Chapter 27 Anticonvulsants and Other Neurologic Therapies in Small Animals
Successful control of seizures with anticonvulsant drugs reflects a balance in achieving seizure control and minimizing undesirable drug side effects. Variability in the disposition of anticonvulsants and interactions among them and other drugs are important confounders of successful therapy. This chapter reviews selected anticonvulsants, focusing on drugs most likely to control seizures in small animals However, drugs that have been used historically and selected drugs that are used in humans but for which information is available in dogs or cats are also discussed, in part to explain why they may be less than ideal choices.The proper use of anticonvulsants is discussed, with an emphasis on the differences in individual drug disposition, detection of these differences (e.g., therapeutic drug monitoring), and rational approaches to responding to these differences by dose modification. The primary topic of discussion is treatment of generalized, tonic–clonic seizures, the most common type afflicting small animals. Opinions regarding anticonvulsant therapy vary among clinicians. Treatment of behavioral disorders that might manifest as seizural type activity is discussed in Chapter 26. Some of the comments and recommendations offered in the discussions of selected drugs reflect personal observations obtained in the direction of a therapeutic drug monitoring service or through clinical trials that focus on the use of anticonvulsants either alone or in combination with phenobarbital.
It is important to approach epilepsy as a clinical manifestation of an underlying disease. Thus therapy is more likely to be effective if the underlying disease is treated. Such causes should be identified and appropriately treated—if possible, before chronic anticonvulsant therapy is instituted. Neutering of affected animals (male and female) is strongly encouraged not only for ethical reasons (i.e., precluding perpetuation of contributing genes) but also to minimize the potential adverse effect of circulating sex hormones on neuronal membrane stability. Unfortunately, the underlying cause of epilepsy often cannot be identified (idiopathic epilepsy) or, if identified, cannot be corrected such that seizures are adequately controlled. In such instances, regardless of the cause of seizures, management is based on control with anticonvulsant drugs. Undesirable side effects are often the limiting factor in the use of anticonvulsant drugs, and not all seizures necessarily require treatment.
KEY POINT 27-1
Long-term therapy with anticonvulsant drugs increases the risk of adverse reactions and drug interactions in the epileptic patient.
Certainly immediate, short-term anticonvulsant therapy is indicated for status epilepticus (see later definition) or cluster seizures. Chronic therapy is generally indicated for seizures that last more than 3 minutes, cluster seizures (for which there is no delineable interictal period), or seizures that occur more frequently than once a month. Seizures that are not sufficiently controlled can lead to additional seizuring (kindling) or to the development of a second “mirror” focus of seizure activity. This might be manifested as a decreasing interictal period or a worsening of seizure activity (including duration).
Physiology and Seizure Pathophysiology
Anatomy
Neurons are either layered or clustered (forming nuclei) and classified according to function (sensory, motor, or interneuron), location, or the neurotransmitter synthesized and released by the neuron.1 Neurons are outnumbered by several orders of magnitude by other cells, including macroglia, microglia, and cells of the vascular elements. Microglia are related to macrophage/monocyte lineage and are either resident or recruited during inflammation. The macroglia are the most abundant of the supportive cells and include, among other cells, astrocytes and oligodendroglia. The latter are the myelin-producing cells, whereas astrocytes provide metabolic support and remove extracellular neurotransmitters.
Barriers to Drug Distribution
The blood–brain barrier and blood–cerebrospinal fluid (CSF) barrier form a permeability boundary, rendering the brain a sanctuary by limiting penetration of macromolecules. They also act as selective barriers to inflow and outflow of small, charged molecules, including drugs, neurotransmitters, and their precursors or metabolites. The blood–brain barrier is absent in selected locations, thus allowing the brain to monitor chemicals. These areas include but are not limited to the area postrema, median eminence of hypothalamus, and the posterior pituitary and pineal glands. Cerebral ischemia and inflammation modify the integrity and function of the barriers. The blood–brain barrier comprises blood capillaries that differ in structure compared to other tissues in that they lack fenestrae and are joined by tight junctions similar to the epithelium of other tissues. Microvessels comprise about 95% of the total surface area of the barrier. Astrocyte foot processes encapsulate the capillaries, forming the tight junctions (Figure 27-1). Because pinocytosis also is absent, compounds can enter the brain only by passive diffusion, limiting penetration to small, lipid-soluble, non-ionized molecules. In addition to the physical barrier, function mechanisms exist to preclude central nervous system (CNS) penetration. These include degradative enzymes, which target not only chemicals but also many peptides, and high concentrations of efflux transporters. These include adenosine triphosphate (ATP)–binding cassette proteins such as P-glycoprotein (among the most abundant and most well characterized) and others, resulting in a combined broad substrate specificity that targets many drugs.2 The substrate specificity of the P-glycoprotein transporter is large; its role in breed susceptibility to CNS adverse drug reactions is discussed in Chapters 2 and 3. Interactions involving P-glycoprotein (or other efflux transporters) are among the drug interactions described at the level of the blood–brain barrier.3 In the dog this protein is regulated by only one gene (ABCB1).4
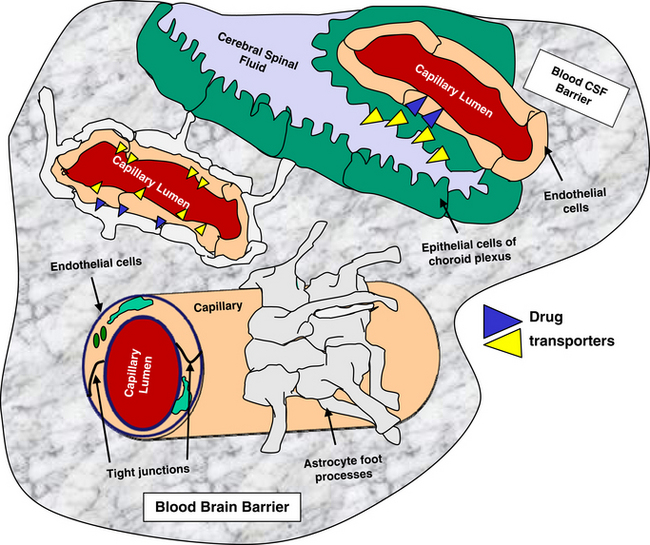
Figure 27-1 Diagrammatic representation of the blood brain (left) and blood–cerebrospinal fluid (right) barriers, structures that limit compound movement from blood into the interstitial fluids of brain parenchyma. The endothelium of the capillaries of the blood–brain barrier lack fenestrae, are joined by tight junctions, and are encapsulated by astrocyte foot processes. Only small, lipid-soluble, non-ionized molecules in the blood stream are likely to move through the endothelium into the brain. The blood–CSF barrier is formed from the epithelium of the choroid plexus, which forms tight junctions. The plexus dips into a double-celled structure made from folding of ependymals cells on themselves. (See text for other mechanisms by which drugs might be excluded from each of these sites.)
The blood–CSF barrier prevents compounds moving from interstitial fluid into brain parenchyma.5 This barrier is located in the epithelium of the choroid plexus, limiting movement of compounds from the blood into the CSF. The choroid plexus consists of the highly vascularized pia mater and cells with microvilli on the CSF side. The plexus dips into pockets formed by folding of ependymal cells onto themselves, forming a double layer structure between the dura and pia (i.e., the arachnoid membrane). This double layer contains the subarachnoid space. Although the capillaries of the choroid plexus are fenestrated, the adjacent choroidal epithelial cells form tight junctions, precluding movement of most macromolecules. Like the blood–brain barrier, the blood–CSF barrier contains organic acid transporters that preclude penetration of many therapeutic agents.6 The role of efflux pumps and P-glycoprotein in particular is discussed later.
The formidable barrier presented to chemical penetration of the brain contributes to therapeutic failure when treating dysfunction of the CNS. A number of strategies have been described to enhance drug delivery to the brain.6 These include manipulation of the drugs such that they are chemically more likely to penetrate the barrier, administration of prodrugs, the combination of drugs with compounds that decrease drug efflux, the design of vectors or other carriers that are able to penetrate, and temporary disruption of the barriers themselves. The use of nanotechnology offers potentially viable options.7 Finally, alternative routes of delivery include intraventricular, intrathecal, olfactory, and direct interstitial routes. None thus far has proved consistently safe or relevant to all therapeutic agents.
Physiology of Neuronal Activation
Neuronal activity can be manipulated through molecular mechanisms at several levels: (1) ion channels, which mediate excitability; (2) neurotransmitters and their receptors; (3) auxiliary intramembranous or cytoplasmic transductive molecules that couple receptor signals to intracellular actions; and (4) neurotransmitter transporters that facilitate their conservation through reaccumulation into the terminal (plasma membrane proteins) and then synaptic vesicles (vesicular transporters) (Figure 27-2). The combined influence of these molecular entities regulate three major cations (Na+, K+ and Ca2+) and the major anion (Cl-). The cations are directly regulated through voltage-dependent Ca2+, N+, and K+ channels, allowing for rapid changes in ion permeability, thus facilitating the excitation–secretion coupling necessary for transmitter release. They also are influenced by ligand-gated ion channels, which in turn are regulated by neurotransmitter binding. These include but are not limited to glutamate receptors and ionophore receptors for acetylcholine (nicotinic), GABAA, and glycine.1
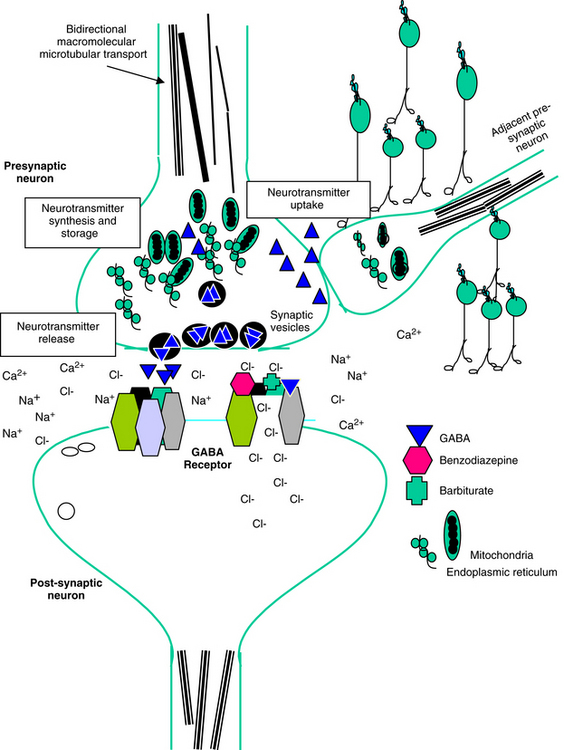
Figure 27-2 Potential therapeutic targets of anticonvulsant and behavior-modifying drugs include the presynaptic nerve terminal and the postsynaptic neuron. Presynaptically, microtubules allow for bidirectional transport of macromolecules to and from the neuronal cell body and distal processes; motors responsible for transport may be subject to drug action. Active uptake, synthesis, storage, and release of neurotransmitters may also be targeted. Postsynaptically, neurotransmitter-receptor interaction can be targeted. The receptor for gamma-aminobutyric acid (GABA) has binding sites for GABA, barbiturates, and benzodiazepines. Interaction between the drug and receptor causes a channel in the receptor to open to chloride flux. Increased chloride concentrations inside the cell increase electronegativity, thus hyperpolarizing the cell. Intracellular targets that express synaptic activity may also be targeted.
Neurons generally release the same substance at each synpaptic terminal in which the neuron is involved. Neurotransmitters generally result in either excitation, reflecting an influx of positively charged ions, depolarizations, and reduced membrane resistance, or inhibition, resulting in hyperpolarization and decreased membrane resistance. Selected transmitters (e.g., monoamines and peptides) may enhance or suppress the classical neuronal response to the neurotransmitter. Neurohormones receive synaptic information from central neurons and respond by secreting transmitters into circulation. Neuromodulators originate from nonsynaptic sites but influence nerve cell excitability. Examples include CO2; ammonia; circulating steroid hormones (neurosteroids); and locally released adenosine, eicosanoids, and nitric oxide. Neuromediators participate in the postsynaptic response and are exemplified by the secondary messengers cyclic AMP or GMP and inositol phosphates.
Neurotransmitters
Amino acid neurotransmitters include the excitatory dicarboxylic amino acids glutamate and aspartate and the inhibitory monocarboxylic ω-amino acids glycine, gamma-aminobutyric acid (GABA), β-alanine, and taurine (Figure 27-3, Table 27-1). Because of their ubiquitous distribution in the brain and their rapid, reversible, and reduntant effects, amino acid neurotransmitters were precluded from initial inclusion as classical neurotransmitters. Glycine, glutamate, and GABA are now considered central neurotransmitters. GABA is enzymatically formed from glutamic acid by glutamic acid decarboxylase. The effects of GABA are experimentally identified in response to picrotoxin and bicuculline. These include inhibitory actions at the level of the local interneuron and potentially presynaptic inhibition in the spinal cord. Convulsant compounds that appear to act through GABA receptors include the selective antagonists penicillin and pentylenetetrazole. Nontherapeutic agents have also been identified that mimic GABA, inhibit its active reuptake, or alter its turnover. Three types of GABA receptors have been identified: A, B, and C, with type A being the most common. It is a ligand-gated, chloride channel ionotropic receptor and is the site of action of many drugs, including benzodiazepines, barbiturates, anesthetic steroids, and volatile anesthetics. The B type GABA receptor is a G protein–coupled receptor (GPCR) that interacts with Gi to inhibit adenylyl cyclase, activate K+ channels, and reduce Ca2+ conductance. The B type receptors act presynaptically as autoreceptors, inhibiting GABA release. Type C GABA receptors are less common, but GABA is much more potent for this receptor. The type C receptor is distinguished by its lack of interaction with several compounds that interact with type A GABA receptors, including baclofen, benzodiazepines, and barbiturates.
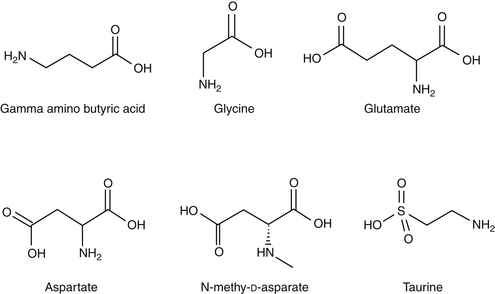
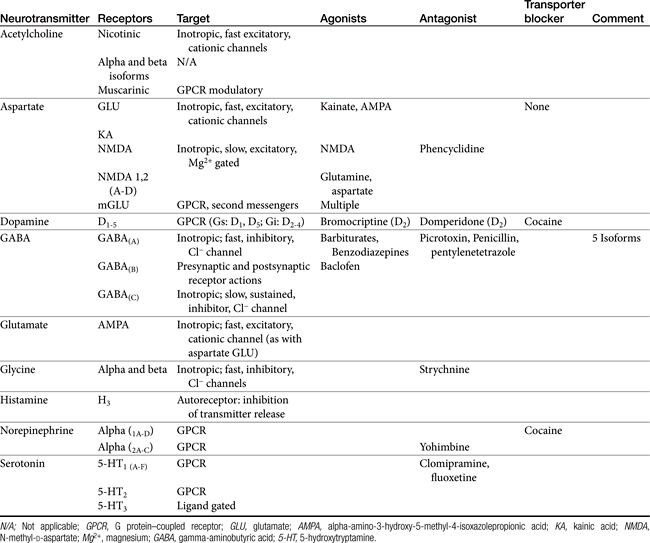
Amino Acids
Glycine receptors are located primarily in the brainstem and spinal cord, where they are inhibitory in nature.
Glutamate and aspartate occur in very high concentrations and are powerful excitatory signals throughout the brain. Glutamate receptors are either ligand-gated ionotropic receptor channels or G protein coupled. Ligand-gated ion channels include N-methyl-d-aspartate (NMDA) and non-NMDA receptors (alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] and kainic acid [KA]).1,8,9 The NMDA receptors, derivatives of aspartic acid, are pH sensitive, and response to endogenous signals, including zinc ions, selected neurosteroids, arachidonic acid metabolites, redox regions, and selected polyamines. Although NDMA receptors are involved in normal synaptic transmission, they appear to be more closely involved with induction of synaptic plasticity. This includes long-term potentiation, which reflects an increase in the magnitude of the postsynaptic response to presynaptic stimulation of a specific strength. The activation of NMDA receptors requires simultaneous firing of at least two neurons, including binding of glutamate released from the synapse and simultaneous postsynaptic depolarization from different neurons. High concentrations of glutamate can cause neuronal death. The mechanism partially, but not solely, reflects excessive activation of NMDA or AMPA/KA receptors and calcium influx. For example, ischemia or hypoglycemia may be associated with massive glutamate release; when coupled with impaired cellular reuptake, cell death may occur. Depletion of Na+ and K+ and increased extracellular Zn2+ may play a role in activation of necrotic or apoptotic cascades.1 The role of these receptors in pain is discussed in Chapter 28.
Monoamines
Acetylcholine is an excitatory neurotransmitter for Renshaw interneurons and other CNS cells. Actions reflect interactions between nicotinic and muscarinic receptors.
Several catecholamines act as neurotransmitters in the CNS. Dopamine comprises more than 50% of CNS catecholamine and is found in high concentrations is selected areas. Its effect depends on the subreceptor targeted, with five thus far identified in the brain (see Table 27-1). Epinephrine is present only in limited areas, and its role has not yet been identified. However, norepinephrine is found throughout the brain but in very large concentrations in the hypothalamus, certain zones in the limbic system, and other regions. All three receptor types (alpha 1, alpha 2, and beta) and subtypes are located in the brain. The nonclassical electrophysiologic synaptic effects of norepinephrine vary with the state but are considered “enabling.” Although excitatory in nature, the effects of norepinephrine (and serotonin) can be anticonvulsant, rather than proconvulsant.10
Biogenic Amines
Serotonin is one of two biogenic amines active in the CNS. Thus far 14 receptor (5-hydroxytryptamine [5-HT]) types or subtypes are found in CNS nuclei that are located in or adjacent to the midline regions of the pons and upper brainstem. Up to five different 5-HT1 receptor subtypes inhibit adenylyl cyclase or regulate K+ and Ca2+ channels. 5-HT2 receptor subtypes (three) are linked to G proteins and phospholipase C. 5-HT2C may modulate CSF production. 5-HT3 receptors are located in the area postrema and solitary tract nucleus, where they cause emesis and antinociception. Members of the 5-HT4 through 5-HT7 receptor types have not yet been studied in the CNS.
Histamine is the second biogenic amine that serves as a neurotransmitter. Four subtypes have been described in the CNS, all acting through G proteins. Most neurons that synthesize histamine are located in the ventral posterior hypothalamus and extend throughout the entire CNS through ascending and descending tracts.1
Synaptic Vesicle Protein
The importance of the synaptic vesicle protein 2 (SV2) emerged as part of the research into the mechanism of action of levetiracetam. Levetiracetam, but not other anticonvulsants, binds to synaptic vesicle (SV) protein through the brain in a steroselective, concentration-dependent manner.11 However, ethosuximide, pentobarbital, and pentylenetetrazole compete with levetiracetam for binding. The distribution of the protein does not follow any pattern consistent with classical receptors, and the protein is located in synaptic vesicled (SVs). Three isoforms exist (A, B, C), with SV2A the most widely distributed. Knockout mice without SV2A die within 3 weeks as a result of spontaneous seizures. The role of SV2A is not clear, but it may modulate exocytosis that precedes neurotransmitter release; disruption of activity appears to result in calcium accumulation and increased neurotransmitter release.11
The Action Potential
The normal resting membrane potential (RMP) of the neuronal cell is 70 mV. The electrical difference across the cell membrane is maintained by a Na+, K+-ATPase pump. Depolarization and the generation of an action potential occur when the RMP becomes sufficiently positive to reach threshold. As with other membranes, the RMP of a neuron is determined by the concentration of negative and positive ions across the membrane. Important ions include Na+, K+, Ca2+, and Cl–. The concentration reflects ion fluxes and thus permeability of the cell membrane to the ions. Fluxes resulting in an increase in positive ions inside the cell relative to the outside hypopolarize the RMP, bringing it closer to threshold and subsequent depolarization. The tendency of a neuron to depolarize reflects, in part, the sum total effect of neurotransmitters (NTs) that interact with the cell membrane. Inhibitory NTs such as GABA render the RMP more negative and less susceptible to depolarization (see Figure 27-2).12 The principal postsynaptic receptor for GABA is the GABAA receptor; activation increases inflow of Cl- ions into the cell, hyperpolarizing the neuron. Glycine and serine are also inhibitory neurotransmitters. Excitatory neurotransmitters, such as acetylcholine and glutamate, elevate the RMP to a more positive status, rendering it more susceptible to reaching the threshold necessary for depolarization.12
Pathology
Conventional evidence indicates that neurons are terminally differentiated cells and do not respond to proliferate stimuli. The presence of neuronal stem cells may, however, offer an avenue of treatment for degenerative conditions or damage. In the absence of proliferation, neurons have developed adaptive functional and structural responses to injury.1
Several animal models have been used to study either seizures or epilepsy. Models intended to study epilepsy ideally include both seizure activity as well as chronic epileptiform behavior (i.e., spontaneous seizures).13 The attributes of animal models are generally based on their relevance to human epilepsy in regard to electrophysiologic activity, etiology, pathologies, signalment, associated behavioral changes, and response to anticonvulsant drugs. That multiple models have been developed for study reflects the more than 100 human seizure or epileptic disorders. Chemical convulsants that cause seizures after systemic or direct administration include KA (a glutamate analog), pilocarpine, NMDA, and GABA antagonists, Pentylenetetrazole is among the most commonly used chemicals; repeated injections mimic electrical kindling. Injections of high doses consistently and predictably cause tonic–clonic seizures, allowing its use as a screen for new drugs. Lower doses induce absencelike seizures. Kindling models are considered epileptic models and involve periodic stimulation of low-intensity signals in the amygdala, resulting in tonic–clonic seizures. The animal remains sensitive to electrical stimulation, with the number of stimuli necessary to induce seizures quantifiable. Genetic models have been either developed or genetically designed;13 for example, deletion of the gene encoding tyrosine kinase prevents the genesis of epilepsy in the kindling model administration of chemoconvulsants.1
Seizures are the clinical results of rapid, excessive neuronal discharge in the brain. Seizures are classified as primary (i.e., genetic) or acquired, and as generalized or focal. Generalized seizures are much more common in small animals; the incidence is greater in dogs than in cats. With seizure onset of a generalized character, convulsive electroencephalographic activity begins simultaneously in all brain regions.14 Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate seizures and by the neurobiologic, cognitive, psychological, and social consequences of this condition. The definition of epilepsy requires the occurrence of at least one epileptic seizure.10
KEY POINT 27-3
Epilepsy is a clinical manifestation of an underlying disorder and is not likely to be cured unless the cause is corrected.
Seizures in epileptic subjects often are attributed to a cortical origin. However, the brainstem, particularly the pontine reticular formation, also can manifest self-sustained seizure discharge and may be important in the generation and expression of generalized tonic convulsions.15 Depression of reticular core activity is an essential characteristic of antiepileptic drugs, suggesting that the reticular formation is involved in the spread and generalization of clinical seizures.16 In the dog the most common form of epilepsy is generalized tonic–clonic or grand mal seizures.17 Epilepsy or seizure disorders of the CNS in the dog may be caused by an acquired organic lesion such as brain tumor, head trauma, toxicosis, electrolyte imbalance, hypoglycemia, renal failure, or hepatic disease (acquired or secondary epilepsy) or may be genetic or inherited (“true,” idiopathic, or primary epilepsy). An autosomal gene associated with a sex-linked suppressor on the X chromosome may explain the higher incidence of seizures in male dogs. Interestingly, in human medicine the role of autoantibodies is emerging as a potential underlying cause of neurotoxicity and associated epileptic seizures.18
Status epilepticus (SE) refers to failure of the patient to recover to a normal alert state between repeated tonic–clonic attacks or episodes that last at least 30 minutes.19 Convulsive or tonic–clonic SE is a medical emergency in which convulsive seizures must be terminated by treatment with anticonvulsant agents. In humans epileptic seizures must not be allowed to persist more than 60 minutes if severe, permanent neurologic injury or death is to be avoided.19 The longer an epileptic seizure persists, the greater the incidence of mortality and morbidity. Hyperthermia caused by continuous muscle contraction may become life threatening during continued seizure activity. Brain damage resulting from hypoxia or the sequelae of hyperthermia is more likely if more than 30 minutes of uninterrupted seizures occur.
During seizures individual neurons depolarize and fire action potentials at high frequencies. Increased excitability reflects increased extracellular potassium and decreased extracellular calcium. Once initiated, the seizure discharge may synchronize with other neurons and propagate or spread to surrounding areas in the brain. Seizures can be initiated by four general mechanisms:
Principles of Anticonvulsant Therapy
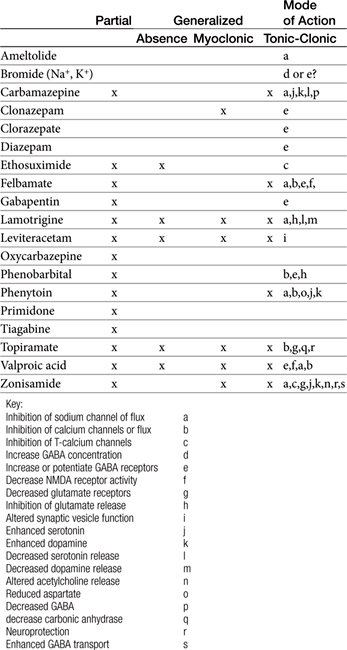
Anticonvulsants block seizure initiation and propagation by blocking either abnormal events in a single neuron or the synchronization of related neurons. Thus a goal of therapy is reduction in the firing frequency. A common mechanism by which this goal might be achieved is prevention of sodium channel recovery from inactivation (i.e., prolongation of the refractory period) (Table 27-2). It is during this period that membranes are nonresponsive to other signals. Example drugs that target this mechanism include carbamazepine, phenytoin, valproic acid, toparimate, and zonisamide.25 Another common mechanism is increased interaction between agonists and GABAA, which facilitates hyperpolarization. Drugs that act as agonists at the GABA(A) receptor include the barbiturates, and the benzodiazepines. At high concentrations these drugs also may also inhibit high-frequency firing. Other drugs include tiagabine, which targets the GABA transporter, GAT-1, decreasing neuronal and glial uptake of GABA and levetiracetam, which targets SV proteins. In general, drugs that target more than one mechanism tend to be most effective. Alternatively, combination therapy with drugs that target different mechanisms of neuronal function may be effective in seizures that do not respond to single-drug therapy.
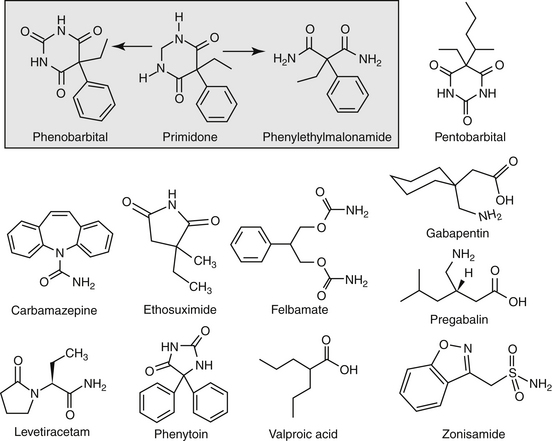
Anticonvulsant drugs target seizures; antiepileptic drugs target epilepsy. For the remainder of this chapter, the terms may be used interchangeably, in part because most antiepileptic drugs are also anticonvulsant drugs. Because successful anticonvulsant therapy may depend on the maintenance of plasma drug concentrations within the therapeutic range, understanding the disposition of each anticonvulsant is paramount to therapeutic success.26,27 Epilepsy is controlled, not cured, with historical rates of success in only 60% to 70% of the cases, although control might be improved to at least 85% if monitoring supports therapy (see later discussion).28 Generally, but not always, treatment for epilepsy involves drug administration for the rest of the animal’s life.29 The most common anticonvulsant drugs that have been used in veterinary medicine are phenobarbital, diazepam, and potassium bromide (Figure 27-4). A number of newer (human) drugs are increasingly being used, including zonisamide, levetiracetam, several benzodiazepines (clorazepate, clonazepam), gabapentin, and (less frequently) felbamate. Older drugs whose historical use has declined include primidone, phenytoin, and valproic acid. Drugs commonly used in humans that are minimally used in dogs or cats include ethosuximide, lamotrigine, and topiramate. Scientific support of efficacy for each of these drugs in dogs or cats generally is limited; differences compared with humans should be anticipated, not only because of differences in the underlying pathophysiology of disease but also in the disposition of the drugs among species and pharmacodynamic response. Even if pharmacokinetic information is available, generally the data were generated in a small sample size of healthy, drug-free animals. Pharmacokinetic differences in anticonvulsant drugs among and within breeds and species can be profound, with drug disposition often affecting the efficacy of the drug. Studies in Beagles or other pure breeds may not reflect the canine population as a whole. Antiepileptic drugs are generally administered chronically. As such an understanding of the determinants of drug disposition (Chapter 1) is paramount to understanding their most effective use, particularly as it pertains to drug toxicity and avoidance of drug fluctuations during a dosing interval that might contribute to either therapeutic failure. The more important points are as follows.
Pharmacokinetic Considerations
Absorption determines the magnitude of, but also the time to, peak anticonvulsant effect. Most anticonvulsants are administered either orally or intravenously, although alternative routes such as nasal and rectal administration are proving increasingly useful for emergency administration (Table 27-3). Most of the anticonvulsants used as antiepileptic drugs are well absorbed after oral administration, with the notable exception of phenytoin. Because food may slow the absorption of anticonvulsant drugs, peak plasma drug concentrations may occur as late as 4 to 6 hours after administration. This generally is not a problem with chronic dosing for drugs that accumulate, but it may affect the time of peak sample collection. Fasting before sample collection might be prudent. Rectal administration has offered a clinically effective alternative for administration for several anticonvulsant drugs. The colonic absorption of anticonvulsant drugs can be predicted to some degree by hydrophilicity. For example, whereas gabapentin is poorly bioavailable compared with phenytoin, maximum plasma concentration of phenytoin after colonic administration equals that after oral administration.30 A potentially emerging problem — suggested by the author’s therapeutic drug monitoring service — are differences in oral bioavailability of human generic products prescribed for animals. The therapeutic equivalence (including bioavailability) that characterizes human generic products may not translate to therapeutic equivalence when that product is used in animals. Pharmacies may change a generic product intermittently as product prices change. Dispensing pharmacists may not understand the potential risk of differences in bioavailability between animals and humans. The prudent client should query pharmacists regarding changes in products, and the discerning clinician might encourage monitoring with new prescriptions, particularly in patients at risk for life-threatening seizures.
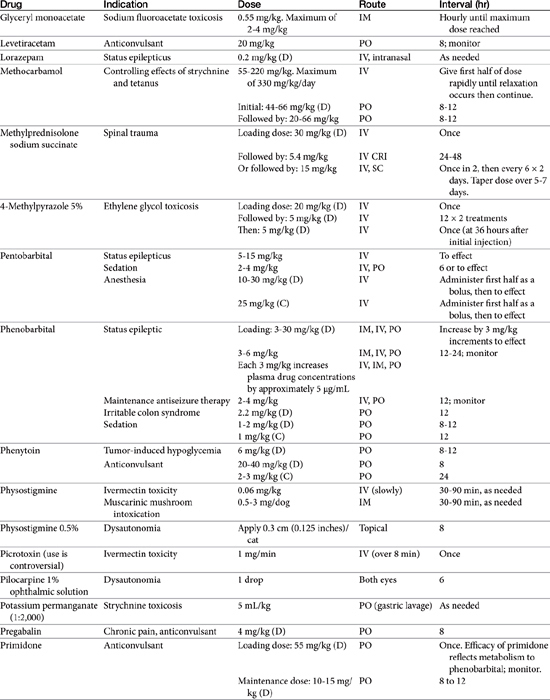
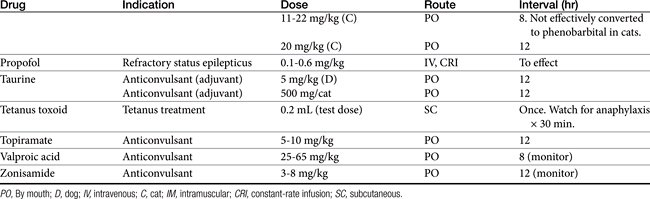
Rectal availability of diazepam has been well demonstrated. Intranasal administration has also been demonstrated as an effective route for selected drugs, particularly benzodiazepines. However, the physical volume that can be administered intranasally may limit practical application.
KEY POINT 27-6
Bioequivalence required of human drugs may not translate to bioequivalence in animals.
Distribution into the CNS is important for all anticonvulsant drugs, although at steady state, each generally distributes sufficiently into the CNS. Indeed, most anticonvulsant drugs are sufficiently lipid soluble that they are distributed to a volume that exceeds total body water (i.e., more than 0.6 L/kg). In contrast to chronic use, for acute situations rate and amount of drug movement into the CNS may not be sufficient for the rapid response necessary to resolve life-threatening seizures. Drug binding to serum proteins may limit the amount and rate of drug moving into the CNS. However, diazepam is the most lipid-soluble anticonvulsant and very rapidly distributes in the CNS despite its 90% protein binding (see Figure 27-4). Phenobarbital is less lipid soluble and is 50% protein-bound, and therapeutic effects may take as long as 15 minutes to be achieved. Löscher and Frey31 described the relative half-times to penetration of anticonvulsant drugs into the CSF of dogs. For diazepam its active metabolites desmethyldiazepam and oxazepam, clonazepam, and ethosuximide, the distribution half-time was 3 to 7 minutes, compared with 12 to 18 minutes for valproic acid, phenytoin, phenobarbital, and carbamazepine. In contrast, distribution half-time for primidone, its metabolite phenylethylmalonamide, and the active metabolite of carbamazepine (i.e., carbamazepine-10, 11-epoxide) was 40 to 50 minutes. Lipophilicity, rather than extent of protein binding or degree of ionization, was the major determinant of the rate of distribution.
The role of P-glycoprotein and other ABC transporters in CNS drug distribution is well recognized, being present at both the blood–brain and blood–CSF barriers. The efflux pumps, often accompanied by drug-metabolizing enzymes, contribute to the poor CNS drug penetrability. However, Mealey and coworkers32 suggest that the impact of P-glycoprotein is very limited at the blood–CSF barrier compared with the blood–brain barrier in dogs, based on similar penetration of a radiolabeled P-glycoprotein substrate into the CSF, but not brain, of normal (wild-type) dogs compared with ABCB1 knockout dogs. The role that efflux pumps contribute toward epilepsy, particularly refractory, is emerging.2 Increased concentrations of Ppg are associated with seizures and refractory in human patients. In animal models of refractory epilepsy, efflux pump inhibitors have been associated with an increase in drug response. Yet several studies have demonstrated that the anticonvulsant drugs often are not substrates for Ppg. Using a canine osteosarcoma cell line, West and Mealey4 demonstrated that diazepam, gabapentin, lamotrigine, levetiracetam, and phenobarbital were weak substrates, whereas carbamazepine, felbamate, phenytoin, topiramate, and zonisamide were not substrates. However, other factors may affect the state of P-glycoprotein. For example, glutamate induces P-glycoprotein expression, which may contribute to a pre-epilpetic condition. Pekcec and coworkers33 demonstrated upregulation of P-glycoprotein in dogs after an episode of SE. Furthermore, a number of other transporters may exist in the brain that might also affect anticonvulsant drugs.
Lipid solubility, which characterizes many anticonvulsants, necessitates hepatic metabolism if the drugs are to be eliminated. Metabolism of anticonvulsant drugs can have a profound effect on therapeutic success. The effect in part depends on the sequelae of phase I metabolism on the particular drug (i.e., inactivation, activation, or generation of toxic compounds). The rate of metabolism of anticonvulsants is variable: Phenobarbital is slowly metabolized, characterized by a long half-life, whereas diazepam is metabolized rapidly and is characterized by a short half-life (Table 27-4). The rate of metabolism may vary markedly among species: Phenytoin is metabolized slowly in humans but is characterized by a half-life of less than 2 hours in dogs. Phase I metabolism often yields an active compound; anticonvulsant efficacy of the metabolite may be equal, greater, or less than that of to the parent drug. Primidone must be metabolized to its active metabolite, phenobarbital before it is effective in dogs. Clorazepate, a benzodiazepine, is a prodrug derivative of a diazepam metabolite that is converted in the stomach to its active form (Figure 27-5). Although diazepam is rapidly metabolized, its duration of pharmacologic effect is prolonged because most of its metabolites have some degree of anticonvulsant effect. The half-life of the metabolites may also be longer than those of the parent compounds.
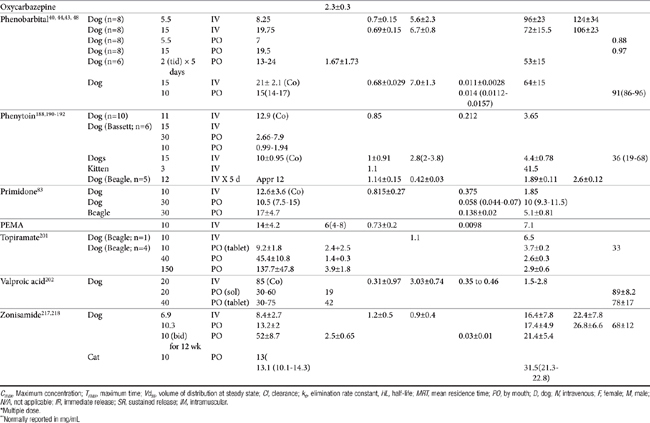
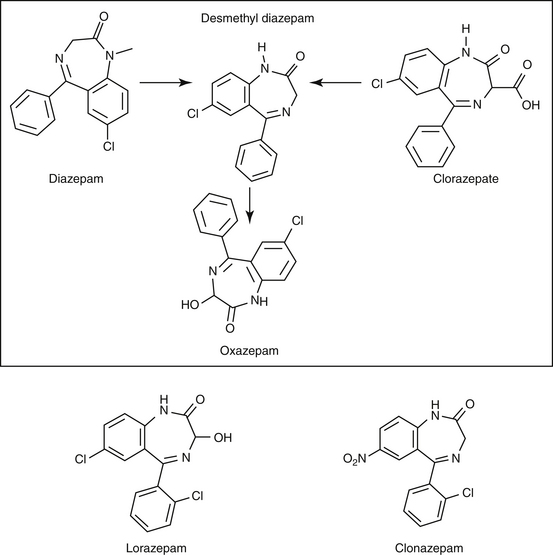
Figure 27-5 Phase I metabolism of diazepam yields metabolites of active, although less potent, metabolites. Clorazepate is metabolized in the stomach. Desmethyl diazepam, nordiazepam.
Hepatic metabolism affects both efficacy and safety, in part because of its influence on fluctuation of drug concentrations during the dosing interval. Although it is clearance (regardless of hepatic or renal) that physiologically affects drug concentrations throughout the dosing interval, half-life is the practical pharmacokinetic measure of its impact (Box 27-1; see also Chapter 1). Neither antiepileptic efficacy nor safety of an anticonvulsant drug should be evaluated until at least 87% of steady-state concentrations has been reached, plus one seizure interval (such that the brain is challenged; see Table 27-1). Every time a dosing regimen is changed, a new steady-state equilibrium should be reached before final reevaluation. The time that must elapse before steady-state concentrations and maximum therapeutic response can occur may be unacceptably long for patients suffering from severe, life-threatening seizures or from unacceptable adverse effects to the initial antiepileptic drug. For such patients a loading dose might be administered such that therapeutic antiepileptic drug concentrations are achieved rapidly. The amount of the loading dose necessary to achieve steady-state concentrations (not equilibrium) increases proportionately with the half-life of the drug to be loaded: The loading dose is a sum of all the daily doses that would have been administered before steady state occurs, less any drug that will be eliminated from the body during that time period. The major disadvantage of a loading dose is the sudden effect of therapeutic concentrations in the CNS; no time is allowed for adaptation, and adverse effects (sedation, ataxia) are more likely than with gradual increases in drug concentrations. The maintenance antiepileptic drug dose that follows the loading dose is designed to maintain concentrations achieved by the loading dose. Both loading and maintenance doses are based on population disposition parameters; monitoring should be used to ensure that dosing regimens achieve targets for the patient. Generally, antiepileptic drug monitoring is indicated immediately after the loading dose is completed (e.g., the next day). Patients should be monitored again one half-life later to ensure that the maintenance dose is able to maintain concentrations achieved with the loading dose. If the maintenance dose does not maintain what is achieved with the loading dose, the majority of the change as new steady state is reached with the new dose will occur during the first half-life of the drug.
Box 27-1 Impact of Dosing Interval and Half-Life
The shorter the half-life of the drug compared with the dosing interval, the more drug concentrations will fluctuate during a dosing interval; the longer the elimination half-life is compared to the dosing interval, the less fluctuation and the more accumulation across time. For example, if the half-life of phenobarbital approximates 72 hours in dogs, 50% of the first dose remains in the body by the sixth dose if the drug is given twice daily. Consequently, phenobarbital accumulates across time and will continue to do so until a steady-state concentration is reached. With a 72-hour half-life, only a small amount of each dose is eliminated during a 12-hour dosing interval—that is, before the next dose is administered. Accordingly, peak and trough concentrations will vary little during a 12-hour dosing interval, as long as the half-life does not dramatically shorten (i.e., due to induction). As steady-state equilibrium is reached (three to five drug half-lives), accumulated phenobarbital concentrations will be much greater than concentrations were after the first dose. The magnitude of accumulation (i.e., maximum drug concentration after the first dose compared with steady-state concentrations) increases as the difference between the dosing interval and half-life increases. Doses for such drugs are designed such that therapeutic concentrations are achieved at steady state. From a clinical standpoint, response to therapy should not be evaluated until steady state is reached; however, by one half-life, concentrations should be at 50% of maximum, which may be enough to affect seizure activity. For such drugs each daily dose contributes little to the total amount of drug in the animal, and therefore adding a single extra dose in a seizuring (or pre-ictal) patient will generally not affect drug concentrations. Rather, a “mini” loading dose generally must be given to rapidly increase drug concentrations sufficient to control seizures. On the other hand, should the patient miss a single dose (and for some drugs, multiple doses), the patient may be “protected” by the slow decline in drug concentrations reflecting the long half-life. In contrast, for drugs with a short half-life compared with the dosing interval, most of each dose is eliminated between doses. Fluctuation between peak and trough concentrations during a dosing interval can be dramatic and dangerous. For example, for a drug with a 12-hour half-life, concentrations will fluctuate 50% during a 12-hour dosing interval and 75% with a 24-hour dosing interval. For such drugs accumulation either does not occur or is minimal. Likewise, steady-state equilibrium does not really apply. Because each dose essentially achieves the maximum (and minimum) concentrations, response to therapy can be assessed rapidly. Likewise, should the patient need rapid control, an additional dose may be helpful because the total amount of drug in the body is generally provided with each dose. However, should a dose be missed for such drugs, a rapid decline in drug concentrations can precipitate seizures, including status epilepticus.
Safety
The safety of anticonvulsant drugs is profoundly affected by metabolism, not only by virtue of short elimination half-lives (for some drugs) but also because of toxicity. Phase I metabolites, by their nature, are reactive. Although intended to progress to phase II metabolism (with subsequent detoxification, particularly by glutathione), some reactive metabolites can interact with and damage surrounding tissues; concentration in the liver can lead to predictable (type A or type I, also referred to as “intrinsic”) hepatotoxicity. Thus the risk of hepatotoxicity is likely to be increased with dose and frequency of administration.However, idiosyncratic toxicity (i.e., neither dose nor duration dependent) may also be the sequelae of a metabolite. Although dogs receiving chronic anticonvulsant therapy often develop abnormalities in serum biochemistries and hepatic function tests,34-36 only about 15% of dogs receiving long-term anticonvulsant therapy have been estimated to be at risk to develop serious hepatotoxicity. This risk is, however, greatly increased if drug concentrations approach the maximum therapeutic range. Primidone is probably most commonly associated with hepatotoxicity in dogs, followed by phenobarbital and then phenytoin in combination therapy.34-36 One of the many reasons that phenytoin is no longer used as an anticonvulsant in dogs is hepatotoxicity. The risk of hepatotoxicity is discussed with each individual anticonvulsant. Toxicity may be enhanced by concurrent administration of drugs (e.g., phenobarbital) that induce drug-metabolizing enzymes and therefore increase the formation of potentially toxic (particularly phenytoin) intermediates.
Selected anticonvulsants (including phenytoin, phenobarbital, and zonisamide) can affect thyroid hormones; testing before initiation of anticonvulsant therapy might be prudent. Mechanisms include displacement from binding proteins (e.g., phenytoin), increased peripheral metabolism (e.g., phenobarbital), or decreased synthesis (e.g., zonisamide).These effects are discussed with the individual drug. Several anticonvulsants have been associated with changes in serum lipids (e.g., phenobarbital, carbamazepine, valproic acid) in both dogs37 and humans.38
Drug Interactions
Most drug interactions involving antiepileptic drugs occur at the level of hepatic drug-metabolizing enzymes. Hepatic metabolism increases the risk of drug interactions; use of any drug metabolized by the liver should be lead to a focus on potential drug interactions. For some anticonvulsants (especially phenobarbital), drug interactions and sequelae have been well described; both induction and inhibition of drug-metabolizing enzymes have been reported. Phenobarbital is the most potent hepatic drug-metabolizing enzyme inducer known and will increase the rate and extend of hepatic metabolism of many drugs, including phenobarbital. Combinations of anticonvulsants can lead to unpredictable effects on drug metabolism. Antiepileptic drugs metabolized by the liver likewise can be affected by other drugs that alter drug-metabolizing enzyme. Cimetidine, ketoconazole, and chloramphenicol are drugs that decrease drug metabolism and have the potential to profoundly increase concentrations of the anticonvulsant. Drug interactions may also involve P-glycoprotein or other efflux transporters.2 The impact of these interactions is an emerging area of research for antiepileptic drugs.
KEY POINT 27-8
The clinician should anticipate that the risk of drug interactions and hepatotoxicity is directly proportional to the proportion of the antiepileptic drug metabolized by the liver.
Drug interactions also can occur for drugs renally excreted, as is exemplified by bromide and drugs that affect urine chloride. Drug–diet interactions also have been described for bromide.
Therapeutic Drug Monitoring
Therapeutic drug monitoring can be a critically impotant tool in the successful control of the difficult epileptic patient. However, not all anticonvulsant drugs can be nor need be monitored (see Chapter 5). For example, a therapeutic range must be established for the drug (and, if established in humans, be relevant for dogs or cats), and response must correlate with plasma drug concentrations. An easy, cost-effective assay that requires minimal sample handling must be available. Among the anticonvulsants to be discussed, automated assays are available for several anticonvulsants whereas more tedious and thus costly assays (e.g., high performance liquid chromoatography) must be implemented for others. Regardless of the methodology,
clinician should confirm that the laboratory has validated any procedure used to quantitate drug in the target species and implements an external quality-assurance program.
In human medicine, the use of therapeutic drug monitoring for newer anticonvulsants (which tend to be safer) is not as well established compared with that for older drugs, in part because of the lack of generally accepted target ranges (Table 27-5). However, Bialer and coworkers27 demonstrated a concentration–response relationship for antiepileptic drugs in humans, justifying monitoring. Controlled clinical trials establishing concentration–response relationships are needed in both human and animals to establish therapeutic ranges. Currently, the range in serum concentration associated with efficacy is large for some drugs and overlaps with toxicity in some patients.39 Nevertheless, therapeutic drug monitoring remains a recognized tool for individualization of drug therapy. The author suggests a “start-up”, “follow-up”, “check-up,” and “what’s-up” approach to monitoring (Box 27-2). Clearly, monitoring should be implemented to avoid toxic concentrations and to establish the minimum effective range for the epileptic patient This latter reason is particularly relevant for previously controlled patients suffering from breakthrough seizures (previously well controlled): a decrease in drug concentrations compared with baseline should lead to a focus on owner compliance, drug interactions, changes in disposition, and so forth, whereas seizures despite no change in concentrations might be interpreted as worsening of underlying disease and the potential need to control seizures more aggressively. therapy not be considered failed until trough concentrations are in the higher end of the therapeutic range. Serum separator tubes should not be used for collection of samples intended for therapeutic drug monitoring.
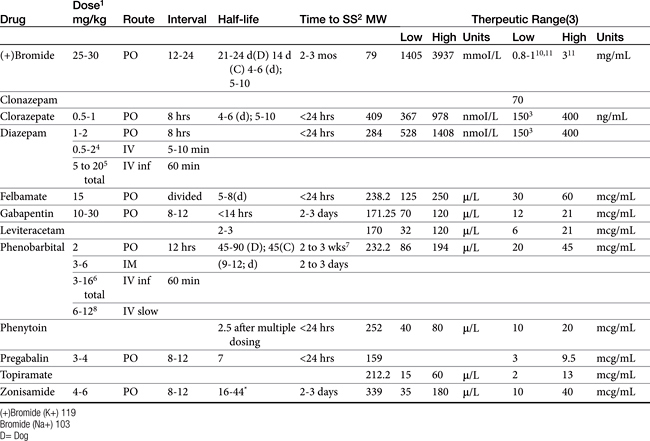
Box 27-2 Therapeutic Drug Monitoring and Antiepileptic Drugs
Start-up: To establish base-line concentrations, and ideally the minimum effective concentration for the patient. This is particularly important in the patient that subsequently has break-through seizures or a sudden change in clinical signs suggestive of side effects or adverse events. Start up monitoring should be designed to establish a minmum effective dose such that the patient is not exposed to higher than necessary concentrations. Start-up is also indicated to establish when a uncontrolled patient has exceeded the maximum therapeutic range and further increases in therapy are not likely to contribute significantly to seizure control. At that point (and ideally only at that point) is combination therapy implemented. Start-up monitoring also should be implemented within a day of a loading dose.
Follow-up: Every time a new dosing regimen is implemented the patient should be remonitored at steady-state (this might be withheld until response is realized). A new dosing regimen is defined by any change in dose or interval, or the appearance of a patient factor that might change drug disposition. This includes transitioning from a loading dose to a maintenance dose or adjusting a maintenance dose. Monitoring also should be implemented if a change is anticipated in the patient that might alter drug half-life and thus drug concentrations. Examples include the addition (or discontinuation) of one or more drugs (antiepileptic drugs or nonantiepileptic drugs that can influence the disposition of the antiepileptic drug), a change in the diet (e.g., for bromide), or a change in patient health such that disposition might change as disease progresses or responds to therapy. Determining the most appropriate time point for monitoring will be complicated in patients in which drug half-life has changed and is no longer predicted by sample population statistics. For such patients more frequent monitoring is suggested.
Check-up: Monitoring proactively is particularly important for patients at risk for therapeutic failure. Monitoring is also important as patients age, are placed on diets, etc. The interval of routine monitoring should be individually determined, ranging from 3 months in patients with proven history of variability in drug concentrations to yearly as part of the annual physical examination.
What’s-up? Monitoring should be implemented if a previously controlled patient begins to experience seizures or clinical signs emerge suggested of side adverse events. Availability of baseline greatly facilitates interpretation. Monitoring helps identify factors which might cause drug concentrations to change (e.g., drug interactions, change in diet, poor owner compliance). Monitoring should also be implemented to help clarify the role of disease in clinical signs.
Loading dose: If a loading dose is used, drug concentrations should be monitored after a loading dose, one drug elimination half-life later and, to establish a new steady state, three to five drug half-lives later. The sample collected at one drug elimination half-life should be equivalent to the loading dose sample; if not, the maintenance dose must be adjusted accordingly. The collection of a sample one half-life after a loading dose is minimally useful unless a postload sample has been collected; because the patient is not yet at steady state with the maintenance dose, plasma drug concentrations are still in a state of flux. No loading dose: If a loading dose is not administered, and the drug is characterized by a long half-life (i.e., several weeks to months until steady state is reached), a sample can be collected at one half-life to allow proactive evaluation of the appropriateness of a chosen dosing regimen: doubling the concentration measured at one half-life predicts baseline steady-state concentrations. If the predicted concentrations are not on target, the maintenance dose can be adjusted. Otherwise, for most drugs, monitoring can begin with a baseline sample collected at three to five drug half-lives.
Number of samples: The need for peak, trough, or both samples depends on the half-life of the drug. If the half-life in the patient is substantially longer than the dosing interval (we recommend greater than 2 to 3 times), peak and trough concentrations will be very similar, and a single trough sample collected just before a dose is sufficient (e.g., bromide, and for the majority of patients, phenobarbital; zonisamide). The trough sample is preferred: not only does it reveal the lowest concentration to which the patient is exposed during a dosing interval, it also is not impact by absorption and distribution and thus is most likely to be consistent for across-time comparisons. For drugs that might fluctuate markedly during a dosing interval (e.g., levetiracetam, gabapentin, pregabalin, and zonisamide in some pateints), both a peak and trough sample should be taken to determine patient half-life. If the half-live is revealed to be long, then subsequent single trough samples are indicated.
Other considerations: Animals should be fasted before samples are collected, particularly for drugs that do not accumulate (i.e., those that are characterized by a short half-life). Special handling preparations generally are unusual, although each laboratory should be called to confirm special handling procedures. Serum separator tubes are contraindicated because the silicon separator can bind anticonvulsant drugs and falsely decrease serum drug concentrations. If used, serum should be immediately harvested after centrifugation.149
Interpretation of therapeutic drug monitoring results can be facilitated by a clinical pharmacologist. Monitoring should be used to identify the therapeutic range for the individual patient.Therapeutic failure should not be considered if a patient is having seizures simply because the therapeutic range has been achieved, nor should a drug be discontinued or the dose increased if a patient is not having seizures despite subtherapeutic concentrations. For a patient that is not sufficiently controlled, doses can be gradually increased until the maximum range has been reached and the risk of adverse affects becomes too great. Many patients require and can tolerate concentrations of less toxic drugs (e.g., bromide) that are well above the maximum.
Anticonvulsant Drugs
Phenobarbital
Mechanism of Action
Phenobarbital sodium (see Figure 27-4) specifically depresses the motor centers of the cerebral cortex, thus enhancing anticonvulsant properties. Electroshock experiments with cats and other species established phenobarbital as one of the most potent anticonvulsants available. It has the widest spectrum of activity in different convulsive seizure patterns. Many AED have been synthesized as structural variants of phenobarbital. For example, primidone is a close congener of phenobarbital.
Phenobarbital is the most effective anticonvulsant to delay progressive intensification of seizure activity that may accompany epilepsy. Phenobarbital both increases the seizure threshold required for seizure discharge and decreases the spread of discharge to surrounding neurons; its primary mechanism is enhancement of responsiveness to the inhibitory postsynaptic effects of GABA. Interaction of GABA opens a chloride channel, resulting in higher intracellular concentrations of chloride and hyperpolarization of the RMP (see Figure 27-3). Phenobarbital also, however, inhibits glutamate activity and probably calcium fluxes across the neuronal membrane. A study that compared CSF concentrations of GABA and glutamate in epileptic Labrador Retrievers versus non–Labrador Retrievers and in non-epileptic Beagles also compared concentrations in those epileptic dogs treated with phenobarbital and those not treated.22 Within the non–Labrador Retriever phenobarbital-treated group, GABA, glutamate, and aspartate CSF concentrations were lower compared with those of their untreated counterparts. This difference was not detected in the Labrador Retrievers treated versus untreated groups; however, only 14 Labrador Retrievers were receiving phenobarbital, which may have limited the power to detect a significant difference.
Phenobarbital might be considered a broad-spectrum anticonvulsant. Despite introduction of new antiepileptic drugs, phenobarbital has generally been the anticonvulsant of choice for the cat and dog.17 It is effective in all types of epileptic seizures observed in cats and dogs.
Disposition
As a weak acid (pKa 7.3), phenobarbital is well absorbed after oral administration, although peak plasma concentrations may not be reached for 4 to 6 hours after administration. The absorption half-life in dogs is 1.27 ± 0.21 hours.40 With an elimination half-life of 0.26 ± 0.18, about 6.4 hours may be required for near complete absorption of phenobarbital from the gastrointestinal tract. Absorption is 88% to 95% complete.41 Phenobarbital is 45% bound to serum protein in dogs.42 The volume of distribution of phenobarbital in dogs is 0.7 ± 0.15 L/kg and 0.7 ± 0.4 in cats.43 Approximately 16 days (8 to 15.5 days) of multiple dosing is necessary to attain steady-state serum concentrations in the normal (uninduced) animal; however, induction may cause this period to be shorter after a change in the dosing regimen of an animal already receiving phenobarbital. Maintenance doses of 5.5 mg/kg daily (in one to three equal doses) administered orally are required to reach an average serum concentration of 20 μg/mL, according to one study.44
KEY POINT 27-10
The efficacy of phenobarbital must be balanced with the risk of liver disease, which increases as drug concentrations increase.
Through microsomal enzyme action, phenobarbital is metabolized by oxidative hydroxylation to form hydroxyphenobarbital. This metabolite has a weak anticonvulsant activity that does not contribute significantly to the action of phenobarbital. Hydroxyphenobarbital is rapidly eliminated from blood by conjugation with glucuronide and excretion in urine of the dog. Up to 25% of the parent drug is eliminated renally in dogs. Individual variability in the rate of phenobarbital elimination is marked owing to differences in hepatic metabolism. Half-life varies not only between and within species but also in the same animal, in part because of induction (discussed later).
Breed differences may exist in phenobarbital clearance. Frey and Löscher42 reported a half-life of phenobarbital at 64 ± 15 hours in largely mongrel dogs, but 32 ± 4.8 in Beagles; clearance was 7.0 ± 1.3 and 13 ± 1.7 mL/kg/min, respectively. Alkalinization of urine (pH >7.5) accelerates excretion of unaltered phenobarbital by reducing back-diffusion (tubular reabsorption) through drug ionization.45 Fukunaga and coworkers46 demonstrated that a twice-daily fed diet containing either 0.6 gof potassium citrate (pH 7.8 to 8.2) or 0.2 g of ammonium chloride (pH approximating 5.9 to 6.5) increased urine clearance of a single dose of phenobarbital compared with a water vehicle (pH 6.8 to 7.5) in Beagles (n = 5; female). However, the effect did not significantly (statistically nor clinically)impact peak plasma drug concentrations (μg/mL: 3.25, 2.96, and 2.97 μg/mL for water placebo, ammonium chloride, or potassium citrate, respectively) or area under the curve. Yet, half-life was affected (52, 59.6, and 51.1 hours, respectively), and as such peak concentrations might be slightly affected if the drug is dosed to steady state.
Phenobarbital is a potent inducer of hepatic drug-metabolizing enzymes and is capable of increasing the rate of clearance of other drugs metabolized by the liver as well as increasing its own rate of metabolism.47 In the dog phenobarbital (2 mg/kg) administered orally 3 times a day for 5 days results in an elimination half-life between 37 and 75 hours, with a mean elimination half-life of 53 ± 15 hours.44 After a single 5-mg/kg intravenous dose, clearance is 5.6 to 6.6 mL/kg per hour, and elimination half-life is 92.6 ± 23.7 hours in dogs40 and 47 ± 3 hours after oral administration (5 mg/kg) in cats. The effects of multiple doses of phenobarbital were documented by Ravis and coworkers.48 After 90 days of treatment (5.5 mg/kg), mean elimination half-life decreased from 88.7 ± 19.6 hours to 47.5 ± 10.7 hours in dogs. In cats 21 days of oral phenobarbital at 5 mg/kg resulted in a half-life of 43 ± 3 hours.43
Preparations
Phenobarbital is available as either an oral or an injectable preparation. Oral tablets contain 0.25, 0.50, or 1 grain (15, 30, and 65 mg, respectively) phenobarbital. An elixir is also available (4 mg/mL) for treatment of very small animals. The injectable form is intended for intravenous use but can be given intramuscularly and rectally. Under the 1970 Controlled Substances Act, phenobarbital is classified as a Schedule IV drug.
Clinical Use
Phenobarbital is reportedly effective in 60% to 80% of canine patients suffering from epilepsy if serum concentrations of the drug are maintained within the recommended therapeutic ranges of 20 to 45 μg/mL.49 This is consistent with the early work of Farnbach,50 who reported that the concentrations of phenobarbital necessary to control seizures (control defined as interictal interval prolonged at least twice that of the pretreatment period). Concentrations considered effective (n = 22) ranged from extremes of 6.5 to 81.3 μg/mL, with others ranging from 14.2 to 46 μg/mL (mean 23 μg/mL); however, concentrations not associated with control (n=20) ranged from 4.7 to 42 μg/mL. Efficacy might be higher if therapeutic failure is not considered until the maximum end of the range (i.e., 35 to 45 μg/mL) has been reached (Figure 27-6.). The author has reported 85% efficacy in eradication of seizures in spontaneous canine epilepsy with phenobarbital concentrations at 25.4 ± 5.7 μg/mL with such an approach.28 In this same study, phenobarbital was found to be superior to bromide for first choice control of seizures in dogs (see the discussion of bromide). Phenobarbital has been used successfully to treat hypersialosis.51
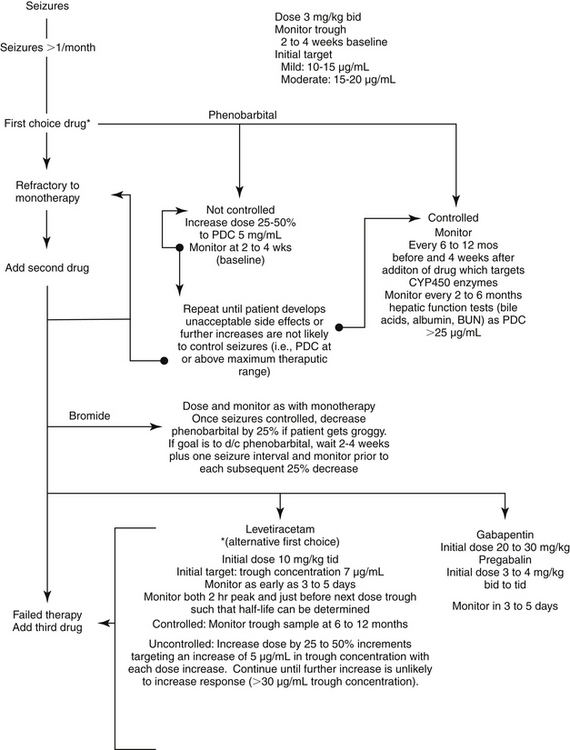
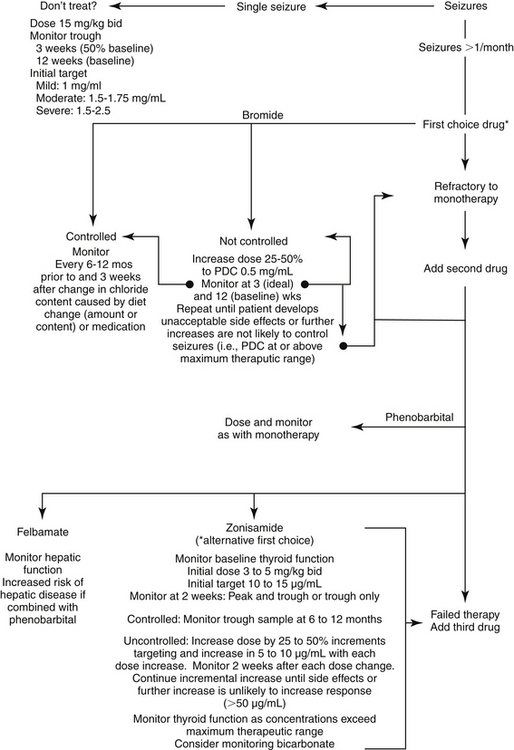
Figure 27-6 Algorithm for modification of anticonvulsant dosing regimens. The goal of monitoring in a stepwise fashion is to achieve adequate control of seizures with the lowest plasma drug concentration possible. This approach minimizes side effects and drug interactions and may allow for the least adaptation by the brain. The highest concentration is generally not initially chosen, particularly for drugs associated with side effects. Combination therapy generally should not be pursued until monotherapy fails (i.e., seizures remain unacceptable despite drug concentrations at the maximum end of the therapeutic range, or in the presence of unacceptable side effects). If initial combination therapy fails, a third drug may be necessary. If the patient responds to combination therapy and if seizure history warrants doing so, the drug that is least effective or associated with the most side effects might be gradually discontinued. Monitoring during withdrawal might be prudent to determine the minimum effective concentration necessary to control seizures should the patient develop breakthrough seizures. Should grogginess emerge with the addition of a second or third anticonvulsant drug, a 25% decrease in the drug most sedative and with the shortest half-life should be considered.
Patients should not considered refractory to phenobarbital therapy until plasma concentrations reach 35 to 40 μg/mL unless unacceptable side effects persist at lower concentrations. However, because of the potential for phenobarbital-induced hepatotoxicity, the author often recommends adjuvant therapy once phenobarbital concentrations exceed 25 μg/mL in an uncontrolled dog. The dose necessary to achieve target concentrations may vary dramatically among dogs and in the same dog across time because of differences or changes in drug disposition. Changes are likely to reflect, in part, induction of drug-metabolizing enzymes; monitoring should occur 1 to 2 months after baseline is established to detect the possible impact of induction. Measurement of both a peak and trough sample can detect a short half-life; for patients in which phenobarbital half-life is 24 hours or less, the same or a slightly higher (e.g., 25% increase) total dose divided into 8-hour rather than 12-hour intervals may minimize fluctuation of plasma drug concentrations. Clearly, once-daily dosing should be avoided in the face of induction that decreases half-life to 36 hours or less. Phenobarbital elimination half-lives of 9 to 12 hours have been documented in the author’s laboratory in some dogs that have been receiving phenobarbital for several months.
Phenobarbital can be administered intravenously for acute control of seizures, although a lag time (i.e., 20 to 30 minutes) may be observed before control of seizures. An intravenous loading dose of 12 mg/kg is designed to achieve therapeutic concentrations (20 μg/mL) immediately; the dose can be decreased proportionately (on the basis of serum phenobarbital concentrations) if the patient is currently receiving phenobarbital. Alternatively, the calculated dose can be given in four to six equal hourly doses. Collection of a postload monitoring sample would be prudent once seizures are controlled; this sample can provide a target for maintenance therapy. A second sample 2 or 3 days later should be used to assess the oral maintenance dose. If used in combination with intravenous diazepam (for its more immediate effects) to prolong the control of seizures, the phenobarbital dose might be administered intramuscularly to avoid respiratory and cardiovascular depression. Once seizures are controlled with phenobarbital alone, monitoring should establish the target concentration for chronic therapy in the patient.
KEY POINT 27-11
The long half-life of phenobarbital precludes rapid response unless a mini “loading” dose is administered.
Limited information is available regarding the use of phenobarbital in cats. Phenobarbital administered at 6 mg/kg daily was useful in suppressing experimentally induced hippocampal generalized seizures in cats; drug concentrations ranged between 15 and 25 μg/mL. After-discharge was totally suppressed in 33% of cats at 35 to 50 μg/mL (12 mg/kg daily). However, phenobarbital was less effective for amygdaloid-kindled seizures.52
Drug Interactions
Hepatic microsomal enzyme activity, especially mixed-function oxidase (cytochrome P450 [CYP]) induction, is accelerated by phenobarbital.53-56 Enzyme induction by phenobarbital appears to be dose related.57 Long-acting barbiturates are better inducers of microsomal enzyme activity than are short-acting compounds. For example, compared on a molar basis, phenobarbital has been described as the most potent enzyme stimulatory agent known,58 whereas pentobarbital and thiopental sodium are less potent inducers of microsomal enzyme activity, in part because duration of therapy dose, and thus impact, is limited. Enzyme induction may take weeks to months and may occur with each dose increase. Induction has been documented in dogs59-62 but apparently not in cats. Although it may occur occur in the latter species, saturation of drug met. Phenobarbital was associated with a twofold increase in CYP protein and increased clearance of the hepatic metabolism marker antipyrine in Beagles treated with 5 mg/kg orally every 12 hours for 35 days. Among the CYP isoenzymes characterized by an increase in activity were CYP1A, CYP2B, CYP2C, and CYP3A; in contrast, CYP2D did not appear to increase.63
Induction does not immediately resolve once a drug is discontinued: For example, once phenobarbital initiates induction, up to 7 months may be required for complete resolution in the dog. Fukunaga and coworkers64 demonstrated that antipyrine clearance was increased in response to phenobarbital at 4 weeks but not 6 weeks in dogs receiving 5 mg/kg of phenobarbital twice daily. Clearance was increased by close to 60% during phenobarbital therapy. Interestingly, volume of distribution increased almost threefold, and half-life increased, as did mean residence time, despite the change in clearance. The investigators did not include a control group that did not receive phenobarbital, complicating interpretation. Clinically, as per the author’s therapeutic drug monitoring laboratory, however, induction often decreases the elimination half-life and thus duration of therapeutic response to other drugs metabolized by those enzymes induced by phenobarbital (see Chapter 2). In the dog, experimental documentation includes digoxin and digitoxin44,65,66 and thiopental.67 However, induction does not appear to affect thiopentone (thiopental) dose requirements in the dog.68 For drugs in which toxic metabolites are produced, the induced liver will produce more metabolites; therefore potentially hepatotoxic drugs should not be used with phenobarbital. N-Acetylcysteine should be used in cases of drug-induced acute hepatopathy, regardless of the cause (exceptions might be made in the presence of hepatic encephalopathy). Longer term support might include s-adenosyl methionine (SAMe) or milk thistle as well as other therapies.
Phenobarbital therapy (2.5 mg/kg orally every 12 hours for 30 days) was associated with a decrease in the peak concentrations of benzodiazepines (diazepam/oxazepam) after single-dose administration of 2 mg/kg either intravenously (about 10%, from 5963 to 5565 ng/mL) or rectally (about 60% decrease, from 630 to 270 ng/mL). However, the target associated with seizure control (150 ng/mL) was achieved in five of six dogs within 8 minutes of rectal administration.69
Side Effects
Behavior
Polyphagia, polydipsia, and polyuria are side effects that occur in animals receiving clinical dosages of phenobarbital. The polyuric effect is apparently due to an inhibitory action in the release of antidiuretic hormone. 70 Identical sedative side effects are observed in the dog after treatment with phenobarbital or primidone (discussed later).17 Dogs appear fatigued and listless after receiving either drug; some are weak in the rear legs, and ataxia occurs. All of these effects may be long lasting and may persist in some cases for the duration of treatment; however, tolerance to these effects generally develops in most dogs 1 to 2 weeks after initiating the dosing regimens.
Bone marrow dyscrasias and others
Phenobarbital can cause what is an apparent allergic reaction manifested as a bone marrow dyscrasia in dogs. Pancytopenia or (more commonly) neutropenia is detected after a complete blood count in animals that presented with a variety of clinical signs. Bone marrow suppression generally resolves rapidly once phenobarbital is discontinued.71 Bone marrow necrosis also has been associated with phenobarbital. 72 Care should be taken to begin an alternative anticonvulsant drug (e.g., bromide) in animals that are at risk for worsening of seizures should phenobarbital be rapidly discontinued. It is likely that a metabolite is the cause of the dyscrasia; as such, an anticonvulsant minimally metabolized by the liver may be a wiser choice for drug replacement. Phenobarbital-induced coagulopathy has been reported in the cat.73 Superficial necrolytic dermatitis has been reported in dogs receiving phenobarbital.74
Thyroid
Phenobarbital can induce tissue (peripheral tissues and the liver) metabolism of thyroid hormones. Serum total thyroxine (T4), total triiodothyronine (T3), free T4, and thyroid-stimulating hormone (TSH) concentrations were compared in epileptic dogs (n = 78) with seizure disorders and treated with phenobarbital (n = 55), phenobarbital and bromide (n = 15), and bromide (n = 8) and clinically normal dogs (n = 150). Whereas T3 and TSH total did not differ among groups, total and free T4 were lower in phenobarbital and phenobarbital plus bromide compared with concentrations in clinically normal dogs. Bromide-treated dogs did not differ from clinically normal dogs. Serum total and free T4 concentrations were lower than normal (i.e., in the range typical for dogs with hypothyroidism) in the dogs treated with phenobarbital.75 In a second study of experimental dogs (n = 12), Gieger and coworkers76 demonstrated that phenobarbital at 4.4 to 6.6 mg/kg adminstered orally twice daily for 27 weeks resulted in significant decreases in serum T4 and free T4 and increased TSH. These changes persisted for up to 4 weeks after discontinuation of therapy.76 Thyroid screens in apparently normal animals that yield results indicative of hypothyroidism do not necessarily indicate the need for treatment; indeed, overtreatment may result in undesirable CNS stimulation. On the other hand, if an animal presents with clinical signs consistent with hypothyroidism, replacement therapy may be indicated.
Osteomalacia
In humans long-term (more than 2 years) treatment of epileptic patients with selected anticonvulsants (phenobarbital and primidone but not phenytoin) has been associated with development of osteomalacia; subnormal serum calcium is seen in such patients. The mechanism is not clear, although accelerated conversion accelerated of vitamin D3 to 25-hydroxycholecalciferol (25-OHD3) but not 25-OHD3 to 1,25-dihydroxycholecalciferol (1,25-(OH)2D3) is a documented effect of phenobarbital.76
Hepatotoxicity
At high plasma drug concentrations (i.e., more than 30 to 40 μg/mL), phenobarbital appears to be hepatotoxic.77 Animals whose liver is induced and thus requires high doses of phenobarbital to maintain drug concentrations in the lower therapeutic range may also be more susceptible to toxicity because of increased formation of metabolites. Phenobarbital will also cause nonpathologic changes in hepatic clinical laboratory tests because of induction of enzymes. Serum alkaline phosphatase (SAP) and the transaminases are likely to increase with chronic therapy.34,36,77-79 These are not necessarily indicative of liver disease. However, Gaskill and coworkers79a suggest that changes in SAP associated with phenobarbital are indicative of hepatopathy rather than hepatic disease. Changes associated with true hepatic pathology appear to be more likely with primidone (see later discussion). Moderate elevations in alanine transaminase and SAP, coupled with changes in hepatic function (e.g., serum bile acids, albumin, and serum [blood] urea nitrogen) are more indicative of hepatic pathology (i.e., liver disease). Hepatic function tests (e.g., serum bile acids) should be studied to monitor the development or progression of liver disease. Bilirubin has not been a sensitive indicator of liver disease induced by phenobarbital in the author’s experience; indeed, it generally increases only with end-stage liver disease, if at all. Interestingly, decreased serum cholesterol levels have occurred relatively early. Liver enzymes should not be monitored until several days after a seizure to avoid the impact of hypoxia and other effects on hepatic leakage enzymes. Consistent increases in serum bile acids and decreased serum urea nitrogen and albumin concentrations are supportive of phenobarbital-induced hepatic disease and the need for rapid but cautious withdrawal of phenobarbital in concert with initiation of an alternative anticonvulsant. Toxicity can occur within several months but appears reversible if the drug is discontinued before fibrotic disease develops.79 The incidence of serious liver toxicity might be reduced by avoiding combinations of drugs in which more than one is characterized by hepatic metabolism, using therapeutic drug monitoring to achieve adequate serum concentrations at the smallest dose possible,and evaluating clinical pathology changes every 4 to 6 months (or more frequently in animals at risk) while the patient is receiving therapy. Although liver disease associated with phenobarbital therapy has been occasionally noted at phenobarbital concentrations ranging from 12 to 25 μg/mL in patients being monitored in the author’s laboratory, the direct cause and effect was not established.
KEY POINT 27-13
Hepatic function tests (serum albumin, urea nitrogen, and bile acids) should be the basis for diagnosis of phenobarbital hepatotoxicity.
Decreasing drug concentrations should also reduce the progression of chronic disease to cirrhotic disease, although what constitutes a safe target phenobarbital concentration in these patients has not been documented. Bromide therapy should be initiated for patients with liver disease (regardless of the cause) who must also receive anticonvulsant therapy. Despite its ability to cause hepatotoxicity, phenobarbital appears to be a safe and effective anticonvulsant if drug concentrations can be maintained well below the recommended maximum.
Hypertriglyceridemia
Phenobarbital is among several anticonvulsant drugs (others being carbamazepine and valproic acid) associated with serum lipid profiles in children.38 Effects of phenobarbital are described as transient. Phenobarbital has been associated with hypertriglyceridemia in dogs.37 Median fasting serum triglyceride (mmol/L) was 0.6 (range 0.9 to 1.6) for phenobarbital (n = 28) and 0.6 (range 1.2 to 3.6 mmol/L) for the combination of phenobarbital and bromide (n = 29); compared to non-epileptic control dogs at 0.4 (n = 57) (range 0.6 to 0.9) not receiving phenobarbital.37 Of the 57 dogs studied, triglycerides were increased in 33% when compared with a reference range established in the non-epileptic dogs. Phenobarbital was higher and more variable in dogs receiving phenobarbital with bromide (109 ± 47 μmole/L) only compared with dogs receiving phenobarbital alone (83 ± 21 μmole/L). Body condition score was significantly related to increased triglycerides. Although no correlation was found between phenobarbital and triglyceride concentrations, a trend was identified between phenobarbital dose and triglycerides. However, the control group consisted of normal healthy animals rather than epileptics not receiving anticonvulsants, and an additional study is warranted to address the potential risk of higher triglycerides in epileptic dogs receiving phenobarbital compared to no phenobarbital.
Phenobarbital may be associated with pancreatitis. Although studies have focused on bromide, studies supporting this association have documented an increased risk in patients receiving both bromide and phenobarbital, as well as phenobarbital alone.80,81
Treatment of Acute Phenobarbital Toxicosis
Treatment of acute phenobarbital toxicosis is generally supportive. Dogs apparently can tolerate marked acute overdosing (in the author’s experience, concentrations approximately 150 μg/mL) without persistent effects, once the drug has been discontinued. Sedation is the most detrimental effect. Artificial respiration with oxygen should be administered to prevent respiratory arrest induced. Although less effective than oxygen, doxapram or other analeptic drugs might stimulate the respiratory center. Alkalinization of the urine accelerates renal excretion of phenobarbital because of ion trapping in the urine, although this is not easily accomplished.45 Activated charcoal effectively accelerates the body clearance of phenobarbital.82 When charcoal is administered in the human, the biological half-life of phenobarbital is decreased from 110 ± 8 to 45 ± 6 hours; it increases the total body clearance of phenobarbital from 4.4 ± 0.2 to 12.0 ± 1.6 mL/kg per hour.82 Assuming drug metabolizing enzymes are not saturated (threshold not known in the dog or cat), the time needed for phenobarbital concentrations to decline such that sedation is no longer present depends on the drug concentrations (i.e., the extent of overdose) and the elimination half-life of the drug in the patient. For example, at 150 μg/mL, approximately three half-lives must elapse before concentrations drop below 35 μg/mL; for a 72-hour half-life, 9 days must elapse. Both the magnitude and half-life (assuming saturation has not occurred) of phenobarbital can be monitored in an overdose patient; a sample should be collected on presentation and again 24 hours later. Timing of sample collections must accompany the samples if a half-life is to be calculated. A follow-up sample 2 or 3 days later can confirm that the original half-life was correct (i.e., saturation has not occurred). In cases indicative of acute hepatopathy, N-acetylcysteine should be administered, as for acetaminophen toxicosis.
Primidone
Primidone is metabolized in the liver to phenylethylmalonic acid (PEMA) and phenobarbital (see Figure 27-4).83 Although all three compounds have anticonvulsant activities, phenobarbital is much more potent and has a longer half-life than primidone and PEMA (and thus accumulates). Thus phenobarbital contributes up to 85% of the anticonvulsant activity, and phenobarbital concentrations, not the parent drug, correlates with primidone efficacy.49 Frey and Löscher42 studied both primidone and PEMA after intravenous and oral administration in dogs and Beagles (see Table 27-4). Primidone is characterized by a short half-life (6 to 12 hours) compared with phenobarbital, although concentrations decrease, presumably as a result of induction, after 14 days of therapy. Phenobarbital reached approximately 65 mmol/mL (11 μg/ml) compared with 50 mol/mL for PEMA, with primidone at 14 mol/mL. The rate of entry into the CNS ranged from 0.01 to 0.023/min for primidone and 0.11 to 0.022/min for PEMA. In 15 epileptic dogs,49 primidone administered at a range of 10.6 to 20.3 mg/kg every 8 hours yielded mean phenobarbital, PEMA, and primidone concentrations were (presumably at steady state) 31.9, 16.5, and 0.9 μg/mL, respectively. As such, phenobarbital should be monitored rather than primidone, using the phenobarbital therapeutic range. Further, all attributes and detractors previously discussed for phenobarbital generally apply to primidone. Primidone has been used in patients refractory to phenobarbital at the maximum therapeutic drug concentration (i.e., 40 μg/mL), but its efficacy in this scenario has not been proved.17,84,85 Efficacy may simply reflect improved conversion to phenobarbital (i.e., animals that are induced may metabolize the drug to greater concentrations of phenobarbital than generated from administration of phenobarbital alone). Farnbach50,86 reported that the concentration of phenobarbital associated with effective anticonvulsant therapy in dogs given primidone (n = 12) ranged from 4.8 to 70.7 μg/mL (median 27; dose 15.2 to 81 mg/kg daily), compared with ineffective concentrations (n = 11), which ranged from 0.3 to 52.8 (dose 5.5 to 83.3 mg/kg). According to Farnbach,50,86 there is no advantage to using primidone rather than phenobarbital for control of epilepsy in most dogs.
The conversion ratio of primidone to phenobarbital is 3.8:1. A patient should receive approximately 65 mg (1 grain) of phenobarbital for each 250 mg of primidone. Because this rate does not reflect the potential effects of phenobarbital (i.e., primidone) induction, however, animals may convert primidone to phenobarbital at different rates. Baseline phenobarbital concentrations should be established before conversion. Conversion should probably be progressive (e.g., 25% change each month) in at-risk patients (i.e., those whose seizure history includes prolonged or cluster seizures). Cats do not metabolize primidone to phenobarbital as efficiently as dogs.87 Phenobarbital, PEMA, and primidone concentrations (μg/mL) were 4.1, 11.5, and 5.1, respectively, in cats (n = 11) receiving 20 mg/kg primidone orally twice daily for up to 90 days. The elimination half-life was 7 hours. Although primidone was considered safe, anticonvulsant concentrations were not therapeutic. Thus the safety of primidone at effective concentrations has not been established in cats, and its use is not recommended for the treatment of feline seizures.
Side Effects
All side effects noted for phenobarbital are caused by primidone. Primidone may induce nystagmus, nausea, drowsiness, and ataxia. According to Schwartz-Porsche et al.,17 polydipsia is more common in dogs treated with primidone. In humans it is recommended that therapeutic plasma concentrations of primidone and its metabolite phenobarbital not exceed 15 and 30 μg/mL, respectively. Megaloblastic anemia is one of the more serious adverse effects of primidone in humans. Pruritis characterized by alopecia, scaling, ulceration, pigmentation, and fissuring has been recorded in dogs.86
KEY POINT 27-14
The use of primidone, a prodrug for phenobarbital, is discouraged because of the increased risk of liver disease.
In the dog primidone induces progressive hepatic injury, as manifested by increases in liver enzyme values.89 In a clinical study, signs of liver toxicity were reported in 14 of 20 dogs.17 Hepatic cirrhosis associated with primidone and phenobarbital after 7 years of use has been reported in a dog.90 Primidone-induced dermatitis has been reported in a dog.88
Drug Interactions
Drug interactions previously described for phenobarbital also occur with primidone. Primidone should not be used concurrently with chloramphenicol, which is a potent inhibitor of the microsomal enzyme system. Severe CNS depression and inappetence occur in the dog after concurrent use of these drugs.91
Pentobarbital Sodium
Pentobarbital sodium (pentobarbitone sodium; Nembutal Sodium), administered intravenously, is considered to be the most efficacious drug for abolishing refractory SE in the dog. Pentobarbital is a general anesthetic (not an anticonvulsant), but it is nonetheless an effective drug for control of nonresponsive seizures; extreme care is required not to overdose. An added advantage of pentobarbital is its ability to scavenge oxygen radicals and decrease cerebral oxygen consumption (see later discussion of increased intracranial pressure). The effective dose varies considerably from one animal to the next. Consequently, pentobarbital is carefully given to effect. Careful monitoring of the cardiovascular and respiratory systems is necessary. Prolonged treatment may be necessary for some patients; some human patients remain anesthetized for up to 9 days before seizures are controlled.
In humans tonic–clonic SE, which is refractory to phenobarbital, phenytoin, or diazepam, may respond to an intravenous infusion of pentobarbital given continuously for 3 days.92 It is then discontinued, and oral phenobarbital, along with other anticonvulsants, is advocated to control recurring epileptic episodes.
Pentobarbital intoxication has been reported after ingestion of drug-tainted tissue. One case report93 described a bitch that became intoxicated after consuming a puppy that had been euthanized with pentobarbital. Other reports have involved zoo animals ingesting food derived from barbiturate-contaminated horse meat.94
Benzodiazepines: Diazepam, Clorazepate, Clonazepam, and Lorazepam
Mechanism of Action
Benzodiazepines enhance the inhibitory effects of GABA in both the brain and spinal cord (see Figure 27-2). Thus they not only decrease seizure spread but also block arousal and centrally depress spinal reflexes (see Chapter 26 for additional discussion). In dogs, tolerance to the anticonvulsant activity of diazepam develops rapidly, within 1 week, and as such, diazepam is not an effective anticonvulsant for chronic therapy in dogs. Intravenous diazepam is, however, the drug of choice for the treatment of SE in both dogs and cats, in part because of its mechanism bt also because it rapidly crosses the blood–brain barrier into the CSF.
Disposition
Diazepam is the prototype benzodiazepine used in small animals. The drug is well absorbed after oral administration but undergoes rapid and extensive hepatic metabolism. Although only 1% to 3% of diazepam is orally bioavailable, 74% to 100% of the drug and all active metabolites are available.42 Diazepam is generally administered intravenously but can also be administered intramuscularly, although absorption is not predictable using this route. In human pediatric and canine patients, diazepam has been administered rectally.95,96
The metabolites of diazepam (nordiazepam [desmethyldiazepam] and oxazepam) are active (see Figure 27-4), although less so (25% to 33%) than the parent compound. 3-Hydroxydiazepam also is a metabolite, although its anticonvulsant activity is not known.96 Although less potent than the parent compound, the half-lives of the metabolites are slightly longer than that of diazepam (3.6 hours for desmethyldiazepam and 5.2 hours for oxazepam, compared with 15 minutes for diazepam).94,96 After oral administration diazepam undergoes rapid first-pass metabolism, with diazepam representing only 1% to 3% of the total (parent and metabolite) area under the curve, compared with 7% to 21% after intravenous administration. Oral bioavailability of all compounds approximates 74% to 100%. After oral administration metabolite concentration surpasses that of the parent compound.96 Using a crossover study in both mixed-breed dogs (n = 6) and Beagles (n = 4), intravenous (0.5 mg/kg) or rectal (2 mg/kg to mixed-breed dogs and 0.5 mg/kg to Beagles) administration, and high-performance liquid chromatography to detect either diazepam and its active metabolites, desmethyldiazepam (nordiazepam) and oxazepam, Papich and Alcorn95 demonstrated that the sum total of metabolites will surpass the parent compound. Löscher and Frey97 reported that the sum bioavailabililty (%) of parent compound and metabolites ranged from 74 to 100 (median 86). Although rectal bioavailaibity of diazepam was only 7.4 ± 5.9% and 2.7 ± 3.2 % for the high and low dose (or mixed breed and Beagles), respectively, bioavailability of diazepam and its two metabolites increased to 79.9 ± 20.7 and 66.0 ± 23.8 for the high dose and low dose, respectively. After intravenous administration, diazepam was characterized by an elimination half-life of approximately of 15 minutes, compared with approximately 2.5 hours disappearance half-life for desmethyldiazepam and 3.8 hours for oxazepam. Peak concentrations of both diazepam and desmethyldiazepam after intravenous administration (0.5 mg/kg) approximated 800 ng/mL; oxazepam peaked at approximately 350 ng/mL. However, whereas diazepam concentrations were below 100 ng/mL within 45 minutes of administration, concentrations were above 300 ng/mL (adjusting for 30% potency of diazepam) for desmethyldiazepam at 8 hours and oxazepam at 1.5 hours. Rectal administration of 2 mg/kg diazepam yielded peak diazepam concentrations of only 75 ng/mL; this compared to peak desmethyldiazepam and oxazepam concentrations of approximately 1600 and 550 ng/mL, respectively. Both desmethyldiazepam and oxazepam concentrations remained above 300 ng/mL at study end (8 hours). As such, rectal administration of diazepam can be expected to yield effective anticonvulsant activity, with concentrations remaining above the minimum recommended for about 8 hours for desmethyldiazepam.95
KEY POINT 27-15
Benzodiazepams tend to be the most rapidly acting anticonvulsants and thus are indicated for acute seizure management.
Diazepam has also been studied in dogs (n=6) after intranasal administration (0.5 mg/kg) either as drops or a commercially available atomizer. Bioavailability approximated 40%. Of the two routes, the atomizer was well tolerated. 98 Another study diazepam administered IN (0.5 mg/kg; drug measured as benzodiazepines) in dogs (n = 6, crossover design) reported mean peak plasma concentration of benzodiazepine was 448 ± 41 ng/mL at 4.5 minutes, compared with 1316 ± 216 at 3 minutes after intravenous administration. Intranasal bioavailability of benzodiazepine was 80 ± 9%. Plasma drug concentrations exceeded the recommended anticonvulsant therapeutic concentration (300 ng/mL).99
The generation of active metabolites complicates the utility of therapeutic monitoring for benzodiazepines as a guide to therapy because anticonvulsant activity is not necessarily correlated with concentration of the parent compound. For diazepam all metabolites and parent drugs should be measured. Methods based on polarized immunofluorescence appear to correlate with total activity relatively well but are likely to underestimate concentrations by 50% or more;94,95 the amount of underestimation does not appear to be predictable but probably reflects failure to detect oxazepam.
Benzodiazepines enter the CSF rapidly, with peak concentrations usually occurring within 15 minutes of intravenous administration. The more lipophilic drugs enter CSF most rapidly. CSF to plasma drug ratios generally are less than unity, although concentrations may stay higher in CSF compared to plasma, prolonging the duration nof effect.100 Both diazepam and desmethyldiazepam rapidly pass into the CSF.96,101
Diazepam and nordiazepam have been studied in cats after intravenous administration of 5, 10, and 20 mg/kg of diazepam and 5 and 10 mg/kg of nordiazepam. Elimination of both drugs was linear over the range of doses covered. Total body clearance of diazepam (4.72 ± 2.45 mL/min/kg) was sixfold greater than that of nordiazepam (0.85 ± 0.25 mL/min/kg). Approximately 50% of an administered dose of diazepam was biotransformed to nordiazepam in the cat.102
Clorazepate is metabolized in the stomach to its active metabolite, nordiazepam (desmethyl diazepam), which is also a major, although less efficacious, metabolite of diazepam. After oral administration of 2 mg/kg, clorazepate reaches peak benzodiazepine concentration of 446 to 1542 ng/mL in dogs; mean residence time was 8.5 hours. After multiple administration, mean residence time was significantly longer (approximately 12 hours).103 Scherkl and coworkers101 described the consequences of long-term use of clorazepate in dogs. After 2 hours of clorazepate infusion, only nordiazepam was detected in CSF. The elimination half-life of nordiazepam ranged from 7.2 to 12 hours and did not change with 6 weeks of oral therapy. Only nordiazepam and oxazepam were in plasma. Oral doses of 2 mg/kg were associated with ataxia and difficulty standing; these signs resolved 3 to 4 days after treatment. Concentrations of 0.180 to 0.780 μg/mL were associated with decreased seizure threshold. Tolerance was demonstrated in two of six dogs, with some, but not total, loss of anticonvulsant activity. Withdrawal also was demonstrated by manifestation of generalized tonic-clonic seizures in two of six dogs rapidly withdrawn from therapy; death occurred in one of the dogs. The remaining four dogs exhibited no signs of withdrawal.
KEY POINT 27-16
Dogs, but not cats, develop tolerance to diazepam and will become refractory to its anticonvulsant effects after approximately 2 weeks.
Clorazepate anecdotally is an alternative anticonvulsant in cats (3.75 to 7.5 mg/cat orally every 8 to 12 hours). However, because it is an active metabolite of diazepam in other species, its role in diazepam-induced hepatotoxicity cannot be ruled out and caution is probably indicated with its use in cats.
Lorazepam is a benzodiazepine derivative used as an anxiolytic in human medicine. It has been studied after intravenous and rectal administration in dogs,104 although its role in the management of seizures has not been identified. After IV administration of 0.2 mg/kg IV, concentrations remained above 190 ng/mL for 20 min in two of three dogs, and above 30 ng/mL for 90 min in all dogs. However, lorazepam was not detectable after rectal administration of 1 mg/kg, presumably due to high first pass metabolism. Lorazepam also has been studied after intranasal administration. Lorezpam is absorbed almost 3 times as rapidly after intranasal administration compared with oral administration, yielding a relative bioavailability that approximates 2.5 times that after oral administration.105 Mariami reported in abstract form that the IN administration of 0.2 mg/kg in dogs yielded peak plasma lorazepam concentrations of 106 ng/mL compared with 165 ng/mL following intravenous administration. Concentrations of lorazepam achieved 30 ng/mL (effective anticonvulsant target in humans) by 9 minutes after IN administration in six of six dogs and remained at or above the target for 60 minutes in three of six dogs. Midazolam also has been studied after IN administration with IN aadminsitration exceeding oral administration by 2.5 fold.106
Clonazepam (Klonopin) is a benzodiazepine derivative and chemically is 5–(o–chlorophenyl)-1,3-dihydro-7-nitro-2H-1,4-benzodiazepine-2-one (see Figure 27-4). It is more potent than diazepam and is used only in the emergency treatment of SE in the dog.42 Clonazepam is given orally or intravenously (0.05-0.2 mg/kg; the intravenous preparation is not available in the United States). Accumulation occurs with continued administration. Tolerance develops within days to weeks after administration, however, as a result of hepatic enzyme induction. Consequently, clonazepam, like diazepam, is unsatisfactory for long-term control of epilepsy in dogs. However, it might be considered for long-term use in cats (0.016 mg/kg/day; target concentration 70 ng/mL). The relationship between clonazepam and liver disease has not been addressed in cats. The disposition of clonazepam has been described in the dog (see Table 27-4).107,108
Preparations
Diazepam is available as both an intravenous and an oral preparation; clorazepate is available as an oral preparation and a sustained-release preparation.109 It is classified as a Schedule IV drug under the 1970 Controlled Substances Act. Clonazepam is available as an intravenous preparation (not in the United States) and as oral tablets.
Safety
Sedation is the most common direct side effect of the benzodiazepines. Adverse effects (sedation, ataxia, increased appetite, and in some cases hyperactivity) are likely to occur if concentrations reach 500 ng/mL. An 8-hour rather than a 12-hour dosing interval may be indicated to avoid both toxic and side effects. Care must be taken not to discontinue benzodiazepines abruptly because of the potential for SE.110
Diazepam may cause hepatotoxicity in cats.111 Reports have focused on cats receiving diazepam as an appetite stimulant rather than as an anticonvulsant. Manifestations include vomiting, depression, jaundice, lethargy, and acute death. Clinical laboratory tests associated with toxicity include increased serum alanine transaminase, aspartate transferase, and alkaline phosphatase activities and increased bilirubin. Toxicity does not appear to be associated with the dose or duration. Toxicity has not been experimentally induced, suggesting that the reaction is idiosyncratic (i.e., nonpredictable).
Physical dependency toward benzodiapenes has been described in dogs, with the risk and severity of signs of withdrawal dependent on dose and drug. Studies that demonstrate dependency often are based on precipitation of clinical signs after administration of a benzodiazepine antagonist; the severity of withdrawal also appears to be dependent on the dose of flumazenil. Both diazepam and lorazepam can be associated with withdrawal in dogs, although only high doses (60 and 1000 mg/kg per day, respectively, for diazaepam and lorazepam) have been studied. Clinical signs included, but were not limited to, tremor, “hot foot walking,” rigidity, and decreased food intake. Lorazepam withdrawal appears to be much less intense but has a shorter latency to onset than the diazepam in manifestation of abstinence syndrome. For diazepam, withdrawal signs include bi-phasic clonic and tonic–clonic convulsions, appearing at 24 and 48 hours. Diazapam syndrome was lethal in two dogs studied experimentally. Administration of diazepam can reduce but not obliterate the major signs of the diazepam withdrawal. The syndrome was worsened by the administration of a benzodiazepine antagonist, although tonic–clonic seizures were not precipitated112 A follow-up study described withdrawal after oral administration of increasing doses of diazepam administered every 8 hours for 5 to 6 weeks: 0.05625, 0.225, 0.5625, 4.5, 9, or 36 mg/kg daily every 8 hours. Abstinence was precipitated by administration of a benzodiazepine antagonist, flumazenil, at 0.66, 2, 6, 18, 36, and 72 mg/kg, but included a placebo. Withdrawal intensity increased proportionately with the dose of diazepam and the dose of flumazenil. The pattern of withdrawal signs varied with dogs receiving high doses of diazepam more sensitive to flumazenil than those receiving lower doses. Seizure activity occurred only in dogs receiving 9 and 36 mg/kg/day of diazepam. Withdrawal signs increased linearly with plasma and brain concentrations of diazepam, oxazepam, and nordiazepam but not with free concentrations of diazepam alone.113
Although desmethyldiazepam (nordiazepam) has been theorized to be the metabolite causing physical dependence in dogs receiving diazepam (in part because the area under the curve for nordiazepam is much greater than that for diazepam),114 follow-up studies using benzodiazepines whose area under the curve is not surpassed by nordiazepam suggest otherwise.115,116
The ability of flumazenil to induce clinical signs of withdrawal, including clonic seizures, in dogs, is well described.114 Flumazenil precipitated withdrawal scores were higher in dogs receiving diazepam (and halazepam, which is converted to nordiazepam) compared with nordiazepam, although clonic seizures were greater in the diazepam- and nordiazepam-dependent dogs, compared with halazepam-dependent dogs. The magnitude of withdrawal does not appear to reflect a predominance of one parent compound or metabolite but rather a combined effect.115 Oral flunitrazepam at 7.6 mg/kg per day caused more severe clinical signs of precipitated withdrawal, compared with diazepam (at 6 to 9 mg/kg every 6 hours) administered in four equally divided doses. However, precipitated signs persisted longer with diazepam than flunitrazepam, leading investigators to conclude that diazepam and flunitrazepam were equivalent in their ability to produce induced signs of withdrawal. Difference in kinetics, including protein-binding, among drugs and metabolites, as well as receptor interactions are likely to play a role in differences in response. For example, the estimated free plasma concentration of flunitrazepam and its metabolites was equal to or greater than that of diazepam and its metabolites; further, affinity of flunitrazepam for the benzodiazepine receptor is greater than that of either diazepam, nordiazepam, or oxazepam.116 The disposition of flumazenil in dogs has been described after oral dosing. Rapid absorption is followed by rapid elimination with concentrations being essentially nondetectable in 4 hours.117 Interestingly, benzodiazepine withdrawal induced by flumazenil was described in one study to occur particularly as plasma concentrations markedly decreased.117
Drug Interactions
Clinically important drug interactions resulting from chronic diazepam therapy have not been reported. Despite reports to the contrary,118 interactions between clorazepate and phenobarbital appear to confound therapy. In our studies clorazepate consistently increases phenobarbital concentrations in patients that have been receiving long-term phenobarbital therapy if doses of clorazepate exceed 1 mg/kg every 8 to 12 hours. The increases are usually evident by the first month of therapy but may take longer. It may be necessary to decrease phenobarbital doses. Yet clorazepate concentrations tend to decrease across time despite no change in dose, presumably because of the inductive effects of phenobarbital. As clorazepate concentrations decrease, phenobarbital concentrations may also decrease, which may put an epileptic at risk, particularly if the dose of phenobarbital has been reduced.
Therapeutic Use
As in humans, efficacy, including rapidity of onset, coupled with lack of toxicity, leads to intravenous diazepam as the first drug of choice for control of SE in humans.45 Likewise, diazepam is the drug of choice for emergency treatment of SE in dogs and cats.42 However, because of its short half-life, diazepam is likely to have to be repeated once or twice during the first 2 hours of stabilization.49 Not surprisingly, various methods of administration have been recommended in an attempt to maintain effective diazepam concentrations necessary to maintain efficacy. Intravenous doses vary, beginning at 5 to 20 mg or 0.5 to 1 mg/kg.42 The dose can be repeated in 1 to 2 minutes if necessary; giving the same dose intramuscularly may provide longer therapeutic concentrations. If a response has not occurred after the second dose of the drug, an intravenous infusion can be implemented (note that the infusion line should first be flushed with the diazepam solution to allow diazepam binding to the polyvinyl). Alternative anticonvulsants might be considered in animals failing to respond (discussed later). Patients that respond to the first or second dosages of diazepam are carefully monitored, and if SE returns within 2 to 4 hours after the initial treatment, the diazepam regimen is repeated. Intravenous diazepam may be replaced with other benzodiazepines, such as clonazepam (0.05 to 0.2 mg/kg), toward which tolerance develops more slowly. For cats an intravenous dose (5 to 10 mg) generally is given to effect. The dose may need to be repeated; up to 20 mg may be necessary. High doses should be injected slowly. Diazepam (0.5 mg/kg using 5 mg/mL solution) has been used successfully by rectal administration for home control of cluster seizures.119
For chronic therapy rapid metabolism coupled with rapid development of tolerance precludes long-term treatment with diazepam and several other benzodiazepines in dogs.29 However, compared with diazepam, tolerance does not appear to develop as readily to the anticonvulsant effects of clorazepate in dogs. Clorazepate has been studied when added to phenobarbital in dogs still seizuring despite phenobarbital concentrations that exceeded 32 μg/ml (author, unpublished data). Seizures were eradicated in about 35% of animals and reduced 50% or more in another 15%. However, its use in combination with phenobarbital was complicated by drug interactions (including marked fluctuations in nordiazepam disappearance half-life), and an apparent increased risk of hepatotoxicity. Drug interactions between phenobarbital and clorazepate may necessitate dose modification in response to changes in drug concentrations. The short half-life (which might be documented with sequential peak and trough plasma drug concentration measurements) may increase the risk of seizures if drug concentrations decrease below effective concentrations in the patient. The therapeutic range of benzodiazepines (including metabolites) in dogs has been extrapolated from human studies and does not reflect combination therapy. For efficacy, trough concentrations should be maintained at least above 100 ng/mL, although monitoring should establish each patient’s range. Dogs receiving phenobarbital are likely to become groggy when peak clorazepate concentrations exceed 300 to 500 ng/mL (varying with the patient).
For chronic control in cats, diazepam can be given orally in doses of 2 to 5 mg 2 or 3 times daily; diazepam may be increased or decreased in increments of 2 mg with or without phenobarbital. Whereas distribution of diazepam in the cat brain reflects blood flow, nordiazepam accumulates to high concentrations throughout all brain matter, which may be the reason for its efficacy with long-term use.120
Bromide
Bromide is an old anticonvulsant that was used in the 1800s for control of seizures. However, it has enjoyed a resurgence in veterinary medicine, proving to be an effective add-on anticonvulsant for refractory (canine) seizures, as well as a sole anticonvulsant for some canine patients.
Mechanism of Action
Bromides mechanism of action is not completely understood.121 The original proposed mechanism—replacement of negatively charged chloride with bromide in the neuron—is unlikely because bromide is less negatively charged than chloride.122 In vitro, bromide enhances GABA-activated currents, leading the authors to conclude that bromide potentiates inhibitory postsynaptic potentials of GABA. Regardless of the mechanism, the anticonvulsant effects of bromide correlate with plasma concentration.123
Disposition
The pharmacokinetics of bromide have been described using either the sodium or potassium salt. In a small study, the half-life in dogs was reported as 21 to 24 days after oral administration, suggesting steady-state concentrations being achieved only at 2.5 to 3 months.124 Distribution is to extracellular fluid,125 yet sufficient quantities penetrate the CNS. Bromide is eliminated slowly (presumably as a result of marked reabsorption) in the kidney. Bromide appears to compete with chloride for renal elimination, with elimination appearing to decrease proportionately to the amount of chloride in the diet.126,127
KEY POINT 27-17
As a renally excreted compound, bromide is not likely to be involved in drug interactions. However, serum bromide concentrations will change proportionately and inversely with the amount of chloride consumed by the patient.
Bromide (20 mg/kg bromide; approximately 26 mg/kg sodium bromide) has been studied in Beagles as the sodium salt after intravenous (n = 4) and oral (n = 4) administration, but only at the daily maintenance dose (which is about 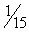 to
to 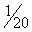 the loading dose).128 Dogs received a diet containing 0.4% chloride on a dry-matter basis. Elimination half-life after intravenous administration was 39 ± 10 days and after oral administration, 46 ± 9 days; clearance (reported in ml/kg/day) was 9 ± 3.9, and apparent volume of distribution (Vd area) was 0.45 ± 0.07 L/kg. Oral bioavailability (calculated from mean area under the curve from intravenous and oral groups) was 46%. A study by the same investigators127 in Beagles measured changes in bromide (sodium; 14 mg/kg orally) disposition in response to dietary chloride content (0.2, 0.4, and 1.3%, dry-matter basis). Mean apparent elimination half-life decreased from 69 ± 22 days (0.2%) to 24 ± 7 days (1.3%). Predicted maximum drug concentrations were quite variable within treatment groups but were profoundly influenced by dietary chloride content, ranging from 1.95 ± 1.1 mg/mL at 0.2% chloride, 1.27 ± 0.36 mg/mL at 0.4% chloride, to a low of 0.3 ± 0.15 mg/mL at 1.3% dietary chloride.
the loading dose).128 Dogs received a diet containing 0.4% chloride on a dry-matter basis. Elimination half-life after intravenous administration was 39 ± 10 days and after oral administration, 46 ± 9 days; clearance (reported in ml/kg/day) was 9 ± 3.9, and apparent volume of distribution (Vd area) was 0.45 ± 0.07 L/kg. Oral bioavailability (calculated from mean area under the curve from intravenous and oral groups) was 46%. A study by the same investigators127 in Beagles measured changes in bromide (sodium; 14 mg/kg orally) disposition in response to dietary chloride content (0.2, 0.4, and 1.3%, dry-matter basis). Mean apparent elimination half-life decreased from 69 ± 22 days (0.2%) to 24 ± 7 days (1.3%). Predicted maximum drug concentrations were quite variable within treatment groups but were profoundly influenced by dietary chloride content, ranging from 1.95 ± 1.1 mg/mL at 0.2% chloride, 1.27 ± 0.36 mg/mL at 0.4% chloride, to a low of 0.3 ± 0.15 mg/mL at 1.3% dietary chloride.
A study of potassium bromide (20 mg/kg per day bromide or approximately 30 mg/kg potassium bromide), mixed as a 20 mg/mL solution in canned dog food, in Beagles (n = 6) receiving 0.55 to 0.72% dietary chloride reported a median bromide concentration at 115 days of 2.45 mg/mL (low of 1.8 and high of 2.7 mg/mL); the time to steady state was not provided, but concentrations were within 90% of steady-state concentrations by 60 days.129 The median elimination half-life, estimated from time to reach steady state, was 15 days (low of 12, high of 20 days). Renal clearance (based on 24-hour urine collection) ranged from 6 to 12 .6 mL/kg per day (median 8.2). The median ratio of bromide in CSF to serum at day 9 was 0.63; this improved to 0.86 (0.82 to 0.99) by day 120. An increase in dose sufficient to generate serum bromide concentrations 3.4 mg/mL (median; does not provide but designed to target 4 mg/mL) resulted in an increase in the CSF: serum ratio to 0.86, suggesting a greater risk than anticipated of CNS side effects. Whereas neurologic side effects were not described at the dose of 60 mg/kg per day, caudal paresis and ataxia developed in two of six dogs. Electrodiagnostic changes were mild even at the high dose; within individual dogs, latency shifts were mild and either progressively increased over time or appeared with doses designed to target 4 mg/mL.
Bromide was studied (report in progress by the author) after rectal administration in normal hound dogs after a loading dose (600 mg/kg) of sterile potassium bromide solution (concentration 250 mg/mL) over 24 hours, either by 6 intrarectal boluses of 100 mg/kg, each every 4 hours, or constant-rate infusion (CRI) throughout the 24-hour period. The average peak serum bromide concentration after intrarectal loading was 0.91 mg/mL (range: 0.81 to 1.11 mg/mL), compared with 1.10 mg/mL after intravenous loading (range: 0.89 to 1.22 mg/mL). The average peak serum K+ concentration during potassium bromide loading was 5.80 mEq/L (range: 5.5 to 6.8 mEq/L) and 6.14 mEq/L (range: 5.6 to 7.7 mEq/L) for intrarectal and intravenous loading, respectively (normal range: 3.5 to 5.5 mEq/L). The mean half-life (t1-2) of intrarectally administered bromide was 488.8 hours, or 20.4 days. A side effect of both intrarectal and intravenous potassium bromide loading was mild sedation, and of intrarectal loading was transient (24 to 48 hours) diarrhea. Bioavailability (F) of intrarectally administered bromide for the 24-hour loading period was calculated to be 107% for the five dogs. In two dogs overall bioavailability (F) of intrarectally administered bromide was calculated to be 57.7%. Results of this study indicate that potassium bromide is well absorbed rectally and that a 24-hour intrarectal loading protocol is safe to administer in normal dogs.
Bromide has been studied in cats after oral administration of the potassium salt at the lower end of the canine dose. After a dose of 30 mg/kg, maximum serum bromide concentration was 1.1 ± 0.2 mg/mL at 8 weeks. Mean disappearance half-life was 1.6 ± 0.2 weeks. Steady state was achieved at a mean of 5.3 ± 1.1 weeks.129a However, the use of bromide in cats is discouraged due to safety reasons.129a
Preparations and Sources
Bromide is available as a potassium or sodium salt. Triple bromide salt preparations (Na, K, and NH4) also may be available through some pharmacies. The accompanying cation does not appear to alter efficacy, although sodium bromide is more difficult to solubilize in water than is potassium bromide. In addition, because potassium weighs less than sodium, 1 g of sodium bromide contains more bromide (78% bromide, or 780 mg) than does 1 g of potassium bromide (67% bromide, or 670 mg). Thus the amount of sodium bromide used to make a solution should be less (211 mg/mL) than potassium bromide (250 mg/mL) to achieve equivalent amounts of bromide in the solution. At the time of publication, a commercial product of potassium bromide is being marketed as a chewable tablet and solution. The product contains B vitamins as well. This product has not been approved by the Food and Drug Administration. It is being marketed under the “generally recognized as safe” category reserved for food of food ingredients; however, this status does not apply to drugs It is important to recognize that the product has not undergone premarket evaluation by any regulatory agency for safety, efficacy, or quality and although its use is not necessarily discouraged, clinicians should be aware that this is a manufactured-compounded product.
Potassium bromide can be compounded by veterinarians or pharmacists on an individual-need basis. It can be purchased through chemical companies (request medicinal or ACS grade [Curtin Matheson]). Chemical companies have refused to sell bromide for medicinal purposes without an investigational new animal drug application (INADA). This application is no longer necessary, however, because the Food and Drug Administration (Division of Drug Compliance) will grant regulatory discretion.
Bromide can be compounded in a syrup or water solution or administered in a gelatin capsule. A number of compounding pharmacies now offer this service, although the compounded solution can be much more costly when compounded by a pharmacist compared with the cost of the ingredients. The solution can be compounded by the clinician with minimal equipment, particularly if monitoring is used to document maintenance of drug concentrations with compounded preparations. If purchased (1-kg bottle) from a chemical company, potassium bromide can be weighed and divided into 250-g (four equal) packets. A liter bottle of distilled water can be purchased from any grocery store. Before the bromide is mixed, a line should be drawn at the 1-L volume mark and approximately 50% of the water removed and set aside. One of the four 250-g packets of bromide is added to the bottle and the bottle shaken well to dissolve the bromide (this may require aggressive shaking and the addition of more water). Once the bromide is dissolved, the volume should be returned to 1 L with either water or a flavored syrup. The final solution will approximate 250 mg/mL. The solution should be stable for at least 6 months, and refrigeration should minimize microbial growth in the solution. Refrigerating the solution may cause the salt to crystallize; warming the solution should cause the drug to redissolve. Note that while this solution can be dispensed from a practice, it cannot legally be sold to others.
Side Effects
Adverse reactions to bromide tend to be dose dependent. They appear to be related to the anticonvulsant or other centrally acting effects of the drug and predominantly affect the CNS (e.g., ataxia, grogginess). This is supported by the recent retrospective report of Rossmeisl and Inzana,130 which described signs of “bromism” in epileptic dogs (n = 31). Clinical signs included ataxia and stupor, with one dog being comatose. Other clinical signs included bilateral mydriasis; head pressing was evident in two dogs. Mean bromide concentration at time of admission was 3.7 ± 0.3 mg/mL compared with the most recent monitoring result before admission (1.9 ± 0.3 mg/mL) and compared with a control group of epileptic dogs without bromism at 1.7 ± 0.1 mg/mL. Of the 31 dogs, 26 dogs also were receiving phenobarbital at a mean concentration of 31 ± 12 μg/mL; control group phenobarbital was 26 ± 1 μg/mL. Causes for the increase in bromide were attributed to reduction in renal function, dosing error, shifting from potassium to sodium bromide, and exposure to bromide in drinking water or alternative medications. Changes in diets, the most common reason for increased bromide in the author’s therapeutic drug monitoring laboratory, was not addressed. Diuresis was implemented in affected dogs (n = 27); four of these dogs had breakthrough seizures. Furosemide therapy was initiated in five dogs; however, it is not clear what impact furosemide has on bromide clearance.
Up to 3 months may be required for accommodation to the sedative effects of bromide. This reflects in part the time necessary to reach steady-state concentrations. Gradual reduction of phenobarbital in 25% increments may resolve some of the side effects and may be preferred to saline diuresis in those patients for which nonthreatening clinical signs of bromism emerge. Alternatively, the bromide dose can be decreased by 25%, although 1 to 2 weeks may elapse before a response is seen. Sodium bromide administered as a loading dose sufficient to achieve steady-state concentrations has been reported to be well tolerated in dogs.131
Fluids containing sodium chloride can be used to treat acute bromide toxicity; monitoring should occur immediately after saline treatment to establish a new baseline. Note that diuresis may increase the risk of breakthrough seizures and SE. Diuresis should be reserved for animals with profound sedation for which a decrease in phenobarbital (if on combination therapy) or bromide will not provide a sufficiently rapid response. Pruritic skin lesions may occur, particularly in patients that had pruritic disorders before starting therapy. A short period of glucocorticoid therapy may control pruritis. Hyperactivity is an occasional side effect and may or may not be dose dependent. Like other anticonvulsants, bromide tends to increase the appetite of dogs. Vomition is not uncommon and appears to reflect the hypertonicity of the salt and direct gastric irritation. Solutions appear to be better tolerated than capsules, although this may vary. Dividing the daily dose into smaller, more frequent doses or feeding before or with medication may decrease gastrointestinal side effects. Sodium bromide may be more tolerated than other bromide salts.
Pancreatitis has been associated with the use of bromide when combined with phenobarbital. A retrospective report found 10% of dogs receiving a combination of potassium bromide and phenobarbital developed clinical signs consistent with pancreatitis, compared with 0.3% of dogs receiving phenobarbital alone.80 Serum pancreatic lipase immunoreactivity was found to be increased in the serum of dogs receiving either bromide or phenobarbital, alone or in combination, indicating that the risk may occur with either drug. 81
Although cats tolerated 8 weeks of oral potassium bromide dosing (experimentally) at 30 mg/kg, clinical patients do not appear to tolerate the drug as well. However, in a retrospective study, bromide (n = 4) or bromide and phenobarbital (n = 3) was associated with eradication of seizures in 7 of 15 cats (bromide dosage range, 1 to 1.6 mg/kg [0.45 to 0.73 mg/lb]); however, bromide administration was associated with adverse effects in 8 of 16 cats. Coughing developed in six of these cats, leading to euthanasia in one cat and discontinuation of bromide administration in two cats. Although somewhat effective in seizure control, the incidence of adverse effects may not warrant routine use of bromide for control of seizures in cats.
Because it is renally eliminated, bromide does not negatively interact with other anticonvulsants metabolized by the liver. However, dietary (or therapeutic) chloride will compete with bromide for renal excretion and can shorten bromide half-life, probably because the kidneys will selectively resorb chloride.127 In addition, laboratory assays may not be able to distinguish among anions; for some, bromide may artificially increase serum chloride measurements.132
Therapeutic Use
Bromide has been used to treat seizures in dogs for over 15 years. Initially, it was used as an adjunct to phenobarbital therapy, an indication that persists.133,134 However, it is increasingly being used as sole therapy.28 Therapeutic ranges vary, with initial studies indicating 0.7 to 2 mg/mL.134 More recently, dogs (n = 122; from 1992-1996) with major motor epilepsy were studied retrospectively to determine the therapeutic range of bromide, either alone or in combination with phenobarbital or primidone. Bromide successfully controlled seizures (≥50% reduction in seizure frequency) in 72% of animals. Phenobarbital or primidone could be discontinued in 19% of animals receiving the combination. For dogs that continued to receive both drugs, 45% remained controlled with a phenobarbital dose below 20 μg/mL. The concentration of bromide necessary to control seizures when given as sole therapy was higher (1.9 mg/mL) than when combined with either barbiturate (1.6 mg/mL). The authors concluded that the range for bromide depends on whether bromide will provide sole anticonvulstant support (0.8 to 2.4 mg/mL) or is combined with a barbiturate (0.88 to 3 mg/mL). If phenobarbital could not be discontinued, the range necessary for control was 9 to 36 μg/mL.135 A distinction in therapeutic ranges as sole or combination therapy of bromide may not be necessary. Concentrations may fall outside or within a range of 1 to 3 mg/mL (based on the gold chloride method of detection) as needed to control the patient’s seizures, regardless of concomitant therapy. Because the side effects of bromide apparently are not associated with cellular toxicity (unlike phenobarbital), the therapeutic range can be surpassed in animals, if tolerated and if necessary to control seizures. Drug concentrations can be increased beyond this concentration if there is no evidence of toxicity. Some animals will present with side effects at concentrations in the lower end of the range, particularly if a loading dose is administered. Therefore it is generally wise to avoid a loading dose if seizure history and drug safety (e.g., phenobarbital) is supportive. Bromide generally should be given once or twice daily, depending on the animal’s gastric tolerance of the drug. Twice-daily administration of bromide offers no benefit over once-daily administration, with the potential exception of decreased gastric irritation. Indeed, an animal can miss several days of dosing with minimal impact on serum drug concentrations. Missed doses in such instances should be given when convenient. On the other hand, if bromide is better tolerated when broken into much smaller, more frequent doses, no disadvantages are apparent with this approach, other than inconvenience to the owner.
KEY POINT 27-19
The long half-life causes the loading dose of bromide (450 mg/kg) to be substantially higher than the daily maintenance dose (30 mg/kg).
In humans bromide has been used to treat intractable seizures in pediatric patients.81,136,137 Bromide’s initial introduction into veterinary medicine was as an add-on anticonvulsant, particularly combined with phenobarbital, for control of refractory seizures in dogs or in patients that have developed liver disease in association with phenobarbital therapy. Increasingly, however, bromide is proving efficacious as a first-choice antiepileptic drug for dogs. For example, bromide has been used as the sole drug for patients whose seizure history is limited to mild seizure episodes.133 Because of its long half-life, bromide may be the drug of choice for dogs whose owners are noncompliant because the plasma drug concentrations are not easily manipulated. The author studied bromide as an add-on anticonvulsant in dogs with refractory epilepsy (n = 20) in which seizures continued despite phenobarbital concentations at 32 μg/ml or more. Bromide eradicated seizures in 60% of the patients and reduced seizure frequency by 50% or more in another 20%.
Two choices should be considered when starting bromide therapy: administration of a maintenance dose, which allows the body to gradually adapt to the presence of the drug, or administration of a loading dose. The maintenance dose approach is indicated when possible to minimize the advent of side effects. A targeted concentration should be selected (lower concentrations for milder seizures; higher concentrations may be necessary for severe seizures). In general, 30 mg/kg per day orally will achieve 1 mg/mL at steady state. For every 0.5 mg/mL increment above 1 mg/mL, another 15 mg/kg per day should be administered. To proactively monitor when using the maintenance dose approach to steady state, one sample should be collected at 3 weeks, or halfway to steady state. Doubling the measured concentrations provides an estimate of steady-state concentrations. However, because kinetics vary among animals, a steady-state sample should be collected to confirm baseline concentrations at 3 months.
The second option for beginning bromide therapy is implementation of loading dose. This approach is recommended in patients for which lack of seizure control can be life threatening (including the risk of euthanasia by a distraught owner), or in patients for whom serum phenobarbital concentrations must be rapidly decreased because of hepatotoxicity or bone marrow suppression. Both loading dose and maintenance dose must be established; the loading dose is designed to target steady-state concentrations, but the patient will not be at steady state until being on the same dose for 2 to 3 months. Thus as the loading dose is discontinued and the maintenance dose is begun, a new time to steady state begins. Drug concentrations may increase or decrease compared with the loading dose as steady-state concentrations are reached. Monitoring can ensure that the maintenance dose will maintain what the loading dose achieved. However, this requires a sample collected after loading (within 1 or 2 days) to document what loading achieved, and another sample one elimination half-life after the maintenance dose is started (about 3 weeks). This time is chosen because if concentrations are not maintained by the loading dose, the majority of change (50%) will occur in the first elimination half-life. If the two samples are not within about 10 to 15% of one another (i.e., postload and 3-week maintenance), the maintenance dose should be increased or decreased accordingly. Generally, an increase at the 3-week point is not problematic (and often is desirable), but a decrease is undesirable. If not curtailed by an increase in the maintenance dose, as steady state is reached, the decrease may be sufficient to lead to seizures 2 to 3 months after the loading dose successfully controlled seizures.
A loading dose should reflect all the doses that would have been given as steady state is reached, less drug eliminated by clearance. Thus the loading dose of bromide is substantially higher than the maintenance dose. To target the lower end of the therapeutic range (1 mg/mL), 450 mg/kg should be given as a loading dose. Thereafter, for every 0.5 mg/mL desired increase in bromide concentrations, the loading dose should be increased by approximately 225 mg/kg. Because animals may not tolerate the loading dose as a single oral dose, it generally is divided into 10 equal doses given twice daily for 5 days. However, note that the maintenance dose should be added to the loading dose if the targeted concentration is to be reached. For example, to target 1.5 mg/mL with a 5-day loading period, the patient should receive 675 mg/kg split over a 5-day period, plus a maintenance dose of 45 mg/kg/day. Thus for 6 days, the patient should receive approximately 135 mg/kg. On day 6 the maintenance dose of 45 mg/kg is begun and a postload sample is collected (assume the loading dose achieved 1.3 mg/mL). A second sample is collected 3 weeks later. Allowing for some variability in analytic error, then the maintenance dose can be adjusted in proportion to the difference between the postload and 3 week sample. For example, if the postload was 1.3 mg/ml, and a 3-week concentration is 0.8 mg/mL, the maintenance dose may need to be increased by about 25% to 30%). If a patient already is receiving bromide and an increase in concentrations is desired (e.g., 0.5 mg/mL), the maintenance dose can be increased 15 mg/kg/day. A third sample can be checked at 3 weeks to make sure drug concentrations are moving in the right direction. If a rapid response is desired in a patient already receiving bromide, a “mini” loading dose of 225 to 250 mg/kg total can be given along with an increase in maintenance dose.
The use of bromide as an add-on anticonvulsant to phenobarbital also is discussed later in this chapter. Monitoring in such patients is important if the goal of the additional bromide is to decrease or discontinue phenobarbital. If the goal is to replace phenobarbital, the bromide target should approximate the current phenobarbital concentration in the patient. For example, if the patient has phenobarbital concentrations of 20 μg/mL, (midtherapeutic range), then the bromide dosing regimen might approximate the midtherapeutic range (i.e., about 1.5 to 2 mg/mL). To prevent excessive grogginess, bromide should be initiated as a maintenance rather than loading dose. At least 3 weeks of dosing (i.e., halfway to steady state) should occur, ideally, to allow a halfway to steady-state sample that confirms the bromide dose is on target before phenobarbital is decreased. However, if the patient becomes unacceptably groggy at any point, assuming the seizure history is appropriate, phenobarbital rather than bromide should be decreased by 25% (not only is bromide safer, response will be more rapid for phenobarbital because its half-life is shorter). If all remains well as the new phenobarbital dose reaches steady state (about 2 weeks), assuming the patient has been challenged by a seizure (thus waiting at least one seizure interval is ideal), then an additional 25% decline in phenobarbital can be considered. Ideally, phenobarbital concentrations are measured at each step. This process can be repeated until phenobarbital is eradicated or the patient seizures again. A much more rapid approach will be necessary in animals that develop a life-threatening adversity to phenobarbital (including bone marrow dyscrasias). In such cases a loading dose is indicated, with immediate (6- to 7-day) and 3-week postload samples being critical to ensure maintenance of bromide concentrations in the targeted range. If the loading dose option is taken, phenobarbital probably can be decreased by 25% to 50% as little as 2 to 3 days into the loading dose. Ideally, phenobarbital concentrations should not be decreased completely unless the patient has a bromide concentration of 1.5 mg/mL. For some patients phenobarbital can be completely discontinued once bromide is begun. For others, a combination may be necessary to control seizures. Unfortunately, for a small percentage of animals, seizures may continue even if both drugs are at the maximum end of the therapeutic range or side effects are untenable.
For life-threatening seizures that have not responded to phenobarbital, bromide also can be given rectally as a loading dose (see the discussion of disposition). Efficacy and safety of bromide versus phenobarbital has been compared in spontaneously epileptic dogs (n=46)28 using a parallel, randomized double-blinded study design. Enrollment was based on seizure history, physical and neurologic examinations, and clinical pathology. Dogs were loaded over a 7-day period to achieve the minimum end of the therapeutic range of the assigned drug phenobarbital (3.5 mg/kg), or bromide (15 mg/kg) was administered every 12 hours. Data (clinical pathology and drug concentrations) were measured at baseline and at 30-day intervals for 6 months. All but three patients completed the study. Seizures initially worsened in three dogs on bromide but not in any phenobarbital-treated patient. Compared with baseline, mean seizure number, frequency, and severity were reduced at 6 months for both drugs; seizure duration was shorter for phenobarbital but not bromide. Seizure activity was eradicated in a greater percentage of phenobarbital-treated patients (85%) compared with bromide-treated patients (65%), and the seizure duration decreased more for those on phenobarbital than for those on bromide, but successful control (at least 50% reduction in seizure number with no unacceptable adverse drug reactions) tended (p = 0.06) to be better only for those in the phenobarbital group. For patients in which seizures were eradicated, mean phenobarbital concentration was 25.4 ± 5.4 (95% confidence interval of 23 to 28 μg/mL) (see Table 27-4) but ranged from 12 μg/mL to 34 μg/mL (dose was 4.1 ± 1.1 mg/kg). For bromide mean concentration associated with eradication of seizures was 1.8 ± 0.5 mg/mL at a range of 0.9 to 3.3 mg/mL (dose of 31 ± 11 mg/kg). Both drugs caused abnormal behaviors. Weight increased by 10% in both groups. Changes in clinical pathology were limited to increased (but within normal) serum alkaline phosphatase and decreased (but within normal) serum albumin at 6 months for phenobarbital compared with baseline and compared with bromide at 6 months. Side effects at 1 month were greater for phenobarbital vs bromide and ataxia (55% vs 22%), polyuria (35% vs 13%), but greater for bromide vs phenobarbital for polyphagia (43% vs 30%), vomiting (57% vs 20%), and hyperactivity (43% vs 35%). At 6 months, most clinical signs had resolved to less than 15% of animals in either group with the exception of vomiting that continued in 21% of dogs in the bromide group (0% in the phenobarbital group). One dog failed phenobarbital therapy because of neutropenia; two dogs failed bromide due to vomiting. This study suggests that both phenobarbital and bromide are reasonable first choices for control of epilepsy in dogs, although phenobarbital may provide better control. Side effects can be expected to be greater in bromide following chronic dosing.
KEY POINT 27-20
If a loading dose is administered, monitoring should occur immediately and then again in 3 weeks. The two concentrations should match if the maintenance dose is correct.
Although bromide has been used in the cat successfully, adverse effects (consistent with bronchial asthma) are sufficiently common that an alternative anticonvulsant should be considered.129a While administration of bromide (n = 4) or bromide and phenobarbital (n = 3) was associated with eradication of seizures in 7 of 15 cats (serum bromide concentration range, 1.0 to 1.6 mg/mL); bromide coughing developed in 6 cats, leading to euthanasia in 1 cat and discontinuation of bromide administration in 2 cats.
Carbamazepine
Carbamazepine is approved for human use as an antiepileptic drug and currently is considered a primary drug for treatment of partial and tonic–clonic seizures. Further, it is a primary choice for treatment of trigeminal neuralgia; this and its efficacy for treatment of manic–depressive disorders may reflect its chemical similarity to tricyclic antidepressants.25 Its mechanism appears to slow recovery of sodium channels, preventing repetitive firing. In humans pharmacokinetics are complex. Slow, erratic oral absorption may take as long as 8 hours and appears to be dose dependent. Carbamazepine is approximately 75% protein bound. Hepatic metabolism, principally by CYP3A4, includes production of an active metabolite, carbamazepine 10,11 epoxide, which may achieve up to 50% of the CNS concentrations of the parent compound. Other metabolites are mostly glucuronidated and excreted in the urine. Carbamazepine induces cytochrome P450 isoenzymes CYP2C and CYP3A. As such, carbamazepine is involved in a number of drug interactions, decreasing the concentrations of several drugs (e.g., valproate, lamotrigine, tiagabine, topiramate, and other nonanticonvulsants). It can in turn be influenced by inducers (phenobarbital, phenytoin, valproic acid) and inhibitors (e.g., erythromycin, cimetidine, fluoxetine) of drug-metabolizing enzymes. Adversities in humans include both acute toxicity (stupor and coma) and increased seizures. Other neurologic signs may emerge with long-term use, potentially reflecting neurotoxicity to which tolerance may develop. Bone marrow suppression and other evidence of hypersensitivity may occur. However, some cases of leukopenia or thrombocytopenia may be only transient, resolving after several weeks. Carbamazepine also reduces antidiuretic hormone but, paradoxically, may cause water retention, a potentially serious complication in patients with cardiac disease. A therapeutic range of 6 to 12 μg/mL (based on the parent compound) has been suggested, but the relationship between parent concentration and response is complex. Side effects have been reported at 9 μg/mL.
Description of the disposition of carbamazepine in dogs is limited, with one report comparing two oral pharmaceutical preparations (see Table 27-4).138 Both the parent compound and active metabolite are characterized by a short half-life (3 hours), which will limit therapeutic use. Carbamazepine has been used intermittently in dogs. A case report139 described successful long-term control of psychomotor seizures in one dog, although parent drug could not be detected in serum. The patient previously had been treated with a variety of anticonvulsants alone or in combination. However, the patient immediately responded to carbamazepine at 7 mg/kg twice daily. The dose was increased to 14 mg/kg. The drug was discontinued because the patient developed leukopenia. Although the leukopenia resolved, clinical signs returned. Therapy was reinstituted, clinical signs resolved, and leukopenia returned but resolved after 3 to 4 months of therapy. In his review Holland139 notes that doses of carbamazepine in dogs have ranged from 4 to 40 mg/kg divided into 2 daily doses.
Oxcarbazepine is an antiepileptic drug that is a keto-analog of carbamazepine approved as a prodrug. Similar to carbamazepine in action, its induction of drug-metabolizing enzymes is less potent than that of carbamazepine, leading to its substitution with combination therapy.
Ethosuximide
Ethosuximide is a succinimide anticonvulsant used to treat absence seizures in humans. Its mechanism is reduced threshold of T-calcium channels in the thalamus. At therapeutic concentrations it is not effective against tonic hind limb extension of electroshock nor kindled seizures.25 Side effects include CNS depression and gastrointestinal upset. Allergic skin reactions have been reported in humans. It has been studied in a limited number of dogs. A concentration of 50 to 70 μg/mL was maintained by oral administration of 15 to 25 mg/kg thrice daily after a loading dose of 40 mg/kg; recommended therapeutic concentrations range from 40 to 100 μg/mL.42,140 The duration of dosing was not provided.
Felbamate
Felbamate was approved in the United States in the late 1990s for treatment of human epilepsy either as the sole drug or in combination with other anticonvulsants. Similar to meprobamate in chemistry, felbamate’s mechanism of action appears to be inhibition of NMDA receptor–mediated calcium or sodium influx (inhibition of excitatory signals) as well as potentiation of GABA receptor–mediated chloride (negative) influx.141 Thus the drug should have a broad mechanism of anticonvulsant activity with an action that might be considered complementary to phenobarbital.
The drug was proved very safe and efficacious in the treatment of partial and generalized seizures in experimental animals142,143 and humans, particularly children.144,145 Initially studied as monotherapy treatment of partial seizures, the drug has since proved useful as monotherapy for other seizures, including generalized seizures.145-148 When added to phenobarbital in refractory canine epileptics in an unpublished clinical report by the author, it eradicated seizures in 35% and decrease seizure numbers by at least 50% or more in another 20%.149
Felbamate is well absorbed after oral administration, although bioavailability in pediatric animals may be as little as 30% of that in adult dogs, necessitating a higher dose.15 The drug is eliminated by hepatic metabolism to metabolites that are largely inactive.150 Initial preclinical toxicity studies in the dog revealed felbamate to be completely absorbed after oral administration of 1.6 to 1000 mg/kg. At each dose Cmax was 12.6 to 168.4 μg/mL, respectively, in dogs with Tmax occurring at 3 to 7 hours, respectively. Plasma elimination half-life was 4.1 to 4.5 hours at both doses. After multiple oral doses of 50 mg/kg, plasma concentrations did not appear to change, and much (at least 50%) of the drug (based on [14C] felbamate) was eliminated in the urine (58% to 87.7%), with at least 7% eliminated in feces and the remainder in bile.151 Volume of distribution was 0.72 L/kg, and binding to plasma proteins was at most 36%. Plasma clearance was 108 mL∗h/kg renal clearance of unchanged drug was between 20% and 35%, and hepatic clearance resulting from metabolism was between 65% and 80% of overall clearance.151
KEY POINT 27-21
The combination of phenobarbital with any other drug eliminated primarily by cytochrome P450 is likely to increase the risk of liver disease.
In adult dogs the half-life of felbamate is 4 to 8 hours (mean of 5.2 hours); the elimination half-life is shorter in pediatric (Beagle) dogs (mean of 2.5 hours) probably.151,152 Felbamate also was studied in adult and pediatric dogs at 60 mg/kg orally once a day for 10 days. Oral bioavailability was less in pediatric dogs compared with adults, apparently because of more rapid clearance. Bioavailability also decreased by day 10 compared with day 1. Safety has been experimentally documented at doses ranging from 15 mg/kg divided twice daily (the starting therapeutic dose) to 300 mg/kg. Oral bioavailability is markedly variable. The oral disposition of felbamate has been described in adult and pediatric male and female dogs after single (see Table 27-4) and multiple dosing. 152 Felbamate was characterized by a lower Cmax (33 and 37 μg/mL for males and females, respectively) and area under the curve and shorter half-life (2.87 ± 0.52 [males] and 2.93 + 0.33 [females] in pediatric animals compared with adults (see Table 27-4). Clearance increased to 0.98 – 0.11 L/hr/kg), resulting in a decrease in half-life from 8.48 and 6.17 hours (males and females, respectively) to 6.1 and 5.1 hr (males and females, respectively) after multiple dosing (10 days).
Sedation, polyuria, polyphagia, and polydipsia, side effects typical of most anticonvulsants, do not appear to occur in dogs. Aplastic anemia caused by bone marrow suppression, however, developed in 1 of 10,000 patients (human) receiving felbamate,153 leading to marked curtailment of the use of the drug in humans. Felbamate has not been sufficiently used in dogs to detect a similar side effect, although it is probably as likely to occur in dogs as in humans. Hepatotoxicy has been reported in dogs receiving both phenobarbital and felbamate and has occurred in 3 of 15 patient treated with combination therapy by the author in an unpublished clinical trial. However, the author has administered felbamate to dogs also receiving phenobarbital at doses as high as 300 mg/kg divided daily for over 6 months with no apparent initial adverse effects. In one of 15 dogs receiving this regimen, progressive liver disease that ultimately proved fatal developed 1.5 years into the combination therapy. Because felbamate is a drug metabolized by the liver, prudence suggests not combining it with phenobarbital, particularly in patients requiring high serum concentrations of phenobarbital to control seizures. In humans interactions have led to increases in phenobarbital by felbamate,154 although this appears to be due to selective inhibition of a cytochrome P450 enzyme in only 25% of the population. Phenobarbital concentrations should be measured to detect any drug interaction that might lead to an increase in drug concentrations and a subsequent increased risk of liver disease. Greater risk may reflect decreased felbamate concentrations resulting from induction by phenobarbital.154 Currently, there is no easy, cost-effective means for assaying felbamate, although selected laboratories may offer the service. Human therapeutic concentrations range from 20 to 100 μg/mL, with trough concentrations ideally remaining in the range of 60 to 80 μg/mL for best efficacy.155
Gabapentin
Gabapentin is an anticonvulsant approved in 1994 for treatment of partial seizures with or without generalization in humans with epilepsy.156-158 It has been used in dogs (and anectodally in cats). It appears to act by a novel mechanism promoting the release of GABA, although the actual mechanism of release is not known. Although gabapentin is absorbed well after oral administration, its absorption appears to be dose dependent, relying on a saturable transport process. This process has been cited as the reason that antiepileptic drug effects last longer than anticipated on the basis of drug half-life, potentially allowing twice-daily administration. The short half-life of gabapentin (in humans) results in steady-state concentrations within 24 to 48 hours. In humans the drug is eliminated entirely by renal elimination, thus avoiding some of the risks of hepatotoxicity and drug interaction. Because the drug is relatively safe, therapeutic drug monitoring is not necessary; rather, the dose is increased as needed to control seizures. Mild dizziness, nausea, and vomiting have occurred in a small percentage of human patients.
Gabapentin studies with animals are limited.159-162 Gabapentin has been studied in the dog after oral administration of 50 mg/kg. Oral bioavailability was 80%, and plasma protein binding was below 3%. Mean intravenous elimination half-life of 2.9 hours has been reported in dogs. Repeated administration did not alter gabapentin pharmacokinetics, nor did gabapentin induce hepatic drug-metabolizing enzymes. In the dog 34% of the dose was metabolized to the inactive N-methyl form. The principal route of excretion was the urine.161,162 A more recent study compared gabapentin (600 mg to Beagles) after oral administration of either an immediate or a slow-release product (see Table 27-4).163 The sustained-release product did not disintegrate, but the release kinetics were not substantially different from those of the immediate-release product.
The addition of gabapentin (35 to 50 mg/kg divided every 8 to 12 hours) to either phenobarbital or bromide was studied in epileptic dogs (n = 17) using an open, uncontrolled design. The interictal period increased, but the number of seizures did not. However, seizures were eradicated in three dogs. Side effects that developed with the addition of gabapentin included sedation, which resolved within several days, and hind limb ataxia, which resolved with a reduction in the bromide dose.164 One of the major disadvantages of this drug is its expense, although several generic preparations are now available. Gabapentin may not be effective for the control of epilepsy when used at doses extrapolated from human patients. Clinical trials are indicated to establish the most appropriate dosing regimen for dogs or cats. Gabapentin is among the drugs for which SE may occur during withdrawal.165
Pregabalin
Pregabalin is the S enantiomer of 3-(aminoethyl)-5-methylhexanoic acid, an analog of GABA. It is also structurally related to the amino acid leucine and gabapentin (Figure 26-4). Pregabalin has been developed for the treatment of neuropathic pain and as adjunctive therapy in the treatment of partial seizures. However, according to manufacturer-generated approval research, its mechanism of action is not clear.166 It does not appear to involve gabaminergic transmission, does not alter binding or responses at GABA-A or GABA-B receptors, and is not a substrate or blocker of GABA transporter GABA transaminase. It decreases central neuronal excitability by binding to an auxiliary subunit (alpha2-delta protein) of a voltage-gated neuronal calcium channel on neurons. It also reduces the release of multiple neurotransmitters (in vitro concentration of 1.6 ng/mL), glutamate, norepinephrine, substance P, and calcitonin gene-related peptide. It serves as a substrate of L-amino acid transporter in neuronal cell membranes, which facilitates pregabalin transport into the cell. In animal models pregabalin controls seizures induced by a variety of methods, including threshold clonic seizures from pentylenetetrazol, and behavioral and electrographic seizures in hippocampal-kindled rats.
Pregabalin appears to be well absorbed after oral administration, minimally bound to plasma proteins, and distributed well to tissues, including the brain. In dogs it is characterized by a volume of distribution of 0.6 L/kg. In most species (e.g., rodents, primates) studied, pregabalin is eliminated principally unchanged in the urine. However, in dogs, while ≥ 80% of a dose is excreted in the urine, approximately 45% of the dose excreted as the N-methyl metabolite. However, pregablin does not appear to affect cytochrome P450 drug-metabolizing enzymes. The disposition of pregabalin has been described in apparently healthy Labrador Retriever–Greyhound cross dogs (four female, two male) after a single oral dose of 4.mg/kg (Table 27-4). The duration that plasma drug concentrations were above the presumed minimum effective concentration of 2.8 μg/mL ranged from 7 to 14.5 hours.167
Dewey and coworkers described the use of pregabalin as an adjunct to anticonvulsant therapy in the control of epilepsy in 11 dogs using an uncontrolled clinical trial.168 Animals had not responded sufficiently to either phenobarbital or bromide, or a combination of the two. Insufficient control was defined as two or more seizures per month despite concentration in the recommended therapeutic range of the appropriate drug. For phenobarbital concentrations ranged from 20 to 40 μg/mL and for bromide 0.2 to 2.81 mg/mL. Dogs whose bromide concentrations were subtherapeutic were receiving phenobarbital in the therapeutic range. Dogs were treated with 3 to 4 mg/kg pregabalin every 8 hours, yielding mean pregabalin concentrations of 6.4+ μg/mL. Therapy resulted in a reduction of seizures by at least 50% (range of 23% to 83%) in seven of nine dogs that completed the study. However, 10 of the 11 dogs developed adverse effects, including ataxia. Increases in liver enzymes were present during the study, but it is not clear if these increases were related to phenobarbital or pregabalin. Although the study provides support for the possible use of pregabalin as an add-on anticonvulsant, a controlled clinical trial is indicated in dogs to assess both efficacy and safety.
Levetiracetam
Levetiracetam is a single (S)-enantiomer acetamide-derivative antiepileptic drug. Its mechanism of action is novel and does not appear to involve any known neurotransmitter, ion channel protein, or receptor. Rather, it appears to interact with synaptic vesicles (SVA), and appears to impede conduction of impulses across the synapse. Levetiracetam was most useful experimentally in blocking seizures caused by pilocarpine and KA and in the kindling model of rats, both models for complex partial seizures with secondary generalization. Food does not impair the extent, but does impair the rate, of oral absorption. In humans close to 70% of the drug is renally excreted; hepatic metabolism of the remainder reflects acetamide hydrolysis, which is not CYP 450 dependent. Levetiracetam is metabolized by plasma B-esterases, which will continue once blood is drawn. As such, serum rather than plasma or whole blood is the desired test tissue of choice.169 The elimination half-life in humans is approximately 7 hours. Drug interactions appear to be minimal; competition for renal tubular secretory proteins may occur.
KEY POINT 27-23
The novel mechanism of action of levetiracetam and its wide therapeutic margin are balanced by its very short half-life.
The disposition of levetiracetam has been described in mongrel dogs (n=6).170,171 Intravenous administration of 20 mg/kg yielded a maximum concentration of approximately 44 μg/mL, volume of distribution of 0.45 ± 0.13 L/kg, clearance of 1.5 mL/min/kg, elimination half-life of 3.6 ± 0.8 hours and mean residence time of 5 ± 1 hours. In a diferent study172 (male and female dogs), oral administration of 54 mg/kg yielded a Cmax of 50 to 65 μg/mL (mean of 53 and 55 μg/mL in male and female dogs, respectively), and an elimination half-life of 2 to 3 hours. Dewey and coworkers173 reported the disposition of levetiracetam in dogs (n = 6) after a single dose (60 mg/kg over 2 min) administered intravenously. The extrapolated peak plasma drug concentration (Co) was 254 ± 81 mg/mL, steady-state volume of distributionof 0.48 L/kg, and clearance of1.4 mL∗min/kg. The elimination half-life was 4 ± 0.82 hours, and mean residence time was 6 ± 0.9 hours. The dose was well tolerated. Patterson and coworkers174 studied levetiracetam after intravenous, intramuscular, and oral (19.5 to 2.6 mg/kg) administration in hound dogs (n = 6). Peak drug concentrations were 37 ± 5, 30.3 ± 3, and 30 ± 4 μg/mL after intravenous (Co:extrapolated peak plasma concentration), intramuscular, and oral administration, respectively. The volume of distribution (beta) was 0.55 L/kg, and clearance was 55 mL/min (not standardized to kg). Elimination half-life was 3 ± 0.3 hours. Bioavailability after intramuscular and oral administration were 113+13% and 100+7%, respectively. No pain was detected with intentional perivascular injection.
In dogs 1200 mg/kg administered intravenously or 2000 mg/kg administered orally was not lethal but was associated with salivation, vomiting, tachycardia, and restlessness; 1200 mg/kg per day for 13 and 52 weeks resulted in transient restlessness and tremor and centrally mediated salivation and vomiting. Long-term administration (≥6 months) in some species was associated with enzyme induction (centrilobular hypertrophy) at 50 mg/kg per day. Liver weight increased, although histopathologic changes did not appear in the liver.
The disposition of levetiracetam has been described in cats (n=10) receiving 20 mg/kg either intravenously or orally as a single dose.175 Cats tolerated dosing well, with no significant adverse effects noted. However, transient mild to moderate hypersalivation occurred with oral dosing. Median peak concentration after intravenous dosing was 37.52 μg/mL (range 28.05 to 51.86 μg/mL), with a median half-life of 2.86 hours (range 2.07 to 4.08 hours) and mean residence time of 4.57 hours (range 3.09 to 6). Clearance was 2 mL/kg/min (range 1.5 to 3.4 mL/kg/min) and steady-state volume of distribution was 0.52 L/kg (range 0.33 to 0.64 L/kg). After oral dosing therapeutic plasma concentrations were achieved in 7 of 10 cats within 10 minutes and remained within the therapeutic range for at least 9 hours. Median peak concentration (Cmax) was 25.54 μg/mL (range 13.22 to 37.11 μg/mL), Tmax was 1.67 hours (range 0.33 to 4 hours), T1/2 was 2.95 hours (range 1.86 to 4.63 hours), and mean residence time was 5.65 hours (range 4.23 to 7.86). Mean oral bioavailabililty was 100%. When levetiracetam is monitored, because of its very short half-life (according to the author’s measurements, as short as 1 hour in dogs and cats), the therapeutic range should be applied to trough rather than peak concentrations. Ideally, both a peak and trough should be measured at least once a year in the patient to determine drug half-life.
Response to levetiracetam of dogs (n=14) with refractory epilepsy was described prospectively.176 Refractoriness was based on monitoring; eligibility required that drug concentrations be in the upper quartile of the recommended range (mean 32 ± 4.6 μg/mL). Levetiracetam was administered at 10 mg/kg orally every 8 hours; the dose was increased to 20 mg/kg thrice daily if seizures did not decline by at least 50%. At 2 months 8 of 10 dogs responded, with seizure number reduced by 73% and number of days or months reduced by 67%. At 6 months 6 of 11 dogs remained classified as responders. However, with long-term follow-up, only three animals remained responders, suggesting that efficacy of levetiracetam declined. Drug concentrations were not measured; as such, the cause of therapeutic failure (i.e., tolerance or worsening disease) was not distinguished from declining drug concentrations.176
The use of levetiracetam in cats has been reported.175,177 Four cats with seizure disorders that were poorly controlled with phenobarbital alone were treated with oral levetiracetam as an add-on drug at a dose regimen of 20 mg/kg body weight, every 8 hours. Levetiracetam serum concentrations were within the reported therapeutic range for people (5 to 30 to 45 μg/mL) for all samples in all cats. The overall average serum levetiracetam level for all cats was 16.5 μg/mL (range: 6.9 to 24.3 μg/mL). The median serum disappearance half-life (t ½) of (based on peak and trough samples) was 5.3 hours. Seizure frequency was reduced by an average of 30.5% in three cats and increased by 33.3% in one cat. The results of this pilot study suggest that levetiracetam is a safe drug for cats that may provide some therapeutic benefit when used as an add-on to phenobarbital.
Phenytoin Sodium
Phenytoin sodium, previously named diphenylhydantoin, depresses motor areas of the cortex (antiepileptic action) without depressing sensory areas. It is approved by the Food and Drug Administration for control of epileptiform convulsions in dogs.
Phenytoin is a hydantoin derivative (see Figure 27-4);45 others of lesser importance are mephenytoin and ethotoin. Hydantoins are five-member ring structures, whereas barbiturates are six-member structures. A major point of difference between the hydantoins and barbiturates is the absence of a C=O group. Phenytoin is not a general anticonvulsant, as is phenobarbital, and is not used for emergency treatment of poisoning by convulsant drugs or tetanic seizures. Oral preparations are available in suspension, capsule, and tablet forms. Phenytoin (50 mg/mL) is also available for human use in a special solvent for intravenous administration. Intravenous injection of the drug causes a marked drop in arterial pressure and is not advised in the dog.178
Phenytoin is no longer used to control seizures in the dog because of its lack of efficacy,179 which may be related to decreased bioavailability and rapid clearance. Phenytoin is much less effective in the dog than either phenobarbital or primidone in the control of epileptic seizures.50 The half-life of phenytoin is too short in the dog to permit maintenance of adequate drug concentrations in plasma and the CNS.17 When administered alone, phenytoin cannot be considered a satisfactory drug for treatment of epilepsy in the dog.29,180 Because of drug interactions and enhanced hepatotoxicity, a combination of phenytoin with phenobarbital is not a viable alternative.
KEY POINT 27-25
Phenytoin is not a preferred drug for treatment of epilepsy because of its short half-life, variability, poor oral bioavailability, and potential for toxicity.
In the cat phenytoin is relatively toxic and generally undesirable as an anticonvulsant.70 The efficacy and safety of phenytoin in cats have not been determined.29
Pharmacologic Activity
Phenytoin produces a stabilizing effect on synaptic junctions that ordinarily allow nerve impulses to be readily transmitted at lower thresholds. Consequently, the level of synaptic excitability that permits impulses to be transmitted easily is reduced or stabilized or both. This effect appears to be associated with active extrusion of Na+ from neurons and decreased posttetanic potentiation or spread of nerve impulses to adjacent neurons. Phenytoin may reduce calcium movement across cell membranes. Further, phenytoin may inhibit activation of protein phosphorylation by the calcium–calmodulin complex.181 Phosphorylation and norepinephrine release in neurons requires calmodulin. Reduction in spread of the “burst” activity associated with epilepsy prevents genesis of the cortical seizure. Phenytoin stabilizes hyperexcitable neurons without causing general depression of the CNS.45
Disposition
Absorption of phenytoin is erratic after intramuscular administration. This may be related to crystallization of the drug at the injection site because of alteration in pH by tissues.45 Administration of phenytoin by the intramuscular route is not advised because considerable necrosis and sloughing at the injection site occur.178 Absorption of the drug from the gastrointestinal tract of the dog is rapid but erratic and incomplete.179 Bioavailability of phenytoin from the tablet formulation averages 36% in the dog.180 Poor oral absorption and differences in product bioavailability contribute to the difficulty in achieving effective serum levels of phenytoin. The generic preparations of phenytoin should not be used.182 Dosing at 30 mg/kg thrice daily for 3 days resulted in concentrations of less than 8.5 μg/mL.179 Dosing at 10 mg/kg thrice daily for 5 to 8 days yielded concentrations less than 2.5 μg/mL.
At therapeutic concentrations (10 to 20 μg/mL), phenytoin is highly bound (75% to 85%) to plasma proteins in animals and humans.183 The high degree of phenytoin binding predisposes this acidic drug to interaction with other drugs by a displacing effect at protein (albumin) binding sites, although the relevance of this in the presence of increased clearance is not known. In uremic patients decreased plasma protein binding is associated with accelerated renal clearance or elimination of the drug. Phenytoin readily crosses the placenta.184 High concentrations of phenytoin are attained in the maternal liver and maternal and fetal hearts. The brain (ostensibly the primary target organ) contains nearly the lowest concentration of the drug.
Phenytoin is metabolized via CYP2C9 and to a lesser degree CYP2C19185 into metahydroxyphenytoin or parahydroxyphenytoin, which are conjugated with glucuronic acid. In humans approximately 60% to 75% of the daily dose of phenytoin is excreted in the glucuronide form;45 the dog also converts a high percentage of phenytoin into this form. In addition, diphenylhydantoic acid, a minor metabolite in some laboratory animals, and dihydrodiol are formed in dogs. Interestingly, high concentrations of diphenylhydantoic acid are found in cat urine. The dihydrodiol metabolite is probably involved in formation of catechol metabolites; these are also formed in most animals.186 Epoxide metabolites are also speculated to be formed in humans. Because phenytoin is not very soluble in water, little of the unmetabolized drug is excreted in urine.
In the dog, despite relatively large single daily oral doses (50 mg/kg), the plasma concentration of the drug is low, perhaps reflecting the short half-life of 6 to 7.8 hours187 compared with 4 to 6 hours after an intramuscular injection (50 mg/kg) as well. The shorter half-life of the second study probably reflects induction after multiple dosing;188 the half-life of phenytoin in the dog dramatically decreases after 7 to 9 days of treatment.179,180. A 2.5-hour half-life was predicted for phenytoin after 14 days of therapy compared with 3.3 hours with single dosing. Other studies have confirmed a short half-life for phenytoin in the dog: After a single intravenous dose (15 mg/kg), a value of 4.5 hours was obtained by Sanders and coworkers,189 and a half-life of 3.65 hours was determined by Pedersoli and coworkers190 after an intravenous bolus of 11 mg/kg. The clearance of phenytoin has been described as dose dependent in dogs.179 More recently, using a parallel design, the disposition of phenytoin was described in Beagles (n=5) after 5 days of intravenuos administration of 12 mg/kg (see Table 27-5).191
After oral administration (10 mg/kg) in the cat, plasma half-life of phenytoin was 24 to 108 hours,188,192 contributing to the prolonged effect of phenytoin observed in the cat more than in other species. This may reflect decreased glucuronide conjugation.
Clinical Use
In humans clinical therapeutic effects and intoxication are related to the blood concentration of phenytoin. A reduction in the number of seizures occurs when phenytoin blood concentrations exceed 10 μg/mL. Recommended therapeutic doses of phenytoin administered orally every 8 hours for control of seizure disorders in the dog indicate considerable variation: 6.6 to 11 mg/kg,178 11 mg/kg,49 and 35 mg/kg.179 However, oral administration of phenytoin (4.4 and 11 mg/kg) every 8 hours fails to reach serum concentrations of 10 μg/mL in the dog. Indeed, serum phenytoin after either single or repeated oral doses of 10 mg/kg does not exceed a concentration of 2 μg/mL.179 To achieve a serum concentration of approximately 10 μg/mL phenytoin, it appears that an oral dose of at least 35 mg/kg given thrice daily is necessary for the adult dog.49,179 According to Pedersoli and coworkers,190 an oral dosage schedule of 20 mg/kg every 8 hours of the phenytoin microcrystalline suspension should be sufficient to reach a serum concentration of 10 μg/mL or higher. This dose, however, will maintain a plasma therapeutic level for only the first 2 or 3 days of treatment.180 Farnbach50 found that only 3 of 77 epileptic dogs receiving phenytoin were controlled; concentrations in all dogs ranged from 0.2 to 13 μg/mL, with a median of 2.3 μg/mL, despite doses ranging from 3 to 129 mg/kg per day (generally divided into twice-daily doses).
Drug Interactions
The combined use of phenytoin and phenobarbital or primidone may lead to increased formation of epoxide metabolites in animals. This could possibly result in cholestatic hepatic injury similar to that reported in three dogs.36 The effect may also reflect induction by phenytoin. Phenytoin must be considered a potent inducer of the hepatic microsomal enzyme system in the dog.180 Between 7 and 9 days after administration of phenytoin, its half-life may be reduced from 5.5 to 1.3 hours. In contrast, the half-life after oral administration in humans averages 22 hours, with a range of 7 to 42 hours. This contrasts to humans, in which phenytoin has a moderate inducing ability in the human. The differences between the two species may help explain the efficacy of phenytoin for control of epileptic seizures in humans compared to dogs. Further, although the combined use of phenytoin and phenobarbital is considered optimal therapy in humans,55 use of both drugs is controversial because both induce hepatic microsomal enzyme activity. This combined induction complicates successful therapy because therapeutic concentrations are difficult to achieve and the increased production of potentially toxic metabolites.178 Metabolism of a number of chemicals or drugs is enhanced by phenytoin. These include digitoxin, dexamethasone, and cortisol.193 In humans the half-life of theophylline decreases by 50% with an increase in clearance of twofold.194
Phenytoin metabolism in humans is inhibited by other drugs prolonging their effect. Examples include dicumarol, chloramphenicol, phenylbutazone, and the phenothiazines. In vitro inhibition has been demonstrated by diazepam and propoxyphene hydrochloride. In the dog the serum half-life of intravenous phenytoin increased from 3 to 15 hours when administered with chloramphenicol, and signs of phenytoin toxicosis reversed within 24 hours after cessation of chloramphenicol treatment.195 Phenylbutazone is also known to increase plasma concentration as a result of inhibition.45 Despite predominant renal elimination, vigabatrin, a drug similar to gabapentin, deceased phenytoin clearance by 31% and increased phenytoin half-life by 45% in Beagles.191 In contrast, gabapentin had no effect.
Serum phenytoin concentration declines in the presence of folic acid therapy, presumably because the hydroxylase enzyme that metabolizes phenytoin is folate dependent. Phenytoin may prolong the prothrombin time.196 Blood coagulation defects similar to that induced by vitamin K deficiency can occur in neonates exposed to phenytoin in utero. The coagulation defect can be reversed by treatment with vitamin K.
Blood Concentrations and Associated Toxicity
In humans mild signs of intoxication such as nystagmus develop with blood levels of 20 μg/mL; patients with levels over 40 μg/mL have marked nystagmus and are uncoordinated and lethargic. Blood levels in the dog would probably have to increase a comparable 100% to 400% as in humans before serious signs of intoxication develop.
Hepatitis, jaundice, and death after clinical use of phenytoin have been reported in one dog that initially received primidone (500 mg daily).197 Toxic hepatopathy and intrahepatic cholestasis associated with phenytoin administration in combination with phenobarbital or primidone (or both) have been reported in three dogs.36,89 Two distinct forms of hepatotoxicity have been ascribed to reflect the sequelae of anticonvulsant phenytoin in dogs.36 The first (type B reaction) is characterized by clinical signs after extended treatment at lower than recommended doses and may result from an (unpredictable) idiosyncratic reaction. Indications are that histologic changes with this form will progress from chronic hepatitis to cirrhosis. The second form is more frequently characterized by intrahepatic cholestasis and is associated with a poor prognosis. This form of liver disease has been associated with high doses of phenytoin in combination with primidone or phenobarbital and may represent an intrinsic hepatotoxicity. Induction of enzymes is likely to increase formation of toxic metabolites, which may contribute to hepatotoxicity. Hepatotoxicity resulting from phenytoin is more likely if the drug is used in combination therapy with either primidone or phenobarbital. Toxicity may be related to generation of toxic metabolites. Two forms of toxicity appear to occur with phenytoin therapy: a dose-independent chronic hepatitis that may progress to cirrhosis and that appears to be reversible after discontinuation of the drug early in the disease and a dose-dependent intrahepatic cholestasis that is accompanied by a poor prognosis.
Other Side Effects
The CNS side effects of phenytoin in the dog are moderate in part because of its action but also because it is rapidly metabolized.49 Transient incoordination and oversedation may occasionally occur after administration of phenytoin. A moderate degree of polyphagia, polydipsia, and polyuria may be seen in animals medicated with this drug. Sialosis, weight loss, and vomiting have been reported after the use of phenytoin in the cat. Inhibition of release of antidiuretic hormone accounts for the polyuria that develops after administration of phenytoin. Insulin secretion also is inhibited.45
Displacement of T4 by highly protein-bound drugs (e.g., phenytoin) from T4-binding globulin increases T4 concentrations, induction of hepatic drug-metabolizing enzymes results in increased clearance of both T4 and T3, and increased conversion of T4 to T3 by peripheral tissues further decreases T4. The latter mechanism has been postulated as the reason that T3 concentrations may remain normal despite increased T3 clearance198 in patients receiving phenytoin. Clinical signs of hypothyroidism may not be apparent in such cases. Note, however, that both serum T4 and free T4 may be decreased in some patients receiving anticonvulsants.
Fosphenytoin is a phosphate ester prodrug of phenytoin, with the latter being released after dephosphorylation by tissue (liver, red blood cells, and others) phosphatases.199 Enzymatic conversion half-life is about 3 minutes in dogs, with equimolar concentrations of phenytoin emerging with fosphenytoin. Similar antiarrhythmic (antiseizure was not reported) effects occurred for fosphenytoin and phenytoin in dogs. Side effects are similar to phenytoin, although local irritation was less with intramuscular administration of fosphenytoin compared with phenytoin. No information could be found regarding use of fosphenytoin in dogs.
Topiramate
Topiramate is a monosaccharide D-fructose anticonvulsant derivative developed in the 1980s.200 Its mechanism of action is not entirely understood but includes enhanced gabaminergic actions at the GABA-A receptor. Sodium channel blockade may also be involved (package insert). Its effects are similar to those of diazepam but are not blocked by flumazenil, suggesting actions through a novel, non-benzodiazepine (BDZ)–sensitive binding site on the GABA-A receptor. It may also inhibit the kainite/AMPA (but not NMDA) glutamate receptor. Topiramate appears to increase the seizure threshold and prevent seizure spread. In selected studies potency is similar to phenytoin and phenobarbital. However, while useful in kindled and ischemia-induced epilepsy, it has variable efficacy for prevention of pentylenetetrazole (a GABA-A receptor antagonist) or picrotoxin-induced seizures. In humans it has been predictably effective in controlling partial and secondary seizures but not absence seizures.
In humans topiramate is eliminated primarily through the kidneys as the parent compound, although up to eight metabolites have been identified.
Topiramate inhibits isoenzymes of carbonic anhydrase; this mechanism appears to be more relevant to side effects than control of seizures. Binding to the enzyme results in a large proportion of the drug being located in erythrocytes at low concentrations. Its oral disposition has been reported in male and female Beagles; intravenous data are available for one dog (see Table 27-4).201 Based on comparison of the area under the curve after 10 mg/kg intravenously and orally, oral bioavailability appears to be about 35% for tabular form but is increased twofold when administered as a solution. Dogs treated with up to 400 mg/kg experienced no lethal side effects, although acute toxicity associated with ataxia, decreased motor activity, tremors, and clonic convulsions occurred. Dosing for 3 and 12 months was not associated with overt indicators of toxicity in dogs. However, increased liver weights (indicative of induction), increased serum gastrin levels and gastric mucosal hyperplasia, and renal and urinary epithelial hypertrophy have been associated with long-term use in other species. Topiramate is available as a sprinkle formulation. Its use apparently has not been reported in dogs or cats.
Valproic Acid
Valproic acid is a vehicle used for other drugs that was serendipitously found to have anticonvulsant properties. It is a simple branched-chain carboxylic acid (see Figure 27-4). Valproic acid is effective against a variety of seizures, including absence seizures. Its mechanism of action appears to be similar to that of phenytoin in that it prolongs recovery of voltage-activated Na+ channels from inactivation. It may also affect Ca2+ fluxes but does not appear to impair GABA. The drug is rapidly absorbed, highly protein bound, and in humans has a half-life of 10 to 15 hours. The half-life can, however, be shortened in the presence of other (inducing) anticonvulsant drugs.
The difficulty in using this drug in dogs probably reflects an inability to achieve therapeutic concentrations. Valproic acid is metabolized in the liver; metabolites can be as potent as the parent compound in controlling seizures, although only one of them enters the CNS to any appreciable extent. Valproic acid can alter liver enzymes (up to 40% of human); increases can be associated with toxicity. Hepatotoxicity is likely to be increased when used in combination with other drugs. It also causes gastrointestinal upset and, like other anticonvulsants, CNS side effects (e.g., sedation, ataxia). Valproic acid has not proved very useful for controlling seizures in dogs. It might be considered in combination with phenobarbital, although drug interactions may complicate therapy.
The disposition of valproic acid has been reported for dogs by several authors (reviewed by Frey and Löscher)42 (see Table 27-4). Peak serum concentration after oral absorption is markedly variable and appears to be greater with solution (30 to 60 μg/mL at 20 mg/kg) compared with tablets (30 to 75 μg/mL at 40 mg/kg). However, bioavailability is not as divergent, reaching 89 ± 8.2 for solution compared with 78 ± 17 for tablets.42 Löscher202 demonstrated that therapeutic drug concentrations could be maintained in part because of decreased protein binding in dogs compared with humans. Half-life of valproic acid did not change over 14 days of treatment at 168 to 180 mg/kg (divided into three equal doses). Löscher attributed some antiepileptic efficacy of valproate to its metabolites, which appear to accumulate in dogs (n = 3). In a study of multiple oral dosing (60 mg/kg thrice daily for 14 days), mean plasma concentrations of valproic acid, 2-en- valproic acid, 3-hydroxy-valproic acid, 3 keto-valproic acid, and “total equivalents” of valproic acid were 20, 3.1, less than 0.4, and 3.7 (23 equivalents) on day 1 of dosing, compared with 8, 16, 0.7, and 9.1 (18 equivalents) at day 14.
The use of valproic acid for treatment in idiopathic epileptic dogs (27 female and 40 male), 29 of which were considered refractory to anticonvulsant drugs was described by Nafe and coworkers.203 The addition of valproic acid (25 to 40 mg/kg per day) resulted in improvement in six of eleven dogs already receiving phenytoin or phenobarbital and one of six dogs receiving primidone (dog was tapered off of primidone). Valproic acid (25 to 105 mg/kg per day) alone markedly improved seizures in 7 of 16 dogs, and the combination of valproic acid and phenobarbital resulted in marked improvement in 14 of 24 dogs. One dog developed noninflammatory alopecia.
Zonisamide
Developed in Japan, zonisamide, 3-sulfamoylmethyl-1,2 benzisoxazole, is a synthetic sulfonamide-based anticonvulsant approved for use in the United States in 1998 for treatment of seizures related to human epilepsy.204 The efficacy of zonisamide for treatment of human epilepsy is similar to phenobarbital and superior to other classic drugs, including valproic acid and phenytoin. Its mechanism is not clear, but it appears to inhibit neuronal voltage-dependent sodium and T-type calcium channels.205,206 It also modulates the dopaminergic system and accelerates the release of GABA from the hippocampus.207,208 An additional potential advantage of zonisamide is free radical scavenging, which protects against the destructive nature of radicals, especially in neuronal membranes.209 Finally, zonisamide blocks the propagation of seizures from cortex to subcortical areas of the brain. Its antiepileptic efficacy has been described as similar to that of phenytoin or valproic acid, thus minimally affecting normal neuronal activity. These multiple mechanisms of action may translate to improved efficacy compared with other anticonvulsant drugs.
The clinical pharmacology of zonisamide has been investigated in humans with similar characteristics in dogs.210-212 Disposition is complicated. Oral absorption tends to be rapid, complete, and minimally impaired by food. After 12 hours of dosing, zonisamide concentrations in the brain are twofold that in plasma. The extent of protein binding is not sufficient to limit the rapid movement into the brain. Binding of zonisamide to erythrocytes (red blood cells) and plasma proteins contributes to complex kinetics. Erythrocyte concentrations in whole blood tend to be twice as high as plasma and serum in humans and are characterized by binding that is both saturable and nonsaturable;213 the saturable portion may reflect binding to carbonic anhydrase in epileptic patients. Accumulation of drug in red blood cells is reversible, and the complex relationship between zonisamide and red blood cells may make therapeutic drug monitoring of plasma or serum advantageous. Metabolism of zonisamide involves both phase I and phase II hepatic metabolism, with cytochrome P450 3A4 being the major isozyme and a glucuronidated compound the major metabolite.214 Enzymes CYP3A4, CYP3A5, and CYP2C19 contribute to metabolism in humans.215 Renal elimination and recovery of zonisamide indicate parent drug recovery of 35%. Using radiolabeled (carbon) zonisamide administered to dogs, 83% of the drug was excreted in 72-hour urine as the either the parent compound or metabolites. The remaining proportion was recovered in feces.210 The terminal half-life of zonisamide in the dog after a single oral dose (20 mg/kg) administration differed depending on the tissue studied, with the shortest being 15 hours for plasma and the longest 42 hours for red blood cells.210 The longer elimination half-life allows a convenient dosing interval while minimizing dramatic fluctuations in zonisamide concentrations that might cause recurrence of seizures. Recommended therapeutic concentrations for zonisamide initially were 10 to 70 μg/mL, with 16.5 to 49.6 μg/mL also suggested when dosed twice daily.216 The author recommends the more commonly accepted 10 to 40 μg/mL; however, monitoring should be based on patient need. Nonlinear pharmacokinetics have been reported in some human patients, particularly with chronic dosing, resulting in disproportionate, and thus unexpected, increases in drug concentrations compared with changes in dose. In dogs undergoing toxicity studies, plasma concentrations never reached steady state over the course of 13 weeks of dosing at 75 mg/kg, compared with proportional steady-state concentrations by week 13 at 10 to 30 mg/ kg.212
KEY POINT 27-26
Its unique mechanism of action, reasonable half-life, and wide therapeutic margin render zonisamide an increasingly useful drug for control of epilepsy in dogs and cats.
Clinical pharmacokinetics of zonisamide have been described in normal dogs (n = 8), four male and four female, ranging from 3 to 4 years of age, using a randomized crossover design after single intravenous and oral administration, 6.85 and 10.25 mg/kg, respectively.217 Zonisamide concentrations differed among blood compartments after single dosing, with oral maximum concentration (Cmax) being greatest in red blood cells (28.73μg/mL) and least (14.36 μg/mL) in plasma. Clearance of zonisamide was 57.55 mL/hr/kg from plasma and 5.06 mL/hr/kg from red blood cells. However, zonisamide concentrations did not differ among blood compartments at the end of multiple dosing, suggesting any blood component can be monitored. The fraction of unbound drug was 60.48 ± 13.4%. Elimination half-life in plasma was 16.4 hours in serum and 57.4 hours in red blood cells. Volume of distribution also differed, being greater (1 L/kg) in plasma and least in (0.4 L/kg) red blood cells. Bioavailability was 126.8% for red blood cells and 189.6% for plasma. After multiple dosing (10.17 mg/kg) twice daily for 8 weeks, the accumulation ratio of zonisamide was 3.5 (plasma) and 4.3 (red blood cells). The resulting mean Cmax at steady state was 56 ± 12 μg/mL, suggesting a beginning dose of 2 to 3 mg/kg twice daily to target the low end of the therapeutic range. The half-life at 8 weeks was 23 ± 6 hours. Plasma drug concentrations varied by 17.2% between 12-hour dosing intervals, which suggests that a 12-hour dosing interval is appropriate. Differences in clinical pathology data occurred at the end of the 8-week study period, although all results remained within normal limits. Serum alkaline phosphatase and calcium increased above baseline, whereas total serum protein and albumin both decreased below baseline.
Zonisamide pharmacokinetics have been described in cats (n = 5) after a single dose of 10 mg/kg (see Table 27-4).218 Safety and adverse reactions were studied during chronic (9 weeks) dosing at 20 mg/kg once daily. Zonisamide was not well tolerated at this dose; 50% of cats exhibited vomiting, diarrhea, and anorexia. Mean peak and trough concentration with chronic dosing in all cats were 46 and 59 μg/mL, respectively, with concentration at 42, 59, and 79 in cats with adversities. Zonisamide appears to be minimally involved in drug interactions typicalof highly protein-bound drugs.210 However, it is involved with interactions involving CYP enzymes. Nakasa and coworkers215 demonstrated that clearance was decreased 31%, 23%, and 17% by ketoconazole, cyclosporine A, and miconazole, respectively; fluconazole inhibited clearance to a lesser degree, but itraconazole appeared to have no effect.
Oral bioavailability of human generic preparations should not be assumed to act similarly in dogs and cats. Accordingly, clinicians and owners both should request that dispensing pharmacists inform the owner or veterinarian if the dispensing pharmacy has changed to a different generic preparation since the last prescription.
Zonisamide does not appear to affect its own metabolism nor the metabolism of other drugs in animals or humans. Phenobarbital will shorten zonisamide half-life. The impact of 35 days of dosing phenobarbital on the disposition of zonisamide was studied in dogs. Unfortunately, all data were pictorially represented, limiting assessment of changes in disposition.219 After 35 days of phenobarbital administration, concentrations appeared to decrease to about 2.75 μg/mL, returning to 3.5 only after approximately 12 weeks after phenobarbital was discontinued. The decrease in half-life appeared to be about 3 hours, or approximately 30%. Phenobarbital shortened the half-life of zonisamide from 27 to 36 hours in humans, resulting in lower plasma drug concentrations.220 The impact of phenobarbital does not appear to be profound, but monitoring is warranted, and collection of both a peak and a trough sample might be warranted in patients receiving phenobarbital with zonisamide. The effect does not appear to warrant starting the drug at higher doses in patients on phenobarbital unless monitoring has confirmed the need.
Dogs appear to tolerate zonisamide well; concentrations in samples submitted for monitoring at the author’s laboratory indicate concentrations that exceed 60, 80, and in some dogs, 100 μg/ml well. Dose increases for zonisamide in dogs may cause disproportionate increases in plasma drug concentrations based on data in the author’s laboratory. It is possible this may reflect saturation of acetylation enzymes in the dogs.
As a sulfonamide, zonisamide inhibits thyroid synthesis of thyroid hormones. Anticonvulsants (phenytoin) may also have a direct negative effect on TSH response to thyrotropin-releasing hormone. Drug-induced changes in T4-binding globulins have also been documented in human patients taking anticonvulsants. Boothe and Perkins217 demonstrated that zonisamide dosed for 8 weeks was associated with a decrease in total T4 below normal limits. Free T4 and TSH were also decreased from pretreatment concentrations, although both were within normal limits. Zonisamide concentrations were higher than the recommended therapeutic range. Thyroxin and TSH concentrations might facilitate diagnosis of hypothyroidism in animals receiving zonisamide.198 Note that thyroid supplementation suppresses response to TSH, and testing should not be performed until supplementation has been discontinued for 4 to 6 weeks. As with carbonic anhydrase inhibitors, zonisamide has been linked to metabolic acidosis in humans. The FDA recommends that bicarbonate be measured before and intermittently during therapy. Renal calculi have formed in a very small number of human patients receiving zonisamide for long periods. Because it does not contain an aryl-amine, allergic responses associated with sulfonamide antimicrobials may not occur in dogs.
Clinical reports of zonisamide use in animals are limited. In one report zonisamide was effective in reduction of seizures in patients with epilepsy that had not sufficiently responded to one or more anticonvulsants (including phenobarbital and/or bromide) in 7 of 12 dogs at doses designed to achieve 10 to 40 μg/mL. Dose reduction or discontinuation of concurrent anticonvulsant was possible in 8 of 12 dogs. Mean concentrations approximated 20 μg/mL; mean dose was 9 mg/kg every 12 hours.221 A second open clinical trial studied zonisamide for treatment of refractory seizures in dogs (n = 13).222 Mean reduction in seizure was 70%, with three dogs relapsing. Drug concentrations were not measured.