Chapter 4 Diseases of the Immune System
See Targeted Therapy available online at studentconsult.com
Immunity refers to protection against infections, and the immune system is the collection of cells and molecules that are responsible for defending the body against the countless pathogenic microbes in the environment. Deficiencies in immune defenses result in an increased susceptibility to infections, which can be life-threatening if the deficits are not corrected. On the other hand, the immune system is itself capable of causing great harm and is the root cause of some of the most vexing and intractable diseases of the modern world. Thus, diseases of immunity range from those caused by “too little” to those caused by “too much or inappropriate” immune activity.
This chapter starts with a brief review of some of the basic concepts of lymphocyte biology and normal immune responses, which establishes a foundation for the subsequent discussions of diseases caused by excessive or inappropriate immune responses, rejection of organ transplants and immune deficiency disorders. The chapter concludes with a discussion of amyloidosis, a disease characterized by the abnormal extracellular deposition of certain proteins (some of which are produced in the setting of immune responses).
Innate and Adaptive Immunity
Defense against microbes consists of two types of reactions (Fig. 4–1). Innate immunity (also called natural, or native, immunity) is mediated by cells and proteins that are always present and poised to fight against microbes, being called into action immediately in response to infection. The major components of innate immunity are epithelial barriers of the skin, gastrointestinal tract, and respiratory tract, which prevent microbe entry; phagocytic leukocytes (neutrophils and macrophages); a specialized cell type called the natural killer (NK) cell; and several circulating plasma proteins, the most important of which are the proteins of the complement system.
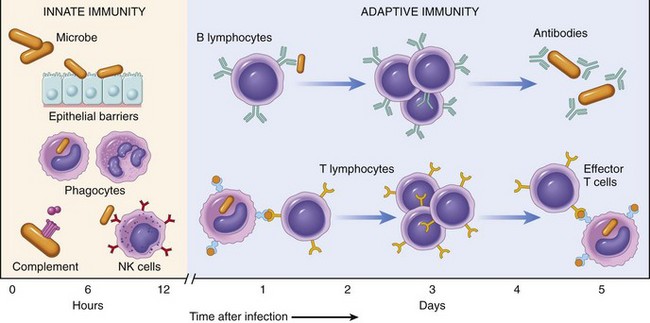
The innate immune response is able to prevent and control many infections. However, many pathogenic microbes have evolved to overcome the early defenses, and protection against these infections requires the more specialized and powerful mechanisms of adaptive immunity (also called acquired, or specific, immunity). Adaptive immunity is normally silent and responds (or “adapts”) to the presence of infectious microbes by becoming active, expanding, and generating potent mechanisms for neutralizing and eliminating the microbes. The components of the adaptive immune system are lymphocytes and their products. By convention, the terms “immune system” and “immune response” refer to adaptive immunity.
There are two types of adaptive immune responses: humoral immunity, mediated by soluble proteins called antibodies that are produced by B lymphocytes (also called B cells), and cell-mediated (or cellular) immunity, mediated by T lymphocytes (also called T cells). Antibodies provide protection against extracellular microbes in the blood, mucosal secretions, and tissues. T lymphocytes are important in defense against intracellular microbes. They work by either directly killing infected cells (accomplished by cytotoxic T lymphocytes) or by activating phagocytes to kill ingested microbes, via the production of soluble protein mediators called cytokines (made by helper T cells). The main properties and functions of the cells of the immune system are described in the next section.
When the immune system is inappropriately triggered or not properly controlled, the same mechanisms that are involved in host defense cause tissue injury and disease. The reaction of the cells of innate and adaptive immunity may be manifested as inflammation. As discussed in Chapter 2, inflammation is a beneficial process, but it is also the basis for many human diseases. Presented later in this chapter is an overview of the ways in which the adaptive immune response triggers pathologic inflammatory reactions.
Cells and Tissues of the Immune System
The cells of the immune system consist of lymphocytes, which recognize antigens and mount adaptive immune responses; specialized antigen-presenting cells (APCs), which capture and display microbial and other antigens to the lymphocytes; and various effector cells, whose function is to eliminate microbes and other antigens. Two remarkable features of the immune system are the specialization of the cells to perform diverse functions, and the precise control mechanisms that permit useful responses when needed and prevent potentially harmful ones.
Lymphocytes
Lymphocytes are present in the circulation and in various lymphoid organs. Although all lymphocytes appear morphologically identical, there are actually several functionally and phenotypically distinct lymphocyte populations. Lymphocytes develop from precursors in the generative lymphoid organs; T lymphocytes are so called because they mature in the thymus, whereas B lymphocytes mature in the bone marrow. Each T or B lymphocyte expresses receptors for a single antigen, and the total population of lymphocytes (numbering about 1012 in humans) is capable of recognizing tens or hundreds of millions of antigens. This enormous diversity of antigen recognition is generated by the somatic rearrangement of antigen receptor genes during lymphocyte maturation, and variations that are introduced during the joining of different gene segments to form antigen receptors. These antigen receptors are rearranged and expressed in lymphocytes but not in any other cell. Therefore, the demonstration of antigen receptor gene rearrangements by molecular methods (e.g., polymerase chain reaction [PCR] assay) is a definitive marker of T or B lymphocytes. Because each lymphocyte has a unique DNA rearrangement (and hence a unique antigen receptor), molecular analysis of the rearrangements in cell populations can distinguish polyclonal (non-neoplastic) lymphocyte proliferations from monoclonal (neoplastic) expansions. Such analyses are used in the diagnosis of lymphoid malignancies (Chapter 11).
T Lymphocytes
Thymus-derived, or T, lymphocytes are the effector cells of cellular immunity and the “helper cells” for antibody responses to protein antigens. T cells constitute 60% to 70% of the lymphocytes in peripheral blood and are the major lymphocyte population in splenic periarteriolar sheaths and lymph node interfollicular zones. T cells do not detect free or circulating antigens. Instead, the vast majority (greater than 95%) of T cells recognize only peptide fragments of protein antigens bound to proteins of the major histocompatibility complex (MHC). The MHC was discovered on the basis of studies of graft rejection and acceptance (tissue, or “histo,” compatibility). It is now known that the normal function of MHC molecules is to display peptides for recognition by T lymphocytes. By forcing T cells to see MHC-bound peptides on cell surfaces the system ensures that T cells can recognize antigens displayed by other cells. T cells function by interacting with other cells—either to kill infected cells or to activate phagocytes or B lymphocytes that have ingested protein antigens. In each person, T cells recognize only peptides displayed by that person’s MHC molecules, which, of course, are the only MHC molecules that the T cells normally encounter. This phenomenon is called MHC restriction. Peptide antigens presented by self MHC molecules are recognized by the T cell receptor (TCR), which is a heterodimer composed of disulfide-linked α and β protein chains (Fig. 4–2, A); each chain has a variable region that participates in binding a particular peptide antigen and a constant region that interacts with associated signaling molecules.
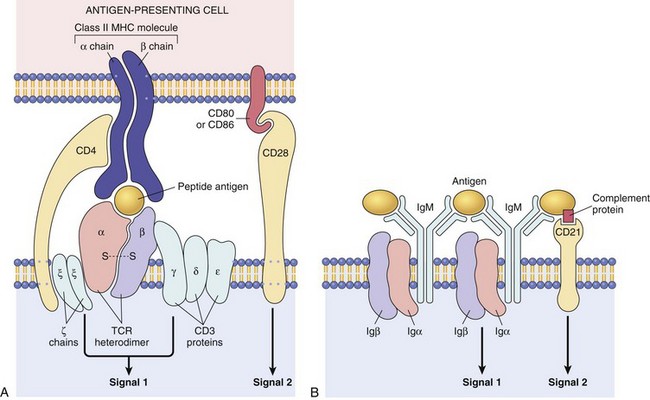
Figure 4–2 Lymphocyte antigen receptors. A, The T cell receptor (TCR) complex and other molecules involved in T cell activation. The TCRα and TCRβ chains recognize antigen (in the form of peptide–MHC complexes expressed on antigen-presenting cells), and the linked CD3 complex initiates activating signals. CD4 and CD28 are also involved in T cell activation. (Note that some T cells express CD8 and not CD4; these molecules serve analogous roles.) B, The B cell receptor complex is composed of membrane IgM (or IgD, not shown) and the associated signaling proteins Igα and Igβ. CD21 is a receptor for a complement component that promotes B cell activation. Ig, immunoglobulin; MHC, major histocompatibilty complex.
TCRs are noncovalently linked to a cluster of five invariant polypeptide chains, the γ, δ, and ε proteins of the CD3 molecular complex and two ζ chains (Fig. 4–2, A). The CD3 proteins and ζ chains do not themselves bind antigens; instead, they are attached to the TCR and deliver intracellular biochemical signals after TCR recognition of antigen. In addition to these signaling proteins, T cells express a number of other invariant molecules that serve diverse functions. CD4 and CD8 are expressed on distinct T cell subsets and serve as coreceptors for T cell activation. During antigen recognition, CD4 molecules on T cells bind to invariant portions of class II MHC molecules (see later) on selected APCs; in an analogous fashion, CD8 binds to class I MHC molecules. CD4 is expressed on 50%–60% of mature T cells, whereas CD8 is expressed on about 40% of T cells. The CD4- and CD8-expressing T cells—called CD4+ and CD8+ cells, respectively—perform different but overlapping functions. CD4+ T cells are “helper” T cells because they secrete soluble molecules (cytokines) that help B cells to produce antibodies (the origin of the name “helper” cells) and also help macrophages to destroy phagocytosed microbes. The central role of CD4+ helper cells in immunity is highlighted by the severe compromise that results from the destruction of this subset by human immunodeficiency virus (HIV) infection. CD8+ T cells can also secrete cytokines, but they play a more important role in directly killing virus-infected or tumor cells, and hence are called “cytotoxic” T lymphocytes (CTLs). Other important invariant proteins on T cells include CD28, which functions as the receptor for molecules that are induced on APCs by microbes (and are called costimulators), and various adhesion molecules that strengthen the bond between the T cells and APCs and control the migration of the T cells to different tissues.
In a minority of peripheral blood T cells and in many of the T cells associated with mucosal surfaces (e.g., lung, gastrointestinal tract), the TCRs are heterodimers of γ and δ chains, which are similar but not identical to the α and β chains of most TCRs. Such γδ T cells, which do not express CD4 or CD8, recognize nonprotein molecules (e.g., bacterial lipoglycans), but their functional roles are not well understood. Another small population of T cells expresses markers of T cells and NK cells. These so-called NKT cells recognize microbial glycolipids, and may play a role in defense against some infections. The antigen receptors of NKT cells are much less diverse than the receptors of “conventional” T cells, suggesting that the former recognize conserved microbial structures.
Another population of T cells that functions to suppress immune responses is that of regulatory T lymphocytes. This cell type is described later, in the context of tolerance of self antigens.
Major Histocompatibility Complex Molecules: The Peptide Display System of Adaptive Immunity
Because MHC molecules are fundamental to T cell recognition of antigens, and because genetic variations in MHC molecules are associated with immunologic diseases, it is important to review the structure and function of these molecules. The human MHC, known as the human leukocyte antigen (HLA) complex, consists of a cluster of genes on chromosome 6 (Fig. 4–3). The HLA system is highly polymorphic; that is, there are several alternative forms (alleles) of a gene at each locus (estimated to number about 3500 for all HLA genes and about 1100 for HLA-B alleles alone). Such diversity provides a system whereby a vast range of peptides can be displayed by MHC molecules for recognition by T cells. As we shall see, this polymorphism also constitutes a formidable barrier to organ transplantation.
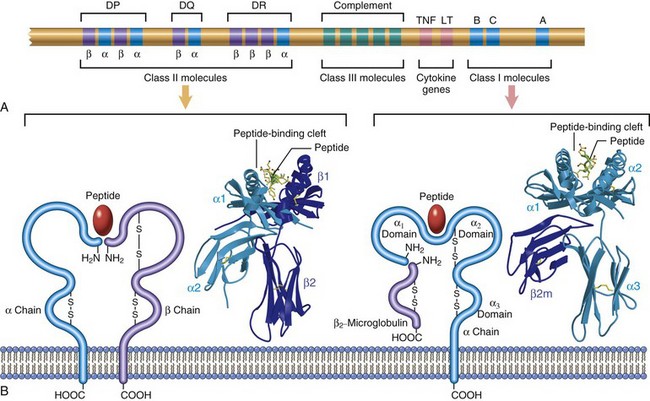
Figure 4–3 The human leukocyte antigen (HLA) complex and the structure of HLA molecules. A, The location of genes in the HLA complex. The sizes and distances between genes are not to scale. The class II region also contains genes that encode several proteins involved in antigen processing (not shown). B, Schematic diagrams and crystal structures of class I and class II HLA molecules. LT, lymphotoxin; TNF, tumor necrosis factor.
(Crystal structures are courtesy of Dr. P. Bjorkman, California Institute of Technology, Pasadena, California.)
On the basis of their chemical structure, tissue distribution, and function, MHC gene products fall into two main categories:
• Class I MHC molecules are encoded by three closely linked loci, designated HLA-A, HLA-B, and HLA-C (Fig. 4–3). Each of these molecules is a heterodimer, consisting of a polymorphic 44-kDa α chain noncovalently associated with an invariant 12-kDa β2-microglobulin polypeptide (encoded by a separate gene on chromosome 15). The extracellular portion of the α chain contains a cleft where the polymorphic residues are located and where foreign peptides bind to MHC molecules for presentation to T cells, and a conserved region that binds CD8, ensuring that only CD8+ T cells can respond to peptides displayed by class I molecules. In general, class I MHC molecules bind and display peptides derived from proteins synthesized in the cytoplasm of the cell (e.g., viral antigens). Because class I MHC molecules are present on all nucleated cells, all virus-infected cells can be detected and eliminated by CD8+ CTLs.
• Class II MHC molecules are encoded by genes in the HLA-D region, which contains at least three subregions: DP, DQ, and DR. Class II MHC molecules are heterodimers of noncovalently linked polymorphic α and β subunits (Fig. 4–3). The extracellular portion of the class II MHC heterodimer contains a cleft for the binding of antigenic peptides and a region that binds CD4. Class II MHC expression is restricted to a few types of cells, mainly APCs (notably, dendritic cells [DCs]), macrophages, and B cells. In general, class II MHC molecules bind to peptides derived from proteins synthesized outside the cell (e.g., those derived from extracellular bacteria) and ingested into the cell. This property allows CD4+ T cells to recognize the presence of extracellular pathogens and to orchestrate a protective response.
• Several other proteins are encoded in the MHC locus, some of which have been called “class III molecules.” These include complement components (C2, C3, and Bf) and the cytokines tumor necrosis factor (TNF) and lymphotoxin. These molecules do not form a part of the peptide display system and are not discussed further.
Each person inherits one HLA allele from each parent; typically, then, two different molecules are expressed for every HLA locus. Cells of a heterozygous person can therefore express six different class I HLA molecules: three of maternal origin and three of paternal origin. Similarly, a given individual expresses maternal and paternal alleles of the class II MHC loci; because some HLA-D α and β chains can mix and match with each other, each class II–expressing cell can have as many as 20 different class II MHC molecules. Different MHC alleles bind to different peptide fragments; the expression of many different MHC molecules allows each cell to present a wide array of peptide antigens.
As a result of the polymorphism at the major HLA loci in the population, a virtually infinite number of combinations of molecules exist, and each person expresses a unique MHC antigenic profile on his or her cells. The combination of HLA alleles for each person is called the HLA haplotype. The implications of HLA polymorphism are obvious in the context of transplantation—because each person has HLA alleles that differ to some extent from every other person’s, grafts from virtually any donor will evoke immune responses in the recipient and be rejected (except, of course, for identical twins). In fact, HLA molecules were discovered in the course of early attempts at tissue transplantation. HLA molecules of the graft evoke both humoral and cell-mediated responses, eventually leading to graft destruction (as discussed later in this chapter). This ability of MHC molecules to trigger immune responses is the reason these molecules are often called “antigens.” It is believed that the polymorphism of MHC genes arose to enable display of and response to any conceivable microbial peptide encountered in the environment.
The role of the MHC in T cell stimulation also has important implications for the genetic control of immune responses. The ability of any given MHC allele to bind the peptide antigens generated from a particular pathogen will determine whether a specific person’s T cells can actually “see” and respond to that pathogen. The inheritance of particular alleles influences both protective and harmful immune responses. For example, if the antigen is ragweed pollen and the response is an allergic reaction, inheritance of some HLA genes may make individuals susceptible to “hay fever,” the colloquial name for ragweed allergy. On the other hand, responsiveness to a viral antigen, determined by inheritance of certain HLA alleles, may be beneficial for the host.
Finally, many autoimmune diseases are associated with particular HLA alleles. We return to a discussion of HLA associations with diseases when we consider autoimmunity.
B Lymphocytes
Bone marrow–derived B lymphocytes are the cells that produce antibodies and are thus the effector cells of humoral immunity. B cells make up 10% to 20% of the circulating peripheral lymphocyte population. They also are present in bone marrow and in the follicles of peripheral lymphoid tissues (lymph nodes, spleen, tonsils, and other mucosal tissues).
B cells recognize antigen by means of membrane-bound antibody of the immunoglobulin M (IgM) class, expressed on the surface together with signaling molecules to form the B cell receptor (BCR) complex (Fig. 4–2, B). Whereas T cells can recognize only MHC-associated peptides, B cells can recognize and respond to many more chemical structures, including soluble or cell-associated proteins, lipids, polysaccharides, nucleic acids, and small chemicals; furthermore, B cells (and antibodies) recognize native (properly folded) forms of these antigens. As with TCRs, each antibody has a unique antigen specificity. The diversity of antibodies is generated during somatic rearrangements of immunoglobulin genes. B cells express several invariant molecules that are responsible for signal transduction and for activation of the cells (Fig. 4–2, B). Some are the signaling molecules attached to the BCR; another example is CD21 (also known as the type 2 complement receptor, or CR2), which recognizes a complement breakdown product that frequently is deposited on microbes and promotes B cell responses to microbial antigens. Interestingly, the ubiquitous Epstein-Barr virus has cleverly evolved to use CD21 as a receptor for binding to B cells and infecting them.
After stimulation, B cells differentiate into plasma cells, which secrete large amounts of antibodies, the mediators of humoral immunity. There are five classes, or isotypes, of immunoglobulins: IgG, IgM, and IgA constitute more than 95% of circulating antibodies. IgA is the major isotype in mucosal secretions; IgE is present in the circulation at very low concentrations and also is found attached to the surfaces of tissue mast cells; and IgD is expressed on the surfaces of B cells but is not secreted. As discussed later, each isotype has characteristic abilities to activate complement or recruit inflammatory cells and thus plays a different role in host defense and disease states.
Natural Killer Cells
Natural killer (NK) cells are lymphocytes that arise from the common lymphoid progenitor that gives rise to T and B lymphocytes. However, NK cells are cells of innate immunity and do not express highly variable and clonally distributed receptors for antigens. Therefore, they do not have specificities as diverse as do T cells or B cells. NK cells have two types of receptors—inhibitory and activating. The inhibitory receptors recognize self class I MHC molecules, which are expressed on all healthy cells, whereas the activating receptors recognize molecules that are expressed or upregulated on stressed or infected cells or cells with DNA damage. Normally, the effects of the inhibitory receptors dominate over those of the activating receptors, thereby preventing activation of the NK cells. Infections (especially viral infections) and stress are associated with reduced expression of class I MHC molecules, thus releasing the NK cells from inhibition. At the same time, there is increased engagement of the activating receptors. The net result is that the NK cells are activated and the infected or stressed cells are killed and eliminated.
Antigen-Presenting Cells
The immune system contains several cell types that are specialized to capture microbial antigens and display these to lymphocytes. Foremost among these APCs are dendritic cells (DCs), the major cells for displaying protein antigens to naive T cells to initiate immune responses. Several other cell types present antigens to different lymphocytes at various stages of immune responses.
Dendritic Cells
Cells with dendritic morphology (i.e., with fine dendritic cytoplasmic processes) occur as two functionally distinct types. Dendritic cells (DCs), sometimes called interdigitating DCs, express high levels of class II MHC and T cell costimulatory molecules and function to capture and present antigens to T cells. DCs reside in and under epithelia, where they are strategically located to capture entering microbes; an example is the Langerhans cell of the epidermis. DCs also are present in the T cell zones of lymphoid tissues, where they present antigens to T cells circulating through these tissues, and in the interstitium of many nonlymphoid organs, such as the heart and lungs, where they are poised to capture the antigens of any invading microbes. One subset of DCs is called plasmacytoid DCs because of their resemblance to plasma cells. These cells are present in the blood and lymphoid organs, and are major sources of the antiviral cytokine type I interferon, produced in response to many viruses.
The second type of cells with dendritic morphology are follicular dendritic cells (FDCs). These cells are located in the germinal centers of lymphoid follicles in the spleen and lymph nodes. FDCs bear receptors for the Fc tails of IgG molecules and for complement proteins and hence efficiently trap antigens bound to antibodies and complement. These cells display antigens to activated B lymphocytes in lymphoid follicles and promote secondary antibody responses, but are not involved in capturing antigens for display to T cells.
Other Antigen-Presenting Cells
Macrophages ingest microbes and other particulate antigens and display peptides for recognition by T lymphocytes. These T cells in turn activate the macrophages to kill the microbes, the central reaction of cell-mediated immunity. B cells present peptides to helper T cells and receive signals that stimulate antibody responses to protein antigens.
Effector Cells
Many different types of leukocytes perform the ultimate task of the immune response, which is to eliminate infections. NK cells are front-line effector cells in that they can rapidly react against “stressed” cells. Antibody-secreting plasma cells are the effector cells of humoral immunity. T lymphocytes, both CD4+ helper T cells and CD8+ CTLs, are effector cells of cell-mediated immunity. These lymphocytes often function in host defense together with other cells. Macrophages, as described in Chapter 2, bind microbes that are coated with antibodies or complement and then phagocytose and destroy these microbes, thus serving as effector cells of humoral immunity. Macrophages also respond to signals from helper T cells, which improves their ability to destroy phagocytosed microbes, thus serving as effector cells of cellular immunity. T lymphocytes secrete cytokines that recruit and activate other leukocytes, such as neutrophils and eosinophils, and together these cell types function in defense against various pathogens.
Lymphoid Tissues
The lymphoid tissues of the body are divided into generative (primary) organs, where lymphocytes express antigen receptors and mature, and peripheral (secondary) lymphoid organs, where adaptive immune responses develop. The generative organs are the thymus and bone marrow, and the peripheral organs are the lymph nodes, spleen, and mucosal and cutaneous lymphoid tissues. Mature lymphocytes recirculate through the peripheral organs, hunting for microbial antigens that they can respond to. An important characteristic of these organs is that T and B lymphocytes are anatomically organized in a manner that facilitates the adaptive immune response, a process that is described later.
 Summary
Summary
Cells and Tissues of the Immune System
• Lymphocytes are the mediators of adaptive immunity and the only cells that produce specific and diverse receptors for antigens.
• T (thymus-derived) lymphocytes express TCRs that recognize peptide antigens displayed by MHC molecules on the surface of APCs.
• B (bone marrow–derived) lymphocytes express membrane-bound antibodies that recognize a wide variety of antigens. B cells are activated to become plasma cells, which secrete antibodies.
• NK cells kill cells that are infected by some microbes or are stressed and damaged beyond repair. NK cells express inhibitory receptors that recognize MHC molecules that are normally expressed on healthy cells, and are thus prevented from killing normal cells.
• APCs capture microbes and other antigens, transport them to lymphoid organs, and display them for recognition by lymphocytes. The most efficient APCs are DCs, which are located in epithelia and most tissues.
• The cells of the immune system are organized in tissues. Some of these tissues are the sites of mature lymphocyte production (the generative lymphoid organs, the bone marrow and thymus), while others are the sites of immune responses (the peripheral lymphoid organs, including lymph nodes, spleen, and mucosal lymphoid tissues).
Overview of Normal Immune Responses
The previous section described the major components of the immune system. This section summarizes the key features of normal immune responses. This overview will serve as a foundation for the subsequent discussions of diseases caused by deficient or uncontrolled immune responses.
The Early Innate Immune Response to Microbes
The principal barriers between hosts and their environment are the epithelia of the skin and the gastrointestinal and respiratory tracts. Infectious microbes usually enter through these routes and attempt to colonize the hosts. The mechanisms of innate immunity operate at every step in a microbe’s attempt to invade. At the site of entry, epithelia serve as physical barriers to infections and eliminate microbes through production of peptide antibiotics and the actions of intraepithelial lymphocytes. If microbes are able to survive and traverse these epithelia, they encounter phagocytes, including neutrophils, which are rapidly recruited from the blood into tissues, and macrophages, which live in tissues under epithelia. The function of these phagocytic cells is to ingest microbes and destroy them by producing microbicidal substances. In response to recognition of microbes, phagocytes, DCs, and many other cell types secrete proteins called cytokines (described later), which promote inflammation and microbial killing and enhance protective immune responses. Cells use several receptors to sense microbes; foremost among these are the Toll-like receptors (TLRs), so named because of homology with the Drosophila Toll protein, that recognize bacterial and viral components (Chapter 2). NK cells kill virus-infected cells and produce the macrophage-activating cytokine IFN-γ. If the microbes enter the blood, many plasma proteins, including the proteins of the complement system, recognize the microbes and are activated, and their products kill microbes and coat (opsonize) the microbes for phagocytosis. In addition to combating infections, innate immune responses stimulate subsequent adaptive immunity, providing signals that are essential for initiating the responses of antigen-specific T and B lymphocytes.
The Capture and Display of Microbial Antigens
Microbes that enter through epithelia, along with their protein antigens, are captured by DCs that are resident in and under these epithelia. Antigen-bearing DCs then migrate to draining lymph nodes (Fig. 4–4). Protein antigens are proteolytically digested in the APCs to generate peptides that are displayed on the surface of the APCs bound to MHC molecules. Antigens in different cellular compartments are presented by different MHC molecules and are recognized by different subsets of T cells. Antigens that are ingested from the extracellular environment are processed in endosomal and lysosomal vesicles and then are displayed bound to class II MHC molecules. Because CD4 binds to class II MHC molecules, CD4+ helper T cells recognize class II–associated peptides. By contrast, antigens in the cytoplasm are displayed by class I MHC molecules and are recognized by CD8+ cytotoxic T cells, because CD8 binds to class I MHC. This segregation of different antigens is key to the specialized functions of CD4+ and CD8+ T cells; as we discuss below, the two classes of T cells are designed to combat microbes that are located in different cellular compartments. Protein antigens, as well as polysaccharides and other nonprotein antigens, can also be recognized directly by B lymphocytes in the lymphoid follicles of the peripheral lymphoid organs.
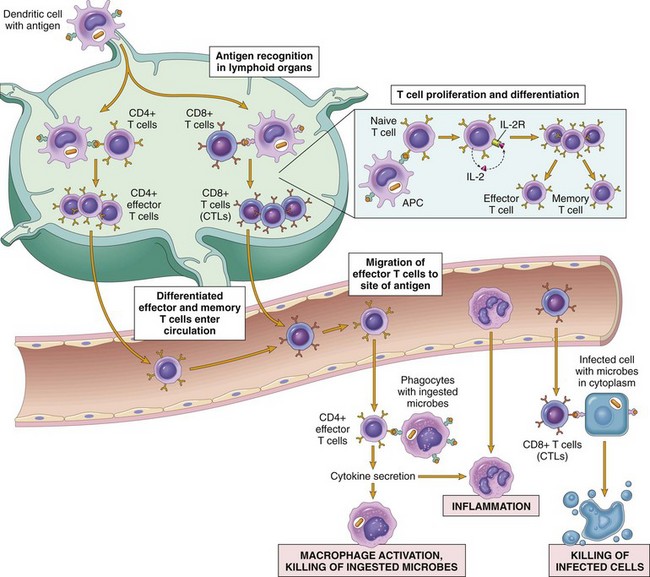
Figure 4–4 Cell-mediated immunity. Naive T cells recognize MHC-associated peptide antigens displayed on dendritic cells in lymph nodes. The T cells are activated to proliferate (under the influence of the cytokine IL-2) and to differentiate into effector and memory cells, which migrate to sites of infection and serve various functions in cell-mediated immunity. Effector CD4+ T cells of the TH1 subset recognize the antigens of microbes ingested by phagocytes and activate the phagocytes to kill the microbes; TH17 effector cells enhance leukocyte recruitment and stimulate inflammation; TH2 cells activate eosinophils. CD8+ CTLs kill infected cells harboring microbes in the cytoplasm. Some activated T cells differentiate into long-lived memory cells. APC, antigen-presenting cell; CTLs, cytotoxic T lymphocytes.
Before being recognized by B and T cells, the microbe elicits an innate immune response. This response activates APCs to express costimulatory molecules and secrete cytokines that stimulate the proliferation and differentiation of T lymphocytes. The principal costimulators for T cells are the B7 molecules (CD80 and CD86) that are expressed on APCs and recognized by the CD28 receptor on naive T cells. The innate immune response to some microbes and polysaccharides also results in the activation of complement, generating cleavage products that enhance the proliferation and differentiation of B lymphocytes. Thus, antigen (signal 1 in Fig. 4–2) and molecules produced during innate immune responses (signal 2 in Fig. 4–2) function cooperatively to activate antigen-specific lymphocytes. The requirement for microbe-triggered signal 2 ensures that the adaptive immune response is induced by microbes and not by harmless substances.
Cell-Mediated Immunity: Activation of T Lymphocytes and Elimination of Cell-Associated Microbes
Naive T lymphocytes are activated by antigen and costimulators in peripheral lymphoid organs, and proliferate and differentiate into effector cells, most of which migrate to any site where the antigen (microbe) is present (Fig. 4–4). Upon activation, T lymphocytes secrete soluble proteins called cytokines, which function as growth and differentiation factors for lymphocytes and other cells, and mediate communications between leukocytes. Because of the important roles of cytokines in both beneficial immune responses and in inflammatory diseases, it is important to understand their properties and actions.
Cytokines: Messenger Molecules of the Immune System
Cytokines are polypeptide products of many cell types (but principally activated lymphocytes and macrophages) that function as mediators of inflammation and immune responses. They were introduced in Chapter 2 in the context of inflammation; here we review their general properties and focus on those cytokines specifically involved in immunity.
Although different cytokines have diverse actions and functions, they all share some common features. Cytokines are synthesized and secreted in response to external stimuli, which may be microbial products, antigen recognition, or other cytokines. Their secretion typically is transient and is controlled by transcription and post-translational mechanisms. The actions of cytokines may be autocrine (on the cell that produces the cytokine), paracrine (on adjacent cells), and, less commonly, endocrine (at a distance from the site of production) (Chapter 2). The effects of cytokines tend to be pleiotropic (one cytokine can have diverse biologic activities, often on many cell types) and redundant (multiple cytokines may have the same activity). Molecularly defined cytokines are called interleukins, referring to their ability to mediate communications between leukocytes.
Cytokines may be grouped into several classes on the basis of their biologic activities and functions.
• Cytokines involved in innate immunity and inflammation, the earliest host response to microbes and dead cells. The major cytokines in this group are TNF and interleukin-1 (IL-1) and a group of chemoattractant cytokines called chemokines. IL-12, IFN-γ, IL-6, IL-23, and several other cytokines also participate in the early innate immune response. Major sources of these cytokines are activated macrophages and DCs, as well as endothelial cells, lymphocytes, mast cells, and other cell types. These were described in Chapter 2.
• Cytokines that regulate lymphocyte responses and effector functions in adaptive immunity. Different cytokines are involved in the proliferation and differentiation of lymphocytes (e.g., IL-2, IL-4), and in the activation of various effector cells (e.g., IFN-γ, which activates macrophages; IL-5, which activates eosinophils). The major sources of these cytokines are CD4+ helper T lymphocytes stimulated by antigens and costimulators. These cytokines are key participants in the induction and effector phases of adaptive cell-mediated immune responses (see later).
• Cytokines that stimulate hematopoiesis. Many of these are called colony-stimulating factors. They function to increase the output of leukocytes from the bone marrow and to thus replenish leukocytes that are consumed during immune and inflammatory reactions.
Effector Functions of T Lymphocytes
One of the earliest responses of CD4+ helper T cells is secretion of the cytokine IL-2 and expression of high-affinity receptors for IL-2. IL-2 is a growth factor that acts on these T lymphocytes and stimulates their proliferation, leading to an increase in the number of antigen-specific lymphocytes. Some of the progeny of the expanded pool of T cells differentiate into effector cells that can secrete different sets of cytokines and thus perform different functions. The best-defined subsets of CD4+ helper cells are the TH1, TH2, and TH17 subsets (Fig. 4–5). TH1 cells produce the cytokine IFN-γ, which activates macrophages and stimulates B cells to produce antibodies that activate complement and coat microbes for phagocytosis. TH2 cells produce IL-4, which stimulates B cells to differentiate into IgE-secreting plasma cells; IL-5, which activates eosinophils; and IL-13, which activates mucosal epithelial cells to secrete mucus and expel microbes, and activates macrophages to secrete growth factors important for tissue repair. TH17 cells produce the cytokine IL-17, which recruits neutrophils and thus promotes inflammation; TH17 cells play an important role in some T cell–mediated inflammatory disorders. These effector cells migrate to sites of infection and accompanying tissue damage. When the differentiated effectors again encounter cell-associated microbes, they are activated to perform the functions that are responsible for elimination of the microbes. The key mediators of the functions of helper T cells are various cytokines and the surface molecule called CD40 ligand (CD40L), which binds to its receptor, CD40, on B cells and macrophages. Differentiated CD4+ effector T cells of the TH1 subset recognize microbial peptides on macrophages that have ingested the microbes. The T cells express CD40L, which engages CD40 on the macrophages, and the T cells secrete the cytokine IFN-γ, which is a potent macrophage activator. The combination of CD40- and IFN-γ–mediated activation results in the induction of potent microbicidal substances in the macrophages, including reactive oxygen species and nitric oxide, leading to the destruction of ingested microbes. TH2 cells elicit cellular defense reactions that are dominated by eosinophils and not macrophages. As discussed later, CD4+ helper T cells also stimulate B cell responses by CD40L and cytokines. Some CD4+ T cells remain in the lymphoid organs in which they were activated and then migrate into follicles, where they stimulate antibody responses; these cells are called follicular helper T cells.
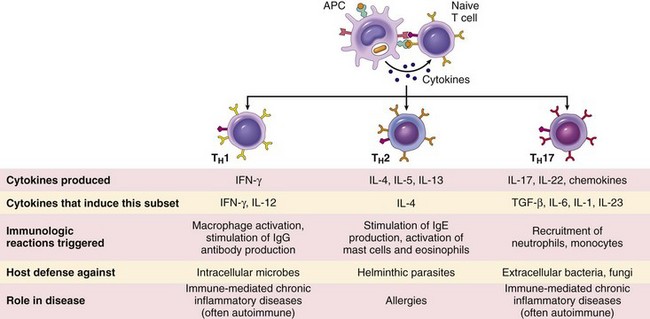
Figure 4–5 Subsets of CD4+ effector T cells. In response to stimuli (mainly cytokines) present at the time of antigen recognition, naive CD4+ helper T cells may differentiate into populations of effector cells that produce distinct sets of cytokines and perform different functions. The types of immune reactions elicited by each subset, and its role in host defense and immunological diseases, are summarized. Two other populations of CD4+ T cells, regulatory cells and follicular helper cells, are not shown.
Activated CD8+ lymphocytes differentiate into CTLs, which kill cells harboring microbes in the cytoplasm. These microbes may be viruses that infect many cell types, or bacteria that are ingested by macrophages but have learned to escape from phagocytic vesicles into the cytoplasm (where they are inaccessible to the killing machinery of phagocytes, which is largely confined to vesicles). By destroying the infected cells, CTLs eliminate the reservoirs of infection.
Humoral Immunity: Activation of B Lymphocytes and Elimination of Extracellular Microbes
Upon activation, B lymphocytes proliferate and then differentiate into plasma cells that secrete different classes of antibodies with distinct functions (Fig. 4–6). There are two major mechanisms of B cell activation.
• T cell–independent. Many polysaccharide and lipid antigens have multiple identical antigenic determinants (epitopes) that are able to engage several antigen receptor molecules on each B cell and initiate the process of B cell activation.
• T cell–dependent. Typical globular protein antigens are not able to bind to many antigen receptors, and the full response of B cells to protein antigens requires help from CD4+ T cells. B cells also can act as APCs—they ingest protein antigens, degrade them, and display peptides bound to class II MHC molecules for recognition by helper T cells. The helper T cells express CD40L and secrete cytokines, which work together to activate the B cells.
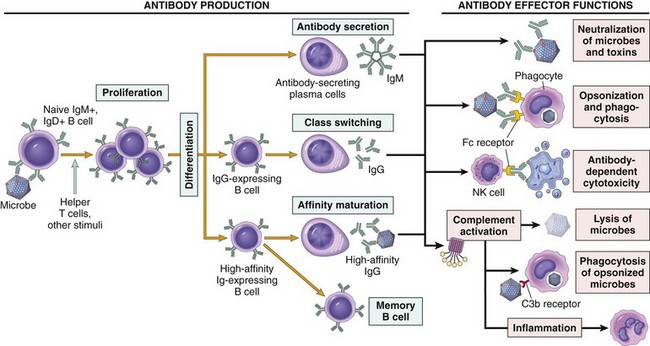
Figure 4–6 Humoral immunity. Naive B lymphocytes recognize antigens, and under the influence of helper T cells and other stimuli (not shown), the B cells are activated to proliferate and to differentiate into antibody-secreting plasma cells. Some of the activated B cells undergo heavy chain class switching and affinity maturation, and some become long-lived memory cells. Antibodies of different heavy chain isotypes (classes) perform different effector functions, shown on the right.
Some of the progeny of the expanded B cell clones differentiate into antibody-secreting plasma cells. Each plasma cell secretes antibodies that have the same specificity as the cell surface antibodies (B cell receptors) that first recognized the antigen. Polysaccharides and lipids stimulate secretion mainly of IgM antibody. Protein antigens, by virtue of CD40L- and cytokine-mediated helper T cell actions, induce the production of antibodies of different classes (IgG, IgA, IgE). This production of functionally different antibodies, all with the same specificity, is called heavy-chain class (isotype) switching; it provides plasticity in the antibody response, allowing antibodies to serve many functions. Helper T cells also stimulate the production of antibodies with higher and higher affinity for the antigen. This process, called affinity maturation, improves the quality of the humoral immune response.
The humoral immune response combats microbes in numerous ways (Fig. 4–6).
• Antibodies bind to microbes and prevent them from infecting cells, thereby “neutralizing” the microbes.
• IgG antibodies coat (“opsonize”) microbes and target them for phagocytosis, since phagocytes (neutrophils and macrophages) express receptors for the Fc tails of IgG molecules.
• IgG and IgM activate the complement system by the classical pathway, and complement products promote phagocytosis and destruction of microbes. Production of most opsonizing and complement-fixing IgG antibodies is stimulated by IFN-γ, typically produced by TH1 helper cells, which respond to many bacteria and viruses, and IgG antibodies are important mechanisms of defense against these microbes.
• IgA is secreted in mucosal tissues and neutralizes microbes in the lumens of the respiratory and gastrointestinal tracts (and other mucosal tissues).
• IgG is actively transported across the placenta and protects the newborn until the immune system becomes mature. This is called passive immunity.
• IgE coats helminthic parasites and functions with mast cells and eosinophils to kill them. As mentioned earlier, TH2 helper cells secrete cytokines that stimulate the production of IgE and activate eosinophils, and thus the response to helminths is orchestrated by TH2 cells.
Circulating IgG antibodies have half-lives of about 3 weeks, which is much longer than the half-lives of most blood proteins, as a consequence of special mechanisms for recycling IgG and reducing its catabolism. Some antibody-secreting plasma cells migrate to the bone marrow and live for years, continuing to produce low levels of antibodies.
Decline of Immune Responses and Immunologic Memory
A majority of effector lymphocytes induced by an infectious pathogen die by apoptosis after the microbe is eliminated, thus returning the immune system to its basal resting state. This return to a stable or steady state, called homeostasis, occurs because microbes provide essential stimuli for lymphocyte survival and activation, and effector cells are short-lived. Therefore, as the stimuli are eliminated, the activated lymphocytes are no longer kept alive.
The initial activation of lymphocytes also generates long-lived memory cells, which may survive for years after the infection. Memory cells are an expanded pool of antigen-specific lymphocytes (more numerous than the naive cells specific for any antigen that are present before encounter with that antigen), and memory cells respond faster and more effectively against the antigen than do naive cells. This is why the generation of memory cells is an important goal of vaccination.
This brief discussion of the normal immune response sets the stage for a consideration of the situations in which immune responses become abnormal, and of how these abnormalities lead to tissue injury and disease.
Summary
Overview of Normal Immune Responses
• The physiologic function of the immune system is defense against infectious microbes.
• The early reaction to microbes is mediated by the mechanisms of innate immunity, which are ready to respond to microbes. These mechanisms include epithelial barriers, phagocytes, NK cells, and plasma proteins (e.g., of the complement system). The reaction of innate immunity is often manifested as inflammation.
• The defense reactions of adaptive immunity develop slowly, but are more potent and specialized.
• Microbes and other foreign antigens are captured by DCs and transported to lymph nodes, where the antigens are recognized by naive lymphocytes. The lymphocytes are activated to proliferate and differentiate into effector and memory cells.
• Cell-mediated immunity is the reaction of T lymphocytes, designed to combat cell-associated microbes (e.g., phagocytosed microbes and microbes in the cytoplasm of infected cells). Humoral immunity is mediated by antibodies and is effective against extracellular microbes (in the circulation and mucosal lumens).
• CD4+ helper T cells help B cells to make antibodies, activate macrophages to destroy ingested microbes, stimulate recruitment of leukocytes, and regulate all immune responses to protein antigens. The functions of CD4+ T cells are mediated by secreted proteins called cytokines. CD8+ CTLs kill cells that express antigens in the cytoplasm that are seen as foreign (e.g., virus-infected and tumor cells).
• Antibodies secreted by plasma cells neutralize microbes and block their infectivity, and promote the phagocytosis and destruction of pathogens. Antibodies also confer passive immunity to neonates.
Hypersensitivity Reactions: Mechanisms of Immune-Mediated Injury
Immune responses that normally are protective are also capable of causing tissue injury. Injurious immune reactions are grouped under hypersensitivity, and the resulting diseases are called hypersensitivity diseases. This term originated from the idea that persons who mount immune responses against an antigen are “sensitized” to that antigen, so pathologic or excessive reactions represent manifestations of a “hypersensitive” state. Normally, an exquisite system of checks and balances optimizes the eradication of infecting organisms without serious injury to host tissues. However, immune responses may be inadequately controlled or inappropriately targeted to host tissues, and in such situations, the normally beneficial response is the cause of disease. In this section we describe the causes and general mechanisms of hypersensitivity diseases and then discuss specific situations in which the immune response is responsible for the disease.
Causes of Hypersensitivity Reactions
Pathologic immune responses may be directed against different types of antigens and may result from various underlying abnormalities.
• Autoimmunity: reactions against self antigens. Normally, the immune system does not react against self-generated antigens. This phenomenon is called self tolerance, implying that the body “tolerates” its own antigens. On occasion, self-tolerance fails, resulting in reactions against the body’s own cells and tissues; collectively, such reactions constitute autoimmunity. The diseases caused by autoimmunity are referred to as autoimmune diseases. We shall return to the mechanisms of self-tolerance and autoimmunity later in this chapter.
• Reactions against microbes. There are many types of reactions against microbial antigens that may cause disease. In some cases, the reaction appears to be excessive or the microbial antigen is unusually persistent. If antibodies are produced against such antigens, the antibodies may bind to the microbial antigens to produce immune complexes, which deposit in tissues and trigger inflammation; this is the underlying mechanism of poststreptococcal glomerulonephritis (Chapter 13). T cell responses against persistent microbes may give rise to severe inflammation, sometimes with the formation of granulomas (Chapter 2); this is the cause of tissue injury in tuberculosis and other infections. Rarely, antibodies or T cells reactive with a microbe cross-react with a host tissue; such cross-reactivity is believed to be the basis for rheumatic heart disease (Chapter 10). In some instances, the disease-causing immune response may be entirely normal, but in the process of eradicating the infection, host tissues are injured. In viral hepatitis, the virus that infects liver cells is not cytopathic, but it is recognized as foreign by the immune system. Cytotoxic T cells try to eliminate infected cells, and this normal immune response damages liver cells.
• Reactions against environmental antigens. Most healthy people do not react strongly against common environmental substances (e.g., pollens, animal danders, or dust mites), but almost 20% of the population are “allergic” to these substances. These individuals are genetically predisposed to make unusual immune responses to a variety of noninfectious, and otherwise harmless, antigens to which all persons are exposed but against which only some react.
In all of these conditions, tissue injury is caused by the same mechanisms that normally function to eliminate infectious pathogens—namely, antibodies, effector T lymphocytes, and various other effector cells. The problem in these diseases is that the response is triggered and maintained inappropriately. Because the stimuli for these abnormal immune responses are difficult or impossible to eliminate (e.g., self antigens, persistent microbes, or environmental antigens), and the immune system has many intrinsic positive feedback loops (amplification mechanisms), once a pathologic immune response starts it is difficult to control or terminate it. Therefore, these hypersensitivity diseases tend to be chronic and debilitating, and are therapeutic challenges. Since inflammation, typically chronic inflammation, is a major component of the pathology of these disorders, they are sometimes grouped under the rubric immune-mediated inflammatory diseases.
Types of Hypersensitivity Reactions
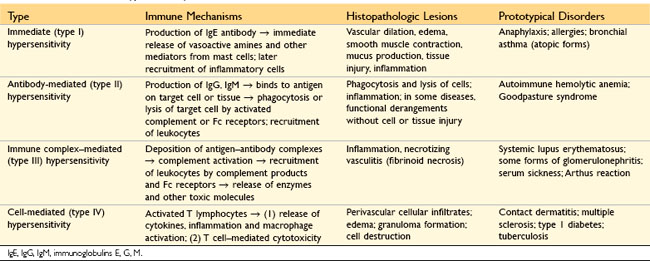
Hypersensitivity reactions are traditionally subdivided into four types based on the principal immune mechanism responsible for injury; three are variations on antibody-mediated injury, whereas the fourth is T cell–mediated (Table 4–1). The rationale for this classification is that the mechanism of immune injury is often a good predictor of the clinical manifestations and may even help to guide the therapy. However, this classification of immune-mediated diseases is not perfect, because several immune reactions may coexist in one disease.
• Immediate (type I) hypersensitivity, often called allergy, results from the activation of the TH2 subset of CD4+ helper T cells by environmental antigens, leading to the production of IgE antibodies, which become attached to mast cells. When these IgE molecules bind the antigen (allergen), the mast cells are triggered to release mediators that transiently affect vascular permeability and induce smooth muscle contraction in various organs, and that also may stimulate more prolonged inflammation (the late-phase reaction). These diseases are commonly called allergic, or atopic, disorders.
• Antibody-mediated (type II) hypersensitivity disorders are caused by antibodies that bind to fixed tissue or cell surface antigens, promoting phagocytosis and destruction of the coated cells or triggering pathologic inflammation in tissues.
• Immune complex–mediated (type III) hypersensitivity disorders are caused by antibodies binding to antigens to form complexes that circulate and deposit in vascular beds and stimulate inflammation, typically as a consequence of complement activation. Tissue injury in these diseases is the result of the inflammation.
• T cell–mediated (type IV) hypersensitivity disorders are caused mainly by immune responses in which T lymphocytes of the TH1 and TH17 subsets produce cytokines that induce inflammation and activate neutrophils and macrophages, which are responsible for tissue injury. CD8+ CTLs also may contribute to injury by directly killing host cells.
Immediate (Type I) Hypersensitivity
Immediate hypersensitivity is a tissue reaction that occurs rapidly (typically within minutes) after the interaction of antigen with IgE antibody that is bound to the surface of mast cells in a sensitized host. The reaction is initiated by entry of an antigen, which is called an allergen because it triggers allergy. Many allergens are environmental substances that are harmless for most persons on exposure. Some people apparently inherit genes that make them susceptible to allergies. This susceptibility is manifested by the propensity of such persons to mount strong TH2 responses and, subsequently, to produce IgE antibody against the allergens. The IgE is central to the activation of the mast cells and release of mediators that are responsible for the clinical and pathologic manifestations of the reaction. Immediate hypersensitivity may occur as a local reaction that is merely annoying (e.g., seasonal rhinitis, or hay fever), severely debilitating (asthma), or even fatal (anaphylaxis).
Sequence of Events in Immediate Hypersensitivity Reactions
Most hypersensitivity reactions follow the same sequence of cellular responses (Fig. 4–7):
• Activation of TH2 cells and production of IgE antibody. Allergens may be introduced by inhalation, ingestion, or injection. Variables that probably contribute to the strong TH2 responses to allergens include the route of entry, dose, and chronicity of antigen exposure, and the genetic makeup of the host. It is not clear if allergenic substances also have unique structural properties that endow them with the ability to elicit TH2 responses. Immediate hypersensitivity is the prototypical TH2-mediated reaction. The TH2 cells that are induced secrete several cytokines, including IL-4, IL-5, and IL-13, which are responsible for essentially all the reactions of immediate hypersensitivity. IL-4 stimulates B cells specific for the allergen to undergo heavy-chain class switching to IgE and to secrete this immunoglobulin isotype. IL-5 activates eosinophils that are recruited to the reaction, and IL-13 acts on epithelial cells and stimulates mucus secretion. TH2 cells often are recruited to the site of allergic reactions in response to chemokines that are produced locally; among these chemokines is eotaxin, which also recruits eosinophils to the same site.
• Sensitization of mast cells by IgE antibody. Mast cells are derived from precursors in the bone marrow, are widely distributed in tissues, and often reside near blood vessels and nerves and in subepithelial locations. Mast cells express a high-affinity receptor for the Fc portion of the ε heavy chain of IgE, called FcεRI. Even though the serum concentration of IgE is very low (in the range of 1 to 100 µg/mL), the affinity of the mast cell FcεRI receptor is so high that the receptors are always occupied by IgE. These antibody-bearing mast cells are “sensitized” to react if the antigen binds to the antibody molecules. Basophils are the circulating counterparts of mast cells. They also express FcεRI, but their role in most immediate hypersensitivity reactions is not established (since these reactions occur in tissues and not in the circulation). The third cell type that expresses FcεRI is eosinophils, which often are present in these reactions and also have a role in IgE-mediated host defense against helminth infections, described later.
• Activation of mast cells and release of mediators. When a person who was sensitized by exposure to an allergen is reexposed to the allergen, it binds to multiple specific IgE molecules on mast cells, usually at or near the site of allergen entry. When these IgE molecules are cross-linked, a series of biochemical signals is triggered in the mast cells. The signals culminate in the secretion of various mediators from the mast cells. Three groups of mediators are the most important in different immediate hypersensitivity reactions (Fig. 4–8):
 Vasoactive amines released from granule stores. The granules of mast cells contain histamine, which is released within seconds or minutes of activation. Histamine causes vasodilation, increased vascular permeability, smooth muscle contraction, and increased secretion of mucus. Other rapidly released mediators include adenosine (which causes bronchoconstriction and inhibits platelet aggregation) and chemotactic factors for neutrophils and eosinophils. Other mast cell granule contents that may be secreted include several neutral proteases (e.g., tryptase), which may damage tissues and also generate kinins and cleave complement components to produce additional chemotactic and inflammatory factors (e.g., C3a) (Chapter 2). The granules also contain acidic proteoglycans (heparin, chondroitin sulfate), the main function of which seems to be as a storage matrix for the amines. Newly synthesized lipid mediators. Mast cells synthesize and secrete prostaglandins and leukotrienes, by the same pathways as do other leukocytes (Chapter 2). These lipid mediators have several actions that are important in immediate hypersensitivity reactions. Prostaglandin D2 (PGD2) is the most abundant mediator generated by the cyclooxygenase pathway in mast cells. It causes intense bronchospasm as well as increased mucus secretion. The leukotrienes LTC4 and LTD4 are the most potent vasoactive and spasmogenic agents known; on a molar basis, they are several thousand times more active than histamine in increasing vascular permeability and in causing bronchial smooth muscle contraction. LTB4 is highly chemotactic for neutrophils, eosinophils, and monocytes. Cytokines. Activation of mast cells results in the synthesis and secretion of several cytokines that are important for the late-phase reaction. These include TNF and chemokines, which recruit and activate leukocytes (Chapter 2); IL-4 and IL-5, which amplify the TH2-initiated immune reaction; and IL-13, which stimulates epithelial cell mucus secretion.
Vasoactive amines released from granule stores. The granules of mast cells contain histamine, which is released within seconds or minutes of activation. Histamine causes vasodilation, increased vascular permeability, smooth muscle contraction, and increased secretion of mucus. Other rapidly released mediators include adenosine (which causes bronchoconstriction and inhibits platelet aggregation) and chemotactic factors for neutrophils and eosinophils. Other mast cell granule contents that may be secreted include several neutral proteases (e.g., tryptase), which may damage tissues and also generate kinins and cleave complement components to produce additional chemotactic and inflammatory factors (e.g., C3a) (Chapter 2). The granules also contain acidic proteoglycans (heparin, chondroitin sulfate), the main function of which seems to be as a storage matrix for the amines. Newly synthesized lipid mediators. Mast cells synthesize and secrete prostaglandins and leukotrienes, by the same pathways as do other leukocytes (Chapter 2). These lipid mediators have several actions that are important in immediate hypersensitivity reactions. Prostaglandin D2 (PGD2) is the most abundant mediator generated by the cyclooxygenase pathway in mast cells. It causes intense bronchospasm as well as increased mucus secretion. The leukotrienes LTC4 and LTD4 are the most potent vasoactive and spasmogenic agents known; on a molar basis, they are several thousand times more active than histamine in increasing vascular permeability and in causing bronchial smooth muscle contraction. LTB4 is highly chemotactic for neutrophils, eosinophils, and monocytes. Cytokines. Activation of mast cells results in the synthesis and secretion of several cytokines that are important for the late-phase reaction. These include TNF and chemokines, which recruit and activate leukocytes (Chapter 2); IL-4 and IL-5, which amplify the TH2-initiated immune reaction; and IL-13, which stimulates epithelial cell mucus secretion.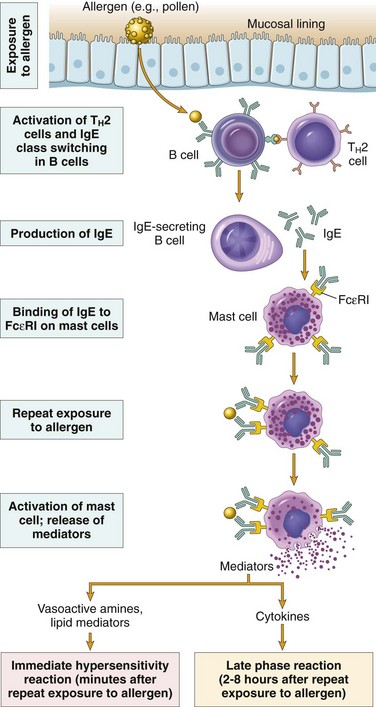
Figure 4–7 Sequence of events in immediate (type I) hypersensitivity. Immediate hypersensitivity reactions are initiated by the introduction of an allergen, which stimulates TH2 responses and IgE production. IgE binds to Fc receptors (FcεRI) on mast cells, and subsequent exposure to the allergen activates the mast cells to secrete the mediators that are responsible for the pathologic manifestations of immediate hypersensitivity.
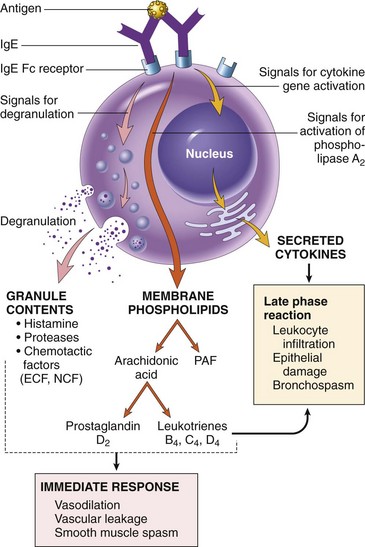
Figure 4–8 Mast cell mediators. Upon activation, mast cells release various classes of mediators that are responsible for the immediate and late-phase reactions. ECF, eosinophil chemotactic factor; NCF, neutrophil chemotactic factor (neither of these has been biochemically defined); PAF, platelet-activating factor.
In summary, a variety of compounds that act on blood vessels, smooth muscle, and leukocytes mediate type I hypersensitivity reactions (Table 4–2). Some of these compounds are released rapidly from sensitized mast cells and are responsible for the intense immediate reactions associated with conditions such as systemic anaphylaxis. Others, such as cytokines, are responsible for the inflammation seen in late-phase reactions.
Table 4–2 Summary of the Action of Mast Cell Mediators in Immediate (Type I) Hypersensitivity
| Action | Mediators |
|---|---|
| Vasodilation, increased vascular permeability | |
| Smooth muscle spasm | |
| Cellular infiltration |
PAF, platelet-activating factor; TNF, tumor necrosis factor.
Often, the IgE-triggered reaction has two well-defined phases (Fig. 4–9): (1) the immediate response, characterized by vasodilation, vascular leakage, and smooth muscle spasm, usually evident within 5 to 30 minutes after exposure to an allergen and subsiding by 60 minutes; and (2) a second, late-phase reaction that usually sets in 2 to 8 hours later and may last for several days and is characterized by inflammation as well as tissue destruction, such as mucosal epithelial cell damage. The dominant inflammatory cells in the late-phase reaction are neutrophils, eosinophils, and lymphocytes, especially TH2 cells. Neutrophils are recruited by various chemokines; their roles in inflammation were described in Chapter 2. Eosinophils are recruited by eotaxin and other chemokines released from TNF-activated epithelium and are important effectors of tissue injury in the late-phase response. Eosinophils produce major basic protein and eosinophil cationic protein, which are toxic to epithelial cells, and LTC4 and platelet-activating factor, which promote inflammation. TH2 cells produce cytokines that have multiple actions, as described earlier. These recruited leukocytes can amplify and sustain the inflammatory response even in the absence of continuous allergen exposure. In addition, inflammatory leukocytes are responsible for much of the epithelial cell injury in immediate hypersensitivity. Because inflammation is a major component of many allergic diseases, notably asthma and atopic dermatitis, therapy usually includes anti-inflammatory drugs such as corticosteroids.
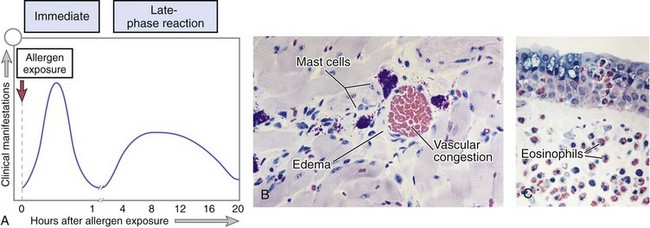
Figure 4–9 Immediate hypersensitivity. A, Kinetics of the immediate and late-phase reactions. The immediate vascular and smooth muscle reaction to allergen develops within minutes after challenge (allergen exposure in a previously sensitized person), and the late-phase reaction develops 2 to 24 hours later. B–C, Morphology: The immediate reaction (B) is characterized by vasodilation, congestion, and edema, and the late-phase reaction (C) is characterized by an inflammatory infiltrate rich in eosinophils, neutrophils, and T cells.
(B and C, Courtesy of Dr. Daniel Friend, Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts.)
Clinical and Pathologic Manifestations
An immediate hypersensitivity reaction may occur as a systemic disorder or as a local reaction. The nature of the reaction is often determined by the route of antigen exposure. Systemic exposure to protein antigens (e.g., in bee venom) or drugs (e.g., penicillin) may result in systemic anaphylaxis. Within minutes of the exposure in a sensitized host, itching, urticaria (hives), and skin erythema appear, followed in short order by profound respiratory difficulty caused by pulmonary bronchoconstriction and accentuated by hypersecretion of mucus. Laryngeal edema may exacerbate matters by causing upper airway obstruction. In addition, the musculature of the entire gastrointestinal tract may be affected, with resultant vomiting, abdominal cramps, and diarrhea. Without immediate intervention, there may be systemic vasodilation with a fall in blood pressure (anaphylactic shock), and the patient may progress to circulatory collapse and death within minutes.
Local reactions generally occur when the antigen is confined to a particular site, such as skin (contact, causing urticaria), gastrointestinal tract (ingestion, causing diarrhea), or lung (inhalation, causing bronchoconstriction). The common forms of skin and food allergies, hay fever, and certain forms of asthma are examples of localized allergic reactions. However, ingestion or inhalation of allergens also can trigger systemic reactions.
Susceptibility to localized type I reactions has a strong genetic component, and the term atopy is used to imply familial predisposition to such localized reactions. Patients who suffer from nasobronchial allergy (including hay fever and some forms of asthma) often have a family history of similar conditions. Genes that are implicated in susceptibility to asthma and other atopic disorders include those encoding HLA molecules (which may confer immune responsiveness to particular allergens), cytokines (which may control TH2 responses), a component of the FcεRI, and ADAM33, a metalloproteinase that may be involved in tissue remodeling in the airways.
The reactions of immediate hypersensitivity clearly did not evolve solely to cause human discomfort and disease. The immune response dependent on TH2 cells and IgE—in particular, the late-phase inflammatory reaction—plays an important protective role in combating parasitic infections. IgE antibodies are produced in response to many helminthic infections, and their physiologic function is to target helminths for destruction by eosinophils and mast cells. Mast cells also are involved in defense against bacterial infections. And snake aficionados will be relieved to hear that their mast cells may protect them from some snake venoms by releasing granule proteases that degrade the toxins. Why these beneficial responses are inappropriately activated by harmless environmental antigens, giving rise to allergies, remains a puzzle.
Summary
Immediate (Type I) Hypersensitivity
• Also called allergic reactions, or allergies
• Induced by environmental antigens (allergens) that stimulate strong TH2 responses and IgE production in genetically susceptible individuals
• IgE coats mast cells by binding to Fcε receptors; reexposure to the allergen leads to cross-linking of the IgE and FcεRI, activation of mast cells, and release of mediators.
• Principal mediators are histamine, proteases, and other granule contents; prostaglandins and leukotrienes; and cytokines.
• Mediators are responsible for the immediate vascular and smooth muscle reactions and the late-phase reaction (inflammation).
• The clinical manifestations may be local or systemic, and range from mildly annoying rhinitis to fatal anaphylaxis.
Antibody-Mediated Diseases (Type II Hypersensitivity)
Antibody-mediated (type II) hypersensitivity disorders are caused by antibodies directed against target antigens on the surface of cells or other tissue components. The antigens may be normal molecules intrinsic to cell membranes or in the extracellular matrix, or they may be adsorbed exogenous antigens (e.g., a drug metabolite). Antibody-mediated abnormalities are the underlying cause of many human diseases; examples of these are listed in Table 4–3. In all of these disorders, the tissue damage or functional abnormalities result from a limited number of mechanisms.
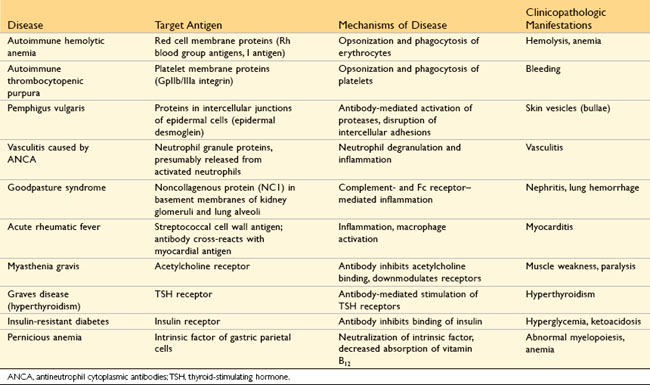
Mechanisms of Antibody-Mediated Diseases
Antibodies cause disease by targeting cells for phagocytosis, by activating the complement system, and by interfering with normal cellular functions (Fig. 4–10). The antibodies that are responsible typically are high-affinity antibodies capable of activating complement and binding to the Fc receptors of phagocytes.
• Opsonization and phagocytosis. When circulating cells, such as erythrocytes or platelets, are coated (opsonized) with autoantibodies, with or without complement proteins, the cells become targets for phagocytosis by neutrophils and macrophages (Fig. 4–10, A). These phagocytes express receptors for the Fc tails of IgG antibodies and for breakdown products of the C3 complement protein, and use these receptors to bind and ingest opsonized particles. Opsonized cells are usually eliminated in the spleen, and this is why splenectomy is of clinical benefit in autoimmune thrombocytopenia and some forms of autoimmune hemolytic anemia.
• Inflammation. Antibodies bound to cellular or tissue antigens activate the complement system by the “classical” pathway (Fig. 4–10, B). Products of complement activation serve several functions (see Fig. 2–18, Chapter 2), one of which is to recruit neutrophils and monocytes, triggering inflammation in tissues. Leukocytes may also be activated by engagement of Fc receptors, which recognize the bound antibodies. This mechanism of injury is exemplified by Goodpasture syndrome and pemphigus vulgaris.
• Antibody-mediated cellular dysfunction. In some cases, antibodies directed against cell surface receptors impair or dysregulate cellular function without causing cell injury or inflammation (Fig. 4–10, C). In myasthenia gravis, antibodies against acetylcholine receptors in the motor end plates of skeletal muscles inhibit neuromuscular transmission, with resultant muscle weakness. Antibodies can also stimulate cellular responses excessively. In Graves disease, antibodies against the thyroid-stimulating hormone receptor stimulate thyroid epithelial cells to secrete thyroid hormones, resulting in hyperthyroidism. Antibodies against hormones and other essential proteins can neutralize and block the actions of these molecules, causing functional derangements.
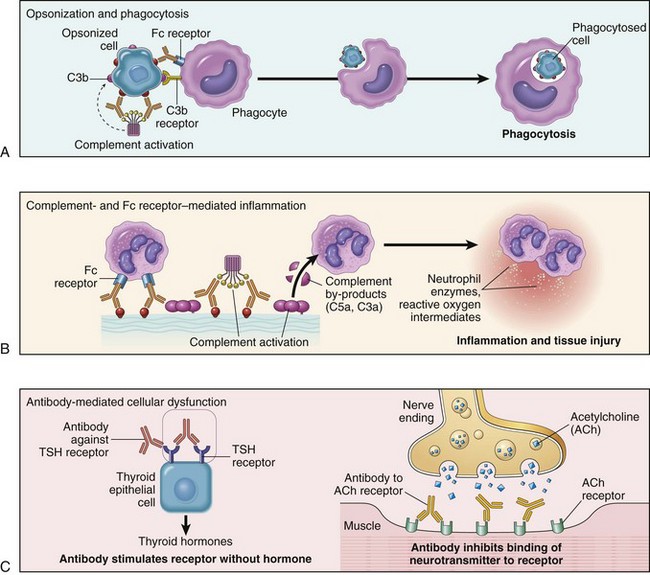
Figure 4–10 Mechanisms of antibody-mediated injury. A, Opsonization of cells by antibodies and complement components, and ingestion of opsonized cells by phagocytes. B, Inflammation induced by antibody binding to Fc receptors of leukocytes and by complement breakdown products. C, Antireceptor antibodies disturb the normal function of receptors. In these examples, antibodies against the thyroid-stimulating hormone (TSH) receptor activate thyroid cells in Graves disease, and acetylcholine (ACh) receptor antibodies impair neuromuscular transmission in myasthenia gravis.
Immune Complex Diseases (Type III Hypersensitivity)
Antigen–antibody (immune) complexes that are formed in the circulation may deposit in blood vessels, leading to complement activation and acute inflammation. The antigens in these complexes may be exogenous antigens, such as microbial proteins, or endogenous antigens, such as nucleoproteins. The mere formation of immune complexes does not equate with hypersensitivity disease; small amounts of antigen–antibody complexes may be produced during normal immune responses and are usually phagocytosed and destroyed. It is only when these complexes are produced in large amounts, persist, and are deposited in tissues that they are pathogenic. Pathogenic immune complexes may form in the circulation and subsequently deposit in blood vessels, or the complexes may form at sites where antigen has been planted (in situ immune complexes). Immune complex–mediated injury is systemic when complexes are formed in the circulation and are deposited in several organs, or it may be localized to particular organs (e.g., kidneys, joints, or skin) if the complexes are formed and deposited in a specific site. The mechanism of tissue injury is the same regardless of the pattern of distribution; however, the sequence of events and the conditions leading to the formation of systemic and local immune complexes are different and are considered separately in the following descriptions. Immune complex diseases are some of the most common immunologic diseases (Table 4–4).
Table 4–4 Examples of Immune Complex–Mediated Diseases
| Disease | Antigen Involved | Clinicopathologic Manifestations |
|---|---|---|
| Systemic lupus erythematosus | Nuclear antigens | Nephritis, skin lesions, arthritis, others |
| Poststreptococcal glomerulonephritis | Streptococcal cell wall antigen(s); may be “planted” in glomerular basement membrane | Nephritis |
| Polyarteritis nodosa | Hepatitis B virus antigens in some cases | Systemic vasculitis |
| Reactive arthritis | Bacterial antigens (e.g., Yersinia) | Acute arthritis |
| Serum sickness | Various proteins (e.g., foreign serum protein such as horse antithymocyte globulin) | Arthritis, vasculitis, nephritis |
| Arthus reaction (experimental) | Various foreign proteins | Cutaneous vasculitis |
Systemic Immune Complex Disease
The pathogenesis of systemic immune complex disease can be divided into three phases: (1) formation of antigen–antibody complexes in the circulation and (2) deposition of the immune complexes in various tissues, thereby initiating (3) an inflammatory reaction in various sites throughout the body (Fig. 4–11).
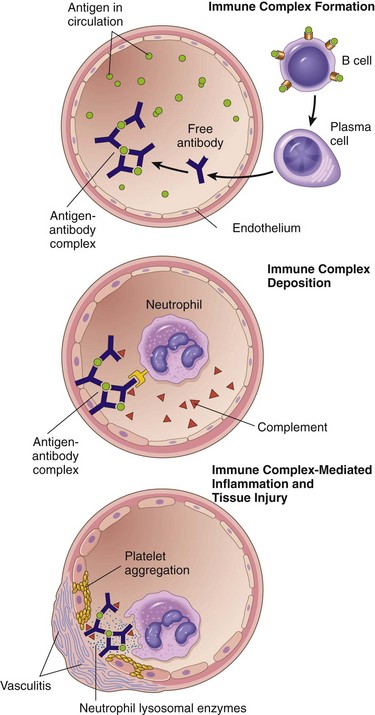
Figure 4–11 Immune complex disease: The sequential phases in the induction of systemic immune complex–mediated diseases (type III hypersensitivity).
Acute serum sickness is the prototype of a systemic immune complex disease. It was first described in humans when large amounts of foreign serum were administered for passive immunization (e.g., in persons receiving horse serum containing antidiphtheria antibody); it is now seen only rarely (e.g., in patients injected with rabbit or horse antithymocyte globulin for treatment of aplastic anemia or graft rejection, or patients with snakebite given anti-venom antibody made in animals). Although serum sickness is no longer common, the study of its pathogenesis sheds light on the mechanisms of human immune complex diseases. Approximately 5 days after the foreign protein is injected, specific antibodies are produced; these react with the antigen still present in the circulation to form antigen–antibody complexes. The complexes deposit in blood vessels in various tissue beds, triggering the subsequent injurious inflammatory reaction.
Several variables determine whether immune complex formation leads to tissue deposition and disease. Perhaps foremost among these factors is the size of the complexes. Very large complexes or complexes with many free IgG Fc regions (typically formed in antibody excess) are rapidly removed from the circulation by macrophages in the spleen and liver and are therefore usually harmless. The most pathogenic complexes are formed during antigen excess and are small or intermediate in size and are cleared less effectively by phagocytes and therefore circulate longer. In addition, the charge of the complex, the valency of the antigen, the avidity of the antibody, and the hemodynamics of a given vascular bed all influence the tendency to develop disease. The favored sites of deposition are kidneys, joints, and small blood vessels in many tissues. Localization in the kidney and joints is explained in part by the high hemodynamic pressures associated with the filtration function of the glomerulus and the synovium. For complexes to leave the circulation and deposit within or outside the vessel wall, an increase in vascular permeability also must occur. This is probably triggered when immune complexes bind to leukocytes and mast cells by means of Fc and C3b receptors and stimulate release of mediators that increase vascular permeability.
Once complexes are deposited in the tissue, the third phase, the inflammatory reaction, ensues. During this phase (approximately 10 days after antigen administration), clinical features such as fever, urticaria, arthralgias, lymph node enlargement, and proteinuria appear. Wherever immune complexes deposit, characteristic tissue damage occurs. The immune complexes activate the complement system, leading to the release of biologically active fragments such as the anaphylatoxins (C3a and C5a), which increase vascular permeability and are chemotactic for neutrophils and monocytes (Chapter 2). The complexes also bind to Fcγ receptors on neutrophils and monocytes, activating these cells. Attempted phagocytosis of immune complexes by the leukocytes results in the secretion of a variety of additional pro-inflammatory substances, including prostaglandins, vasodilator peptides, and chemotactic substances, as well as lysosomal enzymes capable of digesting basement membrane, collagen, elastin, and cartilage, and reactive-oxygen species that damage tissues. Immune complexes can also cause platelet aggregation and activate Hageman factor; both of these reactions augment the inflammatory process and initiate formation of microthrombi, which contribute to the tissue injury by producing local ischemia (Fig. 4–11). The resultant pathologic lesion is termed vasculitis if it occurs in blood vessels, glomerulonephritis if it occurs in renal glomeruli, arthritis if it occurs in the joints, and so on.
Predictably, the antibody classes that induce such lesions are complement-fixing antibodies (i.e., IgG and IgM) and antibodies that bind to phagocyte Fc receptors (IgG). During the active phase of the disease, consumption of complement may result in decreased serum complement levels. The role of complement- and Fc receptor–dependent inflammation in the pathogenesis of the tissue injury is supported by the observation that experimental depletion of serum complement levels or knockout of Fc receptors in mice greatly reduces the severity of lesions, as does depletion of neutrophils.
 Morphology
Morphology
The morphologic appearance of immune complex injury is dominated by acute necrotizing vasculitis, microthrombi, and superimposed ischemic necrosis accompanied by acute inflammation of the affected organs. The necrotic vessel wall takes on a smudgy eosinophilic appearance called fibrinoid necrosis, caused by protein deposition (see Fig. 1–13, Chapter 1). Immune complexes can be visualized in the tissues, usually in the vascular wall (examples of such deposits in the kidney in lupus are shown in Fig. 4–18, E). In due course, the lesions tend to resolve, especially when they were brought about by a single exposure to antigen (e.g., in acute serum sickness or acute poststreptococcal glomerulonephritis) (Chapter 13). However, chronic immune complex disease develops when there is persistent antigenemia or repeated exposure to an antigen. This occurs in some human diseases, such as systemic lupus erythematosus (SLE). Most often, even though the morphologic changes and other findings strongly implicate immune complex disease, the inciting antigens are unknown.
Local Immune Complex Disease
A model of local immune complex diseases is the Arthus reaction, in which an area of tissue necrosis appears as a result of acute immune complex vasculitis. The reaction is produced experimentally by injecting an antigen into the skin of a previously immunized animal (i.e., pre-formed antibodies against the antigen are already present in the circulation). Because of the initial antibody excess, immune complexes are formed as the antigen diffuses into the vascular wall; these are precipitated at the site of injection and trigger the same inflammatory reaction and histologic appearance as in systemic immune complex disease. Arthus lesions evolve over a few hours and reach a peak 4 to 10 hours after injection, when the injection site develops visible edema with severe hemorrhage, occasionally followed by ulceration.
Summary
Pathogenesis of Diseases Caused by Antibodies and Immune Complexes
• Antibodies can coat (opsonize) cells, with or without complement proteins, and target these cells for phagocytosis by macrophages, which express receptors for the Fc tails of IgG molecules and for complement proteins. The result is depletion of the opsonized cells.
• Antibodies and immune complexes may deposit in tissues or blood vessels, and elicit an acute inflammatory reaction by activating complement, with release of breakdown products, or by engaging Fc receptors of leukocytes. The inflammatory reaction causes tissue injury.
• Antibodies can bind to cell surface receptors or essential molecules, and cause functional derangements (either inhibition or unregulated activation) without cell injury.
T Cell–Mediated (Type IV) Hypersensitivity
Several autoimmune disorders, as well as pathologic reactions to environmental chemicals and persistent microbes, are now known to be caused by T cells (Table 4–5). The occurrence and significance of T lymphocyte–mediated tissue injury have been increasingly appreciated as the methods for detecting and purifying T cells from patients’ circulation and lesions have improved. This group of diseases is of great clinical interest because many of the new, rationally designed biologic therapies for immune-mediated inflammatory diseases have been developed to target abnormal T cell reactions. Two types of T cell reactions are capable of causing tissue injury and disease: (1) cytokine-mediated inflammation, in which the cytokines are produced mainly by CD4+ T cells, and (2) direct cell cytotoxicity, mediated by CD8+ T cells (Fig. 4–12). In inflammation, exemplified by the delayed-type hypersensitivity (DTH) reaction, CD4+ T cells of the TH1 and TH17 subsets secrete cytokines, which recruit and activate other cells, especially macrophages, and these are the major effector cells of injury. In cell-mediated cytotoxicity, cytotoxic CD8+ T cells are responsible for tissue damage.
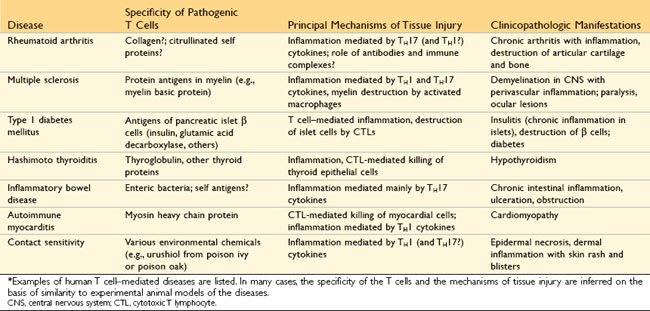
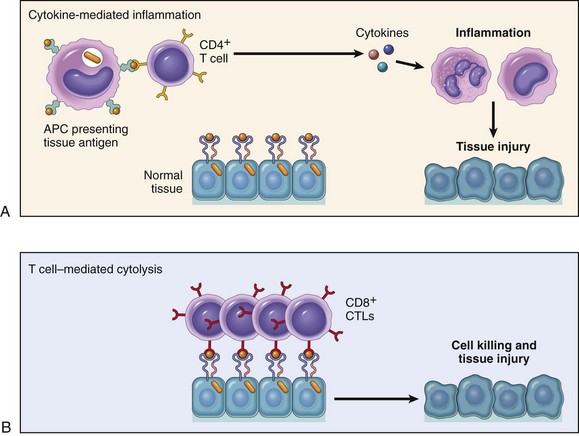
Figure 4–12 Mechanisms of T cell–mediated (type IV) hypersensitivity reactions. A, In cytokine-mediated inflammatory reactions, CD4+ T cells respond to tissue antigens by secreting cytokines that stimulate inflammation and activate phagocytes, leading to tissue injury. B, In some diseases, CD8+ CTLs directly kill tissue cells. APC, antigen-presenting cell; CTLs, cytotoxic T lymphocytes.
Inflammatory Reactions Elicited by CD4+ T Cells
The sequence of events in T cell–mediated inflammatory reactions begins with the first exposure to antigen and is essentially the same as the reactions of cell-mediated immunity (Fig. 4–4). Naive CD4+ T lymphocytes recognize peptide antigens of self or microbial proteins in association with class II MHC molecules on the surface of DCs (or macrophages) that have processed the antigens. If the DCs produce IL-12, the naive T cells differentiate into effector cells of the TH1 type. The cytokine IFN-γ, made by NK cells and by the TH1 cells themselves, further promotes TH1 differentiation, providing a powerful positive feedback loop. If the APCs produce IL-1, IL-6, or IL-23 instead of IL-12, the CD4+ cells develop into TH17 effectors. On subsequent exposure to the antigen, the previously generated effector cells are recruited to the site of antigen exposure and are activated by the antigen presented by local APCs. The TH1 cells secrete IFN-γ, which is the most potent macrophage-activating cytokine known. Activated macrophages have increased phagocytic and microbicidal activity. Activated macrophages also express more class II MHC molecules and costimulators, leading to augmented antigen presentation capacity, and the cells secrete more IL-12, thus stimulating more TH1 responses. Upon activation by antigen, TH17 effector cells secrete IL-17 and several other cytokines, which promote the recruitment of neutrophils (and monocytes) and thus induce inflammation. Because the cytokines produced by the T cells enhance leukocyte recruitment and activation, these inflammatory reactions become chronic unless the offending agent is eliminated or the cycle is interrupted therapeutically. In fact, inflammation occurs as an early response to microbes and dead cells (Chapter 2), but it is greatly increased and prolonged when T cells are involved.
Delayed-type hypersensitivity (DTH), described next, is an illustrative model of T cell–mediated inflammation and tissue injury. The same reactions are the underlying basis for several diseases. Contact dermatitis is an example of tissue injury resulting from T cell–mediated inflammation. It is evoked by contact with pentadecylcatechol (also known as urushiol, the active component of poison ivy and poison oak, which probably becomes antigenic by binding to a host protein). On reexposure of a previously exposed person to the plants, sensitized TH1 CD4+ cells accumulate in the dermis and migrate toward the antigen within the epidermis. Here they release cytokines that damage keratinocytes, causing separation of these cells and formation of an intraepidermal vesicle, and inflammation manifested as a vesicular dermatitis. It has long been thought that several systemic diseases, such as type 1 diabetes and multiple sclerosis, are caused by TH1 and TH17 reactions against self antigens, and Crohn disease may be caused by uncontrolled reactions involving the same T cells but directed against intestinal bacteria. T cell–mediated inflammation also plays a role in the rejection of transplants, described later in the chapter.
Delayed-Type Hypersensitivity
DTH is a T cell–mediated reaction that develops in response to antigen challenge in a previously sensitized individual. In contrast with immediate hypersensitivity, the DTH reaction is delayed for 12 to 48 hours, which is the time it takes for effector T cells to be recruited to the site of antigen challenge and to be activated to secrete cytokines. The classic example of DTH is the tuberculin reaction, elicited by challenge with a protein extract of M. tuberculosis (tuberculin) in a person who has previously been exposed to the tubercle bacillus. Between 8 and 12 hours after intracutaneous injection of tuberculin, a local area of erythema and induration appears, reaching a peak (typically 1 to 2 cm in diameter) in 24 to 72 hours and thereafter slowly subsiding. On histologic examination, the DTH reaction is characterized by perivascular accumulation (“cuffing”) of CD4+ helper T cells and macrophages (Fig. 4–13). Local secretion of cytokines by these cells leads to increased microvascular permeability, giving rise to dermal edema and fibrin deposition; the latter is the main cause of the tissue induration in these responses. DTH reactions are mediated primarily by TH1 cells; the contribution of TH17 cells is unclear. The tuberculin response is used to screen populations for people who have had previous exposure to tuberculosis and therefore have circulating memory T cells specific for mycobacterial proteins. Notably, immunosuppression or loss of CD4+ T cells (e.g., resulting from HIV infection) may lead to a negative tuberculin response even in the presence of a severe infection.
Figure 4–13 Delayed-type hypersensitivity reaction in the skin. A, Perivascular accumulation (“cuffing”) of mononuclear inflammatory cells (lymphocytes and macrophages), with associated dermal edema and fibrin deposition. B, Immunoperoxidase staining reveals a predominantly perivascular cellular infiltrate that marks positively with anti-CD4 antibodies.
(B, Courtesy of Dr. Louis Picker, Department of Pathology, Oregon Health & Science University, Portland, Oregon.)
Prolonged DTH reactions against persistent microbes or other stimuli may result in a special morphologic pattern of reaction called granulomatous inflammation. The initial perivascular CD4+ T cell infiltrate is progressively replaced by macrophages over a period of 2 to 3 weeks. These accumulated macrophages typically exhibit morphologic evidence of activation; that is, they become large, flat, and eosinophilic, and are called epithelioid cells. The epithelioid cells occasionally fuse under the influence of cytokines (e.g., IFN-γ) to form multinucleate giant cells. A microscopic aggregate of epithelioid cells, typically surrounded by a collar of lymphocytes, is called a granuloma (Fig. 4–14, A). The process is essentially a chronic form of TH1-mediated inflammation and macrophage activation (Fig. 4–14, B). Older granulomas develop an enclosing rim of fibroblasts and connective tissue. Recognition of a granuloma is of diagnostic importance because of the limited number of conditions that can cause it (Chapter 2).
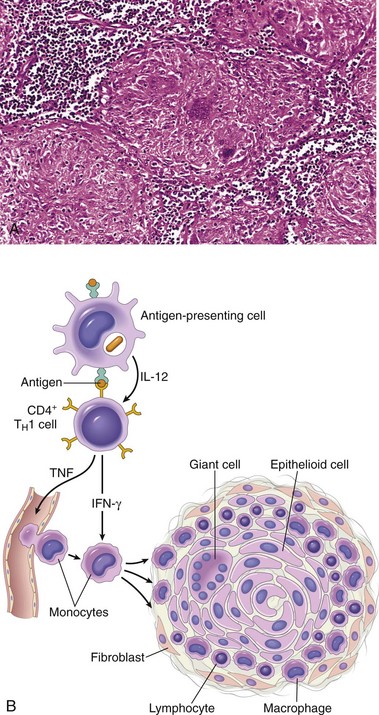
Figure 4–14 Granulomatous inflammation. A, A section of a lymph node shows several granulomas, each made up of an aggregate of epithelioid cells and surrounded by lymphocytes. The granuloma in the center shows several multinucleate giant cells. B, The events that give rise to the formation of granulomas in type IV hypersensitivity reactions. Note the role played by T cell–derived cytokines.
(A, Courtesy of Dr. Trace Worrell, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
T Cell–Mediated Cytotoxicity
In this form of T cell–mediated tissue injury, CD8+ CTLs kill antigen-bearing target cells. As discussed earlier, class I MHC molecules bind to intracellular peptide antigens and present the peptides to CD8+ T lymphocytes, stimulating the differentiation of these T cells into effector cells called CTLs. CTLs play a critical role in resistance to virus infections and some tumors. The principal mechanism of killing by CTLs is dependent on the perforin–granzyme system. Perforin and granzymes are stored in the granules of CTLs and are rapidly released when CTLs engage their targets (cells bearing the appropriate class I MHC–bound peptides). Perforin binds to the plasma membrane of the target cells and promotes the entry of granzymes, which are proteases that specifically cleave and thereby activate cellular caspases. These enzymes induce apoptotic death of the target cells (Chapter 1). CTLs play an important role in the rejection of solid-organ transplants and may contribute to many immunologic diseases, such as type 1 diabetes (in which insulin-producing β cells in pancreatic islets are destroyed by an autoimmune T cell reaction). CD8+ T cells may also secrete IFN-γ and contribute to cytokine-mediated inflammation, but less so than CD4+ cells.
Summary
Mechanisms of T Cell–Mediated Hypersensitivity Reactions
• Cytokine-mediated inflammation: CD4+ T cells are activated by exposure to a protein antigen and differentiate into TH1 and TH17 effector cells. Subsequent exposure to the antigen results in the secretion of cytokines. IFN-γ activates macrophages to produce substances that cause tissue damage and promote fibrosis, and IL-17 and other cytokines recruit leukocytes, thus promoting inflammation.
• T cell–mediated cytotoxicity: CD8+ CTLs specific for an antigen recognize cells expressing the target antigen and kill these cells. CD8+ T cells also secrete IFN-γ.
With the basic mechanisms of pathologic immune reactions as background, we now proceed to a consideration of two categories of reactions that are of great clinical importance: autoimmunity and transplant rejection.
Autoimmune Diseases
Immune reactions to self antigens (i.e., autoimmunity) are the underlying cause of numerous human diseases. Autoimmune diseases currently are estimated to affect 2% to 5% of the population in developed countries, and appear to be increasing in incidence. The evidence that these diseases are indeed the result of autoimmune reactions is more persuasive for some than for others. For instance, in many of these disorders, multiple high-affinity autoantibodies have been identified, and in some cases these antibodies are known to cause pathologic abnormalities (Table 4–6). Similarly, with improving technology, there is growing evidence for the activation of pathogenic self-reactive T cells in some of these diseases. In addition, experimental models have proved very informative, providing circumstantial evidence supporting an autoimmune etiology. Nevertheless, it is fair to say that for many disorders traditionally classified as autoimmune, this etiologic origin is suspected but not proved.
| Organ-Specific | Systemic |
|---|---|
| Diseases Mediated by Antibodies | |
| Autoimmune hemolytic anemia | Systemic lupus erythematosus |
| Autoimmune thrombocytopenia | |
| Autoimmune atrophic gastritis of pernicious anemia | |
| Myasthenia gravis | |
| Graves disease | |
| Goodpasture syndrome | |
| Diseases Mediated by T Cells* | |
| Type 1 diabetes mellitus | Rheumatoid arthritis |
| Multiple sclerosis | Systemic sclerosis (scleroderma) |
| Hashimoto thyroiditis | Sjögren syndrome |
| Crohn disease | |
| Diseases Postulated to Be of Autoimmune Origin† | |
| Primary biliary cirrhosis | Polyarteritis nodosa |
| Autoimmune (chronic active) hepatitis | Inflammatory myopathies |
* A role for T cells has been demonstrated in these disorders, but antibodies also may be involved in tissue injury.
† An autoimmune basis for these disorders is suspected, but the supporting evidence is not strong.
Presumed autoimmune diseases range from those in which specific immune responses are directed against one particular organ or cell type and result in localized tissue damage, to multisystem diseases characterized by lesions in many organs and associated with multiple autoantibodies or T cell–mediated reactions against numerous self antigens. In many of the systemic diseases that are caused by immune complexes and autoantibodies, the lesions affect principally the connective tissue and blood vessels of the various organs involved. Therefore, these diseases are often referred to as “collagen vascular” or “connective tissue” disorders, even though the immunologic reactions are not specifically directed against constituents of connective tissue or blood vessels.
Normal persons are unresponsive (tolerant) to their own (self) antigens, and autoimmunity results from a failure of self-tolerance. Therefore, understanding the pathogenesis of autoimmunity requires familiarity with the mechanisms of normal immunologic tolerance.
Immunologic Tolerance
Immunologic tolerance is unresponsiveness to an antigen that is induced by exposure of specific lymphocytes to that antigen. Self-tolerance refers to a lack of immune responsiveness to one’s own tissue antigens. Billions of different antigen receptors are randomly generated in developing T and B lymphocytes, and it is not surprising that during this process, receptors are produced that can recognize self antigens. Since these antigens cannot all be concealed from the immune system, there must be means of eliminating or controlling self-reactive lymphocytes. Several mechanisms work in concert to select against self-reactivity and to thus prevent immune reactions against the body’s own antigens. These mechanisms are broadly divided into two groups: central tolerance and peripheral tolerance (Fig. 4–15).
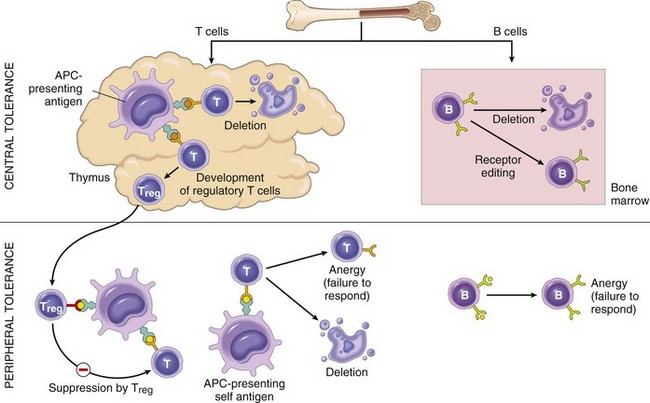
Figure 4–15 Immunologic self-tolerance: The principal mechanisms of central and peripheral self-tolerance in T and B cells.
Central tolerance. The principal mechanism of central tolerance is the antigen-induced deletion (death) of self-reactive T and B lymphocytes during their maturation in central (generative) lymphoid organs (i.e., in the thymus for T cells and in the bone marrow for B cells). In the thymus, many autologous (self) protein antigens are processed and presented by thymic APCs in association with self MHC. Any immature T cell that encounters such a self antigen undergoes apoptosis (a process called deletion, or negative selection), and the T cells that complete their maturation are thereby depleted of self-reactive cells (Fig. 4–15). An exciting advance has been the identification of putative transcription factors that induce the expression of peripheral tissue antigens in the thymus, thus making the thymus an immunologic mirror of self. One such factor is called the autoimmune regulator (AIRE); mutations in the AIRE gene are responsible for an autoimmune polyendocrine syndrome in which T cells specific for multiple self antigens escape deletion (presumably because these self antigens are not expressed in the thymus), and attack tissues expressing the self antigens. Some T cells that encounter self antigens in the thymus are not killed but differentiate into regulatory T cells, as described later.
Immature B cells that recognize self antigens with high affinity in the bone marrow also may die by apoptosis. Some self-reactive B cells may not be deleted but may undergo a second round of rearrangement of antigen receptor genes and then express new receptors that are no longer self-reactive (a process called “receptor editing”).
Unfortunately, the process of deletion of self-reactive lymphocytes is not perfect. Many self antigens may not be present in the thymus, so T cells bearing receptors for such autoantigens can escape into the periphery. There is similar “slippage” in the B cell system as well, and B cells that bear receptors for a variety of self antigens, including thyroglobulin, collagen, and DNA, can be found in healthy persons.
Peripheral tolerance. Self-reactive T cells that escape negative selection in the thymus can potentially wreak havoc unless they are deleted or effectively muzzled. Several mechanisms in the peripheral tissues that silence such potentially autoreactive T cells have been identified (Fig. 4–15):
• Anergy: This term refers to functional inactivation (rather than death) of lymphocytes induced by encounter with antigens under certain conditions. As described previously, activation of T cells requires two signals: recognition of peptide antigen in association with self MHC molecules on APCs, and a set of second costimulatory signals (e.g., through B7 molecules) provided by the APCs. If the second costimulatory signals are not delivered, or if an inhibitory receptor on the T cell (rather than the costimulatory receptor) is engaged when the cell encounters self antigen, the T cell becomes anergic and cannot respond to the antigen (Fig. 4–15). Because costimulatory molecules are not strongly expressed on most normal tissues, the encounter between autoreactive T cells and self antigens in tissues may result in anergy. B cells can also become anergic if they encounter antigen in the absence of specific helper T cells.
• Suppression by regulatory T cells: The responses of T lymphocytes to self antigens may be actively suppressed by regulatory T cells. The best-defined populations of regulatory T cells express CD25, one of the chains of the receptor for IL-2, and require IL-2 for their generation and survival. These cells also express a unique transcription factor called FoxP3. This protein is necessary for the development of regulatory cells, and mutations in the FOXP3 gene are responsible for a systemic autoimmune disease called IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome), which is associated with deficiency of regulatory T cells. Several mechanisms have been proposed to explain how regulatory T cells control immune responses, including secretion of immunosuppressive cytokines (e.g., IL-10, transforming growth factor-β [TGF-β]), which can dampen a variety of T cell responses, and competitive blocking of B7 molecules on APCs.
• Activation-induced cell death: Another mechanism of peripheral tolerance involves apoptosis of mature lymphocytes as a result of self-antigen recognition. One mechanism of apoptosis involves the death receptor Fas (a member of the TNF receptor family), which can be engaged by its ligand coexpressed on the same or neighboring cells. The same pathway is important for the deletion of self-reactive B cells by Fas ligand expressed on T cells. The importance of this pathway of self-tolerance is illustrated by the discovery that mutations in the FAS gene are responsible for an autoimmune disease called the autoimmune lymphoproliferative syndrome (ALPS), characterized by lymphadenopathy and multiple autoantibodies including anti-DNA. Defects in Fas and Fas ligand are also the cause of similar autoimmune diseases in mice. The mitochondrial pathway of apoptosis, which does not depend on death receptors, may also be involved in the elimination of self-reactive lymphocytes.
Mechanisms of Autoimmunity
Proceeding from the foregoing summary of the principal mechanisms of self-tolerance, we can ask how these mechanisms might break down to give rise to pathologic autoimmunity. Unfortunately, there are no simple answers to this question, and the underlying causes of most human autoimmune diseases remain to be determined. As mentioned earlier, certain mutations can compromise one or another pathway of self-tolerance and cause pathologic autoimmunity. Study of these single-gene mutations is extremely informative, and such research helps to establish the biologic significance of the various pathways of self-tolerance. The diseases caused by such mutations are rare, however, and most autoimmune diseases cannot be explained by defects in single genes.
It is believed that the breakdown of self-tolerance and development of autoimmunity result from a combination of inherited susceptibility genes, which influence lymphocyte tolerance, and environmental factors, such as infections or tissue injury, that alter the display of self antigens (Fig. 4–16).
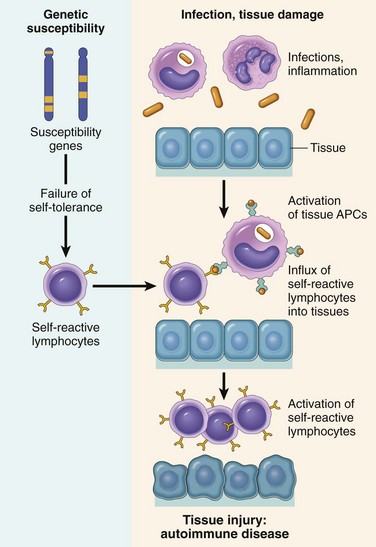
Figure 4–16 Pathogenesis of autoimmunity. Autoimmunity arises from the inheritance of susceptibility genes that may interfere with self-tolerance, in association with environmental triggers (infection, tissue injury, inflammation) that alter the display of self antigens, promote lymphocyte entry into tissues, and enhance the activation of self-reactive lymphocytes.
Genetic Factors in Autoimmunity
There is abundant evidence that susceptibility genes play an important role in the development of autoimmune diseases.
• Autoimmune diseases have a tendency to run in families, and there is a greater incidence of the same disease in monozygotic than in dizygotic twins.
• Several autoimmune diseases are linked with the HLA locus, especially class II alleles (HLA-DR, -DQ). The frequency of a disease in a person with a particular HLA allele, compared with that in people who do not inherit that allele, is called the odds ratio or relative risk (Table 4–7). The relative risk ranges from 3 or 4 for rheumatoid arthritis (RA) and HLA-DR4 to 100 or more for ankylosing spondylitis and HLA-B27. However, how MHC genes influence the development of autoimmunity is still not clear, especially because MHC molecules do not distinguish between self and foreign peptide antigens. It is also worthy of note that most people with a susceptibility-related MHC allele never develop any disease, and, conversely, people without the relevant MHC gene can develop the disease. Expression of a particular MHC gene is therefore but one variable that can contribute to autoimmunity.
• Genome-wide association studies and linkage studies in families are revealing many genetic polymorphisms that are associated with different autoimmune diseases (Table 4–8). Some of these polymorphisms seem to be associated with several diseases, suggesting that the genes involved influence general mechanisms of self-tolerance and immune regulation. Others are disease-specific and may influence end-organ sensitivity or display of particular self antigens. There is great interest in elucidating how these genes contribute to autoimmunity, and many plausible hypotheses have been proposed (Table 4–8), but the actual role of these genes in the development of particular autoimmune diseases is not established.
Table 4–7 Association of Human Leukocyte Antigen (HLA) Alleles with Autoimmune Diseases
| Disease | HLA Allele | Odds Ratio* |
|---|---|---|
| Rheumatoid arthritis (anti-CCP Ab–positive)† | DRB1 | 4–12 |
| Type 1 diabetes | DRB1*0301-DQA1*0501-DQB1*0201 haplotype | 4 |
| DRB1*0401- DQA1*0301-DQB1*0302 haplotype | 8 | |
| DRB1*0301/0401 haplotype heterozygotes | 35 | |
| Multiple sclerosis | DRB1*1501 | 3 |
| Systemic lupus erythematosus | DRB1*0301 | 2 |
| DRB1*1501 | 1.3 | |
| Ankylosing spondylitis | B*27 (mainly B*2705 and B*2702) | 100–200 |
| Celiac disease | DQA1*0501-DQB1*0201 haplotype | 7 |
* The odds ratio (also called relative risk) is the approximate value of the increased risk of the disease associated with the inheritance of particular HLA alleles. The data are from European-derived populations.
† Anti-CCP Ab, antibodies directed against cyclic citrullinated peptides. Data are from patients who tested positive for these antibodies in serum.
Table courtesy of Dr. Michelle Fernando, Imperial College London.
Table 4–8 Selected Non–Human Leukocyte Antigen (HLA) Genes Associated with Autoimmune Diseases
| Putative Gene Involved* | Diseases | Postulated Function of Encoded Protein and Role of Mutation/Polymorphism in Disease |
|---|---|---|
| Genes Involved in Immune Regulation | ||
| PTPN22 | RA, T1D, IBD | Protein tyrosine phosphatase, may affect signaling in lymphocytes and may alter negative selection or activation of self-reactive T cells |
| IL23R | IBD, PS, AS | Receptor for the TH17-inducing cytokine IL-23; may alter differentiation of CD4+ T cells into pathogenic TH17 effector cells |
| CTLA4 | T1D, RA | Inhibits T cell responses by terminating activation and promoting activity of regulatory T cells; may interfere with self-tolerance |
| IL2RA | MS, T1D | α chain of the receptor for IL-2, which is a growth and survival factor for activated and regulatory T cells; may affect development of effector cells and/or regulation of immune responses |
| Genes Involved in Immune Responses to Microbes | ||
| NOD2 | IBD | Cytoplasmic sensor of bacteria expressed in Paneth and other intestinal epithelial cells; may control resistance to gut commensal bacteria |
| ATG16 | IBD | Involved in autophagy; possible role in defense against microbes and maintenance of epithelial barrier function |
| IRF5, IFIH1 | SLE | Role in production of type I IFN, involved in the pathogenesis of SLE (see text) |
AS, ankylosing spondylitis; IBD, inflammatory bowel disease; IFN, interferon; MS, multiple sclerosis; PS, psoriasis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; T1D, type 1 diabetes.
* The probable linkage of these genes with various autoimmune diseases has been defined by genome-wide association studies (GWAS) and other methods for studying disease-associated polymorphisms.
Adapted from Zenewicz L, Abraham C, Flavell RA, Cho J: Unraveling the genetics of autoimmunity. Cell 140:791, 2010.
Role of Infections and Tissue Injury
A variety of microbes, including bacteria, mycoplasmas, and viruses, have been implicated as triggers for autoimmunity. Microbes may induce autoimmune reactions by several mechanisms:
• Viruses and other microbes may share cross-reacting epitopes with self antigens, such that responses may be induced by the microbe but may attack self tissues. This phenomenon is called molecular mimicry. It is the probable cause of a few diseases, the best example being rheumatic heart disease, in which an immune response against streptococci cross-reacts with cardiac antigens. It is not known if more subtle mimicry plays a role in other autoimmune diseases.
• Microbial infections with resultant tissue necrosis and inflammation can cause upregulation of costimulatory molecules on APCs in the tissue, thus favoring a breakdown of T cell anergy and subsequent T cell activation.
There is no lack of possible mechanisms to explain how infectious agents might participate in the pathogenesis of autoimmunity. At present, however, no evidence is available that clearly implicates any microbe in the causation of human autoimmune diseases. Adding to the complexity are recent suggestions (based largely on epidemiologic data) that infections may paradoxically protect affected persons from some autoimmune diseases, notably type 1 diabetes and multiple sclerosis. The possible mechanisms underlying this effect are not understood.
The display of tissue antigens may be altered by a variety of environmental insults, not only infections. As discussed later, ultraviolet (UV) radiation causes cell death and may lead to the exposure of nuclear antigens, which elicit pathologic immune responses in lupus; this mechanism is the proposed explanation for the association of lupus flares with exposure to sunlight. Smoking is a risk factor for RA, perhaps because it leads to chemical modification of self antigens. Local tissue injury for any reason may lead to the release of self antigens and autoimmune responses.
Finally, there is a strong gender bias of autoimmunity, with many of these diseases being more common in women than in men. The underlying mechanisms are still not well understood, and may include the effects of hormones and other factors.
An autoimmune response may itself promote further autoimmune attack. Tissue injury caused by an autoimmune response or any other cause may lead to exposure of self antigen epitopes that were previously concealed but are now presented to T cells in an immunogenic form. The activation of such autoreactive T cells is called “epitope spreading,” because the immune response “spreads” to epitopes that were not recognized initially. This is one of the mechanisms that may contribute to the chronicity of autoimmune diseases.
Summary
Immunologic Tolerance and Autoimmunity
• Tolerance (unresponsiveness) to self antigens is a fundamental property of the immune system, and breakdown of tolerance is the basis of autoimmune diseases.
• Central tolerance: Immature lymphocytes that recognize self antigens in the central (generative) lymphoid organs are killed by apoptosis; in the B cell lineage, some of the self-reactive lymphocytes switch to new antigen receptors that are not self-reactive.
• Peripheral tolerance: Mature lymphocytes that recognize self antigens in peripheral tissues become functionally inactive (anergic), or are suppressed by regulatory T lymphocytes, or die by apoptosis.
• The factors that lead to a failure of self-tolerance and the development of autoimmunity include (1) inheritance of susceptibility genes that may disrupt different tolerance pathways and (2) infections and tissue alterations that may expose self-antigens and activate APCs and lymphocytes in the tissues.
Having discussed the general principles of tolerance and autoimmunity, we proceed to a discussion of some of the most common and important autoimmune diseases. Although each disease is discussed separately, considerable overlap is apparent in their clinical, serologic, and morphologic features. Only the systemic autoimmune diseases are covered in this chapter; the autoimmune diseases that affect single organ systems are more appropriately discussed in the chapters that deal with the relevant organs.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease of protean manifestations and variable clinical behavior. Clinically, it is an unpredictable, remitting and relapsing disease of acute or insidious onset that may involve virtually any organ in the body; however, it affects principally the skin, kidneys, serosal membranes, joints, and heart. Immunologically, the disease is associated with an enormous array of autoantibodies, classically including antinuclear antibodies (ANAs). The clinical presentation of SLE is so variable, with so many overlapping features with those of other autoimmune diseases (RA, polymyositis, and others), that it has been necessary to develop diagnostic criteria for SLE (Table 4–9). The diagnosis is established by demonstration of four or more of the criteria during any interval of observation.
Table 4–9 1997 Revised Criteria for Classification of Systemic Lupus Erythematosus*
| Criterion | Definition |
|---|---|
| 1. Malar rash | Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds |
| 2. Discoid rash | Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions |
| 3. Photosensitivity | Rash occurring as an unusual reaction to sunlight, reported in patient history or as physician observation |
| 4. Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by a physician |
| 5. Arthritis | Nonerosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling, or effusion |
| 6. Serositis | Pleuritis—convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion or |
| Pericarditis—documented by electrocardiogram or rub or evidence of pericardial effusion | |
| 7. Renal disorder | Persistent proteinuria >0.5 g/dL or >3+ if quantitation not performed or |
| Cellular casts—may be red blood cell, hemoglobin, granular, tubular, or mixed | |
| 8. Neurologic disorder | Seizures—in the absence of offending drugs or known metabolic derangements, (e.g., uremia, ketoacidosis, or electrolyte imbalance) or |
| Psychosis—in the absence of offending drugs or known metabolic derangements, (e.g., uremia, ketoacidosis, or electrolyte imbalance) | |
| 9. Hematologic disorder | Hemolytic anemia—with reticulocytosis or |
| Leukopenia—<4.0 × 109/L (4000/mm3) total on two or more occasions or | |
| Lymphopenia—<1.5 × 109/L (1500/mm3) on two or more occasions or | |
| Thrombocytopenia—<100 × 109/L (100 × 103/mm3) in the absence of offending drugs | |
| 10. Immunologic disorder | Anti-DNA antibody to native DNA in abnormal titer or |
| Anti-Sm—presence of antibody to Sm nuclear antigen or | |
| Positive finding of antiphospholipid antibodies based on (1) an abnormal serum level of IgG or IgM anticardiolipin antibodies, (2) a positive test for lupus anticoagulant using a standard test, or (3) a false-positive serologic test for syphilis known to be positive for at least 6 months and confirmed by negative Treponema pallidum immobilization or fluorescent treponemal antibody absorption test | |
| 11. Antinuclear antibody | An abnormal titer of antinuclear antibody by immunofluorescence or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndrome |
* The proposed classification is based on 11 criteria. For the purpose of identifying patients in clinical studies, a person is said to have systemic lupus erythematosus if any 4 or more of the 11 criteria are present, serially or simultaneously, during any period of observation.
From Tan EM, Cohen AS, Fries JF, et al: The revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271, 1982; and Hochberg MC: Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725, 1997.
Incidence and prevalence estimates of SLE vary among racial and ethnic groups; some studies estimate the prevalence to be as high as 0.2% in certain groups. As with many autoimmune diseases, there is a strong (approximately 9 : 1) female preponderance, and the disease affects 1 in 700 women of childbearing age. SLE is more common and severe in black Americans, affecting 1 in 245 women in that group. Onset typically is in the second or third decade of life, but it may manifest at any age, including early childhood.
 Pathogenesis
Pathogenesis
The fundamental defect in SLE is a failure to maintain self-tolerance, leading to the production of a large number of autoantibodies that can damage tissues either directly or in the form of immune complex deposits. As in other autoimmune diseases, the pathogenesis of SLE involves a combination of genetic and environmental factors. Recent studies have revealed interesting clues about the pathogenesis of this enigmatic disorder (Fig. 4–17).
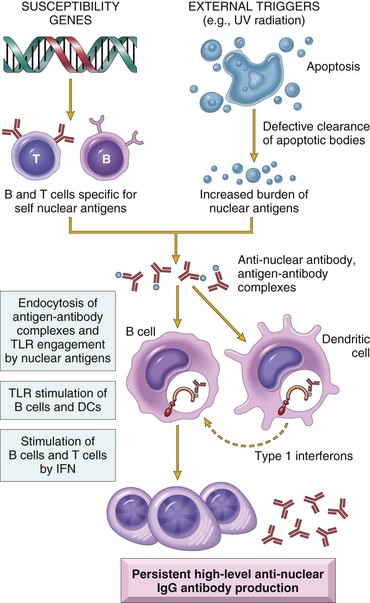
Figure 4–17 Model for the pathogenesis of systemic lupus erythematosus. Genetic susceptibility and exposure result in failure of self-tolerance and persistence of nuclear antigens. Autoantibodies serve to internalize nuclear components, which engage TLRs and stimulate IFN production. IFN may stimulate B and T cell responses to the nuclear antigens. IFN, interferon; IgG, immunoglobulin G; MHC, major histocompatibility complex; TLRs, Toll-like receptors; UV, ultraviolet.
Genetic Factors
Many lines of evidence support a genetic predisposition to SLE.
• Familial association. Family members have an increased risk for the development of SLE, and up to 20% of clinically unaffected first-degree relatives may have autoantibodies. There is a high rate of concordance in monozygotic twins (25%) versus dizygotic twins (1% to 3%).
• HLA association. The odds ratio (relative risk) for persons with HLA-DR2 or HLA-DR3 is 2 to 3, and if both haplotypes are present, the risk is about 5.
• Other genes. Genetic deficiencies of classical pathway complement proteins, especially C1q, C2, or C4, are seen in about 10% of patients with SLE. The complement deficiencies may result in defective clearance of immune complexes and apoptotic cells, and failure of B cell tolerance. A polymorphism in the inhibitory Fc receptor, FcγRIIb, has been described in some patients; this may contribute to inadequate control of B cell activation. Many other genes have been detected by genome-wide association studies, but the role of each of these is not established and their contribution to the development of the disease remains unclear.
Environmental Factors
There are many indications that environmental factors are involved in the pathogenesis of SLE.
• Ultraviolet (UV) radiation (sun exposure) exacerbates the lesions of SLE. A postulated mechanism of this effect is that UV radiation causes apoptosis of host cells, leading to an increased burden of nuclear fragments and inflammatory responses to the products of dead cells.
• Cigarette smoking has been shown to be associated with the development of SLE. Although the mechanism of this is unknown, smoking tobacco may modulate the production of autoantibodies.
• Sex hormones had been thought to exert an important influence on the development of disease, because SLE is 10 times more common in women during reproductive years than in men of similar ages but only 2 to 3 times more common in women during childhood or after the age of 65. However, treatment of women with oral contraceptives containing high doses of estrogen and progesterone did not influence the frequency or severity of disease flares, suggesting that factors other than hormones may account for the increased risk of this disease in women.
• Drugs such as procainamide and hydralazine can induce an SLE-like disease, although typically glomerulonephritis does not develop. These drugs cause demethylation of DNA, which could influence the expression of a variety of genes involved in the development of autoimmunity, or the ability of DNA to activate host cells.
Immunologic Abnormalities in SLE
Studies have implicated several components of the innate and adaptive immune system in the pathogenesis of SLE.
• Type I interferons. Blood cells show a striking molecular signature that indicates exposure to interferon-α (IFN-α), a type I interferon that is produced mainly by plasmacytoid DCs. Some studies have shown that such cells from patients with SLE also produce abnormally large amounts of IFN-α.
• TLR signals. Studies in animal models have shown that TLRs that recognize DNA and RNA, notably the DNA-recognizing TLR9 and the RNA-recognizing TLR7, produce signals that activate B cells specific for self nuclear antigens.
• Failure of B cell tolerance. Studies with B cells from patients with SLE suggest the presence of defects in both central and peripheral tolerance, resulting in a higher frequency of autoreactive B cells than that typical for healthy people.
Based on these clues, a model for the pathogenesis of SLE has been proposed (Fig. 4–17). According to this model, UV irradiation and other environmental insults lead to the apoptosis of cells. Inadequate clearance of the nuclei of these cells, in part because of defects in clearance mechanisms such as complement proteins and receptors, results in a large burden of nuclear antigens. Polymorphisms in various genes, which are susceptibility genes for lupus, lead to defective ability to maintain self-tolerance in B and T lymphocytes, because of which self-reactive lymphocytes remain functional. The self-reactive B cells are stimulated by the self nuclear antigens, and antibodies are produced against the antigens. Complexes of the antigens and antibodies bind to Fc receptors on B cells and DCs and may be internalized. The nucleic acid components engage TLRs and stimulate B cells to produce autoantibodies and activate DCs, particularly plasmacytoid DCs, to produce IFN-α, which further enhances the immune response and causes more apoptosis. The net result is a cycle of antigen release and immune activation resulting in the production of high-affinity autoantibodies.
Spectrum of Autoantibodies in SLE
Antibodies have been identified against a host of nuclear and cytoplasmic components of the cell that are specific to neither organs nor species. Another group of antibodies is directed against surface antigens of blood cells, while yet another is reactive with proteins in complex with phospholipids (antiphospholipid antibodies) (Chapter 3).
• Antinuclear antibodies. ANAs are directed against several nuclear antigens and can be grouped into four categories: (1) antibodies to DNA, (2) antibodies to histones, (3) antibodies to nonhistone proteins bound to RNA, and (4) antibodies to nucleolar antigens. Table 4–10 lists several autoantibodies, including ANAs, and their association with SLE as well as with other autoimmune diseases, to be discussed later. The most widely used method of detecting ANAs is the indirect immunofluorescence assay (IFA), which screens for autoantibodies that bind to a variety of nuclear antigens, including DNA, RNA, and proteins. Four staining patterns are seen with IFA: homogeneous or diffuse, rim or peripheral, speckled, and nucleolar. While each pattern can be suggestive of the presence of specific autoantibodies, the strength of these associations is limited and should not be relied on. ANA testing by IFA is extremely sensitive, as more than 95% of patients with SLE will test positive, but the test’s specificity is quite limited, because patients with other autoimmune diseases, chronic infections, and cancer will test positive as well. Furthermore, ANAs are seen in approximately 5% to 15% of healthy people, and the incidence increases with age. Recently, the IFA has been replaced in many clinical laboratories by multiplex flow cytometry immunoassays that can simultaneously test for multiple specific autoantibodies, but these assays may lack the sensitivity of the IFA. Antibodies to double-stranded DNA (dsDNA) and the so-called Smith (Sm) antigen can be detected by ELISA or multiplex flow methods and are specific for SLE.
• Other autoantibodies. Antibodies against blood cells, including red cells, platelets, and lymphocytes, are found in many patients. Antiphospholipid antibodies are present in 40% to 50% of patients with lupus and react with a wide variety of proteins in complex with phospholipids. Some bind to cardiolipin antigen, used in serologic tests for syphilis, so patients with lupus may have a false-positive test result for syphilis. Antiphospholipid antibodies contribute to coagulation abnormalities, which are described below.
Table 4–10 Selected Autoantibodies Associated with Presumed Autoimmune Diseases
| Autoantibody (Specificity) | Major Disease Association(s) | Likely Role(s) in Disease |
|---|---|---|
| Anti-dsDNA (double-stranded DNA) | SLE* | Formation of immune complexes |
| Anti-Sm (ribonuclear core protein, Sm antigen) | SLE* | Formation of immune complexes |
| Anti-RNP U1 (ribonuclear protein) | SLE, mixed connective tissue disease | Formation of immune complexes |
| Anti–SS-A (Ro), anti–SS-B (La) (ribonucleoproteins) | Sjögren syndrome, SLE | Role in Sjögren syndrome not known |
| Anti–Scl-70 (DNA topoisomerase I) | Systemic sclerosis* | Unknown |
| Anti-histones (histone proteins) | SLE | Formation of immune complexes |
| Anti-centromere (centromere proteins) | Limited scleroderma, systemic sclerosis* | Unknown |
| Antiphospholipid (phospholipid–protein complexes involved in blood coagulation) | Antiphospholipid syndrome, SLE | Thrombotic episodes |
| Anti-Jo1 (histidyl tRNA ligase) | Inflammatory myopathies* | Unknown |
| Anti-mitochondrial | Primary biliary cirrhosis* | Unknown |
| Anti-eTg (transglutaminase) | Dermatitis herpetiformis | Unknown |
| Anti–neutrophil cytoplasmic antibody (ANCA) (proteins in neutrophil cytoplasm) | Various vasculitides* | Formation of immune complexes? |
| Neutrophil degranulation? | ||
| Anti–smooth muscle | Chronic autoimmune hepatitis | Unknown |
Each antibody specificity is detected in 30% to 90% of patients with a particular disease. Asterisks indicate high correlation between the antibody specificity and the disease.
SLE, systemic lupus erythematosus.
Mechanisms of Tissue Injury
Regardless of the exact sequence by which autoantibodies are formed, they are likely to be the mediators of tissue injury, probably through multiple mechanisms.
• Most organ damage in SLE is caused by immune complex deposition. Skin and kidney biopsies from patients with SLE typically demonstrate diffuse and heavy granular deposits of complement and immunoglobulin. Autoantibodies complexed with DNA can be detected as well. These deposits of immune complexes had been thought to cause tissue damage by activating the classical complement pathway (type III hypersensitivity); 75% of patients will have reduced serum levels of C3 and C4 at the time of SLE flares, presumably because complement is being activated and consumed faster than it can be produced. However, people and rodents deficient in C1q are not protected from SLE and actually can spontaneously develop SLE, raising the possibility that complement-independent mechanisms may also contribute to tissue damage.
• Autoantibodies of different specificities contribute to the pathology and clinical manifestations of SLE (type II hypersensitivity). Autoantibodies against red cells, white cells, and platelets opsonize these cells and lead to their phagocytosis, resulting in cytopenias. Autoantibodies against various phospholipids lead to increased thrombosis in patients, with varied clinical consequences, including recurrent spontaneous abortion and thrombotic episodes. These disorders are part of the antiphospholipid syndrome. Paradoxically, these antibodies interfere with clotting tests and are actually called “lupus anticoagulants.” Autoantibodies are also produced against clotting factors such as thrombin, and these too may contribute to clotting disorders. Autoantibodies against central nervous system receptors for various neurotransmitters have been implicated in the neuropsychiatric complications of the disease.
• There is no evidence that ANAs can permeate intact cells. However, if cell nuclei are exposed, the ANAs can bind to them. In tissues, nuclei of damaged cells react with ANAs, lose their chromatin pattern, and become homogeneous, to produce so-called LE bodies or hematoxylin bodies. An in vitro correlate of this is the LE cell, a neutrophil or macrophage that has engulfed the denatured nucleus of another injured cell. When blood is withdrawn and agitated, a number of leukocytes are sufficiently damaged to expose their nuclei to ANAs, with secondary complement activation; these antibody- and complement-opsonized nuclei are then readily phagocytosed. Although the LE cell test is positive in as many as 70% of patients with SLE, it is now largely of historical interest.
Morphology
SLE is a systemic disease with protean manifestations (Table 4–9). The morphologic changes in SLE are therefore extremely variable and depend on the nature of the autoantibodies, the tissue in which immune complexes deposit, and the course and duration of disease. The most characteristic morphologic changes result from the deposition of immune complexes in a variety of tissues.
Blood Vessels
An acute necrotizing vasculitis affecting small arteries and arterioles may be present in any tissue. The arteritis is characterized by necrosis and by fibrinoid deposits within vessel walls containing antibody, DNA, complement fragments, and fibrinogen; a transmural and perivascular leukocytic infiltrate is also frequently present. In chronic stages, vessels show fibrous thickening with luminal narrowing.
Kidneys
Kidney involvement is one of the most important clinical features of SLE, with renal failure being the most common cause of death. The focus here is on glomerular pathology, although interstitial and tubular lesions are also seen in SLE.
The pathogenesis of all forms of glomerulonephritis in SLE involves deposition of DNA–anti-DNA complexes within the glomeruli. These evoke an inflammatory response that may cause proliferation of the endothelial, mesangial, and/or epithelial cells and, in severe cases, necrosis of the glomeruli. Although the kidney appears normal by light microscopy in 25% to 30% of cases, almost all cases of SLE show some renal abnormality if examined by immunofluorescence and electron microscopy. According to the current International Society of Nephrology/Renal Pathology Society morphologic classification, there are six patterns of glomerular disease in SLE (none of which is specific to the disease): class I, minimal mesangial lupus nephritis; class II, mesangial proliferative lupus nephritis; class III, focal lupus nephritis; class IV, diffuse lupus nephritis; class V, membranous lupus nephritis; and class VI, advanced sclerosing lupus nephritis.
• Minimal mesangial lupus nephritis (class I) is rarely encountered in renal biopsies. Immune complexes are present in the mesangium, but there are no concomitant structural alterations detectable by light microscopy.
• Mesangial proliferative lupus nephritis (class II) is seen in 10% to 25% of cases and is associated with mild clinical symptoms. Immune complexes deposit in the mesangium, with a mild to moderate increase in the mesangial matrix and cellularity.
• Focal lupus nephritis (class III) is seen in 20% to 35% of cases. Lesions are visualized in fewer than half the glomeruli, and they may be segmentally or globally distributed within each glomerulus. Active lesions are characterized by swelling and proliferation of endothelial and mesangial cells, infiltration by neutrophils, and/or fibrinoid deposits with capillary thrombi (Fig. 4–18, A). The clinical presentation may range from only mild microscopic hematuria and proteinuria to a more active urinary sediment with red blood cell casts and acute, severe renal insufficiency.
• Diffuse lupus nephritis (class IV) is the most serious form of renal lesions in SLE and is also the most commonly encountered in renal biopsies, occurring in 35% to 60% of patients. It is distinguished from focal lupus nephritis (class III) by involvement of half or more of glomeruli. Most of the glomeruli show endothelial and mesangial proliferation, leading to diffuse hypercellularity of these structures (Fig. 4–18, B) and producing in some cases epithelial crescents that fill Bowman’s space. When extensive, subendothelial immune complexes create a circumferential thickening of the capillary wall, resembling rigid “wire loops” on routine light microscopy (Fig. 4–18, C). Electron microscopy reveals prominent electron-dense subendothelial immune complexes (between endothelium and basement membrane) (Fig. 4–18, D), but immune complexes are also usually present in other parts of the capillary wall and in the mesangium. Immune complexes can be visualized by staining with fluorescent antibodies directed against immunoglobulins or complement, resulting in a granular fluorescent staining pattern (Fig. 4–18, E). In due course, glomerular injury may give rise to scarring (glomerulosclerosis). Most affected patients have hematuria with moderate to severe proteinuria, hypertension, and renal insufficiency.
• Membranous lupus nephritis (class V) occurs in 10% to 15% of cases and is the designation for glomerular disease characterized by widespread thickening of the capillary wall due to deposition of subepithelial immune complexes. Membranous glomerulonephritis associated with SLE is very similar to that encountered in idiopathic membranous nephropathy (Chapter 13). Thickening of capillary walls is caused by increased deposition of basement membrane–like material, as well as accumulation of immune complexes. Patients with this histologic change almost always have severe proteinuria with overt nephrotic syndrome (Chapter 13).
• Advanced sclerosing lupus nephritis (class VI) is characterized by complete sclerosis of greater than 90% of glomeruli and corresponds to clinical end stage renal disease.
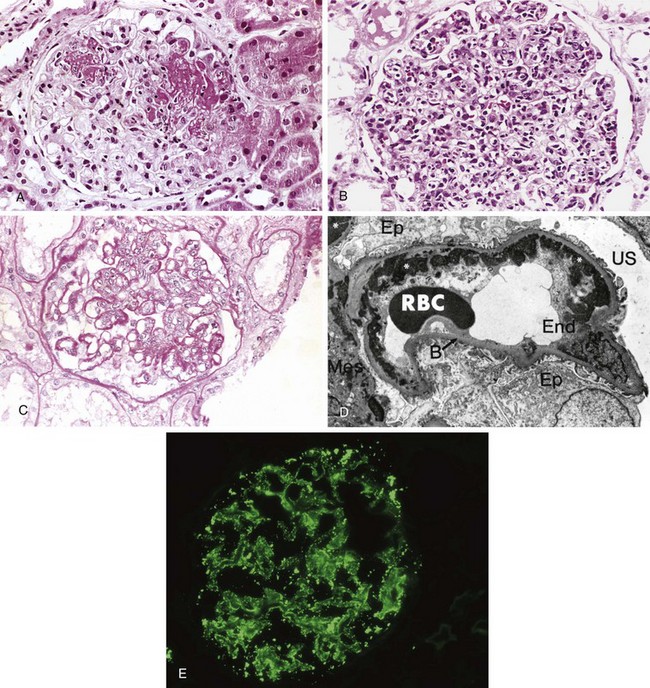
Figure 4–18 Lupus nephritis. A, Focal lupus nephritis, with two necrotizing lesions in a glomerulus (segmental distribution) (H&E stain). B, Diffuse lupus nephritis. Note the marked global increase in cellularity throughout the glomerulus (H&E stain). C, Lupus nephritis showing a glomerulus with several “wire loop” lesions representing extensive subendothelial deposits of immune complexes (periodic acid Schiff stain). D, Electron micrograph of a renal glomerular capillary loop from a patient with SLE nephritis. Confluent subendothelial dense deposits correspond to “wire loops” seen by light microscopy. E, Deposition of IgG antibody in a granular pattern, detected by immunofluorescence. B, basement membrane; End, endothelium; Ep, epithelial cell with foot processes; Mes, mesangium; RBC, red blood cell in capillary lumen; US, urinary space; *, electron-dense deposits in subendothelial location.
(A–C, courtesy of Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts. D, Courtesy of Dr. Edwin Eigenbrodt, Department of Pathology, University of Texas Southwestern Medical School, Dallas. E, Courtesy of Dr. Jean Olson, Department of Pathology, University of California, San Francisco, California.)
Skin
The skin is involved in a majority of patients; a characteristic erythematous or maculopapular eruption over the malar eminences and bridge of the nose (“butterfly pattern”) is observed in approximately half of the cases. Exposure to sunlight (UV light) exacerbates the erythema (so-called photosensitivity), and a similar rash may be present elsewhere on the extremities and trunk, frequently in sun-exposed areas. Histopathologic findings include liquefactive degeneration of the basal layer of the epidermis, edema at the dermoepidermal junction, and mononuclear infiltrates around blood vessels and skin appendages (Fig. 4–19, A). Immunofluorescence microscopy reveals deposition of immunoglobulin and complement at the dermoepidermal junction (Fig. 4–19, B); similar immunoglobulin and complement deposits may also be present in apparently uninvolved skin.
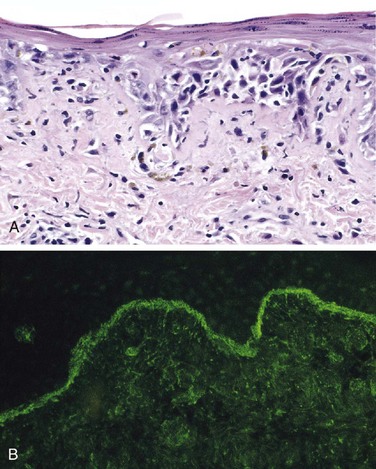
Figure 4–19 Systemic lupus erythematosus involving the skin. A, An H&E-stained section shows liquefactive degeneration of the basal layer of the epidermis and edema at the dermoepidermal junction. B, An immunofluorescence micrograph stained for IgG reveals deposits of immunoglobulin along the dermoepidermal junction. H&E, hematoxylin–eosin; IgG, immunoglobulin G.
(A, Courtesy of Dr. Jag Bhawan, Boston University School of Medicine, Boston, Massachusetts. B, Courtesy of Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas, Texas.)
Joints
Joint involvement is frequent but usually is not associated with striking anatomic changes or with joint deformity. When present, it consists of swelling and a nonspecific mononuclear cell infiltration in the synovial membranes. Erosion of the membranes and destruction of articular cartilage, such as in RA, are exceedingly rare.
CNS
Central nervous system (CNS) involvement also is very common, with focal neurologic deficits and/or neuropsychiatric symptoms. CNS disease often is ascribed to vascular lesions causing ischemia or multifocal cerebral microinfarcts. Small vessel angiopathy with noninflammatory intimal proliferation is the most frequent pathological lesion; frank vasculitis is uncommon. The angiopathy may result from thrombosis caused by antiphospholipid antibodies. Premature atherosclerosis occurs and may contribute to CNS ischemia. Another postulated mechanism for CNS disease is injury from antineuronal antibodies with consequent neurologic dysfunction, but this hypothesis remains unproved.
Other Organs
The spleen may be moderately enlarged. Capsular fibrous thickening is common, as is follicular hyperplasia with numerous plasma cells in the red pulp. Central penicilliary arteries characteristically show thickening and perivascular fibrosis, producing onion-skin lesions.
Pericardium and pleura, in particular, are serosal membranes that show a variety of inflammatory changes in SLE ranging (in the acute phase) from serous effusions to fibrinous exudates that may progress to fibrous opacification in the chronic stage.
Involvement of the heart is manifested primarily in the form of pericarditis. Myocarditis, in the form of a nonspecific mononuclear cell infiltrate, and valvular lesions, called Libman-Sacks endocarditis, also occur but are less common in the current era of aggressive corticosteroid therapy. This nonbacterial verrucous endocarditis takes the form of irregular, 1- to 3-mm warty deposits, seen as distinctive lesions on either surface of the leaflets (i.e., on the surface exposed to the forward flow of the blood or on the underside of the leaflet) (see Chapter 10). An increasing number of patients also show clinical and anatomic manifestations of coronary artery disease. The basis of accelerated atherosclerosis is not fully understood, but the process seems to be multifactorial; certainly, immune complexes can deposit in the coronary vasculature, leading to endothelial damage by that pathway. Moreover, glucocorticoid treatment causes alterations in lipid metabolism, and renal disease (common in SLE) causes hypertension; both of these are risk factors for atherosclerosis (Chapter 9).
Many other organs and tissues may be involved. The changes consist essentially of acute vasculitis of the small vessels, foci of mononuclear infiltrations, and fibrinoid deposits. In addition, lungs may reveal interstitial fibrosis, along with pleural inflammation; the liver shows nonspecific inflammation of the portal tracts.
Clinical Manifestations
SLE is a multisystem disease that is highly variable in clinical presentation. Typically, the patient is a young woman with some, but rarely all, of the following features: a butterfly rash over the face, fever, pain and swelling in one or more peripheral joints (hands and wrists, knees, feet, ankles, elbows, shoulders), pleuritic chest pain, and photosensitivity. In many patients, however, the presentation of SLE is subtle and puzzling, taking forms such as a febrile illness of unknown origin, abnormal urinary findings, or joint disease masquerading as RA or rheumatic fever. ANAs are found in virtually 100% of patients, but an important point is that ANAs are not specific (Table 4–10). A variety of clinical findings may point toward renal involvement, including hematuria, red cell casts, proteinuria, and in some cases the classic nephrotic syndrome (Chapter 13). Laboratory evidence of some hematologic derangement is common, and in some patients anemia or thrombocytopenia may be the presenting manifestation as well as the dominant clinical problem. In still others, neuropsychiatric manifestations, including psychosis or convulsions, or coronary artery disease may be prominent clinical problems. Patients with SLE are also prone to infections, presumably because of their underlying immune dysfunction and treatment with immunosuppressive drugs. Recent strategies include B cell depletion with anti-CD20 antibody (Rituximab) and by blocking growth factors. The course of the disease is variable and unpredictable. Rare acute cases progress to death within weeks to months. More often, with appropriate therapy, the disease is characterized by flareups and remissions spanning a period of years or even decades. During acute flareups, increased deposition of immune complexes and the accompanying complement activation are thought to result in hypocomplementemia. Disease exacerbations usually are treated with corticosteroids or other immunosuppressive drugs. Even without therapy, in some patients the disease may run a benign course with only skin manifestations and mild hematuria for years. The outcome has improved significantly, and a 5-year survival can be expected in approximately 95% of patients. The most common causes of death are renal failure, intercurrent infections, and cardiovascular disease. The incidence of cancer also is increased, particularly B cell lymphomas, an association common to diseases marked by B cell hyperstimulation (e.g., Sjögren syndrome, discussed below). Patients treated with steroids and immunosuppressive drugs incur the usual risks associated with such therapy.
Summary
Systemic Lupus Erythematosus
• SLE is a systemic autoimmune disease caused by autoantibodies produced against numerous self-antigens and the formation of immune complexes.
• The major autoantibodies, and the ones responsible for the formation of circulating immune complexes, are directed against nuclear antigens. Other autoantibodies react with erythrocytes, platelets, and various complexes of phospholipids with proteins.
• Disease manifestations include nephritis, skin lesions and arthritis (caused by the deposition of immune complexes), and hematologic and neurologic abnormalities.
• The underlying cause of the breakdown in self-tolerance in SLE is unknown; it may include excess or persistence of nuclear antigens, multiple inherited susceptibility genes, and environmental triggers (e.g., UV irradiation, which results in cellular apoptosis and release of nuclear proteins).
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic, chronic inflammatory disease affecting many tissues but principally attacking the joints to produce a nonsuppurative proliferative synovitis that frequently progresses to destroy articular cartilage and underlying bone with resulting disabling arthritis. Because the principal lesions affect the joints and bones, this disease, as well as the juvenile form and other inflammatory diseases of joints, is discussed in Chapter 20.
Sjögren Syndrome
Sjögren syndrome is a clinicopathologic entity characterized by dry eyes (keratoconjunctivitis sicca) and dry mouth (xerostomia), resulting from immune-mediated destruction of the lacrimal and salivary glands. It occurs as an isolated disorder (primary form), also known as the sicca syndrome, or more often in association with another autoimmune disease (secondary form). Among the associated disorders, RA is the most common, but some patients have SLE, polymyositis, systemic sclerosis, vasculitis, or thyroiditis.
Pathogenesis
Several lines of evidence suggest that Sjögren syndrome is an autoimmune disease caused by CD4+ T cell reactions against unknown antigens in the ductal epithelial cells of the exocrine glands. There is also systemic B cell hyperactivity, as evidenced by the presence of ANAs and rheumatoid factor (RF) (even in the absence of associated RA). Most patients with primary Sjögren syndrome have autoantibodies to the ribonucleoprotein (RNP) antigens SS-A (Ro) and SS-B (La); note that these antibodies are also present in some SLE patients and are therefore not diagnostic for Sjögren syndrome (Table 4–10). Although patients with high-titer anti-SS-A antibodies are more likely to have systemic (extraglandular) manifestations, there is no evidence that the autoantibodies cause primary tissue injury. A viral trigger also has been suggested, but no causative virus has been identified conclusively. Genetic variables play a role in the pathogenesis of Sjögren syndrome. As with SLE, inheritance of certain class II MHC alleles predisposes to the development of specific RNP autoantibodies.
Morphology
Lacrimal and salivary glands are the primary targets, but other secretory glands, including those in the nasopharynx, upper airway, and vagina, also may be involved. Histologic examination shows an intense lymphocyte (primarily activated CD4+ T cells) and plasma cell infiltrate, occasionally forming lymphoid follicles with germinal centers. There is associated destruction of the native architecture (Fig. 4–20).
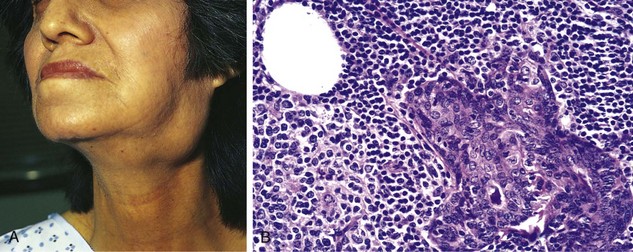
Figure 4–20 Sjögren syndrome. A, Enlargement of the salivary gland. B, Histopathologic findings include intense lymphocytic and plasma cell infiltration with ductal epithelial hyperplasia.
(A, Courtesy of Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas, Texas. B, Courtesy of Dr. Dennis Burns, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)
Lacrimal gland destruction results in a lack of tears, leading to drying of the corneal epithelium, with subsequent inflammation, erosion, and ulceration (keratoconjunctivitis). Similar changes may occur in the oral mucosa as a result of loss of salivary gland output, giving rise to mucosal atrophy, with inflammatory fissuring and ulceration (xerostomia). Dryness and crusting of the nose may lead to ulcerations and even perforation of the nasal septum. When the respiratory passages are involved, secondary laryngitis, bronchitis, and pneumonitis may appear. Approximately 25% of the patients (especially those with anti-SS-A antibodies) acquire extraglandular disease involving the CNS, skin, kidneys, and muscles. Renal lesions take the form of mild interstitial nephritis associated with tubular transport defects; unlike in SLE, glomerulonephritis is rare.
Clinical Course
Approximately 90% of Sjögren syndrome cases occur in women between the ages of 35 and 45 years. Patients present with dry mouth, lack of tears, and the resultant complications described above. Salivary glands are often enlarged as a result of lymphocytic infiltrates (Fig. 4–20). Extraglandular manifestations include synovitis, pulmonary fibrosis, and peripheral neuropathy. About 60% of Sjögren patients have another accompanying autoimmune disorder such as RA. Notably, there is a 40-fold increased risk for developing a non-Hodgkin B cell lymphoma, arising in the setting of the initial robust polyclonal B cell proliferation. These so-called marginal zone lymphomas are discussed in Chapter 11.
Summary
Sjögren Syndrome
• Sjögren syndrome is an inflammatory disease that affects primarily the salivary and lacrimal glands, causing dryness of the mouth and eyes.
• The disease is believed to be caused by an autoimmune T cell reaction against one or more unknown self antigens expressed in these glands, or immune reactions against the antigens of a virus that infects the tissues.
Systemic Sclerosis (Scleroderma)
Systemic sclerosis (SS) is an immunologic disorder characterized by excessive fibrosis in multiple tissues, obliterative vascular disease, and evidence of autoimmunity, mainly the production of multiple autoantibodies. It is commonly called scleroderma because the skin is a major target, but this disorder is better labeled “systemic” because lesions are present throughout the body. Cutaneous involvement is the usual presenting manifestation and eventually appears in approximately 95% of cases, but it is the visceral involvement—of the gastrointestinal tract, lungs, kidneys, heart, and skeletal muscles—that is responsible for most of the related morbidity and mortality.
SS can be classified into two groups on the basis of its clinical course:
• Diffuse scleroderma, characterized by initial widespread skin involvement, with rapid progression and early visceral involvement
• Limited scleroderma, with relatively mild skin involvement, often confined to the fingers and face. Involvement of the viscera occurs late, so the disease in these patients generally has a fairly benign course. This clinical presentation is also called the CREST syndrome because of its frequent features of calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia.
Pathogenesis
The cause of the disease is not known, but genetic and environmental factors probably contribute. A postulated sequence of events is the following (Fig. 4–21).
• Injury to endothelial cells of small arteries by unknown mechanisms leads to endothelial activation, increased expression of adhesion molecules, and migration of activated T cells into the perivascular tissues. The local T cell reaction may cause further activation and injury to endothelial cells.
• T cells respond to some self antigen and produce cytokines. It has been suggested that the dominant T cells are TH2 cells, and their cytokines induce alternative macrophage activation and collagen deposition. The activated T cells and macrophages produce cytokines that activate fibroblasts and stimulate collagen production, resulting in fibrosis. These cytokines include TGF-β, IL-13, platelet-derived growth factor (PDGF), and others.
• Repeated bouts of endothelial damage followed by platelet aggregation lead to endothelial proliferation and intimal fibrosis, which, together with periadventitial fibrosis, narrow the small vessels, with eventual ischemic injury. The subsequent repair reaction may lead to more fibrosis, thus setting up a self-perpetuating cycle.
• B cell activation also occurs, as indicated by the presence of hypergammaglobulinemia and ANAs. Although there is no evidence that humoral immunity plays a significant role in the pathogenesis of SS, two of the ANAs are virtually unique to this disease and are therefore useful in diagnosis (Table 4–10). One of these, directed against DNA topoisomerase I (anti-Scl 70), is highly specific; it is present in as many as 70% of patients with diffuse scleroderma (and in less than 1% of patients with other connective tissue diseases) and is a marker for the development of more aggressive disease with pulmonary fibrosis and peripheral vascular changes. The other ANA is an anticentromere antibody, found in as many as 90% of patients with limited scleroderma (i.e., the CREST syndrome); it indicates a relatively benign course.
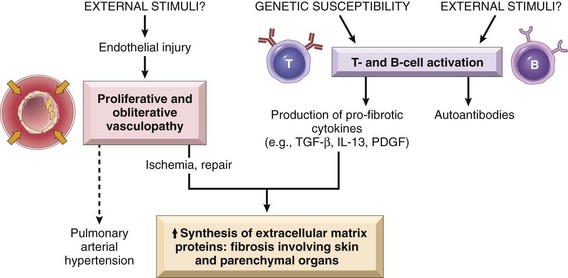
Morphology
Virtually any organ may be affected in SS, but the most prominent changes are found in the skin, musculoskeletal system, gastrointestinal tract, lungs, kidneys, and heart.
Skin
The vast majority of patients have diffuse, sclerotic atrophy of the skin, usually beginning in the fingers and distal regions of the upper extremities and extending proximally to involve the upper arms, shoulders, neck, and face. In the early stages, affected skin areas are somewhat edematous and have a doughy consistency. Histopathologic findings include edema and perivascular infiltrates containing CD4+ T cells. Capillaries and small arteries (as large as 500 µm in diameter) may show thickening of the basal lamina, endothelial cell damage, and partial occlusion. With progression, the edematous phase is replaced by progressive fibrosis of the dermis, which becomes tightly bound to the subcutaneous structures. There is marked increase of compact collagen in the dermis along with thinning of the epidermis, atrophy of the dermal appendages, and hyaline thickening of the walls of dermal arterioles and capillaries (Fig. 4–22, A, B). Focal and sometimes diffuse subcutaneous calcifications may develop, especially in patients with the CREST syndrome. In advanced stages, the fingers take on a tapered, clawlike appearance with limitation of motion in the joints (Fig. 4–22, C), and the face becomes a drawn mask. Loss of blood supply may lead to cutaneous ulcerations and to atrophic changes in the terminal phalanges, including autoamputation.
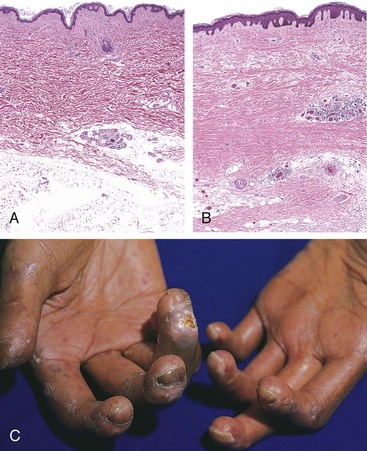
Figure 4–22 Systemic sclerosis. A, Normal skin. B, Extensive deposition of dense collagen in the dermis. C, The extensive subcutaneous fibrosis has virtually immobilized the fingers, creating a clawlike flexion deformity. Loss of blood supply has led to cutaneous ulcerations.
(A–C, Courtesy of Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas, Texas.)
Gastrointestinal Tract
The gastrointestinal tract is affected in approximately 90% of patients. Progressive atrophy and collagenous fibrous replacement of the muscularis may develop at any level of the gut but are most severe in the esophagus, with the lower two thirds often demonstrating an inflexibility not unlike that typical of a rubber hose. The associated dysfunction of the lower esophageal sphincter gives rise to gastroesophageal reflux and its complications, including Barrett metaplasia (Chapter 14) and strictures. The mucosa is thinned and may be ulcerated, and there is excessive collagenization of the lamina propria and submucosa. Loss of villi and microvilli in the small bowel is the anatomic basis for the malabsorption syndrome sometimes encountered in affected patients.
Musculoskeletal System
Synovial hyperplasia and inflammation are common in the early stages; fibrosis later ensues. Although these changes are reminiscent of RA, joint destruction is not common in SS. In a small subset of patients (approximately 10%), inflammatory myositis indistinguishable from polymyositis may develop.
Lungs
The lungs are affected in more than 50% of patients; lung involvement may manifest as pulmonary hypertension and/or interstitial fibrosis. Pulmonary vasospasm from pulmonary vascular endothelial dysfunction is considered important in the pathogenesis of pulmonary hypertension. Pulmonary fibrosis, when present, is indistinguishable from that seen in idiopathic pulmonary fibrosis (Chapter 12).
Kidneys
Renal abnormalities occur in two thirds of patients with SS, most typically associated with thickening of the vessel walls of interlobular arteries (150 to 500 µm in diameter). These show intimal cell proliferation with deposition of various glycoproteins and acid mucopolysaccharides. Although similar to the changes seen in malignant hypertension, the alterations in SS are restricted to vessels 150 to 500 µm in diameter and are not always associated with hypertension. Hypertension does occur in 30% of the patients and, in 20% of those patients, takes an ominously malignant course (malignant hypertension). In hypertensive patients, vascular alterations are more pronounced and are often associated with fibrinoid necrosis involving the arterioles together with thrombosis and infarction. Such patients often die of renal failure, accounting for about half of the deaths attributable to SS. There are no specific glomerular changes.
Heart
Patchy myocardial fibrosis, along with thickening of intramyocardial arterioles, occurs in one-third of the patients; this is putatively caused by microvascular injury and resultant ischemia (so-called cardiac Raynaud). Because of the changes in the lung, right ventricular hypertrophy and failure (cor pulmonale) are frequent.
Clinical Course
SS affects women three times more often than men, with a peak incidence in the 50- to 60-year age group. There is a substantial overlap in presentation between SS and RA, SLE, and dermatomyositis (see later); the distinctive feature of SS is the striking cutaneous involvement. Almost all patients exhibit Raynaud phenomenon, a vascular disorder characterized by reversible vasospasm of the arteries. Typically the hands turn white on exposure to cold, reflecting vasospasm, followed by change to blue as ischemia and cyanosis supervene. Finally, the color changes to red as reactive vasodilation occurs. Progressive collagen deposition in the skin leads to atrophy of the hands, with increasing stiffness and eventually complete immobilization of the joints. Difficulty in swallowing results from esophageal fibrosis and resultant hypomotility. Eventually, destruction of the esophageal wall leads to atony and dilation. Malabsorption may appear if the submucosal and muscular atrophy and fibrosis involve the small intestine. Dyspnea and chronic cough reflect the pulmonary changes; with advanced lung involvement, secondary pulmonary hypertension may develop, leading to right-sided cardiac failure. Renal functional impairment secondary to both the advance of SS and the concomitant malignant hypertension is frequently marked.
The clinical course for diffuse SS is difficult to predict. In most patients the disease pursues a steady, slow, downhill course over the span of many years, although in the absence of renal involvement, life span may be normal. The overall 10-year survival rate ranges from 35% to 70%. The chances of survival are significantly better for patients with localized scleroderma than for those with the usual diffuse progressive disease. Limited scleroderma, or CREST syndrome, frequently has Raynaud phenomenon as its presenting feature. It is associated with limited skin involvement confined to the fingers and face, and these two features may be present for decades before the appearance of visceral lesions.
Summary
Systemic Sclerosis
• SS (commonly called scleroderma) is characterized by progressive fibrosis involving the skin, gastrointestinal tract, and other tissues.
• Fibrosis may be the result of activation of fibroblasts by cytokines produced by T cells, but what triggers T cell responses is unknown.
• Endothelial injury and microvascular disease are commonly present in the lesions of SS, causing chronic ischemia, but the pathogenesis of vascular injury is not known.
Inflammatory Myopathies
Inflammatory myopathies make up a heterogeneous group of rare disorders characterized by immune-mediated muscle injury and inflammation. Based on the clinical, morphologic, and immunologic features, three disorders—polymyositis, dermatomyositis, and inclusion body myositis—have been described. These are discussed in Chapter 21.
Mixed Connective Tissue Disease
The term mixed connective tissue disease refers to a spectrum of pathologic processes in patients who present with clinical features suggestive of SLE, polymyositis, or SS; they also have high titers of antibodies to an RNP antigen called U1RNP. Two other features of mixed connective tissue disease are the paucity of renal disease and an extremely good response to corticosteroids, both of which suggest a favorable long-term prognosis.
Mixed connective tissue disease may manifest as arthritis, swelling of the hands, Raynaud phenomenon, esophageal dysmotility, myositis, leukopenia and anemia, fever, lymphadenopathy, and/or hypergammaglobulinemia. Because of these overlapping features, it is not entirely clear whether mixed connective tissue disease constitutes a distinct clinical entity or if such disorders represent heterogeneous subsets of SLE, systemic sclerosis, and polymyositis; most authorities do not consider it to be a specific entity.
Polyarteritis Nodosa and Other Vasculitides
Polyarteritis nodosa belongs to a group of diseases characterized by necrotizing inflammation of the walls of blood vessels, most likely caused by deposition of immune complexes. The general term noninfectious necrotizing vasculitis differentiates these conditions from those attributable to direct vessel infection (e.g., an abscess) and serves to emphasize that any type of vessel may be involved—arteries, arterioles, veins, or capillaries. A detailed classification and description of vasculitides are presented in Chapter 9.
IgG4-Related Disease
IgG4-related disease (IgG4-RD) is a newly recognized fibroinflammatory condition characterized by a tendency to form tumor-like lesions in several organs. The disorder is often, but not always, associated with elevated serum IgG4 concentrations. However, increased numbers of IgG4-producing plasma cells (or an increased IgG4 to total IgG ratio) in tissue are a sine qua non of this disorder. Although recognized only recently when extra-pancreatic manifestations were identified in patients with autoimmune pancreatitis, IgG4-RD has now been described in virtually every organ system: the biliary tree, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid, pericardium, and skin. The histopathologic features bear striking similarities across organs, regardless of the site of disease. These include a mixed infiltrate of lymphoid cells (T cells, B cells, plasma cells), storiform fibrosis, obliterative phlebitis, and mild to moderate tissue eosinophilia. B cells are typically organized in germinal centers but the T cells—the predominant cell type—are distributed diffusely throughout the lesion. The ratio of IgG4 to IgG-bearing plasma cells (determined by semiquantitative immunohistochemistry) is typically equal to or greater than 50%.
Many medical conditions long viewed as confined to single organs are part of the IgG4-RD spectrum. These include Mikulicz syndrome (enlargement and fibrosis of salivary and lacrimal glands), Riedel thyroiditis, idiopathic retroperitoneal fibrosis, autoimmune pancreatitis, and inflammatory pseudotumors of the orbit, lungs, and kidneys, to name a few.
The role of IgG4 in the pathogenesis of this condition is not fully understood. However, the key role of B cells is supported by initial studies in which depletion of B cells by anti–B cell reagents such as Rituximab provided clinical benefit. It is unclear if the disease is truly autoimmune in nature, and no target autoantigens have been identified.