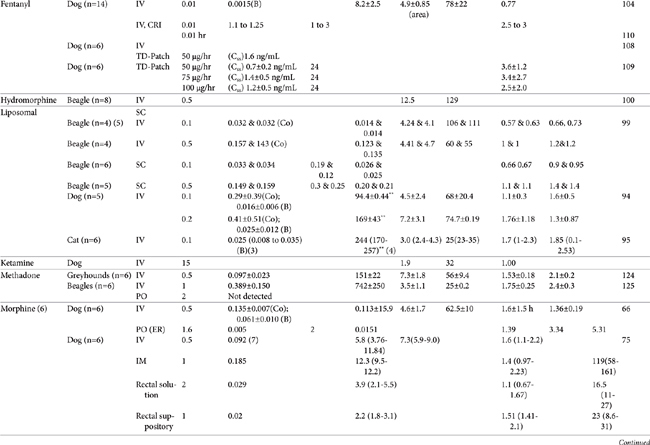
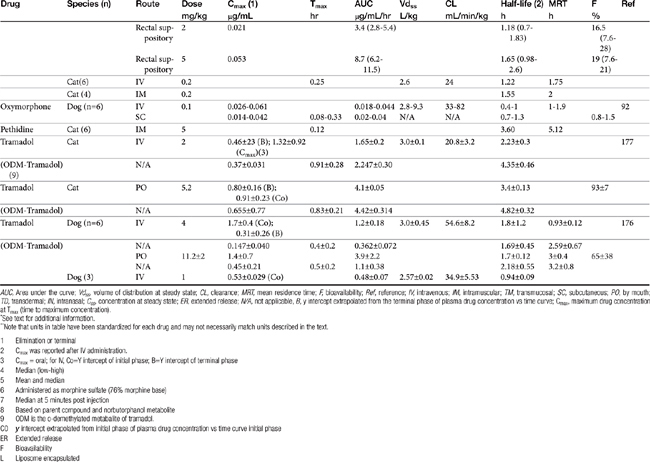
AUC, Area under the curve; Vdss, volume of distribution at steady state; CL, clearance; MRT, mean residence time; F, bioavailability; Ref, reference; IV, intravenous; IM, intramuscular; TM, transmucosal; SC, subcutaneous; PO, by mouth; TD, transdermal; IN, intranasal; Css, concentration at steady state; ER, extended release; N/A, not applicable, B, y intercept extrapolated from the terminal phase of plasma drug concentration vs time curve; Cmax, maximum drug concentration at Tmax (time to maximum concentration).
∗ See text for additional information.
∗∗ Note that units in table have been standardized for each drug and may not necessarily match units described in the text.
| 1 | Elimination or terminal |
| 2 | Cmax was reported after IV administration. |
| 3 | Cmax = oral; for IV, Co=Y intercept of initial phase; B=Y intercept of terminal phase |
| 4 | Median (low-high) |
| 5 | Mean and median |
| 6 | Administered as morphine sulfate (76% morphine base) |
| 7 | Median at 5 minutes post injection |
| 8 | Based on parent compound and norbutorphanol metabolite |
| 9 | ODM is the o-demethylated metabalite of tramadol. |
| C0 | y intercept extrapolated from initial phase of plasma drug concentration vs time curve initial phase |
| ER | Extended release |
| F | Bioavailability |
| L | Liposome encapsulated |
Chapter 28 Control of Pain in Small Animals: Opioid Agonists and Antagonists and Other Locally and Centrally Acting Analgesics
Definition of Pain and its Recognition
Classification of Pain
The International Association for the Study of Pain (IASP) defines pain as an unpleasant sensory and emotional experience associated with actual or potential tissue damage.1 During the past several decades, the importance of pain has evolved from being underestimated, to being recognized for its contributions to morbidity and mortality. In 2000 the Joint Commission on Accreditation of Healthcare Organizations2 mandated that hospitals for humans consider pain as the fifth vital sign, thus ensuring its inclusion in the routine evaluation of all patients.
Pain has been classified in several ways. Severity (mild, moderate, and severe) is among the most common, but this categorization also includes an emotional component of pain. As such, in veterinary medicine this classification requires interpretation by either the clinician or owner. Pain also is classified according to its source. Physiologic pain is a protective mechanism, leading the animal to withdraw from a potentially damaging stimulus. In contrast, pathologic pain reflects tissue damage. Unresolved physiologic pain eventually can lead to pathologic pain by causing neurologic damage (also classified as neuropathic pain). Neuropathic pain is also classified according to the location at which the damage occurs (e.g., neurologic damage), with nociceptive pain reflecting tissue damage. Pain might also be localized to the body site: visceral pain is associated with abdominal or thoracic pain, whereas somatic pain originates from musculoskeletal damage. Either of these locations can be further divided as superficial or deep. Pain also is described by duration. Acute pain is abrupt in onset and resolves in 24 to 72 hours, whereas chronic pain is slow in onset and generally persists for several weeks to months. Acute pain is more protective in nature (compared with chronic pain) and indicates that something is going wrong, whereas chronic pain reflects disturbed homeostasis. Acute pain results from traumatic, surgical, or infectious events and generally is conducive to resolution with analgesics. In contrast, chronic pain is a long-standing physical disorder or emotional distress and includes pain associated with degenerative joint disease and some types of cancer. Chronic pain may not respond to traditional analgesic therapy, with tranquilizers or behavior-modifying drugs combined with environmental manipulation and behavioral conditioning commonly implemented. The transmission of chronic pain increasingly is being understood, with knowledge regarding neural pathways elucidating possible targets of therapy. Accordingly, newer drugs are increasingly being identified for their potential efficacy.3
Transmission of Pain
Nociceptive Pain
Nociception refers to a neural response of nociceptors to a noxious stimulus.1 Nociception is a normal nervous system function that serves as a warning of danger or credible threat. The nociceptive response comprises a nociceptor and three neuron chains, originating in the peripheral tissues and ending in the cerebral cortex. Nociception is unique among sensory nerves because nocicpetors must detect a wide range of stimuli, including physical and chemical signals, heat and cold, and acid and mechanical pressure. Nociceptors are located in every tissue of the body, originating from a group of neuronal bodies in the dorsal root ganglia (Figure 28-1). Nociception varies with location (peripheral versus central) and organ. Nociception in the skin occurs at the surface (i.e., is somatic), resulting in sharp, defined, localized, and limited pain. In contrast, internal, or visceral, nociception is generally diffuse, dull, and often referred.4 The action potential stimulated by a nociceptor crosses the synapse in the dorsal horn and stimulates the second order neurons in the gray matter of the spinal cord. There the signal is transmitted to the brain, where it is processed and interpreted. The transmission of pain from nociceptors is carried by either small, myelinated A delta fibers, smaller unmylelinated C fibers, or A a fibers. A delta fibers are fast, being responsible for sharp and acute pain, and transmit somatic and parietal pain. Because these receptors are discrete, animals can localize this pain. In contrast, C fibers are slow, transmitting dull, aching, burning, or throbbing pain that is difficult to localize. A fibers also are slow, transmitting stimuli associated with vibration, stinging, or tickling. However, because they are not able to discriminate between painful versus nonpainful stimuli, they are not pure nociceptor neurons.
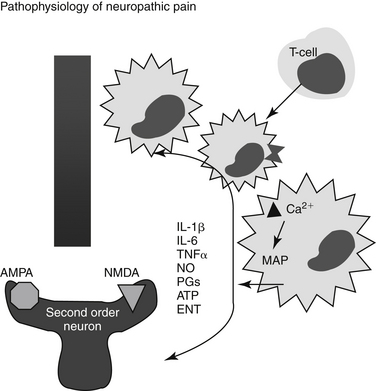
Figure 28-1 Pain pathways and the role of peripheral nerve injury in the emergence of neuropathic pain. See text for the transmission and perception of pain (left). The right side demonstrates mechanisms whereby neuropathic pain might emerge. The lower left diagram delineates the impact of peripheral nerve injury on sensory neurons. Injury is followed by recruitment and proliferation of non-neuronal elements (including Schwann cells, mast cells, macrophages, and T cells). Cells release proinflammatory and other chemicals (TNFα tumor necrosis factor-alpha], IL[interleukin]-1, IL-6, CCL2, PGs (prostaglandins) and NGF [nerve growth factor]). Sensory abnormalities are initiated and maintained either locally at the site of damage or with retrograde transport to cell bodies in the dorsal root ganglion, where neuronal gene expression is influenced. Increased intracellular calcium results in activation of the p38 and MAPK/ERK pathway. Perpetuation of the response is maintained through recruitment of inflammatory cells in general and T-cells in particular. In the spinal cord (upper right), microglia surrounding primary afferent neurons contribute to self-propogation of neuropathic pain after activation by chemicals or the influence of other primary afferent neurons. Subsequent mediator release may presynaptically or postsynaptically modulate pain. Examples include but are not limited to TNF-α, IL-1, IL-6, nitric oxide (NO), ATP, and PGs and excitatory neurotransmitters (ENT). AMPA,amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; NMDA, N-methyl-d-aspartate; MAP, mitogen-activated protein kinases.
Chemical mediators are important components of the nociceptor reflex and offer a target of pharmacologic modulation. These include but are not limited to adrenocorticotropic hormone, glucocorticoids, vasopressin, oxytocin, brain opioids, catecholamines, angiotensin II, endorphins/enkephalins, vasoactive intestinal peptides, substance P (centrally), eicosanoids (prostaglandins, leukotrienes), tissue kininogens (bradykinin), histamine, serotonin, potassium, and proteolytic enzymes.6 Several of these mediators are also associated with stress. Several neurotransmitters associated with nociception and transmission of peripheral pain also function in the dorsal horn, the first site of signal processing. Spinal neurotransmitters include but are not limited to peptides (e.g., substance P, calcitonin gene-related peptide), excitatory and inhibitory amino acids (aspartate, glutamate, gamma-aminobutyric acid [GABA]), nitric oxide, prostaglandins, adenosine-5’-triphosphate (ATP), endogenous opioids, monoamines (serotonin, norepinephrine), protons (acids), and neurotrophins. Opioid peptides are synthesized by interneurons in the superficial dorsal horn; they regulate further neurotransmitter release, probably through decreased calcium conduction. Glutamate, the primary excitatory nociceptor neurotransmitter, is released in response to calcium, following depolarization. Subsequent postsynaptic binding to N-methyl-d-aspartate (NMDA) receptors influences pain transmission, hyperalgesia, allodynia, and neurotoxicity, thus providing the rationale for combination analgesic therapy that includes NMDA-receptor antagonists.
KEY POINT 28-3
N-methyl-d-aspartate receptors and products of cyclooxygenase activity promote several aspects of neuropathic pain.
The IASP definition of pain includes both a sensory and emotional component.2 Indeed, of all the sensory systems, the nociceptive system is most able to elicit an arousal response in the brain. The emotional portion of pain occurs at the level of the limbic system (emotional), cortex (cognitive appraisal), and frontal lobes. Stimulation in the reticular formation results in emotional reaction to pain (anxiety, depression, suffering), whereas stimulation in the cerebral cortex leads to conscious perception and interpretation of pain. Transmission of central pain reflects a “gate control” phenomenon. The dorsal horn cells modulate the patterns of incoming information transmitted to the brain, which ultimately produces response and perception. The brain, in turn, can enhance or counter nociception. For example, when pain is sustained, released opioid peptides bind mu receptors in the brain and spinal cord, decreasing pain. Modulation regulates pain response, thus maintaining homeostasis.7 In addition to central transmission of pain, the first-order neuron can also synapse with neurons that cause a local reflex. The reflex can be myoneural or sympathetic in action (e.g., release of norepinephrine, smooth muscle spasm, vasoconstriction). Voluntary reflexes require conscious pain perception, whereas nociceptive reflexes do not.
Not surprisingly, a variety of mechanisms transduce nociceptive signals.8 At least two classes of channels are involved: One class detects noxious stimuli or products of tissue damage, and another sets the threshold necessary for a nociceptor action potential. Channels that detect noxious stimuli include transient receptor potential (TRP) channels, acid-sensing ion channels (ASICs), and the ATP-gated P2X receptor family. Of these, the TRP channels are emerging as the dominant channels transmitting nociceptor signals. The TRP channels are general receptors, able to sense multiple types of noxious stimuli. Their actions are complex, with channels having different roles in different tissues. Further, each channel is able to respond to more than one stimulus and with overlapping sensitivities. Four of the nine currently described nociceptive TRP channels respond to heat. The TRP receptors are regulated through a number of kinases (e.g., kinase C, kinase A, calcium/calmodulin–dependent kinase). In contrast to the general TRP channels, ASIC channels are specialists, able to detect only acidity, as might occur with bone cancer (high levels of acid leak into surrounding tissues), inflammation (local pH can drop to 5.5), and ischemia (resulting from metabolites such as lactic acid). Because extracellular ATP is a major product of inflammation, P2X channels are also drawing interest for their potential role in inflammatory pain, particularly visceral pain.7
The second type of channels that transmit nociceptor signals set thresholds and include newly discovered voltage-dependent sodium channels. Unlike other sodium channels, these channels are resistant to blockade by tetrodotoxin (TTX), which serves as a method of identifying and classifying the channels. Normally, these channels set the nociceptor threshold very high. However, in the presence of pathology, thresholds are decreased such that nociceptors respond to much weaker stimuli. Thus, unlike most sensory systems, channels responsible for nociception become more sensitive, rather than less sensitive, to stimuli. The TTX-resistant sodium channels tend to be redistributed after tissue injury.
Neuropathic Pain
Pathophysiology
Nociception is not limited simply to transmission of acute (nociceptive) pain; it also contributes to neuropathic pain. Both acute and unrelieved chronic pain can shift from nociceptive to neuropathic pain. Neuropathic pain includes hyperalgesia (overreaction or increased sensitivity to painful stimuli), allodynia (reaction to an innocuous stimulation), or reflex sympathetic dystrophy. The latter is a complex disorder of pain, sensory abnormalities, abnormal blood flow, sweating, and trophic changes in superficial and deep tissues.7 Failure to control development of acute pain and hyperalgesia can lead to chronic pain; progressive and prolonged stimulation can lead to a “wind-up” phenomenon that reflects increased excitation of neurons in the dorsal horn. The wind-up phenomenon is manifested as a pain response outside the site of injury and can persist beyond resolution of the inciting cause (pathologic pain). Dysesthesia is another example of neuropathic pain characterized by an unpleasant spontaneous or provoked sensation. Dysesthesia has been described in humans as a burning or shooting sensation reflecting damage to peripheral nerves. Excess stimulation of pain fibers, or reduced activity of non-nociceptive sensory pathways contribute to an imbalance between painful and nonpainful inputs to the central nervous system (CNS).9 Examples include phantom pain, a centrally mediated pain manifested as an intense burning sensation at the nerve ending of a damaged, paralyzed, or missing extremity. Another example is “stump” pain, a peripheral neuropathic pain secondary to neuroma formation at the site of amputation. These are examples of pain without any obvious noxious input. Further, for some chronic or persistent pain disorders, pain does not necessarily originate at the periphery.4
Heightened pain sensitivity is probably a protective response, reducing tissue exposure to further risk of tissue damage, and normally resolves when the tissue is healed. Both local and spinal changes contribute to neuropathic pain. Tissue damage intensifies the sensation of pain by recruitment of otherwise silent receptors. Chemical mediators are produced and released by both neuronal and non-neuronal cells (e.g., fibroblasts, mast cells, neutrophils), increasing nociceptor sensitivity. Local injury causes release of inflammatory mediators (cyclooxygenase-2 [COX-2], interleukins [ILs], leukotrienes [LTs], H+, K+, histamine, bradykinin), generating a slightly acidic inflammatory milieu that lowers threshold sensitivity (allodynia) by facilitating chemical binding to nociceptors. Activation of the nociceptors initiates neurogenic inflammation, leading to the release of substance P and peptides, which in turn perpetuate inflammation.5 Dysesthesia may reflect spontaneous activity originating in the regenerating, primary, nociceptive neurons (myelinated, small afferent fibers). Changes in dorsal root Na+ channels also have been reported. Dysesthesia may thus reflect a focal inflammatory process as opposed to axonal damage. Further, potential involvement of (fast) Na+ channels that generate ectopic discharges supports the use of local anesthetics (Na+ channel blockers) as part of a combined analgesic approach.9
Heightened sensitivity also involves retrograde signals from the site of injury to the neuronal cell body; signals alter neurotransmitter release and receptor transcription and expression. Among the mediators of hypersensitivity are mitogen-activated protein kinases (MAP: p38, ERK and JNK), which may initiate changes in the microglia of the dorsal horn.6 These changes may persist for weeks. Other mediators released and associated with hypersensitivity in the spinal cord include but are not limited to the proinflammatory cytokines tumor necrosis factor (TNF), IL-1 and IL-6, prostaglandins, and reactive oxygen species. Microglial cell bodies also hypertrophy and multiply, a reaction that, along with release of proinflammatory cytokines, may be inhibited by minocycline. Interestingly, chronic morphine administration may activate microglial cells and thus exacerbate hypersensitivity, an effect that might be reversed by minocycline or pentoxifylline, exemplifying the complexities of hyperalgesia.10
A role for NMDA receptors has not yet been identified in normal spinal cord transmission. However, a role might be suggested in transmission of pain. Both glutamate and aspartate are major excitatory neurotransmitters in the CNS that bind to NMDA receptors. After tissue injury, impulses from sensitized nociceptors of C fibers stimulate glutamate and other chemical release (e.g., neurokinins) in the primary afferent in the spinal cord. Calcium influx and activation of early genes leads to the development and maintenance of hypersensitivity or the wind-up phenomenon and hyperalgesia. Hyperalgesia associated with opioid use may involve NMDA receptors (see later discussion). Another mediator group that may be involved with changes in response to pain is prostaglandins (PGs). Induced COX-2 prostaglandin E (PGE) has been associated with hyperalgesia in either the spinal cord (primary hyperalgesia) or peripherally at nociceptors (secondary hyperalgesia).11 Induction of spinal COX-2 in the dorsal horn has been associated with central sensitization.11-13 Finally, PGs may potentiate the effects of other chemical mediators involved in pain or inflammation (e.g., histamine, bradykinin, substance P, nitric oxide), neurotransmitters (e.g., inhibition of glycine or potentiation of glutamate), or other receptors (e.g., NMDA), although this role in central sensitization and hyperalgesia has yet to be determined.
Nociception may awaken long dormant processes. For example, GABA is normally inhibitory, but it becomes excitatory in the spinal cord after peripheral nerve injury.7 Further, if the inciting cause is sufficient, quiescent nociceptors will be recruited; output of non-nociceptive neurons (e.g., touch receptors) may be interpreted as nociception by second-order neurons in the spinal cord.5
Preemptive control
Successful preemptive analgesia prevents the development of increased central nociceptive pathway responsiveness that is triggered by intense afferent neural activity. Surgery involving limbs exemplifies the importance of preemptive analgesia. For example, local anesthetic blockade of exposed nerves before transaction presumably protects the spinal cord from a sudden, massive impulse surge from nociceptors. Further, ghost phantom pain can be reduced through epidural analgesia or adequate prevention of postoperative pain, particularly during the first 72 hours after surgery.
Meta-analyses of human studies that focused on timing of analgesia in surgical patients warrant review for their implications with regard not only to timing but also to the importance of effective pain control.14,15 One meta-analysis examined randomized, double-blinded, controlled trials that addressed either preemptive or postoperative analgesia for acute or chronic postoperative pain relief. Analgesic protocols that were reviewed studied the following: Twenty of the clinical trials reported the analgesic efficacy of NSAIDs (n=20; diclofenac, ketorolac, ketoprofen, ibuprofen, flurbiprofen, and naproxen [endodontal]), opioids (n=8; morphine, fentanyl, alfentanil, sufentanil, meperidine), and NMDA antagonists (n=8; ketamine, dextromethorphan). Further, epidural, caudal, or intrathecal (n=20) or local anesthetics (n=20) were considered. Outcome measures included comparison of pain scores before and 24 hours after surgery, time to the need for analgesics, and the need for supplemental analgesic therapy. In general, meta-analysis concluded that pre-emptive timing did not alter the magnitude of postoperative pain, although the consumption of analgesia and the time to request additional analgesics were reduced. For NMDA antagonism, preemptive use of ketamine (as part of combination analgesic therapy, generally with opioids) uniformly did not cause a statistical difference in postoperative pain (a finding substantiated by both meta-analyses), whereas dextromethorphan did, although only two trials were reviewed. For epidural analgesia 7 of 11 trials using opioids indicated potential benefits, although the authors concluded that clinical relevance would be improved if epidural analgesia continued for a longer postoperative period. In general, preemptive nonsteroidal antiinflammatory drug (NSAID) therapy provided no difference in pain relief, leading the authors to conclude that the risks associated with preemptive NSAID therapy (e.g., bleeding, renal compromise) may not be justified; the power of the study to detect a significant difference is not known. Only one of the trials addressed the advent of chronic (6 months) pain after a surgical procedure. This trial found chronic pain to be significantly reduced in patients receiving preemptive analgesia. The overall conclusions by the authors was that preemptive analgesia may not be an effective method for reducing postoperative pain. However, the authors also suggested that future studies should not focus on the timing so much as on the approach to prevention of postoperative pain. Specifically, studies might address the importance of aggressive control, including the use of combinations designed to prevent neuropathic pain. Indeed, the lack of evidence of effective preemptive analgesia for control of postoperative pain reported in this study may reflect failed actions that occur in clinical patients. Because even abbreviated sensory events may initiate events leading to central sensitization, effective prevention may be dependent on continuous sensory blockade—that is, throughout the presence of the stimulating nociceptive event. Further, the success of afferent blockade may depend on blockade of input from small nonmyelinated C fibers or spinal, rather than peripheral, analgesia. Indeed, the meta-analysis indicated that neither systemic morphine nor small doses of intrathecal opioids had an effective preemptive impact. However, larger intrathecal doses of morphine were effective. Thorough regional blockade may also be paramount to the success of preemptive operative analgesia.
Pharmacologic Control of Pain
Analgesia reflects the selective interruption of the transmission of injury signals (real or potential) between primary sensory neurons, the spinal cord dorsal horn, the rostroventral medulla, and the cortex opioids.5 The paradigm for the approach of pain control in both human and veterinary medicine has been shifting with the increasing realization that uncontrolled pain is not only bad but also avoidable.16,17 Appropriate pain management is part of the practice of good medicine: For human patients, analgesic control is associated with more rapid clinical recovery, shorter hospital stays, fewer readmits, and improved quality of life.1
Control of Pain
Assessing Response to Pain and Stress
One of the more difficult aspects of acceptable control of pain for the clinician is detection or recognition of pain.19-21 Scientists agree that all animals feel pain, although the level of nociception may vary between vertebrates and invertebrates and among classes of animals.22 Animals may feel pain as easily as human patients; any stimulus that is likely to cause pain in a person will cause pain in animals. Animals differ from people, however, in their response to pain. Indeed, the laws of behavior in wild animals require that abnormal behavior associated with pain be avoided. Avoidance, escape, or control of pain and distress are responses to pain that are important for (wild) animal survival and allow animals to adapt to a new or changed environment. Animals showing weakness, pain, and distress become targets for predators. Ill or injured animals tend to be abandoned by others so that an entire stock is not jeopardized. This evolutionary process makes clinical recognition of pain in animal patients difficult. For example, a critical patient is often unable to manifest pain and unwilling to care for itself; the clinician must be diligent to recognize the likelihood of an underlying disorder in causing pain. Increase in heart rate usually is caused by the underlying pathology, not in response to pain. The more severe the illness, the more important the need for analgesics.
Response to acute and chronic pain varies and includes both physiologic and behavioral changes. If pain is too severe for the animal to accommodate, a state of distress can develop.23 Beyond protection, pain rarely has any useful function and is associated with dramatic and potentially life-threatening physiologic changes.24-26 Physiologic responses to distress include gastrointestinal lesions, immunosuppression, and hypertension. In human patients failure of response to treatment, hospitalization duration, and hospitalization costs can be positively correlated with failure of effective pain control. The sensation of pain can be associated with a marked adrenergic (catecholamine) release, which may cause life-threatening hypertension or cardiac arrhythmias. Response to acute pain includes physiologic changes such as tachycardia, tachypnea, mydriasis, and salivation and behavioral responses such as guarding, protection, vocalization (especially with movement or palpation of painful area), licking, biting or scratching, shaking, restlessness or insomnia, and recumbency.
Failure to control acute pain can stimulate changes at the level of the nociceptor and CNS; nociceptive pain may progress to neuropathic pain. Neuroplastic changes such as hypersensitization or allodynia worsen the stress response that often follows surgery. The response is adaptive and is mediated by behavioral, neural, endocrine, immune, hematologic, and metabolic changes that attempt to restore homeostasis. However, the combined effects can prove detrimental to the patient. Potential sequelae include cardiovascular instability, hypercoagulability, insulin resistance, increased metabolic rate and protein catabolism, and immunosuppression. As such, effective control of pain is paramount in the postoperative or otherwise stressed patient.
Responses to chronic pain include limping; licking, perhaps to the point of self-mutilation (of an associated region if an animal can reach it, an unassociated one if not); reluctance to move; loss of appetite; changes in personality; physiologic dysfunctions such as dysuria, tenesmus, and diarrhea; changes in appearance of hair coat and degree of eye brightness; failure to groom; discharges from eyes and nose; decreased food and water intake; and behavioral changes such as aggression or docility, agitation, cringing, and extreme submissiveness. The well-trained clinician or pet owner can detect subtle changes in gait or posture.
Variability among animals in response to stress also makes diagnosis of pain difficult (Box 28-1).
Box 28-1 Types of Biologic Response to Pain
Types of biologic response to pain vary with genetic, age, or physiologic state or makeup:
The American Veterinary Medical Association reviewed the major consensus concepts generated by the Cross-species Approach to Pain and Analgesia workshop that took place in 2002.22 In addition to recommendations regarding the use of animals in research involving pain, the review provides guidelines for development of a pain assessment tool. The guidelines stipulate that a numeric (1-10) scale, based on observable behaviors and quantifiable biological markers, be used. Factors that are likely to alter assessment include animal factors such as species (and strain), stage of development (including prepartum and postpartum development), gender, previous experience to pain; environmental factors; and the type of pain. Other factors, such as nutrition, drugs, concurrent disease, and owner social status, should also be considered. The American Society of Anesthesiologists developed a pain-scoring system (for humans) based on several categories, each scored from 1 to 15. Included are behavior (depressed = 1, normal = 3, apprehensive = 5, and excited or aggressive = 10), preexisting pain (none = 1, minimal = 5, moderate = 10, and severe = 15), surgically induced trauma or pain (minimal = 5, moderate = 10, and severe = 15), duration of surgery (less than 1 hour = 5, 1 to 2 hours =10, and >3 hours = 15), and patient health (normal = 1, mild disease = 2, severe disease = 3, moribund = 4, and life-threatening = 5).
Appropriate analgesic combinations for any pain group, regardless of score, include opioids, local analgesics, NSAIDS and (for surgical candidates) epidural analgesic therapy. For opioids scores less than 20 might warrant less aggressive control of pain (i.e., mixed agonists–antagonists or partial agonist opioids), whereas scores of 20 to 30 might require more aggressive analgesia (pure opioids) and scores of 30 the most aggressive therapy (e.g., constant-rate infusion [CRI] of pure opioids). The use of NMDA-receptor antagonists should be considered for members of the latter two groups. The prudent clinician might consider assessing pain using a numeric scale at intervals appropriate for the management of the pain and reassess therapy as indicated by the scale. This may be particularly important for prevention or treatment of chronic pain.
Levels of Consciousness
Marked species differences exist in response to drugs intended to control or help control pain. Analgesia is defined as the absence of pain. This may be harder to define in animals than in people. Tranquilization (ataxia, neurolepsis) is a state of behavioral change in which the patient is relaxed and unconcerned by surroundings. Generally, the state is not accompanied by drowsiness, analgesia, or unconsciousness. Targets of tranquilizers include the hypothalamus and reticular activating system. The tranquilized animal feels pain but frequently is indifferent to minor pain. Tranquilizers may act synergistically with analgesics in the control of pain. Sedation reflects a mild degree of central depression. Sedated animals are calm, awake, and possibly drowsy. Sedatives target the cerebral cortex. With anesthesia, light to complete unconsciousness is realized and is accompanied by loss of feeling or sensation. General anesthesia is both a loss of consciousness and a loss of pain; it is also characterized by muscle relaxation, hyporeflexia, and amnesia. However, the loss of pain induced by general anesthesia may not be sufficient to preclude the intraoperative use of analgesics.
Two other states of consciousness may be associated with drugs that also provide analgesia. Hypnosis is a state of artificially induced unconsciousness (sleep) from which the patient can be easily aroused. Akinesia is simply the absence of muscle movements and is generally induced by neuromuscular blocking drugs. Pain may best be controlled with a balanced combination of drugs, each of which targets a different site in the nociception pathway.
Endogenous and Exogenous Pain Control
Endogenous opiates (opiopeptins) provide analgesia when released in high concentrations in selected regions of the brain. These include enkephalins, dynorphins, and endorphins (Figure 28-2). Each opiopeptin is derived from a larger precursor molecule. Each of the precursor molecules has a characteristic anatomic distribution that is not limited to the CNS. The precursor for endorphin (β–endorphin) is pro-opiomelanocortin, which is also the precursor of melanocyte-stimulating hormone as well as adrenocorticotropic hormone and beta-lipotropin, suggesting a strong link between the opioid system and stress hormones.27 Endogenous recognition sites for these chemicals are also the targets of the exogenous drugs. A variety of other neuropeptides also have been implicated in endogenous analgesia (e.g., vasopressin, neurotensin, cholecystokinin, substance P). Some of these act in concert with other chemicals to stimulate nociceptors. Most notable are the eicosanoids (PGs, LTs), substance P, and bradykinin. The inflammatory process involves the release of a number of these chemical mediators either from the tissues at the site of infection or from the inflammatory cells themselves. Control of pain caused by these mediators is often largely dependent on controlling the inflammatory process. Nociceptin (NC, orphanin FQ, or OFQ) is a novel endogenous opioid peptide named for its ability to lower some pain thresholds. The NC/OFQ system is non-opioid, characterized by behavioral and pain modulator actions that differ from the classical opioids.27 Nocistatin is a related protein of the NC/OFQ system.
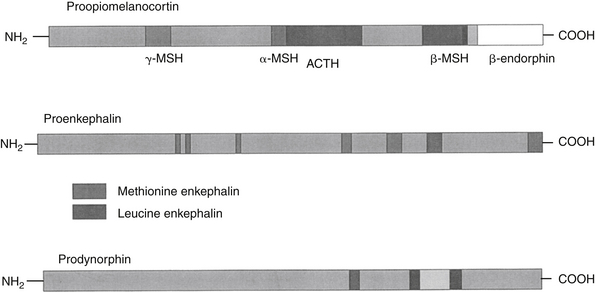
Figure 28-2 Endogenous opiopeptins that provide analgesia (endorphins, enkephalins, and dynorphins) are synthesized from precursor molecules. Each opiopeptin is derived from a larger precursor molecule. Each of the precursor molecules has a characteristic anatomic distribution that is not limited to the central nervous system The precursor for endorphin (β-endorphin) is proopiomelanocortin, which is also the precursor of melanocyte-stimulating hormone (MSH). Endogenous recognition sites for these chemicals are also the targets of the exogenous drugs.
Factors such as emotional state, expectation, attention, blood pressure, stress, counterirritation, and drugs can modulate pain, possibly by activating analgesia systems. In humans national, religious, and cultural backgrounds and ethnicity also influence pain. Emotions also modify pain: Human patients function better when they believe that they have some control over pain, an aspect that may be lost or is difficult to assess in animals.2
Pain may be relieved or its intensity reduced by environmental (e.g., soft bedding) or behavioral (e.g., petting) manipulation and by the administration of drugs (Table 28-1).28 With environmental control, emphasis is placed on the well-being of the animal. For dogs with osteoarthritis, this has centered on controlled exercise, although this method may be an inappropriate anthropomorphism. The single action for dogs most responsible for stress relief appears to be socialization (with humans). Factors such as emotional state, blood pressure, stress, and drugs can modulate pain, possibly by activating endogenous analgesia systems such as the opiates. The endogenous opiates, such as the enkephalins and endorphins, provide analgesia when released in high concentrations in selected regions of the brain.
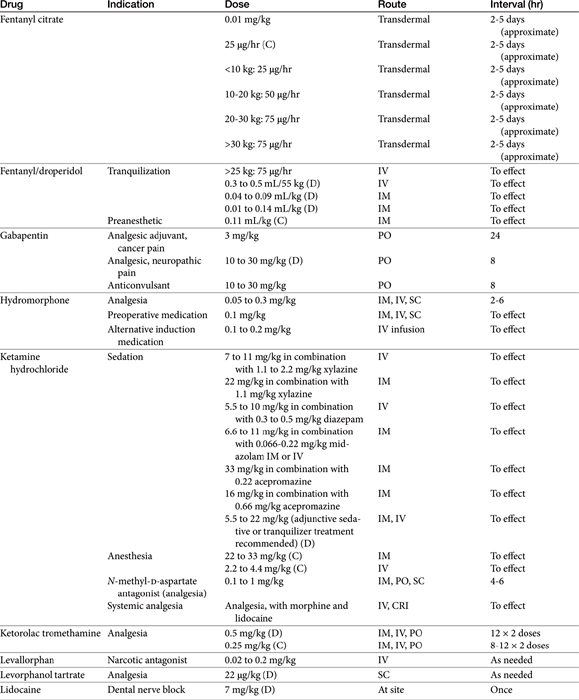
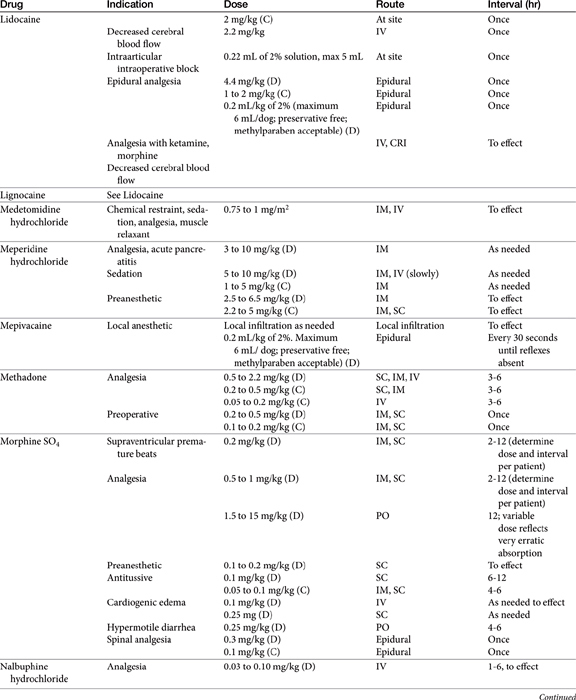
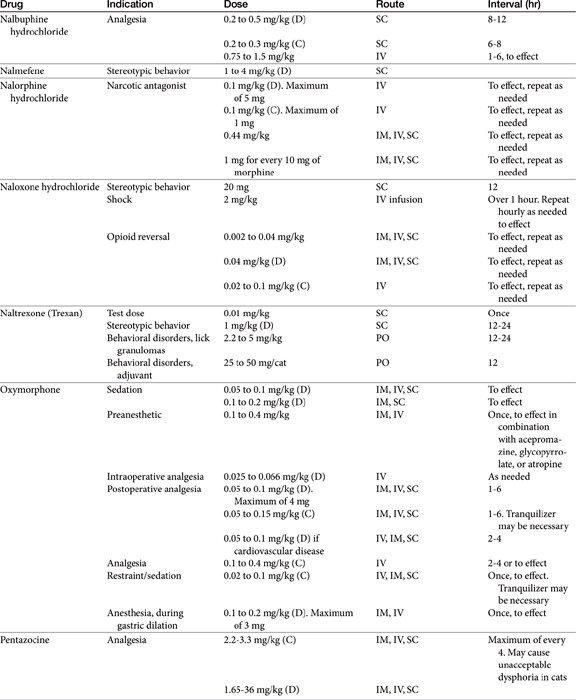
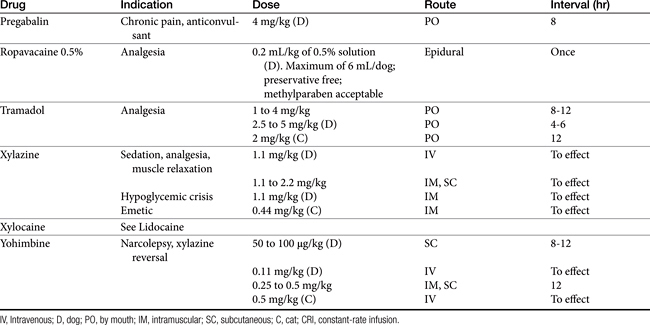
The best treatment for pain is removal of the underlying cause. Control of pain often includes nonpharmacologic modalities. For example, the goal of pain control with chronic wounds in humans includes availability of both local and systemic analgesics but also removal of all nonviable, locally infected tissue and elimination of cellulitis, identification of wound pathogenesis, and assessment of objective improvement through the periodic use of an analgesic scale. Notably, the most important aspect of pain control in humans is objective assessment coupled with assuring the patient that pain will be resolved. The importance of the latter in animals is not known, nor is the ability of veterinary clinicians to provide such assurance.
At one time analgesics were categorized by their major site of action—that is, whether they acted centrally or peripherally. This categorization is limited because of the shared signals between the two locations (see Figure 28-1). Multiple target sites are available for drugs in both the peripheral nervous system and CNS; additionally, a large number of specialized receptors exist in the skin and other tissues that signal chemical, thermal, and mechanical changes. However, redundancies in their activation generally lead to compensatory effects by one system on blockade of another. This is particularly true for TRP channels and ASICs. As such, targeting sites of signal convergence may be more effective. Examples include sodium channels specific to the peripheral nociceptive afferents, glutamate receptors in the spinal cord, and their homologs in the trigeminal system (e.g., NMDA receptors that are uniquely activated by persistent input).4
Opiates
Among the most effective and potent drugs used for controlling pain in animals, particularly acute pain, are the centrally and peripherally acting opioid analgesics.
Definitions
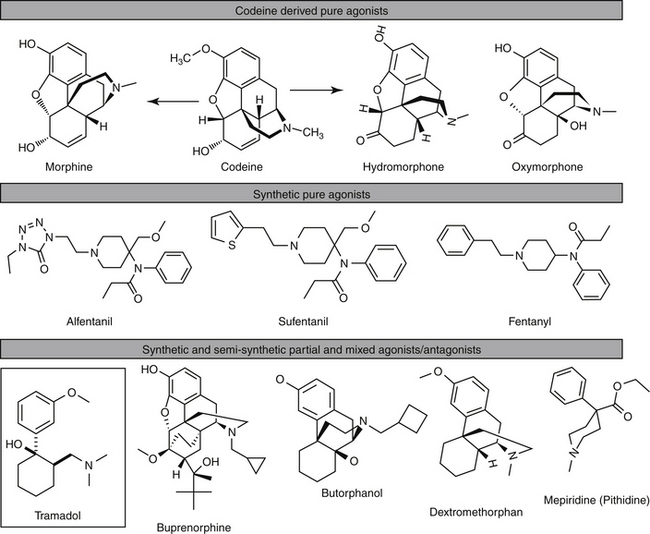
Opiates, including morphine, codeine, and a number of semisynthetic or synthetic derivatives, are drugs derived from opium. A number of drugs are derived from thebaine, a component of opium. Opioids include all drugs that exhibit morphinelike activity, as either agonists or antagonists (Figure 28-3).27 This includes all naturally occurring and synthetic drugs. The term narcotic, from the Greek word for stupor, is most appropriately used for any drug that induces sleep. The term has, however, become more associated with powerful opioid analgesics (which are more likely to be associated with sedation). Interestingly, endogenous opioid peptides exist in many animals, where they act as neurotransmitters and appear to act as modulators of neurotransmission or neurohormones. Their complete physiologic role has yet to be described; however, their existence was demonstrated in part by the ability of naloxone to reverse endogenous analgesia.27 In addition to the endogenous opioids, several opium derivatives found in nature are also found in mammalian cells, usually conjugated or bound to proteins. These include morphine, codeine, and some related compounds.29
Mechanism of Action
Receptors
The existence of multiple opioid receptors was postulated on the basis of in vivo canine studies.30 The pharmacologic effects result from interaction with one or more of three major opioid receptors named based according to the first letter of the first compound that bound to each receptor: mu (μ; morphine), kappa (κ; ketocyclazocine), and delta (δ). Receptors are similar in structure, expressing at least 40 homologies.The NC/OFQ receptor is a fourth member of the class whose similarity to opioid receptors is based on receptor homology. As with sigma receptors, they do not bind to classical opioid ligands. However, changes in as few as four amino acids would allow the nociceptin receptors to bind to classical opioids. Sigma receptors are no longer considered opioid receptors; they bind to and are activated by drugs completely unrelated to opioids. Opioid receptors also are chronologically classified, with OP-1 (δ) discovered first, followed by OP-2(к), and OP-3 (μ) (the most recently discovered). The Committee on Receptor Nomenclature and Drug Classification has indicated a final designation: MOP, DOP, and KOP, reflecting mu (μ), delta (δ), and kappa (κ) opioid receptors, respectively.27 This latter classification will predominate in this chapter. Nociceptor receptors are classified as OP4 or NOP. Other opioid receptors (e.g., epsilon) may reflect splice variants or diamers.
Each of the opioid receptors is subtyped: μ-1, μ-2, к 1-3, and δ1 and δ-2, with differences in structure probably generated after translation. Receptors vary in several characteristics that complicate predictive responses. The μ-1 or MOP1 receptors are described as very high affinity receptors that do not discriminate well between μ and δ. Dimerization among the receptors may play a role in their differential effects; thus far, both δ к and δμ diamers have been identified. Differential affinity of the heterodiamerized receptors compared to homodiamers for selected ligands (endogenous peptides or drugs) markedly alters the pharmacologic response. Further, binding sites of peptides are different from those of alkaloids (drugs): The latter appear to fit completely inside or at the mouth of the receptor core, whereas peptides appear to bind to extracellular loops. Differential binding sites allows for differential ligand effects, influencing conformation and subsequent responses. Opioid receptors differ from many other receptor systems in that a large number of endogenous ligands interact with a small number of receptors, potentially limiting the precise control that characterizes most systems.27 However, mechanisms whereby distinct responses to endogenous opioids might occur despite the lack of apparent substrate specificity include differences in duration of action, activation of multiple receptors or their heterodimers, production of endogenous opioids with unique activation profiles (perhaps in part through differential responses at the level of intracellular signaling), and differences in intracellular trafficking of the receptors themselves (see the discussion of adaptation). These mechanisms offer means by which pharmacologic interventions might be designed to generate the desired opioid response.
Opiate receptors occur throughout the body but are in high density in the dorsal horn of the spinal cord, where they are responsible for modulating pain reception. The NC/OFQ system is distributed in the hippocampus, cortex, and selected sensory sites and is responsible for complex behavior.27 Peripherally, receptors also are located in the spleen, kidney, intestine, vas deferens, and retina.31 These include effects on drug reward and reinforcement, stress responsiveness, and feeding behavior. The system is closely related to stress processes. Lester and Traynor32 demonstrated in vitro that the effect of commonly used opioids correlated to their binding of opioid or NOP receptors throughout the brain and spinal cord of dogs.
Pharmacodynamics
It is likely that species differences in receptor number, location, and specificity or sensitivity to the various drugs are important to differences in response to the opiates. Additionally, the type of interaction between ligand and opioid receptor influences the response. Ligands may act as agonists (which bind and stimulate) or antagonists (which block and inhibit the effect). Endogenous antagonists generally have no direct effect but occasionally may inhibit normal functions (e.g., appetite or release of selected hormones). For example, agonists increase whereas antagonists decrease appetite effects of endogenous opioids. Mixed agonists exhibit variable binding specificities at each receptor type, with some sites being agonistic and other sites antagonistic (e.g., butorphanol is a KOP agonist and MOP antagonist). Partial agonists do the same as mixed agonists, but their positive interaction with the receptors occurs with less than full activity at some of the receptors. The magnitude of each ligand–receptor interaction is likely to vary among species.
KEY POINT 28-6
Predictability in opiod response is confounded by receptor subtype and species differences in receptor type or subtype, tissue distribution, and receptor–drug interactions.
The cellular mechanism and the pharmacologic effects of the opioids probably reflect several different effector mechanisms. All three opioid receptors are coupled to G proteins. Accordingly, they inhibit adenylyl cyclase activity and activate receptor-linked potassium currents while decreasing voltage-gated calcium currents. The proposed, but as yet unproven, analgesic mechanism thus may reflect hyperpolarization of the neuronal resting membrane potential. Further complicating the understanding of their mechanism is their potential to affect other secondary messenger systems. They may activate MAP kinases and phospholipase C (PLC) mediated cascades that lead to formation of inositol triphosphate and diacylglycerol. Other actions include inhibition of neurotransmitter release (acetylcholine, glutamic acid, dopamine, serotonin, substance P [particularly peripherally], norepinephrine), or modulation of a potassium channel. Control of pain appears to occur without diminution of other senses. The NC/OFQ receptors also are linked to G proteins, as has been demonstrated in dogs.31
The pharmacologic effects of opioid derivatives, including the degree of analgesia, depends on the receptor bound, the location of receptor and drug in the body, and the type of interaction between the opioid and the receptor (Table 28-2).29 Opioids act centrally to elevate the pain threshold and alter the psychologic response to pain. The opioids also act peripherally. The primary pharmacologic effects of all opioids are analgesia, euphoria, and sedation (without loss of consciousness).29 MOP receptors give rise to analgesia and sedation above the spinal cord (MOP1) or in the spinal cord (MOP2); interaction with central MOP receptors might provide greater analgesia than interaction with spinal MOP receptors. For example, morphine causes analgesia primarily by way of MOP1 receptors when given systemically.29 However, administration of morphine at both spinal and supraspinal sites results in synergistic analgesic effects, with a tenfold reduction in the total dose of morphine necessary at either site alone.29
Table 28-2 Opioid Receptors, Their Pharmacodynamic Effects, and the Impact of Drug–Receptor Interaction
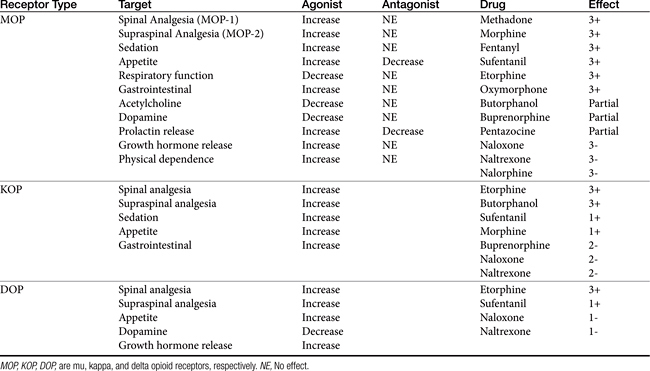
Interaction with MOP receptors also causes euphoria, respiratory depression, and physical dependence.33 MOP receptors located in the gastrointestinal tract mediate the pharmacologic effects characteristic of opiates in this body system. KOP receptors (three subtypes) are responsible for analgesia that is spinal in origin and stimulate miosis and sedation. Interestingly, stimulation of KOP receptors appears to cause effects antagonistic to MOP receptors, including analgesia, tolerance, and reward.33 DOP receptors (two subtypes) are located on smooth muscle and lymphocytes, in addition to the CNS. Interaction with DOP receptors appears to modulate, among other effects, emotional behavior and immunomodulation (see Chapter 26). Sigma receptors interact with a selected synthetic opioids; positive interactions between drugs and these receptors provide no analgesia but do cause adverse events including dysphoria, hallucinations, respiratory stimulation, and some of the vasomotor responses.
The pharmacologic effects of opioids extend beyond control of pain. For example, nonanalgesic pharmacologic effects account for many of the side effects associated with the use of these drugs for control of pain. The effect of opioids on the immune system is discussed in Chapter 31. The cardiovascular system in particular offers an example of the nonanalgesic pharmacodynamic effects of opioids. Opioid receptors regulate the cardiovascular system centrally (hypothalamus and brainstem) and peripherally (cardiac myocytes and blood vessels). In the heart both KOP and DOP, but not MOP, receptors have been identified.34 Appetite is influenced by all three receptor types, with agonists increasing and antagonists decreasing response (see Chapter). DOP opioid receptors interact (by way of the G protein) with several K+ cardiac channels, and KOP opioid receptors interact with calcium cardiac channels.34 Opioid receptors appear to be involved both in the physiology of the normal myocardium and pathophysiology of disease. DOP receptors appear to attenuate adrenergic response and decrease cardiac performance yet also inhibit acetylcholine-induced vagal bradycardia; suppress baroreceptor responses; and increase inotropy, chronotropy, and blood pressure. In contrast, KOP have been associated with arrhythmias. The impact of opioids on the myocardium is, not surprisingly, complex. On the one hand, opioids appear to facilitate arrhythmias and other disturbances associated with circulatory shock, congestive heart failure, and myocardial ischemia and reperfusion injuries. Yet, opioids appear to protect the heart (and several other organs) from hypoxic or ischemic insults. For example, large amounts of endogenous opioids are released in the heart in response to a number of stimuli, including ischemia, leading to a cardioprotective effect. During acute myocardial ischemia, morphine attenuates neutrophils and endothelial activation and reduces adhesion molecules.34 Indeed, opioids (endogenous) appear to be involved in ischemic preconditioning. This phenomenon involves the protective effect of coronary artery occlusion before a prolonged ischemic insult; irreversible tissue damage is prevented, and ATP depletion is reduced.34
Administration of nociceptin (FOQ ligand) causes a broad range of physiologic changes, with the often contradictory responses depending on the site of administration. These include antinociception or pronociception, anxiousness, altered appetite and cardiovascular effects (depression).31 Nociceptin also influences transmission at GABA receptors.35
Pharmacokinetics
Most opiates are well absorbed after oral, subcutaneous, or intramuscular administration (see Table 28-4 later in this chapter). In an attempt to reduce side effects without loss of analgesic activity, while improving the convenience of opioid administration, a number of alternative routes of administration have been studied. Oral, transmucosal (transbuccal), and rectal routes are examples discussed with individual drugs. Additionally, intranasal administration of opioids has been reviewed in humans.36 First-pass metabolism precludes oral as a reasonable route of administration for most opiates. Oral administration can be facilitated with a higher dose (if safe), slow-release products, or generation of active metabolites. For example, morphine is only 25% bioavailable in humans after oral administration. However, it is used effectively through oral administration for control of cancer pain in humans.29 In veterinary medicine use of oral opioids has been limited to codeine (60% bioavailability), hydrocodone, and (at high doses) butorphanol but more recently includes morphine. Morphine is also available as a slow-release oral (and limited injectable) preparation. Buprenorphine (including compounded products) has been used successfully after oral administration. Naltrexone is a pure antagonist, similar in actions to naloxone, which is also orally bioavailable. Selected drugs, including morphine, are available as rectal suppositories.
Drugs for which intranasal pharmacokinetics have been described include fentanyl, pethidine, butorphanol, oxycodone, and buprenorphine. In contrast to other mucosal routes of administration, lipophilicity does not appear to be a major determined of drug absorption through the nasal mucosa; indeed, nasal bioavailability of the opioids studied in humans ranges from a low of 46% for buprenorphine to a high of 71% for fentanyl and butorphonal, with the time to maximum concentrations generally being as little as 5 minutes and less than 50 minutes. Adverse effects were drug- (opioid-) related effects, rather than damage to the nasal mucosa. Limitations to this route of administration include the volume (limited to 150 μl per nostril) and pharmacokinetic variability, which tended to exceed that for intramuscular and subcutaneous administration. Both of these limitations may have reflected the use of the intravenous preparations: administration of an appropriate dose at times required a volume that exceeded that recommended, leading to pharyngeal delivery and subsequent swallowing (oral administration). Current interest in this route as a method of patient-delivered analgesia may lead to approval of products prepared with the intent of intranasal administration.
Transdermal preparations are available for systemic delivery of selected very lipid-soluble products (e.g., fentanyl patch.). In general, transdermal gels have not been successful in the systemic delivery of opioid analgesics, although limited efficacy may ultimately be demonstrated for selected lipid-soluble opioids (e.g., buprenorphine: unpublished work by the author). Epidural or intrathecal administration results in penetration of the spinal cord with limited systemic effects. Drugs that are more lipid soluble (e.g., fentanyl) tend to move rapidly across the dura into the spinal tissue and thus have a rapid, albeit local response. Drugs that are less lipid soluble, such as morphine, do not distribute rapidly into the spinal cord. Therefore they are more likely to diffuse up or down the spinal cord, potentially providing a larger area of analgesia. However, the use of epidural opioids alone for control of postoperative analgesia might be reconsidered for some patients. In children continuous epidural infusion of fentanyl and bupivacaine was found to be superior to intermittent epidural morphine in children undergoing major abdominal or genito-urologic surgery, as is supported by other human studies.37
KEY POINT 28-8
First-pass metabolism that precludes oral absorption of most opioids and a desire to avoid intravenous administration have led to a number of alternative routes of opioid delivery.
Distribution of the opioids from blood into the CNS varies. Most opiates are sufficiently lipid soluble to distribute into the CNS, although the rate into and out of the CNS is variable. Generally, onset of action occurs most rapidly for the highly lipid-soluble drugs (heroin, codeine) but is countered by rapid movement out of the CNS and thus resolution of pharmacologic response. The amphoteric opioids such as morphine move less rapidly, take longer to be effective, and generally act longer. Some opiates, such as loperamide, are designed to poorly penetrate the CNS with the intent of having peripheral (e.g., gastrointestinal) effects only. In the developing fetus, opioid derivatives pass more easily into the CNS because the blood–brain barrier is not fully developed. In humans the developing fetus can suffer severe depression induced by opiates, despite no evidence of depression in the pregnant mother.
Most of the opioids are biotransformed by the liver. Glucuronide conjugation is a common metabolic pathway. In some species metabolitesmay be active, including glucuronide metabolites. Cats may be deficient in some of these pathways, contributing to the increased risk of overdose that occurs with this species. Hepatic elimination for many drugs is “flow limited,” meaning that hepatic blood flow determines the rate of elimination. Thus liver disease, particularly that associated with portal to systemic vascular shunting, renders a patient susceptible to adverse reactions (toxicity). Hepatic metabolites are renally excreted. Elimination of the bile and enterohepatic circulation for some drugs may, however, prolong pharmacologic effects. In general, the opioids are very rapidly eliminated in normal animals, with elimination half-lives ranging from 30 minutes to 2 hours and duration of action being less than 2 hours in many animals for many of the drugs. For selected drugs (morphine, oxymorphone, buprenorphine), pharmacologic effects may remain for up to 6 hours, depending on the type of pain, species, and route of administration. Formation of active metabolites also affects (prolongs) duration of effect. Adherance to receptors may play a role in duration of affect of some drugs (e.g., buprenorphine).
Altered response to the opioids should be expected among species, in very young and very old patients,38 and in patients suffering from hepatic, cardiovascular, or respiratory disease; hypotension; cranial trauma; and (in cats) hyperthyroidism. The duration, but not the extent, of analgesia increases with age in human patients,29 presumably because of changes in hepatic metabolism and hepatic blood flow. Doses are decreased up to 75% in some patients, particularly geriatric patients or those with liver disease. However, prolonging the interval, rather than decreasing the dose, should be a more effective means of compensating for the effects of opioids normally characterized by a short half-life unless volume of distribution changes result in higher plasma concentrations as well. An exception is with CRI, for which doses should be decreased. Both liver disease and renal disease can alter the disposition of opioids, leading to adverse effects. Renal disease affects the elimination of morphine, codeine, and meperidine, in part because of the accumulation of active metabolites.29
Adverse Effects
Central Nervous System
The major disadvantages of opioids reflect general CNS depression, including dose-related respiratory depression and, to a lesser degree, cardiac depression. CNS depression tends to preclude the use of opioids in syndromes such as shock, severe cranial trauma, and diseases associated with respiratory compromise. Synthetic opioids were designed to induce analgesia with minmal undesirable side effects (i.e., sedation, respiratory depression).
Sedation is common with opioid use, depending on the drug and its target receptors, and the species (i.e., MOP and to a lesser degree KOP receptor stimulation in the dog) and can be a disadvantage or an advantage, depending on the clinical situation. Species differences in response to the sedative effects of opioids can be profound. “Morphine mania,” typical of that described in cats is manifested as dysphoria and psychomotor activity. It may reflect sigma receptor stimulation for selected opioids or simply may reflect overdosing. Opioids tend to cause release (rather than inhibition) of some neurotransmitters (e.g., dopamine, acetylcholine), and this may occur in cats with high doses. When opioids are used in combination with phenothiazine derivatives (which block many of the neurotransmitter actions), the incidence of adverse effects appears to be reduced in cats. Dogs also are subject to dysphoria, as is exemplified by a series of three cases reported by Hofmeister and coworkers.39 Dysphoria was manifested as vocalization that began within 5 minutes of administration of either hydrocodone, morphine, or fentanyl in association with postoperative analgesia. Clinical signs abated with administration of naloxone.
KEY POINT 28-9
Synthetic opiods are designed to minimize cardiovascular or respiratory adverse effects without sacrificing analgesic efficacy.
Respiratory depression usually is the cause of death in humans who succumb to opioids. The mechanism is reduction in the responsiveness of the brainstem respiratory centers to carbon dioxide, the primary stimulation for respiration. Centers that regulate respiratory rhythm are also depressed.29 Depression, manifested as a slow respiratory rate, is discernible at doses lower than those associated with sedation and increases as the dose increases. Rate, tidal volume, and minute volume all decrease. Respiratory depression is, however, rarely a clinical concern except in cases of overdose or in the presence of pulmonary dysfunction in cases of standard doses.29 Opioids should be used cautiously in patients with compromised respiratory function. Patients may appear to be handling the drugs well but in fact may be using compensatory mechanisms such as increased respiratory rate.29 Concentrations of CO2 may be increased, and respiratory centers may already be less sensitive to CO2. The administration of an opioid may be dangerous in such situations. Use of opioids in the pregnant animal can lead to marked respiratory depression in the developing fetus, with little to no effect on the mother, because of the underdeveloped blood–brain barrier in the fetus.29
KEY POINT 28-10
“Morphine mania” in cats may simply reflect neurotransmitter release at relatively higher doses.
Opioids have precipitated attacks of asthma in human anesthetized patients, probably as a result of histamine release. The importance of this in animals is not clear.
Opioids increase intracranial pressure as a result of increased concentrations of CO2 and cerebral vasodilation. Cerebrospinal fluid (CSF) pressure also increases.29 These effects may be exaggerated after head injury. The effects on intraocular pressure are not clear and may vary with the species. In humans accommodation is increased, with a decrease in intraocular pressure. Opioids cause miosis in humans, and mydriasis occurs in some species. Mydriasis may impede vision in cats; cats will be sensitive to light until mydriasis resolves.43
Convulsions occur in some species when opioids are administered in high doses. Mechanisms probably include inhibition of GABA and may be more likely with morphinelike drugs.29 The convulsant effects of some opioids can be reversed by naloxone,29 suggesting that drugs with antagonistic actions (e.g., butorphanol, buprenorphine) at some receptors might be preferred in the patient having seizures. The impact of opioids on epileptic patients is not clear.
Morphine and related opioids directly depress the cough center at concentrations lower than that required for analgesia. Respiratory depression and cough suppression do not appear to be related. Thus antitussive opioids do not necessarily cause respiratory depression.29
KEY POINT 28-11
Morphine and related drugs directly suppress cough without causing respiratory depression.
In contrast to depression, opioids directly stimulate the chemoreceptor triggering zone and thus may cause nausea and vomiting. Individual differences in the emetic response to opioids are marked in humans, but the amount of variability is not clear in animals.29 In human patients in whom opioids cause emesis, after subsequent administration, opioids act as antiemetics, blocking further response by the chemoreceptor trigger zone to opioids. Actions at the vestibular apparatus may also be responsible for emesis. The emetic effects that typify administration of opioids as sole agents do not typically occur in the postoperative, sick, or pain-ridden patient. Butorphanol has been used in some species as an antiemetic to control vomiting induced by cisplatin.44 In the awake dog, at doses associated with sedation, MOP agonists (morphine, fentanyl, and methadone) prevent emesis induced by apomorphine and copper sulfate. At lower doses morphine was able to cause emesis. KOP agonists also blocked the apomorphine emetic response at sedative doses. Antiemetic effects were blocked by naloxone. In contrast, DOP agonists did tend to cause emesis, leading the authors to conclude that delta receptor stimulation is associated with emesis, but MOP and KOP receptor stimulation with antiemesis.45 Nausea, vomiting, and salivation are associated with morphine or hydromorphone administration in cats. Buprenorphine, butorphanol, or meperidine are unlikely to be associated with these side effects.43,46 Decreased appetite may occur after several days of continuous opioid treatment in cats.43 Urinary retention has been described for MOP-active opioids in animals, although effects may vary with species.47 Mechanisms may reflect a centrally mediated vasopressin effect on the kidneys. Alternative mechanisms may include opioid-induced hypotension, which is probably more likely with morphine. Anderson and Day47 demonstrated that both fentanyl and morphine decreased urine output in normal and traumatized dogs.
Both long- and short-term use of opioids has paradoxically been associated with hyperalgesia. A biphasic, dose-dependent effect has been described and may reflect an initial increase in excitatory neurotransmitters. The hyperalgesic effects of opioids may involve NMDA receptors. Opioids appear to increase NMDA neural currents, providing a rationale for the combination of opioids with NMDA antagonists for enhanced analgesia. Several types of opioid hyperalgesia have been described both experimentally and clinically. At low doses, probably a reflection of disinhibition, intrathecal opioids induce an excitatory effect, which is otherwise masked with the sedation typical of larger doses. In contrast, very high intrathecal doses stimulate allodynia. With single-dose opioids, increased nociception may appear as the opioid effect diminishes. This may be manifested as an increased postoperative need for analgesia in patients receiving short-acting opioids intraoperatively. Reversal of opioids with naloxone often produces marked hyperalgesia. Hyperalgesia has also been associated with chronic use; among the clinical sequelae might be marked hyperalgesia with opioid withdrawal.48-50
Changes in body temperature reflect altered thermoregulatory response. Hypothermia is more common in dogs, whereas hyperthermia is more common in cats.56 Opioids most commonly associated with hyperthermia in the cat include hydromorphone (at 3 times the dose) and meperidine; buprenorphine is unlikely to be associated with hyperthermia in cats.43,57,58 Experimentally, hydromorphone (1 mg/kg intravenously) increased body temperature (range 104° to 108° F) 1 to 5 hours after anesthesia. Doses at or less than 0.05 mg/kg had no effect, whereas 0.1 mg/kg was associated with increased skin temperature.
Prolonged exposure of opioid receptors to ligands results in adaptation to their presence at multiple cellular levels.30 Cellular tolerance is translated to animal adaptation manifested as tolerance, physical dependence, sensitization, and withdrawal. However, their emergence is not a reason to forgo opioid use. Tolerance refers to a decrease in drug efficacy associated with repeated administration. Acute tolerance (i.e., tachyphylaxis) may reflect short-term receptor desensitization, perhaps owing to phosphorylation of MOP or KOP receptors. Long-term administration of MOP ligands causes superactivation of adenylyl cyclase, the mechanism traditionally assumed to be associated with long-term tolerance. Different ligands appear to initiate acute or chronic tolerance through different mechanisms. Examples include internalization of receptors (MOP or DOP but not MOP), truncation of receptors or other changes. Adaptation by one response may mitigate adaptation by downstream responses; indeed, this sequela (i.e., failure to initiate downstream adaptations) has been proposed as a mechanism of failed desensitization that characterizes some drugs.27 Accordingly, the ability of different ligands to initiate tolerance differs. Changes in nitric oxide or neurotransmitters or their pathways have been implicated as contributors to the development of tolerance.29 Tolerance will most likely develop for analgesia, euphoria, sedation, respiratory depression, nausea or vomiting, and suppression of cough.
KEY POINT 28-12
Opioid therapy may be associated with both tolerance and physical dependence in animals, leading to controlled status for many of the drugs.
Physical dependence occurs when continued administration is necessary to prevent clinical signs characteristic of withdrawal. Physical dependence on opioids occurs as exogenous opioids replace endogenous opiates. Opioids affect numerous physiologic systems that become imbalanced before drug administration. A new balance or equilibrium is established in the presence of the drug,27 and abrupt discontinuation of the drug requires rapid readjustment to a new equilibrium, predisposing the patient to withdrawal. Care must be taken to discriminate between dependence, which is associated with physical withdrawal, and addiction.
Addiction to opioids may reflect attenuation of the inhibition of dopamine release in the nucleus accumbens. This region of the brain has a central role in the reward circuit; dopamine promotes desire. The prevalence of addiction in humans using opoids is high. 51 Abrupt discontinuation of opioids after chronic dosing leads to symptoms of withdrawal, with the severity and duration reflecting the rate of onset and clearance of the opioid. Withdrawal is generally manifested as sign opposite to the original effects caused by the drug and reflects CNS hyperarousal as readaptation to the absence of the drug occurs.27 Symptoms in humans include nausea and diarrhea, coughing, tearing, yawning, sneezing, rhinorrhea, profuse sweating, twitching muscles, abdominal and muscle pain and cramps, piloerection, and dysphoria. Hyperthermia, tachypnea, tachycardia and hypertension also may occur. Pharmacokinetic variables may be helpful in the prediction of withdrawal.27 Morphine dependence has been described in dogs.52 Mixed agonists–antagonists or partial agonists are associated with the least risk of dependence; among the opioids studied, buprenorphine appears to be the least likely to cause dependence in dogs.53 Opioids can be discontinued in drug-dependent human patients without causing signs typical of withdrawal by decreasing the dose 25% to 50% every couple of days. In human patients suffering from withdrawal, clonidine, an α2-adrenergic agonist, minimizes the autonomic symptoms of opioid withdrawal.27
It is the advent of tolerance and physical dependence that has led to the scheduling or classification of most opioids as potential substances of abuse. The actual class designated varies among the states. Pure agonists tend to be scheduled in class II or III; mixed or partial agonists tend to be scheduled in class IV or V, depending on the abuse potential. Both butorphanol and burpenorphine have been rescheduled from class V to class IV.
Cardiovascular System
The effects of opioids in the myocardium have been described. Cardiac depression (particularly bradycardia) is caused by selected opioids; pretreatment with atropine can reduce the incidence. The risk of hypotension is increased with drugs that also cause histamine release.
Those opioids most likely to be associated with histamine release are morphine (but not oxymorphone) and meperidine.40,41 Histamine release caused by buprenorphine has been demonstrated only in vitro; fentanyl, oxymorphone, and butorphanol do not cause histamine release in dogs (as reviewed by Guedes and coworkers).41 Hydromorphone was associated with an apparent type I allergic reaction when used preoperatively in a dog (Chapter 2). However, species differences should be anticipated. Histamine antagonists (H1) partially block morphine-induced hypotension; naloxone completely blocks it in human patients.29,42 Fentanyl and its congeners are less likely to cause hypotension associated with surgery in part because they do not cause histamine release.29 Volume replacement should be instituted before administration of morphine derivatives that causes histamine release in patients that are at risk for hypovolemia. Allergic phenomena in general (including bronchoconstriction) and those manifested in skin may be exacerbated with opioid use if morphinelike drugs are used. In humans opioids cause vasodilation of cutaneous vessels, probably because of histamine release. Pruritis may occur, in part because of histamine but also because of direct effects on neurons.29
Gastrointestinal Tract
In addition to effects at the chemoreceptor trigger zone, opioids have direct effects in the gastrointestinal tract. Hydrochloric acid secretion is generally decreased but occasionally may be increased.29 Tone of smooth muscle in the antral portion of the stomach and upper duodenum will increase, despite decreased gastric peristaltic motility. Passage of endoscopic or other equipment through the stomach can be precluded for up to 12 hours after administration. Drug or food movement through the stomach likewise can be delayed.29 In the small intestine, and particularly in the upper small intestine, resting tone (segmental) is increased, and propulsive activity is markedly decreased.29 Initial stimulation of gastrointestinal motility may result in defecation; subsequent depressed gastric motility may cause constipation with prolonged use.
Opioids also decrease small intestinal secretions, and water absorption increases. In the presence of secretory diarrheas, morphinelike opioids inhibit movement of electrolytes and water into the lumen, probably through inhibition of the stimulatory effects of PGE2, acetylcholine, or vasoactive intestinal peptide.29 Because of increased transit time allowing more complete absorption of luminal contents, opioids may be contraindicated in patients suffering from obstructive gastrointestinal diseases and those associated with bacteria or toxin production. Opioids also decrease biliary and pancreatic secretions.29 Morphinelike opioids, however, cause contraction of the sphincter of Oddi and increased bile duct pressure.29,54,55
Other Effects
Ureteral tone may increase and the voiding reflex of the bladder may be diminished as a result of the increased tone of the external sphincter and increased volume of the urinary bladder. Morphine appears to have antidiuretic effects.29 Inhibitory effects on uterine tone may prolong labor; hyperactivity induced by oxytocin can be normalized with morphine. Neonatal health may be impaired by use of morphinelike drugs during parturition; the neonate appears to be particularly susceptible to respiratory depression induced by opioids, in part because of a poorly developed blood–brain barrier.29
Opioids appear to inhibit cytotoxic activity of natural killer cells. Selected opioid compounds, however, appear to enhance macrophage and killer cell activity, possibly through a novel opioid receptor (see Chapter 31).29
Drug Interactions
The depressant effects of opioids can be exacerbated by other CNS depressants. Several CNS depressants prolong the effects of opioids, including the phenothiazines and tricyclic antidepressants (TCAs). Although some phenothiazines may reduce the amount of opioid necessary for analgesia, others may increase the amount of opioid.29 Among the drug interactions associated with opioids are those at the receptor level (pharmacodynamic) – a interaction often intended for the purposes of reversal of undesireable clinical effect. These interactions are discussed with antagonists.
Drugs
Improvements have altered the state of the interaction between the opioid and the receptor. A useful categorization largely reflects the potency of the opioids at receptors whose ligands control pain: The more powerful drugs tend to be those interacting principally with MOP; as such, they provide the most effective analgesia but the greatest incidence of side effects. Drug companies have attempted to improve the effects of opioids by enhancing the desired pharmacologic effect while minimizing undesirable effects through chemical alteration of the natural opioids. Semisynthetic drugs are made from simple chemical changes of morphine (codeine) or thebaine (e.g., oxycodone, etorphine, and naloxone). Other morphine derivatives include apomorphine (an emetic), hydrocodone, and oxymorphone.29 These changes have allowed for differential effects, perhaps by rendering a full agonist to a partial agonist or antagonist.
Powerful Pure Agonists
Morphine
Morphine is considered the prototypic narcotic. A class II drug, morphine targets primarily MOP and to a lesser degree DOP and KOP receptors. Morphine frequently is studied as morphine sulfate, for which 1 mg will provide 0.76 mg active base.
Morphine causes profound sedation and analgesia for up to 6 hours in the dog. Its effects are reversed with narcotic antagonists. Morphine can cause cardiac depression;59 in addition, hypotension may reflect histamine release or CNS (vasomotor) depression.40,60 Morphine can cause respiratory depression,61 particularly in neonates,62 and can cause acute pulmonary edema resulting from histamine release.60 Therapeutic uses include premedication for surgical anesthesia (reducing the amount of other potentially irreversible CNS depressants) and analgesia. Morphine is also used in cases of acute, fulminating pulmonary edema because of its ability to reduce cardiac preload through hepatic venous constriction and splanchnic pooling.
KEY POINT 28-14
Profound sedation and analgesia associated with morphine is amenable to reversal in the dog.
The minimum effective concentration of morphine associated with analgesia is variable: in humans, it ranges from from 9.1 to 40 ng/mL, but increases to as high as 364 ng/mL with chronic administration (as reviewed by Kunakinch et al).66 A common target for pharmacokinetic studies in dogs is 20 ng/ml. Establishing the minimum target concentration is complicated by the formation of active metabolites. Morphine-6-sulfate (M6S) is metabolite characterized by a thirtyfold greater potency compared with morphine in mouse models, similar to the active morphine metabolite morphine-6 beta-glucuronide (M6G). Its analgesia also is reversed by 3-methoxynaltrexone.63 However, the major metabolite of morphine with clinical significance is morphine-6-β-glucuronide (M6G). It is characterized by pharmacologic activity equal to that of the parent compound in some species. Although more potent than morphine, the metabolite cannot cross the blood–brain barrier as effectively.29 However, with chronic morphine dosing, drug concentrations will accumulate, and the metabolite is likely to contribute more than the parent compound to control of pain in humans. It appears to be associated with fewer gastrointestinal side effects.64
A role for M6G in analgesic efficacy of morphine in dogs is not clearly evident. A study of morphine disposition in dogs66 demonstrated that M6G was not detected (<25 ng/mL) after either intravenous (0.5 mg/kg) or oral (approximately 1.5 mg/kg, extended release) administration of morphine in normal Beagles (n = 6). This is in contrast to Jacqz and coworkers,67 who did detect glucuronidated morphine in dogs. They described a significant (but not exclusive) role of extrahepatic tissues in the formation of the metabolite, which appeared within 5 minutes of administration. Garrett and Jackson68 also detected formation and renal excretion of morphine monoglucuronide in dogs. The differences in metabolite detection between these studies may reflect differences in the actual metabolite itself, and particularly the position of the glucuronide. The impact of the glucuronidated metabolite to analgesic control in dogs may not be clear.
Disposition of Morphine
In contrast to cats, parenteral and epidural administration of morphine is generally well established in dogs and multiple studies are available to describe its disposition; several studies also address its metabolites (Tables 28-3 and 28-4; see also Table 28-1). Numerous studies using variable preparations, routes and doses in dogs are summarized in Table 28-4 and below. 65-75 Multiples studies have focused on more convenient routes of administration (other than IV), including oral (including extended release products), rectal and epidural. Much of the IV data has been generated in studies that focus on alternate routes of delivery or in the context of determining analgesic concentrations.
Table 28-3 Drugs with Potential Epidural Use in Dogs or Cats227
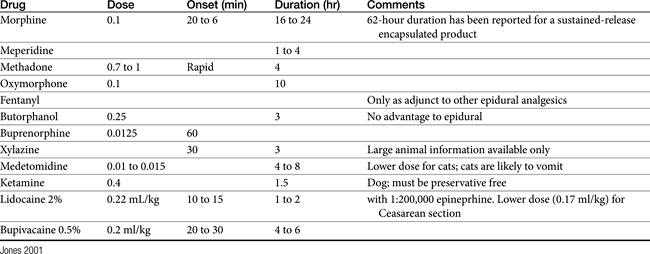
Peak morphine concentration has been determined in several studies after intravenous, IM and SC administration. Morphine clearance appears to be dose dependent in the dog, with an elimination half-life of 83 ± 8 minutes at doses ranging from 7.2 to 7.7 mg/kg versus 37 ± 13 minutes at 0.019-0.07 mg/kg intravenously.68 Bioavailability of intramuscular morphine is high (119%) but variable (57% to 161%), and elimination half-life approximates 90 minutes.
Studies focusing on the nociceptive concentration of morphine revealed an effective 50% concentration of 29.5 ± 5.4 ng/mL in dogs.72 The authors indicated that an intravenous infusion of 0.15 mg/kg/hr or multiple doses of 0.5 mg/kg every 2 hours for 3 doses provided significant nociception. The short half-life of morphine may require administration as a CRI in dogs. Because steady state may require 3 to 5 hours, a loading dose may be indicated. No differences in either analgesia or adverse reactions were detected in dogs undergoing exploratory laporatomy treated with morphine administered either intramuscularly (1 mg/kg at induction and extubation and every 4 hours thereafter) or CRI (0.12 mg/kg/h). Steady-state serum morphine concentration after CRI was 30 ± 2 ng/mL. The power to detect a significant difference was not addressed.
Morphine has been studied in dogs subcutaneously prepared as a solution and as a sustained-release gel (N,O-carboxymethylchitosan polymer) product69 given either subcutaneously or orally70 (it is not clear if the sustained oral preparation is the same as that approved for use in humans). After subcutaneous injection at 1.2 mg/kg, peak concentrations for the solution and sustained product were 488 and 180 ng/mL, respectively, with time to peak concentrations being 10 and 55 minutes, respectively. Variability among animals was large. Elimination half-life was 79 (solution) and 108 (gel) minutes. The duration of analgesia was reported to be between 1 and 2 hours but appeared longer clinically. Emesis should be anticipated.56
Oral administration of morphine in humans generates effective systemic concentrations, although first-pass metabolism requires oral doses that are two to six times parenteral doses. A sustained-release oral morphine preparation (Morphelan) has been approved for use in humans in the United States. The product is designed for both immediate and extended release. Because several days are required for steady-state kinetics to be reached, an initial loading dose of twice the intended subsequent daily dose has been recommended to achieve steady-state concentrations within 2 days of initiation of therapy.71 However, the role of oral administration of morphine in providing effective analgesia in dogs is not clear. The formation of M6G after oral administration of approximately 1.6 mg/kg, morphine sulfate extended release in dogs was previously discussed. In their study, KuKanich and coworkers 66 compared the disposition of intravenous morphine (morphine sulfate 0.5 mg/kg) and orally administered morphine as a commercially available extended-release tablet (1.6 ± 0.1 mg/kg) in Beagles (n=6) (see Table 28-4). One dog vomited after oral administration. Oral absorption of the extended-release tablet was poor (5%) and erratic, achieving a Cmax of morphine of 5.0 ng/ml (variability not provided); this low concentration presumably reflects poor oral absorption rather than high first pass metabolism in that M6G was not detected. This compares to an extrapolated (after distribution) peak concentration of 60 ± 10 ng/ml after IV administration of 0.5 mg/kg. Accordingly, for effective analgesia, a dose of 0.5 mg/kg intravenously every 2 hours was recommended.
More recently, Aragon and coworkers74 reported the disposition of morphine (1 and 2 mg/kg) as either an immediate or oral sustained-release product in dogs (n=14). Although drug concentrations were detectable for 24 hours, concentrations were considered too low and too variable to be of clinical use.
Rectal administration may be a viable alternative route of of administration in dogs. After rectal administration in dogs, morphine is approximately 20% bioavailable,75 achieving peak concentrations of 28 ng/mL when 2 mg/kg is administered as a solution and 20, 21, and 51 ng/mL when administered as a suppository at 1, 2, and 5 mg/kg, respectively. Elimination half-life (minutes) ranged from 65 (solution) to 98 (suppository) (see Table 28-4). Vomiting was common (five of six animals) with a 5-mg/kg rectal suppository.
Morphine was studied in dogs after intramuscular and rectal administration, the latter as either a solution or suppository (see Table 28-4). Median peak concentrations were reported at 5 minutes. The duration that drug concentrations were above a target of 20 ng/mL were 120 minutes for the intramuscular dose, 5 to 30 minutes for the rectal solution, less than 40 minutes for the low (1 mg/kg) and medium (2 mg/kg) suppository dose, and 120 minutes for the high suppository dose (5 mg/kg morphine sulfate).75
Morphine also has been studied in dogs after epidural administration of the solution (0.1 mg/kg)76 and as a sustained-release encapsulated preparation (experimental; 10 and 30 mg in 3 mL saline).77 Both appear to prolong analgesia while avoiding side effects. King and coworkers78 demonstrated that epidural morphine (0.07 mg/kg, containing 0.1% sodium metabisulfate) was not associated with histologic damage to the spinal cord in dogs (n=16). Morphine has been studied in Beagles after repetitive epidural administration (30 mg at 10 mg/mL) as a sustained-release multivesicular liposome preparation (DepoFoam drug delivery system).79 Adverse reactions were limited to moderate, transient behavioral and physiologic changes, consistent with morphine administration, at each injection. A modest effect on cord histopathology was not associated with changes in CSF or neurologic signs.79
Morphine also has been studied intravaginally in humans. When this route was used, pain was effectively reduced in one patient, although close monitoring was necessary.80 Morphine has been studied in both dogs and cats after transdermal administration as a pluronic lethicin organo (PLO) gel. Drug was not quantifiable in either species when administered at 1.6 to 2 mg/kg.65
Adverse Events and Drug Interactions
Adversities of morphine were discussed as a class. Morphine appears to cause more sedation than transdermal fentanyl when used as a postoperative analgesic.76 Guedes and coworkers41 demonstrated that morphine was associated with variable histamine release in conscious dogs at 0.3 and 0.6 mg/kg load followed by 0.17 or 0.34 mg/kg/hr, although the difference from placebo was statistically significant only for the higher dose. Flushing of the skin occurred within 5 minutes of 60% and 100% of dogs receiving the lower and higher dose, respectively. Although no adverse cardiovascular events were detected, adversities might be anticipated in at-risk patients.
Intrathecal administration of morphine at 0.15 mg/kg has been associated with a number of adverse effects in dogs, including bradycardia, hypotension, myoclonus, and urinary retention (as reviewed by Novello and coworkers).81 However, Novello and coworkers81 demonstrated in clinical cases undergoing cervical or thoracolumbar laminectomy that a low intrathecal dose of morphine (0.023 to 0.034 mg/kg) administered at the lumber level 25 to 65 minutes before surgery reduced the dose of fentanyl (1.2 μg/kg/hr) needed to maintain a steady-state heart rate and arterial blood pressure when compared with a placebo group (4.2 μg/kg/hr fentanyl infusion). Ketamine was also administered to all animals (0.5 mg/kg every 60 minutes starting 10 minutes before surgery). Induction and fentanyl protocols differed among animals in the study; it is not clear if both methods were distributed similarly among treatment groups.
Vomiting is relatively consistently associated with morphine administration in dogs. Vomiting occurred after intravenous (0.5 mg/kg) or intramuscular (1 mg/kg) administration in one study (five of six dogs) at a mean morphine concentration of 66 ng/mL, and heart rates were significantly lower.75Drug interactions with morphine appear to be limited. KuKanich and Borum73 found morphine disposition in Greyhounds was not affected by ketoconazole. The simultaneous combination of morphine with the pure antagonists naltrexone did not affect the disposition of either drug in dogs.82
Therapeutic Use
Morphine has been used in the dog and cat effectively as a premedicant or a perioperative analgesic56 using a variety of routes. Lucas and coworkers83 found no differences in the analgesic efficacy of morphine when administered as a CRI (0.12 mg/kg/hr) or intramuscularly at intubation, extubation, and every 4 hours in dogs (n=20) undergoing exploratory laparotomy. Morphine can be administered safely epidurally, although preparations made specifically for epidural administration are preferred. Epidural administration appears to be effective when given either epidurally84,85 or locally (intraarticularly)84 for control of some orthopedic pains.
The postoperative analgesic effects of epidural morphine (0.1 mg/kg) or morphine combined with medetomidine (0.005 mg/kg) have been compared in dogs (n=6/group) undergoing cranial cruciate repair. No differences were detected in control of pain (power of the study not reported), although the combination drug consistently had a lower numeric score. Increasing sample size may have resulted in statistical signification. Likewise, no differences were detected in plasma morphine concentrations between the two treatment groups (peak concentrations of approximately 4 ng/mL).86 The effect of morphine on healing corneal ulcers has been studied in dogs after administration of an ophthalmic preparation. Both MOP and DOP receptors have been identified in the canine cornea. Topical 1% (but not 0.5%) morphine provided good corneal analgesia with no apparent adverse effects.87
Morphine (0.2 mg/kg subcutaneously) was compared with buprenorphine (0.02 mg/kg subcutaneously) and methadone (0.2 mg/kg subcutaneously) in cats (n=8) subjected to mechanical and thermal stimulation. Morphine provided the best analgesia.126
Oxymorphone
Oxymorphone is a semisynthetic drug (class II) that has been used in both dogs and cats, It is characterized with 10 times the potency of morphine. Receptor interaction is similar to that of morphine. Unlike morphine, however, it does not appear to be associated with histamine release in dogs.40 It has been a common component of neuroleptic analgesia. Oxymorphone is effectively antagonized by naloxone and partially antagonized by butorphanol.56,88 Its duration of action is 4 to 6 hours, which may exceed that of its reversal agent, naloxone.89 Like fentanyl, it is associated with marked auditory sensitization, altered thermoregulation, and bradycardia (pretreat with atropine). Decreased heart rate may still occur with epidural administration.90 Oxymorphone is used primarily to induce neuroleptanalgesia or reduce the amount of barbiturate needed for surgical anesthesia. It also, however, has been commonly used to control postoperative pain in small animals, although use has been curtailed by limited availability and increased use of alternative opioids.23 Oxymorphone has been studied with and is approved for use in the dog91 and cat. It remains an excellent analgesic for both young and old patients and for debilitated patients.
Oxymorphone has been studied in normal dogs after intravenous and intramuscular use (Table 28-4).92 Oxymorphone also has been studied in Beagles after administration as a subcutaneously administered slow-release liposome-encapsulated [LE] preparation.93 A dose of 0.5 mg/kg LE preparation yielded concentrations comparable to 0.1 mg/kg of standard morphine achieving a (plasma Cmax of approximately 37 μg/mL using an enzyme-linked immunosorbent assay (ELISA) that measures both parent compound and glucuronide metabolite; however, the elimination phase appeared to be similar (elimination half-life not reported). Sedation and physiologic scores were comparable at these two doses, suggesting no benefit in pharmacologic effects when morphine or its metabolite are delivered as LE preparations compared with standard preparations.
Hydromorphone
Hydromorphone is a pure MOP agonist with a potency (in humans) 5 to 7 times that of morphine suggesting effective concentrations to be 2 to 3 ng/mL. A minimum effective concentration of 4 ng/mL has been suggested (as reviewed by Guedes and coworkers).94 It has been administered for control of pain by a number of routes in humans, including parenteral, transmucosal, and epidural. Hydromorphone (0.1 mg/kg intravenously) disposition and its association with analgesia have been described in the cat.95 Peak plasma concentration was of 94.25 ± 8.40 ng/mL after intravenous administration of 0.1 mg/kg; median concentrations extrapolated from the terminal phase were 25 ng/mL (range 8 to 35). Despite a short elimination half-life (median 100 minutes; range approximately 60 to 140) leading to nondetectable concentrations (less than 1 ng/mL) by 6 hours, analgesia as measured by thermal threshold was maintained for 7.5 hours.95 No behavioral side effects were reported; most cats became nauseated, but none vomited. Mydriasis and “tail tucking” were observed in all cats.
The impact of hydromorphone on nociception has been studied in cats. Hydromorphone at 0.1 mg/kg provided thermal antinociception for 230 minutes (as reviewed by Guedes and coworkers.)94 A dose-dependent effect was evident between 0.025 and 0.1 mg/kg. In contrast to dogs, hyperthermia is associated with hydromorphone treatment in cats. Analgesic effects, based on thermal thresholds, of hydromorphone (0.1 mg/kg intramuscularly), butorphanol (0.4 mg/kg intramuscularly), or the combination have been studied in cats.96 The duration of analgesia was approximately 3 hours for butorphanol, 6 hours for hydromorphone, and 9 hours for the combination. However, the initial intensity of hydromorphone and, to a lesser degree, butorphanol analgesia was substantially reduced when the combination was used. Side effects included dysphoria (butorphanol only) and vomiting (hydromorphone only).97
The pharmacokinetics of hydromorphone have been described in Beagles at 0.1 and 0.5 mg/kg intravenously or subcutaneously (see Table 28-4).99 The short half-life (30 to 60 minutes) led the authors to recommend that treatment should occur at 2-hour dosing intervals for either route of administration or that a CRI be used. Hydromorphone was also studied in Beagles after administration in a controlled-release LE product at 0.5 intravenously or, 1, 2, and 3 mg/kg subcutaneously using a randomized design with a 14- to 28-day washout.100 Serum drug concentrations corrected for dose ranged from 19.41 to 25 ng/mL at 0.19 to 0.27 hour. Concentrations approximated 4 ng/mL for 6 to 72 hours at 2 mg/kg; at 3 mg/kg, concentration exceeded 4 ng/mL for 96 hours.
Guedes and coworkers94 also described the pharmacokinetics (see Table 28-4) and pharmacodyanmics of intravenous hydromorphone (0.1 or 0.2 mg/kg) in dogs (n=5; 2 Beagles) and coupled their studies with pharmacodynamic assessment of analgesia. Antinociceptive effects (based on towel clamp–induced withdrawal) and sedation were compared with those of morphine (0.5 and 1 mg/kg). Hydromorphone clearance surpassed hepatic blood flow reported for dogs, leading the authors to suggest extrahepatic clearance. Both drugs were associated with more antinociception and sedation compared with placebo, but differences were not detected between doses or drugs. The authors suggested that lack of dose dependency may reflect achievement of the maximum effect at the low dose. The lack of differences between drugs was interpreted as confirmation of the potency of hydromorphone compared with morphine. However, the power of the study to detect significant differences was not reported. Analgesic concentrations of 0.5 to 4 ng/mL were suggested on the basis of their work as well as that of others, including studies of humans. Other differences compared with placebo were increased respiratory rate, with return to baseline occurring more rapidly for the hydromorphone compared with morphine group. Body temperature decreased in a dose-dependent manner for both drugs, although changes were significantly different only when compared with placebo. Hydromorphone provided clinically relevant antinociception for at least 2 hours.
Fentanyl
Fentanyl is a synthetic opioid (class II) with a potency 100 times that of morphine and 500 times that of meperidine. Doses are cited in μg/kg rather than mg/kg. Fentanyl is a pure MOP agonist, but it also positively interacts, although to a lesser degree, with DOP and KOP receptors. In contrast to morphine, fentanyl is associated with minimal hemodynamic changes or cardiac suppression. Historically, fentanyl has been recognized as a drug used to induce neuroleptanalgesia (when combined with droperidol). After parenteral administration, however, it causes profound sedation and respiratory depression. Auditory sensitization and altered thermoregulation (leading to panting) may occur. Pretreatment with atropine is indicated when administering the drug parenterally. Because it undergoes redistribution (as with thiobarbiturates), repetitive administration may result in accumulation of the drug in fat and prolonged duration of effect.
Pharmacokinetics and Pharmacodynamics
Fentanyl has been studied in dogs and cats using several different methods of delivery. Using a thermal threshold response, fentanyl concentrations greater than 1 ng/mL have been recommended for therapeutic analgesic effects in cats.101 A dose of 10 μg/kg intravenously provided analgesia (measured by thermal response) for approximately 2 hours in cats.
In normal dogs the elimination half-life approximates 3 hours and the volume of distribution 10 L/kg (see Table 28-4); μg/kg was 1.5 and 2.8 ng/mL, respectively; elimination half-life was approximately 2.5 hours.65 In normal cats the elimination half-life also approximates 3 hours, but volume of distribution appears to be smaller.102. A half-life approximating 2.5 to 3 hours in either species potentially supports the use of a CRI, although a loading dose is indicated. An infusion rate of 0.4 μg/kg/min after a loading dose of 5 μg/kg is recommended in cats. For dogs, fentanyl has been studied after an intravenous loading dose, and an intravenous loading dose (10 μg/kg) followed by CRI (10 μg/kg/hr) for 1 to 3 hours (see Table 28-4).103 Concentrations declined to below 1 ng/mL by 20 minutes after the intravenous loading dose and reached 1 ng/mL again by about 2 hours, with steady-state concentrations at 1, 3, and 4 hours of infusion being 1.0 ± 0.7, 1.11 ± 0.4, and 1.25 ± 0.7 ng/mL, respectively. Variability in concentrations was marked. The elimination half-life of fentanyl after the CRI was discontinued was 151 ± 61 (2.51+1 hours) to 182 ± 68 (3+1 hours).
Fentanyl is among the drugs for which alternative routes of administration have been developed,104 several of which have been studied in dogs and cats. Transdermal delivery of 10 or 30 μg/kg as a PLO gel yielded no detectable plasma fentanyl nor any analgesia in cats in one study.101 However, a second study (unpublished data by author) found a Cmax of 8.8 ± 5 ng/mL at 10 ± 19 hours after transdermal delivery of a PLO gel (5 mg/mL) to cats (n=3) at a mean dose of 0.08 ± 0.03 mg/kg (80 ± 30 μg/kg). Potentially therapeutic concentrations persisted throughout the duration of the study: concentrations of 4 ± 5 ng/mL were measured at 61 hours. In contrast to cats, fentanyl was not detected in dogs after administration of 0.88 mg/kg (880 µg/kg) as a transdermal PLO gel.65
KEY POINT 28-15
Fentanyl is such a potent pure MOP agonist that it is dosed in micrograms rather than milligrams.
Fentanyl (2 μg/kg) has been studied after oral and intranasal administration in cats. Cmax (ng/mL) 5 minutes after oral administration was 0.96 and 2 minutes after intranasal administration, 1.48 ng/mL.101 Although not yet studied in dogs or cats, nn humans, after administration as an aerosol (50 μg/mL solution aerosolized for 5 minutes), fentanyl concentrations were approximately 25% of that achieved after 100 μg intravenously (1.4 ng/mL versus 4.4 ng/mL, respectively), suggesting aerosolization as a possible route of administration.105
Fentanyl has been available for about 10 years as a transdermal drug delivery system intended for control of pain (Figure 28-4). Issues surrounding transdermal drug delivery in animals have been well reviewed. 106 The transdermal patch system has proved safe and effective for delivery of fentanyl to both dogs and cats. The transdermal drug delivery system provides slow, continuous drug delivery that is intended to provide a fairly constant plasma fentanyl concentration. Peak and trough concentrations, which might cause toxicity and therapeutic failure, respectively, are thus avoided. The fentanyl transdermal patch system has been approved for control of cancer pain in humans. The system consists of a patch with an adhesive layer that attaches to the skin. A “release” membrane controls the rate of release of drug from the reservoir. Because the amount of drug that is released is proportional to the size (area) of the patch,107 the dose delivered is the amount of drug (in μg) released per unit time (hour). The system is available in four sizes: 25, 50, 75, and 100 μg/hr. Drug delivery through the stratum corneum can be influenced by a number of factors that vary among patients. These include thickness of the stratum corneum and epidermal layer, skin appendages (e.g., number of sweat glands), body temperature, hydration status, and diseases that might alter the permeability of the stratum corneum.

Figure 28-4 The transdermal fentanyl patch system. The dermis is the site of microcirculation and thus drug absorption.
Species difference mandate that the fentanyl patch system be studied in the target species before use. Peak concentrations occur within 24 hours in the dog and can occur in as few as 3 to 5 hours in the cat. The commercially available fentanyl patch has been studied in both dogs102,108,109 and cats.110 With the 50 μg/hr patch, drug delivery approximated 37 μg/hr (range of 13.7 to 59.8 μg/hr) in dogs, and the patch was found to be an effective means of constant, slow drug delivery.108 A comparison of drug delivery from different patch sizes found the average fentanyl concentration from 24 to 72 hours to be 0.7 ng/mL (50 μg/hr), 1.4 ng/mL (75 μg/hr), and 1.2 ng/mL (100 μg/hr) in dogs weighing approximately 20 kg.109 The elimination (disappearance) half-life ranged from 3.6 hr (50 μg/hr) to a low of 2.5 hr (100 μg/hr). Variability among dogs was marked, making duration of analgesia difficult to predict in individual animals.
Fentanyl delivery has been studied after administration of a 25 μg/hr patch in cats.102 Delivery was approximately 8.5 μg/hr (36% of the predicted) with mean steady-state concentrations of 1.58 ng/mL being achieved at 12 to 100 hr; the maximum concentration of 3.6± 0.8 was achieved at 44±12 hr. Concentrations dropped below 1 ng/mL in four of six cats within 12 hours of patch application and in all cats within 36 hours. The amount of drug delivered through a fentanyl transdermal patch can be reduced by decreasing the surface area contact with the skin. The most accurate approach probably is removal of only a portion of the protective lining cover of the patch, although the proportion of drug delivery will not be exactly proportional to the amount of lining removal. Folding half of a 25 μg/hr patch in cats yielded mean peak concentrations of 1.14 ± 0.9 ng/mL compared with 1.8 ± 0.9 ng/mL (38% reduction) for full patch exposure. 110
Robertson and coworkers101 determined the pharmacodynamic response of fentanyl either intravenously or as a PLO (10 μg/kg) applied to the inner pinna. Plasma fentanyl concentrations greater than 1 ng/mL were required for effective analgesia. The only value reported in the study was Cmax (rather than extrapolated γ intercept) for fentanyl after intravenous administration. However, mean concentrations appeared to be above 1 ng/mL for approximately 60 minutes after intravenous administration. Elimination half-life estimated from the plasma drug concentration versus time curve approximated 15 to 30 minutes. Cats tolerated fentanyl administration well, with most cats exhibiting euphoria. Fentanyl was not detected after PLO administration when administered at 30 μg/kg.
In this same study, Robertson and coworkers101 studied fentanyl after oral or intranasal administration of 2 μg/kg, yielding Cmax values of 0.96 and 1.48 ng/mL at 5 and 2 minutes, respectively. Cats receiving the drug orally or intranasally salivated profusely and were difficult to restrain. Thermal antinociception differed from saline control for approximately 2 hours.
The rate of drug release from the patch and skin can vary with environmental factors that alter delivery through the stratum corneum. Most notable is temperature; fever or a heating pad will increase drug movement. Body temperature was shown to influence fentanyl concentrations when delivered using a fentanyl as a transdermal patch in cats undergoing isoflurane anesthesia. Peak concentrations were 1.8 ± 0.6 ng/mL in normothermic patients compared with 0.6 ± 0.3 ng/mL in hypothermic cats.114 In another study, experimentally induced hypothermia (35°C) was associated with a decline in plasma fentanyl concentrations of 33% (0.6 ng/mL) compared with that at baseline (1.8 ng/mL).117 Patches had been applied 24 hours before anesthesia. In contrast, hyperthermia may increase the risk of fentanyl overdose.115
Therapeutic Use
Only a few clinical trials have compared non-transdermal fentanyl to alternative opioids. Anderson and Day47 compared the effects of morphine (0.12 mg/kg/hr), fentanyl (3 μg/kg/hr) administered by CRI with those of placebo on urine output in healthy (n = 23) and traumatized (n = 18) patients. All animals received a similar rate of fluid infusion. Both fentanyl and morphine decreased urine output in both the trauma and healthy groups (approximately 0.7 to 0.76 mL/kg/hr in all groups compared with 1.2 mL/kg/hr in the untreated group).
Several studies have demonstrated efficacy of the fentanyl transdermal patch system in cats.110,111,113 Fentanyl administered as a 25 μg/hr patch was effective on controlling pain associated with ovariohysterectomy in cats.111 Fentanyl was more effective than butorphanol (0.4 mg/kg intravenously) in cats undergoing ovariohysterectomy or onychectomy.113 A number of studies have documented analgesic efficacy of transdermal fentanyl in dogs. Analgesia did not differ from produced by intramuscular oxymorphone (0.05 mg/kg), but fentanyl was associated with less sedation when used in canine patients undergoing ovariohysterectomy.112 Robinson and coworkers76 compared the efficacy of transdermal fentanyl (100 μg/hr; placed 24 hours before surgery) with that of epidural morphine (0.1 mg/kg) at induction of anesthesia in dogs (n=10) undergoing major orthopedic surgery. Fentanyl provided superior analgesia. The fentanyl patch system was equal to or better than epidural morphine (0.1 mg/kg) in canine patients undergoing orthopedic surgery76.
KEY POINT 28-16
The transdermal fentanyl patch system has proved to be an effective method of analgesia delivery in dogs and cats.
The fentanyl transdermal patch appears to be a reasonable alternative to pain management for dogs and cats, but with several caveats. Patches can cause irritation. Because the time to peak therapeutic concentrations of the drug may be up to 24 hours,109 the need for shorter-term control of pain (e.g., for postoperative pain) should be anticipated. Patches should be applied 12 to 24 hours before the anticipated need for analgesia. Likewise, after the patch is removed, a similar 12- to 24-hour period must elapse before the drug is no longer detectable in the patient. Analgesia is provided for 24 to 72 hours, although variability among animals can be marked, making use of the patch for the individual animal less predictable. Intravenous fentanyl (30 μg/kg has been recommended for dogs) can be given at the time of patch application as a loading dose for patients in need of immediate analgesia. Other opioids also can be given intravenously, intramuscularly, or subcutaneously until the patch is effective. Although a pure opioid (morphine, oxymorphone) is probably preferable, a mixed or partial drug (butorphanol or buprenorphine, respectively) can be given. Because both of the latter drugs have some MOP antagonist effects, however, the MOP effects of fentanyl from the patch will be blocked until the antagonist is eliminated. This may be more problematic with buprenorphine, which has a very tight affinity for MOP receptors.113a
The transdermal delivery of fentanyl appears to be very useful in small animals experiencing pain, including postoperative pain, cancer pain, and pain associated with trauma. The patches can be dosed as follows on the basis of body weight: 5 to 10 kg: 25 μg/hr; 10 to 20 kg: 50 μg/hr; 20 to 30 kg: 75 μg/hr; and more than 30 kg: 100 μg/hr.115 The dose of fentanyl administered by a transdermal patch can be decreased in animals ≤5 kg by folding the adhesive membrane such that only a portion of the patch is exposed to the skin or by cutting away half of the seal (but not the patch itself). However, variability in concentrations will complicate effective use. Exposure of the second half of the patch may not provide equivalent analgesia if applied to a different cat or after all fentanyl has been eliminated. For animals larger than 30 kg, multiple patches can be applied. Patches can be used on an outpatient basis. Heating pads should not be placed at the site of a patch. Patches generally are placed on the dorsum of the neck, which first must be clipped and dried. The patch must come in close, snug contact with the skin. A bandage should be applied to keep the patch in place and prevent the animal or people (especially children) from disturbing the patch.
If the patch is prescribed, veterinarians should apply the patch to, and on completion of analgesia remove it from, the animal rather than allow the client to do so. Clients should be warned that the patches are not approved for use in animals. Patches that are sent home with the patient should be collected when removed from the animal to minimize the risk of drug abuse by pet owners.116 Animals occasionally remove the patch and swallow it; however, the risk of drug toxicity is minimized because it is unlikely that the close contact necessary for drug delivery will occur between the mucosa and the patch. In addition, first-pass metabolism of any fentanyl that is absorbed will limit the amount of drug that reaches systemic circulation. This will not be true if the patch is chewed such that absorption through the buccal mucosa occurs.
Sufentanil
Sufentanil is a thiamylal derivative of fentanyl. It is 5 to 10 times as potent as fentanyl but is apparently associated with fewer cardiac or respiratory side effects. Its half-life is shorter than that of fentanyl, but its onset of action is much more rapid. It has been used as an effective analgesic in dogs.118
Meperidine (Pethidine)
Meperidine (pethidine) (2 to 10 mg/kg intramuscularly every 2 hours [dogs]; 2 to 4 mg/kg intramuscularly every 2 hours [cats]; class II) induces analgesia that is about one fifth that of morphine and lasts approximately 1 to 2 hours. Its sedative properties, however, are useful for preanesthesia. It undergoes marked first-pass metabolism and is eliminated rapidly in dogs. Meperidine reduces the heart rate and systemic arterial pressure in dogs partly through peripheral vasodilation. Bronchoconstriction also occurs in the dog. It is a potent initial stimulant of the gastrointestinal tract. Rapid intravenous injection can cause peripheral vasodilation. Diphenoxylate (the active opioid ingredient in Lomotil) is a derivative of meperidine. In a randomized, blinded, multicenter clinical trial, meperidine (postoperatively, 3.3 mg/kg intramuscularly; n = 57) provided analgesia equal to that of carprofen (preoperatively, 4 mg/kg subcutaneously; n = 59) in cats.119 However, the duration of analgesia was longer for the carprofen group. Millette and coworkers120 studied the effects of meperidine (5 mg/kg intramuscularly) on various nociceptive thresholds in cats (n = 8). Both thermal and mechanical thresholds changed with meperidine.