Chapter 23 Skin
See Targeted Therapy available online at studentconsult.com
The authors thank Drs. Ronald Rapini and Robert Jordan and the Department of Dermatology at the University of Texas Medical School at Houston for many of the clinical photographs in this chapter. The contributions of Dr. George Murphy to this chapter in previous editions are gratefully acknowledged.
Skin diseases are common and diverse, ranging from irritating acne to life-threatening melanoma. Many are intrinsic to the skin, but some are manifestations of diseases involving many tissues, such as systemic lupus erythematosus or genetic syndromes such as neurofibromatosis. In this sense, the skin is a uniquely accessible “window” through which numerous disorders can be viewed and recognized.
Skin is not a mere protective mantle but rather a complex organ that actively participates in regulated cellular and molecular events that govern the body’s interactions with the external environment. It is constantly bathed with microbial and nonmicrobial antigens. These are processed by intraepithelial Langerhans cells, which bear their antigenic cargo to regional lymph nodes and initiate immune responses. Squamous cells (keratinocytes) help maintain skin homeostasis by providing a physical barrier to environmental insults and by secreting a plethora of cytokines that influence both the squamous and dermal microenvironments. The dermis contains both CD4+ helper and CD8+ cytotoxic T lymphocytes, some of which home to the skin by virtue of specialized receptors such as cutaneous lymphocyte antigen. The epidermis contains intraepithelial lymphocytes, including γ/δ T cells, which constitute a component of the innate immune system. Local immune responses involving these immune cells and cytokines account for the microscopic patterns and clinical expressions of cutaneous inflammatory and infectious diseases.
This chapter focuses on a small subset of common and pathogenically illustrative skin diseases. In considering these diseases, it is important to appreciate that the practice of dermatopathology relies on close interactions with clinicians, particularly dermatologists. The clinical history and the gross appearance and distribution of lesions reported by clinicians are often as important as the microscopic findings in arriving at a diagnosis.
Diseases of the skin can be confusing for the student, in part because dermatologists and dermatopathologists use a large and unique lexicon to describe skin lesions. Because knowledge of this vocabulary forms the basis of clear understanding and communication, some of the terms and descriptors that are most commonly used are defined below.
Terms for Macroscopic Lesions
Excoriation: Traumatic lesion breaking the epidermis and causing a red linear mark (i.e., a deep scratch); often self-inflicted.
Lichenification: Thickened and rough skin characterized by prominent skin markings; usually the result of repeated rubbing (see under “Lichen Simplex Chronicus”).
Macule: Flat, circumscribed area, 5 mm or less in diameter, distinguished from surrounding skin by coloration. If greater than 5 mm, referred to as a patch.
Papule: Elevated dome- or flat-topped lesion 5 mm or less in diameter. If greater than 5 mm in diameter, referred to as a nodule.
Plaque: Elevated flat-topped lesion, usually greater than 5 mm in diameter.
Pustule: Discrete, pus-filled raised lesion.
Scale: Dry, horny, platelike excrescence; usually the result of imperfect cornification.
Vesicle: Fluid-filled raised area 5 mm or less in diameter. If greater than 5 mm in diameter, referred to as a bulla. Blister is common term for both vesicles and bullae.
Microscopic Terms
Acantholysis: Loss of intercellular adhesion of keratinocytes.
Acanthosis: Diffuse epidermal hyperplasia.
Dyskeratosis: Abnormal keratinization occurring prematurely within individual cells or groups of cells below the stratum granulosum.
Hyperkeratosis: Hyperplasia of the stratum corneum, often associated with a qualitative abnormality of keratin.
Lentiginous: Linear melanocyte proliferation along the epidermal basal cell layer; can occur as a reactive change or as part of a melanocytic neoplasm.
Papillomatosis: Surface elevation caused by hyperplasia and enlargement of dermal papillae.
Parakeratosis: Keratinization characterized by retention of the nuclei in the stratum corneum. On squamous mucosal membranes, such as buccal mucosa, parakeratosis is normal.
Acute Inflammatory Dermatoses
Thousands of inflammatory dermatoses exist, challenging the diagnostic acumen of even experienced clinicians. In general, acute lesions last from days to weeks and are characterized by inflammation (often marked by mononuclear cells rather than neutrophils and defined as acute due to the limited course of their natural history), edema, and sometimes epidermal, vascular, or subcutaneous injury. Some acute lesions may persist, resulting in transition to a chronic phase, while others are characteristically self-limited.
Urticaria
Urticaria (“hives”) is a common disorder mediated by localized mast cell degranulation, which leads to dermal microvascular hyperpermeability. The resulting erythematous, edematous, and pruritic plaques are termed wheals.
 Pathogenesis
Pathogenesis
In most cases, urticaria stems from an immediate (type 1) hypersensitivity reaction (Chapter 4), in which antigens trigger mast cell degranulation by binding to immunoglobulin E (IgE) antibodies displayed on the mast cell surface. The responsible antigens include pollens, foods, drugs, and insect venom. IgE-independent urticaria may result from exposure to substances that directly incite mast cell degranulation, such as opiates and certain antibiotics. In the vast majority of cases, no clinical cause is discovered despite extensive searching. Hereditary angioedema is caused by an inherited deficiency of C1 esterase inhibitor, which results in uncontrolled activation of complement (Chapter 3). The ensuing urticaria affects the lips, throat, eyelids, genitals, and distal extremities. When the larynx is affected, the condition can be dangerous, since airway patency may be compromised.
 Morphology
Morphology
The histologic features of urticaria often are subtle. There is usually a sparse superficial perivenular infiltrate of mononuclear cells, rare neutrophils, and sometimes eosinophils. Superficial dermal edema creates more widely spaced collagen bundles. Degranulation of mast cells, which normally reside around superficial dermal venules, is difficult to appreciate with routine hematoxylin-eosin (H&E) stains but can be highlighted using a Giemsa stain.
Clinical Features
Urticaria typically affects persons between 20 and 40 years of age, but no age is immune. Individual lesions usually develop and fade within hours, but episodes can persist for days or even months. Persistent lesions sometimes are due to urticarial vasculitis, which is often associated with deposition of complement in dermal venules. Lesions range in size and nature from small, pruritic papules to large, edematous, erythematous plaques. Increased vascular permeability leads to localized dermal edema. Lesions can be confined to a particular part of the body or generalized. In a specific type of urticaria, termed pressure urticaria, lesions are found only in areas exposed to pressure (such as the feet or the buttocks). Although not life-threatening, urticaria can compromise quality of life by causing severe pruritus and social embarrassment. Most cases are treated with antihistamines. Systemic steroids are used in more severe refractory cases.
Acute Eczematous Dermatitis
Eczema is a clinical term that embraces a number of conditions with varied underlying etiologies. New lesions take the form of red papules, often with overlying vesicles, which ooze and become crusted. With persistence, these lesions develop into raised, scaling plaques. The nature and degree of these changes vary among the clinical subtypes, which include the following:
• Allergic contact dermatitis, which stems from topical exposure to an allergen
• Atopic dermatitis, which has traditionally been attributed to allergen exposure, but is now thought to stem from defects in keratinocyte barrier function, many with a genetic basis
• Drug-related eczematous dermatitis, a hypersensitivity reaction to a drug
• Photoeczematous dermatitis, in which eczema appears as an abnormal reaction to UV or visible light
• Primary irritant dermatitis, which results from exposure to substances that chemically, physically, or mechanically damage the skin
In most cases, the skin lesions resolve completely when the offending stimulus is removed or exposure is limited, stressing the importance of investigating the underlying cause. Only the most common form, contact dermatitis, is considered here.
Contact dermatitis is triggered by exposure to an environmental contact sensitizing agent, such as poison ivy, that chemically reacts with self-proteins. The self-proteins modified by the agent are processed by epidermal Langerhans cells, which migrate to draining lymph nodes and present the antigen to naive T cells. This sensitization event leads to acquisition of immunologic memory; on reexposure to the antigen, the activated memory CD4+ T lymphocytes migrate to the affected skin sites. There they release cytokines that recruit additional inflammatory cells and also mediate epidermal damage, as in any delayed-type hypersensitivity reaction (Chapter 4).
Morphology
In the case of contact dermatitis, the pattern of skin involvement is limited to sites of contact with the triggering agent (Fig. 23–1, A), whereas in other forms of eczema, lesions may be widely distributed. Spongiosis, or epidermal edema, characterizes all forms of acute eczematous dermatitis—hence the synonym spongiotic dermatitis. Edema fluid seeps into the epidermis, where it splays apart keratinocytes (Fig. 23–1, B). Intercellular bridges are stretched and become more prominent and are easier to visualize. This change is accompanied by a superficial perivascular lymphocytic infiltrate, edema of dermal papillae, and mast cell degranulation. Eosinophils may be present and are especially prominent in spongiotic eruptions provoked by drugs, but in general the histologic features are similar regardless of cause, emphasizing the need for careful clinical correlation.
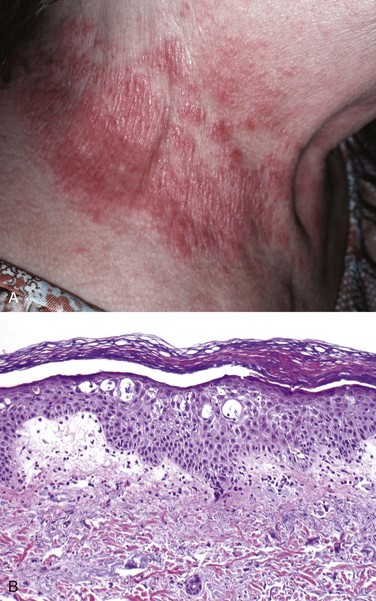
Figure 23–1 Eczematous dermatitis. A, The patterned erythema and scale stems from a nickel-induced contact dermatitis produced by this woman’s necklace. B, Microscopically, there is fluid accumulation (spongiosis) between epidermal cells that can progress to small vesicles if intercellular connections are stretched until broken.
Clinical Features
Lesions of acute eczematous dermatitis are pruritic (itchy), edematous, oozing plaques, often containing vesicles and bullae. With persistent antigen exposure, lesions may become progressively scaly (hyperkeratotic) as the epidermis thickens (acanthosis). Some changes are produced or exacerbated by scratching or rubbing of the lesion (see later under “Lichen Simplex Chronicus”). The clinical causes of eczema are sometimes divided into “inside jobs”—reaction to an internal circulating antigen (such as ingested food or drug)—and “outside jobs”—disease resulting from contact with an external antigen (such as poison ivy).
Susceptibility to atopic dermatitis is often inherited; the disorder is concordant in 80% of identical twins and 20% of fraternal twins. It usually appears in early childhood and in the majority of cases clears in adults. Children with atopic dermatitis often have asthma and allergic rhinitis, termed the atopic triad.
Erythema Multiforme
Erythema multiforme is an uncommon, usually self-limited disorder that appears to be a hypersensitivity response to certain infections and drugs. Antecedent infections include herpes simplex and those caused by mycoplasmas and some fungi. The implicated drugs include sulfonamides, penicillin, salicylates, hydantoins, and antimalarials. Patients present with a wide diversity of lesions (hence called “multiforme”) including macules, papules, vesicles, and bullae, as well as characteristic targetoid lesions consisting of red macules or papules with pale vesicular or eroded centers (Fig. 23–2). The epithelial damage is believed to result from the action of skin-homing cytotoxic T cells that attack the basal cells of the skin and the mucosae, which may display antigens that cross-react with the inciting drug or microbe.
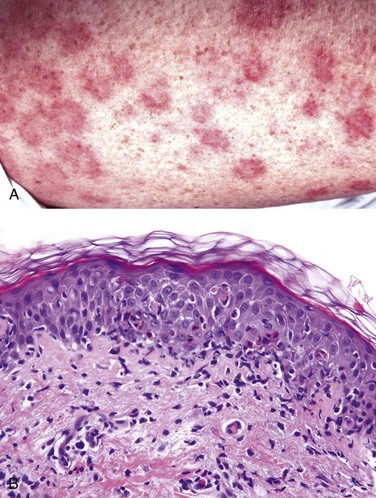
Figure 23–2 Erythema multiforme. A, The target-like lesions consist of a pale central blister or zone of epidermal necrosis surrounded by macular erythema. B, Early lesions show a collection of lymphocytes along the dermoepidermal junction (interface dermatitis) associated with scattered keratinocytes with dark shrunken nuclei and eosinophilic cytoplasm that are undergoing apoptosis.
Morphology
Well-developed lesions have a characteristic “targetoid” appearance (Fig. 23–2, A). Early lesions show a superficial perivascular, lymphocytic infiltrate associated with dermal edema and margination of lymphocytes along the dermoepidermal junction in intimate association with degenerating keratinocytes (Fig. 23–2, B). With time, discrete, confluent zones of basal epidermal necrosis appear, with concomitant blister formation. In the rarer and more severe form of this disease, toxic epidermal necrolysis, the necrosis extends through the full thickness of the epidermis.
Clinical Features
Erythema multiforme exhibits a broad range of severity. The forms associated with infection (most often herpesvirus) are less severe. Erythema multiforme caused by medications can progress to more serious life-threatening eruptions, such as Stevens-Johnson syndrome or toxic epidermal necrolysis. These forms can be life-threatening because they can cause sloughing of large portions of the epidermis, resulting in fluid loss and infections akin to those seen in burn-injured patients. These severe forms most often occur as idiopathic reactions to drugs.
Chronic Inflammatory Dermatoses
Chronic inflammatory dermatoses are persistent skin conditions that exhibit their most characteristic features over many months to years, although they may begin with an acute stage. The skin surface in some chronic inflammatory dermatoses is roughened as a result of excessive or abnormal scale formation and shedding (desquamation).
Psoriasis
Psoriasis is a common chronic inflammatory dermatosis, affecting 1% to 2% of people residing in the United States. Recent epidemiologic studies have shown that psoriasis is associated with an increased risk of heart attack and strokes, a relationship that may be related to a chronic inflammatory state. Psoriasis is also associated in up to 10% of patients with arthritis, which in some cases may be severe.
Pathogenesis
Psoriasis is a multifactorial immunologic disease; both genetic (e.g., human leukocyte antigen [HLA] types) and environmental factors contribute to risk. It is not known if the inciting antigens are self or environmental. Sensitized populations of T cells home to the dermis, including CD4+ TH17 and TH1 cells and CD8+ T cells, and accumulate in the epidermis. These cells secrete cytokines and growth factors that induce keratinocyte hyperproliferation, resulting in the characteristic lesions. Psoriatic lesions can be induced in susceptible persons by local trauma (Koebner phenomenon), which may induce a local inflammatory response that promotes lesion development. GWAS studies have linked an increased risk of psoriasis to polymorphisms in HLA loci and genes affecting antigen presentation, TNF signaling, and skin barrier function.
Morphology
The typical lesion is a well-demarcated, pink to salmon–colored plaque covered by loosely adherent silver-white scale (Fig. 23–3, A). There is marked epidermal thickening (acanthosis), with regular downward elongation of the rete ridges (Fig. 23–3, B). The pattern of this downward growth has been likened to “test tubes in a rack.” Increased epidermal cell turnover and lack of maturation results in loss of the stratum granulosum and extensive parakeratotic scale. Also seen is thinning of the epidermal cell layer overlying the tips of dermal papillae (suprapapillary plates), and dilated and tortuous blood vessels within the papillae. These vessels bleed readily when the scale is removed, giving rise to multiple punctate bleeding points (Auspitz sign). Neutrophils form small aggregates within both the spongiotic superficial epidermis and the parakeratotic stratum corneum. Similar changes can be seen in superficial fungal infections, which need to be excluded with appropriate special stains.
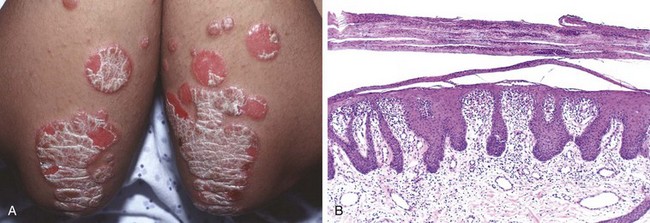
Figure 23–3 Psoriasis. A, Chronic plaques of psoriasis show silvery-white scale on the surface of erythematous plaques. B, Microscopic examination reveals marked epidermal hyperplasia, uniform downward extension of rete ridges (psoriasiform hyperplasia), and prominent parakeratotic scale that is focally infiltrated by neutrophils.
Clinical Features
Psoriasis most frequently affects the skin of the elbows, knees, scalp, lumbosacral areas, intergluteal cleft, and glans penis. Nail changes on the fingers and toes occur in 30% of cases. In most cases, psoriasis is limited in distribution, but it can be widespread and severe. The clinical subtypes are defined by pattern of involvement and severity. Treatment is aimed at preventing the release or actions of inflammatory mediators. Depending upon the disease severity, NSAIDS, immunosuppressive agents such as cyclosporin, and TNF antagonists are used. Newer agents that inhibit TH1 and TH17 immune responses are also being tested.
Lichen Planus
“Pruritic, purple, polygonal, planar papules, and plaques” are the tongue-twisting Ps that describe this disorder of skin and squamous mucosa. The lesions may result from a CD8+ T cell–mediated cytotoxic immune response against antigens in the basal cell layer and the dermoepidermal junction that are produced by unknown mechanisms, perhaps as a consequence of a viral infection or drug exposure.
Morphology
Cutaneous lesions of lichen planus consist of pruritic, violaceous, flat-topped papules, which may coalesce focally to form plaques (Fig. 23–4, A). These papules are often highlighted by white dots or lines called Wickham striae. Hyperpigmentation may result from melanin loss into the dermis from damaged keratinocytes. Microscopically, lichen planus is a prototypical interface dermatitis, so called because the lesions are concentrated at the interface of the squamous epithelium and papillary dermis. There is a dense, continuous infiltrate of lymphocytes along the dermoepidermal junction (Fig. 23–4, B). The lymphocytes are intimately associated with basal keratinocytes, which often atrophy or become necrotic. Perhaps as a response to damage, the basal cells take on the appearance of the more mature cells of the stratum spinosum (squamatization). This pattern of inflammation causes the dermoepidermal interface to assume an angulated, zigzag contour (“sawtoothing”). Anucleate, necrotic basal cells are seen in the inflamed papillary dermis and are referred to as colloid bodies or Civatte bodies. Although these changes bear some similarities to those in erythema multiforme (discussed earlier), lichen planus shows well-developed changes of chronicity, including epidermal hyperplasia, hypergranulosis, and hyperkeratosis.
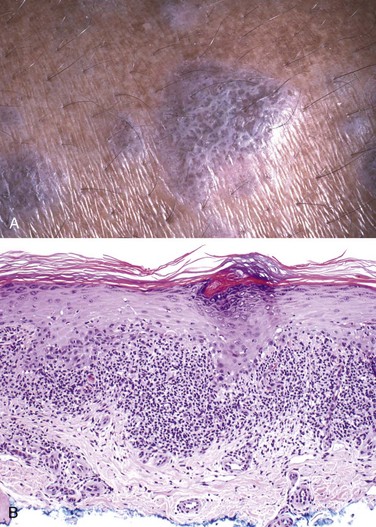
Figure 23–4 Lichen planus. A, This flat-topped pink-purple polygonal papule has white lacelike markings referred to as Wickham striae. B, Microscopic features include a bandlike infiltrate of lymphocytes along the dermoepidermal junction, hyperkeratosis, hypergranulosis, and pointed rete ridges (“sawtoothing”), which results from chronic injury of the basal cell layer.
Clinical Features
Lichen planus is an uncommon disorder that usually presents in middle-aged adults. The cutaneous lesions are multiple and are usually symmetrically distributed, particularly on the extremities, and often occur about the wrists and elbows and on the glans penis. In approximately 70% of cases the oral mucosa is also involved, where the lesions manifest as white papules with a reticulate or netlike appearance. The cutaneous lesions of lichen planus usually resolve spontaneously within 1 to 2 years, but the oral lesions may persist and be of sufficient severity to cause trouble with food intake.
Lichen Simplex Chronicus
Lichen simplex chronicus manifests as roughening of the skin, which takes on an appearance reminiscent of lichen on a tree. It is a response to local repetitive trauma such as continual rubbing or scratching. Nodular forms exist that are referred to as prurigo nodularis. The pathogenesis of lichen simplex chronicus is not understood, but the trauma probably induces epithelial hyperplasia and eventual dermal scarring.
Morphology
Lichen simplex chronicus is characterized by acanthosis, hyperkeratosis, and hypergranulosis. Also seen are elongation of the rete ridges, fibrosis of the papillary dermis, and a dermal chronic inflammatory infiltrate (Fig. 23–5). Of interest, these lesions are similar in appearance to normal volar (palms and soles) skin, in which skin thickening serves as an adaptation to repetitive mechanical stress.
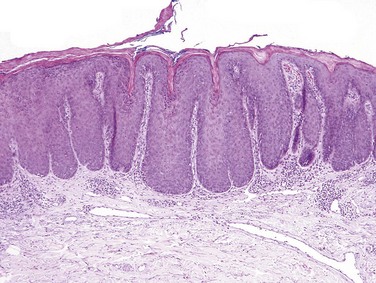
Clinical Features
The lesions often are raised, erythematous, and scaly and can be mistaken for keratinocytic neoplasms. Lichen simplex chronicus can be superimposed on and mask another (often pruritic) dermatosis. It is therefore important to rule out an underlying cause while recognizing that the lesion may be entirely trauma-related.
 Summary
Summary
Inflammatory Dermatoses
• Many specific inflammatory dermatoses exist, which can be mediated by IgE antibodies (urticaria), antigen-specific T cells (eczema, erythema multiforme, and psoriasis), or trauma (lichen simplex chronicus).
• These disorders can be grouped based on patterns of inflammation (e.g., interface dermatitis in lichen planus and erythema multiforme).
• Clinical correlation is essential to diagnose specific skin diseases, since many have overlapping, non-specific histologic features.
Infectious Dermatoses
Bacterial Infections
Numerous bacterial infections occur in skin. These range from superficial infections known as impetigo, to deeper dermal abscesses caused by anaerobes such as Pseudomonas aeruginosa, associated with puncture wounds. The pathogenesis is similar to that for microbial infections elsewhere (Chapter 8). Only impetigo is discussed here.
Morphology
Impetigo is characterized by an accumulation of neutrophils beneath the stratum corneum that often produces a subcorneal pustule. Nonspecific reactive epidermal alternation and superficial dermal inflammation accompany these findings. Bacterial cocci in the superficial epidermis can be demonstrated by Gram stain.
Clinical Features
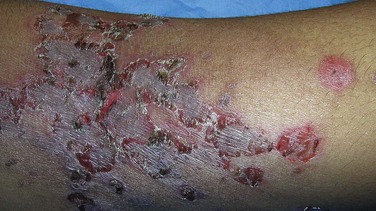
Impetigo, one of the most common bacterial infections of the skin, is seen primarily in children. The causative organism is usually Staphylococcus aureus or, less commonly, Streptococcus pyogenes, and is typically acquired through direct contact with a source. Impetigo often begins as a single small macule, usually on the extremities or the face near the nose or the mouth (Fig. 23–6), which rapidly evolves into a larger lesion with a honey-colored crust of dried serum. Persons colonized by S. aureus or S. pyogenes (usually nasal or anal) are more likely to be affected. Microbiologic culture with assessment of sensitivity to various antibiotics can be useful clinically. A less common bullous form of childhood impetigo may mimic an autoimmune blistering disorder.
Fungal Infections
Fungal infections are varied and range from superficial infections with Tinea or Candida spp. to life-threatening infections of immunosuppressed persons with Aspergillus spp. Fungal infections can be superficial (stratum corneum, hair, and nails), deep (dermis or subcutis), or systemic, the last type arising through hematogenous spread, often in an immunocompromised patient.
Morphology
The histologic appearance varies depending on the organism, host response, and degree of superinfection. Superficial infections are often associated with a neutrophilic infiltrate in the epidermis. Some show a mild eczematous dermatitis with associated intraepidermal neutrophils, while other infections (e.g., Candida) can induce psoriasiform hyperplasia. Deep fungal infections produce greater tissue damage and often elicit a granulomatous response, probably induced by the more vigorous host immune response. Aspergillus can be angioinvasive. Periodic acid–Schiff (PAS) and Gomori methenamine silver stains can be helpful in identifying the fungal organisms.
Clinical Features
Superficial infections usually produce erythematous macules with superficial scale that can be pruritic, while deeper infections such as those seen with Aspergillus spp. in immunocompromised persons are erythematous and often nodular and sometimes show evidence of local hemorrhage. Superficial Candida infections often induce lesions that mimic psoriasis, so it is essential to exclude fungal infections when a new diagnosis of psoriasis is being considered.
Verrucae (Warts)
Verrucae are common lesions of children and adolescents, although they may be encountered in any age group. They are caused by human papillomavirus (HPV). Transmission usually involves direct contact with an infected individual or autoinoculation. Verrucae generally are self-limited, most often regressing spontaneously within 6 months to 2 years.
Pathogenesis
As mentioned earlier, verrucae are caused by HPV. Some members of the HPV family are associated with preneoplastic and invasive cancers of the anogenital region (Chapters 5 and 17). In contrast with HPV-associated carcinomas, most warts are caused by low-risk HPV subtypes that lack transforming potential. Like high-risk viruses, these low-risk viruses express viral E6 and E7 oncoproteins that lead to dysregulated epidermal cell growth and increased survival. Why low-risk viruses cause warts instead of cancer is unclear; subtle functional differences due to structural variation in E6 and E7 proteins that affect interactions with host proteins as well as differences in the abilities of different viral strains to evade the immune response have been proposed.The immune response normally limits the growth of these tumors, and immunodeficiency often is associated with increased numbers and larger sized verrucae.
Morphology
Different kinds of warts are identified on the basis of their gross appearance and location and generally are caused by distinct HPV subtypes. Verruca vulgaris (Fig. 23–7, A), the most common type of wart, can occur anywhere but is found most frequently on the hands, particularly on the dorsal surfaces and periungual areas, where it appears as a gray-white to tan, flat to convex, 0.1- to 1-cm papule with a rough, pebble-like surface. Verruca plana, or flat wart, is common on the face or dorsal surfaces of the hands. These warts are flat, smooth, tan macules. Verruca plantaris and verruca palmaris occur on the soles and palms, respectively. These rough, scaly lesions may reach 1 to 2 cm in diameter and then coalesce to form a surface that can be confused with ordinary calluses. Condyloma acuminatum (venereal wart) occurs on the penis, female genitalia, urethra, and perianal areas (Chapters 17 and 18). Histologic features common to verrucae include epidermal hyperplasia, which is often undulant in character (so-called verrucous or papillomatous epidermal hyperplasia) (Fig. 23–7, B, top panel), and cytoplasmic vacuolization (koilocytosis), which preferentially involves the more superficial epidermal layers, producing halos of pallor surrounding infected nuclei. Infected cells also may demonstrate prominent keratohyalin granules and jagged eosinophilic intracytoplasmic protein aggregates as a result of impaired maturation (Fig. 23–7, B, bottom panel).
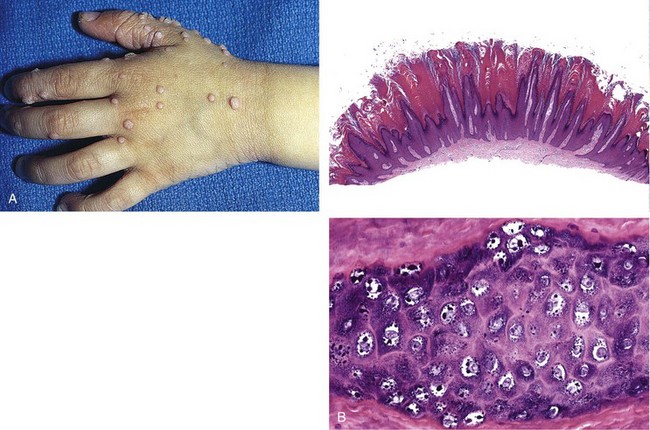
Figure 23–7 Verruca vulgaris. A, Multiple warts, with characteristic rough, pebble-like surfaces. B, Microscopically, common warts contain zones of papillary epidermal proliferation that often radiate symmetrically like the points of a crown (top). Nuclear pallor, prominent keratohyalin granules, and related cytopathic changes are seen at higher magnification (bottom).
Blistering (Bullous) Disorders
Although vesicles and bullae (blisters) occur as a secondary phenomenon in several unrelated conditions (e.g., herpesvirus infection, spongiotic dermatitis), there is a group of disorders in which blisters are the primary and most distinctive feature. Blistering in these diseases tends to occur at specific levels within the skin, a morphologic distinction that is critical for diagnosis (Fig. 23–8).
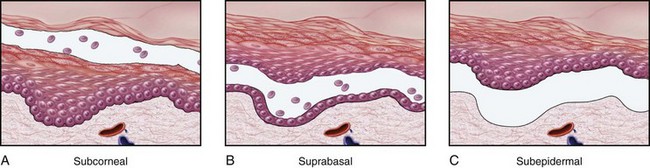
Figure 23–8 Levels of blister formation. A, Subcorneal (as in pemphigus foliaceus). B, Suprabasal (as in pemphigus vulgaris). C, Subepidermal (as in bullous pemphigoid or dermatitis herpetiformis). The level of epidermal separation forms the basis of the differential diagnosis for blistering disorders.
Pemphigus (Vulgaris and Foliaceus)
Pemphigus is a rare autoimmune blistering disorder resulting from loss of normal intercellular attachments within the epidermis and the squamous mucosal epithelium. There are three major variants:
The last entity is associated with internal malignancy and is not discussed here.
Pathogenesis
Both pemphigus vulgaris and pemphigus foliaceus are autoimmune diseases in which the lesions are caused by antibody-mediated (type II) hypersensitivity reactions (Chapter 4). The pathogenic antibodies are IgG autoantibodies that bind to intercellular desmosomal proteins (desmoglein types 1 and 3) of skin and mucous membranes. The antibodies disrupt the intercellular adhesive function of the desmosomes and may activate intercellular proteases as well. The distribution of these proteins within the epidermis determines the location of the lesions. By direct immunofluorescence study, lesional sites show a characteristic fishnet-like pattern of intercellular IgG deposits (Fig. 23–9). As in other autoimmune diseases, pemphigus shows linkage with particular HLA alleles.
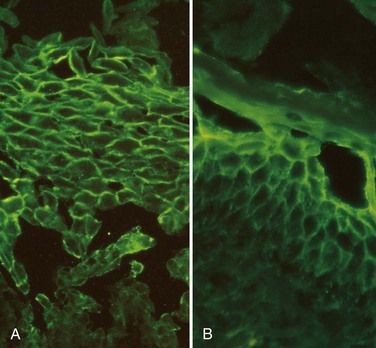
Figure 23–9 Direct immunofluorescence findings in pemphigus. A, Pemphigus vulgaris. There is uniform deposition of immunoglobulin and complement (green) along the cell membranes of keratinocytes in a characteristic “fishnet” pattern. B, Pemphigus foliaceus. Immunoglobulin deposits are confined to superficial layers of the epidermis.
Morphology
Pemphigus vulgaris, by far the most common type, involves both mucosa and skin, especially on the scalp, face, axillae, groin, trunk, and points of pressure. The lesions are superficial flaccid vesicles and bullae that rupture easily, leaving deep and often extensive erosions covered with a serum crust (Fig. 23–10, A). Pemphigus foliaceus, a rare, more benign form of pemphigus, results in bullae confined to skin, with only infrequent involvement of mucous membranes. The blisters in this disorder are superficial, such that more limited zones of erythema and crusting of ruptured blisters are seen (Fig. 23–11, A).
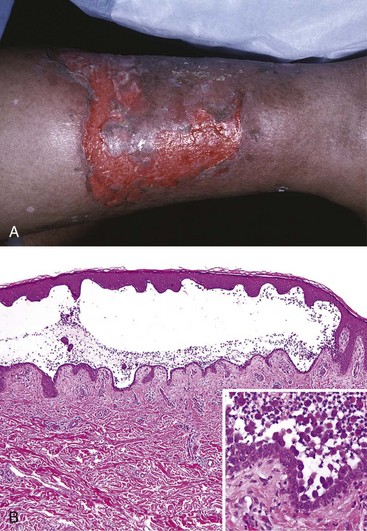
Figure 23–10 Pemphigus vulgaris. A, This erosion on the leg represents a group of confluent, “unroofed” blisters. B, Suprabasal acantholysis results in an intraepidermal blister in which rounded, dissociated (acantholytic) keratinocytes are plentiful (inset).
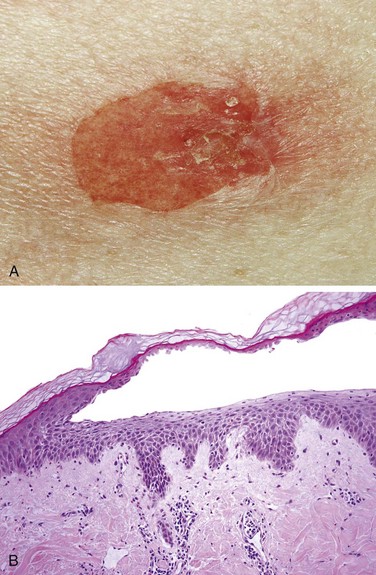
Figure 23–11 Pemphigus foliaceus. A, Gross appearance of a typical blister, which is less severely eroded than those seen in pemphigus vulgaris. B, Microscopic appearance of a characteristic subcorneal blister.
The common histologic denominator in all forms of pemphigus is acantholysis, lysis of the intercellular adhesive junctions between neighboring squamous epithelial cells that results in the rounding up of detached cells. In pemphigus vulgaris, acantholysis selectively involves the layer of cells immediately above the basal cell layer, giving rise to a suprabasal acantholytic blister (Fig. 23–10, B). In pemphigus foliaceus, acantholysis selectively involves the superficial epidermis at the level of the stratum granulosum (Fig. 23–11, B). Variable superficial dermal infiltrates comprised of lymphocytes, macrophages, and eosinophils accompany all forms of pemphigus.
Clinical Features
Pemphigus vulgaris is a rare disorder that occurs most commonly in the elderly and more often in women than men. Lesions are painful, particularly when ruptured, and secondary infections are common. Most cases are associated with oropharyngeal involvement at some point in their course. Most patients require immunosuppressive therapy, sometimes for the remainder of their lives. Medications can cause pemphigus, and when they do, patients most often present with pemphigus foliaceus rather than pemphigus vulgaris. There is also an unusual endemic form of pemphigus foliaceus in South America (fogo selvagem) that is putatively associated with the bite of a black fly.
Bullous Pemphigoid
Bullous pemphigoid is another distinctive acquired blistering disorder with an autoimmune basis.
Pathogenesis
Blistering in bullous pemphigoid is triggered by the linear deposition of IgG antibodies and complement in the epidermal basement membrane (Fig. 23–12, A). Reactivity also occurs in the basement membrane attachment plaques (hemidesmosomes), where most of the bullous pemphigoid antigen (type XVII collagen) is located. This protein normally is involved in dermoepidermal adhesion. IgG autoantibodies to hemidesmosome components fix complement and cause tissue injury by recruiting neutrophils and eosinophils.
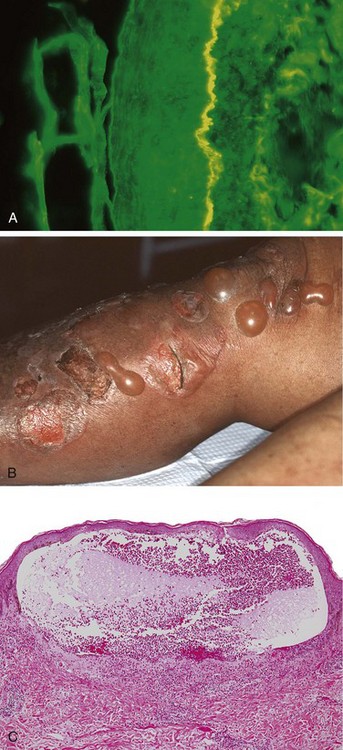
Figure 23–12 Bullous pemphigoid. A, Deposition of IgG antibody detected by direct immunofluorescence as a linear band outlining the subepidermal basement membrane zone (epidermis is on the left side of the fluorescent band). B, Gross appearance of characteristic tense, fluid-filled blisters. C, A subepidermal vesicle with an inflammatory infiltrate rich in eosinophils.
(C, Courtesy of Dr. Victor G. Prieto, Houston, Texas.)
Morphology
Grossly, the lesions of bullous pemphigoid appear as tense bullae, filled with clear fluid, on normal or erythematous skin (Fig. 23–12, B). Bullous pemphigoid is characterized by subepidermal nonacantholytic blisters. Early lesions show a perivascular infiltrate of lymphocytes and variable numbers of eosinophils, occasional neutrophils, superficial dermal edema, and associated basal cell layer vacuolization. The vacuolated basal cell layer eventually gives rise to a fluid-filled blister (Fig. 23–12, C). The blister roof consists of full-thickness epidermis with intact intercellular junctions, a key distinction from the blisters seen in pemphigus.
Clinical Features
The bullae do not rupture as readily as in pemphigus and, if uncomplicated by infection, heal without scarring. The disease tends to follow a remitting and relapsing course and responds to topical or systemic immunosuppressive agents. Gestational pemphigoid (also known as herpes gestationis, a misnomer) is a clinically distinct subtype that appears suddenly during the second or third trimester of pregnancy. It is also caused by autoantibodies against BPAG. It typically resolves after childbirth, but may recur with future pregnancies.
Dermatitis Herpetiformis
Dermatitis herpetiformis is another type of autoimmune blistering disorder characterized by extremely pruritic urticaria and grouped vesicles. The disease affects predominantly males, often in the third and fourth decades of life. In up to 80% of cases, it occurs in association with celiac disease; conversely, only a small minority of patients with celiac disease develop dermatitis herpetiformis. Like celiac disease, dermatitis herpetiformis responds to a gluten-free diet.
Pathogenesis
The strong association of dermatitis herpetiformis with celiac disease provides a clue to its pathogenesis. Genetically predisposed persons develop IgA antibodies to dietary gluten (derived from the wheat protein gliadin) as well as IgA autoantibodies that cross-react with endomysium and tissue transglutaminases, including epidermal transglutaminase expressed by keratinocytes. By direct immunofluorescence, the skin shows discontinuous, granular deposits of IgA selectively localized in the tips of dermal papillae (Fig. 23–13, A). The resultant injury and inflammation produce a subepidermal blister.
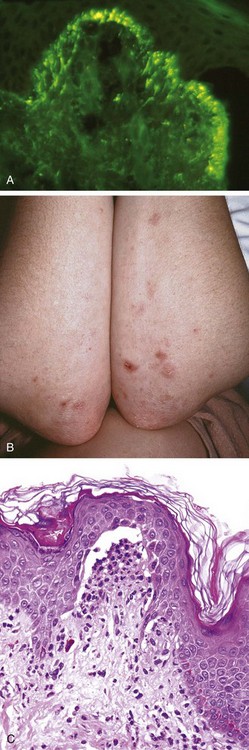
Figure 23–13 Dermatitis herpetiformis. A, Selective deposition of IgA autoantibody at the tips of dermal papillae is characteristic. B, Lesions consist of intact and eroded (usually scratched) erythematous blisters, often grouped (seen here on elbows and arms). C, The blisters are associated with basal cell layer injury, initially caused by accumulation of neutrophils (microabscesses) at the tips of dermal papillae.
(A, Courtesy of Dr. Victor G. Prieto, Houston, Texas.)
Morphology
The lesions of dermatitis herpetiformis are bilateral, symmetric, and grouped and preferentially involve the extensor surfaces, elbows, knees, upper back, and buttocks (Fig. 23–13, B). Initially, neutrophils accumulate selectively at the tips of dermal papillae, forming small microabscesses (Fig. 23–13, C). The basal cells overlying these microabscesses show vacuolization and focal dermoepidermal separation that ultimately coalesce to form a true subepidermal blister.
Summary
Blistering Disorders
• Blistering disorders are classified by the level of epidermal separation.
• These disorders are often caused by autoantibodies specific for epithelial or basement membrane proteins that lead to unmooring of keratinocytes (acantholysis).
• Pemphigus is associated with IgG autoantibodies to various intercellular desmogleins, resulting in bullae that are either subcorneal (pemphigus foliaceus) or suprabasalar (pemphigus vulgaris).
• Bullous pemphigoid is associated with IgG autoantibodies to basement membrane proteins and produces a subepidermal blister.
• Dermatitis herpetiformis is associated with IgA autoantibodies to fibrils that bind the epidermal basement membrane to the dermis, and is also characterized by subepidermal blisters.
Benign and Premalignant Tumors
Benign and Premalignant Epithelial Lesions
Benign epithelial neoplasms are common and probably arise from stem cells residing in the epidermis and hair follicles. These tumors grow to a limited size and generally do not undergo malignant transformation.
Seborrheic Keratosis
These common pigmented epidermal tumors occur most frequently in middle-aged or older persons. They arise spontaneously and are particularly numerous on the trunk, although the extremities, head, and neck also may be sites of involvement.
A significant fraction of these tumors harbor activating mutations in fibroblast growth factor (FGF) receptor 3, which possesses a tyrosine kinase activity that stimulates Ras and the PI3K/AKT pathways. Except for cosmetic concerns, they are usually of little clinical importance. However, in rare patients hundreds of lesions may appear suddenly as a paraneoplastic syndrome (sign of Lesser-Trelat). Patients with this presentation may harbor internal malignancies, most commonly gastrointestinal tract carcinomas, which produce growth factors that stimulate epidermal proliferation.
Morphology
Seborrheic keratoses are round, exophytic, coin-like plaques varying in diameter from millimeters to centimeters that have a “stuck-on” appearance (Fig. 23–14, inset). They are tan to dark brown and have a velvety- to granular-appearing surface. Occasionally, their dark color is suggestive of melanoma, leading to surgical removal.
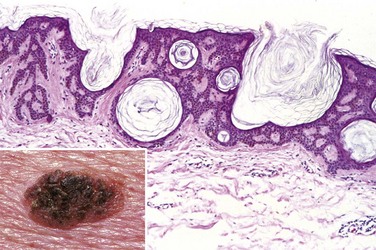
Figure 23–14 Seborrheic keratosis. This roughened, brown, waxy lesion almost appears to be “stuck on” the skin (inset). Microscopic examination shows the lesion to consist of an orderly proliferation of uniform, basaloid keratinocytes that tend to form keratin microcysts (horn cysts).
Microscopically, seborrheic keratoses are composed of monotonous sheets of small cells that resemble the basal cells of the normal epidermis (Fig. 23–14). Variable melanin pigmentation is present within these basaloid cells, accounting for the brown coloration seen grossly. Hyperkeratosis occurs at the surface, and the presence of small keratin-filled cysts (horn cysts) and downgrowth of keratin into the main tumor mass (pseudo–horn cysts) are characteristic features.
Actinic Keratosis
Overt malignancy of the epidermis (squamous cell carcinoma) may be preceded by a series of progressive dysplastic changes. Because such lesions usually are the result of chronic exposure to sunlight and are associated with hyperkeratosis, they are called actinic (sun-related) keratoses. Whether all actinic keratoses evolve to carcinoma with time is conjectural; many lesions regress or remain stable. Enough become malignant, however, to warrant local eradication. A high fraction of these lesions are associated with TP53 mutations caused by UV light–induced DNA damage.
Morphology
Actinic keratoses usually are less than 1 cm in diameter, tan-brown or red in color, and rough (sandpaper-like) to the touch (Fig. 23–15, A). Microscopically, lower portions of the epidermis show cytologic atypia, often associated with hyperplasia of basal cells (Fig. 23–15, B) or with atrophy and diffuse thinning of the epidermal surface. The dermis contains thickened, blue-gray elastic fibers (solar elastosis), the result of chronic sun damage. The stratum corneum is thickened with retained nuclei (parakeratosis). In some lesions full thickness epidermal atypia is seen; such lesions are considered forms of squamous cell carcinoma in situ (Fig. 23–15, C).
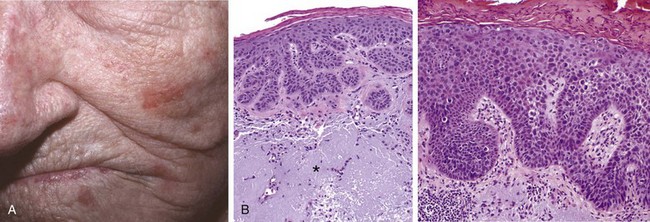
Figure 23–15 Actinic keratosis. A, Most lesions are red and rough (sandpaper-like), owing to excessive scale, as seen in the lesions on the cheek, nose, and chin of this female patient. B, Basal cell layer atypia (dysplasia) with epithelial buds, and associated with marked hyperkeratosis, parakeratosis, and dermal solar elastosis (asterisk). C, More advanced lesions show full-thickness atypia, qualifying as squamous carcinoma in situ.
Clinical Features
Actinic keratoses are very common in fair-skinned people and increase in incidence with age and increasing sun exposure. As would be expected, there is a predilection for sun-exposed areas (face, arms, dorsum of the hands). The lesions can be treated with local cryotherapy (superficial freezing) or topical agents.
Summary
Benign and Premalignant Epithelial Lesions
• Seborrheic keratosis: Round, flat plaques made up of proliferating monotonous epidermal basal cells, which sometimes contain melanin. Hyperkeratosis and keratin-filled cysts are characteristic.
• Actinic keratosis: Present on sun-exposed skin, these lesions show cytologic atypia in lower parts of epidermis and infrequently progress to carcinoma in situ.
Malignant Epidermal Tumors
Squamous Cell Carcinoma
Squamous cell carcinoma is a common tumor arising on sun-exposed sites in older people. These tumors have a higher incidence in men than in women. In addition to sunlight, predisposing factors include industrial carcinogens (tars and oils), chronic ulcers, old burn scars, ingestion of arsenicals, and ionizing radiation. As with squamous cell carcinomas at other sites, those in the skin may be preceded by in situ lesions.
Pathogenesis
The most common exogenous cause of cutaneous squamous cell carcinoma is UV light exposure, which causes DNA damage (Chapter 5). TP53 mutations caused by UV light–induced DNA damage are common, as are activating mutations in HRAS and loss-of-function mutations in Notch receptors, which transmit signals that regulate the orderly differentiation of normal squamous epithelia. In addition to inducing mutations, UV light (UVB in particular) may have a transient immunosuppressive effect on skin by impairing antigen presentation by Langerhans cells. This effect may contribute to tumorigenesis by weakening immunosurveillance. Patients who are immunosuppressed as a result of chemotherapy or organ transplantation, or who have xeroderma pigmentosum, are at increased risk. Tumors in immunosuppressed persons, particularly organ transplant recipients, are likely to be associated with HPV infection. TP53 mutations caused by UV light–induced DNA damage are common, as are activating mutations in HRAS. As with squamous cell carcinomas at other sites, those in the skin may be preceded by in situ lesions.
Morphology
Squamous cell carcinomas in situ appear as sharply defined, red, scaling plaques; many arise from prior actinic keratoses. More advanced, invasive lesions are nodular, show variable scale, and may ulcerate (Fig. 23–16, A). Microscopically, squamous cell carcinoma in situ is characterized by highly atypical cells at all levels of the epidermis, with nuclear crowding and disorganization. Invasive tumors, defined by penetration of the basement membrane (Fig. 23–16, B), show variable degrees of differentiation, ranging from tumors with cells arranged in orderly lobules that exhibit extensive keratinization to neoplasms consisting of highly anaplastic cells with foci of necrosis and only abortive, single-cell keratinization (dyskeratosis).
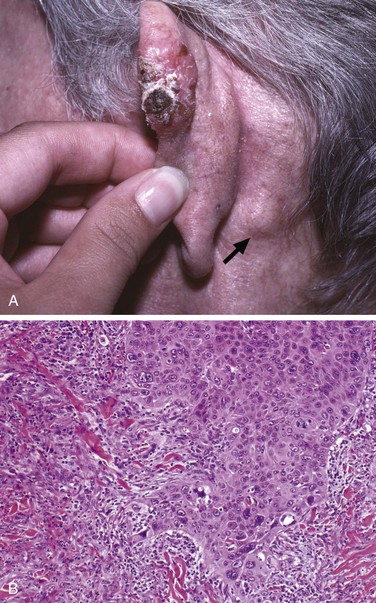
Figure 23–16 Invasive squamous cell carcinoma. A, A nodular, hyperkeratotic lesion occurring on the ear, associated with metastasis to a prominent postauricular lymph node (arrow). B, The tumor invades the dermis infiltrating collagen as irregular projections of atypical squamous cells, which in this case exhibit acantholysis.
Clinical Features
Invasive squamous cell carcinomas of the skin often are discovered while small and resectable. Less than 5% have metastasized to regional nodes at diagnosis. The likelihood of metastasis is related to the thickness of the lesion and degree of invasion into the subcutis. Tumors arising in the context of actinic keratoses may be locally aggressive but generally metastasize only after long periods of time, while those arising in burn scars, ulcers, and non–sun-exposed skin behave less predictably. Mucosal squamous cell carcinomas (oral, pulmonary, esophageal, etc.) generally are much more aggressive.
Basal Cell Carcinoma
Basal cell carcinoma is a common slow-growing cancer that rarely metastasizes. It tends to occur at sites subject to chronic sun exposure and in lightly pigmented individuals.
Pathogenesis
Basal cell carcinoma is associated with dysregulation of the Hedgehog pathway. Inherited defects in the PTCH gene, a tumor suppressor that regulates Hedgehog pathway signaling, cause familial basal cell carcinomas in Gorlin syndrome. The Hedgehog pathway is an important regulator of embryonic development, and subtle developmental anomalies are also often noted in affected persons. Some component of the hedgehog pathway is mutated in the great majority of sporadic basal cell carcinomas as well. Mutations in TP53 are also common in both familial and sporadic tumors.
Morphology
Grossly, basal cell carcinomas manifest as pearly papules, often with prominent, dilated subepidermal blood vessels (telangiectasia) (Fig. 23–17, A). Some tumors contain melanin pigment and thus appear similar to melanocytic nevi or melanomas. Microscopically, the tumor cells resemble the normal epidermal basal cell layer from which they are derived. Because they may arise from either the epidermis or the follicular epithelium, they are not encountered on mucosal surfaces. Two common patterns are seen: multifocal growths originating from the epidermis (superficial pattern), or nodular lesions growing downward into the dermis as cords and islands of variably basophilic cells with hyperchromatic nuclei, embedded in a fibrotic or mucinous stromal matrix (Fig. 23–17, B). Peripheral tumor cell nuclei align in the outermost layer (a pattern termed palisading), which often separates from the stroma, creating a characteristic cleft (Fig. 23–17, C).
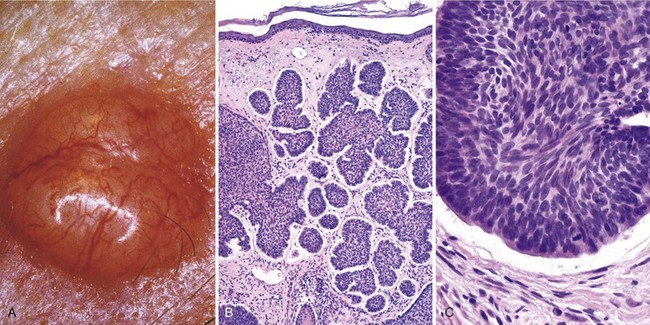
Figure 23–17 Basal cell carcinoma. A, A prototypical pearly, smooth-surfaced papule with associated telangiectatic vessels. B, The tumor is composed of nests of basaloid cells infiltrating a fibrotic stroma. C, The tumor cells have scant cytoplasm and small hyperchromatic nuclei that palisade on the outside of the nest. The cleft between the tumor cells and the stroma is a highly characteristic artifact of sectioning.
Clinical Features
It is estimated that in excess of 1 million basal cell carcinomas are treated in the United States annually. By far the most important risk factor is sun exposure; basal cell carcinoma is more common in warm southern regions of the United States, and its incidence is 40-fold higher in sunny climates near the equator, such as Australia, than it is in Northern European locales. Individual tumors usually are cured by local excision, but approximately 40% of patients will develop another basal cell carcinoma within 5 years. Advanced lesions may ulcerate, and extensive local invasion of bone or facial sinuses may occur if the lesions are neglected for many years.
Summary
Malignant Epidermal Tumors
• The incidence of both basal cell and squamous cell carcinoma is strongly correlated with increasing lifetime sun exposure.
• Cutaneous squamous cell carcinoma can progress from actinic keratoses but also arises from chemical exposure, at thermal burn sites, or in association with HPV infection in the setting of immunosuppression.
• Cutaneous squamous cell carcinoma has potential for metastasis but is much less aggressive than squamous cell carcinoma at mucosal sites.
• Basal cell carcinoma, the most common malignancy worldwide, is a locally aggressive tumor associated with mutations in the Hedgehog pathway. Metastasis is very rare.
Melanocytic Proliferations
Melanocytic Nevi
Strictly speaking, the term nevus denotes any congenital lesion of the skin. Melanocytic nevus, however, refers to any benign congenital or acquired neoplasm of melanocytes.
Pathogenesis
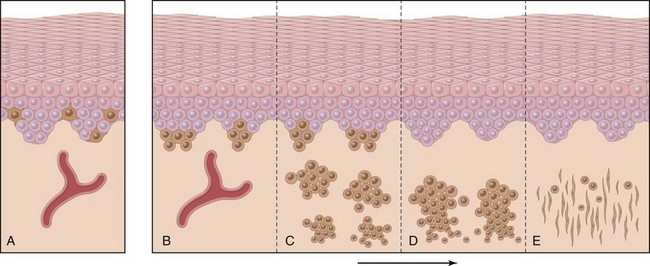
Melanocytic nevi are benign neoplasms derived from melanocytes, highly dendritic, pigment-producing cells that are normally interspersed among basal keratinocytes. Progressive growth and migration of nevus cells from the dermoepidermal junction into the underlying dermis is accompanied by changes that are taken as evidence of cellular senescence (Fig. 23–18). Superficial nevus cells are larger and tend to produce melanin pigment and grow in nests; deeper nevus cells are smaller, produce little or no pigment, and grow in cords or single cells. The deepest nevus cells have fusiform contours and grow in fascicles. This sequence of morphologic changes is of diagnostic importance, since they are absent from melanomas. The majority of benign nevi have an activating mutation in BRAF, which encodes a serine/threonine kinase downstream from RAS in the extracellular regulated kinase (ERK) pathway, or less commonly in NRAS itself. Experimental evidence suggests that unbridled BRAF/RAS signaling initially induces melanocytic proliferation followed by senescence. How these opposing effects are coordinated is unclear, but it is believed that the “brake” on proliferation provided by induced senescence explains why very few nevi transform into malignant melanomas.
Morphology
Common melanocytic nevi are tan-to-brown, uniformly pigmented, small papules (5 mm or less across) with well-defined, rounded borders (Fig. 23–19, A). Early lesions are composed of round-to-oval cells that grow in “nests” along the dermoepidermal junction. Nuclei are uniform and round, and contain inconspicuous nucleoli with little or no mitotic activity. Such early-stage lesions are called junctional nevi. Eventually, most junctional nevi grow into the underlying dermis as nests or cords of cells (compound nevi), and in older lesions the epidermal nests may be lost entirely, creating an intradermal nevi (Fig. 23–19, B).
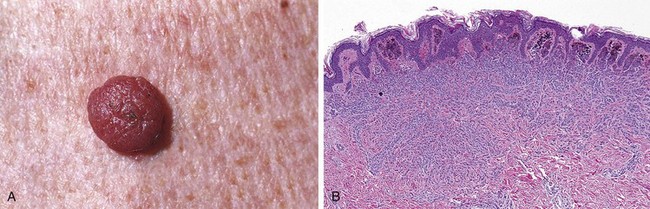
Figure 23–19 Melanocytic nevus. A, Melanocytic nevi are relatively small, symmetric, and uniformly pigmented. B, This nevus shows rounded melanocytes that lose their pigmentation and become smaller and more separated as they extend into the dermis—all signs of cellular senescence that speak to the benign nature of the proliferation.
Clinical Features
There are numerous types of melanocytic nevi, with varied appearances. Although these lesions usually are of only cosmetic concern, they can become irritating or mimic melanoma, requiring their surgical removal. Compound and intradermal nevi often are more elevated than junctional nevi.
Dysplastic Nevus
Dysplastic nevi may be sporadic or familial. The latter are important clinically because they are considered potential precursors of melanoma. As with conventional melanocytic nevi, activating NRAS or BRAF mutations are commonly found in dysplastic nevi and are believed to have a pathogenic role.
Morphology
Dysplastic nevi are larger than most acquired nevi (often more than 5 mm across) and may number in the hundreds (Fig. 23–20, A). They are flat macules to slightly raised plaques, with a “pebbly” surface. They usually have variable pigmentation (variegation) and irregular borders (Fig. 23–20, A, inset).
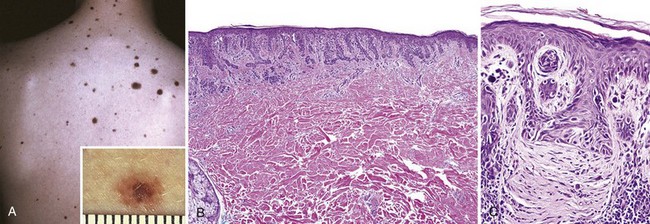
Figure 23–20 Dysplastic nevus. A, Numerous irregular nevi on the back of a patient with the dysplastic nevus syndrome. The lesions usually are greater than 5mm in diameter and have irregular borders and variable pigmentation (inset). B, Compound dysplastic nevi feature a central dermal component with an asymmetric “shoulder” of exclusively junctional melanocytes (lentiginous hyperplasia). The former corresponds to the more pigmented and raised central zone (see A, inset), and the latter, to the less pigmented flat peripheral rim. C, Other important features are cytologic atypia (irregular, dark-staining nuclei) and characteristic parallel bands of fibrosis—part of the host response to these lesions.
Microscopically, dysplastic nevi are mostly compound nevi that exhibit both architectural and cytologic evidence of abnormal growth. Nevus cell nests within the epidermis may be enlarged and exhibit abnormal fusion or coalescence with adjacent nests (bridging). As part of this process, single nevus cells begin to replace the normal basal cell layer along the dermoepidermal junction, producing so-called lentiginous hyperplasia (Fig. 23–20, B). Cytologic atypia consisting of irregular, often angulated, nuclear contours and hyperchromasia is frequently observed (Fig. 23–20, B and C). Associated alterations also occur in the superficial dermis. These consist of a sparse lymphocytic infiltrate, release of melanin pigment that is phagocytosed by dermal macrophages (melanin incontinence), and linear fibrosis surrounding epidermal nests of melanocytes. These dermal changes are elements of the host response to these lesions.
Clinical Features
Unlike ordinary nevi, dysplastic nevi have a tendency to occur on body surfaces not exposed to the sun as well as on sun-exposed sites. Familial dysplastic nevus syndrome is strongly associated with melanoma, as the lifetime risk for the development of melanoma in affected persons is close to 100%. In sporadic cases, only individuals with 10 or more dysplastic nevi appear to be at an increased risk for melanoma. Transformation of dysplastic nevus to melanoma has been documented, both clinically and histologically. However, such cases are the exception, as most melanomas arise de novo and not from a preexisting nevus. Thus, the likelihood that any particular nevus, dysplastic or otherwise, will develop into melanoma is exceedingly low, and these lesions are best viewed as markers of melanoma risk.
Melanoma
Melanoma is less common but much more deadly than basal or squamous cell carcinoma. Today, as a result of increased public awareness of the earliest signs of skin melanomas, most melanomas are cured surgically. Nonetheless, the incidence of these lesions has increased dramatically over the past several decades, at least in part as a result of increasing sun exposure and/or higher detection rates resulting from vigorous surveillance.
Pathogenesis
As with other cutaneous malignancies, sunlight plays an important role in the development of melanoma. The incidence is highest in sun-exposed skin and in geographic locales such as Australia where sun exposure is high and much of the population is fair-skinned. Intense intermittent exposure at an early age is particularly harmful. Recent “deep sequencing” studies have confirmed that tumor genomes contain thousands of acquired mutations, most bearing a signature consistent with UV-induced DNA damage. Sunlight, however, is not the only predisposing factor; hereditary predisposition also plays a role, as already discussed under familial dysplastic nevus syndrome.
As with other cancers, it is believed that melanoma may arise in a stepwise fashion from precursor lesions (Fig. 23–21). Key phases of tumor development are marked by radial and vertical growth. Radial growth describes the initial tendency of a melanoma to grow horizontally within the epidermis (in situ), often for a prolonged period (Fig. 23–21, D). During this stage, melanoma cells do not have the capacity to metastasize, and do not induce angiogenesis. With time, a vertical growth phase supervenes, in which the tumor grows downward into the deeper dermal layers as an expansile mass lacking cellular maturation (Fig. 23–21, E). This event often is heralded by the development of a nodule in a previously flat lesion (Fig. 23-22, A) and correlates with the emergence of a clone of cells with metastatic potential.
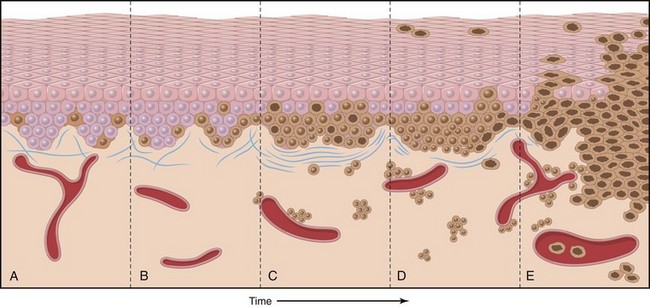
Figure 23–21 Possible steps in development of melanoma. A, Normal skin shows only scattered melanocytes. B, Lentiginous melanocytic hyperplasia. C, Lentiginous compound nevus with abnormal architecture and cytologic features (dysplastic nevus). D, Early or radial growth phase melanoma (large dark cells in epidermis) arising in a nevus. E, Melanoma in vertical growth phase with metastatic potential. Note that no melanocytic nevus precursor is identified in most cases of melanoma. They are believed to arise de novo, perhaps all using the same pathway.
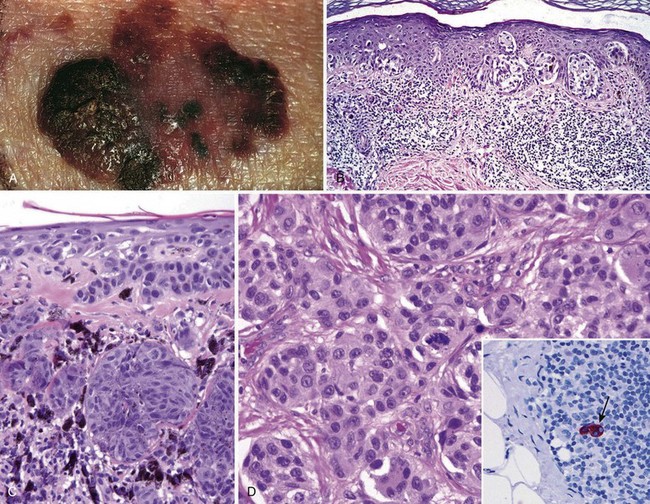
Figure 23–22 Melanoma. A, On clinical evaluation, lesions tend to be larger than nevi, with irregular contours and pigmentation. Macular areas indicate early superficial (radial) growth, while elevated areas often indicate dermal invasion (vertical growth). B, Radial growth phase, with spread of nested and single-cell melanoma cells within the epidermis. C, Vertical growth phase, with nodular aggregates of infiltrating tumor cells within the dermis (epidermis is on the right). D, Melanoma cells have hyperchromatic nuclei of irregular size and shape with prominent nucleoli. Mitoses, including atypical forms such as seen in the center of this field, often are encountered. The inset shows a sentinel lymph node containing a tiny cluster of metastatic melanoma cells (arrow), detected by staining for the melanocytic marker HMB-45.
Most melanomas occur sporadically, but a few are hereditary (with reported rates ranging from less than 5% to 10%). Molecular genetic analysis of familial and sporadic cases has provided important insights into the pathogenesis of melanoma. As with other tumors, malignant transformation of melanocytes is a multistep process that involves activating mutations in proto-oncogenes and loss-of-function mutations in tumor suppressor genes. Germline mutations in the CDKN2A gene (located on 9p21) are found in as many as 40% of the rare individuals who suffer from familial melanoma. This gene encodes the tumor suppressor p16, a cyclin-dependent kinase inhibitor that regulates the G1-S transition of the cell cycle in a retinoblastoma protein–dependent fashion (Chapter 5). The CDNK2A gene also is silenced in some sporadic tumors by methylation. Somatic activating mutations in the proto-oncogenes BRAF or NRAS are observed in a high proportion of melanomas. These mutations, which promote cellular proliferation and survival by activating the extracellular signal–regulated protein kinase (ERK) pathway, generally are mutually exclusive, since BRAF functions downstream of RAS. Also frequently seen is loss of function of the tumor suppressor PTEN, an important negative regulator of PI3K-AKT pathway, which also promotes growth and survival. Some melanomas, particular those arising in acral and mucosal sites, have activating mutations in the c-Kit receptor tyrosine kinase. Agents that selectively inhibit mutant BRAF and c-Kit have produced dramatic responses in patients with metastatic tumors with BRAF and c-Kit mutations, respectively, an encouraging example of molecularly targeted therapy in an otherwise hopeless disease.
Morphology
Unlike benign nevi, melanomas exhibit striking variations in pigmentation, including shades of black, brown, red, dark blue, and gray (Fig. 23–22, A). The borders are irregular and often “notched.”
Microscopically, malignant cells grow as poorly formed nests or as individual cells at all levels of the epidermis (page-toid spread) and in expansile dermal nodules; these constitute the radial and vertical growth phases, respectively (Fig. 23–22, B and C). Of note, superficial spreading melanomas are often associated with a brisk lymphocytic infiltrate (Fig. 23–22, B), a feature that may reflect a host response to tumor-specific antigens. The nature and extent of the vertical growth phase determine the biologic behavior of melanomas. By recording and using these and other variables in aggregate, accurate prognostication is possible.
Individual melanoma cells usually are considerably larger than nevus cells. They have large nuclei with irregular contours, chromatin that is characteristically clumped at the periphery of the nuclear membrane, and prominent “cherry red” eosinophilic nucleoli (Fig. 23–22, D). Immunohistochemical stains can be helpful in identifying metastatic deposits (Fig. 23–22, D, inset).
Clinical Features
Although most of these lesions arise in the skin, they also may involve the oral and anogenital mucosal surfaces, the esophagus, the meninges, and the eye. The following comments apply to cutaneous melanomas.
Melanoma of the skin usually is asymptomatic, although pruritus may be an early manifestation. The most important clinical sign is a change in the color or size of a pigmented lesion. The main clinical warning signs are
1 Rapid enlargement of a preexisting nevus
3 Development of a new pigmented lesion during adult life
These principles are expressed in the so-called ABCs of melanoma: asymmetry, border, color, diameter, and evolution (change of an existing nevus). It is vitally important to recognize melanomas and intervene as rapidly as possible. The vast majority of superficial lesions are curable surgically, while metastatic melanoma has a very poor prognosis.
The probability of metastasis is predicted by measuring the depth of invasion in millimeters of the vertical growth phase nodule from the top of the granular cell layer of the overlying epidermis (Breslow thickness). Metastasis risk also is increased in tumors with a high mitotic rate and in those that fail to induce a local immune response. When metastases occur, they involve not only regional lymph nodes but also liver, lungs, brain, and virtually any other site that can be seeded hematogenously. Sentinel lymph node biopsy (of the first draining node[s] of a primary melanoma) at the time of surgery provides additional information on biologic aggressiveness.
In some cases, metastases may appear for the first time many years after complete surgical excision of the primary tumor, suggesting a long phase of dormancy, during which time the tumor may be held in check by the host immune response. Recognition of the likely role of the host immune response has led to therapeutic trials of immunomodulators. Some impressive responses in patients with advanced melanoma have been seen, especially to antibodies that block endogenous inhibitors of immune responses such as CTLA-4 and PD-1, and thus “release the brakes” on host antitumor immunity.
Summary
Melanocytic Lesions, Benign and Malignant
• Most melanocytic nevi have activating mutations in BRAF or less often NRAS, but the vast majority never undergo malignant transformation.
• Most sporadic dysplastic nevi are best regarded as markers of melanoma risk rather than premalignant lesions. They are characterized by architectural and cytologic atypia.
• Melanoma is a highly aggressive malignancy; tumors only a few millimeters in thickness can give rise to deadly metastases.
• In most cases, melanoma progresses from an intraepithelial (in situ) to an invasive (dermal) form. Characteristics of the dermal tumor such as depth of invasion and mitotic activity correlate with survival.
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135. [A modified classification of melanoma based on both clinical and genetic features. Such molecular classification schemes are critical for progress in targeted therapy.]
Elder DE. Dysplastic nevi: an update. Histopathology. 2010;56:112. [Balanced presentation of the histology and pathogenesis of dysplastic nevi and their relationship to melanoma.]
Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743. [Epidemiology, clinical presentation, molecular pathogenesis, and novel treatment options are succinctly reviewed.]
Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol. 2009;4:551. [The genetic pathways relevant to melanoma suggest future therapeutic interventions.]
Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev. 2004;4:211. [Lymphocytic subtypes and targeting in relation to cutaneous inflammatory diseases providing insight into common pathogenic features.]
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496. [Pathogenesis, clinical features, and new targeted treatment options are discussed.]
Khavari PA. Modelling cancer in human skin tissue. Nat Rev Cancer. 2006;6:270. [Models of human epidermal carcinogenesis indicate that multiple mutations in specific pathways are required for malignant transformation.]
Tsai KY, Tsao H. The genetics of skin cancer. Am J Med Genet. 2004;131C:82. [The genetic bases for skin malignancies are presented along with their associations with predisposing human genetic syndromes that provide insight into their pathogenesis.]
Ujiie H, Shibaki A, Nishie W, Shimizu H. What’s new in bullous pemphigoid. J Dermatol. 2010;37:194. [Recent review of bullous pemphigoid pathogenesis.]
Yokoyama T, Amagai M. Immune dysregulation of pemphigus in humans and mice. J Dermatol. 2010;37:205. [A review of immune disturbances that may underlie pemphigus.]