Chapter 4 Drug-Induced Diseases
Definitions and Predisposing Factors
An adverse drug event (ADE) is any harm caused by a therapeutic or preventive (or diagnostic) intervention (Figure 4-1). The evolution of the definition in human medicine during the past decade has led to further subdivision into medication errors and adverse drug reactions (ADRs).1 Medication errors are ADEs that result from a mistake made by the caregiver, including but not necessarily limited to administration of the wrong drug, dose, interval, or route to the wrong patient. Thus, ADEs are iatrogenic in origin. A potential subcategory of medication errors might be failure to modify a dosing regimen for recognized patient, drug, or disease factors that lead to inappropriately low or high drug concentrations at the site of action.
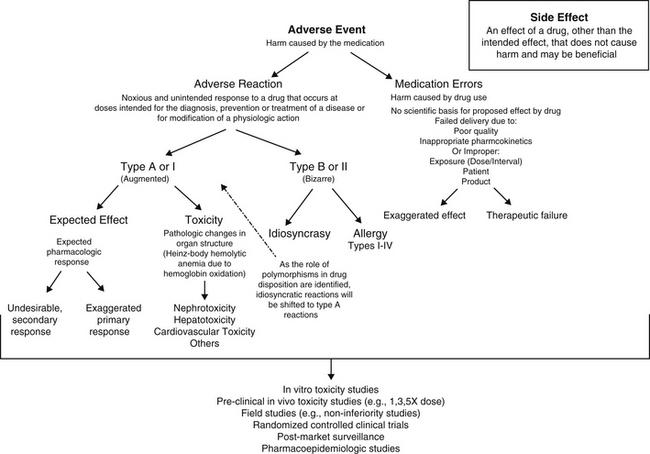
Figure 4-1 Flow chart delineating the definitions of adverse drug events (ADEs), including medication errors and adverse drug reactions (ADRs). ADEs can result in either an exaggerated response or failed response. In contrast to ADR, which implies harm to the patient, side effects do not cause harm and in some cases, may be beneficial. The risk of toxicity or ADR is assessed at several points before and during the approval process, with postmarket surveillance being perhaps the most comprehensive.
In contrast to medication errors, an ADR is a noxious and unintended response to a drug or other medication that occurs at a dose given with the goal of achieving the intended effect of the medication. As such, the term ADR implies a reaction that might cause serious harm to the patient and reflects a patient response to the inherent properties of the drug. An ADR may reflect a pharmacodynamic response or a pharmacokinetic effect. An ADR should be contrasted with the term side effect, which refers to an effect other than the intended effect that does not cause harm. A side effect may not be undesirable and may, in fact, be desirable or inconsequential to the health of the patient. For the purposes of this discussion, the term ADE will be used to refer to ADR with or without an iatrogenic basis. Adverse drug reactions can be further classified as either type A (type I) or type B (type II).2-4 Type A (“augmented”) adverse events generally result from drug concentrations at the site (generally estimated by plasma drug concentrations [PDCs] that either exceed the maximum or drop below the minimum therapeutic range (see Chapter 1). If the clinician is familiar with the drug and the patient, type A reactions are largely predictable and, as such, avoidable. Like type A, side effects are also generally predictable and dose dependent and for the purposes of discussion, will be included with type A adverse events in this chapter.
Generally, type A reactions are manifested as an exaggerated but normal or expected pharmacologic response to the drug (see Figure 4-1).5 This response may be the desired response (e.g., bradycardia in a patient receiving propranolol to slow a sinus tachycardia) but also may reflect an unwanted, secondary response resulting from the drug’s pharmacologic effects (e.g., bronchospasms induced by the beta-blockade effects of propranolol) (Figure 4-2). Some drugs also cause adverse events unrelated to their pharmacologic response. These reactions usually reflect damage to target cells and are referred to in this chapter as cytotoxic adverse reactions. Cytotoxic adverse reactions are exemplified by nephrotoxicity induced by aminoglycosides (Figure 4-3) or hepatic necrosis or methemoglobinemia induced by acetaminophen. Often, it is the metabolite of the drug rather than the drug itself that causes cytotoxicity (see Chapter 2). In such cases, drugs that induce metabolism, particularly in the liver (e.g., phenobarbital, phenytoin; Figure 4-4), may increase the risk of toxicity, whereas drugs that decrease metabolism may reduce the risk of toxicity (e.g., cimetidine).6-8 Cytotoxic drug reactions might be treated with drugs that scavenge radical metabolites (i.e., N-acetylcysteine, a glutathione precursor).
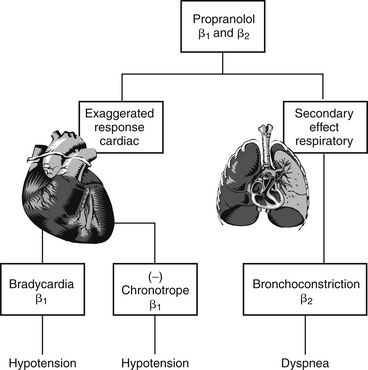
Figure 4-2 Type A drug reactions resulting from overdose. Propranolol offers an example of a type A adverse reaction that reflects both an exaggerated primary response (bradycardia, decreased contractility) and an undesirable secondary response (bronchospasm).
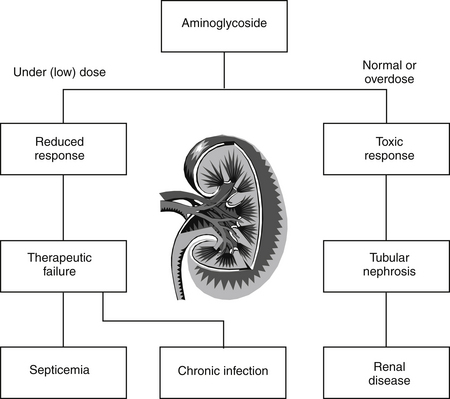
Figure 4-3 Underdose and cytotoxic response. An aminoglycoside such as gentamicin can cause two examples of a type A adverse reaction/therapeutic failure, meaning effective antimicrobial concentrations are not achieved, and a cytotoxic reaction, manifested as nephrotoxicity in response to persistent (rather than very high) plasma drug concentrations.
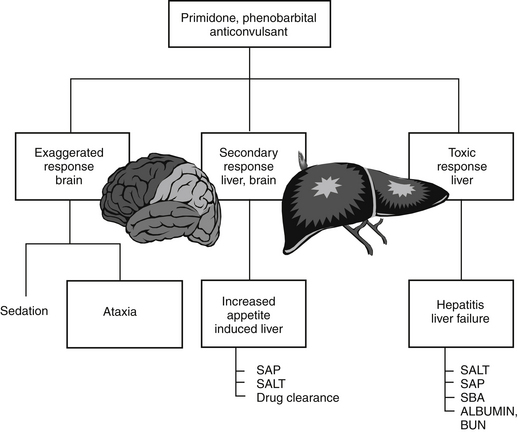
Figure 4-4 Exaggerated and cytotoxic responses. Anticonvulsants metabolized by the liver exemplify a type A adverse reaction manifested as cytotoxicity (hepatic disease). These drugs also can cause an exaggerated (but expected) response (sedation) as well as a secondary undesirable response or side effect that is not necessarily considered severe or life threatening (increased appetite, polyuria, and polydypsia). BUN, Blood urea nitrogen; SALT, serum alanine aminotransferase; SAP, serum alkaline phosphatase; SBA, serum bile acid.
In contrast to type A events, type B (“bizarre”) events are not dose or concentration dependent. As a result, these reactions are not predictable and are largely unavoidable. They occur only in a small percentage of the population receiving the drug; in human medicine they account for approximately 6% to 12% of all ADRs.9 Generally, their incidence—indeed their existence—often is not documented until the drug is in wide use. In addition, because their cause is not well understood, treatment is generally limited to symptomatic therapy. Examples of type B adverse reactions include drug allergies or idiosyncrasies. Many of the idiosyncrasies eventually may be shown to be genetically or otherwise based (e.g., polymorphisms in transport or drug-metabolizing proteins [ivermectin toxicity of Collie-related breeds]), but the cause has yet to be identified and thus the reaction cannot be predicted. As with type A events, type B events may occur in response to the parent drug or its metabolite. This chapter will focus on allergies as type B ADRs.
This chapter discusses the mechanisms and clinical signs of the adverse events caused by selected drugs and methods by which the events might be avoided or treated. Adverse events or side effects that result from the expected pharmacologic action of a drug (e.g., exaggerated pharmacologic effect) are also discussed with each drug in subsequent chapters and generally are not emphasized here. This chapter focuses more on type A than type B events because they are recognized more commonly. The list of drugs included is by no means complete but represents those drugs most commonly recognized, as well as the addition of some that are often overlooked. ADEs are also addressed in chapters that address the use of the drug.
The impact of ADEs on the monetary and health costs in human medicine were assessed in the late 1990s. The implementation of a voluntary, anonymous reporting system has facilitated medication error reporting in human medicine and thus enabled a more accurate assessment of their impact.10 The majority (66%) of medical errors occurred during transfer of patient care within hospitals, and another 19% occurred on discharge. In a human intensive care unit (ICU) environment, 1 out of every 5 doses of medication was associated with an ADE as a result of medication errors. The veterinary profession has yet to implement a mechanism whereby ADEs due to medication errors can be assessed. Yet needs are equally applicable to veterinary medicine.10
Organs most susceptible to type A drug events usually are those subjected to the greatest exposure or concentration of the drug. Thus the organs with the greatest blood flow and those organs capable of drug concentration, such as the liver and kidney, are the most vulnerable to systemic drugs. Highly metabolically active organs are also more likely to manifest toxic effects for two reasons. First, such organs depend on the presence of energy, and anything that impairs acquisition of energy (including blood flow) can lead to malfunction. Second, if the metabolic activity includes metabolism of compounds, the production of potentially reactive metabolites can increase the likelihood of cytotoxicity if these metabolites interact with cellular structures. In contrast to Type A ADEs, the organs most susceptible to damage by type B reactions tend to be the organs that contain tissues that act as haptens for drug-induced allergy (e.g., skin, blood-forming units) or tissues that filter and trap immune complexes (e.g., glomerulus, joints). Organs containing a preponderance of mast cells also are more likely to manifest immune-mediated reactions (i.e., “shock organs”; see the section on allergic reactions). A summary of compounds causing predominantly type A drug reactions and, when available, their antidotes, can be found in Appendix 5. A variety of factors can influence the likelihood of adverse, and particularly toxic, reactions.6-8 Factors that predispose a patient to the development of type A adverse events are discussed in Chapter 2.
Not all adverse reactions are clinically evident. Sometimes the reaction is not detectable unless actively sought. For example, clinical laboratory tests may detect a drug-induced hepatotoxicity (e.g., increased serum alanine transferase activity) that is otherwise clinically silent. Many drugs can directly interfere with or indirectly influence clinical laboratory tests, including endocrine function testing (discussed later).11 Organ predisposition to drug-induced toxicity may show diurnal variations. For example, both aminoglycosides and cisplatin12 exhibit increased renal toxicity in humans when administered in the evening as opposed to in the morning. For the aminoglycosides, safety in the morning has been attributed, in part, to the increase in glomerular filtration rate that occurs in the morning;13 an increased sensitivity to interleukin-6–induced inflammation has been suggested for cisplatin.
Principles of Toxicology Relevant to Pharmacology
Terminology of Toxicity and Safety Assessment
Toxicology refers to the study of poisons and their effect on living organisms.14 A poison is any substance that is injurious to animals; the term is synonymous with toxic substance, toxic chemical, and toxicant. It is important to note that any drug can become a poison (“the dose makes the poison”), and the toxic response to the poison tends to correlate with the dose (or duration). Toxic response refers to the effects manifested by an organism in response to a toxic substance. Toxicity describes the quantitative amount or dose of a poison that produces a toxic response or effect. Acute toxicity generally results from a single dose or exposure or multiple doses in a 24-hour period. Most acute toxicants rapidly interfere with critical cellular processes. Subacute and subchronic toxicity occurs after 1 week to 1 month of exposure. Chronic toxicity occurs after 3 or more months of exposure.14 These latter terms are generally applied to humans, but the relative duration of exposure applies equally to animals. Toxic chemicals can act directly by injuring the cells with which they come in contact or indirectly by injuring a group of cells that subsequently precipitate injury to others. Alternatively, toxins can act indirectly by interfering with a physiologic process on which a group of cells are vitally dependent. Toxins can act systemically (the majority), locally, or a combination of the two.15
An indicator of the assessment of toxicity is the LD50, or the dose per unit weight of a chemical that kills 50% of animals receiving it (the median lethal dose) (Figure 4-5). The LD50 is both drug and species specific. The lethal dose of a drug can be compared with the dose necessary to induce the desired pharmacodynamic response (effective dose, or ED) in a targeted percent of the sample population (see Chapter 1). The ED50 is the dose expected to induce the response in 50% of the population; its more useful companion indicator is the EC50, which describes the concentration at which 50% of individuals respond, thus removing the effects of drug absorption and distribution from consideration. Like the LD50, ED50 and EC50 are generally based on single dosing and do not take into account duration of exposure. The therapeutic index offers a measure of the relative safety of a drug by comparing the dose producing toxicity to that causing the desired effect, or the ratio of the LD50 to ED50 (see Figure 4-5). A related term is margin of safety which is the ratio minimal lethal dose (LD01) and the dose associated with the greatest efficacy (ED99). Caution is recommended if the margin of safety (MOS) is less than 1. Alternatively, rather than measure lethality as the adverse event, the concentration necessary to cause a specific adverse event can be measured and used to calculate the therapeutic index. A second set of terms by which the safety of a compound might be assessed are the lowest observed adverse effect level (LOAEL) and no observed (adverse) effect level (NOEL, or NOAEL). The LOAEL is the lowest concentration of drug associated with observed adverse effects. The NOAEL is defined as the greatest concentration of a compound that causes no detectable adverse effect. When compared with anticipated drug intake, it provides an alternative margin of safety. The absence of these effects can be used to help verify the NOAEL. The NOAEL generally is based on chronic exposure and thus may be more relevant for drugs administered at more than a single dose. NOELs and LOELs (no or lowest observed effect level, respectively) differ in that they may not indicate an adverse effect, but in fact may address a beneficial effect. All four terms are important to risk assessment which addresses exposure and the adverse outcomes that might be associated with exposure (see later discussion).
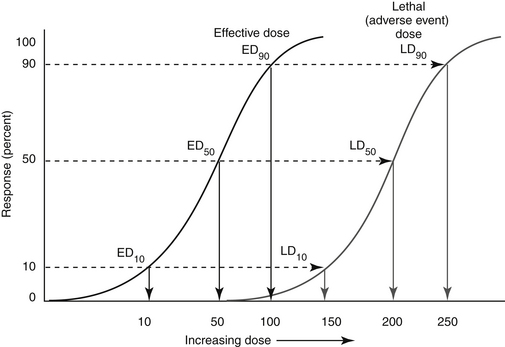
Figure 4-5 The dose necessary to cause the response in 50% of animals tested is referred to as either the lethal dose50 (dose necessary to cause death; LD50 or 200 mg/kg) or effective dose50 (dose necessary to cause targeted pharmacodynamic response; ED50 or 50 mg/kg), respectively. However, to induce the pharmacodynamic response of interest in the target species would require a dose of 50 mg/kg. The therapeutic index for this hypothetical drug would be the LD50/ED50 or 200/50 = 4.
Other toxicologic terms relevant to drugs include teratogen, which is a compound that causes abnormal fetal development. It is important to note that teratogenicity is not the only form of toxicity that may occur in utero; any toxic effect that occurs in the adult is likely to occur in the developing fetus as well. Carcinogenesis refers to the ability of a compound, or a carcinogen, to cause cancer. Cancer cells are cells that have been able to avoid the sophisticated mechanisms that control normal growth, development, and division. Induction of cancer by a compound involves many variables, including duration, dose, and frequency of exposure. Generally, carcinogens take 20 years or more to induce cancer, and the cause-and-effect relationship between the compound and the cancer often is not recognized.15,16 Although some compounds can directly interact with DNA, leading to a cancerous cell, most compounds must first be converted to a reactive metabolite to covalently bond with DNA. Damage may still be avoided if DNA repair occurs before cell division. Cellular damage increases the stimulus for division of adjacent cells, leading to a new cell type with new genotypic and phenotypic properties that can then be transformed to a malignant cell under the correct conditions. A number of compounds are recognized to be initiating agents, targeting molecular DNA, whereas others are considered promoters, acting to increase the incidence of cancer or decrease the latency period without interacting with DNA. These latter compounds must be administered repeatedly and after the initial insult. Endogenous compounds such as growth factors or hormones may act as promoters.15
Many compounds can induce cancer in laboratory animals when they are exposed to extremely high (supratherapeutic) doses for prolonged periods of time. Rarely do these compounds cause cancer in humans, and it is even more unlikely that they will do so in companion animals, in part because the life expectancy of companion animals generally is too short to allow emergence of cancer. Lifestyle changes that increase the risk of drug or toxicant-induced cancer in humans are likely to have the same effect in animals as well. These include but are not limited to exposure to cigarette smoke, exposure to charcoal-cooked food, chronic consumption of alcohol (unlikely in animals), and consumption of foods, many of which contain possible carcinogens.
Two mechanisms by which drugs can cause cell death are apoptosis and necrosis, each with distinct morphologic and biochemical characteristics. Apoptosis is an active process characterized by cell shrinkage, nuclear and cytoplasmic condensation, chromatin fragmentation, and phagocytosis. In contrast, necrosis is a passive process resulting in inflammation associated with cellular and organelle swelling, rupture of the plasma membrane, and spilling of cellular contents into the extracellular milieu.17 Because apoptosis is an active process, sufficient intracellular energy must be maintained; depletion of adenosine triphosphate (ATP) may cause an apoptopic process to become a necrotic process. Not surprisingly, mitochondria appear to play a role in apoptosis. A number of toxicants cause their effects by disrupting mitochondria function and thus ATP production. However, several drugs appear to exert their toxicologic effect through induction of apoptosis (e.g., digoxin, selected chemotherapeutic agents).17
Genetic diversity is increasingly being identified as a cause for individual variation in response to toxins and drugs, leading to the fields of toxicogenetics and pharmacogenetics, respectively.5,16,18 The role of polymorphic cytochrome P450s as a cause of interindividual differences in xenobiotic metabolism and drug toxicity is fairly well established. In humans polymorphism occurs less commonly in those cytochrome P450s responsible for carcinogen activation (CYP1A1 and 2, CYP1B1, CYP2E1, and CYP3A4) compared with those that are primarily responsible for drug metabolism (CYP2C9, CYP2C19, CYP2D6, and CYP3A4). Polymorphisms in drug-metabolizing enzymes have been associated with drug hypersensitivities.6 All hepatic drug-metabolizing P450s are polymorphic with the clinically most important polymorphism in humans occurring with CYP2C9, CYP2C19, and CYP2D6.19 Polymorphisms in cytochrome P450s of animals is now being elucidated but may play a role in adversities in certain breeds (e.g., Beagles, Greyhounds). In addition, polymorphism in P-glycoprotein (P-gp) is a well-established reason for susceptibility to adverse drug events for selected drugs in Collie-related breeds (see Chapter 2).
Postmarket Surveillance
The approval process for human-marketed drugs is designed to identify adverse reactions at several stages preclinical phase (short- and long-term animal testing): through phase III (extended clinical trials) and continuing into phase IV, which includes a mandated postmarket surveillance. However, the drug approval process for animals is not as regimented (see Figure 4-1). The requirements by the Food and Drug Administration’s Center for Veterinary Medicine tend to vary with the drug undergoing approval (See Appendix 1).20 In general, directed toxicity studies that are implemented during the approval process involve a small number (e.g., 2 to 30 but most commonly 4 of each sex) of study animals receiving the drug under conditions of exaggerated use—that is, doses (e.g.,1, 3, 5× the highest labeled dose,) and durations (e.g., 3×) of several magnitudes greater than the anticipated approved intended use. Field trials implemented during the approval process are performed in the approved species, generally as controlled (placebo or positive), randomized clinical trials, under conditions of intended use. Such studies generally involve a much larger sample population (e.g., hundreds of animals) than the directed toxicity studies. However, although toxicity studies do predict a number of adverse events that occur in the target species,21 neither directed toxicity nor field studies are likely to involve a sample size sufficiently large to allow detection of many adverse events that occur in a very small proportion of patients (e.g., 1 in 1000 or more). Detection of such an adverse event is likely to require thousands of subjects. Further, directed studies are generally performed in a sample population of generally healthy animals unaffected by the complexities of host, drug, and disease factors associated with drug use in the target population (see Chapter 2). As such, postmarket surveillance studies and pharmacoepidemiologic studies (which focus on subgroups of target species) are particularly important to the safety assessment of approved drugs used in animal patients.21 For the same reason, if an ADE is suspected as a result of drug therapy, the importance of reporting it (or suspicion of it) cannot be overemphasized. Note that safety assessment of human-marketed drugs are studied in dogs but with an emphasis on human rather than animal safety. Although all approved human-marketed drugs are likely to have been studied in dogs during the preclinical phase, most of this information is not generally available unless specifically requested through Freedom of Information Act mechanisms. If an ADE is suspected, it first should be reported to the manufacturer; by law, animal drug pharmaceutical companies must report adverse events reported in animals to the Food and Drug Administration (FDA). The veterinary profession, probably more so than the human-medicine community, is less likely to report adverse event perhaps because the mechanism by which the information is forwarded to the veterinary health care provider is limited in both scope and distribution. In contrast to drugs, adverse event reporting for animal dietary supplements is entirely voluntary, with no obvious mechanism for reports to be collected, assessed, or returned to the veterinary profession. It is only through postmarket surveillance that more clinically relevant assessments of drug safety can emerge, and subtle differences in the safety of different drugs might become apparent. Assessments include hazard or risk assessment, which express the probability of harm under conditions expected with use of the drug. Hazard is the potential to cause harm—that is, the inherent toxic nature of the compound. Risk is the likelihood that harm will occur. As such, it takes into account both hazard and exposure—that is, the amount of the toxin ingested and thus dose and duration.22 Risk–benefit analysis compares the risk associated with the use of a product and its potential benefits.23 As such, a risk/benefit ratio addresses the acceptability of an adverse reaction by taking into account the importance, frequency, and duration of therapeutic benefit and the adverse reaction. Risk–benefit analysis generally is based on postmarket use of a product and a mechanism of surveillance that detects and assesses adverse events.24
Among the difficulties in assessing risk through postmarket surveillance is the attribution of the adversity to the drug. Information submitted in an ADE report is limited, and care must be taken to not assume a cause–effect relationship The FDA’s Center for Veterinary Medicine has recently reviewed its ADE reporting program.25 It defines an ADE as an undesired or lack of desired response to a drug, medical device, or (in food animals) medicated feed. As such, a distinction is not made between adverse events associated with medication errors and those associated with inherent properties of the drug. In its 2004 report, the FDA indicates that a 6-point scoring system evaluates drug reactions submitted from manufacturers, pet owners, or veterinarians. Information collected includes previous experience with the drug (i.e., historical evidence of adversities from the label information or previously submitted reports), timing of the event in relation to dosing, alternative causes, the role of overdose, and effects of dechallenge or rechallenge. Plans for improved submission include a web-based submission process.
The FDA website (http://www.fda.gov/AnimalVeterinary/SafetyHealth/ProductSafetyInformation/ucm055375.htm; accessed May 2010) can be reviewed for yearly and cumulative adverse event reports that have been reported in animals. Unfortunately, at the time of publication, the reports provide no evidence of frequency of occurrence, other than a ranking. This decision was made in part because of the inability of the FDA to standardize the number of adverse events by the number of doses administered (or units sold).
Predicting Drug Safety
Woodward21 has demonstrated that, to some degree, studies implemented during the approval process tend to predict adversities that emerge in target species, but with marked limitations. Guengerich26 has discussed the role of predictive toxicology based on drug chemistry in assessing clinical safety of drugs. Toxicities are likely to be predictive if the drug is characterized by a high level of intrinsic toxicity; for such drugs chemistry may often predict toxicity. Toxicity is less predictable, although still reasonably so if metabolism plays a role, but is much less predictable if the toxicity is idiosyncratic (i.e., type B or II).
Idiosyncratic Reactions: Allergic Drug Events
The clinical manifestations of idiosyncratic ADRs vary with the type of reaction and the body system targeted. Generally, for allergies, previous exposure to the drug must occur regardless of the type of reaction, or therapy must have been sufficiently long (i.e., at 10 to 14 days for some drugs) for an allergic response to develop. However, exceptions appear to exist, as is exemplified by allergy-based reactions to sulfonamides, which may occur in as early as 5 days (see Chapter 7). Drugs generally are too small in molecular size (<1000 D) to be sufficiently antigenic. As such, drugs that induce an allergic response generally act as haptens, covalently combining with a body tissue that then also becomes antigenic. As a result, the allergic response may be directed toward the drug or tissue.9 The hapten hypothesis is controversial because only a small percentage of persons develop a reaction, possibly because of a failure to develop tolerance. Failed tolerance may occur at one or more proposed sites. First, the role of metabolism in the formation of chemically reactive metabolites as the initial step in mediating idiosyncratic drug responses (including allergies) is increasingly supported by scientific studies. Those individuals that produce more metabolites appear to be more likely to fail to develop immune tolerance to a drug.9 For the same reason, the dose of antigen exposure may determine the type of response. The role of CYP enzymes in idiosyncratic reactions (whether or not allergy based) is increasingly being recognized. Second, the metabolite must bind with an appropriate ligand, one with a high epitope density sufficient to induce an immune response. Third, an allergic response to a drug requires activity of an antigen-processing cell. As such, it may be more likely in the presence of inflammation, such as might be the case with a viral or bacterial infection or after stress or traumatic damage. Molecular signals that activate immune cells may also be more prevalent in the face of large concentrations of reactive metabolites because of their ability to induce oxidative stress.9 Fourth, other factors that may determine emergence of an allergic response (i.e., failed tolerance) may include failed downregulation of regulatory factors. A balance toward protective t-helper (Th1 or 2) response may preclude emergence of an allergic response; a loss of balance may facilitate it. Accordingly, both genetic predisposition and environmental factors contribute to emergent drug allergies.9
Type I allergic reactions (immediate or anaphylactic) are IgE mediated and result from the release of chemical mediators (e.g., histamine, serotonin, eicosanoids) from tissue mast cells or basophils. The reaction occurs within minutes after drug administration regardless of the dose administered (Figure 4-6). Clinical manifestations generally include nausea, vomiting, circulatory collapse, tachycardia, pulmonary edema, and neurologic signs. Urticaria and angioedema may also be evident. Clinical signs may be species dependent, depending on the shock organ of the species. The shock organ generally is the organ in which mast cells occur in greater numbers. In the dog the shock organs tend to be the liver and gastrointestinal tract; in the cat the shock organ generally is the lung, with fulminating pulmonary edema being a clinical manifestation.
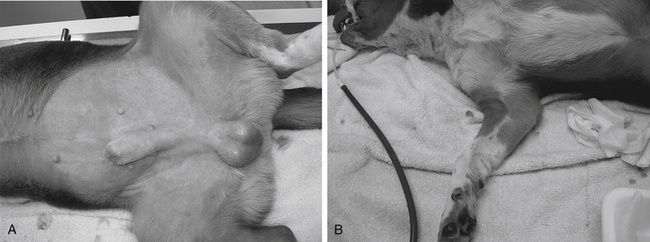
Figure 4-6 An example of dermatologic manifestation of either a type 1 allergic hypersensitivity or an anaphylactoid reaction. A, The former would involve antigen formation; B, whereas the latter would reflect a direct drug-induced histamine release from cutaneous mast cells. Skin lesions appeared 15 minutes after the puppy (undergoing an elective castration) received a preanesthetic dose of hydromorphone.
(Courtesy Harry W. Boothe.)
The exact antigen that causes anaphylaxis may not be known. For example, even though anaphylaxis in microfilaremic dogs after administration of microfilaricides is well documented, the specific antigen released by the effect of the drug is not known.27 When given to microfilaremic dogs, both dimethylcarbamazine and ivermectin can induce shock manifested as peripheral vascular collapse, dyspnea, bloody diarrhea, and other clinical signs and laboratory test results consistent with anaphylaxis.
Treatment of drug-induced anaphylaxis is directed toward prevention of the physiologic response to mediator release (i.e., epinephrine and antihistamines) and prevention of further histamine release (e.g., epinephrine and glucocorticoids; possibly antihistamines). Ideally, antihistamines might also include the newer classes that may decrease mast cell degranulation as well as block H1 receptors associated with histamine-mediated shock. Interestingly, and perhaps disconcertingly, glucocorticoids themselves have been associated with anaphylactic reactions (see Chapter 30).28 Supportive therapy is also indicated. Prophylactic pretreatment in cases of anticipated anaphylaxis helps decrease the manifestations of anaphylaxis by decreasing the mast cell response. Drugs associated with type I allergic reaction in humans include penicillins, angiotensin-converting enzyme inhibitors (particularly in the first 3 weeks of therapy), nonsteroidal antiinflammatories drugs (NSAIDs), and opioids. However, the latter drugs may actually be more associated with an anaphylactic-like reaction, also referred to as an anaphylactoid reaction.
An anaphylactoid reaction is very similar to anaphylaxis but differs in that it is not mediated by an antigen-IgE response and thus is not allergic or immune mediated. Rather, selected drugs or compounds cause direct mast cell degranulation. Generally, these drugs are cationic (basic) and include opioids (particularly morphine [see Figure 4-6], polymyxin, radiographic contrast agents, thiacetarsamide, and amphotericin B). Hyperosmolar solutions such as mannitol can also cause direct mast cell degranulation. Adverse response to rapid intravenous administration of enrofloxacin, which is both hyperosmolar and basic, may represent an anaphlylactoid response or may simply reflect (in cats) stimulation of the chemoreceptor trigger zone. However, ciprofloxacin has been associated with an allergic response in humans,29 and the author is aware of at least one dog treated with ciprofloxacin for which the adverse reaction met the World Health Organization category of “probable” ADR. Anaphylactoid reactions tend to be related to dose. As such, administration of a small test dose may help the clinician determine the likelihood of occurrence. Response is probably more likely with intravenous administration. In addition to the prophylactic measures for anaphylaxis, decreasing the rate of drug administration may help reduce the risk of the adverse response.
Type II reactions (cytotoxic) occur as antibody-bound blood cells become lysed and are removed from circulation. Lysis results from direct binding by either IgG or IgM. Complement may or may not be activated. Either stem cells in the bone marrow or mature circulating cells may be targeted. Targeting red blood cells, leukocytes, and platelets results in, respectively, hemolytic anemia, agranulocytosis and leukopenia, thrombocytopenia, or any combination thereof.
Type III drug reactions (immune complex disease, or serum sickness) are induced by antigen–antibody complexes involving either IgG or IgM and complement activation. Circulating antigen–antibody complexes may be filtered by and lodged in the vasculature of a number of organs, including the kidney, central nervous system (CNS), or peripheral vasculature. Clinical signs generally refer to the predominant organ affected but also include fever and lymphadenopathy. The Arthus reaction is a variation of the type III reaction and is manifested as swelling and pain at the site of drug administration. Among drug reactions in veterinary medicine, the potentiated sulfonamides are probably the most well-recognized cause of type III immune-mediated drug reaction.30 Type IV drug reactions (delayed hypersensitivity, cell mediated) reflect cellular response at the site of the antigen. Lymphocytes and macrophages infiltrate the site and cause mediator release that perpetuates the inflammatory response.
The list of drugs that cause each type of drug-induced allergy is long and probably will remain incomplete. Although some drugs are more likely to cause a specific type of allergic reaction, any drug that causes allergy probably can cause any type of allergy, affecting any body system. Eventually, studies of structure–chemistry relationship involving metabolites and assessment of the immune system’s response might allow identification of the patient at risk of developing an allergic response. Diagnosing an allergic (or any adverse) drug reaction can be very difficult and generally requires dechallenge (i.e., removal of the drug) and rechallenge. Clinical signs generally occur more promptly if the episode reflects reexposure to a previously administered allergen. The ethics of rechallenge (i.e., risk to the patient) may not justify confirmation of a presumed diagnosis. Peripheral eosinophilia and skin lesions often accompany an allergic drug response. ADRs in each of the body systems that have an allergic basis should be noted as such when possible.
Drug-induced allergy can be life threatening. Vasculitis and serum sickness are more likely to become life threatening when the kidney, liver, gastrointestinal tract, and nervous system become involved. Angioedema is life threatening if mucosal edema threatens ventilation.
Among the drugs recognized to be associated with drug allergies in dogs are the sulfonamides. Many medications contain a sulfonamide (a sulfur dioxide [SO2] and nitrogen [N] moiety), including sulfonamide antimicrobials (derivatives of sulfanilamide in which the sulfonamide is attached to an aryl amine; e.g., sulfamethoxazole, sulfadiazine, sulfadimethoxine), “coxib” cyclooxygenase-1–sparing NSAIDs (e.g., deracoxib, firacoxib), carbonic anhydrase inhibitors (e.g., acetazolamide), diuretics (e.g., hydrochlorothiazide, chlorthalidone, furosemide), uricosurics (e.g., probenecid), drugs to treat inflammatory bowel disease (e.g., sulfasalazine), sulfonylureas (e.g., glyburide, glipizide), and selected anticonvulsants (e.g., zonisamide).31 However, it is likely that a metabolite associated with the nitrogen moiety is responsible for the reaction, with reactions being limited to molecules containing an aryl-amine (both the sulfur and an amine moiety are attached directly to a benzene ring; sulfonylarylamine), a structure limited to sulfonamide antimicrobials. The underlying pathophysiology, clinical manifestations, and other aspects of the response have been well described32,33 and are reviewed in Chapter 7.
Adverse Drug Events by Body Systems
Drugs causing adverse events in the various body systems are listed in tabular form in the respective sections on body systems. Discussion of the adverse reaction also can be found in the appropriate chapter. When available, treatments are offered, including tabular presentation (see Appendix 5).
Liver
The liver is vulnerable to drug-induced toxicity for several reasons (Box 4-1).34-37 The potential for hepatotoxicity can be enhanced by dietary imbalance (high fat, low protein), presence of disease concurrent with administration of drugs that alter hepatic drug-metabolizing enzymes or hepatic blood flow,37 and age.38
Box 4-1 Hepatic Drug-Induced Toxicity
The liver is vulnerable to drug-induced toxicity for several reasons:
Drug-induced and chemical-induced liver injury have been classified into two categories.34,35,37 Type I toxins, or intrinsic hepatotoxins, cause type A adverse events, which are predictable, dose and time dependent and occur in most, if not all, subjects exposed to appropriate doses of the substance. Any drug metabolized by the liver probably can cause some degree of type I hepatic disease simply by the production of phase I metabolites, which as a general rule tend to be toxic because of their reactivity. The longer the drug is used and the higher the dose, the more likely the ADE will occur. Type II, or idiosyncratic hepatotoxins, cause type B events, which are unpredictable and dose and time independent (consistent with Type B). Their occurrence is sporadic and not reproducible.
Drug-induced hepatotoxicity (Box 4-2) is associated with a wide range of histologic changes, from acute, reversible, and clinically benign lesions to those that cause fatal massive necrosis, chronic hepatitis, or malignancy.34,35 Some drugs characteristically cause only a single lesion, whereas others cause multiple lesions. The lesions caused by any drug are rarely specific for that drug but can be caused by a variety of drugs or other disorders, often limiting the potential usefulness of biopsy (Figure 4-7).
Box 4-2 Examples of Drugs or Drug Classes Associated with Hepatotoxicity∗†
∗ Many other drugs that are metabolized by the liver are potentially hepatotoxic because of the production of phase I reactive metabolites.
† Microscopic lesions tend to be centrolobular in location, associated with necrosis, but otherwise nonspecific for most drugs.
‡ In the author’s experience, marked increase in serum alkaline phosphatase may occur but is not necessarily associated with hepatic dysfunction.
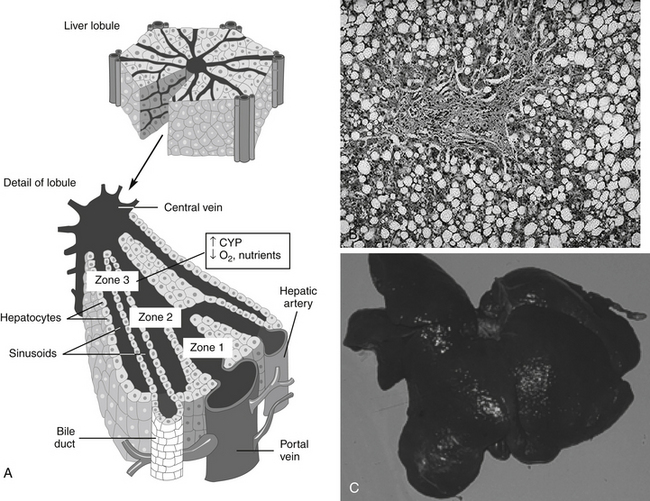
Figure 4-7 A, Drug-induced liver disease is generally nonspecific in presentation. Among the more frequent lesions are necrosis, particularly centrolobular, because hepatocytes in this region (zone 3) contain the most drug-metabolizing enzymes (i.e., production of toxic metabolites) yet receive the least oxygen. Hemorrhage and vacuolization are also common lesions. B, In the persistent presence of the toxin (including drug), the liver will continue to progress to irreversible changes, including the deposition of fibrous tissue as part of the cirrhotic process. C, A liver from a dog that died as a result of end-stage liver disease associated with phenobarbital concentrations above 50 μg/mL for 2 months. Clinical pathologic findings progressed from normal to indicative of end-stage disease within a 3-month period.
(A from Cunningham CC, Van Horn CG: Energy availability and alcohol-related liver pathology, Alcohol Res Health 27(4):281-299, 2003.)
Frequently, drug-induced hepatic injury is limited to select regions or zones (e.g., central, middle, or peripheral) in the lobule.34,35 Various histologic lesions associated with drug hepatotoxicity have been described.34,35 Zonal necrosis usually results from type I or predictable toxins. The production of toxic metabolites may be an important cause of zonal necrosis because drug-metabolizing enzymes predominate in zones most likely to develop necrosis. In most cases of acute injury, the process is either fatal or completely resolved. If exposure is chronic or recurring, however, the lesions may persist and progress, depending on the dose, agent, and health of the patient. Lipid accumulation, usually of triglycerides, may be associated with either minimal alteration of hepatic function or with both clinical and laboratory manifestations of liver dysfunction.
Nonspecific hepatitis is seldom associated with serious or progressive hepatic decompensation or failure and is fully reversible after discontinuation of the drug. Chronic hepatitis usually requires continued exposure and is not the result of self-perpetuation of an acute lesion. In general, prompt and complete resolution of this lesion can occur after timely discontinuation of therapy with the inciting drug. Cirrhosis generally requires prolonged or repeated exposure to the toxin. Silent cirrhosis is a term used to describe the gradual evolution of liver disease to cirrhosis without any clinical illness. Although methotrexate is among the most implicated drugs in humans, it is likely that many drugs that cause progressive liver disease do so “silently” for a long time.
Drug-induced cholestasis is not well understood. Drugs can target bile ducts or canaliculi, causing primarily cholestasis without hepatocellular disease. When accompanied by an inflammatory infiltrate, systemic illness usually occurs, whereas cholestasis without inflammation is associated with no or very mild clinical signs. Recovery usually occurs after discontinuation of drug therapy.34,35 Drugs can also affect primarily sinusoidal or endothelial cells, causing primarily fibrosis or veno-occlusive disease. Veno-occlusive disease tends to be predictable and is most commonly associated in people taking anticancer drugs. An immune basis has been recognized for some drugs causing clinical signs consistent with chronic active hepatitis.
Treatment of drug-induced liver disease is primarily supportive. Because reactive metabolites often either cause or exacerbate disease, however, use of compounds that help prevent metabolite damage to the liver should be considered. Specific examples include N-acetylcysteine, a precursor to intracellular glutathione; ascorbic acid, another type of oxygen radical scavenger; S-adenosylmethionine (SAMe), a compound that contributes to a number of methylation reactions in the body; and the herbal agent silymarin (see Chapter 19). Care must be taken to ensure that the duration of treatment exceeds the duration of activity of the toxicant, as has been demonstrated for N-acetylcysteine for treatment of acetaminophen toxicity.39
Hepatotoxic Drugs
Inhalant Anesthetics
Adverse events to inhalant anesthetics are unusual in veterinary medicine,40,41 in part because duration of anesthetic exposure is limited. Historically, methoxyflurane administration in dogs has occasionally been associated with acute centrilobular necrosis accompanied by a mixed inflammatory infiltrate. Halothane-associated hepatic injury in the dog has not been confirmed, although a clinical case report has described acute hepatic necrosis after its use. In humans the degree and incidence of halothane-induced liver damage do not appear to correlate with the duration or number of exposures and therefore has been suggested to reflect an idiosyncratic hypersensitivity.
Mebendazole and Oxibendazole
The bendazole anthelmintics have been associated with liver pathology. Acute centrilobular hepatic necrosis and fatal fulminating hepatitis have been reported in dogs after the clinical and experimental administration of the anthelmintics mebendazole and oxibendazole.42,43 Clinical signs were evident in as few as 2 days or as many as 10 to 14 days after administration. Although mebendazole was originally thought to be an intrinsic hepatotoxin, other studies suggest that it produces an idiosyncratic reaction.
Sulfonamides
Sulfonamide antimicrobials are associated with toxicity of multiple organs, including the liver.30,32,33,44 Sulfonamides do not appear to differ in their likelihood of causing toxicity. In one report that supports an idiosyncratic reaction, the duration of therapy before hepatotoxicity developed ranged from 4 to 30 days, and the dose ranged from 18 to 53 mg/kg every 12 hours.44 The mechanism of sulfonamide toxicity is discussed in more depth in Chapter 7.
Thiacetarsamide and Melarsomine
Thiacetarsamide (caparsolate) is associated with hepatic injury in humans and animals. Chronic exposures in humans are more likely to cause clinically significant hepatic disease. Hepatotoxicity is, however, a common complication of acute administration of thiacetarsamide for heartworm disease in dogs, although residual effects after therapy is completed are not expected (see Chapter 14). In normal animals melarsomine causes less hepatotoxicity and renal toxicity than thiacetarsamide.45
Bile Acids
Bile acids are hepatotoxic, and they contribute to the development of hepatitis in patients with cholestasis, regardless of the origin. Bile acids are also used therapeutically as choleretics. Among the bile acids present endogenously and used therapeutically, however, those that are lipid soluble (e.g., deoxycholic acid) are more hepatotoxic than those that are water soluble (e.g., ursodeoxycholic acid). Ursodeoxycholic acid rather than deoxycholic acid should be used for therapy. Bile acid therapy should be discontinued in the event of cholecystectomy.
Xylitol
The sugar alcohol xylitol is associated with life-threatening hypoglycemia and hepatic necrosis in dogs. Most cases, reported by the American Society for Prevention of Cruelty to Animals (ASPCA) Poison Control Center, reflect over ingestion of products containing xylitol as a sweetener (a small to average cookie may contain approximately 4 g xylitol). In contrast to humans, xylitol induces a ten-fold increase in insulin secretion in dogs compared to an equivalent amount of glucose. The result is a precipitous drop in serum glucose within 30 to 60 minutes after ingestion of as little as 100 mg/kg. Onset of hypoglycemia may be offset for 12 hr if xylitol-containing gum is ingested. Lethargy, ataxia, collapse, and seizures may occur. Clinical pathology may also reveal hypokalemia as potassium moves into the cell with glucose, and hypophosphatemia as a result of insulin’s effects on cell permeability.45a,b The impact on cats is not clear. More recently, hepatic necrosis has been reported in dogs ingesting xylitol. Enzyme increases within 12 to 24 hours after ingestion of 500 mg/kg or more are followed by clinical signs and sequelae, including coagulopathies consistent with acute hepatopathy. Hyper- rather than hypophosphatemia associated with acute hepatopathy may be a poor prognostic indicator. Sorbitol does not appear to be associated with toxicity in dogs.
Kidney
Like the liver, the kidney is vulnerable to drug-induced toxicity for several reasons (Boxes 4-3 and 4-4).46 Specific cellular or subcellular sites of nephrotoxins frequently are not known. Usually, a toxin affects more than one type of renal tissue because of the high drug concentrations to which the kidney is exposed. The glomerulus is susceptible to direct nephrotoxicity as well as to indirect toxicity such as that caused by immunologic injury.46 Many nephrotoxins cause predominantly proximal tubular damage. This is expected in part because blood flow is greatest in the renal cortex, where the proximal tubules are located. Variations in proximal tubular susceptibility to toxins may reflect different tubular functions.46,47
Box 4-4 Renal Drug-Induced Toxicity
The kidney is vulnerable to drug-induced toxicity for several reasons:
A study in human medicine focused on the use of nephrotoxic drugs in the critical care patient.48 Reductions in renal blood flow associated with hemodynamic responses to a myriad of illnesses predisposes the ICU patient to acute renal failure which occurs in 6% of human ICU patients. Prolonged use of vasopressors increases the risk of renal hypoxia. Additionally, of note, the use of low-dose dopamine (≤3 μg/kg/min) as a nephroprotectant, particularly in patients with acute renal failure (representing at least 6% of ICU patients) was discouraged. Although renal vasodilation and urine flow increase, outcome does not improve, and the increased risk of cardiac or other adversities balances any potential nephroprotection.48 NSAIDs were cited as a particular risk in ICU patients; newer drugs that target COX-2 do not appear to offer an advantage in regard to nephrotoxicity. Nephrotoxicity induced by NSAIDs in the critical care environment occurs rapidly and is manifested as a rapid increase in serum creatinine. If an NSAID must be used in the ICU patient, one with a short half-life is recommended, and use of other nephroactive drugs (those that alter renal blood flow) are discouraged.48 The ICU patient also is at increased risk for aminoglycoside toxicity, with urine enzymes the earliest indicator. Clinical evidence of nephrotoxicity occurs within 5 to 10 days of therapy; once-daily (or less frequent) therapy reduces the risk. Likewise, amphotericin B often is associated with acute renal dysfunction. Sodium-containing fluids and lipid-based products are recommended in the patient at risk. The use of nacetylcysteine to protect the kidney should be considered for selected drugs.
Nephrotoxic Drugs
Most nephrotoxic drugs are discussed in relevant chapters. Methoxyflurane causes a dose-dependent, high-output nephrotoxicity in humans. Toxicity appears to be the result of oxalate metabolites and inorganic fluoride. Oxalate metabolites crystallize in and obstruct the tubules, whereas inorganic fluoride produces tubular necrosis.42 Veterinary reports of methoxyflurane-induced nephrotoxicity are rare, probably because veterinary patients are at a reduced risk of developing nephrotoxicity because exposure (surgery) times are much shorter than in humans.49
Trivalent arsenicals such as thiacetarsamide denature proteins by binding to sulfhydryl groups. The glomerulus is often the first site of arsenical-induced nephrotoxicity, but proximal tubule damage predominates, probably because of the large number of enzymes that are denatured in this region.46 Initial proteinuria is followed by tubular necrosis and degeneration.
Gastrointestinal
Stomatitis may progress to ulcerations with several drugs, particularly antineoplastic agents. Those most likely to cause stomatitis are listed in Table 4-1. A number of drugs are sufficiently caustic that ulcerations occur if the drug remains in contact with the mucosa (see Table 4-1). A number of drugs, including doxycycline and other drugs administered orally as a tablet have been associated with local mucosal damage and subsequent esophageal strictures in cats (see Chapter 19).
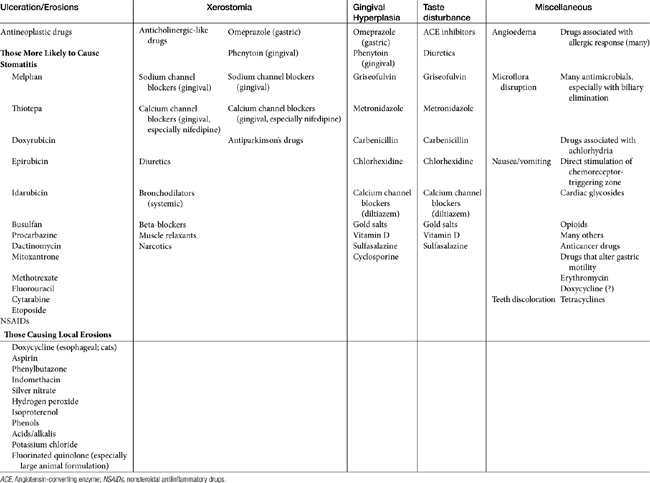
Although not an ADE, several drugs can cause tooth discoloration. Among the most recognized are tetracyclines, which chelate to calcium of either dentin or enamel, resulting in a yellow to brown discoloration. Oxytetracycline causes the least discoloration. The effect occurs during tooth development and is one reason that tetracyclines should not be administered to pregnant animals. The time that must lapse postpartum is not clear in animals. (It is up to 8 years of age in children.) Minocycline can cause discoloration despite animal age, probably as a result of chelation of iron resulting in insoluble complexes. Oral iron solutions can cause transient superficial discoloration of teeth, which can be removed. Other compounds associated with tooth discoloration in humans include isoproterenol, ciprofloxacin (a greenish-yellow discoloration when used in infants), and chlorhexidine (reversible yellowish-brown stains when used as a mouth rinse for more than several days).
Xerostomia (dry mouth) has been associated with anticholinergics and drugs with anticholinergic-like effects (see Table 4-1), as well as other drugs. Taste change is more discernible in humans and is caused by a number of drugs, including several antimicrobials (see Table 4-1).
Most orally administered drugs are probably capable of causing nausea or vomiting simply as a result of irritation or stimulation of the gastrointestinal tract mucosa. Erythromycin, for example, is a prokinetic agent and, as such, may cause upset in up to 50% of animals taking the drug. A patient with disease of the gastrointestinal tract is predisposed to these side effects. Many intravenous drugs also cause nausea or vomiting, particularly if given rapidly because of stimulation of the chemoreceptor–triggering zone. A number of drugs are recognized for their tendency to stimulate this zone regardless of the route of administration. Examples include digoxin, anticancer drugs, and most opioids.
Gingival hyperplasia has been reported for several drugs, including phenytoin and (in humans) calcium or sodium channel blockers.50 The risk of its emergence might be reduced with good dental hygiene; in dogs it responded to metronidazole therapy.51 Gingival hyperplasia is a recognized side effect of cyclosporine administration in dogs and cats (package insert, Atopica); 31% of the dogs developed gingival hyperplasia and gingivitis, which responded to metronidazole and spiramycin.51 However, the more common effect of drugs on the oral mucosa is erosion. A number of drugs cause direct erosion with prolonged contact, especially in the feline esophagus (Table 4-1). Any drug that is antianabolic or inhibits cellular division is potentially toxic to the gastrointestinal tract by impairing the rapid turnover of epithelial cells in the mucosa (see Table 4-1). Tetracyclines and chloramphenicol are antianabolic, although long-term administration is necessary before these drugs affect the gastrointestinal tract.
Anticancer chemotherapeutic drugs best exemplify drugs that decrease epithelial cell turnover. Among the drugs most commonly causing gastrointestinal disease in veterinary medicine are the NSAIDs. These drugs inhibit prostaglandins, which in the gastrointestinal tract mucosa serve to inhibit gastric acid secretion, stimulate bicarbonate and mucus production and epithelialization, and increase blood flow. Among the NSAIDs most likely to cause gastrointestinal tract ulceration are aspirin, which also directly irritates the gastrointestinal tract mucosa, and ibuprofen, whose therapeutic range in the dog appears to be higher than the toxic range. Selected antimicrobials alter the microflora of the gastrointestinal tract and can subsequently cause diarrhea. Achlorhydria induced by a number of drugs can lead to gastrointestinal upset by changing microflora.
Torpet50 has reviewed oral side effects of cardiovascular drugs in humans. The list of possible lesions is impressive, as are the number of drugs associated with lesions, which suggests that the oral mucosa (at least in humans) is sensitive to the effects of many drugs. Lesions include taste disturbances (diuretics, angiotensin-converting enzyme inhibitors), xerostomia (e.g., alpha-agonists and beta or calcium channel blockers, angiotensin-converting enzyme inhibitors), gingival overgrowth (calcium and sodium channel blockers) and ulcerations, as well as a number of syndromes associated with cutaneous lesions indicative of drug allergies, including angioedema (many).
Nervous System
Because of the brain’s role in integrating the body, toxic injury to one of its areas can result in manifestations from another site. Likewise, drugs that cause injury to other systems can result in CNS damage caused by metabolic changes (e.g., hypoglycemia, hypoxia). The high metabolic rate of neurons and their marked need for nutritional support render this system more susceptible than others to damage.15 Neurons are uniquely dependent on the cell body to provide support for the dendrites and axons; the axon, which is devoid of metabolic function, depends on axonal transport for supplies to meet its metabolic needs. Drugs or chemicals that interfere with axonal transport ultimately lead to axonal atrophy.15 The CNS is also uniquely lacking in effective regenerative capacity. Lesions of CNS damage therefore persist, leading to additive effects after subsequent exposures to a toxic compound, as well as delayed manifestations when neuronal reserve can no longer compensate for the abnormalities. Some toxicities may not occur until age-related attrition of neurons causes decompensation, thus prolonging the time between cause and effect and decreasing the likelihood that the relationship between exposure and neurotoxicity will be recognized.15
The blood–brain and blood-CSF barriers limit the incidence of adverse events in the CNS. Increased permeability of this barrier, however, such as might occur in pediatrics or disease, predisposes animals to CNS events. All CNS-active drugs are likely to cause CNS signs if the dose is too high. Drugs that can induce seizures in epileptic patients, and should therefore be avoided, include butyrophenones, metoclopramide, tricyclic antidepressants, and reportedly (although little literature supports this fact) glucocorticoids (Table 4-2). The impact of phenothiazines on seizure activity is less clear (see Chapter 27). A number of antimicrobials can cause seizures. The fluorinated quinolones have received some attention for their possible CNS side effects and, in particular, potentiation of seizures. The mechanism of action appears to be inhibition of gamma-aminobutyric acid–receptor interactions and may (although this has not yet been proven) be facilitated by the presence of NSAIDs.52 High doses are therefore discouraged, particularly in predisposed patients. Several other antimicrobials are associated with CNS toxicity in people.53 These include the beta-lactams, with imipenem and cefazolin being the most epileptogenic (see Table 4-2). Metronidazole also is associated with CNS adverse effects. Clinical signs of metronidazole toxicity in the dog include ataxia, nystagmus, and stumbling. Signs may not occur for 7 to 12 days after therapy is begun (see Chapter 7). Seizures may take up to 2 weeks to resolve; therapy is supportive. Signs may be more dramatic in the cat, including seizures and blindness. Toxicity has been reported at doses as low as 30 mg/kg every 24 hours.54 Seizures respond to diazepam therapy. NSAIDs are associated with CNS side effects, particularly if combined with fluoroquinolones with unsubstituted piperazinyl rings (ciprofloxacin) at position 7.55
Table 4-2 Examples of Drugs Associated with Adverse Drug Reactions in the Central Nervous System
| Drug | Manifestation |
|---|---|
| Beta-lactams (cefazolin) | Hyperexcitability, depression, aggression, seizures |
| Amitraz | Sedation, ataxia, muscle weakness |
| Aminoglycosides | Neuromuscular blockade |
| Antidepressants | Hyperexcitability, depression, aggression, seizures, ataxia |
| Antihistamines | Sedation, excitement |
| Benzyl alcohol | Hypersynthesis, ataxia, aggression, depression, coma (cat) |
| Beta-lactams (cefazolin and imipenem) | Lowered seizure threshold, ataxia |
| Bismuth | Lethargy, somnolence |
| Butyrophenones | Lowered seizure threshold |
| Enrofloxacin | Seizures, exacerbated by coadministration of NSAIDs; dizziness |
| Erythromycin | Seizures, others |
| Glucocorticoids | Lowered seizure threshold with long-term therapy |
| Griseofulvin | Ataxia, seizures |
| Hexachlorophene | Neuropathy |
| Ivermectin | Depression, lethargy, seizures, others |
| Lidocaine | Seizures |
| Metoclopramide | Hyperexcitability, lowered seizure threshold |
| Metronidazole | Ataxia, nystagmus, seizures |
| Milbemycin | Depression, lethargy, seizures, other |
| NSAIDs | Nonseptic meningitis (naproxen), exacerbation of seizures caused by fluorinated quinolones |
| Opioids | General CNS depression |
| Phenobarbital | Hyperexcitability, depression |
| Phenothiazines | Lowered seizure threshold |
| Quinolones | Seizures, other |
| Sulfonamides | Aseptic meningitis |
| Vincristine | Neuropathy |
| Nitrofurantoin | Peripheral neuropathies |
CNS, central nervous system; NSAIDs, Nonsteroidal antiinflammatory drugs.
The CNS toxicity of the avermectins, including ivermectin, selamectin, moxidectin, and milbemycin, results from enhancement of gamma-aminobutyric acid–receptor interactions.56-58 The role of P-gp deficiency in avermectin CNS toxicity in Collies, Australian Shepherds,59 and related breeds has been well established (see Chapters 2 and 3).60 Doses as small as 100 g/kg can cause toxicity in these breeds. Toxicity will, however, also occur in any animal that is sufficiently overdosed. Clinical signs, which may not occur for 2 or 3 days, include emesis, diarrhea, salivation, fever, disorientation, ataxia, trembling, seizures, depression, coma, and blindness. Although picrotoxin or physostigmine (0.06 mg/kg, slow intravenous administration) have been recommended as an antidote, success is not well documented. Treatment with neostigmine (125 μg twice at 6-hour intervals and then daily for 2 days) along with fluid therapy was associated with success in an adult cat receiving 15 mg (16 times the recommended dose) of ivermectin but was unsuccessful in two kittens, each receiving 7.5 or 15 mg of the drug.61 Picrotoxin is associated with toxicities (seizures), and its use is not recommended unless the patient is comatose. One report cites a dose of 1 mg/min (as a 0.1% dilution in 5% dextrose) given as an intravenous drip until clinical response was evident (8 minutes). Seizures in the patient responded to anticonvulsant therapy.62 Supportive therapy is also indicated. Collies and related breeds with the MDR1 deletion mutation may also be at risk of reaction to other CNS-active drugs that serve as a substrate for P-gp (see Chapters 2 and 3). For example, the MDR1 deletion mutation responsible for the toxicity also has been associated with loperamide toxicity in a Collie receiving 0.14 mg/kg twice daily.63
Amitraz has a number of effects. It stimulates alpha-2 receptors in the CNS and alpha-1 and -2 receptors in the periphery, inhibits monoamine oxidases responsible for synaptic removal of monoamines such as dopamine and norepinephrine, and inhibits the synthesis of prostaglandin E2, although the clinical impact of this latter effect is not clear. Sequelae of β2 adrenergic stimulation includes cardiovascular effects such as bradycardia and vasodilation, leading to hypotension, although peripheral alpha stimulation may cause hypertension. CNS effects of sedation, disorientation, and ataxia may progress to coma. Decreased insulin release results in hyperglycemia. Gastrointestinal effects include gastrointestinal stasis, and therefore atropine or other anticholinergics are contraindicated. Clinical signs of amitraz toxicity may reflect the vehicle, which contains xylene and propylene oxide. Signs of acute xylene toxicosis include CNS depression, ataxia, impaired motor coordination, nystagmus, stupor, coma, and episodes of neuroirritability. Treatment is largely supportive and includes alpha antagonists such as yohimbine (0.1 to 0.2 mg/kg, administered subcutaneously), which are used to reverse alpha side effects. For life-threatening hypotension, positive inotropes should be used cautiously. Control of seizures with diazepam has been contraindicated in the veterinary literature but is supported for treatment of amitraz-induced seizures in children.64
Metronidazole can cause CNS derangements in dogs receiving 60 mg/kg or less; duration is dose and duration dependent. Signs may not occur for 7 to 12 days after therapy is begun (see Chapter 7). Clinical signs including ataxia and nystagmus and seizures may take up to 2 weeks to resolve; therapy is supportive.
The potential for phosphate enemas to induce life-threatening CNS derangements has been well documented, particularly in cats. Toxicity is associated with hyperphosphatemia, hypocalcemia, hypernatremia, hyperglycemia, hyperosmolality, and metabolic acidosis. Onset of clinical signs (ataxia, tetany, convulsions, weak pulse, and hypothermia) is rapid and may rapidly progress to death. Treatment is supportive, including (cautious) calcium therapy. A similar phenomenon has been reported after administration of a phosphate-containing urinary acidifier in cats.65
Benzoic acid (alcohol or benzoate) is a preservative commonly added to oral and parenteral drugs at concentrations of 5% or higher. Benzyl alcohol can cause CNS toxicity (characterized by hyperesthesia and depression), particularly in cats. The drug is rapidly metabolized to benzoic acid and subsequently to hippuric acid and benzyl glucuronide. Glucuronide deficiency in cats results in accumulation of benzoic acid. Pharmacists may not be aware that the glucuronide deficiency of cats predisposes them to toxicity with products containing benzoic acid.66 Although the original dose necessary to induce toxicity in the cat was as high as 2 g/kg, clinical cases and experimental studies indicate that even a lower dose can be lethal. Diets containing benzoic acid at 0.2% to 2% (0.2 to 2 gm/dL) have caused clinical toxicities. The death of up to 30 cats in England66 led to experimental studies that determined the maximum tolerable single dose of benzoic acid to be 450 mg/kg; accumulation with multiple doses limits the highest daily dose to 200 mg/kg.67 For example, a product containing 5% benzoic acid contains 5 g/dL or 50 mg/mL (50 mg/g for dry weight products), limiting a single dose to 9 mL/kg, or a daily dose to 4 mL (4 g/kg). Drugs also can be prepared as benzoate salts, although toxicity is less likely because less benzoate is administered on a mg/kg basis. For example, 40% of metronidazole benzoate is benzoate. A dose of 20 mg/kg delivers 12 mg/kg of metronidazole and 8 mg/kg of benzoate. A daily dose of approximately 500 mg/kg of metronidazole benzoate would be necessary to induce benzoate toxicity in the cat, a dose that would be difficult to achieve even with exceeding the recommended dose of 16 mg/kg metronidazole benzoate twice daily.
Tricyclic and other antidepressants can cause a variety of CNS disorders by virtue of their stimulatory effect on several CNS neurotransmitters and potentially inhibitory effects at other sites. Because these transmitters often modulate the normal physiology of multiple body systems, the clinical manifestations of events to these drugs can be diverse and subtle and affect other body systems. Manifestations related to the CNS include seizures, change in behavior, and depression. Many of the side effects caused in people probably cannot be detected in animals (e.g., blurred vision, dizziness, dry mouth). These drugs have not been well studied in animals, but clinical reports suggest up to 25% of animals may show an adverse reaction to these drugs. Clinical signs include increased or decreased appetite, hyperactivity, polydipsia, diarrhea, anxiety, and fear. The disposition of the drugs in humans includes lipid solubility, hepatic metabolism, and high protein binding, all of which are conducive to drug interactions. Toxicity is enhanced when drugs are used in combination. Because the effects of these drugs take several weeks to be realized, doses may be inappropriately increased, further increasing the risk of toxicity.
Peripheral neuropathies have been associated with a number of drugs. In humans peripheral neuropathy associated with nitrofurantoin occurred at doses ranging from 1.5 to 4.5 mg/kg, with a time of onset ranging from 3 weeks to 12 months. Peripheral neuropathy was severe and irreversible in some patients. The aminoglycosides cause peripheral neuromuscular blockade by interfering with calcium-mediated acetylcholine release. This effect is potentiated in the presence of other neuromuscular blockers and anesthetics.
Special Senses
Ocular Toxicity
Ocular ADRs can reflect local or systemic administration.68 Very occasionally, systemic side effects may result from topical administration of ocular drugs.68 Identification of adverse events affecting the eye (Table 4-3) generally depend on postmarketing surveillance systems or case reports; in some cases (e.g., fluoroquinolones in cats), follow-up toxicity studies may document the causal relationship between drug and adversity.69 Adverse events to drugs manifested in the eye will most likely reflect systemic administration, with the retina being the most common site of reaction. Among the more commonly reported adverse events affecting the eye is acute retinal degeneration associated with exposure of the retina to a light source in patients receiving phototoxic drugs.70 A number of drugs used to treat cardiac disease have been associated with a variety of ocular lesions, including changes (increased or decreased) in intraocular pressure.71
Table 4-3 Examples of Drugs Associated with Adverse Drug Reactions Involving the Eye
| Drug or Drug Class | Example Drugs | Lesion |
|---|---|---|
| Cardiac drugs | Hydralazine | Ocular involvement of systemic lupus erythematosus |
| Beta blockers | Photophobia | |
| Reduced tear production | ||
| Edema, conjunctivitis | ||
| Digoxin | Yellow vision | |
| Amblyopia | ||
| Aurothioglucose | Gold deposits leading to vision deficits | |
| Keratopathy | ||
| Cranial neuropathy | ||
| Retinopathy | ||
| Conjunctivitis | ||
| Methotrexate | Retinopathy | |
| Visual disturbances | ||
| Optic neuropathy | ||
| Chlorpromazine | Blurred vision | |
| Cataract | ||
| Pupillary dysfunction | ||
| Retinopathy | ||
| Corneal epithelial damage | ||
| Accommodation dysfunction | ||
| NSAIDS | Ibuprofen | Blurred vision |
| Naproxen | Photophobia | |
| Piroxicam | Retinopathy | |
| Prednisone | Cataracts | |
| Glaucoma (open angle) | ||
| Proptosis | ||
| Exophthalmia | ||
| Interferon | Ischemic retinopathy | |
| Ethambutol | Changes in visual acuity | |
| Isoniazid | Conjunctivitis | |
| Scleral icterus | ||
| Subconjunctival hemorrhage | ||
| (Coagulopathy) | ||
| Sulfonamides | Keratitis sicca | |
| Tetracyclines | Minocycline | Visual disturbances |
| Tetracycline | Scleral pigmentation | |
| Doxycycline | ||
| Drug or Drug Class | Example Drugs | Lesion |
| Quinolones | Hydroxychloroquine | Multiple: corneal deposits, retinal toxicity, |
| Chloroquine | ||
| Fluoroquinolones | Enrofloxacin | Acute retinal degeneration |
| (in order of risk; cats) | Orbifloxacin | |
| Marbofloxacin | ||
| Ciprofloxacin | ||
| Bisphosphonates | Pamidronate, all others | Episcleritis, nerve palsy, ptosis, neuritis |
| Sildenafil (phosphodiesterase 6 inhibitor) | Changes in color and light perception | |
| Blurred vision | ||
| ERG changes | ||
| Photophobia | ||
| Conjunctival hyperplasia | ||
| Topiramate | Acute narrow-angle glaucoma | |
| Uveitis | ||
| Mydriasis | ||
| Phenylephrine∗ | Topical 10% | Systemic hypertension, may be lethal (use 2.5% instead) |
| Beta blockers∗ | Timolol | Aggravation of bronchospasm, congestive heart failure, bradyarrhythmias, sinus arrest |
| Carbonic anhydrase inhibitors∗ | Sulfonamide-based allergic reactions | |
| Prostaglandins analogs∗ | Travoprost | CNS side effects (malaise, etc.) |
| Anticholinergics∗ | Atropine-like (dry mouth, CNS effects) | |
| Glucocorticoids∗ | Adrenal gland suppression | |
| Chloramphenicol∗ | Aplastic anemia (theoretical) |
ERG, Electroretinography; CNS central nervous system.
∗ Indicates a systemic effect from topical drug. This section of the table is from Gray C: Systemic toxicity with topical ophthalmic medications in children, Paediatr Perinat Drug Ther 7(1): 23-27, 2006.
Among the most notable ADEs manifested as ocular toxicity in animals is retinal degeneration in cats after administration of fluorinated quinolones. Although fluoroquinolones do not appear to cause a similar reaction in humans, reversible (corneal epithelial damage) and irreversible macular changes occur with administration of antirheumatic quinolones, chloroquine, and hydrochloroquine.72 Clinical signs of retinal degeneration associated with fluoroquinolone damage in cats include partial, temporary, or total blindness, with damage generally recognized to be irreversible. Although the incidence is rare, toxicity does appear to be predictable and is associated with a higher incidence in special populations, including geriatric cats and feline patients with renal disease. The mechanism of toxicity appears to reflect a deficiency of an efflux transport protein in the feline retina.∗ Fluorinated quinolones are structurally similar to compounds known to cause ocular toxicosis associated with accumulation in lysosomes of retinal pigment cells; additionally, fluoroquinolones have a predilection for pigmented cells of the eye. The fluoroquinolones also have been associated with phototoxicity. The combination of fluoroquinolone with ultraviolet radiation produces both a time- and concentration-dependent ocular toxicity, with a methyl group at position 8 of the quinolone ring reducing the risk.73 Reducing exposure to sunlight (dosing at night or keeping cats indoors) might be prudent for cats receiving fluoroquinolones, particularly if in a high-risk group.25,69 Drugs associated with fluoroquinolone retinal degeneration are discussed in more depth in Chapter 7.
Ototoxicity
Ototoxic drugs can damage both the auditory and the vestibular apparatus (Table 4-4).74,75 Auditory toxicity is often unrecognized, particularly in the older patient, unless complete deafness occurs. Vestibular ototoxicity might be detected as nystagmus or head tilt. Other clinical signs (e.g., tinnitus) are likely to occur in humans, but these side effects largely go unrecognized in animals. Ototoxic drugs generally are associated with loss of hair cells in the organ of Corti, although the biochemical mechanism is seldom known. Ototoxicity can be either reversible or irreversible and is more likely in the presence of a perforated ear drum if drugs are applied topically.
Table 4-4 Examples of Drugs Associated with Adverse Drug Reactions Involving the Ear∗
| Class | Drug |
|---|---|
| Aminoglycoside antimicrobials | Streptomycin |
| Amikacin | |
| Gentamicin | |
| Netilmicin | |
| Kanamycin | |
| Tobramycin | |
| Other antimicrobials | Polymixin |
| Erythromycin | |
| Colistin | |
| Chloramphenicol | |
| Minocycline | |
| Vancomycin | |
| Antiseptics | Ethanol |
| Benzalkonium chloride | |
| Chlorhexidine | |
| Iodine | |
| Iodophors | |
| Diuretics | Furosemide |
| Cancer chemotherapeutic agents | Cisplatin |
| Nonsteroidal antiinflammatories | Salicylates, acetaminophen, naproxen, others |
| Others | Propylene glycol |
| Detergents |
∗ Most drugs administered topically in the ear may be associated with ototoxicity in the presence of a perforated ear drum.
Aminoglycosides are well known for their ototoxic potential. Aminoglycoside-induced ototoxicity generally is irreversible. In contrast to renal tissues, aminoglycosides are not actively accumulated in perilymph, and drug concentrations generally are less in perilymph than in serum. However, because the half-life of the drug is much longer in the perilymph than in serum, surpassing that in serum by days to weeks, exposure of cells to the drug is longer. Proposed biochemical mechanisms of ototoxicity caused by aminoglycosides include impaired glucose metabolism or inhibition of polyphosphoinositide turnover. Although allowing serum drug concentrations to become undetectable does not necessarily prevent ototoxicity, low trough concentrations as should occur with once daily therapy are none-the-less the best means of preventing ototoxicity. Ototoxicity is enhanced by the presence of loop-acting diuretics such as furosemide. The potential for ototoxicity varies among the aminoglycosides. Dihydrostreptomycin was designed as an alternative to streptomycin, which was vestibulotoxic,76 but dihydrostrptomycin also proved to be significantly cochleotoxic and was subsequently withdrawn. Streptomycin and gentamicin are more likely to cause vestibular toxicity, whereas neomycin, kanamycin, tobramycin, and amikacin sulfate are more likely to cause auditory damage. Among these, amikacin is least cochleotoxic; neomycin is so cochleotoxic (and nephrotoxic) that it cannot be used systemically. Netilmicin, the newest of the aminoglycosides, may cause the least ototoxicity. Auditory damage induced by aminoglycosides is initially characterized by the damage to outer cochlear hair cells, with progressive damage targeting inner hair cells. Initial loss impacts high-frequency hearing.76,77 Topical application of 0.1 mL of 3% gentamicin solution to the tympanic bulla was toxic to sensory receptors of the cochlea and the vestibular apparatus in cats.78 Fluorinated quinolones also may cause ototoxicity when given topically.
Other drugs that cause irreversible ototoxicity include the antineoplastics vincristine and vinblastine. The antineoplastic cisplatin also causes irreversible ototoxicity morphologically similar to that caused by aminoglycosides after accumulation of multiple doses, with the effect being both dose and duration independent.77 Toxicity occurs in the hair cells of the organ of Corti and causes predominantly auditory damage. Cisplatin-induced ototoxicity can be unilateral (and hence not always recognized) or bilateral. Occasionally, ototoxic effects are transient. Chloramphenicol also has been associated with ototoxicity in the cat when applied topically with Gelfoam soaked in a concentration of 400 mg/mL.79 Vestibular toxicity caused by chloramphenicol has been documented in humans.
Vehicles and cleansing agents also may be associated with ototoxicity. Propylene glycol is a common vehicle of topical preparations. Its use is associated with granulation and ossification of the auditory bulla and morphologic changes in the organ of Corti. Some disinfectants (e.g., 0.5% chlorhexidine in 70% alcohol) or carrier agents (e.g., propylene glycol) can cause ototoxicity. These drugs cause both vestibular and auditory side effects. Whereas chlorhexidine can cause almost complete destruction of the vestibular and auditory apparatus (in animal models), 70% alcohol does not appear to cause any ototoxicity. Quaternary ammonium disinfectants (e.g., 0.1% benzethonium or benzalkonium chloride) are among the most ototoxic compounds studied. Iodophors also cause ototoxicity, but damage will not be as profound as with quaternary ammonium compounds. Although the mechanism is not known and the extent of ototoxicity is not clear, ceruminolytic agents should not be applied topically in the presence of a perforated ear drum if the label indicates.
Reversible ototoxic ADEs are unusual. Loop-acting diuretics are among the few ototoxic drugs that cause damage that is largely reversible; however, hearing defects may be permanent. Toxicity is limited to the auditory system and reflects morphologic changes in the stria vascularis of the cochlea. The mechanism of ototoxicity is not clear but may reflect acute electrolyte disturbances in the cochlear endolymph, similar to that producing diuresis.77 Furosemide is the least ototoxic of the loop diuretics; presumably, its combination with aminoglycosides (or other ototoxic drugs) would increase the risk of ototoxicity. Several antiseptics produce irreversible ototoxicity, presumably as a result of cell membrane damage similar to that induced in bacteria.
Aspirin, and potentially other NSAIDs, can cause transient hearing loss in humans. Clinical signs generally resolve in 48 to 72 hours. Several possible sites of damage have been recognized, including a vascular basis (i.e., loss of vasoactive prostaglandins) or impaired neurotransmission. Other drugs known to cause ototoxicity include local anesthetics (0.5% lidocaine can cause cochlear damage), tricyclic antidepressants, and, very rarely, beta-blockers. Although not ototoxic by itself, dimethyl sulfoxide should be used cautiously because of its ability to carry other drugs into the inner ear when used as a vehicle.
Integument
The skin is the organ that most commonly manifests ADEs in humans.80 Although the reactions are generally mild, they can become life threatening. The type of lesion varies and includes almost any type of lesion described for the skin. Lesions include wheal and flare reactions, erythema, blisters, lichenoid lesions, purpura, changes in pigmentation, necrosis, pustular lesions, and changes in hair growth. The most common reactions are erythematous macular or papular rashes that resolve in several days even if untreated. These manifestations may also, however, be a precursor to a severe manifestation and thus should be observed closely. As with many organs, because the skin contains drug-metabolizing enzymes, reactions can be due to either the parent compound or its metabolites (or both).
Drug-induced skin events may be a manifestation of an allergic response or an autoimmune disease mediated by the skin. Both type A and type B events occur in the skin. Of the type B events, all types of allergic events (i.e., types I through IV) can involve the skin. Type IV allergic events are best exemplified by contact dermatitis. “Late” events include allergic vasculitis, purpura pigmentosa, and erythema multiforme. A distinct form of allergic (phototoxic) events has been reported in humans and animals, involving the interaction of a drug (or its metabolite) with ultraviolet radiation; the lesion often manifests in light-exposed skin (discussed later). Fixed drug eruptions are not well understood. In humans they are characterized by erythema, often with a central blister, and may occur because regulation of adhesion molecules in the epithelium is disrupted. Drugs are capable of causing autoimmune reactions in the skin, including lupus erythematosus, pemphigus, and pemphigoid skin lesions. Life-threatening drug-induced events that occur in the skin of people include the Stevens–Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), hypersensitivity syndrome, serum sickness, vasculitis, and angioedema.
Lesions of SJS and TEN may be difficult to differentiate; SJS may be a milder form of TEN. Both resemble scalded skin and reflect a cell-mediated cytotoxic reaction against keratinocytes (Figure 4-8). The diseases are characterized by blistering and extensive detachment of the epidermis. The lesions are irregular in shape and are distinguished from erythema multiforme, another drug reaction of the skin, by the irregular shapes and absence of a well-defined border and edematous ring. Both TEN and SJS tend to affect the trunk, whereas erythema multiforme has an affinity for extremities. Mucous membranes are frequently involved, and patients are generally febrile, particularly with TEN. The presence of neutropenia is interpreted as a poor prognosis in human patients suffering from TEN. Treatment of TEN includes a management protocol similar to that for extensive burns; infection with Staphylococcus aureus (which by itself can cause a “scalded skin” lesion) is likely to complicate therapy. Drug events are the primary causes of SJS and TEN; erythema multiforme is caused by selected microorganisms, as well as by drugs. Drugs associated with TEN and SJS in humans include sulfonamides, anticonvulsants, allopurinol, oxicams, and (less frequently) other NSAIDs.
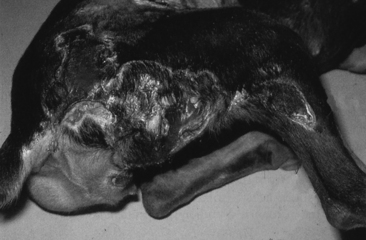
Figure 4-8 Toxic epidermal necrolysis in a Doberman Pinscher treated with chloramphenicol. Although the lesion is several weeks old, damage was evident within several days of intramuscular treatment.
The term hypersensitivity syndrome has been used in the past to refer to any skin ADR. As such, however, it encompasses a wide variety of skin lesions, each treated differently. More recently, the term has been used in human medicine for a syndrome characterized by mucocutaneous eruptions, fever, lymphadenopathy, hepatitis, and eosinophilia. Arthritis or nephritis may also develop. As with SJS and TEN, sulfonamides and anticonvulsants are the most common causes of drug hypersensitivity.
Vasculitis occurs as a result of necrosis and inflammation in blood vessel walls, most commonly in the lower extremities, after antibody interaction with blood vessel walls. Drugs are the primary cause of vasculitis in humans. Penicillins, sulfonamides, thiazide diuretics, phenytoin, and propylthiouracil are the most common interaction-related drugs. Serum sickness–like lesions in the skin also can appear as vasculitis. Lesions result from immune complex deposition in small vessels followed by complement activation and white blood cell infiltration. Lesions appear first as erythema and then progress to more severe eruption. Patients are usually febrile. Drugs that cause serum sickness manifested as dermatologic lesions in humans include selected cephalosporins, minocycline, penicillins, and propranolol.
Dermatologic manifestations of type I hypersensitivities occur at mucocutaneous junctions (including the mucous membranes of the eyes, mouth, nose, lips, or tongue) or present as pruritus, flushing, erythema, and urticaria. Of these, angioedema is the most life threatening because of the risk of upper airway obstruction. Treatment includes epinephrine (for acute respiratory distress), antihistamines, and glucocorticoids.
Skin eruptions have been attributed to a number of drugs in small animals (Table 4-5). Type I skin lesions accompany many cancer chemotherapeutic agents and gold-containing antiarthritic agents. Prednisone and phenytoin can cause alopecia. Most skin reactions reflect type B adverse drug events, however, and as such are largely unpredictable. Alopecia has been reported after oral administration of hetacillin (cat), prednisone (dog), parenteral gold therapy (dog), and phenytoin (dog and cat). Eczematous dermatitis has resulted from oral administration of sulfa drugs (dogs and cats), griseofulvin, diethylcarbamazine, and fluorocytosine. Topical neomycin–triamcinolone preparations and coal tar shampoos can produce generalized eczematous reactions. Generalized exfoliation has resulted from oral administration of quinidine, topical administration of lime sulfur dips, and the use of flea collars. Fixed drug eruptions have resulted from the oral administration of ampicillin and the intravenous administration of sodium thiacetarsamide. Pemphigus vulgaris–like reactions have followed thiabendazole oral therapy, and similar lesions have been reported with gold therapy. Erythematous dermatitis has been reported after the parenteral administration of a phenothiazine derivative. Pruritus has been reported after oral diethylcarbamazine, gold, and bromide (anticonvulsant) therapy. Purpura and lesions typical of TEN have occurred after oral administration of chloramphenicol (see Figure 4-8). Intravenous vitamin K and oral tetracyclines have caused urticaria and angioedema in the dog. Drug eruptions have also been associated with the systemic administration of levamisole. Human recombinant products such as erythropoietin have caused skin or mucocutaneous lesions typical of allergic drug events in dogs.
Table 4-5 Examples of Drugs Associated with Adverse Dermatologic Manifestations
| Drug | Manifestation |
|---|---|
| Ampicillin | Fixed drug eruption |
| Anticancer drugs | Alopecia |
| Bromide | Pruritus |
| Coal tar shampoos | Generalized eczema |
| Chloramphenicol | Purpura, TEN |
| Diethylcarbamazine | Eczematous dermatitis, pruritus |
| Erythropoietin, human recombinant | Skin or mucocutaneous lesions |
| Flea collars | Generalized exfoliation |
| 5-Fluorocytosine | Eczematous dermatitis |
| Glucocorticoids | Alopecia, hyperpigmentation |
| Gold-containing drugs | Alopecia (dog), pruritus, pemphigus vulgaris–like reaction |
| Griseofulvin | Eczematous dermatitis |
| Hetacillin | Alopecia (cat) |
| Levamisole | Drug eruptions |
| Lime sulfur dips | Generalized exfoliation |
| Neomycin (topical) | Generalized eczema |
| Phenothiazine derivatives | Erythematous dermatitis |
| Phenytoin | Alopecia |
| Prednisone | Alopecia (dog) |
| Quinidine | Generalized exfoliation |
| Recombinant products, nontarget species | General skin or mucocutaneous lesions |
| Sulfonamides | Eczematous dermatitis |
| Tetracyclines (oral) | Urticaria, angioedema |
| Thiabendazole | Pemphigus vulgaris–like reaction |
| Thiacetarsamide | Fixed drug eruption |
| Vitamin K (intravenous) | Urticaria, angioedema |
| Cyclosporine | Altered hair coat |
| Many | Other cutaneous manifestations of allergies |
TEN, Toxic epidermal necrolysis
Hormonal therapy is often associated with predictable skin lesions, including bilaterally symmetric alopecia and hyperpigmentation (discussed later in reference to the endocrine system). The effects of glucocorticoids on the skin have been well documented and are often manifested as part of the cushingoid presentation of animals receiving therapy. Cyclosporine has been associated with changes in hair coat.81
Benzoic acid used as a preservative in topical preparations may cause cutaneous erythema, possibly caused by induction of inflammatory prostaglandins.82 Ciprofloxacin was associated in a human patient with the development of erythema multiforme, which histologically was consistent with dermatitis herpetiformis.29 Lesions developed on day 6 of therapy and responded to glucocorticoid therapy.
Photosensitization represents a novel mechanism of toxicity to selected drugs whose treatment occurs during exposure to electromagnetic radiation. Photosensitization occurs if a drug (or biological substrate) is characterized by an abnormally high reactivity to ultraviolet (UV) radiation (artificial or natural). Photosensitization requires the presence of photosensitizers, which induce changes in the drug after the appropriate radiation is absorbed (Box 4-5). Structural requirements of the photosensitizer to induce phototoxicity reflect its ability to absorb radiation wavelengths characterized by effective skin penetration (above 310 nm). As such, the photochemical decomposition to stable photoproducts, free radicals, and/or singlet oxygen is facilitated. The probable role of fluoroquinolone antimicrobials as photosensitizers was discussed previously. Photosensitizers can be found in the cellular content of foods (e.g., flavins and porphyrins), plants or their juices, industrial chemicals (dyes, coal tar, derivatives of chlorinated hydrocarbons), and drugs. Exogenous photosensitizers may enter the body through a variety of routes, including ingestion, inhalation, injection, or direct contact with the skin or mucosa.83 Biological targets subject to photosensitization include cell membranes, cytoplasmic organelles, and the nucleus. Photosensitization may be used therapeutically if it can be directed to targeted tissues or if UV radiation can be applied to selected sites (e.g., cancer therapy). Clinical signs vary with the photosensitizer and the amount of radiation absorbed. Signs range from mild cutaneous reactions (e.g., erythema, pruritus, urticaria and rash) to severe reactions including (in humans) genetic mutations and melanoma. Clinical signs generally occur immediately after exposure to the UV radiation. Variation may also reflect the skin type, as well as physiologic factors such as age and gender (human medicine). Photosensitivity is generally dose dependent but may not happen with the initial drug administration but rather with subsequent administration. The reaction is then referred to as a photoallergy, reflecting binding of the photosensitizer to a skin protein. Occasionally, the response may be delayed and may occur even in the absence of the photosensitizing substrate.
Endocrine System
The impact of xylitol on insulin secretion was discussed with its hepatotoxicity. Mechanisms of drug interference with the thyroid and adrenal axes have been documented in human patients (Table 4-6). Each axis presents several targets for drug interference. Mechanisms that decrease hormone concentrations include suppression of hormone release at each level (i.e., hypothalamus, pituitary, or target organ), often because hormone synthesis is decreased, or altered by peripheral metabolism of the hormone (e.g., thyroid hormones).84 The latter effect is often the result of induction of hepatic drug-metabolizing enzymes. Potent inducers of hepatic drug-metabolizing enzymes include phenobarbital, phenytoin, and rifampin. Whether patients show clinical manifestations of hormone deficiency after induction of metabolizing enzymes remains to be documented. Less commonly, hormone concentrations are physiologically increased by drugs. Again, changes in hepatic metabolism are a common cause. Potent inhibitors of hepatic drug metabolism include cimetidine, chloramphenicol, and ketoconazole.
Table 4-6 Drug-Induced Physiologic Changes in the Adrenocortical and Thryoid Axes∗
| Drug | Comment |
|---|---|
| Cortisol | |
| Increased | |
| Anticonvulsants | |
| Corticotropin | Diagnostic intent |
| Cortisone | For at least 24 hours |
| Estrogen | Increases binding globulin concentrations |
| Fluocinolone | After topical administration |
| Hydrocortisone | For at least 24 hours |
| Insulin | Marked effect with insulin-induced hypoglycemia |
| Lithium | |
| Metoclopramide | After intravenous dosing |
| Opiates | Within 1 hour of intravenous dosing with selected drugs |
| OPPPD (mitotane)† | Therapeutic intent |
| Prostaglandin F2 | Slight effect |
| Vasopressin | Mild increase |
| Decreased | |
| Barbiturates | Preoperative use |
| Beclomethasone | After inhalant administration |
| Clonidine | In growth hormone–deficient children |
| Danazol | Displacement from binding and increased free drug |
| Deoxycorticosterone | After topical administration |
| Dexamethasone† | Diagnostic intent |
| Ephedrine | Accelerated clearance caused by increased hepatic blood flow and enzyme activity |
| Etomidate | Direct suppression of adrenal function |
| Fluocinolone | After topical administration |
| Thyroxine | |
| Increased | |
| Dessicated thyroid | |
| Estrogens | Increased binding capacity of globulin for up to 1 month |
| Fluorouracil | Increased binding capacity |
| Glucocorticoids | Inhibition of conversion |
| Halothane | Increased release from liver |
| Insulin | Increased release from liver |
| Levothyroxine | Suppression of endogenous hormone; exogenous measured |
| Lithium | Report of one patient suffering from presumed drug-induced thyrotoxicosis |
| Phenytoin | |
| Propranolol | Blockage of iodothyronine deiodination in hyperthyroid and euthyroid patients |
| Prostaglandins | Direct effect |
| Tamoxifen | |
| Thyroid | |
| Thyrotropin | |
| Drug | Comment |
| Thyroid-releasing hormone (TRH) | |
| Decreased | |
| Aminosalicylic acid | Prolonged administration may cause hypothyroidism |
| Anabolic steroids | Decreased binding to globulins |
| Androgens | Decreased binding to globulins up to 1 month per administration |
| Anticonvulsants | |
| Asparaginase | |
| Aspirin | Displaces T4 from binding sites to prealbumin |
| Barbiturates | Competition for binding to prealbumin |
| Bromocriptine | In hypothyroidism (response to TRH unchanged) |
| Carbamazepine | Induction of hepatic enzymes; increased extrathyroidal metabolism |
| Chlorpromazine | Increased metabolism by liver |
| Cholestyramine | Decreased intestinal absorption |
| Glucocorticoids | Up to 1 week after therapy |
| Diazepam | Competition for transport proteins |
| Furosemide | Displacement from binding sites and enhanced clearance |
| Growth hormone | Inhibition of thyroid-stimulating hormone (TSH) response to TRH (?) |
| Heparin | |
| Iodides† | |
| Lithium | Reduced thyroidal iodine updake, iodination of tyrosine, release of T4, hepatic metabolism of T4 to T3 |
| Methimazole† | Therapeutic intent |
| Mitotane | Competes with T4 for binding globulin |
| Penicillin | Competes for binding globulin |
| Phenobarbital | Induction of hepatic enzymes |
| Phenylbutazone | Impaired synthesis, competition for binding to albumin |
| Phenytoin | Displacement from binding proteins; induction of hepatic enzymes |
| Potassium iodide | |
| Propylthiouracil | Inhibits synthesis (iodination of tyrosine), therapeutic intent |
| Ranitidine | Slight reduction |
| Salicylate | Competition for transport proteins |
| Somatostatin | Inhibition of TSH release (?) |
| Stanozolol | |
| Sulfonamides† | |
| Terbutaline | Mild decrease |
| Triiodothyronine | |
| Triiodothyronine (Thyronine) | |
| Increased | |
| Estrogens | Increased binding capacity to transport proteins |
| Fluorouracil | Increased binding capacity to transport proteins |
| Heparin | Interference with binding to protein |
| Insulin | 45 minutes after injection; release from liver |
| Phenytoin | |
| Prostaglandins | |
| Tamoxifen | |
| Terbutaline | |
| TRH | Percentage of free T3 unchanged |
| L-Thyroxine | |
| Triiodothyronine | |
| Decreased | |
| Androgens | Decreased binding capacity (diminution of transport proteins) |
| Anticonvulsants | |
| Asparaginase | |
| Aspirin | |
| Carbamazepine | Increased extrathyroidal metabolism |
| Cimetidine | Reduced response to TRH |
| Furosemide | |
| Glucocorticoids | Inhibition of conversion |
| Iodides | Inhibition of conversion |
| Lithium | See under Thyroxine |
| Phenytoin | See under Thyroxine |
| Potassium iodide | |
| Propranolol | Membrane stabilization (see under Thyroxine) |
| Propylthiouracil | |
| Salicylate | See under Thyroxine |
| Somatostatin | See under Thyroxine |
| Stanozolol | See under Thyroxine |
| Sulfonamides† | |
∗ Table reflects serum or plasma values only and is based on information reported by Young (1990).11
† Reported in the veterinary literature.
Drugs can also increase hormone concentrations by competing with and displacing the hormone from carrying proteins. The protein from which hormones are most likely to be displaced is albumin, a nonspecific carrier of many weakly acidic drugs (e.g., NSAIDs). Competition for albumin-binding sites may be less important for those hormones carried by specific carrier proteins, although competition for such binding sites has been documented. In some cases a drug may influence blood hormone concentrations simultaneously at several physiologic sites, complicating interpretation (e.g., the effects of phenytoin on thyroid hormone concentrations). Because animals differ physiologically, extrapolation between species regarding the effect of a drug must be done cautiously. Caution is also advised when extrapolating results of studies in normal animals to the animal suffering from a disease of the endocrine system. For example, propranolol decreases thyroid hormone concentrations in hyperthyroid humans but not in euthyroid dogs.85
In some instances the drug effect on a hormone is well known and is used either diagnostically (e.g., dexamethasone-induced decrease in cortisone or xylazine-induced growth hormone secretion)86 or therapeutically (propranolol or propylthiouracil-induced inhibition of thyroxine [T4]). More commonly, the effect is undesirable. Several examples of undesired, drug-induced physiologic changes in endocrine function have been documented in small animal patients. The example most documented in small animals are the effects of drugs, and particularly glucocorticoids, on the hypothalamic–pituitary–adrenal axis. Interference with this axis can become clinically detrimental. Suppression of the adrenal axis by glucocorticoids is most marked after administration of depot (repositol) forms (e.g., those containing acetate esters).87 Interference has also, however, been documented after administration of a single dose of prednisolone or triamcinolone; multiple doses of methylprednisolone;87 topical administration of triamcinolone;88 and ophthalmic administration of prednisone.89 The impact of other steroids, including dexamethasone and betamethasone, are discussed in Chapter 17.
Glucocorticoids are not the only drugs that interfere with the hypothalamic–pituitary-adrenal axis. The imidazole antifungal drug ketoconazole inhibits the cytochrome P450 enzymes responsible for the synthesis of both sex and adrenal steroids.90 Suppression of testosterone and cortisol has been documented in dogs after oral administration of 10 mg/kg ketoconazole once daily.91 Hormone concentrations are lowered by day 1 and remain low at day 5. Progesterone concentrations increase as testosterone concentrations decrease. The magnitude of testosterone inhibition by ketoconazole apparently resolves, with testosterone concentrations being less predictable 1 month after therapy was started. The inhibitory effect of ketoconazole on testosterone and adrenal steroids has been used therapeutically in the treatment of prostatic cancer and benign prostatic hypertrophy and hyperadrenocorticism, respectively. The newer imidazole antifungal drugs do not appear to inhibit steroid synthesis as effectively as ketoconazole.
Drug interference with evaluation of the thyroid axis is also important because of the prevalence of thyroid dysfunction in small animals. Several drugs, targeting various sites, interfere with thyroid function testing (see Table 4-6).92 Thyroid-stimulating hormone (TSH) response to thyroid-releasing hormone (TRH) is altered by a number of drugs that modulate neurotransmitter (e.g., serotonin, dopamine) concentrations in the brain. Glucocorticoid suppression of TSH response to TRH has been well documented. Higher doses appear to suppress hypophyseal inhibition of TSH, whereas low doses interfere with the hypothalamic response.92 Note, however, that interference of the thyroid axis by glucocorticoids does not preclude simultaneous testing of the thyroid and adrenal axes in healthy dogs.93,94 Antithyroid drugs such as propylthiouracil and methimazole are used therapeutically to block thyroid hormone synthesis; their mechanism occurs, at least in part, at the level of transcription.95
The effects of iodide- and iodine-containing products (including radiographic contrast agents) on thyroid hormone concentrations are well recognized and used therapeutically. Through hypothalamic regulation, iodines cause a rapid increase in TSH response to TRH as T4 and triiodothyronine (T3) concentrations decrease. Sulfonamides can have a profound effect on thyroid function.96 Among the potential mechanisms, direct interference with the conversion of inorganic iodide to diiodotyrosine and thyroxine was demonstrated as early as 1943.97 Decreased concentrations of peripheral hormones are associated with follicular cell hypertrophy and hyperplasia and with decreased colloid formation. Changes are profound in as early as 21 days yet resolve within 3 weeks after therapy is discontinued. These effects occur at high doses that might be used for difficult-to-treat, yet presumably susceptible, higher bacterial or protozoal infections (>60 mg/kg per day but may also occur at lower doses). The effects of sulfonamide NSAIDs and the anticonvulsant zonisamide on thyroid gland function are discussed in Chapters 29 and 27, respectively.
The effects of non-sulfonamide anticonvulsant drugs, especially phenobarbital and phenytoin, on thyroid hormone disposition are less appreciated. Several sites of interference have been identified for anticonvulsant drugs (see Chapter 27).
Hematologic
As with any drug-induced disorder, the lack of universally standardized definitions of what constitutes an adverse reaction complicates recognition of hematologic disorders induced by drugs. The criteria for drug-induced hematologic disorders have been described for humans and are based on cell count, assessment of time to onset after drug exposure and time to resolution of signs after the drug has been discontinued, and the course of the reaction.98 Drug-induced hematologic dyscrasias may reflect a bone marrow response or an effect on peripheral tissues, including blood components (Table 4-7). Bone marrow suppression can result in pancytopenia or affect only a single cell line (i.e., anemia, leukopenia, or thrombocytopenia).99 Both direct bone marrow suppression and toxicity to mature circulating cells may occur.
Table 4-7 Examples of Drugs Associated with Hematologic Disturbances
| Drug | Manifestation |
|---|---|
| Acetaminophen | Methemoglobinemia (especially in cats) |
| Anticancer drugs | Bone marrow suppression∗ |
| Azo dye (urinary antiseptics) | Methemoglobinemia (cats) |
| Benzocaine (and related drugs) | Methemoglobinemia (cats) |
| Chloramphenicol | Bone marrow suppression∗ |
| Cimetidine | Thrombocytopenia |
| Coumarin derivatives | Coagulation dysfunction |
| Erythropoietin (human recombinant) | Anemia |
| Estrogens | Bone marrow suppression∗ |
| Griseofulvin | Bone marrow suppression∗ |
| Heparin | Thrombocytopenia, platelet dysfunction, coagulation dysfunction |
| Methimazole | Methemoglobinemia |
| Methylene blue | Methemoglobinemia (cats) |
| NSAIDs | Platelet dysfunction |
| Phenobarbital | Neutropenia |
| Phenylbutazone | Bone marrow suppression∗ |
| Propylthiouracil | Methemoglobinemia |
| Ranitidine | Anemia |
NSAIDS, Nonsteroidal antiinflammatory drugs.
∗ Bone marrow suppression might be manifested as anemia, leukopenia, thrombocytopenia, or any combination thereof.
Bone marrow and peripheral cells are susceptible to both drugs and their metabolites; reactions may have an immunologic or nonimmunologic basis. Although drug allergies are a well-recognized cause of damage to stem cells of the bone marrow, many drugs are directly toxic. Discerning an immunologic basis can be difficult, however, if the antibodies involved have not been identified. Drugs most commonly associated with nonimmune-mediated bone marrow suppression include most cancer chemotherapeutic agents because of their predictable effects on DNA and cell division. Other drugs associated with nonimmune-mediated bone marrow dyscrasias include phenylbutazone, estrogen derivatives, and chloramphenicol.
Phenobarbital has caused leukopenia and other hematologic disorders when used to treat epilepsy; white cell counts normalize once the drug is discontinued. Whether or not this is an immune-mediated reaction is not clear, but its lack of dose dependency suggests a Type B reaction (See Chapter 27).
Drugs that affect blood components and the manifestations of anemia include all NSAIDs but particularly aspirin (reflecting inhibition of platelet activity), anticoagulants such as warfarin derivatives, and heparin (these generally reflect a relative overdose). Red blood cell malfunction may occur as a result of methemoglobinemia in cats (Figures 4-9 and 4-10). Although acetaminophen clearly causes hepatotoxicity, the feline red blood cell is more sensitive to the presence of radical metabolites compared with the feline liver. Several reasons have been suggested for an apparent increased sensitivity of feline red blood cells to methemoglobin formation: feline hemoglobin may be more sensitive to oxidation; feline erythrocytes may contain lower concentrations of intracellular glutathione; the proportion of subtypes of hemoglobin may differ; and, finally, feline hemoglobin may contain more sulfhydryl groups, which are reactive, than that of other species. Drugs associated with methemoglobinemia in cats include urinary antiseptics containing methylene blue or azodyes, acetaminophen and related compounds, benzocaine, DL-methionine, propylthiouracil, and methimazole.
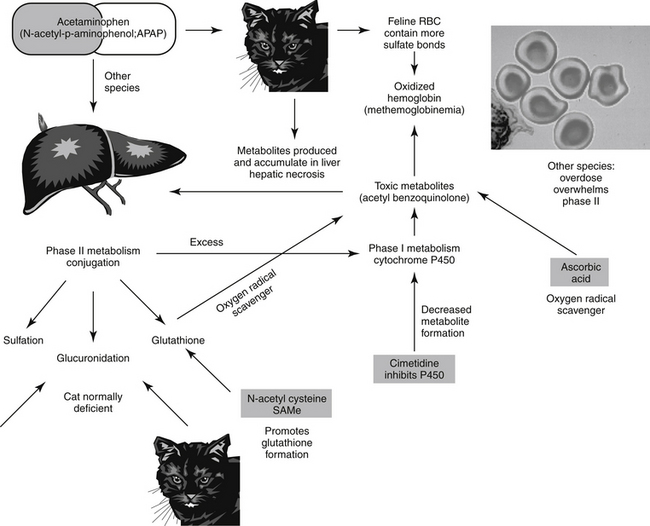
Figure 4-9 Acetaminophen toxicity reflects a cytotoxic type A adverse reaction. Because cats are deficient in glucuronidation, phase II metabolism is easily overwhelmed, and drug is shunted more aggressively back into phase I metabolism. The same process occurs in dogs after an overdose. The products of phase I metabolism are reactive and cause destruction of tissues (liver and red blood cells). Glutathione, an important phase II oxygen-radical scavenger, prevents damage but is easily depleted in cats. Supplementation in the form of N-acetylcysteine can decrease damage. Cimetidine is useful because it decreases phase I metabolism and thus the formation of phase I metabolites.

Figure 4-10 Facial edema in a cat with acetaminophen toxicosis. The mucous membranes of this cat were cyanotic.
Ivermectin was associated with a prolonged prothrombin time and hematomas in humans after a single oral dose, presumably because of a drug-induced vitamin K deficiency.100
Human recombinant erythropoietin and granulopoietin have been used to treat anemias associated with chronic renal disease and leukopenia induced by disease (e.g., parvovirus) or drugs (e.g., anticancer drugs) in dogs and cats. Unfortunately, these proteins are foreign, and antibodies may develop after 10 to 14 days, destroying not only the exogenous drug but also endogenous factors.
Drug-induced hematologic disorders have been described in the human critical care patient.101 Among the challenges in the ICU setting is identifying the drug as the cause, and, in the patient receiving polypharmacy, which drug. It is not clear whether ICU patients are at any greater risk to develop these dyscrasias, but certainly they are at greater risk to react adversely should they occur. Bone marrow under production is manifested as pancytopenia if the pluripotential cell is targeted or monocytopenia if a single hematopoietic cell is targeted. Neutropenia can be manifested within hours (7 to 10), thrombocytopenia in days, and anemia generally in weeks unless exacerbated by hemolysis. Bone marrow aplasia has been associated with chloramphenicol, felbamate, sulfonamides, and NSAIDs. If the effect is direct, clinical signs may resolve once the drug is discontinued, unless the effect is irreversible (e.g., selected chloramphenical reactions in humans). If indirect, immunosuppressive doses of immunomodulators may be indicated. Myelodysplasia as a prelude to acute myelogenous leukemia is associated with high doses or long durations of alkylating or topoisomerase-inhibiting anticancer drugs. Macrocytosis has been associated with drugs that negatively affect metabolic compounds needed for DNA synthesis. Examples include cobalamine, whose absorption is inhibited by neomycin, proton pump blockers, and bifuanide hypoglycemic agents, or folate, whose metabolism is inhibited by methotrexate, phenytoin, and trimethoprim. Drugs that alter the nucleotide pool may cause megablastosis (e.g., hydroxyurea, methotrexate, azathioprine, purine nucleoside analogs). Enzyme deficiencies (thiopurine methytransferase) and drug-induced erythropoietin deficiencies (e.g., cisplatin) also have been reported. Cytopenias resulting from direct effects on cells have been reported. Examples include penicillin-induced thrombocytopenia. Heparin-induced thrombocytopenia reflects binding to PF4 immunoglobulin and subsequent activation of platelets; the risk is lower with low-molecular-weight heparins. Thrombotic microangiopathies (thrombotic thrombocytopenia purpura) has been associated with the antiplatelet agents clopidogrel or ticlopidine and anticancer drugs (mitomycin-C).101
Respiratory System
Although compounds toxic to the lungs occasionally arrive by hematogenous routes, most pulmonary toxicities result from direct exposure of the respiratory tract through the nasopharyngeal or oropharyngeal airways and subsequently the tracheobronchial tract and alveoli.15 Gaseous and particulate toxicants are most common. The airways may also serve as a means of systemic exposure if the toxicant is effectively and rapidly absorbed by the respiratory mucosa. Particulate matter that impacts the airways (generally 5 to 10 μm in size for the tracheobronchial tree and less than 5 μm in size for alveoli) can become trapped in the airways.
The mucociliary apparatus may remove entrapped particulate matter before a toxic response occurs. Some chemicals, however, cause direct injury to upper airways (e.g., chlorine, ammonia, water-soluble gases, and chromium). Compounds depositing in the alveoli can be removed only by blood flow (if the compound is absorbed), biotransformation by Clara cells or type II alveolar cells (which contain cytochrome P450 enzymes), or macrophages.
Pulmonary edema, manifested as an acute respiratory distress syndrome, is caused by severe exposure of the alveoli (in humans) to acute toxicants such as phosgene, chlorine, xylene, and nitrogen oxides. Because the lung is the shock organ in cats, type I allergic reactions may manifest as primarily acute respiratory difficulties. Biotransformation in the lung, as in other tissues, may be a source of a toxic compound as an innocuous chemical is converted into a toxic one (e.g., paraquat, a herbicide metabolized by type II alveolar cells). Macrophage clearance may reflect phagocytosis; macrophage death is accompanied by the release of inflammatory mediators that can damage surrounding cells and contribute to the toxic effects of a drug. Compounds that cause pulmonary injury in humans as a consequence of the inflammatory response include asbestos, beryllium, coal dust, silica, and tungsten.15
Avoiding Adverse Drug Events
Recommendations for avoiding specific toxicities have been given or are described in specific chapters for some of the described drugs. Appendix 5 offers antidotes for many drugs. The incidence of ADEs in general, and ADRs in particular, may be reduced by several proactive actions (Box 4-6), with client education playing an important role. Type B reactions are difficult to avoid because they are unpredictable. An awareness of potential toxicities will make them less likely. Frequent patient monitoring during therapy is the best means of reducing type B adverse reactions.
Box 4-6 Actions to Take to Reduce the Risk of Adverse Drug Events
Reporting Adverse Drug Events
Reporting ADEs is complicated by the difficulty in identifying them. In human medicine, methods have been defined whereby the causal relationship between a suspected reaction and a suspected drug can be assessed. None is universally accepted, but each includes some or all of the following criteria: the time between the administration of the drug and the onset of the reaction or the cessation of the drug and resolution of clinical signs; the course of the reaction, which may vary if the drug is continued or interrupted; the role of the drug and underlying disease being treated as a cause of the reaction; response to readministration of the drug; results of laboratory tests; and the history of previous administration of the drug.98
Reporting of ADEs to the Center for Veterinary Medicine (CVM) of the FDA has markedly increased from 4000 in 1997 to nearly 25,000 a year in the past several years. The increase probably reflects, in part, the increased visibility of the CVM reporting program that accompanied improvements in the collection, analysis, and reporting of the adverse events.25,102 Several avenues are available for reporting an ADE. The animal pharmaceutical company can be directly informed by calling the Medical Affairs officer; by law, these ADEs must be reported to the CVM. The sequelae of reporting an animal adverse event to a human drug is not clear, but mandatory reporting is probably unlikely. In such situations the second route should be pursued: the CVM. The CVM provides directions for reporting ADEs. The form (FORM FDA 1932a, “Veterinary Adverse Experience, Lack of Effectiveness or Product Defect Report”) can be downloaded or printed from http://www.fda.gov/cvm/adereporting.htm or by writing to the following address: ADE Reporting System, Center for Veterinary Medicine, U.S. Food & Drug Administration, 7500 Standish Place, Rockville, MD 20855-2773. Reports also can be reported by telephone (1-888-FDA-VETS). On receipt of an ADE by the CVM, a six-part scoring system is used to evaluate each reaction. The system takes into account previous experience with the drug, alternative causes, timing of the event in the context of dosing, overdosing, and response to drug removal or rechallenge if available.25 Recently, the CVM’s approach to assessing a potential ADE insofar as it’s likelihood to lead to label changes or market withdrawals has come under fire; methods of analysis may undergo reassessment, which will, it is to be hoped, further refine the assessment process and improve accuracy. Results of the ADE reporting program and analysis by the CVM can be reviewed for each drug at the previously cited website. A third alternative to ADE reporting may be the Animal Poison Control Center (APCC) affiliated with the Animal Society for Prevention of Cruelty to Animals (ASPCA) (www.aspcsaapcc.org). The latter organization should be contacted by telephone (888-426-4435) if immediate support is desired for identification and treatment of a potential ADE that may prove life threatening to the animal. Support is likely to entail a $50 consultation fee. Adverse events to animal biologics (vaccines, bacterins, and diagnostic kits) are not handled by the FDA but instead by the U.S. Department of Agriculture (800-752-6255), whereas adverse events to pesticides (topically applied external parasiticides) should be reported to the U.S. Environmental Protection Agency (800-858-PEST). Links to these agencies are available at the FDA website.
Adverse Events to Herbs and Botanicals
Herbal or botanical products, more so than “dietary supplements” of animal origin, may be unsafe for several reasons. Adverse events are more likely when products are used in excess. Five broad classes of active chemicals exist in plants: volatile oils (e.g., catnip, garlic, citrus), fixed oils, resins, alkaloids, and glycosides. Of these, fixed oils, often used as emollients, demulcents, and bases for other agents, are among the least toxic. Resins can be strong gastrointestinal irritants. Alkaloids are among the most pharmacologically active plant chemicals and include a wide range of potentially harmful products. The risk of adverse effects to herbs is increased by the presence of many active ingredients in the same plant (see the discussion of drug interactions in Chapter 2). Indeed, herbalists often used unpurified plant extracts because of the possibility that different chemicals might interact synergistically. The amount of active ingredients may vary dramatically with the portion of the plant (i.e., leaf, flower, stem, root, seed) administered, thus influencing safety. Whereas one portion of the plant might be safe, another portion might not be. Herbalists often administer the whole plant in the belief that, in contrast to the purified extract, toxicity is reduced by a buffering effect of the whole herb. During growth of the plant, environmental contaminants may become unintended residues during the manufacturing process. Microorganisms, including bacteria, fungi, and molds, can either directly contaminate the product or produce contaminating toxins. Bacterial contamination is more likely with root products as opposed to flower or leaf products. Heavy metals such as lead, cadmium, or mercury increasingly are contaminating plants exposed to environmental pollutants. Further, unless the herbal products are grown organically, insecticides and pesticides may contaminate them. Factors during production and storage, such as storage length and conditions, can alter herbal potency and quality. Finally, herbal products might be supplemented with active ingredients (often referred to by the herbal or botanical name or simply not labeled) such as ephedrine, caffeine, or fenfluramine (the latter ingredient being one of the two ingredients in the notorious Fen-phen dietary supplements).
KEY POINT 4-3
The risk of an adverse drug effect with dietary supplements is increased by unsupervised client use; the lack of mandated premarket approval or premarket assessment of quality, safety, and efficacy; the inherent properties of herbal products; and the lack of an effective postmarket surveillance program.
The lack of quality control in labeling of herbals, botanicals, or other novel ingredients (e.g., “nutraceuticals”) may contribute to the advent of adverse effects with these products.103 Many herbal products are not labeled with the concentration per dosing unit of the ingredient of interest. Those that do may contain more or less than the labeled dose, presenting a risk of overdosing or underdosing. Manufacturers may improperly identify plants. Even if properly identified, the consumer may have difficulty in identifying a product as potentially dangerous because an herbal name often is used in place of the more easily recognized chemical name (e.g., guarana for caffeine or ma huang for ephedrine). Further, an herbal agent may be referred to by many different names. The FDA has become more proactive in directing manufacturers to list generic drug names instead of or in addition to herbal names. Proposed sources of quality assurance data for animal dietary supplements include the manufacturer of the product; the Association of American Feed Control Officials; the National Animal Supplement Council; laboratories such as Consumer Lab (www.consumerlab.com); the United States Pharmacopeia (USP) dietary supplement verification program (http://www.usp.org/USPVerified/dietarySupplements); the American Herbal Pharmacopeia (http://www.herbal-ahp.org), which offers the Botanical Safety Handbook and Herbs of Commerce; and Consumer Laboratories, a for-profit organization that provides quality assessment for a fee. However, much of its work is also independent, supported by income generated through its website. For approximately $30 per year, this site provides results of quality testing for a variety of products. Although most of the products are marketed for humans, several veterinary products recently have been evaluated for quality.
A number of other herbs have been associated with adverse effects (Table 4-8). This list is far from inclusive, in part because an effective adverse event reporting system is lacking in either human or veterinary medicine. Although infrequently reported, adverse events have occurred in veterinary patients receiving novel ingredients. The ASPCA APCC published a report of adverse reactions in 47 dogs that ingested a popular weight loss dietary supplement containing guarana (caffeine) and ma huang (ephedrine); 17% of the dogs died after the appearance of clinical signs associated with central and cardioactive compounds.
Table 4-8 Examples of Therapeutic Herbs or Botanicals Associated with Adversities∗
| Ginkgo biloba | Bleeding, altered platelet function |
| St. John’s wort | Gastrointestinal disturbances, allergic reactions, fatigue, dizziness, confusion, dry mouth, photosensitivity |
| Serotonin syndrome (when combined with other similarly acting drugs) | |
| Ephedra (ma huang) | Hypertension, insomnia, arrhythmia, nervousness, tremor, headache, seizure, cerebrovascular event, myocardial infarction, kidney stones |
| Kava | Sedation, oral and lingual dyskinesia, torticollis, oculogyric crisis, exacerbation of Parkinson’s disease, painful twisting movements of the trunk, rash |
| Aconitine | Cardiotoxic |
| Garlic | Altered platelet function |
| Ginger | Altered platelet function, tachycardia and/or hypertension |
| Ginseng | Altered platelet function |
| Feverfew | Altered platelet function |
| Echinacea | Hepatotoxicity |
| Valerian | Hepatotoxicity |
| Goldenseal root | Electrolyte disturbance |
| Licorice | Electrolyte disturbance |
| S-adenosylmethionine | Serotonin syndrome (when administered in combination with other similarly acting drugs) |
∗ Many herbs or botanicals are associated with adversities. The use of various herbal names for each product may preclude detection of adversity. The lack of an effective adverse event reporting system hinders detection and reporting of adversities involving herbs.
Although mechanisms for reporting adverse events in human or veterinary medicine toward herbal or botanical products currently are limited, effective mechanisms are evolving. Among the FDA’s postmarket responsibilities toward (human) dietary supplements is the monitoring of product safety. Note, however, that the FDA may remove a potentially dangerous supplement only when and if the product presents an ‘‘imminent hazard to public health or safety.’’ The burden of proof is on the FDA, not (as with drugs) on the manufacturer. The FDA recently implemented the Adverse Events Reporting System (AERS) to be used as a monitoring tool for dietary supplements as well as other medicinal products. Adverse events also can be reported for human products through the FDA’s MedWatch program. Some recent FDA actions taken to address the safety of dietary supplements can be reviewed at www.consumerlab.com, at the FDA’s Medwatch program, or the FDA’s Center for Drug Evaluation and Research (CDER). Unfortunately, the current FDA sites for reporting adverse events to dietary supplements do not pertain to veterinary products. Reporting to the APCC may continue to be the most effective means of reporting adverse reactions of animals to medicinal agents, particularly those for which a designated watchdog does not exist (e.g., unapproved products), in part because of the APCC’s ability to analyze and quickly report important trends.
Adverse Events to Compounded Preparations
Quality compounding is critically important to the safe and effective administration of drugs to animals, particularly the very small and very large. The role of compounding in small animal medicine was recently reviewed.104 Compounding of animal drugs is specifically legalized by the Animal Medicinal Drug Use Clarification Act (AMDUCA; 21 C.F.R Section 530) (See Appendix 5). However, compounding must be implemented in accordance with the relevant provisions of extralabel drug use. According to the FDA, legal sources of drugs to be compounded are limited to FDA-approved finished forms of either animal or human drugs; the FDA makes no distinction as to which (animal versus human) is the preferred source for companion animal compounding. Because no other source is legalized, all other sources are considered by the FDA to be illegal, including bulk substances (e.g., pure powder) or non–FDA-approved finished drug products obtained outside of the United States. However, formulation of selected compounded prepartions is likely to be easier, yielding a better product, if prepared from pure active ingredient (i.e., bulk). The risk of supporting compounding from bulk includes the temptation to manufacture, rather than compound. Compounding, particularly for products that mimic a commercially available drug, serves as a major disincentive for manufacturers of animal drugs to pursue the approval process. Further, whereas approved animal or human drugs have undergone rigorous, scientific testing to ensure drug safety and efficacy for the patient, compounded products have not. Although pharmacists are directed to compound from written protocols and maintain written records of compounding activities, pharmacists are not currently required to ensure accuracy in product preparation, including product stability. Although a reputable pharmacy may randomly check accuracy of selected drugs, this act currently is voluntary and will be limited to selected drugs and aliquots. Although guidelines exist for establishing expiration dates of compounded products, dates are not necessarily based on scientific data. The United States Pharmacopeia has published a Pharmacist’s Pharmacopeia. Further, the Pharmaceutical Compounding Accreditation Board (PCAB) has recently implemented a robust accreditation program. Adherence to its guidelines will minimize the risk of adverse reactions associated with compounded products (see www.pcab.org).
KEY POINT 4-4
The risk of an adverse drug effect with compounded products is increased by the existence of regulatory oversight and can be minimized by prescribing only through pharmacies accredited by PCAB.
The risks associated with failed delivery (too much or too little) of a compounded product are added to risks associated with the approved finished dosing form of a drug. Adverse events associated with compounded products should be reported to either the (veterinary) pharmaceutical manufacturer or the FDA; if reported to a veterinary manufacturer, the manufacturer is required by law to forward the report to the FDA, although whether this is true for a modified finished dosing form is not clear. Adverse events associated with compounded products may occur at the pharmaceutical phase (during preparation) or, largely because of failed absorption, at the pharmacokinetic phase. The more sophisticated the preparation, the more likely it is that adverse events will occur because of diminished or excessive drug delivery.
Ingredient Errors
Compounding from bulk substances is easier than from approved finished dosing forms because excipients or other materials do not interfere with product preparations. Further, excipients in the finished dosing form will not interfere with dissolution of the drug in the vehicle. However, the use of an approved finished dosing form of a drug for compounding offers a major advantage to use of a bulk substance in that the approved drug has passed stringent tests of analysis regarding drug purity and potency and the absence of contaminants. As such, products formed from bulk substances are associated with greater risks compared with products compounded from approved drugs. In contrast, for bulk substances the burden of purity and accuracy lies with the pharmacist, and there is no mechanism to ensure that this burden has been met. All products, active ingredient or excipients (fillers, preservatives, etc.), domestic or foreign, should either meet USP or equivalent standards or be purchased after FDA inspection. Drugs that are still under U.S. patents are often obtained in this manner. Bulk substances will be accompanied by a credible certificate of analysis. The need for validation of ingredient sources (including all active and inactive substances) is paramount as inexpensive bulk substances increasingly are being acquired from uninspected foreign (particularly Asian) sources.
The active drug in a compounded product might also be substituted for an alternative drug; the substituted drug may not have the same pharmacokinetic or pharmacodynamic characteristics (discussed in more detail in the next section). Veterinarians should indicate on prescriptions that unapproved substitutions are not acceptable for compounded products.
Mathematical Errors
Mathematical errors are probably the most common and potentially the most lethal reason for pharmaceutical compounding errors. Compounding is vulnerable to mathematical mistakes because of its very nature (prescription driven, small volumes) and because much of the equipment and technology that facilitate accuracy and precision of finished dosing forms are not (or should not be) used during compounding. In addition to the source of the ingredient being potentially problematic, pharmacists may substitute drugs without acquiring clinician permission. Mathematical errors may also reflect substitution of the active ingredient. For example, the active drug content may differ, as is demonstrated by metronidazole. The recipe for metronidazole benzoate should contain 1.6 mg for each 1 mg of metronidazole hydrochloride (or the dose must be similarly increased). Bromide offers another example: 1 g of the sodium bromide contains more bromide (774 mg) than the potassium salt (692 mg).
Preparation and Storage Errors
Chemical reactions (oxidation, reduction, hydrolysis) are facilitated by changes in humidity, light, pH, presence of oxidizing trace metals, and increasing environmental temperature. Excipients may enhance instability as a result of changes in pH or the presence of disintegrating agents. Degradation products (drugs or excipients) can cause adverse events. Excipients that are critical to the finished dosing form increase the risk of instability in product compounded from an approved source. Whereas approved products undergo intensive scrutiny with regard to stability and potency, compounded products do not; recipes for compounded preparations rarely are associated with studies that ensure stability or delineate conditions for storage.
Simple syrups (which tend to be acidic), preservatives, combination drugs, or other ingredients can alter drug pH and thus ionization (diffusibility) or stability. The more drugs mixed together in a single preparation, the greater the risk of chemical drug interactions. For example, weak acids and weak bases are likely to chemically inactivate one another. Interactions may occur among the drugs or excipients. As an example, only 54% of a fluorinated quinolone (orbifloxacin) was found to be present when prepared in Lixotinic as a vehicle compared with simpler syrups.
Particle Size
Compounding from approved drugs (legal) is more difficult than from bulk drugs (illegal) because excipients are more likely to result in undissolved macroscopic or microscopic precipitates that indicate undissolved and thus nondiffusible ineffective drug. Sedimentation of undissolved particles may result in caking at the bottom of the drug receptacle; difficulty in shaking or rapid sedimentation (common) after shaking can result in erratic and unpredictable doses. Crushing of any oral tablet may result in unequal particle sizes in the preparation, which in turn will yield different surface areas and different rates of absorption. Fine crushing of the product such that it is no longer a suspension increases the concentration of soluble excipient; chemicals, including those added to the finished dosing form to facilitate degradation, can cause drug instability. Crushing an oral tablet for preparation in a syrup may also lead to unequal distribution of dissolved drug in the finished preparation, and mixing the drug such that it is equally distributed throughout the preparation may not be possible. Repackaging oral tablets or capsules into smaller dosing units may also affect drug efficacy. Diluents such as starch and dextrose might impede oral absorption. Preparation of an oral formulation from an injectable solution to enhance accuracy of dosing is more likely to be inappropriate if the drug salt is different between the preparations. If the injectable product is presented in powder form, the drug is likely to be unstable in liquids and may be destroyed when added to liquid (oral solutions). The addition of flavoring agents to oral products may increase drug instability because of changes in pH or the increased risk of microbial growth (e.g., as with syrups).
Because selected commercial oral preparations have been formulated to alter (slow or facilitate) drug delivery, reformulation of such products is discouraged. Compounding altered release products from bulk substances requires sophisticated techniques not generally available through pharmacists. Enteric-coated or spansule products should not be crushed. Although spansule products might be reformulated without crushing, the amount of drug in each spansule is not necessarily predictable, and random distribution of drug content is likely to yield erratic dosing. Cyclosporine is a complex molecule characterized by poor oral bioavailability; oral absorption requires bile acids or special formulation as a microemulsion product. As such, it is an example of a drug for which compounding should be approached cautiously and be supported by therapeutic drug monitoring. In the author’s drug-monitoring laboratory, cyclosporine blood concentrations were not detectable (two different samples, 2 weeks apart) in one cat receiving a product compounded from an approved microemulsion human product. In keeping with recommendations that the unadulterated animal-approved version be used at the same dose, concentrations expected at the administered dose were detected within 1 week of the change in drug product.
Injectable Products
Administration of injectable products is inherently associated with a higher level of risk compared with administration of topical or oral products because of more rapid drug delivery, risks associated with administration of suspensions rather than solutions, potential impact of impurities (including endotoxin), and need for sterility. Actions taken to ensure sterility and removal of impurities may cause drug degradation. Endotoxin (which is essentially ubiquitous in the environment) is difficult to remove. Without testing, its absence is impossible to document, yet its presence can be lethal. The USP has generated guidelines, and state laws generally delineate regulations specifically for the compounding of injectable products. Veterinarians should be reluctant to prescribe compounded injections, and when doing so, they must be confident that the compounding pharmacist follows these criteria.
Topical Products
Although administration of topical products generally is associated with fewer risks compared with administration of systemic products (the exception is ophthalmic products, which also should be sterile), compounding the proper product can be challenging. The USP has promulgated guidelines for the compounding of topical ingredients, including guidelines designed to ensure drug dissolution and drug movement from the vehicle into the skin. For example, solid ingredients should be reduced to the smallest reasonable particle size, and the active ingredient should then be added to other substances necessary to dissolve the drug so that a uniform liquid or solid dispersion is achieved. Uniformity of dispersion should be demonstrated by spreading a thin film of the finished formulation on a flat transparent surface. Visual examination of a compounded product should be implemented to identify obvious problems with dissolution and so on. Care must be taken to ensure that ingredients are not caustic, irritating, or allergenic. Vehicle selection can be quite difficult: undissolved drug cannot pass into the skin; drug that has too great an affinity for the vehicle will remain in the vehicle. Transdermal gels are examples of products in which particular care must be exercised.
Few published reports exist that delineate adverse events resulting from inappropriate compounding. Despite indications of frequent problems with compounded products, the FDA receives few reports regarding adverse events related to compounded products. This reflects, in part, the lack of mandated reporting of adverse events. However, it also reflects the difficulty in recognizing therapeutic failure resulting from failed delivery. The latter is likely to be detected only if the information is sought and the drug or response to the drug can be easily monitored. Numerous studies have focused on accuracy in labeling of compounded products, particularly in equine medicine. Products found to be mislabeled include omeprazole, ivermectin (both pirated drugs), ketoprofen (one product contained only 50% of the labeled content, whereas 12 of 13 contained close to 100%), amikacin (the percentage of labeled content ranged from 59% to 140%; none was within 10%), and boldenone (all within 15% of labeled content, but two of five contained up to 5% of impurities).
Drug Effects on Clinical Laboratory Tests
A drug-induced disease is often first suspected on the basis of an abnormality in a diagnostic test that cannot be easily attributed to a disease process. Many drugs cause changes in diagnostic tests but are not associated with disease. Confirming the cause-and-effect relationship between drug and abnormalities can be very difficult unless the drug can be discontinued and then readministered.
Drugs interfere with diagnostic tests either directly, at the level of the analytical procedure (in vitro), or by induction of a physiologic change in the patient (in vivo). Of the two levels of interference, it is likely that analytical interferences will occur regardless of the species from which the sample was collected. Thus interferences affecting analytical procedures are better documented in veterinary medicine because they generally can be extrapolated from human analytical testing. Mechanisms of in vitro interference vary. Drugs that interfere with endocrine testing are described as adverse drug events of the endocrine system.
If analytical (in vitro) interference by a drug is suspected, the laboratory should be contacted and questioned. This is particularly important if the patient is receiving drugs structurally similar to the drug being tested. Cross-reactivity between the drug and the test can falsely increase test values. For example, therapeutic corticosteroids cross-react with endogenous corticosteroid hormones, although the percentage of cross-reactivity varies with the assay and the drug. Some drugs cause cytotoxicity (e.g., aminoglycoside-induced nephrotoxicity); some stimulate changes without toxicity (e.g., glucocorticoid-induced alkaline phosphatase); and others interfere with hormones. Drugs can interfere with diagnostic tests in many other ways. The American Association of Clinical Pathologists publishes a handbook that summarizes changes in clinical pathology that might be drug induced. Access to this text or its information may be possible by contacting the appropriate diagnostic laboratory.
Few studies focus on the impact of drugs on clinical laboratory tests. Examples include the impact of bromide (when used as an anticonvulsant) on chloride concentrations when flame ionization is used: the two cannot be distinguished when this method is used, and bromide, being present in much higher concentrations, will artifactually increase chloride. The impact of selected antimicrobials on urine glucose also has been documented in the dog. At 22 mg/kg, cephalexin caused false-positive glucose in 50% of dogs at 6 hours and 33% of dogs at 24 hours when using selected commercially available glucose strips (Chemstrips, Boeringer Ingelheim). A tablet test (Clinitest, Miles Inc.) indicated false-positive results in 100% of dogs (n=6) at 6 and 24 hours after administration of cephalexin at 22 mg/kg; 50% of dogs receiving enrofloxacin at 5 mg/kg and 100% receiving 10 mg/kg also yielded false-positive results.105 Enrofloxacin also caused false negatives at concentrations as low as 20 μg/mL in urine spiked with dextrose at 0.5% and above.
1. Nebeker J.R., Barach P., Samore M.H. Clarifying adverse drug events: a clinician’s guide to terminology, documentation, and reporting. Ann Intern Med. 2004;140:795-801.
2. Lawson D.H., Richard R.M.E. Clinical pharmacy and hospital drug management. London: Chapman & Hall; 1982. pp 211-237
3. Griffin J.P., D’Arcy P.F. A manual of adverse drug interactions, ed 2. Chicago: Billing & Sons; 1979. 3–51
4. Pirmohamed M., Breckenridge A.M., Kitteringham N.R., et al. Adverse drug reactions. Br Med J. 1998;316:1295-1298. 25
5. Meyer U.A., Gut J. Genomics and the prediction of xenobiotic toxicity. Toxicology. 2002:463-466. 181-182
6. Ariens E.J., Simonis A.M., Offermeier J. Introduction to general toxicology. New York: Academic Press; 1976. pp 79-125
7. Mitchell J.R., Smith C.V., Lauferburg B.H., et al. Reactive metabolites and the pathophysiology of acute lethal cell injury. In: Mitchell J.R., Homing M.G., editors. Drug metabolism and drug toxicity. New York: Raven Press; 1984:301-318.
8. Klassen D.C. Principles of toxicology. In: Klassen C.D., Amour M.O., Doull J., editors. Toxicology: the basic science of poisons. ed 3. New York: Macmillan; 1985:11-20.
9. Ju C., Uetrecht J.P. Mechanism of idiosyncratic drug reactions: reactive metabolites formation, protein binding and the regulation of the immune system. Curr Drug Metab. 2002;3:367-377.
10. Schwarz M., Wiskiel R. Medication reconciliation: developing and implementing a program. Crit Care Nurs Clin North Am. 2006;18:502-507.
11. Young D.S. Effects of drugs on clinical laboratory tests, ed 3. Washington, DC: American Association for Clinical Chemistry; 1990.
12. To H., Kikuchi A., Tsuruoka S., et al. Time-dependent nephrotoxicity associated with daily administration of cisplatin in mice. J Pharm Pharmacol. 2000;52(12):1499-1504.
13. Bleyzac N., Allard-Latour B., Laffont A., et al. Diurnal changes in the pharmacokinetic behavior of amikacin. Ther Drug Monit. 2000;22(3):307-312.
14. Osweiler G.D. General toxicological principles. In Peterson M.E., Talcott P.A., editors: Small animal toxicology, ed 2, St Louis: Saunders, 2006.
15. Sipes I.G., Dart R.C. Principles of toxicology. In Wecker L., editor: Human pharmacology: molecular to clinical, ed 5, Philadelphia: Mosby, 2009.
16. Guengerich F.P. Cytochrome P450 and chemical toxicology. Chem Res Toxicol. 2008;21:70-83.
17. Roberston J.D., Orrenius S. Role of mitochondria in toxic cell death. Toxicology. 2002:491-496. 181-182
18. Ingelman-Sundberg M. Polymorphism of cytochrome P450 and xenobiotic toxicity. Toxicology. 2002:447-452. 181-182
19. Martinez M., Modric S., Sharkey M., et al. The pharmacogenomics of P-glycoprotein and its role in veterinary medicine,. J Vet Pharmacol Ther. 2008;31(4):285-300.
20. Food and Drug Administration. Information and requirements for review and approval of new animal drug applications (NADAs). October 14, 2009. at www.fda.gov/AnimalVeterinary/DevelopmentApprovalProcess/NewAnimalDrugApplications/default.htm
21. Woodward K.N. Veterinary pharmacovigilance. Part 6. Predictability of adverse reactions in animals from laboratory toxicology studies. J Vet Pharmacol Ther. 2005;28:213-231.
22. Institute of Medicine. Perspectives, methods, and data challenges, workshop summary. In nutritional risk assessment. Washington, DC: National Academies Press; 2007.
23. Colbert B.L., Biron P. Pharmacovigellence from A to Z: adverse drug event surveillance. Malden, Miss: Blackwell Science; 2002.
24. Hughes D.A., Bayoumi A.M., Pirmohamed M. Current assessment of risk-benefit by regulators: Is it time to introduce decision analyses? Clin Pharmacol Ther. 2007;82:123-127.
25. Hampshire V.A., Doddy F.M., Post L.O. Adverse drug event reports at the United States Food and Drug Administration Center for Veterinary Medicine. J Am Vet Med Assoc. 2004;225:533-536.
26. Guengerich F.P., MacDonald J.S. Applying mechanisms of chemical toxicity to predict drug safety. Chem Res Toxicol. 2007;20:344-369.
27. Kitoh K., Watoh K., Chaga K., et al. Clinical, hematological and biochemical findings in dogs after induction of shock by injection of heartworm extract. Am J Vet Res. 1994;55:1535-1540.
28. Nakamura H., Matsuse H., Obase Y., et al. Clinical evaluation of anaphylactic reactions to intravenous corticosteroids in adult asthmatics. Respiration. 2002;69:309-313.
29. Landor M., Lashinsky A., Waxman J. Quinolone allergy? Ann Allergy Asthma Immunol. 1996;77:273-276.
30. Cribb A. Adverse reactions to sulphonamide and sulphonamide-trimethoprim antimicrobials: clinical syndromes and pathogenesis. Adverse Drug React Toxicol Rev. 1996;15:9-50.
31. Tilles S.A. Practical issues in the management of hypersensitivity reactions: sulfonamides. South Med J. 2001;94(8):817-824.
32. Trepanier L.A., Danhof R., Toll J., et al. Clinical findings of 40 dogs with hypersensitivity associated with administration of potentiated sulfonamides. J Vet Intern Med. 2003;17:647-652.
33. Trepanier L.A. Idiosyncratic toxicity associated with potentiated sulfonamides in the dog. J Vet Pharmacol Ther. 2004;27:129-138.
34. Ockner R.K. Drug-induced liver disease. In: Zakim D., Boyer T.D., editors. Hepatology: a textbook of liver disease. Philadelphia: Saunders; 1982:691-722.
35. Plaa G.L. Toxic responses of the liver. In: Klassen C.D., Amour M.O., Doull J., editors. Toxicology: the basic science of poisons. ed 3. New York: Macmillan; 1985:286-309.
36. Lee W.M. Review article: drug-induced hepatotoxicity. Aliment Pharmacol Ther. 1993;7:4775-4785.
37. Bunch S.E. Hepatotoxicity associated with pharmacologic agents in dogs and cats. Vet Clin North Am Small Anim Pract. 1993;23(3):659-670.
38. Schenkers B. Drug disposition and hepatotoxicity in the elderly. J Clin Gastroenterol. 1994;18:232-237.
39. Smith S.W., Howland M.A., Hoffman R.S., et al. Acetaminophen overdose with altered acetaminophen pharmacokinetics and hepatotoxicity associated with premature cessation of intravenous N-acetylcystein therapy. Ann Pharmacother. 2008;42(9):1333-1339.
40. Ndiritu C.A., Weigel J.W. Hepatorenal injury in a dog associated with methoxyflurane. Vet Med Small Anim Clin. 1977;72:545-550.
41. Grant P.S., Meuten D.J., Pecquet-Croad M.E. Hepatic necrosis associated with the use of halothane in the dog. J Am Vet Med Assoc. 1984;184:478-480.
42. Polzin D.J., Stowe C.M., O’Leary T.P., et al. Acute hepatic necrosis associated with the administration of mebendazole to dogs. J Am Vet Med Assoc. 1981;179:1013-1016.
43. Van Cavteren H., Marsboom R., Vanderberghe J., et al. Safety studies evaluating the effect of mebendazole on liver function in dogs. J Am Vet Med Assoc. 1983;183:93-98.
44. Twedt D.C., Diehl K.J., Lappin M.R., et al. Association of hepatic necrosis with trimethoprim sulfonamide administration in 4 dogs. J Vet Intern Med. 1997;11:20-23.
45. Raynaud J.P. Thiacetarsamide (adulticide) versus melarsomine (RM 340) developed as macrofilaricide (adulticide and larvicide) to cure canine heartworm infection in dogs. Ann Rech Vet. 1992;23:1-25.
45a. Dunayer E.K. New findings on the effects of xylitol ingestion in dogs. Vet Med. 2006:791-797.
45b. Dunayer E.K. Gwaltney-Brant SM: Acute hepatic failure and coagulopathy associated with xylitol ingestion in eight dogs. J Am Vet Med Assoc. 2006;229:1113-1117.
46. Perazella M.A. Drug-induced nephropathy: an update. Expert Opin Drug Saf. 2005;4(4):689-706.
47. Engelhardt J.A., Brown S.A. Drug-related nephropathies. Part II: Commonly used drugs. Compend Contin Educ Pract Vet. 1987;9:281-288.
48. Taber S.S., Mueller B.A. Drug-associated renal dysfunction. Crit Care Clin. 2006;22:357-374.
49. Pedersoli W.M. Serum fluoride concentration, renal and hepatic function test results in dogs with methoxyflurane anesthesia. Am J Vet Res. 1977;38:949-953.
50. Torpet L.A., Kragelund C., Treibel J., et al. Oral adversed drug reactions to cardiovascular drugs. Crit Rev Oral Biol Med. 2004;15(1):28-46.
51. Mouatt J.G. Cyclosporin and ketoconazole interaction for treatment of perianal fistulas in the dog. Aust Vet J. 2002;80(4):207-211.
52. Halliwell R.F., Davey P.G., Labert J.J. The effects of quinolones and NSAIDS upon GABA-evoked currents recorded from rat dorsal root ganglion neurons,. J Antimicrob Chemother. 1991;27:209-218.
53. Thomas R.J. Neurotoxicity of antibacterial therapy. South Med J. 1994;87(9):869-874.
54. Simpson S. Treatment of metronidazole toxicity. Standards of care: emergency and critical care medicine. 2003;5:11.
55. Fillastre J., Leroy A., Borsa-Lebas F., et al. Effects of ketoprofen (NSAID) on the pharmacokinetics of pefloxacin and ofloxacin in healthy volunteers. Drugs Exp Clin Res. 1992;18:487-492.
56. Neer T.M. Drug-induced neurologic disorders. Proc Am Coll Vet Intern Med. 1991;9:261-269.
57. Pullium J.R., Seward R.L., Henry R.T., et al. Investigating ivermectin toxicity in collie dogs. Vet Med. 1985;80:33-40.
58. Tranquili W.J., Paul A.J., Todd K.S. Assessment of toxicosis induced by high dose administration of milbemycin in collies. Am J Vet Res. 1991;52:1170-1172.
59. Mealey K., Pharmacogenetic. Vet Clin North Am Small Anim Pract Pharm. 2006;36:961-973.
60. Mealey K.L., Bentjen S.A., Waiting D.K. Frequency of the mutant MDR1 allele associated with ivermectin sensitivity in a sample population of collies from the northwestern United States. Am J Vet Res. 2002;63:479-481.
61. Muhammad G., Abdul J., Khan M.Z., et al. Use of neostigmine in massive ivermectin toxicity in cats. Vet Hum Toxicol. 2004;46(1):28-29.
62. Sevine F. Letters: picrotoxin, the antidote to ivermectin in dogs? Vet Rec. 1985;116:195-196.
63. Sartor L.L., Bentjen S.A., Trepanier L., et al. Loperamide toxicity in a collie with MDR1 mutation associated with ivermectin sensitivity in Collies. J Vet Intern Med. 2004;18:117-118.
64. Yilmaz H.L., Yildizdas D.R. Amitraz poisoning, an emerging preventive strategies epidemiology, clinical features, management, and preventive strategies. Arch Dis Child. 2003;88:130-134.
65. Fulton R.B., Fruechte L.K. Poisoning induced by administration of a phosphate-containing urinary acidifier in a cat. J Am Vet Med Assoc. 1991;198:883-995.
66. Davidson G. To benzoate or not to benzoate: cats are the question. Int J Pharm Comp. 2001;5:89-90.
67. Bedford P.G.C., Clarke M.A. Suspected benzoic acid poisoning in the cat. Vet Rec. 1971;188:599-601.
68. Fraunfelder F.W. Ocular adverse drug reactions. Expert Opin Drug Saf. 2003;2(4):411-420.
69. Wiebe V., Hamilton P. Fluoroqinolone-induced retinal degeneration in cats. J Am Vet Med Assoc. 2002;221:1568-1571.
70. Mauget-Faysse M., Quaranta M. Incidental retinal phototoxicity associated with ingestion of photosensitizing. Arch Clin Exp Ophthalmol. 2001;239(7):501-508.
71. Patel M. Ocular side effects of systemic drugs. Optometry. 2002;28:33-36.
72. Buckley R., Graham E., Jones S., et al. Oculartoxicity and hydroxychloriquine: guidelines for screening 2009 (replacing the Royal College of Ophthalmologists Guidelines, 2004). April 2004. http://www.library.nhs.uk/guidelinesfinder/ViewResource.aspx?resID=36837
73. Marutani K., Matsumoto M., Otabe Y., et al. Reduced phototoxicity of a fluoroquinolone antibacterial agen with a methoxy group at the 8 position in mice irradiated with long-wave length UV light. Antimicrob Agents Chemother. 1993;37:2217-2222.
74. Huang M.Y., Schacht J. Drug-induced ototoxicity: pathogenesis and prevention. Med Toxicol Adverse Drug Exp. 1989;4:452-467.
75. Griffin J.P. Drug-induced ototoxicity. Br J Audiol. 1988;22:195-210.
76. Matz G., Rybak L., Roland P.S., et al. Ototoxicity of ototopical antibiotic drops in humans. Otolaryngol Head Neck Surg. 2004;130:S79-S82.
77. Humes H.D. Insights into ototoxicity. Analogies to nephrotoxicity. Clin Pharmacokinet. 2000;38(4):367-375.
78. Webster J.C., Carroll R., Benitez I.T., et al. Ototoxicity of topical gentamicin in the cat. J Infect Dis. 1971;124:S138-S144.
79. Roland P.S., Tybak L., Hannley M., et al. Animal ototoxicity of topical antibiotics and the relevance to clinical treatment of human subjects. Otolaryngol Head Neck Surg. 2004;130:S57-S78.
80. Wokenstein P., Revus J. Drug-induced severe skin reactions. Incidence, management and prevention. Drug Saf. 1995;13:56-68.
81. Mouatt J.G. Cyclosporin and ketoconazole interaction for treatment of perianal fistulas in the dog. Aust Vet J. 2002;80(4):207-811.
82. Downard C.D., Roberts L.J.II, Morrow J.D. Topical benzoic acid induces the increased biosynthesis of prostaglandin D2 in human skin in vivo. Clin Pharmacol Ther. 1995;57(4):441-445.
83. Quintero B., Miranda M.A. Mechanisms of photosensitization induced by drugs: A general survey. Ars Pharmaceutica. 2000;41(1):27-46.
84. Boothe D.M. Effects of drugs on endocrine testing. In: Bonagura J., Kirk R.W., editors. Current veterinary therapy (XII), small animal practice. Philadelphia: Saunders; 1995:339-346.
85. Center S.A., Mitchell J., Nachreiner R.F., et al. Effects of propranolol on thyroid function in the dog. J Am Anim Hosp Assoc. 1981;17:813-822.
86. Kemppainen R.J., Sartin J.L. Effects of single intravenous doses of dexamethasone on baseline plasma cortisol concentrations and responses to synthetic ACTH in healthy dogs. Am J Vet Res. 1984;45:742-746.
87. Spencer K.B., Thompson F.N., Clekis T., et al. Adrenal gland function in dogs given methylprednisolone. Am J Vet Res. 1980;41(9):1503-1506.
88. Roberts S.M., Lavach J.D., Macy D.W., et al. Effect of ophthalmic prednisolone acetate on the canine adrenal gland and hepatic function. Am J Vet Res. 1984;45:1711-1713.
89. Zenoble R.D., Kemppainen R.J. Adrenocortical suppression by topically applied corticosteroids in healthy dogs. J Am Vet Med Assoc. 1987;191:685-688.
90. Hostettler K.A., Wrighton S.A., Molowa D.T., et al. Coinduction of multiple hepatic cytochrome P-450 proteins and their mRNAs in rats treated with imidazole antimycotic agents. Mol Pharmacol. 1988;35:279-285.
91. Willard M.D., Nachreiner R., McDonald R., et al. Hormonal and clinical pathologic changes with long-term ketoconazole therapy in the dog and cat. Proc Am Coll Vet Intern Med. 1986;6:13.25-13.27.
92. Wenzel K.W. Pharmacological interference with in vitro tests of thyroid function. Metabolism. 1981;30:717-732.
93. Moriello K.A., Halliwell R.E.W., Oakes M. Determination of thyroxine, triiodothyronine, and cortisol changes during simultaneous adrenal and thyroid function tests in healthy dogs. Am J Vet Res. 1987;48:456.
94. Reimers T.J., Concannon P.W., Cowan R.G. Changes in serum thyroxine and cortisol in dogs after simultaneous injection of TSH and ACTH. J Am Anim Hosp Assoc. 1982;18:923-925.
95. Moriyama K., Tagami T., Usui T., et al. Antithyroid drugs inhibit thyroid hormone receptor-mediated transcription. J Clin Endocrinol Metab. 2007;92(3):1066-1072.
96. Campbell K., Chambers M.D., Davis C.A., et al. Effects of trimethoprim/sulfamethoxazole on thyroid physiology in dogs. Proc Am Coll Vet Dermatol. 1995;11:15-16.
97. Franklin A.L., Chaikoff T.I. The effect of sulfonamides on the conversion in vitro of inorganic iodide thyroxine and diiodotyrosine by thyroid tissue with radioactive iodine as indicator. J Biol Chem. 1943;152:295-301. Accessed Dec. 18, 2009, at http://www.jbc.org/content/152/2/295.full.pdf
98. Benichou C., Celigny P.S. Standardization of definitions and criteria for causality assessment of adverse drug events. Drug-induced blood cytopenias: report of an International Consensus Meeting. Nouv Rev Fr Hematol. 1991;33:257-262.
99. Stroncek D.F. Drug induced immune neutropenia. Transfus Med Rev. 1993;VII:268-274.
100. Homeida M.M., Bagi I.S., Ghalib H.W. Prolongation of prothrombin time with ivermectin. Lancet. 1998;1:1346-1347.
101. Vandendries E.R., Drews R.E. Durg-associated disease: hematologic dysfunction. Crit Care Clin. 2006;22:347-355.
102. Keller W.C., Bataller N., Oeller D.S. Processing and evaluation of adverse drug experience reports at the Food and Drug Administration Center for Veterinary Medicine. J Am Vet Med Assoc. 1998;213:208-211.
103. Committee on Examining the Safety of Dietary Supplements for Horses. Dogs, and Cats; National Research Council: Safety of dietary supplements for horses, dogs, and cats. Washington, DC: National Academies Press; 2008.
104. Boothe D.M. Veterinary compounding in small animals: a clinical pharmacologist’s perspective. Vet Clin North Am Small Anim Pract. 2006;36:1129-1173.
105. Rees C.A., Boothe D.M. Evaluation of the effect of cephalexin and enrofloxacin on clinical laboratory measurements of urine glucose in dogs. J Am Vet Med Assoc. 2004;224:1455-1458.