Small and Large Intestines
The small intestine and colon account for most of the length of the gastrointestinal tract and are the sites of a wide variety of diseases, many of which affect nutrient and water transport. Perturbation of these processes can cause malabsorption and diarrhea. The intestines are also the principal site where the immune system interfaces with a diverse array of antigens present in food and gut microbes. Indeed, intestinal bacteria outnumber eukaryotic cells in the human body by ten-fold. Thus, it is not surprising that the small intestine and colon frequently are involved by infectious and inflammatory processes. Finally, the colon is the most common site of gastrointestinal neoplasia in Western populations.
Intestinal Obstruction
Obstruction of the gastrointestinal tract may occur at any level, but the small intestine is most often involved because of its relatively narrow lumen. Collectively, hernias, intestinal adhesions, intussusception, and volvulus account for 80% of mechanical obstructions (Fig. 14–18), while tumors and infarction account for most of the remainder. The clinical manifestations of intestinal obstruction include abdominal pain and distention, vomiting, and constipation. Surgical intervention usually is required in cases involving mechanical obstruction or severe infarction.
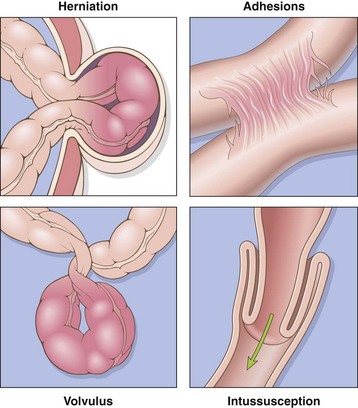
Figure 14–18 Intestinal obstruction. The four major mechanical causes of intestinal obstruction are (1) herniation of a segment in the umbilical or inguinal regions, (2) adhesion between loops of intestine, (3) volvulus, and (4) intussusception.
Hirschsprung Disease
Hirschsprung disease occurs in approximately 1 of 5000 live births and stems from a congenital defect in colonic innervation. It may be isolated or occur in combination with other developmental abnormalities. It is more common in males but tends to be more severe in females. Siblings of patients have an increased risk of Hirschsprung disease.
Patients typically present as neonates with failure to pass meconium in the immediate postnatal period followed by obstructive constipation. The major threats to life are enterocolitis, fluid and electrolyte disturbances, perforation, and peritonitis. Surgical resection of the aganglionic segment with anastomosis of the normal colon to the rectum is effective, although it may take years for patients to attain normal bowel function and continence.
 pathogenesis
pathogenesis
The enteric neuronal plexus develops from neural crest cells that migrate into the bowel wall during embryogenesis. Hirschsprung disease, also known as congenital aganglionic megacolon, results when the normal migration of neural crest cells from cecum to rectum is disrupted. This produces a distal intestinal segment that lacks both the Meissner submucosal plexus and the Auerbach myenteric plexus (“aganglionosis”). Coordinated peristaltic contractions are absent and the subsequent functional obstruction results in dilation proximal to the affected segment. While the mechanisms underlying this defective neural crest cell migration are unknown, heterozygous loss-of-function mutations in the receptor tyrosine kinase RET account for a majority of familial cases and approximately 15% of sporadic cases. However, mutations also occur in other genes, only some of which have been identified, and modifying genes or environmental factors also play a role.
 Morphology
Morphology
Hirschsprung disease always affects the rectum, but the length of the additional involved segments varies. Most cases are limited to the rectum and sigmoid colon, but severe disease can involve the entire colon. The aganglionic region may have a grossly normal or contracted appearance, while the normally innervated proximal colon may undergo progressive dilation as a result of the distal obstruction (Fig. 14–19). Diagnosis of Hirschsprung disease requires demonstrating the absence of ganglion cells in the affected segment.
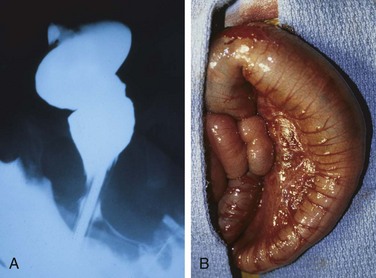
Figure 14–19 Hirschsprung disease. A, Preoperative barium enema study showing constricted rectum (bottom of the image) and dilated sigmoid colon. Ganglion cells were absent in the rectum, but present in the sigmoid colon. B, Corresponding intraoperative appearance of the dilated sigmoid colon.
(Courtesy of Dr. Aliya Husain, The University of Chicago, Chicago, Illinois.)
Abdominal Hernia
Any weakness or defect in the wall of the peritoneal cavity may permit protrusion of a serosa-lined pouch of peritoneum called a hernia sac. Acquired hernias most commonly occur anteriorly, through the inguinal and femoral canals or umbilicus, or at sites of surgical scars. These are of concern because of visceral protrusion (external herniation). This is particularly true of inguinal hernias, which tend to have narrow orifices and large sacs. Small bowel loops are herniated most often, but portions of omentum or large bowel also protrude, and any of these may become entrapped. Pressure at the neck of the pouch may impair venous drainage, leading to stasis and edema. These changes increase the bulk of the herniated loop, leading to permanent entrapment, or incarceration, and over time, arterial and venous compromise, or strangulation, can result in infarction.
 Summary
Summary
Intestinal Obstruction
• Hirschsprung disease is the result of defective neural crest cell migration from cecum to rectum. It gives rise to functional obstruction.
• Abdominal herniation may occur through any weakness or defect in the wall of the peritoneal cavity, including inguinal and femoral canals, umbilicus, and sites of surgical scarring.
Vascular Disorders of Bowel
The greater portion of the gastrointestinal tract is supplied by the celiac, superior mesenteric, and inferior mesenteric arteries. As they approach the intestinal wall, the superior and inferior mesenteric arteries fan out to form the mesenteric arcades. Interconnections between arcades, as well as collateral supplies from the proximal celiac and distal pudendal and iliac circulations, make it possible for the small intestine and colon to tolerate slowly progressive loss of the blood supply from one artery. By contrast, acute compromise of any major vessel can lead to infarction of several meters of intestine.
Ischemic Bowel Disease
Ischemic damage to the bowel wall can range from mucosal infarction, extending no deeper than the muscularis mucosa; to mural infarction of mucosa and submucosa; to transmural infarction involving all three layers of the wall. While mucosal or mural infarctions often are secondary to acute or chronic hypoperfusion, transmural infarction is generally caused by acute vascular obstruction. Important causes of acute arterial obstruction include severe atherosclerosis (which is often prominent at the origin of mesenteric vessels), aortic aneurysm, hypercoagulable states, oral contraceptive use, and embolization of cardiac vegetations or aortic atheromas. Intestinal hypoperfusion can also be associated with cardiac failure, shock, dehydration, or vasoconstrictive drugs. Systemic vasculitides, such as polyarteritis nodosum, Henoch-Schönlein purpura, or Wegener granulomatosis, also may damage intestinal arteries. Mesenteric venous thrombosis can also lead to ischemic disease, but is uncommon. Other causes include invasive neoplasms, cirrhosis, portal hypertension, trauma, or abdominal masses that compress the portal drainage.
Pathogenesis
Intestinal responses to ischemia occur in two phases. The initial hypoxic injury occurs at the onset of vascular compromise and, although some damage occurs, intestinal epithelial cells are relatively resistant to transient hypoxia. The second phase, reperfusion injury, is initiated by restoration of the blood supply and associated with the greatest damage. In severe cases multiorgan failure may occur. While the underlying mechanisms of reperfusion injury are incompletely understood, they involve free radical production, neutrophil infiltration, and release of inflammatory mediators, such as complement proteins and cytokines (Chapter 10). The severity of vascular compromise, time frame during which it develops, and vessels affected are the major variables that determine severity of ischemic bowel disease. Two aspects of intestinal vascular anatomy also contribute to the distribution of ischemic damage:
• Intestinal segments at the end of their respective arterial supplies are particularly susceptible to ischemia. These watershed zones include the splenic flexure, where the superior and inferior mesenteric arterial circulations terminate, and, to a lesser extent, the sigmoid colon and rectum where inferior mesenteric, pudendal, and iliac arterial circulations end. Generalized hypotension or hypoxemia can therefore cause localized injury, and ischemic disease should be considered in the differential diagnosis for focal colitis of the splenic flexure or rectosigmoid colon.
• Intestinal capillaries run alongside the glands, from crypt to surface, before making a hairpin turn at the surface to empty into the postcapillary venules. This configuration allows oxygenated blood to supply crypts but leaves the surface epithelium vulnerable to ischemic injury. This anatomy protects the crypts, which contain the epithelial stem cells that are necessary to repopulate the surface. Thus, surface epithelial atrophy, or even necrosis with consequent sloughing, with normal or hyperproliferative crypts constitutes a morphologic signature of ischemic intestinal disease.
Morphology
Despite the increased susceptibility of watershed zones, mucosal and mural infarction may involve any level of the gut from stomach to anus. Disease frequently is segmental and patchy in distribution, and the mucosa is hemorrhagic and often ulcerated. The bowel wall is thickened by edema that may involve the mucosa or extend into the submucosa and muscularis propria. With severe disease, pathologic changes include extensive mucosal and submucosal hemorrhage and necrosis, but serosal hemorrhage and serositis generally are absent. Damage is more pronounced in acute arterial thrombosis and transmural infarction. Blood-tinged mucus or blood accumulates within the lumen. Coagulative necrosis of the muscularis propria occurs within 1 to 4 days and may be associated with purulent serositis and perforation.
In mesenteric venous thrombosis, arterial blood continues to flow for a time, resulting in a less abrupt transition from affected to normal bowel. However, propagation of the thrombus may lead to secondary involvement of the splanchnic bed. The ultimate result is similar to that produced by acute arterial obstruction, because impaired venous drainage eventually prevents entry of oxygenated arterial blood.
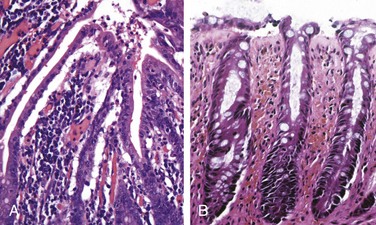
Microscopic examination of ischemic intestine demonstrates atrophy or sloughing of surface epithelium (Fig. 14–20, A). By contrast, crypts may be hyperproliferative. Inflammatory infiltrates initially are absent in acute ischemia, but neutrophils are recruited within hours of reperfusion. Chronic ischemia is accompanied by fibrous scarring of the lamina propria (Fig. 14–20, B) and, uncommonly, stricture formation. In acute phases of ischemic damage, bacterial superinfection and enterotoxin release may induce pseudomembrane formation that can resemble Clostridium difficile–associated pseudomembranous colitis (discussed later).
Clinical Features
Ischemic bowel disease tends to occur in older persons with coexisting cardiac or vascular disease. Acute transmural infarction typically manifests with sudden, severe abdominal pain and tenderness, sometimes accompanied by nausea, vomiting, bloody diarrhea, or grossly melanotic stool. This presentation may progress to shock and vascular collapse within hours as a result of blood loss. Peristaltic sounds diminish or disappear, and muscular spasm creates boardlike rigidity of the abdominal wall. Because these physical signs overlap with those of other abdominal emergencies, including acute appendicitis, perforated ulcer, and acute cholecystitis, the diagnosis of intestinal infarction may be delayed or missed, with disastrous consequences. As the mucosal barrier breaks down, bacteria enter the circulation and sepsis can develop; the mortality rate may exceed 50%.
The overall progression of ischemic enteritis depends on the underlying cause and severity of injury:
• Mucosal and mural infarctions by themselves may not be fatal. However, these may progress to more extensive, transmural infarction if the vascular supply is not restored by correction of the insult or, in chronic disease, by development of adequate collateral supplies.
• Chronic ischemia may masquerade as inflammatory bowel disease, with episodes of bloody diarrhea interspersed with periods of healing.
• CMV infection causes ischemic gastrointestinal disease as a consequence of the viral tropism for and infection of endothelial cells. CMV infection can be a complication of immunosuppressive therapy (Chapter 8).
• Radiation enterocolitis occurs when the gastrointestinal tract is irradiated. In addition to epithelial damage, radiation-induced vascular injury may be significant and produce changes that are similar to ischemic disease. In addition to clinical history, the presence of bizarre “radiation fibroblasts” within the stroma may provide an important clue to the etiology. Acute radiation enteritis manifests as anorexia, abdominal cramps, and a malabsorptive diarrhea, while chronic radiation enteritis or colitis often is more indolent and may present as an inflammatory colitis.
• Necrotizing enterocolitis is an acute disorder of the small and large intestines that can result in transmural necrosis. It is the most common acquired gastrointestinal emergency of neonates, particularly those who are premature or of low birth weight, and occurs most often when oral feeding is initiated (Chapter 6). Ischemic injury generally is considered to contribute to its pathogenesis.
• Angiodysplasia is characterized by malformed submucosal and mucosal blood vessels. It occurs most often in the cecum or right colon, and usually presents after the sixth decade of life. Although the prevalence of angiodysplasia is less than 1% in the adult population, it accounts for 20% of major episodes of lower intestinal bleeding; intestinal hemorrhage may be chronic and intermittent or acute and massive. The pathogenesis is unknown.
Hemorrhoids
Hemorrhoids affect about 5% of the general population. Simply put, hemorrhoids are dilated anal and perianal collateral vessels that connect the portal and caval venous systems to relieve elevated venous pressure within the hemorrhoid plexus. Thus, although hemorrhoids are both more common and less serious than esophageal varices, the pathogenesis of these lesions is similar. Common factors that predispose to hemorrhoids are constipation and associated straining, which increase intra-abdominal and venous pressures, venous stasis of pregnancy, and portal hypertension.
Morphology
Collateral vessels within the inferior hemorrhoidal plexus are located below the anorectal line and are termed external hemorrhoids, while those that result from dilation of the superior hemorrhoidal plexus within the distal rectum are referred to as internal hemorrhoids. On histologic examination, hemorrhoids consist of thin-walled, dilated, submucosal vessels that protrude beneath the anal or rectal mucosa. In their exposed position, they are subject to trauma and tend to become inflamed, thrombosed, and, in the course of time, recanalized. Superficial ulceration may occur.
Clinical Features
Hemorrhoids often manifest with pain and rectal bleeding, particularly bright red blood seen on toilet tissue. Except in pregnant women, hemorrhoids are rarely encountered in persons younger than 30 years of age. Hemorrhoids also may develop as a result of portal hypertension, where the implications are more ominous. Hemorrhoidal bleeding generally is not a medical emergency; treatment options include sclerotherapy, rubber band ligation, and infrared coagulation. In severe cases, hemorrhoids may be removed surgically by hemorrhoidectomy.
Summary
Vascular Disorders of Bowel
• Intestinal ischemia can occur as a result of either arterial or venous obstruction.
• Ischemic bowel disease resulting from hypoperfusion is most common at the splenic flexure, sigmoid colon, and rectum; these are watershed zones where two arterial circulations terminate.
• Systemic vasculitides and infectious diseases (e.g., CMV infection) can cause vascular disease that is not confined to the gastrointestinal tract.
• Angiodysplasia is a common cause of major lower gastrointestinal bleeding in the elderly.
• Hemorrhoids are collateral vessels that form to allow resolution of venous hypertension.
Diarrheal Disease
Malabsorptive Diarrhea
Diarrhea is a common symptom of many intestinal diseases, including those due to infection, inflammation, ischemia, malabsorption, and nutritional deficiency. This section focuses primarily on malabsorption, which manifests most commonly as chronic diarrhea and is characterized by defective absorption of fats, fat- and water-soluble vitamins, proteins, carbohydrates, electrolytes and minerals, and water. Other disorders associated with secretory and exudative types of diarrhea (e.g., cholera and inflammatory bowel disease, respectively) are addressed in separate sections.
Chronic malabsorption causes weight loss, anorexia, abdominal distention, borborygmi, and muscle wasting. A hallmark of malabsorption is steatorrhea, characterized by excessive fecal fat and bulky, frothy, greasy, yellow or clay-colored stools. The chronic malabsorptive disorders most commonly encountered in the United States are pancreatic insufficiency, celiac disease, and Crohn disease. Intestinal graft-versus-host disease is an important cause of both malabsorption and diarrhea after allogeneic hematopoietic stem cell transplantation. Environmental enteropathy (previously known as tropical sprue) is pervasive in some communities within developing countries.
Diarrhea is defined as an increase in stool mass, frequency, or fluidity, typically to volumes greater than 200 mL per day. In severe cases stool volume can exceed 14 L per day and, without fluid resuscitation, result in death. Painful, bloody, small-volume diarrhea is known as dysentery. Diarrhea can be classified into four major categories:
• Secretory diarrhea is characterized by isotonic stool and persists during fasting.
• Osmotic diarrhea, such as that occurring with lactase deficiency, is due to osmotic forces exerted by unabsorbed luminal solutes. The diarrheal fluid is more than 50 mOsm more concentrated than plasma, and the condition abates with fasting.
• Malabsorptive diarrhea caused by inadequate nutrient absorption is associated with steatorrhea and is relieved by fasting.
• Exudative diarrhea is due to inflammatory disease and characterized by purulent, bloody stools that continue during fasting.
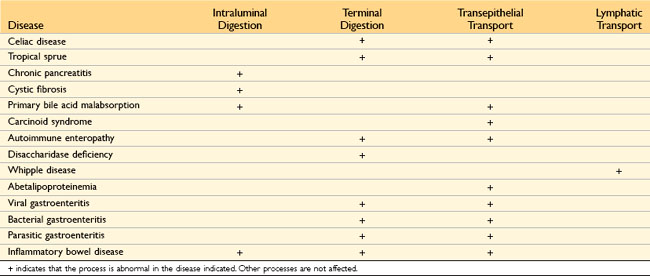
Malabsorption results from disturbance in at least one of the four phases of nutrient absorption: (1) intraluminal digestion, in which proteins, carbohydrates, and fats are broken down into absorbable forms; (2) terminal digestion, which involves the hydrolysis of carbohydrates and peptides by disaccharidases and peptidases, respectively, in the brush border of the small intestinal mucosa; (3) transepithelial transport, in which nutrients, fluid, and electrolytes are transported across and processed within the small intestinal epithelium; and (4) lymphatic transport of absorbed lipids.
In many malabsorptive disorders, a defect in one of these processes predominates, but more than one usually contributes (Table 14–3). As a result, malabsorption syndromes resemble each other more than they differ. Symptoms and signs include diarrhea (from nutrient malabsorption and excessive intestinal secretion), flatus, abdominal pain, and weight loss. Inadequate absorption of vitamins and minerals can result in anemia and mucositis due to pyridoxine, folate, or vitamin B12 deficiency; bleeding due to vitamin K deficiency; osteopenia and tetany due to calcium, magnesium, or vitamin D deficiency; or neuropathy due to vitamin A or B12 deficiency. A variety of endocrine and skin disturbances also may occur.
Cystic Fibrosis
Cystic fibrosis is discussed in greater detail elsewhere (Chapter 6). Only the malabsorption associated with cystic fibrosis is considered here. Owing to the absence of the epithelial cystic fibrosis transmembrane conductance regulator (CFTR), persons with cystic fibrosis have defects in intestinal and pancreatic ductal chloride ion secretion. This abnormality leads to interference with bicarbonate, sodium, and water secretion, ultimately resulting in defective luminal hydration. This failure of hydration can result in meconium ileus, which is present in up to 10% of newborns with cystic fibrosis. In the pancreas, intraductal concretions can begin to form in utero. This leads to obstruction, low-grade chronic autodigestion of the pancreas, and eventual exocrine pancreatic insufficiency in more than 80% of patients. The result is failure of the intraluminal phase of nutrient absorption, which can be effectively treated in most patients with oral enzyme supplementation.
Celiac Disease
Celiac disease, also known as celiac sprue or gluten-sensitive enteropathy, is an immune-mediated enteropathy triggered by the ingestion of gluten-containing cereals, such as wheat, rye, or barley, in genetically predisposed persons. In countries whose populations consist predominantly of white people of European ancestry, celiac disease is a common disorder, with an estimated prevalence of 0.5% to 1%. The primary treatment for celiac disease is a gluten-free diet. Despite the challenges of adhering to such a diet, it does result in symptomatic improvement for most patients.
Pathogenesis
Celiac disease is an intestinal immune reaction to gluten, the major storage protein of wheat and similar grains. Gluten is digested by luminal and brush border enzymes into amino acids and peptides, including a 33–amino acid gliadin peptide that is resistant to degradation by gastric, pancreatic, and small intestinal proteases (Fig. 14–21). Gliadin is deamidated by tissue transglutaminase and is then able to interact with HLA-DQ2 or HLA-DQ8 on antigen-presenting cells and be presented to CD4+ T cells. These T cells produce cytokines that are likely to contribute to the tissue damage and characteristic mucosal histopathology. A characteristic B cell response follows: this includes production of anti-tissue transglutaminase, anti-deamidated gliadin, and, perhaps as a result of cross-reactive epitopes, anti-endomysial antibodies, which are diagnostically useful (see below). However, whether these antibodies contribute to celiac disease pathogenesis or are merely markers remains controversial. In addition to CD4+ cells, there is accumulation of CD8+ cells that are not specific for gliadin. These CD8+ cells may play an ancillary role in causing tissue damage. It is thought that deamidated gliadin peptides induce epithelial cells to produce the cytokine IL-15, which in turn triggers activation and proliferation of CD8+ intraepithelial lymphocytes that can express the MIC-A receptor NKG2D. These lymphocytes become cytotoxic and kill enterocytes that have been induced by various stressors to express surface MIC-A, an HLA class I–like protein that is recognized by NKG2D and, possibly, other epithelial proteins. The damage caused by these immune mechanisms may increase the movement of gliadin peptides across the epithelium, which are deamidated by tissue transglutaminase, thus perpetuating the cycle of disease.
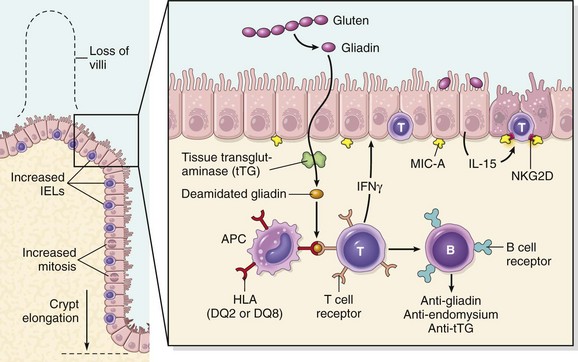
Figure 14–21 Left panel, The morphologic alterations that may be present in celiac disease, including villous atrophy, increased numbers of intraepithelial lymphocytes (IELs), and epithelial proliferation with crypt elongation. Right panel, A model for the pathogenesis of celiac disease. Note that both innate and adaptive immune mechanisms are involved in the tissue responses to gliadin.
While nearly all people eat grain and are exposed to gluten and gliadin, most do not develop celiac disease. Thus, host factors determine whether disease develops. Among these, HLA proteins seem to be critical, since almost all people with celiac disease carry the class II HLA-DQ2 or HLA-DQ8 alleles. However, the HLA locus accounts for less than half of the genetic component of celiac disease. Other genetic contributors are not fully defined. There is also an association of celiac disease with other immune diseases including type 1 diabetes, thyroiditis, and Sjögren syndrome.
Morphology
Biopsy specimens from the second portion of the duodenum or proximal jejunum, which are exposed to the highest concentrations of dietary gluten, generally are diagnostic in celiac disease. The histopathologic picture is characterized by increased numbers of intraepithelial CD8+ T lymphocytes, with intraepithelial lymphocytosis, crypt hyperplasia, and villous atrophy (Fig. 14–22). This loss of mucosal and brush border surface area probably accounts for the malabsorption. In addition, increased rates of epithelial turnover, reflected in increased crypt mitotic activity, may limit the ability of absorptive enterocytes to fully differentiate and contribute to defects in terminal digestion and transepithelial transport. Other features of fully developed celiac disease include increased numbers of plasma cells, mast cells, and eosinophils, especially within the upper part of the lamina propria. With increased serologic screening and early detection of disease-associated antibodies, it is now appreciated that an increase in the number of intraepithelial lymphocytes, particularly within the villus, is a marker of mild forms of celiac disease. Intraepithelial lymphocytosis and villous atrophy are not specific for celiac disease, however, and can be a feature of other disorders, including viral enteritis. The combination of histologic and serologic findings is most specific for diagnosis of celiac disease.
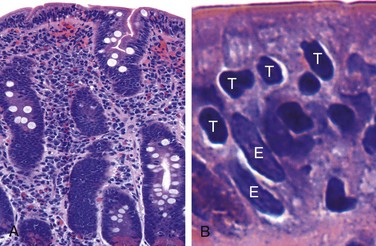
Figure 14–22 Celiac disease. A, Advanced cases of celiac disease show complete loss of villi, or total villous atrophy. Note the dense plasma cell infiltrates in the lamina propria. B, Infiltration of the surface epithelium by T lymphocytes, which can be recognized by their densely stained nuclei (labeled T). Compare with elongated, pale-staining epithelial nuclei (labeled E).
Clinical Features
In adults, celiac disease manifests most commonly between the ages of 30 and 60. However, many cases escape clinical attention for extended periods because of atypical presentations. Some patients have silent celiac disease, defined as positive serology and villous atrophy without symptoms, or latent celiac disease, in which positive serology is not accompanied by villous atrophy. Symptomatic adult celiac disease is often associated with anemia (due to iron deficiency and, less commonly, B12 and folate deficiency), diarrhea, bloating, and fatigue.
Pediatric celiac disease, which affects male and female children equally, may manifest with classic symptoms, typically between the ages of 6 and 24 months (after introduction of gluten to the diet) with irritability, abdominal distention, anorexia, diarrhea, failure to thrive, weight loss, or muscle wasting. Children with nonclassic symptoms tend to present at older ages with complaints of abdominal pain, nausea, vomiting, bloating, or constipation. A characteristic pruritic, blistering skin lesion, dermatitis herpetiformis, also is present in as many as 10% of patients, and the incidence of lymphocytic gastritis and lymphocytic colitis also is increased.
Noninvasive serologic tests generally are performed before biopsy. The most sensitive tests are the presence of IgA antibodies to tissue transglutaminase or IgA or IgG antibodies to deamidated gliadin. Antiendomysial antibodies are highly specific but less sensitive than other antibodies. The absence of HLA-DQ2 or HLA-DQ8 is useful for its high negative predictive value, but the presence of these common alleles is not helpful in confirming the diagnosis.
Patients with celiac disease exhibit a higher than normal rate of malignancy. The most common celiac disease–associated cancer is enteropathy-associated T cell lymphoma, an aggressive tumor of intraepithelial T lymphocytes. Small intestinal adenocarcinoma also is more frequent in persons with celiac disease. Thus, when symptoms such as abdominal pain, diarrhea, and weight loss develop despite a strict gluten-free diet, cancer or refractory sprue, in which the response to a gluten-free diet is lost, must be considered. It is, however, important to recognize that failure to adhere to a gluten-free diet is the most common cause of recurrent symptoms, and that most people with celiac disease do well with dietary restrictions and die of unrelated causes.
Environmental (Tropical) Enteropathy
The term environmental enteropathy refers to a syndrome of stunted growth and impaired intestinal function that is common in developing countries, including many parts of sub-Saharan Africa, such as Gambia; aboriginal populations within northern Australia; and some groups within South America and Asia, such as residents of impoverished communities within Brazil, Guatemala, India, and Pakistan. The impact of environmental enteropathy, which was previously called tropical enteropathy or tropical sprue, cannot be overstated, as it is estimated to affect over 150 million children worldwide. Although malnutrition must contribute to the pathogenesis of this disorder, also referred to as tropical enteropathy, neither supplementary feeding nor vitamin and mineral supplementation are able to fully reverse the syndrome. Repeated bouts of diarrhea suffered within the first 2 or 3 years of life are most closely linked to environmental enteropathy. Many pathogens are endemic in these communities, but no single infectious agent has been linked to these diarrheal episodes. Intestinal biopsy specimens have been examined in only a small number of cases, and reported histologic features are more similar to those of severe celiac disease than to those of infectious enteritis. One hypothesis is that recurrent diarrhea establishes a cycle of mucosal injury, malnutrition, infection, and inflammation. However, this has not been established in part because accepted diagnostic criteria for environmental enteropathy are lacking, as the entity has been defined primarily by epidemiologic assessment of physical and cognitive growth and development.
Lactase (Disaccharidase) Deficiency
The disaccharidases, including lactase, are located in the apical brush border membrane of the villous absorptive epithelial cells. Because the defect is biochemical, biopsies are generally unremarkable. Lactase deficiency is of two types:
• Congenital lactase deficiency is an autosomal recessive disorder caused by a mutation in the gene encoding lactase. The disease is rare and manifests as explosive diarrhea with watery, frothy stools and abdominal distention after milk ingestion. Symptoms abate when exposure to milk and milk products is terminated, thus removing the osmotically active but unabsorbable lactose from the lumen.
• Acquired lactase deficiency is caused by downregulation of lactase gene expression and is particularly common among Native Americans, African Americans, and Chinese populations. Downregulation of lactase occurs in the gut after childhood, perhaps reflecting the fact that, before farming of dairy animals, lactase was unnecessary after children stopped drinking mother’s milk. Onset of acquired lactase deficiency is sometimes associated with enteric viral or bacterial infections.
Abetalipoproteinemia
Abetalipoproteinemia is an autosomal recessive disease characterized by an inability to secrete triglyceride-rich lipoproteins. Although it is rare, it is included here as an example of a transepithelial transport defect that leads to malabsorption. Mutation in the microsomal triglyceride transfer protein renders enterocytes unable to export lipoproteins and free fatty acids. As a result, monoglycerides and triglycerides accumulate within the epithelial cells. Lipid vacuoles in small intestinal epithelial cells are evident by light microscopy and can be highlighted by special stains, such as oil red O, particularly after a fatty meal. Abetalipoproteinemia manifests in infancy, and the clinical picture is dominated by failure to thrive, diarrhea, and steatorrhea. Failure to absorb essential fatty acids leads to deficiencies of fat-soluble vitamins, and lipid defects in plasma membranes often produce acanthocytic red cells (spur cells) in peripheral blood smears.
Irritable Bowel Syndrome
Irritable bowel syndrome (IBS) is characterized by chronic and relapsing abdominal pain, bloating, and changes in bowel habits including diarrhea and constipation. The pathogenesis is poorly defined but involves psychologic stressors, diet, and abnormal gastrointestinal motility. Despite very real symptoms, no gross or microscopic abnormalities are found in most IBS patients. Thus, the diagnosis depends on clinical symptoms. IBS typically manifests between 20 and 40 years of age, and there is a significant female predominance. Variability in diagnostic criteria makes it difficult to establish the incidence, but reported prevalence rates in developed countries typically are between 5% and 10%. In patients with diarrhea, microscopic colitis, celiac disease, giardiasis, lactose intolerance, small bowel bacterial overgrowth, bile salt malabsorption, colon cancer, and inflammatory bowel disease must be excluded (although IBS is common in patients with inflammatory bowel disease). The prognosis for IBS is most closely related to symptom duration, with longer duration correlating with reduced likelihood of improvement.
Microscopic Colitis
Microscopic colitis encompasses two entities, collagenous colitis and lymphocytic colitis. Both of these idiopathic diseases manifest with chronic, nonbloody, watery diarrhea without weight loss. Findings on radiologic and endoscopic studies typically are normal. Collagenous colitis, which occurs primarily in middle-aged and older women, is characterized by the presence of a dense subepithelial collagen layer, increased numbers of intraepithelial lymphocytes, and a mixed inflammatory infiltrate within the lamina propria. Lymphocytic colitis is histologically similar, but the subepithelial collagen layer is of normal thickness and the increase in intraepithelial lymphocytes may be greater, frequently exceeding one T lymphocyte per five colonocytes. Lymphocytic colitis is associated with celiac and autoimmune diseases, including thyroiditis, arthritis, and autoimmune or lymphocytic gastritis.
Graft-Versus-Host Disease
Graft-versus-host disease occurs after allogeneic hematopoietic stem cell transplantation. The small bowel and colon are involved in most cases. Although graft-versus-host disease is secondary to the targeting of antigens on the recipient’s epithelial cells by donor T cells, the lymphocytic infiltrate in the lamina propria is typically sparse. Epithelial apoptosis, particularly of crypt cells, is the most common histologic finding. Intestinal graft-versus-host disease often manifests as a watery diarrhea.
Summary
Malabsorptive Diarrhea
• Diarrhea can be characterized as secretory, osmotic, malabsorptive, or exudative.
• The malabsorption associated with cystic fibrosis is the result of pancreatic insufficiency (i.e., inadequate pancreatic digestive enzymes) and deficient luminal breakdown of nutrients.
• Celiac disease is an immune-mediated enteropathy triggered by the ingestion of gluten-containing grains. The malabsorptive diarrhea in celiac disease is due to loss of brush border surface area and, possibly, deficient enterocyte maturation as a result of immune-mediated epithelial damage.
• Lactase deficiency causes an osmotic diarrhea owing to the inability to break down or absorb lactose.
• Irritable bowel syndrome (IBS) is characterized by chronic, relapsing abdominal pain, bloating, and changes in bowel habits. The pathogenesis is poorly defined.
• The two forms of microscopic colitis, collagenous colitis and lymphocytic colitis, both cause chronic watery diarrhea. The intestines are grossly normal, and the diseases are identified by their characteristic histologic features.
Infectious Enterocolitis
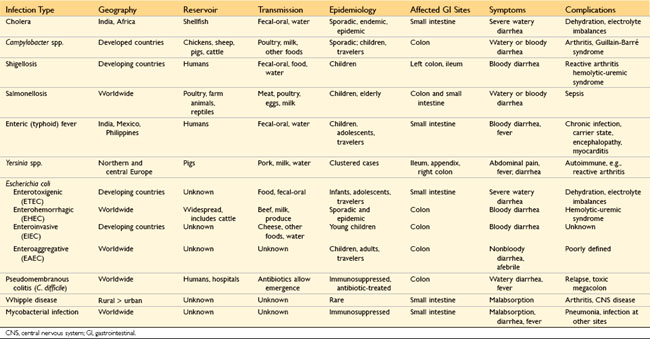
Enterocolitis can manifest with a broad range of signs and symptoms including diarrhea, abdominal pain, urgency, perianal discomfort, incontinence, and hemorrhage. This global problem is responsible for more than 12,000 deaths per day among children in developing countries and half of all deaths before age 5 worldwide. Bacterial infections, such as enterotoxigenic Escherichia coli, frequently are responsible, but the most common pathogens vary with age, nutrition, and host immune status, as well as environmental influences (Table 14–4). For example, epidemics of cholera are common in areas with poor sanitation, as a result of inadequate public health measures, or as a consequence of natural disasters (e.g., the Haiti earthquake of 2010) or war. Pediatric infectious diarrhea, which may result in severe dehydration and metabolic acidosis, commonly is caused by enteric viruses. A summary of the epidemiology and clinical features of selected causes of bacterial enterocolitis is presented in Table 14–4. Representative bacterial, viral, and parasitic enterocolitides are discussed below.
Cholera
Vibrio cholerae organisms are comma-shaped, gram-negative bacteria that cause cholera, a disease that has been endemic in the Ganges Valley of India and Bangladesh for all of recorded history. V. cholerae is transmitted primarily by contaminated drinking water. However, it also can be present in food and causes rare cases of seafood-associated disease. There is a marked seasonal variation in most climates due to rapid growth of Vibrio bacteria at warm temperatures; the only animal reservoirs are shellfish and plankton. Relatively few V. cholerae serotypes are pathogenic, but other species of Vibrio also can cause disease.
Pathogenesis
Despite the severe diarrhea, Vibrio organisms are noninvasive and remain within the intestinal lumen. Flagellar proteins, which are involved in motility and attachment, are necessary for efficient bacterial colonization, and a secreted metalloproteinase that also has hemagglutinin activity is important for bacterial detachment and shedding in the stool. However, it is the preformed enterotoxin, cholera toxin, which causes disease. The toxin, which is composed of five B subunits that direct endocytosis and a single active A subunit, is delivered to the endoplasmic reticulum by retrograde transport. A fragment of the A subunit is transported from the endoplasmic reticulum lumen into the cytosol, where it interacts with cytosolic ADP ribosylation factors to ribosylate and activate the G protein Gsα. This stimulates adenylate cyclase and the resulting increases in intracellular cyclic adenosine monophosphate (cAMP) open the cystic fibrosis transmembrane conductance regulator (CFTR), which releases chloride ions into the lumen. Sodium and bicarbonate absorption are also reduced. Accumulation of these ions creates an osmotic gradient that draws water into the lumen, leading to massive secretory diarrhea. Remarkably, mucosal biopsy specimens show only minimal morphologic alterations.
Clinical Features
Most exposed persons are asymptomatic or suffer only mild diarrhea. Those with severe disease have an abrupt onset of watery diarrhea and vomiting after an incubation period of 1 to 5 days. The rate of diarrheal stool production may reach 1 L per hour, leading to dehydration, hypotension, electrolyte imbalances, muscular cramping, anuria, shock, loss of consciousness, and death. Most deaths occur within the first 24 hours after presentation. Although the mortality rate for severe cholera is 50% to 70% without treatment, fluid replacement can save more than 99% of patients.
Campylobacter Enterocolitis
Campylobacter jejuni is the most common bacterial enteric pathogen in developed countries and is an important cause of traveler’s diarrhea. Most infections are associated with ingestion of improperly cooked chicken, but outbreaks also can be caused by unpasteurized milk or contaminated water.
Pathogenesis
The pathogenesis of Campylobacter infection remains poorly defined, but four major virulence properties contribute: motility, adherence, toxin production, and invasion. Flagella allow Campylobacter to be motile. This facilitates adherence and colonization, which are also necessary for mucosal invasion. Cytotoxins that cause epithelial damage and a cholera toxin–like enterotoxin are also released by some C. jejuni isolates. Dysentery generally is associated with invasion and only occurs with a small minority of Campylobacter strains. Enteric fever occurs when bacteria proliferate within the lamina propria and mesenteric lymph nodes.
Campylobacter infection can result in reactive arthritis, primarily in patients with HLA-B27. Other extraintestinal complications, including erythema nodosum and Guillain-Barré syndrome, a flaccid paralysis caused by autoimmune-induced inflammation of peripheral nerves, are not HLA-linked. Fortunately, Guillain-Barré syndrome develops in 0.1% or less of persons infected with Campylobacter.
Morphology
Campylobacter, Shigella, Salmonella, and many other bacterial infections, including Yersinia and E. coli, all induce a similar histopathology, termed acute self-limited colitis, and these pathogens cannot be reliably distinguished by tissue biopsy. Thus, specific diagnosis is primarily by stool culture. The histology of acute self-limited colitis includes prominent lamina propria and intraepithelial neutrophil infiltrates (Fig. 14–23, A); cryptitis (neutrophil infiltration of the crypts) and crypt abscesses (crypts with accumulations of luminal neutrophils) also may be present. The preservation of crypt architecture in most cases of acute self-limited colitis is helpful in distinguishing these infections from inflammatory bowel disease (Fig. 14–23, B).
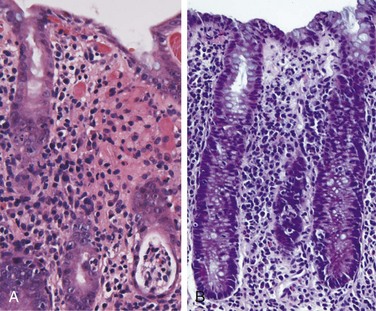
Figure 14–23 Bacterial enterocolitis. A, Campylobacter jejuni infection produces acute, self-limited colitis. Neutrophils can be seen within surface and crypt epithelium and a crypt abscess is present at the lower right. B, Enteroinvasive Escherichia coli infection is similar to other acute, self-limited colitides. Note the maintenance of normal crypt architecture and spacing, despite abundant intraepithelial neutrophils.
Clinical Features
Ingestion of as few as 500 C. jejuni organisms can cause disease after an incubation period of up to 8 days. Watery diarrhea, either acute or with onset after an influenza-like prodrome, is the primary manifestation, and dysentery develops in 15% to 50% of patients. Patients may shed bacteria for 1 month or more after clinical resolution. The disease is self limited and therefore antibiotic therapy generally is not required. Diagnosis is primarily by stool culture since the histologic changes are not specific for Campylobacter colitis.
Shigellosis
Shigella organisms are gram-negative bacilli that are unencapsulated, nonmotile, facultative anaerobes. Although humans are the only known reservoir, Shigella remains one of the most common causes of bloody diarrhea. It is estimated that 165 million cases occur worldwide each year. Shigellae are highly transmissible by the fecal-oral route or through ingestion of contaminated water and food; the infective dose is fewer than 100 organisms and each gram of stool contains as many as 109 organisms during acute phases of the disease.
In the United States and Europe, children in day care centers, migrant workers, travelers to developing countries, and residents of nursing homes are most commonly affected. Most Shigella-associated infections and deaths occur in children younger than 5 years of age; in countries in which Shigella is endemic, it is responsible for approximately 10% of all cases of pediatric diarrheal disease and as many as 75% of diarrheal deaths.
Pathogenesis
Shigella organisms are resistant to the harsh acidic environment of the stomach, which partially explains the very low infective dose. Once in the intestine, organisms are taken up by M (microfold) epithelial cells, which are specialized for sampling and uptake of luminal antigens. After intracellular proliferation, the bacteria escape into the lamina propria. These bacteria then infect small intestinal and colonic epithelial cells through the basolateral membranes, which express bacterial receptors. Alternatively, luminal shigellae can directly modulate epithelial tight junctions to expose basolateral bacterial receptors. The latter is partly mediated by virulence proteins, some of which are directly injected into the host cytoplasm by a type III secretion system. Some Shigella dysenteriae serotypes also release the Shiga toxin Stx, which inhibits eukaryotic protein synthesis and causes host cell death.
Morphology
Shigella infections are most prominent in the left colon, but the ileum may also be involved, perhaps reflecting the abundance of M cells in the epithelium overlying the Peyer’s patches. The histologic appearance in early cases is similar to that in other acute self-limited colitides. In more severe cases, the mucosa is hemorrhagic and ulcerated, and pseudomembranes may be present. Perhaps because of the tropism for M cells, aphthous-appearing ulcers similar to those seen in Crohn disease also may occur. The potential for confusion with chronic inflammatory bowel disease is substantial, particularly if there is distortion of crypt architecture. Confirmation of Shigella infection requires stool culture.
Clinical Features
After an incubation period of 1 to 7 days, Shigella causes self-limited disease characterized by about 6 days of diarrhea, fever, and abdominal pain. The initially watery diarrhea progresses to a dysenteric phase in approximately 50% of patients, and constitutional symptoms can persist for as long as 1 month. A subacute presentation also can develop in a minority of adults. Antibiotic treatment shortens the clinical course and reduces the duration over which organisms are shed in the stool, but antidiarrheal medications are contraindicated because they can prolong symptoms by delaying bacterial clearance.
Complications of Shigella infection are uncommon and include reactive arthritis, a triad of sterile arthritis, urethritis, and conjunctivitis that preferentially affects HLA-B27–positive men between 20 and 40 years of age. Hemolytic uremic syndrome, which typically is associated with enterohemorrhagic Escherichia coli (EHEC), also may occur after infection with shigellae that secrete Shiga toxin.
Escherichia coli
Escherichia coli are gram-negative bacilli that colonize the healthy GI tract; most are nonpathogenic, but a subset cause human disease. The latter are classified according to morphology, mechanism of pathogenesis, and in vitro behavior (Table 14–4). Here we summarize their pathogenic mechanisms:
• Enterotoxigenic E. coli (ETEC) organisms are the principal cause of traveler’s diarrhea, and are spread by the fecal-oral route. They express a heat labile toxin (LT) that is similar to cholera toxin and a heat-stable toxin (ST) that increases intracellular cGMP with effects similar to the cAMP elevations caused by LT.
• Enterohemorrhagic E. coli (EHEC) organisms are categorized as O157:H7 and non-O157:H7 serotypes. Outbreaks of E. coli O157:H7 in developed countries have been associated with the consumption of inadequately cooked ground beef, milk, and vegetables. Both O157:H7 and non-O157:H7 serotypes produce Shiga-like toxins and can cause dysentery. They can also give rise to hemolytic-uremic syndrome (Chapter 13).
• Enteroinvasive E. coli (EIEC) organisms resemble Shigella bacteriologically but do not produce toxins. They invade the gut epithelial cells and produce a bloody diarrhea.
• Enteroaggregative E. coli (EAEC) organisms attach to enterocytes by adherence fimbriae. Although they produce LT and Shiga-like toxins, histologic damage is minimal.
Salmonellosis
Salmonella species, which are members of the Enterobacteriaceae family of gram-negative bacilli, are divided into Salmonella typhi, the causative agent of typhoid fever (discussed in the next section) and nontyphoid Salmonella strains that cause gastroenteritis. Nontyphoid Salmonella infection usually is due to Salmonella enteritidis; more than 1 million cases occur each year in the United States, which result in 2000 deaths; the prevalence is even greater in many other countries. Infection is most common in young children and elderly persons, with peak incidence in summer and fall. Transmission usually is through contaminated food, particularly raw or undercooked meat, poultry, eggs, and milk.
Pathogenesis
Very few viable Salmonella organisms are necessary to cause infection, and the absence of gastric acid, as in persons with atrophic gastritis or those on acid-suppressive therapy, further reduces the required inoculum. Salmonellae possess virulence genes that encode a type III secretion system capable of transferring bacterial proteins into M cells and enterocytes. The transferred proteins activate host cell Rho GTPases, thereby triggering actin rearrangement and bacterial uptake into phagosomes where the bacteria can grow. Salmonellae also secrete a molecule that induces epithelial release of a chemoattractant eicosanoid that draws neutrophils into the lumen and potentiates mucosal damage. Stool cultures are essential for diagnosis.
Typhoid Fever
Typhoid fever, also referred to as enteric fever, is caused by Salmonella typhi and Salmonella paratyphi. It affects up to 30 million individuals worldwide each year. Infection by S. typhi is more common in endemic areas, where children and adolescents are most often affected. By contrast, S. paratyphi predominates in travelers and those living in developed countries. Humans are the sole reservoir for S. typhi and S. paratyphi and transmission occurs from person to person or via contaminated food or water. Gallbladder colonization may be associated with gallstones and a chronic carrier state. Acute infection is associated with anorexia, abdominal pain, bloating, nausea, vomiting, and bloody diarrhea followed by a short asymptomatic phase that gives way to bacteremia and fever with flu-like symptoms. It is during this phase that detection of organisms by blood culture may prompt antibiotic treatment and prevent further disease progression. Without such treatment, the febrile phase is followed by up to 2 weeks of sustained high fevers with abdominal tenderness that may mimic appendicitis. Rose spots, small erythematous maculopapular lesions, are seen on the chest and abdomen. Systemic dissemination may cause extraintestinal complications including encephalopathy, meningitis, seizures, endocarditis, myocarditis, pneumonia, and cholecystitis. Patients with sickle cell disease are particularly susceptible to Salmonella osteomyelitis.
Like S. enteritidis, S. typhi and S. paratyphi are taken up by M cells and then engulfed by mononuclear cells in the underlying lymphoid tissue. Thus, infection causes Peyer’s patches in the terminal ileum to enlarge into plateau-like elevations up to 8 cm in diameter. Mucosal shedding creates oval ulcers oriented along the long axis of the ileum. However, unlike S. enteritidis, S. typhi and S. paratyphi can disseminate via lymphatic and blood vessels. This causes reactive hyperplasia of draining lymph nodes, in which bacteria-containing phagocytes accumulate. In addition, the spleen is enlarged and soft with pale red pulp, obliterated follicular markings, and prominent phagocyte hyperplasia. Randomly scattered small foci of parenchymal necrosis with macrophage aggregates, termed typhoid nodules, are also present in the liver, bone marrow, and lymph nodes.
Pseudomembranous Colitis
Pseudomembranous colitis, generally caused by Clostridium difficile, is also known as antibiotic-associated colitis or antibiotic-associated diarrhea. The latter terms apply to diarrhea developing during or after a course of antibiotic therapy and may be due to C. difficile as well as Salmonella, C. perfringens type A, or S. aureus. However, the latter two organisms produce enterotoxins and are common agents of food poisoning. They do not cause pseudomembranes. Disruption of the normal colonic microbiota by antibiotics allows C. difficile overgrowth. Toxins released by C. difficile cause the ribosylation of small GTPases, such as Rho, and lead to disruption of the epithelial cytoskeleton, tight junction barrier loss, cytokine release, and apoptosis.
Morphology
Fully developed C. difficile–associated colitis is accompanied by formation of pseudomembranes (Fig. 14–24, A), made up of an adherent layer of inflammatory cells and debris at sites of colonic mucosal injury. The surface epithelium is denuded, and the superficial lamina propria contains a dense infiltrate of neutrophils and occasional fibrin thrombi within capillaries. Damaged crypts are distended by a mucopurulent exudate that “erupts” to the surface in a fashion reminiscent of a volcano (Fig. 14–24, B).
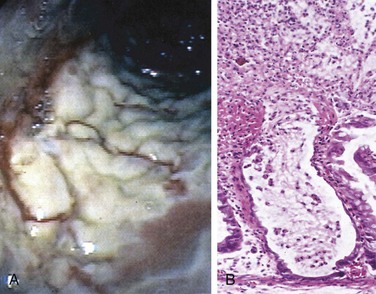
Clinical Features
In addition to antibiotic exposure, risk factors for C. difficile–associated colitis include advanced age, hospitalization, and immunosuppression. The organism is particularly prevalent in hospitals; as many as 20% of hospitalized adults are colonized with C. difficile (a rate 10 times higher than in the general population), but most colonized patients are free of disease. Persons with C. difficile–associated colitis present with fever, leukocytosis, abdominal pain, cramps, hypoalbuminemia, watery diarrhea, and dehydration. Fecal leukocytes and occult blood may be present, but grossly bloody diarrhea is rare. Diagnosis of C. difficile–associated colitis usually is accomplished by detection of C. difficile toxin, rather than culture, and is supported by the characteristic histopathologic findings. Regimens of metronidazole or vancomycin are generally effective treatments, but antibiotic-resistant and hypervirulent C. difficile strains are increasingly common, and the infection may recur in at-risk patients.
Norovirus
Norovirus, previously known as Norwalk-like virus, is a common agent of nonbacterial infectious gastroenteritis. Norovirus causes approximately half of all gastroenteritis outbreaks worldwide and is a common cause of sporadic gastroenteritis in developed countries. Local outbreaks usually are related to contaminated food or water, but person-to-person transmission underlies most sporadic cases. Infections spread easily within schools, hospitals, and nursing homes and, most recently, on cruise ships. After a short incubation period, affected persons develop nausea, vomiting, watery diarrhea, and abdominal pain. Biopsy morphology is nonspecific. The disease is self-limited.
Rotavirus
The encapsulated rotavirus infects 140 million people and causes 1 million deaths each year, making rotavirus the most common cause of severe childhood diarrhea and diarrhea-related deaths worldwide. Children between 6 and 24 months of age are most vulnerable. Protection in the first 6 months of life is probably due to the presence of antibodies to rotavirus in breast milk, while protection beyond 2 years is due to immunity that develops after the first infection. Outbreaks in hospitals and day care centers are common, and infection spreads easily; the estimated minimal infective inoculum is only 10 viral particles. Rotavirus selectively infects and destroys mature (absorptive) enterocytes in the small intestine, and the villus surface is repopulated by immature secretory cells. This change in functional capacity results in loss of absorptive function and net secretion of water and electrolytes that is compounded by an osmotic diarrhea from incompletely absorbed nutrients. Like norovirus, rotavirus produces clinically apparent infection after a short incubation period, manifested by vomiting and watery diarrhea for several days. Vaccines are now available, and their use is beginning to change the epidemiology of rotavirus infection. For unknown reasons, oral rotavirus vaccines have been less effective in developing countries where they are most needed.
Parasitic Disease
Although viruses and bacteria are the predominant enteric pathogens in the United States, parasitic disease and protozoal infections affect over half of the world’s population on a chronic or recurrent basis. The small intestine can harbor as many as 20 species of parasites, including nematodes, such as the roundworms Ascaris and Strongyloides; hookworms and pinworms; cestodes, including flatworms and tapeworms; trematodes, or flukes; and protozoa.
• Ascaris lumbricoides. This nematode infects more than 1 billion people worldwide as a result of human fecal-oral contamination. Ingested eggs hatch in the intestine and larvae penetrate the intestinal mucosa. From here the larvae migrate via the splanchnic circulation to the liver, creating hepatic abscesses, and then through the systemic circulation to the lung, where they can cause Ascaris pneumonitis. In the latter case, larvae migrate up the trachea, are swallowed, and arrive again in the intestine to mature into adult worms.
• Strongyloides. The larvae of Strongyloides live in fecally contaminated ground soil and can penetrate unbroken skin. They migrate through the lungs to the trachea from where they are swallowed and then mature into adult worms in the intestines. Unlike other intestinal worms, which require an ovum or larval stage outside the human, the eggs of Strongyloides can hatch within the intestine and release larvae that penetrate the mucosa, creating a vicious cycle referred to as autoinfection. Hence, Strongyloides infection can persist for life, and immunosuppressed individuals can develop overwhelming infections.
• Necator americanus and Ancylostoma duodenale. These hookworms infect 1 billion people worldwide and cause significant morbidity. Infection is initiated by larval penetration through the skin. After further development in the lungs, the larvae migrate up the trachea and are swallowed. Once in the duodenum, the larvae mature and the adult worms attach to the mucosa, suck blood, and reproduce. Hookworms are the leading cause of iron deficiency anemia in the developing world.
• Giardia lamblia. This flagellated protozoan, also referred to as Giardia duodenalis or Giardia intestinalis, is responsible for the most common pathogenic parasitic infection in humans and is spread by fecally contaminated water or food. Infection may occur after ingestion of as few as 10 cysts. Because cysts are resistant to chlorine, Giardia organisms are endemic in unfiltered public and rural water supplies. In the acid environment of the stomach excystation occurs and trophozoites are released. Secretory IgA and mucosal IL-6 responses are important for clearance of Giardia infections, and immunosuppressed, agammaglobulinemic, or malnourished persons often are severely affected. Giardia evade immune clearance through continuous modification of the major surface antigen, variant surface protein, and can persist for months or years while causing intermittent symptoms. Giardia infection decreases the expression of brush border enzymes, including lactase, and causes microvillous damage and apoptosis of small intestinal epithelial cells. Giardia trophozoites are noninvasive and can be identified in duodenal biopsy specimens by their characteristic pear shape. Giardiasis is clinically characterized by acute or chronic diarrhea and can result in malabsorption.
Summary
Infectious Enterocolitis
• Vibrio cholerae secretes a pre-formed toxin that causes massive chloride secretion. Water follows the resulting osmotic gradient, leading to secretory diarrhea.
• Campylobacter jejuni is the most common bacterial enteric pathogen in developed countries and also causes traveler’s diarrhea. Most isolates are noninvasive. Salmonella and Shigella spp. are invasive and associated with exudative bloody diarrhea (dysentery). Salmonella infection is a common cause of food poisoning. S. typhi can cause systemic disease (typhoid fever).
• Pseudomembranous colitis is often triggered by antibiotic therapy that disrupts the normal microbiota and allows C. difficile to colonize and grow. The organism releases toxins that disrupt epithelial function. The associated inflammatory response includes characteristic volcano-like eruptions of neutrophils from colonic crypts that spread to form mucopurulent pseudomembranes.
• Rotavirus is the most common cause of severe childhood diarrhea and diarrheal mortality worldwide. The diarrhea is secondary to loss of mature enterocytes, resulting in malabsorption as well as secretion.
• Parasitic and protozoal infections affect over half of the world’s population on a chronic or recurrent basis.
Inflammatory Intestinal Disease
Sigmoid Diverticulitis
In general, diverticular disease refers to acquired pseudodiverticular outpouchings of the colonic mucosa and submucosa. Such colonic diverticula are rare in persons younger than 30 years of age, but the prevalence approaches 50% in Western adult populations beyond the age of 60. Diverticula generally are multiple, and the condition is referred to as diverticulosis. This disease is much less common in Japan and nonindustrialized countries, probably because of dietary differences.
Pathogenesis
Colonic diverticula tend to develop under conditions of elevated intraluminal pressure in the sigmoid colon. This is facilitated by the unique structure of the colonic muscularis propria, where nerves, arterial vasa recta, and their connective tissue sheaths penetrate the inner circular muscle coat to create discontinuities in the muscle wall. In other parts of the intestine, these gaps are reinforced by the external longitudinal layer of the muscularis propria, but in the colon, this muscle layer is discontinuous, being gathered into the three bands termed taeniae coli. High luminal pressures may be generated by exaggerated peristaltic contractions, with spasmodic sequestration of bowel segments that may be exacerbated by diets low in fiber, which reduce stool bulk.
Morphology
Anatomically, colonic diverticula are small, flask-like outpouchings, usually 0.5 to 1 cm in diameter, that occur in a regular distribution in between the taeniae coli (Fig. 14–25, A). They are most common in the sigmoid colon, but other regions of the colon may be affected in severe cases. Because diverticula are compressible, easily emptied of fecal contents, and often surrounded by the fat-containing epiploic appendices on the surface of the colon, they may be missed on casual inspection. Colonic diverticula have a thin wall composed of a flattened or atrophic mucosa, compressed submucosa, and attenuated muscularis propria—often, this last component is totally absent (Fig. 14–30, B and C). Hypertrophy of the circular layer of the muscularis propria in the affected bowel segment is common. Obstruction of diverticula leads to inflammatory changes, producing diverticulitis and peridiverticulitis. Because the wall of the diverticulum is supported only by the muscularis mucosa and a thin layer of subserosal adipose tissue, inflammation and increased pressure within an obstructed diverticulum can lead to perforation. With or without perforation, recurrent diverticulitis may cause segmental colitis, fibrotic thickening in and around the colonic wall, or stricture formation. Perforation can lead to formation of pericolonic abscesses, development of sinus tracts, and, occasionally, peritonitis.
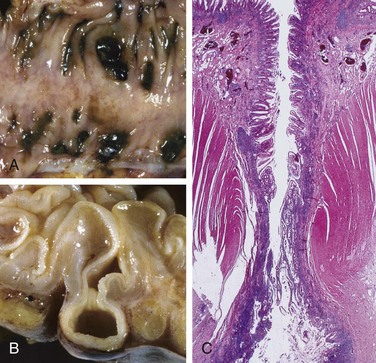
Figure 14–25 Sigmoid diverticular disease.
A, Stool-filled diverticula are regularly arranged. B, Cross-section showing the outpouching of mucosa beneath the muscularis propria. C, Low-power photomicrograph of a sigmoid diverticulum showing protrusion of the mucosa and submucosa through the muscularis propria.
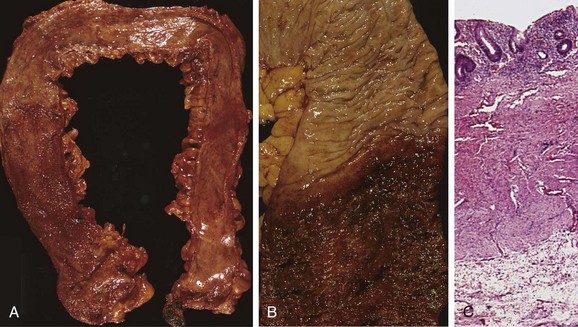
Figure 14–30 Pathology of ulcerative colitis. A, Total colectomy with pancolitis showing active disease, with red, granular mucosa in the cecum (left) and smooth, atrophic mucosa distally (right). B, Sharp demarcation between active ulcerative colitis (bottom) and normal (top). C, This full-thickness histologic section shows that disease is limited to the mucosa. Compare with Figure 14–28, C.
Clinical Features
Most persons with diverticular disease remain asymptomatic throughout their lives. About 20% of those affected develop complaints including intermittent cramping, continuous lower abdominal discomfort, constipation, and diarrhea. Longitudinal studies have shown that while diverticula can regress early in their development they often become more numerous and larger over time. Whether a high-fiber diet prevents such progression or protects against diverticulitis is unclear. Even when diverticulitis occurs, it most often resolves spontaneously or after antibiotic treatment, and relatively few patients require surgical intervention.
Summary
Sigmoid Diverticulitis
• Diverticular disease of the sigmoid colon is common in Western populations over the age of 60. Contributing etiologic factors include low-fiber diets, colonic spasm, and the unique anatomy of the colon. Inflammation of diverticula, diverticulitis, affects a minority of persons with diverticulosis but can cause perforation in its most severe form.
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a chronic condition resulting from inappropriate mucosal immune activation. IBD encompasses two major entities, Crohn disease and ulcerative colitis. The distinction between ulcerative colitis and Crohn disease is based, in large part, on the distribution of affected sites and the morphologic expression of disease at those sites (Fig. 14–26; Table 14–5). Ulcerative colitis is limited to the colon and rectum and extends only into the mucosa and submucosa. By contrast, Crohn disease, which also has been referred to as regional enteritis (because of frequent ileal involvement), may involve any area of the gastrointestinal tract and frequently is transmural.
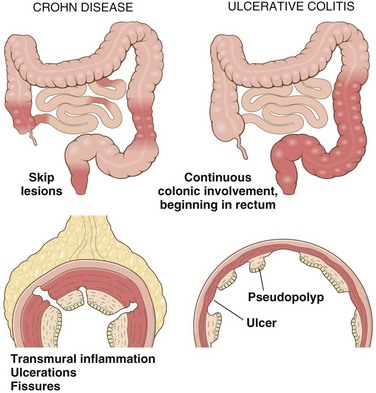
Figure 14–26 Distribution of lesions in inflammatory bowel disease. The distinction between Crohn disease and ulcerative colitis is based primarily on morphology.
Table 14–5 Features That Differ Between Crohn Disease and Ulcerative Colitis
| Feature | Crohn Disease | Ulcerative Colitis |
|---|---|---|
| Macroscopic | ||
| Bowel region affected | Ileum ± colon | Colon only |
| Rectal involvement | Sometimes | Always |
| Distribution | Skip lesions | Diffuse |
| Stricture | Yes | Rare |
| Bowel wall appearance | Thick | Thin |
| Inflammation | Transmural | Limited to mucosa and submucosa |
| Pseudopolyps | Moderate | Marked |
| Ulcers | Deep, knifelike | Superficial, broad-based |
| Lymphoid reaction | Marked | Moderate |
| Fibrosis | Marked | Mild to none |
| Serositis | Marked | No |
| Granulomas | Yes (∼35%) | No |
| Fistulas/sinuses | Yes | No |
| Clinical | ||
| Perianal fistula | Yes (in colonic disease) | No |
| Fat/vitamin malabsorption | Yes | No |
| Malignant potential | With colonic involvement | Yes |
| Recurrence after surgery | Common | No |
| Toxic megacolon | No | Yes |
Note: Not all features may be present in a single case.
Epidemiology
Both Crohn disease and ulcerative colitis are more common in females and frequently present during adolescence or in young adults. In Western industrialized nations, IBD is most common among whites and, in the United States, occurs 3 to 5 times more often among eastern European (Ashkenazi) Jews. This predilection is at least partly due to genetic factors, as discussed next under “Pathogenesis.” The geographic distribution of IBD is highly variable, but it is most prevalent in North America, northern Europe, and Australia. IBD incidence worldwide is on the rise and is becoming more common in regions in which the prevalence was historically low. The hygiene hypothesis suggests that these changes in incidence are related to improved food storage conditions and decreased food contamination. Specifically, it proposes that a reduced frequency of enteric infections due to improved hygiene has resulted in inadequate development of regulatory processes that limit mucosal immune responses early in life. As a result, exposure of susceptible individuals to normally innocuous microbes later in life triggers inappropriate immune responses that may be self-sustaining due to loss of intestinal epithelial barrier function. Although many details are lacking, some data, including some from animal models and the observation in humans that an episode of acute infectious gastroenteritis increases the risk of developing IBD, are consistent with the hygiene hypothesis.
Pathogenesis
The cause(s) of IBD remains uncertain. However, most investigators believe that IBD results from a combination of errant host interactions with intestinal microbiota, intestinal epithelial dysfunction, and aberrant mucosal immune responses. This view is supported by epidemiologic, genetic, and clinical studies as well as data from laboratory models of IBD (Fig. 14–27).
• Genetics. Risk of disease is increased when there is an affected family member, and in Crohn disease, the concordance rate for monozygotic twins is approximately 50%. By contrast, concordance of monozygotic twins for ulcerative colitis is only 16%, suggesting that genetic factors are less dominant in this form of IBD.
• Mucosal immune responses. Although the mechanisms by which mucosal immunity contributes to the pathogenesis of ulcerative colitis and Crohn disease are still being deciphered, immunosuppressive and immunomodulatory agents remain mainstays of IBD therapy. Polarization of helper T cells to the TH1 type is well recognized in Crohn disease, and emerging data suggest that TH17 T cells also contribute to disease pathogenesis. Consistent with this, certain polymorphisms of the IL-23 receptor confer protection from Crohn disease and ulcerative colitis (IL-23 is involved in the development and maintenance of TH17 cells). The protection afforded by IL-23 receptor polymorphisms, together with the recognized effectiveness of anti-TNF therapy in some patients with ulcerative colitis, seems to support roles for TH1 and TH17 cells.
• Epithelial defects. A variety of epithelial defects have been described in Crohn disease, ulcerative colitis, or both. For example, defects in intestinal epithelial tight junction barrier function are present in patients with Crohn disease and a subset of their healthy first-degree relatives. This barrier dysfunction cosegregates with specific disease-associated NOD2 polymorphisms, and experimental models demonstrate that barrier dysfunction can activate innate and adaptive mucosal immunity and sensitize subjects to disease. Interestingly, the Paneth cell granules, which contain antimicrobial peptides that can affect composition of the luminal microbiota, are abnormal in patients with Crohn disease carrying ATG16L1 mutations, thus providing one potential mechanism where a defective feedback loop between the epithelium and microbiota could contribute to disease pathogenesis.
• Microbiota. The quantity of microbial organisms in the gastrointestinal lumen is enormous, amounting to as many as 1012 organisms/mL of fecal material in the colon (50% of fecal mass). This abundance means that, on a per cell level, we are only about 10% human. There is significant inter-individual variation in the composition of this microbial population, which is modified by diet and disease. Despite a growing body of data that suggest that intestinal microbiota contribute to IBD pathogenesis, their precise role remains to be defined. In keeping with this, some antibiotics, such as metronidazole, can be helpful in maintenance of remission in Crohn disease. Ongoing studies suggest that ill-defined mixtures containing probiotic, or beneficial, bacteria also may combat disease in experimental models, as well as in some patients with IBD, although the mechanisms responsible are not well understood.
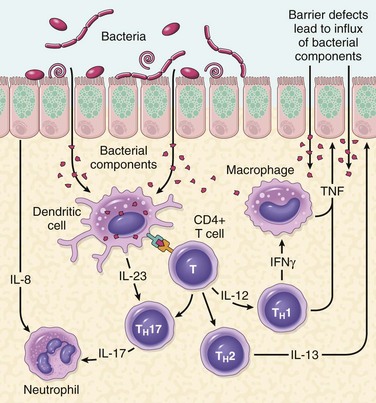
Figure 14–27 A model of pathogenesis of inflammatory bowel disease (IBD). Aspects of both Crohn disease and ulcerative colitis are shown.
One model that unifies the roles of intestinal microbiota, epithelial function, and mucosal immunity suggests a cycle by which transepithelial flux of luminal bacterial components activates innate and adaptive immune responses. In a genetically susceptible host, the subsequent release of TNF and other immune-mediated signals direct epithelia to increase tight junction permeability, which further increases the flux of luminal material. These events may establish a self-amplifying cycle in which a stimulus at any site may be sufficient to initiate IBD. Although this model is helpful in advancing the current understanding of IBD pathogenesis, a variety of factors are associated with disease for unknown reasons. For example, a single episode of appendicitis is associated with reduced risk of developing ulcerative colitis. Tobacco use also modifies the risk of IBD. Somewhat surprisingly, the risk of Crohn disease is increased by smoking, whereas that of ulcerative colitis is reduced.
Crohn Disease
Crohn disease, also known as regional enteritis, may occur in any area of the gastrointestinal tract.
Morphology
The most common sites involved by Crohn disease at presentation are the terminal ileum, ileocecal valve, and cecum. Disease is limited to the small intestine alone in about 40% of cases; the small intestine and the colon both are involved in 30% of patients; and the remainder of cases are characterized by colonic involvement only. The presence of multiple, separate, sharply delineated areas of disease, resulting in skip lesions, is characteristic of Crohn disease and may help in differentiation from ulcerative colitis. Strictures are common (Fig. 14–28, A).
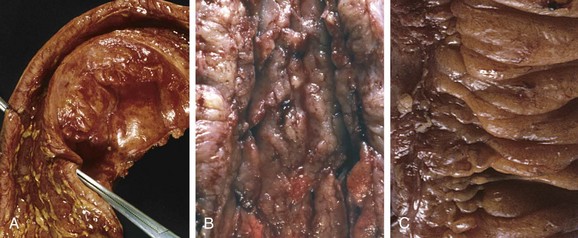
Figure 14–28 Gross pathology of Crohn disease. A, Small intestinal stricture. B, Linear mucosal ulcers and thickened intestinal wall. C, Creeping fat.
The earliest lesion, the aphthous ulcer, may progress, and multiple lesions often coalesce into elongated, serpentine ulcers oriented along the axis of the bowel. Edema and loss of normal mucosal folds are common. Sparing of interspersed mucosa results in a coarsely textured, cobblestone appearance in which diseased tissue is depressed below the level of normal mucosa (Fig. 14–28, B). Fissures frequently develop between mucosal folds and may extend deeply to become sites of perforation or fistula tracts. The intestinal wall is thickened as a consequence of transmural edema, inflammation, submucosal fibrosis, and hypertrophy of the muscularis propria, all of which contribute to stricture formation. In cases with extensive transmural disease, mesenteric fat frequently extends around the serosal surface (creeping fat) (Fig. 14–28, C).
The microscopic features of active Crohn disease include abundant neutrophils that infiltrate and damage crypt epithelium. Clusters of neutrophils within a crypt are referred to as a crypt abscess and often are associated with crypt destruction. Ulceration is common in Crohn disease, and there may be an abrupt transition between ulcerated and normal mucosa. Repeated cycles of crypt destruction and regeneration lead to distortion of mucosal architecture; the normally straight and parallel crypts take on bizarre branching shapes and unusual orientations to one another (Fig. 14–29, A). Epithelial metaplasia, another consequence of chronic relapsing injury, often takes the form of gastric antral-appearing glands (pseudopyloric metaplasia). Paneth cell metaplasia also may occur in the left colon, where Paneth cells normally are absent. These architectural and metaplastic changes may persist even when active inflammation has resolved. Mucosal atrophy, with loss of crypts, may result after years of disease. Noncaseating granulomas (Fig. 14–29, B), a hallmark of Crohn disease, are found in approximately 35% of cases and may arise in areas of active disease or uninvolved regions in any layer of the intestinal wall (Fig. 14–29, C). Granulomas also may be found in mesenteric lymph nodes. Cutaneous granulomas form nodules that are referred to (misleadingly) as metastatic Crohn disease. The absence of granulomas does not preclude a diagnosis of Crohn disease.
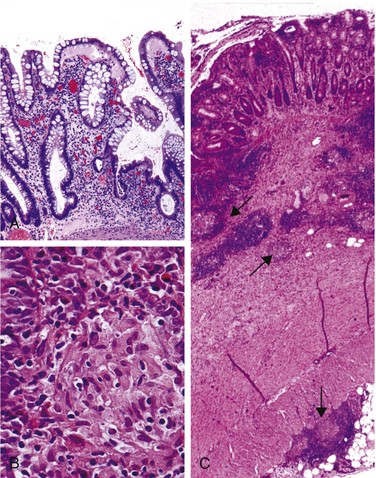
Clinical Features
The clinical manifestations of Crohn disease are extremely variable. In most patients, disease begins with intermittent attacks of relatively mild diarrhea, fever, and abdominal pain. Approximately 20% of patients present acutely with right lower quadrant pain, fever, and bloody diarrhea that may mimic acute appendicitis or bowel perforation. Periods of active disease typically are interrupted by asymptomatic intervals that last for weeks to many months. Disease reactivation can be associated with a variety of external triggers, including physical or emotional stress, specific dietary items, and cigarette smoking.
Iron deficiency anemia may develop in persons with colonic disease, while extensive small bowel disease may result in serum protein loss and hypoalbuminemia, generalized nutrient malabsorption, or malabsorption of vitamin B12 and bile salts. Fibrosing strictures, particularly of the terminal ileum, are common and require surgical resection. Disease often recurs at the site of anastomosis, and as many as 40% of patients require additional resections within 10 years. Fistulas develop between loops of bowel and may also involve the urinary bladder, vagina, and abdominal or perianal skin. Perforations and peritoneal abscesses are common.
Extraintestinal manifestations of Crohn disease include uveitis, migratory polyarthritis, sacroiliitis, ankylosing spondylitis, erythema nodosum, and clubbing of the fingertips, any of which may develop before intestinal disease is recognized. Pericholangitis and primary sclerosing cholangitis also occur in Crohn disease but are more common in ulcerative colitis. As discussed later on, risk of colonic adenocarcinoma is increased in patients with long-standing colonic Crohn disease.
Ulcerative Colitis
Ulcerative colitis is closely related to Crohn disease. However, ulcerative colitis is limited to the colon and rectum. Some extraintestinal manifestations of ulcerative colitis overlap with those of Crohn disease, including migratory polyarthritis, sacroiliitis, ankylosing spondylitis, uveitis, skin lesions, pericholangitis, and primary sclerosing cholangitis.
Morphology
Ulcerative colitis always involves the rectum and extends proximally in a continuous fashion to involve part or all of the colon. Skip lesions are not seen (although focal appendiceal or cecal inflammation occasionally may be present). Disease of the entire colon is termed pancolitis (Fig. 14–30, A). Disease limited to the rectum or rectosigmoid may be referred to descriptively as ulcerative proctitis or ulcerative proctosigmoiditis. The small intestine is normal, although mild mucosal inflammation of the distal ileum, backwash ileitis, may be present in severe cases of pancolitis.
On gross evaluation, involved colonic mucosa may be slightly red and granular-appearing or exhibit extensive broad-based ulcers. The transition between diseased and uninvolved colon can be abrupt (Fig. 14–30, B). Ulcers are aligned along the long axis of the colon but typically do not replicate the serpentine ulcers of Crohn disease. Isolated islands of regenerating mucosa often bulge into the lumen to create small elevations, termed pseudopolyps. Chronic disease may lead to mucosal atrophy and a flat, smooth mucosal surface lacking normal folds. Unlike in Crohn disease, mural thickening is absent, the serosal surface is normal, and strictures do not occur. However, inflammation and inflammatory mediators can damage the muscularis propria and disturb neuromuscular function leading to colonic dilation and toxic megacolon, which carries a significant risk of perforation.
Histologic features of mucosal disease in ulcerative colitis are similar to those in colonic Crohn disease and include inflammatory infiltrates, crypt abscesses, crypt distortion, and epithelial metaplasia. However, skip lesions are absent and inflammation generally is limited to the mucosa and superficial submucosa (Fig. 14–30, C). In severe cases, mucosal damage may be accompanied by ulcers that extend more deeply into the submucosa, but the muscularis propria is rarely involved. Submucosal fibrosis, mucosal atrophy, and distorted mucosal architecture remain as residua of healed disease, but the histologic pattern also may revert to near normal after prolonged remission. Granulomas are not present.
Clinical Features
Ulcerative colitis is a relapsing disorder characterized by attacks of bloody diarrhea with expulsion of stringy, mucoid material and lower abdominal pain and cramps that are temporarily relieved by defecation. These symptoms may persist for days, weeks, or months before they subside, and occasionally the initial attack may be severe enough to constitute a medical or surgical emergency. More than half of the patients have mild disease, and almost all experience at least one relapse during a 10-year period. Colectomy cures intestinal disease, but extraintestinal manifestations may persist.
The factors that trigger ulcerative colitis are not known, but as noted previously, infectious enteritis precedes disease onset in some cases. In other cases the first attack is preceded by psychologic stress, which also may be linked to relapse during remission. The initial onset of symptoms also has been reported to occur shortly after smoking cessation in some patients, and smoking may partially relieve symptoms. Unfortunately, studies of nicotine as a therapeutic agent have been disappointing.
Indeterminate Colitis
Histopathologic and clinical overlap between ulcerative colitis and Crohn disease is common, and it is not possible to make a distinction in up to 10% of patients with IBD. In such cases, termed indeterminate colitis, the small bowel is not involved, and the continuous pattern of colonic disease typically would indicate ulcerative colitis. However, patchy disease, fissures, a family history of Crohn disease, perianal lesions, onset after initiation of cigarette smoking, or findings that are not typical of ulcerative colitis may create uncertainty. Because of extensive overlap in medical management of ulcerative colitis and Crohn disease, patients carrying a diagnosis of indeterminate colitis can be treated effectively. Nevertheless, it is preferable, when possible, to definitively categorize patients, because evolving medical therapies and surgical management differ for ulcerative colitis and for Crohn disease.
Colitis-Associated Neoplasia
One of the most feared long-term complications of ulcerative colitis and colonic Crohn disease is the development of neoplasia. This process begins as dysplasia, which, just as in Barrett esophagus and chronic gastritis, is a step along the road to full-blown carcinoma. The risk of dysplasia is related to several factors:
• Risk increases sharply 8 to 10 years after disease initiation.
• Patients with pancolitis are at greater risk than those with only left-sided disease.
• Greater frequency and severity of active inflammation (characterized by the presence of neutrophils) may increase risk. This is another example of the enabling effect of inflammation on carcinogenesis (Chapter 5).
To facilitate early detection of neoplasia, patients typically are enrolled in surveillance programs approximately 8 years after diagnosis of IBD. The primary exception to this approach is in patients with primary sclerosing cholangitis, who are at markedly greater risk for development of dysplasia and generally are enrolled for surveillance at the time of diagnosis. Surveillance requires regular and extensive mucosal biopsy, making it a costly practice. In many cases, dysplasia occurs in flat areas of mucosa that are not recognized as abnormal on gross evaluation. Thus, advanced endoscopic imaging techniques are beginning to be used experimentally to increase sensitivity of detection in normal-looking tissue.
IBD-associated dysplasia is classified histologically as low-grade or high-grade. High-grade dysplasia can be associated with invasive carcinoma at the same site or elsewhere in the colon and therefore often prompts colectomy, particularly when the changes are multifocal. Low-grade dysplasia may be treated with colectomy or monitored closely, depending on a variety of clinical factors. Colonic adenomas (discussed later on) also occur in patients with IBD, and in some cases these may be difficult to differentiate from a polypoid focus of IBD-associated dysplasia.
Summary
Inflammatory Bowel Disease
• Inflammatory bowel disease (IBD) is an umbrella term for Crohn disease and ulcerative colitis.
• Crohn disease most commonly affects the terminal ileum and cecum, but any site within the gastrointestinal tract can be involved; skip lesions and noncaseating granulomas are common.
• Ulcerative colitis is limited to the colon, is continuous from the rectum, and ranges in extent from only rectal disease to pancolitis; neither skip lesions nor granulomas are present.
• Both Crohn disease and ulcerative colitis can have extraintestinal manifestations.
• The risk of colonic epithelial dysplasia and adenocarcinoma is increased in patients who have had IBD for more than 8 to 10 years.
Colonic Polyps and Neoplastic Disease
Polyps are most common in the colon but may occur in the esophagus, stomach, or small intestine. Those without stalks are referred to as sessile. As sessile polyps enlarge, proliferation of cells adjacent to the polyp and the effects of traction on the luminal protrusion, may combine to create a stalk. Polyps with stalks are termed pedunculated. In general, intestinal polyps can be classified as non-neoplastic or neoplastic. The most common neoplastic polyp is the adenoma, which has the potential to progress to cancer. Non-neoplastic colonic polyps can be further classified as inflammatory, hamartomatous, or hyperplastic.
Inflammatory Polyps
The polyp that forms as part of the solitary rectal ulcer syndrome is an example of the purely inflammatory lesion. Patients present with the clinical triad of rectal bleeding, mucus discharge, and an inflammatory lesion of the anterior rectal wall. The underlying cause is impaired relaxation of the anorectal sphincter, creating a sharp angle at the anterior rectal shelf. This leads to recurrent abrasion and ulceration of the overlying rectal mucosa. Chronic cycles of injury and healing produce a polypoid mass made up of inflamed and reactive mucosal tissue.
Hamartomatous Polyps
Hamartomatous polyps occur sporadically and as components of various genetically determined or acquired syndromes (Table 14–6). As described previously, hamartomas are disorganized, tumor-like growths composed of mature cell types normally present at the site at which the polyp develops. Hamartomatous polyposis syndromes are rare, but they are important to recognize because of associated intestinal and extraintestinal manifestations and the need to screen family members.
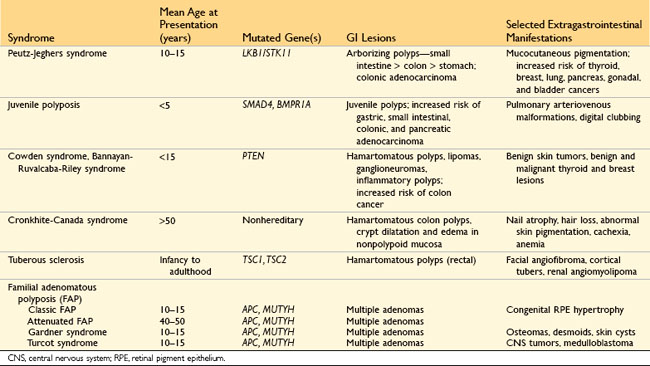
Juvenile Polyps
Juvenile polyps are the most common type of hamartomatous polyp. They may be sporadic or syndromic. In adults, the sporadic form sometimes is also referred to as an inflammatory polyp, particularly when dense inflammatory infiltrates are present. The vast majority of juvenile polyps occur in children younger than 5 years of age. Juvenile polyps characteristically are located in the rectum, and most manifest with rectal bleeding. In some cases, prolapse occurs and the polyp protrudes through the anal sphincter. Sporadic juvenile polyps are usually solitary but in persons with the autosomal dominant syndrome of juvenile polyposis the number varies from 3 to as many as 100. Colectomy may be required to limit the hemorrhage associated with polyp ulceration in juvenile polyposis. Dysplasia occurs in a small proportion of (mostly syndrome-associated) juvenile polyps, and the juvenile polyposis syndrome is associated with increased risk for the development of colonic adenocarcinoma.
Morphology
Individual sporadic and syndromic juvenile polyps often are indistinguishable. They typically are pedunculated, smooth-surfaced, reddish lesions that are less than 3 cm in diameter and display characteristic cystic spaces on cut sections. Microscopic examination shows the spaces to be dilated glands filled with mucin and inflammatory debris (Fig. 14–31, A). Some data suggest that mucosal hyperplasia is the initiating event in polyp development, and this mechanism is consistent with the discovery that mutations in pathways that regulate cellular growth, such as transforming growth factor-β (TGF-β) signaling, are associated with autosomal dominant juvenile polyposis.
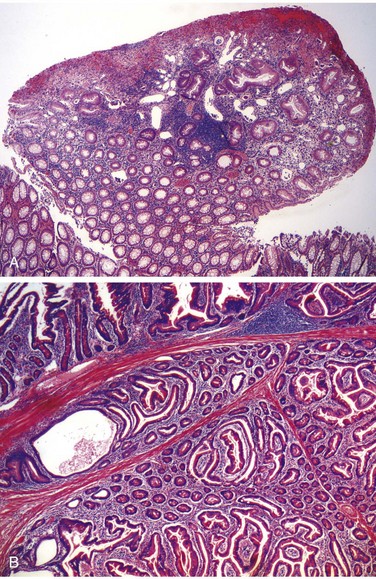
Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome is a rare autosomal dominant disorder defined by the presence of multiple gastrointestinal hamartomatous polyps and mucocutaneous hyperpigmentation that carries an increased risk of several malignancies, including cancers of the colon, pancreas, breast, lung, ovaries, uterus, and testes, as well as other unusual neoplasms. Germ line heterozygous loss-of-function mutations in the gene LKB1/STK11 are present in approximately half of the patients with the familial form of Peutz-Jeghers syndrome, as well as a subset of patients with the sporadic form. Intestinal polyps are most common in the small intestine, although they may also occur in the stomach and colon and, rarely, in the bladder and lungs. On gross evaluation, the polyps are large and pedunculated with a lobulated contour. Histologic examination demonstrates a characteristic arborizing network of connective tissue, smooth muscle, lamina propria, and glands lined by normal-appearing intestinal epithelium (Fig. 14–31, B).
Hyperplastic Polyps
Colonic hyperplastic polyps are common epithelial proliferations that typically are discovered in the sixth and seventh decades of life. The pathogenesis of hyperplastic polyps is incompletely understood, but formation of these lesions is thought to result from decreased epithelial cell turnover and delayed shedding of surface epithelial cells, leading to a “pileup” of goblet cells.
Although these lesions have no malignant potential, they must be distinguished from sessile serrated adenomas, histologically similar lesions that have malignant potential, as described later.
Morphology
Hyperplastic polyps are most commonly found in the left colon and typically are less than 5 mm in diameter. They are smooth, nodular protrusions of the mucosa, often on the crests of mucosal folds. They may occur singly but more frequently are multiple, particularly in the sigmoid colon and rectum. Histologically, hyperplastic polyps are composed of mature goblet and absorptive cells. The delayed shedding of these cells leads to crowding that creates the serrated surface architecture that is the morphologic hallmark of these lesions (Fig. 14–32).
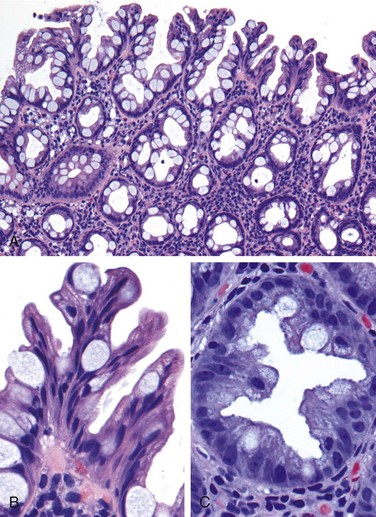
Adenomas
Any neoplastic mass lesion in the gastrointestinal tract may produce a mucosal protrusion, or polyp. The most common and clinically important neoplastic polyps are colonic adenomas, benign polyps that give rise to a majority of colorectal adenocarcinomas. Most adenomas, however, do not progress to adenocarcinoma.
Colorectal adenomas are characterized by the presence of epithelial dysplasia. These growths range from small, often pedunculated polyps to large sessile lesions. There is no gender predilection, and they are present in nearly 50% of adults living in the Western world beginning age 50. Because these polyps are precursors to colorectal cancer, current recommendations are that all adults in the United States undergo surveillance colonoscopy starting at age 50. Because persons with a family history are at risk for developing colon cancer earlier in life, they typically are screened at least 10 years before the youngest age at which a relative was diagnosed. While adenomas are less common in Asia, their frequency has risen (in parallel with an increasing incidence of colorectal adenocarcinoma) as Western diets and lifestyles become more common.
Morphology
Typical adenomas range from 0.3 to 10 cm in diameter and can be pedunculated (Fig. 14–33, A) or sessile, with the surface of both types having a texture resembling velvet (Fig. 14–33, B) or a raspberry, due to the abnormal epithelial growth pattern. Histologically, the cytologic hallmark of epithelial dysplasia (Fig. 14–34, C) is nuclear hyperchromasia, elongation, and stratification. These changes are most easily appreciated at the surface of the adenoma, because the epithelium fails to mature as cells migrate out of the crypt. Pedunculated adenomas have slender fibromuscular stalks (Fig. 14–33, C) containing prominent blood vessels derived from the submucosa. The stalk usually is covered by non-neoplastic epithelium, but dysplastic epithelium is sometimes present.
Figure 14–33 Colonic adenomas. A, Pedunculated adenoma (endoscopic view). B, Adenoma with a velvety surface. C, Low-magnification photomicrograph of a pedunculated tubular adenoma.
Adenomas can be classified as tubular, tubulovillous, or villous on the basis of their architecture. These categories, however, have little clinical significance in isolation. Tubular adenomas tend to be small, pedunculated polyps composed of small, rounded or tubular glands (Fig. 14–34, A). By contrast, villous adenomas, which often are larger and sessile, are covered by slender villi (Fig. 14–34, B). Tubulovillous adenomas have a mixture of tubular and villous elements. Although foci of invasion are more frequent in villous adenomas than in tubular adenomas, villous architecture alone does not increase cancer risk when polyp size is considered.
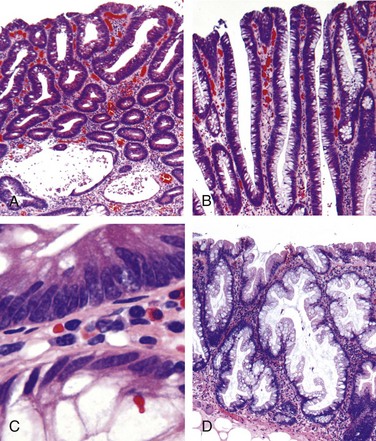
Figure 14–34 Histologic appearance of colonic adenomas. A, Tubular adenoma with a smooth surface and rounded glands. In this case, crypt dilation and rupture, with associated reactive inflammation, can be seen at the bottom of the field. B, Villous adenoma with long, slender projections that are reminiscent of small intestinal villi. C, Dysplastic epithelial cells (top) with an increased nuclear-to-cytoplasmic ratio, hyperchromatic and elongated nuclei, and nuclear pseudostratification. Compare with the nondysplastic epithelium below. D, Sessile serrated adenoma lined by goblet cells without typical cytologic features of dysplasia. This lesion is distinguished from a hyperplastic polyp by involvement of the crypts. Compare with the hyperplastic polyp in Figure 14–32.
The histologic features of sessile serrated adenomas overlap with those of hyperplastic polyps and the typical cytologic features of dysplasia are lacking (Fig. 14–34, D). However, these lesions, which are most common in the right colon, have a malignant potential similar to that of traditional adenomas. The most useful histologic feature that distinguishes sessile serrated adenomas and hyperplastic polyps is the presence of serrated architecture throughout the full length of the glands, including the crypt base, associated with crypt dilation and lateral growth, in the former (Fig. 14–34, D). By contrast, serrated architecture typically is confined to the surface of hyperplastic polyps.
Although most colorectal adenomas are benign lesions, a small proportion may harbor invasive cancer at the time of detection. Size is the most important characteristic that correlates with risk of malignancy. For example, while cancer is extremely rare in adenomas less than 1 cm in diameter, some studies suggest that nearly 40% of lesions larger than 4 cm in diameter contain foci of cancer. In addition to size, high-grade dysplasia is a risk factor for cancer in an individual polyp (but not other polyps in the same patient).
Familial Syndromes
Several syndromes associated with colonic polyps and increased rates of colon cancer have been described. The genetic basis of these disorders has been established and has greatly enhanced the current understanding of sporadic colon cancer (Table 14–7).
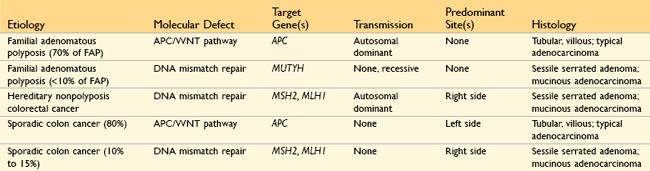
Familial Adenomatous Polyps
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder marked by the appearance of numerous colorectal adenomas by the teenage years. It is caused by mutations of the adenomatous polyposis coli gene (APC). A count of at least 100 polyps is necessary for a diagnosis of classic FAP, and as many as several thousand may be present (Fig. 14–35). Except for their remarkable numbers, these growths are morphologically indistinguishable from sporadic adenomas. Colorectal adenocarcinoma develops in 100% of patients with untreated FAP, often before age 30. As a result, prophylactic colectomy is standard therapy for persons carrying APC mutations. However, patients remain at risk for extraintestinal manifestations, including neoplasia at other sites. Specific APC mutations are also associated with the development of other manifestations of FAP and explain variants such as Gardner syndrome and Turcot syndrome. In addition to intestinal polyps, clinical features of Gardner syndrome, a variant of FAP, may include osteomas of mandible, skull, and long bones; epidermal cysts; desmoid and thyroid tumors; and dental abnormalities, including unerupted and supernumerary teeth. Turcot syndrome is rarer and is characterized by intestinal adenomas and tumors of the central nervous system. Two thirds of patients with Turcot syndrome have APC gene mutations and develop medulloblastomas. The remaining one third have mutations in one of several genes involved in DNA repair and develop glioblastomas. Some patients who have FAP without APC loss have mutations of the base excision repair gene MUTYH. The role of these genes in tumor development is discussed below.
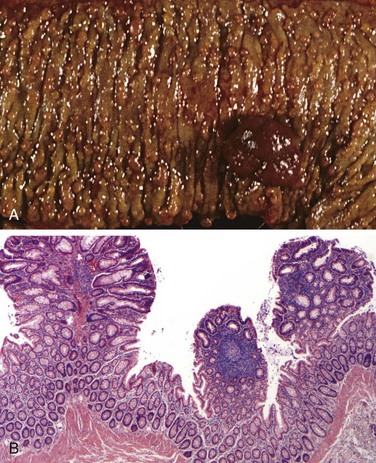
Hereditary Nonpolyposis Colorectal Cancer
Hereditary nonpolyposis colorectal cancer (HNPCC), also known as Lynch syndrome, originally was described as familial clustering of cancers at several sites including the colorectum, endometrium, stomach, ovary, ureters, brain, small bowel, hepatobiliary tract, and skin. Colon cancers in patients with HNPCC tend to occur at younger ages than for sporadic colon cancers and often are located in the right colon (Table 14–7).
Just as identification of APC mutations in FAP has provided molecular insights into the pathogenesis of a majority of sporadic colon cancers, dissection of the defects in HNPCC has shed light on the mechanisms responsible for most of the remaining sporadic cases. HNPCC is caused by inherited germline mutations in genes that encode proteins responsible for the detection, excision, and repair of errors that occur during DNA replication. At least five such mismatch repair genes have been recognized, but a majority of HNPCC cases involve either MSH2 or MLH1. Patients with HNPCC inherit one mutated DNA repair gene and one normal allele. When the second copy is lost through mutation or epigenetic silencing, defects in mismatch repair lead to the accumulation of mutations at rates up to 1000 times higher than normal, mostly in regions containing short repeating DNA sequences referred to as microsatellite DNA. The human genome contains approximately 50,000 to 100,000 microsatellites, which are prone to undergo expansion during DNA replication and represent the most frequent sites of mutations in HNPCC. The consequences of mismatch repair defects and the resulting microsatellite instability are discussed next in the context of colonic adenocarcinoma.
Adenocarcinoma
Adenocarcinoma of the colon is the most common malignancy of the gastrointestinal tract and is a major contributor to morbidity and mortality worldwide. By contrast, the small intestine, which accounts for 75% of the overall length of the gastrointestinal tract, is an uncommon site for benign and malignant tumors. Among malignant small intestinal tumors, adenocarcinomas and carcinoid tumors have roughly equal rates of occurrence, followed by lymphomas and sarcomas.
Epidemiology
Each year in the United States there are more than 130,000 new cases and 55,000 deaths from colorectal adenocarcinoma. This represents nearly 15% of all cancer-related deaths, second only to lung cancer. Colorectal cancer incidence peaks at 60 to 70 years of age, and less than 20% of cases occur before age 50. Males are affected slightly more often than females. Colorectal carcinoma is most prevalent in the United States, Canada, Australia, New Zealand, Denmark, Sweden, and other developed countries. The incidence of this cancer is as much as 30-fold lower in India, South America, and Africa. In Japan, where incidence was previously very low, rates have now risen to intermediate levels (similar to those in the United Kingdom), presumably as a result of changes in lifestyle and diet.
The dietary factors most closely associated with increased colorectal cancer rates are low intake of unabsorbable vegetable fiber and high intake of refined carbohydrates and fat.
In addition to dietary modification, pharmacologic chemoprevention has become an area of great interest. Several epidemiologic studies suggest that aspirin or other NSAIDs have a protective effect. This is consistent with studies showing that some NSAIDs cause polyp regression in patients with FAP in whom the rectum was left in place after colectomy. It is suspected that this effect is mediated by inhibition of the enzyme cyclooxygenase-2 (COX-2), which is highly expressed in 90% of colorectal carcinomas and 40% to 90% of adenomas and is known to promote epithelial proliferation, particularly in response to injury.
Pathogenesis
Studies of colorectal carcinogenesis have provided fundamental insights into the general mechanisms of cancer evolution. The combination of molecular events that lead to colonic adenocarcinoma is heterogeneous and includes genetic and epigenetic abnormalities. At least two distinct genetic pathways APC/β-catenin pathway, have been described. In simplest terms, these are the disturbances of which lead to increased WNT signaling, and the microsatellite instability pathway, which is associated with defects in DNA mismatch repair (see Table 14–7). Both pathways involve the stepwise accumulation of multiple mutations, but the genes involved and the mechanisms by which the mutations accumulate differ. Epigenetic events, the most common of which is methylation-induced gene silencing, may enhance progression along both pathways.
• The APC/β-catenin pathway. The classic adenoma-carcinoma sequence, which accounts for as much as 80% of sporadic colon tumors, typically involves mutation of the APC tumor suppressor early in the neoplastic process (Fig. 14–36). Both copies of the APC gene must be functionally inactivated, either by mutation or epigenetic events, for adenomas to develop. APC is a key negative regulator of β-catenin, a component of the WNT signaling pathway (Chapter 5). The APC protein normally binds to and promotes degradation of β-catenin. With loss of APC function, β-catenin accumulates and translocates to the nucleus, where it activates the transcription of genes, such as those encoding MYC and cyclin D1, which promote proliferation. This is followed by additional mutations, including activating mutations in KRAS, which also promote growth and prevent apoptosis. The conclusion that mutation of KRAS is a late event is supported by the observation that mutations are present in fewer than 10% of adenomas less than 1 cm in diameter, in 50% of adenomas greater than 1 cm in diameter, and in 50% of invasive adenocarcinomas. Neoplastic progression also is associated with mutations in other tumor suppressor genes such as those encoding SMAD2 and SMAD4, which are effectors of TGF-β signaling. Because TGF-β signaling normally inhibits the cell cycle, loss of these genes may allow unrestrained cell growth. The tumor suppressor gene TP53 is mutated in 70% to 80% of colon cancers but is uncommonly affected in adenomas, suggesting that TP53 mutations also occur at late stages of tumor progression. “Loss of function” of TP53 and other tumor suppressor genes often is caused by chromosomal deletions, highlighting chromosomal instability as a hallmark of the APC/β-catenin pathway. Alternatively, tumor suppressor genes may be silenced by methylation of CpG islands, a 5′ region of some genes that frequently includes the promoter and transcriptional start site. Expression of telomerase also increases as lesions become more advanced.
• The microsatellite instability pathway. In patients with DNA mismatch repair deficiency (due to loss of mismatch repair genes, as discussed earlier) mutations accumulate in microsatellite repeats, a condition referred to as microsatellite instability. These mutations generally are silent, because microsatellites typically are located in noncoding regions, but other microsatellite sequences are located in the coding or promoter regions of genes involved in regulation of cell growth, such as those encoding the type II TGF-β receptor and the pro-apoptotic protein BAX (Fig. 14–37). Because TGF-β inhibits colonic epithelial cell proliferation, type II TGF-β receptor mutants can contribute to uncontrolled cell growth, while loss of BAX may enhance the survival of genetically abnormal clones. Mutations in the oncogene BRAF and silencing of distinct groups of genes due to CpG island hypermethylation also are common in cancers that develop through DNA mismatch repair defects. By contrast, KRAS and TP53 typically are not mutated. Thus, the combination of microsatellite instability, BRAF mutation, and methylation of specific targets, such as MLH1, is the signature of this pathway of carcinogenesis.
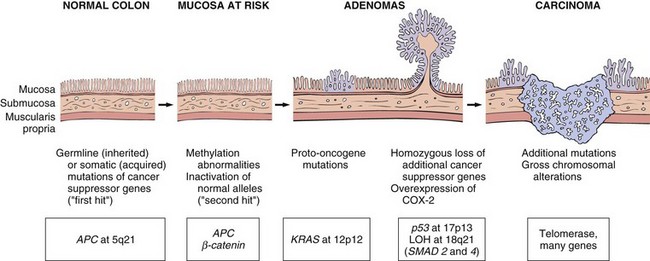
Figure 14–36 Morphologic and molecular changes in the adenoma-carcinoma sequence. It is postulated that loss of one normal copy of the tumor suppressor gene APC occurs early. Persons may be born with one mutant allele, making them extremely prone to the development of colon cancer, or inactivation of APC may occur later in life. This is the “first hit” according to Knudson’s hypothesis. The loss of the intact copy of APC follows (“second hit”). Other mutations involving KRAS, SMAD2, and SMAD4, and the tumor suppressor gene TP53, lead to the emergence of carcinoma, in which additional mutations occur. Although there may be a preferred temporal sequence for these changes, it is the aggregate effect of the mutations, rather than their order of occurrence, that appears most critical.
Figure 14–37 Morphologic and molecular changes in the mismatch repair pathway of colon carcinogenesis. Defects in mismatch repair genes result in microsatellite instability and permit accumulation of mutations in numerous genes. If these mutations affect genes involved in cell survival and proliferation, cancer may develop. LOH, loss of heterozygosity.
Morphology
Overall, adenocarcinomas are distributed approximately equally over the entire length of the colon. Tumors in the proximal colon often grow as polypoid, exophytic masses that extend along one wall of the large-caliber cecum and ascending colon; these tumors rarely cause obstruction. By contrast, carcinomas in the distal colon tend to be annular lesions that produce “napkin ring” constrictions and luminal narrowing (Fig. 14–38), sometimes to the point of obstruction. Both forms grow into the bowel wall over time and may be palpable as firm masses. The general microscopic characteristics of right- and left-sided colonic adenocarcinomas are similar. Most tumors are composed of tall columnar cells that resemble dysplastic epithelium found in adenomas (Fig. 14–39, A). The invasive component of these tumors elicits a strong stromal desmoplastic response, which is responsible for their characteristic firm consistency. Some poorly differentiated tumors form few glands (Fig. 14–39, B). Others may produce abundant mucin that accumulates within the intestinal wall, and these carry a poor prognosis. Tumors also may be composed of signet ring cells that are similar to those in gastric cancer (Fig. 14–39, C).
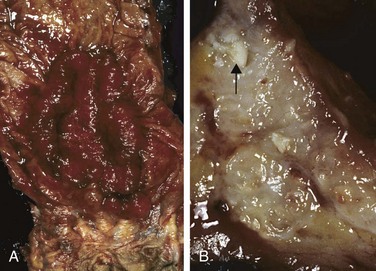
Figure 14–38 Colorectal carcinoma. A, Circumferential, ulcerated rectal cancer. Note the anal mucosa at the bottom of the image. B, Cancer of the sigmoid colon that has invaded through the muscularis propria and is present within subserosal adipose tissue (left). Areas of chalky necrosis are present within the colon wall (arrow).
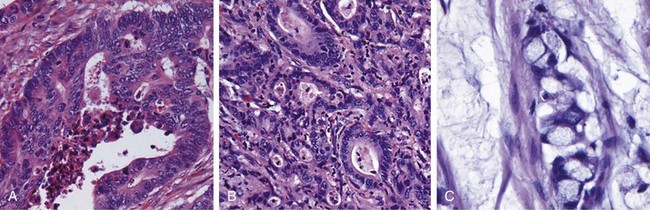
Figure 14–39 Histologic appearance of colorectal carcinoma. A, Well-differentiated adenocarcinoma. Note the elongated, hyperchromatic nuclei. Necrotic debris, present in the gland lumen, is typical. B, Poorly differentiated adenocarcinoma forms a few glands but is largely composed of infiltrating nests of tumor cells. C, Mucinous adenocarcinoma with signet ring cells and extracellular mucin pools.
Clinical Features
The availability of endoscopic screening combined with the recognition that most carcinomas arise within adenomas presents a unique opportunity for cancer prevention. Unfortunately, colorectal cancers develop insidiously and may therefore go undetected for long periods. Cecal and other right-sided colon cancers most often are called to clinical attention by the appearance of fatigue and weakness due to iron deficiency anemia. Thus, it is a clinical maxim that the underlying cause of iron deficiency anemia in an older man or postmenopausal woman is gastrointestinal cancer until proven otherwise. Left-sided colorectal adenocarcinomas may produce occult bleeding, changes in bowel habits, or cramping left lower quadrant discomfort.
Although poorly differentiated and mucinous histologic patterns are associated with poor prognosis, the two most important prognostic factors are depth of invasion and the presence or absence of lymph node metastases. Invasion into the muscularis propria imparts significantly reduced survival that is decreased further by the presence of lymph node metastases (Fig. 14–40, A). These factors were originally recognized by Dukes and Kirklin and form the core of the TNM (tumor-node-metastasis) classification (Table 14–8) and staging system (Table 14–9) from the American Joint Committee on Cancer. Regardless of stage, however, some patients with small numbers of metastases do well for years after resection of distant tumor nodules. This observation once again emphasizes the clinical and molecular heterogeneity of colorectal carcinomas. Metastases may involve regional lymph nodes, lungs (Fig. 14–40, B), and bones, but because of the portal drainage, the liver is the most common site of metastatic lesions (Fig. 14–40, C). The rectum does not drain by way of the portal circulation, and metastases from carcinomas of the anal region often circumvent the liver.
Figure 14–40 Metastatic colorectal carcinoma. A, Lymph node metastasis. Note the glandular structures within the subcapsular sinus. B, Solitary subpleural nodule of colorectal carcinoma metastatic to the lung. C, Liver containing two large and many smaller metastases. Note the central necrosis within metastases.
Table 14–8 AJCC Tumor-Node-Metastasis (TNM) Classification of Colorectal Carcinoma
| Designation | Description |
|---|---|
| Tumor | |
| Tis | In situ dysplasia or intramucosal carcinoma |
| T1 | Tumor invades submucosa |
| T2 | Tumor invades into, but not through, muscularis propria |
| T3 | Tumor invades through muscularis propria |
| T4 | Tumor invades adjacent organs or visceral peritoneum |
| Regional Lymph Nodes | |
| NX | Lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Metastasis in one to three regional lymph nodes |
| N2 | Metastasis in four or more regional lymph nodes |
| Distant Metastasis | |
| MX | Distant metastasis cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis or seeding of abdominal organs |
AJCC, American Joint Committee on Cancer.
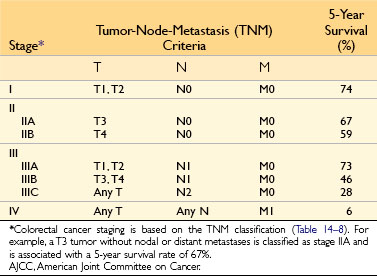
Summary
Colonic Polyps, Adenomas, and Adenocarcinomas
• Intestinal polyps can be classified as non-neoplastic or neoplastic. The non-neoplastic polyps can be further defined as inflammatory, hamartomatous, or hyperplastic.
• Inflammatory polyps form as a result of chronic cycles of injury and healing.
• Hamartomatous polyps occur sporadically or as a part of genetic diseases. In the latter case, they often are associated with increased risk of malignancy.
• Hyperplastic polyps are benign epithelial proliferations most commonly found in the left colon and rectum. They are not reactive in origin, in contrast with gastric hyperplastic polyps; have no malignant potential; and must be distinguished from sessile serrated adenomas.
• Benign epithelial neoplastic polyps of the intestines are termed adenomas. The hallmark feature of these lesions, which are the precursors of colonic adenocarcinomas, is cytologic dysplasia.
• In contrast with traditional adenomas, sessile serrated adenomas lack cytologic dysplasia and share morphologic features with hyperplastic polyps.
• Familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC) are the most common forms of familial colon cancer. FAP is caused by APC mutations, and patients typically have over 100 adenomas and develop colon cancer before the age of 30.
• HNPCC is caused by mutations in DNA mismatch repair genes. Patients with HNPCC have far fewer polyps and develop cancer at an older age than that typical for patients with FAP but at a younger age than in patients with sporadic colon cancer.
• FAP and HNPCC are examples of two distinct pathways of neoplastic transformation, both of which contribute to sporadic colon cancer.
• The vast majority of colonic cancers are adenocarcinomas. The two most important prognostic factors are depth of invasion and the presence or absence of lymph node metastases.