Chapter 16 Quality control
The quality control of crude plant drugs is of paramount importance. In the past, the monographs of national pharmacopoeias adequately covered this aspect for drugs used in the allopathic system of medicine and the British Herbal Pharmacopoeias (1983, 1990, 1996) contained descriptions, tests and quantitative standards for those species commonly employed by medical herbalists. However, there was no control on the plant materials used in the many herbal products manufactured for general retail sale. Under current EU regulations, herbal products can only be manufactured under licence in conformity with the ‘Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2007’, as set out by the Medicines and Healthcare products Regulatory Agency (MHRA) and published by the Pharmaceutical Press, London. Also, the BP/EP 2007 includes a monograph ‘Herbal Drugs’, which gives requirements relating to definition, production, identification, various tests, pesticide residues, heavy metal content, microbiological quality and, where necessary, limits for aflatoxins and radioactive contamination. Quality control personnel are required to have particular expertise in herbal medicinal products in relation to the above.
One possible problem in devising standards for crude drugs concerns the requirement for an assay of the active constituents when the latter may not have been precisely ascertained. Furthermore, one of the tenets of herbal medicine is that the maximum effectiveness of the drug derives from the whole drug or its crude extract rather than from isolated components. In cases where an assay is lacking it is therefore important that the crude drug is properly authenticated, its general quality verified and all formulations of it prepared in accordance with good manufacturing practice. Attention should also be paid to the shelf-life of the crude drug and its preparations.
Although official standards are necessary to control the quality of drugs their use does raise certain problems. Of necessity, to accommodate the considerable variation that occurs between different batches of a natural product it is necessary to set reasonable standards which allow the use of commercial material available in any season. This has resulted in a tendency for producers or manufacturers to reduce all of their material to the lowest requirement; for example, in a good year the majority of the alkaloid-rich leaves of belladonna herb may be removed and used for the economical manufacture of galenicals and the residue of the herb, containing much stem, used to give the powdered drug. Similarly, high-quality volatile oils may be mixed with lower grades and still remain within official limits.
STANDARDS APPLICABLE TO CRUDE DRUGS
There are a number of standards, numerical in nature, which can be applied to the evaluation of crude drugs either in the whole or the powdered condition.
Sampling
Before a consignment of a drug can be evaluated, a sample must be drawn for analysis; considerable care must be exercised to ensure that this sample is truly representative. With large quantities of bulky drugs a different method of sampling is required from that involving broken or powdered drugs. The BP gives no specific instructions for this but the methods of sampling used in the USA are fully described in older editions of the USP. EU guidelines specify that sampling should be carried out by those with particular expertise.
Preliminary examination
In the case of whole drugs the macroscopical and sensory characters are usually sufficient to enable the drug to be identified. The generalappearance of the sample will often indicate whether it is likely to comply with such standards as percentage of seed in colocynth, of ash in valerian or of matter insoluble in alcohol in asafoetida. However, drugs may comply with the descriptions given in the pharmacopoeias and yet be unsatisfactory, as it is often difficult specifically to describe deterioration of drugs owing to faulty harvesting, shipment or storage or deterioration due to age. In such cases the trained worker will be able to infer much of the history of the sample from its appearance. The following examples will serve to indicate the type of evidence to look for.
If leaves and similar structures are baled before being properly dried, much discoloured material may be found in the middle of the bale. Overdrying, on the other hand, makes leaves very brittle and causes them to break in transit. If starch-containing drugs break with a horny fracture, it may usually be inferred that the temperature of drying has been too high and that the starch has been gelatinized. A pale colour in the case of chamomiles indicates that the drug has been collected in dry weather and carefully dried, while the colour of the fractured surface of gentian is a good indication as to whether it has been correctly fermented. Some drugs are particularly liable to deterioration if, during shipment or storage, they become damp (e.g. cascara). Under moist condition moulds readily establish themselves on drugs having a high mucilage content (e.g. psyllium, linseed, squill and cydonia). Evidence of insect attack must also be looked for.
The price of certain drugs depends largely on such factors as size and colour, which are not necessarily related to therapeutic value. This applies to such important drugs as senna leaflets, senna pods, chamomile flowers, ginger, nutmegs and rhubarb.
Foreign matter
The difficulty of obtaining vegetable drugs in an entirely pure condition is fully recognized, and pharmacopoeias contain statements as to the percentage of other parts of the plant or of other organic matter which may be permitted. Table 16.1 gives examples of various official types of limit applicable to specific drugs. Drugs containing appreciable quantities of potent foreign matter, animal excreta, insects or mould should, however, be rejected even though the percentage of such substances be insufficient to cause the rejection of the drug on the percentage of foreign matter.
Table 16.1 Examples of BP limits for foreign matter.
| Drugs | Foreign matter limits |
|---|---|
| Leaves and herbs | |
| Bearberry leaf |  8% Foreign matter of which 5% stems and 3% other foreign matter. Leaves of different colour to official description 10% 8% Foreign matter of which 5% stems and 3% other foreign matter. Leaves of different colour to official description 10% |
| Birch leaf | 3% Fragments of female catkins, 3% other foreign matter |
| Lemon balm | 10% Stems having a diameter 1 mm, 2% other foreign matter |
| Wild thyme | 3% Foreign matter (involves recognition of Thymus vulgaris and T. zygis) |
| Wormwood | 5% Stems with diameter greater than 4 mm, 2% other foreign matter |
| Fruits and seeds | |
| Hawthorn berries | 2% Foreign matter, 5% deteriorated false fruits |
| Juniper berries | 5% Unripe or discoloured cone berries, 2% other foreign matter |
| Psyllium seeds | 1% Foreign matter including greenish unripe fruits. No seeds of other Plantago spp. |
| Inflorescences | |
| Calendula flowers | 5% Bracts, 2% other foreign matter |
| Elder flowers | 8% Fragments of coarse pedicels and other foreign matter, 15% discoloured brown flowers |
| Lime flowers | 2% Foreign matter, absence of other Tilia spp. |
| Rhizomes and roots | |
| Couch grass rhizome | 15% Greyish-black pieces of rhizome in cut drug |
| Marshmallow root | 2% Brown deteriorated root, 2% cork in peeled root |
| Valerian root | 5% Stem bases, 2% other foreign matter |
| Barks | |
| Quillaia bark | 2% Foreign matter |
| Cascara bark | 1% Foreign matter |
In the case of whole drugs a weighed quantity (100–500 g, according to the type of drug), of a carefully taken sample is spread in a thin layer on paper. It is examined at ×6 magnification and the foreign matter is picked out and weighed and the percentage recorded. Details will be found in the appropriate BP Appendix. For foreign organic matter in powdered drugs, see ‘Quantitative Microscopy’.
Moisture content
Not only is the purchase of drugs (e.g. aloes, gelatin, gums) which contain excess water, uneconomical, but also in conjunction with a suitable temperature moisture will lead to the activation of enzymes and, given suitable conditions, to the proliferation of living organisms. As most vegetable drugs contain all the essential food requirements for moulds, insects and mites, deterioration can be very rapid once infestation has taken place.
A large number of methods are now available for moisture determination, many being employed in industries unrelated to pharmacy.
Loss on drying
This is employed in the BP/EP and USP. Although the loss in weight, in the samples so tested, principally is due to water, small amounts of other volatile materials will also contribute to the weight loss. For materials (digitalis, hamamelis leaf, yarrow, hawthorn berries, starch, aloes, fibres) which contain little volatile material, direct drying (100–105 °C) to constant weight can be employed. The moisture balance combines both the drying process and weight recording; it is suitable where large numbers of samples are handled and where a continuous record of loss in weight with time is required. For materials such as balsams which contain a considerable proportion of volatile material, the drying may be accomplished by spreading thin layers of the weighed drug over glass plates and placing in a desiccator over phosphorus pentoxide. Vacuum drying over an absorbent may be utilized, possibly at a specified temperature.
Separation and measurement of moisture
The ‘loss on drying’ methods can be made more specific for the determination of water by separating and evaluating the water obtained from a sample. This can be achieved by passing a dry inert gas through the heated sample and using an absorption train (specific for water) to collect the water carried forward; such methods can be extremely accurate, as shown in their use for the determination of hydrogen in organic compounds by traditional combustion analysis.
Methods based on distillation have been widely used for moisture determination. The sample to be analysed is placed in a flask together with a suitable water-saturated immiscible solvent (toluene, xylene, carbon tetrachloride) and pieces of porous pot and is distilled. The water in the sample has a considerable partial pressure and co-distils with the solvent, condensing in the distillate as an immiscible layer. A simple apparatus (Fig. 16.1A) originally devised by Dean and Stark permits the direct measurement of the water obtained and the less dense solvent (toluene, xylene) is continuously returned to the distillation flask. The method is employed in the USP and in the BP/EP for some volatile oil-containing drugs (Roman chamomile flowers, lovage root, eucalyptus, peppermint and sage leaves) and aniseed and star-anise fruits. To accommodate the loss of water due to solubility in the solvent the BP specifies a preliminary distillation of the solvent with added water (about 2 ml); the exact volume of water separating as a layer is read off and then the drug (sufficient to give a further 2–3 ml water) added to the flask and distillation resumed. Water separated from the drug is calculated from the combined final volume. Heavier-than-water solvents require the receiver shown in Fig. 16.1B. The method is readily applicable to crude drugs and food materials but has the disadvantage that relatively large quantities of the sample (5–10 g) may be required.
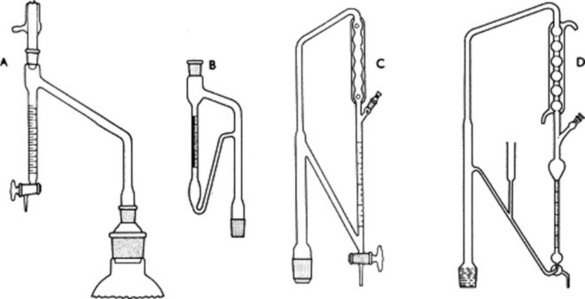
Fig. 16.1 A, Apparatus for the determination of moisture in crude drugs by distillation and for volatile oils heavier than water; B, receiver of apparatus for the determination of water in crude drugs (heavy entrainment) and for volatile oils in drugs; C, receiver for determination of volatile oil in drugs as used by the BP 1980; D, receiver for determination of volatile oil in drugs as used by both the EP and the BP.
Gas-chromatographic methods have become increasingly important for moisture determination because of their specificity and efficiency. The water in the weighed, powdered sample can be extracted with dry methanol and an aliquot submitted to chromatography on a column on either 10% carbowax on Fluoropak 80 or Porapak, a commercial polymer suitable for gas–liquid chromatography (GLC). The water separated by this means is readily determined from the resulting chromatogram. Teflon-6 coated with 10% polyethylene glycol 1500, with n-propanol as an internal standard has also been employed for the determination of moisture in crude drugs.
Chemical methods
The most extensively employed chemical method for water determination is probably the Karl Fischer procedure, which finds use not only in the pharmaceutical, but also in the food, chemical and petrochemical industries. It is used in the BP and is particularly applicable for expensive drugs and chemicals containing small quantities of moisture [very small quantities of water (10 μg to 10 mg) are determined quantitatively by coulometric titration, see below]. Dry extracts of alkaloid-containing drugs, alginic acid, alginates and fixed oils (e.g. arachis, castor, olive and sesame oils for BP parenteral use) may usefully be evaluated. For crude drugs such as digitalis and ipecacuanha the powdered material can first be exhausted of water with a suitable anhydrous solvent (dioxan) and an aliquot taken for titration.
The Karl Fischer reagent consists of a solution of iodine, sulphur dioxide and pyridine in dry methanol. This is titrated against a sample containing water, which causes a loss of the dark brown colour. At the end-point when no water is available, the colour of the reagent persists. The basic reaction is a reduction of iodine by sulphur dioxide in the presence of water. The reaction goes to completion by the removal of sulphur trioxide as pyridine sulphur trioxide, which in turns reacts with the methanol to form the pyridine salt of methyl sulphate, see formulae below.
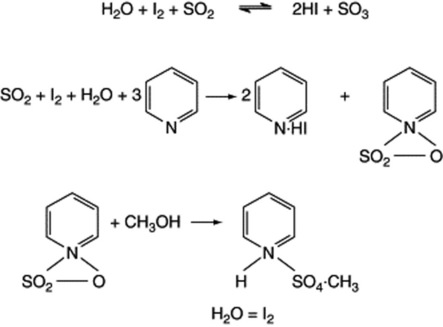
In the absence of methanol, the pyridine sulphur trioxide reacts with another molecule of water. The reagent requires standardization immediately before use and this can be done by employment of a standard solution of water in methanol or by use of a hydrated salt—for example, sodium tartrate (Na2C4H4O6.2H2O). To eliminate interference from atmospheric moisture, the titration is carried out under an atmosphere of dry nitrogen, the end-point being recorded amperometrically. Equipment is now available for a completely automated determination, thus eliminating the manual aspects of sample handling and weighing, introduction to the Karl Fischer cell, titration and data completion. Although the BP Karl Fischer reagent contains pyridine, as above, the latter has been replaced by other bases (e.g. imidazoles) in some commercial reagents. Alternatives to methanol, such as diethylene glycol monoethyl ether, have been introduced to improve reagent stability.
The principal drawbacks of the Karl Fischer method are the instability of the reagent and the possibility of substances in the sample, other than water, which may react with the reagent.
The coulometric method for the quantitative determination of water relies on the same basic reactions as indicated above for the Karl Fischer procedure. However, the iodine is produced electrochemically at the anode by oxidation of iodide and reacts immediately with the sulphur dioxide and water from the sample. When all the water is used up, iodine is produced in excess and this is the electrochemical end-point. If necessary, moisture in a solid sample can be evaporated and passed into the reaction vessel in a stream of dry inert gas. The method is employed for the measurement of the very small amounts of water permissible in fixed oils used for the preparation of parenteral dosage forms; examples include a maximum of 0.1% for soya, olive and evening primrose oils.
Other chemical methods for water determination include treating the sample with various carbides, nitrides and hydrides and measuring the gas evolved; gas chromatography has been employed for the analysis of the liberated gas.
Spectroscopic methods
Water will absorb energy at various wavelengths throughout the electromagnetic spectrum and this fact can be made a basis for its quantitative determination (see later in this chapter for a general discussion on spectroscopy). Measurements can be made in both the infrared and ultraviolet regions; interfering substances must be absent. The method is particularly suitable for very small quantities of water (e.g. trace quantities in gases). Nuclear magnetic resonance (NMR) spectroscopy has been employed for the determination of moisture in starch, cotton and other plant products.
Extractive values
The determination of water-soluble or ethanol-soluble extractive is used as a means of evaluating drugs the constituents of which are not readily estimated by other means. But as suitable assays become available (e.g. with the anthraquinone-containing drugs), some of the previously used extractive tests are no longer required as pharmacopoeial standards. In certain cases extraction of the drug is by maceration, in others by a continuous extraction process. For the latter the Soxhlet extractor is particularly useful and has been in use for many years, not only for the determination of extractives (e.g. fixed oil in seeds) but also for small-scale isolations (Fig. 16.2). A development of the Soxhlet technique is also shown in Fig. 16.2; in this apparatus extraction is by boiling solvent followed by percolation; finally, evaporation yields the extract and the recovered solvent ready for the next sample. Some examples of the types of extractive used are given in Table 16.2.
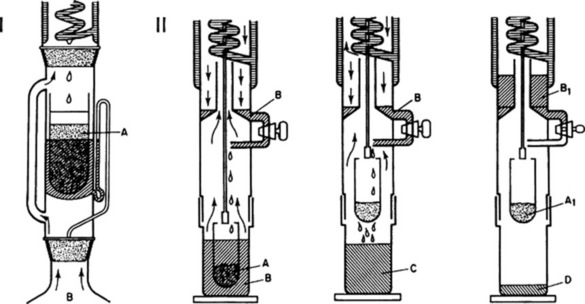
Fig. 16.2 I, Soxhlet continuous extraction apparatus. A, powdered drug for extraction in thimble and plugged with suitable fibre e.g. defatted tow or cotton wool; solvent refluxes into thimble and syphons into flask B, containing boiling solvent, when receiver is full. II, Three-stage continuous extraction and solvent recovery: left, extraction by boiling with solvent; centre, percolation stage; right, removal of solvent. A, sample for extraction; A1, exhausted drug; B, solvent; B1, recovered solvent; C, solvent containing soluble plant constituents; D, final extract. (Soxtec System, Tecator Ltd.)
Table 16.2 Extractives employed for drug evaluation.
| Drug | Method of evaluation |
|---|---|
| Gentian, liquorice, many drugs of BHP | Percentage of water-soluble extractive |
| Quillaia | Percentage of ethanol (45%) extractive |
| Valerian, cocillana, asafoetida | Percentage of ethanol (60%) extractive |
| Ginseng, hop strobiole | Percentage of ethanol (70%) extractive |
| Ginger, ipomoea and jalap | Percentage of ethanol (90%) extractive |
| Benzoin, catechu and Tolu balsam, myrrh | Limits of ethanol-insoluble matter |
| Colocynth | Limit of light petroleum extractive |
| Crushed linseed | Percentage of ether-soluble extractive |
Ash values
When vegetable drugs are incinerated, they leave an inorganic ash which in the case of many drugs (e.g. rhubarb) varies within fairly wide limits and is therefore of little value for purposes of evaluation. In other cases (e.g. peeled and unpeeled liquorice) the total ash figure is of importance and indicates to some extent the amount of care taken in the preparation of the drug. In the determination of total ash values the carbon must be removed at as low a temperature (450 °C) as possible without producing flames. If carbon is still present after heating at a moderate temperature, the water-soluble ash may be separated and the residue again ignited as described in the BP, or the ash may be brokenup, with the addition of alcohol, and again ignited. The total ash usually consists mainly of carbonates, phosphates, silicates and silica.
To produce a more consistent ash, the Pharmacopoeia utilizes a sulphated ash, which involves treatment of the drug with sulphuric acid before ignition, whereby all oxides and carbonates are converted to sulphates. Two methods are given. The first, employed unless otherwise directed, involves moistening a weighed quantity of the drug with (concentrated) sulphuric acid followed by gentle ignition and then repeating the moistening of the charred drug with subsequent firing at 800 °C. The ignition is repeated until a constant weight of ash is achieved. The second method utilizes sulphuric acid R for moistening the drug, followed by gentle heating until the drug is charred; after cooling, a second 1 ml of sulphuric acid R is added and the ignition is continued at 600 °C ± 50 °C until complete incineration is achieved. If the residue exceeds the prescribed limit the process is repeated until a constant weight (within 0.5 mg) is obtained or until the residue complies with the prescribed limit.
If the total ash be treated with dilute hydrochloric acid, the percentage of acid-insoluble ash may be determined. This usually consists mainly of silica, and a high acid-insoluble ash in drugs such as senna, cloves, liquorice, valerian and tragacanth indicates contamination with earthy material. Senna leaf, which may be used directly as the powdered drug, is required to have a low acid-insoluble ash (2.5%); hyoscyamus, however, which unavoidably attracts grit on to its sticky trichomes, is allowed a higher value (12%). Horsetail BP/EP, the dried sterile stems of Equisetum arvense, has a natural content of silica and the acid-insoluble ash value should lie within the limits 10–15%. In the case of ginger a minimum percentage of water-soluble ash is demanded, this being designed to detect the presence of exhausted ginger.
Crude fibre
The preparation of a crude fibre is a means of concentrating the more resistant cellular material of drugs for microscopical examination. It is particularly useful for rhizomes such as ginger which contain relatively large amounts of oleoresin and starch. The technique involves defatting the powder and boiling in turn with standard acid and alkali with suitable washing of the insoluble residue obtained at the different stages (see Chapter 43). The crude fibre so obtained can also be employed quantitatively to assay the fibre content of foods and animal feedstuffs and also to detect excess of certain materials in powdered drugs, e.g. clove stalk in clove. For further details see the 14th edition of this book.
Determination of volatile oil
Minimum standards for the percentage of volatile oil present in a number of drugs are prescribed by many pharmacopoeias. A distillation method is usually employed, and the apparatus first described by Meek and Salvin in 1937 is still widely used in laboratories; the receiver for this apparatus is very similar to that for water estimation by heavy entrainment (Fig. 16.1B). The weighed drug is placed in a distillation flask with water or a mixture of water and glycerin and connected to the receiver (cleaned with chromic acid), which is filled with water and connected to a condenser. On distillation, the oil and water condense and the volatile oil which collects in the graduated receiver as a layer on top of the water is measured. For oils with relative densities around or greater than 1.00, separation from the water is assisted by placing a known volume of xylene in the receiver and reading off the combined oil and xylene. Alternatively, for oils with relative densities greater than water (e.g. clove oil, 1.05), a receiver similar to the type shown in Fig. 16.1A can be used and no xylene is necessary. The BP 1980 employed the apparatus illustrated in Fig. 16.1C; it differs from the above in that the distillate passes through the condenser and so is cooler than with the reflux type. The BP (2007) employs an apparatus similar to that shown in Fig. 16.1D. The time taken to complete the distillation of the oil varies with the nature of the drug and its state of comminution but about 4 h is usually sufficient. Solution of the volatile oil in a fixed oil (e.g. in powdered fruits of the Umbelliferae) may retard distillation. Note that the pharmacopoeial standards for volatile oil contents of powdered drugs are lower than those for the corresponding whole drugs.
Tannin content
A number of drugs (Agrimony, Alchemilla, Hamamelis, Loosestrife, Oak bark, Rhatany, Tormentil) are assayed for their tannin contents (BP 2007). The method refers to those polyphenols adsorbed by hide powder and giving a colour reaction with sodium phosphomolybdotungstate reagent. See individual drugs.
Bitterness value
This standard is relevant to Bogbean leaf, Centaury, Gentian and Wormwood of the BP. These drugs are used for their bitter effect and specific directions for the determination of the standard are given under each monograph. The bitterness value is determined organoleptically by comparison with a quinine hydrochloride solution which acts as the standard.
Swelling index
This is defined in the BP as the volume in millilitres occupied by 1 g of a drug, including any adhering mucilage, after it has swollen in an aqueous liquid for 4 h. The drug is treated with 1.0 ml ethanol (96%) and 25 ml water in a graduated cylinder, shaken every 10 min for 1 h and allowed to stand as specified. In some instances, as with linseed and psyllium seed where the mucilage is in a layer near the surface of the drug, the standard can be determined on the whole drug; in other cases such as marshmallow root where the mucilage is distributed throughout the tissues, the powdered drug is used. Examples are: Agar  10, Cetrarïa 4.5, Fenugreek 6, Fucus 6.0, Ispaghula husk 40 (determined on 0.1 g powder), Ispaghula seed 9, Linseed 4 (whole drug) and 4.5 (powdered drug), Marshmallow root 10, Psyllium 10.0.
10, Cetrarïa 4.5, Fenugreek 6, Fucus 6.0, Ispaghula husk 40 (determined on 0.1 g powder), Ispaghula seed 9, Linseed 4 (whole drug) and 4.5 (powdered drug), Marshmallow root 10, Psyllium 10.0.
Some variations in the method of determination have given rise to other terminology. Thus Skyrme and Wallis in their original work on seeds of Plantago spp. in 1936 used the term swelling factor (24-h standing period) and the BP (1993) cites swelling power in respect of Ispaghula husk (variation in shaking procedure and standing time). Yet again, for Iceland moss the BP 2007 cites swelling value,  .
.
RF values
Pharmacopoeias are increasingly employing thin-layer chromatography as a means for assessing quality and purity. For a discussion of chromatographic analysis, see Chapter 17. It suffices to mention here that the RF value (rate of flow, i.e., distance moved by solute divided by distance moved by solvent front) of a compound, determined under specific conditions, is characteristic and can be used as an aid to identity. RF values vary from 0.0 to 1.00; the hRF (RF × 100) values are in the range 0–100. Quantitative extracts of crude drugs are prepared and compared chromatographically with standard reference solutions of the known constituents. Intensities of the visualized chromatographic spots can be visually compared and the method can be used to eliminate inferior or adulterated drugs. In this way semiquantitative tests for the principles of drugs (peppermint, saffron, German chamomile, digitalis) not rapidly evaluated by other means have been developed.
In an analogous manner, gas chromatographic retention times and peak areas can be employed for the examination of volatile oils and other mixtures.
Microbial contamination
The BP requires a number of drugs (e.g. acacia, agar, pregelatinized starch, sterculia, tragacanth, powdered digitalis etc.) to be free of Escherichia coli in the quantity of material stated; others (e.g. alginic acid, cochineal, guar, tragacanth) are also tested for the absence of Salmonella. Upper limits for total viable aerobic count, commonly 103, 104 microorganisms g−1, are being increasingly applied to crude drugs including the gums, Agar, Tragacanth, Acacia, Guar and Guar Galactomannan. Xanthan gum (q.v.) produced by fermentation has limits of 103 for bacteria and 102 for fungi, g−1. Generally, manufacturers will ensure that, for crude drugs to be taken internally, the limits for bacterial and mould contamination as applied to foodstuffs are adhered to.
In an investigation by Lutomski and Kedzia (Planta Med., 1980, 40, 212) of mould contamination of crude drugs, 246 samples were examined and 24% were contaminated at the level of 10 000 organisms g−1. From 50 crude drugs, 75 Aspergillus and 28 Penicillium strains were isolated; other common genera were Mucor, Rhizopus and Thamnidium. There was no evidence to show that moulds were producing strongly toxic substances in the crude drug and herbal preparations, in contrast to the embryotoxic, teratogenic, mutagenic and carcinogenic substances produced by some species of the above in peanuts, corn, wheat and rice (see Chapter 39). However, in a series of papers Roy and colleagues (Int. J. Crude Drug Res., 1990, 28, 157; Int. J. Pharmacognosy, 1991, 29, 197 and references cited therein) have shown that roots (e.g. Acorus calamus, Picrorrhiza kurroa) and seeds (Neem and Datura) stored under traditional storage conditions in India develop unacceptable levels of mycotoxins, principally aflatoxin B1; the effects of temperature, relative humidity and light on the elaboration of aflatoxin B1 have also been studied. By the use of mass spectrometry, mycotoxins in food products such as cereals, oil seeds and milk are regularly determined at levels of one part per billion and below.
For a review on the microbial contamination of medicinal plants, see W. Kneifel et al., Planta Medica, 2002, 68, 5.
Toxic residues
These may arise in crude drugs as a result of pesticide application during cultivation of the drug and at a later stage from fumigation of the stored product. The problems and the nature of the toxic residues are essentially those encountered in the food industry and in a number of countries regulations exist to cover limits of these residues in foods, cosmetics, drugs and spices. Appendix XI L of the BP 2007 gives the requirements relating to pesticide residues for herbal drugs; specific directions for the sampling of bulk materials are given and the various insecticides and their assays listed. In certain instances it may be necessary to test for aflatoxins and radioactive impurities. It has been reported that many spices, chamomile and valerian obtained commercially contain pesticide residues, but mainly within acceptable levels.
Thin-layer chromatography (TLC) and gas chromatographic methods are available for the determination of organochlorine and urea derivatives, enzymatic methods for organophosphorus compounds, colorimetric methods for urea derivatives, and spectroscopic techniques for paraquat, triazines and heavy metals.
Toxic residues may be substantially reduced or eliminated by the use of infusions of the dried plant material and by the extraction of the useful plant constituents. Storage at 30 °C has been shown to reduce rapidly the ethylene oxide residues in senna pods to tolerable levels. Much research has been devoted to the harmful effects of ethylene oxide which is a very effective insecticide and acaricide; for an in-depth report see Golberg (1986), Hazard Assessment of Ethylene Oxide, Boca Raton, FL., USA: CRC Press.
Heavy metal accumulation
Herbal drugs, like foods, should comply with the WHO guidelines, and the Pharmacopoeial monograph ‘Herbal Drugs’, with respect to heavy metal content. Small quantities of trace elements are invariably present in plant materials and, indeed, some, such as zinc, copper and molybdenum, appear to be necessary microcomponents of a normal diet. However, under certain circumstances the levels of some metals, particularly those of lead, cadmium, copper and mercury, can increase to unacceptable concentrations. This may arise either by the deliberate inclusion of, for example, mercury compounds in a particular herbal formulation or by the natural accumulation of heavy metals in herbs growing under particular environmental conditions. Mercury was once accepted as a common ingredient of Western medicines (see the BPC of 1949) and, more recently, has appeared in Asian medicines exported world-wide to immigrant communities. In the second instance, increased levels of heavy metals can arise from the nature of the soil and via atmospheric pollution. It is of interest to note that one method of prospecting for metals has involved the analysis of the above-ground flora of the area involved; in this respect, some plants are more prone to metal accumulation than others.
The published work on this aspect of herbal drugs is somewhat limited. For recent studies readers are referred to research by V. Rai et al. (Pharm. Biol., 2001, 39, 384) on the accumulation of lead, cadmium, copper and zinc in nine important drugs of Indian medicine. Samples were obtained from various localities in India and consisted of both authenticated material as well as market samples. In most cases, the concentrations of lead and cadmium exceeded the permissible WHO limits; concentrations varied in the same plant obtained from different localities and the authors attributed this to the industrial activity of the region and possible vehicular pollution.
Limitations for particular metals are placed on some products that have been chemically manipulated, for example, nickel in hydrogenated soya and arachis oils. There are pharmacopoeial limits for iron, chromium and zinc in gelatin, cadmium in linseed oil and iron in pilocarpine salts. Determination is by atomic adsorption spectroscopy after acid digestion of the sample with concentrated nitric, hydrochloric and sulphuric acids. Adsorption is measured at the following wavelengths: Cd 228.8 nm, Cu 324.8 nm, Fe 248.3 nm, Ni 232 nm, Pb 283.5 nm and Zn 213.9 nm; separate techniques are given for As 193.7 nm and Hg 253.7 nm.
Test for aristolochic acids
This test is included in the Pharmacopoeia primarily to detect the presence of these acids in unlicensed herbal medicines. They are present in various species of Aristolochia and Asarum, which may be used either as substitutes or adulterants in certain traditional Chinese medicines. Because of the nephrotoxic and carcinogenic properties of aristolochic acid, the use of Aristolochia has been prohibited in the UK since 1991, see ‘Serpentary’; with a further statutory prohibitive order in 2001. Other regulatory orders are in force world-wide. In spite of this, such products periodically appear on the market, a recent example being the availability of Xie Gan Wan pills in South Wales (see MHRA warning, Pharm. J., 2007, 279, 224).
For the pharmacopoeial test, the sample is shaken with 0.1 m sodium hydroxide for at least 2 hours and the filtered solution purified using a solid-phase extraction column. It is then subjected to liquid chromatography and the eluate monitored at 225 nm. No peaks due to aristolochic acid I and arictolochic acid II should appear, as evidenced by comparison with the chromatogram of a reference solution of these two acids.
STANDARDS APPLICABLE TO VOLATILE AND FIXED OILS
Certain standards are particularly appropriate to volatile oils and fixed oils.
Refractive index
The refractive index of a substance is the ratio between the velocity of light in air and the velocity in the substance under test. For light of a given wavelength (it is usual to employ the D line of sodium, which has a doublet of lines at 589.0 nm and 589.6 nm), the refractive index of a material is given by the sine of the angle of incidence divided by the sine of the angle of refraction. The refractive index varies with the temperature, and pharmacopoeial determinations are made at 20 °C.
A convenient instrument is the Abbé refractometer, in which the angle measured is the ‘critical angle’ for total reflection between glass of high refractive index and the substance to be examined. By this means, and by selecting a particular wavelength of light at which to make the measurements, it is possible to calibrate the instrument directly in terms of refractive index. It is the emergent beam that is viewed in the instrument, and the critical angle is indicated by the edge of the dark part of the field of view. In this instrument the need for a monochromatic light source is eliminated by the inclusion of a dispersion ‘compensator’ placed at the base of the telescope tube of the refractometer. This consists of two direct-vision prisms made accurately direct for the D sodium line; the prisms can be made to rotate in opposite directions. The system of variable dispersion which these prisms form can be made to counterbalance the resultant dispersion of the refractometer prism and the material being examined. The temperature of the sample is adjusted by a water jacket.
Automatic refractometers such as the Leica Auto Abbé refractometer are now available. Advantages over the traditional ‘visual’ transmitted light instruments are: (1) they measure refractive index with precision to the fifth decimal place, compared with four decimal places for the visual refractometer (however, current pharmacopoeias require only three decimal places in standards for volatile oils); (2) a reflected light principle is employed meaning that light is not transmitted through the sample and hence problems with dark or coloured samples are avoided; (3) the shadow-line location is determined by the instrument software, eliminating variations in readings caused by individual subjective interpretations of the placement of the shadowline border on a crosswire; (4) no mechanical components are involved, thus reducing wear with time. However, if the increased sensitivity of such instruments is to be fully utilized then additional care and consideration must be given to the measurement of temperature and to the correction for effect of temperature on refractive index.
Measurements of refractive index are particularly valuable for purity assessments of volatile and fixed oils, and many values can be found in the EP, BP, BPC and other pharmacopoeias. Oils of cassia, cinnamon and cinnamon leaf have refractive indices of about 1.61, 1.573–1.600 and about 1.53, respectively, making possible the differentiation of the oils. The refractive index of lemon oil is 1.474–1.476 and that for terpeneless lemon oil 1.475–1.485.
Optical rotation
The optical rotation of a liquid is the angle through which the plane of polarization of light is rotated when the polarized light is passed through a sample of the liquid; this rotation may be either clockwise or anticlockwise. Along with the fundamental effects of the molecules of liquid under investigation, the observed rotation is dependent on the thickness of the layer examined, its temperature and the nature of the light employed. The BP uses the D-line of sodium (λ = 589.3 nm), a layer 1 dm thick and a temperature of 20 °C. With ‘half-shadow’ or ‘triple-shadow’ polarimeters in which the two or three fields are viewed simultaneously and matched to the same intensity, rotations can be measured with an accuracy of at least ± 0.01 degree.
Most volatile oils contain optically active components and the direction of the rotation, and its magnitude, is a useful criterion of purity. Examples to illustrate the range of values found are caraway oil, +74 ° to +80 °; lemon oil, +57 ° to +70 °; terpeneless lemon oil, −5 ° to +2 °; cinnamon oil, 0 ° to −2 °; citronella oil, Java, −5 ° to +2 °; citronella oil, Ceylon, −9 ° to −18 °; nutmeg oil, +8 ° to +18 °; peppermint oil, −10 ° to −30 °; spearmint oil, −45 ° to −60 °.
Many solid materials of natural origin are optically active and the rotation of their solutions (water, ethanol and chloroform are common solvents) can be measured in a similar way to the above. The specific rotation of the solid is given by

where α is the observed rotation in degrees, l is the length of the observed layer in dm, c is the number of grams of substance contained in 100 ml solution, d is the density and p is the number of grams of substance contained in 100 g of solution. The record of the specific rotation of a compound should include the solvent used and the concentration, in addition to the type of light employed (sodium D line of 589.3 nm or mercury green line of 546.1 nm)—for example,[α] 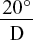 (2.0% in ethanol) = −15 °.
(2.0% in ethanol) = −15 °.
For examples of the use of specific rotation as a physical constant, students can consult the BP or other pharmacopoeial monographs on alkaloidal salts.
The visual polarimeter requires the use of solutions which are not highly coloured or to any extent opaque, and sometimes, with plant extracts, such solutions are difficult to obtain. Because angular rotation falls off linearly, whereas absorption does so exponentially with decrease in path length, the use of short sample tubes has an obvious advantage if it is possible to measure accurately the correspondingly small rotations. Automatic polarimeters employ a sample tube about 1/10th the length of that required by a visual polarimeter. Measurement of small rotations is made possible by utilization of the Faraday electro-optic effect; this involves the rotation of the plane of a polarized beam of light in a magnetic field, the degree of rotation being proportional to the field strength. The instrument is zeroed by means of two solenoids through which passes the polarized beam. The insertion of an optically active solution between the solenoids affords a recordable signal, either (−) or (+), which is generated by a photomultiplier at the end of the light path.
Chiral purity
Optical rotation as described above arises within molecules having at least one asymmetric carbon atom. Such molecules have two possible configurations (enantiomers), which are non-superimposable mirror images of one another and exhibit opposite light-rotational properties [(−) and (+)]. Plants synthesize just one enantiomer, which, under certain conditions, may partially change to the opposite isomer. Equal quantities of both are known as racemic mixtures and have zero rotation; thus, for the alkaloid hyoscyamine, [α]D in 50% ethanol = −22 ° and its racemate atropine is optically inactive. Synthetically produced compounds are normally racemates. The relatively recent introduction of chiral chromatography has provided a method for the quantitative separation of enantiomers and has found use as a standard for volatile oils, which are susceptible to adulteration with the synthetic product. The Pharmacopoeia uses chiral gas chromatography employing fused silica columns 30 m in length with a stationary phase of modified β-cyclodextrine. Chiral purity tests are specified for the volatile oils of caraway, neroli and lavender, and also for carvone.
Quantitative chemical tests
A number of quantitative chemical tests—acid value, iodine value, saponification value, ester value, unsaponifiable matter, peroxide value, anisidine value, acetyl value, volatile acidity—are mainly applicable to fixed oils and are mentioned in Chapter 19. Some of these tests are also useful in the evaluation of resins (acid value, sulphated ash), balsams (acid value, ester value, saponification value), volatile oils (acid value, acetyl value, peroxide value, ester value) and gums (methoxyl determination, volatile acidity).
ASSAYS
A crude drug may be assayed for a particular group of constituents—for example, the total alkaloids in belladonna or the total glycosides of digitalis. Alternatively, it may be necessary to evaluate specific components—for example, the reserpine content, as distinct from the total alkaloid content, of Rauwolfia spp. Biological assays, which can be time-consuming, were at one time employed for the assay of those potent drugs (e.g. digitalis) for which no other satisfactory assay was available. In pharmacopoeias these have now been largely replaced by chemical and physical assays for routine standardization. However, the biological assay remains important for screening plant materials and their fractionated extracts in the search for new drugs. In this respect there is a role for simple biological assays (e.g. brine shrimp toxicity) which can be carried out by the phytochemist without the specialist procedures used by pharmacologists. Some types of assay commonly employed are given in Table 16.3. Often a preliminary purification or fractionation of the active constituents of the drug is required and for this chromatography is finding increasing use. Examples of chromatographic systems employed are listed in Table 16.4 and are more fully explained in Chapter 17. Spectrometric methods, particularly in conjunction with chromatography, are finding increasing use and are dealt with more fully below.
Table 16.3 Types of assay employed for crude drugs.
| Types of assay | Examples |
|---|---|
| Separation and weighing of active constituents | Colchicine in colchicum corm and seed. Resins of podophyllum and of the Convolvulaceae. Crude filicin in male fern. Total balsamic esters of Peru balsam |
| Chemical | ‘Total alkaloids’ of many drugs (e.g. acid–base titrations). Non-aqueous titrations of alkaloid salts. Strychnine in nux vomica. Morphine in opium. Cinnamic aldehyde in oil of cinnamon. Free alcohols in peppermint oil. Carvone in oil of caraway. Assay of fumitory |
| Physical | Cineole in eucalyptus oil (f.p. of o-cresol complex) |
| Spectrometric, including colorimetric and fluorescence | Most groups of active constituents |
| Biological | Cardioactive drugs, antibiotics, vitamins, taenicides, anthraquinone derivatives, mydriatic drugs, saponins, antitumour drugs, antiamoebic drugs, ginkgolides (anti-PAF activity) |
| Radioimmunoassay (RIA) | Hesperidin, limonin and naringin (Citrus), cardenolides (Digitalis lanata), loganin (plant cell cultures), sennosides (Cassia angustifolia), tropane alkaloids (medicinal Solanaceae), morphine and related alkaloids (poppy capsules), lysergic acid derivatives (ergot), quinine (cultured plant tissues), ajmaline (Rauwolfia spp.), vincristine and related alkaloids (Catharanthus roseus), solasodine (Solanum spp.) |
| Enzyme-immunoassay (ELISA) | Quassin, neoquassin, 18-hydroxyquassin (Quassia and Picrasma spp.), podophyllotoxin, tropane alkaloids (Solanaceae), artemisinin in Artemisia annua, pyrrolizidine alkaloids, ergot alkaloids, galanthamine in Leucojum aestivum, saikosaponina in Bupleurum, ginsenosides |
Table 16.4 Chromatographic systems employed in the analysis of drugs.
| Type | Employment |
|---|---|
| Liquid chromatography; BP uses stainless steel columns of varying size; typical packing is chromatographic octadecylsilyl silica gel | Arnica flower, Cola, Devil’s claw, Garlic, Goldenseal root, Opium, Papaveretum, triglycerides of fixed oils (e.g. Refined sesame oil) |
| Gas chromatography | Volatile oils; BP gives chromatographic profiles for some drugs e.g. to aid distinction between Aniseed oil from star-anise and that from Pimpinella anisum. Separation of fatty acids derived from fixed oils. Fatty acid content of fruits, e.g. Saw Palmetto Fruit |
| Thin-layer chromatography | Extensively used in BP and BHP as an identification test and test for purity. Separated constituents can be removed from chromatogram and determined spectrometrically |
Spectroscopic analysis
The electromagnetic vibrations utilized in spectroscopic analysis can be roughly divided, according to wavelength, into the ultraviolet (100–400 nm), the visible (400–800 nm), the near-infrared (800–3000 nm) and the infrared (3–40 μm) regions. The ultraviolet region can be subdivided into three further categories—UVC (100–290 nm), UVB (290–320 nm) and UVA (320–400 nm). These are often quoted in connection with sunlight—all UVC is absorbed by the ozone layer of the atmosphere, UVB is present in small amount but is primarily responsible for major skin damage and UVA, although the major component, is far less damaging. In spectroscopic analysis we are concerned with the capacity of certain molecules to absorb vibrations at specific wavelengths. Thus, the butenolide side-chain of cardiac glycosides is responsible for a strong absorption at 215–220 nm, the conjugated double bonds of lycopene (a pigment of tomatoes and other fruits) give rise to the absorption of light at a wavelength of 470 nm, thus giving a red colour, and the carbonyl group of ketones, carboxylic acids and esters is responsible for a strong absorption in the infrared at about 5.7–6.1 μm. In the ultraviolet and visible regions the characteristic absorption spectrum of a molecule is produced by changes in the electronic energy levels associated with various chromophoric groups within the molecule. These changes involve the absorption of relatively high amounts of energy (in precise quanta), and they are also accompanied by changes in vibrational and rotational energy changes within the molecule. The result is a banded absorption spectrum showing no sharply defined peaks. By comparison, the absorption spectrum of a molecule in the infrared region is much more complex, because here the energies involved are too small to bring about electronic transitions but large enough to produce numerous vibrational and associated rotational energy changes. Each of these changes is associated with a characteristic wavelength and the spectrum shows a much finer structure than in the visible or ultraviolet regions. The infrared spectrum of a molecule can be divided into the ‘fingerprint’ region (7–11 μm), which is characteristic of the molecule under examination but in which it is difficult to assign peaks to specific vibrations, and the remainder of the spectrum, in which many functional groups can be recognized. The BP employs the comparison of infrared spectra of phytochemicals (pilocarpine, physostigmine, etc.) with European Pharmacopoeia Chemical Reference Substances (EPCRS) as a test of identity.
The BP uses ultraviolet absorption characteristics as standards for benzylpenicillin, lanatoside C and a number of alkaloids—for example, morphine, reserpine, cocaine, colchicine and tubocuraine chloride.
If light of a particular wavelength is passed through a solution of a substance, the transmission T = I/I0, where I0 is a measure of the light reaching the detector (a photoelectric cell) when solvent alone is used in the light-path and I is the light reaching the detector when a solution of the substance under investigation is examined. T is measured in experiments but the most useful value is log10(I0/I), the decimal optical density or simply the optical density (E). The optical density, but not the transmission, is proportional to the number of absorbing units in the light-path. For solutions this is Beer’s law. The absorption spectrum of a pure substance under defined conditions of solvent and temperature is a set of values of E observed at different wavelengths in a solution of unit concentration (1 mol l−1) when the thickness of the layer traversed by the light is 1 cm. Alternatively, any other solution of known strength may be used. For example, for a 1% w/v solution with a layer thickness of 1 cm the optical density is indicated by 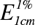 . Such absorption spectra are valuable for the identification, determination of the structure and purity and analysis of compounds. Some substances will absorb ultraviolet light of a certain wavelength and during the period of excitation re-emit light of a longer wavelength and often in the visible region. This is fluorescence, and the fluorescence spectrum is characteristic for those substances which exhibit the phenomenon. The applications of fluorescence analysis are discussed below.
. Such absorption spectra are valuable for the identification, determination of the structure and purity and analysis of compounds. Some substances will absorb ultraviolet light of a certain wavelength and during the period of excitation re-emit light of a longer wavelength and often in the visible region. This is fluorescence, and the fluorescence spectrum is characteristic for those substances which exhibit the phenomenon. The applications of fluorescence analysis are discussed below.
For the quantitative evaluation of a substance, a standard curve is first prepared by measuring the optical densities of a series of standard solutions of the pure compound by use of light of a suitable wavelength, usually that at which the compound gives an absorption maximum. The solutions must be sufficiently dilute to obey Beer’s law. The optical density of the solution to be evaluated is then determined and its composition ascertained from the standard curve. Individual components of a mixture can be determined by ultraviolet absorption, provided that the different compounds exhibit different absorption maxima. Thus, for strychnine and brucine the reported 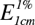 values at the wavelengths (λ) indicated are:
values at the wavelengths (λ) indicated are:
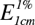 |
||
|---|---|---|
| λ | Strychnine | Brucine |
| 262 nm | 322 | 312 |
| 300 nm | 5.16 | 216 |
By measurement of the extinctions of the mixed alkaloid solution at the above wavelengths, a two-point spectrophotometric assay is available for the determination of both alkaloids. This method, official in the BP (1980) for the assay of nux vomica seeds and preparations, replaced the older, chemical method. A similar type of assay is employed by the EP for quinine-type alkaloids and cinchonine-type alkaloids in cinchona bark; measurements are made at 316 and 348 nm. Occasionally it is useful to examine the ultraviolet spectrum of a more complex mixture; thus, the USP includes an ultraviolet absorbance test for the absence of foreign oils in oils of lemon and orange and the BP an extinction limit test between 268 and 270 nm for castor oil.
In most cases it is essential that no interfering substances are present during the measurements; these can be particularly troublesome in the ultraviolet region, particularly with materials extracted from thin-layer and paper chromatograms. For this reason, if pure solutions are not available for analysis, some form of colorimetric analysis is often preferable, particularly if the reaction used to produce the colour is highly specific for the compound under consideration.
Colorimetric analyses can be carried out with a suitable spectrophotometer—most instruments which operate in the ultraviolet range also have facilities for work in the visible region—but much simpler colorimeters in which suitable filters are used to select the correct wavelengths of light required are quite satisfactory for most purposes. In these instruments a simple light-source is used and, between the lamp and the solution to be analysed, a filter is placed which transmits that range of wavelengths absorbed by the compound under test (i.e. a colour complementary to that of the solution under test). The transmitted light is recorded by a photoelectric cell and the composition of the solution determined by reference to a standard curve. The BP/EP tests the colouring power of Roselle (Hibiscus sabdariffa) by measurement of the absorbance of a water-soluble extract at 520 nm; similarly for an acid extract of Red Poppy Petals at 525 nm.
Characteristic absorption maxima from the more complex infrared spectra can also be utilized in quantitative analysis in the same way as ultraviolet and visible absorptions. Mixtures of substances can also be evaluated; thus, by measurements at 9.80, 9.15 and 9.00 μm it is possible to evaluate separately the 25β- and 25α-epimeric steroidal sapogenins present in plant extracts. A few of the many examples of the application of spectrometric analysis to constituents of drugs are given in Table 16.5.
Table 16.5 Some examples of the application of spectrometric analysis to the constituents of drugs.
| Region of spectrum | Constituents | Wavelength for measurement of optical density |
|---|---|---|
| Ultraviolet | Alkaloids: | |
| Lobeline | 249 nm | |
| Reserpine | 268 nm | |
| Vinblastine | 267 nm | |
| Vincristine | 297 nm | |
| Tubocurarine chloride | 280 nm | |
| Morphine | 286 nm | |
| Colchicine | 350 nm | |
| Cardioactive glycosides with butenolide side-chain | 217 nm | |
| Saponins—glycyrrhizinic acid | 250 nm | |
| Iridoids—harpagoside in Devil’s claw | 278 nm | |
| Quassinoids | 254 nm | |
| Cassia oil—aldehyde content | 286 nm | |
| Bergamot oil—bergapten content | 313 nm | |
| Flavaspidic acid from male fern | 290 nm | |
| Capsaicin | 248 and 296 nm | |
| Vanillin | 301 nm | |
| Allicin—garlic | 254 nm | |
| Vitamin A (cod-liver oil) | 328 nm | |
| Visible | Alkaloids: | |
| Ergot (total alkaloids) | 550 nm by the use of p-dimethylaminobenzaldehyde reagent; 532 nm by the reaction with vanillin in concentrated hydrochloric acid | |
| Morphine | 442 nm by the nitroso reaction | |
| Reserpine | 390 nm by the treatment of alkaloid with sodium nitrite in dilute acid | |
| Tropic acid esters of hydroxytropanes | 555 nm by treatment of alkaloid with fuming nitric acid followed by evaporation to dryness and addition of methanolic potassium hydroxide solution to an acetone solution of the nitrated residue (Vitali–Morin reaction) | |
| Anthraquinones | 500 nm after treatment with alkali (see ‘Senna leaf BP’ for the determination of sennoside; also aloes (512 nm), cascara and rhubarb (515 nm)); 530 nm for the cochineal colour value | |
| Capsaicin in capsicum | 730 nm after reaction with phosphomolybdic acid and sodium hydroxide solution; 505 nm after treatment with diazobenzene-sulphonic acid in 10% sodium carbonate solution | |
| Cardioactive glycosides: | ||
| based on digitoxose-moiety | 590 nm by Keller–Kiliani reaction | |
| based on lactone ring | 620 nm by reaction with m-dinitrobenzene | |
| Ouabain | 495 nm by reaction with alkaline sodium picrate | |
| Cyanogenetic glycosides: | ||
| (cyanide determination) | 630 nm by the pyridine-parazolone colour reaction | |
| Tannins: | ||
| Rhatany, hamamelis leaf | 715 nm using phosphotungstic acid and sodium carbonate solution (see BP) | |
| Procyanidins (as cyanidin chloride) in hawthorn berries | 545 nm | |
| Volatile oils: | ||
| Menthol from peppermint oil | 500–579 nm (green filter) by use of p-dimethylaminobenzaldehyde reagent | |
| Proazulenes (as chamazulene) in yarrow | 608 nm measured on the blue oil obtained by distillation | |
| Flavonoids: | ||
| Birch leaf, calendula flowers (hyperoside), elder flowers (isoquercitrin) | 425 nm with aluminium chloride and glacial acetic acid | |
| Infrared | Alkaloids: | |
| Quinine and strychnine mixtures | 6.2 and 6.06 μm | |
| Steroidal sapogenins | 11.11 μm and 10.85 μm; see text | |
| Volatile oils: | ||
| o-Methoxycinnamaldehyde in cassia oil | Measurements at 7.18 μm and 7.62 μm to distinguish bark oil from leaf and twig oils | |
| Water | 1.9 μm; fairly specific for water without interference from other –OH groups | |
Fluorescence analysis
Many substances—for example, quinine in solution in dilute sulphuric acid—when suitably illuminated, emit light of a different wavelength or colour from that which falls on them. This emitted light (fluorescence) ceases when the exciting light is removed.
Analytical tests based on fluorescence in daylight are not much used, as they are usually unreliable, owing to the weakness of the fluorescent effect. An exception to this is the well-known umbelliferone test, which can be applied to ammoniacum, galbanum and asafoetida. A strongly fluorescent solution of umbelliferone can be prepared by boiling galbanum with acid and filtering into an excess of alcoholic ammonia. Other fluorescent solutions are those of quinine (in dilute acid), aesculin (by infusing horse chestnut bark), chlorophyll (from nettle or parsley leaves), β-naphthol (dissolved in alkali) and aqueous solutions of the dyes eosin and fluorescein.
A very important generalization made by Stokes in 1852 stated that ‘in fluorescence the fluorescent light is always of greater wavelength than the exciting light’. Light rich in short wavelengths is very active in producing fluorescence and for this reason strong ultraviolet light (such as can be obtained from a tungsten arc or mercury vapour lamp) produces fluorescence in many substances which do not visibly fluoresce in daylight. Fluorescence lamps are usually fitted with a suitable filter which eliminates visible radiation from the lamp and transmits ultraviolet radiation of the desired wavelength. Convenient long- and short-wave ultraviolet hand lamps are available for chromatographic observations; it is most important that the eyes are properly protected in the presence of ultraviolet radiation.
For examination, solids may be placed directly under the lamp, whereas liquids may be examined in non-fluorescent dishes or test-tubes or after spotting on to filter paper. Many alkaloids in the solid state show distinct colours—for example, aconitine (light blue), berberine (yellow) and emetine (orange). Pieces of cinchona bark when placed under the lamp show a number of luminous yellow patches and a few light blue ones. If the inner surface of the bark is touched with dilute sulphuric acid the spot immediately turns blue. Ipecacuanha root has a brightly luminous appearance wherever the wood is exposed, while the wood of hydrastics rhizome shines golden yellow. Areca nuts when cut show a light blue endosperm. Slices of calumba appear intensely yellow, with the cambium and phloem distinguished by their dark-green colour. Precipitated and prepared chalks may readily be distinguished from one another.
Most oils, fats and waxes show some fluorescence when examined in filtered ultraviolet light. In general, fixed oils and fats fluoresce least, waxes more strongly, and mineral oils (paraffins) most of all.
Powders may be examined macroscopically as above or microscopically by means of a fluorescence microscope. In connection with powdered drugs may be mentioned the detection of ergot in flour, of cocoa shells in powdered cocoa, and of rumex in powdered gentian. Different varieties of rhubarb may be distinguished from one another. The BP 1973 included a fluorescence test on the entire or powdered drug for the detection of rhapontic rhubarb but this is now replaced by a TLC test.
The location of separated compounds on paper and thin-layer chromatograms by the use of ultraviolet light has been extensively employed. With plant extracts it is often worthwhile to examine the chromatogram in ultraviolet light even if the constituents that one is investigating are not themselves fluorescent. In this way the presence of fluorescent impurities may be revealed which, if otherwise undetected, could interfere with a subsequent absorption analysis. Sometimes fluorescence-quenching can be employed to locate non-fluorescent substances on thin-layer chromatograms. For this, an ultraviolet fluorescent background is produced by the incorporation of a small amount of inorganic fluorescent material into the thin layer. The separated substances cause a local quenching of the background fluorescence and they therefore appear as dark spots on a coloured background.
Quantitative fluorescence analysis
This technique utilizes the fluorescence produced by a compound in ultraviolet light for quantitative evaluation. The instrument employed is a fluorimeter and consists of a suitable ultraviolet source and a photoelectric cell to measure the intensity of the emitted fluorescent light. Within certain limits of concentration the intensity of the fluorescence for a given material is related to its concentration. It is usual to select a narrow range of wavelengths for measurement by inserting a filter between the fluorescing solution and the photoelectric cell. The concentration of a substance in solution is obtained by reference to a standard curve prepared by subjecting standard solutions to the fluorimetric procedure. With plant extracts it is important to ascertain that (1) the substance being determined is the only one in the solution producing a fluorescence at the measured wavelength and (2) there are no substances in the solution which absorb light at the wavelength of the fluorescence. Refined instruments are now available in which the fluorescence spectrum is automatically analysed and in which the wavelength of the incident radiation can also be varied.
Quinine can be conveniently assayed by the measurement of the blue fluorescence (366 nm) produced by irradiation of the alkaloid in a dilute sulphuric acid solution at about 450 nm. The method can be used for the assay of quinine in the presence of other alkaloids (e.g. strychnine). Alexandrian senna has been assayed by measurement of the fluorescence produced in the Bornträger reaction under specified conditions. The hydrastine content of hydrastis root may be determined by oxidizing an extract of the drug with nitric acid and measuring the fluorescence of the hydrastinine produced; by this method berberine and canadine, other alkaloids of hydrastis, are excluded from the assay. Emetine and papaverine may be determined fluorimetrically after oxidation with acid permanganate and noscapine after oxidationwith persulphate. Fluorimetric methods have also been published for the estimation of the ergot and rauwolfia alkaloids, umbelliferone, aflatoxin and a number of drugs in body fluids.
NMR spectroscopy
Although this technique is usually associated with structure-determinations of organic compounds the use of 1H-NMR spectroscopy has been described for the assay of atropine and hyoscine in extracts of belladonna, hyoscyamus and stramonium. It has also been used for the quantitative determination of strychnine and brucine in Strychnos nux-vomica, affording a number of advantages over other methods (M. Frédérich et al., Planta medica, 2003, 69, 1169). Another application has been the classification and correlation of extracts of St John’s wort, involving multivariate data analysis and pharmacological activity (G. Ross et al., Planta medica, 2004, 70, 771).
13C-NMR spectroscopy has been used to distinguish the exudates of various resin-producing families and, together with 1H-nmr spectroscopy, to characterize those of Pinus (Pinaceae) and of other Coniferae families (Cupressaceae, Araucariaceae and Podocarpaceae) (J. B. Lambert et al., J. Nat. Prod., 2005, 68, 625; 2007, 70, 1283).
Immunoassays
Such assays are highly sensitive and usually very specific and have been developed as a powerful analytical tool for the quantitative determination of many compounds in biological fluids.
Radioimmunoassays (RIA)
The assay depends on the highly specific reaction of antibodies to certain antigens. There are various modifications of the technique and it is the saturation method that has been developed for phytoanalysis. Usually the relatively small molecules (below MW 1000) constituting the secondary plant metabolites are not involved in such immunoresponses, but when bound covalently to protein carriers, as haptens, they do become immunogenic. (Haptens are molecules which combine with antibodies but do not stimulate their production unless linked to a carrier molecule.) If such a hapten is prepared in the labelled condition (e.g. 3H- or 125I-labelled) with a known specific activity, mixed with an unknown amount of unlabelled hapten and added to a limited amount of antibody in the form of a serum, then there will be competition between the labelled and unlabelled antigen for the restricted number of binding sites available. This results in some bound and some unbound hapten; these can be separated and a determination of the radioactivity in either fraction, with reference to a standard curve, enables the amount of unlabelled antigen to be calculated. The antiserum is raised in suitable animals (e.g. rabbits).
Following the rapid development of RIA procedures in clinical analyses, and largely owing to the work of Weiler, Zenk and colleagues in Germany since 1976, the method has been satisfactorily applied to a range of plant medicinals as illustrated in Table 16.3.
RIA has the advantage that only small amounts of plant material are required; it is usually specific for a single, or small range of metabolites; relatively crude, unprocessed plant extracts can usually be used; and the process can be mechanized. Thus, it is an efficient tool for the screening of large numbers of plants, some 200–800 specimens being assayed in 1 day. For the application of the method to the selection of high-yielding strains of Digitalis and Solanum, see Chapter 14. With herbarium material, assays can be performed on quantities of sample ranging from 0.5 mg to a few milligrams and in the examination of individual plants, structures as small as anther filaments (e.g. in digitalis) can be accommodated.
Possible disadvantages of the method are the considerable specialized expertise required to set up the assays and the possibility of cross-reactions with components of the plant extract other than those under investigation. Problems arising from the latter need to be ascertained before the assay. The RIA for hyoscine, for example, is highly specific but norhyoscine will react even more strongly; the cross-reaction with 6-hydroxyhyoscyamine is considerably less, and with hyoscyamine, very much less. Similarly, in the assay for solasodine, tomatidine, if present, will cross-react.
Enzyme-linked immunosorbent assays (ELISA)
In this method, competition for an immobilized antibody takes place with a modified form of the compound under analysis that has an enzyme bound to it. Release of the compound–enzyme complex from the binding site and determination of the enzyme activity enables the original solution to be quantified.
Examples of applications to medicinal plants are given in Table 16.3. As with RIAs, the method is very sensitive; thus, for the pyrrolizidine alkaloid retronecine it can be measured in the parts per billion range and one sclerotium of ergot is detectable in 20 kg of wheat.
Tandem mass spectroscopy (MS–MS)
In phytochemistry to date, mass spectroscopy is usually associated with the structure elucidation of compounds rather than with their assay. However, by the simultaneous use of two mass spectrometers in series it is possible to determine quantitatively the amount of a particular targeted compound in complex mixtures, plant extracts or even in dried plant material. Plattner and Powell in their report on maytansinoid identification (J. Nat. Prod., 1986, 49, 475) refer to it as an important analytical tool for ‘needle-in-a-haystack’ analytical problems. Sensitivity to picograms of targeted compounds can be achieved with high specificity and nearly instantaneous response; for sensitivity it compares with RIA but is much more rapidly performed. The method has been used for the analysis of cocaine in plant materials, pyrrolizidine in Senecio and other genera, taxanes from single needles of Taxus cuspidata, aflatoxin B1 in peanut butter, xanthones, steroids and antibiotics. Hoke et al. (J. Nat. Prod., 1994, 57, 277) consider it the best overall method for the determination of taxol, cephalomannine and baccatin in T. brevifolia bark and needle extracts. The chemotaxonomy of the Cactaceae has been investigated by this method.
Quantitative microscopy
Powdered drugs or adulterants which contain a constant number, area or length of characteristic particles/mg (e.g. starch grains, epidermis, trichome ribs respectively) can be determined quantitatively by microscopy using lycopodium spores as an indicator diluent. The method, formerly official in the BP, is described in Chapter 43.