GENES, ENVIRONMENT, AND COMMON DISEASES



Chapter 4 focuses on diseases that are caused by single genes or by abnormalities of single chromosomes. Much progress has been made in identifying specific mutations that cause these diseases, leading to better risk estimates and, in some cases, more effective treatment of the disease. However, these conditions form only a small portion of the total burden of human genetic disease. Most congenital malformations are not caused by single genes or chromosome defects. Many common adult diseases, such as cancer, heart disease, and diabetes, have genetic components, but again they are usually not caused by single genes or by chromosomal abnormalities.1 These diseases, whose treatment collectively occupies the attention of most health care practitioners, are the result of a complex interplay of multiple genetic and environmental∗ factors.
FACTORS INFLUENCING INCIDENCE OF DISEASE IN POPULATIONS
Concepts of Incidence and Prevalence
How common is a given disease, such as diabetes, in a population? Well-established measures are used to answer this question.2 The incidence rate is the number of new cases of a disease reported during a specific period (typically 1 year) divided by the number of individuals in the population. The denominator is often expressed as person-years. The incidence rate can be contrasted with the prevalence rate, which is the proportion of the population affected by a disease at a specific point in time. Prevalence is thus determined by both the incidence rate and the length of the survival period in affected individuals. For example, the prevalence rate of acquired immunodeficiency syndrome (AIDS) is larger than the yearly incidence rate because most people with AIDS survive for several years after diagnosis.
Many diseases vary in prevalence from one population to another. Cystic fibrosis is relatively common among Europeans, occurring about once in every 2500 births. In contrast, it is quite rare in Asians, occurring only once in every 90,000 births. Similarly, sickle cell disease affects approximately 1 in 600 American blacks, but it is rarely seen in whites. Both of these diseases are single-gene disorders, and they vary among populations because disease-causing mutations are more or less common in different populations. (This is in turn the result of differences in the evolutionary history of these populations.) Nongenetic (environmental) factors have little influence on the current prevalence of these diseases.
The picture often becomes more complex with the common diseases of adulthood. For example, colon cancer was until recently relatively rare in Japan, but it is the second most common cancer in the United States. Stomach cancer, on the other hand, is common in Japan but relatively rare in the United States. These statistics, in themselves, cannot distinguish environmental from genetic influences in the two populations. However, because large numbers of Japanese emigrated first to Hawaii and then to the U.S. mainland, we can observe what happens to the rates of stomach and colon cancer among the migrants. It is important that the Japanese émigrés have maintained a genetic identity, marrying largely among themselves. Among first-generation Japanese in Hawaii, the frequency of colon cancer rose several-fold—not yet as high as in the U.S. mainland but higher than in Japan. Among second-generation Japanese on the U.S. mainland, colon cancer rates rose to 5%, equal to the U.S. average. At the same time, stomach cancer has become relatively rare among Japanese-Americans.
These observations strongly indicate an important role for environmental factors in the etiology of cancers of the colon and stomach. In each case, diet is a likely culprit—a high-fat, low-fiber diet in the United States is thought to increase the risk of colon cancer, whereas techniques used to preserve and season the fish commonly eaten in Japan are thought to increase the risk of stomach cancer. It is interesting that the incidence of colon cancer in Japan has increased dramatically during the past several decades as the Japanese population has adopted a more “Western” diet. These results do not, however, rule out the potential contribution of genetic factors in common cancers. Genes also play a role in the etiology of colon and other cancers.
Analysis of Risk Factors
The comparison just discussed is one example of the analysis of risk factors (in this case, diet) and their influence on the prevalence of disease in populations. A common measure of the effect of a specific risk factor is the relative risk. This quantity is expressed as a ratio:
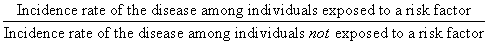
A classic example of a relative risk analysis was carried out in a sample of more than 40,000 British physicians to determine the relationship between cigarette smoking and lung cancer. This study compared the incidence of death from lung cancer in physicians who smoked with those who did not. The incidence of death from lung cancer was 1.66 (per 1000 person-years) in heavy smokers (more than 25 cigarettes daily), but it was only 0.07 in the nonsmokers. The ratio of these two incidence rates is 1.66/0.07, which yields a relative risk of 23.7 deaths. We can thus conclude that the risk of dying from lung cancer increased by about 24-fold in heavy smokers compared with nonsmokers. Many other studies have obtained similar risk figures.
Although cigarette smoking clearly increases one’s risk of developing lung cancer (as well as heart disease, as we will see later), it is equally clear that most smokers do not develop lung cancer. Other lifestyle factors are likely to contribute to one’s risk of developing this disease (e.g., exposure to cancer-causing substances in the air, such as asbestos fibers). In addition, differences in genetic background may be involved. Some studies have suggested that mutations in a gene called FHIT may make some individuals more sensitive to the carcinogenic effects of tobacco smoke.
Many factors can influence the risk of acquiring a common disease such as cancer, diabetes, or high blood pressure. These include age, gender, diet, exercise, and family history of the disease. Usually, complex interactions occur among these genetic and nongenetic factors. The effects of each factor can be quantified in terms of relative risks. The following discussion demonstrates how genetic and environmental factors contribute to the risk of developing common diseases.
PRINCIPLES OF MULTIFACTORIAL INHERITANCE
Traits in which variation is thought to be caused by the combined effects of multiple genes are polygenic (“many genes”). When environmental factors are also believed to cause variation in the trait, which is usually the case, the term multifactorial trait is used.3 Many quantitative traits (those, such as blood pressure, that are measured on a continuous numeric scale) are multifactorial. Because they are caused by the additive effects of many genetic and environmental factors, these traits tend to follow a normal, or bell-shaped, distribution in populations.
An example illustrates this concept. To begin with the simplest case, suppose (unrealistically) that height is determined by a single gene with two alleles, A and a. Allele A tends to make people tall, whereas allele a tends to make them short. If there is no dominance at this locus, then the three possible genotypes (AA, Aa, aa) will produce three phenotypes: tall, intermediate, and short. Assume that the gene frequencies of A and a are each 0.50. If we look at a population of individuals, we will observe the height distribution depicted in Figure 5-1, A.
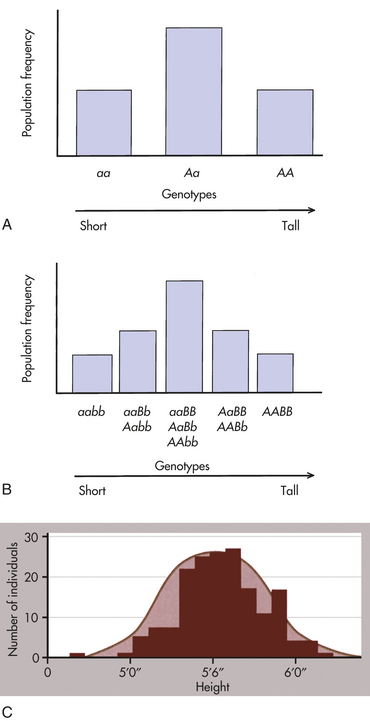
Figure 5-1 Distribution of height. A, Distribution of height in a population, assuming that height is controlled by a single locus with genotypes AA, Aa, and aa. B, Distribution of height, assuming that height is controlled by two loci. Five distinct genotypes are shown instead of three, and the distribution begins to look more like the normal distribution. C, Height is portrayed, realistically, as a trait with a continuous statistical distribution. Because many genes contribute height and tend to segregate independently of one another, the cumulative contribution of different combinations of alleles to height forms a continuous distribution of possible heights, in which the extremes are much rarer than the intermediate values. Variation also can be due to environmental factors such as nutrition. (A and B from Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby; C from Raven PH et al: Biology, ed 8, New York, 2008, McGraw-Hill.)
Now suppose, a bit more realistically, that height is determined by two loci instead of one. The second locus also has two alleles, B (tall) and b (short), and they affect height in exactly the same way as alleles A and a. There are now nine possible genotypes in our population: aabb, aaBb, aaBB, Aabb, AaBb, AaBB, AAbb, AABb, and AABB. An individual may have zero, one, two, three, or four “tall” alleles, so now five distinct phenotypes are possible (Figure 5-1, B). Although the height distribution in our fictional population is still not normal compared with an actual population, it approaches a normal distribution more closely than in the single-gene case just described.
We now extend our example so that many genes and environmental factors influence height, each having a small effect. Then many phenotypes are possible, each differing slightly from the others, and the height distribution of the population approaches the bell-shaped curve shown in Figure 5-1, C.
It should be emphasized that the individual genes underlying a multifactorial trait such as height follow the mendelian principles of segregation and independent assortment, just like any other gene. The only difference is that many of them act together to influence the trait.
Blood pressure is another example of a multifactorial trait. A correlation exists between parents’ blood pressures (systolic and diastolic) and those of their children. The evidence is good that this correlation is partially caused by genes, but blood pressure is also influenced by environmental factors, such as diet, exercise, and stress. Two goals of genetic research are the identification and measurement of the relative roles of genes and environment in the causation of multifactorial diseases.
Threshold Model
A number of diseases do not follow the bell-shaped distribution. Instead, they appear to be either present or absent in individuals, yet they do not follow the inheritance patterns expected of single-gene diseases. A commonly used explanation for such diseases is that there is an underlying liability distribution for the disease in a population (Figure 5-2). Those individuals who are on the “low” end of the distribution have little chance of developing the disease in question (i.e., they have few of the alleles or environmental factors that would cause the disease). Individuals who are closer to the “high” end of the distribution have more of the disease-causing genes and environmental factors and are more likely to develop the disease. For diseases that are either present or absent, it is thought that a threshold of liability must be crossed before the disease is expressed. Below the threshold, an individual appears normal; above it, he or she is affected by the disease.
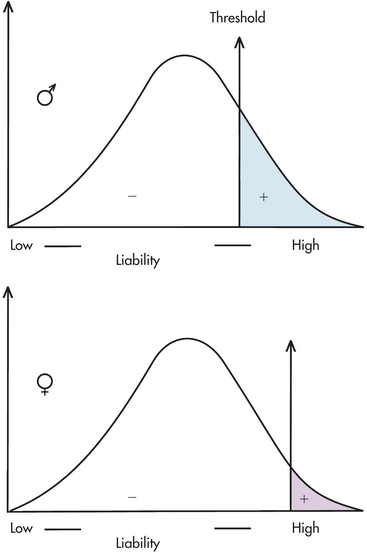
Figure 5-2 A liability distribution in a population for a multifactorial disease. To be affected with the disease, an individual must exceed the threshold on the liability distribution. This figure shows two thresholds, a lower one for males and a higher one for females (as in pyloric stenosis; see text). (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
A disease that is thought to correspond to this threshold model is pyloric stenosis, a disorder that presents shortly after birth and is caused by a narrowing or obstruction of the pylorus, the area between the stomach and intestine. Chronic vomiting, constipation, weight loss, and electrolyte imbalance result from the condition, but it sometimes resolves spontaneously or can be corrected by surgery. The prevalence of pyloric stenosis is about 3 per 1000 live births in whites. It is much more common in males than females, affecting 1 of 200 males and 1 of 1000 females. It is thought that this difference in prevalence reflects two thresholds in the liability distribution—a lower one in males and a higher one in females (see Figure 5-2). A lower male threshold implies that fewer disease-causing factors are required to generate the disorder in males.
The liability threshold concept may explain the pattern of recurrence risks for pyloric stenosis seen in Table 5-1. Note that males, having a lower threshold, always have a higher risk than females. However, the sibling risk also depends on the gender of the proband (i.e., the individual from which the pedigree begins). It is higher when the proband is female than when the proband is male. This reflects the concept that females, having a higher liability threshold, must be exposed to more disease-causing factors than males to develop the disease. Thus a family with an affected female must have more genetic and environmental risk factors, producing a higher recurrence risk for pyloric stenosis in future offspring. It would be expected that the highest risk category would be male relatives of female probands; Table 5-1 shows that this is the case.
Table 5-1
Recurrence Risks (%) for Pyloric Stenosis, Subdivided by Genders of Affected Probands and Relatives∗

∗Note that the risks differ somewhat between the two populations.
Data from Carter CO: Br Med Bull 32(1):21-26, 1976.
A similar pattern has been observed in a study of infantile autism, a behavioral disorder in which the male/female ratio is approximately 4:1. As expected for a multifactorial disorder, the recurrence risks for siblings of male probands (3.5%) is substantially lower than that of siblings of female probands (7%). When the sex ratio for a disease is reversed (i.e., more affected females than males), one would expect a higher recurrence risk when the proband is male.
A number of other congenital malformations are thought to correspond to this model. They include isolated cleft lip and/or cleft palate (CL/P), neural tube defects (anencephaly, spina bifida), clubfoot (talipes), and some forms of congenital heart disease. In this context, isolated means that this is the only observed disease feature (i.e., the feature is not part of a larger constellation of findings, as in CL/P secondary to trisomy 13). In addition, many common adult diseases, such as hypertension, coronary heart disease, stroke, diabetes mellitus (types 1 and 2), and some cancers, are caused by complex genetic and environmental factors and can thus be considered multifactorial diseases.
Recurrence Risks and Transmission Patterns
Whereas recurrence risks can be given with confidence for single-gene diseases (e.g., 50% for typical autosomal dominant diseases, 25% for autosomal recessive diseases), the situation is more complicated for multifactorial diseases. This is because the number of genes contributing to the disease is usually not known, the precise allelic constitution of the parents is not known, and the extent of environmental effects can vary substantially. For most multifactorial diseases, empirical risks (i.e., risks based on direct observation of data) have been derived. To estimate empirical risks, a large series of families is examined in which one child has developed the disease (the proband). Then the siblings of each proband are surveyed to calculate the percentage who also have developed the disease. For example, in the United States about 3% of siblings of individuals with neural tube defects also have neural tube defects (Box 5-1). Thus the recurrence risk for parents who have had one child with a neural tube defect is 3% in the United States. For conditions such as CL/P that are not lethal or severely debilitating, recurrence risks also can be estimated for the offspring of affected parents. Empirical recurrence risks are, of course, specific for each multifactorial disease.
Neural tube defects (NTDs), which include anencephaly, spina bifida, and encephalocele (as well as several other less common forms), are one of the most important classes of birth defects, with a birth prevalence of 1 to 3 per 1000.4 The prevalence of NTDs among different populations varies considerably, with an especially high rate among some northern Chinese populations (as high as 6 or more per 1000 births). For reasons that are not fully known, the prevalence of NTDs has been decreasing in many parts of the United States and Europe during the past 2½ decades.
Normally the neural tube closes at about the fourth week of gestation. A defect in closure, or a subsequent reopening of the neural tube, results in a neural tube defect. Spina bifida (Figure 5-3, A) is the most commonly observed NTD and consists of a protrusion of spinal tissue through the vertebral column (the tissue usually includes meninges, spinal cord, and nerve roots). About 75% of individuals with spina bifida have secondary hydrocephalus, which sometimes in turn produces mental retardation. Paralysis or muscle weakness, lack of sphincter control, and clubfeet are often observed. A study conducted in British Columbia showed that survival rates for people with spina bifida have improved dramatically over the past several decades. Less than 30% of people born between 1952 and 1969 survived to 10 years of age, whereas 65% of those born between 1970 and 1986 survived to this age. Anencephaly (see Figure 5-3, B) is characterized by partial or complete absence of the cranial vault and calvarium and partial or complete absence of the cerebral hemispheres. At least two thirds of newborns with anencephaly are stillborn; term deliveries do not survive more than a few hours or days.
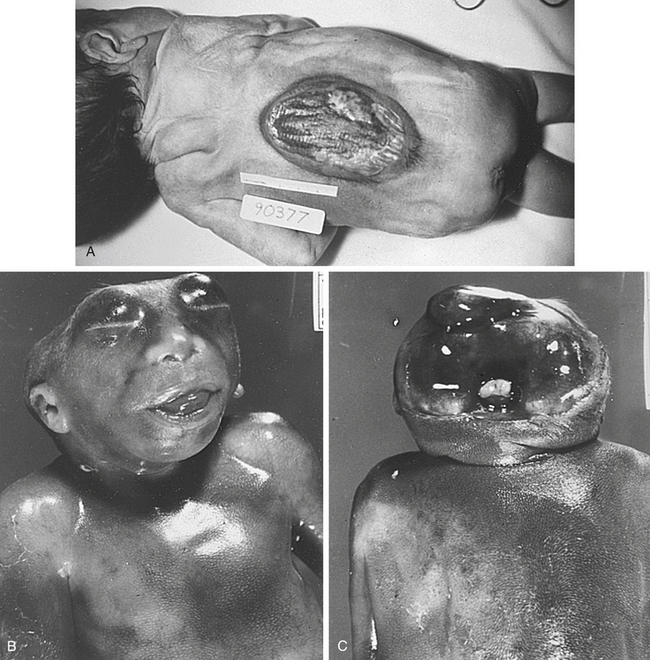
Figure 5-3 Spina bifida and anencephaly. A, Spina bifida in a newborn. B and C, Anencephaly, showing the absence of the cranial vault. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
NTDs are thought to arise from a combination of genetic environmental factors. In most populations surveyed thus far, empirical recurrence risks for siblings of affected people range from 2% to 5%. Consistent with a multifactorial model, the recurrence risk increases with additional affected siblings. Studies conducted in Great Britain showed that the sibling recurrence risk was approximately 5% when one sibling was affected and 10% when two were affected. A Hungarian study showed that the overall prevalence of NTDs was 1 in 300 births and that the sibling recurrence risks were 3%, 12%, and 25% after one, two, and three affected offspring, respectively. Recurrence risks tend to be slightly lower in populations with lower NTD prevalence rates, as predicted by the multifactorial model. Recurrence risk data support the idea that the major forms of NTDs are caused by similar factors. An anencephalic conception increases the recurrence risk for subsequent spina bifida conceptions, and vice versa.
NTDs can usually be diagnosed prenatally, sometimes by ultrasound and usually by an elevation in alpha fetoprotein (AFP) in the maternal serum or amniotic fluid (see Chapter 19). A spina bifida lesion can be either open or closed (i.e., covered with a layer of skin). Fetuses with open spina bifida are more likely to be detected by AFP assays.
A major epidemiologic finding is that mothers who supplement their diet with folic acid at the time of conception are less likely to produce children with NTDs. This result has been replicated in several different populations and thus appears to be well confirmed. It has been estimated that as many as 50% to 70% of NTDs can be avoided simply by dietary folic acid supplementation.5 (Traditional prenatal vitamin supplements have little effect because administration does not usually begin until well after the time that the neural tube closes.) Because mothers would be likely to ingest similar amounts of folic acid from one pregnancy to the next, folic acid deficiency could well account for at least part of the elevated sibling recurrence risk for NTDs. This is an important example of a nongenetic factor that contributes to familial clustering of a disease.
In contrast to most single-gene diseases, recurrence risks for multifactorial diseases can change substantially from one population to another because gene frequencies as well as environmental factors can differ among populations (note the differences between the London and Belfast populations in Table 5-1).
It is sometimes difficult to distinguish polygenic or multifactorial diseases from single-gene diseases that have reduced penetrance or variable expression. Large data sets and good epidemiologic data are necessary to make the distinction. Several criteria are commonly used to define multifactorial inheritance.
First, the recurrence risk becomes higher if more than one family member is affected. For example, the sibling recurrence risk for a ventricular septal defect (VSD), a type of congenital heart defect) is 3% if one sibling has had a VSD but increases to approximately 10% if two siblings have had VSDs.6 In contrast, the recurrence risk for single-gene diseases remains the same regardless of the number of affected siblings. It should be emphasized that this increase does not mean that the family’s risk has actually changed. Rather, it means that we now have more information about the family’s true risk: because they have had two affected children, they are probably located higher on the liability distribution than a family with only one affected child. In other words, they have more risk factors (genetic or environmental) and are more likely to produce an affected child.
Second, if the expression of the disease in the proband is more severe, the recurrence risk is higher. This is again consistent with the liability model because a more severe expression indicates that the affected individual is at the extreme tail end of the liability distribution (see Figure 5-2). His or her relatives are thus at a higher risk for inheriting disease genes. For example, the occurrence of a bilateral (both sides) CL/P confers a higher recurrence risk on family members than does the occurrence of a unilateral (one side) cleft.
Third, the recurrence risk is higher if the proband is of the less commonly affected sex (see the preceding discussion of pyloric stenosis). This is because an affected individual of the less susceptible gender is usually at a more extreme position on the liability distribution.
Fourth, the recurrence risk for the disease usually decreases rapidly in more remotely related relatives (Table 5-2). Whereas the recurrence risk for single-gene diseases decreases by 50% with each degree of relationship (e.g., an autosomal dominant disease has a 50% recurrence risk for siblings, 25% for uncle-nephew relationships, 12.5% for first cousins), it decreases much more quickly for multifactorial diseases. This reflects the fact that many genes and environmental factors must combine to produce a trait. All the necessary risk factors are unlikely to be present in less closely related family members.
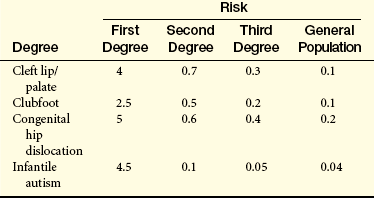
Finally, if the prevalence of the disease in a population is f, the risk for offspring and siblings of probands is approximately 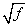 . This does not hold true for single-gene traits because their recurrence risks are independent of population prevalence. It is not an absolute rule for multifactorial traits either, but many such diseases tend to conform to this prediction. Examination of the risks given in Table 5-2 shows that the first three diseases follow the prediction fairly well. However, the observed sibling risk for the fourth disease, infantile autism, is substantially higher than predicted by .
. This does not hold true for single-gene traits because their recurrence risks are independent of population prevalence. It is not an absolute rule for multifactorial traits either, but many such diseases tend to conform to this prediction. Examination of the risks given in Table 5-2 shows that the first three diseases follow the prediction fairly well. However, the observed sibling risk for the fourth disease, infantile autism, is substantially higher than predicted by .
NATURE AND NURTURE: DISENTANGLING THE EFFECTS OF GENES AND ENVIRONMENT
Family members share genes and a common environment. Family resemblance in traits such as blood pressure reflects both genes and environment (“nature” and “nurture,” respectively). For centuries people have debated the relative importance of these two types of factors. It is a mistake, of course, to view them as mutually exclusive. Few traits are influenced only by genes or only by environmental factors. Most are influenced by both. It is useful to try to determine the relative influence of genetic and environmental factors (Figure 5-4). This can lead to a better understanding of disease etiology. It can also help in planning public health strategies. A disease in which the genetic influence is relatively small, such as lung cancer, may be prevented most effectively through emphasis on lifestyle changes (avoidance of tobacco). When a disease has a relatively larger genetic component, as in breast cancer, examination of family history should be emphasized in addition to lifestyle modification.

Figure 5-4 Continuum of genetic diseases. Some diseases (e.g., cystic fibrosis) are strongly determined by genes, whereas others (e.g., infectious diseases) are strongly determined by environmental factors. (Adapted from Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Here, two research strategies are reviewed that often are used to estimate the relative influence of genes and environment: twin studies and adoption studies.
Twin Studies
Twins occur with a frequency of about 1 in 100 births in white populations. They are a bit more common in blacks and a bit less common among Asians. Monozygotic (MZ, identical) twins originate when, for unknown reasons, the developing embryo divides to form two separate but identical embryos. Because they are genetically identical, MZ twins are an example of natural clones. Dizygotic (DZ, fraternal) twins are the result of a double ovulation followed by the fertilization of each egg by a different sperm. Thus dizygotic twins are genetically no more similar than siblings. Because two different sperm cells are required to fertilize the two eggs, it is possible for each DZ twin to have a different father. Whereas MZ twinning rates are constant across populations, DZ twinning rates vary somewhat. DZ twinning increases with maternal age until about 40 years, after which it declines.
Because MZ twins are genetically identical, any differences between them should be caused only by environmental effects.7 MZ twins should thus resemble one another very closely for traits that are strongly influenced by genes. DZ twins provide a convenient comparison because their environmental differences should be similar to those of MZ twins, but their genetic differences are as great as those between siblings. Twin studies thus usually consist of comparisons between MZ and DZ twins.8 If both members of a twin pair share a trait (e.g., a cleft lip), it is said to be a concordant trait. If they do not share the trait, it is a discordant trait. For a trait determined totally by genes, MZ twins should always be concordant, whereas DZ twins should be concordant less often, because they, like siblings, share only 50% of their genes. Concordance rates may differ between opposite-sex DZ twin pairs and same-sex DZ pairs for some traits, such as those that have different frequencies in males and females. For such traits, only same-sex DZ twin pairs should be used when comparing MZ and DZ concordance rates, because MZ twins are necessarily of the same sex.
Table 5-3 gives concordance rates for a number of traits. Note that the concordance rates for contagious diseases such as measles are quite similar in MZ and DZ twins. This is expected because a contagious disease is unlikely to be influenced markedly by genes. On the other hand, the concordance rates are quite dissimilar for schizophrenia and bipolar affective disorder, suggesting a sizable genetic component for these diseases. The MZ correlations for dermatoglyphics (fingerprints), which are determined almost entirely by genes, are close to 1.0.
Table 5-3
Concordance Rates in MZ and DZ Twins for Selected Traits and Diseases∗
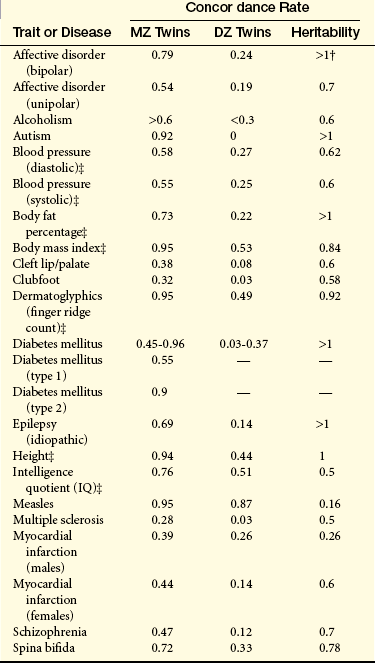
NOTE: Heritability, which is defined as the proportion of the variation in a trait that is due to genetic factors, can be measured as 2(CMZ –CDZ), where CMZ and CDZ are the concordance rates for MZ twins and DZ twins, respectively.
DZ, Dizygotic; MZ, monozygotic.
∗These figures were compiled from a large variety of sources and represent primarily European and U.S. populations.
†Several heritability estimates exceed 1. Because it is impossible for >100% of the variance of a trait to be genetically determined, these values indicate that other factors, such as shared environmental factors, must be operating.
‡Because these are quantitative traits, correlation coefficients are given rather than concordance rates.
At one time, twins were thought to provide a perfect “natural laboratory” in which to determine the relative influences of genetics and environment, but several difficulties arise. One of the most important is the assumption that the environments of MZ and DZ twins are equally similar. As one would expect, MZ twins are often treated more similarly than DZ twins. A greater similarity in environment can make MZ twins more concordant for a trait, inflating the apparent influence of genes. In addition, MZ twins may be more likely to seek the same type of environment, further reinforcing environmental similarity. On the other hand, it has been suggested that MZ twins tend to develop personality differences in an attempt to assert their individuality.
Adoption Studies
Studies of adopted children also are used to estimate the genetic contribution to a multifactorial trait. Children born to parents who have a disease but are then subsequently adopted by parents lacking the disease can be studied to find out whether these children develop the disease. In some cases such children develop the disease more often than a comparative control population (i.e., adopted children who were born to parents who do not have the disease). This provides some evidence that genes may be involved in the causation of the disease, because the adopted children do not share an environment with their affected natural parents. For example, about 8% to 10% of adopted children of a schizophrenic parent develop schizophrenia, whereas only 1% of adopted children of normal parents develop schizophrenia.
As with twin studies, several precautions must be exercised in interpreting the results of adoption studies. First, prenatal environmental influences could have long-lasting effects on an adopted child. Second, children are sometimes adopted after they are several years old, ensuring that some environmental influence would have been imparted by the natural parents. Finally, adoption agencies sometimes try to match the adoptive parents with the natural parents in terms of background, socioeconomic status, and so on. All of these factors could exaggerate the apparent influence of biologic inheritance.
These reservations, as well as those summarized for twin studies, underscore the need for caution in basing conclusions on twin and adoption studies. These approaches do not provide definitive measures of the role of genes in multifactorial disease nor can they identify specific genes responsible for disease. Instead, they serve a useful purpose in providing a preliminary indication of the extent to which a multifactorial disease may be caused by genetic factors. Sophisticated molecular techniques are being used to identify the specific genes that underlie predisposition to multifactorial diseases.
This discussion should make clear that most common diseases are not the result of either genetics or environment. Instead, genetic and nongenetic factors usually interact to influence one’s likelihood of developing a common disease. In some cases a genetic predisposition may interact with an environmental factor to increase the risk of disease to a much higher level than would either factor acting alone. A good example of a gene-environment interaction is given by α1-antitrypsin deficiency, a genetic condition that causes pulmonary emphysema and is greatly exacerbated by cigarette smoking (Box 5-2).
GENETICS OF COMMON DISEASES
Some common multifactorial disorders, the congenital malformations, are by definition present at birth. Others, including heart disease, cancer, diabetes, and most psychiatric disorders, are seen primarily in adolescents and adults. Because these disorders are complex, unraveling their genetics is a daunting task. Nonetheless, significant progress is being made.
Congenital Malformations
Congenital diseases are present at birth. Approximately 2% of newborns present with a congenital malformation; most of these are multifactorial in etiology. Table 5-4 lists some more common congenital malformations. In general, sibling recurrence risks for most of these disorders range from 1% to 5%.
Table 5-4
Prevalence Rates of Common Congenital Malformations in Whites
| Disorder | Prevalence per 1000 Births (Approximate) |
| Cleft lip/palate | 1 |
| Clubfoot | 1 |
| Congenital heart defects | 4-8 |
| Hydrocephaly | 0.5-2.5 |
| Isolated cleft palate | 0.4 |
| Neural tube defects | 1-3 |
| Pyloric stenosis | 3 |
Some congenital malformations, such as CL/P and pyloric stenosis, are relatively easy to repair and thus are not considered to be serious problems. Others, such as the neural tube defects, usually have more severe consequences. Although some cases of congenital malformations occur in the absence of any other problems, it is quite common for them to be associated with other disorders. For example, hydrocephaly and clubfoot are often seen secondary to spina bifida, CL/P is often seen in babies with trisomy 13, and congenital heart defects are seen in children with many other disorders, including Down syndrome.
Environmental factors also cause some congenital malformations. An example is thalidomide, a sedative used during pregnancy in the early 1960s. When ingested during early pregnancy this drug often caused phocomelia (severely shortened limbs) in babies. Maternal exposure to retinoic acid, which is used to treat acne, can cause congenital defects of the heart, ear, and central nervous system. Maternal rubella infection can cause congenital heart defects.
Multifactorial Disorders in the Adult Population
Until quite recently, very little was known about specific genes responsible for common adult diseases. With the more powerful laboratory and analytic techniques now available, this situation is changing. This section reviews recent progress in understanding the genetics of the major common adult diseases. Table 5-5 gives approximate prevalence figures for these disorders in the United States.
Table 5-5
Prevalence of Common Adult Diseases in the United States
| Disease | Number Affected (Approximate) |
| Alcoholism | 14 million |
| Alzheimer disease | 4 million |
| Arthritis | 43 million |
| Asthma | 17 million |
| Cancer | 8 million |
| Cardiovascular disease (all forms) | |
| Coronary artery disease | 13 million |
| Congestive heart failure | 5 million |
| Congenital defects | 1 million |
| Hypertension | 50 million |
| Stroke | 5 million |
| Depression and bipolar disorder | 17 million |
| Diabetes (type 1) | 1 million |
| Diabetes (type 2) | 15 million |
| Epilepsy | 2.5 million |
| Multiple sclerosis | 350,000 |
| Obesity∗ | 60 million |
| Parkinson disease | 500,000 |
| Psoriasis | 3-5 million |
| Schizophrenia | 2 million |
Data from National Center for Chronic Disease Prevention and Health Promotion; American Heart Association (2002 Heart and Stroke Statistical Update); National Institute on Alcohol Abuse and Alcoholism; Office of the U.S. Surgeon General; American Academy of Allergy, Asthma and Immunology; Cown WM, Kandel ER: JAMA 285:594-600, 2001; Flegal et al: JAMA 288:1723–1727, 2002.
Coronary Heart Disease
It is well known that coronary heart disease (CHD) is the leading killer of Americans, accounting for approximately 25% of all deaths in the United States. It is caused by atherosclerosis (narrowing as a result of the formation of lipid-laden lesions) of the coronary arteries. This narrowing impedes blood flow to the heart and can eventually result in a myocardial infarction (destruction of heart tissue caused by an inadequate supply of oxygen). When atherosclerosis occurs in arteries supplying blood to the brain, a stroke can result. Many risk factors for heart disease have been identified, including obesity, cigarette smoking, hypertension, elevated cholesterol level, and positive family history (usually defined as having one affected first-degree relative). Many studies have examined the role of family history in CHD, and they show that an individual with a positive family history is two to seven times more likely to have heart disease than is an individual with no family history (this would be the relative risk of heart disease as a result of a positive family history). Generally, these studies also show that the risk increases if (1) there are more affected relatives; (2) the affected relative or relatives are female (the less commonly affected sex) rather than male; and (3) age of onset in the affected relative is early (before 55 years). For example, one study showed that men between the ages of 20 and 39 years had a relative risk of 3 for CHD if they had one affected first-degree relative. The relative risk increased to 13 if two first-degree relatives were affected with CHD before 55 years of age.11
What part do genes play in the familial clustering of heart disease? Because of the key role of lipids in atherosclerosis, many studies are focusing on the genetic determination of various lipoproteins.12 An important advance in this area has been the isolation and cloning of the gene for the low-density lipoprotein (LDL)–receptor defects that cause familial hypercholesterolemia (see Box 5-3). Many other genes involved in lipid variation, coagulation, and hypertension have been identified, including several genes encoding apolipoproteins (the protein components of lipoproteins) (Table 5-6). Functional analysis of these genes is leading to an increased understanding, and eventually more effective treatment, of CHD.
Box 5-3 Familial Hypercholesterolemia
Autosomal dominant familial hypercholesterolemia (FH) is an important cause of heart disease, accounting for approximately 5% of myocardial infarctions in individuals less than 60 years of age.9 FH is one of the most common autosomal dominant disorders: in most populations surveyed to date, about 1 in 500 people is a heterozygote. Plasma cholesterol levels are approximately twice as high as normal (i.e., about 300 to 400 mg/dl), resulting in substantially accelerated atherosclerosis and distinctive cholesterol deposits in skin and tendons (xanthomas, Figure 5-5). Data compiled from five studies showed that approximately 75% of men with FH developed coronary disease and 50% had a fatal myocardial infarction by 60 years. The corresponding percentages for women were lower (45% and 15%) because women generally develop heart disease at a later age than men.
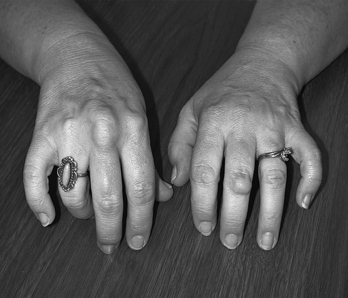
Figure 5-5 Xanthoma. Fatty deposits, referred to as xanthomas as seen here on the knuckles, are often noted in individuals with familial hypercholesterolemia. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Consistent with Hardy-Weinberg predictions, about 1 in 1 million births is homozygous for the FH gene. Homozygotes are much more severely affected, with cholesterol levels ranging from 600 to 1200 mg/dl. Most experience myocardial infarctions before 20 years of age, and a myocardial infarction at 18 months of age has been reported. If untreated, most FH homozygotes die before 30 years of age.
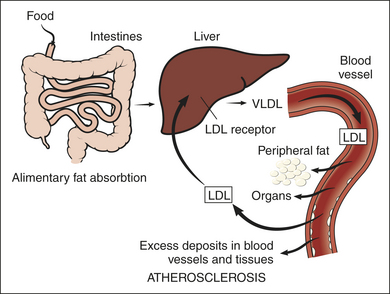
All cells require cholesterol as a component of their plasma membrane. They can either synthesize their own cholesterol, or, preferably, obtain it from the extracellular environment, where it is carried primarily by low-density lipoprotein (LDL). In a process known as endocytosis, LDL-bound cholesterol is taken into the cell via LDL receptors on the cell’s surface (Figure 5-6). FH is caused by a reduction in the number of functional LDL receptors on cell surfaces. Lacking the normal number of LDL receptors, cellular cholesterol uptake is reduced and circulating cholesterol levels increase.
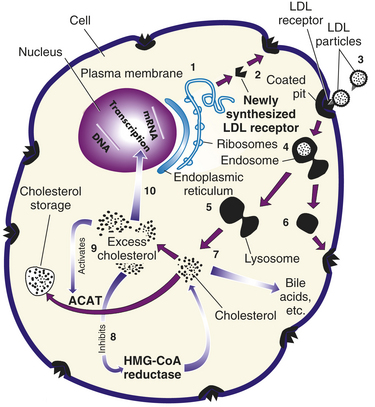
Figure 5-6 Process of receptor-mediated endocytosis. Numbers in parentheses correspond to numbers shown in the figure. (1) The low-density lipoprotein (LDL) receptors, which are glycoproteins, are synthesized in the endoplasmic reticulum of the cell. (2) From here, they pass through the Golgi apparatus to the cell surface, where part of the receptor protrudes outside the cell. (3) The circulating LDL particle is bound by the LDL receptor and localized in cell-surface depressions called coated pits (so named because they are coated with a protein called clathrin). (4) The coated pit invaginates, bringing the LDL particle inside the cell. (5) Once inside the cell, the LDL particle is separated from the receptor, taken into a lysosome, and broken down into its constituents by lysosomal enzymes. (6) The LDL receptor is recirculated to the cell surface to bind another LDL particle (each LDL receptor goes through this cycle approximately once every 10 minutes even if it is not occupied by an LDL particle). (7) Free cholesterol is released from the lysosome for incorporation into cell membranes or metabolism into bile acids or steroids. Excess cholesterol can be stored in the cell as a cholesterol ester or removed from the cell by associating with high-density lipoprotein (HDL). (8) As cholesterol levels in the cell rise, cellular cholesterol synthesis is reduced by inhibition of the rate-limiting enzyme HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A) reductase. (9) Rising cholesterol levels also increase the activity of acyl coenzyme A (acyl CoA): cholesterol acyltransferase (ACAT), an enzyme that modifies cholesterol for storage as cholesterol esters. (10) In addition, the number of LDL receptors is decreased by lowering the transcription rate of the LDL receptor gene itself. This decreases cholesterol uptake. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
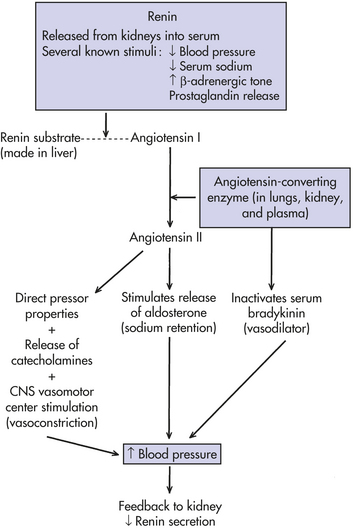
Figure 5-7 Renin-angiotensin-aldosterone system. CNS, Central nervous system. (From Jorde LB et al: Medical genetics, ed 3, St Louis, 2003, Mosby.)
Much of what we know about endocytosis has been learned through the study of LDL receptors. The process of endocytosis and the processing of LDL in the cell are described in detail in Figure 5-6 (endocytosis is discussed in Chapter 1). These processes result in a fine-tuned regulation of cholesterol levels within cells, and they influence the level of circulating cholesterol as well.
The isolation and cloning of the LDL receptor gene in 1984 were critical steps in understanding exactly how LDL receptor defects cause FH. More than 600 different mutations, including missense and nonsense substitutions as well as insertions and deletions, have been identified in the LDL receptor gene. These can be grouped into five broad classes according to their effects on the activity of the receptor.10 Class 1 mutations result in no detectable protein product. Thus heterozygotes would produce only half the normal number of LDL receptors. Class 2 mutations in the LDL receptor gene result in production of the LDL receptor, but it is altered such that it cannot leave the endoplasmic reticulum. It is eventually degraded. Class 3 mutations produce an LDL receptor that is capable of migrating to the cell surface but incapable of normal binding to LDL. Class 4 mutations, which are comparatively rare, produce receptors that are normal except that they do not migrate specifically to coated pits and thus cannot carry LDL into the cell. The final group of mutations, class 5, produces an LDL receptor that cannot dissociate from the LDL particle after entry into the cell. The receptor cannot return to the cell surface and is degraded. Each class of mutations reduces the number of effective LDL receptors, resulting in decreased LDL uptake and hence elevated levels of circulating cholesterol. The number of effective receptors is reduced by about half in FH heterozygotes, and homozygotes have virtually no functional LDL receptors.
Understanding the defects that lead to FH has helped to develop effective therapies for the disorder. Dietary reduction of cholesterol (primarily through the reduced intake of saturated fats) has only modest effects on cholesterol levels in FH heterozygotes. Because cholesterol is reabsorbed into the gut and then recycled through the liver (where most cholesterol synthesis takes place), serum cholesterol levels can be reduced by the administration of bile acid–absorbing resins, such as cholestyramine. The absorbed cholesterol is then excreted. It is interesting that reduced recirculation from the gut causes the liver cells to form additional LDL receptors, lowering circulating cholesterol levels. However, the decrease in intracellular cholesterol also stimulates cholesterol synthesis by liver cells, so the overall reduction in plasma LDL is only about 15% to 20%. This treatment is much more effective when combined with agents such as lovastatin that reduce cholesterol synthesis by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. Decreased synthesis leads to further production of LDL receptors. When these therapies are used in combination, serum cholesterol levels in FH heterozygotes can be reduced to approximately normal levels.
The picture is less encouraging for FH homozygotes. The therapies just discussed can enhance cholesterol elimination and reduce its synthesis, but they are largely ineffective because homozygotes have few or no LDL receptors. Liver transplants, which provide hepatocytes that have normal LDL receptors, have been successful in some cases, but this option is often limited by a lack of donors. Plasma exchange, carried out every 1 to 2 weeks, in combination with drug therapy, can reduce cholesterol levels by about 50%. However, this therapy is difficult to continue for long periods. Somatic cell gene therapy, in which hepatocytes carrying normal LDL receptor genes are introduced into the portal circulation, is now being tested. It may eventually prove to be an effective treatment for FH homozygotes.
The FH story illustrates how medical research has made important contributions both to our understanding of basic cell biology and to advances in clinical therapy. The process of receptor-mediated endocytosis, elucidated largely by research on the LDL receptor defects, is of fundamental significance for cellular processes throughout the body. Equally important is that this research, by clarifying how cholesterol synthesis and uptake can be modified, has led to significant improvements in therapy for this important cause of heart disease.
Illustration form Damjanov I: Pathophysiology for the health-related professions, ed 2, Philadelphia, 2000, Saunders.
Table 5-6
Lipoprotein Genes Known to Contribute to Coronary Artery Disease Risk
| Gene | Chromosome Location | Function of Protein Product |
| Apolipoprotein A-I | 11q | HDL component; LCAT cofactor |
| Apolipoprotein A-IV | 11q | Component of chylomicrons and HDL; may influence HDL metabolism |
| Apolipoprotein C-III | 11q | Allelic variation associated with hypertriglyceridemia |
| Apolipoprotein B | 2p | Ligand for LDL receptor; involved in formation of VLDL, LDL, IDL, and chylomicrons |
| Apolipoprotein D | 2p | HDL component |
| Apolipoprotein C-I | 19q | LCAT activation |
| Apolipoprotein C-II | 19q | Lipoprotein lipase activation |
| Apolipoprotein E | 19q | Ligand for LDL receptor |
| Apolipoprotein A-II | 1p | HDL component |
| LDL receptor | 19p | Uptake of circulating LDL particles |
| Lipoprotein (a) | 6q | Cholesterol transport |
| Lipoprotein lipase | 8p | Hydrolysis of lipoprotein lipids |
| Hepatic triglyceride lipase | 15q | Hydrolysis of lipoprotein lipids |
| LCAT | 16q | Cholesterol esterification |
| Cholesterol ester transfer protein | 16q | Facilitates transfer of cholesterol esters and phospholipids between lipoproteins |
HDL, High-density lipoprotein; IDL, intermediate-density lipoprotein; LCAT, lecithin cholesterol acyltransferase; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.
Adapted in part from King RA, Rotter JI, editors: The genetic basis of common diseases, ed 2, New York, 2002, Oxford University Press.
Environmental factors, many of which are easily modified, are also important causes of CHD. Abundant epidemiologic evidence shows that cigarette smoking and obesity increase the risk of CHD, whereas exercise and a diet low in saturated fats decrease the risk. Indeed, the approximate 50% decline in CHD prevalence in the United States during the past 40 years is usually attributed to a decrease in the proportion of adults who smoke cigarettes, decreased consumption of saturated fats, and an increased emphasis on exercise and a generally healthier lifestyle.
Hypertension
Systemic hypertension, which is seen in at least 15% of the populations of most developed countries, is a key risk factor for heart disease, stroke, and kidney disease. Studies of blood pressure correlations within families indicate that about 20% to 40% of the variation in both systolic and diastolic blood pressure is caused by genetic factors. The fact that this figure is substantially less than 100% indicates that environmental factors also must be important causes of blood pressure variation. The most important environmental risk factors for hypertension are increased sodium intake, decreased physical activity, psychosocial stress, and obesity (but, as discussed later, the latter factor is itself influenced by both genes and environment).
Blood pressure regulation is a highly complex process that is influenced by many physiologic systems, including various aspects of kidney function, cellular ion transport, and heart function. Because of this complexity, it is unlikely that family studies of simple blood pressure will reveal much about genes responsible for hypertension. For this reason most research now focuses on specific components that may influence blood pressure variation, such as angiotensin, angiotensinogen, urinary kallikrein, and sodium-lithium countertransport13 (Figure 5-7). These factors are more likely to be under the control of smaller numbers of genes. For example, studies have implicated the angiotensinogen gene in the causation of both hypertension and preeclampsia (a form of pregnancy-induced hypertension).
Cancer
Cancer is the second leading cause of death in the United States. It is well established that many major types of cancer (e.g., breast, colon, prostate, ovarian) cluster strongly in families. This is caused by both shared genes and shared environmental factors. Although numerous cancer genes are being isolated,14 environmental factors also play an important role in causing cancer. In particular, tobacco use is estimated to account for one third of all cancer cases in the United States, making it the most important known cause of cancer.15
Breast Cancer: Breast cancer is the most common cancer among women, affecting approximately 12% of American women who live to 85 years or more. Formerly the leading cause of cancer death among women, it has been surpassed by lung cancer. Breast cancer aggregates strongly in families. If a woman has one affected first-degree relative, her risk of developing breast cancer doubles. This risk increases if the age of onset in the affected relative is early and if the cancer is bilateral (tumors in both breasts).
An autosomal dominant form of breast cancer accounts for approximately 5% of breast cancer cases in the United States. Genes responsible for this form of breast cancer have been mapped to chromosomes 17 (BRCA1) and 13 (BRCA2). Each of these genes has been cloned, and it is possible to test them for cancer-causing mutations.16 Women who inherit a mutation in BRCA1 or BRCA2 experience a 50% to 80% lifetime risk of developing breast cancer. BRCA1 mutations also increase the risk of ovarian cancer among women (20% to 50% lifetime risk), and they confer a modestly increased risk of prostate and colon cancers. BRCA2 mutations also confer an increased risk of ovarian cancer (10% to 20% lifetime prevalence). Approximately 6% of males who inherit a BRCA2 mutation will develop breast cancer; this represents a 100-fold increase over the risk in the general male population. The evaluation of the BRCA1 and BRCA2 gene products, which are both involved in deoxyribonucleic acid (DNA) repair, is yielding valuable evidence on the etiology of breast cancer in general.
Although BRCA1 and BRCA2 mutations are the most common known causes of inherited breast cancer, this disease also can be caused by inherited mutations in several other tumor suppressor genes (e.g., the CHK2 and TP53 genes). Germline mutations in a tumor suppressor gene called PTEN are responsible for Cowden disease, which is characterized by multiple benign tumors and an increased susceptibility to breast cancer.
Colorectal Cancer: Colorectal cancer is second only to lung cancer in the number of cases occurring annually in the United States, with nearly 154,000 new cases in 2007.17 Approximately 1 in 20 Americans will develop colorectal cancer. Like breast cancer, it clusters in families (in fact, familial clustering of this form of cancer was reported in the medical literature as early as 1881). The risk of colorectal cancer in people with one affected first-degree relative is two to three times higher than in the general population.
This familial aggregation is caused in part by subsets of colorectal cancer cases that are inherited as single-gene traits. Familial adenomatous polyposis occurs in approximately 1 in 8000 whites. The gene responsible for this disorder, APC, was mapped to chromosome 5, and the gene itself was subsequently cloned.18 Identification of the protein product of this gene showed that it functions as a tumor suppressor. Importantly, somatic mutations of APC are found in at least 85% of all colon tumors. Thus although inherited APC mutations play a vital role in relatively rare familial adenomatous polyposis, somatic mutations are involved in the great majority of all common colon cancers.
Hereditary nonpolyposis colorectal cancer, which may account for as many as 5% of colorectal cancer cases, is caused by mutations in any of six genes.19 Cloning of these genes has shown that all of them are involved in the vital process of DNA repair. When this function is compromised, cancer-causing mutations can persist in cells, leading eventually to growth of a tumor.
Other colorectal cancer cases are likely to be caused by a complex interaction of multiple genes. In addition, environmental factors, such as a high-fat, low-fiber diet, are thought to increase the risk of colorectal cancer.
Other Cancers: The genetic basis of various other cancers, including retinoblastoma, has been discussed. Although each of these cancers is relatively rare, study of the causative genes has provided many important insights into the nature of carcinogenesis in general. This will lead to more effective treatment and prevention of all cancers.
Diabetes Mellitus
Like the other disorders discussed in this chapter, the etiology of diabetes mellitus is complex and not fully understood. Nevertheless, progress is being made in understanding the genetic basis of this disorder, which is a leading cause of blindness, heart disease, and kidney failure.20,21 An important advance has been the recognition that diabetes is actually a heterogeneous group of disorders, all characterized by elevated blood sugar. The focus here is on the two major types of diabetes: type 1 (insulin-dependent diabetes mellitus [IDDM]) and type 2 (non–insulin-dependent diabetes mellitus [NIDDM]).
Type 1 Diabetes: Type 1 diabetes, which is characterized by T-cell infiltration of the pancreas and destruction of the insulin-producing beta cells, usually (though not always) presents before age 40. Individuals with type 1 diabetes must receive exogenous insulin to survive. In addition to T-cell infiltration of the pancreas, autoantibodies are formed against pancreatic cells; the latter can be observed long before clinical symptoms occur. These findings, along with a strong association between type 1 diabetes and the presence of several human leukocyte antigen (HLA) class II alleles, indicate that this is an autoimmune disease.
Siblings of individuals with type 1 diabetes face a substantial elevation in risk: approximately 6%, as opposed to a risk of about 0.3% to 0.5% in the general population. The recurrence risk is also elevated when there is a diabetic parent, although this risk varies with the sex of the affected parent. The risk for offspring of diabetic mothers is only 1% to 3%, whereas it is 4% to 6% for the offspring of diabetic fathers (because type 1 diabetes affects males and females in roughly equal proportions in the general population, this risk difference is inconsistent with the sex-specific threshold model for multifactorial traits). Twin studies show that the empirical risks for identical twins of people with type 1 diabetes range from 30% to 50%. In contrast, the concordance rates for dizygotic twins are 5% to 10%. The fact that type 1 diabetes is not 100% concordant among identical twins indicates that genetic factors are not solely responsible for the disorder. There is good evidence that specific viral infections contribute to the causation of type 1 diabetes in at least some individuals, possibly by activating an autoimmune response.
The association of specific HLA class II alleles (see Chapter 21) and type 1 diabetes has been studied extensively, and it is estimated that the HLA system accounts for about 40% of the familial clustering of type 1 diabetes. Approximately 95% of whites with type 1 diabetes have the HLA DR3 and/or DR4 alleles, whereas only about 50% of the general white population has either of these alleles. If an affected proband and a sibling are heterozygous for the DR3 and DR4 alleles, the sibling’s risk of developing type 1 diabetes is nearly 20% (i.e., about 40 times higher than the risk in the general population). In addition, the presence of aspartic acid at position 57 of the DQ chain is strongly associated with resistance to type 1 diabetes. In fact, those who do not have this amino acid at position 57 (and instead are homozygous for a different amino acid) are 100 times more likely to develop type 1 diabetes. The aspartic acid substitution alters the shape of the HLA class II molecule and thus its ability to bind and present peptides to T cells. Altered T-cell recognition may help protect individuals with the aspartic acid substitution from an autoimmune episode.
The insulin gene, which is located on the short arm of chromosome 11, is another logical candidate for type 1 diabetes susceptibility. Polymorphisms within and near this gene have been tested for association with type 1 diabetes. It is estimated that inherited genetic variation in the insulin region accounts for approximately 10% of the familial clustering of type 1 diabetes.
Within the past several years, additional genes have been shown to be associated with susceptibility to type 1 diabetes. The most significant of these are cytotoxic lymphocyte associated-4 (CTLA4), which encodes a protein involved in the regulation of T-cell proliferation, and PTPN22, which encodes a lymphoid-specific tyrosine phosphatase that negatively regulates T-cell activation. It is interesting that variation in the latter gene has been associated with several other autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis, and autoimmune thyroid disease.
Type 2 Diabetes: Type 2 diabetes accounts for more than 90% of all diabetes cases and affects 10% to 20% of the adult populations of many developed countries. A number of features distinguish it from type 1 diabetes. There is nearly always some endogenous insulin production in people with type 2 diabetes, and the disease can often be treated successfully with dietary modification and/or oral drugs. People with type 2 diabetes suffer from insulin resistance (i.e., their cells have difficulty in using insulin). This disease typically occurs among people older than age 40 and, in contrast to type 1 diabetes, is seen more commonly among the obese. The incidence of type 2 diabetes is rising dramatically among adolescents and young adults in developed countries, however, and is strongly correlated with an increased incidence of obesity. Neither HLA associations nor autoantibodies are seen commonly in this form of diabetes. Monozygotic twin concordance rates are substantially higher than in type 1 diabetes, often exceeding 90% (because of age dependence, the concordance rate increases if older subjects are studied). The empirical recurrence risks for first-degree relatives of type 2 diabetes cases are higher than those for type 1, generally ranging from 10% to 15%. The differences between type 1 and type 2 diabetes are summarized in Table 5-7.
Table 5-7
Comparison of Major Features of Types 1 and 2 Diabetes Mellitus
| Feature | Type 1 Diabetes | Type 2 Diabetes |
| Age of onset | Usually <40 yr | Usually >40 yr (except maturity-onset diabetes of the young [MODY]) |
| Insulin production | None | Partial |
| Insulin resistance | No | Yes |
| Autoimmunity | Yes | No |
| Obesity | Not common | Common |
| Monozygotic (MZ) twin concordance | 0.55 | 0.90 |
| Sibling recurrence risk | 1%-6% | 10%-15% |
A significant association has been observed between type 2 diabetes and a common allele of the gene that encodes peroxisome proliferator–activated receptor-γ (PPAR-γ), a transcription factor that is involved in adipocyte differentiation and glucose metabolism. Although this allele confers only a 25% increase in the risk of developing type 2 diabetes, it is found in more than 75% of individuals of European descent. Thus it may help account for a significant proportion of type 2 diabetes cases. Other genes that are significantly associated with type 2 diabetes susceptibility are TCF7L2, which encodes a transcription factor associated with blood glucose homeostasis, and KCNJ11, which encodes a potassium channel that is essential for the regulation of insulin secretion by pancreatic beta cells.
The two most important risk factors for type 2 diabetes are positive family history and obesity; the latter increases insulin resistance. The disease tends to rise in prevalence when populations adopt a diet and exercise pattern typical of U.S. and European populations. Increases have been seen, for example, among Japanese immigrants to the United States and among some native populations of the South Pacific, Australia, and the Americas. Several studies, conducted on male and female subjects, have shown that regular exercise can substantially lower one’s risk of developing type 2 diabetes, even among individuals with a family history of the disease. This is partly because exercise reduces obesity. However, even in the absence of weight loss, exercise increases insulin sensitivity and improves glucose tolerance.
Because of the dramatic increase in obesity in the United States and other developed countries, the prevalence of type 2 diabetes is also rising rapidly, and the average age of onset is decreasing. A small proportion of type 2 diabetes cases occurs early in life, typically before 25 years of age, and typically exhibits autosomal dominant inheritance (unlike most type 2 diabetes). This subset is termed maturity-onset diabetes of the young (MODY). Studies of MODY pedigrees have shown that about half of cases of the disease are caused by mutations in the glucokinase gene. Glucokinase converts glucose to glucose-6-phosphate in the pancreas. In addition to the glucokinase gene, five other genes, all of which are involved in pancreatic development or insulin regulation, have now been shown to be causes of MODY.
Obesity
Obesity is most commonly defined as a body mass index (BMI) greater than 30.∗ Using this criterion, a survey published in 2006 showed that approximately 32% of American adults are obese, and an additional 35% are overweight (BMI greater than 25 but less than 30). The proportion of obese adults and children continues to increase rapidly. Although obesity itself is not a “disease,” it is an important risk factor for several common diseases, including heart disease, stroke, hypertension, and type 2 diabetes.
As one might expect, there is a strong correlation between obesity in parents and their children. This could easily be ascribed to common environmental effects: parents and children usually share similar dietary and exercise habits. However, there is good evidence for genetic components as well. Four adoption studies each showed that the body weights of adopted individuals correlated significantly with their natural parents’ body weights but not with those of their adoptive parents. Twin studies also provide evidence for a genetic effect on body weight, with most studies yielding heritability estimates between 0.60 and 0.80.
Research, aided substantially by mouse models, has shown that several genes each play a role in human obesity. Important among these are the genes that encode leptin (Greek, “thin”) and its receptor. The leptin hormone is secreted by adipocytes (fat storage cells) and binds to receptors in the hypothalamus, the site of the body’s appetite control center. Cloning of the human leptin gene and its receptor led to optimistic predictions that leptin could be a key to weight loss in humans (without the perceived unpleasantness of dieting and exercise). Although mutations in the human leptin gene and its receptor have been identified in a few humans with severe obesity (BMI >40), they both appear to be extremely rare. Clinical trials using recombinant leptin have demonstrated moderate weight loss in a subset of obese individuals. In addition, leptin participates in important interactions with other components of appetite control, such as neuropeptide Y and α–melanocyte-stimulating hormone and its receptor, the melanocortin-4 receptor (MC4R). Mutations in the gene that encodes MCR4 have been found in 3% to 5% of severely obese individuals. Recently, homozygosity for a DNA variant in the FTO gene (which is seen in 16% of whites) has been associated with 40% and 70% increases in the risks of overweight and obesity, respectively. Identification of these human genes is leading to a better understanding of natural weight control in the human, and it could eventually lead to effective treatments for some cases of obesity.
Alzheimer Disease
Alzheimer disease (AD), which is responsible for 60% to 70% of cases of progressive cognitive impairment among older adults, affects approximately 5% to 10% of the population older than 65 years of age and 40% of the population older than 85 years of age. Because of the aging of the population, the number of Americans with AD is predicted to increase substantially during the coming decade. AD is characterized by progressive dementia and memory loss and by the formation of amyloid plaques and neurofibrillary tangles in the brain, particularly in the cerebral cortex and hippocampus. The plaques and tangles lead to progressive neuronal loss, and death usually occurs within 7 to 10 years after the first appearance of symptoms.
The risk of developing AD doubles in individuals who have an affected first-degree relative. Although most cases do not appear to be caused by single loci, approximately 10% follow an autosomal dominant mode of transmission. About 3% to 5% of AD cases occur before age 65 and are considered early onset; these are much more likely to be inherited in autosomal dominant fashion.22
AD is a genetically heterogeneous disorder. Approximately half of early-onset cases can be attributed to mutations in any of three genes, all of which affect amyloid-β deposition.23 Two of the genes, presenilin 1 (PS1) and presenilin 2 (PS2), are very similar to one another, and their protein products are involved in cleavage of the amyloid-β precursor protein (APP). When APP is not cleaved normally, a long form of it accumulates excessively and is deposited in the brain. This is thought to be a primary cause of AD. Mutations in PS1 typically result in especially early onset of AD, with the first occurrence of symptoms in the fifth decade of life.
A small number of cases of early-onset AD are caused by mutations of the gene that encodes APP itself, which is located on chromosome 21. These mutations disrupt normal cleavage sites in APP, again leading to the accumulation of the longer protein product. It is interesting that this gene is present in three copies in trisomy 21 individuals, in which the extra gene copy leads to amyloid deposition and the occurrence of AD in those with Down syndrome (see Chapter 4).
An important risk factor for the more common late-onset form of AD is allelic variation in the apolipoprotein E (APOE) locus, which has three major alleles: ε2, ε3, and ε4. Studies conducted in diverse populations have shown that persons who have one copy of the ε4 allele are at least 2 to 5 times more likely to develop AD, whereas those with two copies of this allele are at least 5 to 10 times more likely to develop AD. The risk varies somewhat by population, with higher ε4-associated risks in Europeans and Japanese and relatively lower risks in Hispanics and blacks. Despite the strong association between ε4 and AD, approximately half of individuals who develop late-onset AD do not have a copy of the ε4 allele, and many who are homozygous for ε4 remain free of AD even at advanced age. The apolipoprotein E protein product is not involved in cleavage of APP but instead appears to be associated with clearance of amyloid from the brain.
Alcoholism
At some point, alcoholism is diagnosed in approximately 10% of adult males and 3% to 5% of adult females in the United States. The national cost of alcoholism, in terms of lost productivity and direct medical costs, is approximately $200 billion per year. More than 100 studies have shown that this disease clusters in families.24 The risk of developing alcoholism among individuals with one affected parent is three to five times higher than for those with unaffected parents.
Most twin studies have yielded concordance rates for DZ twins less than 30% and concordance rates for MZ twins in excess of 60%. Adoption studies have shown that the offspring of an alcoholic parent, even when raised by nonalcoholic parents, have a fourfold increased risk of developing the disorder. To control for possible prenatal effects in an alcoholic mother, some studies have included only the offspring of alcoholic fathers. The results have remained the same. One study showed that the offspring of nonalcoholic parents, when reared by alcoholics, did not have an increased risk of developing alcoholism. These data argue that there may be genes that predispose some people to alcoholism.
It has long been known that an individual’s physiologic response to alcohol can be influenced by variation in the key enzymes responsible for alcohol metabolism (alcohol dehydrogenases [ADH]), which convert ethanol to acetaldehyde, and aldehyde dehydrogenases (ALDH), which convert acetaldehyde to acetate. In particular, an allele of the ALDH2 gene (ALDH2∗2) results in excessive accumulation of acetaldehyde and thus in facial flushing, nausea, palpitations, and lightheadedness. Because of these unpleasant effects, individuals who have the ALDH2∗2 allele are much less likely to become alcoholics. This “protective” allele is common in some Asian populations but is rare in other populations.
More recently genetic studies have implicated genes that encode gamma-aminobutyric acid (GABA) receptors. Because GABA is the brain’s primary inhibitory neurotransmitter, GABA receptor genes are important potential contributors to a genetic susceptibility to alcoholism.
It should be underscored that genes may increase one’s susceptibility to alcoholism. Obviously this is a disease that requires an environmental component, regardless of genetic constitution.
Psychiatric Disorders
The major psychiatric diseases, schizophrenia and affective disorder, have been the subjects of numerous genetic studies.25 Twin, adoption, and family studies have shown that both disorders aggregate in families.
Schizophrenia: Schizophrenia is a severe emotional disorder characterized by delusions, hallucinations, retreat from reality, and bizarre, withdrawn, or inappropriate behavior. (Contrary to popular belief, schizophrenia is not a “split personality” disorder.) The lifetime recurrence risk for schizophrenia among the offspring of one affected parent is approximately 8% to 10%, which is about 10 times higher than the risk in the general population.26 As one might expect, the empirical risks increase when more relatives are affected. For example, an individual with an affected sibling and an affected parent has a risk of about 17%, and an individual with two affected parents has a risk of 46%. The risks decrease when the affected family member is a second- or third-degree relative. Details are given in Table 5-8. On inspection of Table 5-8, it may seem puzzling that the proportion of schizophrenic probands who have a schizophrenic parent is only about 5%, which is substantially lower than the risk for other first-degree relatives (e.g., siblings, affected parents, their offspring). This can be explained by the fact that people with schizophrenia are less likely to marry and produce children than are other individuals. Thus substantial selection against schizophrenia occurs in the population.
Table 5-8
Recurrence Risks for Relatives of Schizophrenic Probands∗

∗Figures are based on multiple studies of Western European populations.
Data from McGue M, Gottesman II, Rao DC: Behav Genet 16(1):75-87, 1986.
Twin and adoption studies also indicate that genetic factors are likely to be involved in schizophrenia. Data pooled from five different twin studies show a 47% concordance rate for MZ twins, compared with a concordance rate of only 12% for DZ twins. When the offspring of a schizophrenic parent are adopted by normal parents, their risk of developing the disease is about 10%, which is approximately the same as the risk when raised by a schizophrenic biologic parent. Although no schizophrenia gene has yet been conclusively identified, promising associations have been uncovered between schizophrenia and several brain-expressed genes whose products interact with glutamate receptors. These include dysbindin 1 (chromosome 6p), neuregulin 1 (chromosome 8p), and G72 (chromosome 13q). Each of these associations has been identified in specific populations, and further studies in other populations will be needed to replicate these findings.
Bipolar Affective Disorder: Bipolar affective disorder, also known as manic-depressive disorder, is a form of psychosis with extreme mood swings and emotional instability. The incidence of the disorder in the general population is approximately 0.5%, but it rises to 5% to 10% among those with an affected first-degree relative. A study using the Danish twin registry yielded concordance rates of 79% and 24% for MZ and DZ twins, respectively.27 The corresponding concordance rates for unipolar disorder (major depression) were 54% and 19%. In general, it appears that bipolar disorder is more strongly influenced by genetic factors than is unipolar disorder.
Comments on Psychiatric Disorders: Large-scale linkage studies involving hundreds of polymorphisms throughout the genome have been carried out for both schizophrenia and bipolar affective disorder. Most of these studies have produced negative results, although a few recent large-scale studies have yielded promising findings. A number of candidate genes have been tested for linkage or association with both diseases. Most of these candidates were chosen on the basis of the known involvement of certain neurotransmitters, receptors, or neurotransmitter-related enzymes in each disease (e.g., schizophrenia can be treated by drugs that block dopamine receptors, and bipolar affective disorder is sometimes treated with lithium). None of the candidate genes tested thus far, including those for sodium-lithium countertransport, various components of the dopaminergic system, and several neurotransmitter-related enzymes (e.g., monoamine oxidase, dopamine-β-hydroxylase, tyrosine hydroxylase), has been shown unequivocally to be linked or associated with either disease.
These results reflect some of the difficulties encountered in doing genetic studies of psychiatric disorders. These disorders are undoubtedly heterogeneous, reflecting the influence of numerous genetic and environmental factors. Also, definition of the phenotype is not always straightforward and it may change through time, significantly complicating genetic analysis.
Other Complex Disorders
The disorders discussed in this chapter represent some of the most common multifactorial disorders and those for which significant progress has been made in identifying genes. Many other multifactorial disorders are being studied as well, and in some cases specific susceptibility genes have been identified. These include, for example, Parkinson disease, hearing loss, multiple sclerosis, amyotrophic lateral sclerosis, epilepsy, asthma, inflammatory bowel disease, and some forms of blindness.
Some General Principles and Conclusions
Some general principles can be deduced from the results obtained thus far on the genetics of complex disorders. First, the more strongly inherited forms of complex disorders generally have an earlier age of onset (e.g., breast cancer, AD, heart disease). Often these represent subsets of cases in which there is single-gene inheritance. Second, when laterality is a component, the bilateral forms are more likely to cluster strongly in families (e.g., breast cancer, CL/P). Third, although the sex-specific threshold model fits some of the complex disorders (e.g., pyloric stenosis, CL/P, autism, heart disease), it fails to fit others (e.g., type 1 diabetes).
A tendency exists, particularly among the lay public, to assume that the presence of a genetic component means that the course of a disease cannot be altered. This is incorrect. Most of the diseases discussed in this chapter have both genetic and environmental components. Thus lifestyle modification (e.g., diet, exercise, stress reduction) often can reduce risk significantly. Such modification may be especially important for individuals with a family history of a disease because they are likely to develop the disease earlier in life. Those with a family history of heart disease, for example, can often add many years of productive living with relatively minor lifestyle alterations. By targeting those who can benefit most from intervention, genetics helps to serve the goal of preventive medicine.
In addition, it should be stressed that the identification of a specific genetic lesion can lead to more effective prevention and treatment of the disease. Identification of mutations that cause autosomal dominant breast cancer may enable early screening and prevention of metastasis. Pinpointing a gene responsible for a neurotransmitter defect in a behavioral disorder such as schizophrenia could lead to the development of more effective drug treatments. In some cases, such as those with familial hypercholesterolemia, gene therapy may prove to be useful in treating the disease. It is important for healthcare practitioners to help individuals understand these facts.
Although the genetics of common disorders is complex and often confusing, the community health effect of these diseases, together with the evidence for hereditary factors in their etiology, demands that genetic studies be pursued. Substantial progress is already being made. The next decade will undoubtedly witness many further advances in the understanding and treatment of these disorders.
SUMMARY REVIEW
Factors Influencing Incidence of Disease in Populations
1. The incidence rate is the number of new cases of a disease reported during a specific period (typically 1 year) divided by the number of individuals in the population.
2. The prevalence rate is the proportion of the population affected by a disease at a specific point in time. This rate, and the incidence rate, can be used to compare population variations in disease frequency.
3. Relative risk is a common measure of the effect of a specific risk factor. It is expressed as a ratio of the incidence rate of the disease among individuals exposed to a risk factor divided by the incidence of the disease among individuals not exposed to a risk factor.
4. Many factors can influence the risk of acquiring a common disease, such as cancer, diabetes, or hypertension. The factors can include age, gender, diet, exercise, and family history of the disease.
Principles of Multifactorial Inheritance
1. Traits in which variation is thought to be caused by the combined effects of multiple genes are polygenic.
2. The term multifactorial is used when environmental factors also are believed to cause variation in the trait.
3. Many quantitative traits (e.g., blood pressure) are multifactorial.
4. Because traits are caused by the additive effects of many genetic and environmental factors, they tend to follow a normal or bell-shaped distribution in populations.
5. Those diseases, however, that do not follow a bell-shaped distribution appear to be either present or absent in individuals. They do not follow the inheritance patterns of single-gene disease. Instead, such diseases may follow an underlying liability distribution. It is thought that a threshold of liability must be crossed before the disease is expressed.
6. Examples of diseases that correspond to the liability model include pyloric stenosis, neural tube defects, CL/P, and some forms of congenital heart disease.
7. Many of the common adult diseases, such as hypertension, coronary heart disease, stroke, diabetes mellitus (types 1 and 2), and some cancers, are caused by complex genetic and environmental factors and are thus multifactorial diseases.
8. For most multifactorial diseases, empirical risks (risks based on direct observation of data) have been derived.
9. In contrast to most single-gene diseases, recurrence risks for multifactorial diseases can change significantly from one population to another because gene frequencies, as well as environmental factors, can differ among populations.
10. Several criteria are used to define multifactorial inheritance: (a) the recurrence risk becomes higher if more than one family member is affected; (b) if the expression of the disease in a proband is more severe, the recurrence risk is higher; (c) the recurrence risk is higher if the proband is of the less commonly affected sex; (d) the recurrence risk for the disease usually decreases rapidly in more remotely related relatives; and (e) if the prevalence of the disease in a population is f, the risk for offspring and siblings of probands is approximately .
Nature and Nurture: Disentangling the Effects of Genes and Environment
1. Family members share genes and a common environment; therefore, resemblance in traits, such as high blood pressure, reflects both genetic and environmental factors (nature and nurture, respectively).
2. Few traits are influenced only by genes or only by environment. Most are influenced by both.
3. When a disease has a relatively larger genetic component, as in breast cancer, examination of family history should be emphasized in addition to lifestyle modification.
4. Two research strategies often are used to estimate the relative influence of genes and environment-lifestyle: twin studies and adoption studies.
5. Monozygotic twins originate when the developing embryo divides to form two separate but identical embryos.
6. Dizygotic twins are the result of a double ovulation followed by the fertilization of each egg by a different sperm.
7. If both members of a twin pair share a trait, they are said to be concordant. If they do not share the same trait, they are discordant.
8. Studies of adopted children also are used to estimate the genetic contribution to a multifactorial trait.
9. A genetic predisposition may interact with an environmental-lifestyle factor to increase the risk of disease; this is called a gene-environment interaction.
1. Congenital diseases are those present at birth. Most of these diseases are multifactorial in etiology.
2. Multifactorial diseases in adults include coronary heart disease, hypertension, breast cancer, colon cancer, diabetes mellitus, obesity, AD, alcoholism, schizophrenia, and bipolar affective disorder.
3. It is incorrect to assume that the presence of a genetic component means that the course of a disease cannot be altered—most diseases have both genetic and environmental aspects.
Concordant trait 170
Congenital disease 172
Discordant trait 170
Dizygotic (DZ, fraternal) twin 170
Empirical risk 167
Gene-environment interaction 171
Incidence rate 164
Liability distribution 166
Monozygotic (MZ, identical) twin 170
Multifactorial trait 165
Phocomelia 172
Polygenic 165
Prevalence rate 164
Quantitative trait 165
Relative risk 165
Threshold of liability 166
REFERENCES
1. King, R.A., Rotter, J.I., Motulsky, A.G. The genetic basis of common diseases,, ed 2. Oxford: Oxford University Press; 2002.
2. Rothman, K.J. Modern epidemiology. New York: Lippincott; 1998.
3. Tiwari, H.K., et al. Multifactorial inheritance and complex diseases. In: Rimoin D.L., et al, eds. Emery and Rimoin’s principles and practice of medical genetics. ed 5. Philadelphia: Churchill Livingstone; 2007:299–306.
4. Kibar, Z., Capra, V., Gros, P. Toward understanding the genetic basis of neural tube defects. Clin Genet. 2007;71(4):295–310.
5. Daly, L.E., et al. Folate levels and neural tube defects: implications for prevention. JAMA. 1995;274(21):1698–1702.
6. Harper, P.S. Practical genetic counseling, ed 5. Oxford: Butterworth Heinemann; 1998.
7. Boomsma, D., Busjahn, A., Peltonen, L. Classical twin studies and beyond. Nat Rev Genet. 2002;3(11):872–882.
8. Neale, M.C., Cardon, L.R. Methodology for genetic studies of twins and families. Dordrecht, The Netherlands: Kluwer; 1992.
9. Marks, D., et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168(1):1–14.
10. Jansen, A.C., et al. Phenotypic variability in familial hypercholesterolaemia: an update. Curr Opin Lipidol. 2002;13(2):165–171.
11. Hunt, S.C., Williams, R.R., Barlow, G.K. A comparison of positive family history definitions for defining risk of future disease. J Chron Dis. 1986;39(10):809–821.
12. Lusis, A.J., Mar, R., Pajukanta, P. Genetics of atherosclerosis. Annu Rev Genomics Hum Genet. 2004;5(1):189–218.
13. Jeunemaitre, X., et al. Molecular basis of human hypertension. In: Rimoin D.L., et al, eds. Emery and Rimoin’s principles and practice of medical genetics. ed 5. Philadelphia: Churchill Livingstone; 2007:1283–1300.
14. Vogelstein, B., Kinzler, K.W. The genetic basis of human cancer, ed 2. New York: McGraw-Hill; 2002.
15. Peto, J. Cancer epidemiology in the last century and the next decade. Nature. 2001;411(6835):390–395.
16. Robson, M., Offit, K. Management of an inherited predisposition to breast cancer. N Engl J Med. 2007;357(2):154–162.
17. American Cancer Society: Cancer statistics, website: www.cancer.org/docroot/STT/stt_0.asp.
18. Galiatsatos, P., Foulkes, W.D. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101(2):385–398.
19. Rowley, P.T. Inherited susceptibility to colorectal cancer. Annu Rev Med. 2005;56:539–554.
20. Owen, K.R., McCarthy, M.I. Genetics of type 2 diabetes. Curr Opin Genet Dev. 2007;17(3):239–244.
21. Daneman, D. Type 1 diabetes. Lancet. 2006;367(9513):847–858.
22. Bird, T.D. Genetic factors in Alzheimer’s disease. N Engl J Med. 2005;352(9):862–864.
23. Blennow, K., de Leon, M.J., Zetterberg, H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403.
24. Edenberg, H.J., Foroud, T. The genetics of alcoholism: identifying specific genes through family studies. Addict Biol. 2006;11(3-4):386–396.
25. Farmer, A., Elkin, A., McGuffin, P. The genetics of bipolar affective disorder. Curr Opin Psychiatry. 2007;20(1):8–12.
26. Norton, N., Williams, H.J., Owen, M.J. An update on the genetics of schizophrenia. Curr Opin Psychiatry. 2006;19(2):158–164.
27. Bertelsen, A., Harvald, B., Hauge, M. A Danish twin study of manic-depressive disorders. Br J Psychiatry. 1977;130:330–351.