1. Pain terminology. International Association for the Study of Pain, IASP Council in Kyoto, November 29-30, 2007.
2. Lanser P., Gesell S. Pain management: the fifth vital sign. Healthc Benchmarks. 2001;8:68-70.
3 Stein C., Clark J.D., Oh U., Vasko M.R., et al. Peripheral mechanisms of pain and analgesia. Brain Res Rev. 2009;60(1):90-113.
4. Dubner R. The neurobiology of persistent pain and its clinical implications. Suppl Clin Neurophysiol. 2004;57:3-7.
5. Heinricher M.M., Tavares I., Leith J.L., et al. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev. 2009;60(1):214-225.
6. Hulsebosch C.E., Hains B.C., Crown E.D., et al. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60(1):202-213.
7. Christianson J.A., Bielefeldt K., Altier C., et al. Development, plasticity and modulation of visceral afferents. Brain Res Rev. 2009;60(1):171-186.
8. Hwang S.W., Oh U. Current concepts of nociception: nociceptive molecular sensors in sensory neurons. Curr Opin Anaesthesiol. 2007;20(5):427-434.
9. Joad A.S.K., Burad J., Mehta C. Intravenous lignocaine infusion for neuropathic pain in cancer patients-A preliminary study. Indian J Anesth. 2002;46(5):360-364.
10. Wawrzczak-Bargiela M.J., Osikowicz A., Makuch M., et al. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav Immun. 2009;23(1):75-84.
11. Kiefer W., Dannhardt G. Novel insights and therapetuical applications in the field of inhibitors of COX-2. Curr Med Chem. 2004;11:3147-3161.
12. Jain N.K., Ishikawa T.O., Spigelman I., et al. COX-2 expression and function in the hyperalgesic response to paw inflammation in mice. Prostaglandins Leukot Essent Fatty Acids. 2008;79(6):183-190.
13. Finnoff J. Differentiation and treatment of phantom sensation, phantom pain, and residual-limb pain. J Am Podiatr Med Assoc. 2001;91(1):23-33.
14. Ong C.K., Lirk P., Seymour R.A., et al. The efficacy of preemptive analgesia for acute postoperative pain management: a meta-analysis. Anesth Analg. 2005;100(3):757-773.
15. Møiniche S., Kehlet H., Dahl J.B. A qualitative and quantitative systematic review of preemptive analgesia for postoperative pain relief. Anesthesiology. 2002;96:725-741.
16. Hogan Q.H. No preemptive analgesia: is that so bad? Anesthesiology. 2002;96(3):526-527.
17. Hellyer P.W. Editorial: a new look at pain. J Vet Intern Med. 2004;18:461-462.
18. Phillips D.M. JCAHO pain management standards are unveiled. J Am Med Assoc. 2000;284:428-429.
19. Johnson J.A. The veterinarian’s responsibility: assessing and managing acute pain in dogs and cats, Part I. Comp Contin Educ Pract Vet. 1991;13:804-807.
20. Kitchell R.L. Problems in defining pain and peripheral mechanisms of pain. J Am Vet Med Assoc. 1987;191:1195-1199.
21. Sackman J.E. Pain. Part II. Control of pain in animals. Comp Contin Educ Pract Vet. 1991;13:181-192.
22. Paul-Murphy J., Ludders J.W., Roberson S.A., et al. The need for a cross-species approach to the study of pain in animals. J Am Vet Med Assoc. 2004;224:692-696.
23. Hansen B., Hardie E. Prescription and use of analgesics in dogs and cats in a veterinary teaching hospital: 258 cases (1983-1989). J Am Vet Med Assoc. 1993;202(9):1485-1494.
24. Cousins M.F. Management of postoperative pain. Int Anesth Res J. 1986;60:32-39.
25. Anthony C. Acute pain in the intensive care unit. In: Shoemaker W.C., Ayers S.M., Grenvic A., et al, editors. Textbook of critical care. ed 3. Philadelphia: Saunders; 1995:1486-1498.
26. Potthoff A. Pain and analgesia in dogs and cats. Comp Contin Educ Pract Vet. 1989;11:887-896.
27. Gutstein H.B., Akil H. Opioid analgesics. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman and Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, 2006. pp 547–590
28. Wright E.M., Marcella K.L., Woodson J.F. Animal pain: evaluation and control. Lab Anim. 1985:20-36. May/June
29. Reisine T., Pasternak G. Opioid analgesics and antagonists. In Hardman J.G., Limbird L.E., editors: Goodman and Gilman’s the pharmacologic basis of therapeutics, ed 9, New York: McGraw-Hill, 1996. pp 521–555
30. O’Brien C.P. Drug addiction and drug abuse. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman and Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, 2006. pp 607–627
31. Johnson E.E., Gibson H., Nicol B., et al. Characterization of nociceptin/orphanin FQ binding sites in dog brain membranes. Anesth Analg. 2003;97(3):741-747.
32. Lester P.A., Traynor J.R. Comparison of the in vitro efficacy of MOP, delta, KOP and ORL1 receptor agonists and non-selective opioid agonists in dog brain membranes. Brain Res. 2006;16:1073-1074. 290–296
33. Pan Z.Z. MOP-opposing actions of the KOP-opioid receptors. Trends Pharmacol Sci. 1998;19:94-98.
34. Schultz J.E.E., Gross G.J. Opioids and cardioprotection. Pharmacol Ther. 2001;89:123-137.
35. Roberto M., Siggins G.R. Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci USA. 2006;103(25):9715-9720.
36. Dale O., Hjortkj Æ R. Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand. 2002;46:759-770.
37. Kart T., Walther-Larsen S., Svejborg T.F., Feilberg V., Eriksen K., Rasmussen M. Comparison of continuous epidural infusion of fentanyl and bupivacaine with intermittent epidural administration of morphine for postoperative pain management in children. Acta Anaesthesiol Scand. 1997;41(4):461-465.
38. Cooper J.W. Reviewing geriatric concerns with commonly used drugs. Geriatrics. 1989;44:79-86.
39. Hofmeister E.H., Herrington J.L., Mazzaferro E.M. Opioid dysphoria in three dogs. J Vet Emerg Crit Care. 2006;16(1):44-49.
40. Robinson E.P., Faggella A.M., Henry D.P., et al. Comparison of histamine release induced by morphine and oxymorphone administration in dogs. Am J Vet Res. 1988;49(10):1699-1701.
41. Guedes A.G.P., Rude E.P., Rider M.A. Evaluation of histamine release during constant rate infusion of morphine in dogs. Vet Anaesth Analg. 2006;33:28-35.
42. Muldoon S., Otto J., Freas W., et al. The effects of morphine, nalbuphine, and butorphanol on adrenergic function in canine saphenous veins. Anesth Analg. 1983;62(1):21-28.
43. Robertson S.A. Managing pain in feline patients. Vet Clin Small Anim. 2005;35:129-146.
44. Schurig J.E., Florczyk A.P., Rose W.C., et al. Antiemetic activity of butorphanol against cisplatin-induced emesis in ferrets and dogs. Cancer Treat Rep. 1982;66(10):1831-1835.
45. Blancquaert J.P., Lefebvre R.A., Willems J.L. Emetic and antiemetic effects of opioids in the dog. Eur J Pharmacol. 1989;128(3):143-150.
46. Robertson S.A., Taylor P.M., Lascelles B.D., et al. Changes in thermal threshold response in eight cats after administration of buprenorphine, butorphanol and morphine. Vet Rec. 2003;153(15):462-465.
47. Anderson M.K., Day T.K. Effects of morphine and fentanyl constant rate infusion on urine output in healthy and traumatized dogs. Vet Anaesth Analg. 2008;35(6):528-536.
48. Xu Xao-Jun. Colpert F, Wiesenfeld-Hallin Z: Opioid hyperalgesia and tolerance versus 5-HT1A receptor-mediated inverse tolerance. Trends Pharmacol Sci. 2003;24(12):634-639.
49. Allen C.P., Zissen M.H. Mechanical allodynia and thermal hyperalgesia upon acute opioid withdrawal in the neonatal rat. Pain. 2004;110:269-280.
50. Sweitzer S.M., Allen C.P., Zissen M.H., Kendig J.J. Mechanical allodynia and thermal hyperalgesia upon acute opioid withdrawal in the neonatal rat. Pain. 2004;110(1-2):269-280.
51. Martell B.A., O’Connor P.G., Kerns R.D., et al. Systematic review: opioid treatment for chronic back pain: prevalence, efficacy, and association with addiction. Ann Intern Med. 2007;146(2):116-127.
52. Martin W.R., Eades C.G., Thompson J.A., et al. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197(3):517-532.
53. Jacob J.J., Michaud G.M., Tremblay E.C. Mixed agonist-antagonist opiates and physical dependence. Br J Clin Pharmacol. 1979;7(Suppl 3):291S-296S.
54. Roebel L.E., Cavanagh R.L., Buyniski J.P. Comparative gastrointestinal and biliary tract effects of morphine and butorphanol (Stadol). J Med. 1979;10(4):225-238.
55. Vieira Z.E., Zsigmond E.K., Duarte B., et al. Double-blind comparison of butorphanol and nalbuphine on the common bile duct by ultrasonography in man. Int J Clin Pharmacol Ther Toxicol. 1993;31(11):564-567.
56. Branson K.R., Gross M.E., Booth N.H. Opioid agonists and antagonists. In: Adams H.R., editor. Veterinary pharmacology and therapeutics. ed 7. Ames, Iowa: Iowa State University Press; 1996:274-310.
57. Robertson S.A., Taylor P.M. Pain management in cats—past, present and future. Part 2. Treatment of pain—clinical pharmacology. J Feline Med Surg. 2004;6(5):321-333.
58. Robertson S.A., Taylor P.M., Sear J.W. Systemic uptake of buprenorphine by cats after oral mucosal administration. Vet Rec. 2003;152(22):675-678.
59. Napier L.D., Stanfill A., Yoshishige D.A., et al. Autonomic control of heart rate in dogs treated chronically with morphine. Am J Physiol. 1998;275(6 Pt 2):H2199-H2210.
60. Hakim T.S., Grunstein M.M., Michel R.P. Opiate action in the pulmonary circulation. Pulm Pharmacol. 1992;5(3):159-165.
61. Cullen L.K., Raffe M.R., Randall D.A., et al. Assessment of the respiratory actions of intramuscular morphine in conscious dogs. Res Vet Sci. 1999;67(2):141-148.
62. Luks A.M., Zwass M.S., Brown R.C., et al. Opioid-induced analgesia in neonatal dogs: pharmacodynamic differences between morphine and fentanyl. J Pharmacol Exp Ther. 1998;284(1):136-141.
63. Zuckerman A., Bolan E., de Paulis T., et al. Pharmacological characterization of morphine-6-sulfate and codeine-6-sulfate. Brain Res. 1999;842(1):1-5.
64. Osborne R., Thompson P., Joel S., Trew D., Patel N., Slevin M. The analgesic activity of morphine-6-glucuronide. Br J Clin Pharmacol. 1992;34(2):130-138.
65. Krotscheck U., Boothe D.M., Boothe H.W. Evaluation of transdermal morphine and fentanyl pluronic lecithin organogel administration in dogs. Vet Ther. 2004;5(3):202-211.
66. KuKanich B., Lascelles B.D.X., Papich M.G. Pharmacokinetics of morphine and plasma concentrations of morphine-6-glucuronide following morphine administration to dogs. J Vet Pharmacol Ther. 2005;28:371-376.
67. Jacqz E., Ward S., Johnson R., et al. Extrahepatic glucuronidation of morphine in the dog. J Pharmacol Exp Ther. 1986;117(1):117-125.
68. Garrett E.R., Jackson A.J. Pharmacokinetics of morphine and its surrogates. III: Morphine and morphine 3-monoglucuronide pharmacokinetics in the dog as a function of dose. J Pharm Sci. 1979;68(6):753-771.
69. Tasker R.A., Ross S.J., Dohoo S.E., et al. Pharmacokinetics of an injectable sustained-release formulation of morphine for use in dogs. J Vet Pharmacol Ther. 1997;20(5):362-367.
70. Dohoo S. Steady-state pharmacokinetics of oral sustained-release morphine sulphate in dogs. J Vet Pharmacol Ther. 1997;20(2):129-133.
71. Eliot L, Cato A, Geiser R, et al: Evaluation of two loading-dose regimens of Morphelan™ in healthy volunteers, Journal of Applied Research in Clinical and Experimental Therapeutics, 2000-2003. Accessed November 24, 2009, at http://www.jarcet.com/articles/Vol2Iss1/Eliot.htm.
72. KuKanich B., Lascelles B.D., Papich M.G. Use of a von Frey device for evaluation of pharmacokinetics and pharmacodynamics of morphine after intravenous administration as an infusion or multiple doses in dogs. Am J Vet Res. 2005;66(11):1968-1974.
73. KuKanich B., Borum S.L. Effects of ketoconazole on the pharmacokinetics and pharmacodynamics of morphine in healthy greyhounds. Am J Vet Res. 2008;69:664-669.
74. Aragon C.L., Read M.R., Gaynor J.S., et al. Pharmacokinetics of an immediate and extended release oral morphine formulation utilizing the spheroidal oral drug absorption system in dogs. J Vet Pharmacol Ther. 2009;32(2):129-136.
75. Barnhart M.D., Hubbell J.A., Muir W.W., et al. Pharmacokinetics, pharmacodynamics, and analgesic effects of morphine after rectal, intramuscular, and intravenous administration in dogs. Am J Vet Res. 2000;61:24-28.
76. Robinson T.M., Kruse-Elliott K.T., Markel M.D. A comparison of transdermal fentanyl versus epidural morphine for analgesia in dogs undergoing major orthopedic surgery. J Am Anim Hosp Assoc. 1999;35(2):95-100.
77. Yaksh T.L., Provencher J.C., Rathbun M.L., et al. Pharmacokinetics and efficacy of epidurally delivered sustained-release encapsulated morphine in dogs. Anesthesiology. 1999;90(5):1402-1412.
78. King F.G., Baxter A.D., Mathieson G. Tissue reaction of morphine applied to the epidural space of dogs. Can Anaesth Soc J. 1984;31(3):268-271.
79. Yaksh T.L., Provencher J.C., Rathbun M.L., et al. Safety assessment of encapsulated morphine delivered epidurally in a sustained-release multivesicular liposome preparation in dogs. Drug Deliv. 2000;7(1):27-36.
80. Ostrop N.J., Lamb J., Reid G. Intravaginal morphine: an alternative route of administration. Pharmacotherapy. 1998;18(4):863-865.
81. Novello L., Corletto F., Rabozzi R., et al. Sparing effect of a low dose of intrathecal morphine on fentanyl requirements during spinal surgery: a preliminary clinical investigation in dogs. Vet Surg. 2008;37:153-160.
82. Langguth P., Khan P.G., Garret E.R. Pharmacokinetics of morphine and its surrogates. XI: Effect of simultaneously administered naltrexone and morphine on the pharmacokinetics and pharmacodynamics in the dog. Biopharm Drug Dispos. 1990;11(5):419-444.
83. Lucas A.N., Firth A.M., Anderson G.A., et al. Comparison of the effects of morphine administered by constant-rate intravenous infusion or intermittent intramuscular injection in dogs. J Am Vet Med Assoc. 2001;218(6):884-891.
84. Day T.K., Pepper W.T., Tobias T.A., et al. Comparison of intra-articular and epidural morphine for analgesia following stifle arthrotomy in dogs. Vet Surg. 1995;24(6):522-530.
85. Pascoe P.J., Dyson D.H. Analgesia after lateral thoracotomy in dogs. Epidural morphine vs. intercostal bupivacaine. Vet Surg. 1993;22(2):141-147.
86. Pacharinsak C., Greene S.A., Keegan R.D., et al. Postoperative analgesia in dogs receiving epidural morphine plus medetomidine. J Vet Pharmacol Therap. 2003;26:71-77.
87. Stiles J., Honda C.N., Krohne S.G., et al. Effect of topical administration of 1% morphine sulfate solution on signs of pain and corneal wound healing in dogs. Am J Vet Res. 2003;64:813-818.
88. Dyson D.H., Doherty T., Anderson G.I., et al. Reversal of oxymorphone sedation by naloxone, nalmefene, and butorphanol. Vet Surg. 1990;19(5):398-403.
89. Copeland V.S., Haskins S.C., Patz J. Naloxone reversal of oxymorphone effects in dogs. Am J Vet Res. 1989;50(11):1854-1858.
90. Torske K.E., Dyson D.H., Conlon P.D. Cardiovascular effects of epidurally administered oxymorphone and an oxymorphone-bupivacaine combination in halothane-anesthetized dogs. Am J Vet Res. 1999;60(2):194-200.
91. Cone E.J., Darwin W.D., Buchwald W.F., et al. Oxymorphone metabolism and urinary excretion in human, rat, guinea pig, rabbit, and dog. Drug Metab Dispos. 1983;11(5):446-450.
92. KuKanich B., Schmidt B.K., Krugner-Higby L.A., et al. Pharmacokinetics and behavioral effects of oxymorphone after intravenous and subcutaneous administration to healthy dogs. J Vet Pharmacol Ther. 2008;31(6):580-583.
93. Smith L.J., Krugner-Higby L., Trepanier L.A. Sedative effects and serum drug concentrations of oxymorphone and metabolites after subcutaneous administration of a liposome-encapsulated formulation in dogs. J Vet Pharmacol Ther. 2004;27:369-372.
94. Guedes A.G.P., Papich M.G., Rude E.P., et al. Pharmacokinetics and physiological effects of intravenous hydromorphone in conscious dogs. J Vet Pharmacol Therap. 2008;31:334-343.
95. Wegner K., Robertson S.A., Kollias-Baker C., et al. Pharmacokinetic and pharmacodynamic evaluation of intravenous hydromorphone in cats. J Vet Pharmacol Ther. 2004;27(5):329-336.
96. Lascelles B.D., Robertson S.A. Antinociceptive effects of hydromorphone, butorphanol, or the combination in cats. J Vet Intern Med. 2004;18(2):190-195.
97. Duncan B., Lascelles X., Robertson S.A. Antinociceptive effects of hydromorphone, butorphanol, or the combination in cats. J Vet Intern Med. 2004;18:190-195.
98. Briggs S.L., Sneed K., Sawyer D.C. Antinociceptive effects of oxymorphone-butorphanol-acepromazine combination in cats. Vet Surg. 1998;27(5):466-472.
99. KuKanich B., Hogan B.K., Krugner-Higby L.A., et al. Pharmacokinetics of hydromorphone hydrochloride in healthy dogs. Vet Anaesth Analg. 2008;35(3):256-264.
100. Smith L.J., KuKanich B., Hogan B.K., et al. Pharmacokinetics of a controlled-release liposome-encapsulated hydromorphone administered to healthy dogs. J Vet Pharmacol Ther. 2008;31(5):415-422.
101. Robertson S.A., Taylor P.M., Sear J.W., et al. Relationship between plasma concentrations and analgesia after intravenous fentanyl and disposition after other routes of administration in cats. J Vet Pharmacol Ther. 2005;28:87-93.
102. Lee D.D., Papich M.G., Hardie E.M. Comparison of pharmacokinetics of fentanyl after intravenous and transdermal administration in cats. Am J Vet Res. 2000;61(6):672-677.
103. Sano T., Nishimura R., Kanazawa H., et al. Pharmacokinetics of fentanyl after single intravenous injection and constant rate infusion in dogs. Vet Anaesth Analg. 2006;33:266-273.
104. Committee on Drugs. Alternative routes of drug administration—advantages and disadvantages (subject review). Pediatrics. 1997;100:143-152.
105. Paut O., Lang P., Massias L. Pharmacokinetics of aerosolized fentanyl in healthy volunteers. Anesthesiology. 2003;99:A490.
106. Riviere J.E., Papich M.G. Potential and problems of developing transdermal patches for veterinary applications. Adv Drug Deliv Rev. 2001;50(3):175-203.
107. Grond S., Radburch L., Lehmann K.A. Clinical pharmcokinetcis of transdermal opioids. Clin Pharmacokinet. 2000;38:50-89.
108. Kyles A.E., Papich M., Hardie E.M. Disposition of transdermally administered fentanyl in dogs. Am J Vet Res. 1996;57(5):715-719.
109. Egger C.M., Duke T., Archer J., et al. Comparison of plasma fentanyl concentrations by using three transdermal fentanyl patch sizes in dogs. Vet Surg. 1998;27(2):159-166.
110. Davidson C., Pettifer G.R., Henry J.D. Plasma fentanyl concentrations and analgesic effects during full or partial exposure of transdermal fentanyl patches in cats. J Am Vet Med Assoc. 2004;224:700-705.
111. Glerum L.E., Egger C.M., Allen S.W., et al. Analgesic effect of the transdermal fentanyl patch during and after feline ovariohysterectomy. Vet Surg. 2001;30(4):351-358.
112. Kyles A.E., Hardie E.M., Hansen B.D., et al. Comparison of transdermal fentanyl and intramuscular oxymorphone on post-operative behaviour after ovariohysterectomy in dogs. Res Vet Sci. 1998;65(3):245-251.
113. Franks J.N., Boothe H.W., Taylor L., et al. Evaluation of transdermal fentanyl patches for analgesia in cats undergoing onychectomy. J Am Vet Med Assoc. 2000;217(7):1013-1020.
114. Hofmeister E.H., Egger C.M. Transdermal fentanyl patches in small animals. J Am Anim Hosp Assoc. 2004;40:468-478.
115. Paddleford R.R. Manual of small animal anesthesia, ed 2. Philadelphia: Saunders; 1999.
116. Barrueto F.Jr., Howland M.A., Hofman, et al. The fentanyl tea bag. Vet Hum Toxicol. 2004;46(1):30-31.
117. Pettifer G.R., Hosgood G. The effect of rectal temperature on perianesthetic serum concentrations of transdermally administered fentanyl in cats anesthetized with isoflurane. Am J Vet Res. 2003;64(12):1557-1561.
118. Carroll G. Personal communication. Texas A&M University; 1992.
119. Balmer T.V., Irvine D., Jones R.S., et al. Comparison of carprofen and pethidine as postoperative analgesics in the cat. J Small Anim Pract. 1998;39(4):158-164.
120. Millette V.M., Steagall P.V., Duke-Novakovski T., et al. Effects of meperidine or saline on thermal, mechanical and electrical nociceptive thresholds in cats. Vet Anaesth Analg. 2008;35(6):543-547.
121. Yeh S.Y., Woods L.A. Excretion of codeine and its metabolites by dogs, rabbits and cats. Arch Int Pharmacodyn Ther. 1971;191(2):231-242.
122. Woods L.A., Meuhlenbeck H.E., Mellett L.B. Plasma levels and excretion of codeine and metabolites in the dog and monkey. J Pharmacol Exp Ther. 1956;117(1):117-125.
123. Davis A.M., Inturrisi C.E. d-Methadone blocks morphine tolerance and N-methyl-D-aspartate-induced hyperalgesia. J Pharmacol Exp Ther. 1999;289(2):1048-1053.
124. KuKanich B., Borum S.L. The disposition and behavioral effects of methadone in Greyhounds. Vet Anaesth Analg. 2008;35(3):242-248.
125. KuKanich B., Lascelles B.D., Aman A.M., et al. The effects of inhibiting cytochrome P450 3A, p-glycoprotein, and gastric acid secretion on the oral bioavailability of methadone in dogs. J Vet Pharmacol Ther. 2005;28(5):461-466.
126. Steagall P.V., Carnicelli P., Taylor P.M., et al. Effects of subcutaneous methadone, morphine, buprenorphine or saline on thermal and pressure thresholds in cats. J Vet Pharmacol Ther. 2006;29(6):531-537.
127. Dohoo S.E., Dohoo I.R. Postoperative use of analgesics in dogs and cats by Canadian veterinarians. Can Vet J. 1996;37(9):546-551.
128. Hubbell J.A., Muir W.W. Evaluation of a survey of the diplomates of the American College of Laboratory Animal Medicine on use of analgesic agents in animals used in biomedical research. J Am Vet Med Assoc. 1996;209(5):918-921.
129. Christie G.J., Strom P.W., Rourke J.E. Butorphanol tartrate: a new antitussive agent for use in dogs. Vet Med Small Anim Clin. 1980;75(10):1559-1562.
130. Houghton K.J., Rech R.H., Sawyer D.C., et al. Dose-response of intravenous butorphanol to increase visceral nociceptive threshold in dogs. Proc Soc Exp Biol Med. 1991;197(3):290-296.
131. Sawyer D.C., Rech R.H., Durham R.A., et al. Dose response to butorphanol administered subcutaneously to increase visceral nociceptive threshold in dogs. Am J Vet Res. 1991;52(11):1826-1830.
132. Pfeffer M., Smyth R.D., Pittman K.A., et al. Pharmacokinetics of subcutaneous and intramuscular butorphanol in dogs. J Pharm Sci. 1980;69(7):801-803.
133. Vaugh D. Pharmacokinetics of albuterol and butorphanol administered intravenously and via buccal patch (Thesis). Texas A&M University; May 2003. p 86
134. Borer L.R., Peel J.E., Seewald W., et al. Effect of carprofen, etodolac, meloxicam, or butorphanol in dogs with induced acute synovitis. Am J Vet Res. 2003;64(11):1429-1437.
135. Mathews K.A., Paley D.M., Foster R.A., et al. A comparison of ketorolac with flunixin, butorphanol, and oxymorphone in controlling postoperative pain in dogs. Can Vet J. 1996;37(9):557-567.
136. Pircio A.W., Buyniski J.P., Roebel L.E. Pharmacological effects of a combination of butorphanol and acetaminophen. Arch Int Pharmacodyn Ther. 1978;35(1):116-123.
137. Troncy E., Cuvelliez S.G., Blais D. Evaluation of analgesia and cardiorespiratory effects of epidurally administered butorphanol in isoflurane-anesthetized dogs. Am J Vet Res. 1996;57(10):1478-1482.
138. Robertson S.A., Lascelles B.D., Taylor P.M., et al. PK-PD modeling of buprenorphine in cats: intravenous and oral transmucosal administration. J Vet Pharmacol Ther. 2005;28(5):453-460.
139. Hosgood G. Pharmacologic features of butorphanol in dogs and cats. J Am Vet Med Assoc. 1990;196:135-136.
140. Sawyer D.C., Rech R.H. Analgesia and behavioral effects of butorphanol, nalbuphine and pentazocine in the cat. J Am Anim Hosp Assoc. 1987;23:439-446.
141. Wells S.M., Glerum L.E., Papich M.G. Pharmacokinetics of butorphanol in cats after intramuscular and buccal transmucosal administration. Am J Vet Res. 2008;69(12):1548-1554.
142. Lascelles B.D., Robertson S.A. Use of thermal threshold response to evaluate the antinociceptive effects of butorphanol in cats. Am J Vet Res. 2004;65(8):1085-1089.
143. Johnson J.A., Robertson S.A., Pypendop B.H. Antinociceptive effects of butorphanol, buprenorphine, or both, administered intramuscularly in cats. Am J Vet Res. 2007;68(7):699-703.
144. Heel R.C., Brogden R.N., Speight T.M., et al. Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1979;17:81-110.
145. Romero D.V., Partilla J.S., Zheng Q.X., et al. Opioid peptide receptor studies. 12. Buprenorphine is a potent and selective MOP/KOP antagonist in the [35S]-GTP-gamma-S functional binding assay. Synapse. 1999;34(2):83-94.
146. Ohtani M., Kotaki H., Sawada Y., et al. Comparative analysis of buprenorphine- and norbuprenorphine-induced analgesic effects based on pharmacokinetic-pharmacodynamic modelling. J Pharamcol Exp Ther. 1995;272(2):505-510.
146a. Boas R.A., Villiger J.W. Clinical actions of fentanyl and buprenorphine. The significance of receptor binding. Br J Anaesth.. 1985;57(2):192-196.
147. Watson A.D., Nicholson A., Church D.B., et al. Use of anti-inflammatory and analgesic drugs in dogs and cats. Aust Vet J. 1996;74(3):203-210.
148. Flecknell P.A. The relief of pain in laboratory animals. Lab Anim. 1984;18(2):147-160.
149. Garrett E.R., Chandran V.R. Pharmacokinetics of morphine and its surrogates. X: analyses and pharmacokinetics of buprenorphine in dogs. Biopharm Drug Dispos. 1990;11(4):311-350.
150. Dahan A., Yassen A., Romberg R., et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anesth. 2006;96(5):627-632.
151. Tracqui A., Tournoud C., Flesch F., et al. Acute poisoning during substitution therapy based on high-dosage buprenorphine, 29 clinical cases—20 fatal cases. Presse Med. 1998;27(12):557-561.
152. Zanette G., Manani G., Giusti F., et al. Paediatric respiratory depression following administration of low dose buprenorphine as postoperative analgesic after fentanyl balanced anaesthesia. Anaesth. 1996;6(5):419-422.
153. Gaulier J.M., Marquet P., Lacassie E., et al. Fatal intoxication following self-administration of a massive dose of buprenorphine. J Forensic Sci. 2000;45(1):226-228.
154. Saarialho-Kere U., Mattila M.J., Paloheimo M., et al. Psychomotor, respiratory and neuroendocrinological effects of buprenorphine and amitriptyline in healthy volunteers. Eur J Clin Pharmacol. 1987;33(2):139-146.
155. Orman J.S., Keating G.M. Buprenorphine/Naloxone: a review of its use in the treatment of opioid dependence. Drugs. 2009;69(5):577-607.
156. Tabuchi Y., Takahashi S. A comparison of fentanyl and buprenorphine in total intravenous anesthesia using propofol during spinal surgery. Masui. 2000;49(7):745-749.
157. Bullingham R.E., McQuay H.J., Dwyer D., et al. Sublingual buprenorphine used postoperatively: clinical observations and preliminary pharmacokinetic analysis. Br J Clin Pharmacol. 1981;12(2):117-122.
158. Taylor P.M., Robertson S.A., Dixon M.J., et al. Morphine, pethidine and buprenorphine disposition in the cat. J Vet Pharmacol Ther. 2001;24(6):391-398.
159. Andaluz A., Moll X., Abellán R., et al. Pharmacokinetics of buprenorphine after intravenous administration of clinical doses to dogs. Vet J. 2009;181(3):299-304.
160. Abbo L.A., Ko J.C., Maxwell L.K., et al. Pharmacokinetics of buprenorphine following intravenous and oral transmucosal administration in dogs. Vet Ther. 2008;9(2):83-93.
161. Krotscheck U., Boothe D.M., Little A.A. Pharmacokinetics of buprenorphine following intravenous administration in dogs. Am J Vet Res. 2008;69(6):722-727.
162. Murrell J.C., Robertson S.A., Taylor P.M., et al. Use of a transdermal matrix patch of buprenorphine in cats: preliminary pharmacokinetic and pharmacodynamic data. Vet Rec. 2007;160(17):578-583.
163. Steagall P.V., Mantovani F.B., Taylor P.M., et al. Dose-related antinociceptive effects of intravenous buprenorphine in cats. Vet J. 2009;182(2):203-209.
164. Bosmans T., Gasthuys F., Duchateau L., et al. A comparison of tepoxalin-buprenorphine combination and buprenorphine forpostoperative analgesia in dogs: a clinical study. J Vet Med A Physiol Pathol Clin Med. 2007;54(7):364-369.
165. Shih A.C., Robertson S., Isaza N., et al. Comparison between analgesic effects of buprenorphine, carprofen, and buprenorphine with carprofen for canine ovariohysterectomy. Vet Anaesth Analg. 2008;35(1):69-79.
166. Slingsby L.S., Taylor P.M., Waterman-Pearson A.E. Effects of two doses of buprenorphine four or six hours apart on nociceptive thresholds, pain and sedation in dogs after castration. Vet Rec. 2006;159(21):705-711.
167. Mathews K.A. Analgesia for the pregnant, lactating and neonatal to pediatric cat and dog. J Vet Emerg Crit Care. 2005;15(4):273-284.
168. Michalis L.L., Hicker P.R., Clark T.A., et al. Ventricular irritability associated with the use of naloxone. Ann Thorac Surg. 1974;18:608-614.
169. Flacke J.W., Flacke W.E., Williams G.D. Acute pulmonary edema following naloxone reversal of high dose morphine anesthesia. Anesthesiology. 1977;47:376-387.
170. Tanaka G.Y. Hypertensive reaction to naloxone. J Am Med Assoc. 1974;228:25-26.
171. Feldberg W., Pyke D.A., Stubbs W.A. Hyperglycaemia, a morphine-like effect produced by naloxone in the cat. J Physiol. 1983;340(1):121-128.
172. Faggella A.M., Aronsohn M.G. Anesthetic techniques for neutering 6- to 14-week-old kittens. J Am Vet Med Assoc. 1993;202(7):1040-1041.
173. Upton R.N., Semple T.J., Macintyre P.E. Pharmacokinetic optimisation of opioid treatment in acute pain therapy. Clin Pharmacokinet. 1997;33:225-244.
174. Tranquilli W.J., Fikes L.L., Raffe M.R. Selecting the right analgesics: indications and dosage requirements. Vet Med. 1989;84:692-697.
175. Kloke M., Rapp M., Bosse B., et al. Toxicity and/or insufficient analgesia by opioid therapy: risk factors and the impact of changing the opioid. A retrospective analysis of 273 patients observed at a single center. Support Care Cancer. 2000;8(6):479-486.
176. KuKanich B., Papich M.G. Pharmacokinetics of tramadol and the metabolite O–desmethyltramadol in dogs. J Vet Pharmacol Ther. 2004;27:239-246.
177. Pypendop B.H., Ilkiw J.E. Pharmacokinetics of tramadol, and its metabolite O-desmethyl-tramadol, in cats. J Vet Pharmacol Ther. 2007;31:52-59.
177a. Sansone RA, Sansone LA: Tramadol: seizures, serotonin syndrome and co-administered antidepressants, Psychiatry 6:17-21, 2009.
178. Steagall P.V., Taylor P.M., Brondani J.T., et al. Antinociceptive effects of tramadol and acepromazine in cats. J Feline Med Surg. 2008;10(1):24-31.
179. Ebert B., Mikkelson S., Thorkildsen C., et al. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rate cortex and spinal cord. Eur J Pharmacol. 1997;333:99-104.
180. Wagner A.E., Walton J.A., Hellyer P.W., et al. Use of low doses of ketamine administered by constant rate infusion as an adjunct for postoperative analgesia in dogs. J Am Vet Med Assoc. 2002;221:72-79.
181. Kaka J.S., Hayton W.L. Ketamine is a non-competitive NMDA receptor antagonist. Biopharm Drug Dispos. 1990;11(5):419-444.
182. Unlügenç H., Gündüz M., Ozalevli M., et al. A comparative study on the analgesic effect of tramadol, tramadol plus magnesium, and tramadol plus ketamine for postoperative pain management after major abdominal surgery. Acta Anaesthesiol Scand. 2002;46(8):1025-1030.
183. Correll G.E., Maleki J., Gracely E.J., et al. Subanesthetic ketamine infusion therapy: a retrospective analysis of a novel therapeutic approach to complex regional pain syndrome. Pain Med. 2004;5(3):263-275.
184. Slingsby L.S., Waterman-Pearson A.E. The post-operative analgesic effects of ketamine after canine ovariohysterectomy-a comparison between pre- or post-operative administration. Res Vet Sci. 2000;69(2):147-152.
185. Anonymous: Committee for Veterinary Medicinal Products, Ketamine: summary report, The European Agency for the Evaluation of Medicine Products Veterinary Medicine Evaluation Unit, December, 1997.
186. Lascelles B.D.X., Gaynor J.S., Smith E.S., et al. Amantadine in a multimodal analgesic regimen for alleviation of refractory osteoarthritis pain in dogs. J Vet Intern Med. 2008;22:53-59.
187. KuKanich B., Papich M.G. Plasma profile and pharmacokinetics of dextromethorphan after intravenous and oral administration in healthy dogs. J Vet Pharmacol Ther. 2004;27:337-341.
188. Dodman N.H., Shuster L., Nesbitt G., et al. The use of dextromethorphan to treat repetitive self-directed scratching, biting, or chewing in dogs with allergic dermatitis. J Vet Pharmacol Ther. 2004;27(2):99-104.
189. Schwarz D.D., Jones W.G., Hedden K.P. Molecular and pharmaoclogical characterization of canine brainstem alphat 2A adrenergic rectpors. J Vet Pharmacol Ther. 1999;22:380-386.
190. Vainio O., Ojala M. Medetomidine, an alpha 2-agonist, alleviates post-thoracotomy pain in dogs. Lab Anim. 1994;28(4):369-375.
191. Kuusela E., Raekallio M., Anttila M., et al. Clinical effects and pharmacokinetics of medetomidine and its enantiomers in dogs. J Vet Pharmacol Ther. 2000;23:15-20.
192. McNamara J.O. Pharmacotherapy of the epilepsies. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis pf therapeutics, ed 11, New York: McGraw-Hill, 2006., pp 501-525
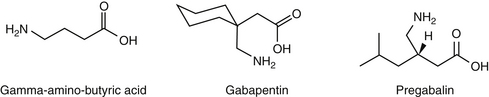
193. Taylor C.P. Mechanisms of analgesia by gabapentin and pregabalin-calciumchannel alpha2-delta [Cavalpha2-delta] ligands. Pain 142(1–2). 2009:13-16.
194. Wiffen P.J., McQuay H.J., Edwards J.E., et al. Gabapentin for acute and chronic pain. Cochrane database of systematic reviews. 2005. Issue 3. Art. No: CD005452. DOI:10.1002/14651858.CD005452
194a. Eckhardt K., Ammon S., Hofmann U., et al. Gabapentin enhances the analgesic effect of morphine in healthy volunteers. Anesth Analg. 2000;91(1):185-191.
194b. Wagner AE, Mich PM, Uhng SR, et al: Clinical evaluation of perioperative administration of gabapentin as an adjunct for postoperative analgesia in dogs undergoing amputation of a forelimb, J Am Vet Med Assoc 236:751-756, 2010.
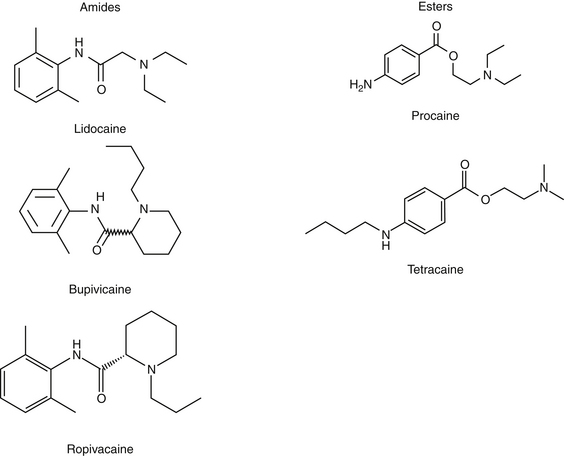
195. Catterall W.A., Mackie K. Local anesthetics. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the pharmacological basis of therapeutics, ed 11, New York: McGraw-Hill, 2006. pp 369-386
196. Wilson D.V., Barnes K.S., Hauptman J.G. Pharmacokinetics of combined intraperitoneal and incisional lidocaine in the dog following overiohysterectomy. J Vet Pharmacol Ther. 2004;27:105-109.
197. Carpenter R.E., Wilson D.V., Evans A.T. Evaluation of intraperitoneal and incisional lidocaine or bupivacaine for analgesia following ovariohysterectomy in the dog. Vet Anaesth Analg. 2004;31:46-52.
198. Ibusuki S., Katsuki H., Takasaki M. The effects of extracellular pH with and without bicarbonate on intracellular procaine concentrations and anesthetic effects in crayfish giant axons. Anesthesiology. 1998;88(6):1549-1557.
199. Gibbon K.J., Cyborski J.M., Guzinski M.V., et al. Evaulation of adverse effects of eMLA (lidocaine/prilocaine) cream for the placement of jugular catheters in healthy cats. J Vet Pharmacol Ther. 2003;26:439-441.
200. Fransson B., Peck K., Smith J., et al. Transdermal absorption of a liposome-encapsulated formulation of lidocaine following topical administration in cats. Am J Vet Res. 2002;63:1309-1312.
201. Weil A.B., Ko J., Inoue T. The use of lidocaine patches. Compend Contin Educ Vet. 2007;29(4):208-210; 214–216.
202. Weiland L., Croubels S., Baert K., et al. Pharmacokinetics of a lidocaine patch 5% in dogs. J Vet Med A Physiol Pathol Clin Med. 2006;53(1):34-39.
203. Ko J., Weil A., Maxwell L., et al. Plasma concentrations of lidocaine in dogs following lidocaine patch application. J Am Anim Hosp Assoc. 2007;43(5):280-283.
204. Ko J.C., Maxwell L.K., Abbo L.A., et al. Pharmacokinetics of lidocaine following the application of 5% lidocaine patches to cats. J Vet Pharmacol Ther. 2008;31(4):359-367.
205. Cassutto B.H., Gfeller R.W. Use of intravenous lidocaine to prevent reperfusion injury and subsequent multiple organ dysfunction syndrome. J Vet Emerg Crit Care. 2003;13(3):137-148.
206. Pypendop B.H., Ilkiw J.E., Robertson S.A. Effects of intravenous administration of lidocaine on the thermal threshold in cats. Am J Vet Res. 2006;67(1):16-20.
207. Muir W.H.III, Weise A.J., March P.A. Effects of morphine, lidocaine, ketamine and morphine-lidocaine-ketamine drug combination on minimum alveolar concentration in dogs anesthetized with isoflurane. Am J Vet Res. 2003;64:1156-1160.
208. Himes R.S.Jr., DiFazio C.A., Burney R.G. Effects of lidocaine on the anesthetic requirements for nitrous oxide and halothane. Anesthesiology. 1977;47(5):437-440.
209. Valverde A., Doherty T.J., Hernández J., et al. Effect of lidocaine on the minimum alveolar concentration of isoflurane in dogs. Vet Anaesth Analg. 2004;31(4):264-271.
210. Smith L.J., Bently E., Shi A., et al. Systemic lidocaine infusion as an analgesic for intraocular surgery in dogs: a pilot study. Vet Anaesth Analg. 2004;31(1):53-63.
211. Pypendop B.H., Ilkiw J.E. Assessment of the hemodynamic effects of lidocaine administered IV in isoflurane-anesthetized cats. Am J Vet Res. 2005;66(4):661-668.
212. Thomasy S.M., Pypendop B.H., Ilkiw J.E., et al. Pharmacokinetics of lidocaine and its active metabolite, monoethylglycinexylidide, after intravenous administration of lidocaine to awake and isoflurane-anesthetized cats. Am J Vet Res. 2005;66(7):1162-1166.
213. Gerard J.L., Edouard A., Berdeaux A., et al. Interaction of intravenous diazepam and bupivacaine in conscious dogs. Reg Anesth. 1989;14(6):298-303.
214. Conzemius M.G., Brockman D.J., King L.G., et al. Analgesia in dogs after intercostal thoracotomy: a clinical trial comparing intravenous buprenorphine and interpleural bupivacaine. Vet Surg. 1994;23(4):291-298.
215. Feldman H., Arthur G., Pitkanen M., et al. Treatment of acute systemic toxicity after the rapid intravenous injection of ropivacaine and bupivacaine in the conscious dog. Anesth Analg. 1991;73:373-384.
216. López-Sanromán J., Cruz J., Santos M., et al. Effect of alkalinization on the local analgesic efficacy of ketamine in the abaxial sesamoid nerve block in horses. J Vet Pharmacol Ther. 2003;26(4):265-269.
217. Terlau H., Olivera B.M. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84(1):41-68.
218. Livett B.G., Sandall D.W., Keays D., et al. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon. 2006;48(7):810-829.
219. Vig M.M. Nicotine poisoning in a dog. Vet Hum Toxicol. 1990;32(6):573-575.
220. Freeman G., Entero H., Brem H. Practical treatment of pain in patients with chronic wounds: pathogenesis-guided management. Am J Surg. 2004;188(Suppl to July):31S-35S.
221. Urch C. The pathophysiology of cancer-induced bone pain: current understanding. Palliat Med. 2004;18:267-274.
222. Portenoy R.K., Farkash A. Practical management of non-malignant pain in the elderly. Geriatrics. 1989;43:29-47.
223. Wall R.T. Use of analgesics in the elderly. Clin Pharm. 1990;6:345-364.
224. Murtaugh R.J., Kaplan P.M. Veterinary emergency and critical care medicine. St Louis: Mosby; 1996.
225. Bullock R., Ward J.D. Management of head trauma. In: Shoemaker W.C., Ayers S.M., Grenvic A., et al, editors. Textbook of critical care. ed 3. Philadelphia: Saunders; 1995:1449-1456.
226. Dewey C.W., Budsberg S.C., Oliver J.E. Principles of head trauma management in dogs and cats: parts I and II. In: Drobatz K., editor. Emergency medicine in small animal practice: the compendium collection. Trenton, NJ: Veterinary Learning Systems; 1997:63-83.
227. Jones R.S. Epidural analgesia in the dog and cat. Vet J. 2001;161:123-131.
228. Bernards C.M. Recent insights into the pharmacokinetics of spinal opioids and the relevance to opioid selection. Curr Opin Anaesthesiol. 2004;17(5):441-447.
229. Sibanda S., Hughes J.M., Pawson P.E., et al. The effects of preoperative extradural bupivacaine and morphine on the stress response in dogs undergoing femoro-tibial joint surgery. Vet Anaesth Analg. 2006;33(4):246-257.
230. Hendrix P.K., Raffe M.R., Robinson E.P., et al. Epidural administration of bupivacaine, morphine, or their combination for postoperative analgesia in dogs. J Am Vet Med Assoc. 1996;209(3):598-607.