Chapter 3 Hemodynamic Disorders, Thromboembolism, and Shock
See Targeted Therapy available online at studentconsult.com
The health of cells and tissues depends on the circulation of blood, which delivers oxygen and nutrients and removes wastes generated by cellular metabolism. Under normal conditions, as blood passes through capillary beds, proteins in the plasma are retained within the vasculature and there is little net movement of water and electrolytes into the tissues. This balance is often disturbed by pathologic conditions that alter endothelial function, increase vascular pressure, or decrease plasma protein content, all of which promote edema—accumulation of fluid resulting from a net outward movement of water into extravascular spaces. Depending on its severity and location, edema may have minimal or profound effects. In the lower extremities, it may only make one’s shoes feel snugger after a long sedentary day; in the lungs, however, edema fluid can fill alveoli, causing life-threatening hypoxia.
Our blood vessels are frequently subject to trauma of varying degrees. Hemostasis is the process of blood clotting that prevents excessive bleeding after blood vessel damage. Inadequate hemostasis may result in hemorrhage, which can compromise regional tissue perfusion and, if massive and rapid, may lead to hypotension, shock, and death. Conversely, inappropriate clotting (thrombosis) or migration of clots (embolism) can obstruct blood vessels, potentially causing ischemic cell death (infarction). Indeed, thromboembolism lies at the heart of three major causes of morbidity and death in developed countries: myocardial infarction, pulmonary embolism, and cerebrovascular accident (stroke).
Hyperemia and Congestion
Hyperemia and congestion both refer to an increase in blood volume within a tissue but they have different underlying mechanisms. Hyperemia is an active process resulting from arteriolar dilation and increased blood inflow, as occurs at sites of inflammation or in exercising skeletal muscle. Hyperemic tissues are redder than normal because of engorgement with oxygenated blood. Congestion is a passive process resulting from impaired outflow of venous blood from a tissue. It can occur systemically, as in cardiac failure, or locally as a consequence of an isolated venous obstruction. Congested tissues have an abnormal blue-red color (cyanosis) that stems from the accumulation of deoxygenated hemoglobin in the affected area. In long-standing chronic congestion, inadequate tissue perfusion and persistent hypoxia may lead to parenchymal cell death and secondary tissue fibrosis, and the elevated intravascular pressures may cause edema or sometimes rupture capillaries, producing focal hemorrhages.
 Morphology
Morphology
Cut surfaces of hyperemic or congested tissues feel wet and typically ooze blood. On microscopic examination, acute pulmonary congestion is marked by blood-engorged alveolar capillaries and variable degrees of alveolar septal edema and intra-alveolar hemorrhage. In chronic pulmonary congestion, the septa become thickened and fibrotic, and the alveolar spaces contain numerous macrophages laden with hemosiderin (“heart failure cells”) derived from phagocytosed red cells. In acute hepatic congestion, the central vein and sinusoids are distended with blood, and there may even be central hepatocyte dropout due to necrosis. The periportal hepatocytes, better oxygenated because of their proximity to hepatic arterioles, experience less severe hypoxia and may develop only reversible fatty change. In chronic passive congestion of the liver, the central regions of the hepatic lobules, viewed on gross examination, are red-brown and slightly depressed (owing to cell loss) and are accentuated against the surrounding zones of uncongested tan, sometimes fatty, liver (nutmeg liver) (Fig. 3–1, A). Microscopic findings include centrilobular hepatocyte necrosis, hemorrhage, and hemosiderin-laden macrophages (Fig. 3–1, B). In long-standing, severe hepatic congestion (most commonly associated with heart failure), hepatic fibrosis (“cardiac cirrhosis”) can develop. Because the central portion of the hepatic lobule is the last to receive blood, centrilobular necrosis also can occur in any setting of reduced hepatic blood flow (including shock from any cause); there need not be previous hepatic congestion.
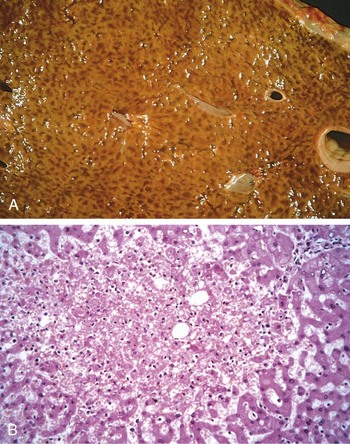
Figure 3–1 Liver with chronic passive congestion and hemorrhagic necrosis. A, In this autopsy specimen, central areas are red and slightly depressed compared with the surrounding tan viable parenchyma, creating “nutmeg liver” (so called because it resembles the cut surface of a nutmeg). B, Microscopic preparation shows centrilobular hepatic necrosis with hemorrhage and scattered inflammatory cells.
(Courtesy of Dr. James Crawford.)
Edema
Approximately 60% of lean body weight is water, two thirds of which is intracellular. Most of the remaining water is found in extracellular compartments in the form of interstitial fluid; only 5% of the body’s water is in blood plasma. As noted earlier, edema is an accumulation of interstitial fluid within tissues. Extravascular fluid can also collect in body cavities such as the pleural cavity (hydrothorax), the pericardial cavity (hydropericardium), or the peritoneal cavity (hydroperitoneum, or ascites). Anasarca is severe, generalized edema marked by profound swelling of subcutaneous tissues and accumulation of fluid in body cavities.
Table 3–1 lists the major causes of edema. The mechanisms of inflammatory edema are largely related to increased vascular permeability and are discussed in Chapter 2; the noninflammatory causes are detailed in the following discussion.
Table 3–1 Pathophysiologic Causes of Edema
| Increased Hydrostatic Pressure |
| Impaired Venous Return |
| Arteriolar Dilation |
| Reduced Plasma Osmotic Pressure (Hypoproteinemia) |
| Lymphatic Obstruction |
| Sodium Retention |
| Inflammation |
Data from Leaf A, Cotran RS: Renal Pathophysiology, 3rd ed. New York, Oxford University Press, 1985, p 146.
Fluid movement between the vascular and interstitial spaces is governed mainly by two opposing forces—the vascular hydrostatic pressure and the colloid osmotic pressure produced by plasma proteins. Normally, the outflow of fluid produced by hydrostatic pressure at the arteriolar end of the microcirculation is neatly balanced by inflow due to the slightly elevated osmotic pressure at the venular end; hence there is only a small net outflow of fluid into the interstitial space, which is drained by lymphatic vessels. Either increased hydrostatic pressure or diminished colloid osmotic pressure causes increased movement of water into the interstitium (Fig. 3–2). This in turn increases the tissue hydrostatic pressure, and eventually a new equilibrium is achieved. Excess edema fluid is removed by lymphatic drainage and returned to the bloodstream by way of the thoracic duct (Fig. 3–2).
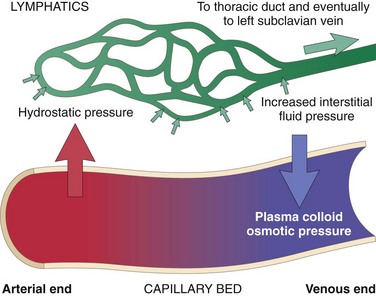
Figure 3–2 Factors influencing fluid movement across capillary walls. Capillary hydrostatic and osmotic forces are normally balanced so there is little net movement of fluid into the interstitium. However, increased hydrostatic pressure or diminished plasma osmotic pressure leads to extravascular fluid accumulation (edema). Tissue lymphatics drain much of the excess fluid back to the circulation by way of the thoracic duct; however, if the capacity for lymphatic drainage is exceeded, tissue edema results.
The edema fluid that accumulates owing to increased hydrostatic pressure or reduced intravascular colloid typically is a protein-poor transudate; it has a specific gravity less than 1.012. By contrast, because of increased vascular permeability, inflammatory edema fluid is a protein-rich exudate with a specific gravity usually greater than 1.020 (see Chapter 2). We will now discuss the various causes of edema.
Increased Hydrostatic Pressure
Local increases in intravascular pressure can result from impaired venous return—for example, a deep venous thrombosis in the lower extremity can cause edema restricted to the distal portion of the affected leg. Generalized increases in venous pressure, with resultant systemic edema, occur most commonly in congestive heart failure (Chapter 10). Several factors increase venous hydrostatic pressure in patients with congestive heart failure (Fig. 3–3). The reduced cardiac output leads to hypoperfusion of the kidneys, triggering the renin-angiotensin-aldosterone axis and inducing sodium and water retention (secondary hyperaldosteronism). In patients with normal heart function, this adaptation increases cardiac filling and cardiac output, thereby improving renal perfusion. However, the failing heart often cannot increase its cardiac output in response to the compensatory increases in blood volume. Instead, a vicious circle of fluid retention, increased venous hydrostatic pressures, and worsening edema ensues. Unless cardiac output is restored or renal water retention is reduced (e.g., by salt restriction or treatment with diuretics or aldosterone antagonists) this downward spiral continues. Because secondary hyperaldosteronism is a common feature of generalized edema, salt restriction, diuretics, and aldosterone antagonists also are of value in the management of generalized edema resulting from other causes.
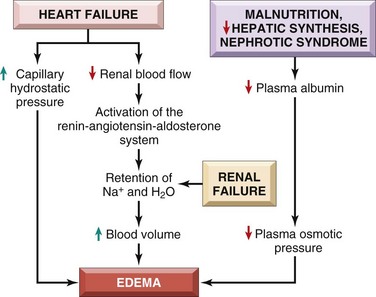
Reduced Plasma Osmotic Pressure
Under normal circumstances albumin accounts for almost half of the total plasma protein. Therefore conditions in which albumin is either lost from the circulation or synthesized in inadequate amounts are common causes of reduced plasma osmotic pressure. In nephrotic syndrome (Chapter 13), damaged glomerular capillaries become leaky, leading to the loss of albumin (and other plasma proteins) in the urine and the development of generalized edema. Reduced albumin synthesis occurs in the setting of severe liver disease (e.g., cirrhosis) (Chapter 15) and protein malnutrition (Chapter 7). Regardless of cause, low albumin levels lead in a stepwise fashion to edema, reduced intravascular volume, renal hypoperfusion, and secondary hyperaldosteronism. Unfortunately, increased salt and water retention by the kidney not only fails to correct the plasma volume deficit but also exacerbates the edema, since the primary defect—low serum protein—persists.
Lymphatic Obstruction
Impaired lymphatic drainage and consequent lymphedema usually result from a localized obstruction caused by an inflammatory or neoplastic condition. For example, the parasitic infection filariasis can cause massive edema of the lower extremity and external genitalia (so-called elephantiasis) by engendering inguinal lymphatic and lymph node fibrosis. Infiltration and obstruction of superficial lymphatics by breast cancer may cause edema of the overlying skin; the characteristic finely pitted appearance of the skin of the affected breast is called peau d’orange (orange peel). Lymphedema also may occur as a complication of therapy. One relatively common setting for this clinical entity is in women with breast cancer who undergo axillary lymph node resection and/or irradiation, both of which can disrupt and obstruct lymphatic drainage, resulting in severe lymphedema of the arm.
Sodium and Water Retention
Excessive retention of salt (and its obligate associated water) can lead to edema by increasing hydrostatic pressure (due to expansion of the intravascular volume) and reducing plasma osmotic pressure. Excessive salt and water retention are seen in a wide variety of diseases that compromise renal function, including poststreptococcal glomerulonephritis and acute renal failure (Chapter 13).
Morphology
Edema is easily recognized on gross inspection; microscopic examination shows clearing and separation of the extracellular matrix elements. Although any tissue can be involved, edema most commonly is encountered in subcutaneous tissues, lungs, and brain.
Subcutaneous edema can be diffuse but usually accumulates preferentially in parts of the body positioned the greatest distance below the heart where hydrostatic pressures are highest. Thus, edema typically is most pronounced in the legs with standing and the sacrum with recumbency, a relationship termed dependent edema. Finger pressure over edematous subcutaneous tissue displaces the interstitial fluid, leaving a finger-shaped depression; this appearance is called pitting edema. Edema due to renal dysfunction or nephrotic syndrome often manifests first in loose connective tissues (e.g., the eyelids, causing periorbital edema). With pulmonary edema, the lungs often are two to three times their normal weight, and sectioning reveals frothy, sometimes blood-tinged fluid consisting of a mixture of air, edema fluid, and extravasated red cells. Brain edema can be localized (e.g., due to abscess or tumor) or generalized, depending on the nature and extent of the pathologic process or injury. With generalized edema, the sulci are narrowed while the gyri are swollen and flattened against the skull.
Clinical Correlation
The effects of edema vary, ranging from merely annoying to rapidly fatal. Subcutaneous edema is important to recognize primarily because it signals potential underlying cardiac or renal disease; however, when significant, it also can impair wound healing or the clearance of infections. Pulmonary edema is a common clinical problem that most frequently is seen in the setting of left ventricular failure but also may occur in renal failure, acute respiratory distress syndrome (Chapter 11), and inflammatory and infectious disorders of the lung. It can cause death by interfering with normal ventilatory function; besides impeding oxygen diffusion, alveolar edema fluid also creates a favorable environment for infections. Brain edema is life-threatening; if the swelling is severe, the brain can herniate (extrude) through the foramen magnum. With increased intracranial pressure, the brain stem vascular supply can be compressed. Either condition can cause death by injuring the medullary centers (Chapter 22).
Hemorrhage
Hemorrhage, defined as the extravasation of blood from vessels, occurs in a variety of settings. As described earlier, capillary bleeding can occur in chronically congested tissues. The risk of hemorrhage (often after a seemingly insignificant injury) is increased in a wide variety of clinical disorders collectively called hemorrhagic diatheses. Trauma, atherosclerosis, or inflammatory or neoplastic erosion of a vessel wall also may lead to hemorrhage, which may be extensive if the affected vessel is a large vein or artery.
Hemorrhage may be manifested by different appearances and clinical consequences.
• Hemorrhage may be external or accumulate within a tissue as a hematoma, which ranges in significance from trivial (e.g., a bruise) to fatal (e.g., a massive retroperitoneal hematoma resulting from rupture of a dissecting aortic aneurysm) (Chapter 9).
Large bleeds into body cavities are given various names according to location—hemothorax, hemopericardium, hemoperitoneum, or hemarthrosis (in joints). Extensive hemorrhages can occasionally result in jaundice from the massive breakdown of red cells and hemoglobin.
• Petechiae are minute (1 to 2 mm in diameter) hemorrhages into skin, mucous membranes, or serosal surfaces (Fig. 3–4, A); causes include low platelet counts (thrombocytopenia), defective platelet function, and loss of vascular wall support, as in vitamin C deficiency (Chapter 7).
• Purpura are slightly larger (3 to 5 mm) hemorrhages. Purpura can result from the same disorders that cause petechiae, as well as trauma, vascular inflammation (vasculitis), and increased vascular fragility.
• Ecchymoses are larger (1 to 2 cm) subcutaneous hematomas (colloquially called bruises). Extravasated red cells are phagocytosed and degraded by macrophages; the characteristic color changes of a bruise are due to the enzymatic conversion of hemoglobin (red-blue color) to bilirubin (blue-green color) and eventually hemosiderin (golden-brown).
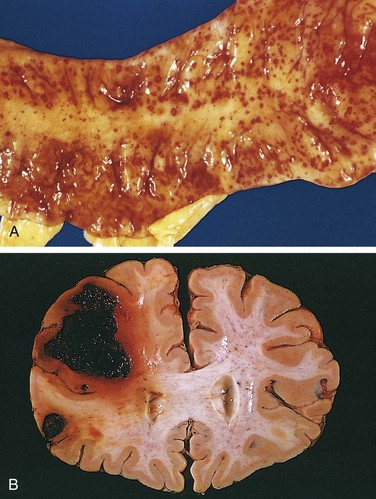
Figure 3–4 A, Punctate petechial hemorrhages of the colonic mucosa, a consequence of thrombocytopenia. B, Fatal intracerebral hemorrhage.
The clinical significance of any particular hemorrhage depends on the volume of blood lost and the rate of bleeding. Rapid loss of up to 20% of the blood volume, or slow losses of even larger amounts, may have little impact in healthy adults; greater losses, however, can cause hemorrhagic (hypovolemic) shock (discussed later). The site of hemorrhage also is important; bleeding that would be trivial in the subcutaneous tissues can cause death if located in the brain (Fig. 3–4, B). Finally, chronic or recurrent external blood loss (e.g., due to peptic ulcer or menstrual bleeding) frequently culminates in iron deficiency anemia as a consequence of loss of iron in hemoglobin. By contrast, iron is efficiently recycled from phagocytosed red cells, so internal bleeding (e.g., a hematoma) does not lead to iron deficiency.
Hemostasis and Thrombosis
Normal hemostasis comprises a series of regulated processes that maintain blood in a fluid, clot-free state in normal vessels while rapidly forming a localized hemostatic plug at the site of vascular injury. The pathologic counterpart of hemostasis is thrombosis, the formation of blood clot (thrombus) within intact vessels. Both hemostasis and thrombosis involve three elements: the vascular wall, platelets, and the coagulation cascade. The discussion here begins with normal hemostasis and its regulation.
Normal Hemostasis
The main steps in the process of hemostasis and its regulation are summarized below and shown in Figure 3–5.
• Vascular injury causes transient arteriolar vasoconstriction through reflex neurogenic mechanisms, augmented by local secretion of endothelin (a potent endothelium-derived vasoconstrictor) (Fig. 3–5, A). This effect is fleeting, however, and bleeding would quickly resume if not for the activation of platelets and coagulation factors.
• Endothelial injury exposes highly thrombogenic subendothelial extracellular matrix (ECM), facilitating platelet adherence, activation, and aggregation. The formation of the initial platelet plug is called primary hemostasis (Fig. 3–5, B).
• Endothelial injury also exposes tissue factor (also known as factor III or thromboplastin), a membrane-bound procoagulant glycoprotein synthesized by endothelial cells. Exposed tissue factor, acting in conjunction with factor VII (see later), is the major in vivo trigger of the coagulation cascade and its activation eventually culminates in the activation of thrombin, which has several roles in regulating coagulation.
• Activated thrombin promotes the formation of an insoluble fibrin clot by cleaving fibrinogen; thrombin also is a potent activator of additional platelets, which serve to reinforce the hemostatic plug. This sequence, termed secondary hemostasis, results in the formation of a stable clot capable of preventing further hemorrhage (Fig. 3–5, C).
• As bleeding is controlled, counterregulatory mechanisms (e.g., factors that produce fibrinolysis, such as tissue-type plasminogen activator) are set into motion to ensure that clot formation is limited to the site of injury (Fig. 3–5, D).
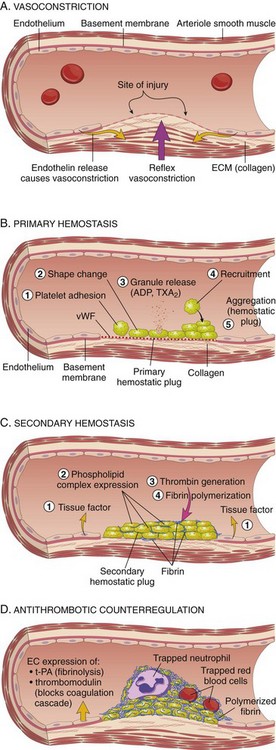
Figure 3–5 Normal hemostasis. A, After vascular injury, local neurohumoral factors induce a transient vasoconstriction. B, Platelets bind via glycoprotein 1b (GpIb) receptors to von Willebrand factor (vWF) on exposed extracellular matrix (ECM) and are activated, undergoing a shape change and granule release. Released adenosine diphosphate (ADP) and thromboxane A2 (TxA2) induce additional platelet aggregation through binding of platelet GpIIb-IIIa receptors to fibrinogen. This platelet aggregate fills the vascular defect, forming the primary hemostatic plug. C, Local activation of the coagulation cascade (involving tissue factor and platelet phospholipids) results in fibrin polymerization, “cementing” the platelets into a definitive secondary hemostatic plug that is larger and more stable than the primary plug and contains entrapped red cells and leukocytes. D, Counterregulatory mechanisms, such as release of t-PA (tissue plasminogen activator, a fibrinolytic product) and thrombomodulin (interfering with the coagulation cascade), limit the hemostatic process to the site of injury.
Discussed next in greater detail are the roles of endothelium, platelets, and the coagulation cascade.
Endothelium
Endothelial cells are central regulators of hemostasis; the balance between the anti- and prothrombotic activities of endothelium determines whether thrombus formation, propagation, or dissolution occurs. Normal endothelial cells express a variety of anticoagulant factors that inhibit platelet aggregation and coagulation and promote fibrinolysis; after injury or activation, however, this balance shifts, and endothelial cells acquire numerous procoagulant activities (Fig. 3–6). Besides trauma, endothelium can be activated by microbial pathogens, hemodynamic forces, and a number of pro-inflammatory mediators (Chapter 2).
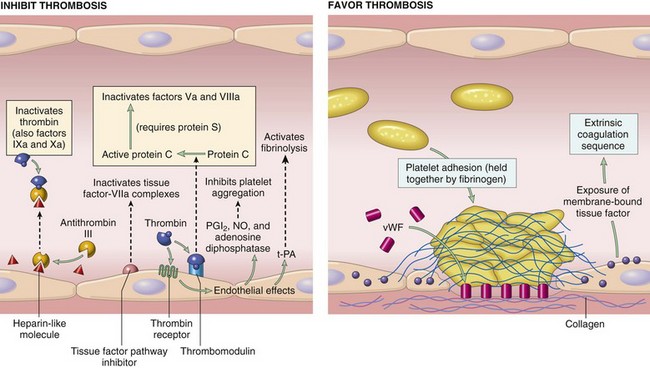
Figure 3–6 Anticoagulant properties of normal endothelium (left) and procoagulant properties of injured or activated endothelium (right). NO, nitric oxide; PGI2, prostaglandin I2 (prostacyclin); t-PA, tissue plasminogen activator; vWF, von Willebrand factor. Thrombin receptors are also called protease-activated receptors (PARs).
Antithrombotic Properties of Normal Endothelium
Inhibitory Effects on Platelets
Intact endothelium prevents platelets (and plasma coagulation factors) from engaging the highly thrombogenic subendothelial ECM. Nonactivated platelets do not adhere to normal endothelium; even with activated platelets, prostacyclin (i.e., prostaglandin I2 [PGI2]) and nitric oxide produced by endothelium impede their adhesion. Both mediators also are potent vasodilators and inhibitors of platelet aggregation; their synthesis by endothelial cells is stimulated by a number of factors (e.g., thrombin, cytokines) produced during coagulation. Endothelial cells also produce adenosine diphosphatase, which degrades adenosine diphosphate (ADP) and further inhibits platelet aggregation (see later).
Inhibitory Effects on Coagulation Factors
These actions are mediated by factors expressed on endothelial surfaces, particularly heparin-like molecules, thrombomodulin, and tissue factor pathway inhibitor (Fig. 3–6). The heparin-like molecules act indirectly: They are cofactors that greatly enhance the inactivation of thrombin (and other coagulation factors) by the plasma protein antithrombin III. Thrombomodulin also acts indirectly: It binds to thrombin, thereby modifying the substrate specificity of thrombin, so that instead of cleaving fibrinogen, it instead cleaves and activates protein C, an anticoagulant. Activated protein C inhibits clotting by cleaving and inactivating two procoagulants, factor Va and factor VIIIa; it requires a cofactor, protein S, which is also synthesized by endothelial cells. Finally, tissue factor pathway inhibitor (TFPI) directly inhibits tissue factor–factor VIIa complex and factor Xa.
Prothrombotic Properties of Injured or Activated Endothelium
Activation of Platelets
Endothelial injury brings platelets into contact with the subendothelial ECM, which includes among its constituents von Willebrand factor (vWF), a large multimeric protein that is synthesized by EC. vWF is held fast to the ECM through interactions with collagen and also binds tightly to Gp1b, a glycoprotein found on the surface of platelets. These interactions allow vWF to act as a sort of molecular glue that binds platelets tightly to denuded vessel walls (Fig. 3–7).
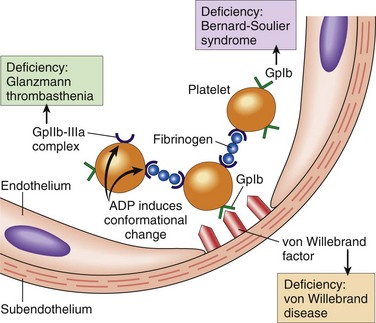
Figure 3–7 Platelet adhesion and aggregation. Von Willebrand factor functions as an adhesion bridge between subendothelial collagen and the glycoprotein Ib (GpIb) platelet receptor. Platelet aggregation is accomplished by fibrinogen binding to platelet GpIIb-IIIa receptors on different platelets. Congenital deficiencies in the various receptors or bridging molecules lead to the diseases indicated in the colored boxes. ADP, adenosine diphosphate.
Activation of Clotting Factors
In response to cytokines (e.g., tumor necrosis factor [TNF] or interleukin-1 [IL-1]) or certain bacterial products including endotoxin, endothelial cells produce tissue factor, the major in vivo activator of coagulation, and downregulate the expression of thrombomodulin. Activated endothelial cells also bind coagulation factors IXa and Xa (see further on), which augments the catalytic activities of these factors.
Antifibrinolytic Effects
Activated endothelial cells secrete plasminogen activator inhibitors (PAIs), which limit fibrinolysis and thereby favor thrombosis.
 Summary
Summary
Endothelial Cells and Coagulation
• Intact, normal endothelial cells help to maintain blood flow by inhibiting the activation of platelets and coagulation factors.
• Endothelial cells stimulated by injury or inflammatory cytokines upregulate expression of procoagulant factors (e.g., tissue factor) that promote clotting, and downregulate expression of anticoagulant factors.
• Loss of endothelial integrity exposes subendothelial vWF and basement membrane collagen, stimulating platelet adhesion, platelet activation, and clot formation.
Platelets
Platelets are anucleate cell fragments shed into the bloodstream by marrow megakaryocytes. They play a critical role in normal hemostasis by forming a hemostatic plug that seals vascular defects, and by providing a surface that recruits and concentrates activated coagulation factors. Platelet function depends on several integrin family glycoprotein receptors, a contractile cytoskeleton, and two types of cytoplasmic granules:
• α granules, which express the adhesion molecule P-selectin on their membranes (Chapter 2) and contain fibrinogen, fibronectin, factors V and VIII, platelet factor-4 (a heparin-binding chemokine), platelet-derived growth factor (PDGF), and transforming growth factor-β (TGF-β)
• Dense bodies (δ granules), which contain adenine nucleotides (ADP and ATP), ionized calcium, histamine, serotonin, and epinephrine
After vascular injury, platelets encounter ECM constituents (collagen is most important) and adhesive glycoproteins such as vWF. This sets in motion a series of events that lead to (1) platelet adhesion, (2) platelet activation, and (3) platelet aggregation (Fig. 3–5, B).
Platelet Adhesion
Platelet adhesion initiates clot formation and depends on vWF and platelet glycoprotein Gp1b. Under shear stress (e.g., in flowing blood), vWF undergoes a conformational change, assuming an extended shape that allows it to bind simultaneously to collagen in the ECM and to platelet Gp1b (Fig. 3–7). The importance of this adhesive interaction is highlighted by genetic deficiencies of vWF and Gp1b, both of which result in bleeding disorders—von Willebrand disease (Chapter 11) and Bernard-Soulier disease (a rare condition), respectively.
Platelet Activation
Platelet adhesion leads to an irreversible shape change and secretion (release reaction) of both granule types—a process termed platelet activation. Calcium and ADP released from δ granules are especially important in subsequent events, since calcium is required by several coagulation factors and ADP is a potent activator of resting platelets. Activated platelets also synthesize thromboxane A2 (TxA2) (Chapter 2), a prostaglandin that activates additional nearby platelets and that also has an important role in platelet aggregation (described below). During activation, platelets undergo a dramatic change in shape from smooth discs to spheres with numerous long, spiky membrane extensions, as well as more subtle changes in the make-up of their plasma membranes. The shape changes enhance subsequent aggregation and increase the surface area available for interaction with coagulation factors. The subtle membrane changes include an increase in the surface expression of negatively charged phospholipids, which provide binding sites for both calcium and coagulation factors, and a conformation change in platelet GpIIb/IIIa that permits it to bind fibrinogen.
Platelet Aggregation
Platelet aggregation follows platelet adhesion and activation, and is stimulated by some of the same factors that induce platelet activation, such as TxA2. Aggregation is promoted by bridging interactions between fibrinogen and GpIIb/IIIa receptors on adjacent platelets (Fig. 3–7). The importance of this interaction is emphasized by a rare inherited deficiency of GpIIb/IIIa (Glanzmann thrombasthenia), which is associated with bleeding and an inability of platelets to aggregate. Recognition of the central role of GpIIb-IIIa receptors in platelet aggregation has stimulated the development of antithrombotic agents that inhibit GpIIb-IIIa function.
Concurrent activation of the coagulation cascade generates thrombin, which stabilizes the platelet plug through two mechanisms:
• Thrombin activates a platelet surface receptor (protease-activated receptor [PAR]), which in concert with ADP and TxA2 further enhances platelet aggregation. Platelet contraction follows, creating an irreversibly fused mass of platelets that constitutes the definitive secondary hemostatic plug.
• Thrombin converts fibrinogen to fibrin (discussed shortly) within the vicinity of the plug, cementing the platelet plug in place.
Red cells and leukocytes are also found in hemostatic plugs. Leukocytes adhere to platelets by means of P-selectin and to endothelium by various adhesion molecules (Chapter 2); they contribute to the inflammatory response that accompanies thrombosis. Thrombin also promotes inflammation by stimulating neutrophil and monocyte adhesion (described later) and by generating chemotactic fibrin split products during fibrinogen cleavage.
Platelet-Endothelial Interactions
The interplay of platelets and endothelium has a profound impact on clot formation. For example, prostaglandin PGI2 (synthesized by normal endothelium) is a vasodilator and inhibits platelet aggregation, whereas TxA2 (synthesized by activated platelets, as discussed above) is a potent vasoconstrictor. The balance between the opposing effects of PGI2 and TxA2 varies: In normal vessels, PGI2 effects dominate and platelet aggregation is prevented, whereas endothelial injury decreases PGI2 production and promotes platelet aggregation and TxA2 production. The clinical utility of aspirin (an irreversible cyclooxygenase inhibitor) in lowering the risk of coronary thrombosis resides in its ability to permanently block TxA2 production by platelets, which have no capacity for protein synthesis. Although endothelial PGI2 production is also inhibited by aspirin, endothelial cells can resynthesize cyclooxygenase, thereby overcoming the blockade. In a manner similar to that for PGI2, endothelium-derived nitric oxide also acts as a vasodilator and inhibitor of platelet aggregation (Fig. 3–6).
Summary
Platelet Adhesion, Activation, and Aggregation
• Endothelial injury exposes the underlying basement membrane ECM; platelets adhere to the ECM primarily through binding of platelet GpIb receptors to vWF.
• Adhesion leads to platelet activation, an event associated with secretion of platelet granule contents, including calcium (a cofactor for several coagulation proteins) and ADP (a mediator of further platelet activation); dramatic changes in shape and membrane composition; and activation of GpIIb/IIIa receptors.
• The GpIIb/IIIa receptors on activated platelets form bridging crosslinks with fibrinogen, leading to platelet aggregation.
• Concomitant activation of thrombin promotes fibrin deposition, cementing the platelet plug in place.
Coagulation Cascade
The coagulation cascade constitutes the third arm of the hemostatic system. The pathways are schematically presented in Figure 3–8; only general principles are discussed here.
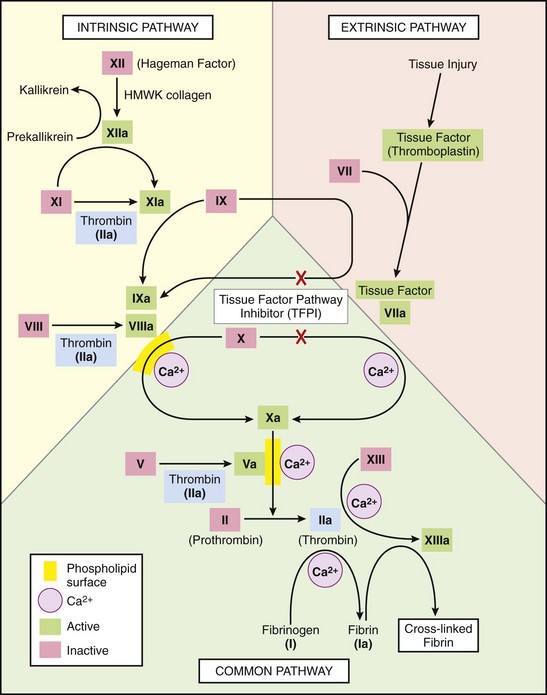
Figure 3–8 The coagulation cascade. Factor IX can be activated by either factor XIa or factor VIIa: In laboratory tests, activation is predominantly dependent on factor XIa, whereas in vivo, factor VIIa appears to be the predominant activator of factor IX. Factors in red boxes represent inactive molecules; activated factors, indicated with a lowercase a, are in green boxes. Note that thrombin (factor IIa) (in light blue boxes) contributes to coagulation through multiple positive feedback loops. The red X’s denote points at which tissue factor pathway inhibitor (TFPI) inhibits activation of factor X and factor IX by factor VIIa. HMWK, high-molecular-weight kininogen; PL, phospholipid.
The coagulation cascade is a successive series of amplifying enzymatic reactions. At each step in the process, a proenzyme is proteolyzed to become an active enzyme, which in turn proteolyzes the next proenzyme in the series, eventually leading to the activation of thrombin and the formation of fibrin. Thrombin has a key role, as it acts at numerous points in the cascade (highlighted in Fig. 3–8). Thrombin proteolyzes fibrinogen into fibrin monomers that polymerize into an insoluble gel; this gel encases platelets and other circulating cells in the definitive secondary hemostatic plug. Fibrin polymers are stabilized by the cross-linking activity of factor XIIIa, which also is activated by thrombin.
Each reaction in the pathway depends on the assembly of a complex composed of an enzyme (an activated coagulation factor), a substrate (a proenzyme form of the next coagulation factor in the series), and a cofactor (a reaction accelerator). These components typically are assembled on a phospholipid surface (provided by endothelial cells or platelets) and are held together by interactions that depend on calcium ions (explaining why blood clotting is prevented by calcium chelators). As shown in Figure 3–9, the sequential cascade of activation can be likened to a “dance” of complexes, with coagulation factors being passed successively from one partner to the next. Parenthetically, the ability of coagulation factors II, VII, IX, and X to bind to calcium requires that additional γ-carboxyl groups be enzymatically appended to certain glutamic acid residues on these proteins. This reaction requires vitamin K as a cofactor and is antagonized by drugs such as coumadin, which is widely used as an anticoagulant.
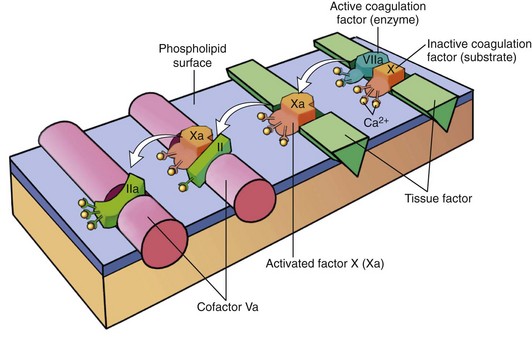
Figure 3–9 Sequential conversion of factor X to factor Xa by way of the extrinsic pathway, followed by conversion of factor II (prothrombin) to factor IIa (thrombin). The initial reaction complex consists of a protease (factor VIIa), a substrate (factor X), and a reaction accelerator (tissue factor) assembled on a platelet phospholipid surface. Calcium ions hold the assembled components together and are essential for the reaction. Activated factor Xa then becomes the protease component of the next complex in the cascade, converting prothrombin to thrombin (factor IIa) in the presence of a different reaction accelerator, factor Va.
Blood coagulation traditionally is divided into extrinsic and intrinsic pathways, converging at the activation of factor X (Fig. 3–8). The extrinsic pathway was so designated because it required the addition of an exogenous trigger (originally provided by tissue extracts); the intrinsic pathway only required exposing factor XII (Hageman factor) to a negatively charged surface (even glass suffices). However, this division is largely an artifact of in vitro testing; there are, in fact, several interconnections between the two pathways. The extrinsic pathway is the most physiologically relevant pathway for coagulation occurring after vascular damage; it is activated by tissue factor, a membrane-bound glycoprotein expressed at sites of injury.
Clinical labs assess the function of the two arms of the pathway using two standard assays.
• Prothrombin time (PT) screens for the activity of the proteins in the extrinsic pathway (factors VII, X, II, V, and fibrinogen). The PT is performed by adding phospholipids and tissue factor to a patient’s citrated plasma (sodium citrate chelates calcium and prevents spontaneous clotting), followed by calcium, and the time to fibrin clot formation (usually 11 to 13 seconds) is recorded. Because factor VII is the vitamin K–dependent coagulation factor with the shortest half-life (roughly 7 hours), the PT is used to guide treatment of patients with vitamin K antagonists (e.g., coumadin).
• Partial thromboplastin time (PTT) screens for the activity of the proteins in the intrinsic pathway (factors XII, XI, IX, VIII, X, V, II, and fibrinogen). The PTT is performed by adding a negatively charged activator of factor XII (e.g., ground glass) and phospholipids to a patient’s citrated plasma, followed by calcium, and recording the time required for clot formation (usually 28 to 35 seconds). The PTT is sensitive to the anticoagulant effects of heparin and is therefore used to monitor its efficacy.
Once thrombin is formed, it not only catalyzes the final steps in the coagulation cascade, but also exerts a wide variety of effects on the local vasculature and inflammatory milieu; it even actively participates in limiting the extent of the hemostatic process (Fig. 3–10). Most of these thrombin-mediated effects occur through protease-activated receptors (PARs), which belong to a family of seven-transmembrane-spanning proteins. PARs are present on a variety of cell types, including platelets, endothelium, monocytes, and T lymphocytes. Thrombin activates PARs by clipping their extracellular domains, causing a conformational change that activates associated G proteins. Thus, PAR activation is a catalytic process, explaining the impressive potency of thrombin in eliciting PAR-dependent effects, such as enhancing the adhesive properties of leukocytes.
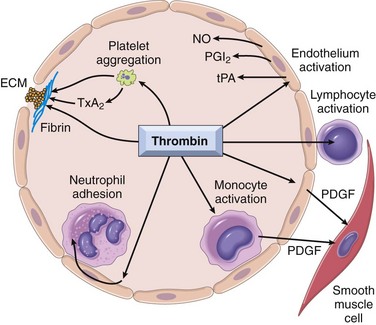
Figure 3–10 Role of thrombin in hemostasis and cellular activation. Thrombin generates fibrin by cleaving fibrinogen, activates factor XIII (which is responsible for cross-linking fibrin into an insoluble clot), and also activates several other coagulation factors, thereby amplifying the coagulation cascade (Fig. 3–8). Through protease-activated receptors (PARs), thrombin activates (1) platelet aggregation and TxA2 secretion; (2) endothelium, which responds by generating leukocyte adhesion molecules and a variety of fibrinolytic (t-PA), vasoactive (NO, PGI2), or cytokine (PDGF) mediators; and (3) leukocytes, increasing their adhesion to activated endothelium. ECM, extracellular matrix; NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostaglandin I2 (prostacyclin); TxA2, thromboxane A2; t-PA, tissue type plasminogen activator. See Figure 3–6 for anticoagulant activities mediated by thrombin via thrombomodulin.
(Courtesy of permission from Shaun Coughlin, MD, PhD, Cardiovascular Research Institute, University of California at San Francisco, San Francisco, California.)
Once activated, the coagulation cascade must be tightly restricted to the site of injury to prevent inappropriate and potentially dangerous clotting elsewhere in the vascular tree. Besides restricting factor activation to sites of exposed phospholipids, clotting also is controlled by three general categories of natural anticoagulants:
• Antithrombins (e.g., antithrombin III) inhibit the activity of thrombin and other serine proteases, namely factors IXa, Xa, XIa, and XIIa. Antithrombin III is activated by binding to heparin-like molecules on endothelial cells—hence the clinical utility of heparin administration to limit thrombosis (Fig. 3–6).
• Protein C and protein S are two vitamin K–dependent proteins that act in a complex to proteolytically inactivate cofactors Va and VIIIa. Protein C activation by thrombomodulin was described earlier; protein S is a cofactor for protein C activity (Fig. 3–6).
• Tissue factor pathway inhibitor (TFPI) is a protein secreted by endothelium (and other cell types) that inactivates factor Xa and tissue factor–factor VIIa complexes (Fig. 3–8).
Clotting also sets into motion a fibrinolytic cascade that moderates the ultimate size of the clot. Fibrinolysis is largely carried out by plasmin, which breaks down fibrin and interferes with its polymerization (Fig. 3–11). The resulting fibrin split products (FSPs or fibrin degradation products) also can act as weak anticoagulants. Elevated levels of FSPs (most notably fibrin-derived D-dimers) can be used for diagnosing abnormal thrombotic states including disseminated intravascular coagulation (DIC) (Chapter 11), deep venous thrombosis, or pulmonary thromboembolism (described in detail later).
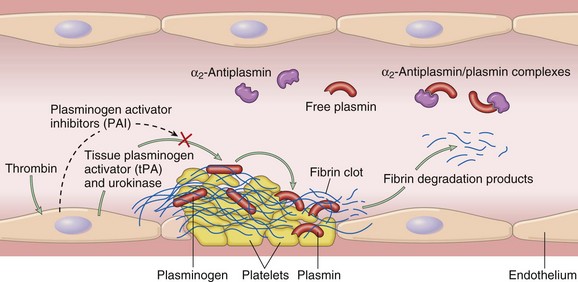
Figure 3–11 The fibrinolytic system, illustrating various plasminogen activators and inhibitors (see text).
Plasmin is generated by proteolysis of plasminogen, an inactive plasma precursor, either by factor XII or by plasminogen activators (Fig. 3–11). The most important of the plasminogen activators is tissue-type plasminogen activator (t-PA); t-PA is synthesized principally by endothelial cells and is most active when attached to fibrin. The affinity for fibrin largely confines t-PA fibrinolytic activity to sites of recent thrombosis. Urokinase-like plasminogen activator (u-PA) is another plasminogen activator present in plasma and in various tissues; it can activate plasmin in the fluid phase. In addition, plasminogen can be cleaved to its active form by the bacterial product streptokinase, which is used clinically to lyse clots in some forms of thrombotic disease. As with any potent regulatory component, the activity of plasmin is tightly restricted. To prevent excess plasmin from lysing thrombi indiscriminately throughout the body, free plasmin rapidly complexes with circulating α2-antiplasmin and is inactivated (Fig. 3–11).
Endothelial cells further modulate the coagulation–anticoagulation balance by releasing plasminogen activator inhibitors (PAIs); these block fibrinolysis and confer an overall procoagulation effect (Fig. 3–11). PAI production is increased by inflammatory cytokines (in particular interferon-γ) and probably contributes to the intravascular thrombosis that accompanies severe inflammation.
Summary
Coagulation Factors
• Coagulation occurs via the sequential enzymatic conversion of a cascade of circulating and locally synthesized proteins.
• Tissue factor elaborated at sites of injury is the most important initiator of the coagulation cascade in vivo.
• At the final stage of coagulation, thrombin converts fibrinogen into insoluble fibrin that contributes to formation of the definitive hemostatic plug.
• Coagulation normally is restricted to sites of vascular injury by
 limiting enzymatic activation to phospholipid surfaces provided by activated platelets or endothelium natural anticoagulants elaborated at sites of endothelial injury or during activation of the coagulation cascade
limiting enzymatic activation to phospholipid surfaces provided by activated platelets or endothelium natural anticoagulants elaborated at sites of endothelial injury or during activation of the coagulation cascadeThrombosis
Having reviewed the process of normal hemostasis, we now turn to the three primary abnormalities that lead to thrombus formation (called Virchow’s triad): (1) endothelial injury, (2) stasis or turbulent blood flow, and (3) hypercoagulability of the blood (Fig. 3–12).
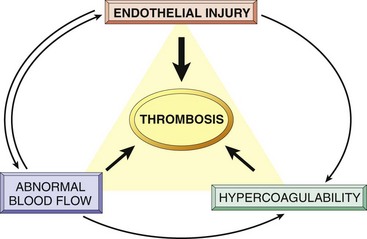
Figure 3–12 Virchow’s triad in thrombosis. Endothelial integrity is the most important factor. Abnormalities of procoagulants or anticoagulants can tip the balance in favor of thrombosis. Abnormal blood flow (stasis or turbulence) can lead to hypercoagulability directly and also indirectly through endothelial dysfunction.
Endothelial Injury
Endothelial injury is an important cause of thrombosis, particularly in the heart and the arteries, where high flow rates might otherwise impede clotting by preventing platelet adhesion or diluting coagulation factors. Examples of thrombosis related to endothelial damage are the formation of thrombi in the cardiac chambers after myocardial infarction, over ulcerated plaques in atherosclerotic arteries, or at sites of traumatic or inflammatory vascular injury (vasculitis). Overt loss of endothelium exposes subendothelial ECM (leading to platelet adhesion), releases tissue factor, and reduces local production of PGI2 and plasminogen activators. Of note, however, endothelium need not be denuded or physically disrupted to contribute to the development of thrombosis; any perturbation in the dynamic balance of the prothrombotic and antithrombotic effects of endothelium can influence clotting locally. Thus, dysfunctional endothelium elaborates greater amounts of procoagulant factors (e.g., platelet adhesion molecules, tissue factor, PAI) and synthesizes lesser amounts of anticoagulant molecules (e.g., thrombomodulin, PGI2, t-PA). Endothelial dysfunction can be induced by a variety of insults, including hypertension, turbulent blood flow, bacterial products, radiation injury, metabolic abnormalities such as homocystinuria and hypercholesterolemia, and toxins absorbed from cigarette smoke.
Abnormal Blood Flow
Turbulence contributes to arterial and cardiac thrombosis by causing endothelial injury or dysfunction, as well as by forming countercurrents and local pockets of stasis. Stasis is a major factor in the development of venous thrombi. Under conditions of normal laminar blood flow, platelets (and other blood cells) are found mainly in the center of the vessel lumen, separated from the endothelium by a slower-moving layer of plasma. By contrast, stasis and turbulent (chaotic) blood flow have the following deleterious effects:
• Both promote endothelial cell activation and enhanced procoagulant activity, in part through flow-induced changes in endothelial gene expression.
• Stasis allows platelets and leukocytes to come into contact with the endothelium when the flow is sluggish.
• Stasis also slows the washout of activated clotting factors and impedes the inflow of clotting factor inhibitors.
Turbulent and static blood flow contribute to thrombosis in a number of clinical settings. Ulcerated atherosclerotic plaques not only expose subendothelial ECM but also cause turbulence. Abnormal aortic and arterial dilations called aneurysms create local stasis and consequently a fertile site for thrombosis (Chapter 9). Acute myocardial infarction results in focally noncontractile myocardium. Ventricular remodeling after more remote infarction can lead to aneurysm formation. In both cases, cardiac mural thrombi are more easily formed due to the local blood stasis (Chapter 10). Mitral valve stenosis (e.g., after rheumatic heart disease) results in left atrial dilation. In conjunction with atrial fibrillation, a dilated atrium is a site of profound stasis and a prime location for the development of thrombi. Hyperviscosity syndromes (such as polycythemia) (Chapter 11) increase resistance to flow and cause small vessel stasis; the deformed red cells in sickle cell anemia (Chapter 11) cause vascular occlusions, and the resultant stasis also predisposes to thrombosis.
Hypercoagulability
Hypercoagulability contributes infrequently to arterial or intracardiac thrombosis but is an important underlying risk factor for venous thrombosis. It is loosely defined as any alteration of the coagulation pathways that predisposes affected persons to thrombosis, and can be divided into primary (genetic) and secondary (acquired) disorders (Table 3–2).
Table 3–2 Hypercoagulable States
| Primary (Genetic) |
| Common (>1% of the Population) |
| Rare |
| Very Rare |
| Secondary (Acquired) |
| High Risk for Thrombosis |
| Lower Risk for Thrombosis |
Primary (inherited) hypercoagulability most often is caused by mutations in the factor V and prothrombin genes:
• Approximately 2% to 15% of whites carry a specific factor V mutation (called the Leiden mutation, after the Dutch city where it was first described). The mutation alters an amino acid residue in factor V and renders it resistant to protein C. Thus, an important antithrombotic counter-regulatory mechanism is lost. Heterozygotes carry a 5-fold increased risk for venous thrombosis, with homozygotes having a 50-fold increased risk.
• A single-nucleotide substitution (G to A) in the 3′-untranslated region of the prothrombin gene is a fairly common allele (found in 1% to 2% of the general population). This variant results in increased prothrombin transcription and is associated with a nearly three-fold increased risk for venous thromboses.
• Less common primary hypercoagulable states include inherited deficiencies of anticoagulants such as antithrombin III, protein C, or protein S; affected patients typically present with venous thrombosis and recurrent thromboembolism in adolescence or early adult life. Congenitally elevated levels of homocysteine contribute to arterial and venous thromboses (and indeed to the development of atherosclerosis) (Chapter 9).
Although the risk of thrombosis is only mildly increased in heterozygous carriers of factor V Leiden and the prothrombin gene variant, these genetic factors carry added significance for two reasons. First, both abnormal alleles are sufficiently frequent that homozygous and compound heterozygous persons are not uncommon, and these individuals are at much higher risk for thrombosis. More importantly, heterozygous individuals are at higher risk for venous thrombosis in the setting of other acquired risk factors, such as pregnancy, prolonged bed rest, and lengthy airplane flights. Consequently, inherited causes of hypercoagulability should be considered in young patients (<50 years of age), even when other acquired risk factors are present.
Secondary (acquired) hypercoagulability is seen in many settings (Table 3–2). In some situations (e.g., cardiac failure or trauma), stasis or vascular injury may be the most important factor. The hypercoagulability associated with oral contraceptive use and the hyperestrogenic state of pregnancy may be related to increased hepatic synthesis of coagulation factors and reduced synthesis of antithrombin III. In disseminated cancers, release of procoagulant tumor products (e.g., mucin from adenocarcinoma) predisposes to thrombosis. The hypercoagulability seen with advancing age has been attributed to increased platelet aggregation and reduced release of PGI2 from endothelium. Smoking and obesity promote hypercoagulability by unknown mechanisms.
Among the acquired thrombophilic states, two are particularly important clinical problems and deserve special mention:
• Heparin-induced thrombocytopenic (HIT) syndrome. This syndrome occurs in up to 5% of patients treated with unfractionated heparin (for therapeutic anticoagulation). It is marked by the development of autoantibodies that bind complexes of heparin and platelet membrane protein (platelet factor-4) (Chapter 11). Although the mechanism is unclear, it appears that these antibodies may also bind similar complexes present on platelet and endothelial surfaces, resulting in platelet activation, aggregation, and consumption (hence thrombocytopenia), as well as causing endothelial cell injury. The overall result is a prothrombotic state, even in the face of heparin administration and low platelet counts. Newer low-molecular-weight fractionated heparin preparations induce autoantibodies less frequently but can still cause thrombosis if antibodies have already formed.
• Antiphospholipid antibody syndrome. This syndrome has protean manifestations, including recurrent thrombosis, repeated miscarriages, cardiac valve vegetations, and thrombocytopenia; it is associated with autoantibodies directed against anionic phospholipids (e.g., cardiolipin) or—more accurately—plasma protein antigens that are unveiled by binding to such phospholipids (e.g., prothrombin). In vivo, these antibodies induce a hypercoagulable state, perhaps by inducing endothelial injury, by activating platelets or complement directly, or by interacting with the catalytic domains of certain coagulation factors. In vitro (in the absence of platelets and endothelium), however, the antibodies interfere with phospholipid complex assembly, thereby inhibiting coagulation (hence the designation lupus anticoagulant). In patients with anticardiolipin antibodies, serologic testing for syphilis will yield a false-positive result, because the antigen in the standard assays is embedded in cardiolipin.
Patients with antiphospholipid antibody syndrome fall into two categories. Many have secondary antiphospholipid syndrome due to a well-defined autoimmune disease, such as systemic lupus erythematosus (Chapter 4). The remainder of these patients exhibit only the manifestations of a hypercoagulable state without evidence of another autoimmune disorder (primary antiphospholipid syndrome). Although antiphospholipid antibodies are associated with thrombotic diatheses, they also occur in 5% to 15% of apparently normal persons; the implication is that their presence may be necessary but not sufficient to cause full-blown antiphospholipid antibody syndrome.
Morphology
Thrombi can develop anywhere in the cardiovascular system. Arterial or cardiac thrombi typically arise at sites of endothelial injury or turbulence; venous thrombi characteristically occur at sites of stasis. Thrombi are focally attached to the underlying vascular surface and tend to propagate toward the heart; thus, arterial thrombi grow in a retrograde direction from the point of attachment, while venous thrombi extend in the direction of blood flow. The propagating portion of a thrombus tends to be poorly attached and therefore prone to fragmentation and migration through the blood as an embolus.
Thrombi can have grossly (and microscopically) apparent laminations called lines of Zahn; these represent pale platelet and fibrin layers alternating with darker red cell–rich layers. Such lines are significant in that they are only found in thrombi that form in flowing blood; their presence can therefore usually distinguish antemortem thrombosis from the bland nonlaminated clots that form in the postmortem state. Although thrombi formed in the “low-flow” venous system superficially resemble postmortem clots, careful evaluation generally reveals ill-defined laminations.
Thrombi occurring in heart chambers or in the aortic lumen are designated mural thrombi. Abnormal myocardial contraction (arrhythmias, dilated cardiomyopathy, or myocardial infarction) or endomyocardial injury (myocarditis, catheter trauma) promote cardiac mural thrombi (Fig. 3–13, A), while ulcerated atherosclerotic plaques and aneurysmal dilation promote aortic thrombosis (Fig. 3-13, B).
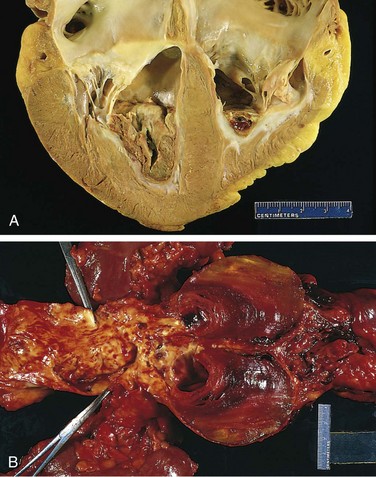
Figure 3–13 Mural thrombi. A, Thrombus in the left and right ventricular apices, overlying white fibrous scar. B, Laminated thrombus in a dilated abdominal aortic aneurysm. Numerous friable mural thrombi are also superimposed on advanced atherosclerotic lesions of the more proximal aorta (left side of photograph).
Arterial thrombi are typically relatively rich in platelets, as the processes underlying their development (e.g., endothelial injury) lead to platelet activation. Although usually superimposed on a ruptured atherosclerotic plaque, other vascular injuries (vasculitis, trauma) can also be causal. Venous thrombi (phlebothrombosis) frequently propagate some distance toward the heart, forming a long cast within the vessel lumen that is prone to give rise to emboli. An increase in the activity of coagulation factors is involved in the genesis of most venous thrombi, with platelet activation playing a secondary role. Because these thrombi form in the sluggish venous circulation, they tend to contain more enmeshed red cells, leading to the moniker red, or stasis, thrombi. The veins of the lower extremities are most commonly affected (90% of venous thromboses); however, venous thrombi also can occur in the upper extremities, periprostatic plexus, or ovarian and periuterine veins, and under special circumstances may be found in the dural sinuses, portal vein, or hepatic vein.
At autopsy, postmortem clots can sometimes be mistaken for venous thrombi. However, the former are gelatinous and due to red cell settling have a dark red dependent portion and a yellow “chicken fat” upper portion; they also are usually not attached to the underlying vessel wall. By contrast, red thrombi typically are firm, focally attached to vessel walls, and contain gray strands of deposited fibrin.
Thrombi on heart valves are called vegetations. Bacterial or fungal blood-borne infections can cause valve damage, leading to the development of large thrombotic masses (infective endocarditis) (Chapter 10). Sterile vegetations also can develop on noninfected valves in hypercoagulable states—the lesions of so-called nonbacterial thrombotic endocarditis (Chapter 10). Less commonly, sterile, verrucous endocarditis (Libman-Sacks endocarditis) can occur in the setting of systemic lupus erythematosus (Chapter 4).
Fate of the Thrombus
If a patient survives an initial thrombotic event, over the ensuing days to weeks the thrombus evolves through some combination of the following four processes:
• Propagation. The thrombus enlarges through the accretion of additional platelets and fibrin, increasing the odds of vascular occlusion or embolization.
• Embolization. Part or all of the thrombus is dislodged and transported elsewhere in the vasculature.
• Dissolution. If a thrombus is newly formed, activation of fibrinolytic factors may lead to its rapid shrinkage and complete dissolution. With older thrombi, extensive fibrin polymerization renders the thrombus substantially more resistant to plasmin-induced proteolysis, and lysis is ineffectual. This acquisition of resistance to lysis has clinical significance, as therapeutic administration of fibrinolytic agents (e.g., t-PA in the setting of acute coronary thrombosis) generally is not effective unless given within a few hours of thrombus formation.
• Organization and recanalization. Older thrombi become organized by the ingrowth of endothelial cells, smooth muscle cells, and fibroblasts into the fibrin-rich thrombus (Fig. 3–14). In time, capillary channels are formed that—to a limited extent—create conduits along the length of the thrombus, thereby reestablishing the continuity of the original lumen. Further recanalization can sometimes convert a thrombus into a vascularized mass of connective tissue that is eventually incorporated into the wall of the remodeled vessel. Occasionally, instead of organizing, the center of a thrombus undergoes enzymatic digestion, presumably because of the release of lysosomal enzymes from entrapped leukocytes. If bacterial seeding occurs, the contents of degraded thrombi serve as an ideal culture medium, and the resulting infection may weaken the vessel wall, leading to formation of a mycotic aneurysm (Chapter 9).
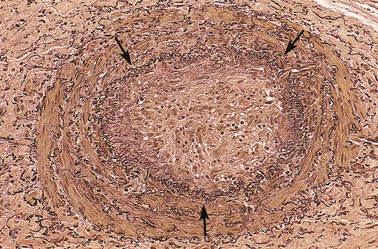
Figure 3–14 Low-power view of a thrombosed artery stained for elastic tissue. The original lumen is delineated by the internal elastic lamina (arrows) and is totally filled with organized thrombus.
Clinical Correlation
Thrombi are significant because they cause obstruction of arteries and veins and may give rise to emboli. Which effect is of greatest clinical importance depends on the site of thrombosis. Thus, while venous thrombi can cause congestion and edema in vascular beds distal to an obstruction, they are most worrisome because of their potential to embolize to the lungs and cause death. Conversely, while arterial thrombi can embolize and cause tissue infarction, their tendency to obstruct vessels (e.g., in coronary and cerebral vessels) is considerably more important.
Venous Thrombosis (Phlebothrombosis)
Most venous thrombi occur in either the superficial or the deep veins of the leg. Superficial venous thrombi usually arise in the saphenous system, particularly in the setting of varicosities; these rarely embolize but can be painful and can cause local congestion and swelling from impaired venous outflow, predisposing the overlying skin to development of infections and varicose ulcers. Deep venous thromboses (“DVTs”) in the larger leg veins at or above the knee joint (e.g., popliteal, femoral, and iliac veins) are more serious because they are prone to embolize. Although such DVTs may cause local pain and edema, the venous obstruction often is circumvented by collateral channels. Consequently, DVTs are entirely asymptomatic in approximately 50% of patients and are recognized only after they have embolized to the lungs.
Lower-extremity DVTs are associated with stasis and hypercoagulable states, as described earlier (Table 3–2); thus, common predisposing factors include congestive heart failure, bed rest and immobilization; the latter two factors reduce the milking action of leg muscles and thus slow venous return. Trauma, surgery, and burns not only immobilize a patient but are also associated with vascular injury, procoagulant release, increased hepatic synthesis of coagulation factors, and reduced t-PA production. Many factors contribute to the thrombotic diathesis of pregnancy; besides the potential for amniotic fluid infusion into the circulation at the time of delivery, pressure produced by the enlarging fetus and uterus can produce stasis in the veins of the legs, and late pregnancy and the postpartum period are associated with hypercoagulability. Tumor-associated procoagulant release is largely responsible for the increased risk of thromboembolic phenomena seen in disseminated cancers, which is sometimes referred to as migratory thrombophlebitis due to its tendency to transiently involve several different venous beds, or as Trousseau syndrome, for Armand Trousseau, who both described the disorder and suffered from it. Regardless of the specific clinical setting, the risk of DVT is increased in persons over age 50.
While the many conditions that predispose to thrombosis are well recognized, the phenomenon remains unpredictable. It occurs at a distressingly high frequency in otherwise healthy and ambulatory people without apparent provocation or underlying abnormality. Equally important is that asymptomatic thrombosis (and presumably subsequent resolution) occurs considerably more frequently than is generally appreciated.
Summary
Thrombosis
• Thrombus development usually is related to one or more components of Virchow’s triad:
hypercoagulability: either primary (e.g., factor V Leiden, increased prothrombin synthesis, antithrombin III deficiency) or secondary (e.g., bed rest, tissue damage, malignancy)• Thrombi may propagate, resolve, become organized, or embolize.
• Thrombosis causes tissue injury by local vascular occlusion or by distal embolization.
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is the sudden or insidious onset of widespread thrombosis within the microcirculation. It may be seen in disorders ranging from obstetric complications to advanced malignancy. The thrombi are generally microscopic in size, yet so numerous as to often cause circulatory insufficiency, particularly in the brain, lungs, heart, and kidneys. To complicate matters, the widespread microvascular thrombosis consumes platelets and coagulation proteins (hence the synonym consumption coagulopathy), and at the same time, fibrinolytic mechanisms are activated. Thus, an initially thrombotic disorder can evolve into a bleeding catastrophe. A point worthy of emphasis is that DIC is not a primary disease but rather a potential complication of numerous conditions associated with widespread activation of thrombin. It is discussed in greater detail along with other bleeding diatheses in Chapter 11.
Embolism
An embolus is an intravascular solid, liquid, or gaseous mass that is carried by the blood to a site distant from its point of origin. The vast majority of emboli derive from a dislodged thrombus—hence the term thromboembolism. Less common types of emboli include fat droplets, bubbles of air or nitrogen, atherosclerotic debris (cholesterol emboli), tumor fragments, bits of bone marrow, and amniotic fluid. Inevitably, emboli lodge in vessels too small to permit further passage, resulting in partial or complete vascular occlusion; depending on the site of origin, emboli can lodge anywhere in the vascular tree. The primary consequence of systemic embolization is ischemic necrosis (infarction) of downstream tissues, while embolization in the pulmonary circulation leads to hypoxia, hypotension, and right-sided heart failure.
Pulmonary Thromboembolism
The incidence of pulmonary embolism is 2 to 4 per 1000 hospitalized patients. Although the rate of fatal pulmonary embolus (PE) has declined from 6% to 2% over the last quarter-century, pulmonary embolism still causes about 200,000 deaths per year in the United States. In greater than 95% of cases, venous emboli originate from thrombi within deep leg veins proximal to the popliteal fossa; embolization from lower leg thrombi is uncommon.
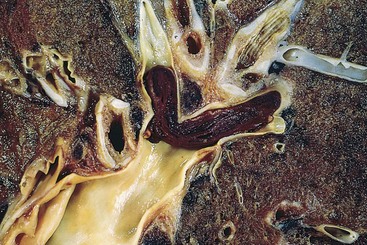
Fragmented thrombi from DVTs are carried through progressively larger channels and usually pass through the right side of the heart before arresting in the pulmonary vasculature. Depending on size, a PE can occlude the main pulmonary artery, lodge at the bifurcation of the right and left pulmonary arteries (saddle embolus), or pass into the smaller, branching arterioles (Fig. 3–15). Frequently, multiple emboli occur, either sequentially or as a shower of smaller emboli from a single large thrombus; a patient who has had one pulmonary embolus is at increased risk for having more. Rarely, an embolus passes through an atrial or ventricular defect and enters the systemic circulation (paradoxical embolism). A more complete discussion of PE is found in Chapter 12; the major clinical and pathologic features are the following:
• Most pulmonary emboli (60% to 80%) are small and clinically silent. With time, they undergo organization and become incorporated into the vascular wall; in some cases, organization of thromboemboli leaves behind bridging fibrous webs.
• At the other end of the spectrum, a large embolus that blocks a major pulmonary artery can cause sudden death.
• Embolic obstruction of medium-sized arteries and subsequent rupture of capillaries rendered anoxic can cause pulmonary hemorrhage. Such embolization does not usually cause pulmonary infarction since the area also receives blood through an intact bronchial circulation (dual circulation). However, a similar embolus in the setting of left-sided cardiac failure (and diminished bronchial artery perfusion) can lead to a pulmonary infarct.
• Embolism to small end-arteriolar pulmonary branches usually causes infarction.
• Multiple emboli occurring over time can cause pulmonary hypertension and right ventricular failure (cor pulmonale).
Systemic Thromboembolism
Most systemic emboli (80%) arise from intracardiac mural thrombi; two thirds are associated with left ventricular infarcts and another 25% with dilated left atria (e.g., secondary to mitral valve disease). The remainder originate from aortic aneurysms, thrombi overlying ulcerated atherosclerotic plaques, fragmented valvular vegetations (Chapter 10), or the venous system (paradoxical emboli); 10% to 15% of systemic emboli are of unknown origin.
By contrast with venous emboli, which lodge primarily in the lung, arterial emboli can travel virtually anywhere; their final resting place understandably depends on their point of origin and the relative flow rates of blood to the downstream tissues. Common arteriolar embolization sites include the lower extremities (75%) and central nervous system (10%); intestines, kidneys, and spleen are less common targets. The consequences of embolization depend on the caliber of the occluded vessel, the collateral supply, and the affected tissue’s vulnerability to anoxia; arterial emboli often lodge in end arteries and cause infarction.
Fat Embolism
Soft tissue crush injury or rupture of marrow vascular sinusoids (long bone fracture) releases microscopic fat globules into the circulation. Fat and marrow emboli are common incidental findings after vigorous cardiopulmonary resuscitation but probably are of little clinical consequence. Similarily, although fat and marrow embolism occurs in some 90% of individuals with severe skeletal injuries (Fig. 3–16, A), less than 10% show any clinical findings. However, a minority of patients develop a symptomatic fat embolism syndrome characterized by pulmonary insufficiency, neurologic symptoms, anemia, thrombocytopenia, and a diffuse petechial rash, which is fatal in 10% of cases. Clinical signs and symptoms appear 1 to 3 days after injury as the sudden onset of tachypnea, dyspnea, tachycardia, irritability, and restlessness, which can progress rapidly to delirium or coma.
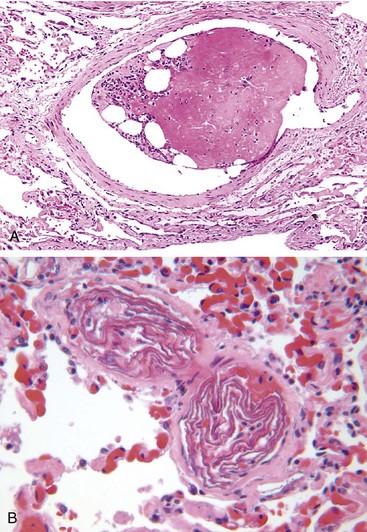
Figure 3–16 Unusual types of emboli. A, Bone marrow embolus. The embolus is composed of hematopoietic marrow and marrow fat cells (clear spaces) attached to a thrombus. B, Amniotic fluid emboli. Two small pulmonary arterioles are packed with laminated swirls of fetal squamous cells. The surrounding lung is edematous and congested.
(Courtesy of Dr. Beth Schwartz, Baltimore, Maryland.)
The pathogenesis of fat emboli syndrome involves both mechanical obstruction and biochemical injury. Fat microemboli occlude pulmonary and cerebral microvasculature, both directly and by triggering platelet aggregation. This deleterious effect is exacerbated by fatty acid release from lipid globules, which causes local toxic endothelial injury. Platelet activation and granulocyte recruitment (with free radical, protease, and eicosanoid release) (Chapter 2) complete the vascular assault. Because lipids are dissolved by the solvents used during tissue-processing, microscopic demonstration of fat microglobules (i.e., in the absence of accompanying marrow elements) requires specialized techniques (frozen sections and fat stains).
Amniotic Fluid Embolism
Amniotic fluid embolism is an uncommon, grave complication of labor and the immediate postpartum period (1 in 40,000 deliveries). The mortality rate approaches 80%, making it the most common cause of maternal death in the developed world; it accounts for 10% of maternal deaths in the United States, while 85% of survivors suffer some form of permanent neurologic deficit. Onset is characterized by sudden severe dyspnea, cyanosis, and hypotensive shock, followed by seizures and coma. If the patient survives the initial crisis, pulmonary edema typically develops, along with (in about half the patients) disseminated intravascular coagulation secondary to release of thrombogenic substances from amniotic fluid.
The underlying cause is entry of amniotic fluid (and its contents) into the maternal circulation via tears in the placental membranes and/or uterine vein rupture. Histologic analysis reveals squamous cells shed from fetal skin, lanugo hair, fat from vernix caseosa, and mucin derived from the fetal respiratory or gastrointestinal tracts in the maternal pulmonary microcirculation (Fig. 13-16, B). Other findings include marked pulmonary edema, diffuse alveolar damage (Chapter 12), and systemic fibrin thrombi generated by disseminated intravascular coagulation.
Air Embolism
Gas bubbles within the circulation can coalesce and obstruct vascular flow and cause distal ischemic injury. Thus, a small volume of air trapped in a coronary artery during bypass surgery or introduced into the cerebral arterial circulation by neurosurgery performed in an upright “sitting position” can occlude flow, with dire consequences. Small venous gas emboli generally have no deleterious effects, but sufficient air can enter the pulmonary circulation inadvertently during obstetric procedures or as a consequence of a chest wall injury to cause hypoxia, and very large venous emboli may arrest in the heart and cause death.
A particular form of gas embolism called decompression sickness is caused by sudden changes in atmospheric pressure. Thus, scuba divers, underwater construction workers, and persons in unpressurized aircraft who undergo rapid ascent are at risk. When air is breathed at high pressure (e.g., during a deep sea dive), increased amounts of gas (particularly nitrogen) become dissolved in the blood and tissues. If the diver then ascends (depressurizes) too rapidly, the nitrogen expands in the tissues and bubbles out of solution in the blood to form gas emboli, which cause tissue ischemia. Rapid formation of gas bubbles within skeletal muscles and supporting tissues in and about joints is responsible for the painful condition called “the bends” (so named in the 1880s because the afflicted person arches the back in a manner reminiscent of a then-popular women’s fashion pose called the Grecian bend). Gas bubbles in the pulmonary vasculature cause edema, hemorrhages, and focal atelectasis or emphysema, leading to respiratory distress, the so-called chokes. A more chronic form of decompression sickness is called caisson disease (named for pressurized underwater vessels used during bridge construction) in which recurrent or persistent gas emboli in the bones lead to multifocal ischemic necrosis; the heads of the femurs, tibiae, and humeri are most commonly affected.
Acute decompression sickness is treated by placing affected persons in a high-pressure chamber, to force the gas back into solution. Subsequent slow decompression permits gradual gas resorption and exhalation so that obstructive bubbles do not re-form.
Summary
Embolism
• An embolus is a solid, liquid, or gaseous mass carried by the blood to a site distant from its origin; most are dislodged thrombi.
• Pulmonary emboli derive primarily from lower-extremity deep vein thrombi; their effects depend mainly on the size of the embolus and the location in which it lodges. Consequences may include right-sided heart failure, pulmonary hemorrhage, pulmonary infarction, or sudden death.
• Systemic emboli derive primarily from cardiac mural or valvular thrombi, aortic aneurysms, or atherosclerotic plaques; whether an embolus causes tissue infarction depends on the site of embolization and the presence or absence of collateral circulation.
Infarction
An infarct is an area of ischemic necrosis caused by occlusion of the vascular supply to the affected tissue; the process by which such lesions form termed infarction, is a common and extremely important cause of clinical illness. Roughly 40% of all deaths in the United States are a consequence of cardiovascular disease, with most of these deaths stemming from myocardial or cerebral infarction. Pulmonary infarction is a common clinical complication, bowel infarction often is fatal, and ischemic necrosis of distal extremities (gangrene) causes substantial morbidity in the diabetic population.
Arterial thrombosis or arterial embolism underlies the vast majority of infarctions. Less common causes of arterial obstruction include vasospasm, expansion of an atheroma secondary to intraplaque hemorrhage, and extrinsic compression of a vessel, such as by tumor, a dissecting aortic aneurysm, or edema within a confined space (e.g., in anterior tibial compartment syndrome). Other uncommon causes of tissue infarction include vessel twisting (e.g., in testicular torsion or bowel volvulus), traumatic vascular rupture, and entrapment in a hernia sac. Although venous thrombosis can cause infarction, the more common outcome is simply congestion; typically, bypass channels rapidly open to provide sufficient outflow to restore the arterial inflow. Infarcts caused by venous thrombosis thus usually occur only in organs with a single efferent vein (e.g., testis or ovary).
Morphology
Infarcts are classified on the basis of their color (reflecting the amount of hemorrhage) and the presence or absence of microbial infection. Thus, infarcts may be either red (hemorrhagic) or white (anemic) and may be either septic or bland.
Red infarcts (Fig. 3–17, A) occur (1) with venous occlusions (such as in ovarian torsion); (2) in loose tissues (e.g., lung) where blood can collect in infarcted zones; (3) in tissues with dual circulations such as lung and small intestine, where partial, inadequate perfusion by collateral arterial supplies is typical; (4) in previously congested tissues (as a consequence of sluggish venous outflow); and (5) when flow is reestablished after infarction has occurred (e.g., after angioplasty of an arterial obstruction).
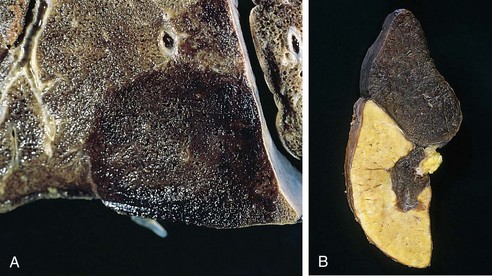
Figure 3–17 Red and white infarcts. A, Hemorrhagic, roughly wedge-shaped pulmonary infarct (red infarct). B, Sharply demarcated pale infarct in the spleen (white infarct).
White infarcts occur with arterial occlusions in solid organs with end-arterial circulations (e.g., heart, spleen, and kidney), and where tissue density limits the seepage of blood from adjoining patent vascular beds (Fig. 3–17, B). Infarcts tend to be wedge-shaped, with the occluded vessel at the apex and the organ periphery forming the base (Fig. 3–17); when the base is a serosal surface, there is often an overlying fibrinous exudate. Lateral margins may be irregular, reflecting flow from adjacent vessels. The margins of acute infarcts typically are indistinct and slightly hemorrhagic; with time, the edges become better defined by a narrow rim of hyperemia attributable to inflammation.
Infarcts resulting from arterial occlusions in organs without a dual circulation typically become progressively paler and sharply defined with time (Fig. 3–17, B). By comparison, hemorrhagic infarcts are the rule in the lung and other spongy organs (Fig. 3–17, A). Extravasated red cells in hemorrhagic infarcts are phagocytosed by macrophages, and the heme iron is converted to intracellular hemosiderin. Small amounts do not impart any appreciable color to the tissue, but extensive hemorrhages leave a firm, brown residuum.
In most tissues, the main histologic finding associated with infarcts is ischemic coagulative necrosis (Chapter 1). An inflammatory response begins to develop along the margins of infarcts within a few hours and usually is well defined within 1 to 2 days. Eventually, inflammation is followed by repair, beginning in the preserved margins (Chapter 2). In some tissues, parenchymal regeneration can occur at the periphery of the infarct, where the underlying stromal architecture has been spared. Most infarcts, however, are ultimately replaced by scar (Fig. 3–18). The brain is an exception to these generalizations: Ischemic tissue injury in the central nervous system results in liquefactive necrosis (Chapter 1).
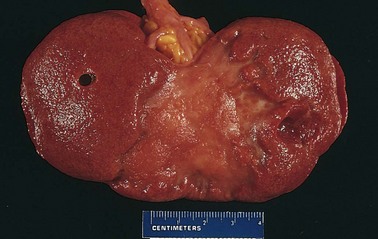
Septic infarctions occur when infected cardiac valve vegetations embolize, or when microbes seed necrotic tissue. In these cases the infarct is converted into an abscess, with a correspondingly greater inflammatory response (Chapter 2).
Factors That Influence Infarct Development
The effects of vascular occlusion range from inconsequential to tissue necrosis leading to organ dysfunction and sometimes death. The range of outcomes is influenced by (1) the anatomy of the vascular supply; (2) the time over which the occlusion develops; (3) the intrinsic vulnerability of the affected tissue to ischemic injury; and (4) the blood oxygen content.
• Anatomy of the vascular supply. The presence or absence of an alternative blood supply is the most important factor in determining whether occlusion of an individual vessel causes damage. The dual supply of the lung by the pulmonary and bronchial arteries means that obstruction of the pulmonary arterioles does not cause lung infarction unless the bronchial circulation also is compromised. Similarly, the liver, which receives blood from the hepatic artery and the portal vein, and the hand and forearm, with its parallel radial and ulnar arterial supply, are resistant to infarction. By contrast, the kidney and the spleen both have end-arterial circulations, and arterial obstruction generally leads to infarction in these tissues.
• Rate of occlusion. Slowly developing occlusions are less likely to cause infarction because they allow time for the development of collateral blood supplies. For example, small interarteriolar anastomoses, which normally carry minimal blood flow, interconnect the three major coronary arteries. If one coronary artery is slowly occluded (e.g., by encroaching atherosclerotic plaque), flow in this collateral circulation may increase sufficiently to prevent infarction—even if the original artery becomes completely occluded.
• Tissue vulnerability to ischemia. Neurons undergo irreversible damage when deprived of their blood supply for only 3 to 4 minutes. Myocardial cells, although hardier than neurons, still die after only 20 to 30 minutes of ischemia. By contrast, fibroblasts within myocardium remain viable after many hours of ischemia.
• Hypoxemia. Understandably, abnormally low blood O2 content (regardless of cause) increases both the likelihood and extent of infarction.
Summary
Infarction
• Infarcts are areas of ischemic necrosis most commonly caused by arterial occlusion (typically due to thrombosis or embolization); venous outflow obstruction is a less frequent cause.
• Infarcts caused by venous occlusion or occurring in spongy tissues typically are hemorrhagic (red); those caused by arterial occlusion in compact tissues typically are pale (white).
• Whether or not vascular occlusion causes tissue infarction is influenced by collateral blood supplies, the rate at which an obstruction develops, intrinsic tissue susceptibility to ischemic injury, and blood oxygenation.
Shock
Shock is the final common pathway for several potentially lethal events, including exsanguination, extensive trauma or burns, myocardial infarction, pulmonary embolism, and sepsis. Regardless of cause, shock is characterized by systemic hypoperfusion of tissues; it can be caused by diminished cardiac output or by reduced effective circulating blood volume. The consequences are impaired tissue perfusion and cellular hypoxia. Although shock initially is reversible, prolonged shock eventually leads to irreversible tissue injury that often proves fatal.
The most common forms of shock can be grouped into three pathogenic categories (Table 3–3):
• Cardiogenic shock results from low cardiac output due to myocardial pump failure. It may be caused by myocardial damage (infarction), ventricular arrhythmias, extrinsic compression (cardiac tamponade) (Chapter 10), or outflow obstruction (e.g., pulmonary embolism).
• Hypovolemic shock results from low cardiac output due to loss of blood or plasma volume (e.g., due to hemorrhage or fluid loss from severe burns).
• Septic shock results from arterial vasodilation and venous blood pooling that stems from the systemic immune response to microbial infection. Its complex pathogenesis is discussed in greater detail next.
Table 3–3 Three Major Types of Shock
| Type of Shock | Clinical Examples | Principal Pathogenic Mechanisms |
|---|---|---|
| Cardiogenic | Myocardial infarction | Failure of myocardial pump resulting from intrinsic myocardial damage, extrinsic pressure, or obstruction to outflow |
| Ventricular rupture | ||
| Arrhythmia | ||
| Cardiac tamponade | ||
| Pulmonary embolism | ||
| Hypovolemic | Hemorrhage | Inadequate blood or plasma volume |
| Fluid loss (e.g., vomiting, diarrhea, burns, trauma) | ||
| Septic | Overwhelming microbial infections | Peripheral vasodilation and pooling of blood; endothelial activation/injury; leukocyte-induced damage; disseminated intravascular coagulation; activation of cytokine cascades |
| Endotoxic shock | ||
| Gram-positive septicemia | ||
| Fungal sepsis | ||
| Superantigens (e.g., toxic shock syndrome) |
Less commonly, shock can result from loss of vascular tone associated with anesthesia or secondary to a spinal cord injury (neurogenic shock). Anaphylactic shock results from systemic vasodilation and increased vascular permeability that is triggered by an immunoglobulin E–mediated hypersensitivity reaction (Chapter 4).
Pathogenesis of Septic Shock
Despite medical advances over the past several decades, septic shock remains a daunting clinical problem. Septic shock kills 20% of its victims, accounts for over 200,000 deaths annually in the United States, and is the number one cause of mortality in intensive care units. The incidence is rising, ironically, in part because of improved life support for critically ill patients, as well as an increase in invasive procedures and the growing numbers of immunocompromised patients (due to chemotherapy, immunosuppression, or HIV infection).
In septic shock, systemic arterial and venous dilation leads to tissue hypoperfusion, even though cardiac output is preserved or even initially increased. The decreased vascular tone is accompanied by widespread endothelial cell activation, often triggering a hypercoagulable state manifesting as disseminated intravascular coagulation. In addition, septic shock is associated with perturbations of metabolism that directly suppress cell and tissue function. The net effect of these abnormalities is hypoperfusion and dysfunction of multiple organs.
At present, gram-positive bacteria constitute the most common cause of septic shock, followed by gram-negative organisms and fungi. Although it was for a time thought that infections had to be disseminated to cause septic shock, infections localized to a specific tissue can trigger sepsis, even without detectable spread to the bloodstream. The ability of diverse flora to precipitate septic shock is consistent with the idea that several different microbial constituents can initiate the process. Most notably, macrophages, neutrophils, dendritic cells, endothelial cells, as well as soluble components of the innate immune system (e.g., complement) recognize and are activated by a variety of substances derived from microorganisms. Once activated, these cells and soluble factors initiate a number of inflammatory responses that interact in a complex, incompletely understood fashion to produce septic shock (Fig. 3–19). As an aside, a similar widespread inflammatory response—the so-called systemic inflammatory response syndrome (SIRS)—can also be triggered in the absence of any apparent underlying infection; causes include extensive trauma or burns, pancreatitis, and diffuse ischemia.
Figure 3–19 Major pathogenic pathways in septic shock. Microbial products activate endothelial cells and cellular and humoral elements of the innate immune system, initiating a cascade of events that lead to end-stage multiorgan failure. Additional details are given in the text. DIC, disseminated intravascular coagulation; HMGB1, high-mobility group box 1 protein; NO, nitric oxide; PAF, platelet-activating factor; PAI-1, plasminogen activator inhibitor-1; PAMP, pathogen-associated molecular pattern; STNFR, soluble tumor necrosis factor receptor; TF, tissue factor; TFPI, tissue factor pathway inhibitor.
Factors contributing to the pathophysiology of septic shock include the following:
• Inflammatory mediators. Cells of the innate immune system express receptors (e.g., Toll-like receptors [TLRs]) (Chapter 2) that recognize a host of microbe-derived substances containing so-called pathogen-associated molecular patterns (PAMPs). Activation of pathogen recognition receptors by PAMPs triggers the innate immune responses that drive sepsis. Upon activation, the inflammatory cells produce TNF and IL-1 (and other cytokines), plus cytokine-like mediators such as high-mobility group box 1 (HMGB1). Reactive oxygen species and lipid mediators such as prostaglandins and platelet-activating factor (PAF) also are elaborated (Chapter 2). These effector molecules activate endothelial cells, resulting in expression of adhesion molecules, a procoagulant phenotype, and secondary waves of cytokine production. The complement cascade also is activated by microbial components, both directly and through the proteolytic activity of plasmin (Chapter 2), resulting in the production of anaphylotoxins (C3a, C5a), chemotaxic fragments (C5a), and opsonins (C3b), all of which can contribute to the pro-inflammatory state.
• Endothelial cell activation and injury. Endothelial activation by microbial constituents or inflammatory cell mediators has three major sequelae: (1) thrombosis; (2) increased vascular permeability; and (3) vasodilation.
The derangement in coagulation is sufficient to produce the formidable complication of disseminated intravascular coagulation in up to half of septic patients. Sepsis alters the expression of many factors ultimately favoring coagulation. Pro-inflammatory cytokines result in increased tissue factor production, while at the same time dampening fibrinolysis by increasing PAI expression. The production of other endothelial anticoagulant factors, such as tissue factor pathway inhibitor, thrombomodulin, and protein C, is also diminished. The procoagulant tendency is further enhanced by decreased blood flow within small vessels, which produces stasis and diminishes the washout of activated coagulation factors. Acting in concert, these effects promote the systemic deposition of fibrin-rich thrombi in small vessels, thus exacerbating tissue hypoperfusion. In full-blown disseminated intravascular coagulation, there is also consumption of clotting factors and platelets, leading to concomitant bleeding and hemorrhage (Chapter 11).
The pro-inflammatory state associated with sepsis leads to widespread vascular leakage and tissue edema, with deleterious effects on both nutrient delivery and waste removal. It appears that inflammatory cytokines loosen endothelial cell tight junctions by causing the adhesion molecule VE-cadherin to be displaced from the junctions. The altered junctions become leaky, resulting in the accumulation of protein-rich exudates and edema throughout the body.
Expression of vasoactive inflammatory mediators (e.g., C3a, C5a, PAF), together with increased NO production, leads to systemic relaxation of vascular smooth muscle, producing hypotension and further reductions in tissue perfusion.
• Metabolic abnormalities. Septic patients exhibit insulin resistance and hyperglycemia. Cytokines such as TNF and IL-1, stress-induced hormones (such as glucagon, growth hormone, and glucocorticoid), and catecholamines all drive gluconeogenesis. At the same time, the pro-inflammatory cytokines suppress insulin release while simultaneously promoting insulin resistance in skeletal muscle and other tissues. Hyperglycemia suppresses neutrophil function—thereby decreasing bactericidal activity—and causes increased adhesion molecule expression on endothelial cells. Although sepsis initially is associated with a surge in glucocorticoid production, this increase is frequently followed by adrenal insufficiency and a relative glucocorticoid deficit. This effect may stem from depression of the synthetic capacity of adrenal glands or frank adrenal necrosis due to disseminated intravascular coagulation (Waterhouse-Friderichsen syndrome) (Chapter 19).
• Immune suppression. The hyperinflammatory state initiated by sepsis can paradoxically lead to a state of immunosuppression. Proposed mechanisms include production of anti-inflammatory mediators (e.g., soluble TNF receptor and IL-1 receptor antagonist), and widespread apoptosis of lymphocytes in the spleen and lymph nodes, the cause of which is uncertain. It is still debated whether immunosuppressive mediators are deleterious or protective in sepsis.
• Organ dysfunction. Systemic hypotension, increased vascular permeability, tissue edema, and small vessel thrombosis all decrease the delivery of oxygen and nutrients to the tissues and contribute to organ dysfunction. High levels of cytokines and secondary mediators can reduce myocardial contractility, thereby blunting cardiac output; increased vascular permeability and endothelial injury in the pulmonary circulation lead to the acute respiratory distress syndrome (ARDS) (Chapter 13). Ultimately, these factors conspire to cause multiorgan failure, particularly of the kidneys, liver, lungs, and heart, culminating in death.
Outcomes in patients with septic shock are difficult to predict; in general those with widespread infections and comorbid diseases have the highest mortality rates, but even young healthy individuals with virulent infections (e.g., meningococcal sepsis) can succumb within hours.
In view of the multiplicity of factors and the complexity of the interactions that underlie sepsis, it is perhaps not surprising that most attempts to intervene therapeutically with inhibitors of specific mediators have been of very modest benefit at best. The standard of care remains treatment with appropriate antibiotics, intensive insulin therapy for hyperglycemia, fluid resuscitation to maintain systemic pressures, and “physiologic doses” of corticosteroids to correct relative adrenal insufficiency. Some promising results have been observed in models of sepsis with treatments directed at restoring endothelial cell integrity.
An additional group of secreted bacterial proteins called superantigens also cause a syndrome similar to septic shock (e.g., toxic shock syndrome). Superantigens are polyclonal T-lymphocyte activators that induce T cells to release high levels of cytokines, which in turn results in a variety of clinical manifestations, ranging from a diffuse rash to vasodilation, hypotension, and death.
Stages of Shock
Shock is a progressive disorder that leads to death if the underlying problems are not corrected. The exact mechanisms of sepsis-related death are still unclear; aside from increased lymphocyte and enterocyte apoptosis, cellular necrosis is minimal. Death typically follows the failure of multiple organs, which usually offer no morphological clues to explain their dysfunction. For hypovolemic and cardiogenic shock, however, the pathways leading to a patient’s demise are reasonably well understood. Unless the insult is massive and rapidly lethal (e.g., exsanguination from a ruptured aortic aneurysm), shock tends to evolve through three general (albeit somewhat artificial) stages. These stages have been documented most clearly in hypovolemic shock but are common to other forms as well:
• An initial nonprogressive stage, during which reflex compensatory mechanisms are activated and vital organ perfusion is maintained
• A progressive stage, characterized by tissue hypoperfusion and onset of worsening circulatory and metabolic derangement, including acidosis
• An irreversible stage, in which cellular and tissue injury is so severe that even if the hemodynamic defects are corrected, survival is not possible
In the early nonprogressive phase of shock, various neurohumoral mechanisms help maintain cardiac output and blood pressure. These mechanisms include baroreceptor reflexes, release of catecholamines and antidiuretic hormone, activation of the renin-angiotensin-aldersterone axis, and generalized sympathetic stimulation. The net effect is tachycardia, peripheral vasoconstriction, and renal fluid conservation; cutaneous vasoconstriction causes the characteristic “shocky” skin coolness and pallor (notably, septic shock can initially cause cutaneous vasodilation, so the patient may present with warm, flushed skin). Coronary and cerebral vessels are less sensitive to sympathetic signals and maintain relatively normal caliber, blood flow, and oxygen delivery. Thus, blood is shunted away from the skin to the vital organs such as the heart and the brain.
If the underlying causes are not corrected, shock passes imperceptibly to the progressive phase, which as noted is characterized by widespread tissue hypoxia. In the setting of persistent oxygen deficit, intracellular aerobic respiration is replaced by anaerobic glycolysis with excessive production of lactic acid. The resultant metabolic lactic acidosis lowers the tissue pH, which blunts the vasomotor response; arterioles dilate, and blood begins to pool in the microcirculation. Peripheral pooling not only worsens the cardiac output but also puts endothelial cells at risk for the development of anoxic injury with subsequent DIC. With widespread tissue hypoxia, vital organs are affected and begin to fail.
In the absence of appropriate intervention, the process eventually enters an irreversible stage. Widespread cell injury is reflected in lysosomal enzyme leakage, further aggravating the shock state. Myocardial contractile function worsens, in part because of increased nitric oxide synthesis. The ischemic bowel may allow intestinal flora to enter the circulation, and thus bacteremic shock may be superimposed. Commonly, further progression to renal failure occurs as a consequence of ischemic injury of the kidney (Chapter 13), and despite the best therapeutic interventions, the downward spiral frequently culminates in death.
Morphology
The cellular and tissue effects of shock are essentially those of hypoxic injury (Chapter 1) and are caused by a combination of hypoperfusion and microvascular thrombosis. Although any organ can be affected, brain, heart, kidneys, adrenals, and gastrointestinal tract are most commonly involved. Fibrin thrombi can form in any tissue but typically are most readily visualized in kidney glomeruli. Adrenal cortical cell lipid depletion is akin to that seen in all forms of stress and reflects increased utilization of stored lipids for steroid synthesis. While the lungs are resistant to hypoxic injury in hypovolemic shock occurring after hemorrhage, sepsis or trauma can precipitate diffuse alveolar damage (Chapter 12), leading to so-called shock lung. Except for neuronal and cardiomyocyte loss, affected tissues can recover completely if the patient survives.
Clinical Course
The clinical manifestations of shock depend on the precipitating insult. In hypovolemic and cardiogenic shock, patients exhibit hypotension, a weak rapid pulse, tachypnea, and cool, clammy, cyanotic skin. As already noted, in septic shock, the skin may be warm and flushed owing to peripheral vasodilation. The primary threat to life is the underlying initiating event (e.g., myocardial infarction, severe hemorrhage, bacterial infection). However, the cardiac, cerebral, and pulmonary changes rapidly aggravate the situation. If patients survive the initial period, worsening renal function can provoke a phase dominated by progressive oliguria, acidosis, and electrolyte imbalances.
Prognosis varies with the origin of shock and its duration. Thus, more than 90% of young, otherwise healthy patients with hypovolemic shock survive with appropriate management; by comparison, septic or cardiogenic shock is associated with substantially worse outcomes, even with state-of-the-art care.
Summary
Shock
• Shock is defined as a state of systemic tissue hypoperfusion due to reduced cardiac output and/or reduced effective circulating blood volume.
• The major types of shock are cardiogenic (e.g., myocardial infarction), hypovolemic (e.g., blood loss), and septic (e.g., infections).
• Shock of any form can lead to hypoxic tissue injury if not corrected.
• Septic shock is caused by the host response to bacterial or fungal infections; it is characterized by endothelial cell activation, vasodilation, edema, disseminated intravascular coagulation, and metabolic derangements.
Akhtar S. Fat embolism. Anesthesiol Clin. 2009;27:533. [Recent overview of the pathogenesis and clinical issues in fat embolism syndrome.]
Coppola A, Tufano A, Cerbone AM, Di Minno G. Inherited thrombophilia: implications for prevention and treatment of venous thromboembolism. Semin Thromb Hemost. 2009;35:683. [Review of the genetic underpinnings of hypercoagulable states in a volume of the journal devoted to various aspects of thrombophilia.]
Crawley J, et al. The central role of thrombin in hemostasis. J Thromb Haemost. 2007;5(Suppl 1):95. [Review of the various pathways impacted by thrombin activation.]
Crawley J, Lane D. The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2008;28:233. [Summary of the physiologic roles of TFPI.]
Cushman M. Epidemiology and risk factors for venous thrombosis. Semin Hematol. 2007;44:62. [Overview of the risk factors and pathophysiology of venous clotting.]
Dahlback B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med. 2005;257:209. [Although slightly older, this is a good one-stop review on normal and abnormal hemostasis.]
Esmon CT, Esmon NL. The link between vascular features and thrombosis. Annu Rev Physiol. 2011. [Up-to-date review of the interactions of endothelium, blood flow, and hemostasis/thrombosis.]
Goldhaber SZ. Advanced treatment strategies for acute pulmonary embolism, including thrombolysis and embolectomy. J Thromb Haemost. 2009;7(Suppl 1):322. [Up-to-date guide to the recognition and therapy of pulmonary embolism.]
Holy EW, Tanner FC. Tissue factor in cardiovascular disease pathophysiology and pharmacological intervention. Adv Pharmacol. 2010;59:259. [Comprehensive review of the roles of tissue factor in hemostasis and potential pathways to be exploited in preventing pathologic thrombosis.]
Hong MS, Amanullah AM. Heparin-induced thrombocytopenia: a practical review. Rev Cardiovasc Med. 2010;11:13. [As characterized by the title, a good, practical review of the mechanisms and therapies for heparin-induced thrombocytopenia.]
Hotchkiss R, Karl I. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138. [Although an older paper, this is extremely well-written, and lays a solid pathogenic foundation for the pathways underlying sepsis.]
Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248. [Excellent and current review of the roles played by platelets in thrombosis and inflammation, as well as possible targets for therapeutic intervention.]
Kwaan HC, Samama MM. The significance of endothelial heterogeneity in thrombosis and hemostasis. Semin Thromb Hemost. 2010;36:286. [Review with newer data regarding the influence of endothelium on hemostasis and thrombosis.]
Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687. [Good general overview of fundamental pathways in coagulation.]
Montagnana M, Franchi M, Danese E, et al. Disseminated intravascular coagulation in obstetric and gynecologic disorders. Semin Thromb Hemost. 2010;36:404. [Review of the mechanisms of DIC, and a good discussion of the pathophysiology of amniotic fluid embolism.]
Munford RS. Severe sepsis and septic shock: the role of gram-negative bacteremia. Annu Rev Pathol. 2006;1:467. [An interesting and provocative view regarding the pathogenesis of septic shock.]
Osinbowale O, Ali L, Chi YW. Venous thromboembolism: a clinical review. Postgrad Med. 2010;122:54. [Good review at a medical student/house officer level.]
Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, et al. The pathogenesis of sepsis. Ann Rev Pathol Mech Dis. 2011;6:19.
Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost. 2009;7:4. [Excellent review of fibrinolytic pathways.]
Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. 2010;376:1498. [Good summary of the anti-phospholipid syndrome with emphasis on diagnosis and therapeutics.]
Wu KK, Matijevic-Aleksic N. Molecular aspects of thrombosis and antithrombotic drugs. Crit Rev Clin Lab Sci. 2005;42:249. [Lengthy, thorough overview of the mechanisms of thrombus formation with emphasis on targets for therapeutic intervention.]
Zwicker J, Furie BC, Furie B. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62:126. [Thorough review of the mechanisms underlying the hypercoagulable state of malignancy.]