Immunity
After studying this chapter, the student will be able to:
1 Define each of the words in the vocabulary list for this chapter.
2 Describe the primary difference between an immune response and an inflammatory response.
3 List and describe the two main types of lymphocytes, their origins, and their activities.
4 List the activities of macrophages.
5 Using the cells involved, describe the difference between antibody-mediated (humoral) immunity and cell-mediated immunity.
6 Describe the difference between passive and active immunity.
7 Give one example of passive immunity and one example of active immunity.
8 List and describe four types of hypersensitivity reactions and give an example of each.
9 Define autoimmunity and describe how it results in disease.
10 Define immunodeficiency and describe how it results in disease.
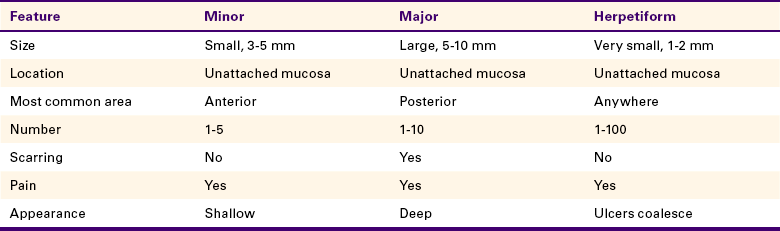
11 Describe and contrast the clinical features of each of the three types of aphthous ulcers.
12 List three systemic diseases associated with aphthous ulcers.
13 Describe and compare the clinical features of urticaria, angioedema, contact mucositis, fixed drug eruption, and erythema multiforme.
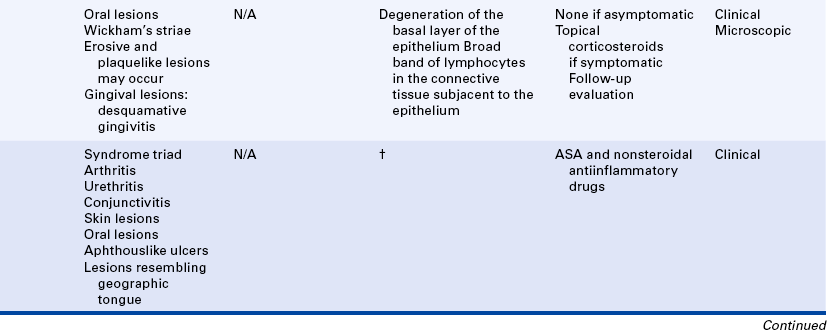
14 Describe the clinical and histologic features of lichen planus.
15 List the triad of systemic signs that comprise Reactive Arthritis (Reiter syndrome) and describe the oral lesions that occur in this condition.
16 Name the two cells that characterize Langerhans cell disease histologically. Describe the acute disseminated form, chronic disseminated form, and chronic localized form and state the names that traditionally have been used for each of these conditions.
17 Describe the oral manifestations of each of the following autoimmune diseases: Lupus erythematosus, pemphigus vulgaris, mucous membrane pemphigoid, and Behçet syndrome.
18 Define desquamative gingivitis, describe the clinical features, and list three diseases in which desquamative gingivitis may occur.
( k″an-thol′ĭ-sis) Dissolution of the intercellular bridges of the prickle cell layer of the epithelium.
k″an-thol′ĭ-sis) Dissolution of the intercellular bridges of the prickle cell layer of the epithelium.
A response of the body to injury that has memory of past exposure to a foreign substance and responds more quickly to a foreign substance when encountered a second time.
(al′er-je) A hypersensitive state acquired through exposure to a particular allergen; reexposure to the same allergen elicits an exaggerated reaction.
(an″ah-fĭ-lak′sis) A severe type of hypersensitivity or allergic reaction in which the exaggerated immunologic reaction results from the release of vasoactive substances such as histamine; the reaction occurs on reexposure to a foreign protein or other substance after sensitization.
(an″tĭ-bod″e) A protein molecule, also called an immunoglobulin, which is produced by plasma cells and reacts with a specific antigen.
(an′′tĭ-jen) Any substance able to induce a specific immune response.
(aw″to-an′tĭ-bod″e) An antibody that reacts against an antigenic constituent of the person's own tissues.
(aw″to-ĭ-mun′dĭ-zēz′) A disease characterized by tissue injury caused by a humoral or cell-mediated immune response against constituents of the body's own tissues.
(B lim′fo-sīt) A lymphocyte, also called a B cell, that matures without passing through the thymus; the B cell can later develop into a plasma cell that produces antibodies.
(sel me′de-a″ted ĭ-mu′nĭ-te) Immunity in which the predominant role is played by T lymphocytes.
(sī′tō-kīn) Cell product produced by the cells involved in the immune response.
(hu′mor-l ĭ-mu′nĭ-te) Immunity in which B lymphocytes and antibodies play the predominant role.
(hi″per-sen″sĭ-tiv′ĭ-te) A state of altered reactivity in which the body reacts to a foreign agent with an exaggerated immune response.
(ĭ-m ū′n kom′pleks) A combination of antibody and antigen.
(ĭ-m″u-no-dĕ-fish′en-se) A deficiency of the immune response resulting from hypoactivity or decreased numbers of lymphoid cells.
(ĭ-m″u-no-glob′u-lin) A protein, also called an antibody, synthesized by plasma cells in response to a specific antigen.
A cell that is a characteristic of lupus erythematosus (LE) and other autoimmune diseases; it is a mature neutrophil that has phagocytized a spherical inclusion derived from another neutrophil.
(lim′foid tish′u) Tissue composed of lymphocytes supported by a meshwork of connective tissue.
(mak′ro-fj) A large tissue-bound mononuclear phagocyte derived from monocytes circulating in blood; macrophages become mobile when stimulated by inflammation and interact with lymphocytes in an immune response.
(mu″ko-sĭ′tis) Mucosal inflammation.
A lymphocyte that is part of the body's initial innate immunity, which by unknown mechanisms is able to directly destroy cells recognized as foreign.
(nĭ-kol′skē sīn) In some bullous diseases such as pemphigus vulgaris and bullous pemphigus the superficial epithelium separates easily from the basal layer on exertion of firm, sliding manual pressure.
(roo′mah-toid fak′tor) A protein, immunoglobulin M (IgM), found in serum and detectable on laboratory tests; it is associated with rheumatoid arthritis and other autoimmune diseases.
(T lim′fo-sīt) A lymphocyte, also called a T cell, that matures in the thymus before migrating to tissues; the T lymphocyte is responsible for cell-mediated immunity and may modulate the humoral immune response.
(thī′mus) A lymphoid organ located high in the chest, which is large in an infant and gradually shrinks in size.
(ze″rof-thal′me-ah) Abnormal dryness of the eyes.
(ze″ro-sto′me-ah) Patient complaint of dryness of the mouth usually caused by decreased salivary flow.
The acute inflammatory response, which is described in Chapter 2, is a rapid first line of defense against tissue injury and disease-producing microorganisms. The acquired immune response, described in this chapter, occurs after the inflammatory response and generally is necessary for complete recovery. This chapter begins with a description of the acquired immune response and includes the oral diseases that result from harmful effects of the immune response. Surveillance against neoplastic cells also involves the immune response. The neoplastic process is described in Chapter 7.
THE ACQUIRED IMMUNE RESPONSE
Like the inflammatory response, the acquired immune response defends the body against injury, particularly from foreign substances such as microorganisms. The immune response differs from the inflammatory response in that it has the capacity to remember past instances of injury and responds more quickly to a foreign substance that is encountered again. It works amid the background of an already activated inflammatory and innate immune response and a working repair process. The innate immune response does not involve memory. It is described along with inflammation in Chapter 2. The acquired immune response involves a complex network of white blood cells. As with inflammation, the immune response may also result in an increased level of injury and disease.
ANTIGENS
Antigens are the foreign substances against which the immune system defends the body. These substances are mainly proteins and are often microorganisms and their toxins. The immune system tolerates the normal components of the body (self) and their normal diversity. Antigens are substances that the immune system recognizes as foreign (nonself). Transformed human cells such as tumor cells or cells infected by viruses can be antigens. Human tissue, as in the case of an organ transplant, tissue graft, or incompatible blood transfusion, can also be an antigen. In diseases called autoimmune diseases, parts of an individual’s own body become antigens. Disease occurs when the immune response identifies components of self as antigen or doesn’t recognize foreign material as antigen.
CELLULAR INVOLVEMENT IN THE IMMUNE RESPONSE
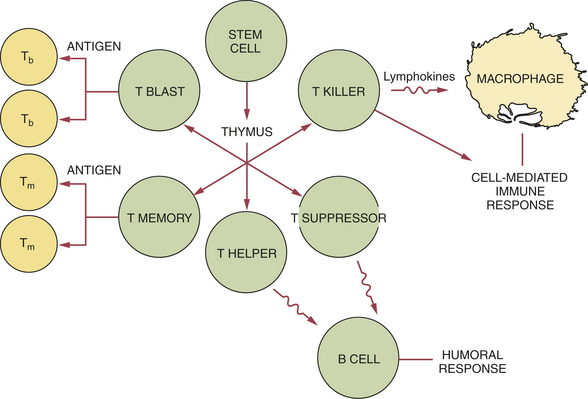
Many cells are involved in the immune response. These cells can produce cell products or cytokines that are active during the immune response. The primary white blood cells involved in the immune response are lymphocytes. These cells are able to recognize and respond to an antigen when it is in contact with receptor sites on the surface of the cell. Lymphocytes, like other white blood cells, are derived from stem cells in the bone marrow. Lymphocytes constitute 20% to 25% of the white blood cell population. They are mobile antigen-sensitive cells with a long life. They have a round nucleus and only a small amount of cytoplasm. Different types of lymphocytes have different functions. The two main types of lymphocyte are called B lymphocytes (B cells) and T lymphocytes (T cells) (Figure 3-1).
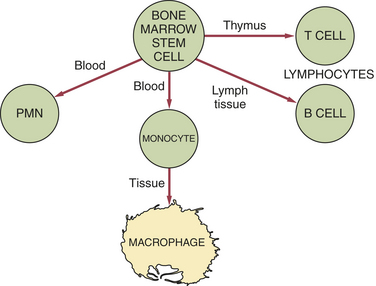
FIGURE 3-1 Differentiation between B and T lymphocytes. The primary cell of the immune response is the lymphocyte. The two main types of lymphocytes are (1) the B lymphocyte and (2) the T lymphocyte. Lymphocytes, like other white blood cells, are derived from stem cells in the bone marrow. PMN, Polymorphonuclear neutrophil.
A third type of lymphocyte is called the natural killer cell (NK cell). This is a large lymphocyte that is part of the body’s innate immunity and not directly involved in acquired immunity. NK cells and their activities are described in Chapter 2. NK cells have the ability to destroy foreign cells early in their appearance since they recognize them as foreign without first having to recognize them as specific antigens. They are usually located within the microcirculation and not in the outlying tissue. This cell seems to be active against viruses and cancer cells. However, in several immunodeficiency diseases, including acquired immunodeficiency syndrome, NK cell function is abnormal.
B LYMPHOCYTES
B lymphocytes develop from the stem cells in the bone marrow and then reside and mature in lymphoid tissue. Lymphoid tissue is found in lymph nodes and many other body locations, including pharyngeal and lingual tonsillar tissues. When antigen stimulates B lymphocytes, they travel to the site of injury. Two main types of B cells develop when a B cell is stimulated by an antigen: the plasma cell and the B memory cell. The B memory cell retains the memory of the antigen. In the presence of an antigen recognized by the memory B, this cell duplicates in a process called clonal selection; all of these newly formed B cells retain the capacity to recognize the previously encountered antigen.
Plasma cells have a round, pin-wheel–shaped nucleus and visible cytoplasm. Plasma cells produce proteins called antibodies. Antibodies, also called immunoglobulins (Igs), are secreted into the blood serum. Five different types of immunoglobulins exist: IgA, IgD, IgE, IgG, and IgM (Table 3-1). They all have variations of the same basic structure (Figures 3-2 and 3-3).
Table 3-1
Known immunoglobulins (antibodies)
| Immunoglobulin | Description |
| IgA | Has two subgroups: serous in the blood and secretory in the saliva and other secretions; aids in defense against proliferation of microorganisms in body fluids |
| IgD | Functions in the activation of B lymphocytes |
| IgE | Involved in hypersensitivity reactions since it can bind to mast cells and basophils and bring about the release of biochemical mediators such as histamine |
| IgG | Major antibody in blood serum; can pass the placental barrier and forms the first passive immunity for the newborn |
| IgM | Involved in early immune responses because of its involvement with IgD in the activation of B cell lymphocytes |
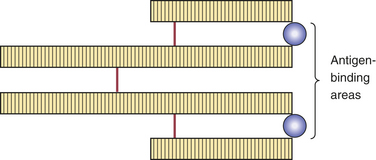
FIGURE 3-2 The basic structure of an antibody or immunoglobulin (Ig) includes four protein chains consisting of two identical heavy polypeptide chains and two identical light chains, shaped to form a Y. The sections that make up the tips of the arms of the Y vary greatly, creating a pocket uniquely shaped to enfold a specific antigen. This is called the variable region. The stem of the Y serves to link the antibody to other participants in the immune response. This area is identical in all antibodies of the same class and is called the constant region.
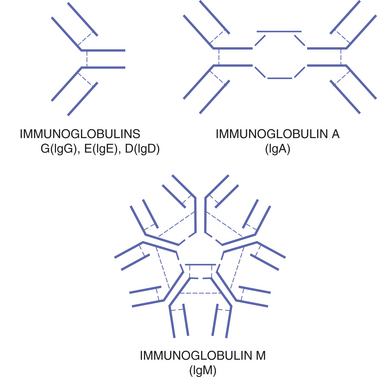
FIGURE 3-3 The five types of antibodies or immunoglobulins include the same basic structure arranged differently. This variation in structure allows them to function differently.
Specific antibodies are produced in response to specific antigens by a specific plasma cell. This specificity is a characteristic function of the immune response. The level of a specific antibody in the blood is called the antibody titer and can be measured by diagnostic laboratory tests. This is helpful in the diagnosis of some infectious diseases.
Antibodies can combine with antigen, forming an immune complex. The portion of the antibody that recognizes and binds to the antigen is called its antigenic determinate. The formation of an immune complex usually renders the antigen inactive. Immune complexes may also be involved in certain disease states.
T LYMPHOCYTES
After they develop from the bone marrow stem cell, the T lymphocytes travel to the thymus and then are processed into mature cells. The thymus is a primary lymphoid organ located high in the chest. It is quite large in infants and shrinks as an individual matures.
T cells depend on unique cell surface molecules called the major histocompatibility complex to help them recognize antigen fragments. Different types of T cells have different functions in the immune response. Some are memory cells similar to those within the B cell population; others are T-helper cells that increase the functioning of the B lymphocytes (enhancing the antibody response) and are easily identifiable by the T4 cell marker (Figure 3-4). T-suppressor cells, which usually carry the T8 marker, suppress or “turn off” the functioning of the B lymphocytes. T-cytotoxic cells, which also carry the T8 marker, are active in surveillance against virally infected cells or tumor cells by directly attacking these cells. T cells within cell-mediated immunity also orchestrate, regulate, and coordinate the overall immune response.
MACROPHAGES
Not only are macrophages present in the connective tissue during inflammation (see Chapter 2 for their role and a description of the cell), but they are also involved in the immune response to an antigen. Macrophages are active in phagocytosis of foreign substances and help both the B cells and T cells during the immune response. After phagocytosis of an antigen at the injury site, the macrophages then act as antigen-processing cells for the lymphocytes. This stimulates the lymphocytes to travel from the lymphoid tissue to the injury site.
Thus macrophages serve as a link between the inflammatory and immune responses since, in addition to phagocytosis, macrophages can also act as antigen-presenting cells. When activated, macrophages can function in many different ways, always amplifying the immune response. Unlike lymphocytes, macrophages do not remember the encountered antigen and need to be reactivated during each encounter.
CYTOKINES
Cytokines are proteins that are made by cells and are able to affect the behavior of other cells. They are produced by the cells of the immune system and play a prominent role in the activation of the immune response. Cytokines are one way that lymphocytes communicate with each other and with other immune system cells. B or T lymphocytes produce cytokines that are called lymphokines. Different cytokines have different functions (Table 3-2). They can activate macrophages and enhance the ability of macrophages to destroy foreign cells. These types of cytokines may also be involved in various other functions concerning leukocytes, fibroblasts, and endothelial cells. Macrophages produce cytokines called monokines. One of the first cytokines to be discovered was interferon. Produced by T cells and macrophages (as well as by cells outside the immune system), interferons are a family of proteins with antiviral properties. Chemokines are a family of small cytokines; their name is derived from their ability to induce directed chemotaxis in nearby responsive cells. They may be involved in controlling infection as part of the immune response such as in an infection with the human immunodeficiency virus.
Table 3-2
Cytokines and their functions during the immune response
| Cytokine | Function |
| Interleukins | Stimulates leukocyte proliferation and other functions |
| Macrophage chemotactic factor | Stimulates macrophage emigration |
| Migration inhibitory factor | Inhibits macrophage activity |
| Macrophage-activating factor | Activates macrophages to produce and secrete lysosomal enzymes |
| Lymphotoxin | Destroys fibroblasts |
| Interferons | Various functions involving leukocytes, fibroblasts, and endothelial cells |
| Tumor necrosis factor | Various functions involving leukocytes and fibroblasts |
MAJOR DIVISIONS OF THE IMMUNE RESPONSE
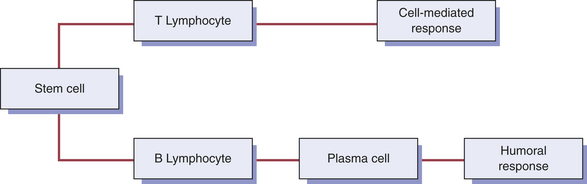
Historically the immune system has been described as two major divisions that differ in their response to an antigen. However, these divisions are interrelated, and present understanding of immune responses recognizes that these divisions are not distinctly separate mechanisms. The immune response to an antigen can take one of two forms (Figure 3-5): humoral immunity and cell-mediated immunity. Humoral (antibody-mediated) immunity involves the production of antibodies with the B lymphocytes as the primary cells. Humoral immunity is responsible for protection against many bacteria and viruses. The other division of the immune system, cell-mediated immunity or cellular immunity, involves lymphocytes, usually T cells, working alone or assisted by macrophages. The cell-mediated division regulates both major responses.
MEMORY AND IMMUNITY
Memory is a characteristic function of the acquired immune response. This contrasts with the inflammatory response, which is not capable of memory. Certain lymphocytes retain the memory of an antigen after an initial encounter. For this reason, the immune response to that antigen is much more rapid and is stronger the next time it is encountered. Immunity is this increased responsiveness that results from the retained memory of an already encountered antigen.

PASSIVE IMMUNITY
Two types of immunity occur: passive and active. Using antibodies produced by another person to protect an individual against infectious disease is called passive immunity. This type of immunity can occur naturally or it can be acquired. Natural passive immunity occurs when antibodies from a mother pass through the placenta to the developing fetus. These antibodies protect a newborn infant from disease while the infant’s own immune system matures.
Passive immunity can also be acquired through an injection of antibodies against a microorganism to which the person has not previously developed antibodies. This is used to confer immediate protection against the disease caused by that microorganism. These antibodies are collected from individuals who have already had the disease and have naturally produced antibodies to the disease-causing microorganism. This type of passive immunity is short-lived but can act immediately. It is used because the unprepared immune system of an individual takes longer to produce antibodies, and in the meantime the disease may develop. Acquired passive immunity, using hepatitis B immunoglobulin, may be provided to dental personnel who do not have immunity to hepatitis B after needlestick or other occupational exposure incidents.
ACTIVE IMMUNITY
Active immunity can occur naturally or be acquired. It occurs naturally when a microorganism causes the disease. Protection against further attack by that microorganism is conferred to the individual if the body recovers from the disease. A less risky way of achieving active immunity is by an acquired or artificial means. A person is injected with or ingests either altered pathogenic microorganisms or products of those microorganisms. The altered microorganism or product cannot produce infection but is able to act as an antigen. This is called a vaccine, and the process is called vaccination. When the pathogenic microorganism is encountered after vaccination, the immune system produces a stronger, faster response and prevents development of the disease. This production of acquired immunity is called immunization.
Immunization lowers the risk of a microorganism causing disease because it safely prepares the immune system to fight future attacks by the disease-causing microorganism. In some cases more than one exposure to the antigen is needed to ensure adequate immunity. A repeated exposure by way of a vaccination is called a booster. Immunization by vaccination is used to protect children and adults against many diseases. Dental personnel should be vaccinated against the hepatitis B virus because of their high risk of occupational exposure to that virus.
IMMUNOPATHOLOGY
Immunopathology is the study of immune reactions involved in disease. The immune response helps defend the body against disease-producing antigens, but it can also malfunction and cause tissue damage. Immunopathology includes diseases caused by malfunctioning of the immune system. Hypersensitivity reactions and autoimmune diseases are examples of conditions in which damage is caused by the immune response.
HYPERSENSITIVITY
Hypersensitivity reactions are also called allergic reactions (allergy) and comprise the same types of reactions that occur when the immune response is fighting microorganisms and protecting the body against disease. However, hypersensitivity reactions are exaggerated immunopathologic responses; tissue destruction occurs as a result of the immune response. Four main types of hypersensitivity reactions occur; they are classified by the nature of the immune response that causes the disease (Table 3-3).
Table 3-3
Examples of hypersensitivity reactions
| Type of Reaction | Examples |
| Type I (anaphylactic type) | Hay fever, asthma, anaphylaxis |
| Type II (cytotoxic type) | Autoimmune hemolytic anemia |
| Type III (immune complex type) | Autoimmune diseases |
| Type IV (cell-mediated type) | Granulomatous disease (e.g., tuberculosis) |
Type I Hypersensitivity
Type I hypersensitivity is a reaction that occurs immediately (within minutes) after exposure to a previously encountered antigen (allergen) such as pollen, latex, or penicillin. In type 1 hypersensitivity plasma cells produce IgE as a response to the antigen. IgE causes mast cells to release their granules containing histamine. This results in edema caused by increased dilation and permeability of blood vessels and in constriction of smooth muscle in the bronchioles of the lungs. This type of hypersensitivity includes hay fever and more serious conditions, including asthma and anaphylaxis. Anaphylaxis can be life threatening because the individual may not be able to breathe as a result of the oropharyngeal tissue swelling and constriction of the bronchioles.
Type II Hypersensitivity
In type II hypersensitivity antibody combines with an antigen that is bound to the surface of tissue cells, usually a circulating red blood cell. Activated complement components, IgG and IgM antibodies in blood, participate in this type of hypersensitivity reaction. The result is the destruction of the tissue that has the antigen on the surface of its cells. This type of reaction occurs in incompatible blood transfusions and in rhesus (Rh) incompatibility. In Rh incompatibility the mother’s antibodies cross the placenta and destroy the newborn’s red blood cells, resulting in hemolytic anemia.
Type III Hypersensitivity
In type III hypersensitivity immune complexes are formed between microorganisms and antibody in the circulating blood. The complexes leave the blood and are deposited in various body tissues or in a localized area. In either case the deposition results in the initiation of an acute inflammatory response. Neutrophils are attracted to the tissues in which the complexes have been deposited. As a result of phagocytosis and death of the neutrophils, lysosomal enzymes are released, causing tissue destruction. This type of hypersensitivity occurs in autoimmune diseases such as systemic lupus erythematosus. Systemic lupus erythematosus is discussed later in this chapter.
Type IV Hypersensitivity
Type IV hypersensitivity involves a cell-mediated immune response rather than a humoral (antibody) response. T lymphocytes that have been introduced to an antigen previously either cause damage to the tissue cells or recruit other cells that cause the damage. This type of hypersensitivity reaction is also called delayed hypersensitivity and is put to use when diagnosing tuberculosis. A visible skin reaction occurs if the individual tested has previously been exposed to the organism that causes tuberculosis. This type of hypersensitivity is also responsible for the rejection of tissue grafts and transplanted organs. New strategies for prevention of rejection, such as therapeutic antibodies against specific T-cell receptors, may produce fewer long-term side effects than the chemotherapies now routinely used.
Hypersensitivity to Drugs
Drugs can act as antigens and cause an immunologically induced inflammatory response. Many factors influence the risk of an allergic (hypersensitivity) reaction to a drug. The route of administration influences how the reaction will be manifested and its severity. Although topical administration of drugs may cause a greater number of reactions than oral (swallowed) or parenteral (administered by injection) routes of administration, the reaction that occurs after parenteral administration may be more widespread and severe because the allergen (the antigen eliciting the response) can be carried quickly to many parts of the body by the circulating blood. The presence of infection may increase the risk of an allergic reaction. Patients with multiple allergies are more likely to have allergic reactions to drugs, and patients with autoimmune diseases such as systemic lupus erythematosus commonly have adverse reactions to medication. Children are less likely than adults to have an allergic reaction to a drug.
Drugs can cause any of the previously described hypersensitivity reactions. Type I allergy to a drug can include anaphylaxis, urticaria (hives), and angioedema (localized swelling). A systemic anaphylactic reaction is more likely to occur with an injected drug but can also occur with a drug administered orally and can be fatal. For example, penicillin may cause a systemic anaphylactic reaction in approximately one in 10,000 patients. It causes about 300 deaths per year in the United States.
The classic example of a type III allergy is called serum sickness. This name was given to a reaction that occurred frequently when patients were given large amounts of horse antitoxin serum to provide passive immunity in the treatment of diphtheria and tetanus. This is no longer the method by which passive immunity to these diseases is provided. This reaction can occur in response to other foreign agents. Penicillin is the single most common cause of serum sickness. However, other drugs such as barbiturates can also cause this reaction. The symptoms of serum sickness include fever, painful swelling of the joints (arthritis), renal disturbance or failure, edema around the eyes, cardiac inflammation, and skin lesions.
Drugs can also cause a type IV hypersensitivity reaction. This T cell–mediated allergic reaction can occur in response to topically applied substances and can produce contact dermatitis and mucositis.
AUTOIMMUNE DISEASES
The immune system learns to differentiate between the body’s own cells and tissues and foreign substances early in embryologic development. This recognition and the nonresponsiveness of the immune system to the body’s own cells and tissues are called immunologic tolerance. In autoimmune diseases, also called connective tissue diseases, the recognition mechanism breaks down, and certain body cells are no longer tolerated. The immune system treats body cells as antigens. An autoimmune disease may involve a single cell type or a single organ or may be even more extensive, involving multiple organs. Tissues and even entire organs may be damaged. An example is the T cells that attack pancreatic cells contributing to diabetes mellitus. Genetic factors may play a role in the predisposition of an individual to autoimmune disease, and viral infection may also be involved. Several autoimmune diseases have oral manifestations. These are described later in this chapter.
IMMUNODEFICIENCY
Immunodeficiency is a type of immunopathologic condition that involves a deficiency in number, function, or interrelationships of the involved white blood cells and their products. This condition may be congenital (present at birth) or acquired (developed after birth). Immunodeficiency may be inherited genetically, or it can be caused by numerous other factors. When a person’s immune system is not functioning adequately, infections and tumors may develop. Studies have shown that stress and depression may be associated with decreased levels of immune function. Acquired immunodeficiency syndrome, which is discussed in Chapter 4, is an example of an immunodeficiency that has numerous oral manifestations.
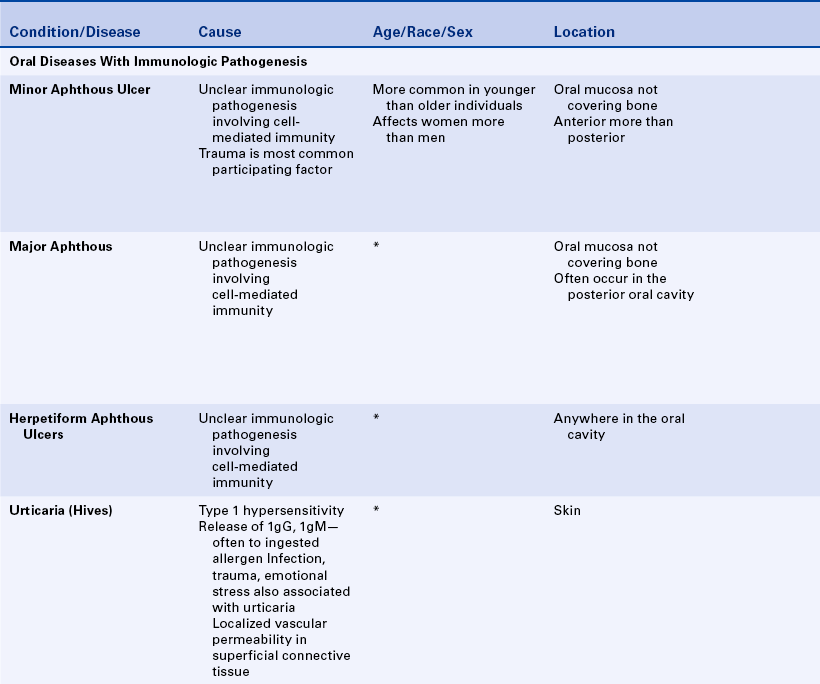
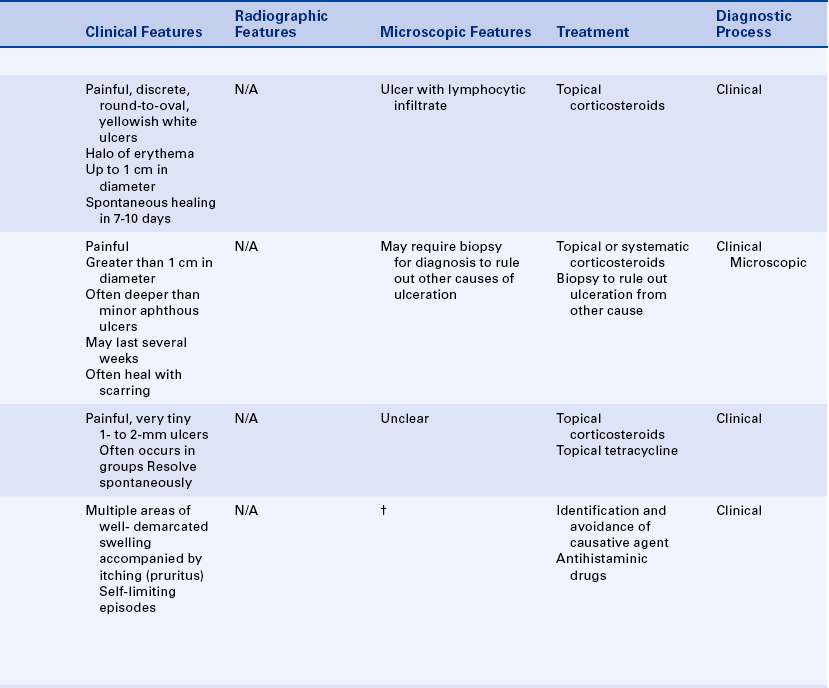
APHTHOUS ULCERS
Aphthous ulcers, also known as canker sores or aphthous stomatitis, are painful oral ulcers for which the cause remains unclear. They frequently recur in episodes. Three forms of recurrent aphthous ulceration exist: (1) minor, (2) major, and (3) herpetiform. The three forms differ in the size and duration of the ulcers. Aphthous ulcers are common ulcers in the oral cavity, occurring in about 20% of the general population. However, a dramatic variation in occurrence takes place, from as high as 57% of a professional school population to 5% of patients studied in a large inner city hospital. The first episode of these ulcers usually occurs in adolescence, and they are somewhat more common in females than in males. The clinical appearance and location are important in establishing the diagnosis.
Trauma is the most commonly reported precipitating factor in the development of aphthous ulcers. They are often reported to occur after trauma to the oral mucosa during dental procedures (e.g., in the area of film placement or at the site of injection of local anesthetics). Some patients may experience aphthous ulcers as a result of manipulation of oral tissues during dental hygiene treatment. Emotional stress has also been suggested as a contributing factor. Some patients associate the initiation of aphthous ulcers with eating certain foods such as citrus fruits. However, it is possible that patients perceive these foods as causative because of the sensitivity of the ulcers to foods with a high acid content. The recurrence of aphthous ulcers has been associated with menstruation, whereas pregnancy has been found to produce a decrease in the episodes. These ulcers occur in association with certain systemic diseases such as:
Substantial evidence indicates that aphthous ulcers have an immunologic pathogenesis. Patients in whom aphthous ulcers develop have slightly elevated levels of antibodies to oral mucous membranes. Histologically an infiltrate of lymphocytes is present in the lesion, suggesting that cell-mediated immunity may be important in development of the ulcers. The infiltrate contains primarily T-helper cells in the prodromal stage, T-cytotoxic cells in the ulcerative phase, and T-helper cells again in the healing stage. The T-cytotoxic cells are probably responsible for the ulceration. However, the specific antigen to which they are responding has not yet been identified.
Minor Aphthous Ulcers
Minor aphthous ulcers are the most commonly occurring type. They appear as discrete, round-to-oval ulcers that can be as large as 1 cm in diameter and exhibit a yellowish-white fibrin surface surrounded by a halo of erythema (Figure 3-6). The ulcers occur on the movable mucosa of the oral cavity (the mucosa not covering bone) and occasionally extend onto the gingiva. They occur on the labial and buccal mucosa, the maxillary and mandibular vestibular mucosa, the ventral and lateral borders of the tongue, and the soft palate and oropharynx. Minor aphthous ulcers are more common in the anterior than the posterior part of the mouth. The ulcers often have a prodromal period of 1 to 2 days, which is characterized by a burning sensation or soreness in the area in which the ulcer will form. Aphthous ulcers are exquisitely painful, and single or multiple lesions may be present. The ulcers heal spontaneously in 7 to 10 days.
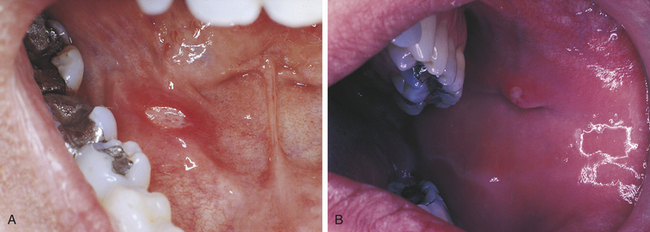
Major Aphthous Ulcers
Major aphthous ulcers (Sutton disease, periadenitis mucosa necrotica recurrens) are larger than 1 cm in diameter and are deeper and last longer than minor aphthous ulcers (Figure 3-7). They are also painful and often occur in the posterior part of the mouth. Major aphthous ulcers may require biopsy for diagnosis to rule out other causes of ulceration. They can take several weeks to heal and frequently result in scarring. Ulcers that resemble major aphthous ulcers have been reported to occur in patients with human immunodeficiency virus infection (see Chapter 4).
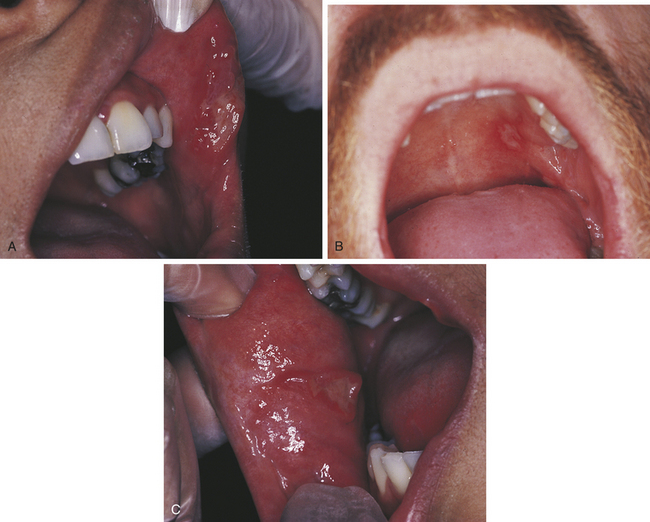
Herpetiform Aphthous Ulcers
Herpetiform aphthous ulcers are very tiny (1 to 2 mm) (Figure 3-8). They are called herpetiform because they resemble ulcers caused by the herpes simplex virus (see Chapter 4). They are painful, may develop anywhere in the oral cavity, and generally occur in groups.
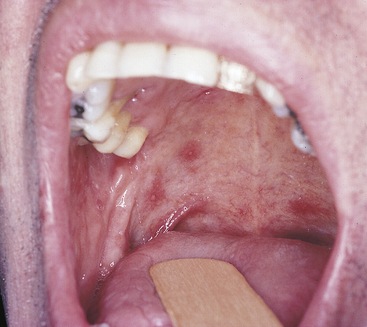
Diagnosis
The diagnosis of minor aphthous ulcers is made on the basis of their distinctive clinical appearance, the location of the lesions, and a complete patient history. Table 3-4 summarizes the clinical features of each of the three types of aphthous ulcers. Laboratory results are not specific for any form of aphthous ulcer.
The location of the ulcers is important in differentiating between recurrent aphthous ulcers and recurrent intraoral ulceration caused by the herpes simplex virus (see Table 4-2). When they occur intraorally, ulcers resulting from the herpes simplex virus appear on keratinized mucosa. This is the mucosa of the palate and gingiva (i.e., the mucosa fixed to bone).
Frequently a biopsy is necessary for diagnosing major aphthous ulcers because they may clinically resemble squamous cell carcinoma or deep fungal infections.
Herpetiform aphthous ulcers may be difficult to distinguish clinically from primary herpetic gingivostomatitis. However, no systemic signs or symptoms exist as in primary herpes simplex infection. Herpetiform aphthous ulcers have been reported to respond to topical application of liquid tetracycline. This response may be helpful in confirming the diagnosis of herpetiform aphthous ulcers because true herpes simplex ulceration does not respond to this treatment.
When a patient develops multiple aphthous ulcers, it is important to consider that they might be associated with a systemic disease. Identification of signs and symptoms that may suggest systemic disease should be considered when reviewing the patient’s medical history. These include the presence of chronic gastrointestinal symptoms such as diarrhea and discomfort (Crohn disease, ulcerative colitis, sprue, intestinal lymphoma), arthritis and skin lesions (Behçet syndrome), and periodic fevers in a child (cyclic neutropenia and PFAFA syndrome).
Treatment
The local application of topical steroids and topical nonsteroidal antiinflammatory agents is helpful in the management of aphthous ulcers. These drugs are most effective when applied very early in the development of the ulcer. Occasionally systemic steroids may be necessary for managing patients with major aphthous ulcers. Topical application of liquid tetracycline is also helpful, particularly in the diagnosis of herpetiform ulcerations when the diagnosis is not clear and the differential diagnosis includes viral infection. The use of topical steroids is contraindicated for viral ulcers; therefore accurate diagnosis is important. Topical anesthetics can help to decrease the pain of aphthous ulcers.
URTICARIA AND ANGIOEDEMA
Urticaria and angioedema are similar lesions. Urticaria, also called hives (Figure 3-9), appears as multiple areas of well-demarcated swelling of the skin, usually accompanied by itching (pruritus). The lesions are caused by localized areas of vascular permeability in the superficial connective tissue beneath the epithelium. Angioedema (Figure 3-10) is a similar lesion that appears as a diffuse swelling of tissue caused by permeability of deeper blood vessels. The skin covering the swelling appears normal, and angioedema is usually not accompanied by itching. Urticaria and angioedema both may occur in acute self-limited episodes. Occasionally chronic or recurrent forms may occur.
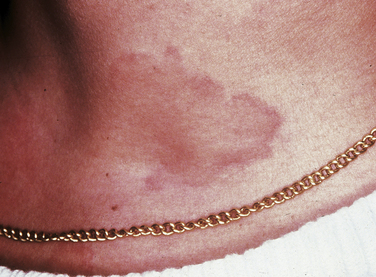
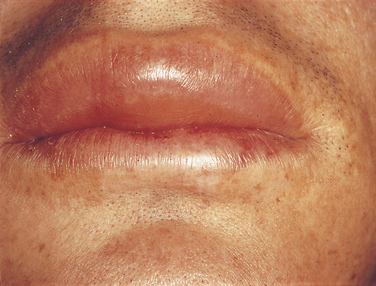
In many cases of urticaria and angioedema the cause cannot be identified. Infection, trauma, emotional stress, and certain systemic diseases have been reported to cause these lesions. Ingested allergens are frequent causes of urticaria.
Several mechanisms are capable of causing the increased vascular permeability that results in urticaria and angioedema, including the release of the chemical mediator histamine from mast cells stimulated by IgE antibodies (type I hypersensitivity) and the activation of IgG or IgM antibodies along with trauma, causing vascular permeability. Acetylsalicylic acid (aspirin) and nonsteroidal antiinflammatory drugs such as ibuprofen can produce a nonspecific effect that can cause vascular permeability. A rare hereditary form of angioedema exists in which uncontrolled activation of the complement cascade occurs, resulting in prolonged vascular permeability.
The diagnoses of urticaria and angioedema are based on the clinical appearance of the lesions and the patient’s history. Avoidance of the causative agent, if identified, is important in managing patients with recurring urticaria and angioedema. Immediate treatment is essential. Antihistaminic drugs are the principal method of treatment. Angioedema involving the larynx and pharynx can cause asphyxiation and therefore may be fatal.
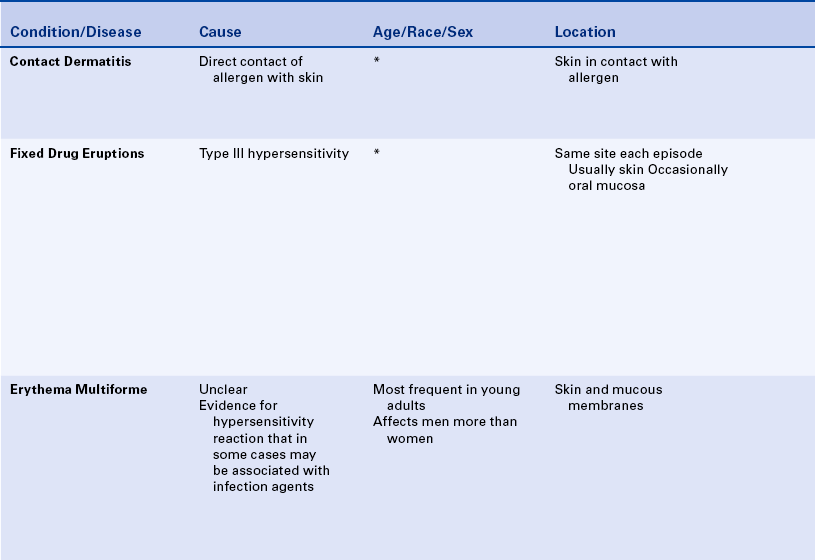
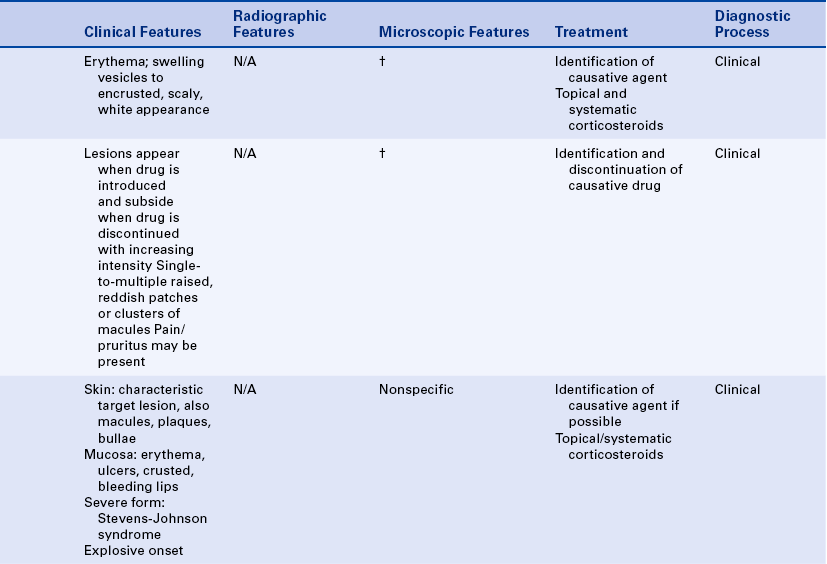
CONTACT MUCOSITIS AND DERMATITIS
Contact mucositis and contact dermatitis are lesions that result from the direct contact of an allergen with the skin or mucosa. The development of these conditions involves a T cell–mediated immune response and is an example of type IV hypersensitivity. The exact underlying immunologic basis is not always clear.
In contact mucositis the mucosa becomes erythematous and edematous, often accompanied by burning and pruritus (Figure 3-11, A). The mucositis occurs at the point at which the offending agent has contacted the mucosa, giving it a smooth, shiny appearance that is firm to palpation. Small vesicles and ulcers may appear in the affected areas. In contact dermatitis the initial lesion may be erythematous, with swelling and vesicles. Later the area becomes encrusted with a scaly, white epidermis.
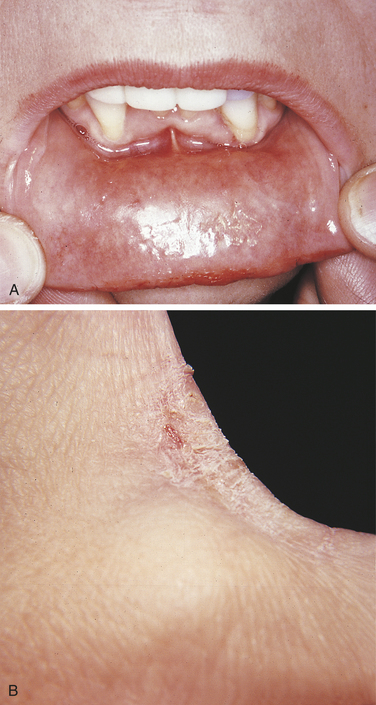
FIGURE 3-11 A, Contact mucositis from acrylic. B, Contact dermatitis from latex gloves (seen here on the skin between the thumb and index finger). (A courtesy Dr. Edward V. Zegarelli.)
Topical antibiotics, antihistamines, preservatives in local anesthetics, and components of topical medication are common causes of allergic reactions. Acrylics, metal-based alloys, epoxy resins, and the flavoring agents in chewing gum and dentifrices all have been reported to cause contact mucositis.
Contact dermatitis, particularly that affecting the hands, may occur in response to the materials used in dentistry. The routine use of gloves in dental care has decreased the incidence of this type of dermatitis, but latex gloves and glove powder have also been responsible for contact dermatitis in some individuals (Figure 3-11, B).
Skin testing for sensitivity to a particular substance can help confirm the agent that caused the lesion. However, first the substance has to be identified. Topical and systemic corticosteroids may be used in the management of these lesions in some patients.
FIXED DRUG ERUPTIONS
Fixed drug eruptions are lesions that appear in the same site each time a drug is introduced. The lesions generally appear suddenly after a latent period of several days and subside when the drug is discontinued. They appear again when the drug is reintroduced, usually with greater intensity. Clinically there may be single or multiple slightly raised, reddish patches or clusters of macules on the skin or rarely the mucous membranes. Pain and pruritus may be associated with these lesions.
The fixed drug eruption is a type of allergic reaction (type III hypersensitivity) in which immune complexes are deposited along the endothelial wall of blood vessels. The ensuing inflammatory reaction causes vasculitis and subsequent damage to the vessel wall, giving rise to erythema and edema of the superficial layers of the skin or mucosa. If possible, the drug causing the reaction should be identified, and its use discontinued.
ERYTHEMA MULTIFORME
Erythema multiforme is an acute, self-limited disease that affects the skin and mucous membranes. The cause is not clear, but some evidence exists that it is a hypersensitivity reaction.
Erythema multiforme most commonly occurs in young adults and affects men more commonly than women. The characteristic skin lesion is called a target, iris, or bull’s eye lesion and consists of concentric rings of erythema alternating with normal skin color. The color is darkest at the center of the lesion (Figure 3-12). The term erythema multiforme refers to the variety of skin lesions that can occur, which range from macules to plaques to bullae. Erythema multiforme can affect the oral mucosa either alone or in association with skin lesions. Skin lesions can occur without oral lesions. The oral lesions are usually ulcers (Figure 3-13, A&B). However, erythematous areas may also occur. The ulcers frequently form on the lateral borders of the tongue. Crusted and bleeding lips are frequently seen in erythema multiforme, and gingival involvement is rare. The disease usually has an explosive onset, and systemic symptoms are mild or lacking. It may be chronic on occasion, or there may be recurrent acute episodes. The most severe form of erythema multiforme is called Stevens-Johnson syndrome (Figure 3-13, C). Mucosal lesions are more extensive and painful, genital mucosa and the mucosa of the eyes may be involved, and lips are generally encrusted and bloody.
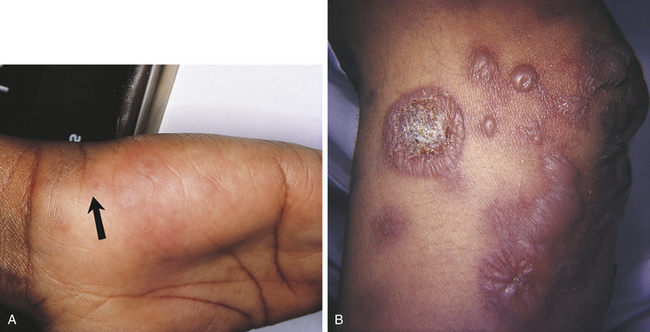
FIGURE 3-12 Skin lesions of erythema multiforme. A, Target lesion (arrow). B, Bullae. (A courtesy Dr. Edward V. Zegarelli.)
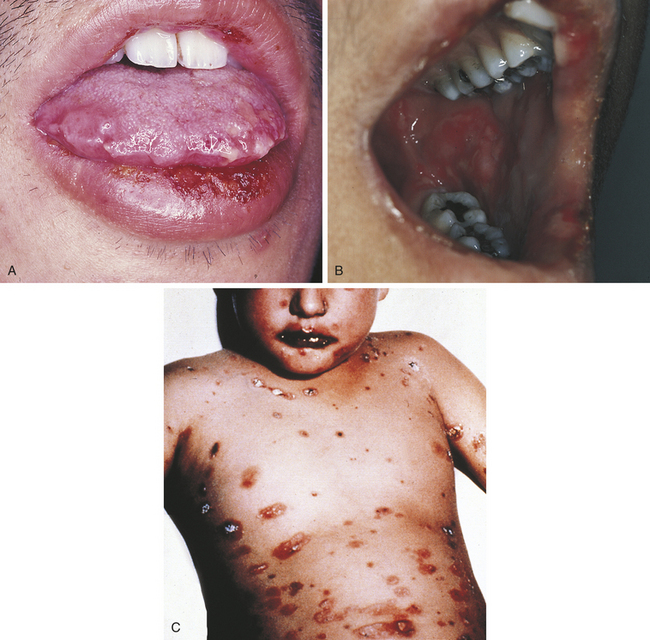
FIGURE 3-13 Oral lesions of erythema multiforme. A, Crusted lip lesions with edema, ulceration, and erythema. B, Erythematous and ulcerated lesions of the lips and buccal mucosa. C, Stevens-Johnson syndrome. (C courtesy Dr. Sidney Eisig.)
Although the cause of erythema multiforme is not clear, in some cases triggering factors can be identified. Infections such as herpes simplex, tuberculosis, and histoplasmosis have been associated with episodes of erythema multiforme, as have malignant tumors and drugs such as barbiturates and sulfonamides.
Diagnosis
The diagnosis of erythema multiforme is made on the basis of the clinical features and the exclusion of other diseases. The microscopic appearance is nonspecific. Biopsy and histologic examinations are sometimes helpful in excluding other diseases with similar clinical features but more distinctive histologic features.
Treatment and Prognosis
Topical corticosteroids may be helpful in mild cases of erythema multiforme, but systemic corticosteroid treatment is usually needed. Eye lesions in Stevens-Johnson syndrome may lead to scarring and blindness. Systemic antiviral medication is sometimes used to attempt to decrease recurrent episodes thought to be stimulated by herpes simplex virus infection.
LICHEN PLANUS
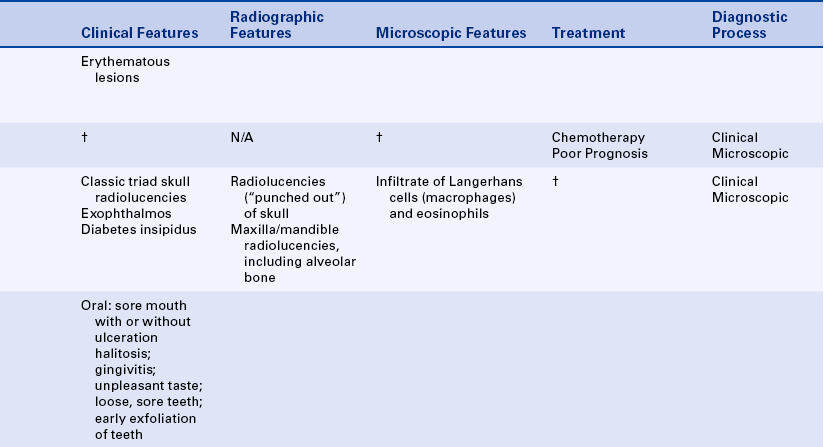
Lichen planus is a benign, chronic disease affecting the skin and oral mucosa. In some patients this disease may affect both the skin and the oral mucosa. In others either oral mucosa or skin alone may be affected. The lesions have a characteristic pattern of interconnecting lines called striae, resembling the pattern of the plant lichen as it grows on rocks and trees.
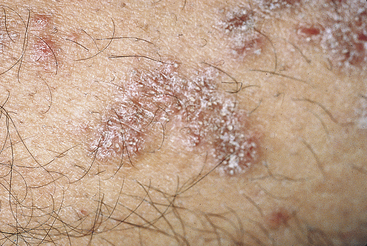
The classic appearance of lichen planus affecting the oral mucosa is an arrangement of interconnecting white lines and circles (Figure 3-14). The slender white lines are called Wickham’s striae. The fundamental lesion is a small, papular, pinhead-sized, domed or hemispheric, glistening white nodule on the mucosa. The clinical appearance depends on the arrangement of these minute papules and striae. The most common location is the buccal mucosa. However, lichen planus also occurs on the tongue, lips, floor of the mouth, and gingiva. Lesions of lichen planus are frequently distributed symmetrically in the oral cavity.
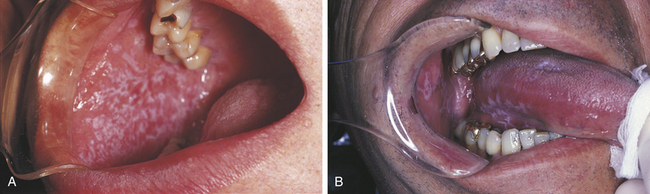
FIGURE 3-14 A and B, Two examples of the oral lesions of lichen planus. (A courtesy Dr. Edward V. Zegarelli.)
The prevalence of lichen planus in the general population of the United States has been reported to be about 1%. In one study of 100 cases, the age of individuals affected ranged from 13 to 78 years. However, the disease is most common in middle age. A slight female predominance has been suggested. In another reported study of 200 cases, most were found to be asymptomatic and were discovered on routine oral examination.
Many factors have been implicated in lichen planus; however, the cause remains unknown. Erosive lesions do tend to worsen with emotional stress. Many drugs and chemicals have been shown to produce lichenoid lesions, but these have a different histologic appearance from true lichen planus.
Types of Lichen Planus
Several forms of lichen planus have been described. The most common is reticular lichen planus. In this form the lesions are composed of Wickham’s striae along with slightly raised white plaquelike areas. Those in which the epithelium separates from the connective tissue and erosions, bullae, or ulcers form are called erosive and bullous lichen planus (Figure 3-15).
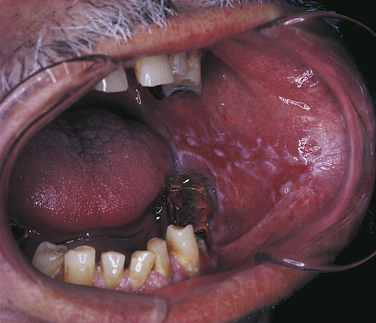
Gingival lesions clinically described as desquamative gingivitis can also be caused by lichen planus (Figure 3-16). In addition to the gingival lesions, striated lesions generally occur elsewhere.
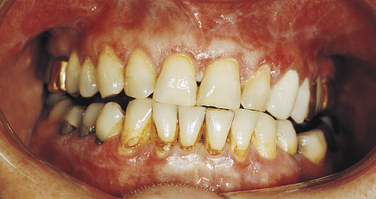
FIGURE 3-16 Lichen planus. The gingival lesions of lichen planus are described clinically as desquamative gingivitis. (Courtesy Dr. Edward V. Zegarelli.)
The skin lesions of lichen planus are 2- to 4-mm papules (Figure 3-17). Wickham’s striae may also be present, as may pruritus. Lesions can occur anywhere on the skin, but the most common sites are the lumbar region, the flexor surfaces of the wrist, and the anterior surface of the ankles. Some patients have only skin lesions, some have only oral lesions, and others have both skin and oral lesions.
Diagnosis
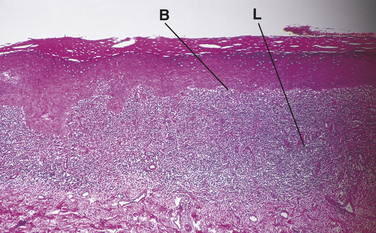
The diagnosis of lichen planus is made on the basis of the distinctive clinical and histologic appearance of biopsy tissue (Figure 3-18). The epithelium is generally parakeratotic and may be either hyperplastic or atrophic. The characteristic microscopic features include degeneration of the basal cell layer of the epithelium in small-to-extensive areas, sawtooth-shaped rete ridges, and a broad band of lymphocytes in the connective tissue immediately subjacent to the epithelium. In erosive areas the separation of the epithelium from the connective tissue occurs at the interface of the epithelium and connective tissue. Epithelial atypia and dysplasia may also occur in lesions that appear clinically as lichen planus, and it has been suggested that these lesions may be premalignant.
Treatment and Prognosis
Lichen planus is a chronic disease. Treatment is indicated only when lesions are symptomatic. Erosive lesions usually respond quickly when topical corticosteroid medication is applied. Improvement of gingival lesions with meticulous oral hygiene has been reported. If a drug can be identified as the causative agent of lichenoid-appearing lesions, discontinuation of the drug should cause the lesions to disappear. However, at times it is not possible to discontinue the drug.
Studies have been reported that suggest that patients with lichen planus may be at increased risk for the development of squamous cell carcinoma. Therefore regular oral soft tissue examination and biopsy of any lesions not consistent with lichen planus are recommended.
REACTIVE ARTHRITIS (REITER SYNDROME)
Reactive arthritis (Reiter syndrome) classically comprises the triad of arthritis, urethritis, and conjunctivitis. However, all the components of the syndrome may not be present. Polyarteritis is generally the most prominent component of this syndrome. (A syndrome is a group of signs and symptoms that occur together. Other syndromes are described elsewhere in this text.) The cause of this syndrome is not clear. An antigenic marker called HLA-B27 is present in most patients with reactive arthritis suggesting a genetic influence. An abnormal immune response to a microbial antigen is considered the most likely mechanism of occurrence. Reactive arthritis is far more prevalent in men (10:1) than in women (15:1). Reactive arthritis characteristically develops 1 to 4 weeks after venereal or gastrointestinal infection and is usually benign and self-limited.
The arthritis of this condition usually involves the joints of the lower extremities (knees and ankles). It may be asymmetric and migratory, chronic and recurrent.
Radiographically a periosteal proliferation can be detected on the heels, ankles, metatarsals, phalanges, knees, and elbows. Fever, malaise, and weight loss may be associated with the arthritis. The urethritis usually precedes the other lesions. No infectious organisms can be identified. The conjunctivitis is usually mild, but some individuals experience iritis (inflammation of the iris).
Skin and mucous membrane lesions are seen in many patients with reactive arthritis. Oral lesions occur almost anywhere in the oral cavity (Figure 3-19). Aphthouslike ulcers, erythematous lesions, and areas of depapillation of the tongue that mimic geographic tongue have been described in patients with reactive arthritis.
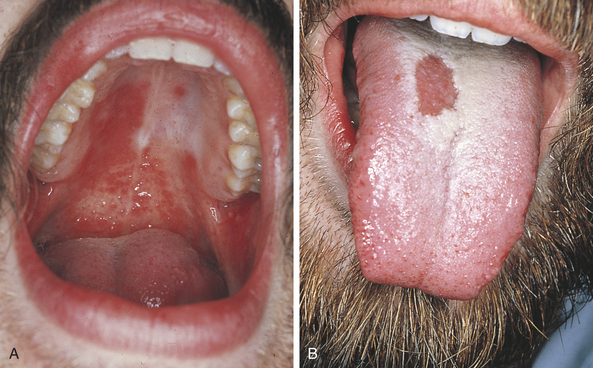
FIGURE 3-19 A and B, Lesions are present on the palate and tongue in this patient with reactive arthritis.
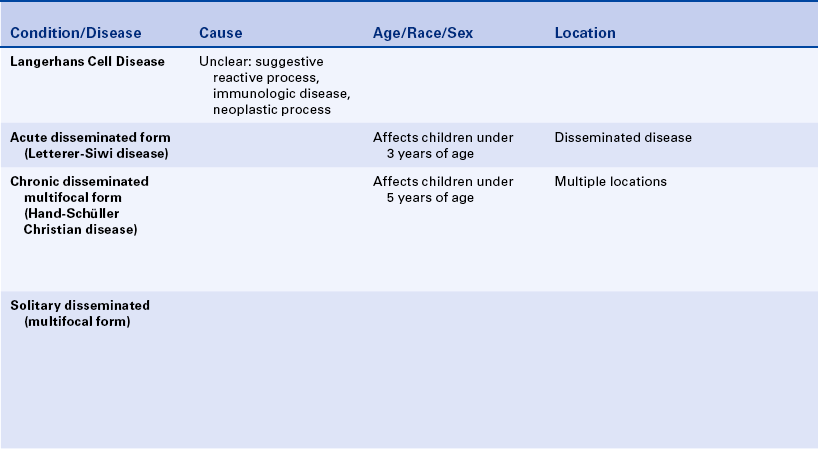
LANGERHANS CELL DISEASE
Langerhans cell disease was formerly called histiocytosis X. Traditionally three entities were grouped under the category of histiocytosis X: (1) Letterer-Siwe disease, (2) Hand-Schüller-Christian disease, and (3) solitary eosinophilic granuloma. Histologically all are characterized by a combination of Langerhans cells and eosinophils (Figure 3-20). Langerhans cells were originally called histiocytes and were responsible for the name histiocytosis X. In all three diseases the proliferating cell is the Langerhans cell. The presence of eosinophils is thought to result from the production of an eosinophilic chemotactic factor produced by the macrophage.
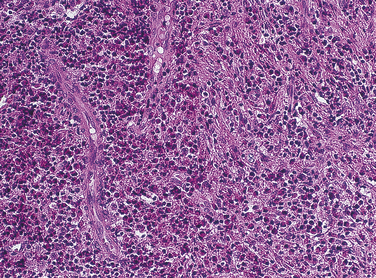
The Langerhans cell (a type of macrophage) is an immunologically competent cell of the mononuclear phagocyte series and participates in cell-mediated immunity. The cause and pathogenesis of Langerhans cell disease remains unclear. A reactive process, a primary immunodeficiency disease, and a neoplastic process have all been suggested.
Letterer-Siwe disease (acute disseminated form) is an acute fulminating disorder that usually affects children younger than 3 years of age. The course of the disease is usually so rapid that significant oral involvement does not often occur. The disease resembles a lymphoma in that it generally has a rapidly fatal course that sometimes responds to chemotherapy.
The chronic disseminated or multifocal form of Langerhans cell disease has been called Hand-Schüller-Christian disease. This form occurs in children, usually younger than 5 years of age. A classic triad is seen in about 25% of patients. All changes are caused by localized collections of Langerhans cells. The triad includes single-to-multiple, well-defined or “punched-out” radiolucent areas in the skull (may also occur in the jawbones); unilateral or bilateral exophthalmos; and diabetes insipidus that is caused by collections of macrophages in the sella turcica area, affecting the pituitary gland.
Oral manifestations of the chronic disseminated form of the disease include sore mouth with or without ulcerative lesions, halitosis, gingivitis, unpleasant taste, loose and sore teeth, early exfoliation of teeth, and nonhealing extraction sites. A characteristic loss of supporting alveolar bone occurs, mimicking advanced periodontal disease (Figure 3-21).
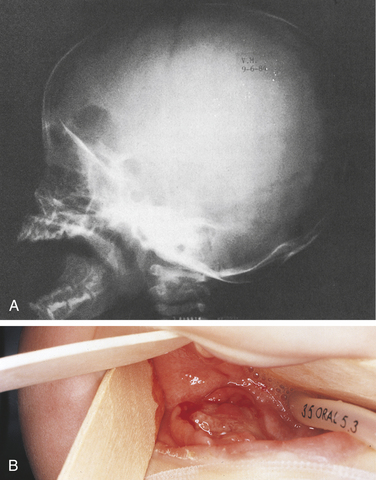
FIGURE 3-21 Langerhans cell disease. Chronic disseminated form also known as Hand-Schüller-Christian disease. A, Skull radiograph. B, Ulcerated lesion of the mandible. (Courtesy Dr. Sidney Eisig.)
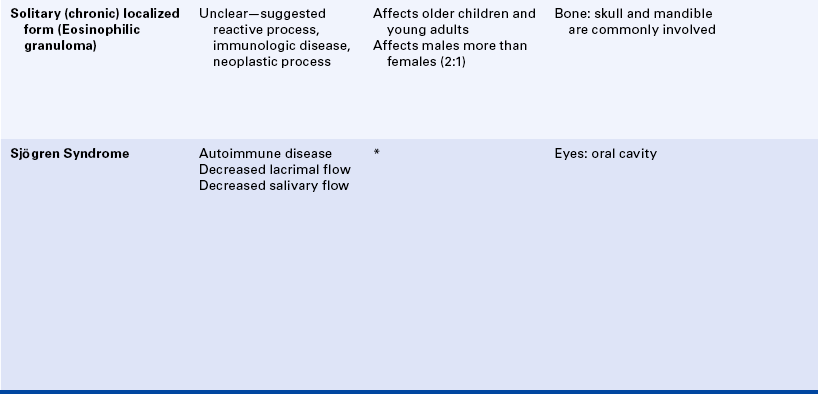
A solitary or chronic localized form has been called eosinophilic granuloma of bone. This form primarily affects older children and young adults. Males are affected twice as commonly as females. The skull and mandible are commonly involved with eosinophilic granuloma. The radiographic appearance varies, and the lesion may resemble periodontal disease or periapical inflammatory disease; or it may appear as a well-circumscribed radiolucency with or without a sclerotic border (Figure 3-22). Cases of multifocal eosinophilic granuloma have also been reported.
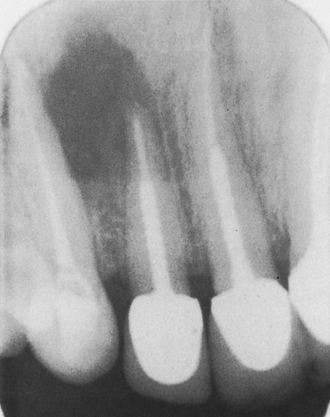
AUTOIMMUNE DISEASES THAT AFFECT THE ORAL CAVITY
Several autoimmune diseases affect the oral cavity (Table 3-5). In this type of disease tissue damage occurs because the immune system treats the person’s own cells and tissues as antigens. An individual with one autoimmune disease has an increased risk of developing another. Although these are systemic diseases, they are included in this chapter because of their relationship to immunity.
Table 3-5
Autoimmune diseases that affect the oral region
| Disease | Oral Manifestation |
| Sjögren syndrome | Xerostomia |
| Lupus erythematosus | White, erosive lesions of the oral mucosa |
| Pemphigus vulgaris | Mucosal ulceration, bullae |
| Benign mucous membrane pemphigoid | Mucosal ulceration, desquamative gingivitis |
| Behçet syndrome | Aphthous ulcers |
| Pernicious anemia∗ | Mucosal atrophy, mucosal ulceration, loss of filiform and fungiform papillae |
∗Described in Chapter 9.
SJÖGREN SYNDROME
Sjögren syndrome is an autoimmune disease that affects the salivary and lacrimal glands, resulting in a decrease in saliva and tears. This combination of dry mouth and dry eyes is sometimes called sicca (dry) syndrome. Decreased salivary flow (hyposalivation) results in dry mouth (xerostomia); decreased lacrimal flow results in dry eyes (xerophthalmia). The eye damage that occurs in Sjögren syndrome is called keratoconjunctivitis sicca. Other secretory glands such as sweat glands and glands that lubricate the vagina can also be affected. The specific cause of Sjögren syndrome is not known. Both cellular and humoral immunity are involved.
In some patients only the mouth and eyes are involved. In others the autoimmune process can be more extensive. Many patients (about 50%) with Sjögren syndrome have another autoimmune disease such as rheumatoid arthritis or systemic lupus erythematosus. Lacrimal and salivary gland involvement without the presence of another autoimmune disease is called primary Sjögren syndrome. When another autoimmune disease accompanies salivary and lacrimal gland involvement, the combination is called secondary Sjögren syndrome.
The oral manifestation of Sjögren syndrome is xerostomia (dry mouth), which occurs as a result of decreased salivary flow and causes the mucosa to become erythematous. Patients complain of oral discomfort. Lack of saliva causes the mouth to feel sticky. Lips are cracked and dry, and a generalized loss of filiform and fungiform papillae occurs on the dorsum of the tongue (Figure 3-23). Patients with xerostomia are at high risk for the development of caries, periodontal disease, and oral candidiasis.
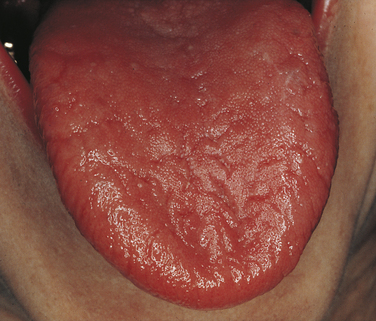
FIGURE 3-23 Sjögren syndrome. This patient had severe xerostomia. The filiform papillae are lacking.
Both major and minor salivary glands are affected. Parotid gland enlargement, usually bilateral and symmetric, occurs in about 50% of patients (Figure 3-24). The characteristic histologic appearance of these enlarged glands consists of replacement of the glands by lymphocytes and the presence of islands of epithelium called epimyoepithelial islands. Biopsy of the minor salivary glands is often performed to confirm a diagnosis of Sjögren syndrome. The minor glands show aggregates of lymphocytes surrounding the salivary gland ducts (Figure 3-25).
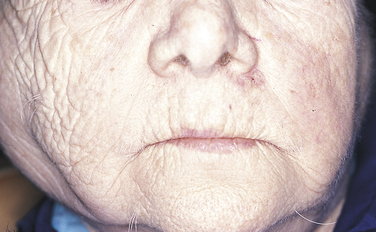
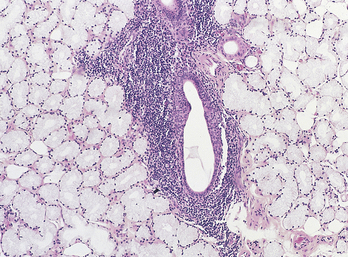
FIGURE 3-25 Microscopy of a minor salivary gland from a patient with Sjögren syndrome. (Courtesy Dr. Harry Lumerman.)
In addition to the oral findings, patients with decreased lacrimal flow that results in xerophthalmia complain of burning and itching of the eyes and photophobia (abnormal visual intolerance to light). Severe eye involvement may lead to ulceration and opacification of the cornea. Twenty percent of patients with Sjögren syndrome also have Raynaud phenomenon, which is a disorder affecting the fingers and toes. Cold and emotional stress trigger the reaction, which is characterized by an initial pallor of the skin that results from vasoconstriction and reduced blood flow. The initial pallor is followed by cyanosis, which occurs because of the decreased blood flow. When the skin is rewarmed, the blood vessels dilate, and the hyperemia results in a reddening of the skin. Finally, in minutes to hours the color returns to normal. Raynaud phenomenon can occur alone or in association with other autoimmune diseases, as well as in Sjögren syndrome. Patients with Sjögren syndrome might also complain of myalgia, arthralgia, and chronic fatigue.
Laboratory abnormalities are commonly found in patients with this syndrome. Ninety percent of patients with Sjögren syndrome have a positive reaction to rheumatoid factor, which is an antibody to IgG that is present in serum. It is an antibody to an antibody. Patients with other autoimmune diseases such as rheumatoid arthritis also react positively to rheumatoid factor. Other autoantibodies, called anti-Sjögren syndrome A and anti-Sjögren syndrome B, are also found in patients with Sjögren syndrome. Other laboratory abnormalities include mild anemia, a decreased white blood cell count, an elevated erythrocyte sedimentation rate, and a diffuse elevation of serum immunoglobulins.
Diagnosis and Management
The diagnosis of Sjögren syndrome is made when at least two of its three components are present: (1) xerostomia, (2) keratoconjunctivitis sicca, and (3) rheumatoid arthritis or another autoimmune disease. Xerostomia can result from conditions other than autoimmune disease. Measurement of stimulated and unstimulated salivary flow and biopsy of minor salivary glands (see Figure 3-25) can be helpful in the diagnosis of Sjögren syndrome. A characteristic pattern is seen in a parotid sialogram (Figure 3-26). The pattern reflects the lack of functioning glandular elements.
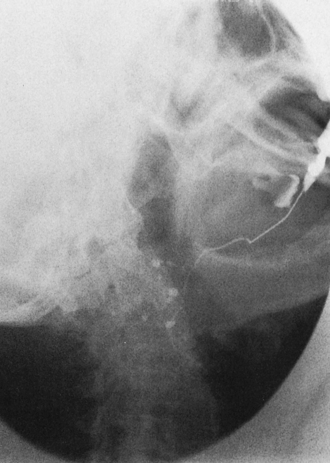
Keratoconjunctivitis is confirmed by eye examination. Lacrimal flow is measured using filter paper. Special examination techniques are necessary to demonstrate the corneal erosions.
For most patients the course of the disease is chronic and benign. However, patients with Sjögren syndrome are at risk for the development of other more serious autoimmune diseases, lymphoma, and Waldenström macroglobulinemia (a disorder characterized by a high concentration of IgM in serum) and therefore should be monitored by a physician.
Treatment
Sjögren syndrome is usually treated symptomatically. Nonsteroidal antiinflammatory agents are used for the arthritis. In severe cases corticosteroids and other immunosuppressive drugs may be necessary. Saliva substitutes are helpful, and for some patients the use of a humidifier at night makes the xerostomia more tolerable. Sugarless gum or lozenges may be used to stimulate saliva production. However, the greater the destruction of salivary gland tissue, the less saliva will be stimulated. An “artificial tears” formula containing methylcellulose helps to protect the eye from the drying effects of the disease. Glasses provide shielding and are helpful to minimize the drying effects of wind. Pilocarpine may also be used in some patients to increase salivary flow.
Maintaining good oral hygiene and using fluoride toothpaste and daily fluoride rinses can minimize the effects of xerostomia. The interval between recall appointments should be sufficiently short to ensure early detection and treatment of root caries. The toothbrush and other oral hygiene aids may need modification if arthritis affects the movement of hands and arms.
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus (SLE) is an acute and chronic inflammatory autoimmune disease of unknown cause. The disorder affects women eight times more frequently than men, predominantly during the childbearing years. It occurs three times more frequently in black women than in white women. SLE is a syndrome rather than a specific disease entity and includes a wide spectrum of disease activity and signs and symptoms that range from lesions confined to the skin such as discoid lupus erythematosus to a widespread, debilitating, life-threatening disease with multiple-organ involvement. SLE is usually chronic and progressive, with periods of remission and exacerbation.
Both cellular and humoral immunity are impaired in SLE. It has been suggested that cellular immunity is primarily affected, resulting in deregulation of the humoral response. Antigen and antibody complexes are deposited in various organs and stimulate an inflammatory response, which may be responsible for the tissue damage that occurs in SLE. Autoantibodies to the patient’s deoxyribonucleic acid (DNA) (antinuclear antibodies) are present in serum. These circulating antibodies are responsible for the positive antinuclear antibody and lupus erythematosus laboratory test results. Production of these antibodies is enhanced by estrogen. Antibodies to lymphocytes are present in some patients. Evidence exists for a genetic component in the pathogenesis of SLE.
Clinical Features
Skin lesions are among the most common signs of the disease, occurring in 85% of individuals (Figure 3-27). The most common skin lesion is an erythematous rash involving areas of the body exposed to sunlight. The classic “butterfly” rash occurs over the bridge of the nose, and there may be erythematous lesions on the fingertips. The lesions can heal with scarring in the center as they continue to spread at the periphery. They tend to worsen when exposed to sunlight. Atrophy and hypopigmentation or hyperpigmentation can follow these lesions. Other skin lesions such as bullae, purpura, a discoid rash, vitiligo, or subcutaneous nodules can also occur.
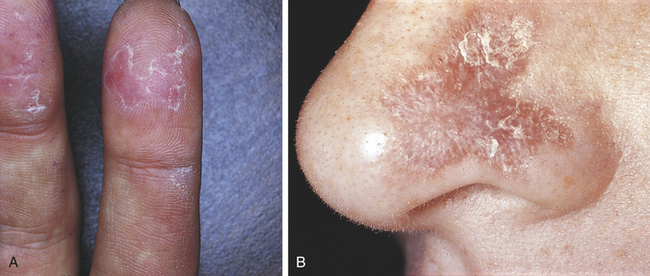
FIGURE 3-27 A and B, Two examples of skin lesions in lupus erythematosus. (B courtesy Dr. Edward V. Zegarelli.)
Arthritis and arthralgia are common manifestations of SLE. Any joint can be involved, and symptoms resemble rheumatoid arthritis but without severe deformities. Raynaud phenomenon occurs in 15% of patients. Myalgia and myositis also occur. A retinal vasculitis causes degeneration of the nerve fiber layer of the retina and can cause loss of vision. Psychoses and depression are signs of central nervous system involvement, and seizures can be present. Involvement of the pleura may cause shortness of breath and chest pain. Pericarditis, cardiac arrhythmias, and endocarditis may be seen late in the disease. Kidney involvement is common. Thrombocytopenia may also occur in patients with SLE.
Oral lesions accompany skin lesions in about 25% of patients with discoid lupus erythematosus, which is the mildest form of this autoimmune disease. Oral lesions appear as erythematous plaques or erosions. White striae radiating from the center of the lesion are usually present. The lesions may resemble lichen planus but are less symmetric in their distribution. The involvement of the oral mucosa in this disease is usually mild (Figure 3-28). Petechiae and gingival bleeding may be present in patients with severe thrombocytopenia. Sjögren syndrome may be present in some patients with SLE.
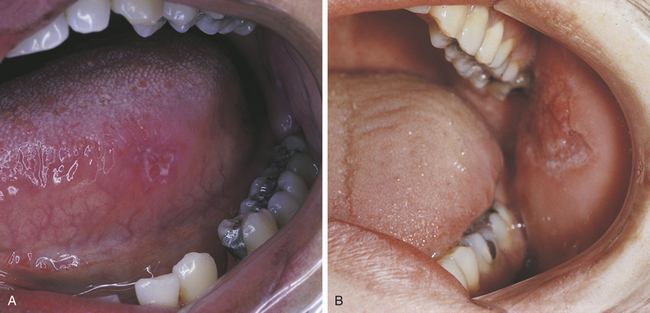
Diagnosis
The diagnosis of systemic lupus erythematosus is usually based on the classic multiorgan involvement and the presence of antinuclear antibodies in serum. LE cells, mature neutrophils that have phagocytized a spherical inclusion derived from another neutrophil, may be identified in circulating blood. The diagnosis of SLE is difficult if the onset is slow and insidious. The histologic appearance of oral lesions resembles that of lichen planus. Microscopically destruction of the basal cells is seen, as in lichen planus. However, the inflammatory infiltrate is distributed around blood vessels in the connective tissue rather than in a subepithelial band. Direct immunofluorescence and immunohistochemical testing of skin and mucosal lesions shows granular and linear deposits of immunoglobulins along the basement membrane area.
Treatment and Prognosis
Once the diagnosis of SLE is made, the decision as to whether treatment is indicated depends on the degree of disease activity. This once-fatal disease is now managed with several drugs. Aspirin and nonsteroidal antiinflammatory agents are used for mild signs and symptoms. Hydroxychloroquine, an antimalarial agent, and corticosteroids combined with other immunosuppressive agents such as azathioprine and cyclophosphamide are also used. If the course of the disease is mild and only a few organs are involved, the prognosis is excellent. The disease can also be fatal. Kidney involvement can be associated with severe hypertension and rapid onset of renal failure, which is a cause of death in patients with SLE. Other common causes of death include hemorrhage secondary to thrombocytopenia, nervous system involvement, and infection.
SLE is an extremely complex disease. A consultation with the patient’s physician should take place before initiating dental treatment. Antibiotic prophylaxis to prevent bacterial endocarditis may be necessary. The extent of systemic involvement and the drugs being used for treatment should be clarified.
PEMPHIGUS VULGARIS
Pemphigus vulgaris is a severe, progressive autoimmune disease that affects the skin and mucous membranes. It is characterized by intraepithelial blister formation that results from breakdown of the cellular adhesion between epithelial cells. This type of epithelial cell separation is called acantholysis. The patient with pemphigus vulgaris has circulating autoantibodies that are reactive against components of the epithelial cell attachment mechanism. The higher the titer of circulating antibodies, the greater the epithelial destruction. These autoantibodies are also found in surrounding epithelial cells. Pemphigus vulgaris is the type of pemphigus seen most frequently. Three other forms of pemphigus are (1) pemphigus vegetans, (2) pemphigus foliaceus, and (3) pemphigus erythematosus. These are much rarer conditions, but all are characterized by epithelial acantholysis. No sex predilection exists. A broad age range has been reported, including both children and elderly individuals. Most cases occur in the fourth and fifth decades of life. Genetic and ethnic factors have been reported. Although it is a relatively rare condition, pemphigus vulgaris is often seen in Ashkenazi Jews.
In more than 50% of cases of pemphigus vulgaris the first signs of disease occur in the oral cavity. Oral lesions can precede cutaneous lesions by many months. The appearance of oral lesions ranges from shallow ulcers to fragile vesicles or bullae.
Because the bullae are so fragile, they rupture soon after they form; the detached epithelium remains as a gray membrane (Figure 3-29). Ulcers are painful and range in size from small to very large. Gentle finger pressure with movement on clinically normal mucosa can produce a cleavage in the epithelium and result in the formation of a bulla. This is called Nikolsky’s sign.
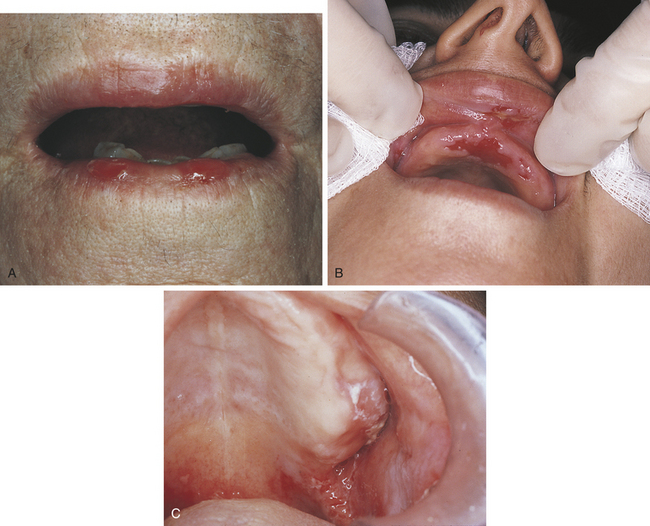
FIGURE 3-29 A to C, Examples of oral lesions in pemphigus vulgaris. (B courtesy Dr. Fariba Younai; C courtesy Dr. Sidney Eisig.)
Skin lesions in pemphigus vulgaris include erythema, vesicles, bullae, erosions, and ulcers. Microscopic examination of tissue shows an intact basal layer of epithelium attached to the underlying connective tissue (Figure 3-30). The loss of attachment between the epithelial cells results in detached cells that appear rounded. These rounded cells are called acantholytic cells or Tzanck cells and are present in the area of separation. Tzanck cells may also be seen on cytologic smear, but the diagnosis must be confirmed by biopsy.
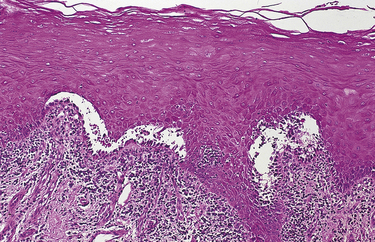
FIGURE 3-30 Pemphigus vulgaris seen by microscopy. The reader should note the acantholysis of the epithelium and the intact basal layer attached to the connective tissue.
Diagnosis
The diagnosis of pemphigus vulgaris is made by biopsy, microscopic examinations, and direct or indirect immunofluorescence. Direct immunofluorescence is performed on biopsy tissue and identifies autoantibodies present in the tissue. The tissue is viewed through a special microscope (in pemphigus vulgaris fluorescence is seen surrounding the cells in the prickle cell layer). In indirect immunofluorescence the patient’s serum is used to detect the presence of circulating autoantibodies. Eighty percent of patients with pemphigus vulgaris have the identifiable circulating autoantibodies.
Treatment and Prognosis
Treatment generally involves high doses of corticosteroids. Other immunosuppressive drugs such as azathioprine and methotrexate are often used in addition to systemic corticosteroids. The amount of autoantibodies present in the patient’s serum correlates with the severity of lesions; therefore this concentration of autoantibodies is used to monitor the success of drug treatment. Pemphigus vulgaris was at one time a life-threatening disease. Today the mortality rate is about 8% to 10% in 5 years and is related to the complications of corticosteroid treatment rather than to the disease itself. The titer of circulating autoantibodies is used to determine disease activity and response to treatment.
Other autoimmune diseases may occur in association with pemphigus vulgaris. Among them are lupus erythematosus, rheumatoid arthritis, and Sjögren syndrome.
MUCOUS MEMBRANE PEMPHIGOID
Mucous membrane pemphigoid is also known as cicatricial pemphigoid and benign mucous membrane pemphigoid. It is a chronic autoimmune disease that affects the oral mucosa, conjunctiva, genital mucosa, and skin. It is not as severe as pemphigus vulgaris. Lesions may heal with scarring. The name cicatricial pemphigoid refers to this healing with scarring. Autoantibodies and complement components have been identified at the basement membrane of the epithelium. Lesions occur as a result of cleavage of the epithelium from the underlying connective tissue in this area.
The most common site for mucous membrane pemphigoid lesions is the gingiva. The gingival lesions have been called desquamative gingivitis (Figure 3-31). The appearance ranges from erythema to ulceration and involves both the free and the attached gingiva. However, desquamative gingivitis is a clinical and descriptive term, and similar lesions can be seen in lichen planus and pemphigus vulgaris. In this disease as in pemphigus vulgaris, a Nikolsky’s sign can be produced on normal-appearing tissue by gentle friction. Bullae, erosions, and ulcers can also occur in other locations in the oral cavity. Bullae are thick walled and much less fragile than those of pemphigus vulgaris, and they may persist for 24 to 48 hours.
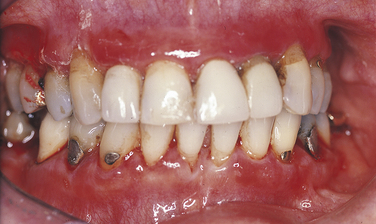
Diagnosis
The diagnosis of mucous membrane pemphigoid is made by biopsy and histologic examination. On microscopic examination the epithelium appears to detach from the connective tissue at the basement membrane (Figure 3-32). No degeneration of the epithelium occurs. An inflammatory infiltrate, usually with prominent plasma cells and eosinophils, is seen in the connective tissue. Direct immunofluorescence shows a linear pattern of fluorescence at the basement membrane. Indirect immunofluorescence (identifying autoantibodies in serum) is usually not helpful in the diagnosis of cicatricial pemphigoid.
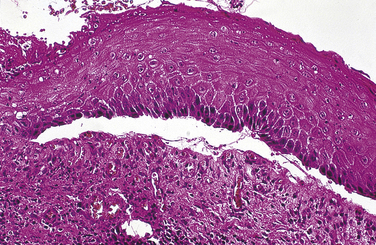
Treatment and Prognosis
Mucous membrane pemphigoid is a chronic disease that follows a benign course. Gingival involvement can occur alone, or more extensive lesions can be present. Topical corticosteroid application is helpful in the management of mild cases. In more severe cases high doses of systemic corticosteroids may be necessary. The disease may be difficult to control and slow to respond to therapy.
Patients may experience episodes of exacerbation followed by periods of remission. An ophthalmologic consultation to rule out the presence of eye lesions should be considered. If eye lesions are present, severe damage to the cornea, conjunctiva, and eyelid can occur.
BULLOUS PEMPHIGOID
Some investigators have suggested that bullous and mucous membrane pemphigoid are variants of a single disease. However, differences exist between the two diseases that make this questionable. Eighty percent of patients with bullous pemphigoid are older than 60 years of age, and no sex predilection exists. Unlike mucous membrane pemphigoid, in bullous pemphigoid circulating autoantibodies are usually detectable; and unlike pemphigus vulgaris, the circulating autoantibodies do not correlate with disease activity. Oral lesions are less common in bullous pemphigoid than in mucous membrane pemphigoid, occurring in only about one third of patients. When they occur, the gingival lesions are very similar to those of mucous membrane pemphigoid, and other mucosal lesions are more extensive and painful. As in mucous membrane pemphigoid, the microscopic appearance of bullous pemphigoid shows a cleavage of the epithelium from the connective tissue at the basement membrane.
High doses of systemic corticosteroids and nonsteroidal antiinflammatory drugs are used in the management of bullous pemphigoid. The disease is chronic with periods of remission and is usually not life threatening.
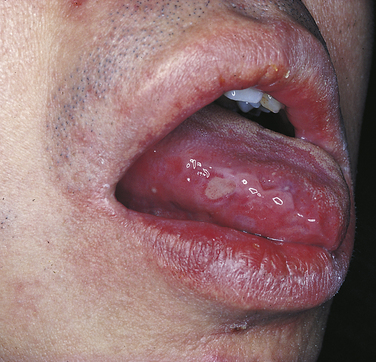
BEHÇET SYNDROME
Behçet syndrome is a chronic, recurrent autoimmune disease consisting primarily of oral ulcers, genital ulcers, and ocular inflammation. No sex predilection exists, and the mean age of onset is 30 years. There is an increased prevalence of this syndrome in individuals from the Mediterranean region and Asia. Evidence for an immunologic pathogenesis includes the identification of antibodies to human mucosa in patients with Behçet syndrome and the association between Behçet syndrome HLA B5 and HLA B51.
The oral ulcers that occur in Behçet syndrome are very similar in appearance to aphthous ulcers (Figure 3-33). They are painful and recurrent. The lesions range in size from a few millimeters to several centimeters. The genital ulcers are usually small and located on the scrotum or base of the penis and the labia majora. The ocular lesions usually begin with photophobia and can develop into conjunctivitis and uveitis. The skin lesions show a papular pattern of pustules and are most common on the trunk and limbs.
Bath-Balogh, M., Fehrenbach, M.J. Illustrated dental embryology, histology, anatomy, ed 2. St Louis: Saunders; 2006.
Fehrenbach, M.J., Herring, S.W. Illustrated anatomy of the head and neck, ed 3. St Louis: Saunders; 2007.
Kumar, V., Cotran, R.S., Robbins, S.L. Robbins basic pathology, ed 8. Philadelphia: Saunders; 2007.
McCance, K., Huether, S. Pathophysiology, ed 5. St Louis: Mosby; 2005.
Neville, B.W., et al. Oral and maxillofacial pathology, ed 3. St Louis: Saunders; 2009.
Parham, P. The immune system. New York: Garland Publishing; 2000.
Al-Johani, K.A., Fedele, S., Porter, S.R. Erythema multiforme and related disorders. Oral Surg Oral Med Oral Pathol Oral Endod. 2007;103:642.
Anderson, K.E., et al. Contact dermatitis: a review. Contact Dermatitis. 1987;16:55.
Antoon, J.W., Miller, R.L. Aphthous ulcers: a review of the literature of etiology, pathogenesis, diagnosis and treatment. J Am Dent Assoc. 1980;101:803.
Ardekian, L., et al. Clinical and radiographic features of eosinophilic granuloma in the jaws: review of 41 lesions treated by surgery and low-dose radiotherapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:238.
Atkinson, J.C., Fox, P.C. Sjögren’s syndrome: oral and dental considerations. J Am Dent Assoc. 1993;124:74.
Barnard, N.A., et al. Oral cancer development in patients with oral lichen planus. J Oral Pathol Med. 1993;22:241.
Barrett, A.W., Scully, C.M., Eveson, J.W. Erythema multiforme involving gingiva. J Periodontol. 1993;64:910.
Baudet-Pommel, M., et al. Early dental loss in Sjögren’s syndrome. Oral Surg Oral Med Oral Pathol. 1994;78:181.
Chaplin, D.D. Part 1. Overview of the human immune response. J Allergy Clin Immunol. 2006;117(2 Suppl Mini-Primer):S430.
Eisenberg, E. Lichen planus and oral cancer: is there a connection between the two? J Am Dent Assoc. 1992;123:104.
Epstein, J.B., et al. Oral lichen planus: progress in understanding its malignant potential and the implications for clinical management. Oral Surg Oral Med Oral Radiol Endod. 2003;96:32.
Escudier, M., Baraw, J., Scully, C. Number VII Behçet’s disease (Adamantiades syndrome). Oral Dis. 2005;12:78.
Eversole, L.R. Immunopathology of oral mucosal ulcerative, desquamative, and bullous diseases. Oral Surg Oral Med Oral Pathol. 1994;77:555.
Feder, H.M. Periodic fever, aphthous stomatitis, pharyngitis, adenitis: a clinical review of a new syndrome. Curr Opin Pediatr. 2000;12:253.
Gawkrodger, D.J., Stephenson, T.J., Thomas, S.E. Squamous cell carcinoma complicating lichen planus: a clinico-pathological study of three cases. Dermatology. 1994;188:36.
Holmstrup, P., Schiotz, A.W., Westergaard, J. Effect of dental plaque control on gingival lichen planus. Oral Surg Oral Med Oral Pathol. 1990;69:585.
Jorizzo, J.L., et al. Oral lesions in systemic lupus erythematosus: do ulcerative lesions represent a necrotizing vasculitis? J Am Acad Dermatol. 1992;27:389.
Katz, R.W., Brahim, J.S., Travis, W.D. Oral squamous cell carcinoma arising in a patient with long-standing lichen planus: a case report. Oral Surg Oral Med Oral Pathol. 1990;70:282.
Krippaehne, J.A., Montgomery, M.T. Erythema multiforme: a literature review and case report. Spec Care Dentist. 1992;12:125.
Loh, H.S., Quah, T.C. Histiocytosis X (Langerhans cell histiocytosis) of the palate: case report. Aust Dent J. 1990;35:117.
Lozada-Nur, F., Gorsky, M., Silverman, S., Jr. Oral erythema multiforme: clinical observations and treatment of 95 patients. Oral Surg Oral Med Oral Pathol. 1989;67:36.
MacPhail, L.A., et al. Recurrent aphthous ulcers in association with HIV infection: description of ulcer types and analysis of T-lymphocyte subsets. Oral Surg Oral Med Oral Pathol. 1991;71:678.
Manton, S.L., Scully, C. Mucous membrane pemphigoid: an elusive diagnosis? Oral Surg Oral Med Oral Pathol. 1988;66:37.
Miller, M.F., Ship, I.I., Ram, C. A retrospective study of the prevalence and incidence of recurrent aphthous ulcers in a professional population, 1958-1971. Oral Surg Oral Med Oral Pathol. 1977;43:532.
Miller, R.L., Gould, A.R., Bernstein, M.L. Cinnamon-induced stomatitis venenata. Oral Surg Oral Med Oral Pathol. 1992;73:708.
Panush, R.S., et al. Retraction of the suggestion to use the term “Reiter’s syndrome”: sixty five years later. Arthritis Rheum. 2007;56:693.
Porter, S.R., Scully, C., Standen, G.R. Autoimmune neutropenia manifesting as recurrent oral ulceration. Oral Surg Oral Med Oral Pathol. 1994;78:178.
Raghoebar, G.M., Brouwer, T.J., Schoots, C.J. Pemphigus vulgaris of the oral mucosa: report of two cases. Quintessence Int. 1991;22:199.
Reiche, E.M., Morimoto, H.K., Nunes, S.M. Stress and depression-induced immune dysfunction: implications for the development and progression of cancer. Int Rev Psychiatry. 2005;17:515.
Rodu, B., Mattingly, G. Oral mucosal ulcers: diagnosis and management. J Am Dent Assoc. 1992;123:83.
Schofield, J.K., Tatnall, F.M., Leigh, I.M. Recurrent erythema multiforme: clinical features and treatment in a large series of patients. Br J Dermatol. 1993;34:63.
Sciubba, J.J. Sjögren’s syndrome: pathology, oral presentation and dental management. Compend Contin Educ Dent. 1994;15:1084.
Spina, A.M., Levine, H.J. Latex allergy: a review for the dental professional. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:5.
Tasher, D., Somekh, E. Dalal: PFAPA syndrome: new clinical aspects disclosed. Arch Dis Child. 2006;91:981.
Vincent, S.D., Lilly, G.E., Baker, K.A. Clinical, historical and therapeutic features of cicatricial pemphigoid: a literature review and open therapeutic trial with corticosteroids. Oral Surg Oral Med Oral Pathol. 1993;76:453.
Vincent, S.D., et al. Oral lichen planus: the clinical, historical and therapeutic features of 100 cases. Oral Surg Oral Med Oral Pathol. 1990;70:165.
Walker, D.M. Oral mucosal immunology: an overview. Ann Acad Med Singapore. 2004;33(4 Suppl):27.
Woo, S.-B., Sonis, S.T. Recurrent aphthous ulcers: a review of diagnosis and treatment. J Am Dent Assoc. 1996;127:1202.
Yazici, H., Barnes, C.G. Practical treatment recommendations for pharmacotherapy of Behçet’s syndrome. Drugs. 1991;42:796.
Zegarelli, D.J. The treatment of oral lichen planus. Ann Dent. 1993;52:3.
REVIEW QUESTIONS
1. The immune system usually defends the body against foreign substances that are called:
2. Memory is an important function of the immune system because:
A It retains the memory of the antibody.
B It allows faster future immune responses.
3. Immunization with a vaccine works by:
A Increasing the risk of an antigen-causing disease.
B Using antibodies produced by another person.
4. A B lymphocyte is a cell in the immune system that:
A Is derived from a precursor stem cell.
B Matures and resides in the thymus.
5. A macrophage is a cell in the immune system that:
A Retains the memory of the encountered antigen.
C Undergoes B cell phagocytosis initially during inflammation.
6. Which statement is true of NK cells?
7. In which type of immunopathologic disease are the cells of the body no longer tolerated and treated by the immune system as antigens?
8. During the anaphylactic type of hypersensitivity reaction, the plasma cells:
9. Which type of hypersensitivity reaction involves activated complement?
10. What type of lymphocyte matures in the thymus, produces lymphokines, and can increase or suppress the humoral immune response?
11. In the immune system antibodies are proteins that are:
12. Which of the following type(s) of immunologic disease involve a decreased number or activity of lymphoid cells?
13. The humoral immune response involves the production of:
14. The measurement of a specific antibody level in the blood is called:
15. Which type of immunity may be provided immediately to dental personnel after needlestick accidents?
16. Which of the following situations would result in the least risk of drug allergy?
17. Which of the following is involved in the regulation of both humoral and cell-mediated immunity?
18. Which of the following is involved in the communication between lymphocytes within the immune system?
19. All of the following are examples of hypersensitivity reactions except:
21. Which one of the following types of hypersensitivity reactions is referred to as delayed hypersensitivity?
22. The “target lesion” on the skin is associated with which of the following diseases?
23. Tzanck cells are seen in which of the following conditions?
24. The oral lesions in Reiter syndrome may resemble:
25. Apththous ulcers are seen in each all of the following systemic diseases EXCEPT:
26. The two cell types that histologically characterize Langerhans cell disease are:
27. Which one of the following is the form of Langerhans cell disease that is characterized by a triad of symptoms?
28. The most benign type of Langerhans cell disease is:
29. The most characteristic oral manifestation of Sjögren syndrome is:
30. Which of the following statements is false?
A The bullae in pemphigus vulgaris are more fragile than those in bullous pemphigoid.
B Acantholysis of the epithelium is seen in pemphigus vulgaris.
C In pemphigoid the separation of the epithelium from the connective tissue occurs at the basement membrane.
31. Which is the most distinct and definitive characteristic that distinguishes pemphigus from pemphigoid?
32. Desquamative gingivitis may be seen in:
33. In angioedema involvement of which of the following tissues could create a life-threatening situation for the patient?
34. Which of the following is a pathologic condition that produces a characteristic butterfly-shaped lesion on the face and oral ulcers, occurs more frequently in females than males, and for which the result of a blood test is important to diagnosis?