Genetics
After studying this chapter, the student will be able to:
1 Define each of the words listed in the vocabulary for this chapter.
2 State the purpose of mitosis.
3 State the purpose of meiosis.
4 Explain what is meant by the Lyon hypothesis and give an example of its clinical significance.
5 Explain what is meant by a gross chromosomal abnormality and give three examples of syndromes that result from gross chromosomal abnormalities.
6 List the four inheritance patterns.
7 Explain what is meant by X-linked inheritance.
8 State the inheritance pattern and describe the oral manifestations and, if appropriate, the characteristic facies for each of the following: cyclic neutropenia, Papillon-Lefèvre syndrome, cherubism, Ellis–van Creveld syndrome, mandibulofacial dysostosis (Treacher Collins syndrome), osteogenesis imperfecta, hereditary hemorrhagic telangiectasia (Osler-Rendu–Parkes Weber syndrome), Peutz-Jeghers syndrome, white sponge nevus (Cannon disease), hypohidrotic ectodermal dysplasia, hypophosphatasia, and hypophosphatemic vitamin D—resistant rickets.
9 State the inheritance pattern, the oral or facial manifestations, and the type and location of the malignancy associated with each of the following syndromes: Gardner syndrome; nevoid basal cell carcinoma syndrome (Gorlin syndrome); multiple mucosal neuromas, medullary carcinoma of the thyroid gland, and pheochromocytoma syndrome (multiple endocrine neoplasia type 2B [MEN 2B]); and neurofibromatosis of von Recklinghausen.
10 State the location and malignant potential of the intestinal polyps in Peutz-Jeghers syndrome and Gardner syndrome.
11 List the four types of amelogenesis imperfecta.
12 Briefly compare and contrast dentinogenesis imperfecta, amelogenesis imperfecta, and dentin dysplasia, including the inheritance patterns, the clinical manifestations, and the radiographic appearance of each.
(ah-lēlz′) Genes that are located at the same level or locus in the two chromosomes of a pair and that determine the same functions or characteristics.
(ah-me′no as′id) Organic compound containing the amino group NH2; amino acids are the main component of proteins.
(aw′to-sōmz) (adjective, autosomal) Nonsex chromosomes, which are identical for men and women.
(bahr bod′e) Condensed chromatin of the inactivated X chromosome, which is found at the periphery of the nucleus of cells in women.
(kar′e-er) In genetics a heterozygous individual who is clinically normal but who can transmit a recessive trait or characteristic; also, a person who is homozygous for an autosomal-dominant condition with low penetrance.
(sen′tro-mēr) Constricted portion of the chromosome that divides the short arms from the long arms.
(kro′mah-tid) Either of the two vertical halves of a chromosome that are joined at the centromere.
(kro′mah-tin) General term used to refer to the material (deoxyribonucleic acid [DNA]) that forms the chromosomes.
(ko′don) Vertical sequence of three bases in DNA that codes for an amino acid.
(kon′′san-gwinĩ-te) Blood relationship; in genetics the term is generally used to describe a mating or marriage between close relatives.
(DNA) (de-ok′′se-ri′′bo-nu-kle′ik as′id) A substance composed of a double chain of polynucleotides; both chains coiled around a central axis form a double helix; it is the basic genetic code or template for amino acid formation.
(dip′loid) Having two sets of chromosomes; the normal constitution of somatic cells.
(dom′ĭ-nant) In genetics a trait or characteristic that is manifested when it is carried by only one of a pair of homologous chromosomes.
(eks′′pres-siv′ĭ-te) Degree of clinical manifestation of a trait or characteristic.
(fa′she-ēz) Appearance of the face.
(gam′ēt) Spermatozoon or ovum.
(je-net′ik het′′er-o-jē-ne′ĭ-te) Having more than one inheritance pattern.
(hap′loid) Cell with a single set of chromosomes; a gamete is haploid.
(het′′er-o-zi′gōt) (adjective, heterozygous) Individual with two different genes at the allele loci.
(ho′′mo-zi′gōt) (adjective, homozygous) Individual having identical genes at the allele loci.
(hi′po-hĭ-dro′sis) Abnormally diminished secretion of sweat.
(hi′po-trĭ-ko′sis) Presence of less than the normal amount of hair.
(kar′e-o-tĭp) Photomicrographic representation of a person′s chromosomal constitution arranged according to the Denver classification.
(lo′kus) (plural, loci) Position occupied by a gene on a chromosome.
(mi-o′sis) Two-step cellular division of the original germ cells, which reduces the chromosomes from 4nDNA to 1nDNA.
(met′ah-f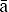 z) Phase of cellular division in which the chromosomes are lined up evenly along the equatorial plane of the cell and in which they are most visible.
z) Phase of cellular division in which the chromosomes are lined up evenly along the equatorial plane of the cell and in which they are most visible.
(mi′′to kon′dre-ĕ) Cytoplasmic organelles that have their own DNA in a circular chromosome.
DNA (mi′′to kon′dre-ĕl DNA) Unique DNA that is maternally inherited.
(mi-to′sis) Way in which somatic cells divide so that the two daughter cells receive the same number of identical chromosomes.
(mū-ta′shun) Permanent change in the arrangement of genetic material.
(o′′o-jen′ĕ-sis) Process of formation of female germ cells (ova).
(o′vum) (plural, ova) Egg; mature feminine germ cell.
(pen′ĕ-trans) Frequency with which a heritable trait is exhibited by individuals carrying the gene or genes that determine that trait.
(fe′noh-tīp) Entire physical, biochemical, and physiologic makeup of an individual; genotype is the genetic composition, and phenotype is its observable appearance.
(re-ses′iv) Trait or characteristic that shows clinically when a double-gene dose (homozygous) exists in autosomic chromosomes or a single-gene dose exists in males if the trait is X linked.
(RNA) (ri′′bo-noo-kle′ik as′id) Single strands of polynucleotides found in all cells; different types of RNA have different functions in the production of proteins by the cell.
(ri′bo-sōm) Cytoplasmic organelles in which proteins are formed on the basis of the genetic code provided by RNA.
(sper′′mah-to-jen′ĕ-sis) Process of formation of spermatozoa (sperm).
(sper′′mah-to-zo′on) (plural, spermatozoa) Mature male germ cell.
(sin′drōm) Set of signs or symptoms (or both) occurring together.
(trans′′lo-ka′shun) Portion of a chromosome attached to another chromosome.
(tri′so-me) Pair of chromosomes with an identical extra chromosome.
Genetics is the science that studies inheritance and the expression of inherited traits. The main objectives of this chapter are to introduce some of the basic concepts of genetics and present the clinical manifestations of some inherited oral disorders of interest to the dental hygienist. The descriptions of many syndromes are included. As explained in previous chapters, a syndrome is a distinctive association of signs and symptoms occurring together in the same patient. The syndromes included in this chapter are inherited. However, other syndromes such as acquired immunodeficiency syndrome (AIDS) are acquired, not inherited. In addition, the fact that an alteration is found as part of a syndrome does not mean that it cannot also occur independently. For example, cleft lip and palate occur as components of several syndromes and can also occur independently. The classification of syndromes is difficult because they are composed of several associated anomalies and the anomalies that compose the syndrome may not be present consistently in all patients with the same syndrome.
The term phenotype is used often in this chapter. It refers to the physical, biochemical, and physiologic traits of an individual. A phenotype can occur as a result of genetic factors or from a combination of genetic factors and environmental influences.
In this chapter, as in previous chapters, basic concepts are discussed first. These concepts are followed by descriptions of specific inherited disorders.
CHROMOSOMES
The hereditary units that are transmitted from one generation to another are called genes. They are found on chromosomes, which are located in the nucleus of the cell. Using a microscope, one can see chromosomes clearly only when the nucleus and cell are dividing (Figure 6-1). At other times the genetic material is dispersed in the nucleus (see Figure 6-1). Each cell of the human body, with the exception of mature germ cells (ova and spermatozoa), has 46 chromosomes. Half of these chromosomes are derived from the father, and the other half from the mother.
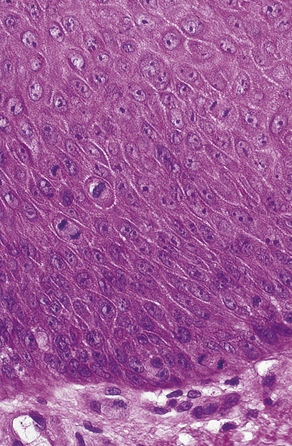
FIGURE 6-1 High-power photomicrograph shows a dividing cell with visible chromosomes and nuclei of cells with scattered chromatin.
Chromosomes contain deoxyribonucleic acid (DNA), which directs the production of amino acids, polypeptides, and proteins by the cell. In addition, DNA has the ability to duplicate itself (self-replication). It creates exact copies of itself; and through the process of cell division cells identical to the original cell are formed.
MITOSIS
All cells in the body, with the exception of ova and spermatozoa, are called somatic cells. Cellular division is achieved by mitosis during a part of the life span of the somatic cell, called the mitotic cycle. The function of mitosis is to create an exact copy of each chromosome and, through division of the original cell, distribute an identical set of chromosomes to each daughter cell. After each cell division is completed and before the next division can occur, the cell enters the gap 1 (G1) phase, which is followed by the S phase, in which replication of the DNA takes place. The gap 2 (G2) phase follows the S phase and ends when mitotic division begins. The cell cycle is illustrated in Figure 6-2.
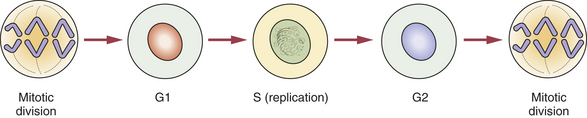
FIGURE 6-2 The mitotic cycle shows the end of mitosis, followed by the G1, S, and G2 phases and the next mitosis.
Stages of Mitosis
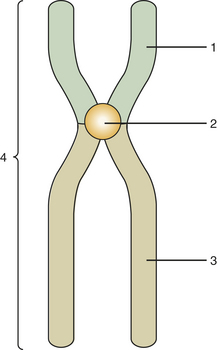
Mitosis is composed of four stages: (1) prophase, (2) metaphase, (3) anaphase, and (4) telophase. In each of these four stages, the chromosomes are distributed in a specific arrangement. In metaphase the chromosomes stain intensely and are arranged almost symmetrically at both sides of the center, or equatorial plane, of the cell. The appearance of a metaphase chromosome resembles the letter X (Figure 6-3), having a pair of “long arms” also known as q arms (see Figure 6-3, 3) and a pair of “short arms” also known as p arms (see Figure 6-3, 1). The size of the chromosome at metaphase and the length of the long and short arms vary from chromosome to chromosome. The constriction present in all chromosomes, which joins the short and long arms, is called the centromere (see Figure 6-3, 2). During metaphase chromosomes are actually formed by two identical vertical halves, each composed of either left or right short and long arms and half of the centromere. Each of these identical halves is called a chromatid (see Figure 6-3, 4). At metaphase each chromatid contains one molecule of DNA; therefore the DNA content of each chromosome is doubled (Figure 6-4). When cell division takes place, each chromosome splits vertically at the centromere; 46 chromatids (which now become chromosomes) form one daughter cell, and the other 46 chromatids form a second daughter cell. During prophase the chromosomes are lining up toward metaphase; in anaphase and telophase the chromatids are in the process of splitting.
MEIOSIS
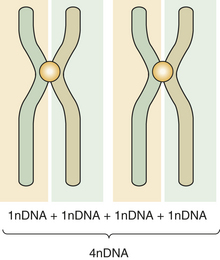
Primitive germ cells (oogonia, spermatogonia) have 46 chromosomes. Mature germ cells (ova, spermatozoa) have 23 chromosomes. Meiosis is a two-step special type of cell division in which the primitive germ cells reduce their chromosome number by half and become mature germ cells. The primitive germ cells have two chromosomes for each pair and are called diploid. The suffix ploid refers to the number of sets of chromosomes, and its prefix refers to the degree of ploidy. In diploid di indicates two. The mature germ cells (or gametes) have half the number of chromosomes and are called haploid. During the period in which the cell is not in division, the DNA content of diploid cells is designated 2nDNA; in metaphase it is double or 4nDNA. After the two stages of meiosis have been completed, the 4nDNA is reduced to 1nDNA. The two steps are called first meiosis and second meiosis. This reduction is necessary to maintain the normal number of human chromosomes. A new embryo must have 46 chromosomes per cell as did its parents. Therefore the union of germ cells needs to result in 46 chromosomes. If two cells with 46 chromosomes were combined, the resulting cell would have 92 chromosomes.
First Meiosis
Before the first meiosis in the primitive germ cells, a replication of DNA occurs that is similar to that observed in the S phase of somatic cells. After replication the members of each pair of chromosomes line up next to each other in an intimate point-by-point relationship (Figure 6-5, A). This pairing does not occur in mitosis. After pairing, the two chromosomes establish actual contact at different locations. These contacts are known as chiasmata (meaning X shaped) and determine points of crossing over (Figure 6-5, B). This crossing over achieves the exchange of chromosome segments between a chromatid of one chromosome and a chromatid of the other chromosome of a pair (between homologous chromosomes) (Figure 6-5, C). This special aspect of the first meiosis takes place at metaphase. After metaphase of the first meiotic division, the chromosomes separate from each other, but no splitting of the centromere occurs. The chromosomes remain intact, and each member of the pair migrates to one of the new cells, each of which contains 23 chromosomes but twice the final amount of DNA. During this migration chromosomes of the paternal and maternal lines segregate at random, thus ensuring diversity of the species by creating a new combination of chromosomes.
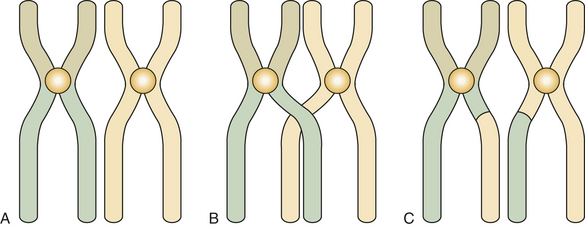
FIGURE 6-5 Homologous autosomal chromosomes line up in first meiosis (A), cross over during metaphase of first meiosis (B), and exchange segments after crossing over (C).
Occasionally the chromosomes that were crossing over do not separate, and both migrate to the same cell. This is known as nondisjunction and results in the formation of a germ cell with an extra chromosome. If this occurs and that cell (either an ovum or a spermatozoon) participates in the formation of an embryo, three chromosomes (trisomy) instead of two result. An example of this type of abnormality is Down syndrome, also called trisomy 21, in which three of chromosome 21 are found instead of two. Trisomy has been reported for several different chromosomes.
In a female embryo oogenesis (ovum development) starts around the third month of prenatal life, and the future ova remain suspended crossing over from about the time of birth until the time ovulation starts. At the beginning of ovulation the first meiosis is completed. Nondisjunction is more prevalent in female oogenesis than in male spermatogenesis. This is probably because of the period of prolonged crossing over; therefore the older the woman, the greater the chance of shedding a trisomic ovum and of bearing a child with Down syndrome or other trisomy.
Second Meiosis
The second stage of meiosis is essentially a mitotic division in which each chromosome splits longitudinally. No replication of DNA occurs before the second meiosis. After the splitting two cells are formed, each containing the right amount of DNA (1nDNA) (Figure 6-6).
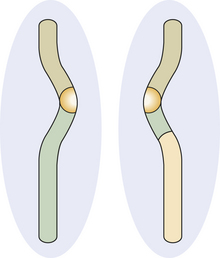
Nondisjunction can occur during the first and second meiosis. If it occurs in second meiosis, a chromosome does not split, and one daughter cell has a full chromosome, and the other has none.
Immediately after fertilization the chromosomes of the ovum and spermatozoon, each having 1nDNA, condense independently and form round structures, each known as a pronucleus. The DNA in these pronuclei replicates, forming a set of 23 full chromosomes for each, the maternal and paternal pronuclei. When the membranes of these pronuclei break, the 46 chromosomes mix at random, initiating the first cellular division (mitosis) that starts the development of the new embryo.
LYON HYPOTHESIS
The sex chromosomes are designated XX in women and XY in men. During the early period of embryonic development (possibly by the end of the second week), the genetic activity of one of the X chromosomes in each cell of a female embryo is inactivated. The inactivation is a random process affecting either the X chromosome derived from the mother or the X chromosome derived from the father. Activated chromosomes are dispersed in the nucleus. The inactivated chromosome remains contracted when the cell is not dividing and forms a structure known as the Barr body.
In female cells the Barr body can be seen easily under the light microscope, especially in cytologic smears, including those obtained from the oral mucosa. The Barr body appears as a dark dot at the periphery of the nucleus (Figure 6-7).
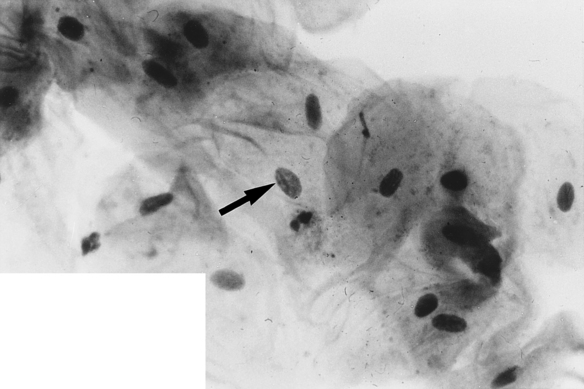
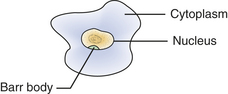
FIGURE 6-7 Buccal smear shows Barr body (arrow points to small dark dot on nuclear membrane) at the periphery of the nucleus of a desquamated epithelial cell from a woman’s buccal mucosa. (Courtesy Dr. Carl J. Witkop.)
This inactivation of one of the X chromosomes in a female embryo was postulated by Mary Lyon and is known as the Lyon hypothesis. This hypothesis has interesting clinical implications for female carriers of conditions caused by genes located on the X chromosome, which are explained later in this chapter.
DEOXYRIBONUCLEIC ACID
Chromosomes contain deoxyribonucleic acid (DNA). DNA contains the basic code or template that carries all genetic information. The basic unit of DNA is called a nucleotide, which is formed by a nitrogen-containing base, a five-carbon sugar (deoxyribose), and a phosphate. Four bases are found in DNA: adenine (A), guanine (G), thymine (T), and cytosine (C). These chains of polynucleotides are coiled to form a structure called a double helix (Figure 6-8). In DNA the base adenine is always bound to the base thymine, and guanine is always bound to cytosine. This is a constant arrangement and is identical in all species from bacteria to humans, with just a few exceptions. Therefore the ratio of adenine to thymine (A/T) is always equal, and the same is true for the ratio of guanine to cytosine (G/C). In humans G/C pairs are about four times greater than A/T pairs. The polynucleotide chains run vertically in opposite directions. Therefore a sequence of adenine, guanine, and cytosine (AGC) is always matched by the sequence of thymine, cytosine, and guanine (TCG).
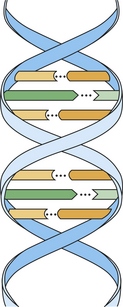
In the linear representation shown the number 1 indicates the nucleotide of one chain, and the number 2 indicates the nucleotide of the other chain. A molecule of hydrogen (H) binds both nucleotides. This arrangement is repeated horizontally to form the polynucleotide double spiral staircase (or helix) appearance of DNA (see Figure 6-8).
The vertical sequence of three bases is called a codon. It codes for an amino acid. Several amino acids form a polypeptide, and one or more polypeptides form a protein. A gene is often equated with the unit that forms a polypeptide.
DNA has the unique capability of self-replication, which is achieved by unwinding its double chain like the plaits of a braid. Each separated chain serves as a blueprint for another chain.
Mitochondrial DNA is found in the circular chromosome of the mitochondria, and it is maternally inherited. This is the DNA present in the cytoplasmic mitochondrial organelles of the ovum. Mitochondrial DNA is passed from the mother to all her offspring, regardless of sex.
RIBONUCLEIC ACID
To produce amino acids, polypeptides, and proteins, the genetic code contained in the DNA is transcribed into ribonucleic acid (RNA), which differs from DNA in that it is a single strand (in its simplest form), its sugar is a ribose (the sugar in DNA is deoxyribose), and the base uracil (U) replaces the thymine (T) in DNA.
Types of Ribonucleic Acid
The four types of RNA are (1) messenger RNA (mRNA), (2) transfer RNA (tRNA), (3) ribosomal RNA (rRNA), and (4) heterogeneous RNA (hnRNA). RNA can be found in both the nucleus and the cytoplasm of a cell.
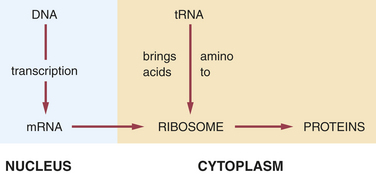
The first type of RNA, mRNA, is a blueprint of the genetic DNA for the coding of proteins. It carries the message for the DNA to ribosomes in the cytoplasm, in which proteins are produced. The second type of RNA, tRNA, transfers amino acids from the cytoplasm to the mRNA, positioning amino acids in the proper sequence to form polypeptides and hence proteins. The third type of RNA, rRNA, combines with several polypeptides to form ribosomes. The fourth type of RNA, hnRNA, is found within the nucleus and is the precursor of mRNA.
In the production of a protein (Figure 6-9) the mRNA carries the genetic code for the formation of that protein to the ribosomes. The tRNA brings amino acids to the ribosomes from the cellular cytoplasm. The amino acid sequence forms proteins according to the genetic code, and these proteins exit the ribosomes as they are formed.
GENES AND CHROMOSOMES
Genes in a chromosome are located in a vertical linear manner. The genes in both members of a pair of chromosomes (homologous chromosomes) govern the same functions or dictate the same characteristics. The genes that are located at the same level (or locus) in homologous chromosomes and that dictate the same functions or characteristics are called alleles.
The manifestations (phenotype) of a gene action are not necessarily the same from one individual to another. This is best explained with the ABO blood group system. The locus can be occupied by either the factor that determines the blood group A or the factor that determines the blood group B. If it is empty, it results in the blood group O. The locus is always the same, and the three genes govern the same function; however, the clinical result is different. In this situation the trait or condition is said to have multiple alleles. For example, if both loci are AA or if they are AO, the person is said to have blood group A. If both loci are BB or BO, the person is said to have blood group B. If the loci are AB or BA, the person is said to have blood group AB. Only if both the loci are empty does the person have blood group O. The locus always controls blood group, but the group depends on the alleles that are present or lacking in each person.
When the allelic genes are identical, the person is said to be homozygous for that gene, or a homozygote. Using the ABO blood group system again as an example, a person with AA, BB, and OO would be homozygous. When the genes are different (for example, AB, AO, or BO), the person is said to be heterozygous for that gene, or a heterozygote. If a gene can express its effect clinically with a single dose (heterozygous), as in the combination AO = blood group A, the characteristic is said to be dominant. If the gene needs a double dose to exhibit its action (homozygous), the resulting characteristic or function is said to be recessive. For example, only the combination OO results in the blood group O.
CHROMOSOMAL ABNORMALITIES
Abnormalities of chromosomes can be divided into two categories: (1) molecular abnormalities and (2) gross abnormalities. Molecular alterations occur at the DNA level and are not detectable microscopically. Most inherited disorders represent examples of molecular changes (mutations) at the level of one or both allelic genes. Examples of these conditions are presented later in this chapter.
Gross chromosomal alterations can be observed in a karyotype. A karyotype (Figures 6-10and 6-11) is a photographic representation of a person’s chromosomal constitution. Clinicians can create a karyotype by culturing cells from blood, skin, or other tissues. One method uses peripheral blood by placing it in a test tube with heparin to avoid coagulation and centrifuging it. After centrifugation the white blood cells (leukocytes) are deposited at the bottom of the tube. The leukocytes are removed and placed in a culture medium containing phytohemagglutinin, which is a substance that enhances mitosis. Because chromosomes are best observed when cell division is arrested at metaphase, colchicine is added to the culture after 72 hours of culture at 37° C to stop mitosis at metaphase. This also prevents the centromere from dividing. A hypotonic solution is then added to the culture to make the cells swell. The cells are then fixed (preserved) and stained and observed under the microscope. Examples of well-defined mitoses are chosen and photographed. The photograph is enlarged, and each chromosome is cut out of the photographic print. When stained, the chromosomes have a bandlike appearance that allows an accurate identification of each. These cutouts are then pasted on a special chart to construct the karyotype.
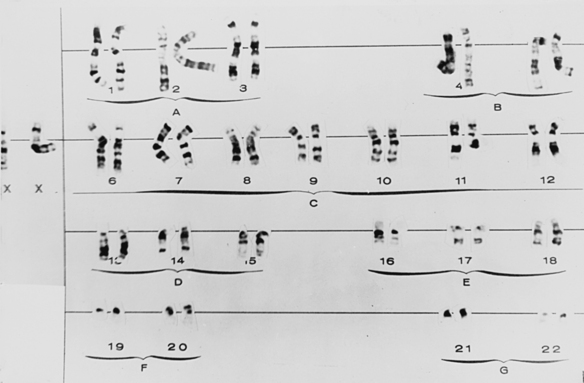
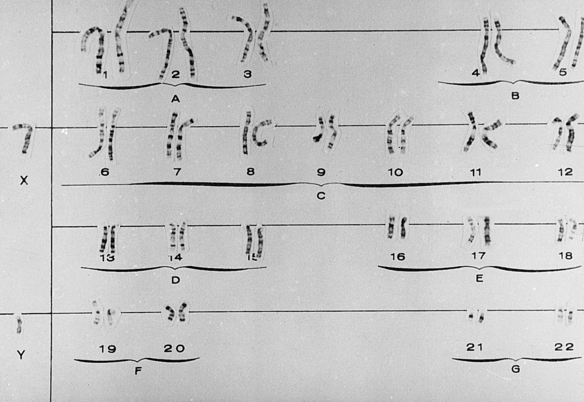
GROSS CHROMOSOMAL ABNORMALITIES
Alterations in Number and Structure of Chromosomes
Gross chromosomal abnormalities are caused by either alterations in chromosome number, which are almost always a result of nondisjunction (explained earlier), or alterations in structure, which develop because of chromosomal breaks or abnormal rearrangements. The following illustrate alterations in number:
 Euploid: A complete second set of chromosomes, the total number being 92. This is incompatible with life.
Euploid: A complete second set of chromosomes, the total number being 92. This is incompatible with life.
Polyploid: Three (triploid) or four (tetraploid) complete sets of chromosomes. This has been described occasionally in humans and is incompatible with life.
Aneuploid: Any extra number of chromosomes that do not represent an exact multiple of the total chromosome complement (e.g., trisomy [a pair with an identical extra chromosome] and monosomy [a missing chromosome from a pair]).
Clinical Syndromes Resulting From Gross Chromosomal Abnormalities
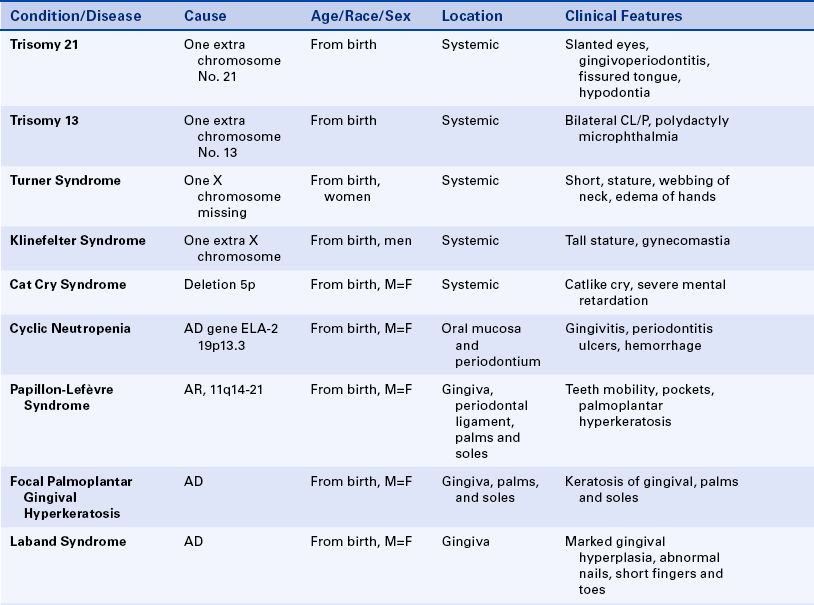
This condition, also known as Down syndrome, is the most frequent of the trisomies. Ninety-five percent of cases of Down syndrome are the result of nondisjunction, mostly associated with late maternal age at the time of conception.
Slanted eyes characterize the facies (the appearance of the face). Patients are generally shorter than normal, and heart abnormalities are present in more than 30% of individuals with trisomy 21. The intelligence level varies from near normal to marked handicap.
Fissured tongue is frequently seen in patients with trisomy 21. Premature loss of teeth, especially the mandibular central incisors, caused by alveolar bone loss is seen frequently. Gingival and periodontal disease has been reported in 90% of affected individuals. Hypodontia (fewer teeth than normal), abnormally shaped teeth, and anomalies in eruption with malposition and crowding of teeth are common findings. Dental hygienists play an important role in the maintenance of oral health in these patients.
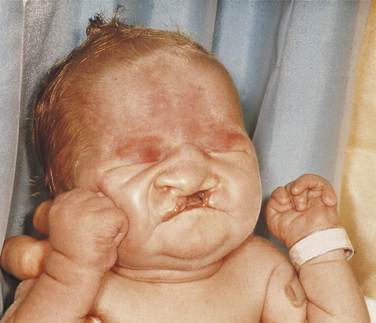
Trisomy 13
This disorder is characterized by multiple abnormalities in various organs. Seventy percent of live-born infants die within the first 7 months of life. Characteristic clinical findings include bilateral cleft lip and palate, microphthalmia or anophthalmia (small eyes or no eyes), superficial hemangioma of the forehead or nape of the neck, growth retardation, severe mental handicap, polydactyly of hands and feet (supernumerary digits), clenching of the fist with the thumb under the fingers, rocker-bottom feet, heart malformations, and several anomalies of the external genitals. The facial appearance is quite striking because of the cleft lip, cleft palate, and ocular abnormalities (Figure 6-12).
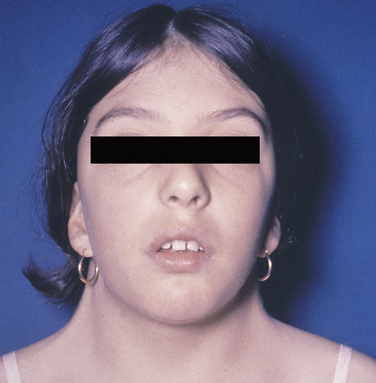
Turner Syndrome
Patients with Turner syndrome have a female phenotype, and in the majority of cases the karyotype has the normal 44 autosomic chromosomes and only one X chromosome. A normal female would have two X chromosomes: one from the mother and one from the father. Most cases of Turner syndrome are the result of nondisjunction of the X chromosome in the paternal gamete. Clinically these women are of short stature and have webbing of the neck and edema of the hands and feet (Figure 6-13). They frequently exhibit a low hairline on the nape of the neck. The chest is broad with wide-spaced nipples. The aorta frequently is abnormal, and body hair is sparse. The external genitals appear infantile, and generally the ovaries are not developed; therefore these individuals have primary amenorrhea (abnormal temporary or permanent cessation of the menstrual cycle). Smears taken from the oral mucosa demonstrate the lack of Barr bodies.
Klinefelter Syndrome
This condition occurs when an ovum carrying two X chromosomes is fertilized by a spermatozoon with a Y chromosome; therefore the fertilized ovum will have two X chromosomes plus a Y chromosome. The majority of cases result from nondisjunction of the X chromosome, generally in the ova of older women. Affected individuals have a male phenotype, and the condition cannot be detected until after puberty. These patients are taller than normal and have wide hips and a female pubic hair distribution. About 50% have gynecomastia (development of female breasts), and intelligence levels are lower than normal in 10% of affected individuals. The penis appears normal, but the testes are smaller and harder than normal and lack seminiferous tubules.
The maxilla is slightly hypoplastic (underdeveloped). Buccal smears reveal the presence of one Barr body.
Variations of Klinefelter syndrome also occur; they are represented by karyotypes containing XXXY or XXXXY. The greater the number of X chromosomes, the more pronounced the clinical manifestations, and the lower the level of intelligence. The maxilla becomes increasingly hypoplastic with increasing number of X chromosomes. Buccal smears show one Barr body for each extra X chromosome.
Cri du chat (Cat Cry) Syndrome and Wolf-Hirschhorn Syndrome
These syndromes are examples of abnormalities caused by deletions. The cri du chat syndrome results from a deletion on the short arm of chromosome 5, and the Wolf-Hirschhorn syndrome results from a deletion on the short arm of chromosome 4. Newborns with a chromosome 5 deletion exhibit a catlike cry at birth and are mentally retarded. No oral abnormalities occur. Most newborns with the deletion in the short arm of chromosome 4 have a cleft palate and intelligence quotients of less than 30.
PATTERNS OF INHERITANCE
Because loci are present in both autosomal and X chromosomes and because of a double- and single-dose effect, four possible inheritance patterns exist. Dominant genes need only a single dose, and recessive genes need a double dose. The inheritance patterns are autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive. Autosomal chromosomes include all chromosomes except those that determine sex (X and Y). The Y chromosome participates only in the differentiation of the masculine gonads.
Autosomal-Dominant Inheritance
A condition having autosomal-dominant inheritance is transmitted vertically from one generation to the next. Males and females are equally affected. When a person has a gene for the condition, the risk of having an affected offspring is 50% for each pregnancy. Genetic risk is always a mathematical estimate of probability governed by chance. Therefore none, less than half, half, more than half, or all of the offspring could be affected by a condition that is transmitted by autosomal-dominant inheritance.
An individual can carry a gene with a dominant effect without presenting any clinical manifestations. This is referred to as lack of penetrance. This situation can be explained partially by the presence of modifying genes in the same or other chromosomes. The clinical manifestations in autosomal-dominant disorders frequently vary among affected individuals. This is known as variable expressivity. Penetrance refers to the number of individuals affected, and expressivity pertains to the degree to which an individual is affected.
Autosomal-Recessive Inheritance
As stated previously, individuals exhibiting an autosomal-recessive trait must be homozygous for the gene. Clinically normal parents of affected children are heterozygous, and both are carriers of the trait. They are not generally recognized as carriers until after the birth of an affected child. If the enzymatic defect is known, carriers can be recognized before the birth of a child by assessing levels of the responsible enzyme in clinically normal members of a family with the trait. For parents who are carriers of the same recessive trait, the risk of having an affected child is 25%, the risk of having a homozygotic normal child is 25%, and the chance of having a heterozygotic carrier is 50% for each pregnancy. As in other inheritance patterns, risk is a mathematical estimate of the probability of an event occurring. If both parents are homozygous (have two of the genes of the trait) for a recessive trait, they would be expected to be affected, and all their children would be affected equally because they would also be homozygous (have two of the same genes) for the trait. In humans this type of situation is quite rare because individuals affected by the same recessive trait usually do not mate.
In genetics consanguinity means a familial relationship and is generally used to describe matings or marriages between close relatives, usually including first cousins. In the United States marriage or mating between very close relatives (parent-offspring, brother-sister, uncle-niece, aunt-nephew) is illegal, and in most states marriage of first cousins is also illegal. However, consanguineous matings occur sporadically. The chance of having deleterious genes in common increases between close relatives. When consanguinity exists between the members of a family, the chance of the offspring being homozygous for a deleterious gene increases. The closer the degree of consanguinity, the greater the risk.
X-Linked Inheritance
Women have two X chromosomes and therefore can be either heterozygous or homozygous for a gene that is located on the X chromosome. Thus in women X-linked traits can be dominant or recessive. Men have only one X and one Y chromosome. If a deleterious gene occurs on the X chromosome in a male, the condition or trait will be seen clinically, regardless of the dominant or recessive behavior of the same gene in women. The man’s X chromosome is transmitted to all of his daughters and to none of his sons. The X chromosome in his sons comes from the mother. Consequently no male-to-male (from father to son) transmission of X-linked traits occurs. Some X-linked dominant traits are lethal in males. Females probably survive because of the action of the allelic normal gene on the second X chromosome. All offspring, male and female, of a woman homozygotic for a dominant X-linked condition will be affected with the condition. This is because all of her X chromosomes contain that gene (and all offspring receive an X chromosome). Because the gene is dominant, only one of the genes is necessary for that condition to occur. A mother who is a carrier of an X-linked recessive trait has a 50% risk of having an affected son and a 50% risk of having a carrier daughter. This is because both daughters and sons have a 50% risk of getting the X chromosome with the gene for that condition. Again the reader should remember that risk is a mathematical prediction. A male affected with an X-linked trait will have no affected sons because he does not give the X chromosome to his sons. All of his daughters will be carriers or affected, depending on the recessive or dominant nature of the trait, because all daughters will receive his X chromosome.
Lyon Hypothesis and X-linked Recessive Traits
As discussed previously, according to the Lyon hypothesis, one of the X chromosomes in the female is genetically cancelled at an early stage of embryonic development. This cancellation affects X chromosomes from both maternal and paternal lines. If a female embryo is a carrier of an X-linked recessive trait, half of the X chromosomes have the normal gene, and the other half have the abnormal gene (allele) for the given trait before cancellation occurs. Classic hemophilia (hemophilia A) is a good example. Hemophilia A is inherited as an X-linked recessive condition. In this disorder blood does not coagulate because of the low or almost nonexistent levels of factor VIII (antihemophilic globulin) in circulating blood. Males with the abnormal gene have a severe coagulation defect. In the female carrier some of the cancelled X chromosomes have the abnormal gene, and others have the normal one. The female carrier is a mosaic (i.e., she has both normal and abnormal X chromosomes). Because cancellation is random, the number of X chromosomes that remain genetically active and contain the normal or abnormal gene will vary. The female carrier’s levels of factor VIII are often reduced; however, this reduction and the length of coagulation time vary, depending on the number of X chromosomes that contain the abnormal gene and remain genetically active. Although female carriers of the gene for hemophilia do not have as severe a problem as males, they tend to bleed more than usual after extraction of teeth or scaling and curettage. The variation in the bleeding problem in female carriers of the gene for hemophilia reflects the Lyon hypothesis. Other inherited disorders that reflect this hypothesis are described in detail later in this chapter. They include the X-linked type of amelogenesis imperfecta and hypohidrotic ectodermal dysplasia.
Genetic Heterogeneity
The term genetic heterogeneity is used when a condition has more than one inheritance pattern and differences in the degree of clinical manifestations for each of the inherited varieties. Amelogenesis imperfecta (described later in the chapter) is a condition that illustrates genetic heterogeneity.
In general, alterations in structural proteins (complex substances formed by one or more polypeptide) are inherited as a dominant trait, whereas alterations in enzymatic proteins (a protein inducing changes in other body substances) are inherited in a recessive manner; however, exceptions to this rule are seen.
Physical characteristics can be inherited as either dominant or recessive traits. The term recessive does not mean deleterious or abnormal. Instead it means that the individual must be homozygous for the trait for it to be seen. For example, in the ABO blood group system blood group O is recessive, whereas groups A and B are dominant. However, no blood group is abnormal.
Genes can be codominant. Again a good example is the ABO blood group. A and B are dominant over O, but when A and B are allelic, the result is blood group AB, in which both genes are exhibited. This is called codominance.
The four inheritance patterns refer to the characteristics that are governed by the action of one gene. This is called single-gene inheritance. Oligogenic inheritance refers to characteristics or traits that are inherited by the participation of several genes. These conditions show greater clinical variation than do those inherited through single-gene action. Characteristics such as tooth shape and form and eye color are determined by oligogenic inheritance. Most physical characteristics are oligogenic. The various genes that participate in determining a trait or condition can be located in the same chromosome or in different chromosomes.
Examples of Molecular Chromosomal Abnormalities
All of the inherited disorders in this section include oral and dental alterations and are discussed by location—gingiva and periodontium, jawbones and facies, oral mucosa, and teeth. Patients affected with these genetic abnormalities should receive routine dental hygiene care to maintain oral health. Specific dental hygiene management considerations are included only if they are unique to the disorder described.
Inherited Disorders Affecting the Gingiva and Periodontium
Most of the disorders included here are rare. However, they exhibit severe gingival or periodontal alterations (or both); therefore the dental hygienist should be aware of the disorders and their clinical manifestations.
Cyclic Neutropenia: The inheritance pattern of cyclic neutropenia is autosomal dominant; the responsible gene has been identified and is called ELA-2 (neutrophil elastase gene). This gene is located on the short arm of chromosome 19. In the chromosome mapping system its location is designated as 19p13.3. The disorder is characterized by a cyclic decrease in the number of circulating neutrophilic leukocytes (neutrophils); a decrease in the number of circulating neutrophils is called neutropenia. The cycles usually occur in intervals of 21 to 27 days, but in some patients the interval may be extended to several months. These episodes of neutropenia generally persist for 2 to 3 days.
The clinical manifestations are related to the decrease in neutrophils. Systemic manifestations of cyclic neutropenia include fever, malaise, sore throat, and occasional cutaneous infections. Oral manifestations consist of severe ulcerative gingivitis or gingivostomatitis (Figure 6-14). In addition to the gingiva, areas of ulceration can also occur on the tongue and surfaces of the oral mucosa. The ulcers are of variable size and have a craterlike appearance. They are very painful and have a bleeding base. Generally the oral lesions are infected secondarily. When neutrophils return to normal, the oral lesions tend to improve.
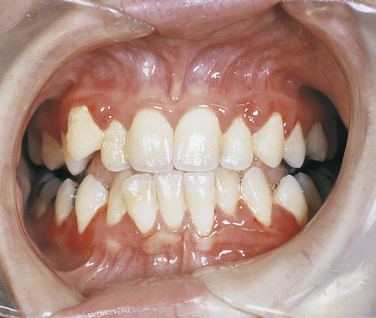
FIGURE 6-14 Hypertrophic gingivitis with areas of gingival recession is exhibited in a patient with cyclic neutropenia.
Patients with cyclic neutropenia are usually managed by first determining the frequency of the cycles through periodic neutrophil counts and then instituting preventive antibiotic therapy to protect against secondary opportunistic infections. Over time episodes of neutropenia and associated ulcerative gingivitis lead to severe periodontal disease, with loss of alveolar bone, tooth mobility, and exfoliation of teeth. Treatment should be initiated when the circulating neutrophil count is normal to reduce the risk of complications such as gingival hemorrhage and secondary infection. Dental hygiene care, including frequent appointments for removal of local irritants and maintenance of optimal oral hygiene, reduces the risk of opportunistic infections in patients with cyclic neutropenia.
Patients with cyclic neutropenia are treated with periodic granulocyte colony-stimulating factor (G-CSF) that reduces the symptoms and results in substantial clinical improvement.
Chronic neutropenia, also known as Kostmann syndrome, is another variety of neutropenia that is inherited as an autosomal recessive. The intraoral manifestations are similar to those of the cyclic variety but are constant. The responsible gene is still unknown, and it is also treated with periodic G-CSF.
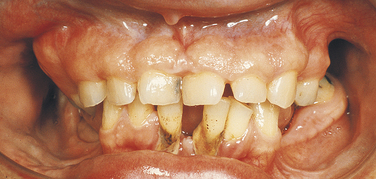
Papillon-Lefèvre Syndrome: Papillon-Lefèvre syndrome has an autosomal-recessive inheritance pattern and is composed of marked destruction of the periodontal tissues (periodontoclasia) of both dentitions with premature loss of teeth and hyperkeratosis of the palms of the hands and soles of the feet (palmar and plantar hyperkeratosis).
The gene for this syndrome has been mapped to the long arm of chromosome 11 regions 14-21 (11q14-21). These patients are normal at birth except for a reddening of the palms of the hands and soles of the feet. Teeth erupt in normal sequence, position, and time. At about 1½ to 2 years of age, a marked gingivoperiodontal inflammatory process develops characterized by edema, bleeding, alveolar bone resorption, and mobility of teeth with consequent exfoliation. A red, scaly keratosis develops on the palms and soles concurrent with these oral lesions and occasionally extends to the dorsal surfaces of the hands and feet. The oral lesions are complicated by superimposed inflammation, and radiographs reveal marked alveolar bone resorption with vertical pockets. Teeth are lost in the same sequence in which they erupted. When the last tooth is lost, the gingiva regains a normal appearance.
The lesions on the hands and feet remain as reddish-white, scaly, thick areas of hyperkeratinization (Figure 6-15). The permanent dentition begins to erupt at the proper time. At about 8 or 9 years of age the gingivoperiodontal destruction is repeated in the same manner as occurred in the primary dentition (Figure 6-16). All permanent teeth are lost before 14 years of age. The gingiva again resumes a normal appearance.
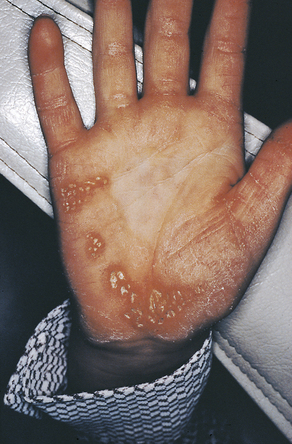
FIGURE 6-15 Areas of hyperkeratinization of the palms are exhibited in a patient with Papillon-Lefèvre syndrome. (From Sedano HO, Sauk JJ, Gorlin RJ: Oral manifestations of inherited disorders, Boston, 1977, Butterworth. Used with permission.)
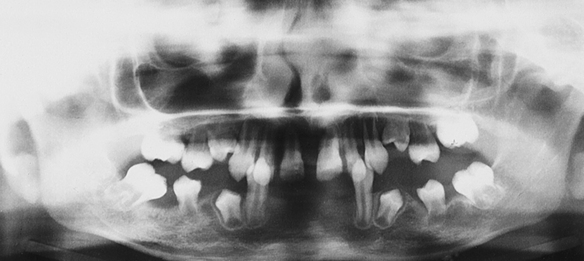
FIGURE 6-16 Panoramic radiograph shows marked periodontal destruction with alveolar bone resorption in the patient in Figure 6-15. (From Sedano HO, Sauk JJ, Gorlin RJ: Oral manifestations of inherited disorders, Boston, 1977, Butterworth. Used with permission.)
Peripheral blood neutrophil is depressed in all patients with Papillon-Lefèvre syndrome. This suggests that the neutrophils may be an important factor in the pathogenesis of severe periodontal disease in these patients. Retinoid therapy markedly improves the skin condition but has no effect on the periodontal disease in patients with this syndrome. To date all therapeutic attempts at preventing the gingival and periodontal destruction and subsequent loss of teeth have been unsuccessful. The basic defect that causes the condition is unknown. Peripheral blood neutrophil chemotaxis has been reported to be depressed in patients with Papillon-Lefèvre syndrome. This decreased chemotaxis suggests that neutrophils may play a role in periodontal destruction in patients with this syndrome. Current research suggests bacterial and viral involvement as the initiating factors for the periodontal destruction. It has been suggested that the genetic component of Papillon-Lefèvre syndrome is a predisposition rather than the main determinant of periodontal disease.
The skin manifestations remain for life, but treatment with retinol (oral etretinate) has proven somewhat effective in controlling the hyperkeratinization. These patients do not have any other abnormalities.
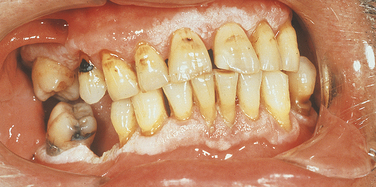
Focal Palmoplantar and Gingival Hyperkeratosis: Areas of hyperkeratinization of the palms and soles and marked hyperkeratinization of the labial and lingual gingiva characterize focal palmoplantar and gingival hyperkeratosis. This syndrome has an autosomal-dominant inheritance pattern. The palmar and plantar hyperkeratosis starts at the tips of the fingers and toes and extends to the surface of the palms and soles. This process increases with age and also becomes localized, with calluses forming on the weight-bearing areas.
The oral hyperkeratinization is bandlike and a few millimeters in width (Figure 6-17). It follows the normal festooned contour of the gingiva. The free gingiva is not affected. Palatal and lingual mucosa occasionally can be affected with areas of hyperkeratinization. These changes start in childhood and increase with age. The rest of the oral cavity is normal.
Gingival Fibromatosis: Gingival fibromatosis is a component of several inherited syndromes. The gingival hypertrophy generally develops early in life, and within a few years the teeth are completely covered. The fibromatosis is composed of very firm tissue with a granular corrugated surface. Generally the color is paler than that of the normal gingiva and results from the marked collagenization of the fibrous connective tissue. The extensive gingival enlargement leads to protrusion of the lips.
In addition to isolated gingival fibromatosis (Figure 6-18), which has an autosomal-dominant inheritance pattern and no other associated abnormalities, gingival fibromatosis is a component of a number of syndromes. Because of the gingival involvement, several of these syndromes, although rare, are described here. However, only those that are well known and occur most frequently are included.
Dental hygiene care can reduce the risk of secondary inflammation and infection in patients with gingival fibromatosis.
Laband Syndrome: The inheritance pattern of Laband syndrome is autosomal dominant. In addition to gingival fibromatosis, patients have dysplastic or absent nails and malformed nose and ears because of soft and pliable cartilage formation, hepatosplenomegaly (enlarged liver and spleen), and hypoplasia of terminal phalanges of the fingers and toes with a resultant froglike appearance.
Gingival Fibromatosis With Hypertrichosis, Epilepsy, and Mental Retardation Syndrome: Gingival fibromatosis with hypertrichosis, epilepsy, and mental retardation syndrome has an autosomal-dominant inheritance pattern. Hypertrichosis (excessive growth of hair), especially of the eyebrows, extremities, genitals, and sacral region, characterize gingival fibromatosis with hypertrichosis, epilepsy, and mental retardation. Epilepsy and mental retardation can also occur in this syndrome but are inconsistent features.
Gingival Fibromatosis With Multiple Hyaline Fibromas: Gingival fibromatosis with multiple hyaline fibromas has an autosomal-dominant inheritance pattern and is also known as the Murray-Puretíc Drescher syndrome. In addition to gingival fibromatosis, it is characterized by hypertrophy of the nail beds and multiple hyaline fibrous tumors developing on the nose, chin, head, back, fingers, thighs, and legs. These tumors on the extremities can produce contractures of various joints, including hips, knees, shoulders, and elbows.
Inherited Disorders Affecting the Jaw Bones and Facies
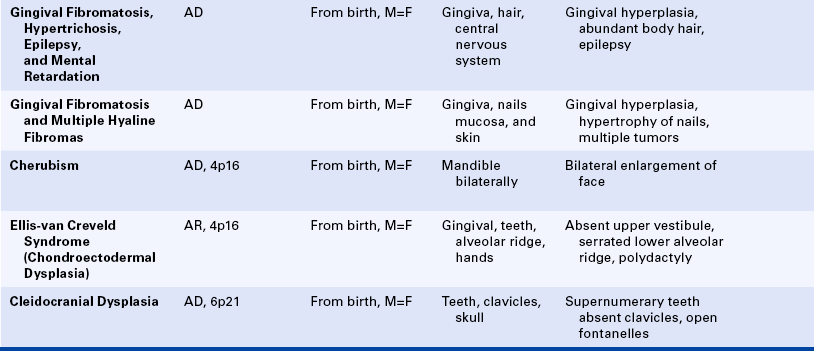
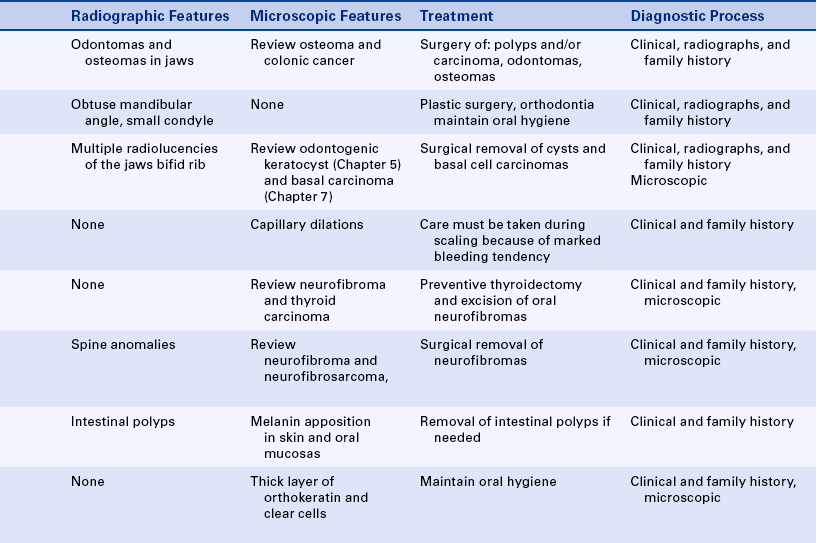
Cherubism: The inheritance pattern of cherubism is autosomal dominant with marked penetrance (when the gene is present, the clinical manifestations are usually seen) in males and variable expressivity and incomplete penetrance in females. The gene for cherubism has been mapped to the short arm of chromosome 4 region 16 (4p16). The first clinical manifestation is a progressive bilateral facial swelling that appears when the patient is between 1½ and 4 years of age. This change can affect either the mandible or the maxilla, but involvement of the mandible is most common. Displacement of the eyes is evident when the maxilla is affected. This, added to the bilateral deformity, produces the characteristic cherubic facial appearance that is responsible for the name of the syndrome. Increased distance between the eyes (ocular hypertelorism) is always present in affected patients. Radiographs of the jaws show a typical “soap-bubble” or multilocular appearance (Figure 6-19), which usually occupies the ascending ramus of the mandible and extends into the molar and premolar areas. Severe cases can involve the full mandible, but the condyle is always spared. When the maxilla is affected, the changes are observed at the level of the tuberosity and involve the antrum.
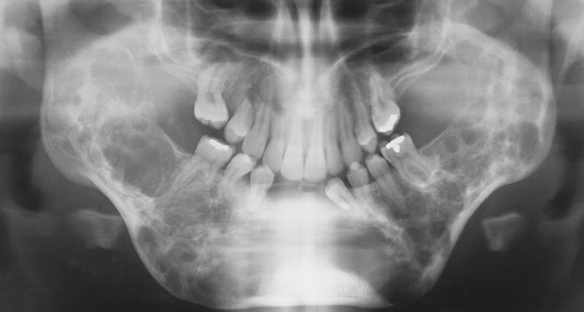
FIGURE 6-19 Panoramic radiograph of the jaws shows typical bilateral soap-bubble image in a patient with cherubism. (From Young WG, Sedano HO: Atlas of oral pathology, Minneapolis, 1981, University of Minnesota Press. Used with permission.)
These areas of bone radiolucency are occupied by fibrous connective tissue containing multinucleated giant cells. The microscopic appearance resembles the central giant cell granuloma (described in Chapter 2). The bone lesions interfere with tooth development and eruption. Most of these patients have pseudoanodontia (teeth falsely appear to be lacking), especially of molars and premolars, because of delayed eruption.
The size of the jaws tends to increase rapidly until about puberty and then generally remains stable. After the individual reaches age 20 or 30 years, radiographs show an almost normal bone appearance, with a few areas of increased bone density. The facial deformity remains for life, and in some patients it can be quite striking.
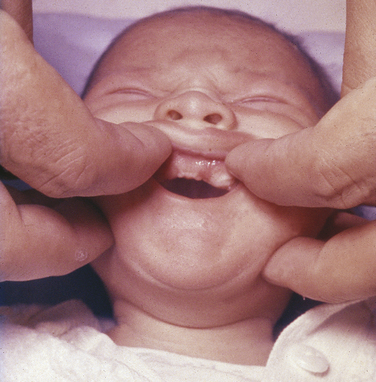
Ellis–Van Creveld Syndrome: Ellis–van Creveld syndrome has an autosomal-recessive inheritance pattern. The gene for this has been mapped to the short arm of chromosome 4 region 16 (4p16). Affected individuals are dwarfs because of distal shortening of the extremities. One third of these patients are mildly mentally handicapped. The hands show polydactyly (supernumerary digits) on the ulnar side, and fingernails and toenails are hypoplastic and deformed. Other skeletal anomalies include curvature of the legs and feet. Fifty percent of affected individuals have congenital heart defects. Anomalies of the external genitals, especially in males, are also observed.
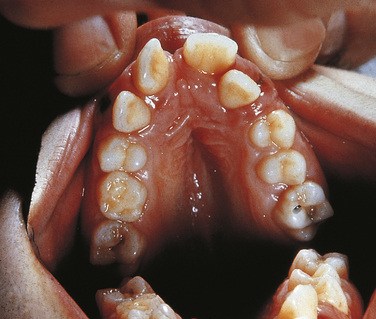
The oral manifestations are constant and characteristic and include fusion of the anterior portion of the maxillary gingiva to the upper lip from canine to canine. Thus the anterior maxillary vestibular sulcus is lacking (Figure 6-20). This anomaly induces a V-notch appearance in the midline of the upper lip. The anterior lower alveolar ridge presents a serrated appearance caused by thick frenula, which start at the vestibular sulcus and traverse the alveolar ridge. The central incisors of both maxilla and mandible are generally lacking and are replaced by a centrally located abnormal tooth. Most of the teeth have a conical shape and exhibit enamel hypoplasia. More than 50% of newborns with this syndrome have natal teeth.
Cleidocranial Dysplasia: The inheritance pattern of cleidocranial dysplasia is autosomal dominant; however, about half of the cases are isolated examples caused by either spontaneous mutation or a gene with poor penetrance. The gene for this syndrome has been mapped to the short arm of chromosome 6 region 21 (6p21). The cranium develops a mushroom shape because the fontanelles remain open. This makes the face appear small. Frontal, parietal, and occipital enlargement is quite noticeable. Skull radiographs reveal the open fontanelles, which are often open for life. The paranasal sinuses are lacking or hypoplastic. The neck is long and narrow because of unilateral or bilateral aplasia (lack of development) or hypoplasia of the clavicles. Affected individuals are able to approximate their shoulders to the midline because of this clavicular alteration. Various other bone anomalies can also be present.
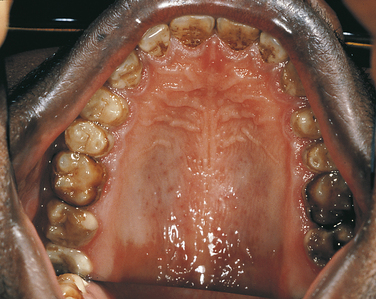
The premaxilla is generally underdeveloped, resulting in pseudoprognathism. These patients have many supernumerary teeth, sometimes even simulating a third dentition (Figure 6-21). They are crowded in the jaws and do not erupt. They also interfere with the eruption of normal teeth, resulting in pseudoanodontia. A lack of cellular cementum has been reported in patients with this syndrome. Multiple cysts can develop in association with the impacted teeth. About 1% of affected patients have cleft lip, cleft palate, or both.
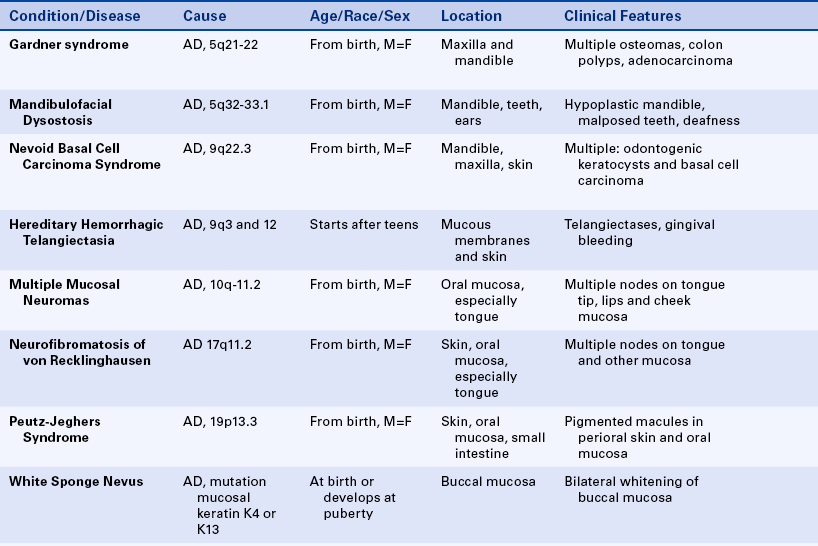
Gardner Syndrome: Gardner syndrome, also known as familial colorectal polyposis, has an autosomal-dominant inheritance pattern with variable expressivity and marked penetrance. The adenomatous polyposis coli gene is responsible for Gardner syndrome and is located on the long arm of chromosome 5 regions 21-22 (5q21-22). One of the basic components is the presence of osteomas in various bones, especially the frontal bones, mandible, and maxilla. When they expand, osteomas of the facial skeleton obliterate the sinuses and induce facial asymmetry. Osteomas can also occur in the long bones of the skeleton but with less frequency than in the bones of the face.
In addition to osteomas, multiple odontomas can occur in the jawbones, especially the mandible (Figure 6-22). Teeth can exhibit hypercementosis and fail to erupt.
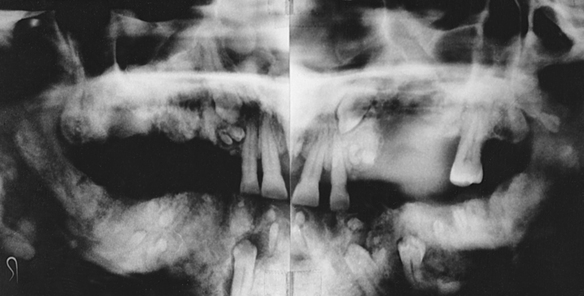
FIGURE 6-22 Panoramic radiograph of a patient with Gardner syndrome shows multiple osteomas and odontomas. (Courtesy Dr. Carl J. Witkop.)
The most serious component of this syndrome is the presence of intestinal polyps, which become malignant at age 30 and after. Polyposis primarily affects the colon and rectum and generally develops before puberty. Some authors advocate intestinal resection when the polyps appear because their malignant transformation into adenocarcinoma is invariable, especially with increasing age.
Mandibulofacial Dysostosis: The inheritance pattern of mandibulofacial dysostosis is autosomal dominant with incomplete penetrance and variable expressivity. The gene for this syndrome has been mapped to the long arm of chromosome 5 regions 32-33.1 (5q32-33.1). The facies shows downward sloping of the palpebral fissures, a hypoplastic nose, hypoplastic malar bones with hypoplasia or absence of the zygomatic process, abnormal and misplaced ears, and a receding chin. The mouth appears fishlike, with downward sloping of the lip commissures. The lower eyelids show a cleft (coloboma) of the outer third, with a lack of lashes medial to it. The ears may exhibit tags, which on occasion can also be seen near the angle of the mouth.
Deafness is a constant feature resulting from a lack of otic ossicles. Mental development is within normal limits; difficulty in learning arises from deafness. Oral manifestations include a markedly hypoplastic mandible with flattened condyles and coronoid processes and an obtuse mandibular angle. Teeth are malposed, and malocclusion with open bite is quite evident. The palate is high, or a cleft is present in about 30% of affected patients (Figure 6-23). Gingival disease is common and is related to the dental abnormalities.
Nevoid Basal Cell Carcinoma Syndrome: The nevoid basal cell carcinoma syndrome, also known as Gorlin syndrome, has an autosomal-dominant inheritance pattern with high penetrance and variable expressivity. The gene for this syndrome maps to the long arm of chromosome 9 region 22.3 (9q22.3). The facies is characterized by mild hypertelorism (increased distance between the eyes) and mild prognathism, with frontal and parietal enlargement and a broad nasal root.
The cutaneous manifestations, which are called nevi, typically are basal cell carcinomas. The term nevus (plural, nevi), as defined here, is a congenital lesion characterized by skin pigmentation. These basal cell carcinomas appear early in life and continue to develop over the nose, eyelids, cheeks, neck, arms, and trunk. They can be flesh colored or pale brown and develop singly or in clusters. Histologically they are basal cell carcinomas. The palms and soles show small pits that become filled with dirt and appear as dark spots on those surfaces.
The oral manifestations of this syndrome consist of multiple cysts of the jaws (Figure 6-24) that (histologically) are odontogenic keratocysts (see Chapter 5). These cysts vary in size; they can be very large and have a marked tendency to recur after surgical removal. Occasionally an ameloblastoma arises in these cysts as part of this syndrome. The cysts develop as early as 5 to 6 years of age in some affected patients and interfere with normal development of the jawbones and teeth.
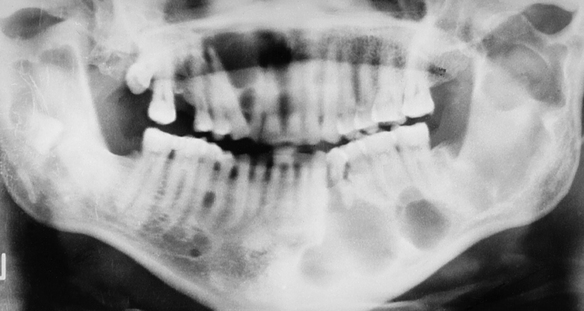
FIGURE 6-24 Multiple mandibular radiolucencies in a patient with nevoid basal cell carcinoma syndrome. These lesions are odontogenic keratocysts.
A great variety of skeletal anomalies have been reported in association with the nevoid basal cell carcinoma syndrome, the most constant being bifurcation or splaying of one or more ribs. Other frequent abnormalities of bone include shortening of the metacarpals, spina bifida occulta (defective closure of the bone encasement of the spinal cord), and kyphoscoliosis (a combination of scoliosis, which is a lateral curving of the spine, and kyphosis, which is an abnormal vertical curvature of the spine or “hunchback”).
Many different neoplasms have been reported in association with this syndrome. Medulloblastoma (brain tumor) has been seen in many cases. Children surviving the medulloblastoma eventually present other manifestations of the syndrome. Other less frequent findings include calcified ovarian fibromas and mesenteric cysts.
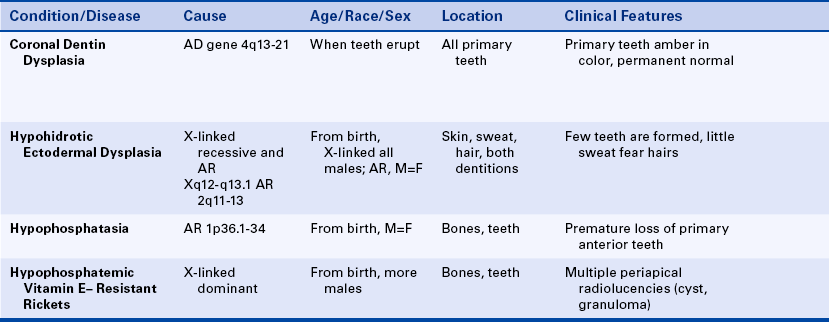
Osteogenesis Imperfecta: Osteogenesis imperfecta is known to have an autosomal-dominant inheritance pattern with variable expressivity. However, only 30% of patients have a family history of this condition. The remaining 70% represent sporadic cases and cases suggesting autosomal-recessive inheritance. The basic defect is produced by different mutations affecting the genes that encode for type I collagen, resulting in abnormally formed bones that fracture easily. The genes responsible for the inherited varieties of osteogenesis imperfecta are mapped as follows: collagen type I alpha 2 is in the long arm of chromosome 7 region 22.1 (7q22.1); collagen type I alpha 1 is in the long arm of chromosome 17 region 21.31-22.05 (17q21.31-22.05). Osteogenesis imperfecta occurs in about 1 in 20,000 births, and major syndromes have been classified in four types: I, II, III, and IV.
The clinical manifestations vary markedly from patient to patient. The sporadic cases tend to be more severe than those with an autosomal-dominant inheritance pattern. The basic defect involves collagen and results in abnormally formed bones that fracture easily. Multiple bone fractures are the main clinical complication of this syndrome. In the congenital form newborns can have several fractured bones at birth. Infants with this condition have experienced fractures just by being moved. In the most severe cases all bones can be affected, presenting a plethora of abnormalities, including bowing of the legs, curvature of the spine (kyphosis and scoliosis), deformity of the skull, shortening of arms and legs, and other abnormalities. In the mildest cases individuals show only blue sclerae (a blue appearance of the white of the eye).
The oral manifestation of this syndrome is a dentinogenesis imperfecta–like condition (a description of dentinogenesis imperfecta follows later in this chapter). Primary teeth are affected in 80% of patients, whereas permanent teeth are affected in only 35% of these individuals (Figure 6-25). The crowns, roots, and pulp chambers generally are smaller than normal. Teeth appear opalescent or translucent at time of eruption, but they darken with age. The enamel is lost because the abnormal dentin cannot provide adequate support.
Torus Mandibularis: Mandibular tori (singular, torus) have an autosomal-dominant inheritance pattern with variable expressivity and marked penetrance. This anomaly can be unilateral or bilateral. These tori occur on the lingual aspect of the mandible in the area of the premolar teeth (Figure 6-26). Occurrence before age 15 years is extremely rare. The size of the torus is variable, and occasionally it can be multilobulated. Mandibular tori are symptomless and generally require no treatment. Surgical removal may be necessary if the patient needs a denture. Intraoral radiographs usually show a radiopacity in the area of the mandibular premolars if mandibular tori are present. (Mandibular tori are also discussed in Chapters 1 and 7.)
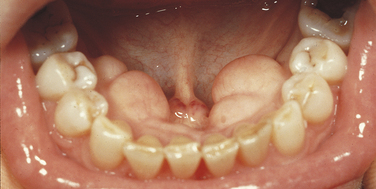
Torus Palatinus: Autosomal dominance with variable expressivity and almost 100% penetrance characterizes torus palatinus. A bony overgrowth occurs at the midline of the hard palate (Figure 6-27). A marked predilection exists for women (2:1), and a higher prevalence exists among Native Americans, including Inuits. Torus palatinus becomes evident around the time of puberty and is only rarely seen in children younger than 14 years of age. The size of this anomaly varies from nearly undetectable to large masses that occupy almost the entire hard palate. Some palatal tori are multilobular. Torus palatinus is symptomless. However, the surface mucosa is thin and easily traumatized. The torus may need to be removed if the patient needs a full denture. (Torus palatinus is also described in Chapters 1 and 7.)
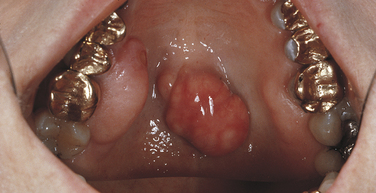
Maxillary Exostosis: Exostoses have an autosomal-dominant inheritance pattern. Maxillary exostoses represent tori that generally develop on the buccal aspect of the maxillary alveolar ridge, usually in the molar and premolar area (Figure 6-28). They are generally symptomless unless traumatized. They may be single, multiple, unilateral, and bilateral and occur less frequently than either palatal or mandibular tori. (Exostoses are also described in Chapter 7.)
Inherited Disorders Affecting the Oral Mucosa
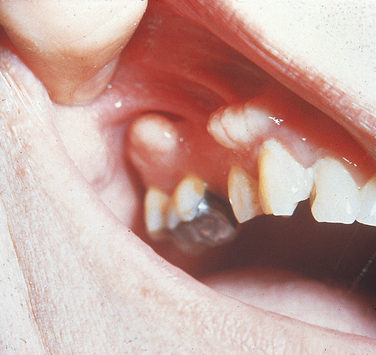
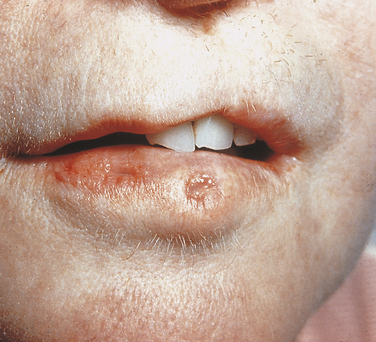
Isolated Cleft Palate and Cleft Lip With or Without Cleft Palate: The majority of cases of facial clefting are multifactorial in origin; they occur in about 1 in 800 births. A large number of inherited syndromes can include cleft lip and palate or isolated cleft palate as a component. When clefting is part of an inherited syndrome, its occurrence within affected families will be in accordance with the inheritance pattern of the syndrome. Most of these syndromes are very rare. Cleft lip-palate and congenital lip pits (Van der Woude syndrome) is the syndrome that is considered to occur most frequently; therefore it is the only one described here. It has an autosomal-dominant inheritance pattern with 80% penetrance for any of its components. In the majority of cases the gene has been mapped to the long arm of chromosome 1 regions 32 to 41 (1q32-41). The labial pits (Figure 6-29) are bilateral and are located near the midline of the vermilion border of the lower lip. They may be 3 mm or more in diameter and generally finish in a blind end. Occasionally they exude saliva because of their association with a labial minor salivary gland. These pits are rarely unilateral, and the clefting is bilateral in about 80% of patients. Some patients without clefting have agenesis (lack of development) of the maxillary lateral incisors or peg lateral incisors. Other oral findings that have been seen include fibrous adhesions between the maxilla and mandible, a cleft uvula, and ankyloglossia.
Hereditary Hemorrhagic Telangiectasia: The inheritance pattern of hereditary hemorrhagic telangiectasia, also known as Osler-Rendu–Parkes Weber syndrome, is autosomal dominant. Recent studies indicate that this syndrome may have two different responsible loci: one at the long arm of chromosome 9 (9q3) and the other in chromosome 12. It is characterized by multiple capillary dilations of the skin and mucous membranes (Figure 6-30). The skin of the face shows numerous pinpoint and spiderlike telangiectases, especially on the lips, eyelids, and around the nose. The scalp and ears are also affected. Similar lesions are present in the nasal mucosa and are responsible for frequent and sometimes serious nosebleeds (epistaxis) that can last for several days. Any organ or mucous membrane can be the site of telangiectasia.
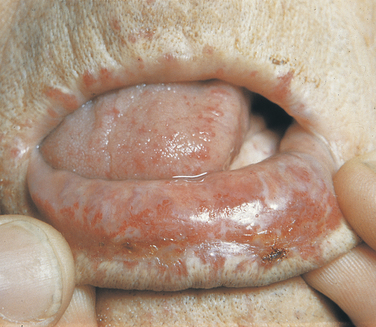
FIGURE 6-30 Multiple telangiectases of lips and tongue in a patient with hereditary hemorrhagic telangiectasia. Gingival bleeding can be profuse in cases like this. (From Sedano HO, Sauk JJ, Gorlin RJ: Oral manifestations of inherited disorders, Boston, 1977, Butterworth. Used with permission.)
Telangiectases of the oral mucosa are especially prominent on the tip and anterior dorsum of the tongue. The palate, gingiva, and buccal mucosa are often affected but to a lesser degree. Hemorrhage from sites in the oral cavity, mainly the lips and tongue, is second in frequency to epistaxis. Gingival bleeding has been reported and is a possible complication of dental hygiene treatment. The risk of gingival hemorrhage should be a concern of the dental hygienist when treating a patient with this syndrome.
Multiple Mucosal Neuroma Syndrome: The combination of multiple mucosal neuromas, medullary carcinoma of the thyroid gland, and pheochromocytoma is identified as multiple endocrine neoplasia, type 2B (MEN 2B). The inheritance pattern is autosomal dominant with decreased penetrance. This syndrome is the result of mutations in the receptor tyrosine kinase that maps to chromosome 10q11.2. Patients are tall with characteristic thick, large lips and often everted upper eyelids. The mucosal neuromas are prominent on the lips and anterior dorsal surface of the tongue (Figure 6-31). They generally appear in the first few years of life. In addition, neuromas can occur on the buccal mucosa and the eyelids. The mucosal neuromas are seen as multiple, mobile, firm lumps covered by normal mucosa. Histologically they are aggregates of nerve tissue.
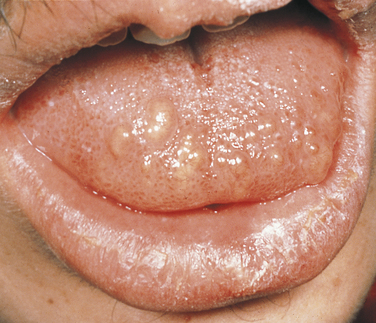
FIGURE 6-31 Patient with MEN 2B syndrome with multiple mucosal neuromas on the tip of the tongue and upper lip. (From Sedano HO, Sauk JJ, Gorlin RJ: Oral manifestations of inherited disorders, Boston, 1977, Butterworth. Used with permission.)
Medullary carcinoma of the thyroid has been diagnosed in more than 75% of patients with this syndrome; it generally develops in the second decade of life. Metastatic lesions develop frequently, and about 20% of patients die as a consequence of metastasis. The thyroid carcinoma produces calcitonin (a hormone normally produced by the C cells of the thyroid gland). The carcinoma can be detected by evaluating plasma levels of this hormone.
Pheochromocytoma is a benign neoplasm that generally develops in ganglia around the adrenal glands. The tumor is often bilateral and is responsible for night sweats, high blood pressure, and episodes of severe diarrhea. It generally develops in the second or third decade of life. The pheochromocytoma induces increased urinary levels of epinephrine and other substances. Other findings in this syndrome have included cutaneous pigmentation and a host of skeletal abnormalities.
Early diagnosis of this syndrome is imperative because of the high malignant potential of the thyroid carcinoma. Some authors recommend preventive thyroidectomy when this syndrome is diagnosed so that the development of carcinoma is avoided. The neuromas of the oral mucosa may be the earliest visible manifestation of this syndrome.
Neurofibromatosis of Von Recklinghausen: Neurofibromatosis of von Recklinghausen, also called von Recklinghausen disease, has an autosomal-dominant inheritance pattern and is probably a disorder of neural crest origin. The gene for this syndrome maps to the long arm of chromosome 17 region 11.2 (17q11.2). Several varieties exist; however, only the classic form is described here.
Multiple neurofibromas, which appear as papules and growths of various sizes, are seen on the facial skin, especially the eyelids. The tumors can arise anywhere, including the oral cavity, and can be present at birth or develop early in life, increasing in number and size at puberty. Malignant transformation of the neurofibromas occurs in an estimated 3% to 15% of patients with neurofibromatosis. Neurofibromas also develop in the central nervous system, eyes, ears, viscera, and intraosseous locations. Mental disability is observed occasionally, and multiple skeletal anomalies are common.
Oral involvement is seen in about 10% of patients and is characterized by single or multiple tumors at any location in the oral mucosa, the most frequent being the lateral borders of the tongue. Gingival neurofibromas can also occur in these patients (Figure 6-32), and intramandibular neurofibromas have been reported and present as radiolucencies in the mandible.
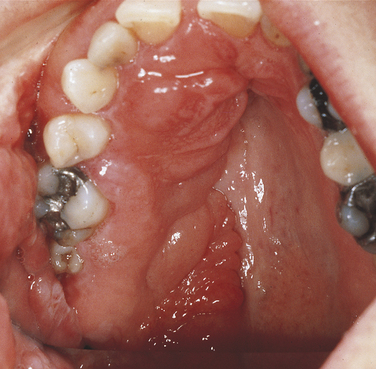
FIGURE 6-32 Multiple neurofibromas of the maxillary gingiva and palate in a patient with neurofibromatosis of von Recklinghausen.
Café au lait (the color of coffee with milk) pigmentation of skin is present from the first decade of life in 90% of patients with neurofibromatosis. This pigmentation is quite marked in the axilla and can also affect other areas of the skin. Café au lait skin pigmentation generally precedes the development of the neurofibromas.
Peutz-Jeghers Syndrome: Autosomal-dominant inheritance characterizes the Peutz-Jeghers syndrome. The gene for this syndrome maps to the short arm of chromosome 19 region 13.3 (19p13.3). It consists of multiple melanotic macular pigmentations of the skin and mucosa, which are associated with gastrointestinal polyposis. The pigmentations occur around the eyes, nose, and mouth (Figure 6-33). These macules are a few millimeters in diameter and vary in number and degree of pigmentation. They tend to diminish with age. Intraorally larger areas of pigmentation are observed on the lips and buccal mucosa of about 98% of affected patients.
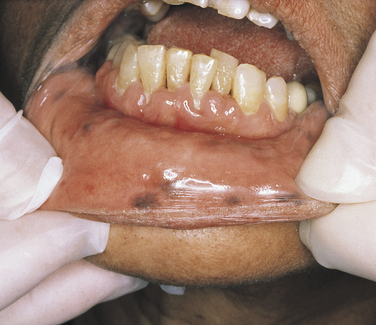
FIGURE 6-33 Multiple small- to medium-size pigmented macules on the labial mucosa of a patient with Peutz-Jeghers syndrome.
Pigmentation of the hands, nasal mucosa, and eyes can also occur. The intestinal polyps are hamartomas (an abnormal growth of normal tissue in its normal location). They develop mostly in the small intestine; only rarely do they undergo malignant transformation.
White Sponge Nevus: Autosomal-dominant inheritance with complete penetrance characterizes white sponge nevus (also called Cannon disease, or familial white folded mucosal dysplasia). The cause of white sponge nevus is a mutation in the mucosal keratin pair K4 or K13. The disorder can be present at birth or develop around puberty. Clinically it is characterized by a white, corrugated, soft, folding oral mucosa. The buccal mucosa is always affected, and in most patients the lesions are bilateral. A thick layer of keratin, which at times desquamates and leaves a raw mucosal surface, produces the whitening. Other areas of the oral mucosa can also be affected, but the free gingiva is spared.
Inherited Disorders Affecting the Teeth
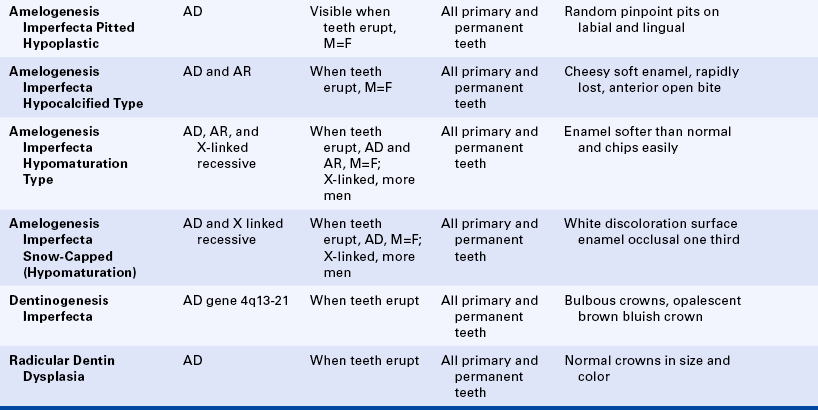
Amelogenesis Imperfecta: A group of inherited conditions affecting the enamel of teeth and having no associated systemic defects characterizes amelogenesis imperfecta. Witkop and Sauk classified amelogenesis imperfecta into four types:
Type I: Hypoplastic amelogenesis imperfecta
Type II: Hypocalcified amelogenesis imperfecta
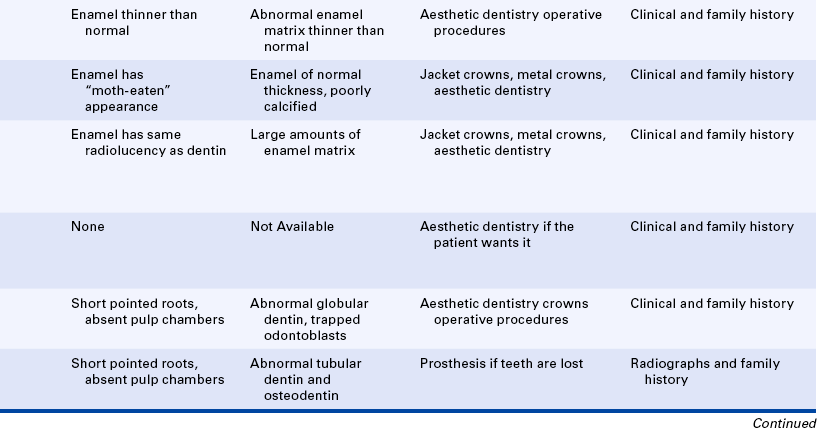
The hypoplastic type of amelogenesis imperfecta is characterized by tooth enamel that does not develop to a normal thickness because of failure of the ameloblasts to lay down enamel matrix properly. On radiographs the abnormal enamel contrasts normally with dentin. Several varieties of this type of amelogenesis imperfecta are further classified according to their clinical presentation: pitted, local, smooth, rough, and enamel agenesis. Combined with the clinical appearance, the classification also includes the inheritance pattern. Thus an autosomal-dominant and an autosomal-recessive variety exist for the local hypoplastic type. Among this group of enamel defects, the most frequent one is the pitted autosomal-dominant variety (Figure 6-34), which is characterized by teeth that have random pits on the enamel. The size of these pits varies from pinpoint to pinhead, and the pits are observed mostly on the labial and buccal surfaces of the permanent teeth. The pits are frequently arranged in rows or columns or both. Occasionally more than one tooth has a normal clinical appearance. The gene for the local hypoplastic autosomal-dominant type of amelogenesis imperfecta maps to the long arm of chromosome 4 (4q11-13).
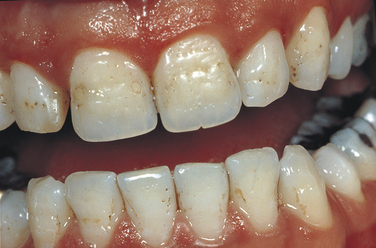
FIGURE 6-34 Pitted autosomal-dominant amelogenesis imperfecta. Multiple pits on the labial surface of the teeth should be noted. Some of the pits have been filled with composite. (From Young WG, Sedano HO: Atlas of oral pathology, Minneapolis, 1981, University of Minnesota Press. Used with permission.)
An enamel of normal thickness that is poorly calcified characterizes the hypocalcified type of amelogenesis imperfecta (Figure 6-35). Two varieties of hypocalcified amelogenesis imperfecta exist: (1) an autosomal-dominant variety, and (2) an autosomal-recessive variety. The autosomal-recessive pattern is more severe in its clinical manifestations. At eruption teeth present yellow-to-orange enamel that is very soft and rapidly lost, leaving exposed dentin. On radiographs the enamel has a moth-eaten appearance and is less radiopaque than dentin. Cervical enamel is better calcified and generally remains on the crown. This type of amelogenesis imperfecta is frequently associated with an anterior open bite.
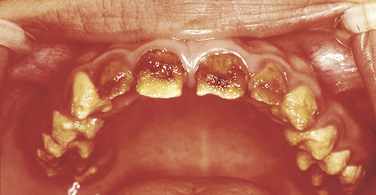
FIGURE 6-35 Loss of enamel is exhibited in the teeth of a patient with hypocalcified amelogenesis imperfecta.
An enamel of mottled appearance but normal thickness characterizes the hypomaturation type of amelogenesis imperfecta. This type of amelogenesis imperfecta is composed of large amounts of enamel matrix; therefore the enamel is softer than normal. With pressure the tip of an explorer will penetrate the enamel. The basic defect seems to be in the enamel rod sheath. The enamel chips easily from the crown. On radiographs it has almost the same radiodensity as dentin. Four varieties of the hypomaturation type of amelogenesis imperfecta exist; however, only the snowcapped type is discussed here.
A type of hypomaturation amelogenesis imperfecta, called snow-capped amelogenesis imperfecta, apparently has an X-linked recessive inheritance pattern in some families and an autosomal-dominant pattern in others. Clinically both varieties are identical and are characterized by a hypomaturation of the surface enamel of the occlusal third of all the teeth of both dentitions. The maxillary teeth are more severely affected with this whitish discoloration (Figure 6-36). The enamel in these areas is of regular hardness and smooth. It does not fracture or chip from the crown.
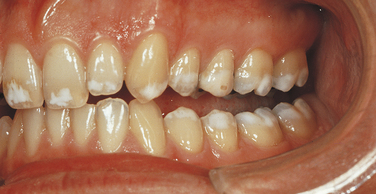
FIGURE 6-36 Uniform whitening of incisal edges and occlusal cusps is exhibited in a case of snow-capped amelogenesis imperfecta.
Its association with taurodontic teeth (described later in this chapter and also in Chapter 5) characterizes the hypoplastic-hypomaturation type of amelogenesis imperfecta. The thin enamel is yellow to brown and pitted. On radiographs the enamel has a radiodensity similar to dentin, and single-rooted teeth have large pulp chambers.
It is generally not easy to diagnose the exact type of amelogenesis imperfecta clinically because of the frequent similarity among different varieties. The mode of inheritance must be kept in mind, and an accurate family history should always be taken. Recently different mutations in several genes have been identified as responsible for some types of amelogenesis imperfecta; the genes are: AMELX, AMELY, ENAM, AMELOBLASTIN, TUFTELIN, ENAMELYSIN, KALLIKREIN, and DLX3.
The prevalence of all forms of amelogenesis imperfecta in the United States has been estimated at 1:15000.
Dentinogenesis Imperfecta: Dentinogenesis imperfecta is usually subdivided into three types. Type one is dentinogenesis imperfecta associated with osteogenesis imperfecta (previously described in this chapter). The other two types are not associated with osteogenesis imperfecta. The teeth in all three types have a similar clinical appearance.
Dentinogenesis imperfecta type II is also known as hereditary opalescent dentin. The inheritance pattern is autosomal dominant, and the gene maps to the long arm of chromosome 4 (4q13-21). Teeth have bulbous crowns with a color that varies from opalescent brown to brownish-blue (Figure 6-37). The primary teeth are usually affected more severely than the permanent teeth. Twenty percent of patients have enamel hypoplasia as well. The dentin is very soft, which produces chipping of enamel that results in tooth attrition. Occasionally this attrition can cause the teeth to be worn down to the alveolar process. Radiographically no pulp chambers or root canals are seen (Figure 6-38). Roots are short and thin with periapical radiolucencies. Patients may lose teeth prematurely because of complications produced by the attrition and the short roots. The basic defect lies with the odontoblasts, which lay down an abnormal matrix and then degenerate. Cells derived from the dental pulp, which lay down abnormal dentin, later replace these odontoblasts.
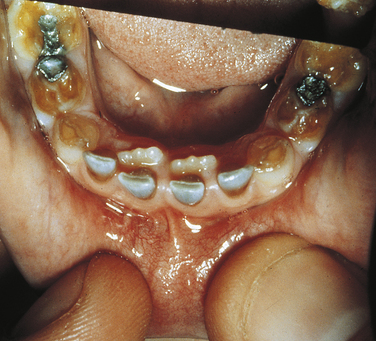
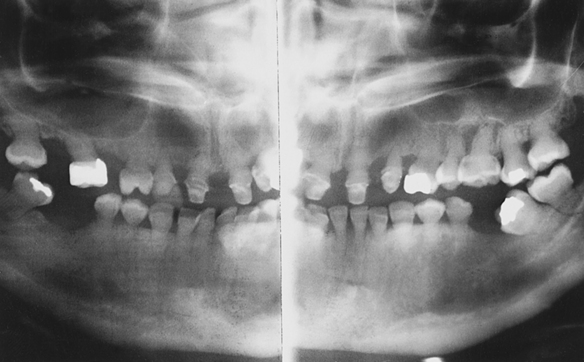
Dentin Dysplasia: Dentin dysplasia is subdivided into type I (radicular dentin dysplasia) and type II (coronal dentin dysplasia).
Radicular Dentin Dysplasia: Teeth with normal crowns, abnormal roots, and an autosomal-dominant inheritance pattern characterize this condition. The basic defect seems to lie in a disturbance in Hertwig’s epithelial root sheath, which guides the formation of the root. Radiographs show total or partial lack of pulp chambers and root canals (Figure 6-39). Primary and secondary dentitions are affected equally. The color of the teeth is normal. Because of the short roots, the teeth generally are exfoliated prematurely, especially in the event of even minor trauma. The pulp chambers of the permanent teeth generally are not obliterated fully and have a half-moon appearance on the radiograph. Occasionally periapical cysts can be associated with this condition.
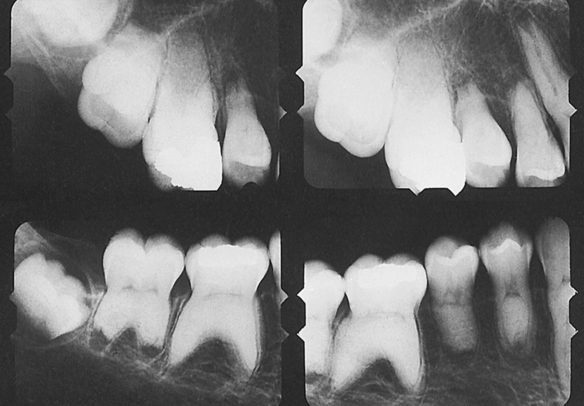
Coronal Dentin Dysplasia: Dentin dysplasia type II has an autosomal-dominant inheritance pattern. The basic defect in this condition is a mutation in the gene termed dentin sialophosphoprotein (DSPP) that maps to the long arm of chromosome 4 (4q13-21). Recently it has been shown that dentinogenesis imperfecta type II and dentin dysplasia type II share the DSPP gene loci and the proteins encoded by that gene. Translucent teeth with an amber color characterize the primary dentition. Radiographs show a lack of pulp chambers and small root canals. Primary teeth are quite similar to those observed in dentinogenesis imperfecta. Permanent teeth present normal crown formation with normal color. Radiographs show thistle-shaped pulp chambers in single-rooted teeth and a bow-tie appearance of the pulp chambers of permanent molars (Figure 6-40). Permanent teeth may or may not have pulp stones.
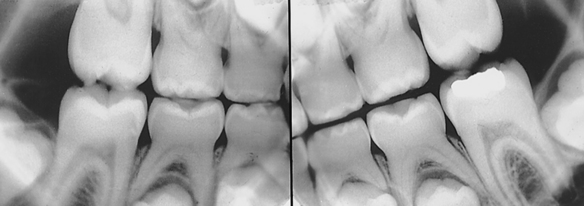
Hypohidrotic Ectodermal Dysplasia: Hypohidrotic ectodermal dysplasia represents a genetic heterogeneity. Although in the majority of families it is inherited as an X-linked recessive trait, in some families it is inherited as an autosomal-recessive trait. The clinical manifestations for both the X-linked and autosomal-recessive forms are identical. The gene for the X-linked variety of this syndrome maps to the long arm of the X chromosome region 12-13.1 (Xq12-13.1). The autosomal-recessive variety maps to the long arm of chromosome 2 (2q11-q13).
This entity represents the most severe form of ectodermal dysplasia. Its major components are hypodontia (partial anodontia), hypotrichosis (less than the normal amount of hair), and hypohidrosis (abnormally diminished secretion of sweat). Affected children are born without lanugo (body hair present at birth), and many have episodes of fevers of unknown causes because of the almost complete lack of sweat glands. Some patients die of hyperthermia (greatly increased body temperature) after prolonged exposure to the sun or heavy exercise. The clinical characteristics may not be apparent until the second year of life.
The facies is quite typical, with marked frontal bossing, depressed nasal bridge (saddle nose), protuberant lips, and almost complete lack of scalp hair. The hair that is present is usually blond, short, fine, and stiff. The skin is soft, thin, and very dry. Sebaceous glands are also lacking. Linear wrinkles and increased pigmentation are seen around the eyes and mouth. The eyelashes and eyebrows are often missing entirely. After puberty the beard generally is normal, but axillary and pubic hair is scanty.
The oral manifestations of hypohidrotic ectodermal dysplasia consist of hypodontia or rarely anodontia. When present, incisors and canines have small, conical crowns (Figure 6-41). The alveolar bone is formed only when teeth are present; therefore patients lacking alveolar processes have loss of vertical dimension with markedly protruding lips. There may also be partial lack and aplasia of minor salivary glands in the buccal, labial, and lower respiratory tract mucosa.
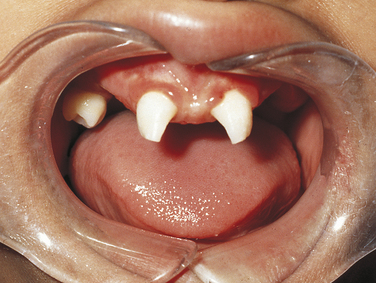
Female carriers of the X-linked form of hypohidrotic ectodermal dysplasia have minor clinical manifestations such as thin and slightly sparse hair, cone-shaped teeth, hypodontia, and variable degrees of reduced sweating. This is in accordance with the Lyon hypothesis of variability of expression.
Hypophosphatasia: The inheritance pattern of hypophosphatasia is autosomal recessive. The gene for this syndrome maps to the short arm of chromosome 1 regions 36.1-34 (1p36.1-34). The basic defect in this condition is a decrease in serum alkaline phosphatase levels with increased urinary and plasma levels of phosphoethanolamine. Alkaline phosphatase participates in the process of calcification of bone and cementum; therefore these two tissues will be altered in patients with hypophosphatasia. Agenesis or abnormal formation of cementum in these patients leads to spontaneous premature shedding of primary teeth, especially mandibular incisors (Figure 6-42). Teeth are exfoliated without evidence of periodontal or gingival disease. The primary molars and permanent teeth are rarely if ever affected, which is probably associated with the greater mechanical fixation resulting from the larger size of their roots. The total lack of cementum observed in exfoliated teeth implies a lack of periodontal fiber attachment, with consequent exfoliation of single-rooted teeth. The most important alteration in this syndrome is the improper formation of mature bone. Therefore individuals who survive the neonatal period experience rachitic-like changes such as bowing of legs and multiple fractures.
Hypophosphatemic Vitamin D–Resistant Rickets: Hypophosphatemic vitamin D–resistant rickets, which is quite common, has an X-linked dominant inheritance pattern and is the result of a mutation in the PHEX gene. It consists of low serum levels of phosphorus, which are produced by low absorption of inorganic phosphate in the renal tubules, rickets or osteomalacia, resistance to treatment with usual doses of vitamin D, and a lack of other abnormalities. Affected individuals generally are of short stature and have bowlegs, especially if the condition is present from childhood. Adult-onset forms also exist, and the clinical manifestations in these patients are minor or even lacking, with the exception of low serum levels of inorganic phosphate.
The characteristic radiographic oral findings are large pulp chambers with very long pulp horns. In addition, the dentin exhibits pronounced cracks that extend to the dentinoenamel junction. These cracks induce fracture of the enamel with microexposure of the pulp and subsequent pulpal infection. Eventually, with the progress of the infectious inflammatory pulpal disease, periapical abscess formation occurs. Gingival abscesses are also frequent. Regional lymphadenitis accompanies these processes.
Pegged Or Absent Maxillary Lateral Incisors: The inheritance pattern of pegged or absent maxillary lateral incisors is generally autosomal dominant with variable expressivity. The lateral incisor can be small, peg shaped, or congenitally lacking, either unilaterally or bilaterally (Figure 6-43). Both primary and secondary dentitions can be affected, but mostly the latter. This condition has a prevalence of 1% to 3% in the white population and about 7% in Asians. In addition, the premolar teeth are congenitally lacking in 10% to 20% of affected individuals.
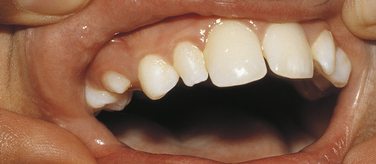
Taurodontism: Taurodontism is a genetic heterogeneous condition with dominant and recessive inheritance. It is characterized by very large, pyramid-shaped molars with large pulp chambers (Figure 6-44). (Taurodontic teeth [bull teeth] are also described in Chapter 5.) Taurodontism is most frequent among Native Americans, including Inuits. The furcation of the roots is displaced apically, and these teeth are classified according to the degree of furcation displacement. It is frequently found in Klinefelter syndrome (XXY) and is associated with many other syndromes as well.
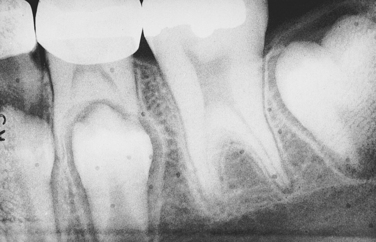
Cummings, M.R. Human heredity: principles and issues, ed 7. Stamford, Conn: Brooks/Cole-Cengage Learning; 2006.
Fitzgerald, M.J.T., Fitzgerald, M. Human embryology. London: Saunders; 1994.
Gorlin, R.J., Cohen, M.M., Hennekam, R.C.M. Syndromes of the head and neck, ed 4. New York: Oxford University Press; 2001.
Neussbaum, R.L., McInnes, R.R., Willard, H.F. Thompson and Thompson genetics in medicine, ed 7. Philadelphia: Saunders; 2007.
Sadler, T.W. Langman’s medical embryology, ed 10. Philadelphia: Lippincott, Williams & Wilkins, 2006.
Sapp, J.P., Eversole, L.R., Wisocki, G.P. Contemporary oral and maxillofacial pathology, ed 2. St Louis: Mosby; 2004.
Witkop, C.J., Jr., Sauk, J.J., Jr. Dental and oral manifestations of hereditary disease. Washington, DC: American Academy of Oral Pathology (monograph); 1971.
Witkop, C.J., Jr., Sauk, J.J., Jr. Heritable defects of enamel. In: Stewart R.E., Prescott G.H., eds. Oral facial genetics. St Louis: Mosby, 1976.
Young, W.G., Sedano, H.O. An atlas of oral pathology. Minneapolis: University of Minnesota Press; 1981.
Barnet, M.L., Friedman, D., Kastner, T. The prevalence of mitral valve prolapse in patients with Down’s syndrome: implications for dental management. Oral Surg Oral Med Oral Pathol. 1988;66:445.
Bozzo, L., Scully, C., Aldred, M.J. Hereditary gingival fibromatosis. Oral Surg Oral Med Oral Pathol. 1994;78:452.
Chadwick, B., et al. Laband syndrome: report of two cases, review of the literature, and identification of additional manifestations. Oral Surg Oral Med Oral Pathol. 1994;78:57.
Crawford, P.J.M., Aldred, M.J. Clinical features of a family with X-linked amelogenesis imperfecta mapping to a new locus (AIH3) on the long arm of the X chromosome. Oral Surg Oral Med Oral Pathol. 1993;76:187.
Ghaffar, K.A., et al. Papillon-Lefèvre syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;88:320.
Koury, M.E., Stella, J.P., Epker, B.N. Vascular transformation in cherubism. Oral Surg Oral Med Oral Pathol. 1993;76:20.
O’Connell, A.C., Marini, J.C. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:189.
Szilágyi, A., et al. Oral manifestations of patients with Turner syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:577.
Yuasa, K., et al. Computed tomography of the jaws in familial adenomatosis coli. Oral Surg Oral Med Oral Pathol. 1993;76:251.
REVIEW QUESTIONS
1. The constriction present in all chromosomes, which joins the short and long arms, is called the:
2. The process by which a primitive germ cell becomes a gamete is called:
B A pair of chromosomes with an identical extra chromosome.
4. The Lyon hypothesis is applied to:
5. Barr bodies are seen at the:
A Periphery of the cytoplasm in all human cells.
B Nuclear periphery of cells in women.
6. The karyotype of a patient with Turner syndrome shows:
7. Hypothetically an autosomal-dominant trait would be clinically present in:
A 25% of the offspring of an affected parent.
B 50% of the offspring of an affected parent.
8. Patients with an X-linked hereditary condition:
9. The major concern for a dental hygienist when treating a patient with cyclic neutropenia should be:
10. A 9-year-old boy exhibits markedly swollen red and bleeding gingiva. In addition, he has tooth mobility, and the intraoral radiographs show marked alveolar bone atrophy with vertical periodontal pockets. Which of the following will be found in this child if he were to have the Papillon-Lefèvre syndrome?
11. In all inherited varieties of gingival fibromatosis, the gingival enlargement is characterized by a marked:
A Collagenization of the connective tissue.
B Hyperplasia of the covering epithelium.
12. A 14-year-old boy is seen in consultation because of bilateral mandibular swelling. Radiographs show a bilateral multilocular lesion in the ascending mandibular rami. The mother of this patient has similar findings. The most likely diagnosis is:
13. A 19-year-old woman is diagnosed with cleidocranial dysplasia. She has absent clavicles and a mushroom-shaped skull. Which of the following conditions is she also most likely to have?
14. Which of the following is the most serious component of Gardner syndrome?
15. Two characteristic clinical components of mandibulofacial dysostosis are:
A Lack of clavicles and delayed teeth eruption.
B Hypoplastic mandible and deafness.
16. Odontogenic keratocysts are a clinical component of:
17. Torus mandibularis and torus palatinus are similar in that both:
A Are inherited as an autosomal-dominant trait.
B Are more prevalent in females.
18. The cause of all forms of labial and palatal clefting is considered to be:
19. The major concern for a dental hygienist when treating a patient with Osler-Rendu–Parkes Weber syndrome should be:
20. The most serious clinical manifestation of the MEN 2B syndrome is considered to be:
21. Which of the following is true for von Recklinghausen disease?
A It is inherited as an autosomal-recessive trait.
B Patients have multiple neurofibromas.
C Patients have gingival fibromatosis.
D Patients experience a generalized whitening of the oral mucosa.
22. Teeth in snowcapped amelogenesis imperfecta have:
23. In dentinogenesis imperfecta type II, teeth have:
24. The characteristic finding in permanent teeth affected with coronal dentin dysplasia is:
25. Patients with hypohidrotic ectodermal dysplasia characteristically have:
26. Patients with hypophosphatasia characteristically have: