Chapter 17 General methods associated with the phytochemical investigation of herbal products
Before about 1800 only slow progress was made in phytochemistry. A few compounds such as cane-sugar, starch, camphor and benzoic acid had long been known, as their preparation was extremely simple; also complex mixtures such as fats, fixed oils, volatile oils, tars and resins had been prepared and used, although virtually nothing was known of their composition. The early scientific workers in the phytochemical field failed to appreciate the extreme complexity of the materials they were trying to investigate and almost entirely lacked the techniques necessary for real progress. Many hundreds of plants were burnt to yield ashes and these early investigators were disappointed to find no significant differences between the ashes of poisonous and those of non-poisonous plants. Expression, aqueous extraction and evaporation had long been used for the preparation of sugar from sugar-cane and the French apothecary Nicholas Leméry (1645–1715) extended the use of extraction processes and made use of alcohol as a solvent. Robert Boyle (1627–91) disposed of the ancient theory of Aristotle that matter was composed of four elements, and although he never isolated an alkaloid, he was obviously moving in the right direction when he treated opium with potassium carbonate and alcohol. In 1747 sucrose was isolated from many plants, including sugarbeet, by the German apothecary A. S. Marggraf (1709–80). K. W. Scheele (1742–86) was highly successful in the phytochemical field and isolated citric, gallic, malic, oxalic, tartaric and prussic acids.
In the nineteenth century progress became more rapid. In 1803 narcotine, the first alkaloid, was isolated; morphine, strychnine, emetine and many others followed rapidly. Between 1813 and 1823 Chevreul elucidated the chemical nature of fats and fixed oils. Until well into the middle of the twentieth century the main emphasis in natural- product chemistry remained the isolation and structure determination of a wide variety of compounds. At this point it became apparent that the principal structural types commonly found in plants had been largely elucidated. Indeed, by this time the attention of natural-product chemists was turning to the elucidation of the actual biosynthetic pathways found in the plant. Such studies were made possible by the introduction of new techniques of separation and analysis. This emphasis has continued until today, when most of the major pathways, including stereochemical aspects, have been studied in some depth. Interest has now moved on to plant biochemistry involving enzymatic and DNA studies related to the biosynthesis of natural products. There has also developed a renewed interest in the patterns of occurrence of compounds in plants (comparative phytochemistry).
Not all the chemical compounds elaborated by plants are of equal interest to the pharmacognosist. Until relatively recently the so-called ‘active’ principles were frequently alkaloids or specific glycosides usually with pronounced pharmacological properties; these therefore received special attention, and in large measure constituted the principal plant drugs of the allopathic system of medicine. It is now realized that many other constituents of plants, particularly those associated with herbal medicine, have medicinal properties which manifest themselves in more subtle and less dramatic ways than the obviously poisonous plants. This has considerably widened the scope of plant metabolites considered worthy of more detailed investigation. Other groups such as carbohydrates, fats and proteins are of dietetic importance, and many such as starches and gums are used in pharmacy but lack any marked pharmacological action. Substances, such as calcium oxalate, silica, lignin and colouring matters, may be of assistance in the identification of drugs and the detection of adulteration.
As a result of the recent interest in the plant kingdom as a potential source of new drugs, strategies for the fractionation of plant extracts based on biological activity rather than on a particular class of compound, have been developed. The chemical examination follows after the isolation of the active fraction.
The phytochemical investigation of a plant may thus involve the following: authentication and extraction of the plant material; separation and isolation of the constituents of interest; characterization of the isolated compounds; investigation of the biosynthetic pathways to particular compounds; and quantitative evaluations. Parallel to this may be the pharmacological assessment of the separated components, which may, in some investigations, precede the characterization.
EXTRACTION OF PLANT MATERIAL
All plant material used should be properly authenticated, as much time and money can be wasted on the examination of material of doubtful origin. The choice of extraction procedure depends on the nature of the plant material and the components to be isolated. Dried materials are usually powdered before extraction, whereas fresh plants (leaves, etc.) can be homogenized or macerated with a solvent such as alcohol. The latter is also particularly useful for stabilizing fresh leaves by dropping them into the boiling solvent. Alcohol is a general solvent for many plant constituents (most fixed oils excepted) and as such may give problems in the subsequent elimination of pigments, resins, etc. Water-immiscible solvents are widely used—light petroleum (essential and fixed oils, steroids), ether and chloroform (alkaloids, quinones). The extraction of organic bases (e.g. alkaloids) usually necessitates basification of the plant material if a water-immiscible solvent is to be used; for aromatic acids and phenols acidification may be required. Extraction itself may be performed by repeated maceration with agitation, percolation or by continuous extraction (e.g. in a Soxhlet extractor, Fig. 16.2). Special methods for volatile oils, such as the enfleurage process, are considered in Chapter 22. Ultrasound may enhance the extraction process for some plant materials and the BP uses this in the preparation of a 50% ethanolic solution of opium for the assay of alkaloids and in the assay procedure of Agnus Castus. Its use has been studied for the extraction of atropine from Hyoscyamus muticus using various solvent systems (A. Djilana and B. Legseir Fitoterapia, 2005, 76, 148).
Spouted bed extraction
In certain instances, as in the production of annatto powder from the seeds of Bixa orellana, the physical removal of the pigment layer of the seed-coat can yield a less impaired product than that produced by solvent extraction. Such methods can involve the use of a ball mill or a spouted bed unit. A development of the latter, the conical spouted bed extractor, has been investigated for annatto production. Basically it consists of a cylinder tapered at both ends and containing the seeds at the lower end through which a jet of hot air is forced. Seeds and pigment-loaded fine particles are propelled into the space above from whence the seeds fall back to be recirculated and the annatto powder moves to a cyclone from which it is collected. For full details see M. L. Passos et al., Drying Technology, 1998, 16, 1855.
Supercritical fluid extraction
The use of supercritical fluids for the extraction of a range of materials including plant products of medicinal, flavouring and cosmetic interest has, during the last decade, become of increasing economic and research interest.
In 1822, Cagniard de la Tour reported that above a certain temperature, and pressure, single substances do not condense or evaporate but exist as a fluid. Under these conditions the gas and liquid phases both possess the same density and no division exists between the two phases. This is the critical state. For water, the critical conditions for temperature (tc) and pressure (pc) are 374°C and 220 atmospheres respectively and for carbon dioxide tc = 31°C and pc = 74 atm. In practice conditions somewhat above the critical temperature and pressure for a particular substance are usually used and these supercritical fluids exhibit properties intermediate between those of the liquid and gaseous phases. In phytochemistry these properties can be exploited to maximize the extraction of plant constituents. For industrial purposes supercritical fluid carbon dioxide has an environmental advantage over many common organic solvents and leaves no solvent residues in the product. It also allows a low temperature process and has proved of value for the extraction of labile expensive fragrances and medicinal phytochemicals. To render it more polar a small amount of modifier, e.g. methanol, may be added to the carbon dioxide. The high pressures, and for some substances the high temperatures, involved in supercritical fluid extraction are the principal disadvantages of the technique.
Pioneer work on medicinal plants was carried out by Stahl and coworkers (Planta Med., 1980, 40, 12, and references cited therein). They studied the use of liquefied and supercritical carbon dioxide and liquefied nitrous oxide for the extraction of various plant constituents, including various types of alkaloids, the pyrethrins and the components of chamomile. With pyrethrum flower extract, the content of pyrethrins is substantially higher (up to 50%) than in commercially available petroleum ether extracts. By a two-step precipitation the active ingredients can be raised to up to 60% without decomposition of the thermolabile pyrethrins.
Further examples involving the extraction of phytochemicals with supercritical carbon dioxide follow:
The use of 10% methanol in CO2 for the extraction of trilactones from ginkgo could be of commercial significance
Piper nigrum muntok (superior aroma of oil; yield 2.8 per cent volatile oil compared with 0.6 per cent by steam distillation); rose petals (product richer in relevant fragrance compounds compared with steam distillation); rosemary (aroma more closely resembles plant fragrance than distilled oil); also studied: angelica root, celery, coriander, Illicium verum, Maytenus illicifolia, pimento
For a review covering the extraction of flavour and fragrance compounds, see M. Gotto et al., Aroma research 2007, 8, 110
For additional information on the method, consult the ‘Further reading’.
Solid phase microextraction
The method is suitable for some volatile oil-containing drugs. T. J. Betts (Planta Medica, 2000, 66, 193), using methyl polysiloxane solid phase microextraction fibres, has extracted the volatile oil from the headspace above fresh cut eucalyptus leaves (37° for 10 min.). The fibres were then desorbed at 200° for capillary gas chromatography of the oil. Not surprisingly, the oil composition differs from that of steam-distilled oils. The method, coupled with gas chromatography and mass spectrometry, has been recently employed for the analysis of the flowers and essential oils from Lavandula angustifolia cultivated in N.E. Italy (C. Da Porto and D. Decorti, Planta Medica, 2008, 74, 182).
SEPARATION AND ISOLATION OF CONSTITUENTS
As the instrumentation for the structure elucidation of organic compounds becomes ever more effective, and allows the use of increasingly small amounts of material, the most difficult operation in phytochemical research becomes that of the isolation and purification of plant constituents. Although the chemical properties of functional groups and moieties contained in compounds such as acids, aldehydes, phenols and alkaloids can be exploited for their separation from other materials, such methods might not fractionate components of the same class; it is in this latter area that new techniques are constantly being developed.
Sublimation
Sublimation may sometimes be possible on the whole drug, as in the isolation of caffeine from tea or for the purification of materials present in a crude extract. Modern equipment employs low pressures with a strict control of temperature.
Distillation
Fractional distillation has been traditionally used for the separation of the components of volatile mixtures; in phytochemistry it has been widely used for the isolation of the components of volatile oils. On a laboratory scale it is not easy by this method to separate minor components of a mixture in a pure state and gas chromatography is now routinely used (q.v.).
Steam distillation is much used to isolate volatile oils and hydrocyanic acid from plant material. The TAS oven (see ‘Thin-layer chromatography’) involves steam distillation on a semi-micro scale for the direct transfer of volatile materials from a powdered drug to a thin-layer plate.
Fractional liberation
Some groups of compounds lend themselves to fractional liberation from a mixture. As an example, a mixture of alkaloid salts in aqueous solution, when treated with aliquots of alkali, will give first the weakest base in the free state followed by base liberation in ascending order of basicity. If the mixture is shaken with an organic solvent after each addition, then a fractionated series of bases will be obtained. A similar scheme can be used for organic acids soluble in water-immiscible solvents; in this case, starting with a mixture of the acid salts, it is possible to fractionally liberate the acids by addition of mineral acids.
Fractional crystallization
A method much used in traditional isolations and still valuable for the resolution of often otherwise intractable mixtures. The method exploits the differences in solubility of the components of a mixture in a particular solvent. Frequently, derivatives of the particular components are employed (picrates of alkaloids, osazones of sugars).
Adsorption chromatography
Of the various methods of separating and isolating plant constituents, the ‘chromatographic procedure’ originated by Tswett is one of the most useful techniques of general application. The use of charcoal for the decolorization and clarification of solutions is well known; coloured impurities are adsorbed by the charcoal and a colourless solution results on filtration. All finely divided solids have the power to adsorb other substances on their surfaces to a greater or lesser extent; similarly, all substances are capable of being adsorbed, some much more readily than others. This phenomenon of selective adsorption is the fundamental principle of adsorption chromatography, the general process of which may be described with reference to one of Tswett’s original experiments.
A light petroleum extract of green leaves is allowed to percolate slowly through a column of powdered calcium carbonate contained in a vertical glass tube. The pigmented contents of the solution are adsorbed on the substance of the column and undergo separation as percolation proceeds. The more strongly adsorbed pigments, xanthophyll and the chlorophylls, accumulate in distinct, characteristically coloured bands near the top of the column, while the less strongly adsorbed pigments, the carotenes, accumulate lower down.
Frequently, complete separation of all the constituents into distinct bands does not result during the first ‘adsorption stage’, but the bands remain crowded together near the top of the column. Such a column may be developed by allowing more of the pure solvent to percolate through the column when the adsorbed materials slowly pass downwards and the separate bands become wider apart. In many cases the process may be rendered more efficient by the use of a different solvent, one from which the substances are less strongly adsorbed. If, for example, light petroleum containing a little alcohol is percolated through the chromatogram obtained in the experiment described above, the bands become wider apart and pass down the column more rapidly than when pure light petroleum is used. As percolation continues, the lower bands reach the bottom of the column and disappear; the pigment is then obtained in the solution leaving the bottom of the column. This process of desorption is termed elution and the solution obtained is the eluate.
It was from such classic experiments of Tswett on the separation of coloured compounds that the term ‘chromatography’ arose and it has remained to describe this method of fractionation although its application to colourless substances is now universal.
Substances are more readily adsorbed from non-polar solvents such as light petroleum and benzene, while polar solvents—alcohol, water and pyridine, for example—are useful eluting media; many substances are adsorbed at one pH and eluted at another.
Various substances may be used as adsorbing materials; alumina is the most common and other materials include kaolin, magnesium oxide, calcium carbonate, charcoal and sugars.
When colourless substances are chromatographed, the zones of adsorbed material are not visible to the eye, although they may, in some cases, be rendered apparent as fluorescent zones when the column is examined under ultraviolet light. Failing this, it becomes necessary to divide the chromatogram into discrete portions and elute or extract each portion separately. Sometimes it is more convenient to collect the eluate from the whole column in fractions for individual examination.
The apparatus required is simple and consists essentially of a vertical glass tube into which the adsorbent has been packed; a small plug of glass wool or a sintered glass disc, at the base of the tube, supports the column. With volatile developing solvents it is usually preferable to use a positive pressure at the head of the column. Numerous modifications of the apparatus are used for large-scale operations, for use with heated solvents and for chromatography in the absence of air or oxygen.
Adsorption chromatography has proved particularly valuable in the isolation and purification of vitamins, hormones, many alkaloids, cardiac glycosides, anthraquinones, etc. It is commonly employed as a ‘clean-up’ technique for the removal of unwanted materials from plant extracts prior to assay.
Thin-layer chromatography with adsorbents such as alumina is an adaptation of the method and is discussed separately in this chapter.
Partition chromatography
Partition chromatography was introduced by Martin and Synge in 1941 for the separation of acetylated amino acids and was first applied to the separation of alkaloids by Evans and Partridge in 1948. The method has now been largely superseded by the more sophisticated HPLC (see below) but it retains the advantage of being inexpensive to set up and operate. The separation of the components of a mixture is, as in counter-current extraction, dependent on differences in the partition coefficients of the components between an aqueous and an immiscible organic liquid.
The aqueous phase is usually the stationary phase and is intimately mixed with a suitable ‘carrier’ such as silica gel, purified kieselguhr or powdered glass and packed in a column as in adsorption chromatography. The mixture to be fractionated is introduced on the column, in a small volume of organic solvent, and the chromatogram is developed with more solvent or successively with different solvents of increasing eluting power. When water is the stationary phase, the solutes undergoing separation travel down the column at different speeds depending on their partition coefficient between the two liquid phases; the use of a buffer solution as aqueous phase widens the scope of the technique, as ionization constants and partition coefficients are exploited in effecting separation.
The separated zones may be located by methods similar to those employed in adsorption chromatography. With water as the aqueous phase, the positions of separated zones of acids or alkalis may be shown by employing a suitable indicator dissolved in the water. This method is clearly not applicable to buffer-, acid- or alkali-loaded columns, and in these cases complete elution (elution development) of the separated zone is often necessary. The eluate is collected in aliquot portions and estimated chemically or physically for dissolved solute. A graph of the analytical figure (titration, optical rotation, optical density, refractive index, etc.) for each fraction of eluate may then be plotted to show the degree of separation of the solutes.
The fractionations obtained in partition chromatography are influenced to a considerable degree by the displacement effect of one solute on another and advantage is taken of this in displacement development, in which the chromatogram is developed with a solution of an acid or a base that is stronger than any in the mixture to be separated. The effect is for the stronger acids or bases to displace the weaker ones, resulting in a rapid clear-cut separation of the constituents. For the elution development of these separated zones it is essential that there is no distortion of the zones, since the front of one band follows immediately on the tail of the preceding less acidic or less basic component.
There have been several theoretical treatments of partition chromatography, all involving certain approximations, since a theory taking into account all known variables would be extremely complicated. For general purposes, one of the most satisfactory treatments of columns loaded with water is that of Martin and Synge, in which the theoretical plate concept of fractional distillation is applied to partition chromatography. In this theory, diffusion from one plate to another is taken as negligible and the partition of solute between two phases is independent of concentration and the presence of other solutes.
Partition chromatography on paper
In 1944 Consden, Gordon and Martin introduced a method of partition chromatography using strips of filter paper as ‘carriers’ for the analysis of amino acid mixtures. The technique was extended to all classes of natural products, and although to a large measure replaced by thin-layer chromatography (TLC), it remains the method of choice for the fractionation of some groups of substances.
The solution of components to be separated is applied as a spot near one end of a prepared filter-paper strip. The paper is then supported in an airtight chamber which has an atmosphere saturated with solvent and water, and a supply of the water-saturated solvent. The most satisfactory solvents are those which are partially miscible with water, such as phenol, n-butanol and amyl alcohol. Either the paper may be dipped in the solvent mixture so that the solvent front travels up the paper (ascending technique) or the trough of solvent may be supported at the top of the chamber, in which case the solvent travels down the paper (descending technique). The BP 2007 gives details of both methods. As the solvent moves, the components also move along the paper at varying rates, depending mainly on the differences in their partition coefficients between the aqueous (hydration shell of cellulose fibres) and organic phases. After the filter-paper strips have been dried, the positions of the separated components can be revealed by the use of suitable developing agents: ninhydrin solution for amino acids; iodine solution (or vapour) or a modified Dragendorff’s reagent for alkaloids; ferric chloride solution for phenols; alkali for anthraquinone derivatives; antimony trichloride in chloroform for steroids and some components of volatile oils; aniline hydrogen phthalate reagent for sugars. The relative positions of the components and the size of the spots depend upon the solvent, and this should be selected to give good separation of the components with well-defined, compact spots. Improved separation of mixtures can often be obtained by adjusting the acidity of the solvent with ammonia, acetic acid or hydrochloric acid or by impregnating the paper with a buffer solution or formamide solution.
For the separation of some substances it is necessary to use a two-dimensional chromatogram: first one solvent is run in one direction, then, after drying of the paper, a second solvent is run in a direction at right angles to the first—this is particularly applicable to mixtures of amino acids.
The ratio between the distance travelled on the paper by a component of the test solution and the distance travelled by the solvent is termed the RF value and, under standard conditions, this is a constant for the particular compound. However, in practice, variations of RF often occur and it is desirable to run reference compounds alongside unknown mixtures.
The quantity of substance present determines the size of the spot with any one solvent and can be made the basis of quantitative evaluation. Also, the separated components of the original mixture can be separately eluted from the chromatogram, by treating the cut-out spots with a suitable solvent, and then determined quantitatively by some suitable method—for example, fluorescence analysis, colorimetry or ultraviolet adsorption. Drugs so evaluated include aloes, digitalis, ergot, hemlock, lobelia, nux vomica, opium, rauwolfia, rhubarb, broom, solanaceous herbs and volatile oils.
High-performance liquid chromatography (HPLC)/high-speed LC
HPLC is a liquid column chromatography system which employs relatively narrow columns (about 5 mm diameter for analytical work) operating at ambient temperature or up to about 200°C at pressures up to 200 atm (20 000 kPa).
The columns are costly and it is usual to employ a small precolumn containing a cartridge of packing material to remove adventitious materials which might otherwise damage the main column. Normal flow rates of eluate are 2–5 ml min−1 but can be up to 10 ml min−1, depending on the diameter of the column and the applied pressure. The apparatus is suitable for all types of liquid chromatography columns (adsorption, partition by the use of bonded liquid phases, reversed phase, gel filtration, ion exchange and affinity). The arrangement of such an apparatus, suitable for use with two solvents and giving graded elution, is illustrated in Fig. 17.1. Detection of the often very small quantities of solute in the eluate is possible by continuous monitoring of ultraviolet absorption, mass spectrum, refractive index, fluorescence and electrical conductance; nuclear magnetic resonance can now be added to this list. To improve detection, solutes may be either derivatized before chromatography (this technique can also be used to improve separations) or treated with reagents after separation (post-column derivatization). A transport system for monitoring is commercially available; in this a moving wire passes through the flowing eluate (coating block) and the dissolved solute, deposited on the wire, is pyrolysed and its quantity automatically recorded. It will be noted that, for any particular fractionation, some detector systems would be selective for certain groups of compounds and others would be universal.
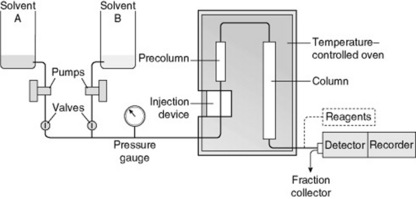
Fig. 17.1 Schematic representation of apparatus for high- performanance liquid chromatography utilizing two solvents.
HPLC can give much improved and more rapid separations than can be obtained with the older liquid chromatography methods and it is therefore finding increasing use in numerous areas. As with GLC apparatus, it is available from many manufacturers and can be completely automated.
Many stationary phases are available, the most widely used being silica based. In these, which consist of porous particles 5–10 μm in diameter, the silanol groups (Si-OH) afford a polar surface which can be exploited in separations using an organic mobile phase as in ordinary adsorption chromatography.
Reversed-phase packing material (Spherisorb ODS) is produced by the bonding of octadecylsilyl groups (C18H37Si–) to silica gel. In the commercial material there appears to be a considerable proportion of residual silanol–OH groups, so that adsorption and partition effects may operate during separation. The hydrocarbon chains probably allow non-polar interaction to take place. The structure of the packing material might be represented as:
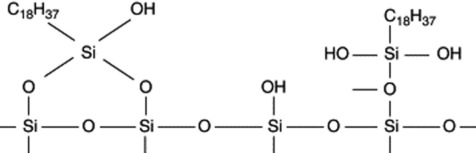
To reduce tailing effects which might be caused by the remaining free hydroxyl groups the latter can be masked by treatment with a short-chain silane, usually trimethylchlorosilane. Silica-based columns are restricted to use in the range pH 3–8 and to overcome this polymer phases operating at pH 1–13 are available; these, however, have the disadvantage of exhibiting a high column back-pressure.
Of relatively recent introduction are chiral stationary phases which are utilized to separate the enantiomers of racemic mixtures. These have great potential for the study of natural products, many of which are optically active, and for isolating the pharmacologically active enantiomer from the racemic mixture of a synthetic drug. Pharmacognostical examples include, among many others, the assay of partially racemized hyoscine extracted from plants, the separation of aromatics and the resolution of mixtures of (+)- and (−)-epicatechin and other proanthocyanidin enantiomers of Cassia fistula and C. javanica. The mode of action of chiral stationary phases is not fully understood; typical materials are cyclodextrins, cellulose- and amino acid-derivatives suitably bonded. Two products are sorbents made of spherical silica gel particles to which β-cyclodextrin (ChiraDex®) or γ-cyclodextrin (Chira-Dex® GAMMA) are covalently bonded via a carbamate bond and long spacer.
For a review on the rapid detection and subsequent isolation of bioactive constituents of crude plant extracts with schemes for LC/UV, LC/MS, LC/MS/MS and LC/NMR see K. Hostettman et al., Planta Medica, 1997, 63, 2.
Supercritical fluid chromatography
This technique, developed over recent years for both the qualitative and quantitative analysis of medicinal products, utilizes supercritical fluids, particularly carbon dioxide, as the mobile phase in liquid chromatography. The low viscosity of the supercritical liquid provides a faster flow rate than standard HPLC and higher diffusivity of the materials to be separated. The plate height in the column, the length of the column and the time required for a particular separation are all reduced compared with the established technique. Sharper elution peaks give increased separation efficiency. Waste-disposal of used solvents is eliminated. Changes to the instrumental set-up have been devised to accommodate the properties of a supercritical liquid.
Counter-current extraction
This is a liquid–liquid extraction process and the principles involved are similar to those of partition chromatography. Developed by Craig in 1944, the extraction machine used (for that time) small amounts of extract and overcame the tedious multiple extraction processes then employed. Now, however, for research purposes, only the smallest of samples for fractionation and analysis are required, making the use of the cumbersome counter-current apparatus largely unnecessary.
Briefly, a lower, stationary phase is contained in a series of tubes and an upper, moving immiscible liquid is transferred from tube to tube along the series, the immiscible liquids being shaken and allowed to separate between each transference. The mixture to be fractionated is placed in the first tube containing the immiscible liquids and the apparatus is agitated and the layers are allowed to separate. The components of the mixture will be distributed between the two layers according to their partition coefficients. The upper phase is moved along to the second tube containing lower phase and more moving phase is brought into contact with the lower phase of tube 1. Shaking and transference again takes place and continues along a sufficient number of tubes to give a fractionation of the mixture.
The applications of counter-current extraction covered many fields of plant chemistry, including alkaloids, amino acids, antibiotics, antitumour compounds, phenols including anthraquinone derivatives, cardiac glycosides, essential oils, fatty acids, plant auxins, prostaglandins, steroids and vitamins.
Other more recent developments involving the counter-current principle are high-speed counter-current chromatography (planet coil centrifugal CCC), droplet counter-current chromatography (DCCC) and centrifugal droplet CCC. Details of these can be found in the 15th edition of this book. For a review (245 refs) giving the background and up-to-date methodology employed in the counter-current separation of natural products see G. F. Pauli et al., J. Nat. Prod., 2008, 71, 1489–1508.
Thin-layer chromatography
In 1958 Stahl demonstrated the wide applicability of TLC, a technique which had been known in principle for many years but was never developed. It has now achieved remarkable success in the separation of mixtures of all classes of natural products and is established as an analytical tool in modern pharmacopoeias.
In outline the method consists of preparing, on a suitable glass plate, a thin layer of material, the sorbent, which may be either an adsorbent as used in column adsorption chromatography or an inert support which holds an aqueous phase as in column partition chromatography. The mixtures to be resolved are dissolved in a suitable solvent and placed as a series of spots on the film towards one end of the plate; this end is then dipped in a suitable solvent mixture and the whole enclosed in an airtight container. The solvent front travels up the film and after a suitable time the plate is removed, the solvent front is marked, the solvent is allowed to evaporate and the positions of the separated compounds are determined by suitable means.
TLC has certain advantages over paper chromatography. Fractionations can be effected more rapidly with smaller quantities of the mixture; the separated spots are usually more compact and more clearly demarcated from one another; and the nature of the film is often such that drastic reagents, such as concentrated sulphuric acid, which would destroy a paper chromatogram, can be used for the location of separated substances.
With adsorption TLC various substances exhibit different adsorptive capacities and any one material may vary in its activity according to the pretreatment. The adsorbent must be chosen in relation to the properties of the solvent and the mixture to be fractionated. In general, for a given substance, if a highly active adsorbent is used, then a solvent with a correspondingly high power of elution for this substance is required. Alumina (acid, basic and neutral) of different activity grades is very commonly employed. In order to produce a film with reasonable handling properties, the adsorbent may be mixed with about 12% of its weight of calcium sulphate (CaSO4.1/2H2O) to act as a binder. Ready-mixed powders are obtainable commercially; they require mixing with a given quantity of water and the slurry needs to be spread by a mechanical device or with a glass rod on to glass plates. The film sets within a few minutes and is then activated by heating at a suitable temperature (105°C for 30 min is common). Commercial ready-spread plates are available. The thickness of the film is characteristically of the order of 250 μm, but for preparative work layers of up to several millimetres thickness are employed. The thick films must be carefully dried to avoid cracks.
Solutions of substances to be examined are applied to the film with the aid of capillary tubes or, for quantitative work, with microsyringes and micrometer pipettes which permit volumes to be read off to ±0.05 μl. A useful innovation for applying steam-volatile components of a powdered drug directly to a thin-layer plate is the Stahl TAS oven. The drug sample is placed in a cartridge together with a suitable propellant (e.g. hydrated silica gel) and inserted in the TAS oven maintained at a predetermined temperature. The tapered exit of the cartridge is situated a short distance from the base-line of a TLC plate and ‘steam-distilled’ components from the drug are deposited ready for immediate development (for illustration see 12th edition). Stahl (Planta Med., 1976, 29, 1) has described a development of this apparatus whereby 18 samples can be simultaneously deposited on a plate under uniform conditions. A more recent application has involved the study of tannin-containing drugs.
The solvents used for running the chromatogram must be pure, and common ones are methanol, ethanol and other alcohols, chloroform, ether, ethyl acetate, n-hexane, cyclohexane, petroleum spirit and mixtures of these. For routine assays automatic multiple development with polarity graduation of the developing solvent can be used. It must be remembered that chloroform ordinarily contains up to 1% of ethanol, which gives it quite different elution properties from those of pure chloroform. Benzene, formerly frequently used as a component of the mobile phase, has now for health reasons been routinely replaced with other non-polar solvents. Similarly, inhalation of chloroform-containing mixtures should be avoided.
TLC, which involves the partition of a substance between two immiscible phases, is again analogous to the column procedure and to paper chromatography. In the latter, the hydration shell of the cellulose fibres forms the stationary phase and thin-layer chromatograms utilizing powdered cellulose give comparable results. Kieselguhr and silica gel are also commonly employed, and their properties as thin layers can be modified by the inclusion of acids, bases and buffer solutions. The thin layers may also include a substance fluorescent in ultraviolet light and this facilitates the detection of solutes which cause a quenching of the background fluorescence. Much used is Kieselgel GF254 ‘Merck’ which gives a background green fluorescence when irradiated with ultraviolet light of wavelength 254 nm. Small quantities of ammonia solution, diethylamine, acetic acid, dimethylformamide and pyridine are often present as constituents of the developing solvents for silica plates. Other layers used include polyamide, which is particularly suitable for phenolic compounds, ‘Sephadex’ (Pharmacia, Uppsala), a cross-linked dextran used as a molecular sieve (gel filtration), and ion exchangers. ‘Reversed-phase’ plates are now available commercially and are stated to be useful in that they will indicate the type of separation which a mixture would undergo by reversed-phase HPLC (q.v.).
As with paper chromatography, the method can be extended to two- dimensional chromatography, to electrophoretic separations, to quantitative evaluations and to work involving radioactive substances.
Compounds resolved on the TLC plate are visualized using either general or specific methods; thus, ultraviolet light will indicate fluorescent compounds (these should be examined in both long- (c. 365 nm) and short- (c. 263 nm) wavelength ultraviolet light. Fluorescence-quenching compounds (a very large number) are detected by the use of impregnated sorbents (see above). Iodine and Dragendorff’s reagents are used in the form of sprays for the general detection of alkaloids although they (iodine in particular) are not absolutely specific for alkaloids. For indole alkaloids, the reagent p-dimethylaminobenzaldehyde is useful for ergot and a phosphomolybdic acid reagent for others. Antimony trichloride in chloroform is used as a spray reagent for steroidal compounds and other terpenoids, similarly anisaldehyde in sulphuric acid; both these require the sprayed chromatograms to be heated at 100°C for varying times (5–20 min) in order to develop the colours. Ammonia vapour can be used for free anthraquinone compounds and Fast Blue Salt B ‘Merck’ for cannabinoids and phloroglucides. Sugars separated by TLC using phosphate buffered amino layers (e.g. precoated plates of silica gel–Merck NH2) can be located by in-situ thermal reaction (150°C for 3–4 min.) and fluorescence monitoring.
The European and British Pharmacopoeias employ thin-layer chromatographic tests for most vegetable drugs; illustrations of the chromatograms of the fatty acids derived from fifteen different fixed oils are given.
Preparative TLC
As mentioned above, thicker layers of sorbent are employed for preparative work and the separated bands of compounds are scraped from the plate and subjected to solvent extraction. With modern spectrometric methods for structure-determination available, this technique generates quantities of material sufficient for a complete analysis.
To speed up separations and to make them on-line for continuous recording, various modifications of preparative TLC have been developed. These include centrifugally accelerated layer chromatography and overpressure layer chromatography. The latter involves the complete covering of the sorbent layer with an elastic membrane under external pressure thus eliminating the vapour phase from the chromato–graphic plate. The mobile phase is forced up the sorbent layer through a special inlet. This method, introduced by Tyihák et al. in 1979–80, was subsequently adapted to on-line overpressure layer chromatography for the preparative separation of a number of natural products (the isolation of frangula-emodin; noscapine and papaverine fractionation; furocoumarin isomers of Heracleum sphondylium (Umbelliferae); the preparation of the secoiridoid glycosides of Gentiana purpurea). See J. Pothier et al. (Fitoterapia, 1997, 68, 42) for the semi-preparative isolation of the alkaloids of Strychnos nux-vomica, opium, Datura stramonium and Lupinus.
Gas–liquid chromatography
The use of a liquid stationary phase and a mobile gaseous phase in chromatography was first suggested by Martin and Synge in 1941 and developed by James and Martin in 1952 for the separation of the lower fatty acids. Gas–liquid chromatography is now extensively used in all branches of analytical chemistry. Many commercial instruments are available, the more sophisticated being completely automated. For a schematic diagram see Fig. 17.2.
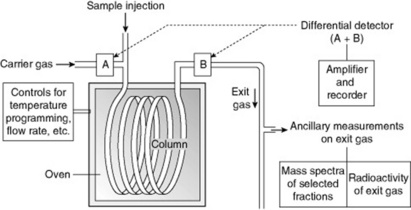
The empty columns are made of glass or metal and are either straight, often up to about 1.3 m in length, or coiled and up to 16 m in length. James and Martin used a tube of 4 mm internal diameter. The liquid stationary phase is held on an inert material, commonly partially fused diatomite. A uniform particle size, with a minimum of dust, is essential, for the inert support and the particles should be as small as possible to give a large surface area but sufficiently large to allow even packing of the column. The choice of stationary phase is governed by the temperature at which the column is to operate and the nature of the material to be fractionated; it should be non-volatile at the operating temperature and should not react with either the stationary and mobile phases or the solutes. Some materials commonly used, with their recommended temperature of operation, include: (1) nonpolar compounds—silicone oils 200–250°C, apiezon oils and greases 275–300°C, silicone gum rubber 400°C, high-boiling-point paraffins such as mineral oil 100°C, squalene 75°C; (2) moderately polar compounds—high boiling point alcohols and their esters 100–225°C; (3) strongly polar compounds—polypropylene glycols and their esters 225°C. Up to 25% by weight of stationary phase is commonly employed on columns; one method of dispersing the stationary phase over the inert support is to dissolve it in a low boiling point solvent such as ether, mix thoroughly with the support and spread out the powder to allow the solvent to evaporate. The powder is then packed into the empty column a little at a time and as evenly as possible and then enclosed in a uniformly heated oven.
A combined support and stationary phase which can replace the above is furnished by cross-linked polymers of specified pore size. These are produced as beads from styrene-like compounds and are marketed under the trade name ‘Porapak’. Advantages of such columns are that they remove undesirable adsorption sites present in diatomite, bleeding of the column (gradual leakage of stationary phase from the column) is reduced, and the rapid elution of water and other highly polar molecules is achieved with little or no tailing.
The operating temperature of the column is critical. Mixtures of low-boiling-point substances can be fractionated at low temperatures; some ethers, for example, can be dealt with at room temperature. Other materials require much higher temperatures—volatile oils 150–300°C, steroids 250°C and pesticides 400°C. Modern instruments can be temperature-programmed so that the column temperature increases as chromatography proceeds. This has the advantage that good separations of mixtures containing compounds with widely different properties can be obtained in one operation and long waits for the emergence of the more strongly retained fractions, with correspondingly less resolution, are lessened.
The mobile phase is a gas which is inert in so far as the other components of the chromatogram are concerned. The choice of gas is dependent on the detector system (see below), and gases commonly used are hydrogen, nitrogen, helium and argon. The flow rate of the gas is important; too high a flow rate will give incomplete separations and too slow a rate will give high retention times and diffuse peaks. Typical flow rates for short columns are 10–50 ml min−1.
By means of a suitable injection device, the sample to be analysed is introduced on to the top of the column; 1.0–5.0 μl is a typical volume but with some detectors it can be considerably less. The measurement of such small volumes is difficult, especially if quantitative results are required, and often the sample is dissolved in a low-boiling-point solvent such as ether; the ether passes rapidly through the column and emerges with the gas front. The mixture to be analysed should volatilize immediately it comes into contact with the stationary phase. Some compounds, not themselves volatile, may be converted into volatile derivatives before chromatography. Thus, sugars, flavonoids including anthocyanins, morphine, codeine and the cardioactive glycosides and aglycones can be chromatographed as their trimethylsiloxy derivatives, which are formed as below:
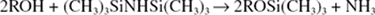
Non-volatile plant acids can first be converted to their methyl esters by treatment with diazomethane.
Along with the analytical columns of the above type, larger preparative columns can be used for the isolation of the separated components in quantities sufficiently large for subsequent examination. Sample sizes of 0.1–20 ml and column lengths of up to 60 m and internal diameters of 1–2 cm illustrate the dimensions involved.
The detector system analyses the effluent gas from the column. It may be of the integral type, in which some property—for example, titration value—of the eluate is recorded or it may be of the differential type, in which some property of the effluent gas is compared with that of a reference gas, often the mobile phase. The latter type is the most commonly used and examples are the katharometer, gas density balance, flame ionization, β-argon ray and electron-capture detectors. For details of these detectors the student is referred to one of the several standard books on gas chromatography. All these differential detectors give an electrical signal which is recorded graphically by a suitable recorder. Because not all detectors give the same relative response to given compounds under the same conditions, some columns are fitted with a double detector system.
The volume of gas that emerges from the column before the arrival of the gas front into which the sample was introduced at the head of the column is termed the ‘hold-up’ volume and it is dependent on the capacity of the column. It is obtained by multiplying the time which the gas front takes to pass through the column by the flow rate; the arrival of the front is often indicated on the recorder by a negative peak caused by the small amount of air injected with the sample. The observed retention volume VR,obs. of a component is calculated from its retention time, and the VR,obs. less the ‘hold-up’ volume is the adjusted retention volume (VR,ad.). By taking into account the pressure drop along the length of the column the net retention volume (VR,net) is obtained. This volume is characteristic for a given compound under the defined conditions of the amount of stationary phase and temperature. Of more universal value is the specific retention volume (VR,sp), which is the VR,net reduced to 0°C g−1 of stationary phase:
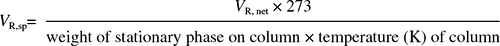
Reference compounds are used to aid the identification of components of a mixture. For quantitative work with differential detectors, the areas enclosed by the peaks are proportional to the quantities of compounds which they represent. To obtain the percentage composition of components within a mixture, without the necessity of placing known amounts of sample on the column, internal standards can be used. These are pure substances, mixed in known proportion with a sample of material to be analysed, which give sharp peaks on the chromatogram not overlapping those of the mixture. Before use, internal standards must be calibrated for detector response against individual components of the mixture.
Sophisticated attachments are available for some equipment. It is thus possible to record the radioactivities and mass spectra of the separated components of a mixture as they emerge from the column. Such instruments have immense potential in biological research. Data storage and retrieval units are now available as standard accessories.
Some pharmacognostical examples of the applications of gas chromatography include the examination of many volatile oils (see, for example the BP assay of Clove Oil), camphor, plant acids, some alkaloids (opium, tobacco and Conium and tropane derivatives), the resins of the Convolvulaceae and of Cannabis, and steroidal compounds such as the sapogenins and cardioactive glycosides and aglycones. The BP test for foreign oils in fixed oils involves the gas-chromatographic separation of the methyl esters of the fatty acids produced by hydrolysis of the sample. The detection and estimation of cocaine and its metabolites in the body is an important forensic application. The estimation of pesticide residues on crops is of utmost importance, and here the sensitivity of detector systems, such as the electron capture detector, has made possible the determination of the chlorinated pesticides down to the parts-per-billion range.
Capillary-column gas chromatography
As the name implies, capillary bore columns are used rather than the standard columns described above. They afford marked improvements in resolving power and in speed of analysis.
The internal diameters of the columns range from about 0.15 mm to about 0.53 mm and the columns can be 1 to 60 m in length. They were originally made of stainless steel and then glass, but fused-silica columns are now considered the obvious choice as they are strong, easy to use, highly inert and give excellent performance. They can be conveniently used in the coiled condition, held, for example, in a 150 mm diameter cage. Such columns hold the stationary phase in a number of ways.(1), Wall-coated open tubular (WCOT) columns have the inner wall of the tube coated with stationary phase up to about 1 μm in thickness. Greater thickness leads to column bleeding in which the stationary phase moves down the column and eventually leaks into the detector. Thicker layers, and hence increased sample capacity, can be achieved with silica columns having specially bonded phases. WCOT columns have the highest efficiency but a low sample capacity. (2), Support-coated open tubular (SCOT) columns have the inner wall lined with a thin layer of support material coated with immobile phase. This has the effect of increasing the available area of immobile phase, affording the column a greater load capacity. The efficiency, while lower than that of the WCOT columns is much higher than that for packed columns. (3) Micropacked columns involve a coated support packed into narrow-bore columns. In all ways they represent a compromise, being more efficient than the normal packed columns but having the same problem in that column length is restricted by the high back-pressure.
Another difference between capillary column chromatography and standard gas chromatography concerns the method of introducing the sample to the column. The volume of sample dissolved in solvent for analysis by the capillary method is too small for a microsyringe and so special injection heads are necessary which either split the sample (e.g. 25:1 with the smaller portion passing to the column) or are of the so-called splitless-injector type which are able to accommodate the relatively large volume of solvent and deliver the dissolved solute to the column.
As with other chromatographic techniques (see HPLC) the introduction of chiral stationary phases has given an added dimension to gas chromatography. Examples include the separation of the two enantiomers of linalool enabling the detection of reconstituted bergamot oil in the genuine oil (A. Cotroneo et al., Flavour Frag. J., 1992, 7, 15) and the detection of added reconstituted lemon oil in the genuine cold-pressed essential oil (G. Dugo et al., J. Essent. Oil Res., 1993, 5, 21). Some volatile oils of the Pharmacopoeia are tested for chiral purity (q.v).
Gel filtration chromatography, gel permeation chromatography (molecular sieves)
These techniques are used for the separation of substances in solution according to their molecular size. The former refers to the use of aqueous mobile phases and the latter to organic mobile phases.
Hydrophilic gels such as those prepared from starch, agar, agarose (a component of agar), polyacrylamide, polyvinylcarbitol and cross-linked dextrans have been used for the fractionation of proteins, peptides, amino acids and polysaccharides.
The particles of these gels possess pores formed by the molecular structure of the gel, and when packed into a column and percolated with a solution, they permit large molecules of solute, which do not enter the pores, to pass rapidly down the column with the solvent via the intergranular interstices. Conversely, small molecules which are able to enter the gel pores become evenly distributed (on equilibrium) across the column and pass more slowly down its length. Thin layers of the sorbent can be used as in TLC.
The dextran gels (Sephadex) are formed by cross-linking dextrans (polymers of glucose in which linkages are almost entirely of the 1,6-α type) with α-epichlorohydrin (Fig. 17.3).
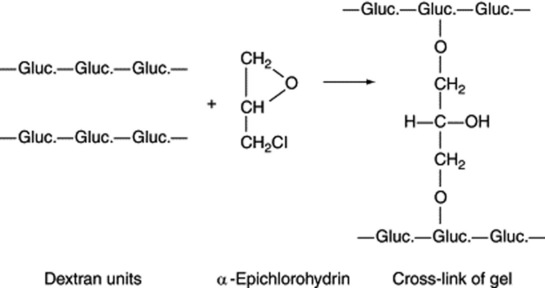
Individual pore sizes are determined by the distance apart of the cross-links, and gels covering a molecular weight range of up to about 200 000 are produced. Each gel type possesses a range of pore sizes, so that below the size limit of complete exclusion of the large molecules, different-sized solute molecules will enter the gel to a greater or lesser extent and so will vary in their elution rates. Similar principles are involved in the use of controlled-pore glass as sorbent; this gives a rigid column with continuous uniform pores. Obviously, the method is most applicable to mixtures containing large molecules of various sizes and to the separation of large molecules from small ones (as in desalting operations on partially hydrolysed proteins). The technique is important in DNA analysis for the separation of those fragments that result from the treatment of DNA with specific restriction enzymes.
Under the heading ‘Size-exclusion chromatography’, the Pharmacopoeia also uses rigid supports as packing material for columns; these may consist of glass, silica or a solvent-compatible cross-linked organic polymer.
Size-exclusion chromatography is used for the determination of those smaller fatty acids (oligomers) that need to be limited in fish oils, such as Fish Oil, Rich in Omega-3-Acids BP/EP, (oligomers, maximum 1.5%).
Electrochromatography
For the electrophoretic separation of mixtures, a filter-paper strip is impregnated with a solution of an electrolyte (usually a buffer solution) and supported in the centre; its two ends are dipped into solutions in which electrodes are immersed. A spot of the material to be fractionated is placed on the paper, the whole apparatus sealed and a potential difference of about 2–10 volts per centimetre applied along the paper. Some separations are carried out at much higher voltages than the above. According to the nature of the charge on the ions of the solute mixture, the solutes will move towards either the anode or the cathode. Thus, the amino acids can be separated either into groups (acid group, neutral group, basic group) or into individual amino acids. The migration velocity for a given substance depends on the magnitude of the ionic charge and the size and shape of the particular molecule. If preferable, paper can be replaced by thin layers of the gels described under ‘Gel filtration’, above. Many alkaloidal mixtures have been separated by this method and also plant acids, the component sugars of cardiac glycosides and anthraquinone derivatives.
A development which combines the advantages of both gel filtration and electrophoresis is that of polyacrylamide gradient gel electrophoresis. It is a two-dimensional electrophoresis system which separates according to mobility of solutes in one direction and according to size in the other.
Capillary electrophoresis is a technique of relatively recent introduction and can give separation efficiencies of the order of 4 × 105 theoretical plates. It provides a more rapid analysis than gel electrophoresis and with detector systems such as the laser-induced fluorescence detector combines a high resolution with a 500-fold increase in sensitivity over UV detection. The method has been used for the analysis of flavonoids.
Affinity chromatography
This method has been developed largely for the resolution of protein mixtures, and it depends on the specific, reversible binding of individual proteins with a particular ligand such as an enzyme substrate or inhibitor. The ligands are coupled with a suitable carrier (cellulose, beaded agarose, controlled-pore glass, polyacrylamide or cross-linked dextrans), possibly with the introduction of a spacer—a suitable chemical moiety such as a hydrocarbon chain—between the ligand and matrix. Excess ligand is removed by washing and the material is packed in a column. A protein mixture in a suitable buffer solution is passed down the column and a protein with sufficient affinity for the bound ligand is retarded and may later be eluted in a purified state by a change in ionic strength or pH of the column buffer. The method has the advantage of preparing in one step a particular component in a high state of purity.
Affinity chromatography has been applied to the purification of enzymes for potential clinical application, for the isolation of certain antibodies and for the specific fractionation of different types of cells (e.g. erythrocytes and lymphocytes).
CHARACTERIZATION OF ISOLATED COMPOUNDS
It is outside the scope of this book to consider in any detail the structure elucidation of natural products. It is sufficient to state that although still utilizing classical chemical methods of degradation, chemists are coming to rely more and more on the use of physical techniques to establish structures of new compounds and to identify known compounds in plant sources. Ultraviolet, infrared, mass and nuclear magnetic resonance spectroscopy together with X-ray crystallographic and optical rotatory dispersion methods have all played a significant role in these developments. Various modifications of mass spectrometry (MS) have become of increasing importance for the structural characterization and determination of the active constituents of plants; these include electron ionization MS, chemical ionization MS, field desorption MS, fast atom bombardment MS and electrospray ionization MS. For an example of the application of electrospray MS combined with sequential tandem mass spectrometry to the investigation of the steroidal saponin mixture of the Chinese and Indian drug Tribulus terrestris, see S. Fang et al., Planta Medica, 1999, 65, 68. Many problems of structure elucidation which 40 years ago were incapable of investigation, either through paucity of material or through lack of suitable chemical methods, can now readily be solved in a standard research laboratory. J. Schmidt et al. (Phytochemistry, 2007, 68, 189), using liquid chromatography and similar combined MS techniques to the above, have demonstrated the importance of such methods for evaluating biosynthetic pathways and for studying the fate of distant natural product precursors in specific plants. They fed [ring-13C-6] tyramine to Papaver somniferum seedlings and elucidated the structures of some twenty alkaloids into which the tyramine was incorporated; the alkaloids included those of the morphinan, benzoisoquinoline, protoberberine, benzo[c]phenanthridine, phthalide, isoquinoline and protopine classes. The routine analytical application of some of these techniques to plant drug analysis has been considered in Chapter 16.
BIOGENETIC INVESTIGATIONS
The living material used in biochemical research is extremely varied. Some work is possible which utilizes the whole organism with a minimum of disturbance—for example, bacteria, yeasts and moulds can be cultivated and investigated biochemically, and with animals, test substances can be added to the food and the blood and excreta analysed. With intact higher plants, however, the ultimate destruction of the plant for analysis is usually necessary. Minces, breis and homogenates are examples of preparations in which the tissues and the cell wall structures have been destroyed but in which the intracellular particles are still intact. The components of such a mixture can be isolated by centrifugation and the biological activity of each fraction can be tested. The penultimate stage in a biogenetic study is the isolation of the enzymes involved in the pathways under consideration and the in vitro demonstration of their properties. Finally, it is becoming increasingly possible to locate and clone the gene responsible for the synthesis of a particular enzyme. Now that the principal overall pathways associated with secondary metabolism have been largely established, it is the enzymic studies that currently receive considerable attention.
The techniques discussed below have been used for the study of secondary metabolism and their application dates from the middle of the last century; they relate principally to the search for the intermediates involved in particular pathways rather than to reaction mechanistics. It must be remembered however that many of the primary metabolic pathways, e.g. the Krebs (TCA) cycle, were established using classical biochemical methods.
TRACER TECHNIQUES
Tracer technology, now widely employed in all branches of science, had its origin in the early part of the last century, when it was realized that elements existed with identical chemical properties but with different atomic weights. Such isotopes may be stable (2H, 13C, 15N, 18O), or the nucleus may be unstable (1H, 14C) and decay with the emission of radiation. If it is possible to detect these isotopes by suitable means, then they can be incorporated into presumed precursors of plant constituents and used as markers in biogenetic experiments.
Radioactive tracers
In biological investigations the use of radioactive carbon and hydrogen, and to a lesser extent and for more specific purposes sulphur, phosphorus and the alkali and alkaline-earth metals, enables the metabolism of compounds to be followed in the living organism. For studies on proteins, alkaloids and amino acids a labelled nitrogen atom may give more specific information than a labelled carbon, but the two available isotopes of nitrogen are both stable, necessitating the use of a mass spectrometer for their use as tracers.
Natural carbon possesses two stable isotopes with mass numbers 12 and 13, the latter having an abundance of 1.10 atoms per cent. Radioactive isotopes of carbon have mass numbers of 10, 11 and 14. 10C has a half-life of 8.8 s and 11C a half-life of 20 min., which limits their usefulness in biological research. However, 14C has an estimated half-life of over 5000 years and in the atomic pile it may be produced by the bombardment of 14N with slow neutrons, the target material usually being aluminium or beryllium nitride.
The immense possibilities in biological research for the use of organic compounds with specific carbon atoms labelled led to the synthesis of many compounds from the inorganic carbon compounds produced in the pile by routes not before commercially utilized. In these syntheses the purity of the product is of great importance, since a small proportion of a strongly radioactive impurity might seriously jeopardize the results of any subsequent experiments.
Many compounds which are most conveniently prepared from natural sources (e.g. certain amino acids by the hydrolysis of proteins) are produced by growing Chlorella in an atmosphere containing 14CO2. All the carbon compounds of the organism thus become labelled, each compound possessing a uniform labelling of its carbon atoms.
Many tritium (3H)-labelled compounds are commercially available. Tritium labelling is effected by catalytic exchange (platinum catalyst) in aqueous media, by irradiation of organic compounds with tritium gas and by hydrogenation of unsaturated compounds with tritium gas. Tritium is a pure β-emitter of low toxicity, half-life 12.43 years, with a radiation energy lower than that of 14C.
Detection and assay of radioactively labelled compounds
When radioactive tracers are used in biogenetic studies, adequate methods for the detection and estimation of the label are essential. For the soft and easily absorbed radiation from 3H-and 14C-labelled compounds the instrument of choice is the liquid scintillation counter. It depends on the conversion of the kinetic energy of a particle into a fleeting pulse of light as the result of its penetrating a suitable luminescent substance. Rutherford successfully used this method in his early studies on radioactivity and he counted the flashes of light produced by bombardment with α-particles on a fluorescent screen prepared from zinc sulphide. The usefulness of the detector was tremendously heightened by the development of the photomultiplier tube, which replaced the human eye in recording the scintillation. Liquid scintillation media, consisting of a solvent in which the excitation occurs and a fluorescent solute which emits the light to actuate the photomultiplier, have also been devised for the purpose of enabling the sample to be incorporated in the same solute, and, hence, attain optimum geometry between sample and scintillator.
Modern instruments are fully automatic (e.g. for 100 samples at a time) and will also measure mixed radiations such as 3H and 14C (this is possible because, although both are β-emitters, they have different radiation energies. With all counters, the instrument is connected to a suitable ratemeter which records the counts over a given time. With 14C, because of its long half-life, no decay corrections are necessary for normal biogenetic experiments. However the half-life is important in carbon dating of old materials. With 3H-labelled material some correction for decay may be necessary if samples are stored for any length of time.
The traditional unit of radioactivity has been the curie, defined as that quantity of any radioactive nuclide in which the number of disintegrations per second is 3.7 × 1010. Subunits are the millicurie (3.7 × 107 disintegrations per second) and the microcurie (3.7 × 104 d.p.s.). The SI unit now used for radioactive disintegration rate is the becquerel (Bq), which has a disintegration rate of 1 s−1, and its multiples include the gigabecquerel (GBq), at 109 s−1 (27.027 millicuries), and the megabecquerel (MBq), at 106 s−1 (27.027 microcuries).
For information on the theoretical basis of radioactive isotope utilization and the regulations governing the use of radioactive substances in universities and research establishments the reader is referred to the standard works and official publications on these subjects.
Autoradiography
A technique used for the location of radioactive isotopes in biological and other material is autoradiography. In this the specimen is placed in contact with a suitable emulsion (e.g. X-ray sensitive film) and after exposure the latter is developed in the usual manner. The resulting autoradiograph gives the distribution pattern of the radioactive substances in the specimen. The method can be applied to whole morphological parts (e.g. leaves) or to histological sections, for which the resulting negative is viewed under a microscope. In a similar manner, radioactive compounds on paper and thin-layer chromatograms can also be detected and the relative amounts of radioactivity in different spots determined by density measurements or by the use of calibrated films.
Precursor–product sequence
For the elucidation of biosynthetic pathways in plants by means of labelled compounds, the precursor–product sequence is commonly invoked. In this a presumed precursor of the constituent under investigation, in a labelled form, is fed to the plant and after a suitable time the constituent is isolated and purified and its radioactivity is determined. If specific atoms of the precursor are labelled, it may be possible to degrade the isolated metabolite and ascertain whether the distribution of radioactivity within the molecule is in accordance with the hypothesis under test. Radioactivity of the isolated compound alone is not usually sufficient evidence that the particular compound fed is a direct precursor, because substances may enter the general metabolic pathways of the plant and from there become randomly distributed through a whole range of products. If this happens, degradation of the isolated constitutent and the determination of the activity of the fragments would probably show that the labelling was random throughout the molecule and not indicative of a specific incorporation of the precursor. Further evidence for the nature of the biochemical incorporation of precursors arises from double- and triple-labelling experiments; either different isotopes, or specific labelling by one isotope at two or more positions in the molecules, are employed. The method has been applied extensively to the biogenesis of many plant secondary metabolites. Leete, in his classical experiment, used two doubly labelled lysines to determine which hydrogen of the lysine molecule was involved in the formation of the piperidine ring of anabasine in Nicotiana glauca. His feeding experiments gave the incorporations shown in Fig. 17.4, indicating the terminal N to be involved.
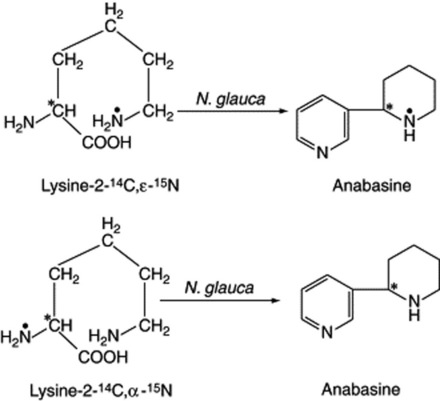
It will be appreciated that with the above lysine precursors it is not necessary to have individual molecules labelled with both 14C and 15N but that the same result is obtained by using standard mixtures of specifically labelled [14C]lysine and [15N]lysine. (Note: 15N is a stable isomer.) Extensive use has been made of 3H/14C ratios in the study of stereospecific hydrogen elimination reactions.
Competitive feeding
If incorporation is obtained, it is still necessary to consider whether this is in fact the normal route of synthesis in the plant and not a subsidiary pathway, invoked as a result of the atypical availability to the plant of the administered compound. Competitive feeding experiments can be of value in determining which of two possible intermediates is normally used by the plant. In its simplest form, without taking into account a number of other factors, competitive feeding could distinguish whether B or B′ was the normal intermediate in the formation of C from A as below:
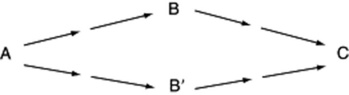
Inactive B and B′ are fed with labelled A to separate groups of plants and a control is performed by feeding labelled A only to another group. If the incorporation of activity into C is inhibited in the plants receiving B, but is unaffected in the group receiving B′, then we may conclude that the pathway from A to C probably proceeds via B. In such experiments involving intact plants, the biological variation between normal plants is often so great that it is difficult to perform a controlled investigation. In comparative studies on the rates of demethylation of codeine and unnatural codeine derivatives in Papaver orientale, Kirkby and colleagues in 1972 overcame this problem by using the same plant simultaneously as the control and as the test plant. They did this by administering to the plant a mixture of 3H-labelled codeine and 14C-labelled unnatural codeine derivative. The products of the conversion of both could be independently followed by their characteristic radiations, even when the metabolites produced were chemically identical. (For the use of competitive feeding in the elucidation of the biogenesis of tropane alkaloids, see Beresford and Woolley, Phytochemistry, 1975, 14, 2209.)
Administration of precursors
Negative results arising from feeding experiments must also be interpreted with caution; thus, the administered precursor may never have reached the necessary site of synthesis in the plant, or the plant may not, at the time of the experiments, have been synthesizing the constituent under investigation. Two examples of the latter situation which, until discovered, were the cause of misleading results in alkaloid studies were the cessation of hordenine production in barley seedlings 15–20 days after germination and the restricted synthesis of hyoscine, as distinct from hyoscyamine, in old plants of Datura stramonium.
Often, the actual weight of labelled material fed to the plant is extremely small and contamination of the solution by microorganisms, during infiltration, can lead to a loss, or even the complete disappearance, of the original compound. This situation is very likely to arise with infiltrations into non-sterile roots—it can, in some instances, be controlled by the use of broad-spectrum antibacterial agents.
A number of methods can be employed to introduce labelled substances into plants. Root feeding is particularly suited to plants which can be grown in hydroponic culture solution and which synthesize the compounds under investigation in the roots. Direct injection of precursor solutions is sometimes possible; this is particularly applicable to plants with hollow stems (Umbelliferae) and to capsules (opium poppy). For the introduction of [1-13C]glucose solution into chamomile flowers K.-P. Adam and J. Zapp (Phytochemistry, 1998, 48, 953) used a microsyringe, injected into the hollow receptacle of the inflorescence. With rigid tissues it is difficult to avoid a loss of solution from the site of injection by this method. Infiltrations can be made into rigid stems by using a wick consisting of a thread drawn through the stem and dipping into the labelled solution; alternatively, a flap can be cut in the stem and this dipped into the solution to be inflitrated.
Sequential analysis
A second method of investigation with 14C is to grow plants in an atmosphere of 14CO2 and, by analysis of the plants at given time intervals, to obtain the sequence in which various related compounds become labelled. From the results obtained, certain biosynthetic routes may become apparent and others rejected. Here, again, degradation of the isolated radioactive compounds is important, because some units of the molecule may become labelled more rapidly than others. This method has been very successfully used in the elucidation of the path of carbon in photosynthesis and also for determining the sequential formation of the opium, hemlock and tobacco alkaloids. Exposure periods to 14CO2 as short as 5 min. have been used to obtain evidence of the biosynthetic sequence piperitone → (−)-menthone → (−)-menthol in Mentha piperita. In a number of instances the pathways suggested by these experiments have been at variance with those obtained by feeding labelled intermediates; it would seem that the latter are therefore examples of non-obligatory intermediates.
Use of stable isotopes
The stable isotopes 2H, 13C, 15N, and 18O, which have a low natural occurrence, can be used in the same way as radioactive elements for labelling compounds to be used as possible intermediates in biosynthetic pathways. The usual methods of detection are mass spectroscopy (15N and 18O) and nuclear magnetic resonance (NMR) spectroscopy (1H and 13C). It is the latter which is becoming of increasing significance for biosynthetic studies (the use of mass spectroscopy and NMR spectroscopy in biogenetic studies should not be confused with their extensive use in structural analysis of organic compounds).
ISOLATED ORGANS, TISSUES AND CELLS
The cultivation of isolated organs and tissues of plants eliminates interference from other parts of the plant which may produce secondary changes in the metabolites. It can be used for feeding experiments in conjunction with labelled compounds and is also useful for the determination of the site of synthesis of particular compounds.
Isolated shoots of plants, when placed in a suitable solution or in water, will usually remain turgid for some days and during this time presumably have a normal metabolism; soon, however, a pathological metabolism commences. The technique can be refined by aseptically connecting the cut end of the shoot to a reservoir of suitable sterile nutrient, when the shoots will remain normal for much longer periods. Such shoots often develop roots at the cut ends—a factor which could invalidate the results of an experiment. The technique has been recently (2007) used for the study of the biogenesis of hyperforin and adhyperforin in Hypericum performatum (q.v.). Isolated leaves can be similarly maintained. Rooted leaves have been used in studies on Nicotiana and Datura. By this method a large quantity of root is obtained with a relatively small amount of aerial parts. It has the advantage that the nutrient solution requires no sugar, as sufficient starch is synthesized in the leaf and consequently bacterial and fungal growth in the nutrient solution is minimized. Petal discs have been used in the investigation of the biosynthesis of oil of rose. Isolated roots have been extensively used. Surface-sterilized seedsare germinated under aseptic conditions, and the root is severed and transferred to a sterile nutrient solution. Under suitable conditions a rapid growth is obtained and subcultures may be produced as necessary. In this way strains of tomato roots have been maintained for many years. For biogenetic and growth studies, selected compounds can be added to the culture as necessary. It has been demonstrated by this method that tropane alkaloids are formed in the roots of a number of Solanaceae. Also, the incorporation of a number of precursors into the alkaloids has been followed.
The technique can be extended to the cultivation of isolated tissues and cells (see Chapter 13), the use of which affords considerable potential in the investigation of biogenetic pathways. They offer the prospect of absolutely uniform plant material, obtainable at all times and manageable under regulated and reproducible conditions— factors rarely possible in work with entire living plants. Furthermore, potential precursors of the metabolite under investigation can easily be added to the system and samples can be taken repeatedly for analysis. The aseptic nature of the culture means that bacterial and fungal modifications of the precursor are eliminated. The method has recently been used to study the bifurcation of the taxoid biosynthetic pathway in Taxus caspidata by the administration of early precursors to the cell culture (R. E. B. Ketchum et al., Phytochemistry, 2007, 68, 335).
GRAFTS
Grafting techniques have considerable use in biosynthetic studies, particularly for the determination of the sites of primary and secondary metabolism of some secondary plant products.
Alkaloid formation by grafted plants has been extensively studied in Nicotiana and the tropane alkaloid-producing Solanaceae. Thus, tomato scions grafted on to Datura stocks accumulate tropane alkaloids, whereas Datura scions on tomato stocks contain only a small amount of tropane alkaloids. This suggests the main site of alkaloid synthesis to be the Datura roots. However, interspecific grafts involving D. ferox and D. stramonium demonstrate that secondary modifications of alkaloids do occur in the aerial parts of these plants. This has been demonstrated conclusively by feeding alkaloid-free scions of D. ferox on Cyphomandra betacea stocks with hyoscyamine; on subsequent analysis, hyoscine was isolated from the leaves. More details of this conversion are given in the appropriate section.
In other genera it has been shown by grafts involving tomato that the alkaloid anabasine is produced in the leaves of Nicotiana glauca, nicotine in the roots of N. tabacum and the pungent principle of capsicum, capsaicin, in the developing fruits. Reciprocal grafts of high and low resin-yielding strains of Cannabis have shown this biochemical character to be determined by the aerial parts.
MUTANT STRAINS
Large numbers of mutant strains of microorganisms have been produced which lack a particular enzyme, which results in their metabolism being blocked at a particular stage. Such an organism may accumulate the intermediate compound immediately before the block and for its survival may require an artificial supply of another intermediate which arises after the block. Such organisms are obviously useful materials in biosynthetic studies and have proved of major importance in some investigations.
A mutant of Lactobacillus acidophilus, by its ability to utilize a constituent of ‘brewer’s solubles’ but not acetate, led to the isolation of mevalonic acid, an important intermediate of the isoprenoid compound pathway.
Ultraviolet-induced mutants of ergot auxotrophic with respect to a number of amino acids have been produced; cultures of these have been used to inoculate rye and the resulting alkaloid contents of the sclerotia have been investigated. Gibberella mutants have been used to obtain novel isoprenoid compounds. With higher plants, in spite of repeated attempts with chemicals and irradiation, less success has been obtained in producing mutants useful for the study of biogenesis of therapeutically active compounds; the production of mature haploid plants from cell cultures of pollen (see Chapter 14) may offer a more satisfactory starting material for future work.
Harborne JB. Phytochemical methods—a guide to modern techniques. Dordrecht, Nl: Chapman and Hall (now under Kluwer Academic Publishers imprint), 1998.
Houghton PJ, Raman A. Laboratory handbook for the fractionation of natural products. Dordrecht, Nl: Chapman and Hall (now under Kluwer Academic Publishers imprint), 1998.
Khaledi MG. High performance capillary electrophoresis. New York, NY: J. Wiley, 1998.
Meyer V. Practical high-performance liquid chromatography, 3rd edn. Chichester, UK: J. Wiley, 1999.
Mukhopadhyay M. Natural extracts using supercritical carbon dioxide. Boca Raton, FL: CRC Press, 2000.
Phillipson JD. Phytochemistry and pharmacognosy. Phytochemistry. 2007;68:2960-2972. Review with 63 references
Reich E, Schibli A. High-performance TLC for the analysis of medicinal plants. Thieme, Stuttgart, 2007.
Taylor LT. Supercritical fluid extraction. New York, NY: J. Wiley and Sons, 1996.
Wagner H, Bladt S, Rickl V. Plant drug analysis. A thin-layer chromatography atlas. Berlin: Springer, 2003.