ALTERATIONS OF NEUROLOGIC FUNCTION IN CHILDREN


Neurologic disorders in children can occur from infancy through adolescence and include congenital malformations, genetic defects in metabolism, brain injuries, infection, tumors, and other disorders that effect neurologic structure and function. The symptoms, diagnosis and management of neurologic disorders in children is often different than that of adults.
STRUCTURE AND FUNCTION OF THE NERVOUS SYSTEM IN CHILDREN
Embryology is a highly complex and often difficult science to understand fully. A basic knowledge of this science is essential because it is the process of embryonic development that explains many of the malformations that occur in children.
Environmental influences also play a significant role in nervous system development. Nutrition, hormones, oxygen levels, and external stimulation all affect normal growth. The proper proportions of essential nutrients are necessary for proliferation of the nervous system tissue (see Nutrition and Disease box). Maternal lifestyle, nutrition, and state of health also have a crucial effect on nervous system development at critical periods of fetal maturation.
The central nervous system (CNS) develops from a dorsal thickening of the ectoderm known as the neural plate. This plate appears around the middle of the third gestational week and unfolds to form a neural groove and neural folds. During the fourth gestational week the neural groove deepens, its folds develop laterally, and it closes dorsally to form the neural tube, epithelial tissue that ultimately becomes the CNS. The neural tube closes first in the cervical region and then “zippers” in two directions—cranially and caudally (Figure 19-1).
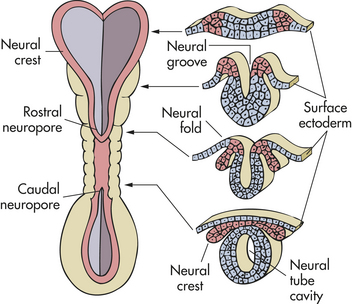
Figure 19-1 Neural tube at 3 weeks of gestation. Neural folds have begun to fuse at the cervical level of the future spinal cord. Right, Cross sections of the neural tube at four different levels; at any given level the embryonic central nervous system (CNS) goes through a series of stages resembling these four cross sections. Total length of neural tube at this time is about 2.5 mm.
In the developmental process, some neuroectodermal cells separate from the neural tube but remain between the tube and the surface ectoderm, creating the neural crest. This cellular band develops into the cranial and spinal ganglia, more commonly referred to as the peripheral nervous system. Other
structures associated with the nervous system arise from mesoderm (somite) and include blood vessels, microglial cells, dural and arachnoid layers of the meninges, the capsule of some peripheral sensory nerve endings, and peripheral nerve coverings.
The cranial end of the neural tube forms the brain, and the remainder develops into the spinal cord. The lumen of the neural tube becomes the ventricles of the brain and the central canal of the spinal cord (Figure 19-2). On either side of the neural tube’s inner surface is a longitudinal groove (sulcus limitans). Anterior to this region (basal plate) the gray matter differentiates into the nuclei of the lower motor neurons. The region posterior to the sulcus (alar plate) differentiates into the sensory nuclei of the spinal cord.
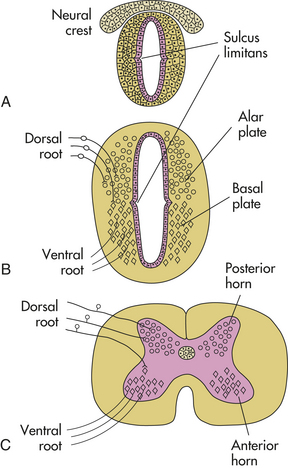
Figure 19-2 Sulcus limitans and alar and basal plates. A, Neural tube during the fourth week of gestation. B, Embryonic spinal cord during the sixth week of gestation; dorsal root ganglion cells, derived from the neural crest, send their central processes into the spinal cord to terminate mainly in alar plate cells; basal plate cells become motor neurons, whose axons exit in the ventral roots. C, Adult spinal cord.
Embryonic development of the nervous system occurs in six stages: (1) dorsal (posterior) induction, (2) ventral (anterior) induction, (3) proliferation, (4) migration, (5) organization, and (6) myelination. (Figure 19-3 summarizes the embryonic development of the nervous system and identifies disorders associated with interference in any of these stages.) Many different events happen simultaneously, and critical periods must pass uninterrupted if the vulnerable fetus is to develop normally.
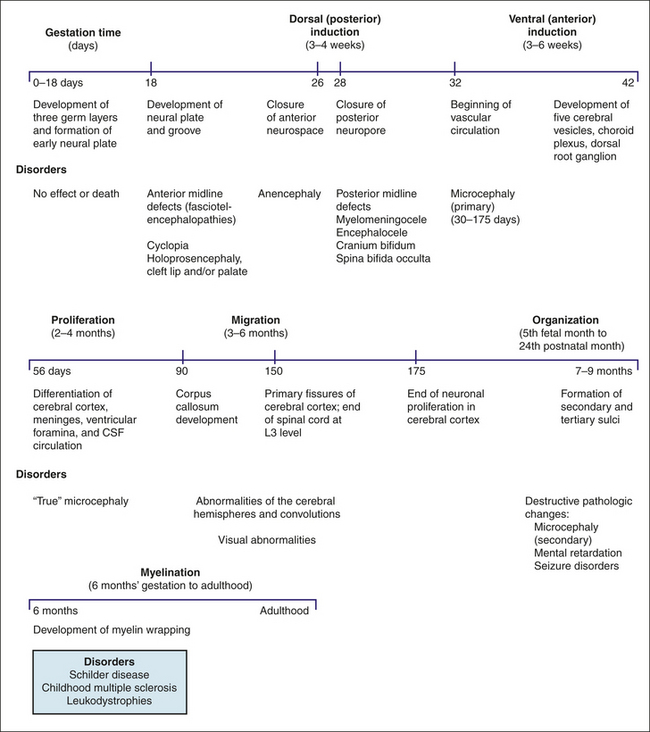
Figure 19-3 Disorders associated with specific stages of embryonic development. CSF, Cerebrospinal fluid.
In the newborn the bones of the skull are separated, but definite sutures (bands of connective tissue) form shortly thereafter. The edges are several millimeters wide to allow for normal growth. At the junctions of the sutures are wider spaces of unossified membranous tissue called fontanels. Sutures and fontanels close as the skull and brain grow and develop.
On average the posterior fontanel closes within the first 3 months of life. By 6 months of age, a fibrous union of suture lines occurs and serrated edges interlock. By approximately 20 to 24 months, the anterior fontanel is closed (Figure 19-4). At approximately 8 years of age, ossification of the cranial bones is complete; the sutures usually are completely fused and cannot be separated by 12 years of age, even in the presence of increased intracranial pressure (ICP).
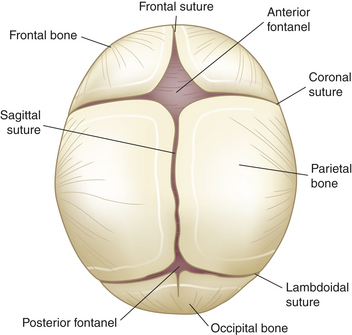
Figure 19-4 Cranial sutures and fontanels in infancy. Fibrous union of suture lines and interlocking of serrated edges (occurs by 6 months; solid union requires approximately 12 years).
Myelin Sheath
Axons are wrapped in concentric layers of myelin, a lipid-protein sheath (see Chapter 14). Specialized connective tissue cells, which in the peripheral nervous system are called Schwann cells, form membranes that wrap around the axon during embryonic development, laying down the lipoprotein lamellae of the myelin sheath. These axons are myelinated, whereas the axons that lack a sheath are thinner, unmyelinated fibers and conduct nerve impulses more slowly. During the first year of life, the presence or absence of various reflexes is indicative of the myelination that has occurred with growth of the infant.
Normal Growth and Development
Human neurologic functioning is primarily at a subcortical level at birth (impulses are handled by the brainstem and spinal cord). Many reflex patterns mediated by brainstem and spinal cord mechanisms are present at birth and disappear at predictable times during infancy. Table 19-1 summarizes the ages at which reflexes appear and disappear.
Table 19-1
| Reflex | Age at Appearance of Reflex | Age at Which Reflex Should No Longer Be Obtainable |
| Stepping | Birth | 6 weeks |
| Moro | Birth | 3 months |
| Sucking | Birth | 4 months awake |
| 7 months asleep | ||
| Rooting | Birth | 4 months awake |
| 7 months asleep | ||
| Palmar grasp | Birth | 6 months |
| Plantar grasp | Birth | 10 months |
| Tonic neck | 2 months | 5 months |
| Neck righting | 4-6 months | 24 months |
| Landau | 3 months | 24 months |
| Parachute reaction | 9 months | Persists for life |
Absence of expected reflex responses at the appropriate age indicates general depression of central or peripheral motor functions. Asymmetric responses may indicate lesions in the motor cortex or may occur with fractures of bones after traumatic delivery or postnatal injury. As the infant matures, the neonatal reflexes disappear in a predictable order as voluntary motor functions supersede them. Abnormal persistence of these reflexes is seen in infants with developmental delays or with central motor lesions.
Several differences between adults and children are important to the pathophysiology of the nervous system in children. First, the head of a normal infant accounts for approximately one fourth of the total height, whereas an adult’s head is one eighth of the total body height. Second, the bones of the infant’s skull are separated at the suture lines, thus forming two fontanels or “soft spots”: one diamond-shaped anterior fontanel and one triangular-shaped posterior fontanel. The posterior fontanel may be open until 2 to 3 months of age; the anterior fontanel normally closes by 18 months. Whereas the adult cranium is a closed cavity with sutures firmly holding the cranial bones together, the infant cranium has room for expansion through the fontanels. An adult’s head size will not expand, regardless of intracranial events such as trauma or increased production of cerebrospinal fluid (CSF). The infant’s head circumference, however, increases in size as a result of normal growth up to 5 years of age. The head is the fastest growing body part during infancy. Abnormal intracranial conditions, such as those characterized by increased ICP, also may result in an increased head circumference in excess of that expected with normal growth. Healthcare providers carefully monitor head growth during the first 5 years of life by measuring head circumference and comparing the results with a standardized growth chart.
STRUCTURAL MALFORMATIONS
Defects of Neural Tube Closure
Neural tube defects (NTDs) are caused by an arrest of the normal development of the brain and spinal cord and occur in about 3000 pregnancies in the United States each year.1 There is a strong association of fetal death with NTDs, reducing their actual prevalence of neural defects at birth.2 Maternal folate deficiency is associated with NTDs, but the specific mechanism that relates to how folate supplements prevent these anomalies is unknown.3 Periconceptional supplementation with folic acid can reduce NTDs by up to 70%.4
Defects of neural tube closure are divided into two categories: posterior defects and anterior midline defects. Posterior defects are more common. These include anencephaly (an, “without”; enkephalos, “brain”) and a group of disorders collectively referred to as the myelodysplasias (dys, “bad”; plassein, “to form”). Although myelodysplasia is defined as a defective formation of the spinal cord, the term is used to refer to anomalies of both the vertebral column and the spine (i.e., spina bifida and myelomeningocele).
Anterior midline defects are less common because the inductive processes occur in a relatively short period (2 to 3 days). These developmental defects may cause brain and skull abnormalities. The most extreme form is cyclopia, in which the child has a single midline orbit and eye with a protruding noselike appendage above the orbit.
Anencephaly
Anencephaly is an anomaly in which the soft, bony component of the skull and part of the brain are missing. This is a relatively common disorder, with an incidence rate of approximately 0.36 per 1000 total live births in the United States each year.5 When development is arrested early in anterior closure of the neural tube, the cerebral hemispheres, diencephalon, mesencephalon, cerebellum, brainstem, or spinal cord may be affected. At birth the infant’s head, viewed face-on, has a froglike appearance. These infants are stillborn or die within a few days after birth. Diagnosis is often made prenatally using ultrasound or evaluating maternal serum alpha fetoprotein (AFP).6
Encephalocele
Encephalocele refers to a herniation or protrusion of brain and meninges through a defect in the skull, resulting in a saclike structure. The incidence rate is approximately 1 in 5000 live births in the United States and Southeast Asia each year.7,8
PATHOPHYSIOLOGY Encephalocele occurs during the first weeks of pregnancy. When the defect contains only meninges, it is referred to as a cranial meningocele. Most encephaloceles occur in the occipital area, with the remainder found in the frontal, parietal, or nasopharyngeal regions.
CLINICAL MANIFESTATIONS Encephalocele usually is seen at birth as a midline skull defect through which a large mass protrudes (Figure 19-5). If the defect is located in the nasopharynx, no external anomaly is visible, but the child may experience nasal airway obstruction. On examination with a nasal speculum, a smooth, round mass will be visible in the nasal passages. A frontal encephalocele may extend into the orbit of the eye and produce proptosis on the affected side.
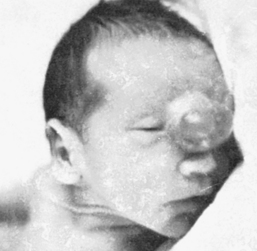
Figure 19-5 Newborn with frontal, nasal, interocular encephalocele. (Courtesy Dr. Charles Linder, Medical College of Georgia.)
EVALUATION AND TREATMENT Diagnosis is based on clinical manifestations and examination of the meningeal sac. With cranial meningocele, surgical repair of the cranial defect affords a good prognosis for most affected infants whose intellectual and motor functioning is normal. An occipital encephalocele may be associated with other findings, such as blindness and cognitive impairment. The size, location, and involvement of the encephalocele help determine a child’s development and intellectual outcome.
Meningocele
Meningocele is a saclike cyst of meninges filled with spinal fluid. It develops during the first four weeks of pregnancy when the neural tube fails to close completely (Figure 19-6).
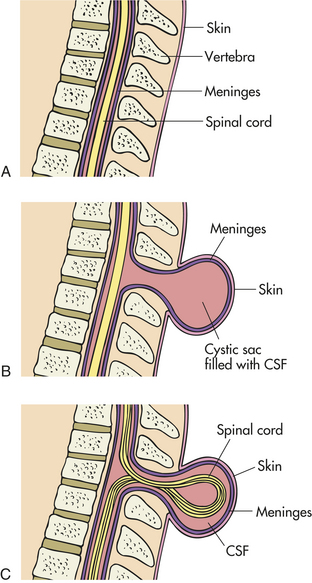
Figure 19-6 Comparison of normal spine and meningocele and myelomeningocele neural tube defects. Diagram depicts section through normal spine (A), spine with meningocele (B), and spine with myelomeningocele (C).
PATHOPHYSIOLOGY Meningocele is a sturctural anomaly of the posterior arch of the vertebra. The cystic dilation of meninges protrudes through the vertebral defect and around the malformed tube. The dura mater may be missing and the arachnoid layer of the meninges bulges beneath the skin. This defect does not involve the spinal cord. Meningoceles occur with equal frequency in the cervical thoracic and lumbar spine areas.
CLINICAL MANIFESTATIONS At birth the infant has a protruding sac on the back at the level of the defect. The sac may be covered by a thin layer of muscle and skin and usually appears as raw, fluid-filled tissue. Abnormal neurologic function sometimes is present. Talipes equinovarus (clubfoot), gait disturbance, bladder dysfunction, and upper extremity weakness also have been associated with meningocele. Hydrocephalus commonly is associated with this diagnosis.
EVALUATION AND TREATMENT The diagnosis is made on the basis of clinical manifestations and examination of the meningeal sac. In an effort to preserve neuronal function and minimize damage that may occur from infection and manipulation of the fragile sac, surgical closure is optimal during the first 72 hours of life. Functional implications depend on the level and severity of the defect (Table 19-2).
Table 19-2
Functional Alterations in Myelodysplasia Related to Level of Lesion
| Level of Lesion | Functional Implications |
| Thoracic | Flaccid paralysis of lower extremities; variable weakness in abdominal trunk musculature; high thoracic level may mean respiratory compromise; absence of bowel and bladder control |
| High lumbar | Voluntary hip flexion and adduction; flaccid paralysis of knees, ankles, and feet; may walk with extensive braces and crutches; absence of bowel and bladder control |
| Midlumbar | Strong hip flexion and adduction; fair knee extension; flaccid paralysis of ankles and feet; absence of bowel and bladder control |
| Low lumbar | Strong hip flexion, extension, and adduction and knee extension; weak ankle and toe mobility; may have limited bowel and bladder function |
| Sacral | Normal function of lower extremities; normal bowel and bladder function |
Data from Farley JA, Dunleavy MJ: Myelodysplasia. In Allen PJ, Vessey JA, editors: Primary care of the child with a chronic condition, ed 4, St Louis, 2004, Mosby.
Myelomeningocele
Myelomeningocele (meningomyelocele; spina bifida cystica) is a hernial protrusion of a saclike cyst (containing meninges, spinal fluid, and a portion of the spinal cord with its nerves) through a defect in the posterior arch of a vertebra. Eighty percent of myelomeningoceles are located in the lumbar and lumbosacral regions, the last regions of the neural tube to close. Myelomeningocele has an incidence rate ranging from 0.5 to 1 per 1000 pregnancies in the United States9 and thus is one of the most common developmental anomalies of the nervous system.
PATHOPHYSIOLOGY A myelomeningocele is the failure of the neural tube to close, resulting in a cystic dilation of meninges and protuberance of the spinal cord through the vertebral defect. This defect occurs during the first 4 weeks of the gestational period; at the end of this time the neural tube is closed anteriorly and posteriorly.
CLINICAL MANIFESTATIONS A myelomeningocele is evident at birth as a pronounced skin defect on the infant’s back (see Figure 19-6). The bony prominences of the unfused neural arches can be palpated at the lateral border of the defect. The defect usually is covered by a transparent membrane that may have neural tissue attached to its inner surface. This membrane may be intact at birth or may leak CSF, thereby increasing the risks of infection and neuronal damage. Until the defect is closed surgically, CSF may accumulate, resulting in further dilation and enlargement of the sac, which may risk more damage to the nervous system.
The actual involvement of the spinal cord has greater implications for the overall function of the infant throughout childhood (see Table 19-2). An absence of neurologic function may occur in some infants with myelomeningocele. Function may be attained if underlying fluid or pus accumulation is prevented from stretching and applying pressure to the neural tissue or if the biochemical alterations do not cause neural tissue to die. Residual neural tissue also may be lost temporarily or permanently at birth because of trauma to the tissue during delivery.
One serious, potentially life-threatening problem associated with myelomeningocele is the Arnold-Chiari type II malformation.10 This deformity involves the downward displacement of the cerebellum, cerebellar tonsils, brainstem, and fourth ventricle (Figure 19-7). (Arnold-Chiari type I malformations also involve downward displacement but are not as great as those in type II; type I is seen more commonly in adults.) Arnold-Chiari II malformation compresses and essentially stretches the posterior region of the cerebellum and brainstem downward through the foramen magnum and into the cervical space. The brainstem houses 10 cranial nerves. Pressure on this region may result in altered function of these nerves or actual palsies.
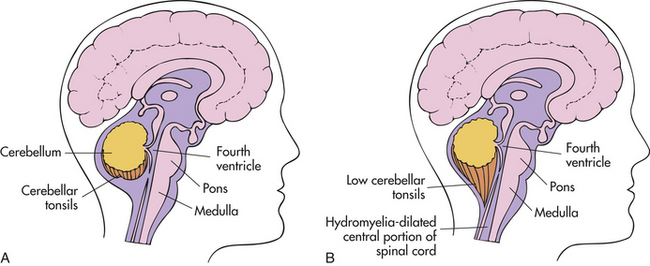
Figure 19-7 Comparison of normal brain and Arnold-Chiari type II malformation. Diagram depicts normal brain (A) and brain with Arnold-Chiari type II malformation (B).
Hydrocephalus occurs in 85% of these infants.11 Seizures also occur in 30% of those with myelodysplasia.12 Visual and perceptual problems, including ocular palsies, astigmatism, and visuoperceptual deficits, are common, particularly with Arnold-Chiari type II malformation.13 Motor and sensory functions below the level of the lesions are altered. This dysfunction may include degrees of weakness, paralysis, spasticity, and bowel and bladder dysfunction. Often these problems worsen as the child grows and the cord ascends within the vertebral canal, pulling primary scar tissue and tethering the cord.14 Several musculoskeletal deformities are related to this diagnosis, including clubfoot, dislocation of hip or hips, and poor spinal alignment. Spinal deformities, such as scoliosis and kyphosis, are common.15
Tethered cord syndrome may develop in children with myelomeningocele, particularly after surgical correction.16 The cord becomes abnormally attached or tethered as a result of scar tissue as it transcends the vertebral canal with growth and impairs oxidative metabolism.17 Symptoms are related to excessive tension on the lumbosacral cord and can include scoliosis, altered gait pattern, changes in muscle strength at or below the lesion, disturbance in urinary and bowel patterns, and back pain.18
EVALUATION AND TREATMENT Diagnosis is based on clinical manifestations and examination of the meningeal sac. However, because the pathophysiology of myelodysplasia is determined early in gestation, prenatal diagnosis is possible through ultrasonography. In addition, the presence of a neural tube defect may result in an elevated amniotic fluid AFP level and subsequent maternal serum AFP levels. Prenatal diagnosis offers the parents the option to terminate the pregnancy or become a candidate for fetal intrauterine repair.19,20 Cesarean delivery may be recommended to minimize trauma to the open myelomeningocele.21
Treatment for the infant with myelomeningocele is early surgical closure of the defect. Intrauterine surgery can be completed.22 Because myelodysplasia affects several other body systems (e.g., renal, gastrointestinal, musculoskeletal), these infants require a lifetime, comprehensive, multidisciplinary approach to treatment. The prognosis depends on the extent of the involvement at birth and the success of prophylactic and acute treatment for potential and actual complications that affect the many body systems. Symptomatic Arnold-Chiari type II malformations require surgical decompression and/or placement of cerebrospinal fluid shunts.11
Spina Bifida and Spina Bifida Occulta
When defects of neural tube closure occur, such as meningocele and myelomeningocele, an accompanying vertebral defect allows the protrusion of the neural tube contents. Such a defect is called spina bifida (split spine). The cause of spina bifida is unknown. Periconceptual maternal folate deficiency and genetic alterations are commonly associated with the defect.23 It also is possible for a defect to occur without any visible exposure of meninges or neural tissue. Because the defect is not apparent to the naked eye (i.e., it is “occult” or hidden), the term spina bifida occulta is used. In spina bifida occulta the posterior vertebral laminae have failed to fuse. This extremely common defect occurs to some degree in 10% to 25% of infants. Approximately 80% of these vertebral defects are located in the lumbosacral regions, most commonly in the fifth lumbar vertebra and the first sacral vertebra, and may be detected prenatally with ultrasonic scanning and AFP testing. About 3% of normal adults have spina bifida occulta of the atlas (cervical vertebra 1). The following cutaneous or subcutaneous abnormalities suggest underlying spina bifida:
• Abnormal growth of hair along the spine, which often is either very coarse or very silky
• A midline dimple with or without a sinus tract
• A cutaneous angioma, usually of the “port-wine” variety
• A subcutaneous mass, usually representing a lipoma or dermoid cyst
Spina bifida occulta usually causes no serious neurologic dysfunctions.24 The spinal cord and spinal nerves generally are anatomically and functionally normal. When dysfunctions occur, the common lumbosacral defects cause gait abnormalities, positional deformities of the feet as a result of muscle weakness, or sphincter disturbances of the bladder and bowel. These dysfunctions become evident during periods of rapid growth. Surgical closure is usually completed in the neonatal period and techniques are being developed for intrauterine closure.25
Cranial Deformities
Skull malformations range from minor, insignificant defects to major defects that are incompatible with life.
Acrania: In acrania the cranial vault is almost completely absent and an extensive defect of the vertebral column often is present. Acrania associated with anencephaly (absence of brain and spinal column) occurs in approximately 1 per 1000 live births and is incompatible with life. The malformation results from a failure of the cranial end of the neural tube to close during the fourth gestational week. Subsequently, the cranial vault fails to form.
Craniosynostosis: Craniosynostosis (craniostenosis) is the premature closure of one or more of the cranial sutures during the first 18 to 20 months of an infant’s life. The incidence of craniosynostosis is 1 in 1800 to 2200 live births.26 Males are affected twice as often as females. Craniosynostosis can occur as part of a syndrome or as an isolated defect. Nonsyndromic craniosynostosis is classified as simple craniosynostosis when only one suture is involved and compound craniosynostosis when two or more sutures are involved. Each suture closure may represent a different disease.27 Craniosynostosis prevents normal skull expansion and causes asymmetric skull growth. Premature closure of a suture causes failure of the growth of the bone located at a right angle to the involved suture. Compensatory growth occurs in regions where the sutures are patent, and this causes the various cosmetic deformities. In the absence of adequate sutures, cerebral growth may exceed the space present. Brain growth may be restricted, and compression may cause neurologic dysfunction from brain damage after 6 months of age (Figure 19-8).
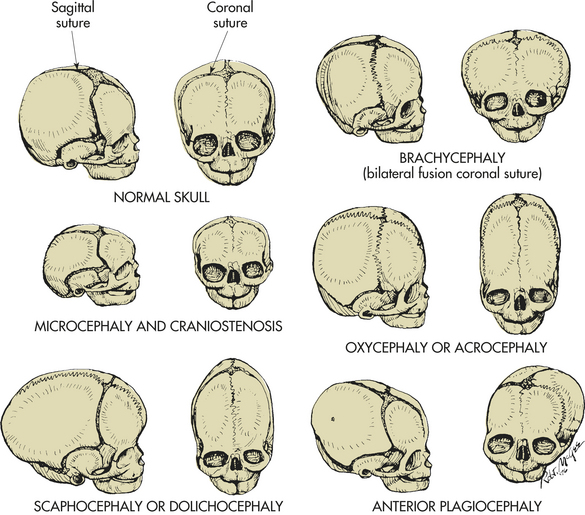
Figure 19-8 Craniosynostosis. Abnormal head configuration resulting from premature closing of cranial sutures. Normal skull, Bones separated by membranous seams until sutures gradually close. Microcephaly and craniostenosis, Microcephaly is head circumference more than 2 standard deviations below the mean for age, gender, race, and gestation and reflects a small brain; craniosynostosis is premature closure of sutures. Scaphocephaly or dolichocephaly (frequency 56%), Premature closure of sagittal suture, resulting in restricted lateral growth. Brachycephaly, Premature closure of coronal suture, resulting in excessive lateral growth. Oxycephaly or acrocephaly (frequency 5.8% to 12%), Premature closure of all coronal and sagittal sutures, resulting in accelerated upward growth and small head circumference. Anterior plagiocephaly (frequency 13%), Unilateral premature closure of coronal suture, resulting in asymmetric growth. (From Hockenberry JH et al: Wong’s nursing care of infants and children, ed 7, St Louis, 2003, Mosby.)
PATHOPHYSIOLOGY The exact causes of craniosynostosis are unknown, but the condition represents more than a single disorder of embryonic development with mutations in multiple cytokine signaling pathyways.28 Syndromic craniosynostosis involves mutations in several genes and accounts for about 30% of cases and chromosomal alterations account for another 10%.29 More than 100 syndromes have been identified, with Apert and Crouzon syndromes as the most common, and involve fibroblast growth factor signaling pathways.30 Most cases of craniosynostosis are nonsyndromic with no identifiable gene mutation.31
CLINICAL MANIFESTATIONS Craniosynostosis is classified according to head contour or suture involvement.31 Final skull contour is determined by the sutures that close, the duration and order of closure, and the ability of other sutures to compensate by expansion. (The frequency and types of craniosynostosis are depicted in Figures 19-8 and 19-9.)

Figure 19-9 Dolichoscaphocephaly in 14-year-old boy. One of the less threatening types of craniosynostosis. (From Dyken PR, Miller MD: Facial features of neurologic syndromes, St Louis, 1980, Mosby.)
Premature closure of the sagittal suture, the most common form of craniosynostosis, causes elongation of the skull in the anteroposterior direction. Other anomalies are seen in 25% of these children. When the coronal suture fuses prematurely, the brain expands in a lateral direction. This type of craniosynostosis is associated with a 33% to 66% incidence of associated anomalies. Approximately half of these children are mentally retarded.
EVALUATION AND TREATMENT Craniosynostosis must be differentiated from the more benign skull deformity known as positional plagiocephaly (flattening of one side of the head).31 Diagnosis is made on the basis of physical examination, head circumference measurements, and radiologic examination. Surgical treatment is indicated when closure of multiple sutures causes chronic increased intracranial pressure. Surgery then limits the extent of brain damage. In children with craniosynostosis of one suture, surgery often is performed for cosmetic purposes to limit the appearance of deformity.32
Microcephaly: Microcephaly is a rare defect in brain growth as a whole (see Figure 19-8). The word microcephaly is derived from the Greek (mikro, “small”; kephale, “head”). Cranial size is significantly below average for the infant’s age, sex, race, and gestation. The small size of the skull reflects a small brain, except in infants with premature closure of the sutures. The condition is not treatable.
True (primary) microcephaly (present at birth) can be caused by genetic alterations, including autosomal dominant, autosomal recessive, or X-linked genes, or various chromosomal abnormalities. Environmental causes include toxin exposure during the period of induction and major cell migration (Box 19-1). Radiation, intrauterine infection, or chemical exposure may be the initiating factor. Secondary microcephaly (develops postnatally) is associated with a variety of causes including infection, trauma, metabolic disorders, maternal anorexia experienced during the third trimester of pregnancy, and the presence of other genetic syndromes.33
Microcephalic brain weight may be as low as 25% of normal, and the number and size of the cortical gyri may be diminished. Growth of the frontal lobes is severely stunted, and the cerebellum often is disproportionately large. In microcephaly caused by perinatal or postnatal disorders, neuronal loss and gliosis may be present in the cerebral cortex. The neurologic manifestations of microcephaly range from decerebrate posture, complete unresponsiveness, and autistic behavior to mild motor impairment, mental retardation, and hyperkinesis.
Congenital Hydrocephalus: Congenital hydrocephalus is characterized by an increased volume of CSF. This increase in volume may be caused by a blockage within the ventricular system in which the CSF flows, an imbalance in production of the CSF, or a reduced reabsorption of the CSF that results in ventricular enlargement and increased ICP. The pressure within the ventricular system pushes and compresses the brain tissue against the skull cavity. When hydrocephalus develops before fusion of the cranial sutures, the skull has the capacity to increase its effort to accommodate this additional space-occupying volume and to preserve neuronal function. The overall incidence of hydrocephalus is approximately 3 per 1000 live births.34 The incidence of hydrocephalus, excluding the hydrocephalus associated with myelomeningocele, is approximately 0.5 to 1 per 1000 live births, with aqueductal stenosis as the cause for approximately one third of these cases.35 (Types of hydrocephalus are discussed in Chapter 16.)
PATHOPHYSIOLOGY Obstructive hydrocephalus is caused most commonly by congenital aqueduct stenosis. The cerebral aqueduct is narrowed or replaced by multiple channels, or “forks,” that end blindly. In a small number of children the stenosis is transmitted as an X-linked recessive trait. The Dandy-Walker malformation is a congenital defect of midline cerebellar structures and fourth ventricle in which hydrocephalus is caused by atresia of the foramina of Luschka or Magendie, leading the ventricular flow of CSF into a “blind pouch.” Other causes of obstructions within the ventricular system that can result in hydrocephalus include brain tumors, cysts, trauma, arteriovenous malformations, blood clots, and infections.
CSF travels throughout the ventricular system, surrounds the brain, and is reabsorbed into the venous system by the arachnoid villi. Blockage of the arachnoid villi may occur in conditions such as bacterial or viral meningitis, intraventricular hemorrhage, and subarachnoid hemorrhage, or may result from congenital malformations within this area. In this instance CSF flows or communicates effectively but is unable to be reabsorbed, resulting in hydrocephalus.
CLINICAL MANIFESTATIONS Congenital hydrocephalus may cause fetal death in utero, or the increased head circumference may require cesarean delivery of the infant. Symptoms of this condition depend directly on the cause and rate of hydrocephalus development. Infants may have no symptoms at birth. During the early weeks of life, the head begins to grow at an abnormal rate. Significant dilation of the ventricles may occur before an abnormal increase in head growth develops. The fontanels enlarge and become full and bulging (Figures 19-10 and 19-11). The separation of the cranial sutures leads to a resonant note when the skull is tapped, a manifestation termed Macewen sign (“cracked-pot” sign). The eyes may assume a staring expression, with sclera visible above the cornea, called sunsetting.
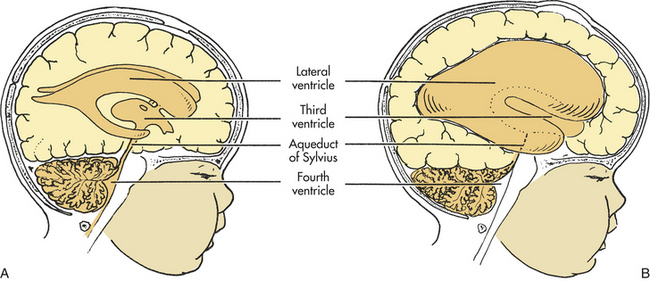
Figure 19-10 Hydrocephalus. A block in the flow of cerebrospinal fluid (CSF). A, Patent cerebrospinal fluid circulation. B, Enlarged lateral and third ventricles caused by obstruction of circulation—stenosis of aqueduct of Sylvius. (From Hockenberry MJ et al: Wong’s essentials of pediatric nursing, ed 8, St Louis, 2009, Mosby.)

Figure 19-11 Child with enlarged head caused by hydrocephalus. (From McLaurin DC: Pediatric neurosurgery, ed 2, Philadelphia, 1989, Saunders.)
The infant may have difficulty holding the head upright. The scalp skin is thin and shiny, and scalp veins may become prominent. The large cranial vault and the face are disproportionate, and frontal bossing may be present. The infant’s cry becomes high pitched as ICP rises; irritability, lethargy, vomiting, and other signs of increased ICP may develop. Dramatic head growth and enlargement, compression of the optic nerves, and optic chiasm occur in chronic, untreated hydrocephalus. However, because of early surgical intervention, these signs of hydrocephalus rarely are seen. When hydrocephalus develops late in childhood, the head may not have the capacity to enlarge, and evidence of increased ICP is present (see Chapter 16).
The relationship between hydrocephalus and mental retardation has been heavily debated. Correlation between the degree of hydrocephalus and impaired cognitive function often is a result of additional complications, such as severe congenital malformations, acute or chronic infections, or progressive brain tumors. Approximately two thirds of children with uncomplicated congenital hydrocephalus treated successfully with shunting may have normal to borderline intelligence.36
EVALUATION AND TREATMENT The definitive diagnostic tool for hydrocephalus is computed tomography (CT), and magnetic resonance imaging (MRI) may add information about the specific cause of the hydrocephalus. In infancy, head circumference measurements also are obtained and monitored. The treatment is surgical placement of a shunt to divert the excess CSF from the ventricular cavity to other areas of the body. Several types of shunts are available.37 The main objective of any shunt system is to decrease ICP and preserve neuronal function. With neurosurgical intervention and follow-up, the 5-year survival usually is greater than 80%. Most deaths that occur within this category result from severe congenital malformations and/or progressive disorders such as brain tumors. Children with hydrocephalus depend on the internal shunt system to maintain safe ICPs and therefore are forever at risk for sudden failure of this system, which leads to acute increased ICP and death.
ENCEPHALOPATHIES
Encephalopathy, a pathology involving the brain, is a general category of syndromes and diseases that include infections, ischemia, and metabolic and toxic causes. Encephalopathies in children are associated with a great variety of known and suspected causes. These disorders may be acute or chronic and static or progressive.
Static Encephalopathies
Brain injury may occur during gestation or birth or at any time during childhood growth and development, causing a static, nonprogressive disorder. The clinical manifestations depend on the site and extent of the injury, as well as the age of the child and stage of development at the time of injury. Varying degrees of impairment may result from diffuse or localized injury to the cortex. For example, cerebral palsy results when the motor areas of the brain are injured. Injury to the occipital lobe of the cerebral cortex early in gestation can interfere with normal cerebral maturation and result in blindness. Cognitive impairment may follow diffuse cerebral injury. Seizures also may develop from cortical injury, particularly if scar tissue remains.
Prenatal factors that affect the developing nervous system may be endogenous or exogenous. The fetus may be affected by impaired embryo implantation, chromosomal abnormalities, infection, trauma, radiation, and toxic substances. Maternal toxemia, diabetes mellitus, and maternal nutritional deficiencies can produce neurologic damage in the fetus. The developing nervous system is most susceptible to injury occurring during the first trimester of pregnancy. Anoxia, trauma, and infections are the most common factors that cause injury to the nervous system in the perinatal period. Infections, metabolic disturbances (acute or a result of inborn errors), trauma, toxins, and vascular disease may injure the nervous system in the postnatal period.
Cerebral Palsy
Cerebral palsy (CP) is the term given to a diverse group of nonprogressive syndromes that affect the brain and cause motor dysfunction beginning in early infancy. Although cerebral palsy is by definition nonprogressive, its clinical manifestations change with growth and maturation of the child.
Cerebral palsy is one of the most common crippling disorders of childhood, affecting nearly 500,000 children in the United States alone. Although the exact incidence is unknown, studies suggest that the incidence is 1 to 2.3 cases of cerebral palsy per 1000 live births.38,39 Causes of cerebral palsy are numerous, and genetic as well as environmental factors may be responsible. These factors can occur during the prenatal (most common), perinatal, or postnatal period (Table 19-3).
Table 19-3
Cerebral Palsy: Predisposing Factors and Known Causes
| Risk Factors | Associated Causes |
| Prenatal | |
| Maternal | Metabolic diseases |
| Nutritional deficiencies (e.g., anemia) | |
| Twin or multiple births | |
| Bleeding | |
| Toxemia | |
| Blood incompatibilities | |
| Exposure to radiation | |
| Infection (e.g., rubella, toxoplasmosis, cytomegalic inclusion disease) | |
| Premature labor | |
| Prematurity | Asphyxia leading to cerebral hemorrhage |
| Genetic factors | Absence of corpus callosum, aqueductal stenosis, cerebellar hypoplasia |
| Congenital anomalies of the brain | Unknown causes not evident on clinical examination |
| Perinatal | Anesthesia or analgesia during labor and delivery |
| Mechanical trauma during delivery | |
| Immaturity at birth | |
| Metabolic disorders (e.g., hyperbilirubinemia, hypoglycemia, amino acid disorders, hyperosmolality) | |
| Electrolyte disturbances (e.g., hypernatremia, hypoglycemia) | |
| Postnatal | Head trauma |
| Infections (e.g., meningitis, encephalitis) | |
| Cerebrovascular accidents | |
| Toxicosis | |
| Environmental toxins (e.g., lead ingestion, methyl mercury ingestion from contaminated fish) |
PATHOPHYSIOLOGY Several factors, alone or in combination, can produce brain damage that leads to cerebral palsy. Prenatal cerebral hypoxia, congenital malformations, and placental pathology can contribute to the systemic degeneration of immature areas of the brain white matter and interfere with cell maturation.40 The severity of the damage depends on the gestational age at the time of the injury and the degree of injury sustained.
Low birth weight and birth asphyxia are commonly identified risk factors for cerebral palsy.41,42 Hypoxia and asphyxia are known to cause edema in the brain. Lack of oxygen and the incorporation of amino acids during protein synthesis lead to acidosis. Carbon dioxide and lactic acid accumulate with acidosis, causing osmotic pressure changes. This condition contributes to generalized cerebral swelling and CNS damage. Magnesium sulfate in preterm newborns and head cooling during or after resuscitation at birth may reduce brain damage and risk of cerebral palsy.43,44
Vascular abnormalities, arterial or venous stasis, and thrombosis can occur as a result of tissue hypoxia or as unrelated structural alterations. These anomalies may result in direct brain trauma that leads to infarction, intraventricular hemorrhage, and subarachnoid hemorrhage. Intraventricular hemorrhage is a common cause of death in newborn infants. Such injuries contribute to CNS damage.
Physical trauma to the central or peripheral nervous system can occur during the birthing process. Linear and depressed fractures are seen in newborns when head molding is extreme, with resultant hemorrhages and tears of the tentorium or falx cerebri. Tearing of the superficial cerebral veins is a relatively common occurrence and causes a thin layer of blood over the cerebral convexity. This blood may irritate the brain and result in CNS dysfunction. The infant’s position during delivery may cause stretching and damage to nerves. Breech deliveries can cause traumatic injury to the brainstem or spinal cord, resulting in a more localized area of impairment. Malformations of the CNS play an important role in brain injury from perinatal trauma and predispose the infant to a greater probability of sustained injury to the CNS. Both faulty maturation of the nervous system and a greater vulnerability to perinatal trauma and hypoxia are responsible for a high incidence of neurologic dysfunction in the preterm infant. Genetic, teratogenic, and early pregnancy influences on the development of cerebral palsy are multifactorial and not yet fully understood.45
CLINICAL MANIFESTATIONS The syndromes associated with cerebral palsy can be classified according to the areas of the brain that are damaged, pyramidal (spastic) and extrapyramidal (nonspastic). Pyramidal/spastic cerebral palsy results from damage or defects in the brain’s corticospinal pathways (upper motor neuron) in either one or both hemispheres and accounts for approximately 70% to 80% of cerebral palsy cases. It is associated with increased muscle tone, prolonged primitive reflexes, exaggerated deep tendon reflexes, clonus, rigidity of the extremities, scoliosis, and contractures. Cognitive impairment occurs in about 30% of cases.46
Extrapyramidal/nonspastic cerebral palsy is caused by damage to cells in the basal ganglia, thalamus, or cerebellum and includes two subtypes: dyskinetic and ataxic. Dyskinetic cerebral palsy is associated with extreme difficulty in fine motor coordination and purposeful movements. Movements are jerky, uncontrolled, and abrupt, resulting from injury to the basal ganglia or thalamus. This form of cerebral palsy accounts for approximately 20% to 25% of cases. Ataxic cerebral palsy is associated damage to the cerebellum and manifests with gait disturbances and instability. The infant with this form of cerebral palsy may have hypotonia at birth, but stiffness of the trunk muscles develops by late infancy. This lack of flexibility exaggerates the infant’s inability to balance body position without support. Persistence of this increased tone in truncal muscles affects the child’s gait and ability to maintain equilibrium and depth perception. This form of cerebral palsy accounts for approximately 5% of cases. A child may have symptoms of each of these cerebral palsy types, which leads to a mixed-variety disorder that accounts for approximately 13% of cases.
Children with cerebral palsy often have associated neurologic disorders, such as seizures (35% to 50%), intellectual impairment ranging from mild to severe (50% to 75%), and visual impairment (50%). Because standardized intelligence tests do not allow for the physical handicaps of cerebral palsy, the incidence of associated cognitive impairment is uncertain. Other associated complications include but are not limited to hearing impairment, communication disorders, respiratory problems, bowel and bladder problems, and orthopedic disabilities.46
EVALUATION AND TREATMENT Diagnosis of cerebral palsy is made on the basis of neurologic examination and history. The management of children with cerebral palsy varies with age, type and severity of involvement, and associated disorders. Thus the scope of care required by the child and family includes ongoing medical, social and educational intervention, and a family-focused multidisciplinary team approach.47
Although the brain injury is static, the clinical picture of cerebral palsy may change with growth and development. The use of intrathecal baclofen pumps, botulinum toxin, and selective dorsal rhizotomy for spasticity has shown improvement in selected children with cerebral palsy.48,49
Inherited Metabolic Disorders of the Central Nervous System
A large number of inherited metabolic disorders have been identified. Because these disorders are inherited, their manifestations usually occur in infancy and childhood. Typically these metabolic disorders damage the entire CNS so extensively that these children do not survive to adulthood. The clinical syndromes of the inherited metabolic disorders depend on the nature of the biochemical defect and the stage of nervous system maturation. (Table 19-4 lists some of these inherited metabolic disorders.) Defects in amino acid and lipid metabolism are more common than rarely occurring defects in carbohydrate metabolism.
Table 19-4
Inherited Metabolic Disorders of the Central Nervous System
| Age of Onset | Disorder |
| Neonatal period | Pyridoxine dependency, galactosemia, maple syrup urine disease and its variant, phenylketonuria (PKU) |
| Early infancy | Tay-Sachs disease and its variants, infantile Gaucher disease, infantile Niemann-Pick disease, Krabbe disease (leukodystrophy), Farber lipogranulomatosis, Pelizaeus-Merzbacher disease and other sudanophilic leukodystrophies, spongy degeneration, Alexander disease, Alpers disease, Leigh disease (subacute necrotizing encephalomyelopathy), congenital lactic acidosis, Zellweger encephalopathy, Lowe disease (oculocerebrorenal disease) |
| Late infancy and early childhood | Disorders of amino acid metabolism, metachromatic leukodystrophy, late infantile GM1 gangliosidosis, late infantile Gaucher and Niemann-Pick diseases, neuroaxonal dystrophy, mucopolysaccharidosis, mucolipidosis, fucosidosis, mannosidosis, aspartylglycosaminuria, amaurotic idiocy (Jansky-Bielschowsky disease, Batten disease, Vogt-Spielmeyer disease, neuronal ceroid lipofuscinosis), Cockayne syndrome |
| Later childhood and adolescence | Progressive cerebellar ataxias of childhood and adolescence, hepatolenticular degeneration (Wilson disease), Hallervorden-Spatz disease, Lesch-Nyhan syndrome and other uremic states, familial calcification of vessels in basal ganglia and cerebellum, familial polymyoclonus, chronic familial leukodystrophy, homocystinuria, Fabry disease |
Defects in Amino Acid Metabolism: Biochemical defects in amino acid metabolism may be classified as (1) those in which the transport of amino acid is impaired, (2) those involving an enzyme or cofactor deficiency, and (3) those grouped around certain chemical components, such as sulfur-containing amino acids.50 Most of the disorders in the literature described to date suggest that the absence of enzymatic activity most often is caused by the genetically determined absence of the enzyme protein.
Diseases caused by an enzymatic deficiency are associated with increased blood concentrations of the amino acid whose degradation pathway is impaired and with the presence of the amino acid in the urine. Because the normal pathway is blocked, small amounts of certain metabolites are found in the blood. Thus in certain diseases, an increase of compromised amino acids and unusual metabolites may appear in blood and urine concentrates.51
Phenylketonuria: Phenylketonuria (PKU) is an inborn error of metabolism characterized by the inability of the body to convert the essential amino acid phenylalanine to tyrosine (Figure 19-12). PKU is caused by phenylalanine hydroxylase deficiency and has a prevalence rate of 1 per 14,000 worldwide.52 Because of its genetic component and distribution, this statistical prevalence varies widely on the basis of geographic and ethnic differences.53 Most natural food proteins contain about 15% phenylalanine, an essential amino acid. Phenylalanine hydroxylase controls the conversion of this essential amino acid to tyrosine in the liver. The body uses tyrosine in the biosynthesis of protein, melanin, thyroxine, and the catecholamines in the brain and adrenal medulla. Phenylalanine hydroxylase deficiency causes an accumulation of phenylalanine in the serum and subsequently in the urinary excretion of abnormal metabolites called phenyl acids. One of these phenyl acids, phenylpyruvic acid, gives the urine a characteristic musty odor and is responsible for the name given to the disorder. Such high blood levels of phenylalanine prevent sufficient neutral amino acid entry into the brain, which contributes to the neuropathologic process of PKU.54 Abnormalities occur, such as anomalous development of the CNS, defective myelination, cystic degeneration of the gray and white matter, and disturbances in cortical layers. Unfortunately, brain damage occurs before the metabolites can be detected in the urine, and damage continues as long as phenylalanine levels remain high.
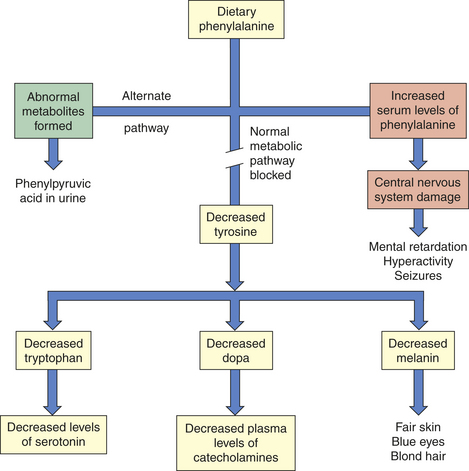
Figure 19-12 Metabolic errors and consequences in phenylketonuria. (Redrawn from Hockenberry MJ et al: Wong’s nursing care of infants and children, ed 8, St Louis, 2007, Mosby.)
Clinical manifestations related to CNS damage range from mild to severe behavioral disturbances, self-abusive tendencies, and seizures. Because of the lack of tyrosine and its relationship to the biosynthesis of melanin, children with PKU have a characteristic phenotype that includes blond hair, blue eyes, and fair skin. Children with genetically darker complexions may be red haired or brunette.
Less severe variants of this disorder are caused by defects in the phenylalanine hydroxylase system rather than the phenylalanine hydroxylase itself. This related disorder, known as hyperphenylalaninemia (HPA), occurs when plasma phenylalanine levels rise above normal but do not rise as high as in PKU.
Nonselective newborn screening is used to detect PKU and HPA in the United States and in more than 30 countries. Such programs are the greatest source of referrals and allow for accurate interpretation and follow-up of test results, including appropriate genetic and nutrition counseling.53 Individuals with PKU also need to be screened for response to BH4 (6-R-L-erythro-5,6,7,8-tetrahydrobiopterin), which significantly decreases blood phenylalanine levels with oral supplements.55
Treatment for PKU involves restriction of phenylalanine in the diet to maintain a nontoxic level and supplement with phenylalanine free L-amino acids. The diet also must be supplemented with adequate sources of energy, protein, and nutrients to allow for optimum growth and brain development. Supplementation of tyrosine also may be required if plasma levels are low. Enzyme replacement and PKU gene therapy are under investigation.56
Defects in Lipid Metabolism: Disorders of lipid metabolism are termed lysosomal storage diseases because each disorder in this group can be traced to a missing lysosomal enzyme. (Lysosomes, the vesicles within the cell whose primary function is to degrade the breakdown products of cellular metabolism, are discussed in Chapter 1.) An estimated 25 to 30 enzymes within the lysosomes participate in the breakdown of lipids, carbohydrates, proteins, and proteolipids A genetic defect results in a missing or defective enzyme and causes an excessive accumulation of a particular cell function. The enzyme defect may occur in the brain, liver, spleen, bone, or lung, thus involving several organ systems. Prenatal diagnosis is available. Therapy has been unsuccessful to date.
Tay-Sachs Disease: Tay-Sachs disease is a fatal autosomal recessive disorder (HexA gene on chromosome 15) caused by deficiency of the lysosomal enzyme hexosaminidase A (HexA), an enzyme that degrades GM2 gangliosides (fatty acids) within nerve cell lysosomes. Consequently there is accumulation of gangliosides (gangliosidosis) with toxicity to nerve cells.57 Approximately 80% of individuals diagnosed are of Jewish ancestry, although sporadic cases appear in the non-Jewish population.51 Prenatal screening is available.
In Tay-Sachs disease the pathologic changes predominate in the CNS, but neurons throughout the body contain characteristic changes in the cytoplasm. With time, neurons become distorted and balloon, and microglial cells, which also are swollen and filled with large granules, proliferate. Cystic degeneration of the cerebral white matter and atrophy of the cerebellar hemispheres often occur. The number of neurons is diminished. Changes in the spinal cord, particularly in the motor cells of the anterior horn of the cord, also are characteristic. Involvement of this region of the spinal cord results in hypotonia, hyporeflexia, and overall weakness.
Onset of this disease usually occurs when the infant is 3 to 6 months old. A loss of milestones is associated with an excessive startle response. Seizures, muscular rigidity, and blindness become prominent after the first year of life, and head size may increase. Death usually occurs by 2 to 5 years of age. No beneficial therapy has been developed. Genetic counseling programs are available, and some states require screening techniques for couples and those at risk who may be carriers.
Seizure Disorders in Children
Epilepsy: The incidence of epilepsy varies greatly with age. The incidence of epilepsy is estimated to be 0.5% to 1% of children with onset during infancy or childhood.58 Infants are particularly susceptible during the first 12 months of life. The incidence decreases with age; 75% to 80% of epilepsy cases initially occur before 20 years of age, with 30% initially occurring within the first 4 years of life. Approximately 181,000 persons in the United States are newly affected each year.58
PATHOPHYSIOLOGY Seizures are the abnormal discharge of electrical activity within the brain. When a sufficient number of neurons become overexcited, they discharge abnormally, which sometimes results in clinical manifestations. If clinical manifestations develop, the specific physical activity that occurs may depend on the origin of the electrical activity and its extent within the brain. Repeated recurrence of seizure activity is known as epilepsy. Seizures may result from an underlying disorder of the CNS or a disorder that directly or indirectly affects normal CNS function (see Table 16-11). Certain types of seizures may have a genetic component or familial predisposition, or they can result from maternal diseases or congenital structural anomalies of the CNS. During the newborn period, asphyxia, intracranial hemorrhage, CNS infections, injury, electrolyte imbalances, and inborn errors of metabolism may cause seizures. Etiologic factors of seizures in older infants and children generally are the same as during the first month of life. Often the cause of seizures is unknown.
Seizures and seizure patterns may change as a child grows and develops. The differences between the immature and mature nervous systems may help explain the changing patterns of clinical seizures with age. The immature nervous system has a reduced capacity for sustaining well-organized seizures. Intracortical connections are poorly developed, and the sending of impulses throughout the cortex is limited. At the cellular level neurons are less capable of firing in repetitive high-frequency bursts. The excitatory output of a seizure focus is further diminished because the affected neurons do not act synchronously. In addition, changing neurotransmitters, immaturity of cells, and ongoing postnatal factors affect seizure expression in children.
CLINICAL MANIFESTATIONS The clinical manifestations at the time of diagnosis vary depending on the primary cause and the extent and involvement of abnormal electrical discharges within the neuronal tissue. Because of the diversities and complexities that seizure activity invariably displays, an international classification system was adopted (see Table 16-9). This classification system groups seizures with similar clinical manifestations (see Table 16-10). Its general purpose is to assist the clinician with the assessment of the clinical course, the identification of the most appropriate treatment, and the evaluation of the individual’s response to treatment.
The international classification system of epilepsy contains three major groupings: (1) partial seizures, (2) generalized seizures, and (3) unclassified epileptic seizures. Each major grouping is then divided into subsets on the basis of clinical manifestations and electroencephalogram (EEG) findings.
Partial seizures are characterized by seizure activity that begins in and usually is limited to one part of either the left or right hemisphere. A simple partial seizure refers to seizure activity that occurs without loss of consciousness. A complex partial seizure refers to seizure activity that occurs with impairment of consciousness. The clinical activity displayed by the individual is contingent on the particular part of the cortex from which the seizure is generated. For example, partial seizures may result in abnormal motor activity, such as twitching or loss of tone, or sensory changes, such as tingling or numbness.
Simple partial seizures generally are confined to one hemisphere, whereas a complex partial seizure involves both cerebral hemispheres. A simple or complex partial seizure may evolve into a generalized tonic-clonic, tonic, or clonic convulsion.
Generalized seizures are those in which the first clinical manifestations indicate that the seizure activity starts in or involves both cerebral hemispheres. Consciousness may be impaired in this grouping of seizures. The clinical manifestations may include convulsive activity (tonic-tonic, tonic, or clonic activity) or nonconvulsive activity (absence seizures). Absence seizures do not follow a mendelian pattern of inheritance that results from a single gene defect but rather have an autosomal dominant inheritance pattern.59 Because both hemispheres are involved, the clinical manifestations almost always are bilateral.
Not all seizure disorders fit neatly into a classified grouping. These seizures are referred to as unclassified epileptic seizures and characteristically have a wide variety of abnormal clinical activity. Examples of this activity include rhythmic eye movements, chewing, and swimming movements. These activities are commonly seen in neonatal seizures.60
In addition to the seizures classified by the international system, there are several types of epileptic syndromes. These are seizure disorders that display a group of signs and symptoms that occur collectively and that characterize or indicate a particular condition. Several syndromes associated with epilepsy occur in infants and children. The three syndromes that occur most often are infantile spasms, Lennox-Gastaut syndrome, and juvenile myoclonic epilepsy.
Infantile spasms (also known as West syndrome) are a severe form of epilepsy characterized by a variety of clinical manifestations.61 The infant may have episodes of sudden flexion or extension movements involving the neck, trunk, and extremities. Mutations in ARX and CDKL5 genes and abnormal interneurons are involved in the pathogenesis of infantile spasms.62 Clinical manifestations of the resulting spasms may range from subtle head nods to violent body contractions, commonly referred to as jackknife seizures. Onset of infantile spasms usually is between 4 and 8 months of age and may be idiopathic or may occur in response to a CNS insult.63 An EEG will display the classic hypsarrhythmic chaotic pattern of epileptic spike and wave discharges on a slow, disorganized background. Infantile spasm manifests a typical clinical course. The “spasms” usually happen in clusters and occur 5 to 150 times per day. They usually are worse when the infant is waking up or falling asleep. Once begun, the seizure activity increases in intensity and severity over time. Invariably a loss of developmental milestones and disability is associated with this syndrome. Infantile spasms are also common (30%) in those with tuberous sclerosis complex (TSC).64 TSC develops from mutations in hamartin (TSC1) and tuberin (TSC2) genes. Tubers are cortical developmental malformations in the brain; they also form in other organs. Epilepsy associated with TSC is often difficult to treat and requires surgical intervention.64
Lennox-Gastaut syndrome is an epileptic syndrome characterized by an onset of seizures early in childhood, usually in males and around 1 to 5 years of age. This syndrome includes a variety of generalized seizures—predominantly tonic-clonic, atonic (drop attacks), akinetic, absence, and myoclonic activity. Mental retardation, delayed psychomotor development, personality disorders, and resistance to treatment are often associated with this syndrome.65
Juvenile myoclonic epilepsy is a primary generalized epilepsy that usually affects adolescents and young adults. Studies have indicated a possible locus on chromosomes 6p11, 15q14, 6q24, and 10q35 as a cause for this type of epilepsy.66 It is a relatively benign form of epilepsy involving myoclonic jerks of the neck, shoulders, and arms. The seizures may occur singularly or repetitively. This form of epilepsy commonly is associated with a normal neurologic examination, normal intelligence, and a positive family history of seizures and is often underdiagnosed.67
EVALUATION AND TREATMENT Diagnosis of epilepsy and seizure classification are based on history, clinical presentation, physical and developmental examination, and the record of milestone achievements. Evaluation and testing include an EEG to isolate the focus or origin and involvement of seizure activity, MRI, or CT scan of the brain to investigate the presence of a lesion or abnormal tissue. A complete metabolic workup must be reviewed to explore the possibility of deficiency or malabsorption.68
Specific treatment for epilepsy is directed at the particular clinical manifestations or syndrome of seizure activity and its underlying causes. Treatment usually begins with the use of anticonvulsant medications. Often the epileptic pattern and clinical course require more than one drug to control the abnormal discharges.69,70 A ketonic diet may be effective as a supplement treatment for epilepsy that is difficult to control with drugs71,72 (see Nutrition & Disease: Ketogenic Diet in Children with Epilepsy).
Surgery provides treatment for some forms of epilepsy that cannot be controlled with drugs. As with medical interventions, surgical therapy focuses on the particular clinical manifestations of seizure activity. Surgical interventions include resection of the epileptogenic zone of brain tissue (i.e., the temporal lobe for partial seizures or partial or complete severing of the corpus callosum for intractable generalized epilepsy). Children with intractable epilepsy have benefited somewhat from using the vagal nerve stimulator.73
Prognosis for epilepsy depends greatly on the type and severity of the disorder, the age of onset, coexisting factors, and the type and success of medical, surgical, and nutritional therapy. Several studies have estimated that approximately 40% to 50% of children diagnosed with epilepsy eventually will be seizure free.
Benign Febrile Seizures: Benign febrile seizures occur in 2% to 5% of children. These seizures usually are brief and self-limited and occur most often between the ages of 6 months and 5 years, with peak age at 14 to 18 months.74,75
PATHOPHYSIOLOGY The pathogenesis of benign febrile seizures is unknown. A familial incidence of benign febrile seizures indicates a genetic predisposition to the problem. Factors that contribute to susceptibility include age, degree and rate of temperature elevation, and nature of the particular fever-inducing illness. Any disorder producing a high fever may provoke benign febrile seizures in susceptible children.76
CLINICAL MANIFESTATIONS Characteristic features distinguish benign febrile seizures from seizures precipitated by fever:
1. Benign febrile seizures are rare before 9 months or after 5 years of age.
2. The convulsion occurs with a rise in temperature greater than 39° C (102.2 °F).
3. An acute respiratory or ear infection usually is present, with no evidence of CNS infection or inflammation.
4. Most seizures occur during the first 24 hours of the illness.
5. The convulsion is short (15 minutes or less), generalized, and predominantly tonic.
7. The seizure usually does not recur during the same infection.
Complex febrile seizures have characteristic features similar to these except that (1) they have a longer duration than do benign febrile seizures, usually longer than 15 minutes; (2) they have focal characteristics; and (3) they usually occur more than once in a 24-hour period. Complex febrile seizures are considered a risk factor for the development of epilepsy and there may be a genetic predisposition.77
EVALUATION AND TREATMENT Reduction of elevated body temperature usually controls benign febrile seizures without anticonvulsant medication. In selected individuals, phenobarbital is the most effective medication for preventing recurrence of benign febrile seizures.
Status Epilepticus: Status epilepticus is defined as the state of continuing or recurring seizure activity in which the recovery from seizure activity is incomplete. Seizure activity is unrelenting and usually lasts for 30 minutes or more. Any one of the seizure activities discussed can evolve into status epilepticus. Status epilepticus is a medical emergency that requires immediate intervention.78
Acute Encephalopathies
Reye syndrome is characterized by encephalopathy and fatty changes in a variety of organs, especially the liver. The incidence of Reye syndrome has declined sharply over the past 20 years, coinciding with increased public awareness of the association between ingestion of aspirin during illness and subsequent development of Reye syndrome.79 Although Reye syndrome is becoming increasingly rare in the population, a brief overview of it is important for the following reasons:
1. It may be considered a prototype for acute hepatic encephalopathies.
2. The potential for recurrence is a factor.
3. The use of acetaminophen over aspirin should be considered important and discussed with the parents when obtaining a history.
PATHOPHYSIOLOGY Reye syndrome usually is associated with influenza B or varicella virus infections in children who have taken aspirin or aspirin-containing products. The exact cause of pathology is unknown, and inborn errors of metabolism can be a contributing factor.80 A distinct clinical syndrome is apparent. The profound hypoglycemia, hypoketonemia, hyperammonemia, and increase in short-chain fatty acids in the serum after liver involvement are responsible for the cerebral manifestations. The liver shows diffuse deposits of lipids and absence of any inflammatory reaction or necrosis. Fatty degeneration of the kidneys leads to azotemia (excess urea in the blood). The brain is extremely edematous.81,82
The development of Reye syndrome has been linked to the administration of salicylates (aspirin). The American Association of Pediatrics has recommended not administering aspirin to children who have varicella or flulike symptoms. A direct relationship clearly exists between this recommendation and the overall decrease in incidence of Reye syndrome. A further reduction has occurred with administration of the varicella vaccine to children between 12 and 18 months of age.83
CLINICAL MANIFESTATIONS Typically Reye syndrome develops in a previously healthy child who is recovering from varicella, influenza B, upper respiratory infection, or gastroenteritis. The various clinical states are as follows:
Stage I—Vomiting, lethargy, drowsiness, nightmares
Stage II—Disorientation, delirium, aggressiveness and combativeness, central neurologic hyperventilation, shallow breathing, hyperactive reflexes, stupor
Stage III—Obtundation, coma, hyperventilation, decorticate rigidity
Stage IV—Deepening coma, decerebrate rigidity, loss of ocular reflexes, large fixed pupils, divergent eye movements
Stage V—Seizures, loss of deep tendon reflexes, flaccidity, respiratory arrest, extremely high blood ammonia levels, death
Death occurs in 30% to 40% of cases from brainstem dysfunction.84
EVALUATION AND TREATMENT According to the Centers for Disease Control and Prevention (CDC), the following conditions must be present for diagnosis of Reye syndrome:
1. Acute, noninflammatory encephalopathy documented by (a) alteration in level of consciousness and, if available, (b) a record of CSF containing leukocytes (8/mm3), and (c) a histologic specimen demonstrating cerebral edema without perivascular or meningeal irritation
2. Hepatomegaly documented by either a liver biopsy or autopsy
3. No more reasonable explanation for cerebral and hepatic abnormalities
The severity of the condition is inversely related to the age of the child at the onset of the illness.
The management of children with Reye syndrome ranges from simple monitoring to extremely complex neurointensive care. Treatment and outcome vary depending on the stage of involvement and the individual child’s symptoms.
Intoxications of the Central Nervous System
Drug-induced encephalopathies always must be considered a possibility in the child with unexplained neurologic changes. Such encephalopathies may result from accidental ingestion, therapeutic overdose, intentional overdose, or ingestion of environmental toxins (the most commonly ingested poisons are listed in Box 19-2). About 2 million childhood poisonings that require medical attention occur each year, 1000 of which result in death.
High blood levels of lead occur in lead poisoning. If lead poisoning is not treated, lead encephalopathy will result and cause serious and irreversible neurologic damage85,86 (Figure 19-13). Those at greatest risk are children 2 to 3 years of age and those prone to picas and living in lead-contaminated environments. Pica is the habitual, purposeful, and compulsive ingestion of nonfood substances such as clay, dirt, and paint chips. Lead intoxication also may occur from long-term exposure to smelters, sniffing of gasoline, lead-based paint, and ingestion of airborne lead.87
An estimated 275,000 to 810,000 U.S. children have excessive amounts of lead in their blood. Black children have a six times greater incidence of symptoms than white children. Most lead exposure is preventable.87
Meningitis
Meningitis is an inflammation of the meningeal coverings of the brain and spinal cord. The origin of such inflammation and acute encephalopathy can be caused by bacteria, viruses or other microorganisms. Aseptic meninigitis has no evidence of bacterial infection but may be associated with viral infection, systemic disease or drugs.
Bacterial Meningitis: Bacterial meningitis is one of the most serious infections to which infants and children are susceptible.88 In the United States there are approximately 6000 cases per year of bacterial meningitis, of which half occur in children younger than 18 years of age. The microorganisms accountable for this illness are Streptococcus pneumoniae (1.1 per 100,000) and Haemophilus influenza type B (Hib) (0.2 per 100,000), especially in children beyond the neonatal period.
Before vaccines, H. influenzae type B was once the most common pathogen of bacterial meningitis in children younger than 5 years. The occurrence has dropped 95% with the advent of the Hib vaccine. Otitis media or sinusitis may be a precursor because the infections are almost always associated with a bacterium in the blood.
S. pneumoniae is the most common microorganism in children 1 to 23 months of age. Staphylococcal or streptococcal meningitis can occur in children of any age but shows a predilection for children who have had neurosurgery, skull fracture, or a complication of systemic bacterial infection. Infections that originate in the middle ear, sinuses, or mastoid cells also may lead to S. pneumoniae infection in children. In addition, this microorganism tends to occur in children with sickle cell disease or splenectomy. One in every 24 children with sickle cell disease develops pneumococcal meningitis by the age of 4 years. This incidence is 36 times greater than that found in the black population without sickle cell disease and 314 times greater than in white children. Escherichia coli and group B beta-hemolytic streptococci are the most common causes of meningitis in the newborn period.89
The second most common microorganism causing bacterial meningitis, particularly in children younger than 4 years, is Neisseria meningitidis (meningococcus) (groups A,B,C,Y and W135).89 Approximately 2% to 5% of healthy children are carriers of N. meningitidis. The risk of meningitis in daycare center contacts of children with meningococcal disease is 1 per 1000.90 During epidemics among military personnel, nasal and oral secretions from as many as 90% of those examined reveal N. meningitidis, suggesting a high rate of infectious transmission.
PATHOPHYSIOLOGY The cause of bacterial meningitis is related to the age of the child and to a number of factors that predispose the child to bacterial infection or that alter the child’s response to an invading microorganism. Meningitis caused by Staphylococcus aureus or Pseudomonas aeruginosa may develop in the child with cystic fibrosis or severe burns. (For further discussion, see Chapter 17.) Infection with N. meningitidis is particularly virulent. Any microorganism may be pathogenic under the appropriate circumstances in a given individual.91
Pathogens enter the nervous system by direct extension from a contiguous (i.e., paranasal sinuses or mastoid cells) or, more commonly, by hematogenous spread (see Figure 17-32). Infection may spread through the blood to the meninges in children with infective endocarditis, pneumonia, thrombophlebitis, or after neurosurgical procedures. Many bacteria are very successful at evading normal defenses (see Chapter 9). Pathogens then cross the blood-brain barrier, enter the cerebrospinal fluid, and multiply. Bacterial toxins increase cerebrovascular permeability, causing alterations in blood flow and edema. Thrombosis and increased ICP can cause neurologic damage. Increased ICP may be increased further by obstruction to the CSF circulation. Thickened meninges and fibrous exudate in the subarachnoid space at the base of the brain obstruct the CSF, resulting in communicating hydrocephalus. Herniation of the brainstem causes death.
CLINICAL MANIFESTATIONS Acute bacterial meningitis often is preceded by an upper respiratory or a gastrointestinal infection. Fever, headache, vomiting, irritability, photophobia, and nuchal and spinal rigidity develop and can progress rapidly to a decreased level of consciousness and seizures. Irritation of the meninges and spinal roots causes pain and resistance to neck flexion (nuchal rigidity), a positive Kernig sign (resistance to knee extension in the supine position with the hips and knees flexed against the body), and a positive Brudzinski sign (flexion of the knees and hips when the neck is flexed forward rapidly). With severe meningeal irritation the child may demonstrate opisthotonic posturing (rigid arching of the back with the head extended). Meningococcal meningitis can produce a characteristic petechial rash.92
EVALUATION AND TREATMENT A definitive diagnosis is made only by examination of CSF obtained from a lumbar puncture. The principles of treatment are similar to those followed for adults (see Chapter 17) and are based on the culture results in which the causative microorganism is identified. The conjugate vaccines against H. influenzae and N. meningitidis (serogroups A, C, Y, and W-135) decrease the rate and spread of this disease. Vaccines for serogroup B N. meningitidis are not yet available.93 Prophylactic antibiotics are effective guided by culture of specific microorganisms.94
The factors that influence outcomes are the age of the child (mortality is highest in infants younger than 1 year), the infective microorganisms (the lowest mortality is in meningococcal meningitis and the highest in meningitis caused by gram-negative enteric microorganisms), and the duration and extent of inflammation before treatment. Approximately 8% of children with H. influenzae meningitis die; 35% of the survivors have serious and permanent sensory or motor dysfunction caused by pressure on the peripheral nerves during the early phases of the illness. Approximately 5% of the children who survive meningitis have hearing deficits; 10% to 15% have cerebral damage, hydrocephalus, motor deficits, or sensory impairments.95
Viral Meningitis: The hallmark of viral meningitis is a mononuclear response in the CSF and the presence of normal blood glucose level. In some cases the findings with aseptic meningitis are consistent with bacterial meningitis. The clinical manifestations are similar to those in bacterial meningitis, although usually milder. Isolation of the virus is difficult and often impossible. Treatment usually begins with aggressive administration of antibiotics (potential bacterial meningitis) until the diagnosis is confirmed. Treatment may include use of antiviral agents.
Human Immunodeficiency Virus Encephalopathy
A particularly vulnerable site of human immunodeficiency virus (HIV) type-1 infection in infants and children is the CNS (see Chapter 9 for details of HIV infection).
PATHOPHYSIOLOGY Progressive HIV encephalopathy is an infrequent and reversible complication of HIV infection as the disease responds to highly active antiretroviral therapy (HAART).96
CLINICAL MANIFESTATIONS The CNS is a distinct reservoir for HIV-1 but the pathogenesis of HIV encephalopathy in children is not clearly understood. The presence of viral products, inflammatory mediators, and overstimulation of the N-methyl-D-aspartate–type receptor system leads to neuronal injury and death.97 (See page 627.)
The 1994 classification from the CDC requires one of the following progressing findings to be present for at least 2 months,98 in the absence of a concurrent illness other than HIV that could explain the findings:
1. Failure to attain or loss of developmental milestones, or loss of intellectual ability, verified by standard developmental scale or neuropsychologic tests
2. Impaired brain growth or acquired microcephaly demonstrated by head circumference measurements or brain atrophy demonstrated by CT or MRI with serial imaging required in children younger than 2 years
3. Acquired symmetric motor deficits manifested by affecting a child 1 month of age or older
The onset of progressive encephalopathy may be a prognostic indicator of a poor outcome and it may be difficult to completely differentiate the effect of HIV infection on the CNS from the effects of prenatal and perinatal exposure. In addition, other insults may accompany HIV in a young child and affect growth and development, such as drug exposure, prematurity, chronic illness, and a chaotic social atmosphere.99,100
EVALUATION AND TREATMENT A definite diagnosis of HIV is made by patient history, viral culture, and clinical manifestations. Monitoring CD8(+) T lymphocytes and monocytes, in addition to CD4(+) T lymphocytes, has been suggested for predicting risk for progressive encephalopathy. Decreases in CD8(+) T lymphocytes diminish defenses against viral infection and facilitate infected monocytes to cross the blood-brain barrier.101 A growing number of investigational protocols are available for treatment of children with HIV. In general, treatment is focused on the preservation and maintenance of the immune system, aggressive response to opportunistic infections, and support and relief of symptomatic occurrences and administration of HAART.97,102 (HIV treatment is discussed in Chapter 9.)
CEREBROVASCULAR DISEASE IN CHILDREN
Cerebrovascular disease in children differs from that in adults in three ways:
1. An absence of predisposing factors, such as high blood pressure and arteriosclerosis
2. Significant differences in the clinical response related to the developing nervous system and thus a greater capacity for the pediatric brain to recover from vascular insult
Cerebrovascular disease can be divided into two categories: occlusive and hemorrhagic.
Occlusive Cerebrovascular Disease
Occlusive cerebrovascular disease is rare in children and may result from embolism, sinovenous thrombosis, or congenital or iatrogenic narrowing of vessels, which leads to a decreased flow of blood and oxygen to areas of the brain. Neonatal arterial ischemic stroke is estimated at 1 in 4000 live births and childhood stroke is about 2 to 8 in 100,000 in the United States. Sickle cell disease and cardiac anomalies are the most common disorders that lead to arterial ischemic stroke.103
Moyamoya disease is a rare, chronic, progressive vascular stenosis of the circle of Willis with obstruction of arterial flow to the brain. Moyamoya, a Japanese term, means “puff of smoke,” which describes its appearance on CT examination. The vascularity may be a congenital anomaly, or it can develop as a result of cranial radiation therapy. Complications are developmental delay, mental retardation, and cerebral hemorrhage.104,105 Treatment is surgical bypass of the occluded region.
Hemorrhagic Cerebrovascular Disease
PATHOPHYSIOLOGY Congenital cerebral arteriovenous malformations are the most common cause of intracranial bleeding and hemorrhagic stroke in children.106 Hemorrhagic disease may result from vascular anomalies that lead to rupture, such as aneurysm, or from congenital arteriovenous malformation. The rupture and symptomatic development of a cerebral aneurysm is rare in children younger than 19 years. Intraventricular hemorrhage associated with premature birth is related to unstable blood pressure, and cerebral blood flow are contributing factors.107
CLINICAL MANIFESTATIONS The extent of the pathologic condition usually is less extensive in children than in adults. Symptoms may include degrees of hemiplegia (flaccid, spastic), weakness, seizures, headache, high fever, nuchal rigidity, hemianopia, sensory changes, facial palsy, and temporary aphasia.
EVALUATION AND TREATMENT Diagnosis of cerebrovascular disease is made through a series of tests, including CT, MRI, magnetic resonance angiography (MRA), angiogram, and echocardiogram.108 History of evolving symptoms and past medical history are of vital importance in attaining an accurate diagnosis. Causative factors in the change in vascular flow often are not determined. Malformations vary in size, location, and symptoms, and these factors determine treatment. Options include surgery, radiation therapy, and embolic occlusion of the malformation109 (see Chapter 17).
Excellent collateral circulation in the child’s brain allows for more rapid recovery of motor function. The developing brain, however, may suffer more global, long-term effects, leading to mental retardation, behavior disorders, and seizures.
CHILDHOOD TUMORS
Brain tumors are the most common solid tumor and the second most common primary neoplasm in children, second only to leukemia. Approximately 50% of solid tumors in children are nonmalignant.110 Overall, brain tumors account for nearly 20% of all childhood cancers, with an annual incidence of 2.4 to 4 per 100,000 in the United States; approximately 2000 cases are diagnosed each year.111 Brain tumors remain the leading cause of death from disease in children ages 1 to 15 years.112
The cause of brain tumors is largely unknown, although genetic, environmental, and immune factors have been implicated in some tumor development. Other factors that have been investigated include familial tendencies, radiation, oncologic viruses, and chemical carcinogens.113 An important area of study has been the relationship of parental occupation to subsequent brain tumors in offspring. Associations have been found with tumors and parental employment, for example, parental exposure to hydrocarbons and employment in the aircraft and paper/pulp industries. Alterations in embryologic development also may play a part in the development of childhood brain tumors. This theory suggests that tumors arise from cells that are “misplaced” during embryonic development, never maturing but later proliferating in this immature form. Chromosomal work has implicated the deletion of chromosomes 22 and 17 in some pediatric brain tumors. Further studies are being conducted that may affect the course of treatment for these children.114 None of these factors, however, has proved significant.
PATHOPHYSIOLOGY Most childhood brain tumors arise from glial tissue, the supportive tissue of the brain. Tumors also may originate in other tissue such as nerve cells, cranial nerves, the pineal and pituitary glands, blood vessels, or neuroepithelium. Brain tumors are classified by the tissue and location from which they arise. Because a uniform pathologic nomenclature has yet to be established, inconsistencies occur when statistical data are compared.
Two thirds of all pediatric brain tumors are found in the posterior fossa region of the brain. This area also may be referred to as infratentorial because it is located below the tentorium. The tentorium is the layer of dura mater that separates the cerebellum from the hemispheres or cerebrum. Thus the area above the tentorium is referred to as the supratentorial region. Approximately one third of childhood brain tumors are located in the supratentorial space, whereas, in the adult population two thirds of brain tumors are located in the supratentorial region and only one third in the infratentorial region. The types and characteristics of childhood brain tumors are summarized in Table 19-5.
Table 19-5
| Type | Characteristics |
| Astrocytoma | Arises from astrocytes, often in the cerebellum or lateral hemisphere |
| Slow growing, solid or cystic | |
| Often very large before diagnosed | |
| Varies in degree of malignancy | |
| Optic nerve glioma | Arises from optic chiasm or optic nerve |
| Slow-growing, low-grade astrocytoma | |
| Medulloblastoma (infiltrating glioma) | Often located in cerebellum, extending into fourth ventricle and spinal fluid pathway |
| Rapidly growing malignant tumor | |
| Can extend outside central nervous system | |
| Brainstem glioma | Arises from pons or myeloencephalon |
| Numerous cell types | |
| Compresses cranial nerves V through X | |
| Ependymoma | Arises from ependymal cells lining ventricles |
| Circumscribed, solid, nodular tumors | |
| Craniopharyngioma | Arises near pituitary gland, optic chiasm, and hypothalamus |
| Cystic and solid tumors that affect vision, pituitary, and hypothalamic functions |
Brain tumors, by virtue of their location, have unique characteristics that distinguish them from tumors found elsewhere in the body. A number of brain tumors in children may be considered histologically benign yet clinically malignant and life threatening because of their location. For example, a tumor located in the brainstem region may appear benign under the microscope, but the clinical presentation threatens and all too often overrides the vital functions of the brainstem.
Types
Medulloblastoma, ependymoma, astrocytoma, brainstem glioma, craniopharyngioma, and optic nerve glioma make up approximately 75% to 80% of all pediatric brain tumors.115 The location of brain tumors in children is illustrated in Figure 19-14; specific characteristics, treatment strategies, and prognoses are listed in Tables 19-5 and 19-6.
Table 19-6
Treatment Strategies for Childhood Brain Tumors
| Tumor Type | Treatment and Prognosis |
| Cerebellar astrocytoma | Surgery; possibly curative |
| Radiation and chemotherapy not proven successful but may delay recurrence | |
| Survival rate of more than 5 years in 50% to 75%; if tumor recurs, it does so very slowly | |
| Medulloblastoma | Surgery, primarily as a partial resection to relieve increased intracranial pressure and “debulk” the tumor |
| Type of treatment is age dependent | |
| Radiation is the primary treatment; may include spinal radiation | |
| Chemotherapy shows promise in conjunction with craniospinal radiation | |
| 35% 5-year survival rate | |
| Brainstem glioma | Surgery, resection occasionally possible |
| Radiation, primarily palliative treatment | |
| Chemotherapy not yet proven beneficial, but new protocols being studied | |
| 20% to 40% 5-year survival rate depending on total resection | |
| Ependymoma | Tumor possibly indolent for many years |
| Surgery rarely curative; risk of resecting an infratentorial tumor is too great | |
| Radiation for palliation (controversy regarding whether local or craniospinal radiation is better) | |
| Chemotherapy used for recurrent disease but with disappointing results | |
| 20% to 80% 5-year survival rate depending on total resection | |
| Craniopharyngioma | Surgery possibly successful when a complete resection is performed (partial resection usually requires further treatment) |
| Radiation after partial surgical resection | |
| Chemotherapy not commonly used | |
| 75% to 85% 5-year survival rate | |
| Optic nerve glioma | Initial treatment controversial |
| Surgery used for diagnosis or relief of hydrocephalus | |
| Radiation useful, particularly if the tumor is not treated by surgery | |
| Cerebral astrocytoma | Surgery used if resection is possible |
| Radiation useful for all grades of astrocytoma | |
| Chemotherapy beneficial in higher-grade tumors, but further study required |
CLINICAL MANIFESTATIONS The actual location of the brain tumor dictates the presenting signs and symptoms (discussed in greater detail with each particular brain tumor type). In addition to tumor location and cell type, the rate of growth of the tumor also determines the presenting signs and symptoms. The ability of the brain and intracranial cavity to compensate for tumor growth is directly related to the rate of its growth. This compensatory mechanism allows the components of the intracranial space (blood, brain, and CSF) to adapt temporarily to slow changes in ICP. Therefore slow-growing tumors can grow to enormous size before signs and symptoms are apparent. Conversely, fast-growing tumors allow little time for compensation of the space-occupying lesion, and clinical symptoms occur quickly.
Signs and symptoms of brain tumors in children vary from generalized and vague to those that are localized and related specifically to the anatomic area. If the tumor is located in the posterior fossa region, the fourth ventricle may become blocked, which leads to hydrocephalus and signs of increased ICP. Increased ICP also may occur because of the additional mass volume within the fixed container of the skull vault. The symptoms of increased ICP include headache, vomiting, lethargy, and irritability. If a young child complains of a headache, a thorough investigation should take place because headache is an uncommon complaint in young children. Headache caused by increased ICP usually is worse in the morning and gradually improves during the day when the child is upright and venous drainage is enhanced. Frequency of headache and other symptoms worsens as the tumor grows. A headache related to increased ICP generally occurs because of expansion of the lateral ventricle and cerebral hemisphere, which causes a stretching of the pain-sensitive dura mater. Irritability or possible apathy and increased somnolence also may result from increased ICP. Like headache, vomiting occurs more commonly in the morning. It frequently is not preceded by nausea and may become projectile, differing from a gastrointestinal disturbance in that the child may be ready to eat immediately after vomiting. Other signs and symptoms that can accompany increased ICP include increased head circumference with bulging fontanel in children younger than 2 years, cranial nerve palsies, and papilledema.
Localized findings relate to the degree of disturbance in physiologic functioning in the area where the tumor is located (see Table 17-9). Infratentorial tumors exhibit localized signs of impaired coordination and balance, including ataxia, gait difficulties, truncal ataxia, and loss of balance. A medulloblastoma is an embroynal tumor and the most common childhood malignant tumor. It occurs as an invasive tumor that develops in the vermis of the cerebellum and may extend into the fourth ventricle.116 Angiogenesis is the hallmark of progressive medulloblastomas. The ependymoma develops in the fourth ventricle and arises from the ependymal cells that line the ventricular system. The histology of the ependymoma varies, which makes the treatment course and prognosis difficult to establish. Because both tumors are located in the posterior fossa region along the midline, presenting signs and symptoms are similar. In addition to those already described, they may obstruct the fourth ventricle, resulting in hydrocephalus and generalized increased ICP, headache, nausea and vomiting, and nystagmus (involuntary eye movement).
In contrast, cerebellar astrocytomas are located on the surface of the right or left cerebellar hemisphere and cause unilateral symptoms (occurring on the same side of the tumor), such as head tilt, limb ataxia, and nystagmus when the eyes are turned toward the tumor.
Brainstem gliomas often cause a combination of cranial nerve involvement, cerebellar signs of ataxia, and corticospinal tract dysfunction. A common clinical pattern includes unilateral paralysis of cranial nerves with contralateral paralysis of the arm and leg, hyperreflexia, and extensor plantar responses. Increased ICP generally does not occur. Children with diffuse tumors usually die within 1 to 2 years.117
The area of the sella turcica, the structure containing the pituitary gland, is the site of several childhood brain tumors, including craniopharyngioma (the most common) and pituitary adenoma (see Chapter 21). These tumors may originate from the pituitary gland or the hypothalamus. They are usually slow growing and may be quite large by the time of diagnosis. Symptoms include headache, seizures, diabetes insipidus, early onset of puberty, and growth delay. Other tumors located in this region of the brain include optic gliomas. Tumors that involve the optic tract may cause complete unilateral blindness and hemianopia of the other eye. Optic atrophy is another common finding.
Supratentorial tumors of the cerebral hemispheres in children are not very common. Tumors located in the cortex may cause focal cerebral dysfunction, weakness, hemiparesis, seizures, and visual changes. Involvement of particular lobes may result in more specific localized symptoms. For example, a tumor located in the frontal lobe may cause changes in affect and behavior, and a tumor in the occipital lobe may cause cortical blindness or blindness in half of the visual field.
EVALUATION AND TREATMENT A child with signs and symptoms of a brain tumor requires a complete workup, including a neurologic, developmental, and ophthalmic examination. CT with contrast enhancement allows direct visualization of the tumor mass. MRI provides advanced, dramatic examination of the brain and neoplasms. Small low-grade tumors not seen on CT may be detected by MRI. Although less commonly used, MRA is very helpful in assessing vascularity of the tumor and its relationship to major blood vessels. Myelographic examination may be used to evaluate tumor dissemination along the spinal column. Lumbar puncture to examine CSF for tumor cells also is an option. Tumors more likely to spread throughout the neuraxis include medulloblastomas and ependymomas.
The most useful treatment for brain tumors is surgical resection.118 Surgery to establish the diagnosis by biopsy or to excise the tumor is part of the initial treatment for most brain tumors. Some brain tumors may be cured with complete resection alone, such as low-grade cerebellar astrocytomas. Contraindications to such interventions are tumors in which surgical resection and biopsy carry a high risk of mortality or serious morbidity (brainstem gliomas). In these instances, diagnosis is made on radiologic evidence and clinical manifestations.
Most brain tumors require additional radiation and chemotherapy.119 Although these treatments are essential for potential eradication of the brain tumor, radiation to the child’s brain is associated with significant morbidity, including acute and long-term sequelae. Prognosis varies according to the type and location of the brain tumor. Historically, survival rates have been low; however, advances have been made with the combination of surgery, radiation therapy, and chemotherapy. Comprehensive care and management of these children and their families are vital. Multidisciplinary teams composed of neurosurgeons, neurologists, neuropathologists, radiation therapists, oncologists, nurses, social workers, physical therapists, and other providers are necessary to provide the continuity and consistency needed to care for these children.
Embryonal Tumors
Neuroblastoma is an embryonal aggressive tumor that originates in neural crest cells that normally give rise to the sympathic nervous system (sympathetic ganglia and the adrenal medulla). The primitive neural crest cells (also called sympathogonia) are pluripotential (i.e., they give rise to several cell types). They mature into sympathetic ganglion cells, pheochromocytes (which are found in the sympathetic nervous system), or neurofibrous tissue. Thus tumors that develop from neural crest cells reflect the varying degrees of differentiation of the cells. Ganglioneuroblastomas are tumors of an intermediate level of cellular differentiation. The most differentiated tumor is a ganglioneuroma, which is considered benign and does not metastasize.
Because neuroblastoma involves a defect of embryonal tissue, it is diagnosed most commonly in young children and infants. Most tumors are diagnosed during the first 2 years of life, and 75% are found before the child is 5 years of age. Occasionally these tumors have been diagnosed at birth with metastasis apparent in the placenta. Neuroblastomas also are known to regress or mature into benign lesions.120 Although it accounts for 8% to 10% of pediatric malignancies, neuroblastoma causes approximately 50% of all solid tumors in children in the first year, and 15% of cancer deaths in children of all ages.121
PATHOPHYSIOLOGY Neuroblastoma is the most primitive, or immature, form of the sympathetic nervous system tumors. Areas of necrosis and calcification often are present in the tumor.
The cause of neuroblastoma is elusive. The tumor has been associated with a number of conditions, including neurofibromatosis and Hirschsprung disease, but most children with neuroblastoma have neither of these conditions. Although familial tendency has been noted in individual cases, a nonfamilial or sporadic pattern occurs in most children with neuroblastoma. Familial cases of neuroblastoma are considered to have an autosomal dominant pattern of inheritance (mechanisms of inheritance are discussed in Chapter 4).
The most common genetic aberration is amplification of N-myc oncogene (myelocytomatosis viral-related oncogene, neuroblastoma derived) and loss of the short arm of chromosome 1, which is believed to represent the loss of a tumor response gene. The greater the number of copies of the N-myc oncogene, the more rapidly progressive and lethal the disease is.122 The human multidrug-resistant gene (MDR1 at chromosomes 3p and 11q1) has been identified and is associated with chemotherapy failure. The MDR1 gene is amplified in some neuroblastoma cells after initial treatment with chemotherapy, and the increased presence of MDR1 is associated with chemotherapy resistance. Chromosome 17q may be overexpressed.123 Other molecular markers (i.e., neurotrophins) for neuroblastoma are being considered to support risk stratification and guidance of treatment.124,125
CLINICAL MANIFESTATIONS The clinical manifestations of neuroblastoma depend on the location of the tumor. Because neuroblastoma originates where there are elements of sympathetic nervous tissue, the tumor can arise in the sympathetic chain (column of sympathetic ganglia that parallels the spinal column), ganglia of effector organs, peripheral ganglia, adrenal medulla, bladder, and inner genitalia. The most common location is in the abdomen (65%) and most often in the adrenal medulla. The tumor is evident as an abdominal mass and may cause anorexia, bowel and bladder alteration, and sometimes spinal cord compression.126
The second most common location of neuroblastoma is the mediastinum (area separating the lungs) (15% of cases). There the tumor may cause dyspnea or infection related to airway obstruction. If the tumor is large, compression of the trachea, bronchi, lymphatic vessels, and mediastinal veins often results. Neck and facial edema may then be caused by superior vena cava syndrome. Less commonly, neuroblastoma may arise from the cervical sympathetic ganglion (3% to 4% of cases). Cervical neuroblastoma often causes Horner syndrome, which consists of miosis (pupil contraction), ptosis (drooping eyelid), enophthalmos (backward displacement of the eyeball), and anhidrosis (sweat deficiency).
The initial signs and symptoms of neuroblastoma often are related to metastatic disease. About half of children present with metastatic disease at the time of diagnosis. Common sites of metastasis include the skin, with characteristic blue or purple nodules; the liver, causing enlargement; bone, causing pain and pathologic fracture; and bone marrow infiltration, occurring in more than 50% of children. A unique but uncommon site of metastasis is the orbit of the eye, causing an ecchymotic discoloration of the upper and lower eyelids and a “raccoon” eye appearance. Opsoclonus-ataxia, also called “dancing eye syndrome,” occurs in about 2% to 4% of cases, and these children have long-term neurologic complications.127
A number of systemic signs and symptoms are characteristic of neuroblastoma, including weight loss, irritability, fatigue, and fever. Intractable diarrhea occurs in 7% to 9% of children and is caused by tumor secretion of the hormone vasoactive intestinal polypeptide (VIP).
Neuroblastoma, more than any other cancer, has been associated with spontaneous remission, commonly in infants who have liver, bone marrow, or skin involvement in addition to the primary site. Remission has been estimated to occur in approximately 7% of cases but may occur much more often. Neuroblastoma in situ (i.e., noninvasive tumor) has been found during autopsies of infants who died of other causes.
More than 90% of children with neuroblastoma have increased amounts of catecholamines and associated metabolites in their urine. High levels of urinary catecholamines and serum ferritin are associated with a poorer prognosis.
EVALUATION AND TREATMENT Initial diagnostic studies are dictated by the location of the primary tumor. Diagnosis begins with a complete physical and neurologic examination. Visualizing examinations, including intravenous pyelogram, CT scan, or MRI of the primary site, provide further information. Investigation of metastatic disease includes skeletal survey, bone scan, liver scan, and bone marrow aspiration and examination. Newer nuclear medicine imaging studies for neuroblastoma, such as 123I-metaiodobenzylguanidine (123I-MIBG) and tumor-specific monoclonal antibody scan, may be helpful. Urinary catecholamine levels are measured by two metabolites: vanillylmandelic acid (VMA) and homovanillic acid (HVA). Other laboratory analyses are likely to include ferritin; serum neuron-specific enolase (NSE), an enzyme produced by neuronal tissues; and gangliosides, lipid molecules that may be shed from the surface of tumor cells.128
The diagnosis of neuroblastoma is confirmed by surgical biopsy and histopathology. A pathology classification system has been developed for neuroblastoma; for many years, however, there was no agreement on a risk staging system for neuroblastoma.129 The International Neuroblastoma Risk Group is completing a consensus document for neuroblastoma risk stratification, which combined with the International Neuroblastoma Staging System, is used to guide treatment for low-, intermediate-, and high-risk patients.129 A special stage (IV-S) is designated for infants who otherwise would be classified as having early stage disease but who also have metastatic disease involving the liver, skin, or bone.128
Treatment is based on the extent of the disease and prognostic markers, such as age, N-myc copy numbers, and high serum ferritin or NSE levels. Low-risk disease is treated surgically. Chemotherapy is used if there is recurrence. Intermediate-risk tumors are treated with surgery and chemotherapy. High-risk neuroblastomas are being treated with high-dose chemotherapy and radiotherapy followed by transplantation of purged autologous bone marrow. Additional treatment may include 13-cis retinoic acid. These combined treatments have increased survival rate in some to 3 years.
Stage IV-S disease requires very little treatment, primarily because of the high rate of spontaneous regression. Low-dose radiation treatment or single-course chemotherapy may be used to reduce large tumors. Approximately 60% of children with high-risk disease will die despite intensive therapy.130
Retinoblastoma
Retinoblastoma is a rare congenital eye tumor of young children that originates in the retina of one or both eyes (Figure 19-15). Retinoblastoma demonstrates both an inherited and an acquired form. Retinoblastoma rarely is diagnosed after the child is 5 years of age. The inherited form of the disease generally is diagnosed during the first year of life and often involves multiple tumors and sometimes both eyes (40%).131 The acquired disease is commonly diagnosed in children 2 to 3 years of age and involves unilateral disease. Although retinoblastoma is the most common pediatric intraocular tumor, the prevalence rate is estimated between 1 in 15,000 and 20,000 live births.131
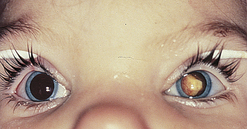
Figure 19-15 Retinoblastoma. The tumor occupies a large portion of the inside of the eye bulbus. (From Damjanov I: Pathology for the health professions, ed 3, St Louis, 2006, Saunders. Courtesy Dr. Walter Richardson and Dr. Jamsheed Khan, Kansas City, Kansas.)
PATHOPHYSIOLOGY Approximately 40% of retinoblastomas are inherited as an autosomal dominant disorder caused by mutations in the RB1 tumor-suppressor gene.132 The remaining 60% are acquired. In the early 1970s Knudson133 proposed the “two-hit” hypothesis to explain the occurrence of hereditary and acquired forms of the disease. This hypothesis predicts that two separate transforming events or “hits” must occur in a normal retinoblast cell to cause the cancer. Further, it proposes that in the inherited form the first hit or mutation occurs in the germ cell (inherited from either parent), and the mutation is contained in every cell of the child’s body. Only a second, random mutation in a retinoblast cell is necessary to transform that cell into cancer. Multiple tumors are observed in the inherited form because these second mutations are likely to occur in several of the approximately 1 to 2 million retinoblast cells. In contrast, the acquired form of retinoblastoma requires that two independent hits or mutations occur in the same somatic cell (after the egg is fertilized) for transformation to cancer. This is much less likely to happen. Figure 19-16 illustrates the two-mutation model for these two patterns of mutation.
Figure 19-16 The two-mutation model of retinoblastoma development. In inherited (familial) retinoblastoma, mutations of the RB locus on chromosome 13q14 lead to neoplastic proliferation of retinal cells. The first mutation is transmitted through the germline of an affected parent. The second mutation occurs somatically in a retinal cell, leading to development of the tumor. In sporadic retinoblastoma, development of a tumor requires two somatic mutations. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, p 184, Philadelphia, 2003, Saunders.)
The gene location in which the initial retinoblastoma mutation occurs is on the long arm of chromosome 13, band q14.134 The gene responsible for retinoblastoma, a tumor suppressor gene, is called the RB gene. The first hit inactivates one allele of the RB gene, and the second hit inactivates the other allele of the gene. Without the normal functioning of the RB gene, production of protein growth regulators that control retinal cell growth is lacking. Because the gene is inactivated, lack of cell growth control results in unregulated proliferation and tumor development.135
The RB gene also has been implicated in other cancers, and survivors of hereditary retinoblastoma may be at increased risk for second cancers, particularly osteosarcoma but also cancer of the lung, breast, prostate, and bladder. Although retinoblastoma occurs in the very young child, second tumors can develop when survivors are in their 20s and 30s; such tumors generally are resistant to therapy.
Retinoblastoma grows as one or more tumors in the retina and extends into the vitreous humor. Free-floating, small tumors in the vitreous humor may attach to the surface of the retina in multiple areas and proliferate (Figure 19-17). The tumor also can invade the optic nerve by infiltrating the cribriform plate of the ethmoid bone or can spread through the sheath around the nerve. In either case the tumor can gain access to the subarachnoid space and the CNS. The tumor spreads into the choroid in 25% of children with retinoblastoma. Because the choroid is highly vascular, metastasis by means of hematogenous spread is possible. When hematogenous spread occurs, metastatic sites include the bone marrow, long bones, lymph nodes, and liver. If the tumor invades the orbit, lymphatic spread is possible. Spontaneous regression occurs, although infrequently, and may be caused by the tumor outgrowing its blood supply.
Figure 19-17 Bilateral retinoblastoma. Presence of white mass consisting of detached retina and neoplastic tissue immediately behind lens in each eye. (From Kissane JM, editor: Anderson’s pathology, ed 8, St Louis, 1985, Mosby.)
CLINICAL MANIFESTATIONS The two most frequent symptoms of retinoblastoma are leukokoria, a white pupillary reflex also called cat’s-eye reflex caused by the mass behind the lens (see Figure 19-15), and strabismus. At that point the tumor is large enough that a light shone into the eye is reflected back by the tumor, making the pupil appear white. Other signs and symptoms include a red, painful eye and limited vision. Any of these signs and symptoms in a child younger than 4 years of age warrants careful ophthalmologic examination of both eyes. Similarly, any newborn with a known genetic risk for retinoblastoma should have routine ophthalmologic examinations.
EVALUATION AND TREATMENT Diagnostic evaluation for retinoblastoma includes documentation of family history; complete ophthalmologic examination; and metastatic studies that include bone marrow aspiration, lumbar puncture for spinal fluid examination, bone scan, and additional ultrasound, radiologic, and CT studies of the orbit and brain. Because of the potential hereditary risk to a child’s siblings, all siblings younger than 4 years also should receive ophthalmologic evaluations.
Retinoblastoma is a treatable tumor, and dual priorities are saving the child’s life and restoring useful vision. Early diagnosis and new chemotherapeutic agents have led to a more conservative approach to treatment. Chemoreduction of the tumor occurs with intravenous chemotherapy to reduce tumor volume. Focal treatments using hyperthermia, laser photocoagulation, or cryotherapy then destroy residual tumor. Large or multiple tumors, indicating more advanced disease, require external beam or plaque radiotherapy and in some cases enucleation (removal) of the eye. Every attempt is made to preserve vision in at least one eye without jeopardizing the child’s survival.136,137
The prognosis for most children with retinoblastoma is excellent, with a greater than 90% long-term survival, although children with bilateral or metastatic disease at diagnosis have a poor prognosis.138 Approximately 75% of children have useful vision in the treated eye.
Acrania 672
Alar plate 666
Anencephaly 669
Arnold-Chiari type II malformation 670
Aseptic meningitis 681
Ataxic cerebral palsy 676
Bacterial meningitis 681
Basal plate 666
Benign febrile seizure 680
Brainstem glioma 687
Cerebellar astrocytoma 687
Cerebral palsy (CP) 675
Congenital hydrocephalus 673
Cranial meningocele 669
Craniopharyngioma 688
Craniosynostosis 672
Cyclopia 669
Dandy-Walker malformation 674
Dyskinetic cerebral palsy 676
Encephalocele 669
Encephalopathy 675
Ependymoma 687
Epilepsy 679
Extrapyramidal/nonspecific cerebral palsy 676
Fontanel 666
Ganglioneuroblastoma 688
Ganglioneuroma 688
Gangliosidosis 678
Generalized seizure 679
Hyperphenylalaninemia (HPA) 677
Infantile spasm 679
Juvenile myoclonic epilepsy 680
Lennox-Gastaut syndrome 680
Lysosomal storage disease 678
Macewen sign (“cracked-pot” sign) 674
Medulloblastoma 687
Meningitis 681
Meningocele 669
Microcephaly 673
Moyamoya disease 685
Myelodysplasia 669
Neural crest 665
Neural fold 665
Neural groove 665
Neural plate 665
Neural tube 665
Neuroblastoma 688
Nonsyndromic craniosynostosis 672
Optic glioma 688
Partial seizure 679
Phenylketonuria (PKU) 677
Pica 681
Pyramidal/spastic cerebral palsy 676
Retinoblastoma 689
Reye syndrome 681
Schwann cell 667
Somite 666
Spina bifida 671
Spina bifida occulta 671
Sulcus limitans 666
Suture 666
Syndromic craniosynostosis 673
Tay-Sachs disease 678
Tethered cord syndrome 671
Tuberous sclerosis complex (TSC) 680
Unclassified epileptic seizure 679
Viral meningitis 684
REFERENCES
1. Centers for Disease Control and Prevention (CDC). Use of supplements containing folic acid among women of childbearing age—United States, 2007. MMWR Morb Mortal Wkly Rep. 2008;57(1):5–8.
2. Kaufman, B.A. Neural tube defects. Pediatr Clin North Am. 2004;51(2):389–419.
3. Anthony, T.E., Heintz, N. The folate metabolic enzyme ALDH1L1 is restricted to the midline of the early CNS, suggesting a role in human neural tube defects. J Comp Neurol. 2007;500(2):368–383.
4. Beaudin, A.E., Stover, P.J. Folate-mediated one-carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Res C Embryo Today. 2007;81(3):183–203.
5. Merserau, P., et al. Spina bifida and ancephaly before and after folic acid mandate—United States 1995-1996 and 1999-2000. MMWR Morb Mortal Wkly Rep. 2004;53(17):362–365.
6. Dashe, J.S., et al. Alpha-fetoprotein detection of neural tube defects and the impact of standard ultrasound. Am J Obstet Gynecol. 2006;195(6):1623–1628.
7. Pollack, I.F. Management of encephaloceles and craniofacial problems in the neonatal period. Neurosurg Clin N Am. 1998;9(1):121–139.
8. Bhattacharjee, A., Chakraborty, A., Purkaystha, P. Frontoethmoidal encephalomeningocoele with colpocephaly: case report and clinical review. J Laryngol Otol. 2008;122(3):321–323.
9. Shaer, C.M., Chescheir, N., Schulkin, J. Myelomeningocele: a review of the epidemiology, genetics, risk facotrs for conception, prenatal diagnosis, and prognosis for affected individuals. Obstet Gynecol Surv. 2007;62(7):471–479.
10. Stevenson, K.L. Chiari type II malformation: past, present and future. Neurosurg Focus. 2004;16(2):E5.
11. Chakraborty, A., et al. Toward reducing shunt placement rates in patients with myelomeningocele. J Neurosurg Pediatr. 2008;1(15):361–365.
12. Reigel, D.H. Infancy through the school years. In: Rowley-Kelly F., Reigel D.H., eds. Teaching the student with spina bifida. Baltimore: Brookes, 1993.
13. Salman, M.S., et al. Smooth ocular pursuit in Chiari type II malformation. Dev Med Child Neurol. 2007;49(4):289–293.
14. Olsson, I., et al. Medical problems in adolescents with myelomeningocele (MMC): an inventory of the Swedish MMC population during 1986-1989. Acta Paediatr. 2007;96(3):446–449.
15. Ko, A.L., et al. Retrospective review of multilevel spinal fusion combined with spinal cord transection for treatment of kyphoscoliosis in pediatric myelomeningocele patients. Spine. 2007;32(22):2493–2501.
16. Yamada, S., Won, D.J. What is the true tethered cord syndrome? Childs Nerv Syst. 2007;23(4):371–375.
17. Yamada, S., et al. Pathophysiology of tethered cord syndrome and similar complex disorders. Neurosurg Focus. 2007;23(2):1–10.
18. Hudgins, R.J., Gilreath, C.L. Tethered spinal cord following repair of myelomeningocele. Neurosurg Focus. 2004;16(2):E7.
19. Bliton, M.J. Ethics: “life before birth” and moral complexity in maternal-fetal surgery for spina bifida. Clin Perinatol. 2003;30(3):449–464.
20. Zambelli, H., et al. Assessment of neurosurgical outcome in children prenatally diagnosed with myelomeningocele and development of a protocol for fetal surgery to prevent hydrocephalus. Childs Nerv Syst. 2007;23(4):421–425.
21. Rintoul, N.E., et al. A new look at myelomeningocele: functional level, vertebral level, and the implication for fetal intervention. Pediatrics. 2002;109(3):409–413.
22. Bruner, J.P. Intrauterine surgery in myelomeningocele. Semin Fetal Neonatal Med. 2007;12(6):471–476.
23. Pitkin, R.M. Folate and neural tube defects. Am J Clin Nutr. 2007;85(1):285S–288S.
24. Behrman, R.E., Kleigman, R.M., Jenson, H.B. Nelson’s textbook of pediatrics, ed 17. Philadelphia: Saunders; 2004.
25. Bruner, J.P., et al. Intrauterine repair of spina bifida: preoperative predictors of shunt-dependent hydrocephalus. Am J Obstet Gynecol. 2004;190(5):1305–1312.
26. Hukki, J., Saarinen, P., Kangasniemi, M. Single suture craniosynostosis: diagnosis and imaging. Front Oral Biol. 2008;12:79–90.
27. Boyadjiev, S.A., et al. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res. 2007;10(3):129–137.
28. Wan, D.C., et al. Current treatment of craniosynostosis and future theraperutic directions. Front Oral Biol. 2008;12:209–220.
29. Passos-Bueno, M.R., et al. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol. 2008;12:107–143.
30. Rice, D.P. Clinical features of syndromic craniosynostosis. Front Oral Biol. 2008;12:91–106.
31. Kimonis, V., et al. Genetics of craniosynostosis. Semin Pediatr Neurol. 2007;14(3):150–161.
32. Clayman, M.A., et al. History of craniosynostosis surgery and the evolution of minimally invasive endoscopic techniques: the University of Florida experience. Ann Plast Surg. 2007;58(30):285–287.
33. Abuelo, D. Microcephaly syndromes. Semin Pediatr Neurol. 2007;14(3):118–127.
34. Garton, H.J., Platt, J.H., Jr. Hydrocephalus. Pediatr Clin North Am. 2004;51(2):305–325.
35. Jackson, P.L. Hydrocephalus. In Jackson P.L., Vessey J.A., eds.: Primary care of the child with a chronic condition, ed 4, St Louis: Mosby, 2003.
36. Kestle, J.R. Pediatric hydrocephalus—current management. Neurol Clin. 2003;21(4):883–895.
37. Beni-Adani, L., et al. The occurrence of obstructive vs absorptive hydrocephalus in newborns and infants: relevance to treatment choices. Childs Nerv Syst. 2006;22(12):1543–1563.
38. Jacobsson, B., Hagberg, G. Antenatal risk factors for cerebral palsy. Best Pract Res Clin Obstet Gynaecol. 2004;18(3):425–436.
39. Murphy, N., Such-Neibar, T. Cerebral palsy diagnosis and management: the state of the art. Curr Probl Pediatr Adolesc Health Care. 2003;33(5):146–169.
40. Nelson, K.B., Chang, T. Is cerebral palsy preventable? Curr Opin Neurol. 2008;21(2):121–135.
41. Lawson, R.D., Badawi, N. Etiology of cerebral palsy. Hand Clin. 2003;19(4):547–556.
42. Rennie, J.M., Hagmann, C.F., Robertson, N.J. Outcome after intrapartum hypoxic ischemia at term. Semin Fetal Neonatal Med. 2007;12(5):398–407.
43. Gunn, A.J., Gluckman, P.D. Head cooling for neonatal encephalopathy: the state of the art. Clin Obstet Gynecol. 2007;50(3):636–651.
44. Degos, V., et al. Neuroprotective strategies for the neonatal brain. Anesth Analg. 2008;106(6):1670–1680.
45. Keogh, J.M., Badawi, N. The origins of cerebral palsy. Curr Opin Neurol. 2006;19(2):129–134.
46. Jones, M.W., et al. Cerebral palsy: introduction and diagnosis (part I). J Pediatr Health Care. 2007;21(3):146–152.
47. Wood, E. The child with cerebral palsy: diagnosis and beyond. Semin Pediatr Neurol. 2007;13(4):286–296.
48. Steinbok, P. Selection of treatment modalities in children with spastic cerebral palsy. Neurosurg Focus. 2006;21(2):e4.
49. Simpson, et al. Botulinum neurotoxin for the treatment of spasticity (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;70(19):1691–1698.
50. Menkes, J.H., Sarnat, H.B. Child neurology, ed 6. Philadelphia: Lippincott Williams & Wilkins; 2000.
51. Swaiman, K.F., Ashwal, S. Pediatric neurology: principles and practice, ed 3. St Louis: Mosby; 1999.
52. Fusetti, F., et al. Structure of tetrameric human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem. 273(27), 1998. [16962–16927].
53. Schmidt, K. Phenylketonuria. In Jackson P.L., Vessey J.A., eds.: Primary care of the child with a chronic condition, ed 4, St Louis: Mosby, 2003.
54. Kaufman, S. An evaluation of the possible neurotoxicity of metabolites of phenylalanine. J Pediatr. 1989;114(5):895–900.
55. Michals-Matalon, K., et al. Response of phenylketonuria to tetrahydrobiopterin. J Nutr. 2007;137(6 Suppl 1):1564S–1567S.
56. Williams, R.A., Mamotte, C.D., Burnett, J.R. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29(1):31–41.
57. Guetta, E., Peleg, L. Rapid detection of fetal Mendelian disorder: Tay-Sachs disease. Meth Mol Biol. 2008;444:147–159.
58. Jarrar, R.G., Buchhalter, J.R. Therapeutics in pediatric epilepsy, Part 1: the new antiepileptic drugs and the ketogenic diet. Mayo Clin Proc. 2003;78(3):359–370.
59. Leppik, E. The expanding role of genetics in epilepsy. Am J Electroneurodiagnostic Tech. 2003;43(2):105–107.
60. Siverstein, F.S., Jensen, F.E. Neonatal seizures. Ann Neurol. 2007;62(2):112–120.
61. Frost, J.D., Jr., Hrachovy, R.A. Pathogenesis of infantile spasms: a model based on developmental desynchronization. J Clin Neurophysiol. 2005;22(1):25–36.
62. Kato, M. A new paradigm for West syndrome based on molecular and cell biology. Epilepsy Res. 2006;70(Suppl 1):S87–S95.
63. Guggenheim, M.A., Frost, J.D., Jr., Hrachovy, R.A. Time interval from a brain insult to the onset of infantile spasms. Pediatr Neurol. 2008;38(1):34–37.
64. Curatolo, P., et al. Management of epilepsy in tuberous sclerosis complex. Expert Rev Neurother. 2008;8(3):457–467.
65. MacAllister, W.S., Schaffer, S.G. Neuropsychological deficits in childhood epilepsy syndromes. Neuropsychol Rev. 2007;17(4):427–444.
66. Grill, M.F., Losey, T.E., Ng, Y.T. The hitchhiker’s guide to the child neurologist’s genetic evaluation of epilepsy. Semin Pediatr Neurol. 2008;15(1):32–40.
67. Renganathan, R., Delanty, N. Juvenile myoclonic epilepsy: under-appreciated and under-diagnosed. Postgrad Med J. 2003;79(298):78–80.
68. Wiebe, S., Tellez-Zenteno, J.F., Shapiro, M. An evidence-based approach to the first seizure. Epilepsia. 2008;49(Suppl 1):50–57.
69. Hadjiloizou, S.M., Bourgeois, B.F. Antiepileptic drug treatment in children. Expert Rev Neurother. 2007;7(2):179–193.
70. Wheless, J.W., et al. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord. 2007;9(4):353–412.
71. Freeman, J.M., Kossoff, E.H., Hartman, A.L. The ketogenic diet: one decade later. Pediatrics. 2007;119(3):535–543.
72. Hartman, A.L., et al. The neuropharmacology of the ketogenic diet. Pediatr Neurol. 2007;36(5):281–292.
73. Buchhalter, J.R., Jarrar, R.G. Symposium on seizures. Therapeutics in pediatric epilepsy, Part 2: epilepsy surgery and vagus nerve stimulation. Mayo Clin Proc. 2003;78(3):371–378.
74. Champi, G., Gaffney-Yocum, P.A. Managing febrile seizures in children. Dimens Crit Care Nurs. 2001;20(5):2–10.
75. Shinnar, S., Glauser, T.A. Febrile seizures. J Child Neurol. 2002;17(Suppl 1):S44–S52.
76. Waruiru, C., Appleton, R. Febrile seizures: an update. Arch Dis Child. 2004;89(8):751–756.
77. Fetveit, A. Assessment of febrile seizures in children. Eur J Pediatr. 2008;167(1):17–27.
78. Neville, B.G., Chin, R.F., Scott, R.C. Childhood convulsive status epilepticus: epidemiology, management and outcome. Acta Neurol Scand. 2007;186(Suppl):21–24.
79. Monto, A.S. The disappearance of Reye’s syndrome—a public health triumph. N Engl J Med. 1999;340(18):1423–1424.
80. Gosalakkal, J.A., Komoji, V. Reye syndrome and Reye-like syndrome. Pediatr Neurol. 2008;39(3):198–200.
81. Belay, E.D., et al. Reye’s syndrome in the United States from 1981 though 1997. New Engl J Med. 1999;340(18):1377.
82. Ward, M.J. Reye’s syndrome: an update. Nurs Pract. 1997;22(12):45.
83. Bhutta, A.T., et al. Reye’s syndrome: down but not out. South Med J. 2003;96(1):43–45.
84. Schror, K. Aspirin and Reye syndrome: a review of the evidence. Paediatr Drugs. 2007;9(3):195–204.
85. Bernard, S.M. Should the Center for Disease Control and Prevention’s childhood lead poisoning intervention level be lowered? Am J Public Health. 2004;94(1):8–9.
86. Meyer, P.A., et al. Surveillance for elevated blood lead levels among children—United States, 1997-2001. MMWR Morb Mortal Wkly Rep. 2003;52(10):1–21.
87. Woolf, A.D., Goldman, R., Bellinger, D.C. Update on the clinical management of childhood lead poisoning. Pediatr Clin North Am. 2007;54(2):271–294.
88. Chang, C.J., et al. Bacterial meningitis in infants: the epidemiology, clinical features and prognostic factors. Brain Dev. 2004;26(3):168–175.
89. Mace, S.E. Acute bacterial meningitis. Emerg Med Clin North Am. 2008;26(2):218–317.
90. Nigrovic, L.E., Kuppermann, N., Malley, R. Development and validation of a multivariable predictive model to distinguish bacterial from aseptic meningitis in children in post–Haemophilus influenzae era. Pediatrics. 2002;110(4):712–719.
91. Milonovich, L.M. Meningococcemia: epidemiology, pathophysiology, and management. J Pediatr Health Care. 2007;21(2):75–80.
92. Feigin, R.D., et al. Bacterial meningitis beyond the neonatal period. In Feigin R.D., et al, eds.: Textbook of pediatric infectious diseases, ed 5, Philadelphia: Saunders, 2003.
93. Broker, M., Fantoni, S. Meningococcal disease: a review on available vaccines and vaccines in development. Minerva Med. 2007;98(5):575–589.
94. Prasad, K., Karlupia, N. Prevention of bacterial meningitis: an overview of Cochrane systematic reviews. Respir Med. 2007;101(10):2037–2043.
95. Ahmed, M.N., Steele, R.W. Meningitis in a young infant. Clin Pediatr. 2004;43(5):495–497.
96. Chirlboga, C.A., et al. Incidence and prevalence of HIV encephalopathy in children with HIV infection receiving highly active anti-retroviral therapy (HAART). J Pediatr. 2005;146(3):402–407.
97. Mitchell, C.D. HIV-1 encephalopathy among perinatally infected children: neuropathogenesis and response to highly active antiretroviral therapy. Ment Retard Dev Disabil Res Rev. 2006;12(3):216–222.
98. , Centers for Disease Control and Prevention: 1994 revised classification system for human immunodeficiency virus infection in children less than 13 years of age. MMWR Morb Mortal Wkly Rep. 1994;43(12):1. Available at www.cdc.gov/mmwr/preview/mmwrhtml/00032890.htm
99. Fahrner, R. Pediatric HIV infections and AIDS. In Jackson P.L., Vessey J.A., eds.: Primary care of the child with a chronic condition, ed 4, St Louis: Mosby, 2003.
100. Tardieu, M. HIV-1 and the developing central nervous system. Dev Med Child Neurol. 1998;40(12):843–846.
101. Petito, C.K., et al. Brain CD8+ and cytotoxic T lymphocytes are associated with, and may be specific for, human immunodeficiency virus type 1 encephalitis in patients with acquired immunodeficiency syndrome. J Neurovirol. 2006;12(4):272–283.
102. McKinney, R.E., Jr., Cunningham, C.K. New treatments for HIV in children. Curr Opin Pediatr. 2004;16(1):76–79.
103. Bernard, T.J., Goldenberg, N.A. Pediatric arterial ischemic stroke. Pediatr Clin North Am. 2008;55(2):323–338. [viii].
104. Horn, P., et al. Arterio-embolic ischemic stroke in children with moyamoya disease. Childs Nerv Syst. 2004;21(2):104–107.
105. Osanai, T., et al. Moyamoya disease presenting with subarachnoid hemorrhage localized over the frontal cortex: case report. Surg Neurol. 2008;69(2):197–200.
106. Jordan, L.C., Hillis, A.E. Hemorrhagic stroke in children. Pediatr Neurol. 2007;36(2):73–80.
107. Whitelaw, A., Odd, D. Postnatal phenobarbital for the prevention of intraventricular hemorrhage in preterm infants. Cochrane Database Syst Rev. (4):2007. [CD001691].
108. Golomb, M.R., et al. Neonatal arterial ischemic stroke and cerebral sinovenous thrombosis are more commonly diagnosed in boys. J Child Neurol. 2004;19(7):493–497.
109. Hartmann, A., et al. Treatment of arteriovenous malformations of the brain. Curr Neurol Neurosci Rep. 2007;7(1):28–34.
110. Rashidi, M., et al. Nonmalignant pediatric brain tumors. Curr Neurol Neurosci Rep. 2003;3(3):200–205.
111. Walter, A.W. Brain tumors in children. Curr Oncol Rep. 2004;6(6):438–444.
112. Kline, M.E. Solid tumors in children. J Pediatr Nurs. 2003;18(2):96–102.
113. Baldwin, R.T., Preston-Martin, S. Epidemiology of brain tumors in childhood—a review. Toxicol Appl Pharmacol. 2003;199(2):118–131.
114. Biegel, J.A. Genetics of pediatric central nervous system tumors. J Pediatr Hematol Oncol. 1997;19(6):492–501.
115. Sklar, C.A. Childhood brain tumors. J Pediatr Endocrinol Metab. 2002;15(Suppl 2):669–673.
116. Gilbertson, R.J. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5(4):209–218.
117. Korones, D.N. Treatment of newly diagnosed diffuse brain stem gliomas in children: in search of the holy grail. Expert Rev Anticancer Ther. 2007;7(5):663–674.
118. Heuer, G.G., et al. Surgical management of pediatric brain tumors. Expert Rev Anticancer Ther. 2007;7(12 Suppl):S61–S68.
119. Partap, S., Fisher, P.G. Update on new treatments and developments in childhood brain tumors. Curr Opin Pediatr. 2007;19(6):670–674.
120. Friedman, G.K., Castleberry, R.P. Changing trends of research and treatment in infant neuroblastoma. Pediatr Blood Cancer. 2007;49(7 Suppl):1060–1065.
121. Castel, V., et al. Molecular biology of neuroblastoma. Clin Transl Oncol. 2007;9(8):478–483.
122. Weinstein, J.L., Katzenstein, H.M., Cohn, S.L. Advances in the diagnosis and treatment of neuroblastoma. Oncologist. 2003;8(3):278–292.
123. Goldstein, L.J., et al. Expression of the multidrug resistance, MDR1, gene in neuroblastomas. J Clin Oncol. 1990;8(1):128.
124. Henry, M.C., Tashjian, D.B., Breuer, C. Neuroblastoma update. Curr Opin Oncol. 2005;17(1):19–23.
125. Tomioka, N., et al. Novel risk stratification of patients with neuroblastoma by genomic signature, which is independent of molecular signature. Oncogene. 2008;27(4):441–449.
126. Golden, C.G., Feusner, J.H. Malignant abdominal masses in children: quick guide to evaluation and diagnosis. Pediatr Clin North Am. 2002;49(6):1369–1392.
127. Mitchell, W.G., et al. Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and neurologic sequelae. Pediatrics. 2002;109(1):86–98.
128. Maris, J.M. Neuroblastoma. Lancet. 2007;369(9579):2106–2120.
129. Park, J.R., Eggert, A., Caron, H. Neuroblastoma: biology, prognosis and treatment. Pediatr Clin North Am. 2008;55(1):97–120. [x, 2008].
130. Goldsby, R.E., Matthay, K.K. Neuroblastoma: evolving therapies for a disease with many faces. Paediatr Drugs. 2004;6(2):107–122.
131. Aerts, I., et al. Retinoblastoma. Orphanet J Rare Dis. 2006;1:31.
132. Leiderman, Y.I., Kiss, S., Mukai, S. Molecular genetics of the RB1-retinoblastoma gene. Semin Ophthalmol. 2007;22(4):247–254.
133. Knudson, A.G. Mutation and cancer: a statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:620.
134. Kivela, T., et al. Retinoblastoma associated with chromosome 13q14 deletion mosaicism. Ophthalmology. 2003;110(10):1983–1988.
135. Rubnitz, J.E., Crist, W.M. Molecular genetics of childhood cancer: implications for pathogenesis, diagnosis, and treatment. Pediatrics. 1997;100(1):101.
136. Munier, F.L., et al. New developments in external beam radiotherapy for retinoblastoma: from lens to normal tissue sparing. Clin Experiment Ophthalmol. 2008;36(1):78–89.
137. Shields, C.L., et al. Continuing challenges in the management of retinoblastoma with chemotherapy. Retina. 2004;24(6):849–862.
138. American Cancer Society: What are the key statistics for retinoblastoma, Cancer Ref Inform, revised July 9, 2008. Available at http://www.cancer.org/docroot/CRI/content/CRI_2_4_1X_What_are_the_key_statistics_for_retinoblastoma_37.asp%3Frnav%3Dcri.