are a number of generic-modified CsA products. However, the therapeutic equivalence that has been established for these products in humans should not be assumed to be therapeutically equivalent to Atopica in animals. Indeed, one preparation made by IVAX Pharmaceuticals and sold by selected chain pharmacies appears to result in much higher concentrations in dogs compared with other preparations on the basis of samples submitted through the author’s laboratory. Monitoring should be the basis for assessing the appropriate dose–concentration relationship for oral human CsA products used in dogs or cats; owners might need to be aware if and when a pharmacy switches from one human generic to another such that prophylactic monitoring might be implemented.
Clinical Pharmacology
The pharmacokinetic behavior of CsA is complex, resulting in marked variability in the relationship between dose and blood concentrations.32 Variability reflects not only differences in both hepatic and intestinal metabolism (by CYP3A4) but also differences in P-glycoprotein among various tissues.32 The disposition of CsA has been well reviewed in the dog.33 Absorption after oral administration is slow and incomplete. The complex lipid nature of CsA complicates absorption by being dependent on bile acids, which generate a microemulsion. In humans oral bioavailability of the capsule ranges from 20% to 50% but is improved by 10% to 20% in the microemulsion form which is not dependent on bile acids.4,34 Peak concentrations occur 1 to 4 hours after oral administration in human beings. When the capsule—but not the microemulsion—is administered with a fatty meal, absorption is slowed. Studies suggest that decreased bioavailability of CsA after oral administration may reflect activity of drug-metabolizing enzymes of the intestinal epithelium35 or P-glycoprotein–mediated drug efflux.
KEY POINT 31-10
The kinetics of cyclosporine A are complicated by its large, liphophilic molecular structure.
The oral bioavailability of microemulsion in dogs is 35% compared with the 20% of the oral preparation.33 Bioavailability of orally administered CsA is approximately 25% to 30% after multiple oral administration in cats (n = 6). Food may impair the absorption of CsA; peak concentrations in fasted dogs were 20% higher than in nonfasted dogs in one study, although time to peak (approximately 1.5 hours) was the same.33 The drug is not predictably absorbed topically after transdermal administration (unpublished data, Boothe 2007). CsA is widely distributed, characterized by a large volume of distribution (13 L/kg in humans). The drug accumulates in erythrocytes (accounting for 50% or more of the drug in humans), and leukocytes (accounting for 10% to 20% of circulating drug in humans).4 Consequently, monitoring of whole blood rather than plasma or serum is recommended. Remaining circulating drug is bound to plasma lipoproteins. Further, concentrations in skin are up to tenfold higher than in blood, although this is based on homogenate data (as reviewed by Guaguère and coworkers33). In contrast, largely because of the influence of adenosine triphosphate–binding transporters P-glycoproteins, especially the ABCB1 cassette (P-glycoprotein, MDR1 gene product), little CsA crosses the blood–brain barrier.
KEY POINT 31-11
In addition to its chemistry, the oral absorption of cyclosporine A is affected by a number of factors, resulting in marked variability. Differences in absorption of human generic or compounded preparations should be anticipated.
CsA is metabolized to a large number of metabolites predominantly by the liver, although sufficient metabolism occurs by intestinal enterocytes that oral bioavailability is affected. Microflorae also may contribute to metabolism.37 CYP3A4 (commonly associated with P-glycoprotein or other efflux pumps) plays a major role in metabolism; many of the drug interactions involving CsA also involve CYP3A4 or associated glycoprotein. More than 25 to 30 metabolites have been documented in humans and dogs.32,38,39 Metabolism is targeted primarily toward the side chains rather than the ring structure and reflects hydroxylation, demethylation, sulfation, and cyclization. The major hydroxylated metabolites (AM1 and AM9) are further metabolized, contributing to the complex metabolic profile. Both AM1 and AM9 may contribute 10 to 20% of CsA activity, with AM1 contributing 27% of trough activity.40 In humans AM1 accumulates with chronic dosing and ultimately may surpass CsA, and monitoring assays might ideally detect the active compounds.39 They are detected to variable degrees by selected immunoassays based on a monoclonal antibody (e.g., Abbott TdX, Architect systems, Seimens Dimension).41,42 Selected laboratories may also offer a high-performance liquid chromatography (HPLC)–based system that, may or may not (generally not) quantitate the metabolites.39 Ultimately metabolism continues until the drug is totally inactivate the drug, although this has not been proved conclusively.4 Metabolites are excreted in bile and feces, with less than 2% of unchanged drug eliminated in the kidneys of dogs. CYP3A4 is largely responsible for metabolism.
The half-life of CsA in dogs is quite variable, depending on the investigator and assay. In blood the half-life ranges from a low of 5 hours (HPLC) to a high of 18 hours (using fluorescent polarized immunoassay [FPIA]); 29 hours was reported for serum.43 That reported for Atopica, the product approved for use in dogs, is 4.5 hours, a length more consistent with that measured in the author’s therapeutic drug monitoring laboratory using FPIA. In a manufacturer-sponsored affinity colony-mediated immunoassay (ACMIA) study, Steffan and coworkers44 reported the disposition of CsA (5 mg/kg) after single-dose oral administration of either capsules or solution in fasting or fed conditions using a randomized crossover design with a 1-week washout between studies (Table 31-3); food decreased peak concentrations and area under the curve by 22%.
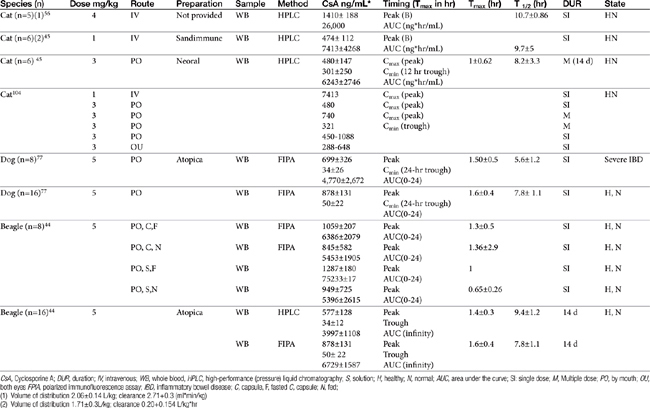
In cats elimination half-life of CsA is 8 hours, which was similar to that after intravenous administration. Liver disease will decrease clearance of CsA substantially. However, for the nonmicroemulsion form, decreased oral absorption in the face of decreased bile may balance decreased clearance. Monitoring is recommended in the presence of hepatic or gastrointestinal disease.
Mehl and coworkers45 reported the disposition of CsA in cats (n = 6 male) after single-dose intravenous (1 mg/kg) and multiple-dose (14 days; 3 mg/kg twice daily) as Neoral (see Table 31-3). Variability in plasma drug concentrations among cats and across time underlines the importance of monitoring if treating life-threatening conditions. After multiple-day (7 to 14 days) oral administration (3 mg/kg), mean (peak) plasma concentrations at 2 hours were 740 ± 326 (7 days) and 655 ± 285 ng/mL (14 days), whereas trough concentrations were 332 ± 237 and 301.5 ± 250 ng/mL, respectively. Oral bioavailability was only 25% to 29%.
Because cats may find the oral preparation unpalatable, the oral solution can be given diluted in olive oil. The intravenous preparation (4 to 6 mg/kg as a 4-hour intravenous infusion) can be given to animals for which oral administration is not possible.46 Ocular administration might serve as an alternative route for systemic delivery of CsA in cats.47 After ocular administration of either an oral preparation or an ocular preparation of CsA in olive oil, (5 mg/kg in each eye as a 10% solution) peak concentrations of 450 to 1033 ng/mL (oral preparation) and 288 to 648 ng/mL (ocular) were achieved, with an absorption lag time ranging from 0 to 1.34 hours for the oral solution and 0.27 to 1.2 hours for ocular preparation. The elimination half-life ranged from 2.41 to 10.04 hours (oral), and 3.09 to 15.75 hours (ocular), respectively. Blood concentrations were sufficient to inhibit lymphocyte activity as measured in vitro, leading the authors to conclude that ocular administration of CsA in an olive oil vehicle might be a reasonable alternative to oral, or even intravenous, administration in cats intolerant to the latter.
Drug Interactions
Novartis48 provides a transplant drug interactions monograph that cites more than 600 references and delineates cyclosporine interactions involving more than 300 drugs. The list of drugs that might interact with CsA is extensive. The manual is available on the Novartis website at no charge and should be consulted when treating patients receiving additional drugs. Selected interactions can be found in Table 31-4. This review will focus on some of the more common interactions, including those used intentionally.
Table 31-4 Cyclosporine Drug Interactions
| Drug | Impact |
|---|---|
| ACE inhibitors | Increased nephrotoxicity |
| Acyclovir | Falsely increased CsA |
| Allopurinol | Increased toxicity |
| Aluminum | Increased nephrotoxicity and CsA bioavailability |
| Aminoglycosides | Increased nephrotoxicity |
| Amlodipine | Increased gingival hyperplasia |
| Antacids | Increased aluminum toxicity |
| Azithromycin | Increased gingival hyperplasia |
| Increased CsA | |
| Bile acids | Increased bioavailability |
| Chloramphenicol | Decreaesed clearance, increased CsA |
| Cimetidine | Delayed absorption |
| No effect (?) on metabolism or absorption | |
| Ciprofloxacin | Antagonizes CsA immunosuppression |
| Cisapride | Increased absorption |
| Clarithromycin | Decreased clearance, increased CsA |
| Cyclophosphamide | Increased hepatotoxiciy |
| Danazol | Decreasd metabolism, increased absorption |
| Dexamethasole | Increased CsA metabolism; synergistic immunosuppression |
| Digoxin | Increased digoxin toxicity |
| Diltiazem | Increased gingival hyperplasia, increased CsA |
| Erythromycin | Increased hepatotoxicity (decreased metabolism?) |
| Famotidine | Decreased clearance, increased CsA |
| Fluconazole | Oral: increased bioavailability |
| Fluoxetine | Decreased clearance, increased CsA |
| Furosemide | Decreased nephrotoxicity |
| Glucocorticoids | Increased metabolism, decreased CsA |
| Itraconazole | Decreased clearance, increased CsA |
| Increased absorption(?) | |
| Ivermectin | Increased neurotoxicity |
| Ketamine | Combined proconvulsant effects |
| Ketaconazole | Decreased clearance, increased CsA |
| Increased absorption | |
| Increased toxicity | |
| Leflunomide | Synergistic immunosuppression |
| Loperamide | Altered absorption |
| Methotrexate | Decreased clearance, increased CsA |
| Increased methotrexate toxicity | |
| Metoclopramide | Increased absorption, increased CsA |
| Metoprolol | Beneficial hemodynamics |
| Mitoxantrone | Increased mitoxantrone concentrations |
| Mycophenolic acid | Synergistic immunosuppression |
| Norfloxacin | Decreased CsA clearance? |
| Omeprazole | Delayed CsA absorption, decreased CsA clearance? |
| Pancuronium | Prolonged duration of pancuronium effect |
| Pentoxifylline | Synergistic inhibition of TNF and other immunosuppression |
| Phenobarbital | Increased CsA clearance, decreased CsA |
| Phenytoin | Increased CsA clearance, decreased CsA |
| Prazosine | Increased afterload |
| Prednisone | Increased risk of hepatotoxicity, hyperlipidemia |
| Primidone | Increased CsA clearance, decreased CsA |
| Propranolol | Antagonism of CsA immunosuppression |
| Decreased propranolol clearance | |
| Ranitidine | Increased risk of hepatotoxicity, thrombocytopenia |
| Rifampin | Increased CsA clearance, decreased CsA |
| Sirolimus | Synergistic immunosuppression |
| Synergistic risk of toxicity | |
| Spironolactone | Hyperkalemia |
| Sucralfate | Increased aluminum |
| Sulfadiazines | Interference with HPLC assay results in increased CsA |
| Tacrolimus | Decreased CsA clearance, increased CsA |
| Terbinafine | Inceased CsA metabolism |
| Ticlopidine | Altered CsA metabolism |
| Trimethoprim | Increased CsA clearance, decreased CsA |
| Ursodeoxycholic acid | Decreased Tmax |
| Vasopressin | Enhanced vasopressin effects |
| Verapamil | Gingival hyperplasia, synergistic immunosuppression |
| Vitamin D3 | Additive suppressive effects |
| Voriconazole | Decreased metabolism? |
ACE, angiotensin-converting enzyme; CsA, cyclosporine A.
From Neoral and Sandimmune Drug Interactions, Novartis package insert, 2007
KEY POINT 31-12
Cyclosporine A is involved with a large number of drug interactions that might cause higher or lower than anticipated plasma drug concentrations.
The enzyme that metabolizes CsA (CYP3A4) is responsible (in humans) for approximately 30% of all drug metabolism; many of CsA drug interactions occur with this enzyme. Further, CsA is a target of P-glycoprotein, another site characterized by a substantial number of CsA–drug interactions.4,49 The risk of drug interactions is another indication of the need for therapeutic drug monitoring as a guide to proper dosing regimens. CsA elimination is accelerated, probably in part because of the induction of drug-metabolizing enzymes, by phenytoin, phenobarbital, rifampin, and sulfamethoxazole–trimethoprim combinations, and others. Dexamethasone also is an inducer of CsA.32 Its elimination is decreased by amphotericin B, erythromycin, and ketoconazole.4 CsA also appears to alter the oral absorption of other drugs.35 CsA use with other immunosuppressive drugs may benefit from these interactions, including glucocorticoids.4
The inhibitory effect of ketoconazole on intestinal epithelial and hepatic drug metabolism and on P-glycoprotein efflux may be of therapeutic benefit in patients receiving CsA by decreasing hepatic clearance and increasing oral bioavailability.35,50-53 Ketoconazole may also decrease the lipoprotein that binds CsA, resulting in higher concentrations of free CsA.51 Studies with ketoconazole in humans found the dose of CsA to be reduced by approximately 70% to 85%.54 The use of ketoconazole in conjunction with CsA as a means of decreasing drug cost has been well accepted in human transplant patients.51 The two drugs have been used safely for up to 47 months in one study reducing the CsA dose as much as 88%.51 Although the effects emerge rapidly, with 62% of the effect is apparent by day 7, the maximum inhibitory effect may not be present for 12 months. This has implications regarding monitoring and supports the need for collection of peak and trough samples such that the impact of ketoconazole on drug elimination half-life might be determined.
The effect of combining ketoconazole with CsA has been studied in dogs.55 Ketoconazole was studied at doses ranging from 1.25 to 20 mg/kg per day, with the magnitude of inhibition increasing with the dose of ketoconazole above but not below 2.5 mg/kg.55 Clearance was reduced by 85% at 10 mg/kg per day. Differences in clearance did not result in significant differences in oral bioavailability. In their review, McNaulty and Lensmeyer56 indicated that in dogs, CsA half-life will increase over twofold and decrease the concentration of active metabolites in response to 2.5 to 10 mg/kg ketoconazole. Up to 75% of the CsA dose could be reduced experimentally in Beagles concurrently receiving a dose of ketoconazole of 13.6 mg/kg per day.57 In a study in dogs with perianal fistulae, Mouatt58 examined the impact of ketoconazole (10 mg/kg once daily) on CsA concentrations (1 mg/kg twice daily). Samples were collected 12 hours after the last dose (duration not clear). Data were not provided on all dogs, but the author noted that at 1.1 mg/kg twice daily of CsA, all dogs exceeded 200 ng/mL at through (12 hours); nine dogs exceeded 900 ng/mL, and two dogs exceeded 1500 ng/mL. Concentrations varied by 10% to 40% in the same dog despite no dose change. This may have reflected variability in time to steady state, which took 2 to 4 weeks. A trough concentration of 200 ng/mL was targeted. Although 8 dogs maintained concentrations of 220 to 520 ng/mL with CsA doses as little as 0.35 to 0.55 mg/kg twice daily, one dog required only 0.16 mg/kg twice daily to maintain 159 to 225 ng/mL, whereas three others required 0.95 to 1.1 mg/kg twice daily to maintain 120 to 235 ng/mL. This marked variability in response to ketaconozole appears to occur in clinical patients as is suggested by samples submitted to the author’s therapeutic drug monitoring laboratory. Marked variability occurs not only in concentrations, but in the time to steady-state, which in turn determines when monitoring should be implemented. For example, the author’s laboratory has demonstrated prolongation of the half-life of CsA in a patient from 4 hours (as reported in normal dogs) to over 150 hour. Steady state was not achieved in this patient until approximately 4 weeks, causing CsA concentrations to continue to increase despite a decrease in dose that was based on monitoring at 1 week. Yet, in other patients, half-life has been less than 12 hours, despite ketoconazole therapy. Detecting these diffrences requires collection of peak and trough samples such that half-life and time to steady-state can be determined in each patient.
The simultaneous administration of ketoconazole with CsA also has been studied in cats. McNaulty and Lensmeyer56 prospectively studied the impact of a two doses of ketoconazole (10 mg/kg at 24 and 0.5 hr) before a single dose of CsA (4 mg/kg intravenously) in cats (n = 5) using a randomized crossover design with a 14-day washout. Concentrations of CsA were detected using HPLC. Clearance was reduced from 2.73 ± 0.3 to 1.22 ± 0.18 mL∗kg/min, resulting in an elimination half-life prolongation from 10 ± 0.9 to 21 ± 3 hours.
Several studies have examined the impact of cimetidine, an inhibitor of CYP3A4, CYP2D6, and CYP1A2 on CsA concentrations. Cimetidine also is a substrate for P-glycoprotein. The results of the studies are contradictory, perhaps reflecting differences in duration of therapy and species. D’Souza and coworkers59 found that 5 days of cimetidine therapy significantly decreased CsA clearance, increased volume of distribution (perhaps by impacting P-glycoprotein in distribution tissues) and prolonged elimination half-life in rabbits, whereas Bar-Meir and coworkers60 found that short-term administration of cimetidine had no effect on CsA metabolism in rats. Shaefer and coworkers61 found that 7 days of dosing with cimetidine or famotidine did not affect CsA pharmacokinetics in healthy men. Daigle62 explored the impact of cimetidine (15 mg/kg orally every 8 hours for 8 days) on the disposition of CsA (5 mg/kg orally per day; the last 3 days of cimetidine administration) in dogs using a randomized crossover design with a 14-day washout. Significant differences could not be demonstrated for Cmax or area under the curve the power to detect a significant change was not addressed. The author’s therapeutic drug monitoring laboratory has identified some patients for which a cimetidine–CsA interaction may be present, but, other patients for which no effect could be measured.
Among the other drugs that impair CsA elimination or compete with efflux pumps include itraconazole, the calcium channel blockers such as diltiazem, and the macrolide antibacterial erythromycin.50 In humans, diltiazem decreases the CsA dose by approximately 30% to 50%.54 Other drugs that influence CYP activity should be expected to potentially affect CsA. For example, in the author’s laboratory, a cat receiving azithromycin (5 mg/kg once daily) along with CsA (5 mg/kg twice daily), exhibited peak concentrations exceeded 4500 ng/mL, presumably in part resulting from an elimination half-life that exceeded 150 hours. Azithromycin also competes for P-glycoprotein. Other drugs that induce P-glycoprotein might decrease CsA concentrations. For example, P-glycoprotein is upregulated in the duodenum of dogs receiving glucocorticoids.62a
Side Effects
CsA is characterized by a narrow therapeutic index in human patients, with renal toxicity the primary adverse effect. Renal tubular cells develop hyperuricemia (worsened by diuretics) and hyperkalemia (a renal tubular and erythrocyte ion channel effect).63 Hepatic injury also occurs. Although less common than renal dysfunction, the risk of severe hepatic damage is markedly increased when CsA is used in combination with cytotoxic drugs. CsA also increases the incidence of gallstones in human patients. However, in contrast to humans, renal and hepatic toxicities do not appear to be in dogs and cats. Risk factors, such as concurrent administration of nephroactive or nephrotoxic drugs, or renal transplantation have not been addressed in dogs or cats. Hyperlipidemia also has been a reported side effect in humans, particularly in patients receiving glucocorticoids. Other side effects reported in humans include neurotoxicity, gastrointestinal upset, and hypertension.4 Development of B cell lymphoma also has been reported in humans; an incidence 10% has been reported in cats undergoing renal transplantation, with mean time to onset of 9 months. 63a Side effects in dogs include gastrointestinal upset and dermatologic or mucosal abnormalities. Vomiting may occur in up to 40% of dogs, although it may be intermittent and short in duration. Diarrhea occurs less commonly (16% to 18%).Vomiting may be more likely when CsA is combined with ketoconazole, but this may reflect higher CsA concentrations. Dermatologic abnormalities reported in dogs include hair loss (possibly reflecting hair growth and pushing of hair from follicles), gingival hyperplasia, and gingivitis in up to 33% of dogs.58 In one study,58 hair loss resolved by 7 weeks, although hairy coats were thinner at study end (see Therapeutic Use). Hyperplasia may reflect an imbalance of fibroblast formation and collagen degradation by collagenase, although stimulation of TGF-β also has been suggested.33 Stimulation of TGF-β also stimulates extracellular matrix and decreases degrading proteases. Treatment with antimicrobials (metronidazole, spiramycin) may be useful for treating hyperplasia.58 Skin healing might be supported rather than inhibited, as occurs with glucocorticoids.67
KEY POINT 31-13
Drugs intended to increase cyclosporine A concentrations are likely to differentially affect animals; both peak and trough concentrations should be measured to determine the full impact.
At the doses used to treat atopy, risk of infection does not appear to be increased in dogs. However, the risk apparently has not been assessed at the higher doses used to treat immunosuppressive diseases. A report of central nervous system (CNS) toxoplasmosis has been reported in two cats receiving CsA (3 mg/kg twice daily for approximately 5 weeks and 6 mg/kg twice daily for approximately 8 weeks, respectively); presenting signs were respiratory in nature. Both had have previously been treated with glucocorticoids for a substantial time before diagnosis. A fatal case of toxoplasmosis was reported in a cat afflicted with eosinophilic granuloma complex receiving CsA at 5 mg/kg per day for a month, which was reduced because of anorexia to every other day. After 6 months of therapy, the patient developed fatal acute hepatic failure when the dose was increased to once daily. Toxoplasmosis was detected histologically in a number of tissues.67 CsA apparently inhibits insulin secretion in a dose-dependent manner in humans.33 Although this does not appear to be true in dogs, prudence dictates close monitoring of insulin needs in diabetic animals receiving CsA. 63b In rare cases neurotoxocity, characterized by reversible cortical blindness, has been reported in human patients receiving CsA after bone marrow transplantation.64
KEY POINT 31-15
The laboratory that will provide monitoring should be consulted regarding their therapeutic range. Therapeutic ranges should not be extrapolated from one laboratory to another.
Mouatt58 reported that 11 of 16 dogs receiving ketoconazole with CsA (Neoral solution) developed hair loss manifested as excessive shedding; five animals also developed metronidazole-responsive gingivitis with hyperplasia. Steffan65 compared adverse events associated with CsA (n = 119; 5 mg/kg per day induction followed by tapering doses) versus methylprednisolone (n = 59; 0.5 to 1 mg/kg/day for 1 week, then every other day for 3 weeks, then tapered) in dogs with atopic dermatitis. The incidence of selected adverse events differed between groups. The most common in the CsA group were gastrointestinal, for a total of 55%. Among these, the most common was vomiting at 37%, which was significantly more than the methylprednisolone group (5%), followed by soft stools and diarrhea (18%). Infections occurred with similar frequency between groups but were more likely to be classified as severe or very severe in the methylprednisolone-treated group and led to more dropouts in that group. Polyuria, polydipsia, and increased appetite were more common in the methylprednisolone-treated group. Gingival hyperplasia was reported in 3% of CsA patients (none in the methylprednisolone-treated group). Serum chemistries indicative of renal function did not change from baseline. Potassium increased by approximately 0.6 mg/dL in the CsA-treated group (and decreased in the methylprednisolone-treated group) but was within normal values.
In their meta-anlysis of clinical trials evaluating the efficacy of CsA for treatment of atopic dermatitis in dogs (10 trials, 672 dogs treated with CsA for a duration of 0.5 to 6 months), Steffan66 and coworkers reported that vomiting occurred in 25% of animals and soft stools or diarrhea in 15%. Remaining side effects occurred in less than 3% of the patients. The incidence of infection was not any greater for CsA-treatment groups and may have decreased in the skin and ear.
The commercial form of CsA intended for intravenous use has been associated with anaphylactoid reactions in the dog. The reactions appear to reflect the solubilizing agent. The vehicle of the intravenous preparation is very irritating and must not extravasate.
Monitoring
Monitoring of CsA concentrations can be an important tool to guide effective yet safe CsA regimens. The intent of monitoring should be to establish and maintain the therapeutic range for the patient, and to avoid toxicity or therapeutic failure, including that associated with drug interactions (see Chapter 5). Several points of controversy exist regarding monitoring. Reasons for monitoring in humans include, but are not necessarily limited to, avoiding nephrotoxicity; minimizing the risk of infection; and avoiding therapeutic failure, for which host-versus-graft rejection is the most important. In veterinary medicine monitoring might be implemented for the same reasons, but the need to avoid toxicity might take second stage to the need to assure effective concentrations in the face of marked variability in blood concentrations versus dose relationships. Identifying the intent of monitoring may influence down timing.
Peak, trough, or midinterval samples are variably collected by veterinarians using the author’s laboratory. Yet, the elimination half-life of CsA is short (approximately 5 to 8 hours). Accordingly, the drug generally does not accumulate or only minimally accumulates with multiple dosing. Therefore in most patients, no single sample can accurately predict the concentration throughout the dosing interval to which the patient is exposed. As such, the more critical the maintenance of effective doses throughout a dosing interval, the more helpful both a peak and trough sample might be. Detecting variability in absorption is best accomplished with peak concentrations whereas trough concentrations are influenced by both variability in absorption and elimination. Peak concentrations might be targeted if toxicity is of concern; trough concentrations might be targeted if a minimum effective dose must be maintained. However, trough concentrations run the risk of being nondetectable and failing to predict the marked variability that characterizes CsA absorption particularly with once daily dosing. The least useful sample is that collected mid-interval unless the patient is known to have a very long half-life. Note that if concentrations are to be compared across time (for example, to confirm that a dose increase resulted in higher concentrations), then care must be taken to collect subsequent samples at the same time point in the dosing interval. A difference of two hours can cause concentrations to fluctuate by 25% or more. The farther apart subsequent samples are and the shorter the elimination half-life of CsA in the patient, the less relevant are comparisons between samples collected across time.
KEY POINT 31-16
In addition to toxicity and efficacy, monitoring should be used to monitor drug interactions and to determine a patient’s therapeutic range once a response has been realized.
Among the important determinants of whether a peak or trough sample is collected is knowing which concentration most accurately predicts response: peak (generally collected at 2 hrs to assure absorption and distribution have been largely completed), trough (just before the next dose, indicating the lowest concentration to which the patient is exposed during an interval) or area under the curve. Timing of sample collection (peak versus trough) is an area of investigation in human medicine. Monitoring traditionally has focused on trough concentrations (discussed later). However, trough (C0, indicating the lowest concentration) correlates poorly to both graft rejection and acute nephrotoxicity.68 Thus peak concentrations (C2, indicating the highest concentrations anticipated at 2 hours) increasingly are becoming more relevant in humans, not only to predict efficacy but also to avoid toxic effects.69 Because toxicity is a lesser concern in animals, trough concentrations might reasonably be preferred to ensure that drug concentrations stay above a minimum effective range throughout the dosing interval. Indeed most animal-based recommendations are based on trough concentrations (see later discussion). However, with the short half-life which characterizes most patients, reaching target concentrations will be easier if a peak sample is consistently collected. This is particularly true if a 24 hour dosing interval is used, for example, when treating atopy. Yet, a half-life of less than 8 hours often results in 24-hour concentrations that are minimally or not detectable. As such, although dose labeled for chronic allergic inflammation (e.g., atopy, asthma), it should not be assumed to be appropriate for autoimmune disorders or graft versus host rejection. For animals in which the CsA half-life is longer than 24 hours, concentrations may not be at steady state. If so, monitoring to establish baseline clearly must be withheld until steady-state concentrations are achieved; monitoring both a peak and trough, however, may be necessary to establish when steady state will occur (see previous discussion).
The impact of drug interactions is another reason for CsA monitoring. If altered clearance is anticipated, elimination half-life is the most likely outcome measure to be impacted that can be monitored. In such instances both a peak and a trough concentration are recommended; if a drug interaction is anticipated (e.g., the addition of ketoconazole), monitoring ideally will occur prior to and approximately 1 week after the inhibiting drug is administered. If a peak and trough concentration cannot be collected, then consistent timing (either a peak or a trough) should be targeted, with a sample before and 1 week after the other drug is changed. Note, however, that if the impact of the drug is to prolong the elimination half-life more than 48 hours, concentrations may not be at steady state at 1 week. The only means by which the duration of time that must elapse before steady state is reached (and thus baseline can be established) is determination of half-life (i.e., both a peak and trough sample). Monitoring should be implemented each time a drug that might alter the disposition of CsA is added or subtracted from the drug armamentarium for the patient.
A second point of concern is the length of time after CsA therapy has begun that monitoring should be implemented. In general, the half-life of CsA is short enough that monitoring can be determined in 3 to 5 days. However, response to CsA may not occur that rapidly. Accordingly, if the goal of monitoring is to establish a therapeutic range in a responding animal, then monitoring is best implemented once a clinical response has been realized This is particularly relevant in the absence of concentration-response data in dogs or cats. The time to response (not resolution) is less clear, but several days to 2 weeks might be reasonable for most targets of therapy. However, some disease may require a longer period for maximum response (e.g., 4 months for perianal fistulae).
KEY POINT 31-17
Recommendations for cyclosporine A concentrations are generally based on either a 2-hour peak or 12-hour trough; mid-interval samples are least helpful. Timing of sample collection should be consistent across time for the patient.
Among the limitations in CsA use in dogs and cats is evidence for the the actual concentration to be targeted, which in turn is closely related to timing of sample collection as well as the method used to detect CsA and the sample submitted. Guidelines offered in human medicine have served as targets for veterinary patients,63 although the appropriateness of extrapolation has not been addressed. The impact of the assay reflects the presence of active metabolites (AM1, AM9) which may contribute up to 10 to 20% of activity in humans (more at trough concentrations); the concentration may be even greater if the metabolites accumulate. Several methods are available for measuring CsA concentrations, including HPLC, which generally detects only the parent compound. However, the contribution of active (and potentially toxic) metabolites has led to methods that detect both parent and metabolites.39 The more common methods are immunoassays based on antibodies directed toward the parent (monoclonal) or parent and metabolites (polyclonal). These include radioimmunoassays (RIAs) and polarized immunofluorescence, which detect parent drug and metabolites. Immunoassays that are monoclonal (e.g., FPIA by Abbott TDx or ACMIA by Seimens) are less likely to detect in activemetabolites compared with polyclonal antibody-based assays. Either type of assay (HPLC versus antibody based) is acceptable; the choice might depend on cost or availability. The advantages of antibody-based assays are enhanced quality assurance and rapidity of turnaround time. The cost also should be lower. Each assay appears to be clinically useful. However, the target concentrations will vary with the methodology as well as the timing of the sample (i.e., peak or trough) and the sample collected (e.g., whole blood, plasma, or serum). Antibody-based assays will be accompanied by higher concentrations, with polyclonal concentrations higher than monoclonal. For example, in dogs blood concentrations measured by TDx assay are 1.5 to 1.8 times higher than those measured with HPLC assay.33,44 However, this increase reflects, in part, detection of active metabolites; as such, monoclonal antibody-based assays that detect the metabolites might be the preferred type. Assays based on HPLC that determine the parent only will have the lowest therapeutic range; HPLC assays that include the metabolites will be higher. A disadvantage of HPLC assays is variability in results among laboratories. Even if similar methods are applied, variability in conditions can result in different concentrations. Thus the importance of using external quality-control programs will be particularly important for laboratories that use HPLC methods.
The sample submitted for CsA quantitation will also influence the concentrations and the therapeutic range. Most CsA in whole blood is located in red blood cells. Accordingly, whole blood is generally the preferred sample. Concentrations based on whole blood will be higher than those measured in plasma or serum. The most important point to be made regarding recommended therapeutic ranges is that they are laboratory specific, varying with sample timing, tissue collection, and assay method. If the laboratory is using an automated system, then the therapeutic ranges of that laboratory will be those established for the instrument. If the laboratory is using HPLC, then the ranges may be specific for that laboratory and only that laboratory. As an example of what might be expected, in humans recommended trough (before next dose) CsA concentrations (ng/mL) are as follows: for HPLC 100 to 300 (whole blood); RIA, monoclonal antibody methodology 150 to 400 (whole blood) or 50 to 125 (plasma or serum); RIA, polyclonal antibody methodology 200 to 800; fluorescent polarized immunoassay 250 to 1000. These numbers are offered as examples of therapeutic ranges. However, therapeutic ranges might also vary with the disease being targeted. A longitudinal replicate study examining differences between laboratories found marked variability within laboratories,70 suggesting that intralaboratory variability is a major contributor to overall variability. Because of these reasons, care should be taken not to extrapolate results based on the methodology unless the laboratory fails to do so. However, the most important point to be made regarding therapeutic ranges is that this population statistic is a guide only. Monitoring should be used to determine the patient’s therapeutic range. Once response has been realized, a sample should be collected, the concentration identified, and subsequent samples collected at that same time during the dosing interval.
Monitoring is generally based on 12 hour dosing. The dosing regimen recommended for treatment of atopy71 should not be assumed to be effective for treatment of immune-mediated diseases. In humans, for which CsA is usually administered by mouth every 12 hours, estimates of drug exposure using the area under the time concentration curve appear to be the most accurate pharmacokinetic measure on which to based dosing in human patients subject to acute rejection episodes. However, the description of area under the curve requires multiple sample collection, which is not only inconvenient but also costly. Two established therapeutic drug-monitoring protocols have been used in humans: the traditional approach, based on trough blood concentrations (C0), and the newer strategy, based on a sample taken 2 hours after oral administration (C2). Because the greatest contributor to interindividual variability in CsA area under the curve in humans is absorption, monitoring that focuses on peak concentrations is evolving as more predictive for CsA. As was noted previously, the risk of graft-versus-host rejection was markedly reduced when CsA dosing was based on peak rather than trough concentrations in humans. However, the use of this parameter as a predictor of CsA exposure is based on a 12-hour dosing interval.72 These recommendations are made regardless of the method of CsA detection (mRIA, EMIT, AxSym, CEDIA, or TDx/Architect) (Abbott FPIA). Mehl and coworkers45 investigated the relationship between peak and trough plasma drug concentrations and area under the curve in cats receiving CsA. After 14 days of dosing, 2-hour peak concentrations correlated better than the 12-hour trough concentration with area under the curve during that same time period, which suggests that a 2-hour peak sample is the preferred sample for monitoring.45 However, this study was based on predicting blood CsA concentrations and not predicting response.
A plethora of literature is dedicated to determining the best peak or trough target concentrations in humans receiving CsA, primarily for transplant patients but also for those with certain chronic allergic inflammatory disorders. For example, 12-hour concentrations, which probably do not accurately predict either rejection or toxicity but are reasonable for maintenance, range from 500 to 800, depending on the author. A review of the literature reveals that the range might also vary during the stage of disease, with initial concentrations for immune-mediated diseases being substantially higher, whereas lower concentrations are acceptable as targets for maintenance once response has been realized. Most recommendations in humans are based on monoclonal antibody assays.72
The consensus among most investigators is that area under the concentration versus time curve is the best predictor of acute or chronic graft rejection and toxicity.69 Mahalati and coworkers68 demonstrated that the initial area under the concentration-time curve (C0 and C4 ; or AUC0-4) was a useful tool: ranges between 4400 and 5500 ng/mL per hour for the first 4 hours of dosing significantly reduced the risk of acute rejection and nephrotoxicity in humans. This parameter is more useful with newer microemulsion products because CsA disposition is more predictable with these products. For this study samples were collected at 0 (just before the next dose), 1,2, 3, and 4 hours. A similar approach might be considered for animals during the early, critical stages of immune-mediated disease, with sample collection at 0 and 4 hours. However, collection of sufficient samples to describe the curve completely is generally not practical in either human or veterinary patients. For single point prediction, 2-hour posttreatment (C2) collection has repeatedly been demonstrated to be the most accurate predictor of area under the curve in humans, although even this time point is controversial. However, Einecke and coworkers69 demonstrated that C2 concentrations were not helpful in identifying patients at risk for rejection, but ranges between 500 and 600 ng/mL (whole blood, CEDIA antibody-based assay) were useful to maintain patients for long periods and avoid toxicity. This study suggests that the target range should vary as treatment moves from the induction to the maintenance mode.
Studies correlating CsA concentrations with efficacy are limited in animals. Perhaps the most supportive is with regard to perianal fistulae. Mouatt58 found that dogs with perianal fistulae (n = 14) responded more rapidly if trough concentrations exceeded 600 ng/mL, but time to resolution (generally 4 weeks) could not be predicted by concentrations in this small group of dogs.
Use of monitoring for atopic dermatitis is less clear. In a manufacturer-sponsored study, Steffan44 and coworkers investigated the relationship between CsA concentrations and response in dogs (n = 97) with atopic dermatitis. Concentrations were measured between 2 and 24 hours of the last dose at 28 days, and this single point concentration was used to predict the time course of CsA, based on a model generated from normal dogs (n = 16). Patients were then grouped into one of five categories of CsA exposure, ranging from very low to very high CsA. Clinical response to therapy was based on scoring of skin lesions (canine atopic dermatitis extent and severity index [CADESI]) and pruritis. Response to therapy (CADESI and pruritis scores) was then compared (not correlated) among groups. The study could not demonstrate a differenc in response among the groups, leading the authors to conclude that concentrations did not correlate with response and monitoring was not indicated. However, this study should not necessary be considered evidence against monitoring in dogs with atopic dermatitis. First, a single time point of CsA was used to predict 24-hour exposure of CsA for clinical patients. Second, the model used to predict CsA was based on normal Beagles, of which only eight had received multiple dosing of CsA. Third, variability in the clinical patients was greater than in normal animals, and the predicted model differed from the observed. The variability in peak CsA, in particular, found in this study is profound, and its impact on therapeutic success should be examined. Variability also occurred in pruritis scores, being equal or greater than the median, which may have impacted the power to detect a significant difference was likely low. Because peak concentrations appear to occur in most animals between 1 and 2 hours, a study that correlates actual peak concentrations with scores might be prudent before the use of CsA monitoring for anticipating response of atopic animals to therapy is set aside. Studies are indicated to determine the best sampling time for dogs receiving CsA. Because concentrations are so low at 24 hours and often undetectable, it is likely that some other sampling period is appropriate for 24-hour dosing intervals.
It is important to emphasize that studies that fail to correlate CsA concentrations with therapeutic response do not prove the lack of or preclude the usefulness of monitoring. In ability to demonstrate a significant difference does not equate to “no treatment” effect. Studies may fail to find a connection because of limitations in study design. In the event that a sufficiently well-designed study (or several studies) should fail to find a correlation, the clinician must remember that the most important reason for monitoring is to establish a therapeutic range for the patient. Once the patient has responded, monitoring might be most useful to maintain that concentration in the patient.
In summary, our laboratory offers the following recommendations for monitoring of patients treated every 12-hours: a 2-hr peak and just before the next dose trough sample is recommended within 3 to 5 days of initiating therapy; the more life threating the target disease, the more important a peak and trough sample may be. For less serious situations, or as treatment shifts from induction to maintenance, a singe 2-hr peak sample or, assuming bid dosing, a single 12 hour trough concentration may be sufficient for establishing and maintaining a target. If therapy is initiated such that CsA disposition might change (whether intentional, such as the addition of ketoconazole, or inadvertent, such as co-treatment with diltiazem or azithromycin or others), a peak and trough sample prior to and 1 week after therapy is initiated is suggested. In situations in which alternatve generic preparations are initiated, a single 2-hr peak concentration before and 3 to 5 days after the switch is recommended. For atopy (24 hr and beyond dosing), a single peak sample should be collected. Likewise, if toxicity is a concern, a single peak sample is indicated. However, in patients with a long half-life, both a peak and trough sample may be necessary to fully assess the risk of toxicity. Monitoring is recommended weekly to biweekly in critical patients, then monthly for the first several months of therapy or until concentrations are stable. For long-term maintenance, the frequency of samply may vary with the stability of the patient but should range from 3 to 6 months. Target concentrations vary with the condition but generally, based on a 12-hr dosing interval using a monoclonal antibody-based assay (i.e., as in the author’s laboratory), a peak concentration of 800 to 1400 ng/mL and a trough concentration of 400 to 600 ng/mL (monoclonal based assay) is recommended for immune mediated diseases. For renal transplantation, trough concentrations of 750 ng/mL are suggested for the first month and 350 to 400 ng/mL, thereafter. For chronic allergic inflammatory disorders, lower concentrations are recommended: 250 ng/mL trough concentrations for chronic inflammatory bowel disorders, and for perianal fistulae, 12 hour trough concentrations at 100 to 600 ng/mL (the higher for induction, the lower for maintenance).
Therapeutic Use
Chronic Allergic Inflammatory Disorders
Atopy and other dermatologic conditions
The use of CsA for treatment of veterinary dermatologic syndromes has been reviewed.73 Olivry and Mueller74 reviewed treatment of atopic dermatitis in dogs in general and found good evidence for efficacy of glucocorticoids and CsA compared with fair evidence for topical triamcinolone, topical tacrolimus, oral pentoxifylline, and oral misoprostol. In the manufacturer-sponsored study, Steffan and coworkers65 prospectively compared the efficacy of CsA (n = 119) and methylprednisolone (n = 59) in dogs with atopic dermatitis using a randomized, blinded (investigators but not owners) parallel study. Dosing began with an induction phase for both drugs, followed by a tapering phase, with the dose determined by CADESI scores. Dosing for CsA was 5 mg/kg once daily for 4 weeks, then tapered on the basis of the CADESI score, and for methylprednisolone, 0.5 to 1 mg/kg daily for 1 week, followed by every other day for 3 weeks, and then tapered on the basis of the CADESI score. Washouts were required for drugs that might influence response. At the end of the 4-month study period, the percentage of reduction in CADESI scores was approximately 45% in both groups. The proportion of responders (CADESI scores reduced by ≥ 50%) was approximatey 58% for methylprednisolone and 66% for CsA; a significant difference could not be demonstrated. Scores worsened in approximately 15% of both groups.
More recently, Steffan and coworkers66 reviewed clinical trials evaluating the efficacy of CsA for treatment of atopic dermatitis in dogs. Ten trials met their standards; 799 dogs were treated with either CsA (n = 672 for a duration of 0.5 to 6 months), placebo (n = 160), oral glucocorticoids (n = 74), or antihistamines (n = 23). Further, safety data were available for 660 dogs. Among the concerns the authors addressed well was publication bias (the study was sponsored by the manufacturer). Treatment success was defined as a 50% or higher reduction in lesion scores after the 4- to 6-week induction period (denoted as high responders). Compared with placebo, but not oral glucocorticoids, the effects of CsA were highly significant. Dosing intervals generally could be prolonged to 48 hours in 40% to 50% of responders after 4 weeks and to twice weekly in 20% to 26% of dogs after 12 to 16 weeks. Predictors of success could not be identified, although initial response was predictive of long-term response. Feeding, age, and body weight did not appear to affect success; the influence of breed could not be assessed. The dose ranged from 2.5 mg/kg twice daily to 5 mg/kg twice weekly. Doses generally begin at 5 mg/kg once daily. The treatment induction period was considered 4 to 6 weeks, although not all studies reviewed treated for that time period. The authors identified those aspects of the meta-analysis that were relevant for clinical practice. They concluded that the severity of skin lesions can be expected to decrease by at least 40% during the initial 4 to 6 weeks of induction therapy at 5 mg/kg. The benefits may be less pronounced in dogs that previously received glucocorticoids. The dose can be reduced, generally by prolonging the interval, after this reduction period if a response has been realized. If clinical signs worsen, the previous dose should be re-instituted. A further reduction in clinical signs may be realized after the 4- to 6-week induction period, with a plateau of 50% to 70% reduction occurring at 2 to 4 months. Unacceptable adverse events (most notably vomiting) should respond to dose reduction if related to the drug. What was not resolved by this meta-analysis is the impact of long-term therapy, as well as the sequelae of discontinuing therapy; response in animals that have not responded to glucocorticoids; and the impact of combination therapies, including topical agents.
Radowica and Power71 retrospectively studied the efficacy of CsA (5 mg/kg per day) when used for a minimum of 6 (range 6 to 30) months for treatment of atopic dermatitis in dogs (n = 51). Continued therapy was necessary to control clinical signs in at least 55% of dogs, with most of these animals receiving the drug 2 to 5 times weekly, but one out of five requiring daily therapy. Therapy could be discontinued in 24% of animals after 6 to 24 months as a result of clinical response. Adverse events occurred in 22% of animals, including oral growths or gingival hyperplasia and hirsutism.
Other dermatologic indications for CsA include, but are not limited to, perianal fistulae, atopy, and eosinophilic granuloma complex. Mathews and coworkers75 found that CsA was effective in eradicating or markedly reducing the size or number of perianal fistulae in 10 of 10 dogs. Initial dosing began at 10 mg/kg orally every 12 hours but was reduced to 5 to 7.5 mg/kg after 1 week because of excessive trough concentrations. Doses were adjusted to a trough measurement of 400 to 600 ng/mL. Duration of therapy ranged from 8 to 12 weeks; remission required another 4- to 6-week trial of therapy in 3 dogs. Remission was persistent in all dogs for at least 6 months and up to 18 months at the time the report was published. A follow-up study of a randomized controlled trial in 20 dogs76 found that fistulae recurred in 7 of 17 dogs treated with CsA and required subsequent treatment or surgical excision. CsA was, however, beneficial presurgically to reduce the extent of excision. The investigators also found that trough CsA concentrations between 100 and 300 ng/mL (HPLC) were effective for treatment of perianal fistulae. The most frequently reported side effect in this study was shedding of hair, which was noticeable by 16 weeks. Older hair coats tended to be replaced with a softer coat.
Dogs (16) with perianal fistulae were prospectively evaluated for the efficacy of CsA (1 mg/kg orally twice daily; administered as Neoral liquid administered in a gelatin capsule) when combined with ketoconazole (10 mg/kg orally once daily); blood concentrations of CsA (based on HPLC) were monitored to maintain trough concentrations above 200 ng/mL (FPIA 300 ng/mL). All dogs showed clinical signs of improvement within 2 weeks of therapy; animals whose concentrations were above 600 ng/mL responded more rapidly, but efficacy did not seem to differ, although the power of the study to detect a relationship between cure rates and drug concentrations was not addressed. Lesions resolved in 93% of the animals within the 16-week treatment period, with 50% of the animals remaining cured at 12 months. Recurrence in the remaining dogs occurred within 1 month (21%) or between 8 and 12 months (21%). Although both medications appeared to be well tolerated, 67% of dogs exhibited excessive hair loss that started at 2 weeks but stopped at 7 weeks; hair coats thinned but were normal by study end. Further, 31% of the dogs developed gingival hyperplasia and gingivitis, which responded to metronidazole and spiramycin.58
Inflammatory bowel disease
Allenspach and coworkers77 studied the efficacy of CsA (Atopica; 5 mg/kg once daily for 10 weeks) for treatment of canine IBD (n = 14) refractory to steroid therapy (prednisolone, the majority receiving 2 mg/kg orally per day for 6 to 14 weeks or more). CsA concentrations were measured in eight dogs with severe disease during a 24-hour period after the first dose; CsA was detected in whole blood using FPIA; a cohort of healthy dogs was available from a previous study (see Table 31-3). Animals were scored (canine IBD activity index [CIBDAI] criteria) on the basis of clinical signs and histopathologic lesions. Of the 14 dogs studied, eight completely responded within 4 weeks and three partially responded (posttreatment score of 2 to 4). Two failed to respond. Five of the 14 dogs with protein-losing enteropathy partially responded (posttreatment median score of 5). Although body weight increased, histopathologic lesions (nine dogs had repeated biopsies) did not. Overall, CsA was considered effective in 78% of the animals. Concentrations of CsA varied more in the diseased dogs (peak occurring at 1-2 hours of 699 ± 326 ng/mL compared ot 878 ± 131 ng/mL in healthy dogs), leading the authors to speculate on the role of previous glucocorticoid therapy, which may have increased P-glycoprotein, thus decreasing absorption. Elimination half-life in the respective diseased versus healthy populations was 5.6 ± 1.2 and 7.8 ± 1.1 hr, respectively. However, the authors did not describe the CsA concentrations in all nonresponders, although one of them had the highest peak and trough concentrations measured in the study. Adverse events not evident before initiation of therapy were vomiting, anorexia (improved by administering with food), gingival ulceration (one dog; resolved after 7 weeks), and alopecia followed by hypertrichosis (full replacement of the hair coat) in the first 2 weeks of therapy.
CsA appeared to act synergistically with dexamethasone for the treatment of IBD in human patients. Each drug appears to target through different mechanisms the chemokines responsible for migration of white blood cells to the site of inflammation.78 In humans a 12-hour trough concentrations of 150 to 250 ng/mL has been recommended for CsA when treating IBD. 79
Asthma
Other potential indications for CsA warrant further investigation. Cats suffering from A. suum–induced airway reactivity had decreased reactivity and remodeling after receiving CsA; differences were noted with 24 hours of therapy.80
CNS Disorders
Adamo and O’Brien84 reported on the successful use of CsA for treatment of granulomatous meningoencephalitis (GME) in a series of three cases. The initial dose of 3 mg/kg twice daily was increased to 6 mg/kg twice daily. Concentrations of CsA (ng/mL) were 235, 337, and 592 (unfortunately, sample timing not provided); variability was assumed to reflect, in part, differences in sampling times, which were not provided. In one dog, the initial dose of 3 mg/kg twice daily yielded CsA of 82 ng/mL (timing not provided); the dose was increased to 10 mg/kg twice daily. This dog did not sufficiently respond, developed azotemia and an E. coli urinary tract infection; on necropsy, disseminated GME was diagnosed. A different dog also was initiated at 3 mg/kg twice daily; CsA was 117 ng/mL, and the dose was increased to 6 mg/kg twice daily, which was subsequently decreased back to 3 mg/kg after 7 months, resulting in CsA at 215 ng/mL. The dose was tapered to 3 mg/kg once daily, yielding CsA at 63 ng/mL. Several attempts to detect CsA in cerebrospinal fluid revealed no detectable drug; however, its large size and the presence of P-glycoprotein is likely to preclude CNS penetration by CsA. However, efficacy of CsA is not likely to depend on penetration of the CNS. On the basis of this series, 6 mg/kg twice daily might be considered until animals with GME are in remission, with the dose tapered to 3 mg/kg twice daily on response. In humans, CsA has proven useful for treatment of chronic inflammatory demyelinated polyradiculoneuropathy; concentrations were not provided.84a
Immune-Mediated Disease
Systemic use of CsA increasingly is being used for treatment of autoimmune disorders, particularly in those that have not responded to traditional immunosuppressive therapy. However, the labeled doses for atopy should not be assumed to be relevant for other immune-mediated diseases, including dermatologic.
Noli and Toma81 reported the successful use of CsA for treatment of immune-mediated adnexal skin disease in a series of cases (n = 3; two cats, one dog). In each case response occurred within 1 month of administration at 5 mg/kg once daily, CsA therapy was successful in treating aplastic anemia in three of four dogs82 when dosed at 5 to 10 mg/kg every 12 hours (unfortunately, CsA concentrations were not provided).
Font and coworkers83 reported a single case of cutaneous lupus erythematosus that was successfully treated with 4 mg/kg CsA once daily along with ketoconazole (4 mg/kg) once daily. Prednisone therapy was continued at 0.2 mg/kg for 10 days. Lesions resolved in 2.5 months of CsA therapy. Concentrations of CsA associated with response at 10 days was 139 ng/mL; the timing of sample collection was not indicated, although it is possible in the presence of ketoconazole, that this concentration did not vary substantially during the dosing interval. Concentrations were 279 ng/mL at the time ketoconazole was stopped. The dog remained in remission at the end of the 18-month report period, receiving 2 mg/kg daily. The use of glucocorticoids, ketoconazole, and lack of information regarding the collection of samples complicates correlating concentratiosn to response. A randomized placebo-controlled study on the use of CsA for treatment of spontaneous glomerulonephritis in dogs46 failed to document a a significant improvement in treatment groups; the power of the study was not addressed. Packed cell volume was lower in the CsA-treated group; in addition, clinical signs compatible with decreasing renal function appeared to be more severe in animals receiving CsA.
With the availability of renal transplantation at several facilities, CsA is increasingly being used to prevent graft-versus-host rejections. Cats respond better than dogs, with renal allografts being maintained at 7.5 mg/kg every 12 hours coupled with prednisolone (0.125 to 0.25 mg/kg every 12 hours).46 Trough concentrations of whole blood should be maintained at 500 ng/mL (HPLC; approximately 750 ng/mL should be expected for FPIA) the first month after transplantation but can be reduced to 250 (375 to 400 FPIA ng/mL) thereafter.
Topical CsA has been used with some success for localized immune-mediated diseases, including pemphigus foliaceus. An ophthalmic preparation is approved for use in dogs to treat keratitis sicca..85 Benefits include both immunomodulation and an increase in tear production. A 0.2% CsA compound in a petrolatum–corn oil ointment is commercially available (Schering-Plough) for keratitis sicca in dogs. Application of one drop of the commercial preparation twice daily should be effective in up to 80% of dogs. The frequency of administration can be decreased to every other day for most dogs.
When compared with dexamethasone and indomethacin, topical CsA was equal in suppressing arachidonic acid–induced inflammation,86 and studies are under way to evaluate its use in canine dermatologic diseases.
Tacrolimus and Related Drugs
Tacrolimus, like CsA, is a macrolide antibiotic, produced by Streptomyces. Like CsA, it inhibits T cell activation, but rather than binding to cyclophilin, it binds to and inhibits an alternative cytosolic protein, FKPB12. Calcineurin-dependent activation of lymphokine expression, apoptosis, and degranulation is inhibited.4 The intracellular receptors for tacrolimus are distinct from those for CsA and it is 500 fold more potent than CsA in its effects. This may reflect, in part, a 3-fold greater uptake in lymphocytes for tacrolimus compared to CsA (however CsA uptake in RBC is 10-fold higher than tacroliumus).86a The drug is available for both oral and intravenous systemic administration. Toxicities are similar to those of CsA in humans, although gingival hyperplasia may be less prevalent. The drug may be more toxic than CsA in dogs, and its systemic use has not been recommended.87 Tacrolimus disposition has been reported in male Beagle dogs receiving 1 to 4 mg/kg.87a Peak and trough concentrations (ng/mL), respectively, ranged from 0.8 to 1.8 ng/mL (at 1 mg/kg) to 3.2 to 5.3 ng/mL (2 mg/kg) and 0.25 to 0.8 ng/mL (1 mg/kg) to 0.2 to 2.3 ng/mL (2 mg/kg). For tacrolimus and sirolimus, trough (before the next 12-hour dose) rather than peak concentrations more effectively predict area under the curve.72 Tacrolimus is also available as a topical ointment. The 0.1% preparation has been used in dogs to treat localized lesions associated with discoid lupus erythematosus as well as two cases of pemphigus erythematosus either as a sole therapy or adjunctive treatment.88 No side effects were reported, and all animals improved after 8 weeks of topical application, with adjuvant therapy being discontinued in the majority of animals. Tacrolimus ointment also has been used effectively for the treatment of canine atopy.89 Using a double-blinded, placebo controlled, cross-over study, dogs (n = 12) were treated with topical 0.1% tacrolimus (maximum dose of 0.1% mL/kg/day) applied topically at sites considered most pruritic for 4 weeks. Following a 2-week washout (based on a 9-hr half-life of tacrolimus in dogs), animals were switched to the alternative treatment. The randomization procedure was not robust (alternating treatment assignments). The mean clinical score improved in both the placebo and the treatment group at 4 weeks compared to baseline, but response was significant for the tacrolimus group. Tacrolimus was detectable in patients (peak concentration at 4 weeks of 1.4 ± 0.2 ng/mL). However, peak concentration of 0.6 ng/mL was also detected at week 4 in the placebo group calling into question the credibility of the report. Tacrolimus may impact intradermal skin testing in dogs, with an effect on the late phase, but not the immediate response. 89a
Griffies and coworkers88 reported on the successful use of tacrolimus (0.1%) topically in a series of cases in dogs. The drug was used as either sole agent or as an adjuvant topically for treatment of localized lesions associated with discoid lupus erythematosus or pemphigus erythematosus. Improvement required approximately 8 weeks of therapy, and adjuvant therapy could be discontinued in some dogs.
In contrast to tacrolimus, sirolimus and its derivative, everolimus, do not inhibit calcineurin; rather, they interact downstream from IL-2, targeting the immunophilin with FKBP12. This complex then inhibits mTOR (mammalian target of rapamycin), which prevents progression from the G1 to S phase transaction in the cell cycle, in a manner that prevents cytokine receptors from activating the cell cycle and subsequent signaling.90 Its use is primarily replacement of CsA to protect renal function in humans.
Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is the morpholinoethyl ester prodrug of mycophenolic acid (MPA), which is a product of several Penicillium species that possess antibacterial, antifungal, antiviral, antitumor, and immunosuppressive properties. MPA is a potent, selective, noncompetitive, reversible inhibitor of inosine monophosphate dehydrogenase (IMPDH). This enzyme is critical for the production of the guanine triphosphate precursor guanine monophosphate, which in turn is necessary for the de novo synthesis of purine nucleotides. As such, the drug is an antimetabolite. Two isoforms of human IMPDH exist. Type I is constitutively expressed in normal, nonreplicating cells, and type II is upregulated in replicating (including neoplastic) cells to the point that it is the predominanting isoform. As IMPDH is inhibited, guanine triphosphate is depleted; failure to make mRNA precludes synthesis of proteins, including cytokines, necessary for cell proliferation. Unlike other anticancer antimetabolites, MMF and its active metabolite MPA are relatively selective for lymphocytes because they are solely dependent on de novo synthesis of purines for DNA synthesis.91,92 MMF is the first drug since CsA to be approved in the United States for prevention of renal allograft rejection; it has proved useful for treatment of steroid-resistant acute liver rejection.93 In addition to its antiproliferative properties toward lymphocytes, MPA also impairs proliferation in nonimmune cells, including smooth muscle cells, renal tubular cells, mesangial cells, and dermal fibroblasts.94 Consequently, MMF might be indicated for non–immune-mediated diseases associated with fibrosis.
A bilateral nephrectomized dog model has been used to study MMF.91 The disposition appears to be characterized by marked variability in dogs. After doses of either 20 or 40 mg/kg, mean peak concentrations of MPA were, respectively, approximately 90 and 130 μg/mL, yet peak concentrations did not statistically differ between the two groups. Based on canine lymphocytes exposed to whole blood containing MPA, 200 μg/mL MPA is necessary to inhibit baseline activity of IMPDH by 50%. The elimination half-life of the drug ranged from 1.45 to 11.09 hours, with a mean of approximately 7 hours, regardless of the dose. The drug also has been studied in normal dogs at approximately 20 mg/kg, Maximum concentrations of MPA after intravenous (2-hour infusion) and oral administration, respectively, were 21 ± 3.6 and 11.8 ± 6.6 μg/mL; oral bioavailability was 54%.95 The elimination half-life of MPA was about 45 minutes, and mean resistance time was approximately 2 hours. The half-life is substantially shorter than that reported in humans (17 to 19 hours). However, suppression of IMPDH lasted for a mean of about 6.5 hours,96 suggesting an 8-hour dosing interval might be appropriate. In human monitoring of trough concentrations has been recommended: trough (predose) concentrations less than 1 μg/mL were associated with acute liver transplantation rejection, whereas concentrations greater than 3.5 μg/mL were associated with a threefold increase in the risk of leukopenia or pneumonia. Therefore trough concentrations between 1 and 3.5 μg/mL are recommended.90 It may be necessary to increase the dose of MMF in patients with low albumin; the dose had to be increased in human patients by approximately twofold if serum albumin was less than 3.5 gm/dL. However, the dose could be decreased by approximately 40% in the presence of renal dysfunction.90
KEY POINT 31-18
Mycophenolic acid targets both T and B lymphocytes and accordingly should be associated with greater immunosuppression compared with cyclosporine A.
The primary side effects in human patients receiving the drug for allograft rejection are leukopenia, gastrointestinal upset, and cytomegalovirus disease. The incidence is, however, small. Gastrointestinal upset is characterized by nausea, diarrhea, vomiting, and abdominal cramping. Gastrointestinal toxicity (and bone marrow suppression) may reflect the need for IMPDH II because of the normally replicating nature of these tissues.97 The drug also appears to be relatively safe in dogs, on the basis of renal and hepatic indices of damage, even when dosed at 80 mg/kg twice daily. At that dose, transient increases in hepatic damage enzymes occurred.98 However, when used chronically at 30 mg/kg orally every 12 hours, severe diarrhea, anorexia, and weight loss occurred in dogs95; at 20 mg/kg orally every 12 hours, signs recurred, although the were less severe, within 1 week of initiating therapy. One dog died acutely with necrotizing pneumonia 7 days after initiation of therapy.96 Dysgranulopoiesis, characterized by pseudo Pelger–Huet anomaly has been reported in a series of human cases receiving MMF for heart–lung transplantation; resolution occurred only after the drug was reduced in dose or discontinued.99
Among the indications for MMF is dose sparing of CsA, which probably is more clinically relevant in humans compared with dogs or cats.100 Initial induction with CsA is followed by the addition of MMF and down titration of CsA. MMF has been used therapeutically in dogs both clinically and experimentally. Clinically, the drug has been used successfully to treat myasthenia gravis,101 although review articles offer anecdotal support for use in other immune-mediated disorders. Experimentally, MMF (10 mg/kg subcutaneously every 12 hours) in combination with CsA (15 mg/kg orally every 12 hours) has been used in dogs to prevent renal allograft rejection.102
Leflunomide
Leflunomide is a recently approved immunomodulatory drug that inhibits dihydroorotate dehydrogenase, an enzyme responsible for the de novo synthesis of pyrimidine. Both T and B cell proliferation is inhibited; accordingly, both cell-mediated and humoral responses are targeted. Cell adhesion also impaired. Other proposed mechanisms are inhibition of cytokine and growth factors mediated by tyrosine kinase. Like MMF, it is a prodrug, with its activity reflecting a malononitrile metabolite (A77 1726). It has been studied for the treatment of rheumatoid arthritis in humans.103 The half-life of the active compound is long in humans, ranging from 1 to several weeks, indicated a long time to steady state. However, a recent disposition study in dogs reveals the elimination half-life of the metabolite to be 14 hrs in the dog. Limited information is available regarding its use in dogs.104 Anecdotal reports suggest that the drug is effective for treating a number of immune-mediated disorders, including immune-mediated hemolytic anemia (IMHA), immune-mediated thrombocytopenia, canine cutaneous histiocytosis, and pemphigus foliaceus (2 to 4 mg/kg orally every 24 hours). Side effects that reportedly occur in dogs include leukopenia, thrombocytopenia, and gastric ulceration. Gregory and coworkers104 reported on the use of the drug (1.5 mg/kg twice daily to 2 to 4 mg/kg once daily) in dogs (n = 29) afflicted with various immune-mediated disorders. These include idiopathic thrombocytopenic purpura (n = 3; good to excellent response), IMHA with idiopathic thrombocytopenic purpura (Evans syndrome), systemic histiocytosis (n = 3; one failure and two total remissions), multifocal nonsuppurative encephalomyelitis (n = 5; good to excellent response), and a variety of other immune-mediated disorders that had not responded to glucocorticoids or azathioprine. Most of the animals had not responded to first- or second-tier immunosuppressants. Side effects included decreased appetite, mild anemia, and bloody vomitus or stools (n = 3). Leflunomide was studied at doses ranging from 2 to 16 mg/kg/day as sole therapy or in combination with CsA (10 mg/kg/day) in dogs undergoing experimental reanl transplantation104a Rejection occurred at 4 mg/kg; mean survival time increased from 9 days (non-immunesuppressed dogs) to 16 days at 4 mg/kg, 28 days at 8 mg/kg. The addition of CsA at 10 mg/kg to leflunomide at 4 mg/kg increased survival time to 68 days. Mean trough leflunomide concentrations ranged from 10 μg/mL (2 mg/kg) to 55 μg/mL (16 mg/kg).104b A recent retrospective study examined the efficacy of leflunomide for treatment of naturally occurring immune-mediated polyarthritis in dogs. 104c At an initial starting dose of 3.0 ± 0.5 mg/kg orally once a day for 1 to 6 weeks, 8 of 14 dogs had complete resolution, and 5 partial resolution. No animal exhibited adverse reactions.
Emerging Drugs
The sphingosine-1-phosphate receptor (S1PR)105 antagonists are a new class of drugs that decrease recirculation of lymphocytes from the lymphatic system to blood and peripheral tissues. These drugs will not be used as monotherapy, but they have shown efficacy in human medicine when combined with CsA, glucocorticoids, and other drugs. Lymphopenia is the most common side effect and is reversible when the drug is discontinued. However, it may also act as a negative chronotrope.
Hormones
Glucocorticosteroids (see Chapter 30) possess both antiinflammatory and immunomodulatory capabilities. Immunomodulation results in actions at a variety of targets in acquired immunodeficiency syndrome. Selected progestational compounds (e.g., megestrol acetate) have been used for their general immunosuppressive effects. Megestrol acetate (discussed in Chapter 19) may have longer and more potent antiinflammatory effects compared with glucocorticoids106; however, in the cat, the species for which these products tend to be used, side effects can be dramatic and life threatening. Thus their use is recommended only for conditions that will not or have not responded to glucocorticoids. Medroxyprogesterone exhibits glucocorticoid-like effects, which reflect, in part, actions at the level of DNA transcription. The impact of glucocorticoids on DNA transcription was previously reviewed (see Chapter 30); those glucocorticoids that target trans (rather than cis) repression (rather than activation) are directed toward desirable effects, avoiding many undesirable metabolic effects. Medroxyprogesterone appears to potentially focus on undesirable effects inflammatory responses, being characterized by a preponderance of transrepression activity rather than transactivation or cis activity.19
Danazol (5 mg/kg 2 to 3 times orally per day) is classified as an androgen but is characterized by weak androgenic activity. In human patients danazol has proved effective in stimulating increased concentrations of complement inhibitor and thus is indicated for treatment of angioneurotic edema.107 Danazol also has immunomodulatory effects that are of benefit in type II immune complex diseases. Danazol apparently decreases the expression of or blocks Fc receptors on macrophages; initial displacement of glucocorticoids owing to competition of binding sites is less likely to be effective as glucocorticoid clearance increases.108-111 The drug has proved useful in a number of type II immune-mediated diseases, including IMHA (autoimmune hemolytic anemia) and immune-mediated thrombocytopenia.11,108,112,113
Cytotoxic Drugs: Antimetabolites and Other Antineoplastic Agents
Antimetabolite and alkylating antineoplastic agents (see Chapter 33) are also used as chemical immunosuppressants by virtue of their effects on actively dividing cells.114,115 Their effects should, however, be regarded as nonspecific. Macrophages, activated T and B cells, and NK cells are the targets of most of the drugs. Those most commonly used for their immunomodulatory effects are cyclophosphamide and azathioprine. In general, neither drug is sufficiently immunomodulatory and safe for use as the sole agent.
Cyclophosphamide is a nitrogen mustard. Its immunomodulatory effects reflect the same mechanism of action as its anticancer effects. Cyclophosphamide alkylates DNA in both proliferating and nonproliferating cells; proliferating cells are more susceptible to alkylation. Both B and T cells are impaired. Because B cells recover more slowly, however, cyclophosphamide inhibits the humoral response more than the cell-mediated response.4 Note that at very high doses cyclophosphamide can actually induce tolerance to an antigen to which the patient has been exposed. Toxicities of cyclophosphamide are typical of drugs that target proliferating cells. In addition, hemorrhagic cystitis has been reported in dogs receiving the drug. Cyclophosphamide therapy should be discontinued if the neutrophil or platelet counts decrease less than 2000/L or less than 100,000/L, respectively.
Azathioprine interacts with nucleophils to form 6-mercaptopurine, which is converted to nucleotides. The nucleotides interfere with purine synthesis or cause DNA damage. The drug is available for both oral and intravenous administration. Co-administration with allopurinol increases the risk of toxicity. Toxicity results from inhibited growth of rapidly growing cells, including cells of the bone marrow and gastrointestinal tract. The association of thiopurine methyltransferase (TPMT) activity and risk of azathioprine-induced neutropenia in dogs is not clear. Difficulty in identifying a relationship is compounded by the low incidence of deficiency (not detected in 470 dogs). The lack of relationship is supported by the finding of severe azathioprine-associated myelosuppression (marked leukopenia, neutropenia, or thrombocytopenia) in six clinically ill dogs classified as nondeficient. Further, an intermediate classification of TPMT (14 to 38 nmol/gHb/hr) in hunting dogs was associated with a decrease in white blood cells after 30 days of azathioprine therapy (2.2 mg/kg orally once daily) but not severe clinical signs of bone marrow depression. As such, a TPMT deficiency may predispose to azathioprine toxicity, but other factors appear to play an important role.116 Cats are much more sensitive to the toxic effects of azathioprine. Although the drug has been used in cats (0.3 mg/kg daily), reformulation may be necessary for accurate dosing. Care must be taken to ensure that the dose is compounded accurately.
Chlorambucil (starting oral dose of 0.1 to 0.2 mg/kg/day) is also a nitrogen mustard, and its cytotoxic effects are similar to those of cyclophosphamide. The drug is available in an oral preparation that allows accurate dosing in cats.106 In contrast to cyclophosphamide, chlorambucil’s myelosuppression is only moderate and gradual. Toxicities are rare in cats.106 The incidence of gastrointestinal side effects can be reduced by alternate-day dosing. In human patients, however, excessive doses can cause hypoplastic bone marrow. Its effects (e.g., in chronic lymphocytic leukemia) are gradual in onset.117 Therapeutic indications include chronic, refractory immune-mediated disorders, including pemphigus foliaceus and refractory cases of feline granuloma complex.
Vincristine is used to treat immune-mediated thrombocytopenia because of its ability to cause the maturation and release of mature thrombocytes from the bone marrow by stimulation of megakaryocyte endomitosis. Other anticancer drugs used for immunomodulatory effects include methotrexate, and vinblastine. The nonspecific effects of anticancer drugs on the immune system and other rapidly dividing cells limit their use on account of host toxicity (primarily bone marrow and gastrointestinal). A major indication for immunomodulation of these drugs is combination with glucocorticoids for treatment of autoimmune diseases. Vinblastine is another vinca alkaloid that has been used to treat type II hypersensitivities, specifically IMHA. Like danazol, vinblastine appears to decrease expression of Fc (IgG) receptors on macrophages.110,111 The effects of vinblastine (weekly doses during induction followed by monthly doses during maintenance) when combined with danazol (given at 2 to 3 up to 10 mg/kg daily) proved effective for treatment of 63% of human patients with chronic idiopathic thrombocytopenic purpura.110
Gold Therapy
Gold has been used for centuries as an elemental agent for the control of pruritis.118 Gold compounds are characterized by gold attached to sulfur (aurothio group) and include the more water-soluble compounds aurothioglucose and gold sodium thiomalate and the lipid-soluble compound auranofin. These compounds suppress or prevent inflammation of the joint and synovium associated with a number of infectious or chemical causes. The mechanism of action is not clear but may be inhibition of the maturation and function of mononuclear phagocytes and T cells. Gold is sequestered in mononuclear phagocytic cells. Other proposed but not generally accepted mechanisms include inhibition of prostaglandin synthesis, collagen linkage, complement activation, and a variety of lysosomal and other enzymes.118
The disposition of the gold compounds has not been studied in animals other than humans; these data provide the basis of discussion. Disposition varies with water solubility. Water-soluble compounds (not prepared in oil) are rapidly absorbed after intramuscular administration, with peak concentrations occurring in 2 to 6 hours. Oral absorption is erratic and not predictable for the water-soluble compounds but is more predictable for auranofin, the hydrophobic compound. Distribution varies with the compound and duration of therapy. The gold is bound (95%) to albumin in blood, but eventually concentrations in selected tissues (inflamed synovium) approximate 10 times that in plasma. For the water-soluble gold compounds, elimination rate constants and half-lives also vary with dose. In humans the plasma half-life is 7 days at a 50-mg total dose, but it increases to several weeks to months with prolonged therapy. Concentrations can be detected in the blood for 60 to 80 days after therapy is discontinued and for up to 1 year in the urine. Concentrations probably are detectable in the liver and skin several to many years after therapy is discontinued. Excretion is predominantly (60% to 90%) renal, with the remaining eliminated in the feces. For auranofin plasma concentrations tend to be lower, and accumulation is much less (20%) than that of the water-soluble compounds, probably because of less tissue binding. Auranofin is eliminated principally in the feces.
Toxicity, manifested as skin or mucocutaneous lesions, occurs in 15% of human patients. Lesions include stomatitis, pharyngitis, tracheitis, colitis, gastritis, and vaginitis. Gray to blue pigmentation may occur in the skin. The likelihood of toxicity is concentration dependent but not based on gold concentration. Whereas proteinuria occurs in up to 50% of human patients receiving gold therapy, renal dysfunction (as a result of proximal tubular damage or gold-induced glomerulonephritis) occurs in 10% or less. Lesions tend to be resolvable if therapy is discontinued before damage is too severe. Thrombocytopenia, which is thought to reflect increased destruction, occurs in a very small percentage of human patients. A number of miscellaneous side or toxic effects that resolve when therapy has been discontinued also have been reported. These include encephalitis, peripheral neuritis, hepatitis, and pulmonary infiltrates. In general, although auranofin is better tolerated than the water-soluble compounds, the incidence of gastrointestinal disturbances (diarrhea, abdominal cramping) is greater. The incidence of side effects and their seriousness can be minimized by regular physical examination. In addition, therapy should be initiated with a small dose that is gradually increased to the maintenance dose. As the dose is increased, treatment should temporarily cease until clinical signs of moderate to severe toxicity resolve. The risk of restarting therapy should be balanced with the need for therapy. Antihistamines or glucocorticoids might decrease the incidence. Dimercaprol, a heavy metal chelating compound, can be used if severe side effects persist after therapy has been discontinued.
The major indication for chrysotherapy in human patients is rheumatoid or other arthritis that has not responded to nonsteroidal antiinflammatory drugs or other therapy. The compounds slow the progression but do not cure disease. The usual human dose is 10 mg of the water-soluble compounds the first week followed by 25 mg the third and fourth weeks and 25 to 50 mg weekly thereafter to a cumulative 1-g dose. Response may take several months. The standard human dose for auranofin is 3 to 6 mg up to 9 mg divided in two to three doses or given once daily. Small animal doses are discussed with dermatologic disorders in this chapter. Wolheim119 has discussed mechanisms by which rheumatic human patients become refractory to gold therapy. Induction of the efflux protein P-glycoprotein appears to play a major role.
Immunostimulant Drugs
Immunostimulants are indicated for immunodeficient animals. As with immunosuppressants, immunostimulants can target either humoral or cell-mediated immunity. The lack of specificity has limited the widespread use of immunostimulants. In general, response to immunostimulants is mild.
Immunostimulants of Microbial Origin
Bacteria and fungal microbes generally nonspecifically stimulate several aspects of the immune response. Macrophage, cytotoxic T cell, Th cell, and B cell activities are enhanced, whereas T cell suppressor activity is decreased. Enhanced activity reflects, in part, increased activity of chemical mediators such as TNFα and IFN. The nonspecific enhancement of immune function can be used therapeutically. Adverse effects can be minimized by administration of killed organisms or selected microbial fractions, which stimulate the response while avoiding infection. A variety of mycobacterial products have been used to nonspecifically stimulate the immune system.23,120 The classic biological response modifiers include Bacillus Calmette–Guérin (BCG) and Propionibacterium acnes (Corynebacterium parvum).
Bacillus Calmette–Guérin
BCG is a live, attenuated strain of Mycobacterium bovis.4 Components of the bacterial cell wall of this organism activate B and primarily T cells; macrophage and neutrophil recruitment in response to lymphokine release results in a granulomatous reaction. The antiviral state induced by BCG appears to be essentially a nonspecific expression of a more or less prolonged stimulation of the immune system. The most likely mechanism for its antiviral activity is direct stimulation of the mononuclear phagocytic system; however, specifically sensitized T lymphocytes that further activate macrophages are generated after a period of time. Stimulated macrophages release colony-stimulating factor, which contributes to macrophage regulation, and IL-1, which promotes lymphocyte proliferation. The antiviral state is thus an indirect benefit of an essentially pathologic, although temporary, situation; the host is characterized by an increased ability to handle viral infections. BCG requires at least 10 days after inoculation to enhance resistance against viral infections. The state of enhanced resistance is, however, long-lasting; repeated administrations (of nonliving product) may lead to a more prolonged and marked effect.120
BCG has been used as an adjuvant in combination with tumor vaccines for dogs and cats. Tumors that undergo remission after BCG therapy may do so as a result of the release of tumor-specific antigens during nonspecific tumor lysis. The use of BCG has been studied in combination with a tumor vaccine. In human patients use of BCG as an adjuvant for anticancer therapy has focused primarily on intravesicular administration in bladder cancer.4
Muramyl Dipeptide
Muramyl dipeptide is a peptidoglycan and represents the smallest immunologically active component of the Mycobacterium cell wall. Like BCG, it nonspecifically enhances B cell, T cell, and macrophage activities. Because it is rapidly eliminated from the body, it is biologically modified to prolong its actions by incorporation into liposomes or conjugation with glycoproteins. Synthetic production has increased accessibility for clinical use.
Mycobacterium
Regressin-V
An emulsion of mycobacterial cell wall fractions, Regressin-V is licensed for the treatment of mixed mammary tumors and mammary adenocarcinoma in dogs and sarcoidosis in horses. The following information was supplied by the manufacturer. Regressin-V contains trehalose dimycolate and muramyl dipetide, which induce IL-1 and tumor necrosis factor-α secretion from monocytes and macrophages and activate T lymphocytes in a variety of species. In a study of seven dogs with mammary adenocarcinoma, complete remission was induced in five, with tumor-free survival times of 3 to 19 months; dogs were then lost to follow-up. There are no data suggesting that Regressin-V had any effect on metastatic disease.
The manufacturer recommends treating canine mammary tumors once 2 to 4 weeks before surgical excision. Fever and malaise may occur after injection. Surgical removal of the tumor creates cosmetic improvement (necrosis and draining of the tumor may be present for weeks); however, survival time is not significantly improved with surgery. If surgery is not performed, therapy can be repeated every 1 to 3 weeks for up to four treatments.
Mycobacterium vaccae
Ricklin Gutzwiller and coworkers121 prospectively studied the impact of a single intradermal injection of heat-killed Mycobaterium vaccae on the severity of atopic dermatitis in dogs (n = 62). A double-blinded, placebo-controlled, parallel multicenter study design was used. Dogs were evaluated for 3 months. Skin lesions were scored on the basis of CADESI. The treatment was well tolerated. Treatment was effective in patients with mild or moderate dermatitis but did not affect animals classified as severe. Interestingly, the CADESI scores decreased by 22% in the placebo group (versus 33% in the treatment group).
Propionibacterium acnes
A killed suspension containing P. acnes (formerly C. parvum: Immunoregulin) has been approved for veterinary use (15 μg/kg intravenously biweekly for two to three treatments). Like BCG, it causes complex, nonspecific immunostimulation. Macrophage and cytotoxic T cell activities are enhanced, as is antibody production by both thymus-dependent and thymus-independent antigens. Cell-mediated immunity is also stimulated. It is indicated for the treatment of clinical signs associated with virus-induced and bacteria-induced immunosuppression, such as that caused by FeLV. The efficacy of P. acnes in the treatment of feline infectious peritonitis virus is questionable. One study was unable to demonstrate a difference in survival rate or mean survival time between untreated and treated animals experimentally infected with a high dose of feline infectious peritonitis virus. Studies investigating the antitumor effects of Propionibacterium acnes in dogs do not support its efficacy against early neoplasia, although it may be more effective in advanced disease when used in combination with other drugs.25 Cats infected with FeLV and afflicted with various non-neoplastic diseases showed some signs of response to treatment with P. acnes (Immunoregulin), although no cat reverted to an FeLV-negative status.23 Studies regarding the efficacy of P. acnes in FeLV-induced tumors apparently have not been done.25
Staphylococcal Protein A
Staphylococcal protein A (SpA) is a cell wall polypeptide of S. aureus Cowan I (SAC) that binds rapidly and with great affinity to immune complexes. Other regions of the molecule initiate T lymphocyte and B lymphocyte proliferation as well as secretion of soluble lymphocytic products such as IFN. Circulating immune complexes have been incriminated as “blocking factors” that aid in a tumor’s escape from immunologic control. Therapeutic trials of the efficacy of SpA have been based on the hypothesis that removal of specific or nonspecific immunosuppressive molecules such as blocking factors will enhance host immune response to the tumor. Other regions of the SpA molecule initiate T lymphocyte and B lymphocyte proliferation and secretion of soluble lymphocyte products such as IFN. Treatment with SpA is achieved by extracorporeal perfusion of host plasma over whole SAC organisms or through filters containing purified SpA. Studies have shown that SpA or SAC treatment reduces tumor size, decreases levels of circulating immune complexes, and induces the appearance of cytotoxic antibodies directed toward neoplastic cells. A study in which SpA was used to treat FeLV-infected cats found that 50% of cats with FeLV-associated disease improved, and 33% of cats afflicted with malignant disease responded with reductions in tumor size as well as in bone marrow and peripheral blood neoplastic cells.23,25,122,123 Viremia was cleared in 28% of treated cats. Circulation of a g-like IFN was demonstrated in some cats that responded to SpA and was followed by the appearance and rising titer of complement-dependent cytotoxic antibody. The antibody reacted with FeLV-infected cells and was specific for the major viral envelope glycoprotein gp70. The ability of cats to persistently remain FeLV negative appeared to correlate with the magnitude of FeLV antibody titer.25
Mixed Bacterial Vaccines
Mixed bacterial vaccines composed of Streptococcus pyogenes and Serratia marcescens have been studied in cats with malignant mammary tumors. Although not statistically significant, survival time was greater (875 days) when surgery was combined with mixed bacterial vaccines versus surgery alone (450 days).
Immunostimulants of Natural Origin
Cytokines
Biological response modifiers of natural original also are discussed in Chapter 32. A number of cytokines produced by leukocytes and related cells actively regulate the immune system. Recombinant DNA technology has led to widespread production and greater application of drugs that modulate cytokines or their receptors or transcription factors in the treatment of immunologically based disease. The cytokines most likely to be used for immunostimulatory effects include the IFNs and selected interleukins.
Passive Immunotherapy: Antibodies
Passive immunotherapy with monoclonal or polyclonal antibodies has been investigated for both the diagnosis and therapy of cancers. Antibodies are directed toward surface antigens expressed by tumor cells. Specificity of antibodies (as long as Igs are derived from the species intended to be treated) should limit host toxicity to those cells expressing the antigen (i.e., tumor cells), thus avoiding systemic toxic effects. The antibodies themselves may be directly toxic to the cell or induce complement-dependent cytotoxicity. In addition, antibodies can be conjugated to drugs, toxins, or radioisotopes toxic to tumor cells, thus limiting the specificity of a variety of pharmaceuticals for tumor cells.
Igs available in plasma obtained from donors generally contain detectable antibodies against bacterial, fungal, and viral pathogens.4 Passive transfer of resistance to the immunodeficient patient can be accomplished with intramuscular or intravenous administration. In human patients the half-life of transferred Igs is approximately 3 weeks.4 Indications for human patients have included congenital or acquired states of immunodeficiency, selected hematologic disorders such as autoimmune hemolytic anemia, infectious viral diseases, and selected neoplastic diseases such as chronic lymphocytic leukemia and multiple myeloma. Potential adverse reactions include anaphylaxis and transfer of infectious organisms.4
Studies investigating the efficacy of passive immunotherapy on feline lymphosarcoma have thus far been limited to the use of polyclonal FeLV-neutralizing antibodies, feline oncornavirus-associated cell membrane antigen (FOCMA) antibodies, or both in cats afflicted with lymphosarcoma and leukemia. In one study five of seven cats treated with both antibodies and six of eight cats treated with FOCMA antibodies alone underwent partial or complete regression of their disease.25 Additional studies by Cotter and coworkers124 suggest that polyclonal anti-FOCMA therapy combined with previously established chemotherapeutic regimens may improve remission time. Monoclonal antibodies have been used to treat canine lymphosarcoma.
Human intravenous immunoglobulin (hIVIG) has been studied in dogs using in vitro (lymphocytes and monocytes) and in vivo (spontaneous immune-mediated diseases) methods. Prepared from plasma of healthy donors, it contains polyspecific IgG and is intended to treat primary immunodeficiencies. In vitro studies125 have shown hIVIG to bind to canine lymphocytes and monocytes and to inhibit through Fc-mediated binding monocyte phagocytosis, including that of antibody-coated red blood cells. It is administered as an intravenous infusion (0.5 to 1.5 g/kg) over 6 to 12 hours, and animals must be closely monitored for signs of anaphylaxis or other adverse reactions (e.g., thromboembolism). Clinical evidence of efficacy exists for treatment of IMHA,126-128 with treated dogs developing an increased hemoglobin and hematocrit concentration up to 4 weeks after therapy. However, 50% of treated animals developed thrombocytopenia and five of seven dogs that died had evidence of thromboembolism. Only 3 of 10 dogs were alive at 12 months, suggesting that long-term survival was not improved. The combination of IgG with IFN α resulted in greater improvement in human patients with hepatitis C.129
Abetimus is a synthetic molecule consisting of four double-stranded oligodeoxyribonucleotides attached to polyethylene glycol (PEG). The nonimmunogenic PEG serves as the carrier. Abetimus induces tolerance to B cells by cross-linking surface antibodies directed toward double-stranded DNA (dsDNA). These antibodies appear to be associated with nephritis associated with systemic lupus erythematosus in humans.130 In a preapproval clinical trial, the number of renal “flares” in patients receiving the drug was less than half of that encountered in patients not receiving the drug. The response was so dramatic that the trial was discontinued before its completion, and the FDA granted orphan drug status in 2000.130
Allergen-Specific Immunotherpy
Allergen-specific immunotherapy (ASIT) involves the administration of gradually increasing amounts of an allergen extract to patients allergic to the allergen to resolve symptoms associated with subsequent exposure to the allergen.131 In their review Colombo and coworkers131 note that when success is defined as a 50% (or greater) increase in the improvement of clinical signs after 4 months or more of therapy, ASIT is successful in 50% to 100% of cases of canine atopic dermatitis. They prospectively evaluated the effect on canine atopic dermatitis of low (1⁄10 the standard) dose induction followed by maintenance ASIT therapy (n = 27) to standard therapy (n = 13). Both groups responded, and neither group emerged as responding better at 9 months. Secondary infections were treated, and use of glucocorticoids was allowed to control severe clinical signs, except during the last 3 months of the study.
Miscellaneous Blood Components
Selective destruction of malignant lymphocytes occurs in several species in response to infusion of blood or blood components.
Antileukemic activity
Antileukemic activity (ALA) of humoral factors in serum, heparinized plasma, and whole blood has been documented in several species.25 Evidence suggests that ALA of blood constituents may be present in high concentrations in cryoprecipitates prepared from heparinized plasma. Physicochemical constituents of the cryoprecipitate that might be the source of ALA include cold-insoluble globulin, fibronectin, cell surface protein, large external transformation-sensitive protein, and opsonic factor.25 A study in 32 cats revealed that of 24 treated with either normal cat serum or whole blood (20 mL/kg), 14 had a complete antileukemic response and 8 had a partial response. Of 10 treated with cryoprecipitate, two completely responded and six had a partial response.132
Fibronectin
Fibronectin injections have resulted in the regression of leukemic nodes in mice, suggesting that fibronectin may be the source of ALA. Fibronectin, a glycoprotein dimer, is a major protein of both blood and tissues; its most important function appears to be tissue remodeling during embryogenesis and wound healing. Antitumor activity may result from immunomodulation and modification of metastasis. Enhancement of opsonization increases the phagocytic ability of macrophages and monocytes, thus enhancing their tumoricidal activity. MacEwen26 studied the efficacy of fibronectin (0.5 to 2 mg/kg intravenously once daily) in 18 cats afflicted with lymphosarcoma. A 50% response rate was reported; one FeLV-positive cat converted to a negative status after treatment.25 A single case of mycosis fungoides in a cat responded to a combination of intravenous and intralesional fibronectin. Intralesional injections caused local epithelial necrosis, which reduced the tumor load by 75% and may have prevented systemic involvement.133
Tumor cell vaccines
Tumor cells are thought to stimulate an immune response. This view is supported by the recognition that some tumors spontaneously regress; tumors are infiltrated with cells of the immune system, and the risk of cancer increases in the patient that is immunosuppressed. The antigenic potential of tumor cells is also the basis for the use of tumor cell vaccines. Tumor cell vaccines are most effective in the presence of residual disease and when combined with nonspecific immunomodulators. Parodi and co workers 133a could not find a statistical improvement in cumulative survival rates in dogs with mammary tumors receiving BCG intralesionally alone (n = 51) or Corynbacterium parvum (killed) (n = 120) Jeglum and coworkers have treated cats afflicted with mammary adenocarcinoma using a combination of an autogenous tumor vaccine and BCG. Although not significantly different, the mean survival time for each treatment group was least for those treated with surgery, BCG, and tumor cell vaccine compared with animals treated with either surgery alone or surgery with BCG. Additional studies with tumor cell vaccines are necessary before their efficacy can be established.
Non-steroidal hormonal agents
Hormonal agents may offer a unique avenue of immunomodulation in viral diseases.23,134 Hormones investigated for their immunomodulatory effects include prostaglandins and thymic proteins. Prostaglandins are local hormones that modulate both T and B lymphocytes. Prostaglandins, however, particularly prostaglandin E2, are also immunosuppressants, and their role in immunopotentiation is limited. Thymosins are a group of endogenous substrates released from the thymus that stimulate the release of several pituitary neuropeptides. Thymic hormones induce maturation of T cell precursors, promote differentiation and proliferation of mature T cells, and thus restore rather than potentiate the immune system. Several synthetic thymic peptides have been synthesized by either chemical means or genetic engineering techniques. The primary clinical application of these proteins is restoration of the immune system in the immunoincompetent (including virally induced) patient.
Acemannan
Acemannan (Acemannan Immunostimulant) is a long-chain polydispersed β1,4-linked mannan-based polysaccharide derived from the aloe vera (barbadensis Miller) plant. It stimulates the release of IL-1, IL-6, TNF-α, IFN-γ, and nitric oxide from macrophages, leading to tumor apoptosis and necrosis.135,136 Other actions include enhancement of macrophage phagocytosis and cytotoxicity and interference with glucosidase I activity, leading to the production of abnormal glycoproteins by neoplastic cells, which appears to be associated with tumor cell death. Direct antiviral activity is associated with modified glycosylation of both virus-infected cells and glycoprotein coats of viruses, leading to inhibition of virus infectivity and replication.
Acemannan is licensed for the treatment of fibrosarcoma in dogs and cats. Intratumoral injection induces tumor encapsulation and necrosis, facilitating surgical excision. The recommended dosage is 2 mg intratumorally and 1 mg/kg intraperitoneally weekly for 6 weeks. No adverse effects of acemannan have been reported at the recommended dosage. In one report eight dogs and five cats with fibrosarcomas were treated with acemannan, surgical excision, and radiation therapy. Seven animals remained tumor-free at 440 to 603 days, with a median survival time of 372 days.137 In an earlier clinical report, a variety of other carcinomas and sarcomas were also reported to respond.138 It is not known what effect acemannan has on feline vaccine-associated sarcomas.
Acemannan has been used to treat cats with FeLV and feline immunovirus infections. Clinically affected cats treated with acemannan had improved quality of life and longer survival times than historical controls. Interestingly, oral administration of acemannan appeared to have the same efficacy as parenteral administration of acemannan, which is similar to cats treated with low-dose oral recombinant human IFN-α.
Synthetic Immunostimulants
Levamisole
Levamisole, a phenylimidazothiazide anthelmintic, has been the subject of intense and controversial research as a biological response modifier.23,120 It is difficult to summarize the experimental and clinical data regarding the immunomodulating capabilities of levamisole. Levamisole has been regarded by some as a chemical agent capable of mimicking hormonal regulation of the immune system. It appears to stimulate recruitment and function of macrophages and T cells but only within a narrow range of doses and duration of administration in either the normal or immunoincompetent patient. Levamisole appears to alter cyclic nucleotide phosphodiesterases, decreasing cyclic guanosine monophosphate degradation and increasing cyclic adenosine monophosphate degradation. Elevated cyclic guanosine monophosphate in lymphocytes enhances proliferation and secretory responses. Chemotaxis, phagocytosis, lymphokine synthesis, and the ratio of helper to suppressor T cells are increased. Levamisole does not appear to have any effect in immunocompetent animals. The modulatory effects of levamisole range from enhancement to inhibition with much strain, sex, age, and antigen variability. T cell stimulation may be mediated by a soluble serum factor. Experimental studies generally have not supported the benefits of levamisole as have clinical studies. Levamisole (2 to 15 mg/kg) was frequently tested prophylactically in experimental trials, however, as opposed to therapeutically (i.e., in infected patients) in clinical trials, suggesting that potential benefits of levamisole result from restoration of immunocompetence after virally induced immunosuppression. Indications for levamisole in human medicine include Hodgkin’s disease and rheumatoid arthritis and as an adjuvant chemotherapy of colorectal cancer.4 The use of levamisole for treatment of cancer in animals is not supported. In two studies involving more than 130 cats with malignant mammary tumors, levamisole (5 mg/kg orally on 3 alternate days per week) did not increase survival time or decrease recurrence rate.139 Levamisole causes adverse reactions typical of excessive cholinergic stimulation.
Cimetidine
Histamine exerts immunomodulatory effects, including suppression of cytotoxic T lymphocytes, downregulation of cytokines, and activation of suppressor T lymphocytes.140 Cimetidine is a histamine (H2) receptor antagonist that experimentally enhances a variety of immunologic functions. Suppressor T lymphocytes possess H2 receptors, which, when blocked, result in potentiation of cell-mediated immunity. Although more studies are indicated, cimetidine may be useful in a variety of conditions associated with immunosuppression. Because it selectively inhibits suppressor function, however, cimetidine also may prove deleterious to patients with autoimmune disorders.
The anticancer effects of cimetidine have been studied in selected human cancers. The H2 receptor blockers have been studied for their effects on gastric cancer cells. Cimetidine, but not famotidine, exhibits antiproliferative effects on gastric cancer cells. Raniditine showed some inhibitory effects.140 Cimetidine also appears to inhibit the growth of colorectal carcinoma;141 lymphocyte infiltration increases in the cancers and is associated with an improved survival rate in patients receiving the drug.
Miscellaneous Immunostimulants
A variety of compounds are capable of inducing IFN; however, the immunomodulating effects of the inducers are variable. For example, double-stranded IFN-inducing polyribonucleotides (poly I:C) cause immunostimulation, whereas tilorone, a simple synthetic IFN inducer, enhances antibody production while depressing cell-mediated immune responses. Many other drugs are in the experimental phases of drug development. The following sections address some of the drugs that may or may not ultimately become approved.
ABPP (Bropirimine)
ABPP (2-amino-5-bromo-6-phenyl-4[3H]-pyrimidinone), approved for use in humans, is a potent inducer of IFN in several species. Hamilton and coworkers142 characterized the kinetics of ABPP and induced IFN in several species. They found that serum levels of IFN after treatment correlated well with serum concentrations of ABPP in the cat. ABPP was lethal in three of eight cats, however, at doses required to achieve minimal detectable levels of IFN. The authors postulated that the toxicity resulted from conversion of ABPP to phenolic derivatives that the cat excretes inefficiently.
Isoniplex
Isoniplex is an antiviral drug (see Chapter 10) that may also be used as an immunopotentiator in viral diseases.23,120 Isoniplex enhances immune responses, including promotion of mitogen-stimulated T-lymphocyte proliferation and augmentation of antibody production and delayed-type hypersensitivity. The suggested mechanism of immunopotentiation by isoniplex begins with penetration of lymphocytes and suppression of viral RNA synthesis. It appears to support lymphocyte function by promoting RNA synthesis and translational ability. Further investigations regarding the use of isoniplex in viral infection should be anticipated because its efficacy results from an ideal combination of antiviral activity and immunopotentiation.
Promodulin
Promodulin is an experimental immunomodulating agent that has been subjected to clinical therapeutic trials for the treatment of cats concurrently infected with FeLV and feline infectious peritonitis.23 Cats were treated with 50 mg/kg (up to 200 mg maximum dose) intravenously once daily for 5 consecutive days. Although promodulin induced rapid remission of clinical signs associated with feline infectious peritonitis, (e.g., anorexia, fever, and serosal effusions), it did not appear to be effective in the treatment of concurrent FeLV infections. Cats that responded did so within 2 weeks of the final injection; the duration of clinical remission appeared to vary between 1 and 3 months. Clinical signs after exacerbation of disease did not respond to a second treatment regimen. Promodulin also was not effective in treating FeLV-induced solid tumors.
Topical immunomodulators
Topical immunomodulators often are characterized by unique mechanisms. These include topical contact sensitizers (e.g., diphencyprone or dinitrochlorobenzene) and induction of mononuclear cell cytokines (IL-12, TNF-α) secretion (imidazoquinolines imiquimod and resiquimod). These latter drugs lead to Th1-domination and a cell-mediated response used in humans to treat local viral infection and cancers.
Imidazoquinolines
Imiquimod and resiquimod (R-848) are members of the family imidazoquinolines. These drugs have proven antiviral and antitumor effects in a variety of animal models. Their immune actions appear to reflect stimulation of several arms of both innate and adaptive immunity, ultimately resulting in an indirect Th-1 dominant response. Their ability to enhance cutaneous-adaptive responses and innate immunologic responses are key to their usefulness. Resiquimod is 10- to 100-fold more potent than imiquimod and is also capable of stimulating granulocyte and macrophage colony-stimulating factors and other mediators. Antigen processing also is improved. Indications include cutaneous viral infections and potentially nonmelanoma skin cancers, with the latter having potential therapeutic applications in animals.143
Treatment of Specific Immune-Mediated Diseases
See also discussion of each syndrome in the appropriate chapter.
Chronic Allergic Diseases
Atopy refers to a genetic tendency toward certain hypersensitivity or allergic diseases. Atopic diseases include asthma, rhinitis, and dermatitis. Chronic allergic disease are generally complex in pathophysiology, with inflammatory disease manifested in the organ in which antigen–tissue interactions are initiated. For example, for asthma, inflammation causes airway narrowing associated with contraction and hypertrophy of bronchial smooth muscle, swelling of mucous membranes, and excessive production of mucus.
Mediators released during inflammation are the major contributors to the pathogenesis of pulmonary disease, particularly feline asthma. Mediators important in the pathogenesis of asthma include preformed mediators histamine and serotonin (particularly in cats); mediators formed in situ, including prostaglandins, leukotrienes, and platelet-activating factor; and reactive oxygen species.
The three atopic syndromes are characterized by common cell and mediators of inflammation in humans (see Figure 31-2).121,144-147 A similar pathophysiology should be anticipated for other allergic diseases. The role of T (CD4+ helper) cells has been well described in the initiation and maintenance of allergic diseases, particularly asthma. In contrast to systemic immune-mediated diseases, in which Th1 cells appear to be the biggest contributors of inappropriate response, Th2 cells appear to be particularly important to allergic diseases. Their cytokines include interleukins and chemotactants that contribute to the inflammatory process. These include IL-4, which increases IgE synthesis; IL-5, which stimulates eosinophil growth and differentiation; IL-9, which causes mast cell differentiation; and in the lungs IL-13, which causes production of mucus and airway hyperactivity. Of these, IL-4, IL-10, and IL-13 in particular, have antiinflammatory activity. Previously, Th1cells were thought to be beneficial by downregulating Th2 cells. However, they are probably proinflammatory and, along with IFN (IFN) γ, contribute to the inflammatory process. The role of leukotrienes, and particularly cyst-LTs, are increasingly being described in atopic diseases.148 In the lungs they are very potent (being 1000 times more so than histamine), causing marked edema, inflammation, and bronchoconstriction. In the gastrointestinal tract, their effects on neutrophils are necessary for ulcer formation; vasoconstriction and platelet aggregation probably contribute further. Senter and coworkers149 found that 50% of atopic dogs responded to zafirlukast, supporting the possible role of leukotrienes as targets of treatment for atopy.
Treatment of inflammatory allergic diseases focuses on control of inflammation and clinical signs at the level of the target tissue. However, treatment of all three atopic disorders increasingly focuses on systemic rather than simply a local allergic disease. In particular, the bone marrow response to allergens and subsequent release of eosinophils are recognized to be an important systemic process in allergic inflammation. A central role of eotaxin and IL-5 has been suggested. Eotaxin released from local tissue cells stimulates, exclusively through CC chemokine receptor 3 (CCR3), flux of eosniophils into the target tissue.150 Both IL-4 and TNF-α stimulate its release. Th2 cells release IL-5; other sources include mast cells and eosinophils. In response to IL-5, eotaxin is released. In human patients with allergic diseases, both are increased in serum. The effects of these signals are not limited to the affected tissue. Rather, the primary significance may be their effects at the level of the bone marrow stroma, another source of IL-5. Among the functions of IL-5 are differentiation and maturation of progenitor cells to eosinophils and basophils, release of mature eosinophils, promotion of their survival, and inhibition of apoptosis. Activated eosinophil progenitor cells contain granulocyte-macrophage colony-stimulating factor and IL-5. Their circulation increases in allergic human patients and in dogs rendered hypersensitive to A. suum.151
Cysteinyl-LTs are necessary for accumulation of eosinophils in target tissues. However, in addition to their local effects, cyst-LTs may also have a role in bone marrow perpetuation of allergic disease. Cyst-LT receptors are expressed on a number of bone marrow progenitor cells and appear to be involved (based on effects of antagonists) in eosinophil– basophil progenitor differentiation. They appear to stimulate eosinphil proliferation in the presence of stimulatory cytokines.152
The commonalities of atopic diseases offer several targets of therapy. The efficacy of glucocorticoids can be appreciated from several aspects, their use is discussed in Chapter 30. However, a number of “second tier” drugs might be used for their glucocorticoid dose-sparing effect or in animals intolerant to or minimally responsive to glucocorticoid therapy (e.g., CsA). Further, the involvement of Th cells supports the potential role of CsA in their treatment. The role of leukotrienes supports use of receptor antagonists or other drugs (e.g., dual-inhibitor nonsteroidal antiinflammatory drugs) that modify their release. In human medicine drugs that target eotaxin, the CCR3, or particularly IL-5 are being explored. Finally, pentoxifylline might be considered for its ability to target TNF. These therapies might be considered for other atopic diseases, including allergic rhinitis and sinusitis perianal fistulae, and others.
Type I Hypersensitivities
Anaphylaxis and Anaphylactoid Reactions
Clinical signs of systemic anaphylaxis include nausea, vomiting, diarrhea, pale mucous membranes, coolness to peripheral extremities, tachycardia, and tachypnea. Localized “anaphylaxis” results in clinical signs referable to the site of localized mast cell degranulation. Examples include angioneurotic edema resulting in swelling of lips, eyelids, and conjuctiva and urticarial lesions (or hives). Treatment is oriented toward preventing further mast cell degranulation, blocking the interaction between histamine (or other mediator) and tissue receptors, and antagonizing the physiologic response to mediators. Drugs that antagonize physiologic response also tend to further decrease mast cell degranulation. The goals of therapy for systemic anaphylaxis include cardiovascular and ventilatory support. Epinephrine is indicated to antagonize bronchoconstriction and provide cardiovascular support. Glucocorticoids (prednisolone sodium succinate) facilitate adrenergic receptor responses and decrease further mast cell mediator release. Histamine 1 (H1) receptor antagonists (e.g., diphenhydramine) are of benefit only in preventing interaction of histamine and its receptors, not in preventing further mast cell degranulation. Therapy may be more effective if administered in anticipation of mast cell degranulation, although this is somewhat controversial. Perioperative anaphylaxis, including drugs, risk factors, and therapies in humans, has been recently reviewed.153
Type II Hypersensitivities: Antigen- and Antibody-Dependent Cytotoxicity
Immune-Mediated Hemolytic Anemia
IMHA occurs as a result of increased red blood cell destruction mediated by the presence of an antibody on the membrane surface. The antigen to which the antibody binds is either of the red blood cell membrane or an exogenous antigen (e.g., drug or microbe) that has adhered to the surface. Igs associated with IMHA in dogs generally are IgG or, less commonly, both IgG and IgM. In cats IgM tends to be more common, with IgG alone causing IMHA in approximately 25% of cases. Complement activation is more likely with IgM-mediated IMHA. Antibody adherence and complement activation that are insufficient to cause erythrocyte lysis will result in damage and subsequent erythrophagocytosis of the deformed red blood cell (e.g., spherocyte). Intravascular hemolysis occurs when complement activation is extensive, leading to erythrocyte lysis. Released hemoglobin binds to serum haptoglobin, preventing glomerular filtration. If haptoglobin becomes saturated, however, free hemoglobin can be filtered. Renal toxicity can accompany intravascular IMHA as a result of antigen–antibody deposition or reaction in the basement membrane; free hemoglobin may also contribute to nephrotoxicity. Direct red blood cell agglutination or intravascular hemolysis increases the risk of thromboembolic disease.
Medical treatment of IMHA focuses on reducing phagocytosis of damaged or antibody-coated erythrocytes by reducing or blocking receptors on phagocytic cells, reducing or preventing the formation of more antibodies, and providing supportive therapy for complications associated with IMHA. Any likely inciting (exogenous) antigen (e.g., drug, microbe) should be removed. Treatment of IMHA is confounded by low survival rates (which range from 10% to 50% discharge survival, with continued patient loss after discharge) and the lack of scientifically based evidence supporting preferred therapies. Several of the clinical trials that address various therapeutic regimens unfortunately are biased in population selection, generally focusing on the worse cases and thus complicating extrapolation of results to less severe cases. Immunosuppressive therapy with glucocorticoids is the cornerstone of therapy; use of immunosuppressants for immune-mediated disorders is reviewed in Chapter 30. Differences of opinion exists regarding the initial route of administration of glucocorticoids, with oral proponents observing that response to glucocorticoids requires 5 to 7 days regardless of route. Although this observation may have some merit (many mechanisms of glucocorticoid require nuclear transcription and protein synthesis; time must elapse before inflammatory cell numbers and function are reduced), many mechanisms (e.g, non-genomic) of glucocorticoid action may occur immediately. Indeed, the importance of glucocorticoid supplementation in shock patients offers support for administration designed to cause immediate response (see Chapter 30). For rapid response dexamethasone (0.1 to 0.2 mg/kg intravenously) or methylprednisolone (11 mg/kg daily for up to 3 days) every 12 to 24 hours is administered initially, followed by oral prednisolone (1 mg/kg every 12 hours) as the animal responds. Choice of prednisone versus prednisolone is addressed in Chapter 30; the latter is strongly encouraged in dogs with life-threatening conditions, and the former should not be used in cats. Further reduction of red blood cell destruction may require administration of CsA, leflunomide or MMF, or their combination; continued failure may require cyclophosphamide or (dogs only) azathioprine. Combination therapy might be considered initially in patients with severe disease (see Chapter 30). Leflunomide doses as high as 8 mg/kg were necessary to prevent renal transplant rejection in dogs. 104a Safety of anticancer drugs can be increased by dosing based on body surface area. Cyclophosphamide can be given as a single intravenous bolus (100 to 250 mg/m2) in patients suffering from intravascular lysis or direct autoagglutination.108 This regimen also is particularly useful for patients that require a blood transfusion. However, efficacy of cyclophosphamide may not be realized until antibodies decline as a result of normal catabolism (generally 1 to 2 weeks).11 Oral administration (50 mg/m2 every 48 hours) is indicated in dogs that do not respond to glucocorticoids or might be considered as part of initial therapy, particularly in severe cases. Using a randomized prospective design, Mason and coworkers154 investigated the impact of cyclophosphamide (50 mg/m2) when combined with prednisone (1 to 2 mg/kg orally every 12 hours; n = 8) compared with prednisone alone (1 to 2 mg/kg orally every 12 hours; n = 10) for treatment of severe (based on clinical signs, less than 1 week in duration and packed cell volume less than 20%) idiopathic IMHA in dogs. The mortality rate did not differ between groups, but the power of the study was limited. Reticulocytosis appeared, and spherocytosis resolved more rapidly in the prednisone-only–treated group (within 1 week), leading the investigators to conclude that the addition of cyclophosphamide offered no advantage to prednisolone alone. Long-term cyclophosphamide will increase the risk of thrombocytopenia and neutropenia.11
Human gammaglobulin (0.5 to 1.5 g/kg intravenously over 6 to 12 hours) may prove useful for dogs that fail to respond to therapy.127,128 Cost may, however, be prohibitive, and use may be limited by development of thrombocytopenia or thrombosis (see previous discussion). CsA may be of benefit in controlling the sequelae of IMHA that reflect tissue (and thus T cell–mediated) damage.11 Blood transfusions are not recommended for patients with IMHA. Dogs with IMHA are predisposed to destruction of transfused red blood cells. Blood substitutes (oxyhemoglobin) may offer a viable alternative to blood transfusion. Although splenectomy should be considered as a surgical adjunct to medical management, removal of the spleen may also result in removal of an important site of extramedullary hematopoiesis. As with glucocorticoids, danazol (5 to 10 mg/kg orally every 12 hours) may block Fc receptors on phagocytic cells, reducing red blood cell destruction.111 However, response may be slow, rendering danazolol lower on the list.
Response to therapy is based on daily hematocrit or platelet counts (or both); initial therapy should be continued or, as needed, improved until the packed cell volume is 15% or more in cats and 20% in dogs or the platelet count is 100, 000/μL or more. Drug therapy should be gradually tapered to avoid relapse or rebound; for example, prednisolone and azathioprine tapered to 0.5 to 1 mg/kg every second day is a reasonable target, with monitoring every 2 weeks as a basis of response. Therapy should be continued for another 3 months or so, with gradual tapering off attempted in animals that have been in remission for 3 to 6 months. For nonresponders, supportive therapy may include transfusions, which are more likely to be effective in patients whose disease reflects bone marrow rather than peripheral cellular targets.
Other supportive therapy should be considered. Anemia and reduced oxygen delivery to the gastrointestinal tract may predispose the patient to gastrointestinal induced ulceration. The importance of cyclooxygenase-2 in gastrointestinal healing warrants consideration of gastroprotectants, particularly in the face of glucocorticoid therapy. As such, antisecretory drugs (e.g., H2-receptor antagonists, omeprazole) and gastroprotectants (e.g., sucralfate) should be considered as long as concerns regarding drug interactions are addressed. The use of heparin, including fractionated products, is controversial but might be considered (Chapter 15).
Immune-Mediated Thrombocytopenia
Immune-mediated thrombocytopenia is a syndrome that occurs more commonly in dogs than cats and in males than females. It can occur in concert with other immune-mediated disorders, including IMHA.11 Thrombocyte numbers can decline as a result of destruction (i.e., antibody/complement-mediated phagocytosis) or, less commonly, decreased formation of mature thrombocytes as a result of antibody/complement-mediated destruction of megakaryocytes.11 As with IMHA, antibody can be directed to an endogenous or exogenous antigen adhered to the platelet or megakaryocyte. The primary clinical signs, which include and reflect inappropriate bleeding, generally do not occur until thrombocyte numbers have dropped below 30,000/μL. Bleeding is more likely with a rapid, as opposed to a gradual, decline.
Medical treatment focuses on prevention of bleeding, decreased destruction of thrombocytes, and restoration of thrombocyte numbers (see also Chapters 15 and 30). Any likely inciting (exogenous) antigen (e.g., drug, microbe) should be removed. Glucocorticoids (dexamethasone, methylprednisolone, or prednisolone) are often the first choice of therapy (see earlier discussion of IMHA) and, in general, cause thrombocyte counts to normalize within 1 week. Use of cyclosporine and leflunoamide should be considered as suggested for other immune-mediated disease. Danazol also can be administered to reduce phagocytosis.112,113 Vincristine (0.75 mg/m2 intravenously) can be administered if platelet numbers fail to increase to sufficient numbers. Once platelet numbers increase, vincristine should be discontinued and glucocorticoids continued for several more weeks.11 Vincristine may also be given after incubation with platelet-rich plasma in cases refractory to glucocorticoids, danazol, and vincristine. Presumably, incubation allows vincristine to bind to platelets. Subsequent phagocytosis destroys the phagocytic cell, causing an overall reduction in platelet destruction.155 If successful, the treatment may need to be repeated as new macrophages are generated. Platelet-rich plasma also can be administered in an attempt to restore platelet numbers to a concentration that is not life threatening (>50,000/μL). The likelihood of relapse of immune-mediated thrombocytopenia also may be reduced after infusion of platelet-rich plasma.11 The use of vinblastine might be considered in relapsing patients (see discussion of cytotoxic drugs) on the basis of its effectiveness in human patients. Splenectomy is a surgical alternative or adjunct therapy that should be considered in refractory cases. Because of the risk of relapse, patients should be monitored periodically. Surgical neutering, particularly of females, may be indicated once platelet numbers have normalized.
Immune-Mediated Neutropenia
As with other immune-mediated hematopoietic diseases, glucocorticoids are indicated for neutropenia.156 Treatment can continue as described for IMHA.
The use of recombinant bone marrow growth factors (see Chapter 15 Chapter 32) to increase bone marrow production of deficient cells is probably not wise unless the factor is derived from the species to be treated. Even in those situations, studies should confirm a lack of immune-mediated reactions when used in patients affected with an immune-mediated disorder.
Dermatologic Disorders
A number of immune-mediated skin diseases reflect a type II hypersensitivity. Included are pemphigus (foliaceus, erythematosus, vulgaris, vegetans), bullous pemphigoid, and dermatomyositis.
Pemphigus Disorders and Bullous Pemphigoid
Pemphigus disorders reflect the reaction of autoantibodies directed toward antigens located in the intercellular spaces between epidermal cells. The definitive antigen is not known but apparently is located in or near the cytoplasmic membrane.11 The various types may reflect variants, crossovers, or altered presentations of the different forms of the disease. In all variants antibody deposition causes the loss of adhesion between epidermal cells, leading to acanthosis. Complement activation results in local mast cell degranulation and an infiltration of inflammatory cells. Clinical signs vary within and among the variants and include visculobullous eruptions, cutaneous ulcerations, exfoliative lesions, and verrucous proliferations of the skin.11 Bullous pemphigoid results from the generation of antibodies toward the lamina lucida of the basement membrane zone. As with pemphigus, complement activation may worsen the inflammatory response. Clinical signs include vesiculobullous lesions at mucocutaneous junctions; in the oral cavity; on footpads; and on the skin of the trunk, groin, axillae, and abdomen.11
Glucocorticoids can be expected to be effective in 40% of canine cases11 of pemphigus. The initial dose should be high (2 to 3 mg/kg orally every 12 hours for 10 to 14 days); the dose can gradually be reduced over 4 weeks (targeting 1 mg/kg orally every 48 hours) if an adequate response has occurred. Failure to respond or inability to decrease the dose of glucocorticoids is an indication for the addition of a second immunosuppressive drug. Generally, azathioprine (2 mg/kg orally every 24 hours) has been the first choice to combine with prednisolone (1 mg/kg orally every 12 hours). Response within 10 to 14 days will allow alternating the drugs each day at the same dose. Continued remission will allow a gradual reduction in the doses of both drugs to 1 mg/kg orally every other day, alternating the drugs daily.11 Cyclophosphamide (50 mg/m2 orally every 24 hours) can be combined with prednisolone (1 mg/kg orally every 12 hours) for 4 consecutive days each week for 2 to 3 weeks. If remission occurs, doses are gradually reduced to 1 mg/kg orally, alternating drugs daily. Chlorambucil (0.1 mg/kg orally every 48 hours) can be used in place of cyclophosphamide; leukopenia and thrombocytopenia are potential side effects of this drug. Use of cyclosporine and leflunoamide should be considered as suggested for other immune-mediated disease.
The third alternative for immunosuppressive chemotherapy is use of aurothioglucose initially in combination with prednisolone (1 to 2 mg/kg orally every 12 to 48 hours). Chrysotherapy should be initiated only after administration of an intramuscular test dose (1 mg for animals less than 10 kg; 5 mg for animals 10 kg or larger) twice, 1 week apart. Toxicity will be manifested as dermatitis, stomatitis, nephrotic syndrome, blood dyscrasias, eosinophilia, thrombocytopenia, and manifestations of allergic reactions. Therapy can be continued at 1 mg/kg weekly intramuscularly until remission; however, continued therapy should be based on an acceptable complete blood count. At that time, the interval is decreased to alternate weeks and finally monthly. Prednisolone therapy might be gradually phased out.
Pemphigus foliaceus in cats can be treated with chlorambucil as the first choice.106 Daily therapy (0.1 to 0.2 mg/kg/day) should be continued until lesions have markedly reduced, which may take 4 to 8 weeks. Alternate-day therapy should be implemented when approximately 75% improvement occurs and continued for several weeks. Complete blood counts should be monitored every 2 weeks of chlorambucil therapy.106
Dermatomyositis
Dermatomyositis might be classified as an inflammatory muscle syndrome. Unlike polymyositis, which appears to involve a cell-mediated, antigen-specific response, dermatomyositis appears to reflect an abnormality of the humoral response, resulting in vasculitis. In human patients treatment with low-dose methotrexate has proved beneficial114,115 Dermatomyositis in veterinary medicine is an inherited idiopathic inflammatory syndrome affecting Collies, Shetland Sheepdogs, and their crosses. Skin lesions include erythema, scaling, and crusting, particularly around the eyes, tips of the ears and tail, digits, and carpal and tarsal regions. Pentoxifylline, a methylxanthine derivative, has been recommended157,158 on the basis of its potential efficacy in humans.159 Studies of the drug in dogs suggest a higher dose than that for humans, and a twice- to thrice-daily dosing interval should be used. Clinically, response may take several weeks. The drug appears to be well tolerated by dogs. Clinical trials in Collies with dermatomyositis are currently under way.
Feline Eosinophilic Granuloma Complex
Eosinophilic granulomas may respond to glucocorticoids. Direct lesional injection of methylprednisolone acetate (2 mg/kg, minimum of 20 mg) every 2 weeks is the preferred method of administration. High oral doses of prednisolone may be an effective alternative.160 Drugs that target leukotrienes should be considered. Response to chlorambucil has been reported104 within 6 weeks of therapy (0.1 to 0.2 mg/kg per day). Response to therapy should be followed by a 12-week period during which the dose of chlorambucil is decreased until discontinued. Megestrol acetate is an undesirable alternative unless the patient has proved refractory to other modalities, including chlorambucil. Urine should be monitored for glucose to detect the development of diabetes mellitus. Levamisole (2.2 mg/kg orally every 48 hours) has been reported to cause some (but incomplete) remission in certain cases.160,161 Additionally, accurate dosing is difficult because of the large tablet size (184 mg). Cats often react adversely, with transient anorexia, vomiting, and hypersalivation being the most common side effects. Bone marrow suppression can be marked and is characterized by a long recovery period.
Type III Hypersensitivities: Immune Complex Disease
Systemic Lupus Erythematosus
The deposition of circulating autoantibodies or autoantibody–antigen complexes in the endothelium, particularly that of the glomerulus, appears to initiate complement-mediated inflammation. The inflammatory site is infiltrated by immune cells. In the glomerulus response includes proliferation of capillary cells, thickening of the basement membrane, and scarring. Other vascular beds affected include the skin, serous membranes, synovial tissues, and cutaneous and visceral blood vessels.114,115 Soluble immune complexes are more problematic than large immune complexes, which precipitate and are rapidly phagocytized. Soluble complexes are able to penetrate deep into vascular endothelial channels, activate complement, and stimulate an inflammatory response. In humans intravenous cyclophosphamide has been established as the treatment of choice for lupus-induced nephritis. Bolus cyclophosphamide has also, however, proved effective for lupus affecting other body systems, including the CNS, lungs, and arteries. Azathioprine and weekly methotrexate are effective for treating human lupus that does not involve major organs, such as rashes, serositis, and arthritis, or in combination with glucocorticoids to reduce the glucocorticoid dose. CsA apparently has not been studied for treatment of systemic lupus erythematosus, although clinical response has been reported.114,115
Because systemic lupus erythematosus can be polysystemic in its presentation, the sequelae of immune complex deposition associated with systemic lupus erythematosus can affect a number of body systems, resulting in the need for medical management of secondary disease. Examples include but are not limited to glomerulonephritis, arthritis, and vasculitis. The reader is referred to the chapters that address these specific body systems for information on management of the sequelae of inflammatory disease. Treatment for the immune-mediated aspect of the disease is the same as for pemphigus skin disorders. Use of cyclosporine and leflunoamide should be considered as suggested for other immune-mediated disease.
Cutaneous Discoid Lupus Erythematosus
Considered a mild form of systemic lupus erythematosus, cutaneous discoid lupus erythematosus is not accompanied by systemic involvement. The most common form presents as a nasal dermatitis; skin surrounding the eyes, pinnae, lips, and feet may also be involved. Treatment includes immunosuppressive doses of glucocorticoids, as described for other immune-mediated diseases, although a lower initial dose may be effective (1 mg/kg orally twice daily). Vitamin E (400 IU orally 2 hours before or after a meal every 12 hours; acetate or succinate) may be effective as the sole agent; however, a 30- to 60-day lag time to efficacy mandates that initial therapy include glucocorticoids. Topical glucocorticoids may be effective in mild cases. Minimizing exposure to the sun, including use of topical sunscreen, or other ultraviolet light also will be helpful, particularly in animals with depigmentation. Niacin or tetracycline (5 to 12 mg/kg orally every 8 hours) was reportedly effective in 70% of cases in one study. Use of cyclosporine and leflunoamide should be considered as suggested for other immune-mediated disease; tacrolimus also might be considered.
Rheumatoid Arthritis and Other Arthritides
The rheumatoid factor is an antibody that reacts with IgG that has bound to antigen and subsequently undergone a conformation change. The reason for selectivity in joints is not understood. In humans drugs used to treat rheumatoid arthritis include gold compounds or penicillamine and cytotoxic drugs, including azathioprine, cyclophosphamide, and methotrexate. Since the late 1980s, low-dose weekly methotrexate has become the preferred medication.114,115 Methotrexate has proved more rapid in onset. Because it is safer than traditional cytotoxic drugs, treatment generally can progress for a longer period of time. Because functional disabilities and progress of the disease occur rapidly in the first years of disease in humans, cytotoxic drugs are begun early. Drug combinations have been advocated because of the possibility of synergistic effects. Examples include methotrexate with sulfasalazine, CsA, or biological agents. Treatment in animals has not been as well investigated and focuses on control of inflammation with glucocorticoids and, if necessary, aspirin. The use of glucocorticoids is supported by the rapid clinical improvement and decrease in IL-6 activity documented in dogs with juvenile polyarthritis syndrome.162 The use of other immunosuppressive drugs in animals (azathioprine, cyclophosphamide) has been reserved for severe cases. On the basis of findings in humans, however, a more aggressive approach may be warranted. Disease-modifying agents (e.g., glucosamine and chondroitin sulfates) should be used to support cartilage repair.
Feline chronic progressive polyarthritis is associated with feline leukemia virus and feline syncytium-forming virus and most commonly presents as osteopenia and periosteal bone proliferation around affected joints. Less commonly, joints are characterized by subchondral marginal erosions similar to those of rheumatoid arthritis. Rheumatoid factor cannot be identified, however. Treatment includes immunosuppressive doses of corticosteroids (1 to 3 mg/kg orally every 12 hours). Nonresponders may require treatment with chlorambucil, cyclophosphamide, or azathioprine. As previously suggested, disease-modifying agents may prove beneficial.
In a retrospective study of dogs (n = 39) with immune-mediated polyarthritis, 56% of dogs were cured after drug therapy. Response to prednisolone alone was 50% (overall 33%), although response to prednisolone (1 to 2 mg/kg every 24 hours or divided every 12 hours) by itself or combined with another drug was 81%. Duration of therapy was generally 4 to 16 weeks, although continuous medication was necessary in 10% to 18%. Response of animals to leflunomide was previously discussed.104a Drugs combined with prednisolone to which animals responded included antimicrobials and immunosuppressive drugs such as cyclophosphamide or azathioprine.161 Treatment with levamisole or CsA as sole therapy generally was not successful.
Type IV Hypersensitivities: the Delayed Response
Allergic Contact Dermatitis
Allergic contact dermatitis is the most common type IV hypersensitivity recognized in small animals, being responsible for up to 10% of dermatologic cases.11 The syndrome is initiated when the skin comes in contact with the inciting antigen or chemical. Actual chemicals that cause contact dermatitis are not known, but it is likely that the chemical acts as a hapten that subsequently covalently bonds to a protein. The location of the protein is not clear but is probably associated with the class II molecule of an APC, which in the skin is the Langerhans cell. Sensitization generally requires 4 to 10 days; subsequent exposure to the chemical results in a marked T cell–mediated response.11 Treatment is best implemented by removal of the inciting antigen and short-term (7-day) administration of prednisolone (0.5 to 1 mg/kg every 12 to 24 hours).
Inflammatory Myopathies
In humans polymyositis appears to reflect cell-mediated, antigen-specific cytotoxicity, with azathioprine being the only cytotoxic drug to be of benefit in controlled studies. Low-dose methotrexate or azathioprine with high-dose glucocorticoid therapy has become the standard therapy. Combinations of methotrexate and either cyclophosphamide or CsA may be effective and are being studied.114,115
Inflammatory Bowel Disease
Inflammatory bowel disease also is discussed in Chapter 19. The use of immune-modifying therapy for human patients with IBD became popular only in the 1990s.114,115,164,165 Controlled clinical trials in humans have focused on azathioprine, 6-mercaptopurine, CsA, and methotrexate. Azathioprine or 6-mercaptopurine has been effective for treatment of Crohn’s disease, although efficacy depends on duration of therapy; at least 13 and more often 17 weeks of therapy is required before the drugs reach their full effects.
Intravenous administration of azothioprine may decrease the time to response. CsA has been studied at low doses (<1 mg/kg per day) or high doses (>5 mg/kg per day) to minimize the risk of nephrotoxicity (less of a concern in veterinary patients), but this has proved of little benefit in Crohn’s disease. It has been more effective for treatment of ulcerative colitis at high doses (8 to 10 mg/kg per day), although only a few patients have been studied. CsA concentrations in responding patients were above 250 ng/mL. Methotrexate appears to be useful for both induction of remission and steroid sparing in patients with Crohn’s disease and ulcerative colitis. Combinations of drugs also have been studied. Azathioprine and 6-mercaptopurine should not be used in combination with methotrexate because of the increased risk of toxicity. CsA and methotrexate have been used in human patients with IBD with some success. Drugs that target leukotrienes should be considered. Pentoxyfylline for treatment of human IBD is discussed in Chapter 19.
Amyloidosis
Primary (or AL) amyloidosis reflects the extracellular deposition of abnormally formed protein associated with a clonal plasma cell dyscrasia.166 The protein is a monoclonal Ig light chain fragment secreted in an abnormal insoluble fibrillar form; the underlying plasma cell dyscrasia is generally insidious and nonproliferating. The AL amyloid fibrils are derived from the N-terminal region of monoclonal Ig light chains and consist of the whole or part of the variable (VL) domain. The propensity for Igs to form amyloid fibrils is inherent to their structure; unfortunately, they cannot be identified. Those able to form myeloid are capable of existing in partially unfolded states because of the loss of tertiary or higher structure. Aggregation and retention of b-sheet secondary results in the formation of protofilaments and fibrils; initiation of the process, or seeding, appears to facilate exponential progress of the amyloid template by “capturing” precursor molecules. Amyloid deposits exist in a state of dynamic turnover. Accumulation of amyloid progressively disrupts the normal tissue structure and ultimately leads to organ failure, frequently including, but not limited to, the kidneys, heart, liver, and peripheral nervous system. Hereditary forms of amyloidosis also exist, as do secondary forms (e.g., chronic immunologic stimulation). Treatment of the latter is best based on removal of the inciting cause. Because no therapy has successfully targeted amyloid deposition, therapy targets suppression of the underlying plasma cell dyscrasia as well as treatment intended to preserve organ function. A variety of low- and high-dose drug therapies, sometimes as sole agents but more commonly as combinations, have been recommended in humans. Low-dose single agents include melphalan or cyclophosphamide, (with or without prednisolone). However, clinical benefit occurred in only about 20% to 30% of human patients, after a median of 12 months’ treatment. Intermediate dose regimens consist of a monthly course of VAD (vincristine, adriamycin, dexamethasone) or similar regimes or intravenous intermediate-dose melphalan 25 mg/m2 with or without dexamethasone. High-dose therapy consists of intravenous melphalan (100 to 200 mg/m2) but with stem cell rescue. Other approaches include a pulsed high-dose dexamethasone or thalidomide (with or without dexamethasone). However, thalidomide is a drug of restricted distribution in the United States and therefore cannot be prescribed by veterinarians. In veterinary medicine colchicine has been recommended for prevention in dogs predisposed to amylodids. Presumably, colchicine may block the deposition of amyloid. The dose is based on that used to treat hepatic fibrosis (0.03mg/kg every 24 hours for 2 weeks) with an increase (0.025 to 0.03mg/kg orally every 12 hours) if the drug is well tolerated during the initial therapy. In patients with renal amyloidosis, dimethyl sulfoxide has been recommended, although its efficacy is unsubstantiated.
1. Aggarwal B.B., Puri R.K. Common and uncommon features of cytokines and cytokine R eceptors: an overview. In: Aggarwal B.B., Puri R.K., editors. Human cytokines: their role in disease and therapy. Cambridge: Blackwell Science; 1995:3.
2. Hilton D.J. An introduction to cytokine receptors. In: Nicola N.A., editor. Guidebook to cytokines and their receptors. Oxford, England: Oxford University Press; 1994:8.
3. Tizard I.R. Interferons. In: Myers M.J., Murtaugh M.P., editors. Cytokines in animal health and disease. New York: Marcel Dekker; 1995:1.
4. Krensky A.M., Vincenti F., Bennett W.M. Immunosuppressants, tolerogens, and immunostimulants. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, pp 1405–1431, 2006.
5. Pfitzner E., Kliem S., Baus D., et al. The role of STATs in inflammation and inflammatory diseases. Curr Pharm Des. 2004;10:2839-2850.
6. Feizy V., Ghobadi A. Atopic dermatitis and systemic autoimmune diseases: a descriptive cross-sectional study. Dermatol Online J. 2006;12(3):3.
7. Bochner B.S., Hamid Q. Advances in mechanisms of allergy. J Allergy Clin Immunol. 2003;111:S819-S823.
8. Menzies-Gow A., Flood-Page P., Sehmi R., et al. Anti-IL-5 (mepolizumab) therapy induces bone marrow eosinophil maturational arrest and decreases eosinophil progenitors in the bronchial mucosa of atopic asthmatics. J Allergy Clin Immunol. 2003;111:714-719.
9. Shadidi K.R. New drug targets in rheumatoid arthritis: focus on chemokines. Biodrugs. 2004;18(3):181-187.
10. Pascher A., Klupp J. Biologics in the treatment of transplant rejection and ischemia/reperfusion injury: new applications for TNFα inhibitors? BioDrugs. 2005;19(4):211-231.
11. Gorman N., Immunology. Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 4. 1995. Saunders. Philadelphia. 1978-2002.
12. Boyaka P.N., Tafaro A., Fischer R., et al. Therapeutic manipulation of the immune system: enhancement of innate and adaptive mucosal immunity. Curr Pharm Des. 2003;9:1965-1972.
13. Hiemstra P.S., Ferni-King B.A., McMichael P.J., et al. Antimicrobial peptides: mediators of innate immunity as templates for the development of novel anti-infective and immune therapeutics. Curr Pharm Des. 2004;10:2891-2905.
14. Linde A., Ross C.R., Davis E.G., et al. Innate immunity and host defense peptides in veterinary medicine. J Vet Intern Med. 2008;22(2):247-265.
15. Rabin B.S. Measurement of immune function in PNI research: relevance of immune measures to health and disease. Proc PNI Workshop. May 1998.
16. Eggert M., Zetta U.K., Klueter An., et al. Transcription factors in autoimmune diseases. Curr Pharm Des. 2004;10:2787-2796.
17. Gibbons M: A Cytokine storm? Why are younger people who usually fight off flu succumbing to swine flu? Advance for Nurses, May 27, 2009. Accessed January 28, 2010, at http://nursing.advanceweb.com/Editorial/Content/Editorial.aspx?CC=200143.
18. Bacher S., Schmitz L.M. The NF-B pathway as a potential target for autoimmune disease therapy. Curr Pharm Des. 2004;10:2827-2837.
19. Dostert A., Heinzel T. Negative glucocorticoid receptor response elements and their role in glucocorticoid action. Curr Pharm Des. 2004;10(23):2807-2816.
20. Schulz M., Eggert M. Novel ligands: fine tuning the transcriptional activity of the glucocorticoid receptor. Curr Pharm Des. 2004;10:2817-2826.
21. Mellon R.D., Bayer B.M. Evidence for central opioid receptors in the immunomodulatory effects of morphine: review of potential mechanisms of action. J Neuroimmunol. 1998;83:19-28.
22. Carr D., Rogers T.J., Weber R.J. The relevance of opioids and opioid receptors on immunocompetence and immune homeostasis. Proc Soc Exp Biol Med. 1996;213:248-257.
23. Deluca H.F., Cantorna M.T., Vitamin D. its role and uses in immunology. FASEB J. 2001;15(14):2579-2585.
24. Ford R.B. Biological response modifiers in the management of viral infection. Vet Clin North Am Small Animal Prac. 1986;16:1191-1204.
25. Carrasco L. Shugar D., editor. Viral chemotherapy. The replication of animal viruses. vol 1. 1984. Pergamon. New York. 111-148.
26. MacEwen E.G. Approaches to cancer therapy using biological response modifiers. Vet Clin North Am Small Anim Pract. 1985;15:667-688.
27. Theilen G.H., Hills D. Comparative aspects of cancer immunotherapy: immunological methods used for treatment of spontaneous cancer in animals. J Am Vet Med Assoc. 1982;181:1134-1137.
28. Pendse S., Sayegh M.H., Frank M.H. P-glycoprotein: a novel therapeutic target for immunomodulation in clinical transplantation and autoimmunity? Curr Drug Targets. 2003;4:469-476.
29. Krensky A.M., Vincenti F., Bennett W.M. Immunosuppressants, tolerogens, and immunostimulants. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, 2006.
30. Halloran P.F. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715-2729.
31. Brazis P., Barandica L., Garcia F., et al. Dermal microdialysis in the dog: in vivo assessment of the effect of cyclosporin A on cutaneous histamine and prostanglandin D2 release. Eur Soc Vet Derm. 2006;17:169-174.
32. Kelly P., Kahan B.D. Review: metabolism of immunosuppressant drugs. Curr Drug Metab. 2002;3:275-287.
33. Guaguère E., Steffan J., Olivry T., Cyclosporin A. a new drug in the field of canine dermatology. Vet Dermatol. 2004;15:61-74.
34. Friman S., Backman L. A new microemulsion formulation of cyclosporin: pharmacokinetic and clinical features. Clin Pharmacokinet. 1996;30:181-193.
35. Gomez D.Y., Wacher V.J., Tomlanovich S.J., et al. The effects of ketoconazole on the intestinal metabolism and bioavailability of cyclosporine. Clin Pharmacol Ther. 1995;58:15-19.
36. Takaya S., Iwatsuki S., Nogughi T., et al. The influence of liver dysfunction of cyclosporine pharmacokinetics: a comparison between 70 percent hepatectomy and complete duct ligation in dogs. Jpn J Surg. 1989;19:49-56.
37. Ohta K., Agematu H., Yamada T., et al. Production of human metabolites of cyclosporin A, AM1, AM4N and AM9, by microbial conversion. J Biosci Bioeng. 2005;99(4):390-395.
38. Vickers A.E.M., Fischer V., Connors S., et al. Cyclosporine A metabolism in human liver, kidney and intestine slices: comparison to rat and dog slices and human cell lines. Drug Metab Dispos. 1992;120:802-809.
39. Khoschsorur G., Erwa W., Fruehwirth F., et al. High-performance liquid chromatographic method of the simultaneous determination of cyclosporine A and its four major metabolites in whole blood. Talanta. 2005;65:638-643.
40. Maurer G., Loosli H.R., Shreier E., et al. Disposition of cyclosporine in several animal species and man I. Structural elucidation of its metabolites. Drug Metab Dispos. 1984;12:120-126.
41. Wallemace P.E., Alexandre K. Evaluation of the new AxSYM cyclosporine assay: comparison with TDx monoclonal whole blood and emit cyclosporine assays. Clin Chem. 1999;45(3):432-435.
42. Yatscoff R.W., Copeland K.R., Faraci C.J. Abbott TDx monoclonal antibody assay evaluated for measuring cyclosporine in whole blood. Clin Chem. 1990;36(11):1969-1973.
43. Sangalli L., Bortolotti A., Jiritano L., et al. Cyclosporine pharmacokinetics in rats and interspecies comparison in dogs, rabbits, rats, and humans. Drug Metab Dispos. 1988;16(5):749-753.
44. Steffan J., Strehlau G., Maurer M., et al. Cyclosporin A pharmacokinetics and efficacy in the treatment of atopic dermatitis in dogs. J Vet Pharmacol Ther. 2004;27(4):231-238.
45. Mehl M.L., Kyles A.E., Craigmill A.L., et al. Disposition of cyclosporine after intravenous and multi-dose oral administration in cats. J Vet Pharmacol Therap. 2003;26:349-354.
46. Vaden S.L. Cyclosporine. In: Bonagura J.D., editor. Kirk’s current veterinary therapy small animal practice (XII). Philadelphia: Saunders; 1995:73-77.
47. Gregory C.R., Hietala S.K., Pedersen N.C., et al. Cyclosporine pharmacokinetics in cats following topical ocular administration. Transplantation. 1989;47(3):516-519.
48. Novartis: Transplant Drug Interactions Monograph, 2007.(http://www.novartis-transplant.com/hcp/about/interactions.jsp?usertrack.filter_applied=true&NovaId=7852773784371409923)
49. Terao T., Hisanaga E., Sai Y., et al. Active secretion of drugs from the small intestinal epithelium in rats by P-glycoprotein functioning as an absorption barrier. J Pharm Pharmacol. 1996;48:1083-1089.
50. McLachlan A.J., Tett S.E. Effect of metabolic inhibitors on cyclosporine pharmacokinetics using a population approach. Drug Monit. 1998;20:390-395.
51. Keogh A., Spratt P., McCosker C., et al. Ketoconazole to reduce the need for cyclosporine after cardiac transplantation. N Engl J Med. 1995;333:628-633.
52. Lown K.S., Mayo R.R., Leichtman A.B., et al. Role of intestinal P-glycoprotein (MRD1) in interpatient variation in the oral bioavailability of cyclosporine. Clin Pharmacol Ther. 1997;62:248-260.
53. Daigle J.C. More economical use of cyclosporine through combination drug therapy. J Am Anim Hosp Assoc. 2002;38:205-208.
54. Martin J.E., Daoud A.J., Schroeder T.J., et al. The clinical and economic potential of cyclosporin drug interactions. Pharmacoeconomics. 1999;15(4):317-337.
55. Myre S.A., Schoeder T.J., Grund V.R., et al. Critical ketoconazole dosage range for cyclosporine clearance inhibition in the dog. Pharmacology. 1991;43:233-241.
56. McNaulty J.F., Lensmeyer G.L. The effects of ketoconazole on the pharmacokinetics of cyclosporine A in cats. Vet Surg. 1999;28:448-455.
57. Dahlinger J., Gregory C., Bea J. Effect of ketoconazole on cyclosporine dose in healthy dogs. Vet Surg. 1998;27:64-68.
58. Mouatt J.G. Cyclosporin and ketoconazole interaction for treatment of perianal fistulas in the dog. Aust Vet J. 2002;80(4):207-811.
59. D’Souza M.J., Pollock S.H., Solomon H.M. Cyclosporine-cimetidine interaction. Drug Metab Dispos. 1988;16(1):57-59.
60. Bar-Meir S., Bardan E., Ronen I., et al. Cimetidine and omeprazole do not affect cyclosporine disposition by the rat liver. Eur J Drug Metab Pharmacokinet. 1993;18(4):355-358.
61. Shaefer M.S., Rossi S.J., McGuire T.R., et al. Evaluation of the pharmacokinetic interaction between cimetidine or famotidine and cyclosporine in healthy men. Ann Pharmacother. 1995;29(11):1088-1091.
62. Daigle J.C., Hosgood G., Foil C.S., et al. Effect of cimetidine on pharmacokinetics of orally administered cyclosporine in healthy dogs. Am J Vet Res. 2001;62(7):1046-1050.
62a. Allenspach K., Bergman P.J., Sauter S., et al. P-glycoprotein expression in lamina propria lymphocytes of duodenal biopsy samples in dogs with chronic idiopathic enteropathies. J Comp Pathol. 2006;134(1):1-7.
63. Kahan B.D. Optimization of cyclosporine therapy. Transplant Proc. 1993;25:5-9.
63a. Wooldridge J.D., Gregory C.R., Mathews K.G., et al. The prevalence of malignant neoplasia in feline renal-transplant recipients. Vet Surg. 2002;31(1):94-97.
63b. Kneteman N.M., Marchetti P., Tordjman, et al. Effects of cyclosporine on insulin secretion and insulin sensitivity in dogs with intrasplenic islet autotransplants. Surgery. 1992;111(4):430-437.
64. Edwards L.L., Wszolek Z.K., Normand M.M. Neurophysiologic evaluation of cyclosporine toxicity associated with bone marrow transplantation. Acta Neurol Scand. 1995;92(5):423-429.
65. Steffan J., Alexander D., Brovedani F., et al. Comparison of cyclosporine A with methylprednisolone for treatment of canine atopic dermatitis: a parallel, blinded, randomized controlled trial. Vet Dermatol. 2003;14:11-22.
66. Steffan J., Favrot C., Mueller R. A systematic review and meta-analysis of the fficacy and safety of cyclosporin for the treatment of atopic dermatitis in dogs. Vet Dermatol. 2006;17:3-16.
67. Last R.D., Suzuki Y., Manning T., et al. A case of fatal systemic toxoplasmosis in a cat being treated with cyclosporin A for feline atopy. Vet Dermatol. 2004;15:194-198.
68. Mahalati K., Belitsky P., West K., et al. Approaching the therapeutic window for cyclosporine in kidney transplantation: a prospective study. J Am Soc Nephrol. 2001;12:828-833.
69. Einecke G., Mai I., Fritsche L., et al. The value of C2 monitoring in stable renal allograft recipients on maintenance immunosuppression. Nephrol Dial Transplant. 2004;19:215-222.
70. Steele B.W., Wang E., Soldin S.J., et al. A longitudinal replicate study of immunosuppressive drugs. Arch Pathol Lab Med. 2003;127:283-288.
71. Radowica S.N., Power H.T. Long-term use of cyclosporine in the treatment of canine atopic dermatitis. Vet Dermatol. 2005;16:81-86.
72. Levy G.A. C2 monitoring strategy for optimising cyclosporin immunosuppression from the Neoral-1 Formulation. BioDrugs. 2001;15(5):279-290.
73. Robson D.C., Burton G.G. Cyclosporin: applications in small animal dermatology. Vet Dermatol. 2003;14:1-9.
74. Olivry T., Mueller R.S. Evidence-based veterinary dermatology: a systematic review of the pharmacotherapy of canine atopic dermatitis. Vet Dermatol. 2003;14:121-146.
75. Mathews K.A., Ayres S.A., Tano C.A., et al. Cyclosporin treatment of perianal fistulas in dogs. Can Vet J. 1997;38:39-41.
76. Mathews K.A., Sukhiani H.R. Randomized controlled trial of cyclosporine for treatment of perianal fistulas in dogs. J Am Vet Med Assoc. 1997;211:1249-1253.
77. Allenspach K., Rufenacht S., Sauter S. Pharmacokinetics and clinical efficacy of cyclosporine treatment of dogs with steroid-refractory inflammatory bowel disease. J Vet Intern Med. 2006;20:239-244.
78. van Deventer S.J. Review article: chemokine production by intestinal epithelial cells: a therapeutic target in inflammatory bowel disease? Aliment Pharmacol Ther. 1997;11(Suppl 3):116-120. discussion, 120-121
79. Pham C.Q., Efros C.B., Berardi R.R. Cyclosporine for severe ulcerative colitis. Ann Pharmacother. 2006;40(1):96-101.
80 Padrid P.A., Cozzi P., Leff A.R. Cyclosporin A inhibits airway reactivity and remodeling after chronic antigen challenge in cats. Am J Respir Crit Care Med. 1996;154:1812-1818.
81. Noli C., Toma S. Three cases of immune-mediated adnexal skin disease treated with cyclosporin. Vet Dermatol. 2006;17:85-92.
82. Barth T., Mischke R., Nolte I. Cyclosporin A in aplastic anemia in dogs: first results. Berl Munch Tierarztl Wochenschr. 1997;110:60-67.
83. Font A., Bardag M., Mascort J., et al. Treatment with oral cyclosporin A of a case of vesicular cutaneous lupus erythematosus in a rough collie. Eur Soc Vet Derm. 2006;17:440-442.
84. Adamo F.P., O’Brien R.T. Use of cyclosporine to treat granulomatous meningoencephalitis in three dogs. J Am Vet Med Assoc. 2004;225(8):1211-1216.
84a. Visudtibhan A., Chiemchanya S., Visudhiphan P. Cyclosporine in chronic inflammatory demyelinating polyradiculoneuropathy. Pediatr Neurol. 2005;33(5):368-372.
85. Williams D.L. A comparative approach to topical cyclosporine therapy. Eye. 1997;11(Pt 4):453-464.
86. Puignero V., Queralt J. Effect of topically applied cyclosporin A on arachidonic acid (AA)‑ and tetradecanoylphorbol acetate (TPA)-induced dermal inflammation in mouse ear. Inflammation. 1997;21:357-369.
86a. Takada K., Katayama N., Kiriyama A., et al. Distribution characteristics of immunosuppressants FK506 and cyclosporin A in the blood compartment. Biopharm Drug Dispos. 1993;14(8):659-671.
87. Vaden S.L. Cyclosporine and tacrolimus. Semin Vet Med Surg Small Anim. 1997;12:161-166.
87a. Venkataramanan R., Watty V.S., Zemaitis M.A., et al. Bioopharmaceutical aspects of FK-506. Transplant Proc. 1987;19(5 Suppl 6):30-35.
88. Griffies J.D., Mendelsohn C.L., Rosenkrantz W.S., et al. Topical 0.1% tacrolimus for the treatment of discoid lupus erythematosus and pemphigus erythematosus in dogs. J Am Anim Hosp Assoc. 2004;40(1):29-41.
89. Marsella R., Nicklin C.F., Saglio S., et al. Investigation on the clinical efficacy and safety of 0.1% tacrolimus ointment (Protopic) in canine atopic dermatitis: a randomized, double-blinded, placebo-controlled, cross-over study. Vet Dermatol. 2004;15:294-303.
89a. Marsella R., Nicklin C.F., Saglio S. Investigation on the effects of topical therapy with 0.1% tacrolimus ointment (Protopic) on intradermal skin test reactivity in atopic dogs. Vet Dermatol. 2004;15:218-224.
90. Tredger J.M., Brown N.W., Adams J. Monitoring mycophenolate in liver transplant recipients: toward a therapeutic range. Liver Transpl. 2004;10(4):492-502.
91. Langman L.J., Shapiro A.M.J., Lakey J.R.T., et al. Pharmacodynamic assessment of mycophenolic acid-induced immunosuppression by measurement of inosine monophosphate dehydrogenase activity in a canine model. Transplantation. 1996;61:87-92.
92. Pirsch J.D., Sollinger H.W. Mycophenolate mofetil: clinical and experimental experience. Ther Drug Monit. 1996;19:357-361.
93. Pfitzmann R., Klupp J., Langrehr J.M., et al. Mycophenolate mofetil for immunosuppression after liver transplantation: a follow-up study of 191 patients. Transplantation. 2003;76(1):130-136.
94. Morath C., Zeier M. Review of the antiproliferative properties of mycophenolate mofetil in non-immune cells. Int J Clin Pharmacol Ther. 2003;41(10):465-469.
95. Boothe D.M., Dewey C., Neerman M. Disposition and oral bioavailability of mycophenolic acid following administration of mycofenolate mofatil in normal dogs. Proc ACIM. 2003.
97. Neerman M.F., Boothe D.M. A possible mechanism of gastrointestinal toxicity posed by mycophenolic acid. Pharmacol Res. 2003;47(6):523-526. (Review. No abstract available. Erratum in: Pharmacol Res 48(4):415, 2003.)
98. Platz K.P., Sollinger H.W., Hullett D.A., et al. RS-61443—a new, potent immunosuppressive agent. Transplantation. 1991;51:27-31.
99. Kennedy G.A., Kay T.D., Johnson D.W., et al. Neutrophil dysplasia characterised by a pseudo-Pelger-Huet anomaly occurring with the use of mycophenolate mofetil and ganciclovir following renal transplantation: a report of five cases. Pathology. 2002;34(3):263-266.
100. Angermann C.E., Störk S., Costard-Jäckle A., et al. Reduction of cyclosporine after introduction of mycophenolate mofetil improves chronic renal dysfunction in heart transplant recipients—the IMPROVED multi-centre study. Eur Heart J. 2004;25(18):1626-1634.
101. Dewey C.W., Boothe D.M., Rinn K.L., et al. Treatment of a myasthenic dog using mycophenolate mofetil. J Vet Emerg Crit Care. 2000;10:177-187.
102. Zaucha J.M., Yu C., Zellmer E., et al. Effects of extending the duration of postgrafting immunosuppression and substituting granulocyte-colony-stimulating factor-mobilized peripheral blood mononuclear cells for marrow in allogeneic engraftment in a nonmyeloablative canine transplantation model. Biol Blood Marrow Transplant. 2001;7(9):513-516.
103. Smolen J.S., Kalden J.R., Scott D.L., et al. Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: a double-blind, randomised, multicentre trial, European Leflunomide Study. Lancet. 1999;353:259-266.
104. Gregory C.R., Stewart A., Sturges B., et al. Leflunomide effectively treats naturally occurring immune-mediated and inflammatory diseases of dogs that are unresponsive to conventional therapy. Transplant Proc. 1998;30:4143-4148.
104a. McChesney L.P., Xiao F., Sankary H.N., et al. An evaluation of leflunomide in the canine renal transplantation model. Transplantation. 1994;57(12):1717-1722.
104b. Colopy S.A., Baker T.A., Muir P. Efficacy of leflunomide for treatment of immune-mediated polyarthritis in dogs: 14 cases (2006-2008). J Am Vet Med Asso. 2010;236(3):312-338.
105. Marsolais D., Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discov. 2009;8(4):297-307.
106. Rhodes K.H. Feline immunomodulators. In: Bonagura J.D., editor. Kirk’s current veterinary therapy small animal practice (XII). Philadelphia: Saunders; 1995:581-584.
107. Snyder P.J. Androgens Brunton L.L., Lazo J.S., Parker K.L., editors. Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, pp 1573–1585, 2006.
108. Ward H. Immune-mediated hematopoietic diseases. Oncology and hematology. 20th Annual Waltham/OSU Symposium for the Treatment of Small Animal Diseases. 1996:99-104.
109. Stadtmauer E.A., Cassileth P.A., Edelstein M., et al. Danazol treatment of myelodysplastic syndromes. Br J Haematol. 1991;77:502-508.
110. Choudhry V.P., Kashyap R., Ahlawat S., et al. Vinblastine and danazol therapy in steroid resistant childhood chronic idiopathic thrombocytopenic purpura. Int J Hematol. 1995;61:157-162.
111. Schreiber A.D., Chien P., Tomaski A., et al. Effect of danazol in immune thrombocytopenic purpura. N Engl J Med. 1987;316:503-508.
112. Bloom J.C., Meunier L.D., Thiem P.A., et al. Use of danazol for treatment of corticosteroid-resistant immune-mediated thrombocytopenia in a dog. J Am Vet Med Assoc. 1989;194:76-78.
113. Holloway S.A., Meyer D.J., Mannella C. Prednisolone and danazol for treatment of immune-mediated anemia, thrombocytopenia, and ineffective erythroid regeneration in a dog. J Am Vet Med Assoc. 1990;197:1045-1048.
114. Langford C.A., Klippel J.H., Balow J.E., et al. Use of cytotoxic agents and cyclosporine in the treatment of autoimmune disease. Part I: rheumatologic and renal diseases. Ann Intern Med. 1998;128:1021-1028.
115. Langford C.A., Klippel J.H., Balow J.E., et al. Use of cytotoxic agents and cyclosporine in the treatment of autoimmune disease. Part II: Inflammatory bowel disease, systemic vasculitis and therapeutic toxicity. Ann Intern Med. 1998;129:49-58.
116. Rodriguez D.B., Mckin A., Easley R., et al. Relationship between red blood cell thiopurine tethyltransferase activity and myelotoxicity in dogs receiving azathioprine. Vet Intern Med. 2004;18:M339-M345.
117. Chabner B.A., Amrein P.C., Druker B.J., et al. Antineoplastic agents. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, pp 1315–1403, 2006.
118. Burke A., Smyth E., FitzGerald G.A. Analgesic-antipyretic and antiinflammatory agents; pharmacotherapy of gout. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, pp 671–715, 2006.
119. Wollheim F.A. Multiple drug resistance and rheumatology, Cutting Edge. Novartis Pharma Basel; 2003.
120. Werner G.H., Zerial A. Shugar D., editor. Viral chemotherapy. Immunopotentiating substances with antiviral activity. vol 1. 1984. Pergamon. New York. 511-559.
121. Ricklin Gutzwiller M.E., Reist M., Peel J.E., et al. Intradermal injection of heat-killed Mycobacterium vaccae in dogs with atopic dermatitis: a multicentre pilot study. Vet Dermatol. 2007;18:87-93.
122. Engelman R.W., Trang L.Q., Good R.A. Clinicopathologic responses in cats with feline leukemia virus–associated leukemia-lymphoma treated with staphylococcal protein A. Am J Pathol. 1985;118:367-378.
123. Engelman R.W., Good R.A., Day N.K. Clearance of retroviremia and regression of malignancy in cats with leukemia-lymphoma during treatment with staphylococcal protein A. Cancer Detect Prevent. 1987;10:435-444.
124. Cotter S.M., Essex M., McLane M.F., et al. Chemotherapy and passive immunotherapy in naturally occurring feline mediastinal lymphoma. In: Hardy W.D., Essex M., McClelland A.J., editors. Feline leukemia virus. Holland: Elsevier North Holland; 1980:219-225.
125. Reagan W.J., Scott-Moncrieff J.C., Christian J., et al. Effects of human intravenous immunoglobulin on canine monocytes and lymphocytes. Am J Vet Res. 1998;59:1568-1574.
126. Scott-Moncrieff Reagan W.J., Glickman L.T., et al. Treatment of nonregenerative anemia with human gamma globulin in dogs. J Am Vet Med Assoc. 1995;206:1895-1900.
127. Scott-Moncrieff J.C., Reagan W.J., Snyder P.W., et al. Intravenous administration of human immune globulin in dogs with immune-mediated hemolytic anemia. J Am Vet Med Assoc. 1997;10:1623-1627.
128. Scott-Moncrieff J.C., Reagan W.J. Human intravenous immunoglobulin therapy. Semin Vet Med Surg. 1997;12:178-185.
129. Malaguarnera M., Guccione N., Musumeci S., et al. Intravenous immunoglobulin plus interferon-in autoimmune hepatitis C. Biodrugs. 2004;18(1):63-70.
130. Anonymous. ADIS research and development profile: Abetimus, Abetimus sodium, LJP 394. Biodrugs. 2003;17(3):212-215.
131. Colombo S., Hill P.B., Shaw D.J., et al. Effectiveness of low dose immunotherapy in the treatment of canine atopic dermatitis: a prospective, double-blinded, clinical study. Vet Dermatol. 2005;16:162-170.
132. Hayes A.A., MacEwen E.G., Matus R.E., et al. Antileukemic activity of plasma cryoprecipitate therapy in the cat. In: Hardy W.D., Essex M., McClelland A.J., editors. Feline leukemia virus. Holland: Elsevier North Holland; 1980:245-251.
133. Caciolo P.L., Hayes A.A., Patnaik A.K., et al. A case of mycosis fungoides in a cat and literature review. J Am Anim Hosp Assoc. 1983;19:505-512.
133a. Parodi A.L., Misdorp W., Mialot J.P., et al. Intratumoral BCG and Corynebacterium parvum therapy of canine mammary tumours before radical mastectomy. Cancer Immunol Immunother. 1983;15(3):172-177.
134. Rosenthal R.C. Hormones in cancer therapy. Vet Clin North Am Small Anim Pract. 1982;12:67-77.
135. Marshall G.D., Gibbone A.S., Pamell L.S. Human cytokines induced by acemannan. J Allergy Clin Immunol. 1993;91:295.
136. Ramamoorthy L., Kemp M.C., Tizard I.R. Acemannan, a beta-(1,4)-acetylated mannan, induces nitric-oxide production in macrophage cell-line Raw-264.7. Mol Pharmacol. 1996;50:878.
137. King G.K., Yates K.M., Greenlee P.G., et al. The effect of acemannan immunostimulant in combination with surgery and radiation therapy on spontaneous canine and feline fibrosarcomas. J Am Anim Hosp Assoc. 1995;31:439.
138. Harris C., Pierce K., King G., et al. Efficacy of acemannan in treatment of canine and feline spontaneous neoplasms. Mol Biother. 1991;3:207.
139. MacEwen E.G., Hays A.A., Mooney S., et al. Evaluation of effect of levamisole on feline mammary cancer, short communication. J Biol ResponseModif. 1984;5:541-546.
140. Hahm K.B., Park I.S., Kim H.C., et al. Comparison of antiproliferative effects of 1‑histamine-2 receptor antagonists, cimetidine, ranitidine, and famotidine, in gastric cancer cells. Int J Immunopharmacol. 1996;18:393-399.
141. Adams W.J., Morris D.L. Pilot study—cimetidine enhances lymphocyte infiltration of human colorectal carcinoma: results of a small randomized control trial. Cancer. 1997;80:15-21.
142. Hamilton R.D., Wynalda M.A., Fitszpatick F.A., et al. Comparison between circulating interferon and drug levels following administration of 2-amino-5-bromo-6-phenyl-4(3H)-pyrimidone (ABPP) to different animal species. J Interferon Res. 1981;2:317-327.
143. Hengge U.H., Benninghoff B., Ruzicka T., et al. Topical immunomodulators-progress towards treating inflammation, infection, and cancer. Lancet Infect Dis. 2001;1:189-198.
144. Denburg J.A., Schmi R., Saito H., et al. Systemic aspects of allergic disease: bone marrow responses. J Allergy Clin Immunol. 2000;106(Suppl 5):S242-S246.
145. Menzies-Gow A.N., Flood-Page P.T., Robinson D.S., et al. Effect of inhaled interleukin-5 on eosinophil progenitors in the bronchi and bone marrow of asthmatic and non-asthmatic volunteers. Clin Exp Allergy. 2007;37(7):1023-1032.
146. Simon D., Braathen. Simon H-U: Eosinophils and atopic dermatitis. Allergy. 2004;59:561-571.
147. Al-Haddad S., Riddell R.H. The role of eosinophils in inflammatory bowel disease. Gut. 2005;54(12):1674-1675.
148. Kanaoka Y., Boyce J.A. Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J Immunol. 2004;173:1503-1510.
149. Senter D.A., Scott D.W., Miller W.H.Jr. Treatment of canine atopic dermatitis with zafirlukast, a leukotriene-receptor antagonist: a single-blinded, placebo-controlled study. Can Vet J. 2002;43(3):203-206.
150. Pease J.E., Williams T.J. Eotaxin and asthma. Curr Opin Pharmacol. 2001;1:248-253.
151. Denburg J.A. Bone marrow in atopy and asthma: hematopoietic mechanisms in allergic inflammation. Immunol Today. 1999;20(3):111-113.
152. Braccioni F., Dorman S.C., O’Byrne P.M., et al. The effect of cysteinyl leukotrienes on growth of eosinophil progenitors from peripheral blood and bone marrow of atopic subjects. J Allergy Clin Immunol. 2002;110:96-101.
153. Dewachter P., Mouton-Faivre C., Emala C.W. Anaphylaxis and anesthesia: controversies and new insights. Anesthesiology. 2009;111(5):1141-1150.
154. Mason N., Duval K., Shofer F.S. Cyclophosphamide exerts no beneficial effect over prednisone alone in the initial treatment of acute immune-mediated hemolytic anemia in dogs: a randomized controlled clinical trial. J Vet Intern Med. 2003;17:206-212.
155. Helfand S., Jain N.C., Paul M. Vincristine-loaded platelet therapy for idiopathic thrombocytopenia in a dog. J Am Vet Med Assoc. 1984;185:224.
156. Brown C.D., Parnell N.K., Brown D., et al. Steroid responsive neutropenia in dogs: 11 cases (1990-2002). ProcAm Coll Vet Int Med abstract. 2003:226.
157. Rees C.A., Boothe D.M., Boeckh A., et al. Dosing regimen and hematologic effects of pentoxifylline and its active metabolites in normal dogs. Vet Ther. 2003;4(2):188-196.
158. Rees C.A., Boothe D.M. Therapeutic response to pentoxifylline and its active metabolites in dogs with familial canine dermatomyositis. VetTher. 2003;4(3):234-241.
159. Asanuma Y., Yamada H., Matsuda T., et al. Successful treatment of interstitial pneumonia with lipo-PGE1 and pentoxifylline in a patient with dermatomyositis. Ryumachi. 1997;37:719-726.
160. Rosenkrantz W.S. Eosinophilic granuloma confusion. In: August J.R., editor. Consultations in feline internal medicine. Philadelphia: Saunders; 1991:121-124.
161. Messinger L.M. Therapy for feline dermatoses. Vet Clin North Am Small Anim Pract. 1995;25:981-1005.
162. Hogenesch H., Snyder P.W., Scott-Moncrieff J.C., et al. Interleukin-6 activity in dogs with juvenile polyarteritis syndrome: effect of corticosteroids. Clin Immunol Immunopathol. 1995;77:107.
163. Clements D.N., Grear R.N., Tattrsall J., et al. Type I immune-mediated polyarthritis in dogs: 39 cases (1997-2002). J Am Vet Med Assoc. 2004;224:1323-1327.
164. Sandborn W.J. Cyclosporine therapy for inflammatory bowel disease: definitive answers and remaining questions. Gastroenterology. 1995;109:1001-1003.
165. Sandborn W.J. A review of immune modifier therapy for inflammatory bowel disease: azathioprine, 6-mercaptopurine, cyclosporine, and methotrexate. Am J Gastroenterol. 1996;91:423-433.
166. Bird J. Guidelines on the diagnosis and management of AL amyloidosis. United Kingdom Myeloma Forum. 2005;125(6):681-700.