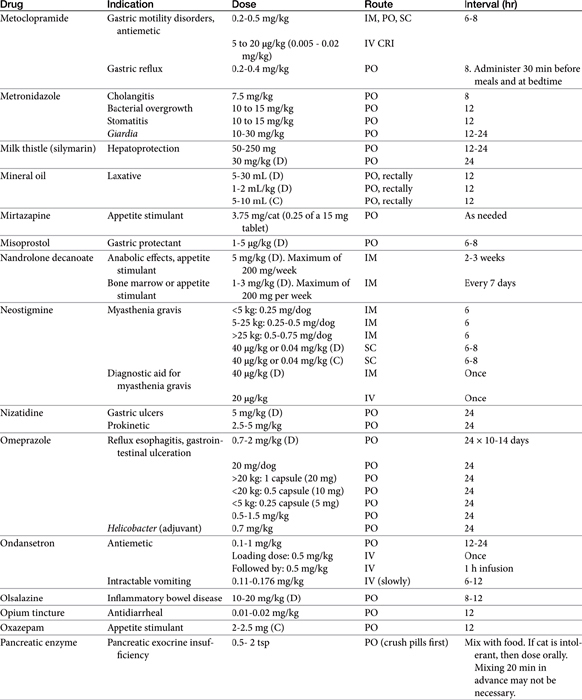
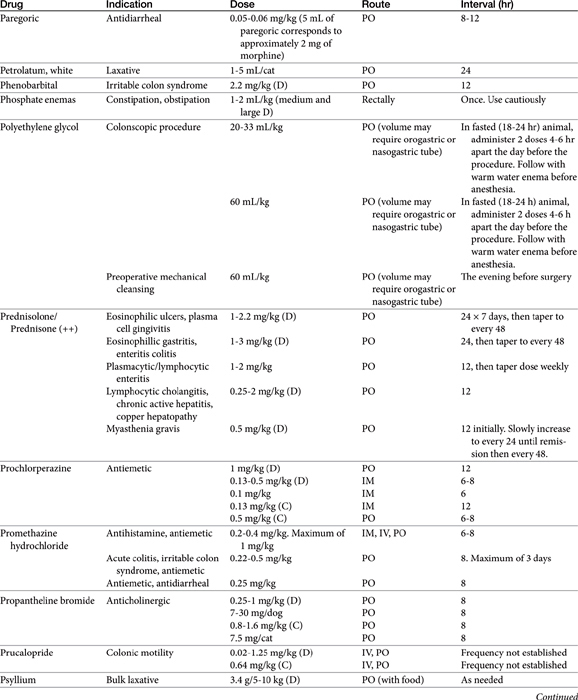
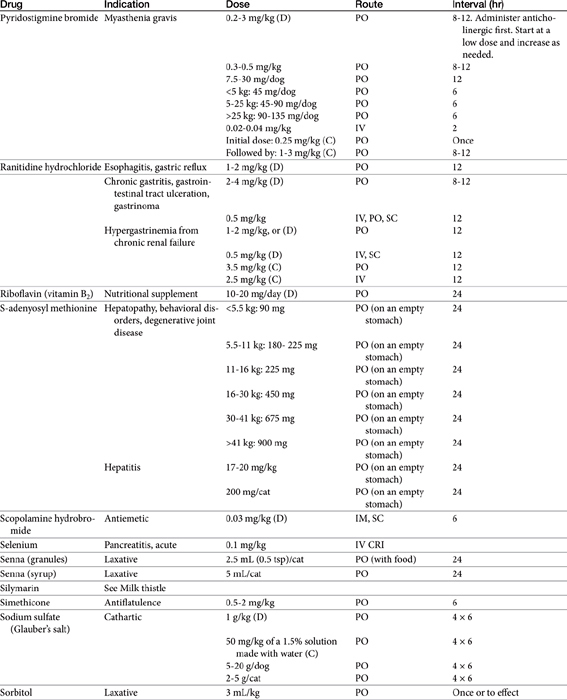
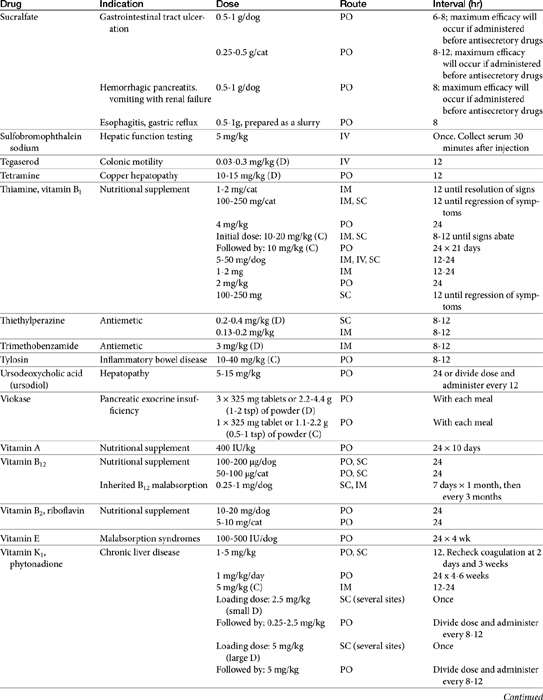
Chapter 19 Gastrointestinal Pharmacology
Drugs that Target Appetite or Caloric Balance
Drugs that alter body weight play can be important in the the prevention or treatment of disease in dogs and cats. Targets of pharmacologic management of weight include decreased energy intake (e.g., appetite, caloric absorption), altered energy partitioning, and increased energy expenditure.
Appetite is controlled primarily, but not exclusively, by the ventral and lateral nuclei of the hypothalamus. The nuclei respond to both short- and long-term signals.1 Hypothalamic directives are influenced by energy, which in turn is influenced by ingestion, absorption, metabolism, and storage. Chemical signals mediating these directives act locally or distantly, often balancing one another. Signals include hormones, neuropeptides, cytokines and neurotransmitters, several of which are pharmacologic targets.
Although a discussion of energy utilization is too complex and extensive for this text, understanding of the role of body fat in appetitic control and body weight has been markedly advanced in the last decade and warrants review of those aspects relevant to drug therapy. In the arcuate nucleus, primary neurons detect concentrations of metabolites and secondary neurons synchronize signals and coordinate vagally mediated responses. Among these neurons are two distinct populations of primary neurons and neuropeptides that control food intake and energy expenditure. Orexigenic peptides increase food intake and include neuropeptide Y (NPY) and agouti-related protein (AgRP); gamma amino butyric acid (GABA) is also released as an orexigenic signal. The NPY/AgRP neurons direct the effects of leptin and ghrelin and stimulate feeding during states of fasting. They also influence the anorexigenic peptides, which decrease food intake. Anorexigenic peptids include the pro-opiomelanocortin (POMC) neurons which produce alpha-melanocyte hormone (α-MSH), a powerful appetite suppressant, and the β-endorphins.. The POMC extensively communicates with hypothalamic neurons as well as other regions that regulate energy. Both orexigenic and anorexigenic neurons have receptors for balancing signaling molecules, which often cause opposing effects by interacting at the same ligand. NPY and α-MSH, located in the ventromedial nucleus of the appetite center, are considered balancing hormones. Other stimulatory mediators include norepinephrine (α2-receptors), dopamine (possibly D1 receptors), and opiate and pancreatic polypeptides. Although GABA also stimulates appetite, its effect may vary with route of administration.1 Other inhibitory mediators include serotonin (5-hydroxytryptamine [5-HT]), calcitonin, cholecystokinin, and corticotrophin-releasing factor. In addition to their central roles, several of these mediators influence energy metabolism peripherally.
Among the major target tissues for signals modulating energy metabolism is adipose tissue, which is now a recognized endocrine organ (Figure 19-1). Hormones play a critical role in energy homeostasis, insulin sensitivity, and lipid and carbohydrate metabolism. The system appears to be more sensitive to preventing starvation rather than obesity. Leptin and insulin appear to be the predominant mediators that signal adiposity: both circulate in proportion to body fat, enter the central nervous system (CNS) in proportion to plasma concentrations, and interact with CNS receptors that influence energy regulation, as reviewed by Havel.2 Other important peripheral mediators include acetylation-stimulating protein (ASP) and adiponectin, each being principally regulated by host nutritional status.
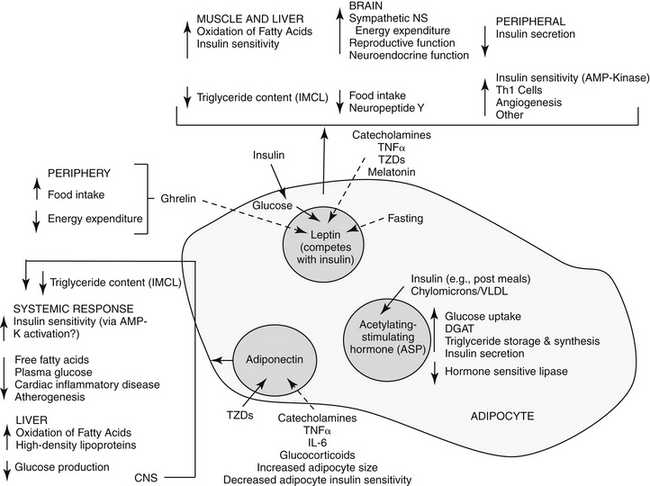
Figure 19-1 The complex interactions between adipose and other organs. The adipocyte is influenced by a number of factors and responds with paracrine, autocrine, and endocrine responses. Leptin is produced by many tissues, but particularly white adipose tissue. It inhibits food intake and increases energy expenditure while increasing insulin sensitivity (by way of activation of AMP kinase). Central influences include reproductive and neuroendocrine functions. In the adipocyte leptin is influenced by changes in adipocyte glucose metabolism as insulin secretion responds to food intake. Adiponectin also increases insulin sensitivity (through activation of AMP kinase), resulting in increased fatty acid oxidation in the muscle and liver and decreased hepatic triglycerides. Acetylation-stimulating protein also reduces hepatic glucose production. Solid arrows, stimulate; dashed arrows, inhibit; DGAT, diglycerol acyltransferase; TZD, thiazolidinediones.
KEY POINT 19-1
Adipose tissue has emerged as one of the biggest endocrine organs, affecting appetite and energy homeostasis.
Leptin was discovered in obese mice that randomly emerged as mutants among normal populations. At least six leptin receptors (“LepR”, a through f) are associated with the ob gene (Ob-R) (obesity gene), suggesting a complex role for this hormone. 3 Leptin receptors are similar to class I cytokine receptors, perhaps linking cytokines to diseases associated with obesity disorders.3
Currently, little information appears to be available regarding leptin content in dogs or cats and information must be drawn from other species. Although most organs produce leptin, white adipose tissue is the primary source, with the amount produced varying among species: more leptin is produced in subcutaneous compared with omental fat in humans, but the opposite is true in rats.3 In humans, leptin content is greatest in the heart, liver, small intestine, prostate, and ovaries versus the lung and kidney in mice. Each tissue may have a particular receptor isoform, allowing differential roles among tissues. Leptin plays an important, albeit complex, role in the peripheral regulation of adipose tissues. Its primary function is as a lipostat, communicating to other tissues the current status of body fat reserves. As such, leptin mediates fuel movement and use (see Figure 19-1) and energy expenditure. 3 It is more effective in signaling deficient rather than excessive energy reserves.2 Activated leptin receptors induce satiety in the appetite center (inhibition of NPY, stimulation of alpha-MSH neurons). Leptin also appears to influence thermogenesis. Peripherally, leptin appears to increase hematopoiesis, angiogenesis, wound repair, and puberty; its role in reproduction is particularly complex. Leptin is primarily regulated by food-induced responses to insulin, with influences being more dramatic during fasting and characterized by diurnal variation.2 In normal animals, insulin appears to directly regulate leptin gene expression and excretion in adipose tissue; it also indirectly influences it through adipocyte glucose use and oxidative metabolism. Both hormones increase in concert.3 Both leptin and insulin share inhibitory signal transduction pathways in response to food.2 Leptin, in turn, appears to inhibit insulin secretion, and, peripherally competes with insulin; however conflicting results also suggest that leptin has no effect on insulin.2,3 Glucocorticoids appear to stimulate leptin gene expression, although it is not clear if this effect is direct (gene expression) or indirect (altered food intake or insulin concentrations). Leptin is downregulated by melatonin at night; accordingly, humans with short sleep cycles may be predisposed to obesity. Depletion of body stores is not detected in hypoleptimemic animals, and as such, is more dangerous than hyperleptinemia. In humans, hypoleptinemia is associated with neuroendocrine, reproductive, metabolic, and immunologic imbalances,and in rodents, marked insulin reistance and hyperlipidemia.2 Leptin concentrations in persons with eating disorders characterized by anorexia are similar to or lower than concentrations in persons without eating disorders.3. Its role in chronic liver disease increasingly is being recognized.4 Many of these abnormalities can be normalized in humans with recombinant human leptin. Differences in genes (including mutations) regulating leptin also are linked to human obesity with hyperleptinemia indicating leptin resistance.
KEY POINT 19-3
Adiponectin influences lipid and carbohydrate metabolism and is necessary for normal insulin action.
The second major hormone influencing energy metabolism, ASP, is produced from complement factor C. Locally, ASP paracrine actions in adipose tissue include increased glucose uptake and diacylglycerol acyltransferase activity and decreased hormone-sensitive lipase (see Figure 19-1). As such, adipocyte triglyceride synthesis and storage increase after eating, resulting in increased free fatty acids and triglyceride clearance. Serum ASP concentrations are increased by lipids, and increase in proportion to body fat in obese human patients; they decrease during states of fasting. Insulin may control ASP in reponse to eating or fasting; ASP in turn may directly stimulate insulin secretion.
Adiponectin, a large protein secreted by adiopocytes, is a third hormone influencing energy metabolism. Its biological effect varies with its state of diamerization.2 Adiponectin appears to influence lipid and carbohydrate metabolism both directly and indirectly (see Figure 19-1). Adiponectin appears to be necessary for normal insulin actions. Concentrations are reduced in patients with type 2 diabetes compared with nondiabetic humans.2 The effect of adiponectin on decreased circulating glucose are independent of fat content and occurs without influencing insulin secretion. Mechanisms may include decreased hepatic glucose formation and increased tissue glucose use by decreasing insulin resistance. Circulating adiponectin is negatively correlated with the content of body fat, particularly visceral (e.g., omental) rather than subcutaneous in humans.2 Adiponectin may reduce ectopic fat in the liver and muscle through increased fat oxidation. Thus, low visceral fat may reduce adiponectin production, contributing to insulin resistance associated with obesity.2 The impact of insulin on adiponectin is not clear. Among the emerging effects of adiponectin is protection against inflammatory cardiovascular disease (including atherosclerosis), which may explains the relationship between body fat and cardiac disease. Cytokines, catecholamines, and glucocorticoids decrease adiponectin production, which may contribute to their characteristic incease in insulin resistance.
A final signal able to centrally influence energy metabolism is ghrelin. Located in peripheral tissues, ghrelin is released by an empty stomach, appears to oppose leptin in the hypothalamus, and has been shown to increase food intake and decrease energy expenditure.
Control of appetite through pharmacologic manipulation of orixogenic and anorexigenic signals has proven difficult. Obvious targets which might suppress appetite (e.g., α-MSH) have profound impacts in multiple body systems, increasing the risk of adverse reactions. The complexties of regulation contribute to the risk of adversity. For example, alternate pathways of synthesis or response tend to emerge with blockade of a target signal.
Appetite Stimulants
Studies regarding the pharmacologic control of appetite have traditionally focused on decreased, rather than increased, food intake, particularly in humans. However, the role of cachexia associated with weight loss and anorexia in human patients with cancer has stimulated a renewed interest in appetite stimulants.5 Drugs that inhibit gluconeogenesis, such as hydrazine sulfate, or promote gastric emptying, such as metoclopramide, have been used successfully to stimulate food intake in some human patients.5 The use of steroids, including megestrol acetate, in treatment of cachexia is discussed later in this chapter.5 Both glucocorticoids and B vitamins have been used to nonspecifically stimulate appetite in animals. Drugs used to treat depression and psychosis in human patients are associated with appetite increase and weight gain.6 They antagonize a variety of receptors, although their clinical potency is often related to increased serotonin, which may, in fact, decrease appetite in some patients.
Mirtazapine is a piperazino-azepine antidepressant characterized by serotonergic activity as a result of 5-HT-1 agonistic activity and inhibition of serotonin reuptake.7 Sympathetic (norepinephrine) actions reflect antagonism of α -2 autoreceptors as well as influences by other receptors. As a behavior-modifying drug, mirtazapine is discussed in greater depth in Chapter 26. Anecdotally, mirtazapine has been used to stimulate appetite in either dogs or cats, the latter at 3 mg/cat every 72 hours.
The benzodiazepines diazepam (Valium) and oxazepam, a metabolite of diazepam (Serax), have successfully induced appetite in cats, probably through gabaminergic effects and central inhibition of the satiety center in the hypothalamus (Table 19-1).8 Diazepam is administered intravenously or orally, whereas oxazepam is administered orally. Of the two drugs, diazepam may be more effective, although sedation is greater. The benzodiazepines do not stimulate appetite in the dog as effectively as in the cat. Hepatotoxicity associated with diazepam therapy when used as an appetite stimulant has been reported in cats9,10 and is discussed in more depth in Chapter 27. Toxicity appears to be idiosyncratic and thus may not be predictable; it is not likely to happen in a large percentage of animals receiving the drug.
Cyproheptadine, an antihistamine with antiserotonin properties, has caused weight gain in geriatric human patients and in adults and younger patients afflicted with eating disorders. Its mechanism probably reflects inhibition of serotonergic receptors that control appetite. Serotonin antagonists also increase food intake in cats,1 and cyproheptadine has been used clinically to stimulate the appetite of some anorexic cats. Cyproheptadine kinetics have been reported in the cat. Oral bioavailability of the tablet is 100%, and the elimination half-life approximates 13 hours.11 Cats tolerated a dose of 8 mg orally with no adverse effects, although impact on appetite was not described. Based on this study, once- to twice-daily dosing of 8 mg appears to be safe.
Cachexia
Cachexia is an involuntary state characterized by loss of more than 5% of body weight. In humans it occurs over a defined period, generally of 2 to 6 months. Ultimately, it is a condition of starvation characterized by depletion of body mass, particularly muscle, but to a lesser degree, adipose tissue.12 Cachexia develops in 50% of human patients with cancer, contributing to substantially shortened survival times. Cachexia also is a manifestation of other chronic diseases, including (in humans), acquired immune-deficiency syndrome (AIDS), heart failure, rheumatoid arthritis, Crohn’s disease, chronic obstructive pulmonary disease, and chronic renal disease.12 It appears to be a cytokine-driven process, with key components including anorexia and a state of hypercatabolism. The principle cytokines appear to include TNF-alpha, interleukins 1 and 6, and interferon-γ Of these, TNF-alpha is presumed to stimulate mechanisms that lead to severe cachexia.12 Muscle wasting may reflect inhibition of myogenic differentiation; an energy sink may result from increased concentrations of mitochondrial uncoupling proteins. Lipoproteinase activity is inhibited by TNF-alpha. Interleukins contribute to CNS-mediated anorexia and decreased albumin synthesis in the liver. Megestrol acetate is the only treatment approved by the Food and Drug Administration for cancer or AIDS-related cachexia syndromes in humans.12 Other less commonly used drugs are glucocorticoids, anabolic steroids, antiseratonergic drugs, dronabinol, and prokinetic drugs.
Megestrol is a synthetic derivative of progesterone.12 As such, megestrol acetate targets cachexia by directly and indirectly stimulating appetite and antagonizing the catabolic metabolic effects of cytokines. As with other steroidal hormones, the effects of megestrol (and progesterone) involve passive diffusion into the cell and binding to specific intracellular progesterone receptors A or B (PR-A or -B) and heat shock proteins. The drugs move into the nucleus, bind to progesterone response elements on target genes, and influence transcription through inhibition (PR-A) or stimulation (PR-B) of other steroid response elements. Although the majority of progesterone effects are genomic, nongenomic actions also occur. Metabolic effects of progesterone include increased basal insulin concentration and increased response to carbohydrate load; increased lipoprotein lipase with altered fat deposition, plasma lipid, and lipoprotein concentrations; and modulation of body temperature.12 In humans and animal models of cancer, megestrol stimulates appetite, increases caloric intake, induces a sense of well-being, and causes weight gain, particularly of fat. Fat is the preferred weight gain because it provides more kilocalories per gram than proteins or carbohydrate and helps stabilize body temperatures.12 Megestrol decreases the effects, sometimes by inhibiting formation of TNF-alpha, interleukin-1 (IL-1), and interleukin-6 (IL-6). Centrally, megestrol appears to modulate neurotransmitters responsible for appetite regulation such as NPY, which in turn stimulates the release of other mediators. Megestrol may also stabilize declining concentrations of β-endorphins in the cerebrospinal fluid.
KEY POINT 19-4
Megestrol acetate targets cachexia either directly and indirectly, stimulating appetite and antagonizing the catabolic metabolic effects of cytokines.
Effects of megestrol are dose and (particularly for fat gain) duration dependent. Initial weight gain requires high concentrations (>300 ng/mL) for more than 40% of a 24-hour dosing interval. In humans this requires administration of the tablet four times daily. Megestrol acetate is used rather than other progestationals, which must be given parenterally.12 Bioavailabilty of megestrol acetate is variable, being greater with the oral solution compared to the tablet. With tablets, peak concentrations may vary 6 fold; variability is much less with the oral solution. Disposition is complex. Hepatic metabolism is necessary to free the steroid from acetate, with the steroids subsequently conjugated with glucuronic acid before elimination. Elimination also varies, with half-lives that range from 13 to 105 hours. In humans, a single daily dose of the suspension (800 mg) achieves peak concentrations between 1500 and 3000 ng/mL. Not surprisingly, the oral solution is associated with a much higher rate of response compared with the tablet. A micronized (nanocrystal) preparation is currently under investigation for human use.
Limited information is available regarding use of megestrol acetate in animals. The disposition of megestrol acetate has been described in Beagles as part of the preclinical assessment in humans. Four preparations were studied for 72 hours after administration of 10 mg/kg (by oral gavage) either in the fasted or the fed (high-fat meal) state. The preparations included two different nanocrystal oral solutions and two commercially available oral suspensions (Par Pharmaceutical and Bristol-Myers). After the high-fat meal, peak concentrations (1600 to 2200 ng/mL) and area under the curve (AUC) were higher with the nanocrystal oral suspensions compared with the commercially available oral suspensions (both approximating 300 ng/mL). Although the elimination half-life was not reported, the disappearance half-life of megestrol appeared to be between 10 and 20 hours.12
Megestrol acetate appears to be better tolerated in humans compared with animals. The primary adverse events in humans are thromobembolic, reflecting increased thrombin receptors in smooth muscles. Venous distention and capacitance increase, contributing to reduced blood flow and stasis. Addison’s disease and glucose intolerance are sporadically reported, reflecting its intrinsic corticosteroid activities.12
Anecdotally, megesterol acetate has been effective in dogs to treat chemotherapy-induced nausea and inappetence. However, in humans a prospective randomized controlled clinical trial in cachectic human cancer patients compared the efficacy of an anabolic steroid (fluoxymesterone [10 mg, 0.142 mg/k] twice daily), megestrol acetate tablets (800 mg [11.4 mg/kg] once daily), and a glucocorticoid (dexamethasone, 0.75 mg [0.01 mg/kg] four times daily) as appetite stimulants. Of the three, the anabolic steroid was least (significantly) and megestrol (nonsignificantly) most clinically effective. Glucocorticoids usually were discontinued because of side effects, although megestrol acetate was associated with the most thromboembolic events.13
Appetite Suppressants and Anti-Obesity Drugs
Obesity is a physiologic disorder of energy balance in which energy intake exceeds energy expenditures. Excessive energy is stored as fat. In rodent models and humans, leptin deficiency or leptin resistance can result in obesity caused by hyperphagia and decreased energy expenditure.3 Other characteristics of obesity in humans include non–insulin-dependent diabetes mellitus, severe insulin resistance, hypothermia and cold intolerance, infertility, and decreased lean body mass.3
Anti-obesity drugs might target mediators of appetite or satiety. However, differences in response to drugs that affect appetite limit extrapolation of studies among species. Hypoleptinemia has been associated with obesity. However, obesity appears to be accompanied by resistance to leptin or leptinlike drugs. Alternative targets for treatment of obesity might include mediators of leptin actions, including NPY, Y1 and Y5 receptor agonists, and melanocortin MC4 receptors. Central chemical targets for drugs that might influence appetite include all mediators, their receptors, or upstream regulators of mediator release. These include anorexigenic signals such as α-MSH, opioids, and serotonin (specifically the 5-HT2C receptor) or mediators of satiety that emerge after food ingestion, such as cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), ghrelin, peptide YY (PYY), bombesin-like peptides, enterostatin, oxyntomodulin, and apolipoprotein IV (apoAVI).
Among the drugs studied for central suppressant effects are cannabinoids, the manipulation of which may influence consumption of highly palatable foods. For example, rimonabant is a cannabinoid receptor type 1 (CB1) receptor antagonist that decreases intake of palatable foods, leading to decreased body weight in rodents. However, dogs (and other species) respond differently to cannabinoids. For example, although CB1 antagonists decrease food intake, causing weight loss in dogs, appetite suppression is attenuated after several weeks. Dogs also appear more sensitive to side effects associated with CB1 antagonists, exhibiting vomiting, diarrhea, and pruritus at doses necessary to decrease appetite. Although cats tolerate antagonists better than dogs, they respond only at doses associated with severe pruritus, panting, agitation, and CNS stimulation.14 Selective 5-HT2C receptor antagonists appear to be effective in decreasing food intake in rodent models but not in dogs or cats. Higher doses that might be more effective are associated with adverse effects.14 The human pancreatic lipase inhibitor orlistat is associated with modest weight loss in dogs. However, significant increase in food intake, presumably in response to caloric loss, is accompanied by markedly increased fecal fat, leading to uncontrolled leakage and perianal and abdominal soiling.14
KEY POINT 19-5
Anti-obesity drugs target mediators of appetite or satiety or alter energy intake, expenditure, or use.
Anti-obesity drugs might target altered energy intake. Dirlotapide is a selective microsomal triglyceride transfer protein inhibitor that blocks both assembly and release of lipoprotein particles into the bloodstream (package insert [PI]). However, the mechanism by which weight gain is controlled is not clear. Appetite suppression reflects local GI effects, including decreased fat absorption. Subsequent lipid filling of enterocytes appears to stimulate a satiety signal. Fecal fat is increased. Efficacy does not appear to correlate with serum concentrations. The impact of dirlotapide on serum or gastric mucosal leptin, ASP, or adiponectin apparently has not been addressed.
In dogs, dirlotapide (Slentrol) is systemically bioavailable after oral administration, although absorption is markedly variable. Elimination of absorbed drug reflects hepatic metabolism that follows nonlinear kinetics, with concentrations increasing disproportionately with dose. Mean half-life varies between 5 to 18 hours at the clinical dose but appears to increase with dose and duration of dosing ((package insert).
Safety of dirlotapide when administered in dogs for 1 year has been established in dogs. Adverse effects, should they occur, generally emerge within the first month of therapy. According to the package insert, the incidence of vomiting and diarrhea was greater in dirlotapide-treated (25% and 12%, respectively) compared with control-treated (corn oil) animals (22% and 7%, respectively). Serum chemistry changes occurred early, including mild to moderately increased serum hepatic transaminase, although concentrations remained within the normal range and decreased over the 4-month treatment period. Mean cholesterol and high-density lipoprotein also decreased (below reference range for cholesterol during the treatment period); however, triglycerides did not change. Serum total protein, albumin, and blood urea nitrogen levels also decreased compared with those of control animals, although all were within normal ranges. Enterocytes were characterized by lipid vacuolization and the liver by mild periportal fatty changes.
Absorption of fat-soluble vitamins might be affected by dilortapide. Plasma vitamins A and E concentrations were lower in treated compared with control dogs, but concentrations appeared to increase during weight stabilization (second through sixth months), reaching control concentrations after treatment was discontinued (PI). A study in 72 obese Labrador Retrievers (n = 48) receiving dilortapide at the labeled dose for 52 weeks was reported in materials obtained through the Freedom of Information Act. Weight loss during the initial and retraining phases was 18.4% and 5% to 6%, respectively. In addition to vomiting and diarrhea, ophthalmic abnormalities were found at study end. These included focal or multifocal retinopathies or diffuse retinal degeneration in eight dogs receiving the drug for 12 months and two dogs for 6 months. Generalized progressive retinal atrophy occurred in one dog treated for 12 months and one dog treated for 6 months, respectively. Cataracts were observed in four dogs receiving the drug for 12 months and one dog receiving the drug for 6 months. Pretreatment ophthalmic exams were not available, and abnormalities were not reported in control dogs, although it is not clear if they did not occur or simply were not recorded. However, a follow-up 9-month study of 34 dogs of different breeds, in which a different formulation was used that yielded pharmacokinetics similar to the commercial preparation, found no ocular lesions, leading investigators to conclude that previous ocular abnormalities reflected breed predisposition.
Use of dilortapide in dogs must be accompanied by a weight loss program. Loss of appetite will not last more than several days after therapy is discontinued. Dosing is complex and is based on body weight (see package insert). A total of 11% to 13% of body weight was lost in patients studied during drug approval (PI), an amount considered to contribute positively to animal health. At study end mean final dose was 0.26 to 0.56 mg/kg. Dilortapide should not be administered to humans or cats. Adverse reactions in humans include abdominal pain, distention, diarrhea, flatulence, nausea, and vomiting.
Several drugs may affect appetite secondary to their intended therapeutic effect. Propofol (1-2 mg/kg IV) was reported in a research abstract15 to be an effective appetite stimulant, presumably through stimulation of GABA-A and NYP and inhibition of serotonin receptors. Anecdotally, omega-3 fatty acids (EPA and DHA) may stimulate appetite by inhibiting cytokines responsible for anorexia.
Emetics and Antiemetics
The Vomiting Reflex
Emesis is a complex protective reflex that is not well developed in all species but does occur in both dogs and cats.16-18 Although several afferent pathways may be responsible for initiating emesis, all signals are coordinated by the emetic center. Located in the lateral reticular formation in the mid brainstem, the emetic center is in close proximity to the nucleus tractus solitarius of the vagus nerve and the chemoreceptor trigger zone (CTZ), the latter of which is located adjacent to the area postrema in the bottom of the lateral ventricle. The latter coordinates vomiting associated with blood-borne chemicals (Figures 19-2 and 19-3). The emetic center coordinates vomiting associated with afferent peripheral and central (neural) signals. Among the signals coordinating vomiting is the tachykinin neuropeptid, ≥substance P. Drugs that cause or ameliorate vomiting generally do so by modifying afferent or efferent neurotransmitters responsible for transmission of the signal from various afferent sites. The emetic center is protected by the blood–brain barrier, whereas the CTZ is not; therefore, the CTZ is able to monitor the presence of emetics in the blood or cerebrospinal fluid. However, drug penetrability to each site varies, affecting both drug safety and efficacy.16-20
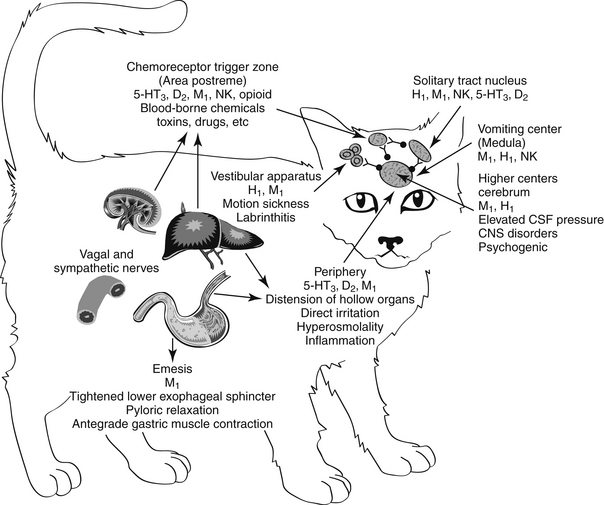
Figure 19-2 Sites that mediate the emetic reflex. The secondary neurotransmitters at each site are in parentheses. Stimuli that mediate emesis at each site are listed below the neurotransmitter. CNS, Central nervous system; CTZ, chemoreceptor trigger zone; CSF, cerebrospinal fluid; LES, lower esophageal sphincter.
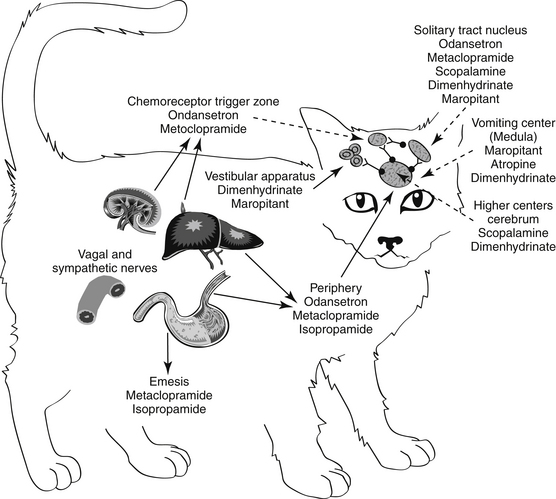
Figure 19-3 Antiemetic drugs effective at each site of the emetic reflex. CTZ, Chemoreceptor trigger zone.
KEY POINT 19-6
Drugs that inhibit emetic signals at the vomition center have the potential to be the broadest in efficacy.
Several sites in the vomiting reflex are targeted by drugs. Ideally, drugs that target the emetic center would be characterized by the broadest spectrum and potentially the best efficacy. Historically, because such drugs must be able to penetrate the blood–brain barrier, they tend to be characterized by increased risk of side effects. This risk is minimized if the drug targets a mediator whose effects are limited to the vomiting center only. In addition to integration of the emetic reflex, impulses integrated by the center include afferent signals from higher centers such as the cerebral cortex and limbic system. For example, psychogenic vomiting, or vomiting induced by visual and olfactory stimuli, originates in the cerebral cortex, whereas head injuries and increased intracranial pressure initiate emesis by way of the limbic pathways. The solitarius nucleus contains receptors for enkephalin, histamine, serotonin (5-HT3), and acetylcholine (ACh). ACh is a major afferent neurotransmitter in the higher centers, with histamine acting as a secondary transmitter by way of H1 receptors.19 However, substance P, a member of the tachykinin family of neuropeptides, has recently been identified as a key neurotransmitter associated with emesis in higher centers, including the emetic center.21 Substance P targets neurokinin (NK1) receptors located throughout the emetic center (as reviewed by Wu20), including the nucleus tractus solitarius, the area postrema, and the dorsal motor nucleus of the vagus.22 The emetic center also involves cannabinoid receptors (also located in the CTZ), although their role is not clear.
Blood-borne chemical compounds stimulate the CTZ.17,19 Examples include circulating toxins associated with disease (uremia, pyometra, liver disease, endotoxemia), radiation sickness, and drugs (e.g., opioids, cardiac glycosides, anticancer chemotherapeutic agents). Signals in the CTZ are mediated by dopaminergic (D2)19 and serotonergic (5-hydroxytryptamine; 5-HT3) receptors23 and neurokinin receptors. Histamine by way of H1 receptors acts as a secondary neurotransmitter at the CTZ. Neurokinin receptors in humans are responsible for the delayed phase of vomiting associated with cisplatin anticancer chemotherapy. Alpha-2 receptors associated with the area postrema also induce emesis in dogs, cats, and other species.24-26 The CTZ also is rich in opioid receptors. The safety of CTZ-active drugs may be increased by selectively targeting the subreceptor types for each neurotransmitter.
Emetic impulses originating from the semicircular canals of the vestibular apparatus are transmitted by the eighth cranial nerve to the vestibular nuclei and then by way of the CTZ and the uvula and nodulus of the cerebellum to the emetic center. This pathway, mediated by histaminergic (subtype H1) receptors, is responsible for eliciting the emesis that accompanies motion sickness and labyrinthitis.27
Peripheral impulses cause emesis that arises from stimulation of the pharynx and fauces; the signals are transmitted by afferent nerves in the ninth cranial nerve to the emetic center. Other peripheral afferent pathways include those arising from stimulation (i.e., irritation or distention) of various visceral organs and tissues. Impulses may be carried by sympathetic or vagal afferents from the heart, stomach, duodenum, small intestine, liver, gallbladder, peritoneum, kidneys, ureter, urinary bladder, and uterus. ACh is the primary neurotransmitter mediating the afferent limb of the emesis reflex from peripheral causes. Muscarinic receptors initiate the impulse that travels to the emetic center by way of the vagus nerve.
Efferent signals that stimulate the emetic reflex travel back to the stomach by the tenth cranial (vagus) nerve. ACh also acts as the primary efferent neurotransmitter in the vagus and in the smooth muscle of the stomach. In the stomach, dopamine receptors (D2) appear to inhibit gastric motility, during nausea and vomiting. In addition, dopamine receptors contribute to reflexes that allow relaxation of the upper stomach and delayed gastric emptying associated with gastric distention caused by food.24 Finally, serotonin (by way of 5-HT3 receptors) contributes afferent pathways from the stomach and small intestine.24
Emetics
Clinically, emesis is pharmacologically induced to empty the anterior portion of the digestive tract. Indications include preparation for induction of general anesthesia in animals that may have food in the stomach (e.g., use of hydromorphone) or treatment of ingested, noncorrosive poisons.
Peripherally Acting (Reflex) Emetics
Although their efficacy and safety vary, a number of substances induce emesis by either distending the pharynx, esophagus, stomach, or duodenum (hollow organs) or irritating the epithelium of the GI tract. Distention with warm water or saline can induce the emetic response. In addition, in the case of toxin ingestion, administration of warm water by stomach tube may help dilute poisons. Emesis can be induced in dogs by oral administration of a solution of warm saturated (strong) sodium chloride or pharyngeal placement of a small amount of plain table salt or neutral salt crystals, such as sodium carbonate. Orally administered hydrogen peroxide (3%) often induces emesis rapidly in cats and dogs, although fatal aspiration of hydrogen peroxide foam is possible. Ipecac syrup is an over-the-counter emetic commonly recommended to induce emesis in human pediatric patients. It contains the alkaloid emetine, which increases lacrimation, salivation, and bronchial secretions. Emesis usually, but not consistently, occurs as a result of both peripheral and central stimulation. If repeated use fails to induce emesis, however, gastric lavage may be indicated to remove potentially toxic doses of the drug. Although ipecac syrup or powder has been used as an emetic for many years for cats, adverse effects include death, and its use in cats is discouraged.
Centrally Acting Emetics
The central effects of ipecac were discussed in the previous section. Although a number of drugs are capable of stimulating the CTZ centrally, certain opiates, particularly apomorphine, are indicated for their emetic effect. Apomorphine hydrochloride is a synthetic derivative of morphine but is characterized with only marginal depressant activity. Its emetic activity reflects stimulation of dopamine receptors and more readily than other morphinelike actions. Emesis will occur regardless of the route of administration, although oral doses are higher than those by other routes to compensate for reduced oral bioavailability. Emesis generally occurs in 2 to 10 minutes after subcutaneous or conjunctival administration. Although apomorphine stimulates vomiting at the CTZ, it also directly depresses the emetic center, and subsequent doses are not likely to induce emesis if the first dose was not successful. Excessive doses of apomorphine can depress the CNS, particularly the respiratory center, and are contraindicated in the presence of existing central depression.Apomorphine is currently available as an injectable preparation.
Xylazine is an α2-agonist historically used for sedative analgesia. Emesis in dogs is not as consistent as in cats. Emesis mediated by α2 stimulation occurs in cats at doses lower than that recommended for sedation (0.05 mg/kg).26 Emetogens were evaluated in cats in anticipation of an antiemetic clinical trial.22 Three emetogens were tested, with xylazine (0.44 mg/kg intramuscularly) reliably causing emesis. In contrast, neither apomorphine (0.04 mg/kg intravenously) nor syrup of ipecac (0.5 mL/kg) predictably caused emesis. Syrup of ipecac causes anorexia for several days. The use of medetomidine to induce emesis has not been reported, although its actions are similar to those of xylazine.
Antiemetics
Antiemetics control emesis by either a central or a peripheral action (see Figures 19-2 and 19-3). Both actions depend on and can be correlated with blockade of neurotransmission at receptor sites.27,28 Centrally acting antiemetics block impulses at higher centers and at the emetic center and include muscarinic anticholinergics and drugs that target neurokinin receptors; antidopaminergics and antiserotonergics, which block dopaminergic receptors at the CTZ; and antihistaminergics, which primarily block H1 receptors at the vestibular apparatus but secondarily at multiple central centers. Antiemetic agents possess either a limited or a broad effect, depending on which signals and centers are inhibited.
Centrally Acting Antiemetics
Vomiting center
Maropitant (Cerenia) is a neurokinin (NK1) receptor antagonist that blocks the actions of substance P in the area postrema and nucleus solatarius; Aprepitant is a human drug in the same classused as rescue anti-emetic therapy in cancer patients nonresponsive to 5HT3–dexamethasone combinations.29 Approved as an oral or subcutaneous preparation for dogs for the prevention of acute vomiting and motion sickness, maropitant has proved efficacious in the control of vomiting associated with many central and peripheral causes (Table 19-2).
In dogs bioavailability is greater after subcutaneous (91%) compared with oral (24%) administration, probably because of first-pass metabolism (PI). Relevant pharmacokinetic parameters include Cmax (ng/mL) of 92 and 81 ng/mL at 0.75 and 2 hours, respectively, after administration of 1 mg/kg subcutaneously and 2 mg/kg orally, respectively. It is highly (99.5%) bound to plasma proteins. Maropitant is metabolized in dogs by CYP2D15 and CYP3A12. Elimination half-life in dogs approximates 9 hours after subcutaneous administration and 4 to 5 hours after oral administration. However, saturation of drug-metabolizing enzymes (probably CYP2D15) results in nonproportional increases in drug concentrations as the dose is increased up to 16 mg/kg orally; proportionality returns at 20 to 50 mg/kg orally. The injectable product remains potent for 28 days when prepared and stored according to labeled directions (amber vial at room temperature). Pain on injection may be common. Side effects delineated on the package insert include bone marrow hypoplasia in puppies younger than 11 (but not greater than 15) weeks of age.
KEY POINT 19-7
As a neurokinin antagonist, the specificity of maropitant results in safe and effective control of motion sickness and vomiting associated with both central and peripheral causes of vomiting.
Maropitant kinetics have been described in the cat after single subcutaneous and oral dosing and multiple subcutaneous dosing.22 The drug was well tolerated in cats (n = 6) during 15 days of subcutaneous administration at doses ranging from 0.5 to 5 mg/kg; one cat developed tremors at the 5 mg/kg dose. No changes occurred in clinical laboratory tests. Plasma maropitant concentrations increased proportionately with dose. After single intravenous dosing, the volume of distribution of maropitant in cats was 6.2 L/kg. Maximum drug concentration after oral and subcutaneous administration of 1 mg/kg in cats were 156 ng/mL and 269 ng/mL (with 50% coefficient of variabilility), respectively. The elimination half-life in cats was 13 to 17 hours; oral bioavailability varied from 50% to 117%. A dose of 1 mg/kg administered intravenously, orally, or subcutaneously prevented emesis induced by xylazine (0.44 mg/kg intramuscularly) and experimentally induced motion sickness.
Maropitant was approved for use in animals in Europe before the United States. It has proved effective for control of vomiting associated with drugs such as cisplatin, apomorphine, and morphine derivatives. Maropitant has been compared with metoclopramide (0.33 mg/kg every 8 hours subcutaneously in study one, 0.5 to 1 mg/kg/day in study two) in two multicenter, prospective, randomized, positively controlled clinical trials.30 Dogs (n = 64 in study one, 77 in study two) were at least 8 weeks of age and had been vomiting for at least 24 hours. Maropitant as studied at 1 mg/kg once daily subcutaneously in study one, and 0.5 to 1 mg/kg subcutaneously in study two with oral dosing of either maropitant or metoclopramide continued in study two until vomiting stopped or for up to 5 days. Vomiting caused by toxin ingestion or in patients with clinical signs indicating the need for acute surgical treatment were excluded. Causes of vomiting were multiple, including metabolic disorders, neoplasia, drug-induced reactions, food intolerance, and parvovirus. In both studies, maropitant was associated with a greater antiemetic response (discontinuation of vomiting) compared with metoclopramide.31
The comparative efficacy and safety of maropitant have been recently described for dogs on the basis of manufacturer-sponsored studies. It was compared (1 mg/kg) to placebo, metoclopramide (0.5 mg/kg subcutaneously), chlorpromazine (0.5 mg/kg subcutaneously), or ondansetron (0.5 mg/kg intravenously) in prevention of apomorphine-induced (0.1 mg/kg intravenously) vomiting. Efficacy in controlling vomiting either central or peripheral in origin was superior to that of chlorpromazine or metoclopramide but did not differ from ondansetron.32 Both safety and efficacy for prevention of emesis associated with motion sickness were assessed in dogs (n = 198) 16 weeks or older. Dogs received approximately 8 mg/kg orally. Data were collected from 26 different clinics in two different crossover randomized, placebo-controlled double-blinded trials with a 14-day washout period between trials. Vomiting was prevented in 86% or 77% of dogs dosed at 2 or 10 hours before a 60-minute car ride. No adverse events were described.
Vestibular Apparatus
Vomiting caused by motion sickness or inner ear disease is mediated by the vestibular apparatus (see Table 19-2). Motion sickness in dogs and cats can be controlled for several (8 to 12) hours by administration of antihistamines such as cyclizine hydrochloride, meclizine hydrochloride, or diphenhydramine hydrochloride (Figure 19-4; see also Figure 19-3). Effects may reflect, in part, sedative effects. In addition to direct effects on neural pathways arising in the vestibular apparatus, actions may also be independent of antihistaminic effects. These may include anticholinergic effects. Those drugs able to penetrate the blood–brain barrier may thus have effects at the vomiting center but generally only at higher doses. Drowsiness and xerostomia (dry mouth) are typical side effects that occur with use of this group of drugs in humans. Although phenothiazine antiemetics may be used to treat motion sickness (e.g., acepromazine), efficacy may reflect sedative rather than direct effects. Centrally active maropitant is approved for use in dogs to treat motion sickness (see preceding discussion) and has been used successfully in cats as well.
Drugs Active at the Chemoreceptor Trigger Zone
Phenothiazines
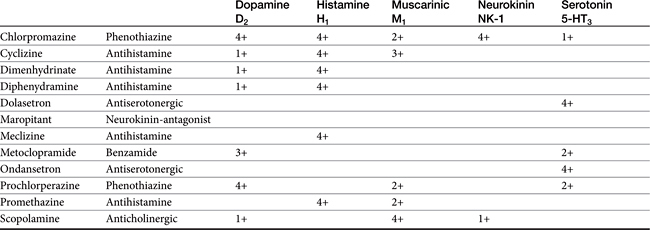
Phenothiazines are broad-spectrum antiemetics that control emesis induced by most central causes other than labyrinthine stimulation (see Figure 19-4). Their classification as broad reflects the variety of signals that serve as primary, secondary, and tertiary mediators. Phenothiazines block emesis mediated by the CTZ at low doses because of their antidopaminergic (D2) and, secondarily, antihistaminergic effects. Several phenothiazines are also characterized by weak antiserotinergic activity (see Table 19-2).24 At higher (perhaps nonpharmacologic) doses, their anticholinergic effects may also act at other central sites, including the vomiting center. A variety of phenothiazine derivatives (e.g., chlorpromazine, prochlorperazine, triflupromazine, perphenazine, trifluoperazine, and mepazine) are used in small animals as antiemetics. The primary adverse effects associated with their use as antiemetics are sedation (which contributes to their efficacy for motion sickness) and hypotension due to peripheral α-blockade. Selection of a particular phenothiazine may be based on avoidance of adverse reactions. Fluid replacement therapy should be instituted if necessary before use of a phenothiazine. The impact of phenothiazine derivatives on the seizure threshold and in epileptic dogs is discussed in more depth in see Chapter 27. In general, their use in epileptic animals may require caution but do not appear to be contraindicated.
Butyrophenone derivatives
Haloperidol (Haldol) and droperidol (Inapsine), which are also used as major tranquilizers, are potent antiemetics because of their antidopaminergic activity. These drugs are rarely used as antiemetics because of their side effects (similar to those encountered with the phenothiazine group but perhaps more profound).
Metoclopramide
Metoclopramide (see Figure 19-4) effectively blocks emesis mediated by the CTZ. Although its potent antagonism of dopamine was thought to be solely responsible for inhibition at the CTZ, metoclopramide is also a mixed 5-HT3 receptor antagonist/5-HT4 receptor agonist; emesis at high doses probably reflects 5-HT3 receptor antagonism.33 Metoclopramide effectively antagonizes apomorphine-induced emesis34 and is 20 times as potent as phenothiazines (although differences in efficacy have not been documented).35 The peripheral effects of metoclopramide on emesis resulting from prokinesis are discussed with the prokinetic drugs. Metoclopramide is indicated for control of emesis induced by a wide variety of blood-borne and peripheral causes.36,37 High doses of metoclopramide, particularly when combined with dexamethasone, have been used to treat emesis associated with cancer chemotherapy in human patients.38-40
Serotonin antagonists
Serotonin antagonists are useful for their antiemetic effects mediated at the CTZ, particularly those induced by chemotherapeutic agents (see Table 19-2).41 Unlike most other antiemetic drugs, antiserotonergics have no effects at other receptors, thus increasing the safety of those drugs selective for serotonin receptors. Ondansetron is a potent antiemetic and affects human cancer patients undergoing chemotherapy.23,33,42 It has also been used in small animals suffering from refractory vomiting that have not responded to other antiemetics. The efficacy of ondansetron reflects, in part, its active metabolite dolasetron, which is also available as an orally administered as well as an intravenous product. Dolasetron also is metabolized (reduced) to a metabolite characterized by greater activity for 5-HT3receptors compared with dolasetron.43 The pharmacokinetics have been reported for dolasetron and its reduced metabolite in dogs (n = 3).43 After intravenous administration of 2 mg/kg, clearance of the parent compound was 109 ± 41 mL/min/kg and volume of distribution was 0.83 ± 0.23 L/kg. After an oral dose of 5 mg/kg, Cmax of the parent compound was 219 ± 149 ng/mL at 0.17 hours. The elimination half-life was 0.15 ± 0.11 hour. The active metabolite appears to potentially double the AUC of active compound. Although oral bioavailability of the parent compound is less than 10%, it is probable that first-pass metabolism results in formation of the active, reduced metabolite: oral administration of 5 mg/kg radioactive dolasetron results in a Cmax of 700 ng/mL radioactivity (i.e., a combination of both radioactive parent and metabolite). Example uses of ondasetron include presurgical preparation, chemotherapy, and treatment of parvovirus infection; vomiting induced by hepatic lipidosis or GI irritation is less likely to respond. Ogilvie44 has reviewed the use of dolasetron in dogs.
KEY POINT 19-9
Side effects of antiserotinergic antiemetics active at the CTZ are limited by their selectivity at the site.
Cyproheptadine is an anthistaminergic antiserotoninergic drug that has been used to control vomiting and diarrhea (the latter associated with spasticity) in humans. Its use in animals might be limited to more chronic control of vomiting when combined with other compounds or as part of combination therapy to treat vomiting and diarrhea associated with inflammatory bowel disease (IBD).
Miscellaneous Antiemetics
Sedatives such as the barbiturates (phenobarbital) and the benzodiazepines have been used to control psychogenic and behavioral vomiting. Glucocorticoids and in particular dexamethasone are characterized by antiemetic effects, although the antiemetic mechanism of action is not understood.24 An antiinflammatory mechanism has been proposed. Glucocorticoids also appear to act in an additive or synergistic fashion when combined with other antiemetics. Both dexamethasone and methylprednisolone have been used in human patients to control vomiting associated with chemotherapy.
Natural extractions or synthesis of Cannabis sativa cannaboids inhibit vomiting, probably through stimulation of CB1 receptors. Dronabinol is an example of an antiemetic used in humans prophylactically to prevent chemotherapy-induced vomiting. It is characterized by complex kinetics that include high protein binding, extensive first pass metabolism to active and inactive metabolites. Side effects are similar to sympathomimetic drugs. Because dogs and cats respond differently to drugs which target cannanboid receptors, use should be based only on scientific evidence in the target species.
Peripherally Acting Antiemetics
Protectants
Drugs that locally protect the GI epithelium from further irritation may help prevent vomiting. Drugs that modulate gastric acid secretion might also provide antiemetic effects; these drugs are discussed later with the antiulcer drugs. Demulcents, antacids, and protectants such as kaolin, pectin, and bismuth salts are of limited benefit in the control of emesis that is gastric in origin. Distention or initial irritation of the stomach by these agents may exacerbate emesis. Antacids may be effective in certain cases. Other peripherally acting antiemetics include drugs that affect gastric motility, including anticholinergic drugs, and prokinetic drugs such as metoclopramide and domperidone (discussed later with modulators of GI motility).
Anticholinergics
Anticholinergic drugs that block muscarinic receptors in the emetic center also inhibit peripheral cholinergic transmission. Those anticholinergic drugs that do not cross the blood–brain barrier well are essentially peripheral in action and include glycopyrrolate, propantheline, isopropamide, and methscopolamine (which should not be used for cats). The ability of anticholinergics to suppress emesis is probably related to inhibition of afferent vagal impulses, relief of GI smooth muscle spasms, and inhibition of gastroenteric secretions. Delayed gastric emptying caused by these drugs may itself cause emesis, and anticholinergics should not be used for more than 3 days by the vomiting patient. Because of their anticholinergic properties, these drugs should not be used in combination with drugs whose actions depend on cholinergic activity in ganglion or smooth muscle. These include metoclopramide, cisapride, and the opioids.
Prokinetics
Prokinetics, and specifically metoclopramide, are peripherally acting antiemetics because of their prokinetic effects on the GI tract. Metoclopramide physiologically antagonizes emesis by virtue of its actions on the upper gastroduodenal area: increased esophageal sphincter tone, duodenal pyloric relaxation, and antegrade contraction of the gastric antrum. The prokinetic effects of metoclopramide are discussed later with other drugs that modify GI motility.
Antiulcer Drugs
Pathophysiology of Gastrointestinal Ulceration
Gastroduodenal Ulceration
The events leading to gastroduodenal ulceration are complex and reflect interactions between acid-secreting and defense mechanisms of the GI mucosa.45,46 Regardless of the cause of GI erosion or ulceration, the basic pathologic mechanism is similar. Gastric acid secretion is a prerequisite for damage to the GI mucosa46,47 with luminal damage not occurring unless luminal pH is less than 7. Pepsin and bile acids can contribute to mucosal damage. Even though these chemicals are inherently caustic, mucosal damage generally does not occur in the face of normal mucosal cytoprotective mechanisms. These include but are not limited to secretion of bicarbonate and mucus and rapid epithelial turnover. Deceased mucosal blood flow can have a profound effect on the ability of the injured mucosa to heal itself. Drugs used to control or treat GI erosion and ulceration include those that inhibit gastric acid secretion or provide or facilitate other cytoprotective effects. The role of Helicobacter sp. in the pathogenesis of gastroduodenal ulceration in human patients has been well established, but its role in disease in animals is less well documented (see later discussion; e.g., IBDs).
Physiology of Gastric Acid Secretion
Gastric acid secretion occurs in four phases. The first three phases—referred to as cephalic, gastric, and intestinal—are stimulated by food and mediated by gastrin, which is the most potent secretagogue.48 Secretion is persistent during these phases, and gastric pH progressively decreases as nutrients traverse the GI tract. Gastrin secretion is inhibited as gastric pH declines to 3.5 and is completely inhibited at a pH of 1.5, to begin again only when pH approximates 3 to 3.5. The fourth phase of gastric acid secretion is basal and occurs in the absence of external stimuli. The amount of basal secretion varies among animals. In humans basal secretion follows a circadian rhythm, reaching a peak at midnight and a nadir at 7 am.49 As a model for the study of antisecretory drugs, information can be found regarding basal and responsive gastric acid secretion in dogs.50,51
In fasted Beagle dogs (n = 8), gastric pH (collected by stomach tube) fluctuated from 2.7 to 8.3. Basal pH tended to be 7.0; treatment with a placebo (500 mg lactose) reduced pH almost the same magnitude as pentagastrin (3 and 2, respectively) at 1-2 hrs post treatment.50
Gastric acid secretion at the cellular level involves the generation and subsequent secretion of hydrogen ions by the parietal (oxyntic) cells of the gastric mucosa. Responses are controlled through chemical signals interacting with corresponding receptors located on the basolateral membrane of parietal cells. Central and peripheral signals stimulating gastric acid secretion include endocrine (gastrin: CCK), paracrine (histamine: H2), and neuronal (ACh: M3) (Figure 19-5).49 In addition to its direct effects, acetycholine indirectly increases release of gastrin from G cells and histamine from enterochromaffin cells. Of the mediators increasing gastric acid secretion, gastrin is the most potent, although its effects are mediated indirectly through stimulation of histamine receptors, particularly on enterochromaffin cells.48 Somatostatin, which inhibits gastric acid secretion, is released from D cells when gastric pH is less than 3. In humans the effects of Helicobacter spp. may reflect, in part, the ability of these organisms to decrease D cells. The hydrogen ion pump, located at the apical membrane and associated with the smooth endoplasmic reticulum, is unique in that it is a hydrogen–potassium ATPase exchange system. Three distinct pathways are capable of stimulating gastric acid. Each acts through chemical mediators that in turn interact with receptors on the parietal cell membrane.49 H2 receptors are linked to adenylyl cyclase and cyclic AMP.48 Of these, the ACh pathway appears to be less important in small animals.
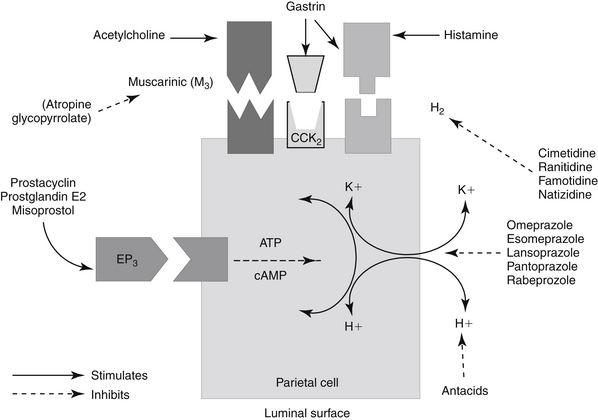
Figure 19-5 Receptor interactions that mediate gastric acid secretion by the parietal cell include acetylcholine with muscarinic receptors and histamine with H2 receptors. Gastrin may interact with either receptor. Receptor stimulation activates the K+, H+-ATPase pump and exchange of potassium for hydrogen into the lumen. Prostaglandin E1 (PGE) modulates gastric acid secretion by inhibiting cyclic adenosine monophosphate (cAMP). ATP, Adenosine triphosphate.
KEY POINT 19-12
Prevention and treatment of gastrointestinal ulceration focuses on prevention of hydrochloric acid secretion and promotion of cytoprotection.
Intracellular messengers mediating gastric acid secretion vary with the receptor stimulated. Histamine increases cAMP production, which subsequently activates the adenylyl cyclase cAMP-dependent protein kinases. Gastrin and muscarinic stimulation by cholinergic drugs increased cytosolic calcium, through inositol phosphate pathways. Both pathways activate an H+, K+-ATPase proton pump that exchanges hydrogen and potassium across the parietal cell membrane. Prostaglandins of the E series serve to modulate these effects, inhibiting gastric acid secretion by blocking cAMP production through EP3 receptors, also on parietal cells.48,49 The impact of opioid receptors on gastric acid secretion is discussed with drugs targeting intestinal secretion.
Mucosal Defenses
The primary mucosal defense of the esophagus reflects increased lower esophageal sphincter tone. Defenses of the GI mucosa require sufficient mucosal blood flow and act to prevent or repair GI ulceration (Figure 19-6).52-54 These include (1) secretion of bicarbonate into the lumen and neutralization of hydrochloric acid in the lumen; (2) secretion of a thick, insoluble, alkaline mucus that traps and neutralizes inward-moving hydrogen ions and protects against macromolecules such as pepsin; (3) a gastric epithelial barrier composed of active phospholipids, a lipoprotein cell membrane, and tight junctional complexes, all of which prevent hydrogen ion back diffusion; (4) mucosal blood flow, which first provides nutrients and oxygen to mucosal cells and second removes hydrogen ions that have penetrated the gastric barrier; (5) rapid replication of mucosal epithelial cells; and (6) production of cytoprotective agents. Many of these effects reflect local secretion of prostaglandin E2 and I2, important defense mechanisms. They modulate hydrochloric acid secretion, increase bicarbonate and mucus production, and enhance mucosal blood flow and epithelialization.55,56 Sulfhydryls also produced locally may act as scavengers of oxygen and other tissue-damaging radicals.57
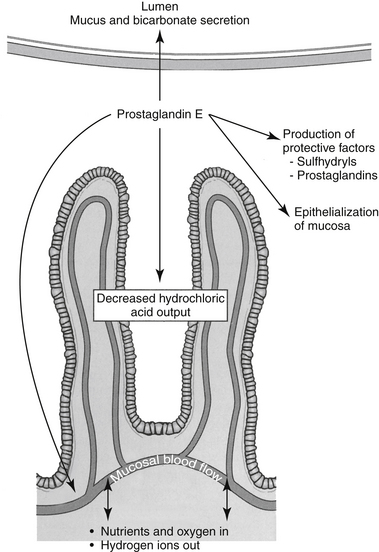
Figure 19-6 Protective mechanisms mediated by prostaglandin E against gastroduodenal ulceration provide targets for drug therapy. Bicarbonate secretion acts to neutralize gastric acid; mucus protects against hydrochloric and bile acids. The rapid turnover of epithelial cells is paramount for rapid healing if damage occurs. Mucosal blood flow not only provides critical oxygen and nutrients necessary for epithelialization but also removes H+ ions that have penetrated the protective barrier. Other protective factors include mechanisms to control gastric hydrochloric acid secretion and the production of protective factors that scavenge mediators capable of cell damage.
Gastric Antisecretory Drugs
Drugs used to prevent or modulate gastric acid secretion include anticholinergics, H2-receptor antagonists, proton pump inhibitors, and prostaglandin E2.49,55,58,59 Despite the role of muscarinic receptors in gastric acid secretion, anticholinergics have not proved effective for the control of GI ulceration in animals and are not discussed. Drugs that modify gastric acid (e.g., antisecretory drugs or antacids) are discussed with cytoprotectants. All drugs that modify gastric pH can cause complications of achlorhydria when used chronically. Although both gastric acid and pepsin are required for hydrolysis of proteins and other foods, achlorhydria is rarely accompanied by malabsorption unless bacterial overgrowth occurs. Achlorhydria can lead to malabsorption of certain nutrients, among them vitamin B12 and iron, as well as decreased absorption of some (weakly acidic) drugs. Although the advent of antihistaminergic antisecretory drugs represented a landmark change in the approach to medical management of GI ulcers in humans, their use is increasingly being replaced with proton pump inhibitors.
H2-Receptor antagonists
H2-receptor antagonists are reversible, competitive inhibitors that reduce both the amount and the hydrogen ion content of gastric secretion and the amount of pepsin60 induced by a variety of secretagogues.61 Secretion of intrinsic factor also is reduced, although this effect does not appear to be clinically relevant.24,48 Each antagonist is a congener of histamine, containing a bulky side chain (see Figure 19-4).24,48 Cimetidine; ranitidine; and, to a lesser degree, famotidine have been used to control gastric acid secretion in animals. Nizatidine is the most recent of the approved drugs and has been used least in dogs and cats. Each drug varies in potency, duration of action, disposition, and drug interactions.62 Ranitidine is 5 to 12 times more potent as an inhibitor of gastric acid secretion than cimetidine, whereas famotidine is nine times more potent than ranitidine and 32 times more potent than cimetidine. Famotidine (see Figure 19-4) has the longest duration of action.40 In a cat model, famotidine was 4.5 times as potent as ranitidine; effects of famotidine were reversible at the highest dose studied (0.01-0.32 μmol kg/hr) supporting the need for higher doses or twice-daily dosing for conditions in which histamine-mediated high gastric acid output is mediated.63 In animal models, including dogs, nizatidine is more potent than cimetidine.64 In a Beagle model, ranitidine (50 mg IV) reduced resulted in a mean gastric pH of 7.8 by 1 hr; however, basal gastric pH was high as well. The pH was maintained for the 4-hour duration of the study.50 Although the H2-receptor antagonists have variable prokinetic actions, they appear to have inconsistent effects on the rate of gastric emptying or lower esophageal sphincter pressure.48
Disposition
The disposition of antisecretory antihistamines has not been well studied in animals, with information drawn largely from human data. Cimetidine, the oldest of the clinically used H2-receptor antagonists, is rapidly absorbed from the GI tract, although food will delay the process. The drug undergoes hepatic metabolism and is about 70% bioavailable after oral administration. It is excreted in the urine in both the unchanged and conjugated forms. The plasma half-life is about 1 hour but may be prolonged in the presence of liver or kidney disease.
Ranitidine is less bioavailable (50%) than cimetidine after oral administration. Its elimination half-life is approximately 2.5 hours. Absorption is not impaired by food as with cimetidine. It is minimally protein bound (15%). Hepatic elimination is responsible for 30% of an intravenous dose and 73% of an oral dose.65
Famotidine is only 37% bioavailable after oral administration reflecting decreased oral absorption; however, this is compensated for somewhat by increased potency. In contrast, nizatidine is rapidly and completely absorbed.60 Both drugs are largely eliminated unchanged in urine.60 Nizatidine is almost exclusively eliminated by renal excretion, which suggests that it might be the preferred H2-receptor antagonist for patients with hepatic disease. Its efficacy apparently has not been studied clinically in animals, although its safety has been established in healthy dogs.62 Famotidine renal clearance appears to be saturable in the dog, albeit at suprapharmacologic doses.66
Drug interactions
The antisecretory antihistamines can be involved in a number of drug interactions, with cimetidine being best characterized.48,67 Cimetidine, like all antisecretory drugs, impairs the oral absorption of a number of drugs (generally weak acids) through the alteration of GI pH. Cimetidine also directly impairs the absorption of many drugs by directly binding to the drugs. These effects might be balanced by competition for P-glycoprotein, for which cimetidine is a substrate. Cimetidine is a potent microsomal enzyme inhibitor and will decrease the hepatic metabolism of concurrently administered drugs.68,69 Enzymes targeted in humans include CYP1A2, CYP2C9, and CYP2D6. Occasionally, this effect may be clinically useful, as in the prevention of acetaminophen intoxication in cases of accidental overdose.70 However, impaired metabolism of other drugs can also lead to clinically relevant toxicity of other drugs metabolized by the liver. The impact of cimetidine on cyclosporine elimination has been studied in dogs. Although one study indicated a longer half-life, several other studies have demonstrated “no effect” of cimetidine on the disposition of cyclosporine (see Chapter 31). It is likely that the impact varies among animals, indicating a need to monitor. Cimetidine also reduces hepatic blood flow by about 20% and has been shown to reduce the clearance of flow-limited drugs such as propranolol and lidocaine.71 Unlike cimetidine, the other antihistamines have limited to no effects on hepatic blood flow. Although ranitidine also inhibits CYP, its affinity for the enzymes is only about 10% of that for cimetidine. Famotidine and nizatidine have limited to no effect on the metabolism of other drugs (or endogenous compounds). Famotidine is a potent inhibitor of transport of cationic drugs, although the clinical relevance of this is not clear.
Adverse reactions
The side effects seen with any of the H2-receptor antagonists are generally minor even at relatively high doses. Thrombocytopenia has been reported. Although there have been a number of reported side effects for ranitidine in humans, limited experience to date in animals has not indicated any serious toxic manifestations from ranitidine. Famotidine and nizatidine are devoid of many of the side effects of cimetidine.64
A clinically important disadvantage of H2-receptor antagonists described in humans is relapse of gastroduodenal ulceration during or after H2-receptor antagonist therapy is discontinued. Although several explanations for relapse have been offered, rebound hypersecretion of gastric acid appears to be most plausible.72-74 Suppression of gastric acid by H2-receptor antagonists results in increased plasma gastrin concentrations as early as 3 hours after a single dose. Subsequent stimulation of gastric mucosal G cells results in gastric acid hypersecretion that becomes evident when the drugs are discontinued. The likelihood of hypersecretion is compounded by increased parietal cell receptor sensitivity, which apparently characterizes (human) patients afflicted with ulcers.75 Among the H2 receptors studied, cimetidine seems to be the most likely and famotidine or nizatidine the least likely to cause rebound gastric acid hypersecretion.74-76 Rebound hypersecretion can be minimized by tapering the dose as the drug is discontinued. Tolerance to the antisecretory effect of antihistaminergic antisecretory drugs also occurs, being well described in humans. Tolerance appears within 3 days of therapy and may not respond to increasing doses.48
Clinical use
The principal therapeutic uses of H2-receptor antagonists include uremic gastritis, gastric and duodenal ulcers, stress-related erosive gastritis, and hypersecretory conditions such as gastrinoma or systemic mastocytosis. Although H2-receptor antagonists can be used to treat drug-induced (e.g., nonsteroidal antiinflammatory drug [NSAID]) ulceration, their efficacy is controversial and other, more specific antidotes (e.g., PGE1) or more effective antisecretory drugs (e.g., proton pump inhibitors) should first or also be administered.77 On the other hand, the drugs have proved beneficial in providing protection against gastric ulceration induced by a number of etiologic agents, including aspirin and stress.48 Their combination with proton-pump inhibitors is discussed in the following section. When treating drug-induced ulcers, antisecretory drugs that inhibit drug metabolizing enzymes should be avoided. H2-receptor antagonists also appear to be effective in controlling upper GI bleeding when hemorrhage is not due to erosion of major blood vessels. H2-receptor antagonists have also been used in gastroesophageal reflux disorders, esophagitis, and duodenal gastric reflux. In exocrine pancreatic insufficiency, cimetidine or ranitidine (and presumably famotidine), if given about 30 minutes before feeding, may decrease enzymatic and acid hydrolysis of replacement pancreatic enzymes added to food on their contact with gastric secretions, thus improving the efficacy and decreasing the cost of their use. Patients suffering from short bowel syndrome may benefit from long-term H2-receptor therapy to decrease the hyperacidity associated with this syndrome. The H2-receptor antagonists are sufficiently safe that high doses can be given to humans to maintain pharmacologic effects with once- to twice-daily dosing.48 A meta-analysis in humans studied the impact of renal disease on the disposition of H2-receptor blockers. Declining renal function is associated with a concomitant reduction in the renal clearance of those drugs renally eliminated. Appropriate dose reduction was associated with decrease in cost, as well as decrease in adverse events, with the major adversity being mentation disorders.78
Proton Pump Inhibitors
The substituted benzimidazole proton pump inhibitors are the most potent antisecretory drugs, reducing gastric acid secretion by 80% to 95%.48 Each is a potent and irreversible antagonist of the H+, K+-ATPase proton pump, the final step in gastric acid secretion stimulated by any secretagogue. No differences in antisecretory efficacy have been demonstrated among these drugs. Omeprazole will be discussed as the model drug.
Mechanism of action
Omeprazole (Prilosec) (see Figures 19-4 and 19-5), was the first of the commercially available drugs. It is sold as a racemic mixture. Other proton pump inhibitors currently approved for use in the United States include esomeprazole (Nexium), the S-isomer of omeprazole (cleared more slowly than the R isomer in some species), lansoprazole (Prevacid), rabeprazole (Aciphex), and pantoprazole (Protonix). Omeprazole is approximately 30 times more potent as an antacid than is cimetidine.79 Secretory volume is not as affected as is acidity.79
Pharmacokinetics
As a weak base, omeprazole is unstable in an acid environment and thus is formulated as encapsulated enteric-coated granules.79 Drug dissolution occurs in the more alkaline environment of the small intestine. Acidity degrades (inactivates) the drug. Consequently, the drugs are generally prepared as enteric-coated products or combined with antacids (e.g., sodium bicarbonate). Compounded products must be made with attention to formulation, including pH, to ensure pharmaceutical efficacy. Oral bioavailability increases with environmental intestinal pH, and plasma drug concentrations tend to increase the first 4 to 5 days of therapy.79 The complicated nature of proton pump inhibitors has limited the availability of parenteral preparations; however, pantoprazole and lansoprazole (and, in Europe, esomeprazole) are available for intravenous administration.
Once absorbed, the acidic environment (pH 0.8 to 1) of the GI tract causes omeprazole to selectively partition into the secretory canniculi of parietal cells compared with other cells (pH 5). In the acidic environment, the drug is protonated, trapped, and subsequently further transformed to the active inhibitor. As such, proton pump inhibitors are prodrugs. Ideally, the drugs are administered about 30 minutes before a meal; other antisecretory drugs do not appear to affect proton pump activity.80 Indeed, antihistaminergic antisecretory drugs might be given in combination with proton pump inhibitors for a rapid response because of the slower onset of action of the proton pump inhibitors.81 Once in the canniculi, omeprazole covalently and irreversibly binds to sulfhydryl groups of potassium-adenosine triphosphate (H+, K+-ATPase), thus inhibiting the energy source for the proton pump.48,79 Because the enzyme is permanently inhibited, secretion of HCl will resume only after new molecules have been formed in the luminal membrane, which generally requires 24 to 48 hours.48 Therefore the duration of action of proton pump inhibitors is much longer than their plasma half-life. Drug accumulation in parietal cells and alternating activity of parietal cells or pumps result in a lag time of up to 3 to 5 days before maximum effect (generally 70% of pumps are inhibited at steady state) is realized.79,82 Consequently, alternative drugs may need to be considered if a rapid response is desired. In addition, efficacy will be maintained at low plasma drug concentrations and for some time after the drug is discontinued. Because of these characteristics, omeprazole can be administered once daily.82 In humans omeprazole is highly (96%) bound to serum albumin and α1-acid glycoprotein. Its apparent volume of distribution is 0.31 L/kg.79 Clearance is accomplished through hepatic metabolism; in humans the major enzymes are CYP2C19, for which genetic polymorphisms have been reported (decreased activity in Asians), and CYP 3A4, an enzyme characterized by broad substrate specificity. Also in humans drug elimination depends on hepatic metabolism to inactive metabolites, and elimination half-life is short (52 minutes).79 Omeprazole has been studied in dogs.83 Oral bioavailability is reduced, although therapeutic concentrations can be achieved.
Dosing at 0.17 mg/kg orally once a day for five years was well tolereated in Beagles (n = 10). Changes in disposition were not detected across time. The AUC was similar to that measured in humans receiving 20 mg (approximately 0.28 mg/kg) daily. omeprazole daily. Mean inhibition of acid secretion by omeprazole 4-7 hours after dosing approximalted 50%.
Drug interactions
Partial inhibition of drugs eliminated by selected cytochrome P450 enzymes have been reported for omeprazole.84 However, compared with cimetidine, omeprazole may be less likely to be involved in drug interactions. All proton pump inhibitors appear to inhibit a variety of CYPS (including CYP3A4), with clinically relevant drug interactions described for warfarin, cyclosporine, and diazepam. Omeprazole (more so than other proton pump inhibitors) appears to inhibit CYP2C19 and induce CYP1A2 (metabolism of several trycyclic or other behavior-modifying drugs); its S isomer is characterized by less in hibition. Other interactions are likely to exist, indicating caution when combining proton pump inhibitors with other drugs metabolized by the liver. Lansoprazole may be involved with fewer cytochrome P450–based drug interactions. Drug interactions appear to occur at the level of P-glycoprotein; omeprazole, lansoprazole, and pantoprazole appear to be substrates.85 Because of the delay in onset of action, the use of more rapidly acting antihistaminergic drugs might be considered as proton pump therapy has begun. That the latter does not appear to have the efficacy of the former has been demonstrated.80,81
Adverse reactions
Adverse reactions caused by omeprazole are limited because the drug is selective for the H+, K+-ATPase pump. An exception is the sequelae of achlorhydria. Diarrhea and transient fluctuations in liver enzymes have been reported. Hypergastrinemia has been documented in human patients79 after therapy with omeprazole, and is more severe compared with that associated with use of antihistaminergic antisecretory drugs and may be associated with gastric hyperplasia or an increase in gastric tumors. Rebound hypersecretion of gastric acid as described for antihistaminergic drugs should be anticipated, and discontinuation of proton pump inhibitors should occur gradually, with tapering or substitution of alternative drugs in at-risk patients.48 Hypertrophy of gastric mucosa has been reported. A marked increase in gastric acid secretory capacity has been detected after omeprazole treatment, presumably owing to proliferation of an enterochromaffin-like cell mass.86 However, in contrast to H2-receptor blockers, proton pump inhibitors may not cause tolerance because their inhibition is distal to histamine-mediated secretion targeted by increased gastrin. Chronic use may decrease absorption of vitamin B12, although clinical relevance is not clear.48
Clinical use
Proton pump inhibitors are indicated to support gastroduodenal healing, prevention or treatment of gastroesophogeal reflux, treatment of hypersecretory conditions, and other conditions that benefit from reduced hydrochloric acid secretion (e.g., IBDs). Omeprazole is the drug of choice for the treatment of the Zollinger–Ellison syndrome. Omeprazole has been used to control gastric acid secretion that has not responded to H2-receptor antagonists, although its superiority to these and other antacid drugs has not been firmly established. Generally, however, studies support the superiority of omeprazole over cimetidine for treatment of GI ulceration, including response of pain.79 Lansoprazole is often preferred in humans for prevention of NSAID-induced ulceration but does not appear to offer an benefit in terms of long-term efficacy.
Bersenas and coworkers87 studied normal fasting and postprandial intragastric pH and its response to a variety of antisecretory drugs in Beagles (n = 12). Antisecretory drugs included ranitidine (2 mg/kg intravenously every 12 hours), famotidine (0.5 mg/kg intravenously every 12 hours), pantoprazole (1 mg/kg intravenously every 24 hours), and omeprazole (1 mg/kg orally every 24 hours), or saline placebo. Intragastric pH was recorded on days 0, 2, and 6. Outcome measures included median 24-hour intragastric pH, percentage of time pH was at or above 3, and percentage of time pH was at or above 4. All outcome measures were better (pH higher for longer) in the fasting compared with the postprandial state. Only famotidine, pantoprazole, and omeprazole suppressed gastric acid secretion, compared with placebo. The investigators also found that a suspension of omeprazole (1 mg/kg orally every 12 hours; n = 6) but not famotidine (0.5 mg/kg intravenously every 8 hours; n = 6) suppressed gastric acid secretion; information was not available regarding quality assurance of the suspensions. The authors concluded that only famotidine (at the doses and intervals studied), pantoprazole, and omeprazole significantly suppressed gastric acid secretion and the only suspension that was anticipated to be effective in treating gastric acid-related diseases (in normal dogs), based on outcome measures targeted in humans, was omeprazole; the power of the study may have impacted interpretation. This study suggests that ranitidine at 2 mg/kg, administered intravenously, is not an effective antisecretory drug in dogs, and famotidine is indicated at 12-hour intervals.
Prostaglandin Analogs
In addition to blockade of receptors that mediate secretion, gastric acid might also be modulated by prostaglandins of the E series. Their actions appear to be mediated by interaction with a basolateral membrane receptor. Intracellular concentrations of cAMP decrease, which in turn decreases protein kinase activity and hydrogen ion concentration (see Figure 19-6).49 Misoprostol is a methyl ester analog of prostaglandin E1. As such, it is pharmacologically active after oral administration, with effects lasting longer than those of endogenous prostaglandins.88 Food delays its time of onset, which generally occurs within 30 minutes. Effects in humans tend to last for 3 hours, reflecting, in part, a short half-life of 20 to 40 minutes. The effects of misoprostol tend to be restricted to the local environment, with systemically absorbed drug rapidly metabolized by the liver.89 Misoprostol does not appear to alter serum gastrin levels, and rebound acid hypersecretion has not been reported.83 Basal, nocturnal, and food-induced gastric acid secretion is inhibited by misoprostol, which appears to be the primary cytoprotective effect. Although 75% to 85% of basal acid secretion may be inhibited, high doses at frequent intervals (four times daily in humans) may be necessary. The drug is not likely to be as effective as H2-receptor or proton pump antagonists in decreasing intraluminal pH and appears less effective in controlling pain associated with hydrochloric acid secretion. Unabsorbed drug that reaches the intestine can cause intestinal secretion, smooth muscle contraction, and thus diarrhea (occurring in up to 30% of human patients), but these side effects may be resolved after several days.88 Misoprostol can exacerbate clinical signs associated with inflammatory bowel disease and should be avoided in patients at risk.48 Misoprostol can induce uterine contractions and is contraindicated in pregnancy; clients who are of childbearing age should be warned of this potential side effect. The primary indication for misoprostol is prevention or treatment of NSAID-induced ulceration, although more convenient drugs tend to be used for prevention. Its use might be considered in diseases associated with marked mast cell influx (e.g., mastocytosis) because of its inhibitory effect on mast cell degranulation.90
Cytoprotective Drugs
Antacids
Antacids have largely been replaced by drugs that more effectively prevent deleterious effects of gastric acid. Nonetheless, antacids continue to have a role in the treatment or prevention of mucosal disorders associated with gastric acid or other caustic agents. As with many GI-active drugs, information regarding the effects of antacids largely is extrapolated from data from studies on other species. Antacids chemically neutralize HCl present in the gastric lumen such that gastric luminal pH is increased to an acceptable level (pH of 3 or 4, at minimum) without causing systemic alkalosis.91 Inactivation of pepsin and binding of bile salts by some products (e.g., aluminum hydroxide) are also important. Finally, some products (e.g., aluminum hydroxide) also induce the local synthesis of mucosal protectants (e.g., prostaglandins and sulfhydryls).57,92 Some antacids are indicated for their impact on electrolyte (e.g., phosphorus) absorption. Factors influencing rational antacid therapy are rate of acid secretion, duration of time the antacid remains in the stomach, the potency of the antacid, and adverse effects.93
The relative efficacy of antacids is based on the number of milliequivalents of acid-neutralization available (the volume of 1N HCl, or milliequivalents of HCl, that can be titrated to a pH of 3.5 within 15 minutes). The major nonsystemic antacids used in veterinary medicine are salts of aluminum, magnesium, and calcium used either alone or in combination with each other or with various protectants, adsorbents, and astringent. In general, in vitro, 1 gram of these antacid compounds generally neutralize 20 to 35 mEq of acid. Neutralization of (reaction with) HCl generates chlorides, water, and carbon dioxide48; release of CO2 from carbonates can cause abdominal distention and belching.48 In the fasting state the action of gastric antacids is usually transient and lasts only 1 to 2 hours. In general, antacids are cleared from the stomach within 30 minutes. Neutralization of acid in the stomach antrum removes negative feedback control of gastrin release, which in turn leads to elevated gastrin levels and enhanced HCl secretion, with increased tone of the lower esophageal sphincter. In the past antacid administration was recommended at 4- to 6-hour intervals to minimize rebound hypersecretion. The presence of food, which increases gastric pH to about 5,48 will prolong the effects of antacids for up to 2 to 3 hours. In human patients, however, administration with each meal has proved more convenient yet equally efficacious. Time to onset of action is different among antacids. Calcium and sodium carbonate are considered fast acting, magnesium salts moderate to rapid acting, and aluminum salts slow acting.
KEY POINT 19-21
Antacids containing a combination of aluminum and magnesium hydroxide provide the best balance in efficacy and avoidance of side effects.
Aluminum hydroxide is a good adsorbent (of bile acids and pepsin), as well as an antacid. Although slow in action, it tends to provide prolonged antacid effects.48 Because it is slow acting, it is often combined with more rapidly acting magnesium salts. An advantage of aluminum hydroxide compared to other antacids is its stimulatory effect on local prostaglandin production in the intestinal mucosa.92 Aluminum preparations tend to cause constipation and are often mixed with magnesium salts (laxative inaction) to prevent this side effect. However, this combination is not always effective, with emergence of constipation (because of aluminum) and diarrhea (because of magnesium) emerging in some patients. Aluminum hydroxide also decreases phosphate absorption by forming insoluble aluminum phosphates in the intestine and is used to control serum phosphorus in patients with renal disease. Note that prolonged administration with meals may cause hypophosphatemia in patients.
Magnesium-containing products can raise gastric pH higher than aluminum-containing antacids (as high as 9 versus 4).91 Magnesium hydroxide is the most commonly used form of magnesium. Magnesium salts tend to be laxative and are often found in combination with aluminum and calcium salts. Their cathartic effects result from soluble but unabsorbed magnesium salts that remain in the intestine and retain water. The neutralizing effect of magnesium hydroxide is prompt and prolonged. Up to 20% of the magnesium is absorbed in normal circumstances, and in the presence of renal dysfunction repeated administration can result in hypermagnesia. Combination antacid products containing both aluminum and magnesium are often used to balance the adverse effects of each cation or bowel function.93 Magaldrate is a hydroxymagnesium–aluminate hydroxide complex that rapidly dissociates in the gastric acid to magnesium or aluminum hydroxide, providing a sustained effect.
KEY POINT 19-22
Sucralfate must bind to damaged tissues and therefore is ineffective until damage occurs. However, prophylactic use should still be considered.
Calcium carbonate is a rapid-acting, potent antacid with a prolonged duration. Slowly developing metabolic alkalosis, gastric acid rebound, hypercalcemia, and calciuria with metastatic calcification and urolithiasis, hypophosphatemia, and constipation are, however, potential side effects that may occur after chronic administration of calcium carbonate.93 In addition, interference with calcium-dependent processes may lead to excessive gastrin and HCl secretion.48 Release of carbon dioxide may increase gastrin release owing to distention.
Antacids may be involved in a number of drug interactions, despite their largely local effects. Antacids may alkalinize the urine, generally increasing it 1 pH unit. The rate of elimination of renally eliminated weak acids will be increased (e.g., NSAIDs, phenobarbital), whereas that of weak bases is decreased.48 Drug interactions of antisecretory drugs reflecting increased gastric pH may also occur with antacids. Oral absorption of a number of other drugs94 may be affected as a result of changes in gastric pH. Further, magnesium and aluminum in particular are likely to chelate other drugs, precluding their absorption. Therefore antacids should be given approximately 2 hours before or after other orally administered drugs.
In addition to gastric distention (carbonates) and altered electrolytes that vary with each compound, side effects to antacids are not common. Antacids may increase the risk of food allergies based on a mouse model in which proteins extracted from caviar induced caviar-specific IgE antibodies, T-cell reactivity, and increased GI eosinophil and mast cells in mice simultaneously receiving ranitidine or sucralfate. The authors identified the aluminum hydroxide component of sucralfate as being responsible for the increased risk of allergic response. The rationale for the increased risk was incomplete protein digestion.95
Gastric hyperacidity, peptic ulcer, gastritis, reflux esophagitis, and chronic renal failure (uremia) are the more common indications for antacid preparations in veterinary medicine. Pyloric and duodenal peptic ulcers that may be related to gastric hyperacidity have been reported in dogs. Antacids intended to treat gastroduodenal ulceration generally are administered orally 1 and 3 hours after meals and at bedtime. For severe conditions or to treat uncontrolled reflux, doses in humans are repeated as often as every 30 to 60 minutes. Suspensions have greater neutralizing capacity than do powder or tablet dosage forms; in humans tables must be thoroughly chewed for maximum effect. A number of acid products intended to treat “acid indigestion” in humans contain silmethicone (see the discussion of antiflatulence agents), a surfactant that decreases esophageal reflux. Whereas the choice of over-the-counter preparations containing a combination of antacids and silmethicone is reasonable, products that are combined with aspirin should be avoided.
Sucralfate
Sucralfate (see Figure 19-4) is an orally administered disaccharide (octasulfate of sucrose) aluminum hydroxide product that binds to and protects damaged epithelial cells from acid, bile, and pepsin activity.96-99 In the acid environment of the stomach, sucrose is freed from aluminum hydroxide, cross-polymerizes, and binds to exposed (damaged) anions of GI epithelial cell membranes (Figure 19-7).100 The maximum protective effects of sucralfate depend on an acid environment (pH <4) for activation; therefore it should be administered at least 30 minutes before antacids100 and on an empty stomach, 1 hour before feeding.48 Binding occurs in the base of ulcer craters and is greater in duodenal than in gastric ulcers. Sucralfate also binds to and inactivates bile acids, thus promoting its use in biliary disorders, and inhibits pepsin-mediated hydrolysis of mucosal proteins.101 In addition to binding and protection of cells, the polymerized sucrose prevents exudation of protein and electrolytes into the gastric lumen. Effects of sucralfate may last up to 6 hours, supporting a four-times-daily dosing regimen.48 The amount of aluminum hydroxide released from sucralfate may not effectively neutralize gastric acidity, although this may be controversial.102 Sucralfate appears to stimulate production of local mediators that protect the gastric mucosa. These include prostaglandins99,101,103 sulfhydryl ions or other oxygen radical scavengers, and epidermal growth factor.104 Sucralfate effects on epidermal growth factor cause its accumulation in ulcerated lesions.99,105 Sucralfate also increases mucosal blood flow either by inducing local nitric oxide or prostaglandin production106 or by directly stimulating mucosal angiogenesis.107
Figure 19-7 An endoscopic view of sucralfate bound to damaged gastrointestinal epithelium. Binding not only protects the damaged epithelium from further damage but also prevents loss of critical nutrients and fluids.
KEY POINT 19-23
In addition to its mechanical protection, sucralfate also increases local prostaglandin protection and promotes angiogenesis and epithelialization.
Drug interactions with sucralfate are limited to local effects. Sucralfate binds to a number of drugs, with the prototypic example being cimetidine. Because cimetidine also increases gastric pH (thus potentially reducing activation of sucralfate), these two drugs probably should be alternated (i.e., administer sucralfate 1 to 2 hours before cimetidine) in patients receiving both drugs. In addition to direct binding to drugs, sucralfate may influence drug absorption through formation of a thick mucus layer. Thus, in general, other drugs should be administered at least 2 hours before sucralfate.
Sucralfate is minimally absorbed after oral administration and is associated with few, if any, side effects. Sucralfate is recognized to be the safest drug available for treatment of gastroduodenal ulcers.101 Constipation occurs in up to 2% of humans. Currently, sucralfate is recommended for treatment of gastroduodenal erosion ulceration, regardless of the cause. Its prophylactic use is also recommended for illnesses associated with ulceration such as renal or liver disease, mastocytosis, and IBDs for which prolonged use of antiprostaglandin is indicated. However, sucralfate does not prevent the formation of the ulcer; rather, it protects damaged tissue once it occurs. Sucralfate appears beneficial prophylactically for stress-induced ulcers but is less effective prophylactically in patients receiving NSAID therapy.100 Sucralfate is effective for treatment of acid-induced esophagitis,108 although antisecretory drugs such as omeprazole or H2-receptor antagonists are probably superior.48 In critical patients at risk for nosocomial pneumonia, sucralfate may be preferred to antisecretory drugs such that increased gastric pH might be prevented.48 Sucralfate has been administered rectally (in humans) for treatment of rectal ulcerative disease.
Modulators of Gastrointestinal Motility
Normal Physiology
The GI tract functions to ensure the metabolic survival of the host and in doing so sorts ingested materials as to nutritional, toxic, or pathologic status. To accomplish its activities, the alimentary tract has its own enteric neurologic system (ENS) composed of vagal and sympathetic nerves (Figure 19-8). The network communicates through a large variety of chemicals, including neurotransmitters and neuropeptides. Primary signals include ACh, the tachykinins (substance P, neurokinin A), nitric oxide, adenosine triphosphate (ATP), vasoactive intestinal polypeptide, opioid peptides, neuropeptide Y, and 5-hydroxytryptamine.109 Circuits of the ENS include intrinsic primary afferent neurons; interneruons; and either excitatory or inhibitory neurons that innervate effector motor, secretomotor, or vasodilator neurons. Additionally, the ENS integrates with the CNS, forming a brain–gut axis. The interactions of the integrated signals are complex and often not well characterized and thus are difficult to correct in the face of dysfunction, whether induced by disease or drugs.
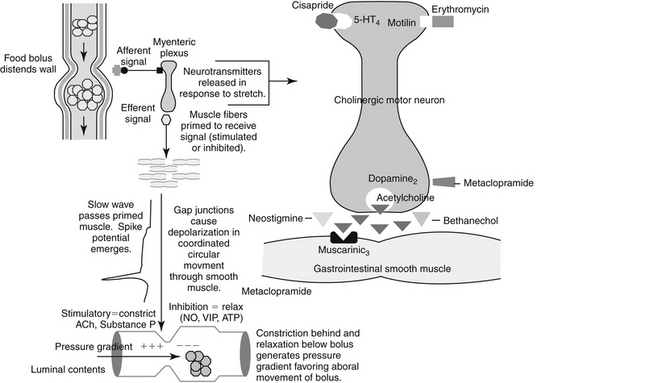
Figure 19-8 The integrated neurologic systems of the intestinal tract. See text for details. Metoclopramide antagonizes dopamine receptors, which antagonize release of acetylcholine. Cisapride is an agonist of 5-HT4 receptors, which are excitatory in the enteric nervous system. Erythromycin acts as an agonist at excitatory motilin receptors. Ach, Acetylcholine, NO, nitric oxide; VIP, vasoactive intestinal peptide.
Regulation of electrical and mechanical, activities of GI smooth muscle is a complex but ordered activity involving hormonal, myogenic, and neurogenic factors and sensory, relay and effector functions.24 Integration occurs at the level of the CNS at both spinal and supraspinal levels110 and locally through the ENS, including intrinsic and interneuronal nerves and ganglia are located between the longitudinal and smooth muscles and at least 10 different classes of receptors located on the smooth muscle cell. Intrinsic innervation includes the Auerbach (or myenteric) plexus, which forms a neural sympathetic and parasympathetic network connecting longitudinal and circular smooth muscle and the secondary Meissner (or submucosal) plexus, which parasympathetically innervates circular muscle. Both systems are secretory and motor in effect, although the Meissner plexuses predominantly modify mucosal absorption and secretion. Coordination of inhibitory and excitatory muscle movements results in two primary movements: mixing and aboral propulsion of liquefied contents. Mixing reflects rhythmic segmentations caused by simple nonprogressive contractions of circular muscle. Electrical activity for propulsion occurs initially as a “slow waves” whose crest is followed by “spike potentials.” Slow waves keep the membrane potential in a constant state of fluctuation, with the frequency of contraction waves ranging between 10 to 20 times per minute, being fastest in the small intestine. Slow waves coordinate synchronous muscle contraction by releasing neurotransmitters from the myenteric plexus such that smooth muscle fibers are “primed” to respond to the spike potential that follows. Gap junctions between circular and longitudinal fibers ensure that electrical stimuli (i.e., depolarization) spread outward in a circular fashion such that an entire ring of circular smooth muscle contracts in a coordinated fashion (Figure 19-9).
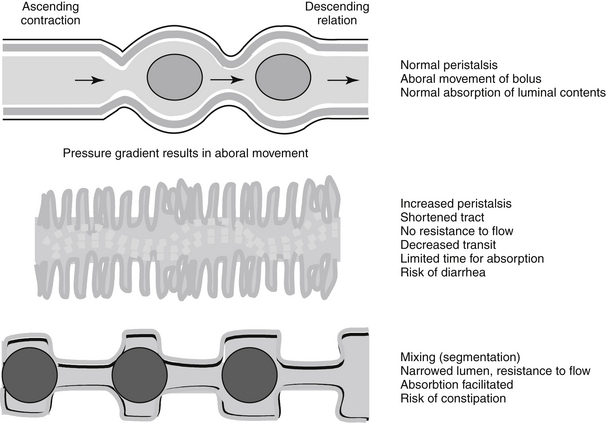
Figure 19-9 The sequelae of smooth muscle contraction in the intestines vary with the type of muscle. Longitudinal muscle contraction shortens the gastrointestinal tract; peristalsis thus forces expulsion of luminal contents. Contraction of circulatory muscle causes segmentation or resistance to outflow. This effect predominates with opioids. Anticholinergic drugs impair both types of muscle activity.
Peristalsis is the major propulsive movement, occurring principally in the esophagus and intestines, involving both longitudinal and circular smooth muscles and reflecting signals originating in the myenteric plexus without being influenced by signals peripheral to the ENS. Peristalsis reflects an ascending excitatory reflex of circular smooth muscle causing contraction on the oral side of a bolus, with ACh and substance P as the primary interneuronal mediators, and a descending inhibitory aboral reflex that causes relaxation, with nitric oxide, vasoactive intestinal peptide, and ATP as mediators.24 Luminal contents follow the net pressure gradient. Contraction of longitudinal smooth muscle in the descending (aboral) phase and relaxation in the ascending oral phase facilitates peristaltic movements and aboral movement of intestinal contents.111 Although peristaltic activity is mainly propulsive, it also ensures mixing and successful absorption. Wilson and coworkers review the physiology of the gastroesophageal sphincter.112 Relaxation is mediated by noncholinergic and nonadrenergic signals. Absorption of luminal contents is facilitated as intestinal luminal contents are impeded by narrowing of the intestinal luminal diameter.
Receptors and Signals
Most smooth muscle activity ultimately reflects response to ACh through cholinergic receptors. However, activity of cholinergic receptors is modulated by a variety of other receptors (see Figure 19-8). Muscarinic (M2) receptors regulate phasic bowel movements during fasting. Several types of muscarinic receptors have been identified pharmacologically (M1-4) in the GI tract. Of these, M1 or M3 receptors interact with G protein and mobilize intracellular calcium; M2 and M41`receptors inhibit adenylyl cyclase and regulate ion channels. M1 receptors present in the myenteric plexus may inhibit motility by way of gabaminergic mechanisms. M2 receptors, located presynaptically and postsynaptically, mediate presynaptic inhibition of ACh release. M3 receptors appear to be located on smooth muscle cells, and although less abundant than M2 by a ratio of 4:1, they are more important because they increase intracellular calcium.24,110
KEY POINT 19-24
Regulation of gastrointestinal motility is complex, with muscarininic (M2 and M3 subtypes) and 5-HT4 being the major role players.
A number of other receptor types influence GI motility. Adrenergic receptors, including α1, and α2 postsynaptically and α2, presynaptically regulate ACh release from the myenteric plexus. Generally, cholinergic neurons stimulate and adrenergic neurons inhibit gastric motility. Both H1 and H2 receptors have been identified in the GI tract. They are located both prejunctionally, where they control ACh release, and postjunctionally. Stimulation of H1 receptors induces, whereas H2-receptors inhibit, smooth muscle contraction.113 The role of dopamine and serotonin in gastric motility is complex and not well elucidated. Among the effects of dopamine are reduction in esophageal sphincter and intragastric pressures. Dopamine, by way of D2 receptors, inhibits smooth muscle of the stomach, duodenum, and colon and has been implicated as a mediator of receptive relaxation in dogs (see Figure 19-8).24,34 Dopamine exerts its inhibitory effect through inhibition of ACh at myenteric motorneurons.34
Over 90% of serotonin in the body is located in the GI tract, indicating its importance in this system. The majority of serotonin is produced by enterochromaffin cells that rapidly release serotonin in response to a variety of mechanical or chemical stimuli. Its effects generally are stimulatory by way of 5-HT3 and 5-HT4 receptors and inhibitory by way of 5-HT1P receptors. The result is a peristaltic reflex, mediated by 5-HT1P and 5-HT4 receptors at the myenteric plexuses and 5-HT3 receptors at extrinsic vagal and spinal sensory neurons.24 Serotonin often exerts its effects through secondary chemicals. For example, the inhibitory effects of 5-HT1P receptors reflect release of nitric oxide, which reduces muscle tone, whereas the stimulatory effects of 5-HT4 receptors reflect enhanced ACh release. However, the effects of 5-HT4 receptor stimulation appear to vary with species and origin. In the guinea pig ileum, effects are stimulatory, but in human and canine colons, 5-HT4 receptor stimulation causes relaxation of smooth muscle.114 In the dog, 5-HT4 receptors appear to mediate relaxation in gastric cholinergic neurons where gastric contraction facilitates gastric emptying, and jejunal and ileal mucosa and circular colonic smooth muscle cells, where activation appears to cause relaxation. Conflicting studies may reflect the different roles that 5-HT4 receptors have in stimulating contraction and peristalsis: a positive effect may occur with mucosal stimulation but not luminal distention.114 5-HT4 receptors appear to be potent secretagogues in most species, again with effects variable among species and location. These differences among species and tissues suggest that extrapolation of therapeutic indications and doses should be based on scientific studies in the target species. The effects of serotonin are ameliorated, in part, by reuptake. However, uptake can be inhibited by CNS-active serotonin reuptake inhibitors, although the sequelae may cause diarrhea.
Other sites are targeted by motility-modifying drugs. Activation of the noncholinergic, nonadrenergic inhibitory neurons results in bowel relaxation.113 Prostanoids and specifically prostaglandin E (PgE) receptors have been identified in the gastric fundus and ileum. PgE receptors may modulate motility from the esophagus to the colon.113 Generally, PgE inhibits mechanical activity of circular smooth muscle, whereas prostaglandins of the D and F series are stimulatory. At high doses, PgE stimulates peristaltic activity, although this may represent mechanical response to excess watery fluid in the intestinal lumen stimulated by PgE.113 Motilin is a peptide hormone located in M and enterochromaffin cells of the GI tract. Motilin appears to enhance and potentially induce phase III activity.
Smooth muscle activity in the large bowel varies from that in the small bowel. In the small bowel, slow waves generally occur continuously and propagate aborally. In the colon slow waves are sometimes absent, and propagation may be variable.110
Prokinetic Drugs
Prokinetics enhance the transit of intraluminal contents.34 The mechanisms of action of these drugs are varied and are not completely understood. Their effects on intestinal functions generally reflect either promotion of an agonist, such as ACh by muscarinic drugs, or inhibition of an inhibitory signal, such as dopamine (see Figure 19-8).34 Organ-specific and species-specific differences complicate our comprehension of these drugs.34
Cholinergics
Clinically, the use of cholinergics is limited by their tendency to cause systemic effects. Bethanechol is an ester derivative of choline that acts as a cholinergic agonist almost exclusively at muscarinic (M2) receptors.113 Bethanechol will enhance the amplitude of contractions throughout the GI tract, including the lower esophageal sphincter (Figure 19-10; see also Figure 19-8).34,113 Its effects on the coordination of small intestinal contraction may be minimal, however, and thus it is often not considered to be a prokinetic agent.34 Adverse effects reflect direct enhanced parasympathomimetic stimulation and include abdominal cramps, diarrhea, salivation, and bradycardia.34
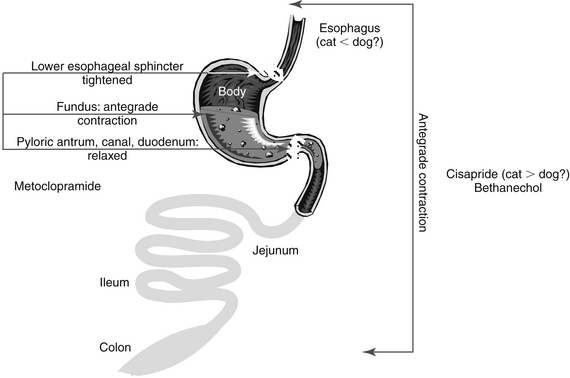
Figure 19-10 The sites of actions of two clinically useful prokinetic drugs. In vivo, at standard doses, metoclopramide’s actions are limited to the lower esophagus (feline more than canine), stomach, and upper duodenum, where it physiologically antagonizes vomiting. Cisapride appears to increase motility throughout the gastrointestinal tract.
Metoclopramide
Metoclopramide is a lipid-soluble derivative of para-aminobenzoic acid. It is structurally related to procainamide, a cardiac antiarrhythmic (see Figure 19-4).34 In addition to its central antidopaminergic (antiemetic) effects, metoclopramide acts peripherally as both an antidopaminergic and as a direct and indirect stimulator of cholinergic receptors.34,35
The effect of metoclopramide on canine gastromotility was described as early as 1969.115 Compared with saline, metoclopramide decreased transit time and volume but did not affect flow of intestinal contents. Clinically, its effects appear to be limited to the upper intestinal tract (see Figure 19-10).35,37,116,117 The peripheral effects of metoclopramide apparently reflect enhanced release of ACh from intrinsic cholinergic neurons. These effects are completely inhibited by pretreatment with anticholinergics such as atropine.34,37,118 The peripheral effects, however, appear to be mediated by effects on other (noncholinergic) local neurotransmitters, particularly dopamine. Serotonin receptors also may play a role.34 The prokinetic activity of metoclopramide reflects muscarinic activity, D2 receptor antagonist activity, and 5-HT4 receptor agonist activity.34 Because of its effects on the stomach, metoclopramide physiologically antagonizes emesis by increasing the tone in the lower esophageal sphincter, increasing the force and frequency of gastric antral contractions (gastrokinetic effect), relaxing the pyloric sphincter, and promoting peristalsis in the duodenum and jejunum, resulting in accelerated gastric emptying and upper intestinal transit.35
KEY POINT 19-25
Clinically, the prokinetic effect of metoclopramide is limited to the stomach, lower esophagus, and pylorus.
Metoclopramide is well absorbed orally but undergoes significant first-pass metabolism with a bioavailability in the 50% to 70% range. Tissue distribution is rapid, and excretion is both renal and hepatic. Because the plasma half-life in the dog is only 90 minutes, metoclopramide has a short duration of action.35 Dose-dependent CNS side effects include nervousness, restlessness, listlessness, depression, and disorientation.34,35,119 Extrapyramidal antidopaminergic effects include tremors and motor restlessness. Tremors in cats, particularly after intravenous administration, are not unusual. Gynecomastia caused by enhanced release of prolactin has been reported in humans.34 GI disorders may also be observed, with constipation being common with long-term use.
Metoclopramide has a demonstrated impact on the oral bioavailability of several drugs, with the effect varying with the drug. For example, co-administration with the oral hypoglycemic agent ciglitazone decreased Cmax by 16% and AUC 8%,120 but co-administration with cephalexin increased cephalexin Cmax by 21% and AUC by 36%.121 It is not clear if the latter effect reflected increased gastric emptying or interference with intestinal transport proteins. As an antiemetic, the main indications for metoclopramide include severe and intractable emesis caused by chemotherapy or other blood-borne toxins as well as nausea and vomiting associated with delayed gastric emptying, gastroesophageal reflux, reflux gastritis, and peptic ulceration. As a prokinetic, metoclopramide is indicated for treatment of a variety of gastric motility disorders, including gastric dilation, volvulus, postoperative ileus, gastric ulceration, and idiopathic gastroparesis.37 Metoclopramide is contraindicated in GI obstruction or perforation, potentially in epilepsy, and for patients receiving neuroleptics. Because of their anticholinergic effects, atropine and the opioid analgesics antagonize the action of metoclopramide.
Domperidone
Domperidone is a dopamine antagonist whose prokinetic properties are similar to those of metoclopramide.122 However, unlike metaclopramide, it has no cholinergic activity and is not inhibited by atropine. Domperidone does not cross the blood–brain barrier as readily as metoclopramide. Like metoclopramide, however, domperidone can affect central dopamine receptors and thus modulate temperature control, prolactin secretion, and activity at the CTZ.34 Extrapyramidal side effects are rare. Domperidone acts peripherally to coordinate antroduodenal contractions. Its peripheral effects accelerate small intestinal transit, but colonic activity is apparently unaffected.119
Cisapride
Cisapride has the broadest spectrum of action of the prokinetic agents.123 It causes dose-dependent increased activity at all GI sites, including the esophagus, stomach, jejunum, ileum, small intestine, and colon34 (see Figure 19-10). Because 75% of the feline esophagus is skeletal muscle, esophageal prokinetic effects are likely to be less in cats than in dogs. The prokinetic actions of cisapride appear to reflect indirect stimulation of cholinergic nerves. Serotonin appears to mediate this effect through 5-HT4 receptors (see Figure 19-8). Stimulation of colonic contractions reflects 5-HT2a. Because secretion is not enhanced, stimulation probably occurs at the level of the myenteric plexus.34
Well absorbed after oral administration, cisapride undergoes first-pass metabolism, with oral bioavailability in humans being 50%. Metabolites are apparently inactive. Volume of distribution is large (2.4 L/kg) in humans, and elimination half-life is 10 hours. Cisapride kinetics have been reported in the cat.124 Oral administration is characterized by 30% bioavailability, and the elimination half-life approximates 5 hours. A dose of 1 mg/kg every 8 hours or 1.5 mg/kg every 12 hours is recommended on the basis of these data. Elimination may be prolonged in the presence of liver disease.123 Indications for use of cisapride are similar to those for metoclopramide (excluding dopaminergic effects). Indications in humans include any disorder associated with impaired gastric emptying as well as gastroesophageal reflux. Unlike metacolopramide, cisapride does not interact with central dopamine receptors, and its use is not associated with extrapyramidal side effects.113 However, stimulation of 5-HT4 receptors by cisapride (and other drugs) causes blockade of the rapid component of potassium influx through the delayed rectifier potassium channel. Prolongation of the QT interval results in torsades de pointes and the potential for sudden cardiac death. Subsequently, cisapride has been withdrawn from the human-medicine market, mandating the need for compounding for veterinary use. Myocardial side effects have not been reported in peer-reviewed literature but have been reported anecdotally. Further, in vitro studies have revealed a similar effect in the canine myocardium. The risk is greater when cisapride is combined with other drugs that inhibit the cytochrome P450 enzyme responsible for cisapride metabolism in humans (e.g., clarithromycin, itraconazole).125
Miscellaneous and Pending Prokinetic Drugs
Erythromycin stimulates GI motility at low doses not associated with antimicrobial effects (0.5-1 mg/kg every 8 hours). Its effects appear to mimic those of motilin (see Figure 19-8). Lower esophageal sphincter pressure will be increased. However, contraction in the stomach is not coordinated and may result in “dumping” of food from the stomach into the intestines before maceration is complete. Increased small bowel motility has led to its clinical use in humans with small bowel dysmotilities, but clinical use has not been reported in animals. Doses greater than 3 mg/kg may actually cause spastic contractions and cramps. The prokinetic effects of erythromycin may be responsible, in part, for GI disturbances that occur in up to 50% of patients receiving this drug. However, tolerance develops rapidly, probably because of downregulation of motilin receptors. Other macrolides will also stimulate motility to variable degrees.
Among the H2- receptor antagonists, ranitidine and nizatidine inhibit anticholinesterase activity and thus are prokinetic at antisecretory doses.126 Comparison between cisapride and ranitidine, an H2-receptor antagonist, for the treatment of gastroesophageal reflux reveals both to be effective.127 Nizatidine was demonstrated to have prokinetic effects similar to those of cisapride owing to noncompetitive inhibition of ACh at a level comparable to that of neostigmine.128
Misoprostol, a PgE analog, also is associated with prokinetic properties in the colons of dogs and cats. At least two prokinetic agents have been in various stages of approval in human medicine: prucalopride (R093877; Janssen Pharmaceutical) and tegaserod. Both are potent, partial agonists of 5-HT4 receptors. Prucalopride (benzamide) is void of activity at other serotonin receptors or cholinesterase enzyme activity. Although pharmacokinetics have not been reported in dogs, prucalopride causes a dose-dependent (0.01-0.16 mg/kg [dog]) increase in gastric emptying and increased giant migrating contractions with defecation in dogs (0.02-1.25 mg/kg) or cats (0.64 mg/kg).129 Fecal consistency does not appear to be affected. In contrast to prucalopride, tegaserod is a nonbenzamide. In addition to effects at 5-HT4 receptors, it acts as a weak agonist at 5-HT1D receptors. Like prucalopride, tegaserod acts as a prokinetic in the canine colon, with effects apparent within 1 hour of an intravenous dose (0.03-0.3 mg/kg). Prokinesis also appears to occur in the intestine on the basis of normalization of transit time in opioid-induced bowel dysfunction in dogs (3-6 mg/kg orally). Because the colonic effects of tegaserod do not appear to be dose dependent, prokinetic effects in the colon may reflect an alternative mechanism of action. The impact of tegaserod on the canine stomach or in the cat have yet to be described. Tegaserod was approved for human use in the United States in 2002 (Zelnorm).
The Impact of Nongastrointestinal Drugs on Gastrointestinal Motility
Benzodiazepines influence gastrointestinal smooth muscle activity and secretion. In fasted dogs, the diazepam (0.5 mg/kg, IV) was associated with disruption of migrating myoelectric complexes and increased conctractility in the jejunum. In humans, diazepam (approximately 0.15 mg/kg PO) suppressed basal gastric acid secretion for 5 hours posttreatment.129a,b A variety of drugs alter esophageal sphincter function. Anesthetic drugs that reduce gastroesophageal sphincter tone include acepromazine, diazepam, morphine, halothane, isoflurane, xylazine, and atropine. The effect of intravenous morphine and butorphanol on GI motility has been studied in dogs. Morphine (0.1 mg/kg) was associated with a dose-dependent increase in duodenal smooth muscle activity and a dose-dependent decrease in bile duct flow, with the effects described as spasmogenic. In contrast, butorphanol administered at a dose described as equianalgesic (0.025 mg/kg intravenously) had little or no effect on the biliary or GI smooth muscle in dogs.130
The effect of xylazine (1 mg/kg) or the combination of acepromazine (0.1 mg/kg) and butorphanol (0.05, 0.2, or 1 mg/kg) on GI motility as it influenced GI radiocontrast studies was studied prospectively in healthy dogs (n = 6) using a randomized crossover saline–placebo-controlled design.131 Total gastric emptying time (gastric and intestinal) was prolonged by xylazine and the combination of acepromazine and butorphanol (all doses). However, nonmanual restraint was facilitated and the combination protocol with butorphanol at 0.05 mg/kg provided sufficient chemical restraint to allow functional morphologic examination of the GI tract (within 5 hours).131 It is unclear how this combination would affect the study in an animal with GI disease.
The impact of selected drugs on feline gastroduodenoscopy has been reported in normal cats receiving ketamine intramuscularly followed by isoflurane maintenance. Each of eight cats was studied four times, once per drug. No differences were detected in difficulty or time to pass the endoscope through the cardiac or pyloric sphincters for hydromorphone, with or without glycopyrrolate, medetomidine, or butorphanol. Passage into the stomach required only 16 seconds, and the drug took at most 2 minutes (generally 20 seconds) to pass into the duodenum. Although sample size may have precluded detection of significant differences among drugs, the authors concluded that none of the drugs impeded endoscopy.132
Drugs Affecting the Intestinal Tract
Physiology, Pathophysiology, and Motility
The primary clinical sign reflecting intestinal disease is diarrhea; accordingly, drugs targeting the intestinal tract frequently are intended to control diarrhea. Drug therapy for diarrhea tends to be nonspecific, preferably targets the underlying disease rather than the GI tract, but occasionally might target the physiologic (rather than pathologic) cause of diarrhea. Diarrheas can be classified as inflammatory or infectious, osmotic (including malabsorption), and secretory. The goal of antidiarrheal therapy generally is to reduce the discomfort and inconvenience of frequent bowel movements and, when indicated, to replace fluids or electrolytes lost with diarrhea. Rehydration followed by oral replacement therapy often is the preferred treatment for diarrheas associated with infectious agents.133 Suitable solutions contain K+, HCO+, Na+, and glucose in sufficient quantities to replace stool losses.133 Substantial evidence exists linking GI secretion with GI motility, with increased motility usually being accompanied by increased fluid and electrolyte secretion.134 The physiology of intestinal motility was previously discussed.
Absorption and Secretion
Increased amounts of fecal water reflect either diminished absorption or a net secretion (accumulation) of fluid into the lumen of the intestine. In all diarrheal states, increased fecal water loss is associated with an overall secretion of electrolytes and water in selected segments of the GI tract. The absorptive capacity of the alimentary canal is overwhelmed distal to the site of secretion. Sodium-absorbing cells are present predominantly on the villi, and chloride-secreting cells are located primarily in the crypts.
Absorption in the small intestine occurs by passive sodium absorption across the luminal membrane and by active secretion of sodium across the basolateral membrane. Water follows the osmotic draw of sodium into the lateral intracellular space. The electrochemical gradient caused by sodium movement facilitates chloride diffusion into the cell.133 A specific brush border carrier for NaCl co-transport accomplishes absorption. Nutrients such as glucose and other organic solutes, however, facilitate solvent drag of water and electrolytes as they enter cells.133 Secretion in crypt cells of the intestinal epithelium is initiated by intracellular signaling (cAMP) or calcium. Increased chloride conductance into the lumen results in sodium recycling, first through the lateral intercellular space and second into the lumen. Although NaCl co-transport can be inhibited, NaCl movement can still occur as a result of solvent drag (i.e., that mediated by nutrients).133
Increased intestinal cell cyclic adenosine monophosphate, cyclic guanosine monophosphate, and Ca2+ (through calmodulin) all diminish sodium absorption and increase chloride secretion, with a net efflux of water into the lumen. Cholera enterotoxin is the best-known intestinal secretagogue,133 but several hormones, including vasoactive intestinal peptide, gastric inhibitory peptide, CCK, secretin, glucagon, and PGE1, and infectious agents (e.g., Escherichia coli, Staphylococcus spp.) are associated with net fluid accumulation.133 Several laxative agents such as bile acids and ricinoleic acid may act through this mechanism. The exact role of intestinal motility in the alteration of fluid and electrolyte movement and the role of mucosal permeability or mucosal damage in the genesis of fluid accumulation within the gut lumen remain unclear.
Modulators of Intestinal Motility and Secretions
Anticholinergic Agents
Parasympatholytic or antimuscarinic agents diminish motor and secretory activity of the GI tract. Tone and propulsive movements are decreased (see Figure 19-9), and these agents will often relax spasm of visceral smooth muscle. Such antimuscarinic drugs are thus known as antispasmodics or spasmolytics. Although cholinolytic agents are commonly used as spasmolytics in antidiarrheal mixtures, the absence of segmentation may lead to severe forms of diarrhea with intestinal paralysis or ileus induced by the cholinergics. The main benefit of anticholinergic agents may be related to their ability to reduce intestinal secretions.
KEY POINT 19-27
A disadvantage of anticholinergic drugs is decreased gastrointestinal motility, loss of segmentation, and severe diarrhea.
Antimuscarinic agents used as spasmolytics include the belladonna alkaloids (atropine and hyoscine), their congeners (atropine methonitrate, homatropine methobromide, hyoscine butylbromide,), and synthetic cholinolytic drugs (aminopentamide, dicyclomine, glycopyrrolate, mepenzolate, oxyphenonium, propantheline). Many of the belladonna alkaloid derivatives are substituted tertiary amines and thus may have undesirable CNS and other systemic effects. The synthetic groups are mostly substituted quaternary amines and are devoid of CNS effects. Xerostomia, loss of lens accommodation, urinary retention, constipation, tachycardia, and CNS stimulation are potential side effects that may be encountered when parasympatholytics are administered.
Opioids
Receptors and peptides
Opiates have been used since antiquity to control diarrhea, and they remain the cornerstone of nonspecific antidiarrheal therapy for humans.48 Opioids appear to influence normal GI physiology.135 Opioids may stimulate GI motility both locally and by central effects in the brain or spinal cord.113 Both opioid peptides and opioid receptors have been identified throughout the GI tract. The specific location of receptors has been based on the physiologic effects of opioid agonists and antagonists.135-137 Both in vitro and in vivo preparations have been studied, often with conflicting results. Confounding interpretation is the variability in number and receptor type among species. High-affinity, reversible, and saturable binding of opioid receptors has been identified in longitudinal and circular smooth muscles and the myenteric and submucosal plexuses. Binding has also been noted in the muscularis mucosae. Although multiple opioid receptors have been identified, μ-type (OP3) and δ-type (OP1) opioid-binding sites appear to predominate among the species. Morphine acts to stimulate μ-receptors of the myenteric plexus, thus inducing migrating motor activity in the duodenum and jejunum. The relative importance of different receptor types in the control of intestinal peristalsis has not been established.
Not surprisingly, in addition to opioid recetpors, endogenous opioids are also present in high concentrations in the intestinal wall. Biosynthesis of enkephalins has been demonstrated in the myenteric plexus of some species. In addition, antral G cells are thought to be capable of synthesis of enkephalins or endorphins. β-endorphin and dynorphin have also been demonstrated. In cats enkephalin has been detected in both myenteric and submucosal plexuses. In contrast to other species, the predominant location of enkephalin neurons in dogs is the submucosal plexus. Opioid nerve fibers have also been documented in the lower esophagus, pyloric junction, and cardiac and ileocecal regions. Specific degradative enzymes for the opioid peptides have also been identified in similar locations.135
Impact of opioids on gastrointestinal motility
Much of the research regarding opioids and the GI tract reflects an attempt to understand the mechanisms of GI adversities resulting from opioid analgesic therapy. In vitro studies indicate that endogenous opioids in the GI tract modulate normal GI motility and gastrin release. Exogenous opioids depress the normal peristaltic reflex (see Figure 19-9), as has been repetitively demonstrated with the use of the pure antagonist naloxone, which consistently increases peristaltic activity. As such, among the side effects of opioids is constipation, associated with an inhibitory effect on smooth muscle motility. The intestinal opioid mechanism occurs in vitro throughout the intestinal tract, with function apparently increasing distally from duodenum to ileum. Endogenous opioids may thus be partially responsible for the gradient of intestine, a term that describes the oral to aboral phenomenon of decreasing frequency of peristaltic waves and decreasing sensitivity to distention stimuli.135 GI opioids appear to be subject to feedback control.135 The widely accepted mechanism of opioid actions in the GI tract is principally presynaptic inhibition of ACh (and potentially other mediators, such as serotonin) release.109 Postsynaptic modulation of the effects of ACh already released may also be important for the effects of opioids on peristalsis. Intracellular mechanisms may involve increased calcium-dependent potassium conductance and hyperpolarization. Opioids also reduce calcium entry during the action potential and deplete neurons of calcium. In addition to inhibition of propulsion, tonic spasmogenic effects may occur in some species. The spasmogenic effect caused by opioids is antagonized by atropine. The effect of opioids on GI sphincters varies, with the effect appearing to vary with dose. For example, excitation (contraction) of the choledochoduodenal junction in dogs occurs with some drugs at low doses and inhibition at higher doses. Species differences in sites of action, receptor populations, and motility may be more quantitative than qualitative.135
Impact of opioids on intestinal secretion
As with motility, the effects of opioids on gastric acid secretion vary with the study, species, and opioid. Opioids enhance gastric acid secretion mediated by histamine using in vitro studies, but the effect on ACh-induced secretion varies among in vivo studies. Dose dependence may account for some of the variability, with excitation or enhancement of basal secretion occurring at lower doses and inhibition at higher doses. A dual effect on stimulated gastric acid secretion appears to be mediated both peripherally and centrally.135
In contrast to gastric secretion, the effect of opioids on intestinal secretion appears to be consistent among the species.135 Opioids stimulate the net absorption of water and electrolytes in enterocytes of both small and large intestines in a variety of species. In vitro studies indicate that these peripheral effects are mediated by δ-receptors. Receptor types may, however, vary with the site. These effects, which may reflect facilitated absorption or inhibited secretion, are largely responsible for the antidiarrheal properties of the opioids. Several mediators, acting centrally and peripherally, may mediate opioid antisecretory effects, with the impact varying with the chemical mediator. Presynaptic inhibition of ACh release and inhibition of prostaglandin-mediated adenylate cyclase activity have been implicated as the target signals, with sodium, but not chloride, the targeted ion that is negatively influenced. Opioids also centrally decrease intestinal secretions, perhaps through the sympathetic nervous system. Antisecretory effects may involve norepinephrine and its effects on vasoinhibitory peptide, PgE, or ACh.113 Decreased intracellular free calcium also has been implicated as a possibility. In contrast to water and electrolyte secretion, opioids act to increase bicarbonate secretion from the gastric and duodenal mucosa.135
Drugs
Diphenoxylate hydrochloride is a meperidine derivative used specifically to control diarrhea. It is often administered in combination with atropine-like compounds, whose bitter taste and drying effect on salivary secretions are added as a deterrent to substance abuse. The actions of diphenoxylate largely depend on a direct peripheral effect on the GI wall. Because diphenoxylate can penetrate the blood–brain barrier, systemic opiate effects may occur. The potential for drug abuse has led to its designation as a Schedule V drug. The use of opioids in cats is generally reasonable, as long as accommodation is made for the increased sensitivity that appears to characterizes their response.
Loperamide hydrochloride, a butyramide derivative, is an orally active and effective antidiarrheal agent used for symptomatic control of acute and chronic nonspecific diarrhea. Unlike diphenoxylate, systemic opiate agonist effects do not appear to occur after oral administration of loperamide, and there are few side effects. Although loperamide has some structural similarities to diphenoxylate, it does not cross the blood–brain barrier, and it differs both qualitatively and quantitatively from diphenoxylate and difenoxin in its pharmacologic actions.138 Intestinal transit time and intestinal luminal capacity increase after treatment with loperamide.113
KEY POINT 19-29
Because loperamide does not penetrate the blood–brain barrier, it has fewer side effects than diphenoxylate.
Miscellaneous Antisecretory Drugs
There are a number of potentially useful drugs that have not been extensively studied but for which there is some evidence of clinical benefit. Glucocorticoids have been found to be beneficial in treating refractory chronic diarrheal disease as well as chronic inflammatory diseases of the intestinal tract. Glucocorticoids stimulate active sodium absorption in the jejunum, ileum, cecum, and colon. However, because of many undesirable side effects when used chronically, the glucocorticoids should not be used on a routine basis to treat diarrhea.
Adrenergic agents appear to act predominantly by increasing basal fluid absorption and do so at very low concentrations. The mechanisms involved are unclear. Clonidine and other α2-adrenergic agonists are potentially useful in this regard. Calcium/calmodulin antagonists may act by stimulating active absorption as well as by inhibiting intestinal secretion, but the precise mode of action of these drugs is not clear. Several drugs with this effect have been found to be useful in the control of certain forms of secretory diarrhea. Examples include chlorpromazine and trifluoperazine. NSAIDs such as aspirin, indomethacin, flunixin, and the subsalicylate of bismuth subsalicylate inhibit the cyclooxygenase pathway of arachidonic acid metabolism and thereby suppress the formation of prostaglandin mediators. The role of various prostaglandins in intestinal motility as well as in absorption and secretory processes is complex, and the inhibition of prostaglandin synthesis will not consistently influence secretory diarrheal states. The NSAIDs may, however, prove to be therapeutically beneficial in some acute and chronic diarrheal syndromes as long as care is taken regarding their gastrointestinal advesre effects.
Asulfidine (sulfasalazine) is a sulfapyridine-5-aminosalicylic acid (5-ASA; mesalazine) compound joined by a diazo bond that is broken by colonic microbes.139 It is the 5-ASA component that is beneficial, with the sulfapyridine simply carrying the active drug to the colon.139 This finding led to the development of 5-ASA products without the sulfonamide component. Compounds equally efficacious to sulfasalzine include osalazine, balsalazide, and mesalazine. An advantage of these products is efficacy above the colon (i.e., in the absence of microbial metabolic activation). A variety of preparations are available such that oral absorption is prevented and specific sites of delivery (e.g., jejunem versus ileum versus colon) might be targeted, with the intent of topical, rather than systemic, delivery. These include pH-dependent (e.g., Asacol) and slow continuous release (Pentasa) mesalazine (also known as mesalamine), diazo bond delivery (osalazine, a diamer of mesalazine), or balsalazide (a prodrug).140 The exact mechanism of action of mesalazine is not known, but presumed activities include inhibition of cytokines, leukotrienes, and nuclear factor kappa B. Mesalazine is metabolized by both epithelial and hepatic acetylation. Although these newer 5-ASA products have not been proved to be more efficacious than sulfasalazine in treatment of IBD in humans, they are better tolerated. Because they are better tolerated than glucocorticoids, they are the cornerstone of therapy for some human IBDs.
As with all aspirin-containing compounds, caution is indicated when a sulfasalazine is used in the cat because salicylic acid released in the colon can be subsequently absorbed. For example, the aspirin component of mesalamine (and thus osalazine) may be 30% or more bioavailable (demonstrated in humans), and care should be taken to avoid aspirin toxicity, especially in cats. Mesalamine enema may contain sufficient sodium benzoate (as a preservative), which also should be used cautiously in cats (see Chapter 4).
Gastrointestinal Protectants and Absorbents
Compounds that are not absorbed from the GI tract and either line the mucosal surface or adsorb toxic compounds are often incorporated into antidiarrheal mixtures. The protectants seemingly produce a coating of the GI epithelium that prevents irritation or erosion by potentially harmful substances. The adsorbents physically bind chemical compounds, which precludes their absorption, and they are then eliminated in the feces. Use of these two therapeutic classes is obviously directed at potentially harmful agents of either inorganic or organic nature. Adsorbents will also, however, bind concurrently administered drugs used for therapeutic purposes. Care must be taken with the use of over-the-counter products. Active ingredients may change under the same trade name. Although over-the-counter products often are safe, some may present a health risk to certain patients (e.g., bismuth subsalicylate and cats).
Many protectants and adsorbents possess both properties to varying degrees. Those most frequently used are magnesium trisilicate, hydrated magnesium aluminum trisilicate (activated attapulgite), kaolin (natural hydrated aluminum silicate), aluminum hydroxide and phosphate, bismuth salts, calcium carbonate, pectin (natural polygalacturonic acids), and activated charcoal.
The combination kaolin–pectin product is dissolved in 20 parts water. Described as a demulcent and adsorbent, the drug supposedly binds and removes bacteria and their metabolic products and toxins. These effects are controversial. Although stool consistency may improve, studies do not indicate that fluid and electrolyte imbalance is corrected, nor is the course of disease shortened.141
KEY POINT 19-30
Bismuth subsalicylate is an effective antidiarrheal because of its adsorptive capacity and effects on secretion.
The insoluble bismuth salts have been used for more than 400 years.142 Products include bismuth subcarbonate, bismuth subnitrate, and bismuth subsalicylate. Bismuth subsalicylate is a crystalline 1:1 trivalent bismuth and salicylate compound. It is chemically transformed throughout the GI tract to bismuth and salicylate. The drug has been shown to have both antisecretory and antimicrobial effects in several species.142 The subsalicylate fraction has been shown to have antiprostaglandin synthetase effects, which would enhance its action in controlling diarrheal syndromes.133 In humans and cats, nearly all the salicylate is systemically available.142,143 Caution is recommended in order to prevent salicylate toxicity in cats receiving this drug.
Activated charcoal has primarily adsorbent properties. Because of its broad spectrum of adsorptive activity and its rapidity of action, it is one of the most valuable agents for emergency treatment of certain cases of poisoning. It forms a stable complex with many substances and permits their evacuation from the body. Charcoal preparations vary according to the source of base material, surface area, capacity for drug binding and affinity, and avidity of drug binding.144 Source materials are usually lignite, wood, or peat. Activation forms more pores and enlarges the surface area. Activation time is directly correlated with the molecular size of the compounds adsorbed. Because most drugs are of an intermediate molecular weight, charcoals with pore sizes between 10 to 20 Å are most appropriate.141 Administration with a cathartic, such as sorbitol, is a common practice and facilitates rapid movement of the charcoal–toxin complex.144 Activated charcoal loses its efficacy as the time interval between treatment and toxin ingestion increases. The optimal dose and interval for administration of activated charcoal have not been well established,144 although a charcoal to toxicant ratio of 10:1 has been recommended.141 One source suggests treatment at 6-hour intervals.144 Powders are superior to tablets.141 Food generally decreases the efficacy of these products. In the common domestic species, 20 to 120 mg/kg powered activated charcoal is usually administered as a drench after mixing with water. An activated charcoal suspension may be used for gas lavage in simple-stomached animals.
Cholestyramine is a basic anion exchange resin that binds to acidic side chains such as those occurring in bile acids. Endotoxin also is bound. To increase the number of basic binding sites, cholestyramine is attached to a polystyrene matrix that can act as a nonspecific adsorbent. As bile salts are bound in the GI tract, lipoproteins, cholesterol, and neutral fat absorption is also decreased. Although specifically indicated in humans for pruritis associated with increased bile acids (hypercholesterolemic syndromes) cholestyramine has also been used to symptomatically treat diarrhea, particularly that which is intractable. Nausea, constipation, steatorrhea, and decreased fat-soluble vitamin absorption are reported undesirable effects. The product should be administered in food or water.141
Laxatives and Cathartics
Laxatives and cathartics promote defecation by increasing frequency of defecation or fecal volume or consistency.119,145-147 Laxatives (or aperients) promote elimination of a soft-formed stool, whereas cathartics (or purgatives) tend to produce a more fluid evacuation. The difference between these two effects may be just a matter of dose, but in some instances laxatives are only capable of increasing the hydration or softness of the fecal mass without ever inducing catharsis. The enhanced intestinal transit times that occur with use of some of these cathartics are usually due to intrinsic local myenteric reflexes within the visceral smooth muscle or to stimulation of the cholinergic receptors of the extrinsic parasympathetic nervous system. Although a traditional classification of the group is presented here, it should be noted that many cathartics alter intestinal electrolyte transport to increase fecal water excretion, so the grouping of these compounds should perhaps more logically follow their effects on intestinal electrolyte movement.
A number of deleterious effects may occur with excessive or constant use of cathartics. Severe, continuous diarrhea and abdominal colic, leading to dehydration and even shock, may follow overdosage. Other potentially harmful effects include decreased sensitivity of the intestinal mucosa, megacolon, flatulence, loss of electrolytes (especially sodium, potassium, chloride, and bicarbonate), secondary aldosteronism, melanosis coli (anthraquinones), steatorrhea, protein-losing gastroenteropathy, excessive calcium loss with resultant osteomalacia, and exacerbation of inflammatory intestinal disease. Several drugs can also distribute into milk and adversely affect suckling young.
Emollient Laxatives
The emollient laxatives (lubricant laxatives, mechanical laxatives, fecal softeners) act unchanged. They are not absorbed to any appreciable extent and simply soften and lubricate the fecal mass, which in turn facilitates expulsion. Although not always reliable, particularly in the ruminant, they are used in all species.
Mineral oil (liquid paraffin) is a commonly used lubricant laxative. It is bland and generally safe, but chronic administration may impair absorption of fat-soluble vitamins, other nutrients, and co-administered therapeutic agents. Decreased irritability of the intestinal mucosa may develop with protracted use and, paradoxically, cause chronic constipation. White or yellow soft paraffins are used most commonly for small animals as lubricant laxatives (e.g., feline hairballs). Several anionic surfactants are employed as fecal softeners. Examples include docusate sodium, previously called dioctyl sodium sulfosuccinate and dioctyl calcium sulfosuccinate.
Simple Bulk Laxatives
The simple bulk laxatives are hydrophilic and not digested. As such, they adsorb water and swell, forming an emollient gel. Increased volume or bulk causes distention and reflex contraction, leading to peristaltic activity. Feces remain soft and hydrated. Methylcellulose, carboxymethylcellulose sodium, and plantago seed (psyllium seed) are examples of simple bulk purgatives. Wheat bran, prunes, and other fruits also belong in this group. In addition to their bulk action, celluloses and hemicelluloses will be fermented in the hind gut by bacteria to produce volatile fatty acids and other products that exert an osmotic effect, which enhances their laxative action. Meteorism and a very fluid stool often result from the use of simple bulk laxatives.
Osmotic Cathartics
Osmotic cathartics (saline purgatives) consist of salts or compounds that are either partially and slowly absorbed or not absorbed. Water is osmotically retained or attracted into the intestinal lumen, although enhanced mucosal secretion of fluid may also occur. Drinking water must be freely available, and use is contraindicated with dehydration. Effects are realized in monogastric animals generally in 3 to 12 hours. Magnesium salts are frequently used as saline purgatives. Magnesium ions also cause release of CCK, which increases peristaltic activity. Magnesium sulfate (Epsom salts), isotonic in a 6% solution, magnesium hydroxide, magnesium oxide (milk of magnesia), and magnesium citrate are the magnesium salts most commonly employed. The solutions need not be hypertonic to produce an effect. About 20% of the magnesium ions are absorbed when magnesium sulfate is dosed orally, and if purgation does not occur, additional amounts of magnesium may be absorbed with subsequent depression of the excitable tissues in the body. This is more likely to occur if renal function is impaired. Salts such as sodium sulfate (Glauber’s salt), sodium phosphate, potassium sodium tartrate (Rochelle salt), and even large quantities of sodium chloride are effective saline purgatives. Saline purgatives are often combined with polyethylene glycol (PEG) for whole bowel irrigation implemented for bowel preparation before lower bowel procedures such as surgery or colonoscopy.The safety and efficacy of sodium phosphate has been described in healthy dogs (n = 8) when used to prepare for colonoscopy. Orogastric sodium phosphate (1 mL/kg in 2 mL/kg water followed by 2 mL/kg) was less effective as a preparatory agent compared with PEG (66 mL/kg) and bisacodyl (10 mg/dog; dog weight not provided). Most (five of eight) dogs receiving sodium phosphate vomited during or after administration. In contrast, dogs receiving PEG tended to regurgitate. Sodium phosphate enemas were associated with transient hyperphosphatemia and hypocalcemia that were not considered clinically relevant in these normal dogs. Other electrolyte concentrations statistically varied but remained within normal limits. The protocols for colonoscopy preparation included 20 mL/kg warm water enemas; treatments were repeated at 4 hours and enemas at 24 hours (before procedures). Enemas may have contributed to vomiting because of distention. Dogs receiving PEG were also pretreated (20 minutes) with metoclopramide (0.3 mg/kg subcutaneously). The authors concluded that sodium phosphate enemas should not be considered as preparative agents for colonoscopy.148
The sugar alcohols mannitol and sorbitol will also induce an osmotic catharsis, as will the synthetic disaccharide lactulose, which is not digested in the small intestine because no specific enteric enzyme is present. It passes to the large intestine, where saccharolytic microflora ferment lactulose to produce acetic, lactic, and other organic acids, which in turn lower the pH of the colonic content and exert an osmotic effect. Water is attracted, the fecal mass softens, and colonic peristalsis ensues. Lactulose is used for chronic constipation and treatment of hepatic encephalopathy. Acidification of the contents of the large intestine favors a greater formation of the ionized and thus nonabsorbable ammonium ion rather than the readily absorbable ammonia molecule, which requires detoxification in the liver by the urea cycle. Hyperammonemia is thus decreased. Absorption of other toxic amines from the hind gut is also reduced by acidification of the contents. Some meteorism may be evident after administration of lactulose. Lactitol is an alternative osmotic cathartic. It is less sweet than lactulose and may be better tolerated.
Irritant Cathartics
Contact or irritant purgatives were thought to stimulate the mucosal lining of the GI tract and thereby initiate local myenteric reflexes that would enhance intestinal transit. However, they also activate secretion. Irritant cathartics act either directly or indirectly, depending on whether the compound must be metabolized to its active product. Some purgatives are so highly irritating that they may cause severe colic and superpurgation.
Several bland vegetable oils act as irritant purgatives. Their action is based on hydrolysis by pancreatic lipase in the small intestine and subsequent formation of sodium and potassium salts of the released fatty acids, which act as irritant soaps. They differ in potency depending on the oil used. Castor oil produces highly irritant ricinoleates; raw linseed oil leads to formation of less irritant linoleates; and olive oil leads to rather mild oliveates. The response to castor oil is prompt, and evacuation of the whole intestinal tract occurs, leading to an almost complete emptying. Moist bulky feeds are necessary after purgation with castor oil. It is used mainly in nonruminants and often employed in calves and foals. The effect occurs in 4 to 8 hours in small animals.
The diphenylmethane cathartics appear to have a greater effect on the large intestine. Their precise mechanism of action is unclear. An effect is usually seen within 6 to 8 hours, and excessive catharsis may occur with overdosage. Bisacodyl also is a diphenylmethane cathartic that inhibits glucose absorption and Na+, K+ ATP activity, as well as altering motor activity of the visceral smooth muscle. Only about 5% of any dose of bisacodyl is absorbed. This agent is used both orally and by enema.
Enemas
Introduction of solutions or suppositories into the rectum to initiate the defecation reflex is a useful and simple method to correct or prevent constipation. Many preparations have successfully served as enemas, including soapy water (soft anionic soap), isotonic or hypertonic sodium chloride solutions, sorbitol, glycerol, surfactants such as sodium lauryl sulfoacetate, mineral oil, and olive oil. Enema preparations that contain phosphate should not be used indiscriminately because they can precipitate potentially fatal hyperphosphatemia, hypocalcemia, and hypernatremia in cats149 or debilitated animals.
Agents Promoting Gastrointestinal Functions
Several preparations are used therapeutically to control specific GI diseases by promoting digestive or other metabolic processes. These compounds generally consist of normal microbiota or digestive enzymes or related substances that are used for replacement therapy in deficiency states.
Digestive Enzymes
Pancreatic extracts that stimulate pancreatic exocrine secretions are of therapeutic benefit in cases of chronic pancreatitis and pancreatic hypoplasia, in which glandular function is diminished or destroyed. Pancreatin (Panteric, Stamyl, Viokase) obtained from hog pancreas is the major ingredient of most commercial pancreatic enzyme preparations. Enteric-coated preparations to prevent destruction of pepsin in the stomach are generally thought to be better than noncoated preparations. In a few instances, enteric-coated preparations are the most effective in dogs with pancreatic insufficiency; they are added to food and must be provided with each meal. Dosage is adjusted to obtain a normal stool. Simultaneous administration of nonsystemic alkalinizing agents to maintain an optimal pH range for enzyme activity has not proved successful clinically. However, the administration of cimetidine about half an hour before dosing with pancreatic extract does limit gastric inactivation of the enzymes. Proper dietary control is also essential for the successful management of animals suffering from pancreatic insufficiency. Premixing in food does not appear to be necessary for efficacy of these products.
Bile acids and their salts promote absorption of long-chain fatty acids and fat-soluble vitamins. They also act as choleretics (discussed previously). Examples include dehydrocholic acid (Decholin) and chenodiol (previously called chenodeoxycholic acid).
Diastases are amylolytic enzymes obtained from malt and Aspergillus oryzae and are used for replacement of pancreatic α-amylase and to control flatulence caused by gas produced from soluble carbohydrates by bacterial flora.
Antiflatulence Drugs
Simethicone is an inert over-the-counter mixture of siloxane polymers stabilized with silicon dioxide.24 It acts as an antifoaming agent by covering bubbles with a thin layer that facilitates bubble collapse. Reduction in foam may reduce gas volume, although therapeutic efficacy is not clear.24
Biotherapeutics: Prebiotics, Probiotics, and Synbiotics
The concept that microbes might be beneficial rather than simply detrimental to health enjoys a long history of anecdotal evidence. Among the oldest observations is the Old Testament recording of Abraham ingesting sour milk (Genesis 18:8). The purported health benefits of microbial products are varied, but they are more often supported by testimonials than by scientific evidence. However, hypothesis-driven research is increasingly generating evidence-based examples of the therapeutic benefits of biotherapy.
KEY POINT 19-32
The normal intestinal microbiota plays an important role in nutrition, metabolism, pathogen control, and immunity.
Product Definitions
The term probiotic was originally coined in 1965 to refer to substances secreted by one microorganism that stimulate the growth of another.150 The term has been modified at least five times and continues to vary with the author. Most recent modifications are intended to encompass nondairy products (e.g., plant-based products), recognizing the importance of organism load to ensure a therapeutic effect; to allow for transient, rather than prolonged, effects that would otherwise require transplantation (implying colonization), and to allow for multiple and diverse benefits, including those manifested beyond the GI tract.150 Perhaps the easiest working definition of probiotics is that offered by the World Health Organization: live microorganisms that, when administered in adequate amounts, confer a health benefit on the host.150
In contrast to probiotics, prebiotics are nondigestible food ingredients (e.g., dietary fiber) that beneficially affect the bacterial population. They differ from other fermentable carbohydrates in that they interact with selective microorganisms.151 They presumably are most beneficial to those microbes targeted for their therapeutic benefit, thus shifting the composition of the intestinal microbiota toward the beneficial organisms. Examples include fructooligosaccharides, inulin, transgalactosylated oligosaccharides, and soybean oligosaccharides that selectively promote the growth of bifidobacteria. Metabonomics is the scientific discipline that studies compounds formed from prebiotics. Synbiotics contain both prebiotics and probiotics, with the prefix syn implying a synergistic effect of the prebiotic on the probiotic portion of the combination products. To be a symbiotic, the prebiotic portion should have demonstrated positive effects on the specific probiotic component of the combination product.150 Some products sold as probiotics are actually the fermentation products produced by target microbe; presumably, the products are those imparting the therapeutic benefits of the microbes.
Normal Microbiota
Normal Flora
Understanding the impact of biotherapeutics requires an appreciation of the normal GI microflora. The total body microbiota (human and presumably dogs or cats) outnumbers other cells by approximately tenfold. Up to 30% of dry fecal matter represents microbes. The number of microbial species in the GI tract (again in humans) is estimated to be between 400 and 500 based on 16S rRNA sequencing and metagenomic. However, only a fraction (approximately 10%) are conducive to cultures.152,153 Whereas some of these unculturable microbes are closely related to those already identified, others represent totally new species. Therefore very little is known regarding the microbiota of the GI tract. To further complicate our understanding, the number and diversity of microbes increases with a number of factors. Differences owing to location in the GI tract reflect, in part, differences in pH and transit time. Peristalsis and low pH lower microbial counts in the stomach, duodenum, and jejunum,152 whereas numbers and diversity increase in the ileum (104-8 CFU or colony-forming units/mL) and colon. Rapid transit decreases numbers because bacteria colonizing sites with a high transit rate (e.g., lower ileum) must be able to effectively adhere to the mucosal epithelium; thus organisms such as lactobacilli must compete with pathogenic organisms for adherence receptors if they are to colonize these regions. Organisms that do not have to adhere are likely to be present in much higher numbers in the colon. Higher colonic pH (5 to 6) leads to slower epithelial turnover, which also increases bacterial counts. More rapid microbial growth results in a lower redox potential.
KEY POINT 19-33
The normal microbiota consists of close to 500 organisms, most of which have not been cultured or otherwise specifically identified.
Intestinal bacterial by-products produced under low redox conditions contribute to a higher short-chain fatty acid concentration. Even within the colon, bacterial growth and fermentation products are diverse among regions, with growth and carbohydrate fermentation greater proximally and protein fermentation greater distally. Diet has a profound impact on microbiota. Nondigestible foods are a source of energy and carbon for the microbiota and may influence metabolites that positively or negatively influence the population. Food also affects host GI function and health and thus indirectly affects the microbiota.152
The microbiota of the human GI tract was recently reviewed and may provide insight into the complexities of microbiota in other species.152 The number of colony-forming units per mL increased from the stomach and duodenum (103) to the colon (1014). The composition increases in complexity with age such that the microbiota is relatively simple in newborn and very old subjects. Nutritional and physical requirements are sufficiently sophisticated that the conditions for optimal growth simply are not known for up to 70% of the bacterial population.151 However, four microhabitats have been described: the lumen; the mucous layer of epithelial cells; the surface of epithelial cells; and the crypts of the ileum, cecum, and colon (which generally are colonized with motile, spiral-shaped organisms). In humans a central core of microbiota might be partitioned into five bacterial groups: Clostridium leptum; Clostridium coccoides; Bacteroides; and, in smaller part, Bifidobacterium.151
The GI flora of dogs has been described and compared to that in humans,154,155 (Table 19-3) and the microbiota of the cat has been described.154 Selective media often do not support the growth of target microbes in dogs, contributing to the list of unculturable bacteria.154 However, because the dog is a carnivore, its GI tract is shorter than that of humans. Similarities between human and dog include the proportion of gram-negative to anaerobic organisms, the makeup of gram-negative isolates, and floral behavior in response to probiotics or prebiotics.154,156 Genera that are numerous in both the GI tract of restricted-access dogs and humans include Lactobacillus, Bifidobacterium, Eubacterium, Bacteroides, and Peptostreptococcus. The major difference between dogs and humans appears to be the proportion of bifidobacteria;154 however, a study examining floral changes in response to different housing environments demonstrated that the number of bifidobacteria dramatically increase under conditions in which exposure to the environment is controlled.156 Under such conditions major differences between the two species among culturable bacteria are limited and include the following: (1) Bacteroides and Streptococcus are the most common isolates in the ileum and colon of dogs; however, these isolates, although not the predominant isolate in humans, are present in a large proportion of humans; (2) Fusobacterium is not as numerous in dogs as in humans. In the cat, bifidobacteria appear to be even less numerous than in the dog.
Table 19-3 Canine Normal Microbiota
| Region | Human (%) | Dog (log CFU/g) |
|---|---|---|
| Gram-Negative Aerobes | ||
| Escherichia coli | N/A | |
| Enterobacter | 1% | |
| Pseudomonas | N/A | |
| Gram-Negative Rods | Jejunum (4)∗ | |
| Feces(6)∗ | ||
| Gram-Positive Aerobes | ||
| Enterococcus | Colon (6-7)† | |
| Streptococcus | Colon (8-9)† | |
| Staphyloccoccus | Colon (8-9)† | |
| Combined | Jejunum (3)∗ | |
| Feces (9)∗ | ||
| Gram-Positive Cocci | Jejunum (3)∗ | |
| Feces(4)∗ | ||
| Corynebacterium | N/A | |
| Gram-Positive Rods | Jejunum (3)∗ | |
| Feces (4)∗ | ||
| Gram-Negative Anaerobes | ||
| Bacteroides | 20% to 42% | Colon (10+) |
| Jejunum (2)∗ | ||
| Feces (6)∗ | ||
| Porphyromonas | N/A | |
| Prevotella | 6% | |
| Fusobacterium | Colon (8-9)† | |
| Jejunum (3)∗ | ||
| Feces (6)∗ | ||
| Bifidobacterium | 1%-7% | Colon (6-8)† |
| Veillonellaceae | N/A | |
| Gram-Negative Rods, other | Jejunum (3)∗ | |
| Feces (6)∗ | ||
| Gram-Positive Anaerobes | ||
| Gram-Positive Cocci | Jejunum (3)∗ | |
| Feces (10)∗ | ||
| Peptostreptococcus | N/A | |
| Peptococcus | N/A | |
| Gram-Positive Rods | Jejunum (3)∗ | |
| Feces (7)∗ | ||
| Lactobacillus | Colon (7-10)† | |
| Clostridium | 22%-30% | Colon (6-8)† |
| Jejunum (3)∗ | ||
| Feces (10)∗ | ||
| Eubacterium | N/A | |
CFU, Colony-forming units. Eubacterium and Corynebacterium are pleomorphic.
∗ Data from Mentula S, Harmoinen J, Heikkilä M et al: Comparison between cultured small-intestinal and fecal microbiotas in beagle dogs, Appl Environ Microbiol 71(8):4169-75, 2005.
† Data from Rastall RA: Bacteria in the gut: friends and foes and how to alter the balance, J Nutr 134:2022S-2026S, 2004.
Gastrointestinal Microbe–Host Interactions
The sophisticated interactions among the intestinal mucosa, host immune cells, the microbiota, and food have been described,157 and the impact of probiotics on host immunity has been reviewed.158 It is beyond the scope of this chapter to discuss the interactions, but it is notable that the mechanisms by which commensals are recognized as such and fail to stimulate an inflammatory response by the GI tract are not yet known. Interactions are modulated in part through pattern recognition receptors (PRRs) that recognize microbial motifs referred to as pathogen-associated molecular patterns (PAMPs). The PAMPs are evolutionarily highly conserved, not varying within microbes in the same class. Consequently, mammalian cells are able to recognize all microorganisms with only a few PRRs. Receptor families that identify PAMPS include Toll-like receptors located either on the cell surface or intracellularly and nucleotide-binding oligomerization domain (NOD) receptors. Thus far, 11 mammalian Toll-like receptors have been identified.157 Whether colonization is necessary for immunomodulation to occur is not clear.158 However, microbial adhesion to the epithelial cells or mucus is generally recognized to be necessary. Activation of the immune system may reflect microbial alteration of epithelial tight junctions with subsequent bacterial translocation.
Impact of Normal Flora on Gastrointestinal Health and Disease
In the last decade, the role of normal microbiota in GI physiology has been recognized as critical, protecting against invasion of pathogenic strains of bacteria, facilitating maturation and maintenance of the immune system, facilitating normal bowel smooth muscle function, supporting digestion of certain foods, and contributing to nutrition through the production of vitamins (vitamin K; vitamin B in some species) and other nutrients (e.g., short-chain fatty acids). Energy salvage is facilitated through conversion of nutrients to short-chain fatty acids.152,159 Known effects of commensal intestinal bacteria include GI epithelial cell proliferation, energy capture, and production of metabolites; the latter, in particular, also can result in detrimental health effects.152
By-products of microbial metabolism may be either beneficial or detrimental. When included with probiotics, by-products are intended to support beneficial organisms or ameliorate negative effects of by-products. However, organisms themselves are chosen for their anticipated by-products. Among the organisms most commonly cited as beneficial are Lactobacillus and Bifidobacterium spp. Major sources of food for microbes include resistant starches; dietary fibers such as cellulose, hemicellulose, pectin, and inulin; and unabsorbed sugars and sugar alcohols. Substrates also include dietary protein (host or diet) and endogenous materials such as pancreatic enzymes, GI secretions, the mucoid layer, and sloughed GI epithelial cells. By-products of carbohydrates are formed primarily in the proximal colon; products include monosaccharides, disaccharides, and oligosaccharides. Further metabolism of these by-products results in short-chain fatty acids (including butyrate), as well as lactic, succinic, and formic acid. Butyrate is the major fuel source of colonic epithelial cells. However, butyrate purportedly also reduces the risk of colon cancer and inhibits proinflammatory cytokines. Intestinal microbes may metabolize other compounds into active or inactive metabolites, which may be subsequently absorbed. Examples include flavonoids, isoflavonoids and plant lignans.152 Benefits of microbial fermentation of plant products include the production of enterolignans associated with estrogenic and antioxidant effects. Oxalobacter formigenes transforms oxalates; its absence increases the risk of oxalate stones in humans.152 The production of potentially beneficial by-products often requires cooperative actions among different populations of microbes.
Bacterial by-products have a number of adverse effects. Protein by-products, produced primarily in the distal colon, include ammonia and amines resulting from deaminations; these products are associated with procarcinogenic effects (e.g., nitrosamines). Cysteine and methionine degradation yields sulfides, which inhibit colonic use of butyrate. Anaerobic colonic fermentation of aromatic amino acids tyrosine to phenols and tryptophan to indoles and their subsequent metabolism yield several procarcinogenic compounds. Many of these determinental compounds also play a role in hepatic encephalopathy. Bacterial deconjugation and dehydroxylation of bile acids contribute to their enterohepatic circulation; other compounds conjugated by compounds such as taurine, glycine, and sulfate may likewise be recycled. The general effect of recycling is longer exposure to potentially toxic compounds.
Probiotics as Therapeutic Agents
The science behind the use of probiotics is profoundly complicated by the unknown nature of the normal microbiota; the variables that influence it, including diets, diseases, and other patient factors such as breed, age, or gender; a lack of understanding of the pathophysiology of targeted disease; and issues regarding the quality of the products. Further hampering our understanding are the studies themselves. A variety of in vitro and in vivo models have been used (as reviewed by de Vrese and Schrezenmeir160 and Lenoir-Wijnkoop and coworkers151), but poor scientific design mars the credibility of many.
KEY POINT 19-35
Evidence for the beneficial effects of probiotics will be difficult to establish because of the impact of multiple (including unknown) variables on response.
In humans probiotics have been demonstrated to have an impact on a large number of potential health problems (Table 19-4).151 Although the data were drawn from humans, a review of those indications relevant to dogs and cats is reasonable; this discussion is limited to those indications for which sufficient evidence exists to support potential to probable therapeutic benefit. This review summarizes human reviews of well-designed clinical trials.151,161,162
Table 19-4 Examples of Probiotic Indications for Treatment of (Human) Disease161
Gingival disease
Theoretically, probiotic microbes able to adhere to dental tissues occupy spaces that otherwise would be occupied by pathogens. For example, dairy products containing Lactobacillus or Bifidobaceriumorganisms might compete with cariogenic microbes such as salivary Streptococcus mutans or others. Several clinical trials have demonstrated a reduction in the number of dental caries in humans who ingest probiotics containing Lactobacillus.151
Antibiotic-induced diarrhea
Diarrhea as a side effect of selected commonly used antibiotics may reflect colonic overgrowth of Clostridium difficile or other organisms. Strategies for treatment (discontinuation of the inciting antibiotic, immediate retreatment with a new antibiotic) are not always successful, and one of the most commonly selected antibiotic in humans (oral vancomycin) to treat overgrowth of Clostridium organisms tends to be associated with adverse events. Although other antibiotics may be as effective as vancomycin, use of probiotics is a reasonable approach.
Based on a review of 10 clinical trials in close to 2000 children ranging in age from less than 1 year up to 18 years of age, probiotics were beneficial in the prevention of pediatric antibiotic-associated diarrhea.162 Probiotic strains that showed the most promise included Lactobacillus GG, Lactobacillus sporogenes, and Saccharomyces boulardii when 5 to 40 billion colony-forming units were administered daily. Other organisms studied included members of Lactobacillus spp., Bifidobacterium spp., Streptococcus spp., and S. boulardii alone or in combination, including other treatments that might prevent antibiotic-associated diarrhea. The impact of age (e.g., infants versus older children) or duration of antibiotic therapy could not be assessed. Probiotics tended to be well tolerated after 2 to 12 weeks of therapy. Although the review concluded that routine treatment of probiotics for the prevention of pediatric antibiotic-associated diarrhea could not yet be recommended, further studies are warranted.
Infectious diarrhea
Probiotics also appear to be beneficial when used as an adjunct to rehydration fluids in the treatment of acute, infectious diarrhea in adults and children, although further research is indicated to determine the most effective probiotic and dosing regimen. In contrast, the use of probiotics as an adjunct to antibiotic therapy for C. difficile colitis could not be supported by a review of relevant clinical trials. However, the number of studies was small.
Liver disease
A review of clinical trials studying the use of probiotics for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis found insufficient evidence to support or refute treatment. However, the use of probiotics and synbiotics to support liver function or treat liver disease has been reviewed by Lenoir-Wijnkoop and coworkers151 and is discussed with regard to the management of liver disease.
Hyperlipidemia
Several species of Lactobacillus may bind to cholesterol in the presence of bile acids and low redox potential.161
Inflammatory bowel disease
Because of the complex relationship between the intestinal microbiota and the immune response, the use of probiotics as adjunct therapy for treatment of IBD is reasonable.163 Probiotics have been used successfully to reduce Helicobacter pylori colonization in humans; severity of mucosal inflammation also is reduced in a mouse model. Clinical trials investigating the efficacy of probiotics in the treatment of H. pylori often are conflicting, ranging from ineffective as sole therapy to effective if used in conjunction with standard therapy antimicrobial and antiulcer therapy. One study in humans (n = 138) found that equal numbers of Lactobacillus acidophilus, Lactobacillus bulgaricus, Streptococcus thermophilus, and Bifidobacterium lactis (total of 109 organisms/mL) combined with quadruple therapy (amoxicillin, metronidazole, bismuth subcitrate, and omeprazole) eradicated infection in 85% of humans compared with 71% receiving quadruple therapy alone (as reviewed by Lenoir-Wijnkoop and coworkers151).
Probiotic therapy with conventional therapy does not appear to provide any benefit compared with conventional therapy alone in patients with mild to moderate ulcerative colitis. Although limited evidence suggests that probiotics may reduce clinical signs, support was not sufficient for recommendations. Likewise, evidence was not sufficient to support a role for probiotics in the treatment of Crohn’s disease.
Probiotics do not appear to be effective for prevention of allergic diseases or food hypersensitivity in pediatric patients.
Urinary tract infection
The use of probiotics in the treatment of urinary tract infections in human patients was reviewed by Lenoir-Wijnkoop.151 GI bacteria are a major source of urinary tract infections. As in animals, those causing UTI in humans are predominantly caused by uropathogenic E. coli, gram-negative organisms, and Enterococcus faecalis. In general, Lactobacillus sp. is the most common probiotic recommended for treatment of UTI. However, selected species are likely to emerge as more effective than others. A meta-analysis of 25 studies in human medicine found probiotics (lactobacilli) to be of benefit in treatment of bacterial vaginosis. However, a clear benefit could not be demonstrated for UTI (five studies); conclusions were difficult to draw because of limitations, including differences in study design and limited sample size.163a
Oxalate urolithiasis
The use of probiotics in the treatment of renal oxalate stones in human patients was reviewed by Lenoir-Wijnkoop and coworkers.151 The absence of O. formigenes from fecal microbiota increases the risk of kidney stones. Both animal and human studies have documented that O. formigenes is able to become established in the GI tract, and establishment reduces urinary oxalate concentration. At least two studies have demonstrated a potential benefit of probiotics containing O. formigenes in the prevention or treatment of oxalate crystalluria.164,165 Weese and coworkers166 demonstrated that fecal samples containing lactic acid bacteria and several bacteria isolated from dogs or cats degraded oxalate in vitro. The addition of selected prebiotics (e.g., guar gum) increased the degradation.166
Allergic diseases
The use of probiotics in the treatment of allergic diseases (including atopic allergic disease and asthma in human patients) was reviewed by Lenoir-Wijnkoop and coworkers.151 The rationale for efficacy reflects the immunomodulatory effects of probiotics. Based on clinical trials in humans, response has been limited to younger patients with severe disease. Similarly, probiotics have had only a limited, if any, effect on allergic rhinitis and asthma in humans.
Nosocomial infections
A meta-analysis examined the impact of probiotics on the prevention and treatment of nosocomial C. difficile–associated diarrhea and hospital-associated pneumonia in humans.167 Although the prevention of antibiotic-induced diarrhea was a recognized beneficial effect, no evidence was found to support the theory that probiotics prevent nosocomial infection; clinical trials were recommended.
Irritable bowel syndrome
A meta-analysis168 identified the limitations of drawing conclusions given differences in study design, including organisms, doses, and the definition of irritable bowel syndrome (IBS). Clinical efficacy was reported in several studies, with improvement manifested as decreased bloating, gas, and pain. Lactobacillus spp. and Bifidobacterium were targeted treatments.
Prebiotics (as opposed to probiotics) also have been associated with therapeutic benefits in humans with effects extending beyond local (GI) sites. The list is not much different than that for probiotics. Those conditions for which scientific evidence supports the potential use of prebiotics include allergies, immunomodulation (gut-associated lymphoid tissue as well as cellular Th1/Th2 ratio) that improves resistance to infections, constipation, irritable bowl syndrome, mineral metabolism (particularly strengthening of bone), prevention of cancer, and treatment of H. pylori infections. In addition, prebiotics have been scientifically associated with weight loss, presumably as a result of increased satiety mediated through GI suppression of ghrelin. Feed conversion efficiency has been described in food production animals. Interestingly, fermentation of prebiotics purportedly increases bacterial biomass in which nitrogen is fixed, thus decreasing the ammonia load in patients with hepatic encephalopathy.
Scientific data regarding the use of biotherapeutics in dogs and cats are slowly emerging. The use of prebiotics and probiotics in dogs has been reviewed by Rastall.154 L. acidophilus DSM 13241 fed (2 × 109 colony-forming units daily per dog) in 15 healthy dogs decreased the number of culturable Clostridum spp. and was associated with an increase in indices indicative of immunomodulation (increased serum IgG and monocytes and decreased plasma nitric oxide). Prebiotics that have been studied154 include lactosucrose and fructooligosaccharides. In their report, the authors noted that most prebiotics are enzymatically synthesized and that the technology can be modified such that enzymes are obtained from the target microbe, thus yielding highly specific probiotics.
Feeding 1.5 g lactusucrose daily for 2 weeks to healthy dogs (n = 8) increased bifidobacteria and decreased Clostridium spp. In another study, Bifidobacterium and Lactobacillus organisms also increased, and Clostridium and Enterobacteriaceae organisms decreased in cats receiving 0.75 g daily. A decrease in toxin levels and odor also was described for both dogs and cats. Fructooligosaccharides fed at a rate of 4 g/day to adult health dogs (n = 20) increased Bifidobacterium and Lactobacillus organisms and decreased Clostridium spp. Although both lactate and butyrate increased, ammonia, dimethylsulfide, and hydrogen sulfide also increased. Fructooligosaccharides at a rate of 0.75% in the diet of healthy adult cats for 2 weeks decrease Clostridium spp. and E. coli. Rastall154 also described the formulation of a symbiotic containing Lactobacillus organisms cultured from one dog, following the tradition of probiotics being based on microbes isolated from the target species. Among the isolates cultured, Lactobacillus mucosae, L. acidophilus, and Lactobacillus reuteri were subjected to prebiotic carbohydrates. Baillon and coworkers169 prospectively studied the viability of L. acidophilus added to a dry dog food at a concentration of 10(9) colony-forming units. Dogs (n = 15) were fed control and treated diet for 2 weeks each. Supplementation of the diet resulted in fecal recovery of organisms during but not after feeding of the treated diet, indicating colonization had occurred. The number of clostridial organisms was decreased.
Probiotics might comprise wild-type microbes associated with natural microbiota or genetically mutated forms of otherwise pathogenic but normal microbiota. Different strains of probiotic bacteria are presumed to impart differential effects, with variability representing habitat preferences of the microbe, specific capabilities of the microorganism, and differential enzymes. The number of colony-forming units ingested as probiotics is minor compared with the normal microbiota.151 However, they travel through regions of the GI tract that are sparsely populated and therefore may transiently become the dominant microbe, potentially muting pathogens as a result of competitive exclusion. Rinkinen and coworkers170 demonstrated the ability of lactic acid bacteria to competitively inhibit adhesion of selected canine pathogens (including C. perfringens) in vivo using isolated canine jejunal mucosa. However, Enterococcus faecium actually facilitated adhesion and colonization of Campylobacter jejuni, supporting the need for controlled clinical trials before assumptions are made regarding the impact of probiotics.
Probiotics and related compounds are not approved drugs and undergo no premarket approval with regard to efficacy, safety, or quality. Data, if available, represent efforts of the manufacturer or the scientific community. Consumer Laboratories (www.consumerlab.com) reviewed issues specifically related to quality assurance of a probiotic product. Many of these factors should be addressed on the label, including the following:
KEY POINT 19-36
Because probiotics and related compounds are not approved products, they undergo no premarket approval with regard to efficacy, safety, or quality; extra care should be taken to find a high-quality product.
Consumer Laboratories might also be solicited to identify superior products. Of 27 products (23 human, 4 pet) reviewed by Consumer Laboratories (as reviewed by the author January 2010), 10 (9 human, 1 pet) failed to contain the labeled amount of microbes and 5(2 human, 3 pet) failed to provide at least 109 colony-forming units per serving (although dogs and cats may require less than 1 billion). Marked variability was found in the dose delivered among pet products. No product failed as a result of microbial contamination (with mold). Weese and coworkers166 also examined the quality of probiotics, including eight veterinary products. The contents of only two of thirteen products matched labeled descriptions, with five veterinary products not providing specific content information. Some products did not contain all labeled products while some products contained additional bacteria, some of which were potentially pathogenic. Organisms in several products were not viable. These studies emphasize the importance of selecting a probiotic that meets the standards of quality assurance reviews.