Chapter 10 Heart
See Targeted Therapy available online at studentconsult.com
The heart is a truly remarkable organ, beating more than 40 million times a year and pumping over 7500 liters of blood a day; in a typical lifespan, the cumulative volume would fill three “supertanker” ships. The cardiovascular system is the first organ system to become fully functional in utero (at approximately 8 weeks of gestation); without a beating heart and vascular supply, further development cannot occur, and fetal demise is inevitable. When the heart fails postnatally, the results are equally catastrophic. Indeed, cardiovascular disease remains the leading contributor to mortality worldwide and accounts for nearly 40% of all U.S. deaths—approximately 1 death every 30 seconds, or 750,000 deaths each year (accounting for 50% greater mortality than for all forms of cancer combined). The annual economic impact of ischemic heart disease, the most prevalent form of heart disease, is in excess of $100 billion. Moreover, almost a third of these deaths are “premature,” occurring in persons younger than 75 years of age; thus, an additional economic burden is imposed through lost years of productivity.
Overview of Heart Disease
Although a host of diseases can affect the cardiovascular system, the pathophysiologic pathways that result in a “broken” heart distill down to six principal mechanisms:
• Failure of the pump. In the most common situation, the cardiac muscle contracts weakly and the chambers cannot empty properly—so-called systolic dysfunction. In some cases, the muscle cannot relax sufficiently to permit ventricular filling, resulting in diastolic dysfunction.
• Obstruction to flow. Lesions that prevent valve opening (e.g., calcific aortic valve stenosis) or cause increased ventricular chamber pressures (e.g., systemic hypertension or aortic coarctation) can overwork the myocardium, which has to pump against the obstruction.
• Regurgitant flow. Valve lesions that allow backward flow of blood create conditions that add increased volume workload to the affected chambers with each contraction.
• Shunted flow. Defects (congenital or acquired) that divert blood inappropriately from one chamber to another, or from one vessel to another, lead to pressure and volume overloads.
• Disorders of cardiac conduction. Uncoordinated cardiac impulses or blocked conduction pathways can cause arrhythmias that reduce contraction frequency or diminish effective cardiac output.
• Rupture of the heart or major vessel. Loss of circulatory continuity (e.g., gunshot wound through the thoracic aorta) leads to exsanguination, hypotensive shock, and death.
Heart Failure
Heart failure generally is referred to as congestive heart failure (CHF). CHF is the common end point for many forms of cardiac disease and typically is a progressive condition that carries an extremely poor prognosis. In the United States alone, nearly 5 million persons are affected, resulting in more than 1 million hospitalizations and 300,000 deaths each year, with a financial burden in excess of $18 billion. Most cases of heart failure are due to systolic dysfunction—inadequate myocardial contractile function, characteristically a consequence of ischemic heart disease or hypertension. Alternatively, CHF also can result from diastolic dysfunction—inability of the heart to adequately relax and fill, such as in massive left ventricular hypertrophy, myocardial fibrosis, amyloid deposition, or constrictive pericarditis. Indeed, heart failure in elderly persons, diabetic patients, and women may be more commonly attributable to diastolic dysfunction. Various studies suggest that 40–60% of cases of CHF may be due to diastolic dysfunction. Finally, heart failure also can be caused by valve dysfunction (e.g., due to endocarditis) or can occur in normal hearts suddenly burdened with an abnormal load (e.g., with fluid or pressure overload).
CHF occurs when the heart cannot generate sufficient output to meet the metabolic demands of the tissues—or can only do so at higher-than-normal filling pressures; in a minority of cases, heart failure can be a consequence of greatly increased tissue demands, as in hyperthyroidism, or poor oxygen carrying capacity as in anemia (high-output failure). CHF onset can be abrupt, as in the setting of a large myocardial infarct or acute valve dysfunction. In many cases, however, CHF develops gradually and insidiously owing to the cumulative effects of chronic work overload or progressive loss of myocardium.
In CHF, the failing heart can no longer efficiently pump the blood delivered to it by the venous circulation. The result is an increased end-diastolic ventricular volume, leading to increased end-diastolic pressures and, finally, elevated venous pressures. Thus, inadequate cardiac output—called forward failure—is almost always accompanied by increased congestion of the venous circulation—that is, backward failure. As a consequence, although the root problem in CHF typically is deficient cardiac function, virtually every other organ is eventually affected by some combination of forward and backward failure.
The cardiovascular system attempts to compensate for reduced myocardial contractility or increased hemodynamic burden through several homeostatic mechanisms:
• The Frank-Starling mechanism. Increased end-diastolic filling volumes dilate the heart and cause increased cardiac myofiber stretching; these lengthened fibers contract more forcibly, thereby increasing cardiac output. If the dilated ventricle is able to maintain cardiac output by this means, the patient is said to be in compensated heart failure. However, ventricular dilation comes at the expense of increased wall tension and amplifies the oxygen requirements of an already-compromised myocardium. With time, the failing muscle is no longer able to propel sufficient blood to meet the needs of the body, and the patient develops decompensated heart failure.
• Activation of neurohumoral systems:
 Release of the neurotransmitter norepinephrine by the autonomic nervous system increases heart rate and augments myocardial contractility and vascular resistance. Activation of the renin-angiotensin-aldosterone system spurs water and salt retention (augmenting circulatory volume) and increases vascular tone. Release of atrial natriuretic peptide acts to balance the renin-angiotensin-aldosterone system through diuresis and vascular smooth muscle relaxation.
Release of the neurotransmitter norepinephrine by the autonomic nervous system increases heart rate and augments myocardial contractility and vascular resistance. Activation of the renin-angiotensin-aldosterone system spurs water and salt retention (augmenting circulatory volume) and increases vascular tone. Release of atrial natriuretic peptide acts to balance the renin-angiotensin-aldosterone system through diuresis and vascular smooth muscle relaxation.• Myocardial structural changes, including augmented muscle mass. Cardiac myocytes cannot proliferate, yet can adapt to increased workloads by assembling increased numbers of sarcomeres, a change that is accompanied by myocyte enlargement (hypertrophy) (Fig. 10–1).
In pressure overload states (e.g., hypertension or valvular stenosis), new sarcomeres tend to be added parallel to the long axis of the myocytes, adjacent to existing sarcomeres. The growing muscle fiber diameter thus results in concentric hypertrophy—the ventricular wall thickness increases without an increase in the size of the chamber. In volume overload states (e.g., valvular regurgitation or shunts), the new sarcomeres are added in series with existing sarcomeres, so that the muscle fiber length increases. Consequently, the ventricle tends to dilate, and the resulting wall thickness can be increased, normal, or decreased; thus, heart weight—rather than wall thickness—is the best measure of hypertrophy in volume-overloaded hearts.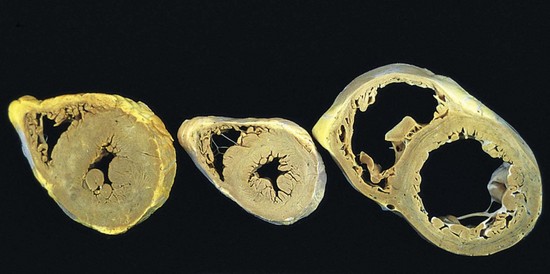
Figure 10–1 Left ventricular hypertrophy, with and without dilation, viewed in transverse sections. Compared with a normal heart (center), the pressure-overloaded heart (left) has an increased mass, a thick wall, and a smaller lumen. The volume-overloaded heart (right) has an increased mass, larger lumen, and enlarged size, but a normal wall thickness.
(Reproduced by permission from Edwards WD: Cardiac anatomy and examination of cardiac specimens. In Emmanouilides GC, Allen HD, Riemenschneider TA, Gutgesell HP [eds]: Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adults, 5th ed. Philadelphia, Williams & Wilkins, 1995, p 86.)
Compensatory hypertrophy comes at a cost to the myocyte. The oxygen requirements of hypertrophic myocardium are amplified owing to increased myocardial cell mass. Because the myocardial capillary bed does not expand in step with the increased myocardial oxygen demands, the myocardium becomes vulnerable to ischemic injury. Hypertrophy also typically is associated with altered patterns of gene expression reminiscent of the fetal myocytes, such as changes in the dominant form of myosin heavy chain produced. Altered gene expression may contribute to changes in myocyte function that lead to increases in heart rate and force of contraction, both of which improve cardiac output, but which also lead to higher cardiac oxygen consumption. In the face of ischemia and chronic increases in workload, other untoward changes also eventually supervene, including myocyte apoptosis, cytoskeletal alterations, and increased extracellular matrix (ECM) deposition.
Pathologic compensatory cardiac hypertrophy is correlated with increased mortality; indeed, cardiac hypertrophy is an independent risk factor for sudden cardiac death. By contrast, the volume-loaded hypertrophy induced by regular aerobic exercise (physiologic hypertrophy) typically is accompanied by an increase in capillary density, with decreased resting heart rate and blood pressure. These physiologic adaptations reduce overall cardiovascular morbidity and mortality. In comparison, static exercise (e.g., weight lifting) is associated with pressure hypertrophy and may not have the same beneficial effects.
Left-Sided Heart Failure
Heart failure can affect predominantly the left or the right side of the heart or may involve both sides. The most common causes of left-sided cardiac failure are ischemic heart disease (IHD), systemic hypertension, mitral or aortic valve disease, and primary diseases of the myocardium (e.g., amyloidosis). The morphologic and clinical effects of left-sided CHF stem from diminished systemic perfusion and the elevated back-pressures within the pulmonary circulation.
 Morphology
Morphology
Heart
The gross cardiac findings depend on the underlying disease process, for example, myocardial infarction or valvular deformities may be present. With the exception of failure due to mitral valve stenosis or restrictive cardiomyopathies (described later), the left ventricle usually is hypertrophied and can be dilated, sometimes massively. Left ventricular dilation can result in mitral insufficiency and left atrial enlargement, which is associated with an increased incidence of atrial fibrillation. The microscopic changes in heart failure are nonspecific, consisting primarily of myocyte hypertrophy with interstitial fibrosis of variable severity. Superimposed on this background may be other lesions that contribute to the development of heart failure (e.g., recent or old myocardial infarction).
Lungs
Rising pressure in the pulmonary veins is ultimately transmitted back to the capillaries and arteries of the lungs, resulting in congestion and edema as well as pleural effusion due to an increase in hydrostatic pressure in the venules of the visceral pleura. The lungs are heavy and boggy, and microscopically show perivascular and interstitial transudates, alveolar septal edema, and accumulation of edema fluid in the alveolar spaces. In addition, variable numbers of red cells extravasate from the leaky capillaries into alveolar spaces, where they are phagocytosed by macrophages The subsequent breakdown of red cells and hemoglobin leads to the appearance of hemosiderin-laden alveolar macrophages—so-called heart failure cells—that reflect previous episodes of pulmonary edema.
Clinical Features
Dyspnea (shortness of breath) on exertion is usually the earliest and most significant symptom of left-sided heart failure; cough also is common as a consequence of fluid transudation into air spaces. As failure progresses, patients experience dyspnea when recumbent (orthopnea); this occurs because the supine position increases venous return from the lower extremities and also elevates the diaphragm. Orthopnea typically is relieved by sitting or standing, so patients usually sleep in a semiseated position. Paroxysmal nocturnal dyspnea is a particularly dramatic form of breathlessness, awakening patients from sleep with extreme dyspnea bordering on feelings of suffocation.
Other manifestations of left ventricular failure include an enlarged heart (cardiomegaly), tachycardia, a third heart sound (S3), and fine rales at the lung bases, caused by the opening of edematous pulmonary alveoli. With progressive ventricular dilation, the papillary muscles are displaced outwards, causing mitral regurgitation and a systolic murmur. Subsequent chronic dilation of the left atrium can cause atrial fibrillation, manifested by an “irregularly irregular” heartbeat. Such uncoordinated, chaotic atrial contractions reduce the ventricular stroke volume and also can cause stasis. The stagnant blood is prone to form thrombi (particularly in the atrial appendage) that can shed emboli and cause strokes and manifestations of infarction in other organs.
Systemically, diminished cardiac output leads to decreased renal perfusion that in turn triggers the renin-angiotension-aldosterone axis, increasing intravascular volume and pressures (Chapter 3). Unfortunately, these compensatory effects exacerbate the pulmonary edema. With further reduction in renal perfusion, prerenal azotemia may supervene, with impaired excretion of nitrogenous wastes and increasing metabolic derangement. In severe CHF, diminished cerebral perfusion can manifest as hypoxic encephalopathy with irritability, diminished cognition, and restlessness that can progress to stupor and coma.
Right-Sided Heart Failure
Right heart failure usually is the consequence of left-sided heart failure, since any pressure increase in the pulmonary circulation inevitably produces an increased burden on the right side of the heart. Isolated right-sided heart failure also can occur in a few diseases. The most common of these is severe pulmonary hypertension, resulting in right-sided heart pathology termed cor pulmonale. In cor pulmonale, myocardial hypertrophy and dilation generally are confined to the right ventricle and atrium, although bulging of the ventricular septum to the left can cause left ventricular dysfunction. Isolated right-sided failure also can occur in patients with primary pulmonic or tricuspid valve disease, or congenital heart disease, such as with left-to-right shunts causing chronic volume and pressure overloads.
The major morphologic and clinical effects of pure right-sided heart failure differ from those of left-sided heart failure in that engorgement of the systemic and portal venous systems typically is pronounced and pulmonary congestion is minimal.
Morphology
Liver and Portal System
The liver usually is increased in size and weight (congestive hepatomegaly). A cut section displays prominent passive congestion, a pattern referred to as nutmeg liver (Chapter 3); congested centrilobular areas are surrounded by peripheral paler, noncongested parenchyma. When left-sided heart failure is also present, severe central hypoxia produces centrilobular necrosis in addition to the sinusoidal congestion. With long-standing severe right-sided heart failure, the central areas can become fibrotic, creating so-called cardiac cirrhosis.
Right-sided heart failure also leads to elevated pressure in the portal vein and its tributaries (portal hypertension), with vascular congestion producing a tense, enlarged spleen (congestive splenomegaly). Chronic passive congestion of the bowel wall with edema can be severe enough to interfere with absorption of nutrients and medications.
Pleural, Pericardial, and Peritoneal Spaces
Systemic venous congestion due to right heart failure can lead to transudates (effusions) in the pleural and pericardial spaces, but usually does not cause pulmonary parenchymal edema. Pleural effusions are most pronounced when there is increase in pulmonary venous as well as systemic venous pressures, as occurs in combined right and left heart failure. When large (e.g., 1 L or more), pleural effusions can cause atelectasis, and, very uncommonly, substantial pericardial effusions (greater than 500 mL) can limit cardiac filling and cause cardiac failure (due to tamponade). A combination of hepatic congestion (with or without diminished albumin synthesis) and portal hypertension leads to peritoneal transudates (ascites) The effusions into the various body cavities typically are serous, with a low protein content, and lack inflammatory cells.
Subcutaneous Tissues
Peripheral edema of dependent portions of the body, especially ankle (pedal) and pretibial edema, is a hallmark of right heart failure. In chronically bedridden patients, the edema may be primarily presacral. In particularly severe cases, generalized massive edema (anasarca) may be seen.
Clinical Features
Unlike left-sided heart failure, pure right-sided heart failure typically is associated with very few respiratory symptoms. Instead, the clinical manifestations are related to systemic and portal venous congestion, including hepatic and splenic enlargement, peripheral edema, pleural effusion, and ascites. Venous congestion and hypoxia of the kidneys and brain due to right heart failure can produce deficits comparable to those caused by the hypoperfusion caused by left heart failure.
Of note, in most cases of chronic cardiac decompensation, patients present with biventricular CHF, encompassing the clinical syndromes of both right-sided and left-sided heart failure. As congestive heart failure progresses, patients may become frankly cyanotic and acidotic, as a consequence of decreased tissue perfusion resulting from both diminished forward flow and increasing retrograde congestion.
 Summary
Summary
Heart Failure
• CHF occurs when the heart is unable to provide adequate perfusion to meet the metabolic requirements of peripheral tissues; inadequate cardiac output usually is accompanied by increased congestion of the venous circulation.
• Left-sided heart failure is most commonly secondary to ischemic heart disease, systemic hypertension, mitral or aortic valve disease, or primary diseases of the myocardium; symptoms are mainly a consequence of pulmonary congestion and edema, although systemic hypoperfusion can cause renal and cerebral dysfunction.
• Right-sided heart failure is due most often to left heart failure and, less commonly, to primary pulmonary disorders; signs and symptoms are related chiefly to peripheral edema and visceral congestion.
Congenital Heart Disease
Congenital heart diseases are abnormalities of the heart or great vessels that are present at birth. They account for 20% to 30% of all birth defects and include a broad spectrum of malformations, ranging from severe anomalies incompatible with intrauterine or perinatal survival, to mild lesions that produce only minimal symptoms at birth, or are entirely unrecognized during life. Congenital heart disease affects 6 to 8 of every 1000 liveborn infants, and the incidence is higher in premature infants and in stillborns; roughly 40,000 children are born each year in the United States with clinically significant cardiac malformations, and another 40,000 have subclinical disease. Defects that permit maturation and live birth usually involve only single chambers or regions of the heart. Twelve entities account for 85% of cases of congenital heart disease; their frequencies are shown in Table 10–1.
Table 10–1 Frequency of Congenital Cardiac Malformations*
| Malformation | Incidence per 1 Million Live Births | % |
|---|---|---|
| Ventricular septal defect | 4482 | 42 |
| Atrial septal defect | 1043 | 10 |
| Pulmonary stenosis | 836 | 8 |
| Patent ductus arteriosus | 781 | 7 |
| Tetralogy of Fallot | 577 | 5 |
| Coarctation of aorta | 492 | 5 |
| Atrioventricular septal defect | 396 | 4 |
| Aortic stenosis | 388 | 4 |
| Transposition of great arteries | 388 | 4 |
| Truncus arteriosus | 136 | 1 |
| Total anomalous pulmonary venous connection | 120 | 1 |
| Tricuspid atresia | 118 | 1 |
| total | 9757 |
* Summary of 44 published studies. Percentages do not add to 100% because of rounding.
Data from Hoffman JI, Kaplan S: The incidence of congenital heart disease. J Am Coll Cardiol 39:1890, 2002.
Thanks to surgical advances, the number of patients surviving with congenital heart disease is increasing rapidly, including over 1 million persons in the United States alone. Although surgery may correct the hemodynamic abnormalities, the repaired heart may not be completely normal, since the myocardial hypertrophy and cardiac remodeling brought about by the congenital defect may be irreversible; in addition, virtually all cardiac surgery results in some degree of myocardial scarring. Such changes lead secondarily to arrhythmias, ischemia, and myocardial dysfunction, which occasionally appear many years after surgical correction.
 Pathogenesis
Pathogenesis
In most instances, congenital heart disease arises from faulty embryogenesis during gestational weeks 3 through 8, when major cardiovascular structures develop; the cause is unknown in almost 90% of cases. Of the known etiologic factors, environmental causes, including congenital rubella infection, teratogens, and maternal diabetes, and genetic factors are the best characterized. The contribution of specific genetic loci has been demonstrated in familial forms of congenital heart disease and by well-defined associations with certain chromosomal abnormalities (e.g., trisomies 13, 15, 18, and 21, and Turner syndrome).
Cardiac morphogenesis involves multiple genes that work together to choreograph a complex series of tightly regulated events. Key steps include commitment of progenitor cells to the myocardial lineage, formation and looping of the heart tube, segmentation and growth of the cardiac chambers, cardiac valve formation, and connection of the great vessels to the heart. Proper orchestration of these remarkable transformations depends on networks of transcription factors and several signaling pathways and molecules, including the Wnt, vascular endothelial growth factor (VEGF), bone morphogenetic protein (BMP), transforming growth factor-β (TGF-β), fibroblast growth factor, and Notch pathways. Also essential for cardiac morphogenesis is the mechanical force imparted by flowing pulsatile blood, which is somehow sensed by the cells of the developing heart and vessels.
Since crafting a normal heart involves many steps, even subtle perturbations can adversely influence the outcome. Most of the known genetic defects are autosomal dominant mutations causing loss (or sometimes gain) of function of a particular factor (Table 10–2). Several mutations involve transcription factors. For example, atrial and ventricular septal defects (ASDs and VSDs, respectively) and/or conduction defects may be caused by transcription factor mutations, such as TBX5 mutations in the Holt-Oram syndrome and NKX2.5 or GATA4 mutations in sporadic, nonsyndromic cases. Other disorders (e.g., Noonan syndrome) are associated with mutations in intracellular signaling cascades that cause constitutive activation. microRNAs, as well as epigenetic changes (e.g., DNA methylation), also are increasingly recognized as important contributors. It is likely that even transient environmental stresses at critical junctures early in pregnancy can cause subtle changes in transcription factor activity, intracellular signaling, or morphogenic gradients that may recapitulate the defects produced by heritable mutations.
Table 10–2 Selected Examples of Gene Defects Associated With Congenital Heart Disease*
| Disorder | Gene(s) | Gene Product Function |
|---|---|---|
| Nonsyndromic | ||
| ASD or conduction defects | NKX2.5 | Transcription factor |
| ASD or VSD | GATA4 | Transcription factor |
| Tetralogy of Fallot | ZFPM2 or NKX2.5 | Transcription factors |
| Syndromic† | ||
| Alagille syndrome—pulmonary artery stenosis or tetralogy of Fallot | JAG1 or NOTCH2 | Signaling proteins or receptors |
| Char syndrome—PDA | TFAP2B | Transcription factor |
| CHARGE syndrome—ASD, VSD, PDA, or hypoplastic right side of the heart | CHD7 | Helicase-binding protein |
| DiGeorge syndrome—ASD, VSD, or outflow tract obstruction | TBX1 | Transcription factor |
| Holt-Oram syndrome—ASD, VSD, or conduction defect | TBX5 | Transcription factor |
| Noonan syndrome—pulmonary valve stenosis, VSD, or hypertrophic cardiomyopathy | PTPN11, KRAS, SOS1 | Signaling proteins |
ASD, atrial septal defect; CHARGE, posterior coloboma, heart defect, choanal atresia, retardation, genital and ear anomalies; PDA, patent ductus arteriosus; VSD, ventricular septal defect.
* Note that different mutations can cause the same phenotype, and that mutations in some genes can cause multiple phenotypes (e.g., NKX2.5). Many of these congenital lesions also can occur sporadically, without specific genetic mutation.
† Only the cardiac manifestations of the syndrome are listed; the other skeletal, facial, neurologic, and visceral changes are not.
Clinical Features
The various structural anomalies in congenital heart disease can be assigned to three major groups based on their hemodynamic and clinical consequences: (1) malformations causing a left-to-right shunt; (2) malformations causing a right-to-left shunt (cyanotic congenital heart diseases); and (3) malformations causing obstruction.
A shunt is an abnormal communication between chambers or blood vessels. Depending on pressure relationships, shunts permit the flow of blood from the left to the right side of the heart (or vice versa).
• With right-to-left shunt, a dusky blueness of the skin (cyanosis) results because the pulmonary circulation is bypassed and poorly oxygenated blood enters the systemic circulation.
• By contrast, left-to-right shunts increase pulmonary blood flow and are not associated (at least initially) with cyanosis. However, they expose the low-pressure, low-resistance pulmonary circulation to increased pressures and volumes; these conditions lead to adaptive changes that increase lung vascular resistance to protect the pulmonary bed, resulting in right ventricular hypertrophy and—eventually—failure. With time, increased pulmonary resistance also can cause shunt reversal (right to left) and late-onset cyanosis.
• Some congenital anomalies obstruct vascular flow—by narrowing the chambers, valves, or major blood vessels; a malformation characterized by complete obstruction is called an atresia. In some disorders (e.g., tetralogy of Fallot), an obstruction (pulmonary stenosis) can be associated with a shunt (right-to-left, through a VSD).
The altered hemodynamics of congenital heart disease usually lead to chamber dilation or wall hypertrophy. However, some defects result in a reduced muscle mass or chamber size; this is called hypoplasia if it occurs before birth and atrophy if it develops postnatally.
Left-to-Right Shunts
Left-to-right shunts are the most common type of congenital cardiac malformation. They include atrial septal defects (ASDs), ventricular septal defects (VSDs), and patent ductus arteriosus (PDA) (Fig. 10–2). ASDs typically increase only right ventricular and pulmonary outflow volumes, while VSDs and PDAs cause both increased pulmonary blood flows and pressures. Manifestations of these shunts range in severity from no symptoms at all to fulminant heart failure.
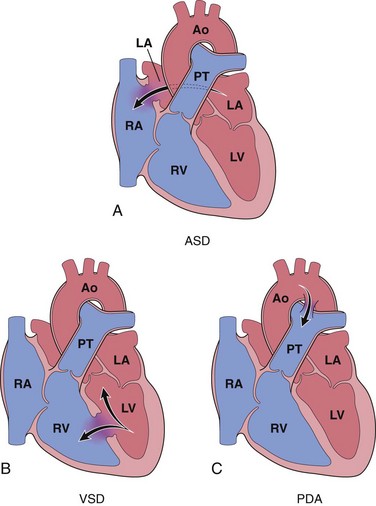
Figure 10–2 Common congenital left-to-right shunts (arrows indicate direction of blood flow). A, Atrial septal defect (ASD). B, Ventricular septal defect (VSD). C, Patent ductus arteriosus (PDA). Ao, aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle.
Cyanosis is not an early feature of these defects. However, prolonged left-to-right shunting with volume and pressure overloads eventually causes pulmonary hypertension and secondarily right-sided pressures that exceed those on the left; at that point, reversal of blood flow occurs, with resultant right-to-left shunting, and the development of cyanosis. Such reversal of flow and shunting of unoxygenated blood into the systemic circulation is called Eisenmenger syndrome. Once significant pulmonary hypertension develops, the structural defects of congenital heart disease are considered irreversible. This is the rationale for early surgical (or even nonsurgical) intervention.
Atrial Septal Defects and Patent Foramen Ovale
During normal cardiac development patency is maintained between right and left atria by a series of ostia (primum and secundum) that eventually become the foramen ovale; this arrangement allows oxygenated blood from the maternal circulation to flow from the right to the left atrium, thereby sustaining fetal development. At later stages of intrauterine development, tissue flaps (septum primum and septum secundum) grow to occlude the foramen ovale, and in 80% of cases, the higher left-sided pressures in the heart that occur at birth permanently fuse the septa against the foramen ovale. In the remaining 20% of cases, a patent foramen ovale results; although the flap is of adequate size to cover the foramen, the unsealed septa can potentially allow transient right-to-left blood flow. Paradoxical embolism, defined as venous emboli (e.g., from deep leg veins) that enter the systemic arterial circulation, may also occur if right-sided atrial pressures increase, such as with pulmonary hypertension or a Valsalva maneuver during sneezing or bowel movements.
In contrast to a patent foramen ovale, an ASD is an abnormal fixed opening in the atrial septum that allows unrestricted blood flow between the atrial chambers. A majority (90%) of ASDs are so-called ostium secundum defects in which growth of the septum secundum is insufficient to occlude the second ostium.
Morphology
Ostium secundum ASDs (90% of ASDs) typically are smooth-walled defects near the foramen ovale, usually without other associated cardiac abnormalities. Hemodynamically significant lesions are accompanied by right atrial and ventricular dilation, right ventricular hypertrophy, and dilation of the pulmonary artery, reflecting the effects of a chronically increased volume load. Ostium primum ASDs (accounting for 5% of these defects) occur at the lowest part of the atrial septum and can be associated with mitral and tricuspid valve abnormalities, reflecting the close relationship between development of the septum primum and the endocardial cushions. In more severe cases, additional defects may include a VSD and a common atrioventricular canal. Sinus venosus ASDs (accounting for another 5% of the cases) are located high in the atrial septum and often are accompanied by anomalous drainage of the pulmonary veins into the right atrium or superior vena cava.
Clinical Features
A majority of ASDs are asymptomatic until adulthood. Although VSDs are the most common congenital malformations at birth (Table 10–1), many close spontaneously. Consequently, ASDs—which are less likely to spontaneously close—are the most common defects to be first diagnosed in adults. ASDs initially cause left-to-right shunts, as a consequence of the lower pressures in the pulmonary circulation and the right side of the heart. In general, these defects are well tolerated, especially if they are less than 1 cm in diameter; even larger lesions do not usually produce any symptoms in childhood. Over time, however, chronic volume and pressure overloads can cause pulmonary hypertension. Surgical or intravascular ASD closures are thus performed to reverse the hemodynamic abnormalities and preempt the development of heart failure, paradoxical embolization, and irreversible pulmonary vascular disease. Mortality is low, and postoperative survival is comparable to that for a normal population.
Ventricular Septal Defects
Defects in the ventricular septum allow left-to-right shunting and constitute the most common congenital cardiac anomaly at birth (Table 10–1 and Fig. 10–3). The ventricular septum normally is formed by the fusion of a muscular ridge that grows upward from the apex of the heart to a thinner membranous partition that grows downward from the endocardial cushions. The basal (membranous) region is the last part of the septum to develop and is the site of approximately 90% of VSDs. Although more common at birth, most VSDs close spontaneously in childhood, so that the overall incidence in adults is lower than that for ASDs. Only 20% to 30% of VSDs occur in isolation; most are associated with other cardiac malformations.
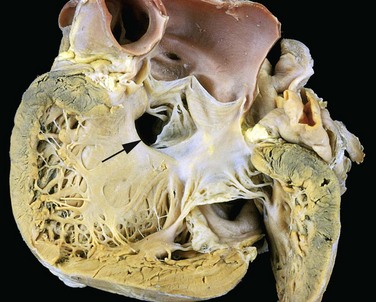
Figure 10–3 Ventricular septal defect of the membranous type (arrow).
(Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, Minnesota.)
Morphology
The size and location of VSDs are variable (Fig. 10–3), ranging from minute defects in the membranous septum to large defects involving virtually the entire interventricular wall. In defects associated with a significant left-to-right shunt, the right ventricle is hypertrophied and often dilated. The diameter of the pulmonary artery is increased, owing to the greater volume ejected by the right ventricle. Vascular changes typical of pulmonary hypertension are common (Chapter 12).
Clinical Features
Small VSDs may be asymptomatic, and roughly half of those in the muscular portion of the septum close spontaneously during infancy or childhood. Larger defects, however, result in chronic severe left-to-right shunting, often complicated by pulmonary hypertension and congestive heart failure. Progressive pulmonary hypertension, with resultant reversal of the shunt and cyanosis, occurs earlier and more frequently with VSDs than with ASDs. Early surgical correction is therefore indicated for such lesions. Small or medium-sized defects that produce jet lesions in the right ventricle—which can cause endothelial damage—also increase the risk for development of infective endocarditis.
Patent Ductus Arteriosus
The ductus arteriosus arises from the left pulmonary artery and joins the aorta just distal to the origin of the left subclavian artery. During intrauterine life, it permits blood flow from the pulmonary artery to the aorta, thereby bypassing the unoxygenated lungs. Shortly after birth in healthy term infants, the ductus constricts and is functionally closed after 1 to 2 days; these changes occur in response to increased arterial oxygenation, decreased pulmonary vascular resistance, and declining local levels of prostaglandin E2. Complete obliteration occurs within the first few months of extrauterine life, leaving only a strand of residual fibrous tissue known as the ligamentum arteriosum. Ductal closure often is delayed (or even absent) in infants with hypoxia (related to respiratory distress or heart disease). PDAs account for about 7% of congenital heart lesions (Table 10–1 and Fig. 10–2), and the great majority of these (90%) are isolated defects.
Clinical Features
PDAs are high-pressure left-to-right shunts that produce harsh, “machinery-like” murmurs. A small PDA generally causes no symptoms, although larger defects eventually can lead to Eisenmenger syndrome with cyanosis and congestive heart failure. The high-pressure shunt also predisposes affected patients to development of infective endocarditis. While there is general agreement that isolated PDAs should be closed as early in life as is feasible, preservation of ductal patency (by administering prostaglandin E) can be lifesaving when a PDA is the only means to sustain systemic or pulmonary blood flow (e.g., in infants with aortic or pulmonic atresia).
Right-to-Left Shunts
Cardiac malformations associated with right-to-left shunts are distinguished by early cyanosis. This occurs because poorly oxygenated blood from the right side of the heart flows directly into the arterial circulation. Two of the most important conditions associated with cyanotic congenital heart disease are tetralogy of Fallot and transposition of the great vessels (Fig. 10–4). Clinical consequences of severe, systemic cyanosis include clubbing of the tips of the fingers and toes (hypertrophic osteoarthropathy), polycythemia, and paradoxical embolization.
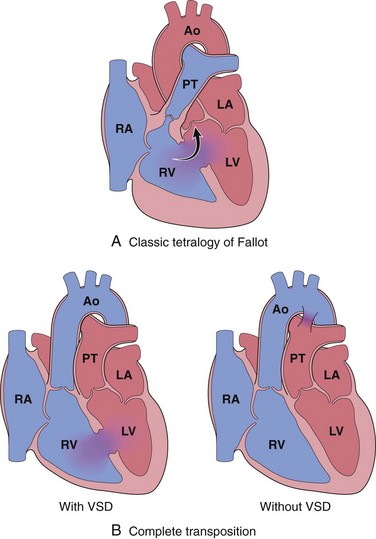
Figure 10–4 Common congenital right-to-left shunts (cyanotic congenital heart disease). A, Tetralogy of Fallot (arrow indicates direction of blood flow). B, Transposition of the great vessels with and without VSD. Ao, aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle.
Tetralogy of Fallot
Tetralogy of Fallot is the most common cause of cyanotic congenital heart disease and accounts for about 5% of all congenital cardiac malformations (Table 10–1). The four cardinal features are (1) VSD; (2) right ventricular outflow tract obstruction (subpulmonic stenosis); (3) overriding of the VSD by the aorta; and (4) right ventricular hypertrophy (Fig. 10–4, A). All of the features of tetralogy of Fallot result from anterosuperior displacement of the infundibular septum leading to abnormal septation between the pulmonary trunk and the aortic root.
Morphology
The heart is large and “boot-shaped” as a consequence of right ventricular hypertrophy; the proximal aorta is dilated, while the pulmonary trunk is hypoplastic. The left-sided cardiac chambers are of normal size, while the right ventricular wall is markedly hypertrophied, sometimes even exceeding the thickness of the left ventricle. The VSD usually is large and lies in the vicinity of the membranous portion of the interventricular septum; the aortic valve lies immediately over the VSD (overriding aorta) and is the major site of egress for blood flow from both ventricles. The obstruction of the right ventricular outflow most often is due to narrowing of the infundibulum (subpulmonic stenosis) but also can be caused by pulmonary valve stenosis or complete atresia of the valve and the proximal pulmonary arteries. In such cases, a persistent PDA or dilated bronchial arteries may be the only route for blood to reach the lungs.
Clinical Features
The hemodynamic consequences of tetralogy of Fallot are right-to-left shunting, decreased pulmonary blood flow, and increased aortic volumes. The clinical severity largely depends on the degree of the pulmonary outflow obstruction; even untreated, some patients can survive into adult life. Thus, if the pulmonic obstruction is mild, the condition resembles an isolated VSD because the high left-sided pressures cause only a left-to-right shunt with no cyanosis. More commonly, more severe degrees of pulmonic stenosis cause early cyanosis. Moreover, as the child grows and the heart increases in size, the pulmonic orifice does not expand proportionately, leading to progressive worsening of functional stenosis. Fortuitously, the pulmonic outflow stenosis protects the pulmonary vasculature from pressure and volume overloads, so that pulmonary hypertension does not develop, and right ventricular failure is rare. Nevertheless, patients develop the typical sequelae of cyanotic heart disease, such as polycythemia (due to hypoxia) with attendant hyperviscosity and hypertrophic osteoarthropathy; right-to-left shunting also increases the risk for infective endocarditis and systemic embolization. Complete surgical repair is possible with classic tetralogy of Fallot but is more complicated in the setting of pulmonary atresia.
Transposition of the Great Arteries
Transposition of the great arteries is a discordant connection of the ventricles to their vascular outflow. The embryologic defect is an abnormal formation of the truncal and aortopulmonary septa so that the aorta arises from the right ventricle and the pulmonary artery emanates from the left ventricle (Fig. 10–4, B). The atrium-to-ventricle connections, however, are normal (concordant), with the right atrium joining the right ventricle and the left atrium emptying into the left ventricle.
The functional outcome is separation of the systemic and pulmonary circulations, a condition incompatible with postnatal life unless a shunt such as a VSD exists for adequate mixing of blood and delivery of oxygenated blood to the aorta. Indeed, VSDs occur in a third of cases and provide stable shunts (Fig. 10–4, B). There is marked right ventricular hypertrophy, since that chamber functions as the systemic ventricle; the left ventricle is atrophic, since it pumps only to the low-resistance pulmonary circulation. Some patients with transposition of the great arteries have a patent foramen ovale or PDA that allows oxygenated blood to reach the aorta, but these tend to close; as a result, such infants typically require emergent surgical intervention within the first few days of life.
Clinical Features
The dominant manifestation is cyanosis, with the prognosis depending on the magnitude of shunting, the degree of tissue hypoxia, and the ability of the right ventricle to maintain systemic pressures. Without surgery (even with stable shunting), most patients with uncorrected transposition of the great arteries die within the first months of life. However, improved surgical techniques now permit definitive repair and such patients often survive into adulthood.
Obstructive Lesions
Congenital obstruction to blood flow can occur at the level of the heart valves or more distally within a great vessel. Obstruction can also occur proximal to the valve, as with subpulmonic stenosis in tetralogy of Fallot. Relatively common examples of congenital obstruction are pulmonic valve stenosis, aortic valve stenosis or atresia, and coarctation of the aorta.
Aortic Coarctation
Coarctation (narrowing, or constriction) of the aorta is a common form of obstructive congenital heart disease (Table 10–1). Males are affected twice as often as females, although females with Turner syndrome frequently have coarctation. There are two classic forms (Fig. 10–5): (1) an “infantile” form featuring hypoplasia of the aortic arch proximal to a PDA and (2) an “adult” form consisting of a discrete ridgelike infolding of the aorta, adjacent to the ligamentum arteriosum. Coarctation can occur as a solitary defect, but in more than half of the cases it is accompanied by a bicuspid aortic valve. Aortic valve stenosis, ASD, VSD, or mitral regurgitation also can be present.
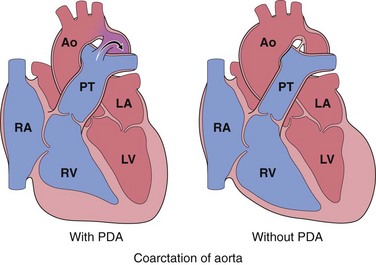
Figure 10–5 Coarctation of the aorta with (“infantile” or preductal form) and without a patent ductus arteriosus (PDA) (“adult” or postductal form); arrow indicates direction of blood flow. Ao, aorta; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle.
Morphology
“Infantile” (preductal) coarctation is characterized by circumferential narrowing of the aortic segment between the left subclavian artery and the ductus arteriosus; the ductus typically is patent and is the main source of (unoxygenated) blood delivered to the distal aorta. The pulmonary trunk is dilated to accommodate the increased blood flow; because the right side of the heart now perfuses the body distal to the narrowed segment (“coarct”), the right ventricle typically is hypertrophied.
In the more common “adult” (postductal) coarctation, the aorta is sharply constricted by a tissue ridge adjacent to the nonpatent ligamentum arteriosum (Fig. 10–6). The constricted segment is made up of smooth muscle and elastic fibers that are continuous with the aortic media. Proximal to the coarctation, the aortic arch and its branch vessels are dilated and the left ventricle is hypertrophied.
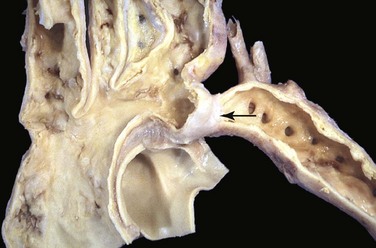
Figure 10–6 Coarctation of the aorta, postductal type. The coarctation is a segmental narrowing of the aorta (arrow). Such lesions typically manifest later in life than preductal coarctations. The dilated ascending aorta and major branch vessels are to the left of the coarctation. The lower extremities are perfused predominantly by way of dilated, tortuous collateral channels.
(Courtesy of Sid Murphree, MD, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Clinical Features
Clinical manifestations depend almost entirely on the severity of the narrowing and the patency of the ductus arteriosus.
• Preductal coarctation with a PDA usually presents early in life, classically as cyanosis localized to the lower half of the body; without intervention, most affected infants do not survive the neonatal period.
• Postductal coarctation without a PDA usually is asymptomatic, and the disease may remain unrecognized well into adult life. Classically, there is upper extremity hypertension paired with weak pulses and relative hypotension in the lower extremities, associated with symptoms of claudication and coldness. Exuberant collateral circulation “around” the coarctation often develops through markedly enlarged intercostal and internal mammary arteries; expansion of the flow through these vessels can lead to radiographically visible “notching” of the ribs.
In most cases, significant coarctations are associated with systolic murmurs and occasionally palpable thrills. Balloon dilation or surgical resection with end-to-end anastomosis (or replacement of the affected aortic segment by a prosthetic graft) yields excellent results.
Summary
Congenital Heart Disease
• Congenital heart disease represents defects of cardiac chambers or the great vessels; these either result in shunting of blood between the right and left circulation or cause outflow obstructions. Lesions range from relatively asymptomatic to rapidly fatal. Environmental (toxic or infectious) and genetic causes both contribute.
• Left-to-right shunts are the most common and typically are associated with ASDs, VSDs, or a PDA. These lesions result in chronic right-sided pressure and volume overloads that eventually cause pulmonary hypertension with reversal of flow and right-to-left shunts with cyanosis (Eisenmenger syndrome).
• Right-to-left shunts most commonly are caused by tetralogy of Fallot or transposition of the great arteries. These lesions cause early-onset cyanosis and are associated with polycythemia, hypertrophic osteoarthropathy, and paradoxical embolization.
• Obstructive lesions include forms of aortic coarctation; the clinical severity of these lesions depends on the degree of stenosis and the patency of the ductus arteriosus.
Ischemic Heart Disease
Since cardiac myocytes generate energy almost exclusively through mitochondrial oxidative phosphorylation, cardiac function is strictly dependent upon the continuous flow of oxygenated blood through the coronary arteries. Ischemic heart disease (IHD) is a broad term encompassing several closely related syndromes caused by myocardial ischemia—an imbalance between cardiac blood supply (perfusion) and myocardial oxygen and nutritional requirements. Despite dramatic improvements in therapy in the past quarter-century, IHD in its various forms remains the leading cause of mortality in the United States and other developed nations, accounting for 7 million deaths worldwide each year.
In more than 90% of cases, IHD is a consequence of reduced coronary blood flow secondary to obstructive atherosclerotic vascular disease (Chapter 9). Thus, unless otherwise specified, IHD usually is synonymous with coronary artery disease (CAD). In most cases, the syndromes of IHD are the late manifestations of coronary atherosclerosis that has been gradually building for decades (beginning even in childhood or adolescence).
Less frequently, IHD can result from increased demand (e.g., with increased heart rate or hypertension); diminished blood volume (e.g., with hypotension or shock); diminished oxygenation (e.g., due to pneumonia or CHF); or diminished oxygen-carrying capacity (e.g., due to anemia or carbon monoxide poisoning).
The manifestations of IHD are a direct consequence of the insufficient blood supply to the heart. The clinical presentation may include one or more of the following cardiac syndromes:
• Angina pectoris (literally, “chest pain”): Ischemia induces pain but is insufficient to cause myocyte death. Angina can be stable (occurring predictably at certain levels of exertion), can be caused by vessel spasm (Prinzmetal angina), or can be unstable (occurring with progressively less exertion or even at rest).
• Acute myocardial infarction (MI): The severity or duration of ischemia is sufficient to cause cardiomyocyte death.
• Chronic IHD with CHF: Progressive cardiac decompensation after acute MI, or secondary to accumulated small ischemic insults, eventually precipitates mechanical pump failure.
• Sudden cardiac death (SCD): This can occur as a consequence of tissue damage from MI, but most commonly results from a lethal arrhythmia without myocyte necrosis (see later under “Arrhythmias”).
The term acute coronary syndrome is applied to any of the three catastrophic manifestations of IHD—unstable angina, acute MI, and SCD.
Epidemiology
Nearly a half-million Americans die annually of IHD. As troubling as this toll is, it represents a spectacular advance over previous eras; since peaking in 1963, the mortality related to IHD in the United States has declined by 50%. The improvement can be largely attributed to interventions that have diminished cardiac risk factors (behaviors or conditions that promote atherosclerosis) (Chapter 9), in particular smoking cessation programs, hypertension and diabetes treatment, and use of cholesterol-lowering agents. To a lesser extent, diagnostic and therapeutic advances have also contributed; these include aspirin prophylaxis, better arrhythmia control, coronary care units, thrombolysis for MI, angioplasty and endovascular stenting, and coronary artery bypass graft surgery. Maintaining this downward trend in mortality will be particularly challenging given the predicted longevity of “baby boomers,” as well as the epidemic of obesity that is sweeping the United States and other parts of the world.
Pathogenesis
IHD is primarily a consequence of inadequate coronary perfusion relative to myocardial demand. This imbalance occurs as a consequence of the combination of preexisting (“fixed”) atherosclerotic occlusion of coronary arteries and new, superimposed thrombosis and/or vasospasm.
Atherosclerotic narrowing can affect any of the coronary arteries—left anterior descending (LAD), left circumflex (LCX), and right coronary artery (RCA)—singly or in any combination. Clinically significant plaques can be located anywhere but tend to occur within the first several centimeters of the LAD and LCX, and along the entire length of the RCA. Sometimes, secondary branches also are involved (i.e., diagonal branches of the LAD, obtuse marginal branches of the LCX, or posterior descending branch of the RCA).
Fixed obstructions that occlude less than 70% of a coronary vessel lumen typically are asymptomatic, even with exertion. In comparison, lesions that occlude more than 70% of a vessel lumen—resulting in so-called critical stenosis—generally cause symptoms in the setting of increased demand; with critical stenosis, certain levels of exertion predictably cause chest pain, and the patient is said to have stable angina. A fixed stenosis that occludes 90% or more of a vascular lumen can lead to inadequate coronary blood flow with symptoms even at rest—one of the forms of unstable angina (see later discussion).
Of importance, if an atherosclerotic lesion progressively occludes a coronary artery at a sufficiently slow rate over years, remodelling of other coronary vessels may provide compensatory blood flow for the area at risk; such collateral perfusion can subsequently protect against MI even if the vessel eventually becomes completely occluded. Unfortunately, with acute coronary blockage, there is no time for collateral flow to develop and infarction results.
The following elements contribute to the development and consequences of coronary atherosclerosis:
• Inflammation plays an essential role at all stages of atherosclerosis, from inception to plaque rupture (Chapter 9). Atherosclerosis begins with the interaction of endothelial cells and circulating leukocytes, resulting in T cell and macrophage recruitment and activation. These cells drive subsequent smooth muscle cell accumulation and proliferation, with variable amounts of matrix production, all overlying an atheromatous core of lipid, cholesterol, calcification, and necrotic debris. At later stages, destabilization of atherosclerotic plaque occurs through macrophage metalloproteinase secretion.
• Thrombosis associated with a disrupted plaque often triggers the acute coronary syndromes. Partial vascular occlusion by a newly formed thrombus on a disrupted atherosclerotic plaque can wax and wane with time and lead to unstable angina or sudden death; alternatively, even partial luminal occlusion by thrombus can compromise blood flow sufficiently to cause a small infarction of the innermost zone of the myocardium (subendocardial infarct). Organizing thrombi produce potent activators of smooth muscle proliferation, which can contribute to the growth of atherosclerotic lesions. Mural thrombus in a coronary artery can also embolize; indeed, small emboli can be found in the distal intramyocardial circulation (along with associated microinfarcts) at autopsy of patients who have had unstable angina. In the most serious extreme, completely obstructive thrombus over a disrupted plaque can cause a massive MI.
• Vasoconstriction directly compromises lumen diameter; moreover, by increasing local mechanical shear forces, vessel spasm can potentiate plaque disruption. Vasoconstriction in atherosclerotic plaques can be stimulated by (1) circulating adrenergic agonists, (2) locally released platelet contents, (3) imbalance between endothelial cell–relaxing factors (e.g., nitric oxide) and –contracting factors (e.g., endothelin) due to endothelial dysfunction, and (4) mediators released from perivascular inflammatory cells.
Acute Plaque Change
Onset of myocardial ischemia depends not only on the extent and severity of fixed atherosclerotic disease but also on dynamic changes in coronary plaque morphology. In most patients, unstable angina, infarction, and often sudden cardiac death occur because of abrupt plaque change followed by thrombosis—hence the term acute coronary syndrome (Fig. 10–7).
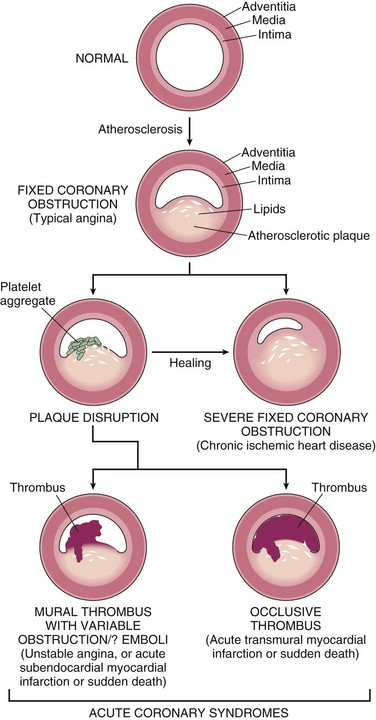
Figure 10–7 Diagram of sequential progression of coronary artery lesions leading to various acute coronary syndromes.
(Modified and redrawn from Schoen FJ: Interventional and Surgical Cardiovascular Pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989, p 63.)
The initiating event typically is sudden disruption of partially occlusive plaque. More than one mechanism of injury may be involved: Rupture, fissuring, or ulceration of plaques can expose highly thrombogenic constituents or underlying subendothelial basement membrane, leading to rapid thrombosis. In addition, hemorrhage into the core of plaques can expand plaque volume, thereby acutely exacerbating the degree of luminal occlusion.
Factors that trigger acute plaque change are believed to act by increasing the lesion’s susceptibility to disruption by mechanical stress. Both intrinsic aspects of plaque composition and structure (Chapter 9) and extrinsic factors, such as blood pressure and platelet reactivity, may contribute as follows:
• Plaques that contain large atheromatous cores, or have thin overlying fibrous caps are more likely to rupture, and are therefore termed “vulnerable.” Fissures frequently occur at the junction of the fibrous cap and the adjacent normal plaque-free arterial segment, where the mechanical stresses are highest and the fibrous cap is thinnest. Fibrous caps also are continuously remodeling; their overall balance of collagen synthesis versus degradation determines mechanical strength and plaque stability. Collagen is produced by smooth muscle cells and degraded by the action of metalloproteases elaborated by macrophages. Consequently, atherosclerotic lesions with a paucity of smooth muscle cells or large numbers of inflammatory cells are more vulnerable to rupture. Of interest, statins (inhibitors of hydroxymethylglutaryl Co-A reductase, a key enzyme in cholesterol synthesis) may be of additional benefit in CAD and IHD by reducing plaque inflammation and increasing plaque stability, beyond their cholesterol-lowering effects.
• Influences extrinsic to plaque also are important. Adrenergic stimulation can put physical stress on the plaque by causing hypertension or local vasospasm. Indeed, the surge in adrenergic stimulation associated with awakening and rising may underlie the observation that the incidence of acute MI is highest between 6 am and 12 noon. Intense emotional stress also leads to adrenergic stimulation, explaining the association of natural catastrophes such as earthquakes and floods with secondary waves of MIs in susceptible individuals.
In a majority of cases, the vulnerable “culprit lesion” in patients who suffer an MI was not critically stenotic or even symptomatic before its rupture. As noted previously, anginal symptoms typically occur with fixed lesions exhibiting greater than 70% chronic occlusion. Pathologic and clinical studies show that two thirds of ruptured plaques are 50% stenotic or less before plaque rupture, and 85% exhibit initial stenotic occlusion of 70% or less. Thus, the worrisome conclusion is that a large number of asymptomatic adults are at significant risk for a catastrophic coronary event. At present, it is impossible to predict plaque rupture in any given patient.
Plaque disruption and ensuing nonocclusive thrombosis also are common, repetitive, and often clinically silent complications of atheromas. The healing of such subclinical plaque disruption and overlying thrombosis is an important mechanism by which atherosclerotic lesions progressively enlarge (Fig. 10–7).
Angina Pectoris
Angina pectoris is an intermittent chest pain caused by transient, reversible myocardial ischemia. The pain probably is a consequence of the ischemia-induced release of adenosine, bradykinin, and other molecules that stimulate the autonomic afferents. Three variants are recognized:
• Typical or stable angina is predictable episodic chest pain associated with particular levels of exertion or some other increased demand (e.g., tachycardia). The pain is classically described as a crushing or squeezing substernal sensation, that can radiate down the left arm or to the left jaw (referred pain). The pain usually is relieved by rest (reducing demand) or by drugs such as nitroglycerin, a vasodilator that increases coronary perfusion.
• Prinzmetal or variant angina occurs at rest and is caused by coronary artery spasm. Although such spasms typically occur on or near existing atherosclerotic plaques, completely normal vessel can be affected. Prinzmetal angina typically responds promptly to vasodilators such as nitroglycerin and calcium channel blockers.
• Unstable angina (also called crescendo angina) is characterized by increasingly frequent pain, precipitated by progressively less exertion or even occurring at rest. Unstable angina is associated with plaque disruption and superimposed thrombosis, distal embolization of the thrombus, and/or vasospasm; it is often the harbinger of MI, caused by complete vascular occlusion.
Myocardial Infarction
Myocardial infarction (MI), also commonly referred to as “heart attack,” is necrosis of heart muscle resulting from ischemia. Roughly 1.5 million people per year in the United States suffer an MI; of these, one third die—half before they can get to the hospital. The major underlying cause of IHD is atherosclerosis; while MIs can occur at virtually any age, the frequency rises progressively with increasing age and with increasing atherosclerotic risk factors (Chapter 9). Nevertheless, approximately 10% of MIs occur before age 40, and 45% occur before age 65. Blacks and whites are equally affected. Men are at significantly greater risk than women, although the gap progressively narrows with age. In general, women tend to be remarkably protected against MI during their reproductive years. However, menopause—with declining estrogen production—is associated with exacerbation of coronary artery disease and IHD is the most common cause of death in elderly women.
Pathogenesis
The vast majority of MIs are caused by acute coronary artery thrombosis (Fig. 10–7). In most instances, disruption of preexisting atherosclerotic plaque serves as the nidus for thrombus generation, vascular occlusion, and subsequent transmural infarction of the downstream myocardium. In 10% of MIs, however, transmural infarction occurs in the absence of occlusive atherosclerotic vascular disease; such infarcts are mostly attributable to coronary artery vasospasm or to embolization from mural thrombi (e.g., in the setting of atrial fibrillation) or valve vegetations. Occasionally, especially with infarcts limited to the innermost (subendocardial) myocardium, thrombi or emboli may be absent. In such cases, severe diffuse coronary atherosclerosis leads to marginal perfusion of the heart. In this setting, a prolonged period of increased demand (e.g., due to tachycardia or hypertension) can lead to ischemic necrosis of the myocardium most distal to the epicardial vessels. Finally, ischemia without detectable atherosclerosis or thromboembolic disease can be caused by disorders of small intramyocardial arterioles, including vasculitis, amyloid deposition, or stasis, as in sickle cell disease.
Coronary Artery Occlusion
In a typical MI, the following sequence of events takes place:
• An atheromatous plaque is suddenly disrupted by intraplaque hemorrhage or mechanical forces, exposing subendothelial collagen and necrotic plaque contents to the blood.
• Platelets adhere, aggregate, and are activated, releasing thromboxane A2, adenosine diphosphate (ADP), and serotonin—causing further platelet aggregation and vasospasm (Chapter 3).
• Activation of coagulation by exposure of tissue factor and other mechanisms adds to the growing thrombus.
• Within minutes, the thrombus can evolve to completely occlude the coronary artery lumen.
The evidence for this scenario derives from autopsy studies of patients dying of acute MI, as well as imaging studies demonstrating a high frequency of thrombotic occlusion early after MI. Angiography performed within 4 hours of the onset of MI demonstrates coronary thrombosis in almost 90% of cases. When angiography is performed 12 to 24 hours after onset of symptoms, however, evidence of thrombosis is seen in only 60% of patients, even without intervention. Thus, at least some occlusions clear spontaneously through lysis of the thrombus or relaxation of spasm. This sequence of events in a typical MI also has therapeutic implications: Early thrombolysis and/or angioplasty can be highly successful in limiting the extent of myocardial necrosis.
Myocardial Response to Ischemia
Loss of the myocardial blood supply leads to profound functional, biochemical, and morphologic consequences. Within seconds of vascular obstruction, aerobic glycolysis ceases, leading to a drop in adenosine triphosphate (ATP) and accumulation of potentially noxious metabolites (e.g., lactic acid) in the cardiac myocytes. The functional consequence is a rapid loss of contractility, which occurs within a minute or so of the onset of ischemia. Ultrastructural changes (including myofibrillar relaxation, glycogen depletion, cell and mitochondrial swelling) also become rapidly apparent. These early changes are potentially reversible. Only severe ischemia lasting at least 20 to 40 minutes causes irreversible damage and myocyte death leading to coagulation necrosis (Chapter 1). With longer periods of ischemia, vessel injury ensues, leading to microvascular thrombosis.
Thus, if myocardial blood flow is restored before irreversible injury occurs, cell viability can be preserved; this is the rationale for early diagnosis of MI, and for prompt intervention by thrombolysis or angioplasty to salvage myocardium at risk. As discussed later, however, reperfusion also can have untoward effects. In addition, despite timely reperfusion, in the postischemic state, myocardium remains profoundly dysfunctional for at least several days. This defect is caused by persistent abnormalities in cellular biochemistry that result in a non-contractile state (stunned myocardium). Such stunning can be severe enough to produce transient but reversible cardiac failure.
Myocardial ischemia also contributes to arrhythmias, probably by causing electrical instability (irritability) of ischemic regions of the heart. Although massive myocardial damage can cause a fatal mechanical failure, sudden cardiac death in the setting of myocardial ischemia most often (in 80% to 90% of cases) is due to ventricular fibrillation caused by myocardial irritability.
Irreversible injury of ischemic myocytes first occurs in the subendocardial zone (Fig. 10–8). This region is especially susceptible to ischemia because it is the last area to receive blood delivered by the epicardial vessels, and also because it is exposed to relatively high intramural pressures, which act to impede the inflow of blood. With more prolonged ischemia, a wavefront of cell death moves through other regions of the myocardium, with the infarct usually achieving its full extent within 3 to 6 hours; in the absence of intervention, the infarct can involve the entire wall thickness (transmural infarct). Clinical intercession within this critical window of time can lessen the size of the infarct within the “territory at risk.”
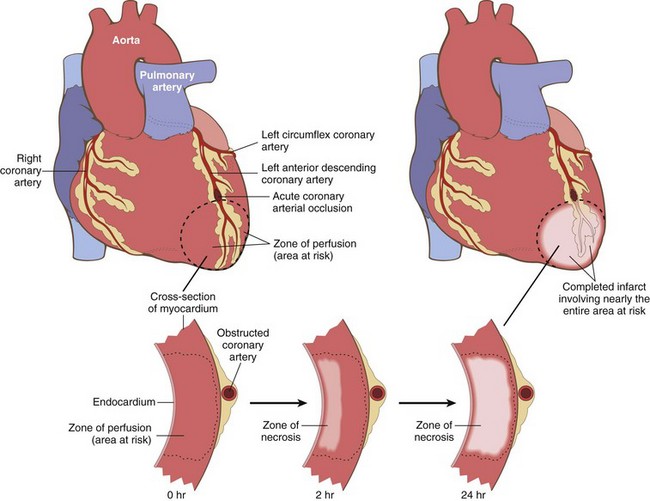
Figure 10–8 Progression of myocardial necrosis after coronary artery occlusion. A transmural segment of myocardium that is dependent on the occluded vessel for perfusion constitutes the area at risk (outlined). Necrosis begins in the subendocardial region in the center of the ischemic zone and with time expands to involve the entire wall thickness. Note that a very narrow zone of myocardium immediately beneath the endocardium is spared from necrosis because it can be oxygenated by diffusion from the ventricle.
Patterns of Infarction
The location, size, and morphologic features of an acute myocardial infarct depend on multiple factors:
• The size and distribution of the involved vessel (Fig. 10–9)
• The rate of development and the duration of the occlusion
• Metabolic demands of the myocardium (affected, for example, by blood pressure and heart rate)
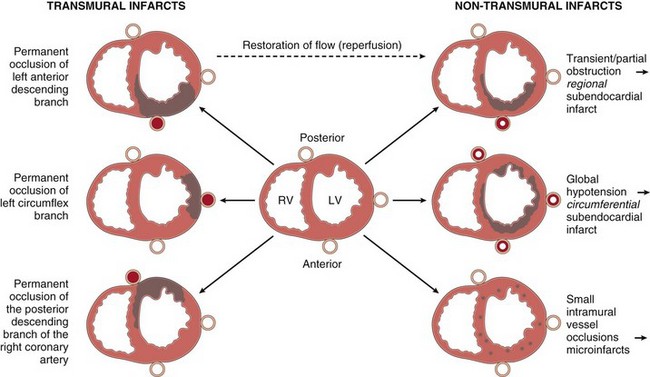
Figure 10–9 Dependence of myocardial infarction on the location and nature of the diminished perfusion. Left, Patterns of transmural infarction resulting from major coronary artery occlusion. Right ventricle may be involved with occlusion of right main coronary artery (not depicted). Right, Patterns of infarction resulting from partial or transient occlusion (top), global hypotension superimposed on fixed three-vessel disease (middle), or occlusion of small intramyocardial vessels (bottom).
Acute occlusion of the proximal left anterior descending (LAD) artery is the cause of 40% to 50% of all MIs and typically results in infarction of the anterior wall of the left ventricle, the anterior two thirds of the ventricular septum, and most of the heart apex; more distal occlusion of the same vessel may affect only the apex. Similarly, acute occlusion of the proximal left circumflex (LCX) artery (seen in 15% to 20% of MIs) will cause necrosis of the lateral left ventricle, and proximal right coronary artery (RCA) occlusion (30% to 40% of MIs) affects much of the right ventricle. The posterior third of the septum and the posterior left ventricle are perfused by the posterior descending artery. The posterior descending artery can arise from either the RCA (in 90% of people) or the LCX. By convention, the coronary artery—either RCA or LCX—that gives rise to the posterior descending artery and thereby perfuses the posterior third of the septum is considered the dominant vessel. Thus, in a right dominant heart, occlusion of the RCA can lead to left ventricular ischemic injury, while in a left dominant heart, occlusion of the left main coronary artery will generally affect the entire left ventricle and septum. Occasionally coronary occlusions are encountered in the left main coronary artery—a lesion dubbed a “widow maker” because so much myocardial territory is supplied that acute obstructions of the left main coronary artery typically are fatal. Occlusions may also affect secondary branches, such as the diagonal branches of the LAD artery or marginal branches of the LCX artery. By contrast, significant atherosclerosis or thrombosis of penetrating intramyocardial branches of coronary arteries is rare.
Even though the three major coronary arteries are end arteries, these epicardial vessels are interconnected by numerous intercoronary anastomoses (collateral circulation). Although these channels normally are closed, gradual narrowing of one artery allows blood to flow from high to low pressure areas through the collateral channels. In this manner, gradual collateral dilation can provide adequate perfusion to areas of the myocardium despite occlusion of an epicardial vessel. Based on the size of the involved vessel and the degree of collateral circulation, myocardial infarcts may take one of the following patterns.
• Transmural infarctions involve the full thickness of the ventricle and are caused by epicardial vessel occlusion through a combination of chronic atherosclerosis and acute thrombosis; such transmural MIs typically yield ST segment elevations on the electrocardiogram (ECG) and can have a negative Q waves with loss of R wave amplitude. These infarcts are also called ST elevated MIs (STEMIs).
• Subendocardial infarctions are MIs limited to the inner third of the myocardium; these infarcts typically do not exhibit ST segment elevations or Q waves on the ECG tracing. As already mentioned, the subendocardial region is most vulnerable to hypoperfusion and hypoxia. Thus, in the setting of severe coronary artery disease, transient decreases in oxygen delivery (as from hypotension, anemia, or pneumonia) or increases in oxygen demand (as with tachycardia or hypertension) can cause subendocardial ischemic injury. This pattern also can occur when an occlusive thrombus lyses before a full-thickness infarction can develop.
• Microscopic infarcts occur in the setting of small vessel occlusions and may not show any diagnostic ECG changes. These can occur in the setting of vasculitis, embolization of valve vegetations or mural thrombi, or vessel spasm due to elevated catecholamines—either endogenous (e.g., pheochromocytoma or extreme stress), or exogenous (e.g., cocaine).
Morphology
Nearly all transmural infarcts (involving 50% or more of the ventricle thickness) affect at least a portion of the left ventricle and/or interventricular septum. Roughly 15% to 30% of MIs that involve the posterior or posteroseptal wall also extend into the right ventricle. Isolated right ventricle infarcts occur in only 1% to 3% of cases. Even in transmural infarcts, a narrow rim (approximately 0.1 mm) of viable subendocardial myocardium is preserved by diffusion of oxygen and nutrients from the ventricular lumen.
The gross and microscopic appearance of an MI depends on the age of the injury. Areas of damage progress through a highly characteristic sequence of morphologic changes from coagulative necrosis, to acute and then chronic inflammation, to fibrosis (Table 10–3). Myocardial necrosis proceeds invariably to scar formation without any significant regeneration; studies looking at whether tissue stem cells can be used to regenerate functional myocardium are ongoing but have yet to bear fruit.
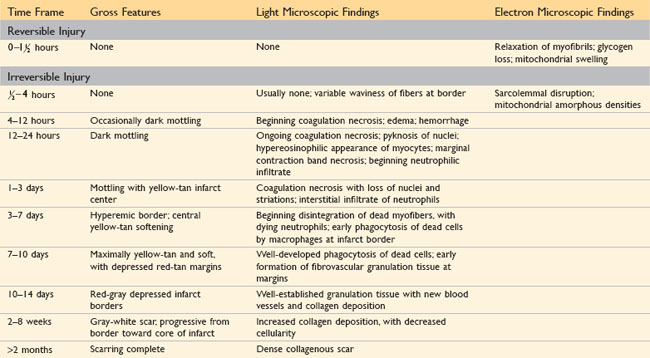
Recognition of very recent myocardial infarcts can be challenging, particularly when death occurs within a few hours. Myocardial infarcts less than 12 hours old usually are not grossly apparent. However, infarcts more than 3 hours old can be visualized by exposing myocardium to vital stains, such as triphenyltetrazolium chloride, a substrate for lactate dehydrogenase. Because this enzyme is depleted in the area of ischemic necrosis (it leaks out of the damaged cells), the infarcted area is unstained (pale), while old scars appear white and glistening (Fig. 10–10). By 12 to 24 hours after MI, an infarct usually can be grossly identified by a red-blue discoloration caused by stagnated, trapped blood. Thereafter, infarcts become progressively better delineated as soft, yellow-tan areas; by 10 to 14 days, infarcts are rimmed by hyperemic (highly vascularized) granulation tissue. Over the succeeding weeks, the infarcted tissue evolves to a fibrous scar.
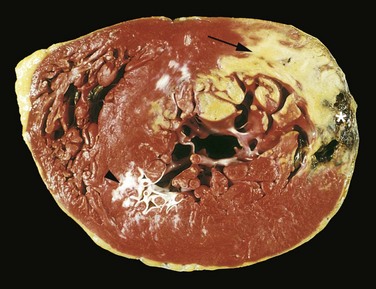
Figure 10–10 Acute myocardial infarct of the posterolateral left ventricle demonstrated by a lack of triphenyltetrazolium chloride staining in areas of necrosis (arrow); the absence of staining is due to enzyme leakage after cell death. Note the anterior scar (arrowhead), indicative of remote infarction. The myocardial hemorrhage at the right edge of the infarct (asterisk) is due to ventricular rupture, and was the acute cause of death in this patient (specimen is oriented with the posterior wall at the top).
The microscopic appearance also undergoes a characteristic sequence of changes (Table 10–3 and Figure 10–11). Typical features of coagulative necrosis (Chapter 1) become detectable within 4 to 12 hours of infarction. “Wavy fibers” also can be present at the edges of an infarct; these reflect the stretching and buckling of noncontractile dead fibers. Sublethal ischemia can also induce intracellular myocyte vacuolization; such myocytes are viable but frequently are poorly contractile.
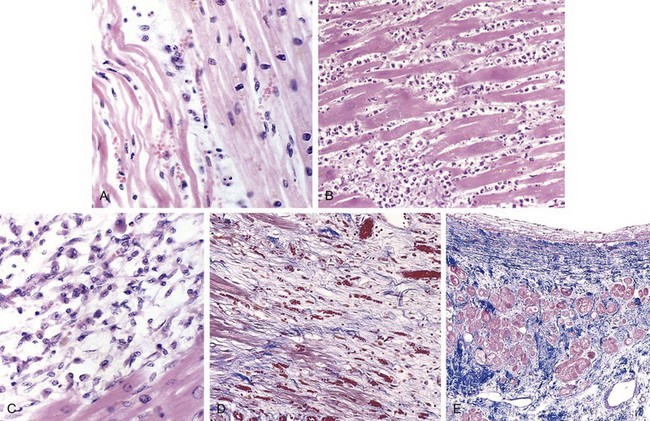
Figure 10–11 Microscopic features of myocardial infarction and its repair. A, One-day-old infarct showing coagulative necrosis and wavy fibers, compared with adjacent normal fibers (at right). Necrotic cells are separated by edema fluid. B, Dense neutrophilic infiltrate in the area of a 2- to 3-day-old infarct. C, Nearly complete removal of necrotic myocytes by phagocytic macrophages (7 to 10 days). D, Granulation tissue characterized by loose connective tissue and abundant capillaries. E, Healed myocardial infarct consisting of a dense collagenous scar. A few residual cardiac muscle cells are present. D and E are Masson’s trichrome stain, which stains collagen blue.
Necrotic myocardium elicits acute inflammation (typically most prominent 1 to 3 days after MI), followed by a wave of macrophages that remove necrotic myocytes and neutrophil fragments (most pronounced by 5 to 10 days after MI). The infarcted zone is progressively replaced by granulation tissue (most prominent 1 to 2 weeks after MI), which in turn forms the provisional scaffolding upon which dense collagenous scar forms. In most instances, scarring is well advanced by the end of the sixth week, but the efficiency of repair depends on the size of the original lesion. Healing requires the migration of inflammatory cells and ingrowth of new vessels from the infarct margins. Thus, an MI heals from its borders toward the center, and a large infarct may not heal as fast or as completely as a small one. Once an MI is completely healed, it is impossible to distinguish its age: Whether present for 8 weeks or 10 years, fibrous scars look the same.
Infarct Modification by Reperfusion
The therapeutic goal in acute MI is to salvage the maximal amount of ischemic myocardium; this is accomplished by restoration of tissue perfusion as quickly as possible (hence the adage “time is myocardium”). Such reperfusion is achieved by thrombolysis (dissolution of thrombus by tissue plasminogen activator), angioplasty, or coronary arterial bypass graft. Unfortunately, while preservation of viable (but at-risk) heart can improve both short- and long-term outcomes, reperfusion is not an unalloyed blessing. Indeed, restoration of blood flow into ischemic tissues can incite greater local damage than might otherwise have occurred—so-called reperfusion injury. The factors that contribute to reperfusion injury include: 1) Mitochondrial dysfunction: ischemia alters the mitochondrial membrane permeability, which allows proteins to move into the mitochondria. This leads to swelling and rupture of the outer membrane, releasing mitochondrial contents that promote apoptosis; 2) Myocyte hypercontracture: during periods of ischemia the intracellular levels of calcium are increased as a result of impaired calcium cycling and sarcolemmal damage. After reperfusion the contraction of myofibrils is augmented and uncontrolled, causing cytoskeletal damage and cell death; 3) Free radicals including superoxide anion (•O2), hydrogen peroxide (H2O2), hypochlorous acid (HOCl), nitric oxide–derived peroxynitrite, and hydroxyl radicals (•OH) are produced within minutes of reperfusion and cause damage to the myocytes by altering membrane proteins and phospholipids; 4) Leukocyte aggregation, which may occlude the microvasculature and contribute to the “no-reflow” phenomenon. Further, leukocytes elaborate proteases and elastases that cause cell death; 5) Platelet and complement activation also contribute to microvascular injury. Complement activation is thought to play a role in the no-reflow phenomenon by injuring the endothelium.
The typical appearance of reperfused myocardium in the setting of an acute MI is shown in Figure 10–12. Such infarcts typically are hemorrhagic as a consequence of vascular injury and leakiness. Microscopically, irreversibly damaged myocytes subject to reperfusion show contraction band necrosis; in this pathologic process, intense eosinophilic bands of hypercontracted sarcomeres are created by an influx of calcium across plasma membranes that enhances actin-myosin interactions. In the absence of ATP, the sarcomeres cannot relax and get stuck in an agonal tetanic state. Thus, while reperfusion can salvage reversibly injured cells, it also alters the morphology of irreversibly injured cells.
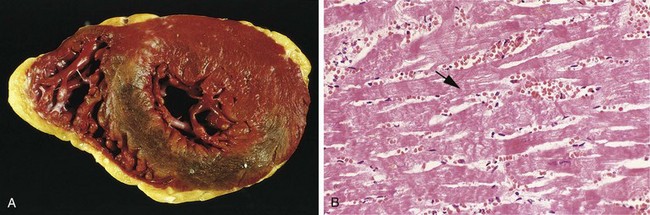
Figure 10–12 Reperfused myocardial infarction. A, The transverse heart slice (stained with triphenyl tetrazolium chloride) exhibits a large anterior wall myocardial infarction that is hemorrhagic because of bleeding from damaged vessels. Posterior wall is at top. B, Hemorrhage and contraction bands, visible as prominent hypereosinophilic cross-striations spanning myofibers (arrow), are seen microscopically.
Clinical Features
The classic MI is heralded by severe, crushing substernal chest pain (or pressure) that can radiate to the neck, jaw, epigastrium, or left arm. In contrast to angina pectoris, the associated pain typically lasts several minutes to hours, and is not relieved by nitroglycerin or rest. However, in a substantial minority of patients (10% to 15%), MIs have atypical signs and symptoms and may even be entirely asymptomatic. Such “silent” infarcts are particularly common in patients with underlying diabetes mellitus (in which autonomic neuropathies may prevent perception of pain) and in elderly persons.
The pulse generally is rapid and weak, and patients are often diaphoretic and nauseous (particularly with posterior wall MIs). Dyspnea is common, attributable to impaired myocardial contractility and dysfunction of the mitral valve apparatus, with resultant acute pulmonary congestion and edema. With massive MIs (involving more than 40% of the left ventricle), cardiogenic shock develops.
Electrocardiographic abnormalities are important for the diagnosis of MI; these include Q waves, ST segment changes, and T wave inversions (the latter two representing abnormalities in myocardial repolarization). Arrhythmias caused by electrical abnormalities in ischemic myocardium and conduction system are common; indeed, sudden cardiac death from a lethal arrhythmia accounts for the vast majority of MI-related deaths occurring before hospitalization.
The laboratory evaluation of MI is based on measuring blood levels of macromolecules that leak out of injured myocardial cells through damaged cell membranes (Fig. 10–13); these molecules include myoglobin, cardiac troponins T and I (TnT, TnI), creatine kinase (CK) (specifically the myocardial isoform, CK-MB), and lactate dehydrogenase. Troponins and CK-MB have high specificity and sensitivity for myocardial damage.
• CK-MB remains a valuable marker of myocardial injury, second only to the cardiac-specific troponins (see next entry). Total CK activity is not a reliable marker of cardiac injury since various isoforms of CK are also found in brain, myocardium, and skeletal muscle. However, the CK-MB isoform—principally derived from myocardium, but also present at low levels in skeletal muscle—is the more specific indicator of heart damage. CK-MB activity begins to rise within 2 to 4 hours of MI, peaks at 24 to 48 hours, and returns to normal within approximately 72 hours.
• TnI and TnT normally are not found in the circulation; however, after acute MI, both are detectable within 2 to 4 hours, with levels peaking at 48 hours and remaining elevated for 7 to 10 days. Although cardiac troponin and CK-MB are equally sensitive markers of the early stages of an MI, persistence of elevated troponin levels for approximately 10 days allows the diagnosis of an acute MI long after CK-MB levels have returned to normal. With reperfusion, both troponin and CK-MB levels may peak earlier owing to more rapid washout of the enzyme from the necrotic tissue.
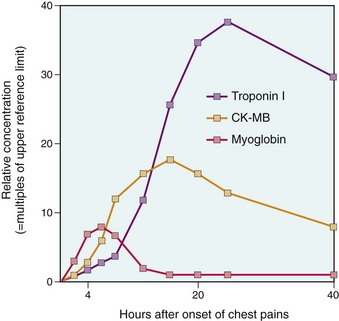
Consequences and Complications of Myocardial Infarction
Extraordinary progress has been made in improving patient outcomes after acute MI; the overall in-hospital death rate for MI is approximately 7%. Unfortunately, out-of-hospital mortality is substantially worse: A third of persons with ST elevation MIs (STEMIs) will die, usually of an arrhythmia within an hour of symptom onset, before they receive appropriate medical attention. Such statistics make the rising rate of coronary artery disease in developing countries with scarce hospital facilities all the more worrisome.
Nearly three fourths of patients experience one or more of the following complications after an acute MI (Fig. 10–14):
• Contractile dysfunction. In general, MIs affect left ventricular pump function in proportion to the volume of damage. In most cases, there is some degree of left ventricular failure manifested as hypotension, pulmonary congestion, and pulmonary edema. Severe “pump failure” (cardiogenic shock) occurs in roughly 10% of patients with transmural MIs and typically is associated with infarcts that damage 40% or more of the left ventricle.
• Papillary muscle dysfunction. Although papillary muscles rupture infrequently after MI, they often are dysfunctional and can be poorly contractile as a result of ischemia, leading to postinfarct mitral regurgitation. Much later, papillary muscle fibrosis and shortening or global ventricular dilation also can cause mitral valve insufficiency.
• Right ventricular infarction. Although isolated right ventricular infarction occurs in only 1% to 3% of MIs, the right ventricle frequently is injured in association with septal or left ventricular infarction. In either case, right-sided heart failure is a common outcome, leading to venous circulation pooling and systemic hypotension.
• Myocardial rupture. Rupture complicates only 1% to 5% of MIs but is frequently fatal when it occurs. Left ventricular free wall rupture is most common, usually resulting in rapidly fatal hemopericardium and cardiac tamponade (Fig. 10–14, A). Ventricular septal rupture creates a VSD with left-to-right shunting (Fig. 10–14, B), and papillary muscle rupture leads to severe mitral regurgitation (Fig. 10–14, C). Rupture occurs most commonly within 3 to 7 days after infarction—the time in the healing process when lysis of myocardial connective tissue is maximal and when much of the infarct has been converted to soft, friable granulation tissue. Risk factors for free wall rupture include age older than 60 years, anterior or lateral wall infarctions, female gender, lack of left ventricular hypertrophy, and first MI (as scarring associated with prior MIs tends to limit the risk of myocardial tearing).
• Arrhythmias. MIs lead to myocardial irritability and conduction disturbances that can cause sudden death. Approximately 90% of patients develop some form of rhythm disturbance, with the incidence being higher in STEMIs versus non-STEMIs. MI-associated arrhythmias include heart block of variable degree (including asystole), bradycardia, supraventricular tachyarrhythmias, ventricular premature contractions or ventricular tachycardia, and ventricular fibrillation. The risk of serious arrhythmias (e.g., ventricular fibrillation) is greatest in the first hour and declines thereafter.
• Pericarditis. Transmural MIs can elicit a fibrinohemorrhagic pericarditis; this is an epicardial manifestation of the underlying myocardial inflammation (Fig. 10–14, D). Heralded by anterior chest pain and a pericardial friction rub, pericarditis typically appears 2 to 3 days after infarction and then gradually resolves over the next few days. Extensive infarcts or severe pericardial inflammation occasionally can lead to large effusions or can organize to form dense adhesions that eventually manifest as a constrictive lesion.
• Chamber dilation. Because of the weakening of necrotic muscle, there may be disproportionate stretching, thinning, and dilation of the infarcted region (especially with anteroseptal infarcts).
• Mural thrombus. With any infarct, the combination of attenuated myocardial contractility (causing stasis) and endocardial damage (causing a thrombogenic surface) can foster mural thrombosis (Fig. 10–14, E), eventually leading to left-sided thromboembolism.
• Ventricular aneurysm. A late complication, aneurysms of the ventricle most commonly result from a large transmural anteroseptal infarct that heals with the formation of a thinned wall of scar tissue (Fig. 10–14, F). Although ventricular aneurysms frequently give rise to formation of mural thrombi, arrhythmias, and heart failure, they do not rupture.
• Progressive late heart failure. Discussed later on as “chronic IHD.”
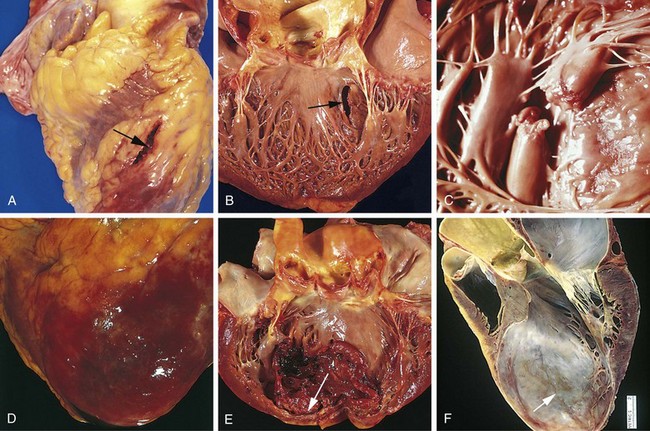
Figure 10–14 Complications of myocardial infarction. A–C, Cardiac rupture. A, Anterior free wall myocardial rupture (arrow). B, Ventricular septal rupture (arrow). C, Papillary muscle rupture. D, Fibrinous pericarditis, with a hemorrhagic, roughened epicardial surface overlying an acute infarct. E, Recent expansion of an anteroapical infarct with wall stretching and thinning (arrow) and mural thrombus. F, Large apical left ventricular aneurysm (arrow).
(A–E, Reproduced by permission from Schoen FJ: Interventional and Surgical Cardiovascular Pathology: Clinical Correlations and Basic Principles. Philadelphia, WB Saunders, 1989; F, Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, Minnesota.)
The risk of developing complications and the prognosis after MI depend on infarct size, site, and type (subendocardial versus transmural infarct). Thus, large transmural infarcts are associated with a higher probability of cardiogenic shock, arrhythmias, and late CHF, and patients with anterior transmural MIs are at greatest risk for free wall rupture, expansion, formation of mural thrombi, and aneurysm formation. By contrast, posterior transmural infarcts are more likely to be complicated by serious conduction blocks, right ventricular involvement, or both; when acute-onset VSDs occur in this area, they are more difficult to manage. Overall, patients with anterior infarcts have a much worse clinical course than those with posterior infarcts. With subendocardial infarcts, thrombi may form on the endocardial surface, but pericarditis, rupture, and aneurysms rarely occur.
In addition to the aforementioned scarring, the remaining viable myocardium attempts to compensate for the loss of contractile mass. Noninfarcted regions undergo hypertrophy and dilation; in combination with the scarring and thinning of the infarcted zones, the changes are collectively termed ventricular remodeling. The initial compensatory hypertrophy of noninfarcted myocardium is hemodynamically beneficial. The adaptive effect of remodeling can be overwhelmed, however, and ventricular function may decline in the setting of expansion and ventricular aneurysm formation.
Long-term prognosis after MI depends on many factors, the most important of which are left ventricular function and the severity of atherosclerotic narrowing of vessels perfusing the remaining viable myocardium. The overall mortality rate within the first year is about 30%, including deaths occurring before the patient reaches the hospital. Thereafter, the annual mortality rate is 3% to 4%.
Chronic Ischemic Heart Disease
Chronic IHD, also called ischemic cardiomyopathy, is essentially progressive heart failure secondary to ischemic myocardial damage. In most instances, there is a history of previous MI. In this setting, chronic IHD appears when the compensatory mechanisms (e.g., hypertrophy) of residual viable myocardium begin to fail. In other cases, severe obstructive CAD can cause diffuse myocardial dysfunction without frank infarction.
Morphology
Patients with chronic IHD typically exhibit left ventricular dilation and hypertrophy, often with discrete areas of gray-white scarring from previous healed infarcts. Invariably, there is moderate to severe atherosclerosis of the coronary arteries, sometimes with total occlusion. The endocardium generally shows patchy, fibrous thickening, and mural thrombi may be present. Microscopic findings include myocardial hypertrophy, diffuse subendocardial myocyte vacuolization, and fibrosis from previous infarction.
Cardiac Stem Cells
Because of the serious morbidity associated with IHD, there is much interest in exploring the possibility of using cardiac stem cells to replace the damaged myocardium. Although cardiac regeneration in metazoans (such as newts and zebrafish) is well described, the myocardium of higher-order animals is classically considered a postmitotic cell population without replicative potential. Increasing evidence, however, points to the presence of bone marrow–derived precursors—as well as a small resident stem cell population within the myocardium—capable of repopulating the mammalian heart. These cells are characterized by the expression of a cluster of cell surface markers that allow their isolation and purification. Besides self-renewal, these cardiac stem cells generate all cell lineages seen within the myocardium. Like all other tissue stem cells, they occur in very low frequency. They have a slow intrinsic rate of proliferation, which is greatest in neonates and decreases with age. Of interest, stem cell numbers and progeny increase after myocardial injury or hypertrophy, albeit to a limited extent, since hearts that suffer an MI clearly do not recover any significant function in the necrotic zone. Nevertheless, the potential for stimulating the proliferation of these cells in vivo is tantalizing because it could facilitate recovery of myocardial function after acute MI or chronic IHD. Conversely, ex vivo expansion and subsequent administration of such cells after an MI is another area of vigorous investigation. Unfortunately, results thus far have been less than exciting. Implanted stem cells may show some cardiomyocyte differentiation, but the durability of this benefit has been limited, and they do not contribute significantly to the restoration of contractile force; moreover, aberrant integration into the conducting system of the host heart carries the risk of formation of autonomous arrhythmic foci.
Summary
Ischemic Heart Disease
• In the vast majority of cases, cardiac ischemia is due to coronary artery atherosclerosis; vasospasm, vasculitis, and embolism are less common causes.
• Cardiac ischemia results from a mismatch between coronary supply and myocardial demand and manifests as different, albeit overlapping syndromes:
Angina pectoris is exertional chest pain due to inadequate perfusion, and is typically due to atherosclerotic disease causing greater than 70% fixed stenosis (so-called critical stenosis). Unstable angina results from a small fissure or rupture of atherosclerotic plaque triggering platelet aggregation, vasoconstriction, and formation of a mural thrombus that need not necessarily be occlusive. Acute myocardial infarction typically results from acute thrombosis after plaque disruption; a majority occur in plaques that did not previously exhibit critical stenosis. Sudden cardiac death usually results from a fatal arrhythmia, typically without significant acute myocardial damage. Ischemic cardiomyopathy is progressive heart failure due to ischemic injury, either from previous infarction(s) or chronic ischemia.• Myocardial ischemia leads to loss of myocyte function within 1 to 2 minutes but causes necrosis only after 20 to 40 minutes. Myocardial infarction is diagnosed on the basis of symptoms, electrocardiographic changes, and measurement of serum CK-MB and troponins. Gross and histologic changes of infarction require hours to days to develop.
• Infarction can be modified by therapeutic intervention (e.g., thrombolysis or stenting), which salvages myocardium at risk but may also induce reperfusion-related injury.
• Complications of infarction include ventricular rupture, papillary muscle rupture, aneurysm formation, mural thrombus, arrhythmia, pericarditis, and CHF.
Arrhythmias
As is well known, the heart contains specialized conduction system consisting of excitatory myocytes that regulate the rate and rhythm of cardiac contraction and are essential for normal cardiac function. This system is influenced by direct neural inputs (e.g., vagal stimulation), adrenergic agents (e.g., epinephrine [adrenaline]), hypoxia, and potassium concentrations (i.e., hyperkalemia can block signal transmission altogether). The components of the conduction system include (1) the sinoatrial (SA) node pacemaker (located at the junction of the right atrial appendage and superior vena cava), (2) the atrioventricular (AV) node (located in the right atrium along the atrial septum), (3) the bundle of His, connecting the right atrium to the ventricular septum, and the subsequent divisions into (4) the right and left bundle branches that stimulate their respective ventricles.
Abnormalities in myocardial conduction can be sustained or sporadic (paroxysmal). Aberrant rhythms can be initiated anywhere in the conduction system, from the SA node down to the level of an individual myocyte; they are typically designated as originating from the atrium (supraventricular) or within the ventricular myocardium. Arrhythmias can manifest as tachycardia (fast heart rate), bradycardia (slow heart rate), an irregular rhythm with normal ventricular contraction, chaotic depolarization without functional ventricular contraction (ventricular fibrillation), or no electrical activity at all (asystole). Patients may be unaware of a rhythm disorder or may note a “racing heart” or palpitations; loss of adequate cardiac output due to sustained arrhythmia can produce lightheadedness (near syncope), loss of consciousness (syncope), or sudden cardiac death (see further on).
Ischemic injury is the most common cause of rhythm disorders, because of direct damage or due to the dilation of heart chambers with consequent alteration in conduction system firing.
Far less common are inherited causes of arrhythmias. These are caused by mutations in genes that regulate various ion channels that regulate depolarization and repolarization of myocardial cells. Such channelopathies are important (but fortunately uncommon) substrates for fatal arrhythmias. They underlie some cases of sudden cardiac death, which is discussed next.
Sudden Cardiac Death
Sudden cardiac death (SCD) most commonly is defined as sudden death, typically due to sustained ventricular arrhythmias in individuals who have underlying structural heart disease which may or may not have been symptomatic in the past. Some 300,000 to 400,000 persons are victims of SCD each year in the United States alone. Coronary artery disease is the leading cause of death, being responsible for 80% to 90% of cases; unfortunately, SCD often is the first manifestation of IHD. Of interest, autopsy typically shows only chronic severe atherosclerotic disease; acute plaque disruption is found in only 10% to 20% of cases. Healed remote MIs are present in about 40% of the cases.
In younger victims of SCD, other, nonatherosclerotic causes are more common, including:
• Hereditary (channelopathies) or acquired abnormalities of the cardiac conduction system
• Congenital coronary arterial abnormalities
• Dilated or hypertrophic cardiomyopathy
• Myocardial hypertrophy. Increased cardiac mass is an independent risk factor for SCD; thus, in some young persons who die suddenly, including athletes, hypertensive hypertrophy or unexplained increased cardiac mass is the only pathologic finding.
The ultimate mechanism of SCD most often is a lethal arrhythmia (e.g., asystole or ventricular fibrillation). Of note, frank infarction need not occur; 80% to 90% of patients who suffer SCD but are successfully resuscitated do not show any enzymatic or ECG evidence of myocardial necrosis—even if the original cause was IHD! Although ischemic injury (and other pathologic conditions) can directly affect the major components of the conduction system, most cases of fatal arrhythmia are triggered by electrical irritability of myocardium distant from the conduction system.
The relationship of coronary artery disease to the various clinical end points discussed earlier is depicted in Figure 10–15.
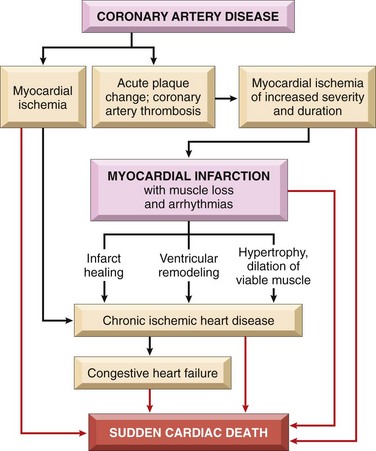
Figure 10–15 Pathways in the progression of ischemic heart disease showing the relationships among coronary artery disease and its major sequelae.
The prognosis for patients vulnerable to SCD is markedly improved by medical intervention, particularly by implantation of automatic cardioverter-defibrillators that sense and electrically counteract episodes of ventricular fibrillation.
Summary
Arrhythmias
• Arrhythmias can be caused by ischemic or structural changes in the conduction system or by myocyte electrical instability. In structurally normal hearts, arrhythmias more often are due to mutations in ion channels that cause aberrant repolarization or depolarization.
• SCD most frequently is due to coronary artery disease leading to ischemia. Myocardial irritability typically results from nonlethal ischemia or from preexisting fibrosis from previous myocardial injury. SCD less often is due to acute plaque rupture with thrombosis that induces a rapidly fatal arrhythmia.
Hypertensive Heart Disease
As discussed in Chapter 9, hypertension is a common disorder associated with considerable morbidity and affecting many organs, including the heart, brain, and kidneys. The comments here will focus specifically on the major cardiac complications of hypertension, which result from pressure overload and ventricular hypertrophy. Myocyte hypertrophy is an adaptive response to pressure overload; there are limits to myocardial adaptive capacity, however, and persistent hypertension eventually can culminate in dysfunction, cardiac dilation, CHF, and even sudden death. Although hypertensive heart disease most commonly affects the left side of the heart secondary to systemic hypertension, pulmonary hypertension also can cause right-sided hypertensive changes—so-called cor pulmonale.
Systemic (Left-Sided) Hypertensive Heart Disease
The criteria for the diagnosis of systemic hypertensive heart disease are (1) left ventricular hypertrophy in the absence of other cardiovascular pathology (e.g., valvular stenosis), and (2) a history or pathologic evidence of hypertension. The Framingham Heart Study established unequivocally that even mild hypertension (above 140/90 mm Hg), if sufficiently prolonged, induces left ventricular hypertrophy. Roughly 25% of the U.S. population suffers from at least this degree of hypertension.
Morphology
As discussed earlier, systemic hypertension imposes pressure overload on the heart and is associated with gross and microscopic changes somewhat distinct from those caused by volume overload. The essential feature of systemic hypertensive heart disease is left ventricular hypertrophy, typically without ventricular dilation until very late in the process (Fig. 10–16, A). The heart weight can exceed 500 g (normal, 320 to 360 g), and the left ventricular wall thickness can exceed 2.0 cm (normal, 1.2 to 1.4 cm). With time, the increased left ventricular wall thickness imparts a stiffness that impairs diastolic filling and can result in left atrial dilation. In long-standing systemic hypertensive heart disease leading to congestive failure, the ventricle typically is dilated.
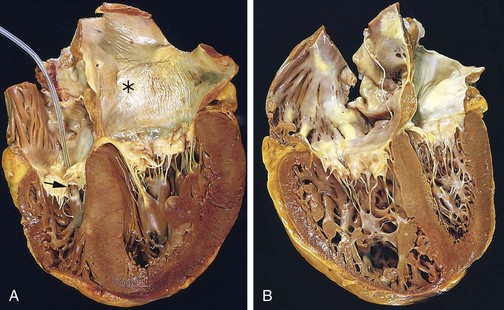
Figure 10–16 Hypertensive heart disease. A, Systemic (left-sided) hypertensive heart disease. There is marked concentric thickening of the left ventricular wall causing reduction in lumen size. The left ventricle and left atrium are on the right in this four-chamber view of the heart. A pacemaker is present incidentally in the right ventricle (arrow). Note also the left atrial dilation (asterisk) due to stiffening of the left ventricle and impaired diastolic relaxation, leading to atrial volume overload. B, Chronic cor pulmonale. The right ventricle (shown on the left side of this picture) is markedly dilated and hypertrophied with a thickened free wall and hypertrophied trabeculae. The shape and volume of the left ventricle have been distorted by the enlarged right ventricle.
Microscopically, the transverse diameter of myocytes is increased and there is prominent nuclear enlargement and hyperchromasia (“boxcar nuclei”), as well as intercellular fibrosis.
Clinical Features
Compensated hypertensive heart disease typically is asymptomatic and is suspected only from discovery of elevated blood pressure on routine physical exams, or from ECG or echocardiographic findings of left ventricular hypertrophy. In some patients, the disease comes to attention with the onset of atrial fibrillation (secondary to left atrial enlargement) and/or CHF. The mechanisms by which hypertension leads to heart failure are incompletely understood; presumably the hypertrophic myocytes fail to contract efficiently, possibly due to structural abnormalities in newly assembled sarcomeres and because the vascular supply is inadequate to meet the demands of the increased muscle mass. Depending on the severity and duration of the condition, the underlying cause of hypertension, and the adequacy of therapeutic control, patients can (1) enjoy normal longevity and die of unrelated causes, (2) develop progressive IHD owing to the effects of hypertension in potentiating coronary atherosclerosis, (3) suffer progressive renal damage or cerebrovascular stroke, or (4) experience progressive heart failure. The risk of sudden cardiac death also is increased. Effective hypertension control can prevent or lead to the regression of cardiac hypertrophy and its attendant risks.
Pulmonary Hypertensive Heart Disease—Cor Pulmonale
Cor pulmonale consists of right ventricular hypertrophy and dilation—frequently accompanied by right heart failure—caused by pulmonary hypertension attributable to primary disorders of the lung parenchyma or pulmonary vasculature (Table 10–4). Right ventricular dilation and hypertrophy caused by left ventricular failure (or by congenital heart disease) is substantially more common but is excluded by this definition.
Table 10–4 Disorders Predisposing to Cor Pulmonale
| Diseases of the Pulmonary Parenchyma |
| Chronic obstructive pulmonary disease |
| Diffuse pulmonary interstitial fibrosis |
| Pneumoconiosis |
| Cystic fibrosis |
| Bronchiectasis |
| Diseases of the Pulmonary Vessels |
| Recurrent pulmonary thromboembolism |
| Primary pulmonary hypertension |
| Extensive pulmonary arteritis (e.g., Wegener granulomatosis) |
| Drug-, toxin-, or radiation-induced vascular obstruction |
| Extensive pulmonary tumor microembolism |
| Disorders Affecting Chest Movement |
| Kyphoscoliosis |
| Marked obesity (pickwickian syndrome) |
| Neuromuscular diseases |
| Disorders Inducing Pulmonary Arterial Constriction |
| Metabolic acidosis |
| Hypoxemia |
| Obstruction to major airways |
| Idiopathic alveolar hypoventilation |
Cor pulmonale can be acute in onset, as with pulmonary embolism, or can have a slow and insidious onset when due to prolonged pressure overloads in the setting of chronic lung and pulmonary vascular disease (Table 10–4).
Morphology
In acute cor pulmonale, the right ventricle usually shows only dilation; if an embolism causes sudden death, the heart may even be of normal size. Chronic cor pulmonale is characterized by right ventricular (and often right atrial) hypertrophy. In extreme cases, the thickness of the right ventricular wall may be comparable with or even exceed that of the left ventricle (Fig. 10–16, B). When ventricular failure develops, the right ventricle and atrium often are dilated. Because chronic cor pulmonale occurs in the setting of pulmonary hypertension, the pulmonary arteries often contain atheromatous plaques and other lesions, reflecting long-standing pressure elevations.
Summary
Hypertensive Heart Disease
• Hypertensive heart disease can affect either the left ventricle or the right ventricle; in the latter case, the disorder is called cor pulmonale. Elevated pressures induce myocyte hypertrophy and interstitial fibrosis that increases wall thickness and stiffness.
• The chronic pressure overload of systemic hypertension causes left ventricular concentric hypertrophy, often associated with left atrial dilation due to impaired diastolic filling of the ventricle. Persistently elevated pressure overload can cause ventricular failure with dilation.
• Cor pulmonale results from pulmonary hypertension due to primary lung parenchymal or vascular disorders. Hypertrophy of both the right ventricle and the right atrium is characteristic; dilation also may be seen when failure supervenes.
Valvular Heart Disease
Valvular disease results in stenosis or insufficiency (regurgitation or incompetence), or both.
• Stenosis is the failure of a valve to open completely, obstructing forward flow. Valvular stenosis is almost always due to a primary cuspal abnormality and is virtually always a chronic process (e.g., calcification or valve scarring).
• Insufficiency results from failure of a valve to close completely, thereby allowing regurgitation (backflow) of blood. Valvular insufficiency can result from either intrinsic disease of the valve cusps (e.g., endocarditis) or disruption of the supporting structures (e.g., the aorta, mitral annulus, tendinous cords, papillary muscles, or ventricular free wall) without primary cuspal injury. It can appear abruptly, as with chordal rupture, or insidiously as a consequence of leaflet scarring and retraction.
Stenosis or regurgitation can occur alone or together in the same valve. Valvular disease can involve only one valve (the mitral valve being the most common target), or more than one valve. Abnormal flow through diseased valves typically produces abnormal heart sounds called murmurs; severe lesions can even be palpated as thrills. Depending on the valve involved, murmurs are best heard at different locations on the chest wall; moreover, the nature (regurgitation versus stenosis) and severity of the valvular disease determines the quality and timing of the murmur (e.g., harsh systolic or soft diastolic murmurs).
The outcome of valvular disease depends on the valve involved, the degree of impairment, the tempo of its development, and the effectiveness of compensatory mechanisms. For example, sudden destruction of an aortic valve cusp by infection can cause massive regurgitation and the abrupt onset of cardiac failure. By contrast, rheumatic mitral stenosis usually progresses over years, and its clinical effects can be well tolerated until late in the course.
Valvular abnormalities can be congenital or acquired. By far the most common congenital valvular lesion is a bicuspid aortic valve, containing only two functional cusps instead of the normal three; this malformation occurs with a frequency of 1% to 2% of all live births, and has been associated with a number of mutations including those affecting proteins of the Notch signaling pathway. The two cusps are of unequal size, with the larger cusp exhibiting a midline raphe resulting from incomplete cuspal separation (Fig. 10–17, B). Bicuspid aortic valves are generally neither stenotic nor incompetent through early life; however, they are more prone to early and progressive degenerative calcification (see further on).
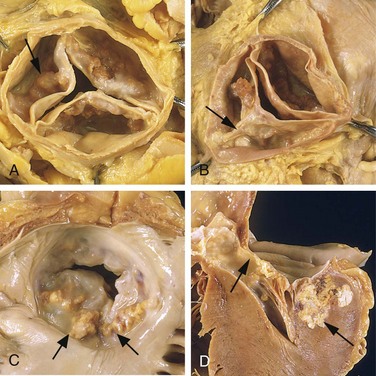
Figure 10–17 Calcific valvular degeneration. A, Calcific aortic stenosis of a previously normal valve (viewed from above the valve). Nodular masses of calcium are heaped up within the sinuses of Valsalva (arrow). Note that the commissures are not fused, as in rheumatic aortic valve stenosis (Fig. 10–19, C). B, Calcific aortic stenosis occurring on a congenitally bicuspid valve. One cusp has a partial fusion at its center, called a raphe (arrow). C and D, Mitral annular calcification, with calcific nodules within the annulus (attachment margin) of the mitral leaflets (arrows). C, Left atrial view. D, Cut section demonstrating the extension of the calcification into the underlying myocardium. Such involvement of adjacent structures near the interventricular septum can impinge on the conduction system.
The most important causes of acquired valvular diseases are summarized in Table 10–5; acquired stenoses of the aortic and mitral valves account for approximately two thirds of all valve disease.
Table 10–5 Etiology of Acquired Heart Valve Disease
| Mitral Valve Disease | Aortic Valve Disease |
|---|---|
| Mitral Stenosis | Aortic Stenosis |
| Mitral Regurgitation | Aortic Regurgitation |
Fen-phen, fenfluramine-phentermine. Data from Schoen FJ: Surgical pathology of removed natural and prosthetic valves. Hum Pathol 18:558, 1987.
Degenerative Valve Disease
Degenerative valve disease is a term used to describe changes that affect the integrity of valvular extracellular matrix (ECM). Degenerative changes include
• Calcifications, which can be cuspal (typically in the aortic valve) (Fig. 11–17, A and B) or annular (in the mitral valve) (Fig. 11–17, C and D). The mitral annular calcification usually is asymptomatic unless it encroaches on the adjacent conduction system.
• Decreased numbers of valve fibroblasts and myofibroblasts
• Alterations in the ECM. In some cases, changes consist of increased proteoglycan and diminished fibrillar collagen and elastin (myxomatous degeneration); in other cases, the valve becomes fibrotic and scarred.
• Changes in the production of matrix metalloproteinases or their inhibitors
Degenerative changes in the cardiac valves probably are an inevitable part of the aging process, because of the repetitive mechanical stresses to which valves are subjected—40 million beats per year, with each normal opening and closing requiring substantial valve deformation.
Calcific Aortic Stenosis
Calcific aortic degeneration is the most common cause of aortic stenosis. Although progressive age-associated “wear and tear” has been the pathologic mechanism most often proposed, cuspal fibrosis and calcification are increasingly viewed as the valvular counterparts to age-related arteriosclerosis. Thus, chronic injury due to hyperlipidemia, hypertension, inflammation, and other factors implicated in atherosclerosis probably play a significant role in the pathogenesis. In most cases, calcific degeneration is asymptomatic and is discovered only incidentally by viewing calcifications on a routine chest radiograph or at autopsy. In other patients, valvular sclerosis and/or calcification can be severe enough to cause stenosis, necessitating surgical intervention.
The incidence of calcific aortic stenosis is increasing with the rising average age for the U.S. population. In anatomically normal valves, it typically begins to manifest when patients reach their 70s and 80s; onset with bicuspid aortic valves is at a much earlier age (often 40 to 50 years).
Morphology
The hallmark of calcific aortic stenosis is heaped-up calcified masses on the outflow side of the cusps; these protrude into the sinuses of Valsalva and mechanically impede valve opening (Fig. 10–17, A and B); commissural fusion (usually a sign of previous inflammation) is not a typical feature of degenerative aortic stenosis, although the cusps may become secondarily fibrosed and thickened. An earlier, hemodynamically inconsequential stage of the calcification process is called aortic valve sclerosis.
Clinical Features
In severe disease, valve orifices can be compromised by as much as 70% to 80% (from a normal area of approximately 4 cm2 to as little as 0.5 to 1 cm2). Cardiac output is maintained only by virtue of concentric left ventricular hypertrophy, and the chronic outflow obstruction can drive left ventricular pressures to 200 mm Hg or more. The hypertrophied myocardium is prone to ischemia, and angina can develop. Systolic and diastolic dysfunction collude to cause CHF, and cardiac decompensation eventually ensues. The development of angina, CHF, or syncope in aortic stenosis heralds the exhaustion of compensatory cardiac hyperfunction and carries a poor prognosis; without surgical intervention, 50% to 80% of patients die within 2 to 3 years of the onset of symptoms like CHF, angina, and syncope.
Myxomatous Mitral Valve
In myxomatous degeneration of the mitral valve, one or both mitral leaflets are “floppy” and prolapse—they balloon back into the left atrium during systole. Mitral valve prolapse is a primary form of myxomatous mitral degeneration affecting some 0.5% to 2.4% of adults; thus, it is one of the most common forms of valvular heart disease in the Western world. Men and women are equally affected. Secondary myxomatous mitral degeneration can occur in any one of a number of settings where mitral regurgitation is caused by some other entity (e.g., IHD).
Pathogenesis
The basis for primary myxomatous degeneration is unknown. Nevertheless, an underlying (possibly systemic) intrinsic defect of connective tissue synthesis or remodeling is likely. Thus, myxomatous degeneration of the mitral valve is a common feature of Marfan syndrome (due to fibrillin-1 mutations) (Chapter 6), and occasionally occurs in other connective tissue disorders. In some patients with primary disease, additional hints of structural abnormalities in the systemic connective tissue, including scoliosis and high-arched palate, may be found. Subtle defects in structural proteins (or the cells that make them) may cause hemodynamically stressed connective tissues rich in microfibrils and elastin (e.g., cardiac valves) to elaborate defective ECM. Secondary myxomatous change presumably results from injury to the valve myofibroblasts, imposed by chronically aberrant hemodynamic forces.
Morphology
Myxomatous degeneration of the mitral valve is characterized by ballooning (hooding) of the mitral leaflets (Fig. 10–18). The affected leaflets are enlarged, redundant, thick, and rubbery; the tendinous cords also tend to be elongated, thinned, and occasionally rupture. In those with primary mitral desease, concomitant tricuspid valve involvement is frequent (20% to 40% of cases); less commonly aortic and pulmonic valves can also be affected. On histologic examination, the essential change is thinning of the valve layer known as the fibrosa layer of the valve, on which the structural integrity of the leaflet depends, accompanied by expansion of the middle spongiosa layer owing to increased deposition of myxomatous (mucoid) material. The same changes occur whether the myxomatous degeneration is due to an intrinsic ECM defect (primary), or is caused by regurgitation secondary to another etiologic process (e.g., ischemic dysfunction).
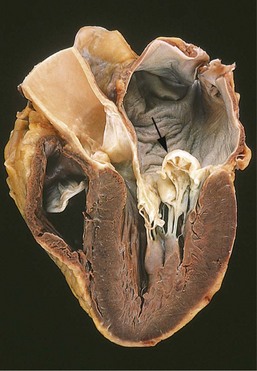
Figure 10–18 Myxomatous degeneration of the mitral valve. There is prominent hooding with prolapse of the posterior mitral leaflet (arrow) into the left atrium; the atrium also is dilated, reflecting long-standing valvular insufficiency and volume overload. The left ventricle is on the right in this four-chamber view.
(Courtesy of William D. Edwards, MD, Mayo Clinic, Rochester, Minnesota.)
Clinical Features
Most patients are asymptomatic, and the valvular abnormality is discovered only incidentally on physical examination. In a minority of cases, patients may complain of palpitations, dyspnea, or atypical chest pain. Auscultation discloses a midsystolic click, caused by abrupt tension on the redundant valve leaflets and chordae tendineae as the valve attempts to close; there may or may not be an associated regurgitant murmur. Although in most instances the natural history and clinical course are benign, approximately 3% of patients develop complications such as hemodynamically significant mitral regurgitation and congestive heart failure, particularly if the chordae or valve leaflets rupture. Patients with primary myxomatous degeneration also are at increased risk for the development of infective endocarditis (see later), as well as sudden cardiac death due to ventricular arrhythmias. Stroke or other systemic infarction may rarely occur from embolism of thrombi formed in the left atrium.
Rheumatic Valvular Disease
Rheumatic fever is an acute, immunologically mediated, multisystem inflammatory disease that occurs after group A β-hemolytic streptococcal infections (usually pharyngitis, but also rarely with infections at other sites such as skin). Rheumatic heart disease is the cardiac manifestation of rheumatic fever. It is associated with inflammation of all parts of the heart, but valvular inflammation and scarring produces the most important clinical features.
The valvular disease principally takes the form of deforming fibrotic mitral stenosis; indeed rheumatic heart disease is essentially the only cause of acquired mitral stenosis. The incidence of rheumatic fever (and thus rheumatic heart disease) has declined remarkably in many parts of the Western world over the past several decades; this is due to a combination of improved socioeconomic conditions, rapid diagnosis and treatment of streptococcal pharyngitis, and a fortuitous (and unexplained) decline in the virulence of many strains of group A streptococci. Nevertheless, in developing countries and economically depressed urban areas in the United States, rheumatic fever and rheumatic heart disease remain important public health problems.
Pathogenesis
Acute rheumatic fever is a hypersensitivity reaction classically attributed to antibodies directed against group A streptococcal molecules that also are cross-reactive with host antigens (see also Chapter 4). In particular, antibodies against M proteins of certain streptococcal strains bind to proteins in the myocardium and cardiac valves and cause injury through the activation of complement and Fc receptor–bearing cells (including macrophages). CD4+ T cells that recognize streptococcal peptides also can cross-react with host antigens and elicit cytokine-mediated inflammatory responses. The characteristic 2- to 3-week delay in symptom onset after infection is explained by the time needed to generate an immune response; streptococci are completely absent from the lesions. Since only a small minority of infected patients develop rheumatic fever (estimated at 3%), a genetic susceptibility is likely to influence the development of the cross-reactive immune responses. The chronic fibrotic lesions are the predictable consequence of healing and scarring associated with the resolution of the acute inflammation.
Morphology
Acute rheumatic fever is characterized by discrete inflammatory foci within a variety of tissues. The myocardial inflammatory lesions—called Aschoff bodies—are pathognomonic for rheumatic fever (Fig. 10–19, B); these are collections of lymphocytes (primarily T cells), scattered plasma cells, and plump activated macrophages called Anitschkow cells occasionally punctuating zones of fibrinoid necrosis. The Anitschkow cells have abundant cytoplasm and central nuclei with chromatin condensed to form a slender, wavy ribbon (so-called caterpillar cells). During acute rheumatic fever, Aschoff bodies can be found in any of the three layers of the heart—pericardium, myocardium, or endocardium (including valves). Hence, rheumatic fever is said to cause pancarditis, with the following salient features:
• The pericardium exhibits a fibrinous exudate, which generally resolves without sequelae.
• The myocardial involvement—myocarditis—takes the form of scattered Aschoff bodies within the interstitial connective tissue.
• Valve involvement results in fibrinoid necrosis and fibrin deposition along the lines of closure (Fig. 10–19, A) forming 1- to 2-mm vegetations—verrucae—that cause little disturbance in cardiac function.
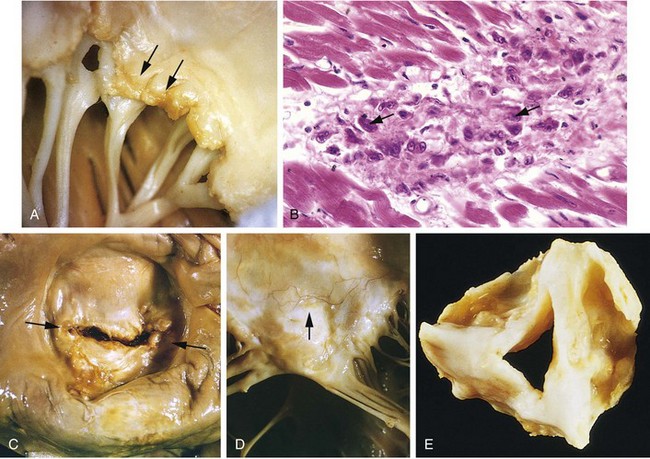
Figure 10–19 Acute and chronic rheumatic heart disease. A, Acute rheumatic mitral valvulitis superimposed on chronic rheumatic heart disease. Small vegetations (verrucae) are visible along the line of closure of the mitral valve leaflet (arrows). Previous episodes of rheumatic valvulitis have caused fibrous thickening and fusion of the chordae tendineae. B, Microscopic appearance of an Aschoff body in acute rheumatic carditis; there is central necrosis associated with a circumscribed collection of mononuclear inflammatory cells, including some activated macrophages with prominent nucleoli and central wavy (caterpillar) chromatin (arrows). C and D, Mitral stenosis with diffuse fibrous thickening and distortion of the valve leaflets, commissural fusion (arrows), and thickening and shortening of the chordae tendineae. There is marked left atrial dilation as seen from above the valve (C). D, Anterior leaflet of an opened rheumatic mitral valve; note the inflammatory neovascularization (arrow). E, Surgically removed specimen of rheumatic aortic stenosis, demonstrating thickening and distortion of the cusps with commissural fusion.
(E, From Schoen FJ, St John-Sutton M: Contemporary issues in the pathology of valvular heart disease. Hum Pathol 18:568, 1967.)
Chronic rheumatic heart disease is characterized by organization of the acute inflammation and subsequent scarring. Aschoff bodies are replaced by fibrous scar so that these lesions are rarely seen in chronic rheumatic heart disease. Most characteristically, valve cusps and leaflets become permanently thickened and retracted. Classically, the mitral valves exhibit leaflet thickening, commissural fusion and shortening, and thickening and fusion of the chordae tendineae (Fig. 10–19, C-E). Fibrous bridging across the valvular commissures and calcification create “fishmouth” or “buttonhole” stenoses (Fig. 10–19, C). Microscopic examination shows neovascularization (grossly evident in Fig. 10–19, D) and diffuse fibrosis that obliterates the normal leaflet architecture.
The most important functional consequence of rheumatic heart disease is valvular stenosis and regurgitation; stenosis tends to predominate. The mitral valve alone is involved in 70% of cases, with combined mitral and aortic disease in another 25%; the tricuspid valve usually is less frequently (and less severely) involved; and the pulmonic valve almost always escapes injury. With tight mitral stenosis, the left atrium progressively dilates owing to pressure overload, precipitating atrial fibrillation. The combination of dilation and fibrillation is a fertile substrate for thrombosis, and formation of large mural thrombi is common. Long-standing passive venous congestion gives rise to pulmonary vascular and parenchymal changes typical of left-sided heart failure. In time, this leads to right ventricular hypertrophy and failure. With pure mitral stenosis, the left ventricle generally is normal.
Clinical Features
Acute rheumatic fever occurs most often in children; the principal clinical manifestation is carditis. Nevertheless, about 20% of first attacks occur in adults, with arthritis being the predominant feature. Symptoms in all age groups typically begin 2 to 3 weeks after streptococcal infection, and are heralded by fever and migratory polyarthritis—one large joint after another becomes painful and swollen for a period of days, followed by spontaneous resolution with no residual disability. Although cultures are negative for streptococci at the time of symptom onset, serum titers to one or more streptococcal antigens (e.g., streptolysin O or DNAase) usually are elevated. The clinical signs of carditis include pericardial friction rubs and arrhythmias; myocarditis can be sufficiently aggressive that cardiac dilation ensues, causing functional mitral insufficiency and CHF. Nevertheless, less than 1% of patients die of acute rheumatic fever.
The diagnosis of acute rheumatic fever is made based on serologic evidence of previous streptococcal infection in conjunction with two or more of the so-called Jones criteria: (1) carditis; (2) migratory polyarthritis of large joints; (3) subcutaneous nodules; (4) erythema marginatum skin rashes; and (5) Sydenham chorea, a neurologic disorder characterized by involuntary purposeless, rapid movements (also called St. Vitus dance). Minor criteria such as fever, arthralgias, ECG changes, or elevated acute phase reactants also can help support the diagnosis.
After an initial attack and the generation of immunologic memory, patients are increasingly vulnerable to disease reactivation with subsequent streptococcal infections. Carditis is likely to worsen with each recurrence, and the damage is cumulative. However, chronic rheumatic carditis usually does not manifest itself clinically until years or even decades after the initial episode of rheumatic fever. At that time, the signs and symptoms of valvular disease depend on which cardiac valve(s) are involved. In addition to various cardiac murmurs, cardiac hypertrophy and dilation, and CHF, patients with chronic rheumatic heart disease often have arrhythmias (particularly atrial fibrillation in the setting of mitral stenosis), and thromboembolic complications due to atrial mural thrombi. In addition, scarred and deformed valves are more susceptible to infective endocarditis. The long-term prognosis is highly variable. In some cases, a relentless cycle of valvular deformity ensues, yielding hemodynamic abnormality, which begets further deforming fibrosis. Surgical repair or replacement of diseased valves has greatly improved the outlook for patients with rheumatic heart disease.
Infective Endocarditis
Infective endocarditis is a serious infection mandating prompt diagnosis and intervention. Microbial invasion of heart valves or mural endocardium—often with destruction of the underlying cardiac tissues—characteristically results in bulky, friable vegetations composed of necrotic debris, thrombus, and organisms. The aorta, aneurysmal sacs, other blood vessels and prosthetic devices also can become infected. Although fungi, rickettsiae (agents of Q fever), and chlamydial species can cause endocarditis, the vast majority of cases are caused by extracellular bacteria.
Infective endocarditis can be classified into acute and subacute forms, based on the tempo and severity of the clinical course; the distinctions are attributable to the virulence of the responsible microbe and whether underlying cardiac disease is present. Of note, a clear delineation between acute and subacute endocarditis does not always exist, and many cases fall somewhere along the spectrum between the two forms.
• Acute endocarditis refers to tumultuous, destructive infections, frequently involving a highly virulent organism attacking a previously normal valve, and capable of causing substantial morbidity and mortality even with appropriate antibiotic therapy and/or surgery.
• Subacute endocarditis refers to infections by organisms of low virulence involving a previously abnormal heart, especially scarred or deformed valves. The disease typically appears insidiously and—even untreated—follows a protracted course of weeks to months; most patients recover after appropriate antibiotic therapy.
Pathogenesis
Infective endocarditis can develop on previously normal valves, but cardiac abnormalities predispose to such infections; rheumatic heart disease, mitral valve prolapse, bicuspid aortic valves, and calcific valvular stenosis are all common substrates. Prosthetic heart valves (discussed later) now account for 10% to 20% of all cases of infective endocarditis. Sterile platelet-fibrin deposits at sites of pacemaker lines, indwelling vascular catheters, or damaged endocardium due to jet streams caused by preexisting cardiac disease all can be foci for bacterial seeding with subsequent development of endocarditis. Host factors such as neutropenia, immunodeficiency, malignancy, diabetes mellitus, and alcohol or intravenous drug abuse also increase the risk of infective endocarditis, as well as adversely affecting outcomes.
The causative organisms differ depending on the underlying risk factors. Fifty percent to 60% of cases of endocarditis occurring on damaged or deformed valves are caused by Streptococcus viridans, a relatively banal group of normal oral flora. By contrast, the more virulent S. aureus (common to skin) can attack deformed as well as healthy valves and is responsible for 10% to 20% of cases overall; it also is the major offender in infections occurring in intravenous drug abusers. Additional bacterial agents include enterococci and the so-called HACEK group (Haemophilus, Actinobacillus, Cardiobacterium, Eikenella, and Kingella), all commensals in the oral cavity. More rarely, gram-negative bacilli and fungi are involved. In about 10% of all cases of endocarditis, no organism is isolated from the blood (“culture-negative” endocarditis) because of previous antibiotic therapy, difficulties in isolating the offending agent, or because deeply embedded organisms within the enlarging vegetation are not released into the blood.
Foremost among the factors predisposing to endocarditis is seeding of the blood with microbes. The mechanism or portal of entry of the agent into the bloodstream may be an obvious infection elsewhere, a dental or surgical procedure that causes a transient bacteremia, injection of contaminated material directly into the bloodstream by intravenous drug users, or an occult source from the gut, oral cavity, or trivial injuries. Recognition of predisposing anatomic substrates and clinical conditions causing bacteremia allows appropriate antibiotic prophylaxis.
Morphology
In both acute and subacute forms of the disease, friable, bulky, and potentially destructive vegetations containing fibrin, inflammatory cells, and microorganisms are present on the heart valves (Figs. 10-20 and 10-21). The aortic and mitral valves are the most common sites of infection, although the tricuspid valve is a frequent target in the setting of intravenous drug abuse. Vegetations may be single or multiple and may involve more than one valve; they can sometimes erode into the underlying myocardium to produce an abscess cavity (ring abscess) (Fig. 10-21, B). Shedding of emboli is common because of the friable nature of the vegetations. Since the fragmented vegetations contain large numbers of organisms, abscesses often develop at the sites where emboli lodge, leading to development of septic infarcts and mycotic aneurysms.
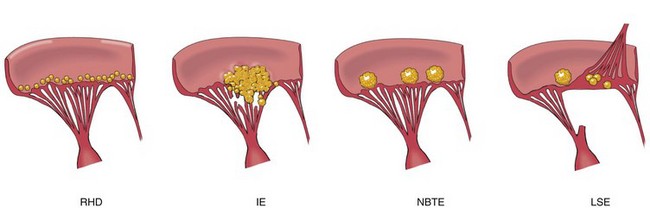
Figure 10–20 The major forms of vegetative endocarditis. The acute rheumatic fever phase of rheumatic heart disease is marked by the appearance of small, warty, inflammatory vegetations along the lines of valve closure; as the inflammation resolves, substantial scarring can result. Infective endocarditis (IE) is characterized by large, irregular, often destructive masses that can extend from valve leaflets onto adjacent structures (e.g., chordae or myocardium). Nonbacterial thrombotic endocarditis (NBTE) typically manifests with small to medium-sized, bland, nondestructive vegetations at the line of valve closure. Libman-Sacks endocarditis (LSE) is characterized by small to medium-sized inflammatory vegetations that can be attached on either side of valve leaflets; these heal with scarring.
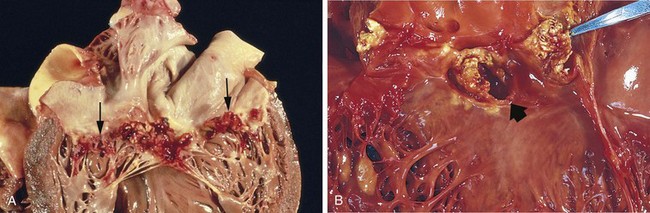
Figure 10–21 Infective endocarditis. A, Subacute endocarditis caused by Streptococcus viridans on a previously myxomatous mitral valve. The large, friable vegetations are denoted by arrows. B, Acute endocarditis caused by Staphylococcus aureus on congenitally bicuspid aortic valve with extensive cuspal destruction and ring abscess (arrow).
Subacute endocarditis typically elicits less valvular destruction than that associated with acute endocarditis. On microscopic examination, the subacute vegetations of infective endocarditis often have granulation tissue at their bases (suggesting chronicity), promoting development of chronic inflammatory infiltrates, fibrosis, and calcification over time.
Clinical Features
Fever is the most consistent sign of infective endocarditis. However, in subacute disease (particularly in the elderly), fever may be absent, and the only manifestations may be nonspecific fatigue, weight loss, and a flulike syndrome; splenomegaly also is common in subacute cases. By contrast, acute endocarditis often manifests with a stormy onset including rapidly developing fever, chills, weakness, and lassitude. Murmurs are present in 90% of patients with left-sided lesions; microemboli can give rise to petechia, nail bed (splinter) hemorrhages, retinal hemorrhages (Roth spots), painless palm or sole erythematous lesions (Janeway lesions), or painful fingertip nodules (Osler nodes); diagnosis is confirmed by positive blood cultures and echocardiographic findings.
Prognosis depends on the infecting organism and on whether or not complications develop. Complications generally begin within the first weeks after onset of the infectious process and can include glomerulonephritis due to glomerular trapping of antigen-antibody complexes, with hematuria, albuminuria, or renal failure (Chapter 13). A septic pathophysiologic picture, arrhythmias (suggesting invasion into underlying myocardium), and systemic embolization bode particularly ill for the patient. Left untreated, infective endocarditis generally is fatal. However, with appropriate long-term (6 weeks or more) antibiotic therapy and/or valve replacement, mortality is reduced. For infections involving low-virulence organisms (e.g., Streptococcus viridans or Streptococcus bovis), the cure rate is 98%, and for enterococci and Staphylococcus aureus infections, cure rates range from 60% to 90%; however, with infections due to aerobic gram-negative bacilli or fungi, half of the patients ultimately succumb. The cure rate for endocarditis arising on prosthetic valves is 10% to 15% lower across the board.
Noninfected Vegetations
Nonbacterial Thrombotic Endocarditis
Nonbacterial thrombotic endocarditis (NBTE) is characterized by the deposition of small (1 to 5 mm in diameter) thrombotic masses composed mainly of fibrin and platelets on cardiac valves. Although NBTE can occur in otherwise healthy persons, a wide variety of diseases associated with general debility or wasting are associated with an increased risk of NBTE—hence the alternate term marantic endocarditis. In contrast with infective endocarditis, the valvular lesions of NBTE are sterile and are nondestructive (Fig. 10–22).
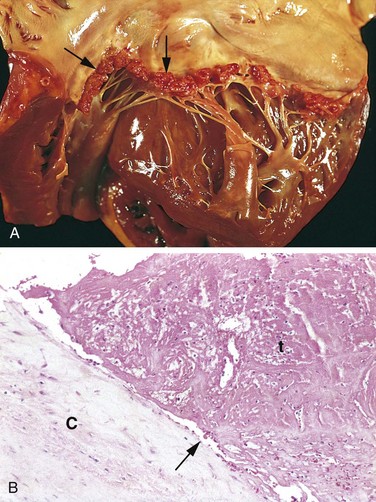
Figure 10–22 Nonbacterial thrombotic endocarditis (NBTE). A, Small thrombotic vegetations along the line of closure of the mitral valve leaflets (arrows). B, Photomicrograph of NBTE lesion, showing bland thrombus, with virtually no inflammation in the valve cusp (C) or the thrombotic deposit (t). The thrombus is only loosely attached to the cusp (arrow).
Valvular damage is not a prerequisite for NBTE; indeed, the condition usually is found on previously normal valves. Rather, hypercoagulable states are the usual precursor to NBTE; such conditions include chronic disseminated intravascular coagulation, hyperestrogenic states, and those associated with underlying malignancy, particularly mucinous adenocarcinomas. This last association probably relates to the procoagulant effect of circulating mucin and/or tissue factor elaborated by these tumors. Endocardial trauma, such as from an indwelling catheter, also is a well-recognized predisposing condition.
Although the local effect on the valve usually is trivial, NBTE lesions can become clinically significant by giving rise to emboli that can cause infarcts in the brain, heart, and other organs. An NBTE lesion also can serve as a potential nidus for bacterial colonization and the consequent development of infective endocarditis.
Libman-Sacks Endocarditis
Libman-Sacks endocarditis is characterized by the presence of sterile vegetations on the valves of patients with systemic lupus erythematosus. The lesions probably develop as a consequence of immune complex deposition and thus exhibit associated inflammation, often with fibrinoid necrosis of the valve substance adjacent to the vegetation; subsequent fibrosis and serious deformity can result in lesions that resemble chronic rheumatic heart disease. These can occur anywhere on the valve surface, on the cords, or even on the atrial or ventricular endocardium (Fig. 10–20). Similar lesions can occur in the setting of antiphospholipid antibody syndrome (Chapter 3).
Carcinoid Heart Disease
The carcinoid syndrome results from bioactive compounds such as serotonin released by carcinoid tumors (Chapter 14); systemic manifestations include flushing, diarrhea, dermatitis, and bronchoconstriction. Carcinoid heart disease refers to the cardiac manifestation caused by the bioactive compounds and occurs in half of the patients in whom the systemic syndrome develops. Cardiac lesions typically do not occur until there is a massive hepatic metastatic burden, since the liver normally catabolizes circulating mediators before they can affect the heart. Classically, endocardium and valves of the right heart are primarily affected since they are the first cardiac tissues bathed by the mediators released by gastrointestinal carcinoid tumors. The left side of the heart is afforded some measure of protection because the pulmonary vascular bed degrades the mediators. However, left-sided heart carcinoid lesions can occur in the setting of atrial or ventricular septal defects and right-to-left flow, or they can arise in association with primary pulmonary carcinoid tumors.
Pathogenesis
The mediators elaborated by carcinoid tumors include serotonin (5-hydroxytryptamine), kallikrein, bradykinin, histamine, prostaglandins, and tachykinins. Although it is not clear which of these is causative, plasma levels of serotonin and urinary excretion of the serotonin metabolite 5-hydroxyindoleacetic acid correlate with the severity of right-sided heart lesions. The valvular plaques in carcinoid syndrome also are similar to lesions that occur with the administration of fenfluramine (an appetite suppressant) or ergot alkaloids (for migraine headaches); of interest, these agents either affect systemic serotonin metabolism or bind to hydroxytryptamine receptors on heart valves.
Morphology
The cardiovascular lesions associated with the carcinoid syndrome are distinctive, glistening white intimal plaquelike thickenings on the endocardial surfaces of the cardiac chambers and valve leaflets (Fig. 10–23). The lesions are composed of smooth muscle cells and sparse collagen fibers embedded in an acid mucopolysaccharide–rich matrix. Underlying structures are intact. With right-sided involvement, typical findings are tricuspid insufficiency and pulmonic stenosis.
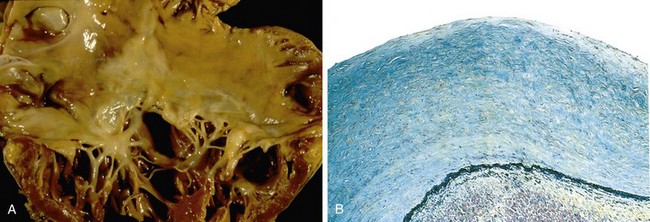
Figure 10–23 Carcinoid heart disease. A, Characteristic endocardial fibrotic lesion “coating” the right ventricle and tricuspid valve, and extending onto the chordae tendineae. B, Microscopic appearance of the thickened intima, which contains smooth muscle cells and abundant acid mucopolysaccharides (blue-green in this Movat stain, which colors the underlying endocardial elastic tissue black).
Prosthetic Cardiac Valves
Although prosthetic heart valves are less-than-perfect substitutes for the native tissues, their introduction has radically altered the prognosis for patients with valve disease. Two types of prosthetic valves are currently used, each with its own advantages and disadvantages:
• Mechanical valves are now most commonly double tilting disk devices made of pyrolytic carbon. They have excellent durability but require chronic anticoagulation, with the attendant risks of hemorrhage or valve thrombosis, if anticoagulation is inadequate. Mechanical aortic valves can also cause significant red cell hemolysis as a consequence of mechanical shear forces (the so-called Waring blender effect) (Chapter 11).
• Bioprosthetic valves are manufactured from glutaraldehyde-fixed porcine or bovine tissues, or cryopreserved human valves. These do not require anticoagulation but are less durable and eventually fail owing to matrix deterioration. Virtually all biologic valve leaflets undergo some degree of stiffening after implantation; the loss of mobility may be sufficient to cause significant stenosis. Calcification of bioprosthetic leaflets is common and can contribute to the stenosis. Bioprosthetic valves also can perforate or tear, resulting in valvular insufficiency.
• All forms of prosthetic valves are susceptible to infection. In mechanical valves, infective endocarditis typically involves the suture line and adjacent perivalvular tissue; the associated tissue changes can cause the valve to detach (paravalvular leak). In bioprosthetic valves, the valve leaflets as well as the perivalvular tissues can become infected.
Summary
Valvular Heart Disease
• Valve pathology can lead to occlusion (stenosis) and/or to regurgitation (insufficiency); acquired aortic stenosis and mitral valve stenosis account for approximately two thirds of all valve disease.
• Valve calcification typically results in stenosis; abnormal matrix synthesis and turnover leads to myxomatous degeneration and insufficiency.
• Inflammatory valve diseases cause postinflammatory neovascularization and scarring. Rheumatic heart disease results from antistreptococcal antibodies that cross-react with cardiac tissues; it most commonly affects the mitral valve and is responsible for 99% of cases of acquired mitral stenosis.
• Infective endocarditis can be aggressive and rapidly destroy normal valves (in the acute form), or can be indolent and minimally destructive of previously abnormal valves (in subacute infective endocarditis). Systemic embolization can produce septic infarcts.
• Nonbacterial thrombotic endocarditis occurs on previously normal valves as a result of hypercoagulable states; embolization is an important complication.
Cardiomyopathies
Most cardiac muscle diseases are secondary to some other condition, e.g., coronary atherosclerosis, hypertension, or valvular heart disease. However, there are also cardiac diseases attributable to intrinsic myocardial dysfunction. Such diseases are termed cardiomyopathies (literally, “heart muscle diseases”); these can be primary—that is, principally confined to the myocardium—or secondary presenting as the cardiac manifestation of a systemic disorder. Cardiomyopathies are thus a diverse group that includes inflammatory disorders (e.g., myocarditis), immunologic diseases (e.g., sarcoidosis), systemic metabolic disorders (e.g., hemochromatosis), muscular dystrophies, and genetic disorders of myocardial fibers. In many cases, the cardiomyopathy is of unknown etiology and thus is termed idiopathic; however, a number of previously “idiopathic” cardiomyopathies have been shown to be the consequence of specific genetic abnormalities in cardiac energy metabolism or in structural and contractile proteins.
Cardiomyopathies can be classified according to a variety of criteria, including the underlying genetic basis of dysfunction; indeed, a number of the arrhythmia-inducing channelopathies that are included in some classifications of cardiomyopathy were alluded to earlier. For purposes of general diagnosis and therapy, however, three time-honored clinical, functional, and pathologic patterns are recognized (Fig. 10–24 and Table 10–6):
• Dilated cardiomyopathy (DCM) (including arrhythmogenic right ventricular cardiomyopathy)
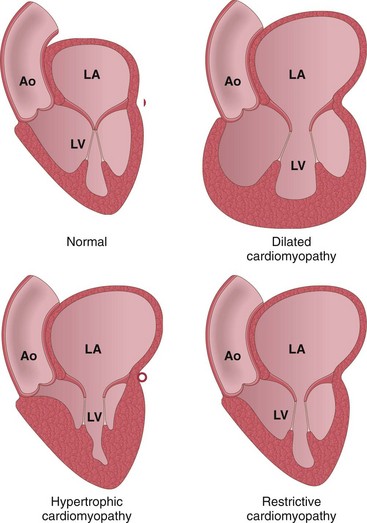
Figure 10–24 The three major forms of cardiomyopathy. Dilated cardiomyopathy leads primarily to systolic dysfunction, whereas restrictive and hypertrophic cardiomyopathies result in diastolic dysfunction. Note the changes in atrial and/or ventricular dilation and in ventricular wall thickness. Ao, aorta; LA, left atrium; LV, left ventricle.
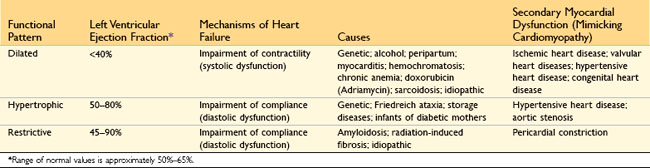
Another rare form of cardiomyopathy is left ventricular noncompaction; it is a congenital disorder characterized by a distinctive “spongy” appearance of the ventricles, associated with CHF and arrhythmias.
Of the three major patterns, DCM is most common (90% of cases), and restrictive cardiomyopathy is the least frequent. Within each pattern, there is a spectrum of clinical severity, and in some cases clinical features overlap among the groups. In addition, each of these patterns can be caused by a specific identifiable cause, or can be idiopathic (Table 10–6).
Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is characterized by progressive cardiac dilation and contractile (systolic) dysfunction, usually with concurrent hypertrophy; regardless of cause, the clinicopathologic patterns are similar.
Pathogenesis
By the time it is diagnosed, DCM has frequently already progressed to end-stage disease; the heart is dilated and poorly contractile, and at autopsy or cardiac transplant, fails to reveal any specific pathologic features. Nevertheless, genetic and epidemiologic studies suggest that at least five general pathways can lead to end-stage DCM (Fig. 10–25):
• Genetic causes. DCM has a hereditary basis in 20% to 50% of cases and over 40 genes are known to be mutated in this form of cardiomyopathy; autosomal dominant inheritance is the predominant pattern, most commonly involving mutations in encoding cytoskeletal proteins, or proteins that link the sarcomere to the cytoskeleton (e.g., α-cardiac actin). X-linked DCM is most frequently associated with dystrophin gene mutations affecting the cell membrane protein that physically couples the intracellular cytoskeleton to the ECM; (different types of dystrophin mutations also underlie Duchenne and Becker muscular dystrophies, Chapter 21). Uncommon forms of DCM are caused by mutations of genes in the mitochondrial genome that encode proteins involved in oxidative phosphorylation or fatty acid β-oxidation, presumably leading to defective ATP generation. Other cytoskeletal proteins that are affected in genetic forms of DCM include desmin (the principal intermediate filament protein in cardiac myocytes), and the nuclear lamins A and C. Since contractile myocytes and conduction fibers share a common developmental pathway, congenital conduction abnormalities also can be a feature of inherited forms of DCM.
• Infection. The nucleic acid “footprints” of coxsackievirus B and other enteroviruses can occasionally be detected in the myocardium from late-stage DCM patients. Moreover, sequential endomyocardial biopsies have documented instances in which infectious myocarditis progressed to DCM. Consequently, many cases of DCM are attributed to viral infections (discussed later), even though inflammation is absent from the end-stage heart. Simply finding viral transcripts or demonstrating elevated antiviral antibody titers may be sufficient to invoke a myocarditis that was “missed” in its early stages.
• Alcohol or other toxic exposure. Alcohol abuse is strongly associated with the development of DCM. Alcohol and its metabolites (especially acetaldehyde) have a direct toxic effect on myocardium. Moreover, chronic alcoholism can be associated with thiamine deficiency, introducing an element of beriberi heart disease (Chapter 7). DCM also can develop after exposure to other toxic agents, particularly doxorubicin (Adriamycin), a chemotherapeutic drug, and cobalt.
• Peripartum cardiomyopathy occurs late in gestation or several weeks to months postpartum. The etiology is likely to be multifactorial, with contributing factors including pregnancy-associated hypertension, volume overload, nutritional deficiency, metabolic derangements (e.g., gestational diabetes), and/or immunologic responses; recent experiments also suggest that a cleavage product of prolactin (which rises late in pregnancy) can induce myocardial dysfunction. Fortunately, approximately half of these patients spontaneously recover normal function.
• Iron overload in the heart can result either from hereditary hemochromatosis (Chapter 15) or from multiple transfusions. DCM is the most common manifestation, and may be attributable to interference with metal-dependent enzyme systems or to injury caused by iron-mediated production of reactive oxygen species.
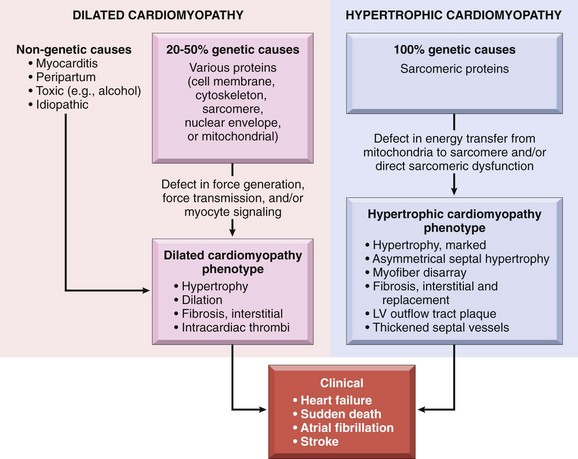
Figure 10–25 Causes and consequences of dilated and hypertrophic cardiomyopathy. A significant fraction of dilated cardiomyopathies—and virtually all hypertrophic cardiomyopathies—have a genetic origin. Dilated cardiomyopathies can be caused by mutations in cytoskeletal, sarcomeric, nuclear envelope, or mitochondrial proteins; hypertrophic cardiomyopathies typically are caused by sarcomeric protein mutations. Although the two forms of cardiomyopathy differ in cause and morphology, they have common clinical end points. LV, left ventricle.
Morphology
The heart in DCM characteristically is enlarged (up to two to three times the normal weight) and flabby, with dilation of all chambers (Fig. 10–26). Because of the wall thinning that accompanies dilation, the ventricular thickness may be less than, equal to, or greater than normal. Mural thrombi are often present and may be a source of thromboemboli. By definition, valvular and vascular lesions that can cause cardiac dilation secondarily (e.g., atherosclerotic coronary artery disease) are absent.
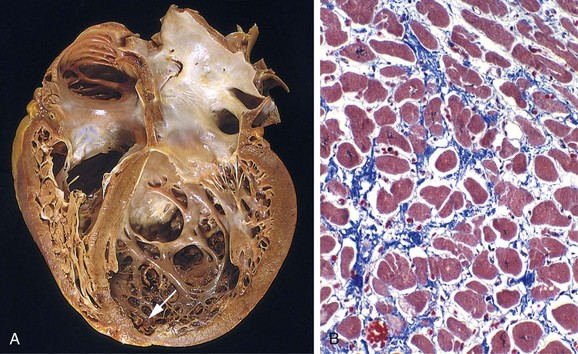
Figure 10–26 Dilated cardiomyopathy (DCM). A, Four-chamber dilation and hypertrophy are evident. A small mural thrombus can be seen at the apex of the left ventricle (arrow). B, The nonspecific histologic picture in typical DCM, with myocyte hypertrophy and interstitial fibrosis (collagen is blue in this Masson trichrome–stained preparation).
The characteristic histologic abnormalities in DCM are nonspecific, and do not typically point to a specific etiologic entity. An exception is DCM secondary to iron overload, in which marked accumulation of intramyocardial hemosiderin is demonstrable by staining with Prussian blue.
In general, the severity of morphologic changes in DCM does not necessarily reflect either the degree of dysfunction or the prognosis. Most myocytes exhibit hypertrophy with enlarged nuclei, but many are attenuated, stretched, and irregular. There is also variable interstitial and endocardial fibrosis, with scattered areas of replacement fibrosis; the latter mark previous myocyte ischemic necrosis caused by hypoperfusion.
Clinical Features
DCM can occur at any age but most commonly is diagnosed between the ages of 20 and 50 years. It typically manifests with signs of slowly progressive CHF, including dyspnea, easy fatigability, and poor exertional capacity, although patients can slip precipitously from a compensated to a decompensated state. The fundamental defect in DCM is ineffective contraction. Thus, in end-stage DCM, the cardiac ejection fraction typically is less than 25% (normal being 50% to 65%). Secondary mitral regurgitation and abnormal cardiac rhythms are common, and embolism from intracardiac (mural) thrombi can occur. Half of the patients die within 2 years, and only 25% survive longer than 5 years; death usually is due to progressive cardiac failure or arrhythmia. Cardiac transplantation is the only definitive treatment. Implantation of long-term ventricular assist devices is being increasingly utilized, however, and in some patients a course of mechanical assistance can produce durable regression of cardiac dysfunction.
Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an autosomal dominant disorder of cardiac muscle with variable penetrance; it classically manifests with right-sided heart failure and rhythm disturbances that can cause sudden cardiac death. Morphologically, the right ventricular wall is severely thinned owing to myocyte replacement by massive fatty infiltration and lesser amounts of fibrosis (Fig. 10–27). Many of the mutations involve genes encoding desmosomal junctional proteins at the intercalated disk (e.g., plakoglobin), as well as proteins that interact with the desmosome (e.g., the intermediate filament desmin).
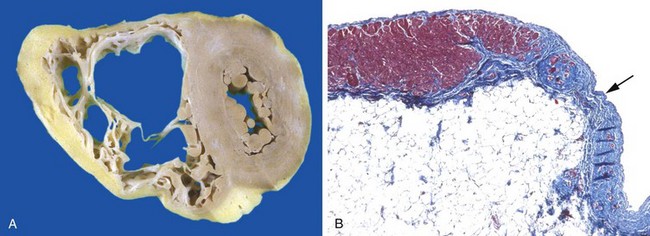
Figure 10–27 Arrhythmogenic right ventricular cardiomyopathy. A, The right ventricle is markedly dilated with focal, almost transmural replacement of the free wall by adipose tissue and fibrosis. The left ventricle has a grossly normal appearance in this heart; it can be involved (albeit to a lesser extent) in some instances. B, The right ventricular myocardium (red) is focally replaced by fibrous connective tissue (blue, arrow) and fat (Masson trichrome stain).
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by myocardial hypertrophy, defective diastolic filling, and—in a third of cases—ventricular outflow obstruction. The heart is thick-walled, heavy, and hypercontractile, in striking contrast with the flabby, poorly contractile heart in DCM. Systolic function usually is preserved in HCM, but the myocardium does not relax and therefore exhibits primary diastolic dysfunction. HCM needs to be distinguished clinically from disorders causing ventricular stiffness (e.g., amyloid deposition) and ventricular hypertrophy (e.g., aortic stenosis and hypertension).
Pathogenesis
Most cases of HCM are caused by missense mutations in one of several genes encoding proteins that form the contractile apparatus. In most cases, the pattern of transmission is autosomal dominant, with variable expression. Although more than 400 causative mutations in nine different genes have been identified, HCM is fundamentally a disorder of sarcomeric proteins. Of these, β-myosin heavy chain is most frequently affected, followed by myosin-binding protein C and troponin T. Mutations in these three genes account for 70% to 80% of all cases of HCM.
The diverse mutations underlying HCM have one unifying feature: they all affect sarcomeric proteins and increase myofilament activation. This results in myocyte hypercontractility with concomitant increase in energy use and net negative energy balance. Some of the genes mutated in HCM are also mutated in DCM (e.g., beta-myosin) but in DCM the (allelic) mutations depress motor function as opposed to gain of function in HCM.
Morphology
HCM is marked by massive myocardial hypertrophy without ventricular dilation (Fig. 10–28, A). Classically, there is disproportionate thickening of the ventricular septum relative to the left ventricle free wall (so-called asymmetric septal hypertrophy); nevertheless, in about 10% of cases of HCM, concentric hypertrophy is seen. On longitudinal sectioning, the ventricular cavity loses its usual round-to-ovoid shape and is compressed into a “banana-like” configuration. An endocardial plaque in the left ventricular outflow tract and thickening of the anterior mitral leaflet reflect contact of the anterior mitral leaflet with the septum during ventricular systole; these changes correlate with functional left ventricular outflow tract obstruction.
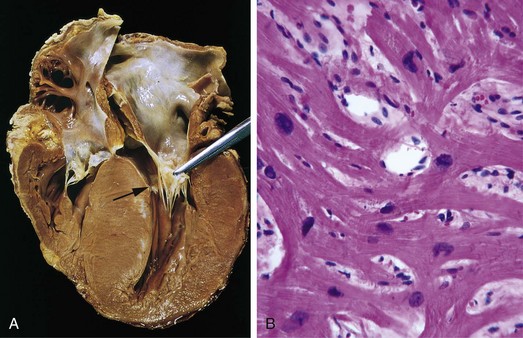
Figure 10–28 Hypertrophic cardiomyopathy with asymmetric septal hypertrophy. A, The septal muscle bulges into the left ventricular outflow tract, giving rise to a “banana-shaped” ventricular lumen, and the left atrium is enlarged. The anterior mitral leaflet has been moved away from the septum to reveal a fibrous endocardial plaque (arrow) (see text). B, Histologic appearance demonstrating disarray, extreme hypertrophy, and characteristic branching of myocytes, as well as interstitial fibrosis.
The characteristic histologic features in HCM are marked myocyte hypertrophy, haphazard myocyte (and myofiber) disarray, and interstitial fibrosis (Fig. 10–28, B).
Clinical Features
Although HCM can present at any age it typically manifests during the postpubertal growth spurt. The clinical symptoms can be best understood in the context of the functional abnormalities. It is characterized by a massively hypertrophied left ventricle that paradoxically provides a markedly reduced stroke volume. This condition occurs as a consequence of impaired diastolic filling and overall smaller chamber size. In addition, roughly 25% of patients have dynamic obstruction to the left ventricular outflow by the anterior leaflet of the mitral valve. Reduced cardiac output and a secondary increase in pulmonary venous pressure cause exertional dyspnea, with a harsh systolic ejection murmur. A combination of massive hypertrophy, high left ventricular pressures, and compromised intramural arteries frequently leads to myocardial ischemia (with angina), even in the absence of concomitant CAD. Major clinical problems include atrial and ventricular fibrillations with mural thrombus formation, infective endocarditis of the mitral valve, CHF, and sudden death. Most patients are improved by therapy that promotes ventricular relaxation; partial surgical excision or controlled alcohol-induced infarction of septal muscle also can relieve the outflow tract obstruction. As mentioned earlier, HCM is an important cause of sudden cardiac death. In almost one third of the cases of sudden cardiac death in athletes under the age of 35, the underlying cause is HCM.
Restrictive Cardiomyopathy
Restrictive cardiomyopathy is characterized by a primary decrease in ventricular compliance, resulting in impaired ventricular filling during diastole (simply put, the wall is stiffer). Because the contractile (systolic) function of the left ventricle usually is unaffected, the functional state can be confused with constrictive pericarditis or HCM. Restrictive cardiomyopathy can be idiopathic or associated with systemic diseases that also happen to affect the myocardium, for example radiation fibrosis, amyloidosis, sarcoidosis, or products of inborn errors of metabolism.
Morphology
The ventricles are of approximately normal size or only slightly enlarged, the cavities are not dilated, and the myocardium is firm. Biatrial dilation commonly is due to poor ventricular filling and pressure overloads. Microscopic examination reveals variable degrees of interstitial fibrosis. Although gross morphologic findings are similar for restrictive cardiomyopathy of disparate causes, endomyocardial biopsy often can reveal a specific etiologic disorder.
Three forms of restrictive cardiomyopathy merit brief mention:
• Amyloidosis is caused by the deposition of extracellular proteins with the predilection for forming insoluble β-pleated sheets (Chapter 4). Cardiac amyloidosis can occur with systemic amyloidosis or can be restricted to the heart, particularly in the case of senile cardiac amyloidosis. In the latter instance, deposition of normal (or mutant) forms of transthyretin (a liver-synthesized circulating protein that transports thyroxine and retinol) in the hearts of elderly patients results in a restrictive cardiomyopathy. Four percent of African Americans carry a specific mutation of transthyretin that is responsible for a four-fold increased risk of isolated cardiac amyloidosis in that population.
• Endomyocardial fibrosis is principally a disease of children and young adults in Africa and other tropical areas; it is characterized by dense diffuse fibrosis of the ventricular endocardium and subendocardium, often involving the tricuspid and mitral valves. The fibrous tissue markedly diminishes the volume and compliance of affected chambers, resulting in a restrictive physiology. Endomyocardial fibrosis has been linked to nutritional deficiencies and/or inflammation related to helminthic infections (e.g., hypereosinophilia); worldwide, it is the most common form of restrictive cardiomyopathy.
• Loeffler endomyocarditis also exhibits endocardial fibrosis, typically associated with formation of large mural thrombi, but without geographic predilection. Histologic examination typically shows peripheral hypereosinophilia and eosinophilic tissue infiltrates; release of eosinophil granule contents, especially major basic protein, probably engenders endo- and myocardial necrosis, followed by scarring, layering of the endocardium by thrombus, and finally thrombus organization. Of interest, some patients have an underlying hypereosinophilic myeloproliferative disorder driven by constitutively active platelet derived growth factor receptor (PDGFR) tyrosine kinases (Chapter 11). Treatment of such patients with tyrosine kinase inhibitors can result in hematologic remission and reversal of the endomyocardial lesions.
Myocarditis
Myocarditis encompasses a diverse group of clinical entities in which infectious agents and/or inflammatory processes primarily target the myocardium. It is important to distinguish these conditions from those, such as IHD, in which the inflammatory process is a consequence of some other cause of myocardial injury.
Pathogenesis
In the United States, viral infections are the most common cause of myocarditis, with coxsackieviruses A and B and other enteroviruses accounting for a majority of the cases. Cytomegalovirus (CMV), human immunodeficiency virus (HIV), influenza virus, and others are less common pathogens. Offending agents occasionally can be identified by nucleic acid footprints in infected tissues, or by serologic studies showing rising antibody titers. While some viruses cause direct cytolytic injury, in most cases the injury results from an immune response directed against virally infected cells; this is analogous to the damage inflicted by virus-specific T cells on hepatitis virus–infected liver cells (Chapter 15). In some cases viruses trigger a reaction against cross-reacting proteins such as myosin heavy chain.
The nonviral infectious causes of myocarditis run the entire gamut of the microbial world. The protozoan Trypanosoma cruzi is the agent of Chagas disease. Although uncommon in the northern hemisphere, Chagas disease affects up to half of the population in endemic areas of South America, with myocardial involvement in the vast majority. About 10% of the patients die during an acute attack; others can enter a chronic immune-mediated phase with development of progressive signs of CHF and arrhythmia 10 to 20 years later. Toxoplasma gondii (household cats are the most common vector) also can cause myocarditis, particularly in immunocompromised persons. Trichinosis is the most common helminthic disease with associated cardiac involvement.
Myocarditis occurs in approximately 5% of patients with Lyme disease, a systemic illness caused by the bacterial spirochete Borrelia burgdorferi (Chapter 8). Lyme myocarditis manifests primarily as self-limited conduction system disease, frequently requiring temporary pacemaker insertion.
Noninfectious causes of myocarditis include lesions associated with systemic diseases of immune origin, such as systemic lupus erythematosus and polymyositis. Drug hypersensitivity reactions (hypersensitivity myocarditis) also can occur with exposure to any of a wide range of agents; such reactions typically are benign and only in rare circumstances lead to CHF or sudden death.
Morphology
In acute myocarditis, the heart may appear normal or dilated; in advanced stages, the myocardium typically is flabby and often mottled with pale and hemorrhagic areas. Mural thrombi can be present.
Microscopically, active myocarditis is characterized by edema, interstitial inflammatory infiltrates, and myocyte injury (Fig. 10–29). A diffuse lymphocytic infiltrate is most common (Fig. 10–29, A), although the inflammatory involvement is often patchy and can be “missed” on endomyocardial biopsy. If the patient survives the acute phase of myocarditis, lesions can resolve without significant sequelae or heal by progressive fibrosis.
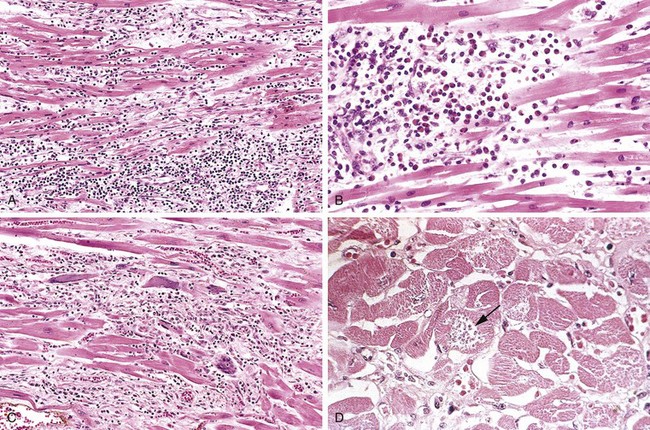
Figure 10–29 Myocarditis. A, Lymphocytic myocarditis, with edema and associated myocyte injury. B, Hypersensitivity myocarditis, characterized by perivascular eosinophil-rich inflammatory infiltrates. C, Giant cell myocarditis, with lymphocyte and macrophage infiltrates, extensive myocyte damage, and multinucleate giant cells. D, Chagas myocarditis. A myofiber distended with trypanosomes (arrow) is present, along with mononuclear inflammation and myofiber necrosis.
In hypersensitivity myocarditis, interstitial and perivascular infiltrates are composed of lymphocytes, macrophages, and a high proportion of eosinophils (Fig. 10-29, B). Giant cell myocarditis is a morphologically distinctive entity characterized by widespread inflammatory cellular infiltrates containing multinucleate giant cells (formed by macrophage fusion). Giant cell myocarditis probably represents the aggressive end of the spectrum of lymphocytic myocarditis, and there is at least focal—and frequently extensive—necrosis (Fig. 10-29, C). This variant carries a poor prognosis.
Chagas myocarditis is characterized by the parasitization of scattered myofibers by trypanosomes accompanied by an inflammatory infiltrate of neutrophils, lymphocytes, macrophages, and occasional eosinophils (Fig. 10-29, D).
Clinical Features
The clinical spectrum of myocarditis is broad; at one end, the disease is asymptomatic, and patients recover without sequelae. At the other extreme is the precipitous onset of heart failure or arrhythmias, occasionally with sudden death. Between these extremes are many levels of involvement associated with a variety of signs and symptoms, including fatigue, dyspnea, palpitations, pain, and fever. The clinical features of myocarditis can mimic those of acute MI. Clinical progression from myocarditis to DCM occasionally is seen.
Summary
Cardiomyopathy
• Cardiomyopathy is intrinsic cardiac muscle disease; there may be specific causes, or it may be idiopathic.
• The three general pathophysiologic categories of cardiomyopathy are dilated (accounting for 90% of the cases), hypertrophic, and restrictive (least common).
• DCM results in systolic (contractile) dysfunction. Causes include myocarditis, toxic exposures (e.g., alcohol), and pregnancy. In 20% to 50% of cases, mutations affecting cytoskeletal proteins are responsible.
• HCM results in diastolic (relaxation) dysfunction. Virtually all cases are due to autosomal dominant mutations in the proteins that make up the contractile apparatus, in particular β-myosin heavy chain.
• Restrictive cardiomyopathy results in a stiff, noncompliant myocardium and can be due to depositions (e.g., amyloid), increased interstitial fibrosis (e.g., due to radiation), or to endomyocardial scarring.
• Myocarditis is myocardial damage caused by inflammatory infiltrates secondary to infections or immune reactions. Coxsackieviruses A and B are the most common pathogens in the United States. Clinically, myocarditis can be asymptomatic, give rise to acute heart failure, or evolve to DCM.
Pericardial Disease
Pericardial disorders include effusions and inflammatory conditions, sometimes resulting in fibrous constriction. Isolated pericardial disease is unusual, and pericardial lesions typically are associated with a pathologic process elsewhere in the heart or surrounding structures or are secondary to a systemic disorder.
Pericarditis
Primary pericarditis is uncommon. It most often is due to viral infection (typically with concurrent myocarditis), although bacteria, fungi, or parasites may also be involved. In most cases, pericarditis is secondary to acute MI, cardiac surgery, radiation to the mediastinum, or processes involving other thoracic structures (e.g., pneumonia or pleuritis). Uremia is the most common systemic disorder associated with pericarditis. Less common secondary causes include rheumatic fever, systemic lupus erythematosus, and metastatic malignancies. Pericarditis can (1) cause immediate hemodynamic complications if it elicits a large effusion (resulting in cardiac tamponade) (see further on), (2) resolve without significant sequelae, or (3) progress to a chronic fibrosing process.
Morphology
In patients with acute viral pericarditis or uremia, the exudate typically is fibrinous, imparting an irregular, shaggy appearance to the pericardial surface (so-called “bread and butter” pericarditis). In acute bacterial pericarditis, the exudate is fibrinopurulent (suppurative), often with areas of frank pus (Fig. 10–30); tuberculous pericarditis can exhibit areas of caseation. Pericarditis due to malignancy often is associated with an exuberant, shaggy fibrinous exudate and a bloody effusion; metastases can be grossly evident as irregular excrescences or may be grossly inapparent, especially in the case of leukemia. In most cases, acute fibrinous or fibrinopurulent pericarditis resolves without any sequelae. With extensive suppuration or caseation, however, healing can result in fibrosis (chronic pericarditis).
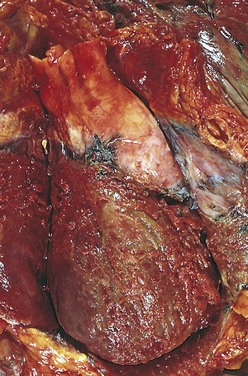
Figure 10–30 Acute suppurative (purulent, exudative) pericarditis, caused by extension from a pneumonia.
Chronic pericarditis may be associated with delicate adhesions or dense, fibrotic scars that obliterate the pericardial space. In extreme cases, the heart is so completely encased by dense fibrosis that it cannot expand normally during diastole—resulting in the condition known as constrictive pericarditis.
Clinical Features
Pericarditis classically manifests with atypical chest pain (not related to exertion and worse in recumbency), and a prominent friction rub. When associated with significant fluid accumulation, acute pericarditis can cause cardiac tamponade, with declining cardiac output and consequent shock. Chronic constrictive pericarditis produces a combination of right-sided venous distention and low cardiac output, similar to the clinical picture in restrictive cardiomyopathy.
Pericardial Effusions
Normally, the pericardial sac contains at most 30 to 50 mL of clear, serous fluid. Serous and/or fibrinous effusions in excess of this amount occur most commonly in the setting of pericardial inflammation. Other types of pericardial effusions and their causes include
• Serous: congestive heart failure, hypoalbuminemia of any cause
• Serosanguineous: blunt chest trauma, malignancy, ruptured MI or aortic dissection
The consequences of pericardial accumulations depend on the volume of fluid and the ability of the parietal pericardium to stretch; the latter depends largely on how fast the effusion accumulates. Thus, slowly accumulating effusions—even as large as 1000 mL—can be well-tolerated. By contrast, rapidly developing collections of as little as 250 mL (e.g., ruptured MI or ruptured aortic dissection) can so restrict diastolic cardiac filling as to produce potentially fatal cardiac tamponade.
Cardiac Tumors
Metastatic Neoplasms
Tumor metastases constitute the most common malignancy of the heart; metastatic cardiac lesions occur in about 5% of patients dying of cancer. Although any malignancy can secondarily involve the heart, certain tumors have a higher predilection for cardiac metastases. In descending order these are lung cancer, lymphoma, breast cancer, leukemia, melanoma, hepatocellular carcinoma, and colon cancer.
Primary Neoplasms
Primary cardiac tumors are uncommon; moreover, most also are (fortunately) benign. The five most common have no malignant potential and account for 80% to 90% of all primary heart tumors. In descending order of frequency, these are myxomas, fibromas, lipomas, papillary fibroelastomas, and rhabdomyomas. Angiosarcomas constitute the most common primary malignant tumor of the heart. Only the myxomas and rhabdomyomas merit further mention here.
Myxomas are the most common primary tumors of the adult heart (Fig. 10–31). Roughly 90% are atrial, with the left atrium accounting for 80% of those.
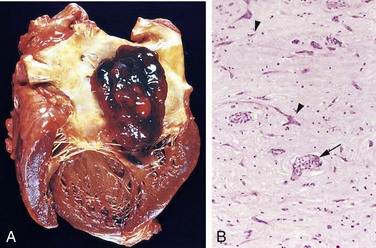
Figure 10–31 Atrial myxoma. A, A large pedunculated lesion arises from the region of the fossa ovalis and extends into the mitral valve orifice. B, Abundant amorphous extracellular matrix contains scattered multinucleate myxoma cells (arrowheads) in various groupings, including abnormal vascular formations (arrow).
Rhabdomyomas are the most frequent primary tumors of the heart in infants and children; they frequently are discovered owing to valvular or outflow obstruction. Cardiac rhabdomyomas occur with high frequency in patients with tuberous sclerosis caused by mutations in the TSC1 or TSC2 tumor suppressor genes; loss of TSC-1 and -2 activity leads to myocyte overgrowth. Because they often regress spontaneously, rhabdomyomas are best considered to be hamartomas rather than true neoplasms. Like certain other tumors that appear in very young children (e.g., neuroblastoma), rhabdomyomas often regress spontaneously for unknown reasons.
Morphology
Myxomas are almost always single, classically arising in the region of the fossa ovalis (atrial septum). They can be small (less than 1 cm in diameter) to massive (up to 10 cm across), sessile or pedunculated masses (Fig. 10–31, A), most often manifesting as soft, translucent, villous lesions with a gelatinous appearance. Pedunculated forms often are sufficiently mobile to swing into the mitral or tricuspid valve during systole, causing intermittent obstruction or exerting a “wrecking ball” effect that damages the valve leaflets.
Histologically, myxomas are composed of stellate, frequently multinucleated myxoma cells (typically with hyperchromatic nuclei), admixed with cells showing endothelial, smooth muscle, and/or fibroblastic differentiation (undifferentiated cells also are present); all of the cell types arise from differentiation of multipotential mesenchymal tumor cells. The cells are embedded in an abundant acid mucopolysaccharide ground substance (Fig. 10–31, B). Hemorrhage, poorly organizing thrombus, and mononuclear inflammation also are usually present.
Rhabdomyomas are gray-white masses up to several centimeters in diameter that protrude into the ventricular chambers. Histologic examination shows a mixed population of cells; most characteristic, however, are large, rounded, or polygonal cells containing numerous glycogen-laden vacuoles separated by strands of cytoplasm running from the plasma membrane to the centrally located nucleus, so-called spider cells.
Clinical Features
The major clinical manifestations are due to valvular “ball-valve” obstruction, embolization, or a syndrome of constitutional signs and symptoms including fever and malaise. This syndrome is attributable to tumor elaboration of the cytokine interleukin-6, a major mediator of the acute-phase response. Echocardiography is the diagnostic modality of choice, and surgical resection is almost uniformly curative.
Other Cardiac Tumors
• Lipomas are localized, poorly encapsulated masses of adipose tissue; these can be asymptomatic, create ball-valve obstructions (as with myxomas), or produce arrhythmias.
• Papillary fibroelastomas usually are only incidentally identified lesions, although they can embolize. Generally located on valves, they form distinctive clusters (up to 1 cm in diameter) of hairlike projections that grossly resemble sea anemones. Histologic examination shows myxoid connective tissue containing abundant mucopolysaccharide matrix and laminated elastic fibers, all surrounded by endothelium.
• Cardiac angiosarcomas and other sarcomas are not clinically or morphologically distinctive from their counterparts in other locations and therefore merit no additional comment here.
Cardiac Transplantation
Although permanent ventricular assist device implantation is increasingly an option for management of end-stage heart disease, cardiac transplantation remains the treatment of choice for patients with intractable heart failure. Without transplantation, medically managed end-stage heart failure carries a 50% 1-year mortality rate, and less than 10% of the patients survive 5 years. Roughly 3000 heart transplantation procedures are performed annually worldwide, mostly for DCM and IHD. Even so, the need far outstrips the number of available organs, and many more patients die while on a waiting list (estimated at 50,000 per year) than undergo successful transplantation.
Beyond the issues of supply and demand, the major complications of cardiac transplantation are acute cardiac rejection and allograft arteriopathy (Fig. 10–32). The immunosuppression required for allograft survival also increases the risk of opportunistic infections and certain malignancies (e.g., Epstein-Barr virus–associated lymphoma).
• Rejection is suspected clinically in the setting of fever, reduced cardiac ejection fraction, unexplained arrhythmia, or an edematous, thickened ventricular wall on cardiac ultrasound examination. It is diagnosed by endomyocardial biopsy of the transplanted heart. Rejection is characterized by an interstitial lymphocytic inflammation, associated myocyte damage (Fig. 10–32, A), and a histologic pattern similar to that seen in viral myocarditis (Fig. 10–29, A). In both instances, T cell–mediated killing and local cytokine production compromise cardiac function. Increasingly, antibody-mediated injury also is recognized as an important mechanism in allograft rejection. When myocardial injury is not extensive, the “rejection episode” can be reversed by augmented immunosuppressive therapy. Advanced rejection can be irreversible and fatal.
• Allograft arteriopathy is the single most important long-term limitation for cardiac transplantation. It is a condition of late, progressive, diffusely stenosing intimal proliferation in the coronary arteries (Fig. 10–32, B), leading to ischemic injury. Within 5 years of transplantation, significant arteriopathy has developed in 50% of patients, and virtually all patients have lesions within 10 years. The pathogenesis of this disorder involves immunologic responses that induce local production of growth factors, which in turn promote intimal smooth muscle cell recruitment and proliferation with ECM synthesis. Allograft arteriopathy is a particularly vexing problem because it can lead to silent MI (transplant recipients have denervated hearts and do not experience angina), progressive CHF, or sudden death.
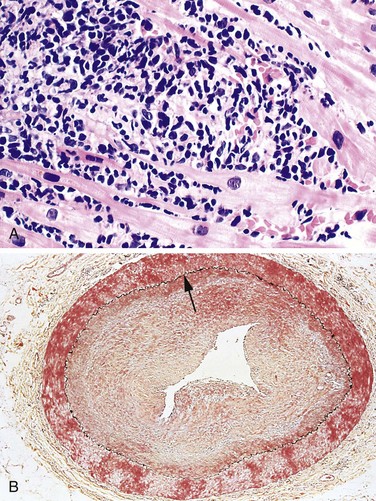
Figure 10–32 Rejection of cardiac allografts. A, Acute cardiac allograft rejection, typified by a lymphocyte infiltrate associated with cardiac myocyte damage. Note the similarity of rejection and viral myocarditis (Fig. 10–29, A). B, Allograft arteriopathy, with severe concentric intimal thickening producing critical stenosis. The internal elastic lamina (arrow) and media are intact. (Movat pentachrome stain.)
(B, Reproduced by permission from Salomon RN, Hughes CC, Schoen FJ, et al: Human coronary transplantation-associated arteriosclerosis. Evidence for chronic immune reaction to activated graft endothelial cells. Am J Pathol 138:791, 1991.)
Despite these problems, the outlook for transplant recipients generally is good, with a 1-year survival rate of 80% and a 5-year survival rate more than 60%.
Azaouagh A, Churzidse S, Konorza T, Erbel R. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a review and update. Clin Res Cardiol. 2011;100:383. [Excellent up-to-date look at this entity and its genetic causes.]
Bhattacharyya S, Davar J, Dreyfus G, Caplin ME. Carcinoid heart disease. Circulation. 2007;116:2860. [Good review of the pathophysiology, diagnosis, and treatment of this entity.]
Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. N Engl J Med. 2000;342:256. 334 [A nice two-part review of congenital cardiac malformations; despite the date, this is still a useful and highly accessible summary of the various conditions.]
Cannon RO3rd. Mechanisms, management and future directions for reperfusion injury after acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2005;2:88. [Great review of the mechanisms and therapeutic approaches to limiting reperfusion injury after MI.]
Cerrone M, Priori SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J. 2011;32:2109. [Up-to-date, well-organized description of the known ion channel disorders that cause sudden cardiac death.]
Cooper LTJr. Myocarditis. N Engl J Med. 2009;360:1526. [A nice review of etiology, pathogenesis, and clinical features.]
Guilherme L, Köhler KF, Kalil J. Rheumatic heart disease: mediation by complex immune events. Adv Clin Chem. 2011;53:31. [A well-written and scholarly discussion of the pathogenic mechanisms regarding rheumatic heart disease.]
Hill EE, Herijgers P, Herregods MC, Peetermans WE. Evolving trends in infective endocarditis. Clin Microbiol Infect. 2006;12:5. [Good, clinically oriented overview of the developments in microorganisms, diagnosis, and therapies for infective endocarditis.]
Huang JB, Liu YL, Sun PW, et al. Molecular mechanisms of congenital heart disease. Cardiovasc Pathol. 2010;19:e183. [Comprehensive review of the genes and pathways underlying congenital heart disease.]
Li C, Xu S, Gotlieb AI. The response to valve injury. A paradigm to understand the pathogenesis of heart valve disease. Cardiovasc Pathol. 2011;20:183. [Nice overview of pathologic concepts in valvular disease.]
Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481. [A well-written review of the pathways, as well as the diagnostic and therapeutic implications of atherosclerotic coronary disease.]
MacGrogan D, Nus M, de la Pompa JL. Notch signaling in cardiac development and disease. Curr Top Dev Biol. 2010;92:333. [A scholarly review of the role of Notch in cardiac development.]
Mitchell RN. Graft vascular disease: immune response meets the vessel wall. Annu Rev Pathol. 2009;4:19. [Comprehensive overview of allograft arteriopathy, including animal models, pathogenic mechanisms, clinical diagnosis, and therapy.]
New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381. [A good overview of the mechanisms leading to degenerative calcification on valves and vessels.]
Ovize M, Baxter GF, Di Lisa F, et al. Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406. [A good overview of the mechanisms and potential therapeutic interventions for ischemia-reperfusion injury and for ischemic pre-conditioning in limiting infarct size.]
Rasmussen TL, Raveendran G, Zhang J, Garry DJ. Getting to the heart of myocardial stem cells and cell therapy. Circulation. 2011;123:1771. [A well-written overview of the challenges and current state of the art regarding stem cell therapies in heart disease.]
Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743. [A well-written and authoritative overview of the genetics and pathophysiology of hypertrophic cardiomyopathy from one of the leading groups in the world.]
Sliwa K, Hilfiker-Kleiner D, Petrie MC, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail. 2010;12:767. [A definitive overview and position paper.]
Watkins H, Houman A, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364:1643. [An excellent review of the molecular basis of cardiomyopathies.]
Wu JC, Child JS. Common congenital heart disorders in adults. Curr Probl Cardiol. 2004;29:641. [Exhaustively thorough overview of the congenital heart disorders seen in the adult population, often as a consequence of improved pediatric therapies.]
Zipes, DP, Libby, P, Bonow, RO, Braunwald, E. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 9th ed, Philadelphia: Saunders, 2011. [An outstanding and authoritative text, with excellent sections on heart failure and atherosclerotic cardiovascular disease.]