Chapter 6 Genetic and Pediatric Diseases
See Targeted Therapy available online at studentconsult.com
Genetic Diseases
The completion of the human genome project has been a landmark event in the study of human diseases. It has now been established that humans have only about 25,000 protein-coding genes, far fewer than the 100,000 previously estimated and almost half the number in the lowly rice plant (Oryza sativa)! The unraveling of this “genetic architecture” promises to unlock secrets of inherited as well as acquired human disease, since ultimately all diseases involve changes in gene structure or expression. Powerful technologies now allow applications of the human gene sequences to the analysis of human diseases. For example, the human genome project cost approximately 3 billion dollars and many years to complete; current high-throughput sequencing technologies can do the same work in a few weeks for less than $10,000. The speed and reduced costs of DNA sequencing are increasingly facilitating the application of “personalized medicine” to the treatment of cancer and other diseases with a genetic component.
Because several pediatric disorders are of genetic origin, developmental and pediatric diseases are discussed along with genetic diseases in this chapter. However, it must be borne in mind that not all genetic disorders manifest in infancy and childhood, and conversely, many pediatric diseases are not of genetic origin. To the latter category belong diseases resulting from immaturity of organ systems. In this context it is helpful to clarify three commonly used terms: hereditary, familial, and congenital. Hereditary disorders, by definition, are derived from one’s parents, are transmitted in the gametes through the generations, and therefore are familial. The term congenital simply implies “present at birth.” Of note, some congenital diseases are not genetic (e.g., congenital syphilis). On the other hand, not all genetic diseases are congenital; the expression of Huntington disease, for example, begins only after the third or fourth decade of life.
Nature of Genetic Abnormalities Contributing to Human Disease
There are several types of genetic abnormalities that affect the structure and function of proteins, disrupting cellular homeostasis and contributing to disease.
Mutations in Protein-Coding Genes
As is well recognized, the term mutation refers to permanent changes in the DNA. Those that affect germ cells are transmitted to the progeny and may give rise to inherited diseases. Mutations in somatic cells are not transmitted to the progeny but are important in the causation of cancers and some congenital malformations.
Details of specific mutations and their effects are discussed along with the relevant disorders throughout this book. Cited here are some common examples of gene mutations and their effects:
• Point mutations result from the substitution of a single nucleotide base by a different base, resulting in the replacement of one amino acid by another in the protein product. The mutation in the β-globin chain of hemoglobin giving rise to sickle cell anemia is an excellent example of a point mutation that alters the meaning of the genetic code. Such mutations are sometimes called missense mutations.
• By contrast, certain point mutations may change an amino acid codon to a chain termination codon, or stop codon. Such “nonsense” mutations interrupt translation, and in most cases RNAs are rapidly degraded, a phenomenon called nonsense mediated decay, such that little or no protein is formed.
• Frameshift mutations occur when the insertion or deletion of one or two base pairs alters the reading frame of the DNA strand.
• Trinucleotide repeat mutations belong to a special category, because these mutations are characterized by amplification of a sequence of three nucleotides. Although the specific nucleotide sequence that undergoes amplification varies with different disorders, all affected sequences share the nucleotides guanine (G) and cytosine (C). For example, in fragile X syndrome, prototypical of this category of disorders, there are 200 to 4000 tandem repeats of the sequence CGG within a gene called FMR1. In normal populations, the number of repeats is small, averaging 29. The expansions of the trinucleotide sequences prevent normal expression of the FMR1 gene, thus giving rise to mental retardation. Another distinguishing feature of trinucleotide repeat mutations is that they are dynamic (i.e., the degree of amplification increases during gametogenesis). These features, discussed in greater detail later in this chapter, influence the pattern of inheritance and the phenotypic manifestations of the diseases caused by this class of mutations.
Alterations in Protein-Coding Genes Other Than Mutations
In addition to alterations in DNA sequence, coding genes also can undergo structural variations, such as copy number changes (amplifications or deletions), or translocations, resulting in aberrant gain or loss of protein function. As with mutations, structural changes may occur in the germline, or be acquired in somatic tissues. In many instances, pathogenic germ line alterations can involve a contiguous portion of a chromosome rather than a single gene, such as in the 22q microdeletion syndrome, discussed later on. With the widespread availability of array technology for assessing genome-wide DNA copy number variation at very high resolution, pathogenic structural alterations have now been discovered in common disorders such as autism. Cancers often contain somatically acquired structural alterations, including amplifications, deletions, and translocations. The so-called Philadelphia chromosome—translocation t(9;22) between the Bcr and Abl genes in chronic myelogenous leukemia (Chapter 11)—is a classic example.
Sequence and Copy Number Variations (Polymorphisms)
A surprising revelation from the recent progress in genomics is that, on average, any two individuals share greater than 99.5% of their DNA sequences. Thus, the remarkable diversity of humans is encoded in less than 0.5% of our DNA. Though small when compared to the total nucleotide sequences, this 0.5% represents about 15 million base pairs. The two most common forms of DNA variations (polymorphisms) in the human genome are single-nucleotide polymorphisms (SNPs) and copy number variations (CNVs).
• SNPs represent variation at single isolated nucleotide positions and are almost always biallelic (i.e., one of only two choices exist at a given site within the population, such as A or T). Much effort has been devoted to making SNP maps of the human genome. These efforts have identified over 6 million SNPs in the human population, many of which show wide variation in frequency in different populations. SNPs may occur anywhere in the genome—within exons, introns, or intergenic regions—but less than 1% of SNPs occurs in coding regions. These coding sequence variations are important, since they could alter the gene product and predispose to a phenotypic difference or to a disease. Much more commonly, however, the SNP is just a marker that is co-inherited with a disease-associated gene as a result of physical proximity. Another way of expressing this is to say that the SNP and the causative genetic factor are in linkage disequilibrium. There is optimism that groups of SNPs could serve as reliable markers of risk for multigenic complex diseases such as type II diabetes and hypertension, and that by identifying such variants, strategies for disease prevention could be developed (discussed later).
• CNVs are a recently identified form of genetic variation consisting of different numbers of large contiguous stretches of DNA from 1000 base pairs to millions of base pairs. In some instances these loci are, like SNPs, biallelic and simply duplicated or deleted in a subset of the population. In other instances there are complex rearrangements of genomic material, with multiple alleles in the human population. Current estimates are that CNVs are responsible for between 5 and 24 million base pairs of sequence difference between any two individuals. Approximately 50% of CNVs involve gene-coding sequences; thus, CNVs may underlie a large portion of human phenotypic diversity. There is a significant overrepresentation of certain gene families in regions affected by CNVs; these include genes involved in the immune system and in the nervous system. It is assumed that copy number diversity in such gene families has been subject to strong evolutionary selection, since they would enhance human adaptation to changing environmental factors.
Epigenetic Changes
Epigenetic changes are those involving modulation of gene or protein expression in the absence of alterations in DNA sequence (i.e., mutation) or structure of the encoding gene. Epigenetic regulation is of critical importance during development, as well as in homeostasis of fully developed tissues. One central mechanism of epigenetic regulation is by alterations in the methylation of cytosine residues at gene promoters—heavily methylated promoters become inaccessible to RNA polymerase, leading to transcriptional silencing. Promoter methylation and silencing of tumor suppressor genes (Chapter 5) commonly are observed in many human cancers, leading to unchecked cell growth and proliferation. Another major player in epigenetic regulation of transcription involves the family of histone proteins, which are components of structures called nucleosomes, around which DNA is coiled. Histone proteins undergo a variety of reversible modifications (e.g., methylation, acetylation) that affect secondary and tertiary DNA structure, and hence, gene transcription. As expected, abnormalities of histone modification are observed in many acquired diseases such as cancer, leading to transcriptional deregulation. Physiologic epigenetic silencing during development is called imprinting, and disorders of imprinting are discussed later on.
Alterations in Non-Coding RNAs
It is worth noting that until recently the major focus of gene hunting has been discovery of genes that encode for proteins. Recent studies indicate, however, that a very large number of genes do not encode proteins. Instead, the non-encoded products of these genes—so-called “non-coding RNAs (ncRNAs)”—play important regulatory functions. Although many distinct families of ncRNAs exist, here we will only discuss two examples: small RNA molecules called microRNAs (miRNAs), and long non-coding RNAs (lncRNAs) (the latter encompasses ncRNAs >200 nucleotides in length). The miRNAs, unlike messenger RNAs, do not encode proteins but instead inhibit the translation of target mRNAs into their corresponding proteins. Posttranscriptional silencing of gene expression by miRNA is preserved in all living forms from plants to humans and is therefore a fundamental mechanism of gene regulation. Because of their profound influence on gene regulation, miRNAs are assuming central importance in efforts to elucidate normal developmental pathways, as well as pathologic conditions, such as cancer. Andrew Fire and Craig Mello were awarded the Nobel prize in physiology or medicine in 2006 for their work on miRNAs.
By current estimates, there are approximately 1000 genes in humans that encode miRNAs. Transcription of miRNA genes produces primary miRNA transcript (pri-miRNA), which is processed within the nucleus to form another structure called pre-miRNA (Fig. 6–1). With the help of specific transporter proteins, pre-miRNA is exported to the cytoplasm. Additional “cutting” by an enzyme, appropriately called Dicer, generates mature miRNAs that are about 21 to 30 nucleotides in length (hence the designation micro-). At this stage the miRNA is still double-stranded. Next, the miRNA unwinds, and single strands of this duplex are incorporated into a multiprotein complex called RNA-induced silencing complex (RISC). Base pairing between the miRNA strand and its target mRNA directs the RISC to either cause mRNA cleavage or repress its translation. In this way, the gene from which the target mRNA was derived is silenced (at a post-transcriptional state). Given that the numbers of miRNA genes are far fewer than those that encode proteins, it follows that a given miRNA can silence many target genes. All mRNAs contain a so-called seed sequence in their 3′ untranslated region (UTR), which determines the specificity of miRNA binding and gene silencing.
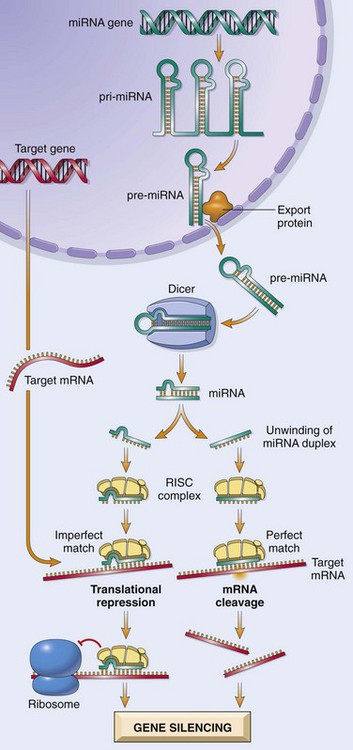
Figure 6–1 Generation of microRNAs and their mode of action in regulating gene function. pri-miRNA, primary microRNA transcript; pre-miRNA, precursor microRNA; RISC, RNA-induced silencing complex.
Another species of gene-silencing RNA, called small interfering RNAs (siRNAs), works in a manner quite similar to that of miRNA. Unlike miRNA, however, siRNA precursors are introduced by investigators into the cell. Their processing by Dicer and functioning via RISC are essentially similar to that described for miRNA. Synthetic siRNAs have become powerful tools for studying gene function in the laboratory, and are being developed as possible therapeutic agents to silence specific genes, such as oncogenes, whose products are involved in neoplastic transformation.
Recent studies have elucidated an untapped universe of lncRNAs (by some calculations, the number of lncRNAs may exceed coding mRNAs by ten-fold to twenty-fold), and their putative functions in the human genome might explain why humans are at the apex of the evolutionary pyramid despite the relatively modest number of coding genes. lncRNAs modulate gene expression in many ways; for example, they can bind to regions of chromatin, restricting access of RNA polymerase to the encompassed coding genes within the region. One of the best known examples of lncRNAs is XIST, which is transcribed from the X-chromosome, and plays an essential role in physiologic X chromosome inactivation (see later). XIST itself escapes X inactivation, but forms a repressive “cloak” on the X chromosome from which it is transcribed, resulting in gene silencing. Emerging studies are highlighting the roles of lncRNAs in various human diseases, from atherosclerosis to cancer.
With this brief review of the nature of abnormalities that contribute to the pathogenesis of human diseases, we can turn our attention to the three major categories of genetic disorders: (1) those related to mutant genes of large effect, (2) diseases with complex multigenic inheritance (sometimes known as multifactorial disorders), and (3) those arising from chromosomal aberrations. The first category, sometimes referred to as mendelian disorders, includes many uncommon conditions, such as the storage diseases and inborn errors of metabolism, all resulting from single-gene mutations of large effect. Most of these conditions are hereditary and familial. The second category includes some of the most common disorders of humans, such as hypertension and diabetes mellitus. Multifactorial, or complex, inheritance implies that both genetic and environmental influences condition the expression of a phenotypic characteristic or disease. The third category includes disorders that are the consequence of numeric or structural abnormalities in the chromosomes.
To these three well-known categories, it is necessary to add a heterogeneous group of genetic disorders that, like mendelian disorders, involve single genes but do not follow simple mendelian rules of inheritance. These single-gene disorders with nonclassic inheritance patterns include those resulting from triplet repeat mutations, those arising from mutations in mitochondrial DNA, and those in which the transmission is influenced by an epigenetic phenomenon called genomic imprinting. Each of these four categories is discussed separately.
Mendelian Disorders: Diseases Caused by Single-Gene Defects
Single-gene defects (mutations) follow the well-known mendelian patterns of inheritance (Tables 6-1 and 6-2). Although individually each is rare, altogether they account for approximately 1% of all adult admissions to hospitals and about 6% to 8% of all pediatric hospital admissions. Listed next are a few important tenets and caveats of relevance in a consideration of mendelian disorders:
• Mutations involving single genes follow one of three patterns of inheritance: autosomal dominant, autosomal recessive, or X-linked.
• A single-gene mutation may lead to many phenotypic effects (pleiotropy), and conversely, mutations at several genetic loci may produce the same trait (genetic heterogeneity). For example, Marfan syndrome, which results from a basic defect in connective tissue, is associated with widespread effects involving the skeleton, eye, and cardiovascular system, all of which stem from a mutation in the gene encoding fibrillin, a component of connective tissues. On the other hand, retinitis pigmentosa, an inherited disorder associated with abnormal retinal pigmentation and consequent visual impairment, can be caused by several different types of mutations. Recognition of genetic heterogeneity not only is important in genetic counseling but also facilitates understanding of the pathogenesis of common disorders such as diabetes mellitus (Chapter 19).
• It is now increasingly being recognized that even known “single-gene” diseases are influenced by inheritance at other genetic loci, which are called modifier genes. As discussed later in the section on cystic fibrosis, these modifier loci can affect the severity or extent of the disease.
• The use of proactive prenatal genetic screening in high-risk populations (e.g., persons of Ashkenazi Jewish descent) has significantly reduced the incidence (Table 6–1) of certain genetic disorders such as Tay-Sachs disease.
Table 6–1 Estimated Prevalence of Selected Mendelian Disorders Among Liveborn Infants
| Disorder | Estimated Prevalence |
|---|---|
| Autosomal Dominant Inheritance | |
| Familial hypercholesterolemia | 1 in 500 |
| Polycystic kidney disease | 1 in 1000 |
| Hereditary spherocytosis | 1 in 5000 (Northern Europe) |
| Marfan syndrome | 1 in 5000 |
| Huntington disease | 1 in 10,000 |
| Autosomal Recessive Inheritance | |
| Sickle cell anemia | 1 in 500 (U.S. African Americans)* |
| Cystic fibrosis | 1 in 3200 (U.S. Caucasians) |
| Tay-Sachs disease | 1 in 3500 (U.S. Ashkenazi Jewish; French Canadians) |
| Phenylketonuria | 1 in 10,000 |
| Mucopolysaccharidoses—all types | 1 in 25,000 |
| Glycogen storage diseases—all types | 1 in 50,000 |
| Galactosemia | 1 in 60,000 |
| X-Linked Inheritance | |
| Duchenne muscular dystrophy | 1 in 3500 (U.S. males) |
| Hemophilia | 1 in 5000 (U.S. males) |
* The prevalence of heterozygous sickle cell trait is 1 in 12 for U.S. African Americans.
Table 6–2 Biochemical Basis and Inheritance Pattern for Selected Mendelian Disorders
| Disease | Abnormal Protein | Protein Type/Function |
|---|---|---|
| Autosomal Dominant Inheritance | ||
| Familial hypercholesterolemia | Low-density lipoprotein receptor | Receptor transport |
| Marfan syndrome | Fibrillin | Structural support: extracellular matrix |
| Ehlers-Danlos syndrome* | Collagen | Structural support: extracellular matrix |
| Hereditary spherocytosis | Spectrin, ankyrin, or protein 4.1 | Structural support: red blood cell membrane |
| Neurofibromatosis, type 1 | Neurofibromin-1 (NF-1) | Growth regulation |
| Adult polycystic kidney disease | Polycystin-1 (PKD-1) | Cell–cell and cell–matrix interactions |
| Autosomal Recessive Inheritance | ||
| Cystic fibrosis | Cystic fibrosis transmembrane regulator | Ion channel |
| Phenylketonuria | Phenylalanine hydroxylase | Enzyme |
| Tay-Sachs disease | Hexosaminidase | Enzyme |
| Severe combined immunodeficiency | Adenosine deaminase | Enzyme |
| α- and β-Thalassemias† | Hemoglobin | Oxygen transport |
| Sickle cell anemia† | Hemoglobin | Oxygen transport |
| X-linked Recessive Inheritance | ||
| Hemophilia A | Factor VIII | Coagulation |
| Duchenne/Becker muscular dystrophy | Dystrophin | Structural support: cell membrane |
| Fragile X syndrome | FMRP | RNA translation |
†Although full-blown symptoms require biallelic mutations, heterozygotes for thalassemia and sickle cell anemia may present with mild clinical disease. Thus, these disorders sometimes are categorized as “autosomal codominant” entities.
* Some variants of Ehlers-Danlos syndrome have an autosomal recessive inheritance pattern.
Transmission Patterns of Single-Gene Disorders
Disorders of Autosomal Dominant Inheritance
Disorders of autosomal dominant inheritance are manifested in the heterozygous state, so at least one parent in an index case usually is affected; both males and females are affected, and both can transmit the condition. When an affected person marries an unaffected one, every child has one chance in two of having the disease. The following features also pertain to autosomal dominant diseases:
• With any autosomal dominant disorder, some patients do not have affected parents. Such patients owe their disorder to new mutations involving either the egg or the sperm from which they were derived. Their siblings are neither affected nor at increased risk for development of the disease.
• Clinical features can be modified by reduced penetrance and variable expressivity. Some persons inherit the mutant gene but are phenotypically normal. This mode of expression is referred to as reduced penetrance. The variables that affect penetrance are not clearly understood. In contrast with penetrance, if a trait is consistently associated with a mutant gene but is expressed differently among persons carrying the gene, the phenomenon is called variable expressivity. For example, manifestations of neurofibromatosis 1 range from brownish spots on the skin to multiple tumors and skeletal deformities.
• In many conditions, the age at onset is delayed, and symptoms and signs do not appear until adulthood (as in Huntington disease).
• In autosomal dominant disorders, a 50% reduction in the normal gene product is associated with clinical signs and symptoms. Because a 50% loss of enzyme activity can be compensated for, involved genes in autosomal dominant disorders usually do not encode enzyme proteins, but instead fall into two other categories of proteins:
 Those involved in regulation of complex metabolic pathways, often subject to feedback control (e.g., membrane receptors, transport proteins). An example of this mechanism of inheritance is familial hypercholesterolemia, which results from mutation in the low-density lipoprotein (LDL) receptor gene (discussed later).
Those involved in regulation of complex metabolic pathways, often subject to feedback control (e.g., membrane receptors, transport proteins). An example of this mechanism of inheritance is familial hypercholesterolemia, which results from mutation in the low-density lipoprotein (LDL) receptor gene (discussed later).The biochemical mechanisms by which a 50% reduction in the levels of such proteins results in an abnormal phenotype are not fully understood. In some cases, especially when the gene encodes one subunit of a multimeric protein, the product of the mutant allele can interfere with the assembly of a functionally normal multimer. For example, the collagen molecule is a trimer in which the three collagen chains are arranged in a helical configuration. Even with a single mutant collagen chain, normal collagen trimers cannot be formed, so there is a marked deficiency of collagen. In this instance the mutant allele is called dominant negative, because it impairs the function of a normal allele. This effect is illustrated in some forms of osteogenesis imperfecta (Chapter 20).
Disorders of Autosomal Recessive Inheritance
Disorders of autosomal recessive inheritance make up the largest group of mendelian disorders. They occur when both of the alleles at a given gene locus are mutants; therefore, such disorders are characterized by the following features: (1) The trait does not usually affect the parents, but siblings may show the disease; (2) siblings have one chance in four of being affected (i.e., the recurrence risk is 25% for each birth); and (3) if the mutant gene occurs with a low frequency in the population, there is a strong likelihood that the affected patient (the proband) is the product of a consanguineous marriage.
In contrast with the features of autosomal dominant diseases, the following features generally apply to most autosomal recessive disorders:
• The expression of the defect tends to be more uniform than in autosomal dominant disorders.
• Complete penetrance is common.
• Onset is frequently early in life.
• Although new mutations for recessive disorders do occur, they are rarely detected clinically. Because the affected person is an asymptomatic heterozygote, several generations may pass before the descendants of such a person mate with other heterozygotes and produce affected offspring.
• In many cases, enzyme proteins are affected by the mutation. In heterozygotes, equal amounts of normal and defective enzyme are synthesized. Usually the natural “margin of safety” ensures that cells with half of their complement of the enzyme function normally.
X-Linked Disorders
All sex-linked disorders are X-linked. No Y-linked diseases are known as yet. Save for determinants that dictate male differentiation, the only characteristic that may be located on the Y chromosome is the attribute of hairy ears, which is not altogether devastating. Most X-linked disorders are X-linked recessive and are characterized by the following features:
• They are transmitted by heterozygous female carriers only to sons, who of course are hemizygous for the X chromosome.
• Heterozygous females rarely express the full phenotypic change, because they have the paired normal allele; although one of the X chromosomes in females is inactivated (see further on), this process of inactivation is random, which typically allows sufficient numbers of cells with the normal expressed allele to emerge.
• An affected male does not transmit the disorder to sons, but all daughters are carriers. Sons of heterozygous women have one chance in two of receiving the mutant gene.
 Summary
Summary
Transmission Patterns of Single-Gene Disorders
• Autosomal dominant disorders are characterized by expression in heterozygous state; they affect males and females equally, and both sexes can transmit the disorder.
• Enzyme proteins are not affected in autosomal dominant disorders; instead, receptors and structural proteins are involved.
• Autosomal recessive diseases occur when both copies of a gene are mutated; enzyme proteins are frequently involved. Males and females are affected equally.
• X-linked disorders are transmitted by heterozygous females to their sons, who manifest the disease. Female carriers usually are protected because of random inactivation of one X chromosome.
Diseases Caused by Mutations in Genes Encoding Structural Proteins
Marfan Syndrome
In Marfan syndrome, a connective tissue disorder of autosomal dominant inheritance, the basic biochemical abnormality is a mutation affecting fibrillin. This glycoprotein, secreted by fibroblasts, is the major component of microfibrils found in the extracellular matrix. Microfibrils serve as scaffolding for the deposition of tropoelastin, an integral component of elastic fibers. Although microfibrils are widely distributed in the body, they are particularly abundant in the aorta, ligaments, and the ciliary zonules that support the ocular lens; these tissues are prominently affected in Marfan syndrome.
Fibrillin is encoded by the FBN1 gene, which maps to chromosomal locus 15q21. Mutations in the FBN1 gene are found in all patients with Marfan syndrome. However, molecular diagnosis of Marfan syndrome is not yet feasible, because more than 600 distinct causative mutations in the very large FBN1 gene have been found. Since heterozygotes have clinical symptoms, it follows that the mutant fibrillin protein must act as a dominant negative by preventing the assembly of normal microfibrils. The prevalence of Marfan syndrome is estimated to be 1 per 5000. Approximately 70% to 85% of cases are familial, and the rest are sporadic, arising from de novo FBN1 mutations in the germ cells of parents.
While many of the abnormalities in Marfan syndrome can be explained on the basis of structural failure of connective tissues, some, such as overgrowth of bones, are difficult to relate to simple loss of fibrillin. Recent studies indicate that loss of microfibrils gives rise to abnormal and excessive activation of transforming growth factor-β (TGF-β), since normal microfibrils sequester TGF-β, thereby controlling bioavailability of this cytokine. Excessive TGF-β signaling has deleterious effects on vascular smooth muscle development and the integrity of extracellular matrix. In support of this hypothesis, mutations in the TGF-β type II receptor give rise to a related syndrome, called Marfan syndrome type 2 (MFS2). Of note, angiotensin receptor blockers, which inhibit the activity of TGF-β, have been shown to improve aortic and cardiac function in mouse models of Marfan syndrome and currently are being evaluated in clinical trials.
 Morphology
Morphology
Skeletal abnormalities are the most obvious feature of Marfan syndrome. Patients have a slender, elongated habitus with abnormally long legs, arms, and fingers (arachnodactyly); a high-arched palate; and hyperextensibility of joints. A variety of spinal deformities, such as severe kyphoscoliosis, may be present. The chest is deformed, exhibiting either pectus excavatum (i.e., deeply depressed sternum) or a pigeon-breast deformity. The most characteristic ocular change is bilateral dislocation, or subluxation, of the lens secondary to weakness of its suspensory ligaments (ectopia lentis). This abnormality is so uncommon in persons who do not have this genetic disease that the finding of bilateral ectopia lentis should raise the diagnostic possibility of Marfan syndrome. Most serious, however, is the involvement of the cardiovascular system. Fragmentation of the elastic fibers in the tunica media of the aorta predisposes affected patients to aneurysmal dilation and aortic dissection (Chapter 9). These changes, called cystic medionecrosis, are not specific for Marfan syndrome. Similar lesions occur in hypertension and with aging. Loss of medial support causes dilation of the aortic valve ring, giving rise to aortic incompetence. The cardiac valves, especially the mitral valve, may be excessively distensible and regurgitant (floppy valve syndrome), giving rise to mitral valve prolapse and congestive cardiac failure (Chapter 10). Death from aortic rupture may occur at any age, and aortic rupture is in fact the most common cause of death. Less commonly, cardiac failure is the terminal event.
Although the lesions described are typical of Marfan syndrome, they are not seen in all cases. There is much variation in clinical expression, and some patients may exhibit predominantly cardiovascular lesions with minimal skeletal and ocular changes. The variable expressivity is believed to be related to different allelic mutations in the FBN1 gene.
Ehlers-Danlos Syndromes
Ehlers-Danlos syndromes (EDSs) are a group of diseases characterized by defects in collagen synthesis or structure. All are single-gene disorders, but the mode of inheritance encompasses both autosomal dominant and recessive patterns. There are approximately 30 distinct types of collagen; all have characteristic tissue distributions and are the products of different genes. To some extent, the clinical heterogeneity of EDS can be explained by mutations in different collagen genes.
At least six clinical and genetic variants of EDS are recognized. Because defective collagen is the basis for these disorders, certain clinical features are common to all variants.
As might be expected, tissues rich in collagen, such as skin, ligaments, and joints, frequently are involved in most variants of EDS. Because the abnormal collagen fibers lack adequate tensile strength, the skin is hyperextensible and joints are hypermobile. These features permit grotesque contortions, such as bending the thumb backward to touch the forearm and bending the knee upward to create almost a right angle. Indeed, it is believed that most contortionists have one of the EDSs; however, a predisposition to joint dislocation is one of the prices paid for this virtuosity. The skin is extraordinarily stretchable, extremely fragile, and vulnerable to trauma. Minor injuries produce gaping defects, and surgical repair or any surgical intervention is accomplished only with great difficulty because of the lack of normal tensile strength. The basic defect in connective tissue may lead to serious internal complications, including rupture of the colon and large arteries (vascular EDS); ocular fragility, with rupture of the cornea and retinal detachment (kyphoscoliotic EDS); and diaphragmatic hernias (classical EDS), among others.
The molecular bases for three of the more common variants are as follows:
• Deficiency of the enzyme lysyl hydroxylase. Decreased hydroxylation of lysyl residues in types I and III collagen interferes with the formation of cross-links among collagen molecules. As might be expected, this variant (kyphoscoliotic EDS), resulting from an enzyme deficiency, is inherited as an autosomal recessive disorder.
• Deficient synthesis of type III collagen resulting from mutations affecting the COL3A1 gene. This variant, the vascular type, is inherited as an autosomal dominant disorder and is characterized by weakness of tissues rich in type III collagen (e.g., blood vessels, bowel wall), predisposing them to rupture.
• Deficient synthesis of type V collagen due to mutations in COL5A1 and COL5A2 is inherited as an autosomal dominant disorder and results in classical EDS.
Summary
Marfan Syndrome
• Marfan syndrome is caused by a mutation in the FBN1 gene encoding fibrillin, which is required for structural integrity of connective tissues.
• The major tissues affected are the skeleton, eyes, and cardiovascular system.
• Clinical features may include tall stature, long fingers, bilateral subluxation of lens, mitral valve prolapse, aortic aneurysm, and aortic dissection.
• Clinical trials with drugs that inhibit TGF-β signaling such as angiotensin receptor blockers are ongoing, as these have been shown to improve aortic and cardiac function in mouse models.
Ehlers-Danlos Syndromes
• There are six variants of Ehlers-Danlos syndromes, all characterized by defects in collagen synthesis or assembly. Each of the variants is caused by a distinct mutation.
• Clinical features may include fragile, hyperextensible skin vulnerable to trauma, hypermobile joints, and ruptures involving colon, cornea, or large arteries. Wound healing is poor.
Diseases Caused by Mutations in Genes Encoding Receptor Proteins or Channels
Familial Hypercholesterolemia
Familial hypercholesterolemia is among the most common mendelian disorders; the frequency of the heterozygous condition is 1 in 500 in the general population. It is caused by a mutation in the LDLR gene that encodes the receptor for low-density lipoprotein (LDL), the form in which 70% of total plasma cholesterol is transported. A brief review of the synthesis and transport of cholesterol follows.
Normal Cholesterol Metabolism
Cholesterol may be derived from the diet or from endogenous synthesis. Dietary triglycerides and cholesterol are incorporated into chylomicrons in the intestinal mucosa, which drain by way of the gut lymphatics into the blood. These chylomicrons are hydrolyzed by an endothelial lipoprotein lipase in the capillaries of muscle and fat. The chylomicron remnants, rich in cholesterol, are then delivered to the liver. Some of the cholesterol enters the metabolic pool (to be described), and some is excreted as free cholesterol or bile acids into the biliary tract. The endogenous synthesis of cholesterol and LDL begins in the liver (Fig. 6–2). The first step in the synthesis of LDL is the secretion of triglyceride-rich very-low-density lipoprotein (VLDL) by the liver into the blood. In the capillaries of adipose tissue and muscle, the VLDL particle undergoes lipolysis and is converted to intermediate-density lipoprotein (IDL). In comparison with VLDL, the content of triglyceride is reduced and that of cholesteryl esters enriched in intermediate-density lipoprotein (IDL), but IDL retains on its surface two of the three VLDL-associated apolipoproteins B-100 and E. Further metabolism of IDL occurs along two pathways: Most of the IDL particles are directly taken up by the liver through the LDL receptor described later; others are converted to cholesterol-rich LDL by a further loss of triglycerides and apolipoprotein E. In the liver cells, IDL is recycled to generate VLDL.
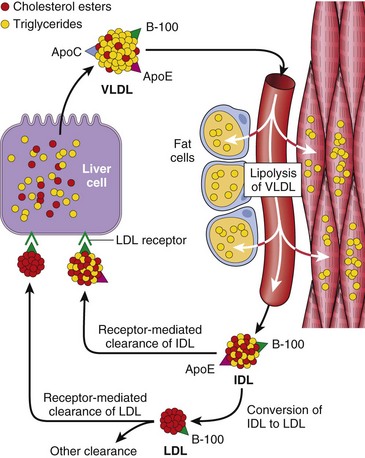
Figure 6–2 Low-density lipoprotein (LDL) metabolism and the role of the liver in its synthesis and clearance. Lipolysis of very-low-density lipoprotein (VLDL) by lipoprotein lipase in the capillaries releases triglycerides, which are then stored in fat cells and used as a source of energy in skeletal muscles. IDL (intermediate-density lipoprotein) remains in the blood and is taken up by the liver.
Two thirds of the resultant LDL particles are metabolized by the LDL receptor pathway, and the rest is metabolized by a receptor for oxidized LDL (scavenger receptor), to be described later. The LDL receptor binds to apolipoproteins B-100 and E and thus is involved in the transport of both LDL and IDL. Although the LDL receptors are widely distributed, approximately 75% are located on hepatocytes, so the liver plays an extremely important role in LDL metabolism. The first step in the receptor-mediated transport of LDL involves binding to the cell surface receptor, followed by endocytotic internalization inside so-called “clathrin-coated pits” (Fig. 6–3). Within the cell, the endocytic vesicles fuse with the lysosomes, and the LDL molecule is enzymatically degraded, resulting ultimately in the release of free cholesterol into the cytoplasm. The cholesterol not only is used by the cell for membrane synthesis but also takes part in intracellular cholesterol homeostasis by a sophisticated system of feedback control:
• It suppresses cholesterol synthesis by inhibiting the activity of the enzyme 3-hydroxy-3-methylglutaryl–coenzyme A reductase (HMG-CoA reductase), which is the rate-limiting enzyme in the synthetic pathway.
• It stimulates the formation of cholesterol esters for storage of excess cholesterol.
• It downregulates the synthesis of cell surface LDL receptors, thus protecting cells from excessive accumulation of cholesterol.
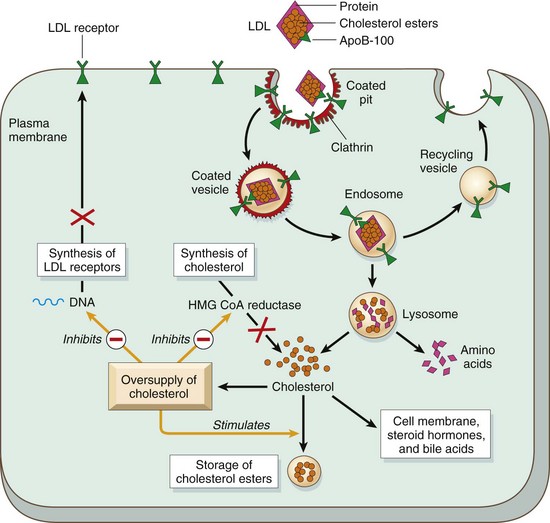
Figure 6–3 The LDL receptor pathway and regulation of cholesterol metabolism. The yellow arrows show three regulatory functions of free intracellular cholesterol: (1) suppression of cholesterol synthesis by inhibition of HMG-CoA reductase, (2) stimulating the storage of excess cholesterol as esters, and (3) inhibition of synthesis of LDL receptors. HMG-CoA reductase, 3-hydroxy-3-methylglutaryl–coenzyme A reductase; LDL, low-density lipoprotein.
The transport of LDL by the scavenger receptors, alluded to earlier, seems to take place in cells of the mononuclear-phagocyte system and possibly in other cells as well. Monocytes and macrophages have receptors for chemically modified (e.g., acetylated or oxidized) LDLs. The amount catabolized by this “scavenger receptor” pathway is directly related to the plasma cholesterol level.
 Pathogenesis of Familial Hypercholesterolemia
Pathogenesis of Familial Hypercholesterolemia
In familial hypercholesterolemia, mutations in the LDL receptor protein impair the intracellular transport and catabolism of LDL, resulting in accumulation of LDL cholesterol in the plasma. In addition, the absence of LDL receptors on liver cells also impairs the transport of IDL into the liver, so a greater proportion of plasma IDL is converted into LDL. Thus, patients with familial hypercholesterolemia develop excessive levels of serum cholesterol as a result of the combined effects of reduced catabolism and excessive biosynthesis (Fig. 6–2). In the presence of such hypercholesterolemia, there is a marked increase of cholesterol traffic into the monocyte-macrophages and vascular walls mediated by the scavenger receptor. This accounts for the appearance of skin xanthomas and premature atherosclerosis.
Familial hypercholesterolemia is an autosomal dominant disease. Heterozygotes have a two- to three-fold elevation of plasma cholesterol levels, whereas homozygotes may have in excess of a five-fold elevation. Although their cholesterol levels are elevated from birth, heterozygotes remain asymptomatic until adult life, when they develop cholesterol deposits (xanthomas) along tendon sheaths and premature atherosclerosis resulting in coronary artery disease. Homozygotes are much more severely affected, developing cutaneous xanthomas in childhood and often dying of myocardial infarction before the age of 20 years.
Analysis of the cloned LDL receptor gene has revealed that more than 900 different mutations can give rise to familial hypercholesterolemia. These can be divided into five categories. Class I mutations are uncommon, and they are associated with complete loss of receptor synthesis. With class II mutations, the most prevalent form, the receptor protein is synthesized, but its transport from the endoplasmic reticulum to the Golgi apparatus is impaired due to defects in protein folding. Class III mutations produce receptors that are transported to the cell surface but fail to bind LDL normally. Class IV mutations give rise to receptors that fail to internalize within clathrin pits after binding to LDL, while class V mutations encode receptors that can bind LDL and are internalized but are trapped in endosomes because dissociation of receptor and bound LDL does not occur.
The discovery of the critical role of LDL receptors in cholesterol homeostasis has led to the rational design of the statin family of drugs that are now widely used to lower plasma cholesterol. They inhibit the activity of HMG-CoA reductase and thus promote greater synthesis of LDL receptor (Fig. 6–3).
Summary
• Familial hypercholesterolemia is an autosomal dominant disorder caused by mutations in the gene encoding the LDL receptor.
• Patients develop hypercholesterolemia as a consequence of impaired transport of LDL into the cells.
• In heterozygotes, elevated serum cholesterol greatly increases the risk of atherosclerosis and resultant coronary artery disease; homozygotes have an even greater increase in serum cholesterol and a higher frequency of ischemic heart disease. Cholesterol also deposits along tendon sheaths to produce xanthomas.
Cystic Fibrosis
With an incidence of 1 in 3200 live births in the United States, cystic fibrosis (CF) is the most common lethal genetic disease that affects white populations. It is uncommon among Asians (1 in 31,000 live births) and African Americans (1 in 15,000 live births). CF follows simple autosomal recessive transmission, and does not affect heterozygote carriers. There is, however, a bewildering compendium of phenotypic variation that results from diverse mutations in the CF-associated gene, the tissue-specific effects of loss of this gene’s function, and the influence of newly recognized disease modifiers. It is, fundamentally, a disorder of epithelial transport affecting fluid secretion in exocrine glands and the epithelial lining of the respiratory, gastrointestinal, and reproductive tracts. Indeed, abnormally viscid mucous secretions that block the airways and the pancreatic ducts are responsible for the two most important clinical manifestations: recurrent and chronic pulmonary infections and pancreatic insufficiency. In addition, although the exocrine sweat glands are structurally normal (and remain so throughout the course of this disease), a high level of sodium chloride in the sweat is a consistent and characteristic biochemical abnormality in CF.
Pathogenesis
The primary defect in CF is abnormal function of an epithelial chloride channel protein encoded by the CF transmembrane conductance regulator (CFTR) gene at chromosomal locus 7q31.2. The changes in mucus are considered secondary to the disturbance in transport of chloride ions. In normal epithelia, the transport of chloride ions across the cell membrane occurs through transmembrane proteins, such as CFTR, that form chloride channels. Mutations in the CFTR gene render the epithelial membranes relatively impermeable to chloride ions (Fig. 6–4). However, the impact of this defect on transport function is tissue-specific. The major function of the CFTR protein in the sweat gland ducts is to reabsorb luminal chloride ions and augment sodium reabsorption through the epithelial sodium channel (ENaC). Therefore, in the sweat ducts, loss of CFTR function leads to decreased reabsorption of sodium chloride and production of hypertonic (“salty”) sweat (Fig. 6–4, top). In contrast with that in the sweat glands, CFTR in the respiratory and intestinal epithelium forms one of the most important avenues for active luminal secretion of chloride. At these sites, CFTR mutations result in loss or reduction of chloride secretion into the lumen (Fig. 6–4, bottom). Active luminal sodium absorption through ENaCs also is increased, and both of these ion changes increase passive water reabsorption from the lumen, lowering the water content of the surface fluid layer coating mucosal cells. Thus, unlike the sweat ducts, there is no difference in the salt concentration of the surface fluid layer coating the respiratory and intestinal mucosal cells in normal persons and in those with CF. Instead, the pathogenesis of respiratory and intestinal complications in CF seems to stem from an isotonic but low-volume surface fluid layer. In the lungs, this dehydration leads to defective mucociliary action and the accumulation of concentrated, viscid secretions that obstruct the air passages and predispose to recurrent pulmonary infections.
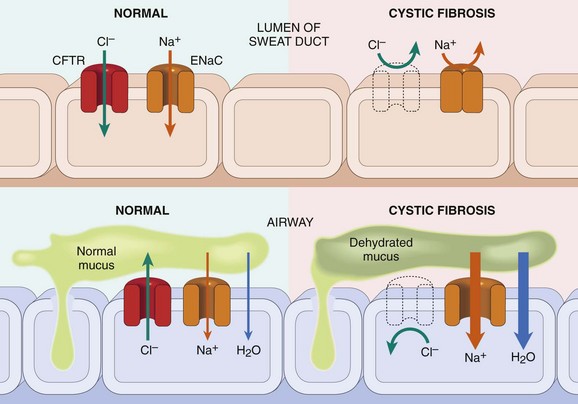
Figure 6–4 Top, In cystic fibrosis (CF), a chloride channel defect in the sweat duct causes increased chloride and sodium concentration in sweat. Bottom, Patients with CF have decreased chloride secretion and increased sodium and water reabsorption in the airways, leading to dehydration of the mucus layer coating epithelial cells, defective mucociliary action, and mucous plugging. CFTR, cystic fibrosis transmembrane conductance regulator; ENaC, epithelial sodium channel responsible for intracellular sodium conduction.
Since the CFTR gene was cloned in 1989, more than 1300 disease-causing mutations have been identified. They can be classified as severe or mild, depending on the clinical phenotype: Severe mutations are associated with complete loss of CFTR protein function, whereas mild mutations allow some residual function. The most common severe CFTR mutation is a deletion of three nucleotides coding for phenylalanine at amino acid position 508 (ΔF508). This causes misfolding and total loss of the CFTR. Worldwide, ΔF508 mutation is found in approximately 70% of patients with CF. Since CF is an autosomal recessive disease, affected persons harbor mutations on both alleles. As discussed later, the combination of mutations on the two alleles influences the overall phenotype, as well as organ-specific manifestations. Although CF remains one of the best-known examples of the “one gene–one disease” axiom, there is increasing evidence that other genes modify the frequency and severity of organ-specific manifestations. One example of a candidate genetic modifier is mannose-binding lectin, a key effector of innate immunity involved in phagocytosis of microorganisms. In the setting of CF, polymorphisms in one or both mannose-binding lectin alleles that produce lower circulating levels of the protein are associated with a three-fold higher risk of end-stage lung disease, due to chronic bacterial infections.
Morphology
The anatomic changes are highly variable and depend on which glands are affected and on the severity of this involvement. Pancreatic abnormalities are present in 85% to 90% of patients with CF. In the milder cases, there may be only accumulations of mucus in the small ducts, with some dilation of the exocrine glands. In more advanced cases, usually seen in older children or adolescents, the ducts are totally plugged, causing atrophy of the exocrine glands and progressive fibrosis (Fig. 6–5). The total loss of pancreatic exocrine secretion impairs fat absorption, so avitaminosis A may contribute to squamous metaplasia of the lining epithelium of the ducts in the pancreas, which are already injured by the inspissated mucus secretions. Thick viscid plugs of mucus also may be found in the small intestine of infants. Sometimes these cause small bowel obstruction, known as meconium ileus.
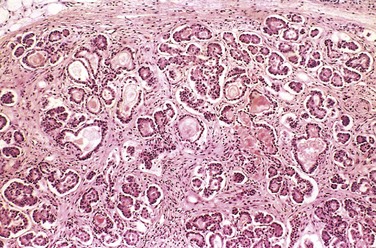
Figure 6–5 Mild to moderate changes of cystic fibrosis in the pancreas. The ducts are dilated and plugged with eosinophilic mucin, and the parenchymal glands are atrophic and replaced by fibrous tissue.
The pulmonary changes are the most serious complications of this disease (Fig. 6–6). These changes stem from obstruction and infection of the air passages secondary to the viscous mucus secretions of the submucosal glands of the respiratory tree. The bronchioles often are distended with thick mucus, associated with marked hyperplasia and hypertrophy of the mucus-secreting cells. Superimposed infections give rise to severe chronic bronchitis and bronchiectasis. Development of lung abscesses is common. Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms responsible for lung infections. Even more sinister is the increasing frequency of infection with another pseudomonad, Burkholderia cepacia. This opportunistic bacterium is particularly hardy, and infection with this organism has been associated with fulminant illness (“cepacia syndrome”). The liver involvement follows the same basic pattern. Bile canaliculi are plugged by mucinous material, accompanied by ductular proliferation and portal inflammation. Hepatic steatosis is a common finding in liver biopsies. Over time, cirrhosis develops, resulting in diffuse hepatic nodularity. Such severe hepatic involvement is encountered in less than 10% of patients. Azoospermia and infertility are found in 95% of the affected males who survive to adulthood; bilateral absence of the vas deferens is a frequent finding in these patients. In some males, this may be the only feature suggesting an underlying CFTR mutation.
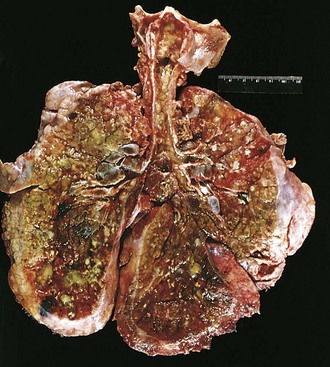
Figure 6–6 Lungs of a patient who died of cystic fibrosis. Extensive mucous plugging and dilation of the tracheobronchial tree are apparent. The pulmonary parenchyma is consolidated by a combination of both secretions and pneumonia; the greenish discoloration is the product of Pseudomonas infections.
(Courtesy of Dr. Eduardo Yunis, Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania.)
Clinical Course
In few childhood diseases are clinical manifestations as protean as those of CF (Table 6–3). The signs and symptoms are extremely varied and range from mild to severe, from presence at birth to onset years later, and from confinement to one organ system to involvement of many. Approximately 5% to 10% of the cases come to clinical attention at birth or soon after because of an attack of meconium ileus. Exocrine pancreatic insufficiency occurs in a majority (85% to 90%) of patients with CF and is associated with “severe” CFTR mutations on both alleles (e.g., ΔF508/ΔF508), whereas 10% to 15% of patients with one “severe” and one “mild” CFTR mutation, or two “mild” CFTR mutations, retain sufficient pancreatic exocrine function that enzyme supplementation is not required—the pancreas-sufficient phenotype. Pancreatic insufficiency is associated with malabsorption of protein and fat and increased fecal loss. Manifestations of malabsorption (e.g., large, foul-smelling stools; abdominal distention; poor weight gain) appear during the first year of life. The faulty fat absorption may induce deficiency states of the fat-soluble vitamins, resulting in manifestations of avitaminosis A, D, or K. Hypoproteinemia may be severe enough to cause generalized edema. Persistent diarrhea may result in rectal prolapse in as many as 10% of children with CF. The pancreas-sufficient phenotype usually is not associated with other gastrointestinal complications, and in general, these patients demonstrate excellent growth and development. “Idiopathic” chronic pancreatitis occurs in a subset of patients with pancreas-sufficient CF and is associated with recurring episodes of abdominal pain with life-threatening complications.
Table 6–3 Clinical Features and Diagnostic Criteria for Cystic Fibrosis
Adapted with permission from Rosenstein BJ, Cutting GR: The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 132:589, 1998.
Cardiorespiratory complications, such as chronic cough, persistent lung infections, obstructive pulmonary disease, and cor pulmonale, constitute the most common cause of death (accounting for approximately 80% of fatalities) in patients who receive follow-up care in most CF centers in the United States. By 18 years of age, 80% of patients with classic CF harbor P. aeruginosa, and 3.5% harbor B. cepacia. With the indiscriminate use of antibiotic prophylaxis against Staphylococcus, there has been an unfortunate resurgence of resistant strains of Pseudomonas in many patients. Recurrent sinonasal polyps can occur in as many as 10% to 25% of patients with CF; accordingly, children who present with such polyps should be tested for abnormalities of sweat chloride. Significant liver disease occurs late in the natural history of CF and is foreshadowed by pulmonary and pancreatic involvement; with increasing life expectancy, liver disease is now the third most common cause of death in patients with CF (after cardiopulmonary and transplant-related complication).
In most cases, the diagnosis of CF is based on persistently elevated sweat electrolyte concentrations (often the mother makes the diagnosis because her infant “tastes salty”), characteristic clinical findings (sinopulmonary disease and gastrointestinal manifestations), or a family history. Sequencing the CFTR gene is, of course, the standard modality for diagnosis of CF. Therefore, in patients with clinical findings or family history (or both) suggestive of this disorder, genetic analysis may be warranted. Advances in management of CF have meant that more patients are now surviving to adulthood; the median life expectancy is now 36 years and continues to increase. Clinical trials with gene therapy in humans are still in their early stages but provide a source of encouragement for millions of patients with CF worldwide.
Summary
Cystic Fibrosis
• CF is an autosomal recessive disease caused by mutations in the CFTR gene encoding the CF transmembrane regulator.
• The principal defect is of chloride ion transport, resulting in high salt concentrations in sweat and in viscous luminal secretions in respiratory and gastrointestinal tracts.
• CFTR mutations can be severe (ΔF508), resulting in multisystem disease, or mild, with limited disease extent and severity.
• Cardiopulmonary complications constitute the most common cause of death; pulmonary infections, especially with resistant pseudomonads, are frequent. Bronchiectasis and right-sided heart failure are long-term sequelae.
• Pancreatic insufficiency is extremely common; infertility caused by congenital bilateral absence of vas deferens is a characteristic finding in adult patients with CF.
• Liver disease, including cirrhosis, is increasing in frequency due to improved survival.
Diseases Caused by Mutations in Genes Encoding Enzyme Proteins
Phenylketonuria
There are several variants of phenylketonuria (PKU), an inborn error of metabolism that affects 1 in 10,000 live-born white infants. The most common form, referred to as classic phenylketonuria, is quite common in persons of Scandinavian descent and is distinctly uncommon in African American and Jewish populations.
Homozygotes with this autosomal recessive disorder classically have a severe lack of the enzyme phenylalanine hydroxylase (PAH), leading to hyperphenylalaninemia and PKU. Affected infants are normal at birth but within a few weeks exhibit a rising plasma phenylalanine level, which in some way impairs brain development. Usually, by 6 months of life, severe mental retardation becomes all too evident; less than 4% of untreated phenylketonuric children have intelligence quotients (IQs) greater than 50 or 60. About one third of these children are never able to walk, and two thirds cannot talk. Seizures, other neurologic abnormalities, decreased pigmentation of hair and skin, and eczema often accompany the mental retardation in untreated children. Hyperphenylalaninemia and the resultant mental retardation can be avoided by restriction of phenylalanine intake early in life. Hence, several screening procedures are routinely performed to detect PKU in the immediate postnatal period.
Many female patients with PKU who receive dietary treatment beginning early in life reach child-bearing age and are clinically normal. Most of them have marked hyperphenylalaninemia, because dietary treatment is discontinued after they reach adulthood. Between 75% and 90% of children born to such women are mentally retarded and microcephalic, and 15% have congenital heart disease, even though the infants themselves are heterozygotes. This syndrome, termed maternal PKU, results from the teratogenic effects of phenylalanine or its metabolites that cross the placenta and affect specific fetal organs during development. The presence and severity of the fetal anomalies directly correlate with the maternal phenylalanine level, so it is imperative that maternal dietary restriction of phenylalanine be initiated before conception and continued throughout pregnancy.
The biochemical abnormality in PKU is an inability to convert phenylalanine into tyrosine. In normal children, less than 50% of the dietary intake of phenylalanine is necessary for protein synthesis. The remainder is converted to tyrosine by the phenylalanine hydroxylase system (Fig. 6–7). When phenylalanine metabolism is blocked because of a lack of PAH enzyme, minor shunt pathways come into play, yielding several intermediates that are excreted in large amounts in the urine and in the sweat. These impart a strong musty or mousy odor to affected infants. It is believed that excess phenylalanine or its metabolites contribute to the brain damage in PKU. Concomitant lack of tyrosine (Fig. 6–7), a precursor of melanin, is responsible for the light color of hair and skin.
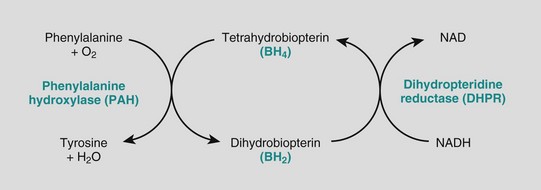
Figure 6–7 The phenylalanine hydroxylase system. NADH, nicotinamide adenine dinucleotide, reduced form.
At the molecular level, approximately 500 mutant alleles of the PAH gene have been identified, only some of which cause a severe deficiency of the enzyme. Infants with mutations resulting in a lack of PAH activity present with the classic features of PKU, while those with approximately 6% residual activity present with milder disease. Moreover, some mutations result in only modest elevations of blood phenylalanine levels without associated neurologic damage. This latter condition, referred to as benign hyperphenylalaninemia, is important to recognize, because affected persons may well have positive screening tests but do not acquire the stigmata of PKU. Because of the numerous disease-causing alleles of the phenylalanine hydroxylase gene, molecular diagnosis is not feasible, and measurement of serum phenylalanine levels is necessary to differentiate benign hyperphenylalaninemia from PKU; the levels in the latter disorder typically are five times (or more) higher than normal. Once a biochemical diagnosis is established, the specific mutation causing PKU can be determined. With this information, carrier testing of at-risk family members can be performed.
While 98% of cases of PKU are attributable to mutations in PAH, approximately 2% arise from abnormalities in synthesis or recycling of the cofactor tetrahydrobiopterin (Fig. 6–7). Clinical recognition of these variant forms of PKU is important to establish a prognosis, because the patients cannot be treated by dietary restriction of phenylalanine.
Galactosemia
Galactosemia is an autosomal recessive disorder of galactose metabolism that affects 1 in 60,000 live-born infants. Normally, lactase splits lactose, the major carbohydrate of mammalian milk, into glucose and galactose in the intestinal microvilli. Galactose is then converted to glucose in several steps, in one of which the enzyme galactose-1-phosphate uridyltransferase (GALT) is required. Lack of this enzyme, due to homozygous mutations in the encoding gene GALT, is responsible for galactosemia. As a result of this transferase deficiency, galactose-1-phosphate and other metabolites, including galactitol, accumulate in many tissues, including the liver, spleen, lens of the eye, kidney, and cerebral cortex.
The liver, eyes, and brain bear the brunt of the damage. The early-onset hepatomegaly is due largely to fatty change, but in time widespread scarring that closely resembles the cirrhosis of alcohol abuse may supervene (Chapter 15). Opacification of the lens (cataract) develops, probably because the lens absorbs water and swells as galactitol, produced by alternative metabolic pathways, accumulates and increases its tonicity. Nonspecific alterations appear in the central nervous system (CNS), including loss of nerve cells, gliosis, and edema. There is still no clear understanding of the mechanism of injury to the liver and brain.
Almost from birth, affected infants fail to thrive. Vomiting and diarrhea appear within a few days of milk ingestion. Jaundice and hepatomegaly usually become evident during the first week of life. Accumulation of galactose and galactose-1-phosphate in the kidney impairs amino acid transport, resulting in aminoaciduria. Fulminant Escherichia coli septicemia occurs with increased frequency. The diagnosis of galactosemia can be suspected from demonstration in the urine of a reducing sugar other than glucose, but tests that directly identify the deficiency of the transferase in leukocytes and red cells are more reliable. Antenatal diagnosis is possible by assay of GALT activity in cultured amniotic fluid cells or determination of galactitol level in amniotic fluid supernatant.
Many of the clinical and morphologic changes of galactosemia can be prevented or ameliorated by early removal of galactose from the diet for at least the first 2 years of life. Control instituted soon after birth prevents the cataracts and liver damage and permits almost normal development. Even with dietary restrictions, however, it is now established that older patients frequently are affected by a speech disorder and gonadal failure (especially premature ovarian failure) and, less commonly, by an ataxic condition.
Summary
Phenylketonuria
• PKU is a disorder of autosomal recessive inheritance caused by a lack of the enzyme phenylalanine hydroxylase and consequent inability to metabolize phenylalanine.
• Clinical features of untreated PKU may include severe mental retardation, seizures, and decreased pigmentation of skin, which can be avoided by restricting the intake of phenylalanine in the diet.
• Female patients with PKU who discontinue dietary treatment can give birth to children with malformations and neurologic impairment resulting from transplacental passage of phenylalanine metabolites.
Galactosemia
• Galactosemia is caused by an inherited lack of the GALT enzyme, leading to accumulation of galactose-1-phosphate and its metabolites in tissues.
• Clinical features may include jaundice, liver damage, cataracts, neural damage, vomiting and diarrhea, and E. coli sepsis. Dietary restriction of galactose can prevent at least some of the more severe complications.
Lysosomal Storage Diseases
Lysosomes, the digestive system of the cells, contain a variety of hydrolytic enzymes that are involved in the breakdown of complex substrates, such as sphingolipids and mucopolysaccharides, into soluble end products. These large molecules may be derived from the turnover of intracellular organelles that enter the lysosomes by autophagy, or they may be acquired from outside the cell by phagocytosis. With an inherited lack of a lysosomal enzyme, catabolism of its substrate remains incomplete, leading to accumulation of the partially degraded insoluble metabolites within the lysosomes (Fig. 6–8). Approximately 40 lysosomal storage diseases have been identified, each resulting from the functional absence of a specific lysosomal enzyme or proteins involved in their function. Traditionally, lysosomal storage disorders are divided into broad categories based on the biochemical nature of the substrates and the accumulated metabolites, but a more mechanistic classification is based on the underlying molecular defect (Table 6–4). Within each group are several entities, each resulting from the deficiency of a specific enzyme. Despite this complexity, certain features are common to most diseases in this group:
• Autosomal recessive transmission
• Patient population consisting of infants and young children
• Storage of insoluble intermediates in the mononuclear phagocyte system, giving rise to hepatosplenomegaly
• Frequent CNS involvement with associated neuronal damage
• Cellular dysfunctions, caused not only by storage of undigested material but also by a cascade of secondary events triggered, for example, by macrophage activation and release of cytokines
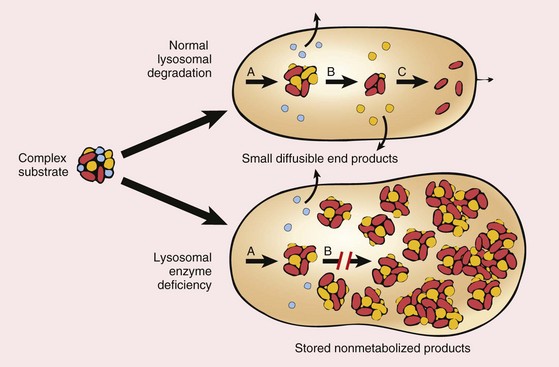
Figure 6–8 Pathogenesis of lysosomal storage diseases. In this example, a complex substrate is normally degraded by a series of lysosomal enzymes (A, B, and C) into soluble end products. If there is a deficiency or malfunction of one of the enzymes (e.g., B), catabolism is incomplete, and insoluble intermediates accumulate in the lysosomes.
Table 6–4 Lysosomal Storage Disorders
| Disease Category | Disease | Deficiency |
|---|---|---|
| Primary lysosomal hydrolase defect | Gaucher disease | Glucocerebrosidase |
| GM1 gangliosidosis | GM1-β-galactosidase | |
| Tay-Sachs disease | Hexosaminidase, α subunit | |
| Sandhoff disease | Hexosaminidase, β subunit | |
| Fabry disease | α-Galactosidase A | |
| Krabbe disease | Galactosylceramidase | |
| Niemann-Pick disease types A and B | Sphingomyelinase | |
| Posttranslational processing defect of lysosomal enzymes | Mucosulfatidosis (juvenile sulfatidosis) | Multiple sulfatases |
| Inefficient targeting of synthesized hydrolase to the lysosome | Mucolipidosis types II and III alpha/beta | N-acetyl glucosamine-1-phosphotransferase |
| Defect in lysosomal enzyme protection | Galactosialidosis | Protective protein cathepsin A (β-galactosidase and neuraminidase) |
| Defect in soluble nonenzymatic lysosomal proteins | GM2 activator protein deficiency, variant AB | GM2 activator protein |
| Sphingolipid activator protein deficiency | Sphingolipid activator protein | |
| Transmembrane (nonenzymatic) protein deficiency | Niemann-Pick disease type C (NPC) | NPC1 and NPC2 |
| Salla disease (free sialic acid storage) | Sialin |
Data from Jeyakumar M, Dwek RA, Butters TD, Platt FM: Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci 6:1, 2005.
Fortunately for the potential victims of the diseases, most of these conditions are very rare, and their detailed description is better relegated to specialized texts and reviews. Only a few of the more common conditions are considered here. Type II glycogen storage disease (Pompe disease), also a lysosomal disorder, is discussed later in the chapter.
Tay-Sachs Disease (GM2 Gangliosidosis: Deficiency in Hexosaminidase β Subunit)
Gangliosidoses are characterized by accumulation of gangliosides, principally in the brain, as a result of a deficiency of a catabolic lysosomal enzyme. Depending on the ganglioside involved, these disorders are subclassified into GM1 and GM2 categories. Tay-Sachs disease, by far the most common of all gangliosidoses, is characterized by a mutation in and consequent deficiency of the β subunit of the enzyme hexosaminidase A, which is necessary for the degradation of GM2. More than 100 mutations have been described; most affect protein folding or intracellular transport. The brain is principally affected, because it is most involved in ganglioside metabolism. The storage of GM2 occurs within neurons, axon cylinders of nerves, and glial cells throughout the CNS. Affected cells appear swollen and sometimes foamy (Fig. 6–9, A). Electron microscopy reveals whorled configurations within lysosomes composed of onion-skin layers of membranes (Fig. 6–9, B). These pathologic changes are found throughout the CNS (including the spinal cord), peripheral nerves, and autonomic nervous system. The retina usually is involved as well, where the pallor produced by swollen ganglion cells in the peripheral retina results in a contrasting “cherry red” spot in the relatively unaffected central macula.
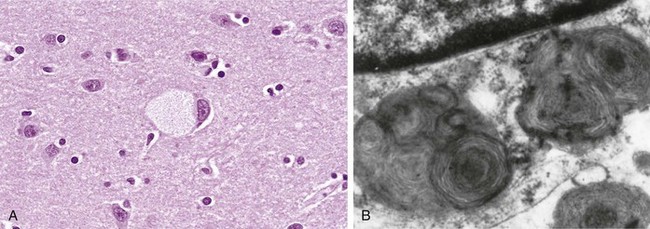
Figure 6–9 Ganglion cells in Tay-Sachs disease. A, Under the light microscope, a large neuron has obvious lipid vacuolation. B, A portion of a neuron under the electron microscope shows prominent lysosomes with whorled configurations. Part of the nucleus is shown above.
(A, Courtesy of Dr. Arthur Weinberg, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas. B, Courtesy of Dr. Joe Rutledge, Children’s Regional Medical Center, Seattle, Washington.)
The molecular basis for neuronal injury is not fully understood. Because in many cases the mutant protein is misfolded, it induces the so-called “unfolded protein” response (Chapter 1). If such misfolded proteins are not stabilized by chaperones, they trigger apoptosis. These findings have spurred clinical trials of molecular chaperone therapy for this and similar lysosomal storage diseases. Such therapy involves use of small molecules that increase chaperone synthesis or reduce degradation of misfolded proteins by the proteosomes.
In the most common acute infantile variant of Tay-Sachs disease, infants appear normal at birth, but motor weakness begins at 3 to 6 months of age, followed by neurologic impairment, onset of blindness, and progressively more severe neurologic dysfunctions. Death occurs within 2 or 3 years. Tay-Sachs disease, like other lipidoses, is most common among Ashkenazi Jews, among whom the frequency of heterozygous carriers is estimated to be 1 in 30. Heterozygote carriers can be reliably detected by estimation of the level of hexosaminidase in the serum or by DNA analysis.
Niemann-Pick Disease Types A and B
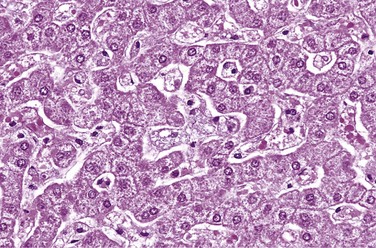
Type A and type B Niemann-Pick disease are related entities characterized by a primary deficiency of acid sphingomyelinase and the resultant accumulation of sphingomyelin. In type A, characterized by a severe deficiency of sphingomyelinase, the breakdown of sphingomyelin into ceramide and phosphorylcholine is impaired, and excess sphingomyelin accumulates in all phagocytic cells and in the neurons. The macrophages become stuffed with droplets or particles of the complex lipid, imparting a fine vacuolation or foaminess to the cytoplasm (Fig. 6–10). Electron microscopy confirms that the vacuoles are engorged secondary lysosomes that often contain membranous cytoplasmic bodies resembling concentric lamellated myelin figures, sometimes called “zebra” bodies. Because of their high content of phagocytic cells, the organs most severely affected are the spleen, liver, bone marrow, lymph nodes, and lungs. The splenic enlargement may be striking. In addition, the entire CNS, including the spinal cord and ganglia, is involved in this tragic, inexorable process. The affected neurons are enlarged and vacuolated as a result of the storage of lipids. This variant manifests itself in infancy with massive visceromegaly and severe neurologic deterioration. Death usually occurs within the first 3 years of life. By comparison, patients with the type B variant have organomegaly but no neurologic manifestations. Estimation of sphingomyelinase activity in the leukocytes or cultured fibroblasts can be used for diagnosis of suspected cases, as well as for detection of carriers. Antenatal diagnosis is possible by enzyme assays or DNA probe analysis.
Niemann-Pick Disease Type C
Although previously considered to be related to type A and type B Niemann-Pick disease, type C (NPC) is quite distinct at the biochemical and molecular levels and is more common than types A and B combined. Mutations in two related genes, NPC1 and NPC2, can give rise to the disorder, with NPC1 being responsible for a majority of cases. Unlike most other lysosomal storage diseases, NPC is due to a primary defect in lipid transport. Affected cells accumulate cholesterol as well as gangliosides such as GM1 and GM2. Both NPC1 and NPC2 are involved in the transport of free cholesterol from the lysosomes to the cytoplasm. NPC is clinically heterogeneous: The most common form manifests in childhood and is marked by ataxia, vertical supranuclear gaze palsy, dystonia, dysarthria, and psychomotor regression.
Gaucher Disease
Gaucher disease results from mutation in the gene that encodes glucocerebrosidase. There are three autosomal recessive variants of Gaucher disease resulting from distinct allelic mutations. Common to all is variably deficient activity of a glucocerebrosidase that normally cleaves the glucose residue from ceramide. This deficit leads to an accumulation of glucocerebroside, an intermediate in glycolipid metabolism, in the mononuclear phagocytic cells and their transformation into so-called Gaucher cells. Normally the glycolipids derived from the breakdown of senescent blood cells are sequentially degraded by the phagocytic cells of the body particularly in the liver, spleen, and bone marrow. In Gaucher disease, the degradation stops at the level of glucocerebrosides, which accumulate in the phagocytes. These phagocytes—the Gaucher cells—become enlarged, with some reaching a diameter as great as 100 µm, because of the accumulation of distended lysosomes, and acquire a pathognomonic cytoplasmic appearance characterized as “wrinkled tissue paper” (Fig. 6–11). No distinct vacuolation is present. It is evident now that Gaucher disease is caused not just by the burden of storage material but also by activation of the macrophages. High levels of macrophage-derived cytokines, such as interleukins (IL-1, IL-6) and tumor necrosis factor (TNF), are found in affected tissues.
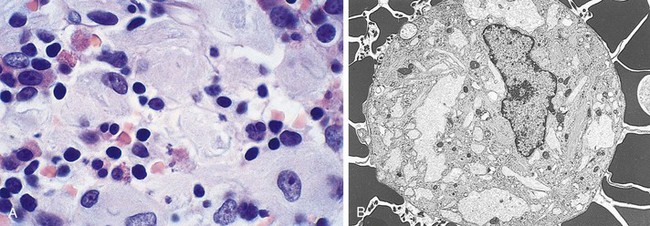
Figure 6–11 Gaucher disease involving the bone marrow. A, Gaucher cells with abundant lipid-laden granular cytoplasm. B, Electron micrograph of Gaucher cells with elongated distended lysosomes.
(Courtesy of Dr. Matthew Fries, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas.)
One variant, type I, also called the chronic non-neuronopathic form, accounts for 99% of cases of Gaucher disease. It is characterized by clinical or radiographic bone involvement (osteopenia, focal lytic lesions, and osteonecrosis) in 70% to 100% of cases. Additional features are hepatosplenomegaly and the absence of CNS involvement. The spleen often enlarges to massive proportions, filling the entire abdomen. Gaucher cells are found in the liver, spleen, lymph nodes, and bone marrow. Marrow replacement and cortical erosion may produce radiographically visible skeletal lesions, as well as a reduction in the formed elements of blood. Bone changes are believed to be caused by the aforementioned macrophage-derived cytokines. Type I is most common in Ashkenazi Jews; unlike other variants, it is compatible with long life. Types II and III variants are characterized by neurologic signs and symptoms. In type II, these manifestations appear during infancy (acute infantile neuronopathic form) and are more severe, whereas in type III, they emerge later and are milder (chronic neuronopathic form). Although the liver and spleen also are involved, the clinical features in types II and III are dominated by neurologic disturbances, including convulsions and progressive mental deterioration. The level of glucocerebrosides in leukocytes or cultured fibroblasts is helpful in diagnosis and in the detection of heterozygote carriers.
Current therapy is aimed at lifelong enzyme replacement by infusion of recombinant glucocerebrosidase. A newer form of therapy involves reducing the substrate (glucocerebroside) by oral administration of drugs that inhibit glucocerebroside synthase. Since glucosylceramide is reduced, its accumulation also is reduced. Recent clinical trials in humans have shown considerable promise for this modality of therapy, with decrease in splenomegaly and improvements in skeletal disease. On the horizon is glucocerebrosidase gene therapy involving infusion of autologous hematopoietic stem cells transfected with the normal gene.
Mucopolysaccharidoses
Mucopolysaccharidoses (MPSs) are characterized by defective degradation (and therefore excessive storage) of mucopolysaccharides in various tissues. Recall that mucopolysaccharides form a part of ground substance and are synthesized by connective tissue fibroblasts. Most of the mucopolysaccharide is secreted into the ground substance, but a certain fraction is degraded within lysosomes. Multiple enzymes are involved in this catabolic pathway; it is the lack of these enzymes that leads to accumulation of mucopolysaccharides within the lysosomes. Several clinical variants of MPS, classified numerically from MPS I to MPS VII, have been described, each resulting from the deficiency of one specific enzyme. The mucopolysaccharides that accumulate within the tissues include dermatan sulfate, heparan sulfate, keratan sulfate, and (in some cases) chondroitin sulfate.
Hepatosplenomegaly, skeletal deformities, lesions of heart valves, and subendothelial arterial deposits, particularly in the coronary arteries, and lesions in the brain, are common threads that run through all of the MPSs. In many of the more protracted syndromes, coronary subendothelial lesions lead to myocardial ischemia. Thus, myocardial infarction and cardiac decompensation are important causes of death. Most cases are associated with coarse facial features, clouding of the cornea, joint stiffness, and mental retardation. Urinary excretion of the accumulated mucopolysaccharides often is increased. With all of these disorders except one, the mode of inheritance is autosomal recessive; the exception, Hunter syndrome, is an X-linked recessive disease. Of the seven recognized variants, only two well-characterized syndromes are discussed briefly here.
MPS type I, also known as Hurler syndrome, is caused by a deficiency of α-l-iduronidase. In Hurler syndrome, affected children have a life expectancy of 6 to 10 years, and death is often due to cardiac complications. Accumulation of dermatan sulfate and heparan sulfate is seen in cells of the mononuclear phagocyte system, in fibroblasts, and within endothelium and smooth muscle cells of the vascular wall. The affected cells are swollen and have clear cytoplasm, resulting from the accumulation of material positive for periodic acid–Schiff staining within engorged, vacuolated lysosomes. Lysosomal inclusions also are found in neurons, accounting for the mental retardation.
The other well-characterized variant, MPS type II or Hunter syndrome, differs from Hurler syndrome in its mode of inheritance (X-linked), the absence of corneal clouding, and often its milder clinical course. As in Hurler syndrome, the accumulated mucopolysaccharides in Hunter syndrome are heparan sulfate and dermatan sulfate, but this results from a deficiency of l-iduronate sulfatase. Despite the difference in enzyme deficiency, an accumulation of identical substrates occurs because breakdown of heparan sulfate and dermatan sulfate requires both α-l-iduronidase and the sulfatase; if either one is missing, further degradation is blocked.
Summary
Lysosomal Storage Diseases
• Tay-Sachs disease is caused by an inability to metabolize GM2 gangliosides due to lack of the β subunit of lysosomal hexosaminidase. GM2 gangliosides accumulate in the CNS and cause severe mental retardation, blindness, motor weakness, and death by 2 to 3 years of age.
• Niemann-Pick disease types A and B are caused by a deficiency of sphingomyelinase. In the more severe, type A variant, accumulation of sphingomyelin in the nervous system results in neuronal damage. Lipid also is stored in phagocytes within the liver, spleen, bone marrow, and lymph nodes, causing their enlargement. In type B, neuronal damage is not present.
• Niemann-Pick disease type C is caused by a defect in cholesterol transport and resultant accumulation of cholesterol and gangliosides in the nervous system. Affected children exhibit ataxia, dysarthria, and psychomotor regression.
• Gaucher disease results from lack of the lysosomal enzyme glucocerebrosidase and accumulation of glucocerebroside in mononuclear phagocytic cells. In the most common, type I variant, affected phagocytes become enlarged (Gaucher cells) and accumulate in liver, spleen, and bone marrow, causing hepatosplenomegaly and bone erosion. Types II and III are characterized by variable neuronal involvement.
• Mucopolysaccharidoses result from accumulation of mucopolysaccharides in many tissues including liver, spleen, heart, blood vessels, brain, cornea, and joints. Affected patients in all forms have coarse facial features. Manifestations of Hurler syndrome include corneal clouding, coronary arterial and valvular deposits, and death in childhood. Hunter syndrome is associated with a milder clinical course.
Glycogen Storage Diseases (Glycogenoses)
An inherited deficiency of any one of the enzymes involved in glycogen synthesis or degradation can result in excessive accumulation of glycogen or some abnormal form of glycogen in various tissues. The type of glycogen stored, its intracellular location, and the tissue distribution of the affected cells vary depending on the specific enzyme deficiency. Regardless of the tissue or cells affected, the glycogen most often is stored within the cytoplasm, or sometimes within nuclei. One variant, Pompe disease, is a form of lysosomal storage disease, because the missing enzyme is localized to lysosomes. Most glycogenoses are inherited as autosomal recessive diseases, as is common with “missing enzyme” syndromes.
Approximately a dozen forms of glycogenoses have been described in association with specific enzyme deficiencies. On the basis of pathophysiologic findings, they can be grouped into three categories (Table 6–5):
• Hepatic type. Liver contains several enzymes that synthesize glycogen for storage and also break it down into free glucose. Hence, a deficiency of the hepatic enzymes involved in glycogen metabolism is associated with two major clinical effects: enlargement of the liver due to storage of glycogen and hypoglycemia due to a failure of glucose production (Fig. 6–12). Von Gierke disease (type I glycogenosis), resulting from a lack of glucose-6-phosphatase, is the most important example of the hepatic form of glycogenosis (Table 6–5).
• Myopathic type. In striated muscle, glycogen is an important source of energy. Not surprisingly, most forms of glycogen storage disease affect muscles. When enzymes that are involved in glycolysis are deficient, glycogen storage occurs in muscles and there is an associated muscle weakness due to impaired energy production. Typically, the myopathic forms of glycogen storage diseases are marked by muscle cramps after exercise, myoglobinuria, and failure of exercise to induce an elevation in blood lactate levels because of a block in glycolysis. McArdle disease (type V glycogenosis), resulting from a deficiency of muscle phosphorylase, is the prototype of myopathic glycogenoses.
• Type II glycogenosis (Pompe disease) is caused by a deficiency of lysosomal acid maltase and so is associated with deposition of glycogen in virtually every organ, but cardiomegaly is most prominent. Most affected patients die within 2 years of onset of cardiorespiratory failure. Therapy with the missing enzyme (glucosidase) can reverse cardiac muscle damage and modestly increase longevity.
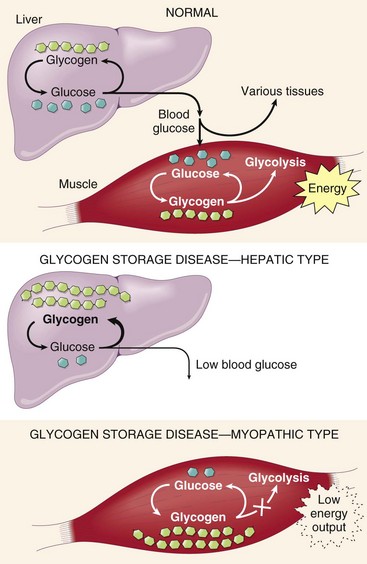
Figure 6–12 Top, A simplified scheme of normal glycogen metabolism in the liver and skeletal muscles. Middle, The effects of an inherited deficiency of hepatic enzymes involved in glycogen metabolism. Bottom, The consequences of a genetic deficiency in the enzymes that metabolize glycogen in skeletal muscles.
Summary
Glycogen Storage Diseases
• Inherited deficiency of enzymes involved in glycogen metabolism can result in storage of normal or abnormal forms of glycogen, predominantly in liver or muscles or in all tissues.
• In the hepatic form (von Gierke disease), liver cells store glycogen because of a lack of hepatic glucose-6-phosphatase. There are several myopathic forms, including McArdle disease, in which muscle phosphorylase lack gives rise to storage in skeletal muscles and cramps after exercise. In Pompe disease there is lack of lysosomal acid maltase, and all organs are affected, but heart involvement is predominant.
Diseases Caused by Mutations in Genes Encoding Proteins That Regulate Cell Growth
As detailed in Chapter 5, two classes of genes, proto-oncogenes and tumor suppressor genes, regulate normal cell growth and differentiation. Mutations affecting these genes, most often in somatic cells, are involved in the pathogenesis of tumors. In approximately 5% to 10% of all cancers, however, mutations affecting certain tumor suppressor genes are present in all cells of the body, including germ cells and hence can be transmitted to the offspring. These mutant genes predispose the offspring to hereditary tumors, a topic discussed in greater detail in Chapter 5.
Complex Multigenic Disorders
Complex multigenic disorders—so-called multifactorial or polygenic disorders—are caused by interactions between variant forms of genes and environmental factors. A genetic variant that has at least two alleles and occurs in at least 1% of the population is called a polymorphism. According to the common disease–common variant hypothesis, complex multigenic disorders occur when many polymorphisms, each with a modest effect and low penetrance, are co-inherited. Two additional important facts have emerged from studies of common complex disorders such as type 1 diabetes:
• While complex disorders result from the collective inheritance of many polymorphisms, different polymorphisms vary in significance. For example, of the 20 to 30 genes implicated in type 1 diabetes, 6 or 7 are most important, and a few HLA alleles contribute more than 50% of the risk (Chapter 19).
• Some polymorphisms are common to multiple diseases of the same type, while others are disease-specific. This observation is well illustrated in immune-mediated inflammatory diseases (Chapter 4).
Several normal phenotypic characteristics are governed by multigenic inheritance, such as hair color, eye color, skin color, height, and intelligence. These characteristics (also known as quantitative trait loci [QTLs]) show a continuous variation within, as well as across, all population groups. Environmental influences, however, significantly modify the phenotypic expression of complex traits. For example, type 2 diabetes mellitus has many of the features of a complex multigenic disorder. It is well recognized clinically that affected persons often first exhibit clinical manifestations of this disease after weight gain. Thus, obesity as well as other environmental influences, unmasks the diabetic genetic trait. Assigning a disease to this mode of inheritance must be done with caution. Such attribution depends on many factors but first on familial clustering and the exclusion of mendelian and chromosomal modes of transmission. A range of levels of severity of a disease is suggestive of a complex multigenic disorder, but as pointed out earlier, variable expressivity and reduced penetrance of single mutant genes also may account for this phenomenon. Because of these problems, sometimes it is difficult to distinguish between mendelian and multifactorial disorders.
Cytogenetic Disorders
Chromosomal abnormalities occur much more frequently than is generally appreciated. It is estimated that approximately 1 in 200 newborn infants has some form of chromosomal abnormality. The figure is much higher in fetuses that do not survive to term. It is estimated that in 50% of first-trimester spontaneous abortions, the fetus has a chromosomal abnormality. Cytogenetic disorders may result from alterations in the number or structure of chromosomes and may affect autosomes or sex chromosomes.
Before embarking on a discussion of chromosomal aberrations, it is appropriate to review karyotyping as the basic tool of the cytogeneticist. A karyotype is a photographic representation of a stained metaphase spread in which the chromosomes are arranged in order of decreasing length. A variety of techniques for staining chromosomes have been developed. With the widely used Giemsa stain (G banding) technique, each chromosome set can be seen to possess a distinctive pattern of alternating light and dark bands of variable widths (Fig. 6–13). The use of banding techniques allows identification of each chromosome, and can detect and localize structural abnormalities large enough to produce changes in banding pattern (described later).
Figure 6–13 G-banded karyotype from a normal male (46,XY). Also shown is the banding pattern of the X-chromosome with nomenclature of arms, regions, bands, and sub-bands.
(Karyotype courtesy of Dr. Stuart Schwartz, Department of Pathology, University of Chicago, Chicago, Illinois.)
Numeric Abnormalities
In humans, the normal chromosome count is 46 (i.e., 2n = 46). Any exact multiple of the haploid number (n) is called euploid. Chromosome numbers such as 3n and 4n are called polyploid. Polyploidy generally results in a spontaneous abortion. Any number that is not an exact multiple of n is called aneuploid. The chief cause of aneuploidy is nondisjunction of a homologous pair of chromosomes at the first meiotic division or a failure of sister chromatids to separate during the second meiotic division. The latter also may occur during mitosis in somatic cells, leading to the production of two aneuploid cells. Failure of pairing of homologous chromosomes followed by random assortment (anaphase lag) can also lead to aneuploidy. When nondisjunction occurs at the time of meiosis, the gametes formed have either an extra chromosome (n + 1) or one less chromosome (n − 1). Fertilization of such gametes by normal gametes would result in two types of zygotes: trisomic, with an extra chromosome (2n + 1), or monosomic (2n − 1). Monosomy involving an autosome is incompatible with life, whereas trisomies of certain autosomes and monosomy involving sex chromosomes are compatible with life. These, as we shall see, are associated with variable degrees of phenotypic abnormality. Mosaicism is a term used to describe the presence of two or more populations of cells with different complements of chromosomes in the same individual. In the context of chromosome numbers, postzygotic mitotic nondisjunction would result in the production of a trisomic and a monosomic daughter cell; the descendants of these cells would then produce a mosaic. As discussed later, mosaicism affecting sex chromosomes is common, whereas autosomal mosaicism is not.
Structural Abnormalities
Structural changes in the chromosomes usually result from chromosomal breakage followed by loss or rearrangement of material. Such changes usually are designated using a cytogenetic shorthand in which p (French, petit) denotes the short arm of a chromosome, and q, the long arm. Each arm is then divided into numbered regions (1, 2, 3, and so on) from centromere outward, and within each region the bands are numerically ordered (Fig. 6–13). Thus, 2q34 indicates chromosome 2, long arm, region 3, band 4. The patterns of chromosomal rearrangement after breakage (Fig. 6–14) are as follows:
• Translocation implies transfer of a part of one chromosome to another chromosome. The process is usually reciprocal (i.e., fragments are exchanged between two chromosomes). In genetic shorthand, translocations are indicated by t followed by the involved chromosomes in numeric order—for example, 46,XX,t(2;5)(q31;p14). This notation would indicate a reciprocal translocation involving the long arm (q) of chromosome 2 at region 3, band 1, and the short arm of chromosome 5, region 1, band 4. When the entire broken fragments are exchanged, the resulting balanced reciprocal translocation (Fig. 6–14) is not harmful to the carrier, who has the normal number of chromosomes and the full complement of genetic material. However, during gametogenesis, abnormal (unbalanced) gametes are formed, resulting in abnormal zygotes. A special pattern of translocation involving two acrocentric chromosomes is called centric fusion type, or robertsonian, translocation. The breaks typically occur close to the centromere, affecting the short arms of both chromosomes. Transfer of the segments leads to one very large chromosome and one extremely small one (Fig. 6–14). The short fragments are lost, and the carrier has 45 chromosomes. Because the short arms of all acrocentric chromosomes carry highly redundant genes (e.g., ribosomal RNA genes), such loss is compatible with survival. However, difficulties arise during gametogenesis, resulting in the formation of unbalanced gametes that could lead to abnormal offspring.
• Isochromosomes result when the centromere divides horizontally rather than vertically. One of the two arms of the chromosome is then lost, and the remaining arm is duplicated, resulting in a chromosome with two short arms only or two long arms only. The most common isochromosome present in live births involves the long arm of the X chromosome and is designated i(Xq). When fertilization occurs by a gamete that contains a normal X chromosome, the result is monosomy for genes on Xp and trisomy for genes on Xq.
• Deletion involves loss of a portion of a chromosome. A single break may delete a terminal segment. Two interstitial breaks, with reunion of the proximal and distal segments, may result in loss of an intermediate segment. The isolated fragment, which lacks a centromere, almost never survives, and thus many genes are lost.
• Inversions occur when there are two interstitial breaks in a chromosome, and the segment reunites after a complete turnaround.
• A ring chromosome is a variant of a deletion. After loss of segments from each end of the chromosome, the arms unite to form a ring.
General Features of Chromosomal Disorders
• Chromosomal disorders may be associated with absence (deletion, monosomy), excess (trisomy), or abnormal rearrangements (translocations) of chromosomes.
• In general, loss of chromosomal material produces more severe defects than does gain of chromosomal material.
• Excess chromosomal material may result from a complete chromosome (as in trisomy) or from part of a chromosome (as in robertsonian translocation).
• Imbalances of sex chromosomes (excess or loss) are tolerated much better than are similar imbalances of autosomes.
• Sex chromosomal disorders often produce subtle abnormalities, sometimes not detected at birth. Infertility, a common manifestation, cannot be diagnosed until adolescence.
• In most cases, chromosomal disorders result from de novo changes (i.e., parents are normal, and risk of recurrence in siblings is low). An uncommon but important exception to this principle is exhibited by the translocation form of Down syndrome (described later).
Some specific examples of diseases involving changes in the karyotype are presented next.
Cytogenetic Disorders Involving Autosomes
Three autosomal trisomies (21, 18, and 13) and one deletion syndrome (cri du chat syndrome), which results from partial deletion of the short arm of chromosome 5, were the first chromosomal abnormalities identified. More recently, several additional trisomies and deletion syndromes (such as that affecting 22q) have been described. Most of these disorders are quite uncommon, but their clinical features should permit ready recognition (Fig. 6–15).
Only trisomy 21 and 22q11.2 deletion occur with sufficient frequency to merit further consideration.
Trisomy 21 (Down Syndrome)
Down syndrome is the most common of the chromosomal disorders. About 95% of affected persons have trisomy 21, so their chromosome count is 47. As mentioned earlier, the most common cause of trisomy, and therefore of Down syndrome, is meiotic nondisjunction. The parents of such children have a normal karyotype and are normal in all respects. Maternal age has a strong influence on the incidence of Down syndrome. It occurs in 1 in 1550 live births in women younger than 20 years, in contrast with 1 in 25 live births in women older than 45 years. The correlation with maternal age suggests that in most cases the meiotic nondisjunction of chromosome 21 occurs in the ovum. Indeed, in 95% of cases the extra chromosome is of maternal origin. The reason for the increased susceptibility of the ovum to nondisjunction is not fully understood. No effect of paternal age has been found in those cases in which the extra chromosome is derived from the father.
In about 4% of all patients with trisomy 21, the extra chromosomal material is present not as an extra chromosome but as a translocation of the long arm of chromosome 21 to chromosome 22 or 14. Such cases frequently (but not always) are familial, and the translocated chromosome is inherited from one of the parents, who typically is a carrier of a robertsonian translocation. Approximately 1% of patients with trisomy 21 are mosaics, usually having a mixture of 46- and 47-chromosome cells. These cases result from mitotic nondisjunction of chromosome 21 during an early stage of embryogenesis. Clinical manifestations in such cases are variable and milder, depending on the proportion of abnormal cells.
The diagnostic clinical features of this condition—flat facial profile, oblique palpebral fissures, and epicanthic folds (Fig. 6–15) —are usually readily evident, even at birth. Down syndrome is a leading cause of severe mental retardation; approximately 80% of those afflicted have an IQ of 25 to 50. Ironically, these severely disadvantaged children may have a gentle, shy manner and may be more easily directed than their more fortunate normal siblings. Of interest, some mosaics with Down syndrome have mild phenotypic changes and often even have normal or near-normal intelligence. In addition to the phenotypic abnormalities and the mental retardation already noted, some other clinical features are worthy of mention:
• Approximately 40% of the patients have congenital heart disease, most commonly defects of the endocardial cushion, including atrial septal defects, atrioventricular valve malformations, and ventricular septal defects (Chapter 10). Cardiac problems are responsible for a majority of the deaths in infancy and early childhood. Several other congenital malformations, including atresias of the esophagus and small bowel, also are common.
• Children with trisomy 21 have a 10- to 20-fold increased risk of developing acute leukemia. Both acute lymphoblastic leukemias and acute myeloid leukemias occur (Chapter 11).
• Virtually all patients with trisomy 21 older than age 40 develop neuropathologic changes characteristic of Alzheimer disease, a degenerative disorder of the brain (Chapter 22).
• Patients with Down syndrome demonstrate abnormal immune responses that predispose them to serious infections, particularly of the lungs, and to thyroid autoimmunity (Chapter 19). Although several abnormalities, affecting mainly T cell functions, have been reported, the basis for the immunologic disturbances is not clear.
Despite all of these problems, improved medical care has increased the longevity of persons with trisomy 21. Currently the median age at death is 47 years (up from 25 years in 1983). Although the karyotype of Down syndrome has been known for decades, the molecular basis for this disease remains elusive. Data from the human genome project indicate that chromosome 21 carries about 500 annotated genes, including approximately 170 that are conserved protein-coding genes and 5 miRNAs. It is unclear whether the phenotype of Down syndrome arises as a consequence of increased gene dosage of protein coding genes on chromosome 21 itself or of the effects of deregulated miRNA expression on target genes located on other chromosomes (as described previously, miRNAs act through inhibition of target gene expression). Two chromosome 21 candidate genes, DYRK1A, which codes for a serine-threonine kinase, and RCAN1 (regulator of calcineurin 1), which codes for a protein that inhibits a critical cellular phosphatase enzyme called calcineurin, have emerged as the “top culprits” in the pathogenesis of Down syndrome.
22q11.2 Deletion Syndrome
The 22q11.2 deletion syndrome encompasses a spectrum of disorders that result from a small interstitial deletion of band 11 on the long arm of chromosome 22. The clinical features of this deletion syndrome include congenital heart disease affecting the outflow tracts, abnormalities of the palate, facial dysmorphism, developmental delay, thymic hypoplasia with impaired T cell immunity (Chapter 4), and parathyroid hypoplasia resulting in hypocalcemia (Chapter 19). Previously, these clinical features were believed to represent two different disorders: DiGeorge syndrome and velocardiofacial syndrome. However, it is now known that both are caused by 22q11.2 deletion. Variations in the size and position of the deletion are thought to be responsible for the differing clinical manifestations. When T cell immunodeficiency and hypocalcemia are the dominant features, the patients are said to have DiGeorge syndrome, whereas patients with the so-called velocardiofacial syndrome have mild immunodeficiency but pronounced dysmorphology and cardiac defects. In addition to these malformations, patients with 22q11.2 deletion are at particularly high risk for psychoses such as schizophrenia and bipolar disorder. The molecular basis for this syndrome is not fully understood. The affected region of chromosome 11 encodes many genes. Among these, a transcription factor gene called TBX1 is suspected to be responsible, since its loss seems to correlate with the occurrence of DiGeorge syndrome.
The diagnosis of this condition may be suspected on clinical grounds but can be established only by detection of the deletion by fluorescence in situ hybridization (FISH) (Fig. 6–37, B).
Summary
Cytogenetic Disorders Involving Autosomes
• Down syndrome is associated with an extra copy of genes on chromosome 21, most commonly due to trisomy 21 and less frequently from translocation of extra chromosomal material from chromosome 21 to other chromosomes or from mosaicism.
• Patients with Down syndrome have severe mental retardation, flat facial profile, epicanthic folds, cardiac malformations, higher risk of leukemia and infections, and premature development of Alzheimer disease.
• Deletion of genes at chromosomal locus 22q11.2 gives rise to malformations affecting the face, heart, thymus, and parathyroids. The resulting disorders are recognized as (1) DiGeorge syndrome (thymic hypoplasia with diminished T cell immunity and parathyroid hypoplasia with hypocalcemia) and (2) velocardiofacial syndrome (congenital heart disease involving outflow tracts, facial dysmorphism, and developmental delay).
Cytogenetic Disorders Involving Sex Chromosomes
A number of abnormal karyotypes involving the sex chromosomes, ranging from 45,X to 49,XXXXY, are compatible with life. Indeed, phenotypically normal males with two and even three Y chromosomes have been identified. Such extreme karyotypic deviations are not encountered with the autosomes. In large part, this latitude relates to two factors: (1) lyonization of X chromosomes and (2) the small amount of genetic information carried by the Y chromosome. The consideration of lyonization must begin with Mary Lyon, who in 1962 proposed that in females, only one X chromosome is genetically active. X inactivation occurs early in fetal life, about 16 days after conception: Either the paternal or the maternal X chromosome is randomly inactivated in each of the primitive cells representing the developing embryo. Once inactivated, the same X chromosome remains genetically neutralized in all of the progeny of these cells. Moreover, all but one X chromosome is inactivated, and so a 48,XXXX female has only one active X chromosome. This phenomenon explains why normal females do not have a double dose (compared with males) of phenotypic attributes encoded on the X chromosome. The Lyon hypothesis also explains why normal females are in reality mosaics, containing two cell populations: one with an active maternal X, the other with an active paternal X.
Although essentially accurate, the Lyon hypothesis subsequently has been somewhat modified. Most important, the initial presumption that all of the genes on the inactive X are “switched off” has been revised as more recent studies suggest that 21% of genes on Xp, and a smaller number (3%) on Xq, escape X inactivation. This possibility has implications for monosomic X chromosome disorders, or Turner syndrome, as discussed later on.
Extra Y chromosomes are readily tolerated because the only information known to be carried on the Y chromosome seems to relate to male differentiation. Of note, whatever the number of X chromosomes, the presence of a Y invariably dictates the male phenotype. The gene for male differentiation (SRY, sex-determining region of Y chromosome) is located on the short arm of the Y.
Described briefly next are two disorders, Klinefelter syndrome and Turner syndrome, that result from aberrations of sex chromosomes.
Klinefelter Syndrome
Klinefelter syndrome is best defined as male hypogonadism that develops when there are at least two X chromosomes and one or more Y chromosomes. Most affected patients have a 47,XXY karyotype. This karyotype results from nondisjunction of sex chromosomes during meiosis. The extra X chromosome may be of either maternal or paternal origin. Advanced maternal age and a history of irradiation in either parent may contribute to the meiotic error resulting in this condition. Approximately 15% of the patients show mosaic patterns, including 46,XY/47,XXY, 47,XXY/48,XXXY, and variations on this theme. The presence of a 46,XY line in mosaics usually is associated with a milder clinical condition.
Klinefelter syndrome is associated with a wide range of clinical manifestations. In some persons it may be expressed only as hypogonadism, but most patients have a distinctive body habitus with an increase in length between the soles and the pubic bone, which creates the appearance of an elongated body. Also characteristic is eunuchoid body habitus. Reduced facial, body, and pubic hair and gynecomastia also are frequently seen. The testes are markedly reduced in size, sometimes to only 2 cm in greatest dimension. In keeping with the testicular atrophy, the serum testosterone levels are lower than normal, and urinary gonadotropin levels are elevated.
Klinefelter syndrome is the most common cause of hypogonadism in males. Only rarely are patients fertile, and presumably such persons are mosaics with a large proportion of 46,XY cells. The sterility is due to impaired spermatogenesis, sometimes to the extent of total azoospermia. Histologic examination reveals hyalinization of tubules, which appear as ghostlike structures on tissue section. By contrast, Leydig cells are prominent, as a result of either hyperplasia or an apparent increase related to loss of tubules. Although Klinefelter syndrome may be associated with mental retardation, the degree of intellectual impairment typically is mild, and in some cases, no deficit is detectable. The reduction in intelligence is correlated with the number of extra X chromosomes. Klinefelter syndrome is associated with a higher frequency of several disorders, including breast cancer (seen 20 times more commonly than in normal males), extragonadal germ cell tumors, and autoimmune diseases such as systemic lupus erythematosus.
Turner Syndrome
Turner syndrome, characterized by primary hypogonadism in phenotypic females, results from partial or complete monosomy of the short arm of the X chromosome. With routine cytogenetic methods, the entire X chromosome is found to be missing in 57% of patients, resulting in a 45,X karyotype. These patients are the most severely affected, and the diagnosis often can be made at birth or early in childhood. Typical clinical features associated with 45,X Turner syndrome include significant growth retardation, leading to abnormally short stature (below the third percentile); swelling of the nape of the neck due to distended lymphatic channels (in infancy) that is seen as webbing of the neck in older children; low posterior hairline; cubitus valgus (an increase in the carrying angle of the arms); shieldlike chest with widely spaced nipples; high-arched palate; lymphedema of the hands and feet; and a variety of congenital malformations such as horseshoe kidney, bicuspid aortic valve, and coarctation of the aorta (Fig. 6–16). Cardiovascular abnormalities are the most common cause of death in childhood. In adolescence, affected girls fail to develop normal secondary sex characteristics; the genitalia remain infantile, breast development is minimal, and little pubic hair appears. Most patients have primary amenorrhea, and morphologic examination reveals transformation of the ovaries into white streaks of fibrous stroma devoid of follicles. The mental status of these patients usually is normal, but subtle defects in nonverbal, visual-spatial information processing have been noted. Curiously, hypothyroidism caused by autoantibodies occurs, especially in women with isochromosome Xp. As many as 50% of these patients develop clinical hypothyroidism. In adult patients, a combination of short stature and primary amenorrhea should prompt strong suspicion for Turner syndrome. The diagnosis is established by karyotyping.
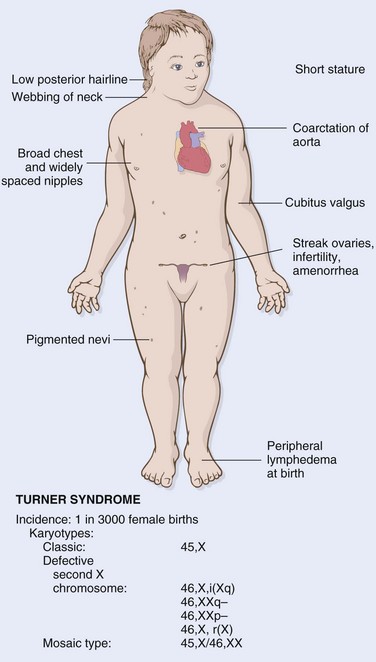
Approximately 43% of patients with Turner syndrome either are mosaics (one of the cell lines being 45,X) or have structural abnormalities of the X chromosome. The most common is deletion of the short arm, resulting in the formation of an isochromosome of the long arm, 46,X,i(X)(q10). The net effect of the associated structural abnormalities is to produce partial monosomy of the X chromosome. Combinations of deletions and mosaicism are reported. It is important to appreciate the karyotypic heterogeneity associated with Turner syndrome because it is responsible for significant variations in the phenotype. In contrast with the patients with monosomy X, those who are mosaics or have deletion variants may have an almost normal appearance and may present only with primary amenorrhea.
The molecular pathogenesis of Turner syndrome is not completely understood, but studies have begun to shed some light. As mentioned earlier, both X chromosomes are active during oogenesis and are essential for normal development of the ovaries. During normal fetal development, ovaries contain as many as 7 million oocytes. The oocytes gradually disappear so that by menarche their numbers have dwindled to a mere 400,000, and when menopause occurs fewer than 10,000 remain. In Turner syndrome, fetal ovaries develop normally early in embryogenesis, but the absence of the second X chromosome leads to an accelerated loss of oocytes, which is complete by age 2 years. In a sense, therefore, “menopause occurs before menarche,” and the ovaries are reduced to atrophic fibrous strands, devoid of ova and follicles (streak ovaries). Because patients with Turner syndrome also have other (nongonadal) abnormalities, it follows that some genes for normal growth and development of somatic tissues also must reside on the X chromosome. Among the genes involved in the Turner phenotype is the short stature homeobox (SHOX) gene at Xp22.33. This is one of the genes that remain active in both X chromosomes and is unique in having an active homologue on the short arm of the Y chromosome. Thus, both normal males and females have two copies of this gene. One copy of SHOX gives rise to short stature. Indeed, deletions of the SHOX gene are noted in 2% to 5% of otherwise normal children with short stature. Whereas one copy of SHOX can explain growth deficit in Turner syndrome, it cannot explain other important clinical features such as cardiac malformations and endocrine abnormalities. Clearly, several other genes located on the X chromosome also are involved.
Summary
Cytogenetic Disorders Involving Sex Chromosomes
• In females, one X chromosome, maternal or paternal, is randomly inactivated during development (Lyon hypothesis).
• In Klinefelter syndrome, there are two or more X chromosomes with one Y chromosome as a result of nondisjunction of sex chromosomes. Patients have testicular atrophy, sterility, reduced body hair, gynecomastia, and eunuchoid body habitus. It is the most common cause of male sterility.
• In Turner syndrome, there is partial or complete monosomy of genes on the short arm of the X chromosome, most commonly due to absence of one X chromosome (45,X) and less commonly from mosaicism, or from deletions involving the short arm of the X chromosome. Short stature, webbing of the neck, cubitus valgus, cardiovascular malformations, amenorrhea, lack of secondary sex characteristics, and fibrotic ovaries are typical clinical features.
Single-Gene Disorders with Atypical Patterns of Inheritance
Three groups of diseases resulting from mutations affecting single genes do not follow the mendelian rules of inheritance:
• Diseases caused by triplet repeat mutations
• Diseases caused by mutations in mitochondrial genes
• Diseases associated with alteration of imprinted regions of the genome
Triplet Repeat Mutations: Fragile X Syndrome
Fragile X syndrome is the prototype of diseases in which the causative mutation occurs in a long repeating sequence of three nucleotides. Other examples of diseases associated with trinucleotide repeat mutations are Huntington disease and myotonic dystrophy. About 40 diseases are now known to be caused by this type of mutation, and all disorders discovered so far are associated with neurodegenerative changes. In each of these conditions, amplification of specific sets of three nucleotides within the gene disrupts its function. Certain unique features of trinucleotide repeat mutations, described later, are responsible for the atypical pattern of inheritance of the associated diseases.
Fragile X syndrome results from a mutation in the FMR1 gene, which maps to Xq27.3. The syndrome gets its name from the karyotypic appearance of the X chromosome in the original method of diagnosis: Culturing patient cells in a folate-deficient medium typically revealed a discontinuity of staining or constriction in the long arm of the X chromosome. This method has now been supplanted by DNA-based analysis of triplet repeat size as discussed later. With a frequency of 1 in 1550 for affected males and 1 in 8000 for affected females, fragile X syndrome is the second most common genetic cause of mental retardation, after Down syndrome. Clinically affected males have moderate to severe mental retardation. The characteristic physical phenotype includes a long face with a large mandible, large everted ears, and large testicles (macroorchidism). Although characteristic of fragile X syndrome, these abnormalities are not always present or may be quite subtle. The only distinctive physical abnormality that can be detected in at least 90% of postpubertal males with fragile X syndrome is macroorchidism.
As with all X-linked diseases, fragile X syndrome predominantly affects males. Analysis of several pedigrees, however, reveals some patterns of transmission not typically associated with other X-linked recessive disorders (Fig. 6–17). These include the following:
• Carrier males: Approximately 20% of males who, by pedigree analysis and by molecular tests, are known to carry a fragile X mutation are clinically and cytogenetically normal. Because carrier males transmit the trait through all their daughters (phenotypically normal) to affected grandchildren, they are called normal transmitting males.
• Affected females: From 30% to 50% of carrier females are affected (i.e., mentally retarded), a number much higher than that for other X-linked recessive disorders.
• Anticipation: This term refers to the phenomenon whereby clinical features of fragile X syndrome worsen with each successive generation, as if the mutation becomes increasingly deleterious as it is transmitted from a man to his grandsons and great-grandsons.
Figure 6–17 Fragile X pedigree. X and Y chromosomes are shown. Note that in the first generation, all sons are normal and all females are carriers. During oogenesis in the carrier female, premutation expands to full mutation; hence, in the next generation, all males who inherit the X with full mutation are affected. However, only 50% of females who inherit the full mutation are affected, and often only mildly.
(Based on an original sketch courtesy of Dr. Nancy Schneider, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
These unusual features have been related to the dynamic nature of the mutation. In the normal population, the number of repeats of the sequence CGG in the FMR1 gene is small, averaging around 29, whereas affected persons have 200 to 4000 repeats. These so-called full mutations are believed to arise through an intermediate stage of premutations characterized by 52 to 200 CGG repeats. Carrier males and females have premutations. During oogenesis (but not spermatogenesis), the premutations can be converted to full mutations by further amplification of the CGG repeats, which can then be transmitted to both the sons and the daughters of the carrier female. These observations provide an explanation for why some carrier males are unaffected (they have premutations), and certain carrier females are affected (they inherit full mutations). Recent studies indicate that premutations are not so benign after all. Approximately 30% of females carrying the premutation have premature ovarian failure (before the age of 40 years), and about one third of premutation-carrying males exhibit a progressive neurodegenerative syndrome starting in their sixth decade. This syndrome, referred to as fragile X–associated tremor-ataxia, is characterized by intention tremors and cerebellar ataxia and may progress to parkinsonism. It is clear, however, that the abnormalities in permutation carriers are milder and occur later in life.
Pathogenesis
The molecular basis for fragile X syndrome is beginning to be understood and relates to silencing of the product of the FMR1 gene, familial mental retardation protein (FMRP). The normal FMR1 gene contains CGG repeats in its 5′ untranslated region. When the number of trinucleotide repeats in the FMR1 gene exceeds approximately 230, the DNA of the entire 5′ region of the gene becomes abnormally methylated. Methylation also extends upstream into the promoter region of the gene, resulting in transcriptional suppression of FMR1. The resulting absence of FMRP is believed to cause the phenotypic changes. FMRP is widely expressed in normal tissues, but higher levels are found in the brain and the testis. Current evidence suggests that FMRP is an RNA-binding protein that is transported from the cytoplasm to the nucleus, where it binds specific mRNAs and transports them to the axons and dendrites (Fig. 6–18). It is in the synapses that FMRP–mRNA complexes perform critical roles in regulating the translation of specific mRNAs. The absence of this finely coordinated “shuttle” function seems to underlie the causation of fragile X syndrome.
Figure 6–18 A model for the action of familial mental retardation protein (FMRP) in neurons. FMRP plays a critical role in regulating the translation of axonal proteins from bound RNAs. These locally produced proteins, in turn, play diverse roles in the microenvironment of the synapse.
(Adapted from Hin P, Warren ST: New insights into fragile X syndrome: from molecules to neurobehavior. Trends Biochem Sci 28:152, 2003.)
As noted, in addition to fragile X syndrome, many other neurodegenerative diseases related to trinucleotide repeat expansions are recognized. Some general principles follow:
• In all cases, gene functions are altered by an expansion of the repeats, but the precise threshold at which premutations are converted to full mutations differs with each disorder.
• While the expansion in fragile X syndrome occurs during oogenesis, in other disorders such as Huntington disease, premutations are converted to full mutations during spermatogenesis.
• The expansion may involve any part of the gene, and the range of possibilities can be divided into two broad categories: those that affect untranslated regions (as in fragile X syndrome) or coding regions (as in Huntington disease) (Fig. 6–19). When mutations affect noncoding regions, there is “loss of function,” since protein synthesis is suppressed (e.g., FMRP). By contrast, mutations involving translated parts of the gene give rise to misfolded proteins that interfere with function of normal proteins (e.g., Huntington disease). Many of these so-called toxic gain-of-function mutations involve CAG repeats that encode polyglutamine tracts, and the resultant diseases are sometimes referred to as “polyglutamine diseases,” affecting primarily the nervous system. Accumulation of misfolded proteins in aggregates within the cytoplasm is a common feature of such diseases.
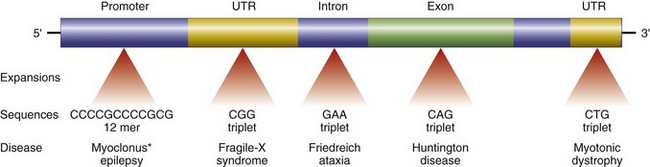
Figure 6–19 Sites of expansion and the affected sequence in selected diseases caused by nucleotide repeat mutations. UTR, untranslated region.
*Though not strictly a trinucleotide-repeat disease, progressive myoclonus epilepsy is caused, like others in this group, by a heritable DNA expansion. The expanded segment is in the promoter region of the gene.
Summary
Fragile X Syndrome
• Pathologic amplification of trinucleotide repeats causes loss-of-function (fragile X syndrome) or gain-of-function mutations (Huntington disease). Most such mutations produce neurodegenerative disorders.
• Fragile X syndrome results from loss of FMR1 gene function and is characterized by mental retardation, macroorchidism, and abnormal facial features.
• In the normal population, there are about 29 CGG repeats in the FMR1 gene. The genomes of carrier males and females contain premutations with 52 to 200 CGG repeats that can expand to 4000 repeats (full mutations) during oogenesis. When full mutations are transmitted to progeny, fragile X syndrome occurs.
Diseases Caused by Mutations in Mitochondrial Genes
Mitochondria contain several genes that encode enzymes involved in oxidative phosphorylation. Inheritance of mitochondrial DNA differs from that of nuclear DNA in that the former is associated with maternal inheritance. The reason for this peculiarity is that ova contain mitochondria within their abundant cytoplasm, whereas spermatozoa contain few, if any, mitochondria. The mitochondrial DNA complement of the zygote is therefore derived entirely from the ovum. Thus, only mothers transmit mitochondrial genes to all of their offspring, both male and female; however, daughters but not sons transmit the DNA further to their progeny.
Diseases caused by mutations in mitochondrial genes are rare. Because mitochondrial DNA encodes enzymes involved in oxidative phosphorylation, diseases caused by mutations in such genes affect organs most dependent on oxidative phosphorylation (skeletal muscle, heart, brain). Leber hereditary optic neuropathy is the prototypical disorder in this group. This neurodegenerative disease manifests itself as progressive bilateral loss of central vision that leads in due course to blindness.
Diseases Caused by Alterations of Imprinted Regions: Prader-Willi and Angelman Syndromes
All humans inherit two copies of each gene (except, of course, the sex chromosome genes in males), carried on homologous maternal and paternal chromosomes. It was long assumed that there was no difference between normal homologous genes derived from the mother and those from the father. Indeed, this is true for many genes. It has now been established, however, that functional differences exist between the paternal and the maternal copies of some genes. These differences arise from an epigenetic process called genomic imprinting, whereby certain genes are differentially “inactivated” during paternal and maternal gametogenesis. Thus, maternal imprinting refers to transcriptional silencing of the maternal allele, whereas paternal imprinting implies that the paternal allele is inactivated. At the molecular level, imprinting is associated with methylation of the gene promoter, as well as related events such as modification of DNA-binding histone proteins, the sum total effect of which is to silence the gene. Imprinting occurs in ovum or sperm and is then stably transmitted to all somatic cells derived from the zygote.
Genomic imprinting is best illustrated by considering two uncommon genetic disorders: Prader-Willi syndrome and Angelman syndrome.
Prader-Willi syndrome is characterized by mental retardation, short stature, hypotonia, obesity, small hands and feet, and hypogonadism. In 60% to 75% of cases, an interstitial deletion of band q12 in the long arm of chromosome 15—del(15)(q11;q13)—can be detected. In many patients without a detectable cytogenetic abnormality, FISH analysis reveals smaller deletions within the same region. It is striking that in all cases, the deletion affects the paternally derived chromosome 15. In contrast with Prader-Willi syndrome, patients with the phenotypically distinct Angelman syndrome are born with a deletion of the same chromosomal region derived from their mothers. Patients with Angelman syndrome also are mentally retarded, but in addition they present with ataxic gait, seizures, and inappropriate laughter. Because of the laughter and ataxia, this syndrome has been called the happy puppet syndrome. A comparison of these two syndromes clearly demonstrates the “parent-of-origin” effects on gene function. If all the paternal and maternal genes contained within chromosome 15 were expressed in an identical fashion, clinical features resulting from these deletions would be expected to be identical regardless of the parental origin of chromosome 15.
The molecular basis of these two syndromes can be understood in the context of imprinting (Fig. 6–20). A set of genes on the maternal chromosome at 15q12 is imprinted (and hence silenced), so the only functional alleles are provided by the paternal chromosome. When these are lost as a result of a deletion (in the paternal chromosome), the patient develops Prader-Willi syndrome. Conversely, a distinct gene that also maps to the same region of chromosome 15 is imprinted on the paternal chromosome. Only the maternally derived allele of the gene normally is active. Deletion of this maternal gene on chromosome 15 gives rise to the Angelman syndrome. Molecular studies of cytogenetically normal patients with Prader-Willi syndrome have revealed that in some cases, both of the structurally normal copies of chromosome 15 are derived from the mother. Inheritance of both chromosomes of a pair from one parent is called uniparental disomy. The net effect is the same (i.e., the patient does not have a functional set of genes from the [nonimprinted] paternal chromosome 15).
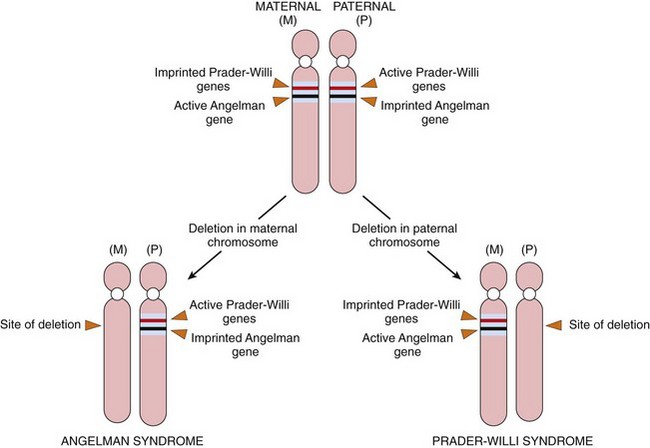
Angelman syndrome, as might be expected, also can result from uniparental disomy of parental chromosome 15. The Angelman syndrome gene (imprinted on the paternal chromosome) is now known to encode a ligase that has a role in the ubiquitin-proteasome proteolytic pathway (Chapter 1). This gene, called, somewhat laboriously, UBE3A, is expressed primarily from the maternal allele in specific regions of the normal brain. In Angelman syndrome, UBE3A is not expressed in these areas of the brain—hence the neurologic manifestations.
Prader-Willi syndrome, unlike Angelman syndrome, probably is caused by the loss of function of several genes located on chromosome 15 between q11 and q13. These genes are still being fully characterized.
Summary
Genomic Imprinting
• Imprinting involves transcriptional silencing of the paternal or maternal copies of certain genes during gametogenesis. For such genes only one functional copy exists in the individual. Loss of the functional allele (not imprinted) by deletions gives rise to diseases.
• Prader-Willi syndrome results from deletion of paternal chromosomal region 15q12 and is characterized by mental retardation, short stature, hypotonia, obesity, and hypogonadism.
• Angelman syndrome results from deletion of maternal chromosomal region 15q12 and is characterized by mental retardation, ataxia, seizures, and inappropriate laughter.