ALTERATIONS OF HORMONAL REGULATION



Function of the endocrine system involves complex interrelationships and interactions that maintain dynamic steady-states, provide growth and reproductive capabilities, and allow for adoptive changes in times of stress. Dysfunction of the endocrine system initially was described in terms of excessive or insufficient function of the endocrine gland with alterations in hormone levels. Alterations in function were thought to be caused by either hypersecretion or hyposecretion of the various hormones, leading to abnormal hormone concentrations in the blood. Techniques for studying the various components of the endocrine system have improved, and evidence has shown that dysfunction may also result from abnormal receptor function or from altered intracellular response to the hormone-receptor complex.
MECHANISMS OF HORMONAL ALTERATIONS
Significantly elevated or depressed hormone levels may result from a variety of causes (Table 21-1). Dysfunction of an endocrine gland may involve the gland’s failure to produce adequate amounts of biologically free or active hormone forms. This failure may occur when the secretory cells are unable to produce or obtain an adequate quantity of required hormone precursors or when they are unable to convert the precursors to the active hormone. A gland also may synthesize or release excessive amounts of hormone. In addition, feedback systems that recognize the need for a particular hormone may fail to function properly or may respond to inappropriate signals (see Chapter 20). Once hormones are released into the circulation, they may be degraded at an altered rate or they may be inactivated by antibodies before reaching the target cell. Ectopic sources of hormones (hormones produced by nonendocrine tissues) may result also in abnormally elevated hormone levels; i.e., bronchopulmonary tumors that release adrenocortictropic hormone (see page 765). This mechanism operates without benefit of the normal feedback system for hormone control. In these cases the ectopic hormone production is said to be autonomous.

Research has been directed toward understanding causes for the failure of the target cell to respond to its hormone (hormone insensitivity). The general types of abnormal target cell responses currently recognized are receptor-associated disorders and intracellular disorders. Receptor-associated disorders have been identified primarily in water-soluble hormones, such as insulin. These types of disorders usually involve one of the following: (1) a decrease in the number of receptors, leading to decreased or defective hormone-receptor binding; (2) impaired receptor function, resulting in insensitivity to the hormone; (3) presence of antibodies against specific receptors that either reduce available binding sites or mimic hormone action, exaggerating target cell response; or (4) unusual expression of receptor function, as occurs in some tumor cells with abnormal receptor activity.
Intracellular disorders may involve inadequate synthesis of the second messenger, such as cyclic adenosine monophosphate (cAMP), needed to transduce the hormonal signal into intracellular events. The target cell for water-soluble hormones may have a faulty response to hormone-receptor binding and thus fail to generate the required second messenger. The cell also may have an abnormal response to the second messenger if levels of intracellular enzymes or proteins are altered. (Second messengers for various hormones are listed in Table 20-4.) Both of these pathogenic mechanisms result in failure of the target cell to express the usual hormonal effect.
Pathogenic mechanisms affecting target cell response for lipid-soluble hormones, such as thyroid hormone or glucocorticoids, either occur less often or are recognized less often than those affecting the water-soluble hormones. These hormone-resistant states have been generally linked to mutations in the nuclear receptor for the hormone or, in some instances, to alterations in nuclear co-regulators.1 The number of receptors may be decreased, or those receptors may have an altered affinity for hormones.2 Both mechanisms would affect hormone-receptor binding. Alterations in generation of new messenger ribonucleic acid (mRNA) or absence of substrates for new protein synthesis also may occur, resulting in altered target cell response.3,4
ALTERATIONS OF THE HYPOTHALAMIC-PITUITARY SYSTEM
Documenting abnormal release of hypothalamic-releasing hormones has been difficult because of the relative inaccessibility of the hypothalamic-pituitary unit in the brain and the short half-life and small concentrations of the hypothalamic hormones. Perhaps the most common cause of apparent hypothalamic dysfunction is interruption of the pituitary stalk caused by destructive lesions, rupture after head injury, surgical transection, or tumor. In these cases, interruption of the physical connections between the hypothalamus and the pituitary gland causes apparent pituitary disease. For example, diabetes insipidus (antidiuretic hormone [ADH] insufficiency) may result, depending on the location at which the infundibular stem is interrupted. If the lesion is close to the hypothalamus, diabetes insipidus is likely; the farther away the lesion is from the hypothalamus, the less likely is the occurrence of diabetes insipidus.
The absence of hypothalamic releasing or inhibiting hormones (Figure 21-1) causes a variety of manifestations. For example, if there is an absence of gonadotropin-releasing hormone (GnRH) from the hypothalamus, then there is a lack of stimulation of gonadotropin follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary, thus the menses cease in women and spermatogenesis is impaired in men.
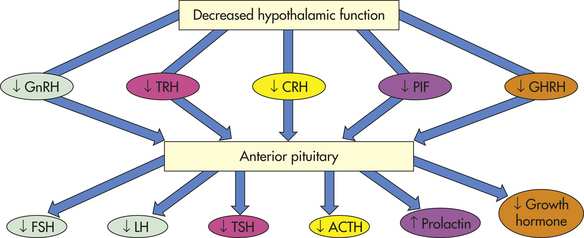
Figure 21-1 Loss of hypothalamic hormones. ACTH, Adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; FSH, follicle-stimulating hormone; GHRH, growth hormone-releasing hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; PIF, prolactin-releasing inhibiting factor (likely dopamine); TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
Diseases of the Posterior Pituitary
Diseases of the posterior pituitary that cause clinically significant alterations in hormone function usually are related to abnormal secretion of antidiuretic hormone (ADH, arginine vasopressin). An excess amount of this hormone results in water retention and a hypoosmolar state, whereas deficiency in the amount or response to ADH results in serum hyperosmolarity. These complex pathophysiologic states not only have significant clinical effects on the modulation of body fluids and electrolytes, but also affect cognitive and emotional responses to stress.5
Syndrome of Inappropriate Antidiuretic Hormone Secretion
Syndrome of inappropriate antidiuretic hormone (SIADH) secretion is characterized by high levels of ADH in the absence of normal physiologic stimuli for its release. SIADH can complicate malignancies, pulmonary disorders, central nervous system disorders, surgical procedures and the use of certain medications.
The most common cause of elevated levels of ADH is ectopically produced ADH. SIADH is associated with some forms of cancer, apparently because of the ectopic secretion of ADH by tumor cells. Tumors that have been reported in association with SIADH include small cell carcinoma of the lung, duodenum, stomach, and pancreas; cancers of the bladder, prostate, and endometrium; lymphomas; and sarcomas.6 Pulmonary disorders associated with SIADH include pneumonia (e.g., tuberculosis), asthma, cystic fibrosis, and respiratory failure requiring mechanical ventilation. Central nervous system disorders that may cause SIADH include encephalitis, meningitis, intracranial hemorrhage, tumors, and trauma.6
Any surgery can result in postoperative fluid volume shifts that result in increased ADH secretion for as long as 5 to 7 days after surgery. The precise mechanism is uncertain but is likely related to fluid and volume changes following surgery, the amount and type of intravenous fluids given, and the use of narcotic analgesics. Transient SIADH is especially common after pituitary surgery because stored ADH is released in an unregulated fashion. Medications are an important cause of SIADH, especially in older adults. These include hypoglycemic medications (chlorpropamide), antidepressants, antipsychotics, narcotics, general anesthetics, chemotherapeutic agents, nonsteroidal anti-inflammatory drugs, and synthetic ADH analogs.6 These drugs serve either to simulate ADH release or enhance the physiologic effects of ADH or have a biologic action similar to ADH.
PATHOPHYSIOLOGY The cardinal features of SIADH are the result of enhanced renal water retention. Water retention results from the action of ADH on renal collecting ducts, where it increases their permeability to water thus increasing water reabsorption by the kidneys. (Renal function is discussed in Chapter 35.) This results in an expansion of extracellular fluid volume that leads to dilutional hyponatremia (low serum sodium), hypoosmolarity, and urine that is inappropriately concentrated with respect to serum osmolarity.6,7
CLINICAL MANIFESTATIONS The symptoms of SIADH are primarily the result of hypotonic (dilutional) hyponatremia. The severity and rapidity of onset of the hyponatremia determine the extent of the symptoms. Thirst, impaired taste, anorexia, dyspnea on exertion, fatigue, and dulled sensorium occur when the serum sodium decreases rapidly from 140 to 130 mEq/L. Peripheral edema is usually absent. Symptoms resolve with correction of hyponatremia. Severe gastrointestinal symptoms, including vomiting and abdominal cramps, occur with a drop in sodium from 130 to 120 mEq/L. With a serum sodium level below 115 mEq/L, confusion, lethargy, muscle twitching, and convulsions may occur. Even if hyponatremia develops slowly, serum sodium levels below 110 to 115 mEq/L are likely to cause severe and sometimes irreversible neurologic damage.
EVALUATION AND TREATMENT A diagnosis of SIADH requires the following signs: (1) serum hypoosmolality (<280 mOsm/kg) and hyponatremia (serum sodium <135 mEq/l); (2) urine hyperosmolarity (i.e., the osmolality of the urine is greater than expected for the concomitant serum osmolality); (3) urine sodium excretion that matches sodium intake; (4) normal renal, adrenal, and thyroid function; and (5) absence of conditions that can alter volume status (e.g., recent diuretic use, heart failure, hypervolemia from any cause, or renal insufficiency).6,8 In order to make the diagnosis of SIADH, the individual should have both normal adrenal and thyroid function because thyroid hormone and glucocorticoids are essential for free water clearance by the kidneys.6
The treatment of SIADH involves the correction of the underlying causal problems; emergency correction of severe hyponatremia by administration of hypertonic saline; and, most importantly, fluid restriction to 600 to 800 ml/day. Careful monitoring is important. If hyponatremia is too rapidly corrected, a severe neurologic syndrome, central pontine myelinolysis, can ensue.6 Resolution usually occurs within 3 days, with a 2- to 3-kg weight loss resulting from enhanced free water clearance. No drug therapy is available to suppress ectopically produced ADH; however, demeclocycline, which causes the renal tubules to develop resistance to ADH, may be used to treat resistant or chronic SIADH. An ADH-receptor agonist, conivaptan, has been approved for the treatment of hospitalized individuals with hyponatremia caused by ADH excess.6,9 Oral forms of ADH receptor antagonists are being developed.9,10
Diabetes Insipidus
Diabetes insipidus (DI) is an insufficiency of ADH, leading to polyuria (frequent urination) and polydipsia (frequent drinking).11,12 There are two forms: neurogenic (central), and nephrogenic (renal). Neurogenic DI is the form encountered most often in clinical practice and is caused by insufficient amounts of ADH.13 The nephrogenic form is caused by an inadequate renal response to ADH.14
Neurogenic DI occurs when any organic lesion of the hypothalamus, pituitary stalk, or posterior pituitary interferes with ADH synthesis, transport, or release.12,13 Causative lesions include primary or secondary brain tumors, hypophysectomy, aneurysms, thrombosis, infections, and immunologic disorders. DI is a well-recognized complication of closed-head trauma.15 Genetic mutations have been identified as a cause of central DI, including those that affect ADH genes directly and those that affect ADH copeptides.12 Uncommonly, central DI can be a hereditary disorder (1% to 2% of cases) characterized by structural changes in the pituitary gland.
Nephrogenic DI is associated with an insensitivity of the renal collecting tubules to ADH. The nephrogenic form of DI can be genetic or acquired.16 Several genetic abnormalities that affect the vasopressin receptor have been noted in DI.12,14,17,18 One of the best described is a mutation in the gene that codes for aquaporin-2, which is one of the four water transport channels in the renal tubule.18,19 Acquired nephrogenic DI is generally related to disorders and drugs that damage the renal tubules or inhibit the generation of cAMP in the tubules. These disorders include pyelonephritis, amyloidosis, destructive uropathies, polycystic disease, and intrinsic renal disease, all of which lead to irreversible DI. Drugs that may induce a reversible form of nephrogenic DI include lithium carbonate, colchicines, amphotericin B, loop diuretics, general anesthetics such as methoxyflurane, and demeclocycline.12,14,16
Psychogenic (primary) polydipsia may be confused with a partial deficiency of ADH. It is caused by the chronic ingestion of extremely large quantities of fluid that wash out the renal medullary concentration gradient, which results in a partial resistance to ADH (see Chapter 36). This condition resolves with effective management of water ingestion.
PATHOPHYSIOLOGY Individuals with DI have partial or total inability to concentrate urine. In neurogenic DI, insufficient ADH is produced. In nephrogenic DI, ADH levels are normal but the collecting ducts do not respond to ADH stimulation. Both of these conditions lead to an inability of the kidney to increase permeability to water. This causes excretion of large volumes of dilute urine, leading to an increase in plasma osmolality. In conscious individuals the thirst mechanism is stimulated and induces polydipsia. For unknown reasons the person usually craves cold drinks. The urine output is varied but can increase from the normal output of 1 to 2 L/day to as much as 8 to 12 L/day. The urine specific gravity is low, from 1.00 to 1.005, which is consistent with the failure to reabsorb water. Dehydration develops rapidly without ongoing fluid replacement. If the individual with DI cannot keep up with the urinary loss of water, serum hypernatremia and hyperosmolality occur. Other serum electrolytes generally are not affected.
CLINICAL MANIFESTATIONS The signs and symptoms of DI include polyuria, nocturia, continuous thirst, and polydipsia. Untreated individuals with long-standing DI may develop a large bladder capacity and hydronephrosis (see Chapter 36).
Idiopathic neurogenic DI usually has an abrupt onset, and many individuals can specifically recall the date of onset of their symptoms. Those with posttraumatic or postneurosurgical DI may develop a classic three-phase syndrome.15,20 Initially, significant diuresis occurs, apparently as a result of acute damage to the hypothalamic centers involving ADH secretion.13 The second phase is one of antidiuresis, which may represent necrosis of denervated tissue of the posterior pituitary with release of ADH into the circulation. The final phase is one of polyuria and polydipsia, reflecting a permanent loss of the ability to secrete adequate amounts of ADH, which does not have to be completely absent for polyuria and polydipsia to occur. Nephrogenic DI usually has a more gradual onset.
EVALUATION AND TREATMENT DI must be distinguished from other polyuric states, including diabetes mellitus, osmotically induced diuresis, and psychogenic polydipsia. The basic criteria for the diagnosis of DI includes polyuria, polydipsia, low urine specific gravity (<1.010), low urine osmolality (<200 mOsml/kg), hypernatremia, high serum osmolality (300 mOsm or more depending on adequate water intake), and continued diuresis despite a serum sodium of 145 mEq/L or greater.11–13
The diagnosis of DI is generally established through water deprivation testing and by correlating the clinical presentation with serum osmolarity and plasma ADH levels.14 Water restriction is a useful test because people without DI respond with a rapid decrease in urine volume and an increase in urine osmolality. People with DI have no decrease in urine volume or increase in urine osmolarity, thus serum osmolality is always higher than urine osmolality in DI after 8 hours of water deprivation. In individuals with severe DI, water deprivation testing can be hazardous. If the individual loses more than 3% of the pretest body weight, circulatory collapse and shock can ensue. The diagnosis of psychogenic polydipsia can be extremely difficult, and differentiation from nephrogenic DI (caused by the washout of renal concentrating gradient) is based on plasma ADH levels.
Treatment for neurogenic DI is based on the extent of the ADH deficiency and on individual variables such as age, endocrine and cardiovascular status, and lifestyle. Individuals who have a urine output in excess of 9 L/day and a urine osmolality of less than 100 mOsm/kg after a dehydration or water restriction test generally require ADH replacement. Replacement therapy for symptomatic neurogenic DI includes administration of the synthetic vasopressin analog desmopressin acetate (DDAVP) given intranasally or orally. Drugs that potentiate the action of otherwise insufficient amounts of endogenous ADH, such as chlorpropamide, carbamazapine, and clofibrate, may be used in individuals with incomplete ADH deficiency.12
Treatment for nephrogenic DI requires treatment of any reversible underlying disorders, discontinuation of etiologic medications, and correction of associated electrolyte disorders. Although the use of thiazide diuretics has been implicated as a cause for DI, they improve salt and water absorption at the proximal tubule and may be helpful in moderate DI.12,14,16
Diseases of the Anterior Pituitary
Disorders of the anterior pituitary may involve either hypofunction or hyperfunction of the gland. Hypopituitarism can range in presentation from the absence of selective pituitary trophic hormones to complete failure of hormonal functions of the anterior pituitary. Hypopituitarism results from either an inadequate supply of hypothalamic-releasing hormones, damage to the pituitary stalk, or an inability of the gland to produce hormones.21–23 Spontaneous mutations of the prophet of pituitary transcription factor (PROP-1) gene involved in early embryonic pituitary development leads to combined hormonal deficiencies.24,25 The hormones include thyroid-stimulating hormone (TSH), growth hormone (GH), adrenocorticotropic hormone (ACTH), and prolactin and cause failure to thrive and short stature in children. Hyperfunction of the anterior pituitary usually results from an adenoma composed of secretory pituitary cells or may, rarely, result from the ectopic production of hypothalamic-releasing peptides.
Hypopituitarism
The most common causes of hypopituitarism lie within the pituitary gland itself. Anterior pituitary hypofunction may result from infarction of the gland, removal or destruction of the gland, or space-occupying lesions such as pituitary adenomas or aneurysms. Adenomas and aneurisms may compress otherwise normal secreting pituitary cells and lead to compromised hormonal output.21,22
One cause of hypopituitarism is pituitary infarction (death of tissue). Infarction may be seen in conjunction with Sheehan syndrome (ischemic pituitary necrosis) caused by severe postpartum hemorrhage. Pituitary infarction is also seen with shock, pituitary apoplexy, sickle cell disease, and during pregnancy in women with diabetes mellitus. Other more common causes of hypopituitarism are genetic abnormalities, head trauma, pituitary tumors, infections (e.g., meningitis, syphilis, tuberculosis), vascular malformations, subarachnoid hemorrhage, surgical ablation related to tumor removal, and granulomatous lesions.22,26
PATHOPHYSIOLOGY The pituitary gland is highly vascular and is therefore extremely vulnerable to ischemia and infarction. In addition, the pituitary relies heavily on portal blood flow from the hypothalamus. In traumatic brain injury, disruption of blood flow can cause infarction with subsequent necrosis and fibrosis of pituitary tissue.21,26 After tissue necrosis, edema with swelling of the gland occurs. Expansion of the pituitary within the fixed compartment of the sella turcica further impedes blood supply to the pituitary. Over time the pituitary undergoes shrinkage, and symptoms of hypopituitarism develop.27
The likelihood of infarction is increased during pregnancy when there is increased size and vasculature of the gland and a rare condition known as Sheehan syndrome may develop. In 1961 Sheehan and Stanfield proposed that the primary pathologic mechanism in postpartum pituitary infarction is vasospasm of the artery supplying the anterior pituitary. A commonly identified cause is some event that leads to circulatory collapse (such as postpartum hemorrhage) and compensatory vasospasm. If vasospasm is sustained for more than several hours, tissue necrosis occurs. The pituitary gland may be particularly susceptible to necrosis because its blood supply, through the hypophyseal system, is already partially deoxygenated and, especially in the hyperplastic pituitary of pregnancy, oxygen demands are increased. A second mechanism that may be involved in pituitary infarction in the postpartum woman with Sheehan syndrome is an increased risk for intravascular coagulation. In such individuals, excessive fibrin is deposited in the pituitary vessels, predisposing the woman to decreased blood supply and infarction of the pituitary.
CLINICAL MANIFESTATIONS The signs and symptoms of hypofunction of the anterior pituitary are highly variable and depend on the affected hormones. If all hormones are absent (a condition termed panhypopituitarism), the individual experiences cortisol deficiency from lack of ACTH, thyroid deficiency from lack of TSH, and gonadal failure and loss of secondary sex characteristics from absence of FSH and LH. A decrease in GH and, consequently, insulin-like growth factor-1 results in delayed growth in children (Figure 21-2) and a vague, multisymptom syndrome in adults.21,22 Children also have dwarfism with GH insensitivity (Laron syndrome), in which the GH receptor is altered.28 Menses may cease from absence of FSH and LH. In addition, postpartum women are unable to lactate because of the absence of prolactin.
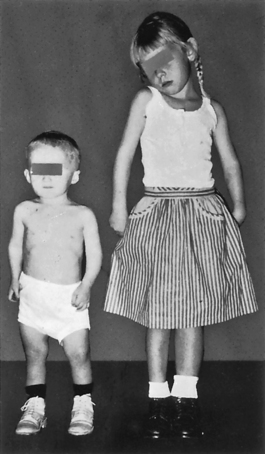
Figure 21-2 Hypopituitary dwarfism. A 4-year-old boy whose height is 25 inches. The girl is also 4 years old and has a normal height of 39 inches. Dwarf has a normal face, as well as head, trunk, and limbs of approximately normal proportions. (From Brashear HR, Raney RB: Shand’s handbook of orthopaedic surgery, ed 10, St Louis, 1986, Mosby.)
ACTH deficiency is a potentially life-threatening disorder because cortisol is required for many aspects of cellular metabolism. ACTH deficiency is usually encountered with generalized pituitary hypofunction and rarely occurs as an isolated event. Within 2 weeks of the complete absence of ACTH, symptoms of cortisol insufficiency develop, including nausea, vomiting, anorexia, fatigue, and weakness. Hypoglycemia is caused by increased insulin sensitivity, decreased glycogen reserves, and decreased gluconeogenesis associated with hypocortisolism. In women, loss of body hair and decreased libido may be caused by decreased adrenal androgen production. ACTH deficiency has a limited effect on aldosterone secretion.
TSH deficiency also is rarely seen in isolation but most often occurs in conjunction with other pituitary hormone deficiencies. The effects of decreased TSH levels may become apparent 4 to 8 weeks after the onset of hypothyrotropinemia. Cold intolerance, dryness of skin, mild myxedema, lethargy, and decreased metabolic rate occur as a result of hypothyroidism induced by decreased TSH levels. The symptoms are usually less severe than those associated with primary hypothyroidism, in which lack of thyroxine is related to disease in the thyroid gland (see p. 739).
The onset of FSH and LH deficiencies in women of reproductive age is associated with amenorrhea and atrophic vagina, uterus, and breasts. In postpubertal males, atrophy of the testes and decreased beard growth occur. Men as well as women experience a decrease in body hair and diminished libido. FSH and LH deficiencies often occur as a result of pressure on the gonadotropes from other sources, such as tumors. If there is enlargement caused by tumor, symptoms may include headache and visual disturbances with blurring and field defects from pressure on the optic chiasm.
GH deficiency occurs in children and adults. In children it may be genetic or it may be the result of tumors such as craniopharyngiomas.28,29 Several genetic defects have been identified in the GH axis that account for impaired GH action.30 The more common type is a recessive mutation in the GHRH gene resulting in a failure of GH secretion. A rare mutation, loss of the GH gene itself, has been observed. Mutations that cause GH insensitivity also have been reported. These mutations may involve the GH receptor, insulin-like growth factor 1 (IGF-1) biosynthesis, IGF-1 receptors, or defects in GH signal transduction.30,31 Individuals with GH insensitivity do not respond normally to exogenously administered GH. Lastly, structural lesions of the pituitary or hypothalamus also may cause GH deficiency and may be associated with other anterior pituitary hormone deficiencies. In adults, GH deficiency is most often caused by structural or functional abnormalities of the pituitary. A decline in GH production is an inevitable consequence of aging and the significance of this phenomenon is poorly understood.32
GH deficiency in children is manifested by growth failure, but not all children with short stature have GH deficiency. Other causes of growth failure not related to GH deficiency include systemic illness, hypothyroidism, malnutrition, and emotional deprivation. Another feature of GH deficiency in children is fasting hypoglycemia, likely due to impaired substrate mobilization for gluconeogenesis and enhanced insulin sensitivity.
An adult GH deficiency syndrome has been described in those who have complete or even partial failure of the anterior pituitary. Symptoms of adult GH deficiency syndrome are vague and include social withdrawal, fatigue, loss of motivation, and a diminished feeling of well-being. Several studies also have documented increased mortality in adults who are GH deficient. Osteoporosis and alterations in body composition (i.e., reduced lean body mass) are common concomitants of adult GH deficiency.
GH replacement therapy has become relatively simple with the introduction of recombinant human growth hormone. In children, GH replacement therapy is monitored by measuring linear growth and IGF-1 levels.28 GH replacement in adults is much more controversial and is generally reserved for those with symptomatic hypopituitarism.21,33
EVALUATION AND TREATMENT The diagnostic evaluation of suspected pituitary disease is often challenging and must be carefully interpreted together with the individual’s signs and symptoms. Simultaneous measurements of the tropic hormones from the pituitary and target endocrine glands are crucial and, in some cases, dynamic testing of the various axes is indicated.21,22 Radiographic assessment of the pituitary (magnetic resonance imaging [MRI] or computed tomography [CT] scans) may demonstrate enlargement of the pituitary, abnormal areas of enhancement suggestive of an adenoma, deviation of the pituitary stalk, or evidence of a locally aggressive tumor. However, some radiographic findings may be nonspecific and require clinical correlation to establish a diagnosis.
In general, treatment of hypopituitarism involves replacing target gland hormone(s) that are deficient because of lack of tropic anterior pituitary hormones.21,22,34 In cases of circulatory collapse, immediate therapy with glucocorticoids and intravenous fluids is critical. Thyroid and cortisol replacement therapy must be maintained. Gender-specific sex steroid replacement therapy is also initiated to improve general well-being and to prevent osteoporosis.
Hyperpituitarism: Primary Adenoma
Pituitary adenomas are usually benign slow-growing tumors that arise from cells of the anterior pituitary, most commonly those that secrete GH and prolactin. The molecular pathogenesis of pituitary adenomas is not clearly understood. The incidence of pituitary adenomas may be as high as 22%, but most of these are microadenomas found incidentally on high-resolution MRI scanning and are asymptomatic.35 The vast majority of pituitary microadenomas are hormonally silent and do not pose significant hazards to the individual.36 More significant adenomas are associated with morbidity and mortality attributable to alterations in hormone secretion or to invasion or impingement of surrounding structures. Primary pituitary carcinomas are rare, representing about 0.2% of all pituitary tumors.37
PATHOPHYSIOLOGY Local expansion of pituitary adenomas may cause both neurologic and secretory defects. Neurologically, the tumor may impinge on the optic chiasm if it extends upward from the sella turcica. This causes a variety of visual disturbances, depending on the area of the optic chiasm that is compressed. If the tumor is locally aggressive, it may invade the cavernous sinus and cause cavernous sinus thrombosis with impairment of the function of the oculomotor, trigeminal, trochlear, and abducens cranial nerves, evoking symptoms relative to their function. Extension also may involve the hypothalamus, disturbing hypothalamic control of wakefulness, thirst, appetite, and temperature.
The adenomatous tissue secretes the hormone of the cell type from which it arose, without regard to physiologic needs and without benefit of regulatory feedback mechanisms. GH-, LH-, and FSH-secreting cells in the pituitary are most sensitive to pressure from expanding tumors within the rigid sella turcica, and as a consequence, hyposecretion of these hormones is most often seen in people with a large pituitary gland.
CLINICAL MANIFESTATIONS The clinical manifestations of pituitary adenomas are related to tumor growth and hormone hypersecretion or hyposecretion. Effects from an increase in tumor size include such nonspecific complaints as headache and fatigue. Visual changes produced by pressure on the optic chiasm include visual field impairments (occasionally beginning in one eye and progressing to the other) and temporary blindness. If the tumor infiltrates other cranial nerves, neurologic function is affected.
Pituitary adenomas arise from hormone-producing cells of the pituitary, and most often are associated with increased secretion of GH and prolactin. Paradoxically, the pressure produced by a pituitary adenoma is also associated with decreased function of neighboring anterior pituitary cells, which results in hyposecretion of other anterior pituitary hormones. For example, gonadotropic hyposecretion often results in menstrual irregularity in women, decreased libido, and receding secondary sex characteristics in men and women. If the tumor exerts sufficient pressure, thyroid and adrenal hypofunction may occur because of lack of TSH and ACTH. These result in the symptoms of hypothyroidism and hypocortisolism.
EVALUATION AND TREATMENT Diagnosis of pituitary adenoma involves physical and laboratory evaluations, including pertinent hormone assays and radiographic examination of the skull. This may be accomplished by CT scanning or dynamic MRI used in conjunction with contrast material. Dynamic MRIs provide superior imaging and greater sensitivity for small lesions in comparison with CT scans.38
The goal of treatment is to protect the individual from the effects of tumor growth and to control hormone hypersecretion or hyposecretion while minimizing damage to appropriately secreting portions of the pituitary. Depending on the tumor size and type, individuals may be treated with specific medications to suppress tumor growth, transsphenoidal tumor resection, or radiation therapy.39
Hypersecretion of Growth Hormone: Acromegaly
Acromegaly occurs in adults who are exposed to continuously excessive levels of GH and concomitant elevation of IGF-1.40 In children and adolescents whose epiphyseal plates have not yet closed, the effect of increased GH levels on long bone growth is termed giantism (Figure 21-3).

Figure 21-3 Giantism. A pituitary giant and dwarf contrasted with normal-size men. Excessive secretion of growth hormone by the anterior lobe of the pituitary gland during the early years of life produces giants of this type, whereas deficient secretion of this substance produces well-formed dwarfs. (From Patton K, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
Acromegaly is a rare disease with an estimated prevalence of 70 persons per million.40 Approximately 15% of all pituitary tumors release excessive GH. The most common cause of acromegaly is a primary autonomous GH-secreting pituitary adenoma.40,41 Acromegaly occurs more often in women than men and is diagnosed most often in adults in their 40s and 50s, although the disease is usually present for years preceding the diagnosis.
Acromegaly is a slowly progressive disease that if untreated is associated with a decreased life expectancy. The increased number of deaths associated with acromegaly are caused by cardiac hypertrophy, hypertension, atherosclerosis, and type 2 diabetes mellitus that lead to coronary artery disease.42 Malignancies, including colon, breast, and lung cancer, are also more common in individuals with acromegaly.40,41
PATHOPHYSIOLOGY With a GH-secreting adenoma, the usual GH baseline secretion pattern is lost, as are sleep-related GH peaks. A totally unpredictable secretory pattern ensues. With only slight elevations of GH, IGF-1 levels increase, stimulating growth. In the adult, epiphyseal closure has occurred and increased amounts of GH and IGF-1 cannot stimulate further long bone growth. Instead, these elevations cause connective tissue proliferation, an increase in the extracytoplasmic matrix, and bony proliferation. Thickening of the articular cartilage with fibrosis is followed by narrowing of the joint spaces and formation of osteophytes, leading to chronic arthritis. These changes affect large joints and the vertebrae, limiting mobility and causing pain.40
GH acts on the renal tubules to increase phosphate reabsorption, leading to mild hyperphosphatemia. The metabolic effects of GH hypersecretion include impaired carbohydrate tolerance and increased metabolic rate. Hyperglycemia may be seen as a result of GH’s inhibition of peripheral glucose uptake and increased hepatic glucose production, followed by insulin resistance and, finally, compensatory hyperinsulinism. Not surprisingly, because of the aforementioned changes in glucose use, approximately one third of people with GH abnormalities have glucose intolerance and half of those individuals develop type 2 diabetes mellitus.40 Type 2 diabetes mellitus occurs when the pancreas is unable to secrete enough insulin to offset the effects of GH.
CLINICAL MANIFESTATIONS As a result of connective tissue proliferation, individuals with acromegaly have enlarged tongues, interstitial edema, increase in the size and function of sebaceous and sweat glands (leading to increased body odor), and coarse skin and body hair. The coarse skin condition becomes very apparent when procedures such as inserting an intravenous needle are performed; the skin is very thick and difficult to penetrate. Bony proliferation results in large joint arthropathy with swelling and decreased range of motion and periosteal vertebral growth, which causes kyphosis.40 Enlargement of the facial bones and the bones of the hands and feet result in protrusion of the lower jaw and forehead and a need for increasingly larger sizes of shoes, hats, rings, and gloves (Figures 21-4 and 21-5).

Figure 21-4 Acromegaly. Chronologic sequence of photographs showing slow development of acromegaly. (From Belchetz P, Hammond P: Mosby’s color atlas and text of diabetes and endocrinology, Edinburgh, 2003, Mosby.)
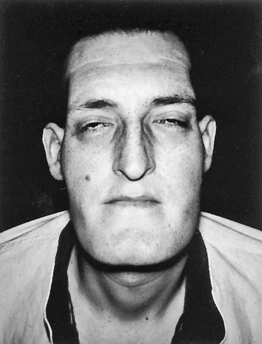
Figure 21-5 Acromegaly. Note large head, forward projection of jaw, and protrusion of frontal bone. (From Thibodeau GA: Anatomy & physiology, St Louis, 1987, Mosby.)
Because IGF-1 stimulates cartilaginous growth, the increased IGF-1 levels cause elongation of ribs at the bone-cartilage junction, leading to a barrel-chest appearance and increased proliferation of cartilage in joints. This in turn causes backache, arthralgia, and arthritis. These are early manifestations of acromegaly. When shaking hands with an individual with acromegaly, one can palpate the large soft tissues. With bony and soft tissue overgrowth, entrapment of nerves may occur, leading to peripheral nerve damage as manifested by weakness, muscular atrophy, footdrop, and sensory changes in the hands.
Although the associated pathophysiology is not clearly understood at present, hypertension and left heart failure are seen in one third to one half of individuals with acromegaly. Cardiomyopathy associated with progressive and unrestrained myocardial growth is a significant factor.40 Headache occurs in 50% to 87% of cases and does not appear related to GH levels, size of the tumor, or presence of hypertension. Because of a space-occupying lesion, central nervous system symptoms of headache, seizure activity, visual disturbances (e.g., bitemporal hemianopia from compression of the optic chiasm), papilledema, and compression hypopituitarism may occur.
If compression hypopituitarism does occur because of a large GH-secreting adenoma, the secretion of the gonadotropins may be affected. This causes amenorrhea in women and loss of libido and erectile dysfunction in men because of pituitary stalk compression. Dopamine delivery to the anterior pituitary is impaired in 30% to 40% of individuals with acromegaly resulting in hyperprolactinemia. In addition, co-secretion of GH and prolactin by the same neoplastic cell line has been documented.40
EVALUATION AND TREATMENT Diagnosis of acromegaly is accomplished by documenting GH suppression during oral glucose tolerance testing and elevated IGF-1 levels.41 The goals of treatment are to normalize GH and IGF-1 serum levels, restoring normal pituitary function and relieving or preventing complications related to tumor expansion. The treatment of choice for acromegaly is transsphenoidal surgical removal of the GH-secreting adenoma.43 Treatment by radiation therapy may be effective when rapid control of GH levels is not essential, when the individual is not a good surgical candidate, or when hyperfunction persists after subtotal resection. Octreotide, octreotide long acting and lanreotide are somatostatin analogs that have been shown to be effective in lowering elevated GH levels, reversing many of the clinical manifestations of the disease, and causing tumor shrinkage in nearly half of individuals.40,43,44 Pegvisomant is an effective drug that induces tissue insensitivity to GH.40,45
Hypersecretion of Prolactin: Prolactinoma
Pituitary tumors that secrete prolactin are called prolactinomas and are the most common of the hormonally active pituitary tumors encountered in clinical medicine.46,47 The physiologic actions of prolactin include breast development during pregnancy, postpartum milk production, and suppression of ovarian function in nursing women.
In addition to pituitary tumors, many conditions or medications can elevate prolactin in the absence of pituitary pathologic condition. For example, renal failure, polycystic ovarian disease (see Chapter 22), primary hypothyroidism, breast stimulation, or even venipuncture can increase prolactin levels.47,48 Prolactin is under tonic inhibitory hypothalamic control through the secretion of dopamine (prolactin inhibitor factor [PIF]).46 Thus medications that block the effects of dopamine at the pituitary can increase prolactin and stimulate proliferation of prolactin-secreting cells (lactotrophies). These include antipsychotics (risperidone, chlorpromazine), metoclopramide, tricyclic antidepressants, methyldopa, and estrogens.46 Any process that interferes with the delivery of dopamine from the hypothalamus to the lactotrophies (pituitary stalk tumor, pituitary stalk transection, or compressive pituitary tumor) also results in hyperprolactinemia. Because thyrotropin-releasing hormone (TRH) stimulates prolactin secretion in addition to enhancing TSH release, prolactin may be elevated in individuals with primary hypothyroidism.
PATHOPHYSIOLOGY The hallmark of a prolactinoma is sustained increases in serum prolactin. Indeed, tumor size roughly correlates with the degree of prolactin elevation. Hyperprolactinemia has several reproductive consequences. Prolactin suppresses GnRH pulses at the hypothalamus, impairs pulsatile pituitary gonadotropin release, and blunts the gonadal responsiveness to gonadotropins. In estrogen- and progesterone-primed breasts, milk production is stimulated.
CLINICAL MANIFESTATIONS Pathologic elevation of prolactin in women results in amenorrhea, nonpuerperal milk production (galactorrhea), hirsutism (excessive body hair in a masculine distribution pattern), and osteopenia caused by estrogen deficiency.46,47 Menstrual abnormalities and galactorrhea are alarming symptoms in women, and, as a result, women generally present earlier in the course of the illness and are found to have microadenomas (less than 1 cm in size). Men, on the other hand, are more likely to have larger tumors at the time of diagnosis with associated compressive or impingement symptoms such as headache or visual impairment. Hyperprolactinemia in men causes hypogonadism, erectile dysfunction, impaired libido, oligospermia, and diminished ejaculate volume.47
EVALUATION AND TREATMENT The diagnostic evaluation of hyperprolactinemia starts with a careful history to exclude medications that may cause elevations in prolactin. Symptoms of hypothyroidism should be elicited, and screening with a serum TSH is mandatory. A careful search for a nonpituitary cause should be pursued if prolactin is less than 50 ng/ml. Prolactin levels more than 200 ng/ml are usually associated with a prolactinoma and are an indication for MRI scanning of the pituitary.46,47
Dopaminergic agonists (bromocriptine, cabergoline, and pergolide) are the treatment of choice for prolactinomas, and their use is often associated with both a rapid reduction in the size of the tumor and a reversal of the gonadal effects of hyperprolactinemia.46,47 Restoration of fertility in previously anovulatory women is common. Although there is an association between valvular heart disease and cabergoline or pergolide used in the treatment of parkinsonism, those individuals receive much higher doses of these medications than those used for prolactinoma. Studies on the long-term safety of cabergoline in treatment of prolactinoma are underway.49 Alternatives to dopaminergic agonists are being developed including somatostatin analogs and prolactin receptor antagonists.47 In individuals resistant or intolerant to these medications, transsphenoidal surgery and radiotherapy are options.47,48
ALTERATIONS OF THYROID FUNCTION
Disorders of thyroid function develop as a result of primary dysfunction or disease of the thyroid gland or, secondarily, as a result of pituitary or hypothalamic alterations. Primary thyroid disorders result in alterations of thyroid hormone (TH) levels with secondary feedback effects on pituitary TSH. For example, when there are primary elevations in TH, TSH will secondarily decrease because of negative feedback. When TH is decreased because of a condition affecting the thyroid gland, TSH will be elevated. Secondary disorders of the thyroid gland are related to disorders of pituitary gland TSH production. When there is excessive TSH production, TH is elevated secondary to the primary elevation of TSH. The reverse is true with inadequate TSH production.
Hyperthyroidism
PATHOPHYSIOLOGY Thyrotoxicosis is a condition that results from any cause of increased levels of circulating TH. The terms thyrotoxicosis and hyperthyroidism are often used interchangeably. The prevalence of hyperthyroidism is estimated to be 1.2% in the United States, of which 0.7% is subclinical.50 Thyrotoxicosis has a variety of causes. Identifying the cause is important because the treatment and expected outcome vary accordingly. Primary hyperthyroidism is a form of thyrotoxicosis in which excess TH is synthesized and secreted by the thyroid gland. Specific diseases that can cause primary hyperthyroidism include Graves disease, toxic multinodular goiter, solitary hyperfunctioning nodules, and, very rarely, follicular thyroid carcinoma. Thyrotoxicosis also can occur transiently in subacute thyroiditis (viral, postpartum, painless, or de Quervain thyroiditis) because of the release of preformed TH; however, this is not considered a cause of true hyperthyroidism because an abnormal amount of TH synthesis does not occur, and therefore the thyrotoxicosis is not sustained.50 Another cause of thyrotoxicosis is ingestion of excess TH medication, sometimes called thyrotoxicosis factitia (Figure 21-6). Secondary hyperthyroidism is very rare and is caused by TSH-secreting pituitary adenomas. Although each of these conditions is associated with specific pathophysiology and manifestations, all forms of thyrotoxicosis share some common characteristics.
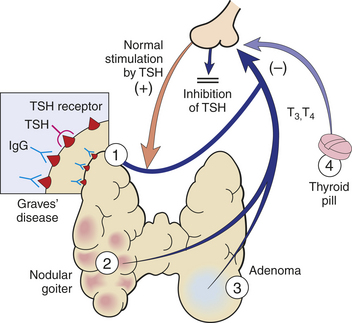
Figure 21-6 Hyperthyroidism may have several causes, among them: 1, Grave disease; 2, toxic multinodular goiter; 3, follicular adenoma; 4, thyroid medication. (From Damjanov I: Pathology for the health professions, St Louis, 2006, Saunders.)
CLINICAL MANIFESTATIONS The clinical features of thyrotoxicosis are attributable to the metabolic effects of increased circulating levels of TH (Figure 21-7). This usually results in an increased metabolic rate with heat intolerance and increased tissue sensitivity to stimulation by the sympathetic division of the autonomic nervous system. The major manifestations are summarized in Table 21-2. Enlargement of the thyroid gland (goiter) is common in hyperthyroid conditions caused by stimulation of TSH receptors.
Table 21-2
Systemic Manifestations of Hyperthyroidism
| System | Clinical Manifestations | Mechanisms Underlying Clinical Manifestations |
| Endocrine | Enlarged thyroid gland (goiter) (97%-99% of cases); systolic or continuous bruit over thyroid; increased cortisol degradation; hypercalcemia and decreased PTH secretion; diminished sensitivity to exogenous insulin | Hyperactivity of the thyroid gland; excess bone resorption leading to hypercalcemia and a disruption of PTH-regulating mechanisms; increased insulin degradation |
| Reproductive | Oligomenorrhea or amenorrhea; erectile dysfunction and decreased libido; increased serum estradiol and estrone but lower than normal levels of free estradiol and estrone | Menstrual cycle alterations that may be related to hypothalamic or pituitary disturbances; increase in sex hormone–binding globulin |
| Gastrointestinal | Weight loss; increased peristalsis leading to less formed and more frequent stools; nausea, vomiting, anorexia, abdominal pain; increased use of hepatic glycogen stores and of adipose and protein stores; decrease in serum lipid levels (including triglycerides, phospholipids, and cholesterol); changes in vitamin metabolism leading to decrease in tissue stores of vitamins | Increased catabolism leading to the body’s inability to meet its metabolic needs; increased glucose absorption; increase in cholesterol excretion in feces and cholesterol conversion to bile salts; impaired conversion of B vitamins to their coenzymes, causing increased need for water-soluble and fat-soluble vitamins |
| Integumentary | Excessive sweating, flushing, and warm skin; heat intolerance; hair fine, soft, and straight; temporary hair loss; nails that grow away from nail beds, palmar erythema | Hyperdynamic circulatory state |
| Sensory (eyes) | Ocular manifestations including elevated upper eyelid leading to decreased blinking and a staring quality; fine tremor of lid; infiltrative ocular changes associated with Graves disease | Overactivity of Mueller muscle; inflammation of retroorbital contents |
| Cardiovascular | Increased cardiac output and decreased peripheral resistance; tachycardia at rest; loud heart sounds; supraventricular dysrhythmias, left ventricular dilation and hypertrophy | Hypermetabolism and need to dissipate heat |
| Nervous | Restlessness; short attention span; compulsive movement; fatigue; tremor; insomnia; increased appetite; emotional lability | Not clearly defined; alterations in cerebral metabolism resulting from excess thyroid hormone |
| Pulmonary | Dyspnea; reduced vital capacity | Weakness of respiratory muscles |
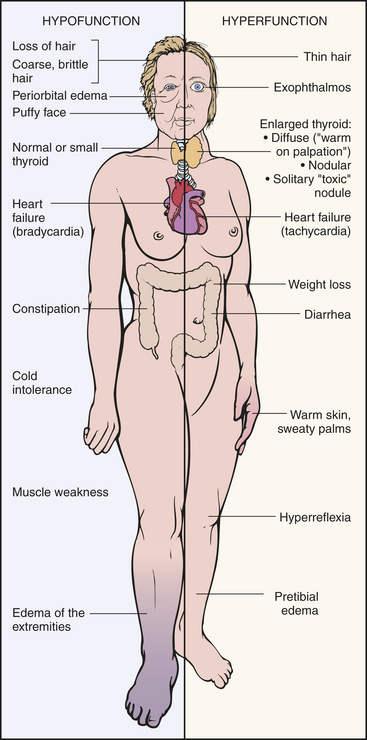
Figure 21-7 Clinical manifestations of hyperthyroidism and hypothyroidism. (From Damjanov I: Pathology for the health professions, St Louis, 2006, Saunders.)
EVALUATION AND TREATMENT The diagnosis of thyrotoxicosis is based on symptoms of TH excess and documentation of increased circulating thyroid hormone levels. Elevated serum free thyroxine (T4) and triiodothyronine (T3) are found in all forms of thyrotoxicosis. In primary hyperthyroidism, TSH is decreased and in secondary hyperthyroidism it is increased. Radioactive iodine uptake (RAIU) can be used in evaluating the etiology of thyrotoxicosis50 (Figure 21-8).
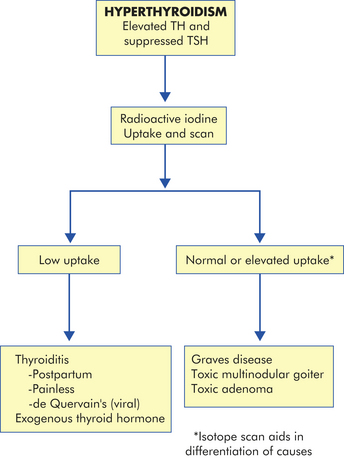
Figure 21-8 Evaluation of hyperthyroidism. Radioactive iodine is used in the differential diagnosis of hyperthyroidism. TH, Thyroid hormone; TSH, thyroid-stimulating hormone.
Treatment is directed at controlling excessive TH production, secretion, or action. The major types of therapy currently used to achieve these goals include antithyroid drug therapy (methimazole or propylthiouracil), radioactive iodine therapy, and surgery. One of the major complications of both radioactive iodine and surgical treatment of hyperthyroidism is excessive ablation of the gland, resulting in hypothyroidism.
Hyperthyroid Conditions
Graves Disease: Graves disease is an autoimmune disease that results in stimulation of the thyroid gland and resultant hyperthyroidism. It is the underlying cause of 50% to 80% of cases of hyperthyroidism and has a prevalence of approximately 0.5% in the U.S. population.51 It occurs more commonly in women. This disease is characterized as a multisystem syndrome consisting of one or more of the following: (1) hyperthyroidism, (2) diffuse thyroid enlargement (goiter), (3) ophthalmopathy, and (4) dermopathy.
Genetic factors interacting with environmental triggers play an important role in the pathogenesis of autoimmune thyroid disease.50–52 Variants in several major histocompatibility complex (MHC) genes have been associated with Graves disease, and it has a concordance rate of 35% for monozygotic twins.51,52 Triggers for the onset of Graves symptoms include stressful life events, recent childbirth, and infection.51
The pathology of Graves disease indicates that normal regulatory mechanisms are overridden by abnormal immunologic mechanisms. T lymphocytes are sensitized to thyroid antigens and stimulate B cells to produce immunoglobulin G (IgG) antibodies that bind to TSH receptors in the thyroid gland and stimulate the synthesis and secretion of excess TH. These autoantibodies are called thyroid-stimulating immunoglobulins (TSI) (also called thyroid-stimulating antibodies [TSAb] or thyroid receptor antibodies [TRAb]) and are found in more than 95% of people with Graves disease.50 The hyperfunction of the thyroid gland leads to suppression of TSH and TRH because of the normal negative feedback from elevated levels of TH. The hyperfunction of the thyroid gland is reflected in a dramatically increased iodide uptake and increased rate of thyroid gland metabolism, which may in turn contribute to hypervascularity and enlargement of the gland (goiter). There is a disproportionate increase in T3 production that reflects long-term hyperstimulation of the thyroid gland.
A small number of individuals with Graves disease and very high levels of TSI experience pretibial myxedema (Graves dermopathy), characterized by subcutaneous swelling on the anterior portions of the legs and by indurated and erythematous skin.53 Thyroid-associated dermopathy is associated with thyrotropin receptor antigens on fibroblasts and recruited T lymphocytes. These manifestations occasionally appear on the hands giving the appearance of clubbing of the fingers (thyroid acropachy).54
Many individuals with Graves disease experience ocular manifestations (Figure 21-9). Two categories of ocular manifestations are associated with Graves disease: (1) functional abnormalities resulting from hyperactivity of the sympathetic division of the autonomic nervous system and (2) infiltrative changes involving the orbital contents with enlargement of the ocular muscles.55 Functional abnormalities occur in most individuals with Graves disease. These abnormalities include a lag of the globe on upward gaze or a lag of the upper lid on downward gaze and are caused by overactivity of Müeller (eyelid) muscles. This manifestation does not affect ocular function and resolves with treatment for hyperthyroidism.56
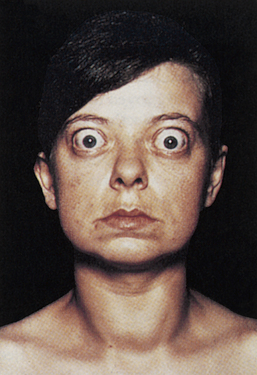
Figure 21-9 Thyrotoxicosis (Graves disease). Note large and protruding eyeballs in association with a large goiter. (From Seidel et al: Mosby’s guide to physical examination, ed 4, St Louis, 1999, Mosby.)
Infiltrative ophthalmopathy occurs in 50% to 70% of individuals with Graves disease. It is characterized by orbital fat accumulation and inflammation with edema of the orbital contents resulting in protrusion of the globe (exophthalmos).55 These changes result in extraocular muscle weakness leading to diplopia (double vision). The individual also may experience irritation, pain, lacrimation, photophobia, and blurred vision. Occasionally, decreased visual acuity, papilledema (edema of the optic nerve), visual field impairment, exposure keratopathy, and corneal ulceration may occur.
Therapy for Graves disease includes antithyroid drugs (propylthiouracil and methimazole), radioactive iodine, or surgery. Unfortunately, current treatment for Graves disease does not reverse the infiltrative ophthalmopathy or the pretibial myxedema. An experienced oculoplastic surgeon and glucocorticoids can help many individuals with progressive ophthalmopathy.56 Skin lesions rarely require treatment, but if they are symptomatic they may respond to topical glucocorticoids.53
Hyperthyroidism Resulting from Nodular Thyroid Disease: The thyroid gland normally enlarges in response to an increased secretion of TSH that may occur in puberty, pregnancy, or iodine deficiency. The increased number of follicles is a compensatory mechanism in response to increased TSH levels. When the condition requiring increased TH resolves, TSH secretion normally subsides and the thyroid gland returns to its original size. Irreversible changes may have occurred in some follicular cells, however, so that such cells then function autonomously and produce excessive amounts of TH. On the other hand, some of these clusters of cells may cease to function. The balance between the amount of TH produced by hyperfunctioning nodules and that produced by the remainder of the gland determines whether an individual becomes euthyroid or hyperthyroid. Once thyrotoxicosis results, the condition generally is termed toxic multinodular goiter; however, only one nodule may become hyperfunctioning and is termed toxic adenoma. Mutations of the TSH receptor have been found in most of the solitary, hyperfunctioning thyroid adenomas.50
Manifestations of hyperthyroidism resulting from toxic multinodular goiter or a toxic adenoma are similar to those of Graves disease, although infiltrative ophthalmopathy and myxedema do not occur. The symptoms usually develop slowly and appear over time. The incidence of malignancy in toxic nodular goiter is estimated to be as high as 9%, so most individuals should undergo MRI or fine-needle aspiration biopsy prior to treatment.57,58 Treatment consists of a combination of antithyroid drugs, radioactive iodine, and surgery.59
Thyrotoxic Crisis: Thyrotoxic crisis (thyroid storm) is a rare but dangerous worsening of the thyrotoxic state, in which death can occur within 48 hours without treatment. The condition may develop spontaneously, but it occurs most often in individuals who have undiagnosed or partially treated severe hyperthyroidism and who are subjected to excessive stress from other causes. These causes may include infection, pulmonary or cardiovascular disorders, trauma, burns, seizures, surgery (especially thyroid surgery), obstetrical complications, emotional distress, or dialysis.60
The systemic symptoms of thyrotoxic crisis include hyperthermia; tachycardia, especially atrial tachydysrhythmias; high-output heart failure; agitation or delirium; and nausea, vomiting, or diarrhea contributing to fluid volume depletion.61–63 Treatment includes (1) the use of drugs that block TH synthesis (i.e., propylthiouracil), (2) the use of beta-blockers for control of cardiovascular symptoms, and (3) supportive care.60,61
Hypothyroidism
Hypothyroidism is the most common disorder of thyroid function and affects between 0.1% and 2% of individuals in the United States.64 Hypothyroidism is caused by a deficient production of TH by the thyroid gland. Hypothyroidism may be primary or secondary. Primary hypothyroidism causes include (1) defective hormone synthesis resulting from autoimmune thyroiditis, endemic iodine deficiency, or iatrogenic loss of thyroid tissue after surgical or radioactive treatment for hyperthyroidism; and (2) congenital defects.64 Secondary (central) hypothyroidism, which is much less common, includes conditions that cause either pituitary or hypothalamic failure with failure to stimulate normal thyroid function.65
PATHOPHYSIOLOGY In primary hypothyroidism the loss of functional thyroid tissue leads to a decreased production of TH (see Chapter 20). Without the negative feedback of TH on the pituitary, there is an increased secretion of TSH that may lead to goiter. On the other hand, the cellular infiltration that occurs in autoimmune thyroiditis also may cause thyroid enlargement independently of the trophic actions of TSH. Secondary hypothyroidism is caused most commonly by failure of the pituitary to synthesize adequate amounts of TSH, thus it is characterized by low TH levels in association with inappropriately low TSH or TRH levels (Figure 21-10). Pituitary adenomas that compress surrounding pituitary cells or as a result of their treatment are the most common causes of secondary hypothyroidism. Other causes include traumatic brain injury, subarachnoid hemorrhage, or Sheehan syndrome.65
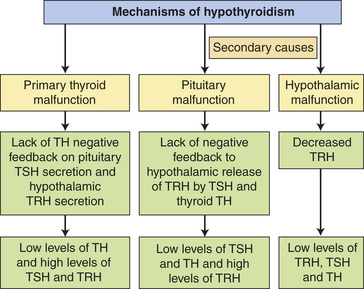
CLINICAL MANIFESTATIONS Hypothyroidism generally affects all body systems, with the extent of the symptoms closely related to the degree of TH deficiency (see Figure 21-7). The onset is usually insidious over months or years. The lowered levels of TH result in decreased energy metabolism and heat production. The individual develops a low basal metabolic rate, cold intolerance, lethargy, tiredness, and slightly lowered basal body temperature. Many organ systems are affected (Table 21-3). The decrease in TH leads to increases in TSH production and may cause goiter.
Table 21-3
Systemic Manifestations of Hypothyroidism
| System | Clinical Manifestations | Mechanisms Underlying Clinical Manifestations |
| Neurologic | Confusion, syncope, slowed speech and thinking, memory loss; lethargy, headaches, hearing loss, night blindness; slow, clumsy movements; cerebellar ataxia; slow alpha-wave activity and loss of amplitude in EEG; reduced cAMP response to epinephrine, glucagons, and PTH stimulation; decreased appetite | Decreased cerebral blood flow leading to cerebral hypoxia; reduced intracellular processes caused by decreased β-adrenergic activity that may be related to a decrease in the number of β-adrenergic receptor sites |
| Endocrine | Increased TSH production in primary hypothyroidism; enlarged pituitary thyrotropes, increase in serum prolactin levels with galactorrhea; decreased rate of cortisol turnover but with normal serum cortisol levels | Impaired TH synthesis or defects in iodide trapping leading to compensatory TSH production; chronic overstimulation of thyrotropes of TRH and by TSH synthesis; stimulation of lactotropes by TRH related to increased prolactin levels; decreased deactivation of cortisol |
| Reproductive | Decreased androgen secretion in men, increased estriol formation in women; low total hormone values but with increased amounts of unbound hormone; anovulation, decreased libido, and a high incidence of spontaneous abortion in women; erectile dysfunction, decreased libido, and oligospermia in men | Altered metabolism of estrogens and androgens; decreased levels of sex hormone–binding globulin |
| Hematologic | Decrease in red cell mass leading to normocytic, normochromic anemia; macrocytic anemia associated with vitamin B12 deficiency and inadequate folate or iron absorption in the gastrointestinal tract | Decreased basal metabolic rate and reduced oxygen requirements, decreased production of erythropoietin, possible relationship between TH and optimal hematologic response to vitamin B12 |
| Cardiovascular | Reduction in stroke volume and heart rate causing lowered cardiac output; increased peripheral vascular resistance to maintain systolic blood pressure; normal response to exercise but with alterations in circulatory system at rest (prolonged circulation time and decreased blood flow to tissues); cool skin and cold tolerance; enlarged heart; decreased intensity of heart sounds and variety of ECG changes (sinus bradycardia, prolonged PR interval, depressed P waves, flattened or inverted T waves, and low-amplitude QRS complexes); cardiac tamponade (although rare) (see Chapter 30) | Decreased metabolic demands and loss of regulatory and rate-setting effects of TH; protein-mucopolysaccharide-rich fluid in the pericardial sac associated with enlarged heart; pericardial effusions associated with heart sounds and ECG changes |
| Pulmonary | Dyspnea; myxedematous changes in respiratory muscles leading to hypoventilation and carbon dioxide retention, which contribute to myxedema coma | Pleural effusions associated with dyspnea, although effusions may be asymptomatic |
| Renal | Reduced renal blood flow and glomerular filtration rate leading to decreased renal excretion of water; increase in total body water and dilutional hyponatremia; reduced production of erythropoietin | Hemodynamic alterations associated with reduced blood flow and filtration; increased total body water related to decreased excretion and mucinous deposits in tissue |
| Gastrointestinal | Constipation, weight gain, and fluid retention; decreased absorption of most nutrients; decreased protein metabolism leading to retarded skeletal and soft-tissue growth and slightly positive nitrogen balance; edema; decreased glucose absorption and delayed glucose uptake; elevated serum lipid values | Reduced intake and reduced peristaltic activity that may progress to fecal impaction; water absorption related to prolonged transit time; fluid retention associated with myxedematous changes; edema associated with high concentrations of exchangeable albumin in the extravascular space caused by increased capillary permeability to proteins; depressed insulin degradation; depressed lipid synthesis and degradation |
| Musculoskeletal | Muscle aching and stiffness; slow movement and slow tendon jerk reflexes; decreased bone formation and resorption, increased bone density; aching and stiffness in joints | Decreased rate of muscle contraction and relaxation contributing to slow movement and reflexes |
| Integumentary | Dry, flaky skin; dry, brittle head and body hair; reduced growth of nails and hair, slow wound healing | Reduced sweat and sebaceous gland secretion |
| Myxedema | Accumulation of hyaluronic acid, which binds water and causes a puffy appearance | |
| Cool skin | Decreased circulation to skin |
cAMP, Cyclic adenosine monophosphate; ECG, electrocardiogram; EEG, electroencephalogram; PTH, parathyroid hormone; TH, thyroid hormone; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
The characteristic sign of severe or long-standing hypothyroidism is myxedema, which is histologically similar to the pretibial myxedema deposits that often occur with Graves disease. Myxedema is a result of an alteration in the composition of the dermis and other tissues. The connective fibers are separated by an increased amount of protein and mucopolysaccharides.
This protein-mucopolysaccharide complex binds water, producing nonpitting, boggy edema, especially around the eyes, hands, and feet and in the supraclavicular fossae (Figure 21-11). Myxedema is also responsible for thickening of the tongue and the laryngeal and pharyngeal mucous membranes. This results in thick, slurred speech and hoarseness, both of which are common in hypothyroidism.
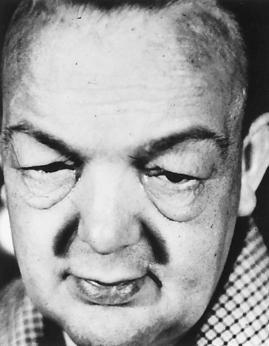
Figure 21-11 Myxedema. Note edema around eyes and facial puffiness. (From Thibodeau GA: Anatomy & physiology, St Louis, 1987, Mosby.)
Myxedema coma, a medical emergency, is a diminished level of consciousness associated with severe hypothyroidism.60,66 Signs and symptoms include hypothermia without shivering, hypoventilation, hypotension, hypoglycemia, and lactic acidosis. Older individuals with severe vascular disease and with moderate or untreated hypothyroidism are particularly at risk for developing myxedema coma. It also may occur after overuse of narcotics or sedatives or after an acute illness in hypothyroid individuals.
EVALUATION AND TREATMENT The diagnosis of primary hypothyroidism is made by documentation of the clinical symptoms of hypothyroidism, and measurement of increased levels of TSH and decreased TH (total T3 and both total and free T4). When hypothyroidism is caused by pituitary deficiencies, serum TSH levels are decreased or are inappropriately normal in the face of low levels of TH.65 Hormone replacement therapy is the treatment of choice for hypothyroidism.67 TH is available as a synthetic hormone (levothyroxine), which is preferred over the crude extract from animal thyroid glands (desiccated thyroid). Treatment of myxedema coma with TH combined with circulatory and ventilatory support is usually effective; however, mortality can be as high as 40% in severe cases.66,68 Subclinical hypothyroidism is estimated to occur in 4% to 8% of U.S. adults and is defined as an elevation in TSH with normal levels of circulating TH.64,69 Treatment of subclinical hypothyroidism remains controversial.67,70
The restoration of normal TH levels should be timed appropriately; a regimen of hormonal therapy depends on the individual’s age, the duration and severity of the hypothyroidism, and the presence of other disorders, particularly cardiovascular disorders. The goal is maximal metabolic restoration consistent with the individual’s overall well-being and normalization of TSH levels in individuals with primary hypothyroidism.67
Hypothyroid Conditions
Primary Hypothyroidism: There are several causes of primary hypothyroidism. Some are associated with spontaneous recovery and resultant euthyroidism, whereas others are linked to permanent hypothyroidism. Iodine deficiency (endemic goiter) is the most common cause of hypothyroidism worldwide, but is relatively rare in the United States. The most common cause of hypothyroidism in the United States is autoimmune thyroiditis (Hashimoto disease, chronic lymphocytic thyroiditis), which results in gradual inflammatory destruction of thyroid tissue by infiltration of lymphocytes and circulating thyroid autoantibodies (antithyroid peroxidase and antithyroglobulin antibodies).71 Variants in MHC antigens have been associated with autoimmune thyroiditis that are different from those found in Graves disease.52,72 Hashimoto disease occurs in genetically predisposed individuals and is associated with high iodine intake, selenium deficiency, smoking, and chronic hepatitis C.73 In addition to thyroid autoantibodies, autoreactive T lymphocytes, antibody activation of natural killer cells (antibody dependent cell mediated cytotoxicity), cytokines, and induction of apoptosis also are involved in the tissue destruction seen in Hashimoto thyroiditis.71,74,75 Goiter formation is commonly observed.
Spontaneous recovery of thyroid function is seen in three conditions: subacute thyroiditis, painless thyroiditis, and postpartum thyroiditis. Subacute thyroiditis is a nonbacterial inflammation of the thyroid often preceded by a viral infection. It is accompanied by fever, tenderness, and enlargement of the thyroid.76 The inflammatory process initially results in elevated levels of TH caused by release of stored thyroglobulin, then is associated with transient hypothyroidism before the gland recovers normal activity.50 Symptoms may last for 2 to 4 months and nonsteroidal anti-inflammatory agents, beta-blockers, and, possibly, TH supplementation may be required during the course of the illness. Painless thyroiditis has a course similar to subacute thyroiditis but is pathologically identical to Hashimoto disease. Iatrogenic hypothyroidism results from radioiodine thyroid ablation, thyroidectomy, and medications (lithium and amiodarone). Postpartum thyroiditis generally occurs within 6 months of delivery, occurs in up to 7% of all women, and has a course similar to painless thyroiditis. Pathologic specimens suggest it is related to Hashimoto disease. Spontaneous recovery is seen in most women affected with this form of thyroiditis; however, persistent hypothyroidism does occur.77
Congenital Hypothyroidism: Congenital hypothyroidism, classified as a rare form of primary hypothyroidism, occurs in infants as a result of absent thyroid tissue (thyroid agenesis) and hereditary defects in TH synthesis. Thyroid agenesis occurs more often in female infants, with permanent abnormalities in 1 of every 3000 to 4000 live births. There is evidence that the incidence of congenital hypothyroidism is increasing in the United States, although the cause of this increase is not known.78
TH is essential for embryonic growth, particularly of brain tissue. The infant will be mentally retarded if there is no T4 during fetal life, but this can be significantly reversed with administration of T4 immediately after birth.
Clinical manifestations of hypothyroidism may not be evident until after 4 months of age. Signs and symptoms include difficulty eating, hoarse cry, and protruding tongue caused by myxedema of oral tissues and vocal cords; hypotonic muscles of the abdomen with constipation, abdominal protrusion, and umbilical hernia; subnormal temperature; lethargy; excessive sleeping; slow pulse; and cold, mottled skin. Skeletal growth is stunted because of impaired protein synthesis, poor absorption of nutrients, and lack of bone mineralization. The individual will become dwarfed, with short limbs, if not treated (cretinism) (Figure 21-12). Dentition is often delayed. Mental retardation is a function of the severity of hypothyroidism and the delay before initiation of treatment.

Figure 21-12 Adult cretin. Note characteristic facial features, dwarfism (44 inches), absent axillary and scant pubic hair, poorly developed breasts, potbelly, and small umbilical hernia. (From Schneeberg NG: Essentials of clinical endocrinology, St Louis, 1970, Mosby.)
Hypothyroidism is difficult to identify at birth, but high birth weight, hypothermia, delay in passing meconium, and neonatal jaundice are suggestive signs. Cord blood can be examined in the first days of life for T4 and TSH levels.79 Treatment is administration of T4. The probability of normal growth and intellectual function is high if treatment is started before the child is 3 or 4 months old. The earlier TH replacement is initiated, the better the child’s outcome. Recent studies suggest that persistent problems with health-related quality of life are common among adolescents treated for congenital hypothyroidism.80,81
Thyroid Carcinoma: Thyroid carcinoma is the most common endocrine malignancy but is relatively rare, accounting for 37,200 estimated new cases and 1630 estimated cancer deaths in 2008 in the United States.82 The most consistent causal risk factor in the development of thyroid cancer appears to be exposure to ionizing radiation, especially exposure during childhood or puberty. Iodine deficiency also affects incidence.83 Papillary and follicular thyroid carcinomas are the most frequent, and medullary and anaplastic thyroid carcinomas are less common. Most tumors are well differentiated.
Most individuals with thyroid carcinoma have normal T3 and T4 levels and are therefore euthyroid. Thyroid cancer typically is discovered as a small thyroid nodule or as a metastatic tumor most commonly occurring in the regional lymph nodes, lungs, brain, or bone. Changes in voice and swallowing and difficulty in breathing are related to tumor growth impinging on the trachea or esophagus. The diagnosis of thyroid carcinoma is generally made by fine-needle aspiration of a thyroid nodule.83 Ultrasonography and radioisotope scanning may be helpful in assessing the malignant potential of a thyroid nodule; however, ultrasound-guided aspiration biopsy of small (less than 1 cm) thyroid nodules is very helpful in providing an earlier diagnosis and earlier institution of therapy.
Treatment for well-differentiated thyroid carcinoma remains somewhat controversial mainly because of its protracted nature and the relatively low mortality regardless of the method of treatment. Treatment of well-differentiated tumors includes a near-total or total thyroidectomy, postoperative radioactive iodine, and suppression of TSH with levothyroxine. Anaplastic thyroid carcinoma carries a grave prognosis, and palliation with surgical debulking, external beam radiotherapy, or chemotherapy may be offered.83,84
ALTERATIONS OF PARATHYROID FUNCTION
Hyperparathyroidism is characterized by a greater than normal secretion of parathyroid hormone (PTH). The causes of hyperparathyroidism are classified as either primary or secondary, and their associated pathophysiologic mechanisms are somewhat different.
PATHOPHYSIOLOGY Primary hyperparathyroidism is characterized by inappropriate excess secretion of PTH by one or more of the parathyroid glands.85–87 It is one of the most common endocrine disorders: 80% to 85% of cases are caused by parathyroid adenomas, another 10% to 15% result from parathyroid hyperplasia, and approximately 1% are caused by parathyroid carcinoma.85 In primary hyperparathyroidism, normal feedback mechanisms, such as elevated serum levels of ionized calcium, fail to normally inhibit PTH secretion by the parathyroid gland.
The cause of primary hyperparathyroidism is unknown; however, recent data suggest that there are two mechanisms for the development of this condition. The first is a clonal proliferation of parathyroid cells with a higher threshold for calcium feedback, and the second is generalized growth of parathyroid tissue. The former is most likely the cause of adenomas, and the latter is probably the cause for hyperplasia. There is also a familial form of the disease that includes a wide range of inherited endocrine disorders such as multiple endocrine neoplasia type 1 (MEN-1).85 Hypercalcemia and hypophosphatemia are the hallmarks of primary hyperparathyroidism. The effects of excessive PTH secretion and primary hyperparathyroidism on various organ systems are summarized in Table 21-4.
Table 21-4
Manifestations of Primary Hyperparathyroidism
| Symptoms | Responsible Derangements | Mechanisms |
| Renal colic, nephrolithiasis, recurrent urinary tract infections, renal failure | Hypercalciuria, hyperphosphaturia, proximal renal tubular bicarbonate leak, urine pH >6 | Calcium phosphate salts precipitate in alkaline urine, renal pelvis, and collecting ducts; calcium oxalate stones also formed |
| Abdominal pain, peptic ulcer disease | Hypercalcemia-stimulated hypergastrinemia | Elevated hydrochloric acid secretion |
| Pancreatitis | Hypercalcemia | Etiology of relationship unknown |
| Bone disease, osteitis fibrosa and osteitis cystica, osteoporosis | PTH-stimulated bone resorption, metabolic acidosis | Osteoporosis now more commonly encountered, but other disorders are more specific for hyperparathyroidism |
| Muscle weakness, myalgia | PTH excess, possible direct effect on striated muscle and on nerves | Characteristic myopathic changes in muscle histology (neuropathy of type I and type II muscle fibers) |
| Neurologic and psychiatric problems (impaired memory, confusion, stupor, coma) | Hypercalcemia | Neuropathy; electroencephalographic changes present |
| Polyuria, polydipsia | Hypercalcemia | Direct effect on renal tubule to decrease responsiveness to antidiuretic hormone |
| Constipation | Hypercalcemia | Decreased peristalsis of gastrointestinal tract |
| Anorexia, nausea, and vomiting | Hypercalcemia | Central stimulation of vomiting center |
| Hypertension | Renal disease, direct effect of calcium on arterial smooth muscle, pheochromocytoma | Plasma rennin activity elevated or normal |
| Arthralgia and arthritis | Gout, pseudogout, periarticular classification | Hyperuricemia, chronic renal failure with high calcium × phosphate product |
From Harden RH et al, editors: William’s textbook of endocrinology, ed 10, Philadelphia, 2002, Saunders.
Secondary hyperparathyroidism is caused by an increase in PTH secondary to a chronic disease state, such as chronic renal failure or intestinal malabsorption, which causes a decrease in serum ionized calcium levels (hypocalcemia). Hypercalcemia does not occur in secondary hyperparathyroidism because the parathyroid tissue is not autonomous and is only responding to a physiologic stimulus (hypocalcemia).
The most common cause of secondary hyperparathyroidism is chronic renal failure (with failure of glomerular filtration), which results in hyperphosphatemia, reduced levels of activated vitamin D, and hypocalcemia, which stimulates PTH secretion. Disturbances in calcium and vitamin D metabolism that arise in chronic renal disease diminish activation of the parathyroid calcium-sensing receptor leading to increases in PTH secretion.88 Because vitamin D metabolism is impaired in renal failure, eucalcemia cannot be restored unless vitamin D supplements are administered.
Other causes of secondary hyperparathyroidism include dietary deficiency in vitamin D or calcium; decreased intestinal absorption of vitamin D or calcium; and ingestion of drugs, such as phenytoin, phenobarbital, and laxatives, which either accelerates the metabolism of vitamin D or decreases intestinal absorption of calcium.
Two other conditions must be differentiated from primary and secondary hyperparathyroidism. Pseudohypoparathyroidism is an inherited condition that presents with increased PTH levels and hypocalcemia. These individuals are resistant to PTH and cannot produce cAMP in response to PTH. Familial hypocalciuric hypercalcemia (FHH) is a condition that can mimic hyperparathyroidism and is characterized by a high serum calcium, low serum phosphate, and low urine calcium excretion. It is caused by a mutation in the calcium-sensing receptor in the parathyroid gland. It can be differentiated from primary hyperparathyroidism by measurement of 24-hour urine calcium excretion.
CLINICAL MANIFESTATIONS Hypersecretion of PTH in primary and secondary hyperparathyroidism causes excessive osteoclastic activity, resulting in bone resorption. (Bone resorption is discussed in Chapter 41.) Pathologic bone changes include pathologic fractures, kyphosis (curvature) of the dorsal spine, and compression fractures of the vertebral bodies.
In primary hyperparathyroidism, hypercalcemia affects proximal renal tubular function, causing hypercalciuria, metabolic acidosis, and production of an abnormally alkaline urine.87 PTH also enhances the renal excretion of phosphate, which results in hypophosphatemia (low serum phosphate) and hyperphosphaturia (increased urine phosphate). The combination of these three variables—hypercalciuria, alkaline urine, and hyperphosphaturia—predisposes the individual to the formation of calcium stones.87 Kidney stones are often formed in the renal pelvis or in the renal collecting ducts and may be associated with infections. Kidney stones and renal infection may lead to impaired renal function. Hypercalcemia also impairs the concentrating ability of the renal tubule by decreasing its response to ADH. Chronic hypercalcemia of hyperparathyroidism is associated with mild insulin resistance, necessitating increased insulin secretion to maintain normal glucose levels. Hypercalcemia also affects the muscular, nervous, and gastrointestinal systems.87 (The clinical symptoms of primary hyperparathyroidism are summarized in Table 21-4.)
Secondary hyperparathyroidism caused by renal disease presents clinically not only with bone resorption but also the symptoms of hypocalcemia and hyperphosphatemia. Hypocalcemia can cause many significant clinical problems (see Chapter 3). Hyperphosphatemia can cause deleterious effects on the cardiovascular system.89
EVALUATION AND TREATMENT The diagnosis of hyperparathyroidism is relatively straightforward. The concurrent findings of increased ionized calcium in the face of elevated or inappropriately normal intact PTH (which documents an abnormal feedback mechanism) are suggestive of primary hyperparathyroidism. Sestamibi (a radioisotope) scanning, CT, or ultrasound is used to localize adenomas prior to surgery.85,86 The definitive treatment of primary hyperparathyroidism is surgery. Surgery is generally reserved for individuals with documented complications of hyperparathyroidism (osteoporosis, nephrolithiasis, or gastrointestinal or neuropsychiatric complications), severely elevated serum calcium levels (more than 1 mg/dl above the upper limit or normal for the laboratory), marked hypercalciuria (more than 400 mg/24 hr), or individuals younger than 50 years of age. In those individuals who fail surgery, other treatments such as bisphosphonates, corticosteroids, and a new class of calcium-lowering drugs, called calcimimetics (e.g., cinacalcet), may be considered.86,90
If hypercalcemia is documented but PTH levels are low, the differential diagnosis shifts to hypercalcemia of malignancy, granulomatous diseases (sarcoidosis), excessive calcium ingestion, or to hypervitaminosis A or D. Treatment of these conditions depends on the underlying cause.
If serum calcium is low but PTH is elevated, secondary hyperparathyroidism is likely. Evaluation for renal function frequently documents chronic renal disease. Treatment for secondary hyperparathyroidism in chronic renal disease requires calcium replacement, dietary phosphate restriction and phosphate binders, and vitamin D replacement.91 Treatment also may include calcimimetics that work to increase the parathyroid calcium receptor sensitivity, thus lowering PTH levels.92,93
Hypoparathyroidism
Hypoparathyroidism (abnormally low PTH levels) most commonly is caused by damage to the parathyroid glands during thyroid surgery.94 Postoperative hypoparathyroidism occurs in approximately 0.5% to 6.6% of all individuals undergoing thyroid surgery.95 This is caused by the anatomic proximity of the parathyroid gland to the thyroid gland. Hypoparathyroidism also is associated with genetic syndromes, including familial hypoparathyroidism and DiGeorge syndrome (velocaridofacial syndrome).94,96 Hypomagnesemia also can cause a decrease in PTH secretion and function.94 An idiopathic or autoimmune form of hypoparathyroidism also is recognized.
PATHOPHYSIOLOGY In hypoparathyroidism a lack of circulating PTH causes a depressed serum calcium level and an increased serum phosphate level. In the absence of PTH the abilities to resorb calcium from bone and to regulate calcium reabsorption from the renal tubules are impaired. The phosphaturic effects of PTH are lost, resulting in hyperphosphatemia.
The effects of hypomagnesemia on the peripheral metabolism and clearance of PTH are not clearly understood. Once serum magnesium levels return to normal, however, PTH secretion returns to normal, as does peripheral tissues’ responsiveness to PTH. Hypomagnesemia may be related to chronic alcoholism, malnutrition, malabsorption, increased renal clearance of magnesium caused by the use of aminoglycoside antibiotics or certain chemotherapeutic agents, or prolonged magnesium-deficient parenteral nutritional therapy.
CLINICAL MANIFESTATIONS Symptoms associated with hypoparathyroidism are related to hypocalcemia. Hypocalcemia causes a lowering of the threshold for nerve and muscle excitation so that a nerve impulse may be initiated by a slight stimulus anywhere along the length of a nerve or muscle fiber. This is manifested as muscle spasms, hyperreflexia, tonic-clonic convulsions, laryngeal spasms, and in severe cases, death from asphyxiation. Chvostek and Trousseau signs may be used to evaluate for neuromuscular irritability. Chvostek sign is elicited by tapping the cheek resulting in twitching of the upper lip. Trousseau sign is elicited by sustained inflation of a sphygmomanometer placed on the upper arm to a level above the systolic blood pressure with resultant painful carpal spasm.94
Other symptoms of hypocalcemia are caused by mechanisms that are not yet understood. These symptoms include dry skin, loss of body and scalp hair, hypoplasia of developing teeth, horizontal ridges on the nails, cataracts, basal ganglia calcifications (which may be associated with a parkinsonian syndrome), and bone deformities, including brachydactyly and bowing of the long bones.
Phosphate retention caused by increased renal reabsorption of phosphate is associated also with hypoparathyroidism. Hyperphosphatemia is associated with inhibition of the renal enzyme necessary for the conversion of vitamin D to its most active form. This enzyme, 25-OH vitamin D 1α-hydroxylase also is required by PTH. This tends to depress serum calcium levels further by reducing gastrointestinal absorption of calcium.
EVALUATION AND TREATMENT A low serum calcium level and high phosphorus level in the absence of renal failure, intestinal disorders, or nutritional deficiencies suggest hypoparathyroidism. Intact PTH levels are low in hypoparathyroidism, and measurement of serum magnesium and urinary calcium excretion can help in diagnosis.94 There is an inherited condition associated with hypocalcemia but with normal levels of PTH, called pseudohypoparathyroidism (see p. 743).
The treatment of hypoparathyroidism is directed toward the alleviation of hypocalcemia. In acute states this involves parenteral administration of calcium, which allows correction of serum calcium within minutes. Maintenance of serum calcium is achieved with pharmacologic doses of an active form of vitamin D and oral calcium. Injectable human PTH (teriparatide) has been explored in several recent studies, and results are encouraging.94,97 As serum calcium levels return to normal, phosphaturia usually is stimulated. This leads to a return to normal serum phosphate levels. In some individuals, however, the absence of the phosphaturic effect of PTH causes a persistent hyperphosphatemia. Significant elevations of phosphorus should be treated with drugs that inhibit gastrointestinal absorption of phosphate (phosphate binders).94
DYSFUNCTION OF THE ENDOCRINE PANCREAS: DIABETES MELLITUS
Diabetes mellitus is not a single disease but a group of clinically heterogeneous disorders that have glucose intolerance in common. It encompasses many causally unrelated diseases and includes many different etiologies of disturbed glucose tolerance. The term diabetes mellitus is used to describe a syndrome characterized by chronic hyperglycemia and other disturbances of carbohydrate, fat, and protein metabolism. The American Diabetes Association classifies four categories of diabetes mellitus (Table 21-5):
Table 21-5
Classification and Characteristics of Diabetes Mellitus

Data from American Diabetes Association (Committee Report): Diabetic Care 26(Suppl. 1):S5-S20, 2003; Agency for Healthcare Quality and Research, U.S. Preventive Services Task Force (USPSTF): Screening for Gestational Diabetes Mellitus Recommendation Statement May 2008. Available at http://www.ahqr.gov/clinic/uspstf08/gestdiab/gdrs.htm
1. Type 1 (absolute insulin deficiency)
2. Type 2 (insulin resistance with an insulin secretory deficit)
Types 1 and 2 diabetes are the most common and are discussed in greatest detail in this text.
The criteria for the diagnosis of diabetes include symptoms, elevated fasting plasma glucose (FPG) concentration, and/or abnormal oral glucose tolerance test (OGTT)98 (Box 21-1). Two conditions associated with a high risk for diabetes, impaired fasting glucose (IFG) and impaired glucose tolerance (IGT), are considered prediabetes.98
A normal fasting glucose is less than 100 mg/dl. IFG is defined as a fasting glucose greater than or equal to 100 mg/dl but less than 126 mg/dl. Glucose tolerance is normal if the 2-hour postload glucose level is less than 140 mg/dl. IGT is defined as a 2-hour postload glucose level greater than or equal to 140 but less than 200 mg/dl. IGT results from reduced suppression of hepatic glucose output and reduced pancreatic islet cell function.
Any abnormality of glucose tolerance has potentially serious consequences. Numerous epidemiologic studies have shown an increased risk of cardiovascular disease and premature death in individuals with glucose intolerance. In addition, individuals with abnormal glucose tolerance have a 3% to 7% yearly risk of developing overt diabetes. In comparison, a healthy individual’s lifetime risk of acquiring diabetes is around 7% to 10%.
Types of Diabetes Mellitus
Diabetes mellitus is the most common pediatric chronic disease and affects 0.17% of U.S. children.99 The common form of type 1 diabetes mellitus is the result of an autoimmune-mediated specific loss of beta cells in the pancreatic islets. The incidence of the condition is increasing in some areas, with other areas showing no change in incidence.99 Table 21-6 summarizes the epidemiology of diabetes mellitus.
Table 21-6
Epidemiology and Etiology of Diabetes Mellitus in the United States
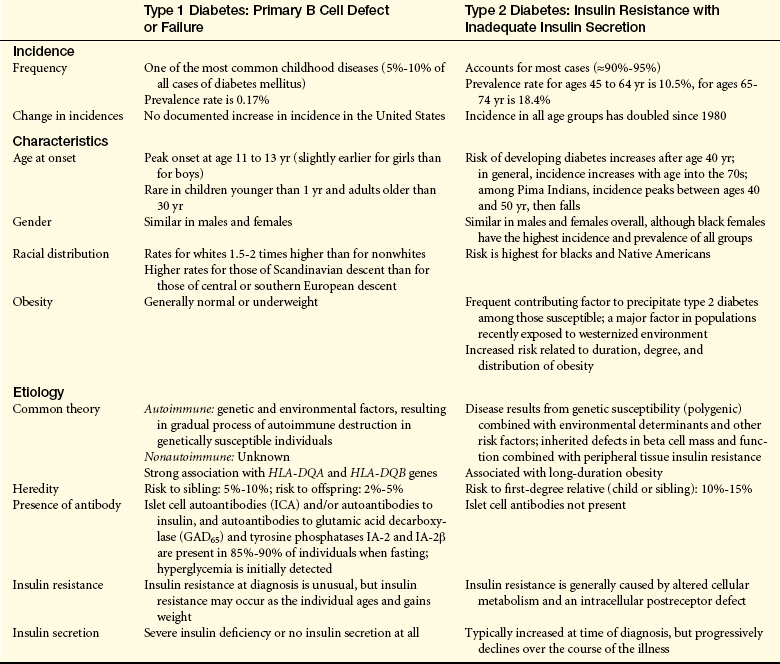
Data from American Diabetes Association: Diabetic Care 30(Suppl 1):S42-S47, 2007.
Type 1 diabetes mellitus is thought to be the result of a genetic-environmental interaction. Between 10% and 13% of individuals with newly diagnosed type 1 diabetes have a first-degree relative (parent or sibling) with type 1 diabetes. Diagnosis has a seasonal distribution, with more cases reported during autumn and winter in the northern hemisphere. Diagnosis is rare during the first 9 months of life and peaks at 12 years of age.
PATHOPHYSIOLOGY Two distinct types of type 1 diabetes have been identified: autoimmune and nonimmune. In autoimmune-mediated diabetes mellitus, environmental-genetic factors are thought to trigger cell-mediated destruction of pancreatic beta cells. Autoimmune type 1 diabetes is called type 1A. Nonimmune type 1 diabetes is far less common than immune. It occurs secondary to other diseases, such as pancreatitis, or to a more fulminant disorder termed idiopathic (type 1B) diabetes. Type 1B diabetes occurs mostly in people of Asian or African descent and affected individuals have varying degrees of insulin deficiency.100
Genetic Susceptibility: The exact nature of genetic susceptibility to type 1A diabetes is not clearly understood. The strongest association is with MHC (histocompatibility leukocyte antigen [HLA]) class II alleles HLA-DQ and HLA-DR).100–102 The HLA-DR marker is associated with other autoimmune disorders, such as Graves, Hashimoto, and Addison diseases.101 The risk of developing type 1 diabetes increases 5 to 8 times when one of those specific markers is present. When the individual is heterogeneous for HLA-DR3 and HLA-DR4, the risk is 20 to 40 times higher than that of the general population. Specific human antigens also are thought to decrease the risk of developing type 1 diabetes. For example, HLA-DR2 is associated with an unusually low risk. Current theories of causation hold that islet cell destruction occurs predominantly in genetically susceptible people.
Environmental Factors: Environmental factors are thought to have a significant contribution to the development of type 1 diabetes mellitus. Some types of viral infections have been implicated with autoimmune damage to beta cells, including congenital rubella, cytomegaloviruses, mumps, and Epstein-Barr virus.102,103 Bovine serum albumin, a major constituent of cow’s milk, may be involved in triggering beta cell autoantibodies.102 Stress may advance development of type 1 diabetes mellitus by stimulating secretion of counterregulatory hormones and affecting immune responses (see Chapter 10). Specific environmental factors linked to type 1 diabetes mellitus are presented in Box 21-2.
Immunologically Mediated Destruction of Beta Cells: Type 1 diabetes mellitus is a slowly progressive autoimmune T cell–mediated disease that occurs in genetically susceptible individuals (Figure 21-13 and see Chapter 8). Environmental mechanisms are thought to play a role in triggering an autoimmune response to beta cells. The destruction of beta cells progresses through the following stages:
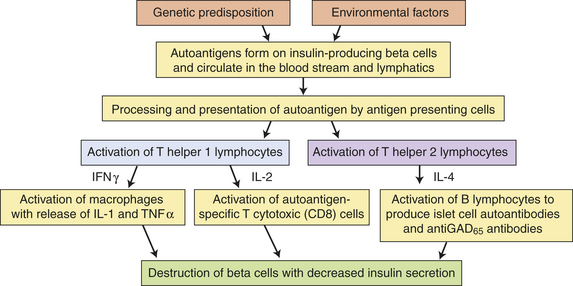
Figure 21-13 Pathophysiology of type 1 diabetes mellitus. GAD65, glutamic acid decarboxylase; INF-γ Interferon-gamma; IL, interleukin; TNF-α, tumor necrosis factor-alpha.
1. Lymphocyte and macrophage infiltration of the islets resulting in inflammation (insulinitis) and islet beta cell death. Autoantigens are expressed on the surface of pancreatic islet cells and circulate in the bloodstream and lymphatics (see Figure 21-13). Circulating autoantigens are ingested by antigen-presenting cells that activate CD4+ T helper lymphocytes.100,104 The activated T helper lymphocytes secrete interleukin 2 (IL-2) that activates beta cell autoantigen-specific T cytotoxic lymphocytes, causing them to proliferate and attack many more islet cells through secretion of toxic perforins and granzymes.104,105 The T helper lymphocytes also secrete interferon that activates macrophages and stimulates the release of inflammatory cytokines (including IL-1 and tumor necrosis factor [TNF]), which cause further B-cell destruction and apoptosis.100,104,106
2. Production of autoantibodies against islet cells, insulin, glutamic acid decarboxylase (GAD), and other cytoplasmic proteins. T helper lymphocytes also produce IL-4, which stimulates B lymphocytes to proliferate and produce antibodies (see Figure 21-13). Islet cell autoantibodies (ICAs) precede evidence of beta cell deficiency and can be found in the serum years before symptoms occur.100,102,106,107 Antiglutamic acid decarboxylase (antiGAD65) antibodies (an enzyme in beta cells that is involved in glucagon synthesis) are more persistent which makes them clinically useful in differentiating the etiology of diabetes in a given individual.100 Autoantibodies against insulin (insulin autoantibodies [IAAs]) also have been noted. It is likely that IAAs may form during the process of active islet cell and beta cell destruction.
Another mechanism being explored in the pathogenesis of type 1 diabetes is a relative inactivity of T regulatory cells. These T lymphocytes normally serve to inhibit the immune response. Mutations affecting T regulatory cells have been found in a rare form of diabetes called neonatal diabetes, leading investigators to hypothesize about a role for T regulatory cell dysfunction in type 1A diabetes.101,108
Over time these immune mechanisms lead to a decrease in beta cell mass and insulin production. C-peptide, a component of proinsulin cleaved during insulin synthesis, declines. C-peptide may play a protective role in preventing beta cell destruction; therefore, decreasing amounts of this important peptide may accelerate the decline in beta cells.109 C-peptide deficiency also has been implicated in the glomerular and neuronal complications of diabetes.110–113 There may be further effects on beta cell decline as the individual ages. For example, the “accelerator hypothesis” suggests that increased body weight and associated proinflammatory state are contributors to worsening hyperglycemia in type 1 and type 2 diabetes.114
Hyperglycemia, Glucagon, and Hyperketonemia: Before hyperglycemia occurs, 80% to 90% of the function of the insulin-secreting beta cells in the islet of Langerhans must be lost. Beta cell abnormalities are present long before the acute clinical onset of type 1 diabetes.
A disequilibrium of hormones produced by the islets of Langerhans occurs in diabetes mellitus. Normally the paracrine action of insulin suppresses secretion of glucagon. Alpha and beta cell functions are abnormal, and a lack of insulin and a relative excess of glucagon (produced by alpha cells) exist in type 1 diabetes. The ratio of insulin to glucagon in the portal vein controls hepatic glucose and fat metabolism. Considerable data have documented that high levels of glucagon relative to insulin levels contribute to the generation of hyperglycemia and hyperketonemia. Relative hyperglucagonemia occurs in every form of diabetes mellitus. Thus the full metabolic syndrome seen in diabetes is caused by both hormones, a finding that ultimately may provide an entirely new therapeutic approach to its management.
Insulin normally stimulates lipogenesis and inhibits lipolysis, thus preventing fat catabolism. With insulin deficiency, lypolysis is enhanced and there is an increase in the amount of nonesterified fatty acids delivered to the liver. The consequence is increased glyconeogenesis contributing to hyperglycemia and production of ketone bodies (acetoacetate, hydroxybutyrate, and acetone) by the mitochondria of the liver at a rate that exceeds peripheral use. Accumulation of ketone bodies causes a drop in pH and triggers the buffering system associated with metabolic acidosis. Diabetic ketoacidosis (DKA), caused by increased levels of circulating ketones in the absence of the antilipolytic effect of insulin, may occur (p. 755).
CLINICAL MANIFESTATIONS Historically, type 1 diabetes mellitus has been thought to have an abrupt onset. More recently, however, prospective studies show a distinctive natural history involving a long preclinical period with gradual destruction of beta cells, eventually leading to insulin deficiency and hyperglycemia. Generally, this latent period is longer in older individuals and often results in misclassification of an older type 1 individual as having type 2 diabetes.
Type 1 diabetes mellitus affects the metabolism of fat, protein, and carbohydrates. Glucose accumulates in the blood and appears in the urine as the renal threshold for glucose is exceeded, producing an osmotic diuresis and symptoms of polyuria and thirst. Wide fluctuations in blood glucose levels occur. In addition, protein and fat breakdown occur because of the lack of insulin, resulting in weight loss (Table 21-7).
Table 21-7
Clinical Manifestations and Rationale for Type 1 Diabetes Mellitus
| Manifestations | Rationale |
| Polydipsia | Because of elevated blood sugar levels, water is osmotically attracted from body cells, resulting in intracellular dehydration and hypothalamic stimulation of thirst |
| Polyuria | Hyperglycemia acts as an osmotic diuretic; the amount of glucose filtered by the glomeruli of the kidneys exceeds that which can be reabsorbed by the renal tubules; glycosuria results, accompanied by large amounts of water lost in the urine |
| Polyphagia | Depletion of cellular stores of carbohydrates, fats, and protein results in cellular starvation and a corresponding increase in hunger |
| Weight loss | Weight loss occurs because of fluid loss in osmotic diuresis and the loss of body tissue as fat and proteins are used for energy as a result of the effects of insulin deficiency |
| Fatigue | Metabolic changes result in poor use of food products, contributing to lethargy and fatigue; sleep loss from severe nocturia also contributes to fatigue |
EVALUATION AND TREATMENT The diagnosis of diabetes is not difficult when the symptoms of polydipsia, polyuria, polyphagia, weight loss, and hyperglycemia are present in fasting and postprandial states. Nearly half of children ages 4 years and younger and nearly one quarter of those between the ages of 5 and 15 with type 1 diabetes are first diagnosed when they present with the signs and symptoms of DKA.100 Acidosis causes a compensatory increase in the respiratory rate and depth known as Kussmaul respirations. Acetone (a ketone body) is blown off, giving the breath a sweet or fruity odor.
If the diagnosis is equivocal, based on FPG, then an OGTT may be needed. In nonpregnant women, an OGTT consists of the administration of a 75-g oral glucose load after a 10-hour fast followed by measurement of plasma glucose 2 hours later. Another mechanism that can be used to identify the plasma glucose concentration over time is the measurement of glycosylated hemoglobin or, more precisely, hemoglobin A1c (HgbA1c). In the normal 120-day life span of a red blood cell, glucose molecules join hemoglobin, forming glycosylated hemoglobin. In individuals with persistent hyperglycemia, increases in the quantities of three glycosylated hemoglobins (A1a, A1b, and A1c) are noted. A buildup of glycosylated hemoglobin within the red cell reflects the average level of glucose to which the cell has been exposed during its life cycle (approximately 120 days). HgbA1c can be used for screening chronic hyperglycemia and to assess the effectiveness of therapy by monitoring long-term serum glucose regulation.
C-peptide, a component of proinsulin released during insulin production, can be measured in the serum as a surrogate for insulin levels and is indicative of residual beta cell mass and function. Other important aspects of evaluation include looking for evidence of the acute and chronic complications of type 1 diabetes, including DKA and renal, nervous system, cardiac, peripheral vascular, retinal, and bony tissue damage (see pp. 754 and 758).
Many different kinds of therapies are being tested to prevent the autoimmune destruction of beta cells that is characteristic of type 1 diabetes. These include immunosuppression with antirejection drugs (e.g., mycophenolate mofetil, rituximab, monoclonal antibodies to CD3, and cyclosporine), immunomodulation therapies (e.g., nicotinamide, bacille Calmette-Guérin, vitamin D, and DiaPep277), and oral or intranasal insulin.101,107,115 Some of these studies are ongoing; however, the results so far have shown some potential for slowing disease progression but not in preventing diabetes. Many new clinical trials are planned.
Treatment regimens are designed to achieve optimal glucose control (as measured by the HgbA1c) without causing episodes of significant hypoglycemia. The Diabetes Control and Complications Trial (DCCT) compared individuals whose blood sugars were tightly controlled (with blood glucose checks four times per day, three or more insulin injections daily or insulin pump, and meal planning) with those who received standard treatment. Intensively treated individuals who achieve near-normal glucoses (HgbA1c less than 7%) can expect a 50% to 75% reduction in the risk of developing or progression of retinopathy, neuropathy, and nephropathy after 8 to 9 years.116 Although long-term complications were decreased in the tightly controlled group, achieving near-normal glucose levels was accompanied by risks, such as severe hypoglycemia and weight gain. However, the benefits of tight glucose control in the intensively treated group continued to be evident for more than 20 years.100,117,118 Successful management requires individual planning according to type of disease, age, and activity level. All individuals with type 1 diabetes require some combination of insulin, meal planning, exercise, and self-monitoring of blood glucose.100,118,119 Several different types of insulin preparations are available, as well as new technologies for more physiologic insulin delivery systems.120 Blood glucose monitoring also is an essential part of management for which there are numerous types of monitoring devices. Finally, islet cell and whole pancreas transplantation has been successful in selected individuals (see What’s New? Islet Transplant and Type 1 Diabetes Mellitus). Individuals should be screened at least yearly for complications of diabetes.
Type 2 Diabetes Mellitus
In the United States, type 2 diabetes mellitus affects 10.5% of those ages 45 to 64 years (up from 5.6% in 1985) and 18.4% of those ages 65 to 74 years (up from 10.2% in 1985).99 The incidence of diabetes has doubled in all adult age groups in the past two decades. Prevalence varies by ethnic group and gender. Although the increase in overall prevalence of diabetes has increased the most in white men (116% increase since 1980), the condition remains most common in black women with an overall prevalence of 8.8% and a prevalence of 34% in those ages 65 to 74.99 There also is an increased prevalence of type 2 diabetes in children, especially in Native American children ages 15 to 1999 and in obese children121–123 (see Table 21-6).
An environmental-genetic interaction appears to be responsible for type 2 diabetes. The most well-recognized risk factors are age, obesity, hypertension, physical inactivity, and family history. The metabolic syndrome is a constellation of disorders (central obesity, dyslipidemia, prehypertension, and a fasting blood glucose more than or equal to 100 mg/dl) that together confer a high risk of developing type 2 diabetes and associated cardiovascular complications (see What’s New? Metabolic Syndrome and Diagnosis). Other novel risk factors being investigated include elevated C-reactive protein, decreased adiponectin, increased leptin, and increased IL-6.124 A unique manifestation of insulin resistance in women of reproductive age is polycystic ovary syndrome (PCOS), which is associated with a risk of diabetes seven times the average risk for women without PCOS.
PATHOPHYSIOLOGY Many genes have been identified that are associated with type 2 diabetes, including those that code for beta cell mass, beta cell function (ability to sense
blood glucose levels, insulin synthesis, and insulin secretion), proinsulin and insulin molecular structure, insulin receptors, hepatic synthesis of glucose, glucagon synthesis, and cellular responsiveness to insulin stimulation.125–128 These genetic abnormalities combined with environmental influences, such as obesity, result in the basic pathophysiologic mechanisms of type 2 diabetes: insulin resistance and decreased insulin secretion by beta cells (Figure 21-14). Both of these mechanisms are essential to the development of type 2 diabetes. Although many individuals with risk factors for type 2 diabetes (including obesity, metabolic syndrome, and hypertension) are insulin resistant, only those individuals who are genetically predisposed to beta cell dysfunction (and therefore a relative deficiency in insulin) will develop type 2 diabetes.129–130
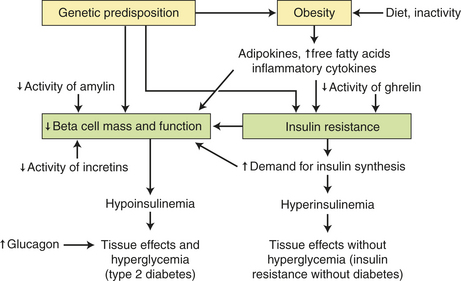
Insulin resistance is defined as a suboptimal response of insulin-sensitive tissues (especially liver, muscle, and adipose tissue) to insulin. Several mechanisms are involved in abnormalities of the insulin signaling pathway and contribute to insulin resistance. These include an abnormality of the insulin molecule, high amounts of insulin antagonists, down-regulation of the insulin receptor, decreased or abnormal activation of postreceptor kinases, and alteration of glucose transporter (GLUT) proteins.131 Obesity is present in 60% to 80% of those with type 2 diabetes and is a major contributor to insulin resistance through several important mechanisms:
1. Adipokines are hormones produced in adipose tissue. A nuclear receptor, called peroxisome proliferator-activated receptor gamma (PPARγ), is highly expressed in adipose cells and is responsible for modulating the changes in adipokines, including increased serum levels of leptin (leptin resistance) and resistin and decreased levels of adiponectin.128,130–133 These changes are associated with decreased insulin sensitivity. Although the specific mechanisms by which these adipokines alter insulin sensitivity are still being explored, a group of insulin-sensitizing drugs, called the thiazolidinediones
that modulate PPARγ activity, have been used in the treatment of type 2 diabetes for many years.
2. Elevated serum free fatty acids (FFAs) and intracellular deposits of triglycerides and cholesterol are also found in obese individuals who have what has been termed “metabolic overload” (high caloric and lipid intake). These changes interfere with intracellular insulin signaling and thus decrease tissue responses to insulin. Increases in fatty acids also cause alterations in insulin secretion within the beta cell.131,133–135
3. Inflammatory cytokines (TNF-α, IL-6) are released from intra-abdominal adipocytes or adipocyte-associated mononuclear cells and induce insulin resistance through a postreceptor mechanism.132,135–138
4. Obesity is correlated with hyperinsulinemia and decreased insulin receptor density.
Compensatory hyperinsulinemia prevents the clinical appearance of diabetes for many years. Eventually, however, beta cell dysfunction develops and leads to a relative deficiency of insulin activity. The islet dysfunction may be caused by a decrease in beta cell mass, abnormal function of the beta cells, or some combination. A progressive decrease in the weight and number of beta cells occurs in type 2 diabetes, and several different mechanisms have been implicated. Beta cells are extremely sensitive to high levels of glucose and free fatty acids, and under these so-called glucolipotoxic conditions, beta cells undergo apoptotic cell death.128,130,132,139 The adipokine leptin decreases insulin synthesis in the beta cell. A variety of inflammatory cytokines, including TNF-α and IL-1β, have also been shown to be toxic to beta cells.128,130,132,140 Thus many of the obesity-related causes of insulin resistance (elevated free fatty acids [FFA] hyperglycemia, adipokines, and inflammatory cytokines) also promote programmed cell death in B cells. Beta cell “exhaustion” from increased demand for insulin biosynthesis, associated with intracellular oxidative stress and endoplasmic reticulum dysfunction, also has been implicated in beta cell apoptosis.132,141
Glucagon is a hormone produced by the alpha cells of the pancreas and acts primarily in the liver to increase blood glucose by stimulating glycogenolysis and gluconeogenesis. Glucagon acts as an antagonist to insulin. In healthy individuals, high glucose levels cause glucagon release to be inhibited. In type 2 diabetes, pancreatic alpha cells are less responsive to glucose inhibition resulting in increased glucagon secretion.142,143 These abnormally high levels of glucagon have long been known to play a role in the increased hepatic production of glucose and resultant hyperglycemia seen in type 2 diabetes.
Amylin is a hormone co-secreted with insulin by the beta cells. A deficiency of amylin in type 1 and type 2 diabetes parallels the reduction in insulin secretion. Amylin inhibits glucagon secretion, and problems with glycemic control may be related to altered glucagon control or assimilation of nutrients in relation to the deficit of amylin. Drugs aimed at improving amylin function are being studied.144 Amyloid deposition in the pancreas resulting in islet cell destruction also may be related to changes in amylin function.132,145,146 However, the role of amyloid accumulation in the pathogenesis of type 2 diabetes is unclear.
The incretins are a class of peptides released from the gastrointestinal tract in response to food intake that increase the sensitivity of beta cells to circulating glucose levels, thus improving insulin responsiveness to meals. Glucagon-like peptide 1 (GLP-1) is cleaved from proglucagon in the intestinal mucosa. Another incretin is glucose-dependent insulinotropic polypeptide (GIP), which is synthesized in the duodenum and jejunum. These incretins bind to receptors on beta cells and increase the synthesis and secretion of insulin in response to glucose levels.147 They are then inactivated by the enzyme dipeptidyl peptidase IV (DPP-IV). Currently one GLP-1 agonist and one DPP-IV inhibitor are approved for use in the United States and others are being evaluated for the treatment of type 2 diabetes147–150 (see What’s New? Incretin Hormones for Diabetes Mellitus Therapy).
Ghrelin is a peptide produced in the stomach and pancreatic islets that stimulates GH receptors. Decreased levels of circulating ghrelin have been associated with insulin resistance and increased fasting insulin levels; its use as a potential treatment for type 2 diabetes is being investigated.151
CLINICAL MANIFESTATIONS Clinical manifestations of type 2 diabetes are often nonspecific. Although younger people may develop the condition, it generally affects those older than 30 years. The individual often is overweight, dyslipidemic, hyperinsulinemic, and hypertensive. The individual with type 2 diabetes may show some classic symptoms of diabetes, such as polyuria and polydipsia, but more often will have nonspecific symptoms such as fatigue, pruritus, recurrent infections, visual changes, or symptoms of neuropathy (paresthesias or weakness) (Table 21-8).
Table 21-8
Clinical Manifestations and Rationale for Type 2 Diabetes Mellitus
| Manifestation | Rationale |
| Recurrent infections (e.g., boils and carbuncles; skin infections) and prolonged wound healing | Growth of microorganisms is stimulated by increased glucose levels; impaired blood supply hinders healing |
| Genital pruritus | Hyperglycemia and glycosuria favor fungal growth; candidal infections, resulting in pruritus, are a common presenting symptom in women |
| Visual changes | Blurred vision occurs as water balance in the eye fluctuates because of elevated blood glucose levels; diabetic retinopathy is another cause of visual loss |
| Paresthesias | Paresthesias are common manifestations of diabetic neuropathies |
| Fatigue | Metabolic changes result in poor use of food products, contributing to lethargy and fatigue |
EVALUATION AND TREATMENT The diagnostic criteria for type 2 diabetes are similar to that of type 1 (see Box 21-1, p. 747). As with type 1 diabetes, the goal of treatment for individuals with type 2 diabetes is the restoration of
near-euglycemia (a normal blood glucose level) and correction of related metabolic disorders.
Dietary measures, including restriction of the total caloric intake, are of primary importance in both the prevention and treatment of type 2 diabetes.119,121,152 As the obese individual loses weight, the body’s resistance to insulin often diminishes so that weight loss results in improved glucose tolerance. Nonobese individuals with type 2 diabetes should consume calories consistent with their ideal weight and pattern of activity. The emphasis of medical nutrition therapy (MNT) in type 2 diabetes mellitus should be focused on achieving glucose, lipid, and blood pressure goals (see Nutrition & Disease: Medical Nutrition Therapy [MNT] for Prevention and Treatment of Diabetes).
Exercise is an important aspect of prevention and treatment of type 2 diabetes.119,121 Exercise reduces postprandial blood glucose levels, diminishes insulin requirements, lowers triglyceride and cholesterol levels, and increases the level of high-density lipoprotein (HDL) cholesterol. In addition, exercise is a valuable adjunct to weight loss for the overweight individual. Hypoglycemia may result, however, when the exercising individual receives sulfonylurea or insulin therapy.
In those individuals with morbid obesity unresponsive to diet and exercise interventions, bariatric surgery may be indicated. Recent studies suggest that gastric bypass surgery is associated with a decrease in the incidence of type 2 diabetes and marked improvements in glycemic control in those with established diabetes.152–155
Although the first approach to treatment of the individual with type 2 diabetes is appropriate meal planning and exercise, medications are usually needed for optimal management. Sulfonylurea, biguanide, thiazolidinediones, DPP-IV inhibitors, and α-glucosidase inhibitors are useful in treating some individuals with type 2 diabetes (Table 21-9). Use of oral hypoglycemic agents requires a pancreas capable of secreting insulin. Sulfonylureas acutely augment beta cell insulin secretion. Biguanides (metformin) inhibit hepatic glucose production and increase the sensitivity of peripheral tissue to insulin. α-Glucosidase inhibitors decrease postprandial hyperglycemia through delaying carbohydrate digestion and absorption. The thiazolidinedione class of insulin-sensitizing compounds activate a nuclear receptor termed the peroxisome proliferator-activated receptor (PPARγ), which in turn regulates cellular carbohydrate and lipid metabolism. As discussed previously, DPP-IV inhibitors increase GLP-1 levels that in turn help augment endogenous insulin secretion. Insulin therapy may be needed in the later stages of type 2 diabetes because of loss of beta cell function.156 There is some evidence that exogenous insulin may help prevent further beta cell apoptosis.157 Because the pathogenesis of type 2 diabetes involves a combination of insulin resistance and a relative insulin deficiency, it is common to combine therapeutic agents from different classes of oral agents in order to achieve acceptable glycemic control.
Other Specific Types of Diabetes Mellitus and Gestational Diabetes Mellitus
As described in Table 21-5 (p. 746), the American Diabetes Association (ADA) classification of diabetes mellitus includes not only the most common forms of diabetes (type 1 and type 2) but also “other specific types of diabetes mellitus” and “gestational diabetes mellitus.”98 Other specific types of diabetes include genetic defects in beta cell function, genetic defects in insulin action, diseases of the exocrine pancreas, endocrinopathies, drug- or chemical-induced beta cell dysfunction, infections, and other uncommon autoimmune and inherited disorders that are associated with diabetes.
The most well-described of these other specific types of diabetes is termed maturity-onset diabetes of youth (MODY). MODY includes six specific autosomal dominant mutations including genes for hepatocyte nuclear factor-1α (HNF-1α; MODY 3), glucokinase (MODY 2), HNF-4α (MODY 1), insulin promoter factor-1 (IPF-1; MODY 4), HNF-1β (MODY 5), and NeuroD1 (MODY 6).158–160 Individuals with these genetic defects have a strong family history of diabetes mellitus, are of normal weight, and are usually diagnosed before age 25. MODY was previously classified as a form of type 2 diabetes because insulin levels are often in the normal range yet are inappropriately low for the degree of hyperglycemia. However, weight gain is not a prominent feature of MODY, whereas it is an important contributor to type 2 diabetes. Interestingly, the gene mutations found in MODY are not commonly found in type 2 diabetes.159–161 Diagnosis and management are similar to type 2 diabetes.
Gestational diabetes mellitus (GDM) is defined as any degree of glucose intolerance with onset or first recognition during pregnancy.98 GDM complicates approximately 4% of all pregnancies and represents approximately 90% of pregnancies complicated by diabetes.98,162 The exact mechanism of GDM is unknown, but insulin resistance and inadequate insulin secretion are contributing factors. Diagnosis of GDM is based on the presence of risk factors (older age, family history, history of glucose intolerance, obesity, membership in certain ethnic or racial group, and history of poor obstetric outcomes, infant weighing greater than 9 pounds), plus the measurement of an elevated fasting or casual FPG. OGTT can also be used to confirm the diagnosis. Often there are no symptoms and careful glucose control prenatally, during pregnancy, and after delivery are essential to both the short- and long-term health of mother and baby.162–164 There is an increased risk for type-2 diabetes later in life in women who develop gestational diabetes.
Acute Complications of Diabetes Mellitus
Hypoglycemia is a lowered plasma glucose level. Its causes may be exogenous, endogenous, or functional (Tables 21-10, 21-11, and 21-12 contain summaries of causes). In general, hypoglycemia occurs when blood glucose levels are less than 35 mg/dl in newborns for the first 48 hours of life and less than 45 to 60 mg/dl in children and adults. Some individuals may become symptomatic before glucose levels decrease to 60 mg/dl if the decrease is relatively rapid. Hypoglycemia occurs most often in individuals with diabetes mellitus treated with insulin (see Table 21-10 for a list of both endogenous and exogenous causes of hypoglycemia). It occurs in more than 90% of those with type 1 diabetes (especially in those on multiple daily injections of insulin) and limits the management of the disease.100,165 Hypoglycemia in diabetes is sometimes called insulin shock or insulin reaction. Individuals with type 2 diabetes are at less risk for hypoglycemia than those with type 1 diabetes because they retain relatively intact glucose counterregulatory mechanisms.166 However, hypoglycemia does occur in type 2 diabetes when treatment involves insulin secretagogues (e.g., sulfonylureas) or exogenous insulin.156,166–168 Hypoglycemia also can occur as a result of many functional causes unrelated to diabetes (see Table 21-11).
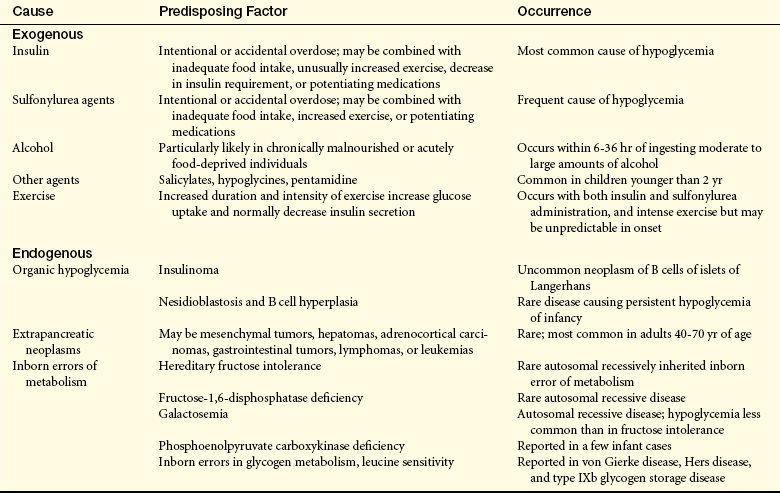
Table 21-11
Functional Causes of Hypoglycemia
| Dysfunction | Precipitating Factor | Occurrence |
| Alimentary hypoglycemia | Rapid dumping of carbohydrates into the upper small intestine | Postgastrectomy |
| Spontaneous reactive hypoglycemia | Syndrome of unknown cause with symptoms such as diaphoresis, tachycardia, tremulousness, headache, fatigue, drowsiness, and irritability | Rarely diagnosed throughout the world; widely diagnosed in United States, prompting American Diabetes Association and Endocrine Society to issue statement that entity is probably overdiagnosed; it is a benign condition |
| Alcohol-promoted reactive hypoglycemia | Drinking on an empty stomach | More common with drinks containing both alcohol and glucose or saccharin (e.g., beer, gin and tonic, rum and cola, whisky and ginger ale) |
| Posthyperalimentation hypoglycemia | Rapid discontinuation of total parenteral alimentation | Easily prevented by gradually reducing parenteral administration (alimentation) |
| Endocrine-deficiency states | Glucocorticoid deficiency | A danger for any person with adrenal insufficiency |
| Growth hormone deficiency | Particularly during a prolonged fast in children | |
| Severe liver deficiency | Insufficient glucose output by the liver | Fasting hypoglycemia |
| Lack of body stores for protein, fat, and carbohydrates | Profound malnutrition | Frequent; also found with relative frequency in kwashiorkor |
| Prolonged muscular exercise | Metabolism of energy-producing substances | Occurs if exercise is too prolonged or severe or if nutritional intake and carbohydrate stores are insufficient |
| Functional or transient hypoglycemia in infancy | Transient neonatal hypoglycemia | Occurs in 10% of live births, during first 3 days of life |
| Maternal diabetes | Caused by beta cell hyperplasia and possibly relative hypoglucagonemia | |
| Erythroblastosis fetalis | Frequently associated with erythroblastosis fetalis | |
| Leucine-induced hypoglycemia | Generally in infants younger than 6 months of age; severe hypoglycemia attacks may occur postprandially or after short periods of fasting | |
| Ketotic or ketogenic hypoglycemia | One of the most common forms of hypoglycemia in childhood, occurs after food deprivation in children 1-8 yr old; generally, spontaneous recovery before 10 yr old |
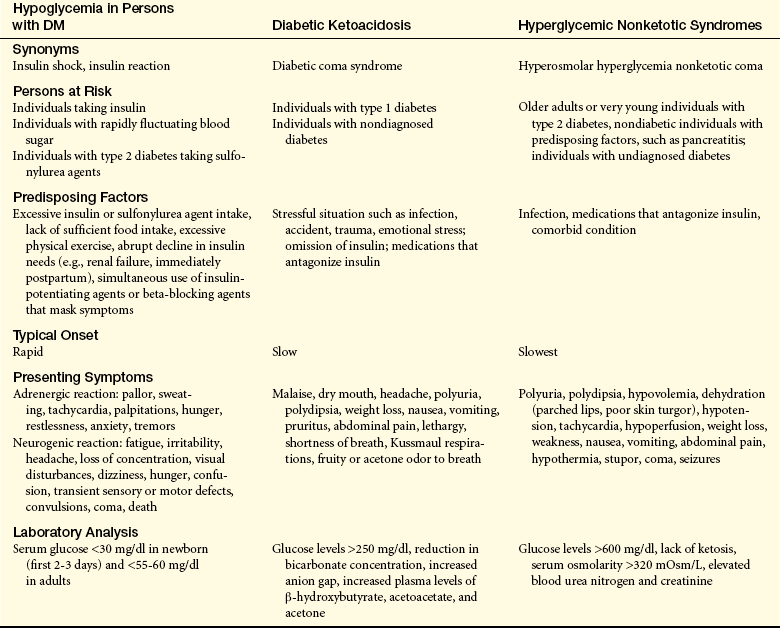
Symptoms of hypoglycemia result from either activation of the sympathetic nervous system (adrenergic symptoms) or from an abrupt cessation of glucose delivery to the brain (neuroglycopenic symptoms) or both.60 Symptoms commonly vary among individuals but tend to be consistent for each person. Adrenergic reactions occur when the decrease in blood glucose is rapid with tachycardia, palpitations, diaphoresis, tremors, pallor, and arousal anxiety.169 The response is probably generated when the hypothalamus senses decreased glucose levels. Reduced substrate delivery to the brain (neuroglycopenia) causes changes in neuronal kinase activity and firing rates,170 thus producing further symptoms including headache, dizziness, irritability, fatigue, poor judgment, confusion, visual changes, hunger, seizures, and coma. Hypoglycemia unawareness is a phenomenon that occurs in individuals without appropriate autonomic warning symptoms before development of neuroglycopenia. These individuals should be advised to raise their glycemic targets to avoid severe hypoglycemia.119 If an individual is receiving a beta-blocking medication, the autonomic symptoms may be blunted, and recovery from hypoglycemia may be delayed because of impaired glycogenolysis and hampered delivery of gluconeogenic substrates to the liver.
When hypoglycemic symptoms are nonspecific, the safest treatment is to provide some form of glucose because failure to provide glucose may precipitate convulsions, coma, and death. The ADA recommends that glucose (15 to 20 g) be given to a conscious patient with hypoglycemia, and that at-risk individuals and caregivers should be instructed in glucagon administration.119 Prevention of hypoglycemia episodes through alternate therapeutic regimens, individualizing target blood glucose levels, frequent self-monitoring of blood glucose, and proper education should be the goal.118,166–168,171
Diabetic Ketoacidosis
Ketoacidosis, a serious complication of diabetes mellitus, is a common cause for hospital admissions. Although average mortality rates throughout the United States are less than 2%, the highest rates of mortality are observed in older adults and those with other severe underlying illnesses.172 Diabetic ketoacidosis (DKA) develops when there is an absolute or relative deficiency of insulin. This is most common in individuals with type 1 diabetes but can occur in those with type 2 diabetes as well.60,172–174 In addition, a syndrome called ketosis-prone diabetes (KPD) has been described in which affected individuals do not fit into the traditional ADA categories for diabetes and are more likely to be persons of African, African American, or Hispanic origin.175 The most common precipitating factor for DKA is intercurrent illness, such as infection, trauma, surgery, or myocardial infarction. Interruption of insulin administration also may result in DKA. In 20% to 30% of cases, no precipitating factors are noted. Emotional factors and stress, particularly in children, are thought to contribute to the development of DKA. The frequency of DKA peaks in adolescence.
PATHOPHYSIOLOGY In a state of relative insulin deficiency there is an increase in insulin counterregulatory hormones including catecholamines, cortisol, glucagon, and GH. Catecholamines, cortisol, glucagon, and GH antagonize insulin by increasing glucose production. In addition, these hormones decrease use of glucose. Profound insulin deficiency results in decreased glucose uptake, increased fat mobilization with release of fatty acids, and accelerated gluconeogenesis and ketogenesis (Figure 21-15). Relatively increased glucagon levels also contribute to activation of the gluconeogenic (glucose-forming) and ketogenic (ketone-forming) pathways in the liver. Because of the insulin deficiency, hepatic overproduction of β-hydroxybutyrate and acetoacetic acids causes increased ketone concentrations.173 Ordinarily ketones are used by tissues as an energy source to regenerate bicarbonate. This balances the loss of bicarbonate, which occurs when the ketone is formed. Hyperketonemia (increased blood ketone levels) may be a result of impairment in the use of ketones by peripheral tissue, which permits strong organic acids to circulate freely. Bicarbonate buffering then does not occur, and the individual develops a metabolic acidosis.
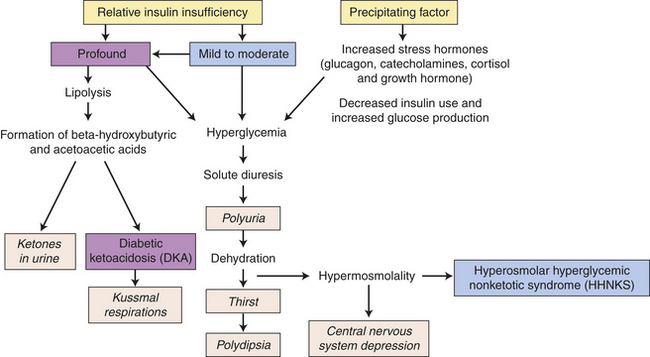
CLINICAL MANIFESTATIONS The signs and symptoms of DKA are fairly nonspecific. Polyuria and dehydration result from the osmotic diuresis associated with hyperglycemia. Here the plasma glucose level is higher than the individual’s renal threshold, allowing much glucose to be lost in the urine. Sodium, phosphorus, and magnesium deficits are common. The most important electrolyte disturbance, however, is a marked deficiency in total body potassium. Although the serum potassium may appear normal or elevated because of volume contraction and a shift of potassium out of the cell and into the blood caused by metabolic acidosis, the total body deficiency of potassium may reach 3 to 5 mEq/kg. Symptoms of diabetic ketoacidosis include Kussmaul respirations (hyperventilation in an attempt to compensate for the acidosis), postural dizziness, central nervous system depression, ketonuria, anorexia, nausea, abdominal pain, thirst, and polyuria.
EVALUATION AND TREATMENT The diagnosis of ketoacidosis is suggested when individuals have symptoms of vomiting, abdominal pain, dehydration, an acetone odor on the breath, and change in sensorium. The ADA criteria for diagnosis of DKA include a serum glucose of more than 250 mg/dl, a serum bicarbonate of less than 18 mg/dl, a serum pH of less than 7.30, presence of an anion gap, and presence of urine and serum ketones.176
Treatment of DKA involves continual administration of low-dose insulin to decrease glucose levels.60,172 Fluids are administered to replace lost fluid volume, and electrolytes—particularly sodium, potassium, and phosphorus—are administered as needed. Fluids and electrolytes should be monitored closely. Electrolyte deficits become apparent as fluid volume is replaced. After the administration of insulin, the concentration of β-hydroxybutyrate promptly begins to decrease and after a slight increase, acetoacetate also begins to decrease. A persistent ketonuria may be observed for several days after treatment. Continuous monitoring of the individual is essential to ensure an uncomplicated recovery from DKA. Cerebral edema is the most common cause of morbidity and mortality during the first day of treatment for DKA in children. The mechanisms are poorly understood.177 Health teaching emphasizes predisposing factors and strategies for avoiding DKA.
Hyperosmolar Hyperglycemic Nonketotic Syndrome
Hyperosmolar hyperglycemic nonketotic syndrome (HHNKS) is a life-threatening emergency most often precipitated by infections, medications, nonadherence to diabetes treatment, or coexisting disease.174,176 HHNKS is more commonly seen with type 2 diabetes. It can also occur in individuals with pancreatic destruction from other causes such as chronic pancreatitis.
PATHOPHYSIOLOGY HHNKS differs from DKA in the degree of insulin deficiency (which is more profound in DKA) and the degree of fluid deficiency (which is more marked in HHNKS) (see Figure 21-15).176 Levels of free fatty acids in HHNKS are consistently lower than those found in DKA. HHNKS is characterized also by a lack of ketosis. Because the amount of insulin required to inhibit fat breakdown is less than that needed for effective glucose transport, insulin levels are sufficient to prevent excessive lipolysis but not to use glucose properly. Glucose levels are considerably higher in HHNKS than in DKA because of volume depletion.
CLINICAL MANIFESTATIONS The clinical features of HHNKS include a serum glucose of more than 600 mg/dl, a serum pH greater than 7.30, a serum bicarbonate greater than15 mg/dl, a serum osmolarity greater than 320 mOsm/L, and either absent or small ketones in the urine and serum.176 Glycosuria and polyuria in HHNKS result from the extreme serum glucose elevation. As much as 19 g of glucose per hour may be lost in diuresis, which also causes severe volume depletion, increased serum osmolarity, intracellular dehydration, and loss of electrolytes including potassium. Neurologic changes, such as stupor, correlate with the degree of hyperosmolarity and are more common in HHNKS than in DKA.60
EVALUATION AND TREATMENT The serum ketone concentration is normal or only mildly elevated and only minimal acidosis is seen in HHNKS, otherwise DKA and HHNKS show considerable overlap in symptoms and treatment. Insulin infusion should be combined with fluid repletion over 24 hours.60,176 An important distinction, however, is that the dehydration in HHNKS is far more severe than that in DKA. Thus fluid replacement, with both crystalloids and colloids, is more rapid. Potassium deficits may be so extreme in HHNKS that more than 1 week of potassium repletion may be needed to correct the total body deficits. Phosphorus and sodium also may be needed. HHNKS is a significant risk factor for venous thrombosis and recent studies suggest that anticoagulant prophylaxis should be considered.60,178 Mortality is also high in HHNKS, and is related to the age of the individual and comorbid conditions including the severity of the precipitating illness (see Table 21-12 for a comparison of the three acute complications described thus far).
Somogyi Effect
The Somogyi effect is a unique combination of hypoglycemia followed by rebound hyperglycemia. The problem is more common in individuals with type 1 diabetes mellitus, particularly in children, and should be investigated whenever fluctuations in blood sugar levels are serious.179
PATHOPHYSIOLOGY The Somogyi effect occurs when hypoglycemia stimulates glucose counterregulation, including epinephrine, GH, cortisol, and glucagon release.180 These hormones serve to increase blood glucose by gluconeogenesis (formation of glucose from nonglucose sources) and glycogenolysis (breakdown of glycogen into glucose). They mobilize fatty acids and proteins while inhibiting peripheral glucose use. These hormones may cause insulin resistance for 12 to 48 hours. Commonly, excessive carbohydrate intake may be a major contributor to rebound hyperglycemia. Also hypoglycemia generally occurs during the peak of injected insulin; therefore, as counterregulatory hormones are activated and carbohydrate is consumed by the individual, insulin levels are on the decline, which contributes to the subsequent hyperglycemia.180 The frequency of this phenomenon is debated, and recent studies suggest that it is much less common than previously reported.
CLINICAL MANIFESTATIONS In addition to fluctuating glucose levels, subtle symptoms of hypoglycemia occur. If an individual has nocturnal hypoglycemia, there may be complaints of nightmares and early morning headaches. Ketonuria may occur if the mobilization of energy sources overshoots the body’s need for glucose and exogenous insulin is depleted.
EVALUATION AND TREATMENT If the individual has nocturnal hypoglycemia, diagnosis involves the measurement of plasma glucose during the night using monitors capable of continuous glucose sensing.179 Treatment consists of decreasing insulin dosage or changing the time of administration.
Dawn Phenomenon
The dawn phenomenon is an early morning rise in blood glucose concentration with no hypoglycemia during the night. It appears to be related to nocturnal elevations of GH, a counterregulatory hormone that causes hyperglycemia by decreasing peripheral (other than liver) glucose uptake. Increased clearance of plasma insulin also may be involved. Altering the time and dose of insulin manages the problem.181 Treating dawn phenomenon may result in the Somogyi effect and vice versa.
Chronic Complications of Diabetes Mellitus
A number of serious complications are associated with any type of diabetes mellitus and include microvascular (e.g., retinopathy, nephropathy, and neuropathies) and macrovascular disease (e.g., coronary artery disease, stroke, and peripheral vascular disease), and infection. Most complications are associated with metabolic alterations, primarily hyperglycemia. Strict control of blood glucose significantly reduces complications. Five metabolic events are associated with the tissue-damaging effects of chronic hyperglycemia and the pathogenesis of diabetic complications: shunting of glucose to the polyol pathway, activation of protein kinase C, induction of reactive oxygen species (oxidative stress), production of advanced glycation end products, and accumulation of hexosamines.182–184
Hyperglycemia and the Polyol Pathway
Tissues that do not require insulin for glucose transport, such as kidney, red blood cells (RBCs), blood vessels, eye lens, and nerves, use an alternate metabolic pathway for glucose metabolism known as the polyol pathway. With hyperglycemia, glucose is shunted to this pathway and is converted to sorbitol (a polyol) by the enzyme aldose reductase. Sorbitol is then slowly converted to fructose by sorbitol dehydrogenase.185 The accumulation of sorbitol and fructose increases intracellular osmotic pressure and attracts water, leading to cell injury. This is particularly evident in the lens of the eye and leads to swelling with visual changes and cataracts. In nerves, sorbitol interferes with ion pumps, damages Schwann cells, and disrupts nerve conduction. RBCs become swollen and stiff and interfere with perfusion. Aldose reductase inhibitors may slow or prevent some diabetic complications, particularly neuropathies, although their effectiveness has been limited.186
Protein Kinase C
Protein kinase C (PKC) is an enzyme that is inappropriately activated in different tissues by hyperglycemia, particularly the diacylglycerol (DAG)-PKC pathway. Various consequences have been observed, including insulin resistance, production of extracellular matrix and cytokines, vascular cell proliferation, enhanced contractility, and increased permeability.187 These effects may contribute to the macrovascular, microvascular, and neurologic complications of diabetes. Specific PKC inhibitors are under investigation and have shown some promise in stabilizing or preventing retinopathy, nephropathy, and neuropathy.188
Hyperglycemia and Nonenzymatic Glycosylation
Nonenzymatic glycosylation is the reversible attachment of glucose to proteins, lipids, and nucleic acids without the action of enzymes. With recurrent or persistent hyperglycemia, glucose becomes irreversibly bound to collagen and other proteins in red blood cells (e.g., glycated hemoglobin), blood vessel walls, and interstitial tissue. The products of this binding are known as advanced glycosylation end product (AGE) and their receptor (RAGE) have a number of properties that may cause tissue injury or pathologic conditions associated with diabetes189–192:
1. Cross-linking and trapping of proteins, including albumin, low-density lipoprotein (LDL), immunoglobulin, and complement, with thickening of the basement membrane or increased permeability in blood vessels and nerves
2. Binding to cell receptors, such as macrophages and glomerular mesangial cells, and inducing release of cytokines and growth factors that stimulate cellular proliferation in the glomeruli, smooth muscle of blood vessels, and collagen synthesis with fibrosis
3. Induction of lipid oxidation, oxidative stress, and inflammation
4. Inactivation of nitric oxide with loss of vasodilation and diminished endothelial function
5. Procoagulant changes on endothelial cells with promotion of platelet adhesion and reduced fibrinolysis
Pharmacologic agents that inhibit AGE formation or block their receptor (RAGE) are being evaluated.189
Hyperglycemia and Oxidative Stress
Chronic hyperglycemia increases the production of reactive oxygen species (ROS) and the detrimental effects of oxidative stress. AGEs, defects in the polyol pathway, uncoupling of nitric oxide synthase, xanthine oxidase, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase generate ROS, which damage large and small vessels and contribute to atherogenesis, cardiovascular disease, nephropathy, and neuropathy. Direct cellular injury, as well as the formation of gene products that cause cellular injury, contribute to these late complications of diabetes mellitus.193,194
Hyperglycemia and the Hexosamine Pathway
Chronic hyperglycemia causes shunting of excess intracellular glucose into the hexosamine pathway and leads to O-linked glycosylation of several enzymes and proteins with alteration in signal transduction pathways and oxidative stress. The O-linked attachment of N-acetylglucosamine (O-GlcNAc) on serine and threonine residues of nuclear and cytoplasmic proteins is associated with insulin resistance and cardiovascular complications of diabetes mellitus.195,196
Microvascular Disease
Diabetic microvascular complications are a leading cause of blindness, end-stage renal failure, and various neuropathies.197 Thickening of the capillary basement membrane, endothelial cell hyperplasia, thrombosis, and pericyte degeneration are characteristic of diabetic microangiopathy and emerges over a period of 1 to 2 years. The thickening eventually results in decreased tissue perfusion. Hyperglycemia is a prerequisite for these microvascular changes and may be related to glycation of structural proteins, which results in the accumulation of AGEs. The frequency of the lesions appears to be proportional to the duration of the disease and blood glucose levels. In the DCCT study, individuals with tightly controlled blood glucose were half as likely to have renal and eye complications as those who received standard treatment.198 Hypoxia and ischemia of various organs may result from microangiopathy. Three areas often affected are the retina, the kidney, and nerves.
Retinopathy: The retina is the most metabolically active structure per weight of tissue in the body. Thus the retina is a vulnerable target for microvascular disease in diabetes mellitus. Diabetic retinopathy appears to be a response to retinal ischemia resulting from blood vessel changes and red blood cell aggregation (Figure 21-16). Low-grade inflammation and poorly controlled hypertension is a risk factor for worsening of retinopathy. Vascular endothelial growth factors and GH appear to play a role in developing retinopathy, and therapeutic strategies have been developed to exploit this phenomenon.199 The prevalence and severity of the retinopathy are strongly related to the age of the individual and duration of the diabetes and glycemic control. Retinopathy may be present in individuals at the time of diagnosis of type 2 diabetes as a result of the long preclinical latency of this form of diabetes. The vast majority of individuals with diabetes mellitus have some degree of retinopathy, and retinopathy is closely associated with systemic vascular complications including nephropathy, cardiovascular complications, and stroke.200
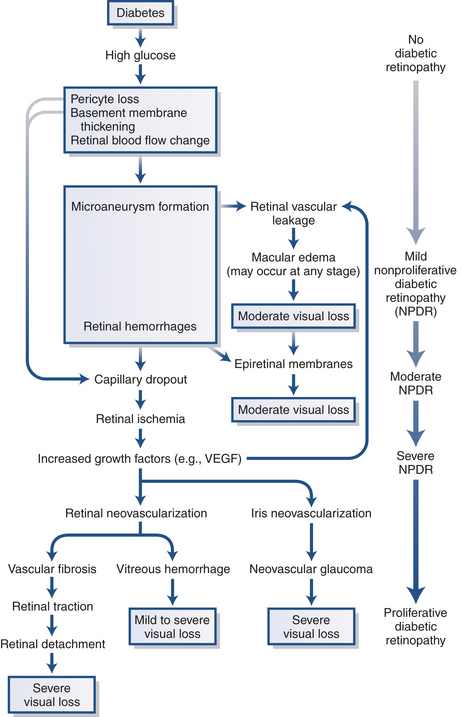
Figure 21-16 Diabetic retinopathy. VEGF, Vascular endothelial growth factor. (From Kronenberg et al: Williams textbook of endocrinology, ed 11, Philadelphia, 2008, Saunders.)
The three stages of retinopathy are described in Table 21-13. Nonproliferative retinopathy (stage I) is characterized by an increase in retinal capillary permeability, vein dilation, microaneurysm formation, superficial (flame-shaped) and deep (blot) hemorrhages, cotton wool spots from nerve damage, and macular edema. Preproliferative retinopathy (stage II) is a progression of retinal ischemia with areas of poor perfusion that culminate in infarcts. Proliferative diabetic retinopathy (stage III) is the result of neovascularization and fibrous tissue formation within the retina or optic disc. Traction of the new vessels on the vitreous humor may cause retinal detachment or hemorrhage into the vitreous humor.201

Maculopathy is a progressive process that may accompany the increased retinal capillary permeability, vessel occlusion, and ischemia. If formation of exudates, edema, or ischemia occurs near the fovea, serious loss of vision may result. Laser treatments are used to reduce the rate of vision loss from diabetic macular edema and neovascularization. Vitrectomy is a surgical procedure used to treat an intravitreal hemorrhage secondary to rupture of a neovascular capillary tuft. Cataracts, optic neuropathy and defects in eye muscle function are also associated with the chronic complications of hyperglycemia and diabetes mellitus.
Diabetic Nephropathy: Diabetes is the most common cause of end-stage renal disease in the Western world. Without appropriate management, approximately 30% of individuals with type 1 and 40% of those with type 2 diabetes develop nephropathy.202 The early phases of nephropathy are asymptomatic and begin to develop after 10 years in type 1 diabetes or 5 to 8 years in type 2 diabetes. There are some differences in renal lesions in type 1 and type 2 diabetes, with glomerular changes in type 1 being most prominent; however, hyperglycemia is the major initiating factor in both types.203
The exact process responsible for destruction of kidneys in diabetes is unknown. Multiple mechanisms contribute to nephropathy, including hyperglycemia, systemic hypertension, hyperperfusion, hyperfiltration, increased blood viscosity, increased glomerular pressure, albuminuria, protein kinase C, growth factors, advanced glycation end products, inflammatory cytokines, oxidative stress, the renin-angiotensin-aldosterone system, and hypercholesterolemia.204–206 Genetic factors may confer susceptibility or resistance to diabetic nephropathy in type 1 diabetes.207 The glomeruli are injured by at least two mechanisms: protein denaturation by high glucose levels and adverse effects of intraglomerular microcirculatory hypertension. Renal glomerular changes can occur early in diabetes mellitus and occasionally may precede the overt manifestations of the disease (Figure 21-17). Glomerular enlargement and glomerular basement membrane (GBM) thickening, resulting in diffuse intercapillary glomerulosclerosis, develop during the first few years of diabetes. The Kimmelstiel-Wilson nodule, with thickening at the center of the glomerular lobules and thickening of the peripheral basement membrane, is distinctive in individuals with diabetes.208 increased mesangial matrix occurs contributing to resistance of glomerular capillary blood flow and decreased glomerular filtration rates. The tubular basement membrane also thickens in parallel with GBM thickening and mesangial expansion.
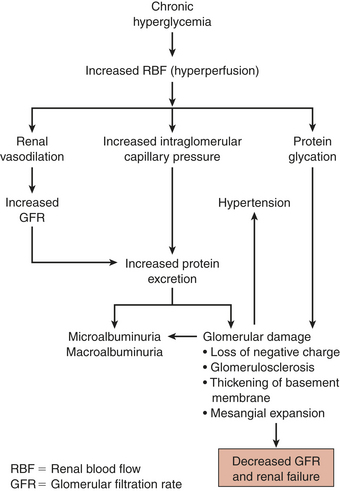
Microalbuminuria is the first manifestation of renal dysfunction (30 to 300 mg/day). Continuous untreated proteinuria generally heralds a life expectancy less than 10 years. Microalbuminuria is also an independent risk factor for cardiovascular disease.209 The determinants of proteinuria in diabetic nephropathy are not completely understood. Glycated albumin generates ROS and directly damages glomerular membrane epithelial cells, vascular smooth muscle, and mesangial cells. Glomerular endothelial dysfunction also promotes albuminuria.210,211 As renal failure progresses, extensive vascular and extravascular changes occur.
Before the development of clinical proteinuria (more than 300 mg/day), no clinical signs or symptoms of progressive glomerulosclerosis are likely to be evident. Later, hypoproteinemia, reduction in plasma oncotic pressure, fluid overload, anasarca (generalized body edema), and hypertension may occur.212 Microalbuminuria and clinical proteinuria are risk factors for cardiovascular disease and progressive renal impairment.213
As renal function continues to deteriorate, individuals with type 1 diabetes may experience hypoglycemia, which necessitates a decrease in insulin therapy. The hypoglycemia occurs because the kidney’s ability to metabolize insulin is lost along with other renal functions. As the glomerular filtration rate drops below 10 ml/minute, uremic signs such as nausea, lethargy, acidosis, anemia, and uncontrolled hypertension occur (see Chapter 36 for a discussion of renal failure). Impaired kidney function also accelerates retinopathy and cardiovascular disease.214
The development of more sensitive tests has permitted the detection of small amounts of urinary albumin, microalbuminuria. Earlier intervention with tight glucose control and angiotensin-converting enzyme (ACE) inhibitors or angiotensin II receptor blockers has reduced proteinuria and slowed the progression of nephropathy. Aggressive treatment of hypertension is another therapeutic intervention definitively shown to slow the progression of established renal disease.215
Diabetic Neuropathies: Diabetic neuropathy is the most common cause of neuropathy in the Western world and is probably the most common complication of diabetes. Nerves do not require insulin for glucose transport and are particularly vulnerable to the pathologic effects of hyperglycemia. The prevalence of neuropathy is similar for type 1 and type 2 diabetes.216 Neuropathy in type 1 diabetes has more severe structural and functional changes and more rapid progression.217 Neuropathy and other long-term complications are thought to result from the interaction of multiple metabolic, genetic, and environmental factors. The underlying pathophysiology is complex and related to chronic hyperglycemia. A combination of mechanisms is likely, including microangiopathy, oxidative stress, growth factor deficiency, abnormal signaling from AGE-RAGE interaction, polyol flux, and inflammation218 (Figure 21-18). Neuropathy is classified into two stages: subclinical and clinical. In the subclinical stage, there is electromyographic (EMG) evidence of peripheral nerve dysfunctions, such as slowed motor and sensory nerve conduction, without clinical signs. In the clinical stage, symptoms or clinically detectable neurologic deficits are present.
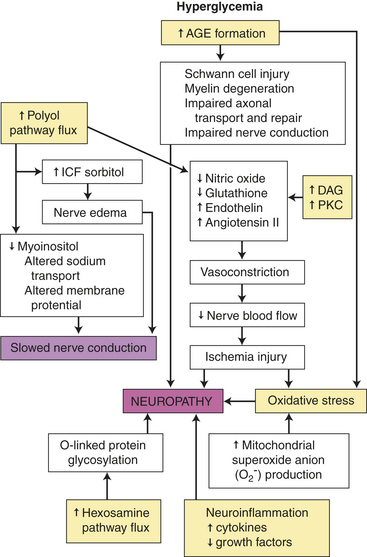
Figure 21-18 Multifactorial pathogenesis of diabetic neuropathy. The yellow boxes represent the consequences of chronic hyperglycemia in the development of neuropathy. AGE, Advanced glycosylation end product; DAG, diacylglycerol; ICF, intracellular fluid; PKC, protein kinase C.
Diabetic neuropathy is a form of “dying back” neuropathy, in which the distal portions of the neurons are initially and eventually more severely affected. The earliest morphologic change in both the peripheral nerves and central nervous system is axonal degeneration that preferentially involves sensory nerve fibers, particularly the smaller polymodal unmyelinated peripheral C fibers and the larger myelinated A-delta fibers. Metabolic activity of Schwann cells is disturbed, causing segmental loss of myelin and a characteristic pattern of demyelination and remyelination observed in long-term diabetic neuropathy. The location of the pathologic condition can include the spinal cord, the posterior root ganglia, or the peripheral nerves. These changes may occur alone or in combination. Nerve degeneration begins in the periphery. Sensory nerve injury generally precedes motor nerve injury.219
Distal symmetric polyneuropathy (sensory, autonomic, and motor nerve involvement) is the most common neuropathy with involvement of both large and small nerve fibers. Loss of small nerve fiber function includes neuropathic pain, loss of sensation, and carries high risk for development of foot ulceration with subsequent gangrene and amputation. Large nerve fiber involvement results in sensory loss of proprioception and vibration with ataxia, loss of coordination, and risk for falls and fractures.220
Involvement of the autonomic nervous system can also occur early. Multiple alterations can develop affecting gastrointestinal enteric nerves (nausea, bloating, gastroparesis, diarrhea, or constipation), bladder and sexual function (loss of bladder sensation, urine retention, recurrent infection, erectile dysfunction), sweating, and body temperature regulation. Cardiovascular autonomic neuropathy may occur early particularly in type 1 diabetes with heart rate variability, changes in baroreceptor reflexes, postural hypotension, dysrhythmias, exercise intolerance, and painless myocardial infarction.221,222
Alterations in cognitive function and increased risk for dementia may accompany long-term complications in the brain particularly in type 2 diabetes. Cognitive alterations in type 1 diabetes are more controversial.223,224
Alterations in sensory perception and motor nerve conduction velocity and electromyography have shown abnormalities at the onset of diabetes and there are varying manifestations of diabetic neuropathies involving both the somatic and autonomic nerves (Table 21-14). Some of the neuropathic syndromes are progressive, but many—such as painful peripheral neuropathy, mononeuropathy (wristdrop, footdrop), diabetic amyotrophy, diabetic neuropathic cachexia, and visceral manifestations associated with autonomic neuropathy (e.g., diabetic diarrhea and orthostatic hypotension)—may spontaneously improve. Charcot neuroarthropathy (Charcot joint) is the progressive degeneration and structural disorganization of a joint, particularly in the foot of people with long-term diabetes. The pathogenesis is not clear but may be related to loss of sensation or neurally mediated alterations in osteoclastic bone resorption, or both.225 The DCCT demonstrated a 60% reduction in results related to the appearance of clinical neuropathy and parameters of subclinical nerve dysfunction in the intensive insulin therapy cohort.198 Similar results were seen in the Kumamoto trial226 and the United Kingdom Prospective Diabetes Study.227 Much investigation regarding the pathophysiology and progression of diabetic neuropathies remains to be done.

Macrovascular Disease
Macrovascular disease is a major cause of morbidity and mortality, particularly among individuals with type 2 diabetes mellitus. Children with poorly controlled type 2 diabetes have high risk for macrovascular disease within one or two decades.228 The premature atherosclerosis of diabetes has many contributing factors, including hyperinsulinemia (insulin resistance), hyperglycemia, hypertriglyceridemia, low HDL, high LDL, lipoprotein oxidation, inflammation, vascular consequences of AGEs and their endothelial receptors (RAGEs), and altered endothelial function. The fibrous plaques of atherosclerosis are associated with the proliferation of subendothelial smooth muscle in the arterial wall. Other factors in the serum of individuals with diabetes also stimulate this proliferation (Figure 21-19).
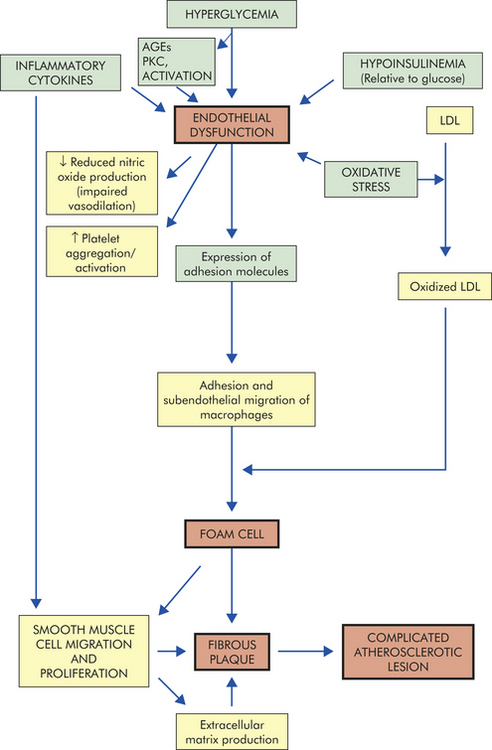
Figure 21-19 Diabetes mellitus and atherosclerosis. Diabetes with its associated hyperglycemia, relative hypoinsulinemia, oxidative stress, and proinflammatory state contributes to atherogenesis by causing arterial endothelial dysfuncton (impaired vasodilation, adhesion of inflammatory cells and alteration in permeability), dyslipidemia and smooth muscle proliferation resulting in atherosclerotic lesions. AGEs, Advanced glycosylation end products; LDL, low-density lipoprotein; PKC, protein kinase C.
In addition, deposition of lipids in vascular lesions may be facilitated in individuals with diabetes. Triglyceride elevations with low levels of the protective HDL cholesterol are common in individuals with type 2 diabetes mellitus in association with increased quantities of small, dense (very atherogenic) LDL cholesterol and endothelial cell and platelet abnormalities.229 Increased levels of the atherogenic oxidized LDL also are seen in hyperglycemic individuals. Hypertension can increase capillary wall pressure, damage endothelium, increase capillary permeability, decrease nitric oxide synthesis, and decrease autoregulation of blood flow.230,231 Further work is needed to clarify the complexities of macrovascular complications.
Coronary Artery Disease: The risk of coronary artery disease (CAD) for those individuals with type 2 diabetes is higher than for the general population even when hypertension and hyperlipidemia are taken into account. CAD is the most common cause of death in individuals with type 2 diabetes and is common in those with type 1.232 In general, the prevalence of CAD increases with the duration but not the severity of diabetes.
Myocardial infarction (death of heart muscle as a result of coronary artery occlusion) is the cause of death in 20% of those with diabetes, and individuals with diabetes mellitus have a higher mortality during the acute phase of myocardial infarctions than do nondiabetic individuals. Increased platelet adhesion and decreased fibrinolysis promote thrombus formation and vascular occulsion.233 In addition, the incidence of cardiomyopathy (myocardial dysfunction in the absence of coronary artery disease [CAD] and hypertension) is higher in individuals with diabetes. The reason is unclear but may be related to the presence of increased amounts of collagen in the ventricular wall, which reduces the mechanical compliance of the heart during filling, inflammation, and changes in calcium handling. Diastolic dysfunction is the earliest symptom.234
Stroke: Stroke is twice as common in those with diabetes as in the nondiabetic population.235 Ischemic stroke is more common than hemorrhagic stroke. The survival rate for an individual with diabetes after a massive stroke is typically shorter than for a person without diabetes. Hypertension, hyperglycemia, and dyslipidemia are definite risk factors (see Chapter 30), and aggressive management of blood pressure, hyperglycemia, and lipidemia in individuals with diabetes has been shown to reduce the incidence of stroke.236
Peripheral Arterial Disease: The increased incidence of peripheral arterial disease (PAD), neuropathy, gangrene, and amputation in diabetic persons has been documented in many studies, particularly in individuals with type 2 diabetes.237,238 Many individuals with type 2 diabetes have evidence of peripheral vascular disease at the time of their initial diagnosis. The atherosclerotic process in diabetic persons is more common, appears at a younger age, advances more rapidly than vascular changes in nondiabetic persons, and increases the risk of cardiac death.239 The prevalence of PAD is nearly equal in males and females with diabetes. Age, duration of diabetes, glycemic control, genetics, and additional risk factors influence the development of PAD.
Because of occlusions of the small arteries and arterioles, most of the gangrenous changes of the lower extremities occur in patchy areas of the feet and toes.240 Smaller vessels often have more advanced disease than larger vessels in the same individuals. Figure 21-20 illustrates how foot lesions of diabetes can lead to amputation. Fifty percent of nontraumatic amputations in the United States are performed on individuals with diabetes. Hospital mortality for individuals with diabetes who undergo major amputation is between 10% and 23%. The survival rate after surgery is only about 40% at the end of 5 years.237
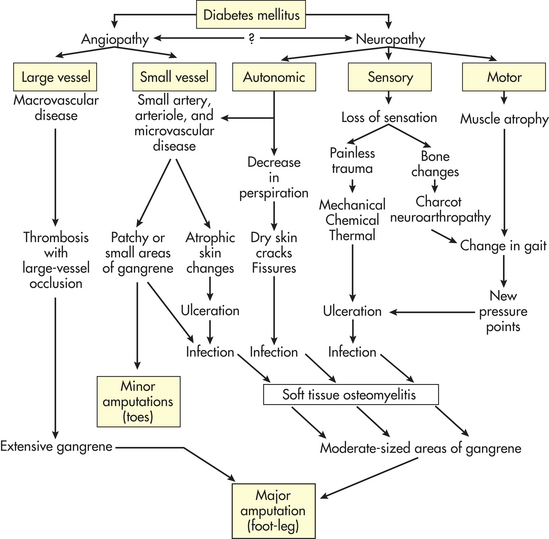
Infection
Increased morbidity and mortality from infectious agents have been documented in those with diabetes.241 The individual with diabetes is at increased risk for infection throughout the body for several reasons:
1. Impaired vision caused by retinal changes and impaired touch caused by neuropathy diminish the prevention of breaks in the skin by decreasing the early warning systems. Once breaks in skin integrity occur, tissues may have increased susceptibility to infection because of hypoxia, a second reason for susceptibility to infection.
2. Microvascular and macrovascular complications cause decreased oxygen supply to tissues. In addition, the increased content of glycosylated hemoglobin in the red blood cell impedes the release of oxygen to tissues.
3. Pathogens are able to multiply rapidly once they have gained access to the tissues. Some pathogens proliferate rapidly because the increased glucose in body fluids provides an excellent source of energy.
4. Decreased blood supply resulting from vascular changes decreases the supply of white blood cells to the affected area.
5. Function of the white cells is impaired by ischemia and hyperglycemia. Chemotaxis is abnormal, and phagocytosis is defective.
6. Inflammatory responses to microbial invasion are diminished and clinical signs of infection may be absent.
7. Sensory neuropathy leads to loss of protective sensation with injury and repeated trauma that leads to open wounds and soft tissue or osseous infection.
The risk of infection is especially high for individuals undergoing surgery and for those taking immunosuppressant medications.242,243