ADAPTIVE IMMUNITY
GENERAL CHARACTERISTICS OF ADAPTIVE IMMUNITY
GENERATION OF CLONAL DIVERSITY



The third line of defense in the human body is adaptive (acquired) immunity, often called the immune response, or immunity. Once external barriers have been compromised and inflammation (see Chapter 6) has been activated, the adaptive immune response is called into action. The molecules and cells of the immune response are closely integrated with those of the innate response. Many components of the innate response facilitate the development of the adaptive immune response. Conversely, products of the adaptive immune response use many components of the inflammatory response. Thus both systems are essential for complete protection against infectious disease: inflammation is relatively rapid, nonspecific, and short-lived, whereas adaptive immunity is slower acting, specific, and very long-lived. Because many inflammatory processes are triggered or affected by immune processes and vice versa, an understanding of both systems is necessary for a complete appreciation of how pathogenic infections are combated. Chapter 8 discusses medically relevant aberrations in both inflammation and immunity, including allergies, diseases that involve unwanted immunologic destruction of healthy tissue, and diseases that are caused by a deficiency in the normal immune or inflammatory responses. Chapter 9 presents an overview of infection and Chapter 10 discusses the connection between stress and disease and the interrelatedness of the immune, nervous, and endocrine systems.
GENERAL CHARACTERISTICS OF ADAPTIVE IMMUNITY
The immune system of the normal adult is continually challenged by a spectrum of substances that it may recognize as foreign, or “non-self.” These substances, called antigens, are often associated with pathogens such as viruses, bacteria, fungi, or parasites, although they are also found on noninfectious environmental agents such as pollens, foods, and bee venom, and still others are associated with clinically derived drugs, vaccines, transfusions, and transplanted tissues (Table 7-1). Unlike inflammation, which is nonspecifically activated by cellular damage as well as pathogenic microorganisms, the immune response is primarily designed to afford long-term specific protection (i.e., immunity) against particular invading microorganisms, that is, it has a “memory” function.1 The products of the adaptive immune response include a type of serum protein—immunoglobulins, or antibodies—and a type of blood cell—lymphocytes (Figure 7-1).

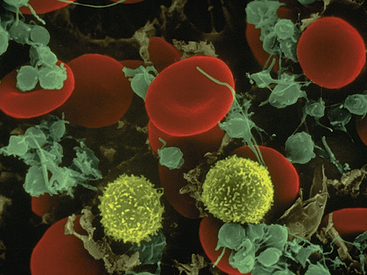
Figure 7-1 Scanning electron micrograph showing lymphocytes (yellow), red blood cells, and platelets. (Copyright Dennis Kunkel Microscopy, Inc.)
Specificity and memory are the primary characteristics that differentiate the immune response from other protective mechanisms. This chapter first discusses the nature of that specificity by defining the various types of antigens that may be seen by the immune system, how they are recognized by antibodies and lymphocytes, and the specific intercellular recognition molecules that are necessary for effective immune responses. After the recognition molecules are defined, the development of the immune response is discussed. An immune response can be divided into two phases (Figure 7-2). Before birth, humans produce a large population of T lymphocytes (T cells) and B lymphocytes (B cells) that have the capacity to recognize almost any foreign antigen found in the environment. Each individual T or B cell, however, specifically recognizes only one particular antigen, but the sum of the population of lymphocyte specificities may represent millions of foreign antigens. This process is called the generation of clonal diversity and occurs in specialized (primary) lymphoid organs; the thymus for T cells and the bone marrow for B cells. While passing through these tissues, the lymphocytes mature and undergo changes that commit them to becoming either B or T cells. Lymphocytes are released from these organs into the circulation as immature cells that have the capacity to react with antigen (immunocompetent). These cells migrate to other (secondary) lymphoid organs in the body in preparation for exposure to antigen (Figure 7-3).
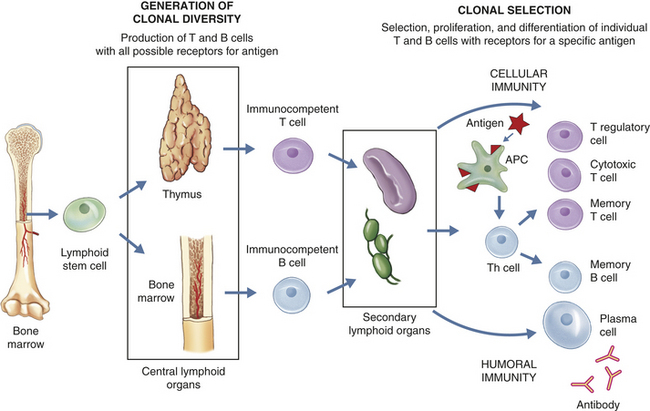
Figure 7-2 Overview of immune response. The immune response can be separated into two phases: the generation of clonal diversity and clonal selection. During the generation of clonal diversity, lymphoid stem cells from the bone marrow migrate to the central lymphoid organs (the thymus or regions of the bone marrow), where they undergo a series of cellular division and differentiation stages resulting in either immunocompetent T cells from the thymus or immunocompetent B cells from the bone marrow. (This process is outlined in more detail in Figures 7-10 and 7-12.) These cells are still naive in that they have never encountered foreign antigen. The immunocompetent cells enter the circulation and migrate to the secondary lymphoid organs (e.g., spleen and lymph nodes), where they take up residence in B- and T-cell–rich areas. The clonal selection phase is initiated by exposure to foreign antigen. The antigen is usually processed by antigen-presenting cells (APCs) for presentation to helper T cells (Th cells) (more detail in Figure 7-16). The intercellular cooperation among APCs, Th cells, and immunocompetent T and B cells results in a second stage of cellular proliferation and differentiation (more details in Figures 7-19 and 7-22). Because antigen has “selected” those T and B cells with compatible antigen receptors, only a small population of T and B cells undergo this process at one time. The result is an active cellular immunity or humoral immunity, or both. Cellular immunity is mediated by a population of “effector” T cells that can kill targets (cytotoxic T cells) or regulate the immune response (T regulatory cells), as well as a population of memory cells (memory T cells) that can respond more quickly to a second challenge with the same antigen. Humoral immunity is mediated by a population of soluble proteins (antibodies) produced by plasma cells and by a population of memory B cells that can produce more antibody rapidly to a second challenge with the same antigen.
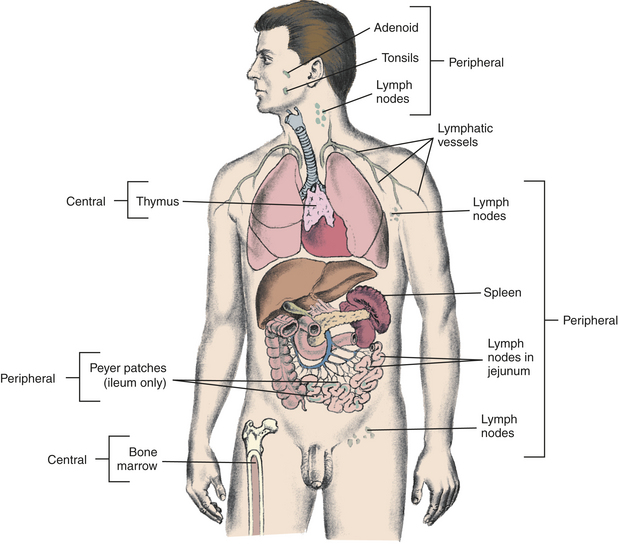
Figure 7-3 Lymphoid tissues: sites of B-cell and T-cell differentiation. Immature lymphocytes migrate through central (primary) lymphoid tissues: the bone marrow (central lymphoid tissue for B lymphocytes) and the thymus (central lymphoid tissue for T lymphocytes). Mature lymphocytes later reside in the T- and B-lymphocyte–rich areas of the peripheral (secondary) lymphoid tissues.
The lymphocytes remain dormant until antigen initiates the second phase of the immune response, clonal selection. This process involves a complex interaction among cells. To initiate an effective immune response, most antigens must be “processed” because they cannot react directly with cells of the immune system but must be shown or “presented” to the immune cells in a very specific manner. This is the job of antigen-processing (antigen-presenting) cells, generally referred to as APCs. In general, three groups of cells must cooperate to make an immune response. The APCs interact with subpopulations of T cells that facilitate immune responses (helper T cells), and immunocompetent B or T cells, resulting in differentiation of B cells into active antibody-producing cells (plasma cells) and T cells into effector cells, such as cytotoxic T cells. The last portion of this chapter discusses how these products (antibody and T cells) protect against infection, including how they interact with components of the inflammatory process.
Humoral and Cell-Mediated Immunity
The immune response has two arms: antibody and T cells, both of which protect against infection. Antibody circulates in the blood and binds to antigens on infectious agents. This interaction can result in direct inactivation of the microorganism or activation of a variety of inflammatory mediators (e.g., complement, phagocytes) that will destroy the pathogen. Antibody is primarily responsible for protection against many bacteria and viruses. This arm of the immune response is termed humoral immunity.
T cells also undergo differentiation during an immune response and develop into several subpopulations of cells that react directly with antigen on the surface of infectious agents. Some develop into T cells that can stimulate the activities of other leukocytes via cell-to-cell contact or through the secretion of cytokines. Others develop into cytotoxic T cells (Tc cells) that attack and kill targets directly. Targets for Tc cells include cells infected by a variety of viruses, as well as cells that have become cancerous. This arm of the immune response is termed cellular immunity. As discussed in this chapter, the humoral and cellular immune responses are interdependent at many levels. In the end, the success of an acquired immune response depends on the functions of both the humoral and cellular responses, as well as the appropriate interactions between them. Additionally, both arms produce specialized subpopulations of memory cells that are long-lived and capable of “remembering” the antigen and responding more rapidly and efficiently on subsequent exposure to the same antigen.2 On reexposure, memory cells do not require much further differentiation and will therefore rapidly become new plasma cells or effector T cells.
Active vs. Passive Immunity
Adaptive immunity can be either active or passive, depending on whether the antibodies or T cells are produced by the individual in response to antigen or are administered directly. Active acquired immunity (active immunity) is produced by an individual after either natural exposure to an antigen or after immunization, whereas passive acquired immunity (passive immunity) does not involve the host’s immune response at all. Rather, passive immunity occurs when preformed antibodies or T lymphocytes are transferred from a donor to the recipient. This can occur naturally, as in the passage of maternal antibodies across the placenta to the fetus, or artificially, as in a clinic using immunotherapy for a specific disease.3 Unvaccinated individuals who are exposed to particular infectious agents (e.g., hepatitis A virus, rabies virus) often will be given immunoglobulins that are prepared from individuals who already have antibodies against that particular pathogen. Whereas active acquired immunity is long-lived, passive immunity is only temporary because the donor’s antibodies or T cells are eventually destroyed.
RECOGNITION AND RESPONSE
The foundation of any successful immune response is the specific recognition of antigen by antibody or receptors on the surface of B or T cells, followed by a set of complex intercellular communications among a variety of antigen-presenting cells and lymphocytes. To fully understand the immune response, it is necessary to initially understand the basis for that recognition. Many of the molecules discussed in this chapter are part of a nomenclature that uses the prefix “CD” followed by a number (e.g., CD1 or CD2) (Table 7-2). The definition of the CD (cluster of differentiation) format has changed over time. It was originally used to describe proteins found on the surface of lymphocytes. Currently, CD is the accepted format for labeling a very large family of proteins found on the surface of many cells. Many have alternative names, which may be used in this chapter. The list of identified molecules is constantly increasing (the number of molecules with a CD designation is probably in excess of 250). In a similar fashion, the list of known cytokines is continually growing, with more than 100 having been identified so far. A large number of CD molecules and cytokines contribute to the acquired immune response. We have attempted to focus on a small number of highly important examples to illustrate the immensely complicated, but highly effective, interactions that take place to produce a protective immune response.
Table 7-2
Select CD Molecules and Their Functions
| CD Molecules | Primary Location | Functions |
| CD1 | APCs | Presents lipid antigens |
| CD2 | All T cells, NK cells | T-cell marker; adhesion molecule that binds to CD58 (LFA-3) and provides a co-stimulatory signal |
| CD3 | All T cells | Associated with TCR and provides intracellular signaling |
| CD4 | Th cells | Binds to MHC class II as co-receptor with the TCR |
| CD8 | Tc cells | Binds to MHC class I as co-receptor with the TCR |
| CD19 | B cells | Complexes with CD21 to form a co-receptor for B cells |
| CD20 | B cells | Major regulator of B-cell function |
| CD21 | B cells | A receptor for complement that complexes with CD19 to form a co-receptor for B cells |
| CD25 | Activated T cells | α-chain of IL-2 receptor |
| CD28 | T cells | Adhesion molecule that binds to CD80 to provide co-stimulatory signal for Tc cells |
| CD40 | B cells, macrophages | Adhesion molecule that binds to CD154 to provide co-stimulatory signal for B cells |
| CD45 | All lymphocytes | Has multiple types; augments antigen signal |
| CD58 (LFA-3) | Most cells | Adhesion molecule that binds to CD2 to provide a co-stimulatory signal |
| CD80 (B7-1) | APCs | Adhesion molecule that binds to CD28 to provide a co-stimulatory signal |
| CD154 (CD40L) | Th2 cells | Adhesion molecule that binds to CD40 to provide a co-stimulatory signal |
APCs, Antigen-presenting cells; IL, interleukin; MHC, major histocompatibility complex; NK, natural killer; Tc, cytotoxic;
Antigens and Immunogens
An antigen is a molecule that can react with antibodies or antigen receptors on B and T cells. Most, but not all, antigens are also immunogens. An antigen that is immunogenic will induce an immune response resulting in the production of antibodies or functional T cells. Although the terms antigen and immunogen commonly are used as synonyms, there are some differences between the two, so a substance may be antigenic yet not be immunogenic.
To function as an antigen, at least a portion of a molecule’s chemical structure must be recognized by and bound to an antibody and/or to specific receptors on a lymphocyte. The precise portion of the antigen that is configured for recognition and binding is called its antigenic determinant, or epitope. The matching portion on the antibody or the lymphocyte receptor is sometimes referred to as the antigen-binding site, or paratope. The size of an antigenic determinant is relatively small, perhaps just a few amino acids or sugar residues on the surface of a large molecule (Figure 7-4). Therefore, macromolecules (e.g., proteins, polysaccharides, nucleic acids) usually contain multiple and diverse antigenic determinants, and the immune response against the macromolecule will usually consist of a mixture of specific antibodies against several of these determinants.
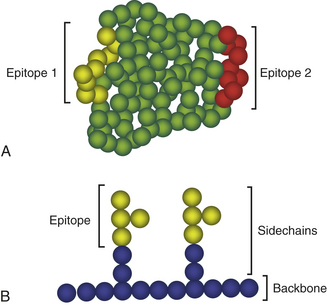
Figure 7-4 Antigenic determinants (epitopes). Shown are generic examples of epitopes on protein (A) and polysaccharide (B) molecules. In A, an antigenic protein may have multiple different epitopes (epitopes 1 and 2) that react with different antibodies. Each sphere represents an amino acid with the yellow spheres representing epitope 1 and the red spheres representing epitope 2. Individual epitopes may consist of 8 or 9 amino acids. In B, a polysaccharide is constructed of a backbone with branched side chains. Each sphere represents an individual carbohydrate with the yellow spheres representing the carbohydrates that form the epitope. In this example, two identical epitopes are shown that would bind two identical antibodies.
Certain criteria influence the degree to which an antigen is immunogenic. These include (1) foreignness to the host, (2) appropriateness in size, (3) having an adequate chemical complexity, and (4) being present in a sufficient quantity.
Foremost among the criteria for immunogenicity is the antigen’s foreignness. A self-antigen that fulfills all these criteria except foreignness does not normally elicit an immune response. Thus most individuals are tolerant to their own antigens. The immune system has an exquisite ability to distinguish self (self-antigens) from non-self (foreign antigens). Tolerance, once thought to be a state of nonresponsiveness in which the immune system passively allowed self-antigens to persist, is now known to have a variety of mechanisms. In some cases, a state of central tolerance exists, in which lymphocytes with receptors against self-antigens have been eliminated. In other cases, tolerance is peripheral tolerance and part of the adaptive immune response. Rather than merely tolerating some self-antigens, the immune system actively prevents their recognition by lymphocytes and antibodies. The response to self-antigens may be actively regulated by specialized T lymphocytes called T regulatory (Treg) cells (see Figure 7-2). Some pathogens have a survival advantage by their capacity to mimic self-antigens and avoid inducing an immune response.
Molecular size also contributes to an antigen’s immunogenicity. In general, large molecules (those bigger than 10,000 daltons), such as proteins, polysaccharides, and nucleic acids, are most immunogenic. Low-molecular-weight molecules such as amino acids, monosaccharides, fatty acids, and the purine and pyrimidine bases, tend to be unable to induce an immune response. Many small molecules can function as haptens: antigens that are too small to be immunogens by themselves but become immunogenic in combination with larger molecules that function as carriers for the hapten. For example, the antigens of penicillin and poison ivy are haptens, but they initiate allergic responses only after binding to large-molecular-weight proteins in the allergic individual’s blood or skin. Antigens that induce an allergic response are also called allergens.
Chemical complexity affects immunogenicity. The best immunogens contain a diversity of chemically different components. For instance, a large synthetic protein consisting only of alanine amino acids would not be very immunogenic, despite its size and foreignness. However, if other amino acids, such as tyrosine, tryptophan, or phenylalanine, were inserted into the structure, the degree of immunogenicity would increase greatly.
Finally, antigens that are present in extremely small or large quantities may be unable to elicit an immune response and therefore by definition are also nonimmunogenic. In many cases, high or low extremes of antigen quantities may induce a state of tolerance rather than immunity.
Even if an antigen fulfills all these criteria, the quality and intensity of the immune response may still be affected by a variety of additional factors. For example, the route and vehicle of antigenic entry or administration are critical to the immunogenicity of some antigens. This has important clinical implications. The most common routes for clinical administration of antigen, such as vaccines, are intravenous, intraperitoneal, subcutaneous, intranasal, and oral. Each route preferentially stimulates a different set of lymphocyte-containing (lymphoid) tissues and therefore results in the induction of different types of cell-mediated or humoral immune responses. For some vaccines, the route may affect the protectiveness of the immune response so that the individual is protected if immunized by one route, but may remain susceptible to infection if administered through a different route. Immunogenicity of an antigen also may be altered by being delivered along with substances that stimulate the immune response; these substances are known as adjuvants. Finally, the genetic makeup of a host can play a critical role in the immune system’s ability to respond to many antigens; some individuals appear to be unable to respond to immunization with a particular antigen, whereas they respond well to other antigens. For instance, a small percentage of the population fails to produce a measurable immune response to the most common vaccines, despite multiple injections. Many other factors can modulate the immune response. These include the individual’s age, nutritional status, genetic background, and reproductive status, as well as exposure to traumatic injury, concurrent disease, or the use of immunosuppressive medications.
Molecules That Recognize Antigen
Antigen is directly recognized by three molecules: circulating antibody and antigen receptors on the surface of B lymphocytes (B-cell receptor, or BCR) and T lymphocytes (T-cell receptor, or TCR) (Figure 7-5).
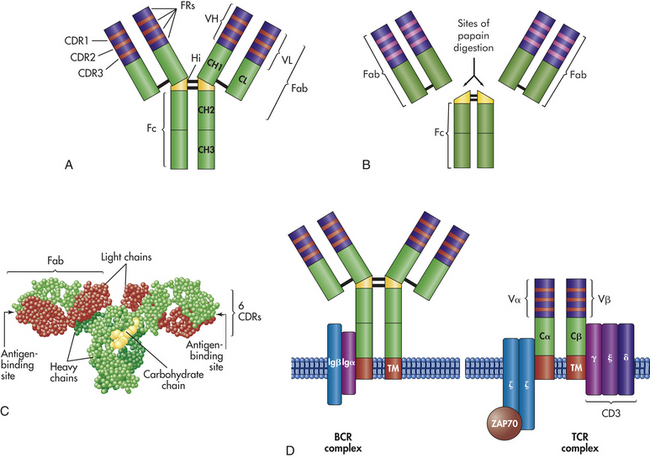
Figure 7-5 Antigen-binding molecules. Antigen-binding molecules include soluble antibody (A, B, C) and cell-surface receptors (D). A, The typical antibody molecule consists of two identical heavy chains and two identical light chains connected by interchain disulfide bonds (− between chains in the figure). Each heavy chain is divided into three regions with relatively constant amino acid sequences (CH1, CH2, and CH3) and a region with a variable amino acid sequence (VH). Each light chain is divided into a constant region (CL) and a variable region (VL). The hinge region (Hi) provides flexibility in some classes of antibody. Within each variable region are three highly variable complementary-determining regions (CDR1, CDR2, CDR3) separated by relatively constant framework regions (FRs) B, Fragmentation of the antibody molecule by limited digestion with the enzyme papain has identified three important portions of the molecule: an Fc and two identical Fab fragments. Both Fab fragments bind antigen. As the antibody folds (C), the CDRs are placed in proximity to form the antigen-binding site. D, The antigen receptor on the surface of B cells (BCR complex) is a monomeric antibody with a structure similar to circulating antibody, with an additional hydrophobic transmembrane region (TM) that anchors the molecule to the cell surface. The active BCR complex contains molecules (Igα and Igβ) that are responsible for intracellular signaling after the receptor has bound antigen. The T-cell receptor (TCR) consists of an α- and a β-chain joined by a disulfide bond. Each chain consists of a constant region (Cα and Cβ) and a variable region (Vα and Vβ). Each variable region contains CDRs and FRs in a structure similar to that of antibody. The active TCR is associated with several molecules that are responsible for intracellular signaling. These include CD3, which is a complex of γ (gamma), ε (epsilon), and δ (delta) subunits and a complex of two ζ (zeta) molecules. The ζ molecules are attached to a cytoplasmic protein kinase (ZAP70) that is critical to intracellular signaling. (C from Patton KT, Thibodeau GA: Anatomy & physiology, ed 7, St Louis, 2010, Mosby.)
Antibody
An antibody, or immunoglobulin, is a serum glycoprotein produced by plasma cells in response to a challenge by an immunogen. The term immunoglobulin is used to denote all molecules that are known to have specificity for antigen, whereas the term antibody is generally used to denote one particular set of immunoglobulins with specificity against a known antigen. There are five molecular classes of immunoglobulins (IgG, IgA, IgM, IgE, and IgD) that are characterized by antigenic, structural, and functional differences (Figure 7-6). Within two of the immunoglobulin classes are several distinct subclasses including four subclasses of IgG and two subclasses of IgA.
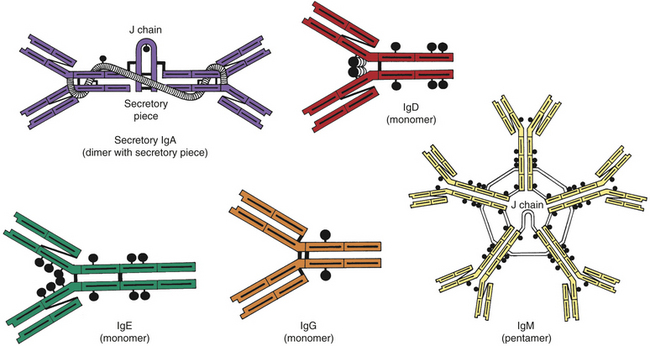
Figure 7-6 Structure of different immunoglobulins. Secretory IgA, IgD, IgE, IgG, and IgM. The black circles attached to each molecule represent carbohydrate residues.
Classes: IgG is the most abundant class of immunoglobulins; they constitute 80% to 85% of those circulating in the body and account for most of the protective activity against infections (Tables 7-3 and 7-4). As a result of selective transport across the placenta, maternal IgG is also the major class of antibody found in blood of the fetus and newborn. Four subclasses of IgG have been described: IgG1, IgG2, IgG3, and IgG4.
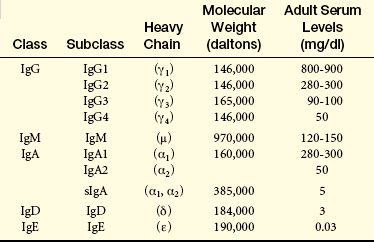
Table 7-4
Biologic Properties of Immunoglobulins
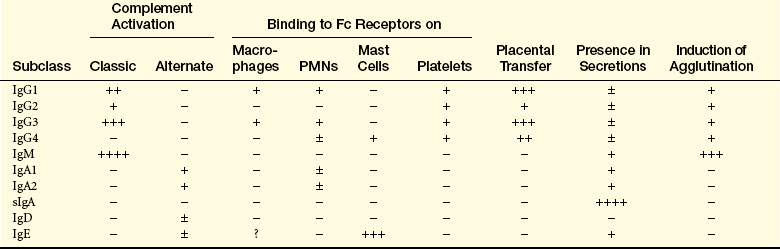
Fc, Crystalline fragment; Ig, immunoglobulin; PMN, polymorphonuclear neutrophil; sIgA, secretory immunoglobulin A; −, lack of activity; +, relative degree of activity.
IgA can be divided into two subclasses, IgA1 and IgA2. IgA1 molecules are found predominantly in the blood, whereas IgA2 is the predominant class of antibody found in normal body secretions. The IgA molecules found in bodily secretions are dimers anchored together through a J chain and “secretory piece.” This secretory piece is attached to the IgAs inside mucosal epithelial cells and may function to protect these immunoglobulins against degradation by enzymes also found in the secretions.
IgM is the largest of the immunoglobulins and usually exists as a pentamer that is stabilized by a J (joining) chain. It is the first antibody produced during the initial, or primary, response to antigen. IgM is synthesized early in neonatal life, and its synthesis may be increased as a response to infection in utero.
Information on the role of IgD is limited. This class of immunoglobulins is found in very low concentrations in the blood, where they do not appear to have a known function. IgD is located primarily on the surface of developing B lymphocytes, where they function as one type of B-cell antigen receptors.
IgE is the least concentrated of any of the immunoglobulin classes in the circulation. It appears to have very specialized functions as a mediator of many common allergic responses (see Chapter 8) and in the defense against parasitic infections.
Molecular Structure: Structural analysis of immunoglobulins began with Porter’s early studies on the effects of the enzyme papain on IgG.4 The nomenclature of antibody structure originated from that work. Limited papain digestion cleaved IgG into three fragments, two of which were identical. The two identical fragments were found to retain the ability to bind antigen, and each was termed an antigen-binding fragment (Fab). The third fragment crystallized when separated from the Fab portions and was termed the crystalline fragment (Fc) (see Figure 7-5).
What Porter learned about the structure of IgG still applies not only to this class of immunoglobulins but also to each of the other classes. The Fab portions of an immunoglobulin contain the recognition sites (receptors) for antigenic determinants and confer the molecule’s specificity toward a particular antigen. The Fc portion is responsible for most of the biologic functions of antibodies, including activation of the complement cascade and opsonization by binding to Fc receptors on the surface of the cells of the innate immune system.
The basic structure of the antibody molecule consists of four polypeptide chains—two identical light (L) chains and two identical heavy (H) chains (see Figure 7-5). Within the same molecule, the two heavy chains are identical and the two light chains are identical. The class of antibody is determined by which heavy chain is used: gamma (IgG), mu (IgM), alpha (IgA), epsilon (IgE), or delta (IgD). The light chains of an antibody molecule are of either the kappa (κ) or lambda (λ) type. The light and heavy chains are held together by two major forces: noncovalent bonds and disulfide linkages. A set of disulfide linkages between the heavy chains occurs in the hinge region and in some instances lends a degree of molecular flexibility at that site so that the Fab regions can move.
Light and heavy chains are further subdivided into constant (C) and variable (V) regions. The constant regions have relatively stable amino acid sequences within a particular immunoglobulin class or subclass. Thus the amino acid sequence of the constant region of one IgG1 should be almost identical with the sequence of the same region of another IgG1, even if they react with different antigens. Conversely, among different antibodies, the sequences of the variable regions are characterized by a large number of amino acid differences. Therefore, two IgG1 molecules against different antigens may have many differences in the amino acid sequence of their variable regions. The variable region can be further subdivided because most of the region’s viability in amino acid sequence is localized into three areas of the variable region. These three areas were once called hypervariable regions, but are now called complementary-determining regions (CDRs). The four regions separating the CDRs have relatively stable amino acid sequences and are called framework regions (FRs).
Antigen Binding: The combined amino acid sequences of the variable regions of both the heavy (VH) and light (VL) chains determine the conformation of the antigen-binding site and therefore the antigenic specificity of the immunoglobulin molecule.5 Most proteins will naturally fold and take on secondary or tertiary structures. As the immunoglobulin molecules fold, the FRs control the accuracy of folding in the variable region, and the CDRs in both variable regions are moved into proximity, resulting in an antigen-binding site formed by the three CDRs of the heavy chain and the three CDRs of the light chain. The chemical nature of the particular amino acids in those sites, as well as the topography of the site, determine specificity toward a particular antigen. The antigen that will bind most strongly must have complementary chemistry and topography with the binding site formed by the antibody. The antigen fits into this binding site with the specificity of a key into a lock and is held there by noncovalent chemical interactions (Figure 7-7). In some cases the substitution of a single critical amino acid in a CDR may have a significant effect on the shape of the binding site and thus the specificity of the antibody molecule.
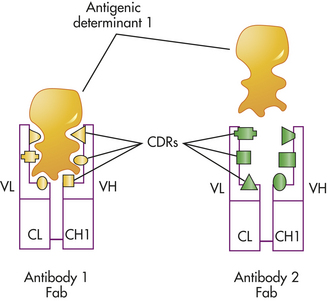
Figure 7-7 Antigen-antibody binding. The specificity required for antibody binding with an antigen is determined by the shape and chemistry of the six complementary-determining regions (CDRs) in the combining site on the variable region of the antibody. This figure indicates two different antibodies (Fab portions of antibody 1 and antibody 2) that have different sets of CDRs and therefore different specificities. As indicated, the antigenic determinant that reacts well with antibody 1 is unable to react with antibody 2 because of differences in the antibody combining site. Fab, Antigen-binding fragment.
Because the heavy and light chains are identical within the same antibody molecule, the two binding sites are also identical and have specificity for the same antigen. The number of functional binding sites is called the antibody’s valence. Most antibody classes (i.e., IgG, IgE, IgD, and circulating IgA) have a valence of 2, but secretory IgA has a valence of 4. IgM, being a pentamer, has a theoretical valence of 10, but can simultaneously use only about five binding sites because a large antigen binding to one site blocks antigen binding to another site.
B-Cell Receptor Complex
The B-cell receptor (BCR) complex is located on the surface of B lymphocytes (see Figure 7-5). Its role is to recognize antigen, but unlike circulating antibody, the receptor must communicate that information to the cell’s nucleus.6 Therefore, the BCR complex consists of antigen-recognition molecules and accessory molecules involved in intracellular signaling (Igα and Igβ). BCRs on the surface of immunocompetent B cells are membrane-associated IgM and IgD immunoglobulins that are produced from the same genes that are used by plasma cells to produce soluble antibodies. As a BCR, however, IgM is a monomer rather than the pentamer primarily found in the blood.
The BCR signaling complex consists of two Igα and Igβ heterodimers that are closely associated with the BCR and contain tyrosine kinase signaling activity. The antibody portion of the BCR complex is responsible for recognition and binding to an antigen, but by itself cannot provide the intracellular signals required to activate the B cell and complete its maturation and the production of antibodies. That message is conveyed by the Igα and Igβ heterodimers.
T-Cell Receptor Complex
T lymphocytes use a similar but distinct array of proteins in their recognition and response to antigens. The T-cell receptor (TCR) complex is composed of an antibody-like transmembrane protein (TCR) and a group of accessory proteins (collectively referred to as CD3) that are involved in intracellular signaling (see Figure 7-5).7 Similar to activation of the B lymphocyte, the TCR is responsible for recognition and binding to the antigen, whereas the accessory proteins are responsible for the intracellular signaling necessary for activation and differentiation of the T cell. Each of the individual components of the TCR complex is important, and several severe defects in the T-cell immune response have been related to mutations in individual components of the complex (see Chapter 8).
Molecules That Present Antigen
For an effective immune response, most antigens must be processed within cells and expressed on the surface of those cells in a very specific manner. Some types of antigen are managed only by highly specialized cells: antigen-presenting cells, or APCs. Other types of antigens can be processed and presented by almost any type of cell. Several sets of cell-surface molecules have the responsibility for appropriately presenting antigen. These molecules are described below.
Major Histocompatibility Complex
An essential set of recognition molecules are members of the major histocompatibility complex (MHC). Most antibody and cellular immune responses are dependent on antigen presentation by APCs. Additionally, the role of cytotoxic T cells in killing virally infected cells depends on presentation of the viral antigen on the infected cell’s surface. Antigen presentation is the primary role of molecules of the MHC.8
MHC molecules are glycoproteins found on the surface of all human cells except red blood cells. They are divided into two general classes, class I and class II, based on their molecular structure, distribution among cell populations, and function in antigen presentation. MHC class I molecules are heterodimers composed of a large α-chain along with a smaller chain called β2 microglobulin. MHC class II molecules are also heterodimers with both α- and β-chains. The general properties of each of the MHC classes are summarized in Figure 7-8.
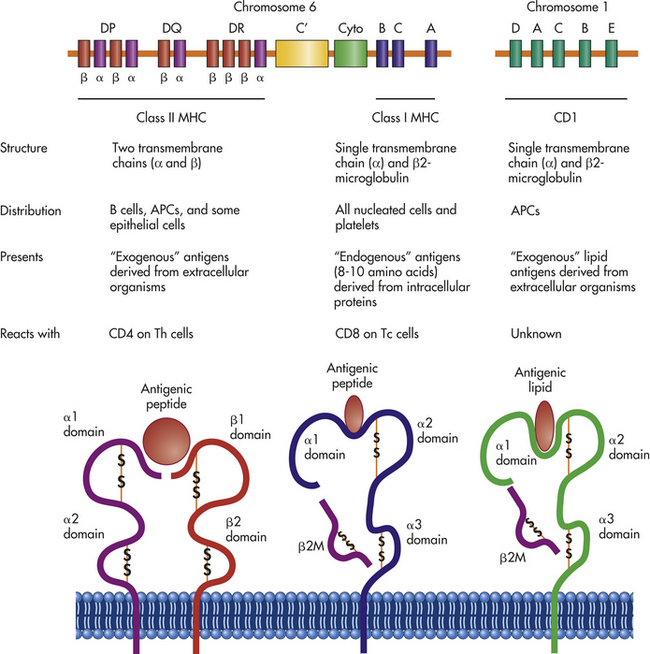
Figure 7-8 Genetics and structure of antigen-presenting molecules. Three sets of molecules are primarily responsible for antigen presentation: MHC class I, MHC class II, and CD1. The MHC molecules are encoded from the MHC region on chromosome 6, which contains information for class I and class II molecules, as well as for several other molecules that participate in the innate or immune responses. These include several complement proteins (C′) and cytokines (cyto), which are referred to as MHC class III molecules. Three principal class I molecules, HLA-A, HLA-B, and HLA-C, are presented here, but this region contains information for the α-chains of several other molecules, including HLA-E, HLA-F, and HLA-G. The MHC class I products complex with β2-microglobulin, which is encoded by a gene on chromosome 15. The MHC class I molecules present small peptide antigens in a pocket formed by the α1 and α2 domains of the α-chain. The conformation of the molecule is stabilized by β2-microglobulin as well as by intrachain disulfide bonds (-S-S-). The α- and β-chains of class II molecules are also encoded in this region: HLA-DR, HLA-DP, and HLA-DQ. In some cases, multiple genes for α- and β-chains are available. The MHC class II molecules present peptide antigens in a pocket formed by the α1 domain of the α-chain and β1 domain of the β-chain. The genes for CD1 molecules are encoded on chromosome 1, which contains genes for five α-chains (CD1A-E), and the α-chains complex with β2-microglobulin to present lipid antigens in a pocket formed by the α1 and α2 domains. All three sets of antigen-presenting molecules are anchored to the plasma membrane by hydrophobic regions on the ends of the α- and β-chains. MHC, major histocompatibility complex.
Molecules of the two MHC classes are encoded from different genetic loci that are located as a large complex of genes on the short arm of human chromosome 6 (see Figure 7-8). The MHC also contains other genes that control the quality and quantity of an immune response, which are commonly referred to as class III MHC genes. The primary MHC class I genes consist of three closely linked loci on this chromosome labeled A, B, and C. The primary MHC class II genes are located within the D region, which actually consists of three separate and independent loci, DR, DP, and DQ.
The class I and class II MHC loci are the most genetically diverse (polymorphic) of any human genetic loci. Within the human population, the numbers of possible different alleles (i.e., forms of the gene) expressed by each locus is astounding: 649 at the A locus, 1029 at the B locus, 350 at the C locus, 643 at the DR locus (α and β), 125 at the DQ locus (α and β), and 154 at the DP locus (α and β). These numbers are based on the polymorphism of observed DNA sequences and may not reflect differences in function. Clearly, not every allele is expressed in the same individual. Humans have two copies of each MHC locus (one inherited from each parent) that are codominant so that molecules encoded by each parent’s genes are expressed on the cell surface. Within an individual, each locus will be expressing only one allele. For instance, each person will have only two different A proteins (one from each parent). However, with the tremendous number of possible alleles that can be expressed, it is likely that any two unrelated individuals will have different sets of MHC molecules on their cell surfaces so that each of us is distinct.
Transplantation: The diversity of MHC molecules becomes clinically relevant during organ transplantation. Cells in transplanted tissue or organs from one individual will have a different set of MHC surface antigens than those of the recipient; therefore, the recipient can mount an immune response against the foreign MHC antigens, resulting in rejection of the transplanted tissue. As a result of studies of transplantation, the human MHC molecules are also referred to as human leukocyte antigens (HLAs), and the different MHC genetic loci are commonly called HLA-A, HLA-B, HLA-C, HLA-DR, HLA-DQ, and HLA-DP. To minimize the chance of tissue rejection, the donor and recipient are often tissue typed beforehand to identify differences in HLA antigens.9 The more similar two individuals are in their HLA tissue type, the more likely a transplant from one to the other will be successful.
Although a large number of alleles exist at the molecular level, the diversity is considerably less at the antigenic level: there are approximately 67 different HLA-A antigens, 149 HLA-B antigens, and 39 HLA-C antigens. Because of the large number of different alleles, it is highly unlikely that a perfect “match” can be found in the general population between a potential donor and the recipient.
The specific combination of alleles at the six major HLA loci on one chromosome (A, B, C, DR, DQ, and DP) is termed a haplotype. Each individual has two HLA haplotypes, one from the paternal chromosome 6 and another from the maternal chromosome. Because the different HLA loci within the MHC are in such close proximity to one another, haplotypes are not usually disrupted by recombination and are thus inherited intact. Each parent passes on one HLA haplotype to each of his or her offspring, meaning that children usually share one haplotype with each parent (Figure 7-9). Odds dictate that children will share one haplotype with half of their siblings and either no haplotypes or both haplotypes with a quarter of their siblings. Thus the chance of finding a match among siblings is much higher (25%) than the general population.
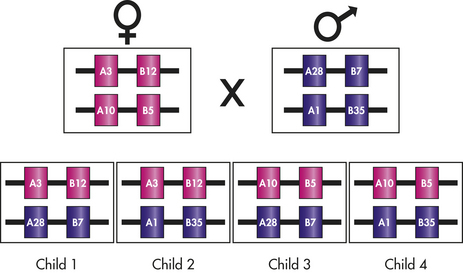
Figure 7-9 Inheritance of HLA. HLA alleles are inherited in a codominant fashion so that both maternal and paternal antigens are expressed. Specific HLA alleles are commonly given numbers to indicate different antigens. In this example, the mother has linked genes for HLA-A3 and HLA-B12 on one chromosome 6 and genes for HLA-A10 and HLA-B5 on the second chromosome 6. The father has HLA-A28 and HLA-B7 on one chromosome and HLA-A1 and HLA-B35 on the second chromosome. On one particular chromosome, the HLA antigens are firmly linked, with crossovers occurring in only 1% of individuals. The children from this pairing may have one of four possible combinations of maternal and paternal HLA. HLA, Human leukocyte antigen.
It should be noted, however, that although HLA alleles are the primary contributor to rejection of a transplant, a number of other antigens also have a role in determining tissue compatibility. Some of these are encoded on other chromosomes and are inherited independently of HLA antigens. This means that although two people have the same HLA makeup, a graft or transplant still may be rejected because of differences between other antigens. It is preferable to obtain a graft or transplant from a closely related individual, such as a sibling, because the chance of sharing both the same HLA antigens and other undetermined antigenic differences encoded outside the MHC is much greater.
CD1
Another set of antigen-presenting molecules are members of the CD1 group.10 CD1 molecules have very low genetic polymorphism, a structure similar to MHC class I, and are found primarily on APCs and cells in the thymus. Unlike MHC molecules that present proteins, the CD1 molecules appear to specialize in presenting lipid antigens contained in lipoproteins, glycolipids, and other molecules.11 These antigens are commonly important factors in infections with bacteria of the Mycobacterium spp. (e.g., Mycobacterium tuberculosis that causes tuberculosis and Mycobacterium leprae that causes leprosy), which have a very large amount of lipid in their cell membranes.
Molecules That Hold Cells Together
The efficient development of an immune response requires several antigen-independent interactions between cells. The interactions between specific cellular receptors and their ligands result in intracellular signaling events that are independent of the TCR or BCR complexes but are necessary complements to the antigen-specific signal. Several of these molecules are listed in Box 7-1.
Cytokines and Their Receptors
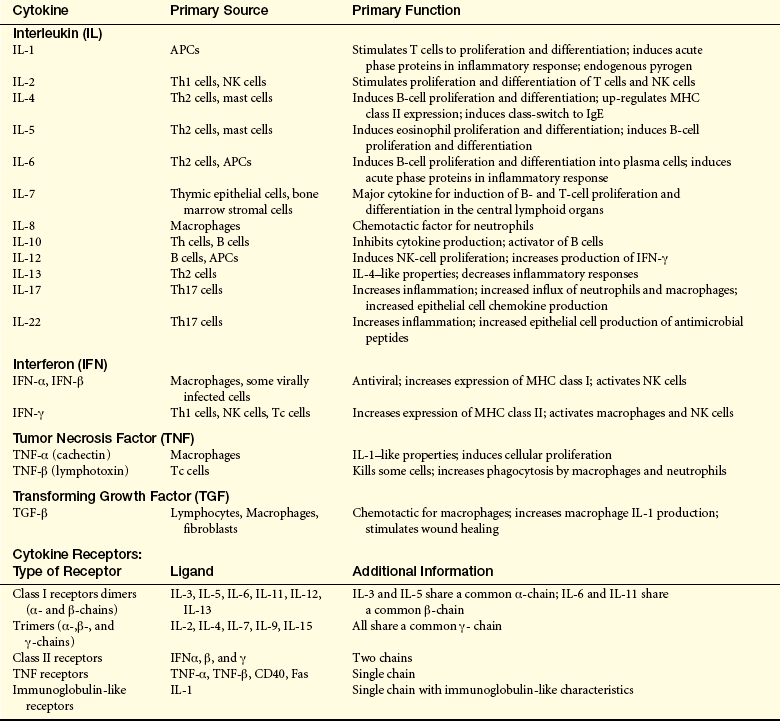
As discussed in Chapter 6, cytokines are low-molecular-weight proteins, or glycoproteins, that function as chemical signals between cells.12 A large number of cytokines are secreted by APCs and lymphocytes and provide both positive and negative regulation of the immune response. The effects of particular cytokines depend on binding to specific cellular receptors, which are linked to intracellular signaling pathways. The lymphocyte may respond in many ways. One of the most common responses is an increase in the production of proteins, many of which are other cytokines or cytokine receptors. Many cytokines also cause a lymphocyte to proliferate and differentiate. The participation of cytokines is essential to the development of an adequate immune response, and in general, the precise combination of cytokines influences the ultimate response of a given cell. Specific deficiencies in the immune response that result from genetic mutations that lead to defective cytokine production or defective cytokine receptors are discussed in Chapter 8. Table 7-5 provides information about key cytokines and receptors that are known to influence the immune response.
GENERATION OF CLONAL DIVERSITY
It has been suggested that more than 108 different antigenic determinants may be recognized by receptors on an individual’s immunocompetent B cells. A similar number may be recognized by T-cell receptors. However, each T or B cell has only a single receptor specificity that recognizes only one antigen, and each is present before that individual is ever exposed to foreign antigen. Thus before the individual is exposed to any foreign antigen, millions of different T- and B-cell antigen receptors must be constructed to recognize any potential antigenic determinant.
Several theories were proposed to explain how such a great diversity of recognition could be produced. The process occurs in two phases: the generation of clonal diversity, during which all the necessary receptor specificities are produced, and clonal selection, during which antigen selects those lymphocytes with compatible receptors, expands their population, and causes differentiation into antibody-secreting plasma cells or mature T cells (Table 7-6).13,14 The generation of clonal diversity takes place in the primary (central) lymphoid organs (i.e., thymus and bone marrow), is driven by hormones, does not require foreign antigen, and results in the generation of immature but immunocompetent T and B cells with receptors that can recognize virtually any antigenic molecule. Both T and B cells are derived from common precursor cells (lymphoid stem cells) that arise in either the liver (in the fetus) or in the bone marrow (of a child or adult). These precursor cells are distinct from the precursor cells that give rise to cells of the innate immune system. The immunocompetent T and B cells migrate from the primary lymphoid organs to secondary (peripheral) lymphoid organs (e.g., spleen, lymph nodes, adenoids, tonsils, Peyer patches), where they await antigen. Clonal selection is initiated by antigen and results in a mature and specific immune response against that antigen.
Table 7-6
Generation of Clonal Diversity vs. Clonal Selection
| Generation of Clonal Diversity | Clonal Selection | |
| Purpose? | To produce large numbers of T and B lymphocytes with the maximum diversity of antigen receptors | Select, expand, and differentiate clones of T and B cells against a specific antigen |
| When does it occur? | Primarily in the fetus | Primarily after birth and throughout life |
| Where does it occur? | Central lymphoid organs: thymus for T cells, bone marrow for B cells | Peripheral lymphoid organs, including lymph nodes, spleen, and other lymphoid tissues |
| Is foreign antigen involved? | No | Yes, antigen determines which clones of cells will be selected |
| What hormones/cytokines are involved? | Thymic hormones, IL-7, others | Many cytokines produced by Th cells and APCs |
| Is tolerance induced? | Central tolerance induced as autoreactive cells are deleted | Peripheral tolerance induced as autoreactive cells are regulated |
| Final product? | Immunocompetent T and B cells that can react with antigen but have not seen antigen, and migrate to the secondary lymphoid organs | Plasma cells that produce antibody, effector T cells that help (Th), kill targets (Tc), or regulate immune responses (Treg); memory B and T cells |
APCs, Antigen-presenting cells; IL, interleukin; Tc, cytotoxic T cells; Th, helper T cells; Treg, regulatory T cells.
Although generation of clonal diversity primarily occurs in the fetus, it probably continues to a low degree throughout most of adult life. Clonal selection usually begins at birth and proceeds throughout the life of the individual as new antigens are encountered, although it can begin as early as the eighth week of gestation in humans.
As a result of this process, T and B lymphocytes have the capacity to react against virtually any antigen found in nature. This endless array of possible antibodies and TCRs certainly cannot be constructed from the amount of DNA that is in the nucleus of a human lymphocyte. The enormous repertoire of specificities is instead made possible by rearrangement of existing deoxyribonucleic acid (DNA) during T and B cell development in the primary lymphoid organs. Loci in the DNA that encode for the variable regions of immunoglobulins and TCRs are recombined in a unique way to generate receptors that collectively can recognize and bind to any possible antigen. The DNA in the nucleus of a developing T and B cell is actually cut and spliced (repaired), a process known as somatic recombination, so that after this manipulation, the progeny of a single lymphocyte will synthesize identical immunoglobulins or TCRs. Those variable regions, however, are cut and spliced differently from those of another lymphocyte, making each cell unique and therefore able to react with different antigens. The particular process for B and T cells is discussed following.
T-Cell Maturation
The central lymphoid organ for T-cell development is the thymus, which is an organ located near the heart. Precursor cells (lymphoid stem cells) arise in early embryonic life from the yolk sac and fetal liver and later from the bone marrow. They migrate to the thymus and enter in the subcapsular region.15–17 As the cells move through the thymic cortex to the medulla, they are instructed by interactions with various thymic cells (epithelial cells, macrophages, and dendritic cells) and thymic hormones to undergo proliferation and progressive development of the characteristics of immunocompetent T cells (Figure 7-10).18 Changes include the development of the T-cell receptor complex and expression of characteristic surface molecules. Many T cells randomly develop TCRs against self-antigens, but are deleted during this process. The final antigen-reactive T cells are released into the blood and take up residence in the secondary lymphoid organs to await antigen.
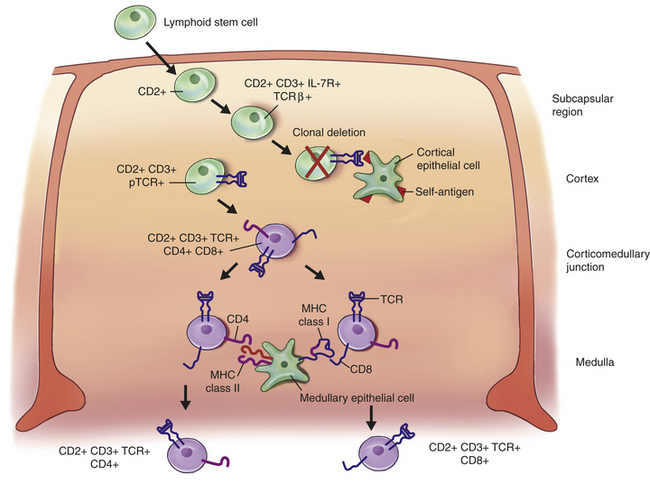
Figure 7-10 T-cell development in the thymus. During the generation of clonal diversity in the fetus, lymphoid stem cells undergo several stages of cellular division and differentiation in a central lymphoid organ (the thymus) under the control of hormones but without the influence of foreign antigen. A simplified scheme for that process is presented here. The differentiation process is characterized by the up-regulation of many important surface molecules (only some of which are shown) and the random development of a huge number of different T-cell receptors against all possible antigens that the adult may encounter. The lymphoid stem cell enters the subcapsular region of the thymus, where it begins to undergo differentiation. One of the first surface changes is the appearance of the molecule CD2, which is a marker for all T cells. In the cortex of the thymus, the developing cell encounters epithelial cells that guide most of the early differentiation process. The pre–T cell begins expressing the surface receptor for the cytokine IL-7, which is produced by the epithelial cell along with other thymic hormones to drive the T-cell differentiation process. At this stage the T cell begins constructing the T-cell receptor (TCR) by first rearranging and expressing the TCR β-chain (more detail is provided in Figure 7-11) and expressing CD3 molecules. Although the TCR α-chain has not yet been produced, the β-chain is expressed on the surface as a pre-TCR (pTCR) using a protein that acts as a surrogate for the α-chain. Because of the randomness of the process, some pTCRs are produced with specificities toward self-antigens. Many of these undergo negative selection and are deleted (clonal deletion) by apoptosis induced through interactions with self-antigens presented by the epithelial cells. Survivors of negative selection move toward the thymic cortex and begin expressing the TCR α-chain, the normal TCR, and both CD4 and CD8 on their surfaces. These CD4+, CD8+ “double-positive” cells encounter medullary epithelial cells that express both MHC class I and class II molecules. The phenotype of the developing T cell is positively selected so that interaction between CD4 and MHC class II selects for retention of CD4 expression, whereas interaction between CD8 and MHC class I favors the CD8 phenotype. Thus two populations of “single-positive” immunocompetent T cells leave the thymus: one cell is CD4+, CD8− (destined to be a helper T [Th] cell) and the other is CD4−, CD8+ (destined to be a cytotoxic T [Tc] cell).
Production of the T-Cell Receptor
Like antibody, the TCR reacts with antigen (see Figure 7-5). Although the structure of the TCR closely resembles a Fab portion of antibody, the TCR uses different protein chains than are used for antibody. The most common TCR contains α- and β-chains, each of which has a variable region and a constant region. Within each variable region are three CDR regions separated by FR regions.
The great amount of variable region diversity necessary for identifying the huge number of antigens found in nature is produced by random recombination of multiple genes to encode the variable regions of both the α- and β-chains. In the germline genes, the information for the amino acid sequence of the α-chain variable region is found on chromosome 14 in two separated, but closely associated, locations: a set of V region genes and a set of J region genes (Figure 7-11). The TCR α-chain locus has multiple (at least 50) V genes and multiple (at least 50) J genes. During somatic recombination in a developing T cell, one of the possible V genes is randomly selected and spliced to one of the J genes, with the intervening DNA being removed. This DNA rearrangement process is controlled by two enzymes produced by the genes RAG-1 and RAG-2 (recombination activating genes). These enzymes cut double-stranded DNA at specific recognition sites (recombinant signal sequences); then repair the break resulting in excision of the DNA between the selected V and J genes. At transcription, the genetic information for the α-chain variable region is still separated from the gene for the α-chain constant region. This product is transcribed into messenger ribonucleic acid (mRNA) that contains information for the variable region (VJ) separated by a span of RNA from the information for the α-chain constant region. An RNA-processing step removes the intervening span, bringing the message for the variable and constant regions together into a final mRNA product that is translated into the intact α-chain protein. The random selection and pairing of 50 V and 50 J genes by a large number of developing T cells can result in more than 2500 possible α-chains.
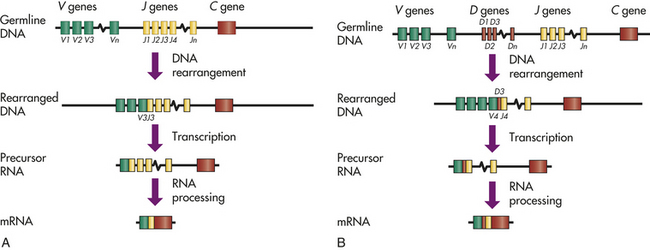
Figure 7-11 DNA rearrangement of genes for antigen-binding molecules. During the generation of clonal diversity, a tremendous number of different antigen-binding molecules are produced. These include the B-cell receptor (BCR), which consists of a membrane-bound antibody molecule, and the T-cell receptor (TCR). The process by which receptor diversity is created is identical for all antigen-binding molecules and is summarized in this figure. Maximum diversity with minimum use of DNA is accomplished by random rearrangement of sets of genes that encode different portions of the variable regions. A, The variable regions of the light chain of antibody and the α-chain of the TCR independently rearrange two sets of genes: V region genes and J region genes. The light chain uses its own set of genes, and the α-chain uses a completely different set. In neither case is the exact number of V or J region genes known; therefore, in this figure they are numbered from 1 to an unknown value (n). In a particular cell’s DNA, one V gene is randomly selected and moved to a position immediately adjacent to a randomly selected J gene. In this example, V3 and J3 were selected. The DNA between the selected genes is enzymatically removed and the DNA repaired, so that the rearranged DNA in this example is missing the portion found in the germline DNA between V3 and J3. This product is transcribed into a precursor ribonucleic acid (RNA) that contains information for the rearranged VJ pair, a span containing other unselected J regions, and information for the appropriate constant region (C gene) of the molecule. The RNA between the VJ and the C regions is not translated; therefore, it is removed by RNA processing to produce a messenger RNA (mRNA) that is translated. B, The variable region of the antibody heavy chain and the TCR β-chain results from a similar DNA rearrangement, with the added diversity contributed by a group of D region genes. The joining of D and J occurs first, with the removal of intervening DNA. In this example, D3 and J4 were chosen. This is followed by rearrangement of the V gene (e.g., V4) and formation of a VDJ region in the rearranged DNA. The precursor RNA contains information for the VDJ, the intervening portion of DNA, and the appropriate constant region. After RNA processing, an mRNA is formed for the intact antibody heavy chain or the TCR β-chain. Once the DNA is rearranged and spliced in a given B or T cell, all of the antigen receptors produced by that cell employ the same V, D, and J segments and have the same specificity.
In a similar fashion, the TCR β-chain locus on chromosome 7 has three sets of genes that rearrange to encode the variable region of that chain: at least 20 V genes, 13 J genes, and 2 intervening and relatively short D genes that add further diversity. Using the RAG-1 and RAG-2 enzymes, a developing T cell randomly selects a set of V, D, and J genes for DNA recombination. The VDJ rearranged segment is transcribed with a β-chain constant region, the intervening RNA is removed during processing, and the final mRNA is translated into an intact β-chain.
The α- and β-chains are joined by that cell and inserted into the membrane to make an antigen-specific TCR. The enormous number of possible combinations of α-chain V and J regions along with the β-chain V, D, and J regions enables the generation of a population of T cells with a large diversity of TCRs (estimated at 1.3 x 105 possible combinations). For both chains, the V region genes encode the amino acid sequences that include CDR1 and CDR2 and their appropriate FR regions. The J regions contain information for CDR3 and FR4. The TCR β-chain D regions encode a short amino acid sequence found in the CDR3 and greatly increases the diversity of the β-chain CDR3. Imprecise joining increases the diversity of the CDR3 regions of both the α- and β-chains even further. For example, the sites of VJ and VDJ joining may shift slightly resulting in an amino acid being inserted or deleted from the protein.
Although the αβ TCR is the preferred antigen receptor, some T cells use alternative genes: gamma (γ) (chromosome 7) and delta (δ) (chromosome 14, in the middle of α-chain genes). T cells with γδ TCRs appear to migrate to unique areas of the body (the epithelial areas in the skin, reproductive tract, intestine, respiratory tract) and have different and less well understood functions than the T cells with αβ TCRs.
Changes in Characteristic Surface Markers
Differentiation of T cells in the thymus also results in changes in a variety of important surface molecules. As the developing T cells move through the thymic cortex, they initiate the expression of the molecule CD2 on the cell surface. CD2 is a marker for T cells and is expressed on virtually every subpopulation of cells that have undergone development in the thymus. Within the cortex, the cells begin rearranging the variable region genes necessary for forming a functional T-cell receptor. The T-cell receptor undergoes several stepwise changes until the final αβ TCR is formed. Concurrently, the TCR accessory molecules (collectively called CD3) are expressed. The cell also begins making two important surface proteins, CD4 and CD8, which are concurrently expressed on the developing cell’s surface at this stage. These CD4+, CD8+ cells are often called “double-positive” cells. Much of T-cell development is controlled by hormones and cytokines in the thymus, and an early step in maturation is expression of the receptor for interleukin (IL)-7 (IL-7R), which is a major cytokine that drives the differentiation process. After entering the medulla of the thymus, the double-positive cells become “single-positive.” That is, some of the cells suppress production of the CD8 molecule and remain only CD4+, whereas others suppress CD4 production and remain CD8+. This branch in the differentiation pathway leads to two groups of cells with different functional characteristics: CD4 cells tend to recognize antigen presented by MHC class II molecules and develop into helpers in the later clonal selection process (helper T cells), whereas CD8 cells recognize antigen presented by MHC class I molecules and become mediators of cell-mediated immunity and kill other cells directly (cytotoxic T cells).
Central Tolerance
During the random rearrangement of VJ and VDJ genes to produce the T-cell receptor, some combinations result in specificities that recognize self-antigens. If some of these autoreactive T cells were allowed to progress further in development and leave the thymus, a severe immunologic reaction against the individual’s own tissues could result. One stage at which tolerance for self-antigens is maintained is the deletion of autoreactive T cells in the thymus, which is referred to as central tolerance.
A variety of self-antigens are expressed by thymic cells. Many thymic cells express MHC class I or MHC class II molecules. During the T cell’s double-positive stage, if a TCR strongly reacts with MHC class I or class II, the T cell will undergo apoptosis, referred to as clonal deletion. A large spectrum of other self-antigens is expressed on the surface of thymic macrophages, dendritic cells, and especially epithelial cells. If a developing T cell’s TCR binds strongly with a self-antigen, it is deleted. Although this process of negative selection induces more than 95% of T cells to undergo apoptosis in the thymus, a limited number of autoreactive clones persist and must be controlled by other means in the peripheral lymphoid organs (peripheral tolerance).
The destiny of the double-positive cells with TCRs specific for foreign antigens (which are not expressed in the thymus) is determined by their interaction in the thymus with MHC antigens. If their surface CD4 molecules bind to MHC class II molecules on the thymic cells, the T cell will become CD4 single-positive. However, if their surface CD8 reacts with MHC class I molecules, the cells will become CD8 single-positive. This positive selection process results in about 60% of immunocompetent T cells being CD4+ and 40% being CD8+ when they leave the thymus.
B-Cell Maturation
Although the thymus is the central lymphoid organ for T-cell development, humans do not appear to have a discrete organ for B-cell development. In chickens, B lymphocytes undergo differentiation in an organ called the bursa of Fabricius. In humans, portions of the bone marrow function as a bursal-equivalent tissue for B-cell development.19
Regardless of the lack of a discrete organ, B-cell differentiation undergoes a very similar process to that described above for T cells. Lymphoid stem cells in the bone marrow interact with stromal cells through a variety of intercellular adhesion molecules (Figure 7-12). As the stem cell begins to mature, it progressively develops a variety of necessary surface markers, the earliest being CD45R and the IL-7 receptor. IL-7, produced by the stromal cells, is critical in driving the further differentiation and proliferation of the B cell. The next stage in development is formation of the B-cell receptor.
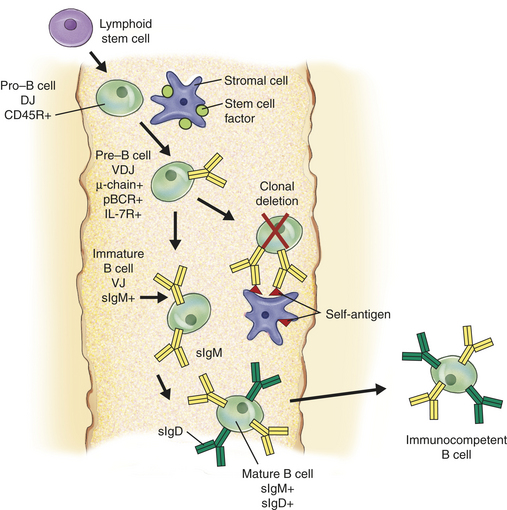
Figure 7-12 B-cell development in the bone marrow. During the generation of clonal diversity, lymphoid stem cells enter portions of the bone marrow that serve as the central lymph organ for B-cell development. Interactions with a series of bone marrow stromal cells guide the proliferation and differentiation process through direct cell-to-cell contact and the production of cytokines and hormones by the stromal cells, but without the presence of foreign antigen. A simplified scheme for that process is presented here. As with T-cell development, the differentiation process of B cells is characterized by the up-regulation of many important surface molecules (only some of which are shown) and the random development of a huge number of different B-cell receptors. The early B cell (pro–B cell) binds to a membrane-bound cytokine (stem cell factor) on the stromal cell and initiates expression of the surface molecule CD45R and begins to rearrange the DJ regions of the antibody heavy-chain gene. As the cell progresses to the pre–B-cell stage, it concludes DNA rearrangement of the heavy chain (VDJ) and begins expressing cytoplasmic mu (μ) heavy chain. The μ- chain is incorporated into a pre–B-cell receptor (pBCR) using a surrogate protein in place of the light chain. The cell also up-regulates the IL-7 receptor (IL-7R), which interacts with IL-7 produced by the stromal cells to drive the remaining steps in differentiation. Some pBCRs have specificities toward self-antigen. Many of these encounter self-antigen expressed on the stromal cells and undergo negative selection (clonal deletion). The surviving cells (immature B cells) rearrange the light chain DNA (VJ) and express a BCR consisting of light chain and the μ-heavy chain (surface IgM [sIgM]). In the mature B cell, changes in processing of the heavy-chain precursor RNA results in co-expression of sIgM and IgD (sIgD) (see Figure 7-13 for more details).
Production of the B-Cell Receptor
The BCR is an antibody that is anchored to the plasma membrane. The process by which BCR diversity is generated is virtually identical to the process in T cells and also requires the genetic rearrangement of V, D, and J genes.20 The segments of DNA that encode either kappa (κ) (chromosome 2) or lambda (λ) (chromosome 22) light chains contain about 70 V and 5 J segments, whereas the heavy chain locus on chromosome 14 contains about 80 V, 30 D, and 6 J regions. The locus for the antibody heavy chain also contains multiple sequential regions for different constant regions, with the gene for the mu (μ) constant region being closest to the VDJ region, and the delta (δ) constant region gene being next in sequence (Figure 7-13). These are followed by the constant region genes for other classes and subclasses. In the developing B cell, the initial RNA transcript contains information for the VDJ recombination, the μ constant region, and the δ constant region. Transcription is signaled to stop immediately after the δ constant region. During the following RNA processing step to form a final mRNA product, the cell can alternatively process one mRNA to retain the μ constant region only or process another mRNA molecule to remove the μ constant region and retain the δ constant region. Thus one cell can use multiple mRNA molecules and alternative RNA processing to simultaneously produce two different heavy chains, μ and δ, both of which have the same variable region.
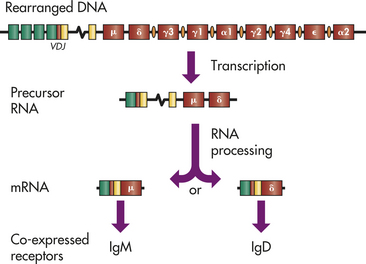
Figure 7-13 Genetics of the B-cell receptor. Most mature immunocompetent B cells express both surface IgM and IgD as the B-cell receptor. In the germline DNA, the heavy chain gene complex consists of a series of V, D, J, and constant region genes. In humans, each class and subclass of antibody has a unique constant region gene arranged in the indicated order. Switch regions occur preceding every constant region gene, except mu (μ) (IgM) and delta (δ) (IgD). After successful DNA rearrangement of the VDJ regions, a ribonucleic acid (RNA) molecule is transcribed that contains the information from the VDJ, intervening DNA, the μ constant region, and the δ constant region. Precursor RNA molecules are alternatively processed to produce messenger RNAs (mRNAs) containing either μ or δ. Initially, RNA processing favors the μ chain and production of surface IgM (see Figure 7-12), but as the B cell matures, both mRNA molecules are produced.
The developing B cell rearranges and expresses the heavy chain, which is followed by the rearrangement of either the κ or λ light chain so that only one type is produced. The light chains are assembled with two μ heavy chains to form a monomeric IgM antibody or with two δ chains to form an IgD antibody. Because each heavy chain used the same VDJ rearrangement and the same light chain, the variable regions and therefore the specificities of the IgM and IgD are identical. At this stage of B-cell development, both antibodies have hydrophobic, or sticky, “tails” that results in insertion into the plasma membrane and the co-expression of IgM and IgD receptors on the cell surface.
Changes in Characteristic Surface Markers
As with T cells, B-cell differentiation is also characterized by the development of a variety of important surface molecules. These include CD21 (a complement receptor) and CD40 (adhesion molecule required for later interactions with Th).
Central Tolerance
During formation of the BCR in the bone marrow, a large number of autoreactive B cells are eliminated if exposed to self-antigen.21 It is estimated that more than 90% of developing B cells are induced to undergo apoptosis.
INDUCTION OF AN IMMUNE RESPONSE: CLONAL SELECTION
As described in the previous chapter, successful invasion by a pathogen will initially elicit an inflammatory response as a host attempts to destroy and clear the invading microorganism. In addition to carrying out their roles as inflammatory effector cells, some of the cells involved in innate immunity are responsible for communicating with immature B and T lymphocytes to initiate specific and longer-acting acquired immunity. This intercellular communication occurs via direct cellular contact in peripheral lymphoid tissues and is essential for the specificity of the adaptive immune response.
Secondary Lymphoid Organs
The secondary lymphoid organs include the spleen, lymph nodes, adenoids, tonsils, Peyer patches (intestines), and the appendix (see Figure 7-3). Immunocompetent lymphocytes enter the secondary lymphoid organs through the blood and enter specialized small veins, called high endothelial venules (HEVs), where they bind to the endothelium through a family of adhesion molecules.22 The lymphocytes migrate from the vessels into the lymphoid tissues, which contain B- and T-cell–rich areas. B lymphocytes that encounter antigen in the secondary lymph organs usually undergo a process of differentiation and proliferation that results in the formation of specialized germinal centers in these organs (Figure 7-14).23
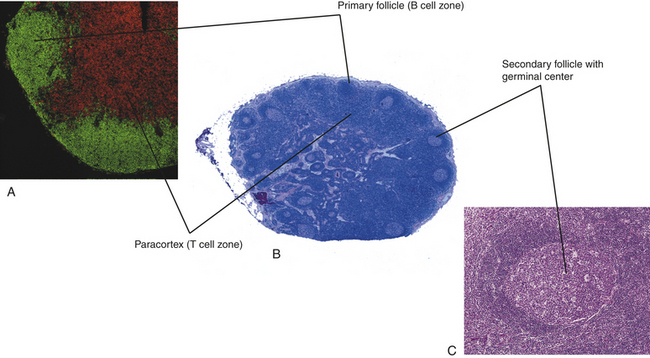
Figure 7-14 Histology of a secondary lymphoid organ. A, The lymph node contains areas (primary follicles) that are rich in immunocompetent B cells (stained green), and T cells (stained red) in the paracortex. B, A lymph node is organized into an outer cortex and an inner medulla. C, In response to antigen, B cells undergo proliferation, resulting in the formation of secondary follicles with germinal centers. (Modified from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Antigen Processing and Presentation
Most antigens do not react directly with T or B cells, but require processing and presentation in the appropriate fashion.24 This is the duty of APCs.
Pathogens that penetrate the external barriers and enter the tissues or bloodstream encounter a variety of phagocytic cells and are therefore likely to be ingested and destroyed. If the infectious agent is in the tissues, they may elicit an inflammatory response that results in the infiltration of macrophages into the site. Additionally, the infectious agent or fragments of the microorganism may be removed by the lymphatics, which drain to the lymph nodes. The lymph nodes are extremely rich in dendritic cells and macrophages, which phagocytose the material and function as APCs for T and B lymphocytes in the lymph nodes. Pathogens entering through the blood stream may be removed by phagocytic cells in the spleen and other lymphoid tissues. In either case, the phagocytic cells that digest invading pathogens are also responsible for processing antigens from the pathogen and displaying or presenting those antigens on the phagocyte’s surface to neighboring lymphocytes in order to initiate the adaptive immune response against that specific pathogen.
Many cells have the capacity to present antigen to some degree, but dendritic cells, macrophages, and B lymphocytes are so efficient at antigen presentation that they are considered “professional” APCs. Each of these three APCs is responsible for the presentation of antigens of different types and from different sources. B cells present antigen to Th cells that facilitate development of the humoral immune response. Macrophages are very effective in presenting antigen to memory Th cells in order to initiate a rapid response to antigens (i.e., secondary immune response). The dendritic cells are perhaps the most effective in presenting antigen to naive immunocompetent Th cells.25 Dendritic cells develop from bone marrow precursor cells, either of myeloid or lymphoid lineage (at least two populations of dendritic cells have been described). They migrate to the peripheral tissues (e.g., skin, intestinal tract) and to the secondary lymphoid organs. Immature dendritic cells at a site of inflammation function as phagocytes, and the process of phagocytosis can initiate differentiation and directed migration to the secondary lymphoid organs, particularly the lymph nodes (Figure 7-15). Thus dendritic cells can carry processed antigen from a site of inflammation to the T-cell–rich areas of the lymph nodes.
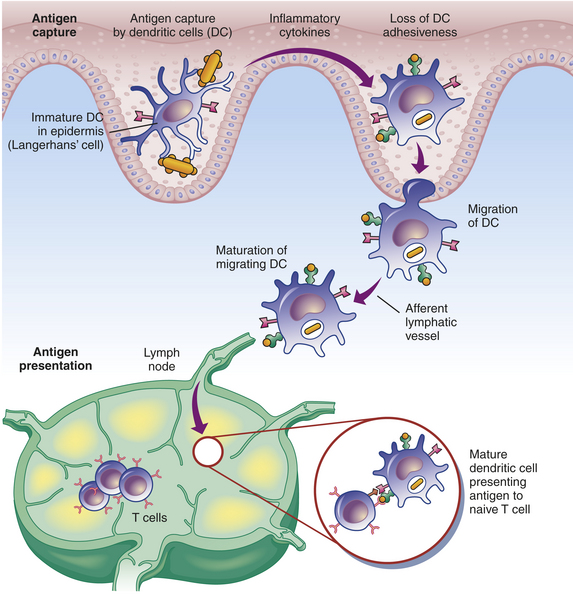
Figure 7-15 The role of the dendritic cell in capturing antigen. Immature dendritic cells in the tissues encounter and phagocytose antigen, which results in the production of inflammatory cytokines and a loss of adhesive interactions with neighboring cells. The maturing dendritic cell migrates through the lymphatic vessels to a regional lymph node, where it presents the antigen to immunocompetent T cells to initiate the clonal selection process. (Redrawn from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Both antigen processing and presentation are necessary for an adaptive immune response to occur. Although B and T lymphocytes are immunocompetent before they have “seen” an antigen on the surface of an APC, they are considered “naive” until they have actually done so. The processing and presentation of antigens to naive lymphocytes result in activation of an acquired immune response only if (1) the antigen is of the appropriate type; (2) the lymphocytes are prepared to recognize the presented antigen; and (3) the antigen is presented appropriately.
Pathways of Antigen Processing
In general, the immune system responds to two types of antigens: exogenous and endogenous.26 Using infection as a model, exogenous antigens are carried on microorganisms that are trapped and killed by phagocytic cells; therefore, they come from outside the cell. Endogenous antigens are synthesized within a cell. These include viral antigens because viruses infect cells and use the normal cellular protein-synthesizing machinery to translate the viral genes into viral proteins. Endogenous antigens also may include those uniquely produced by cancerous cells. When many cells undergo malignant change, they begin producing unique proteins that are specific to cancer cells and are presented as foreign antigens on the cell surface.
Exogenous and endogenous antigens are preferentially presented by different classes of MHC molecules: class I MHC molecules generally present endogenous antigens, and class II molecules prefer exogenous antigens (Figure 7-16). Because class I MHC molecules are expressed on all cells, except red blood cells, any change in that cell due to viral infection or malignancy may result in foreign antigen being presented by MHC class I on that cell’s surface. Class II MHC molecules are co-expressed with MHC class I on a more limited number of cells that have APC function, including macrophages, dendritic cells, B lymphocytes, activated T lymphocytes, and some endothelial cells.
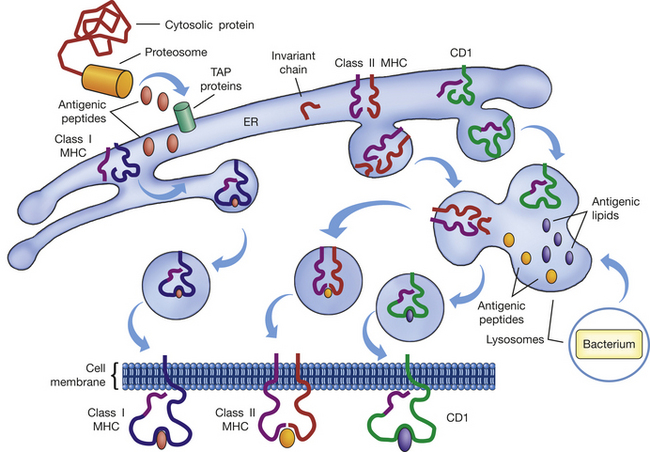
Figure 7-16 Antigen processing. Antigen processing and presentation are required for initiation of most immune responses. Foreign antigen may be either endogenous (cytosolic protein) or exogenous (e.g., bacterium). Endogenous antigenic determinants (antigenic peptides) are produced by cellular proteasomes and transported by TAP proteins into the endoplasmic reticulum (ER) where the MHC and CD1 molecules are being assembled. In the ER, antigenic peptides bind to the α-chains of the MHC class I molecule, and the complex is transported to the cell surface. In the ER, the α- and β-chains of the MHC class II molecules are also being assembled, but the antigen-binding site is blocked by a small molecule (invariant chain) to prevent interactions with endogenous antigenic peptides. The MHC class II–invariant chain complex is transported to lysosomes, where exogenous antigenic fragments have been generated as a result of phagocytosis. In the lysosomes the invariant chain is digested and replaced by exogenous antigenic peptides, after which the MHC class II–antigen complex is inserted into the cell membrane. CD1 is also assembled in the ER, but its antigen-binding site is specific for lipid antigenic determinants and does not bind endogenous antigenic peptides. The CD1 molecule is transported to the lysosomes and may encounter and bind antigenic lipids produced by phagocytic digestion of engulfed bacteria. The CD1-antigen complex is transported to the cell membrane and presents lipid antigens. MHC, Major histocompatibility complex; TAP, transporter associated with antigen processing.
Thus the term antigen processing relates to the process by which exogenous and endogenous antigens are linked with the appropriate MHC molecules. Endogenous antigens are usually components of proteins synthesized in the cytosol. They are degraded in the cytosol by proteasomes into small peptides and transported by TAP (transporter associated with antigen processing) proteins (TAP-1 and TAP-2) into the endoplasmic reticulum, where MHC class I and class II molecules are assembled.27 The class I MHC molecules have open antigen-binding sites so that antigen, the class I MHC α-chain, and a β2-microglobulin molecule form a stable complex that is transported through the Golgi apparatus to the plasma membrane. The antigenic peptides presented by class I MHC are usually very small, 8 to 10 amino acids in length.
MHC class II molecules are also assembled in the endoplasmic reticulum but do not bind with endogenous antigen because the antigen-binding site is blocked by a small protein called invariant chain. Exogenous antigens are internalized by phagocytosis and small antigenic molecules produced by digestion in the lysosomes. The MHC class II complexes of the class II α- and β-chains, with invariant chain, are transported to the lysosomes containing exogenous antigens. In the lysosomal environment, the invariant chain is digested and replaced by antigenic molecules that are usually slightly larger (in excess of 12 amino acids in length) than those presented by MHC class I.
CD1 presents a variety of lipid-containing antigens that are usually derived from phagocytosis and digestion of infectious microorganisms with very high lipid content in their cell membranes. Therefore, CD1 complexes with antigen in the lysosomes, in a fashion similar to MHC class II. The “pocket” that holds antigen for presentation by CD1 is generally more narrow and deeper than described for MHC molecules, and it is lined with many hydrophobic amino acids that interact with lipid.
Helper T Lymphocytes
Regardless of whether an antigen primarily induces a cellular or humoral immune response, a subpopulation of T lymphocytes, helper T cells (Th cells), is usually necessary for the process.28,29 As indicated by the name, this group of T cells helps the antigen-driven maturation of both B and T cells. They perform this task by facilitating and magnifying the interaction between APCs and the immunocompetent lymphocytes. This extremely important role involves three distinct steps: (1) the Th cell directly interacts with the APC through a variety of antigen-specific and antigen-independent receptors; (2) the Th cell undergoes a differentiation process during which a variety of cytokine genes are activated; and (3) depending on the pattern of cytokines expressed, the mature Th cell interacts with either immunocompetent B or T cells to enhance their response to antigen, which results in differentiation into either plasma cells or effector T cells, such as cytotoxic T cells. Th cells are critical to most immune responses, and a variety of major Th-cell defects that lead to severely diminished immune responses are discussed in later chapters.
APC-Th Cooperation
Cells that are destined to become Th cells emerge from the thymus with characteristic cell surface markers. They have a functional αβ TCR complex and express the surface molecule CD4 and lack CD8. These are generally referred to as precursor Th cells, or sometimes Thp cells (Figure 7-17). As described previously, the TCR recognizes antigen, and the CD4 molecule confines antigen presentation to MHC class II molecules, thus CD4+ cells are class II restricted. In order to undergo maturation, the Th cell must receive three independent signals; antigen binding through the combined interaction of the TCR complex and CD4, co-stimulatory signals through a variety of intercellular adhesion molecules, and activation of specific cytokine receptors.30 If the appropriate signaling pathways are activated, the cell will differentiate through multiple intermediate stages into functional Th cells.
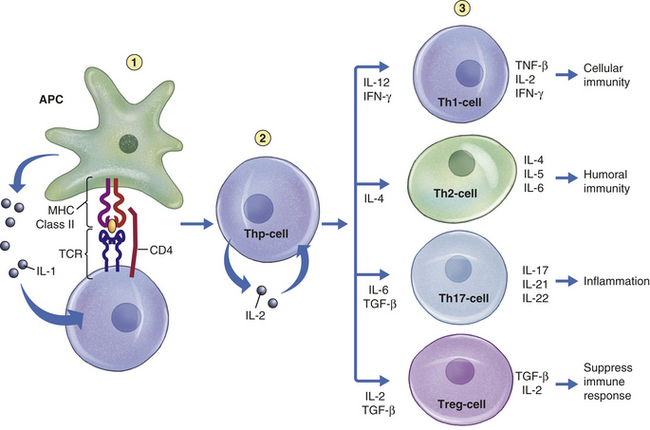
Figure 7-17 Development of T-cell subsets. The most important step in clonal selection is the production of populations of helper T (Th) cells (Th1, Th2, and Th17) and regulatory T (Treg) cells that are necessary for the development of cellular and humoral immune responses. In this model, APCs (probably multiple populations) may influence whether a precursor Th cell (Thp cell) will differentiate into a Th1, Th2, Th17, or Treg cell. Differentiation of the Thp cell is initiated by three signaling events. The antigen signal is produced by the interaction of the T-cell receptor (TCR) and CD4 with antigen presented by MHC class II molecules. A set of co-stimulatory signals is produced from interactions between adhesion molecules (e.g., CD80 and CD28) (not shown). A third signal is produced by the interactions of cytokines (particularly interleukin [IL]-1) with appropriate cytokine receptors (IL-1R) on the Thp cell. The Thp cell up-regulates IL-2 production and expression of the IL-2 receptor (IL-2R), which act in an autocrine fashion to accelerate Thp-cell differentiation and proliferation. Commitment to a particular phenotype results from the relative concentrations of other cytokines. IL-12 and IFN-γ produced by some populations of APCs favor differentiation into the Th1-cell phenotype; IL-4, which is produced by a variety of cells, favors differentiation into the Th2-cell phenotype; IL-6 and TGF-β (T-cell growth factor) facilitate differentiation into Th17 cells; IL-2 and TGF-β induce differentiation into Treg cells. The Th1 cell is characterized by the production of cytokines that assist in the differentiation of cytotoxic T (Tc) cells, leading to cellular immunity, whereas the Th2 cell produces cytokines that favor B-cell differentiation and humoral immunity. Th1 and Th2 cells affect each other through the production of inhibitory cytokines: IFN-γ will inhibit development of Th2 cells, and IL-4 will inhibit the development of Th1 cells. Th17 cells produce cytokines that affect phagocytes and increase inflammation. Treg cells produce immunosuppressive cytokines that prevent the immune response from being excessive. APC, Antigen presenting cell; IFN, interferon; MHC, major histocompatibility complex; TGF, transforming growth factor.
The complex of an antigenic peptide presented by an MHC class II molecule is recognized by multiple molecules on the Th-cell surface. The TCR binds directly to the antigen, whereas CD4 independently binds to a different site on the MHC class II β-chain. This co-recognition of the MHC/antigen complex by the TCR and CD4 brings CD4 into proximity with the CD3 components of the TCR complex, which initiates a series of enzymatic interactions among other molecules associated with the cytoplasmic portions of CD3 and CD4, such as the protein kinases p56lck and ZAP-70. These molecules activate a signaling pathway from the TCR to the Th-cell nucleus.
The antigenic signal alone is inadequate and may even inactivate the Th cell if co-stimulatory signals are not present. Co-stimulatory molecules are necessary for proper differentiation to occur. A variety of molecular interactions have been described, but the most critical appears to involve B7 on the APC and CD28 on the Th cell. Other interactions occur between CD48 on the APC and CD2 on the Th cell and between a variety of other adhesion molecules. In each case, the Th-cell molecule sends an activation signal to the nucleus. An additional signal is provided by cytokine. At this early stage of Th-cell differentiation, IL-1 secreted by the APC provides this signal through interaction with the IL-1 receptor on the Th cell.
The initial differentiation response by the Th cell includes the production of the cytokine IL-2 and up-regulation of IL-2 receptors. IL-2 is secreted and acts in an autocrine (self-stimulating) fashion to induce further maturation and proliferation of the Th cell. Without IL-2 production, the Th cell cannot efficiently mature into a functional helper cell. At this point, Th cells undergo one of several different differentiation pathways into Th subsets.
Th Subsets
The most clearly characterized Th-cell subsets are Th1 and Th2 cells, and the newly described Th17 cells (see Figure 7-17). These subsets have different functions: Th1 cells help develop cellular immunity, Th2 cells help develop humoral immunity, and Th17 cells increase the inflammatory response. A fourth subset, Treg cells, is discussed later in this chapter. The Th subsets differ considerably in the spectrum of cytokines produced by each, as well as the expression of surface cytokine receptors and intercellular adhesion molecules. Th1 cells produce IL-2, tumor necrosis factor (TNF)-β, and IFN-γ; Th2 cells produce IL-4, IL-5, IL-6, and IL-13; and Th17 cells produce IL-17, IL-21, and IL-22. As will be seen in the next portions of this chapter, the Th1 cytokines affect Tc cell development, and the Th2 cytokines are needed for B cell maturation, including class-switch. Th17 cell cytokines affect inflammatory response, particularly neutrophil and macrophage infiltration and production of antimicrobial proteins and chemokines by epithelial cells.31 Members of the IL-17 cytokine family induce epithelial cell chemokines and neutrophil infiltration, and IL-22 more specifically affects epithelial cell antimicrobial protein production.29,32 Thus Th17 cells control many aspects of inflammation, including chronic inflammation.33,34 Th1, Th2, and Th17 cells also have different cytokine receptors, so that IFN-γ produced by Th1 cells will bind to receptors on Th2 and Th17 cells and suppress their function. Likewise, Th2 cells produce IL-4, which suppresses Th1 and Th17 cells through their IL-4 receptors. Thus in some instances the immune response favors antibody formation, with suppression of a cell-mediated response, whereas in other instances the opposite is true. For example, antigens derived from viral or bacterial pathogens and those derived from cancer cells are hypothesized to induce a greater number of Th1 cells relative to Th2 cells, whereas antigens derived from multicellular parasites and allergens are hypothesized to result in production of more Th2 cells.29 Many antigens (e.g., tetanus vaccine), however, produce excellent humoral and cell-mediated responses simultaneously.
How a Th cell is guided into becoming a Th1, Th2, or Th17 cell is not fully known. Some evidence indicates that different subpopulations of APCs influence the choice by secreting different profiles of cytokines that may favor one route of differentiation over another (see Figure 7-17).29
B-Cell Activation: The Humoral Immune Response
When an immunocompetent B cell encounters an antigen for the first time, only those cells with specific BCRs complementary to that antigen’s determinant sites are stimulated to proliferate and differentiate (clonal selection), resulting in multiple copies of that particular B cell. The differentiated B cell becomes a plasma cell and can be found in the blood, secondary lymphoid organs (primarily spleen and lymph nodes), and some inflammatory sites. Each plasma cell is a factory for antibody production and is dedicated to the secretion of a single class or subclass of antibody with one variable region and therefore specificity against one antigenic determinant.
Primary and Secondary Immune Responses
The immune response to antigenic challenge has classically been divided into two phases—the primary and secondary responses—that can be most easily demonstrated by serologic tests that measure plasma concentrations of antibody over time (Figure 7-18). On initial exposure to most antigens, there is a latent period, or lag phase, during which B-cell differentiation and proliferation occur. After approximately 5 to 7 days, IgM antibody specific for that antigen can be detected in the circulation. The lag phase is a result of the time necessary for clonal selection, including antigen processing and presentation, induction of Th cells, interactions between immunocompetent B cells and Th cells, and the maturation and proliferation of the B cells into plasma cells and memory cells.
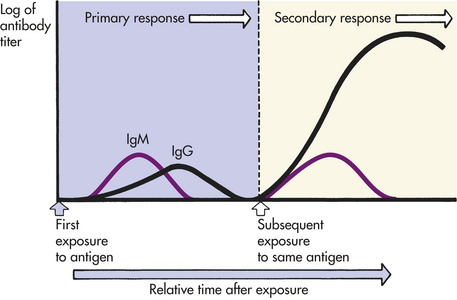
Figure 7-18 Primary and secondary immune responses. Antigen responses are dominated by two classes of immunoglobulins, IgM and IgG. IgM predominates on initial exposure to the antigen in the primary response, with IgG appearing later. After the host’s immune system is primed, another challenge by the same antigen induces the secondary response in which some IgM and larger amounts of IgG are produced.
This is the initial response, or primary immune response. Typically, IgM will be produced first, followed by IgG against the same antigen. The quantity of IgG may be about equal to or less than the amount of IgM production. If no further exposure to the antigen occurs, the circulating antibody is catabolized (broken down) and measurable quantities fall. The individual’s immune system, however, has been primed. A second challenge by the same antigen results in the secondary (anamnestic) immune response, which is characterized by the more rapid production of a larger amount of antibody than the primary response. The rapidity of the secondary immune response is the result of the presence of memory cells that do not require further differentiation. IgM may be transiently produced in the secondary response and the quantity may be about the same as that produced in the primary response. IgG production is increased considerably, making it the predominant antibody class of the secondary response. It is often present in concentrations several times larger than those of IgM, and levels of circulating IgG specific for that antigen may remain elevated for an extended period of time. If the antigenic challenge is in the form of a vaccine or occurs through natural infection, the level of protective IgG may remain elevated for decades.
The existence of a prolonged and protective secondary immune response explains how vaccinations provide protection against certain pathogenic microorganisms. Edward Jenner, an English physician of the late eighteenth century, performed the first well-documented vaccine trial.35,36 Although some of the stories about Jenner’s experiments are fanciful, it is known that Jenner recognized that milkmaids were protected from the deadly smallpox virus if they had previously developed cowpox, a bovine equivalent of smallpox that causes only mild disease in humans. Jenner took material from a cowpox pustule on the hand of an infected milkmaid and injected it into the arm of an 8-year-old boy. After the boy’s initial inflammatory reaction to the injection subsided, Jenner injected him again, this time with material from a smallpox pustule. Fortunately, the experiment was a success because Jenner is reported to have reinjected smallpox virus into the boy at least 20 times without the child becoming ill. In Jenner’s experiment, the antigens on the cowpox virus and the smallpox virus were sufficiently similar that the cowpox antigen functioned as an altered or attenuated smallpox antigen. The antibodies and lymphocytes that recognized and destroyed cowpox also were able to recognize the smallpox virus, thereby protecting the immunized child against smallpox. In 1798, Jenner used the term vaccination (vacca = cow) to describe his technique.
Cellular Interactions
As with most aspects of immunity, a sequence of cellular interactions is required to produce an effective antibody response (Figure 7-19).37 The immunocompetent B cell is also an APC and expresses surface IgM and IgD BCRs. Unlike the T-cell receptor that can only “see” processed and presented antigen, the BCR can react with soluble antigen. Antigen binding to the BCR complex activates intracellular kinases, in a fashion similar to the TCR receptor complex. In many instances, circulating antigen, either on macromolecules or the surface of a pathogen, will have activated the complement system through the alternative or lectin pathways. Thus complement receptors on the B cell, such as CD19 and CD21, act as co-receptors to bind antigen. As a result of signaling from the BCR complex and other surface co-receptors, the antigen-bearing macromolecule is internalized, broken down in the lysosomes, and complexes with MHC class II molecules for presentation on the cell surface, where it is recognized by a Th2 cell through the TCR and CD4. The intercellular bridge created through antigen induces the Th2 cell to up-regulate additional surface receptors and secrete cytokines. Direct interaction between CD40 on the B-cell surface and the CD40 ligand (CD40L, also called CD154) on the Th2 cell, as well as the interaction of B7 on the B cell and CD28 on the Th cell and exposure of the B cell to Th2-cell cytokines (particularly IL-4) induces proliferation of the B cell and maturation into a plasma cell. A major component of maturation is class switch.
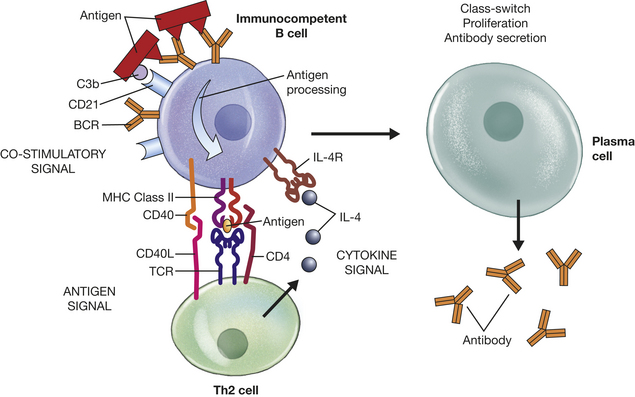
Figure 7-19 B-cell clonal selection. Immunocompetent B cells undergo proliferation and differentiation into antibody-secreting plasma cells. Three signals are necessary. The antigen signal is provided by the B cell itself. A B cell can recognize soluble antigen directly through the B-cell receptor and co-receptors, such as complement receptors (CD21), which usually involve accessory molecules such as CD19 (not shown). Antigen is internalized and processed for presentation by MHC class II molecules, which interact with the T-cell receptor (TCR) and CD4 on Th2 cells. Co-stimulatory signals are provided through adhesion molecules, particularly CD40 and CD40L (CD154). The cytokine signal is provided by Th2 cytokines (particularly IL-4) binding to appropriate cytokine receptors (IL-4R) on the B cell. Additional cytokines influence switch to particular classes or subclasses of antibody. MHC, Major histocompatibility complex.
Class-Switch
The immunocompetent B cell uses IgM and IgD as receptors. During the clonal selection process, however, each B cell has the option of changing the class of antibody to a secreted form of one of the four IgG subclasses, one of the two IgA subclasses, or IgE, or continuing to produce IgM but changing to a secreted form, usually a pentamer. This process is called class- or isotype-switch. During this process the variable region of the antibody heavy chain is conserved, and the light chain remains unchanged from that used in the BCR; therefore, the antigenic specificity also remains unchanged.
The mechanism of class-switch involves another DNA rearrangement, during which the VDJ region encoding the heavy chain’s variable region is moved to another site on the DNA that is adjacent to the gene for a different constant region under the control of activation-induced cytidine deaminase (AICD) (Figure 7-20).38 The DNA is cut and mended with removal of the DNA that was between the VDJ site and the new constant region. Specific recognition sites (switch regions) precede each constant region gene, and the particular constant region chosen for class-switch appears to be, at least partially, under the control of specific Th2 cytokines. For instance, IL-4 and IL-13 appear to preferentially stimulate switch to IgE, and transforming growth factor-β (TGF-β) and IL-5 appear to play major roles in class-switch to IgA.
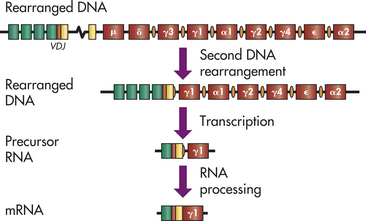
Figure 7-20 Genetics of class-switch. During clonal selection, most B cells switch from expression of surface IgM and IgD to a different class or subclass of antibody. A first set of DNA rearrangements during the generation of clonal diversity resulted in formation of the VDJ region. The class-switch process involves a second DNA rearrangement during which the VDJ region is moved to a switch region (orange ovals) immediately preceding the new class/subclass of antibody. In this example, the B cell undergoes class-switch to a γ1 heavy chain and secretion of an IgG1 antibody. The intervening DNA between the VDJ and the selected switch region is excised, and the DNA is repaired (DNA after second rearrangement) and transcribed into a precursor ribonucleic acid (RNA). The RNA is processed to a messenger RNA (mRNA) with information for the new heavy chain.
A few antigens can bypass the need for Th cells and can directly stimulate B-cell maturation and proliferation. These are called T-independent antigens (Figure 7-21). They are mostly bacterial products that are large and are likely to have repeating antigenic determinants (multiple identical antigenic determinant sites) that bind and crosslink several B-cell receptors. The accumulated intracellular signal is adequate to induce differentiation to a plasma cell but is not adequate to induce class-switch. The CD40-CD40L interaction is a necessary component of the signal that leads to class-switch. Therefore, T-independent antigens usually induce a relatively pure IgM primary and secondary immune response.
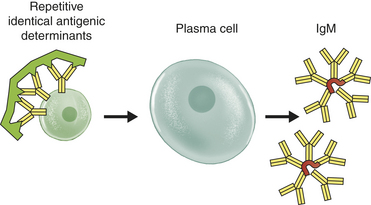
Cellular Differentiation
During the clonal selection process, B cells differentiate into antibody-producing plasma cells and into a set of long-lived memory cells. During B cells’ differentiation into plasma cells, the CDR portions of the antibody variable region are prone to somatic point mutations that lead to changes in single amino acids. Some of these changes produce better antibodies that bind more strongly (higher affinity) to the antigen. The presence of antigen creates a positive selective pressure toward the developing B cells that express the higher-affinity antibody, which results in a process called affinity maturation, in which the quality of the circulating antibody improves over time.
The memory cells remain inactive until subsequent exposure to the same antigen. On reexposure, these memory cells do not require much further differentiation and will therefore differentiate rapidly into new plasma cells.39
T-Cell Activation: The Cellular Immune Response
Activation of the cell-mediated arm of the immune response begins with the binding of antigen to specific T-cell receptors. Through a variety of intercellular collaborations that are mediated by specific cellular receptors and cytokines, the naive T cell proliferates and differentiates into a functional (effector) T cell. The two main effector functions of activated T cells are (1) direct killing of foreign and/or abnormal cells and (2) assistance and/or activation of other cells, such as macrophages. The first function is carried out by a subclass of T cells termed cytotoxic T lymphocytes (Tc cells, or CTLs). Activation of macrophages is performed by a special subset of Th cells. Additional T cells develop into cells that regulate the immune response in order to avoid inadvertently attacking self-antigens or to avoid overactivation of the immune response. This mixed population of cells is termed T regulatory (Treg) cells. Finally, memory T cells are also produced to help induce secondary cell-mediated immune responses.
Cellular Interactions
During the clonal selection phase of the cell-mediated immune response, immunocompetent T cells in the peripheral lymphoid organs must recognize antigen that has been processed and presented by MHC class I molecules (Figure 7-22).40 The antigen is usually an endogenous antigen expressed on the surface of cells infected with a virus or that have become malignant. The T cells have a functional αβ TCR complex and express the surface molecule CD8, rather than CD4. The presence of the CD8 molecule confines antigen recognition to MHC class I molecules, therefore CD8+ T cells are class I restricted. The TCR binds directly to the antigenic peptide, whereas CD8 independently binds to a different site on the MHC class I α-chain. This co-recognition of the MHC/antigen complex by the TCR and CD8 brings CD8 into proximity with the CD3 components of the TCR complex, which initiates a series of enzymatic interactions among other molecules associated with the cytoplasmic portions of CD3 and CD4, as was described for Th-cell activation. These molecules activate a signaling pathway from the TCR to the T-cell nucleus.
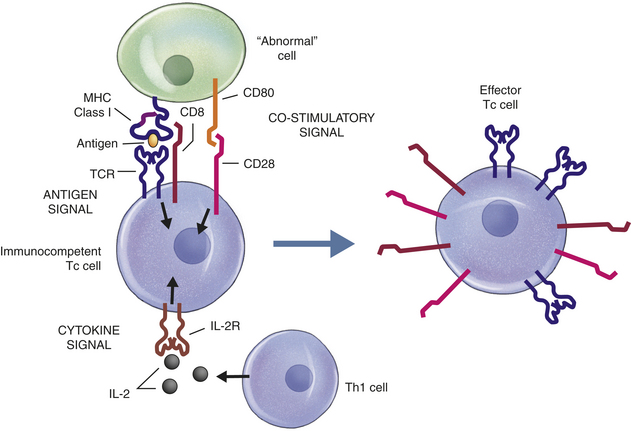
Figure 7-22 Tc-cell clonal selection. The development of effector cytotoxic T (Tc) cells during clonal selection results from three cooperative signaling events provided by antigen, co-stimulatory adhesion molecules, and cytokines. The immunocompetent Tc cell “sees” antigen presented by MHC class I molecules on the surface of a virally infected or cancerous “abnormal” cell. The antigen–MHC class I complex is recognized simultaneously by the T-cell receptor (TCR), which binds to antigen, and CD8, which binds to the MHC class I molecule. The proximity of signaling molecules associated with the cytoplasmic portions of CD8 and the TCR result in intracellular signaling. A separate signal results from the interaction of several groups of adhesion molecules (e.g., CD80 and CD28 in this example). The third signal is provided by the interaction of cytokine, particularly IL-2 from Th1 cells, and the appropriate receptor. MHC, Major histocompatibility complex.
To undergo maturation, the T cell must receive independent signals from a variety of co-stimulatory intercellular adhesion molecules and specific cytokine receptors. If the appropriate signaling pathways are activated, the cell will proliferate and differentiate through multiple intermediate stages into functional Tc cells. The co-stimulatory signals for Tc-cell maturation are virtually the same as has been described for Th-cell maturation: B7 on the cell-presenting antigen and CD28 on the T cell, CD48 on the antigen-presenting cell and CD2 on the T cell, and a variety of other adhesion molecules. Development of Tc cells also requires cytokines, especially IL-2, produced by the Th1 cell.
Cellular Differentiation
The result of these cellular interactions is the production of active Tc cells with the capacity to identify antigens on the surface of infected or malignant cells and then to destroy those cells. As with B cells, some of the T cells that become activated in response to antigen presentation will not become effectors that destroy infected targets, but instead develops into a population of memory T cells. These cells have the capacity to rapidly respond to further exposure to the same antigen.
Superantigens
A group of molecules has the ability to bind the variable portion of the TCR β-chain outside of its normal antigen-specific binding site, as well as the α-chain of MHC class II molecules outside of their antigen-presentation sites (Figure 7-23). Thus these molecules are not digested and processed by an APC to be presented to an immune cell. This binding results in adherence of the TCR and MHC class II molecules, independent of antigen recognition, and provides an activation signal for Th-cell activation and proliferation. The normal antigen-specific recognition between Th cells and APCs results in activation of relatively few cells: only those cells with specific TCRs against that antigen. The type of binding described here results in activation of large populations of T lymphocytes, regardless of antigen specificity.41,42 Thus these molecules have been referred to as superantigens (SAGs).
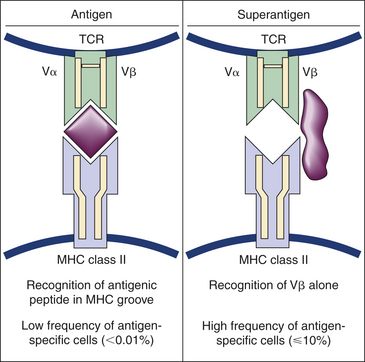
Figure 7-23 Superantigens. The T-cell receptor (TCR) and an MHC class II molecule normally simultaneously interact with a processed antigen to induce T-cell differentiation. Superantigens, such as some bacterial toxins, bind directly to the TCR and the MHC class II molecules. Superantigens activate Th cells independently of TCR antigen specificity. MHC, Major histocompatibility complex.
SAGs induce an excessive production of cytokines, including IL-2, IFN-γ and TNF-α.41,42 The overproduction of inflammatory cytokines results in symptoms of a systemic inflammatory reaction, including fever, low blood pressure, and potentially, fatal shock. Some examples of SAGs are the bacterial toxins produced by Staphylococcus aureus and Streptococcus pyogenes (including the superantigens that cause toxic shock syndrome and food poisoning).43 Some viruses are also able to produce superantigens, although the exact nature of these antigens is unclear.
EFFECTOR MECHANISMS
Protection Against Infection
The chief function of circulating antibodies is to protect the host from infection. Protection can be afforded by antibody in several ways, either directly or indirectly (Figure 7-24). Directly, antibody can cause neutralization (inactivating or blocking the binding of an antigen to a receptor), agglutination (clumping insoluble particles that are in suspension), or precipitation (making a soluble antigen into an insoluble precipitate) of infectious agents or their toxic products. Indirectly, antibodies activate several components of innate immunity, including complement and phagocytes.
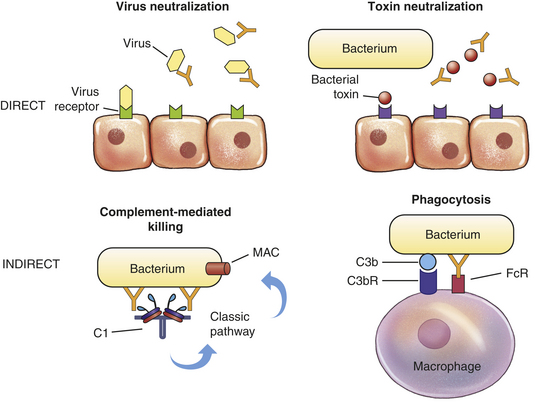
Figure 7-24 Direct and indirect functions of antibody. Protective activities of antibodies can be direct (through the action of antibody alone) or indirect (requiring activation of other components of the innate immune response, usually through the Fc region). Direct means include neutralization of viruses or bacterial toxins before they bind to receptors on the surface of the host’s cells. Indirect means include activation of the classical complement pathway through C1, resulting in formation of the membrane-attack complex (MAC) or by increased phagocytosis of bacteria opsonized with antibody and complement components bound to appropriate surface receptors (FcR and C3bR).
Direct Effects: To cause infection, many pathogens must attach to specific receptors on the host’s cells. For instance, viruses that cause the common cold or the influenza virus must attach to specific receptors on epithelial cells. Some bacteria, such as Neisseria gonorrhoeae that causes gonorrhea, must attach to specific sites on epithelial cells. Antibodies may protect the host against infection by covering the portions of the microorganism that it needs to bind to the host cell. Neutralization, or prevention of attachment to the host cell, thereby prevents infection of the host.
Protection against many viral infections can be elicited effectively by vaccination with inactivated or attenuated (weakened) viruses to induce neutralizing antibody production at the site of typical viral entrance into the body. A good indication of the degree of protection against viral infection is the level of circulating antibodies found in the blood. The level of circulating antibodies is referred to as an antibody titer. However, many viruses (e.g., measles, herpes) are inaccessible to antibodies after initial infection because they do not circulate in the bloodstream but instead remain inside infected cells, spreading by direct cell-to-cell contact. Neutralizing antibodies against this type of virus are most effective in preventing the initial infection. Other viruses, such as polio and influenza, spread through the blood, are more susceptible to the effects of circulating antibodies, and can be controlled by antibodies even after the initial infection.
The symptoms of some infectious diseases result directly from toxins produced by infecting bacteria. For instance, the symptoms of tetanus or diphtheria are mediated by specific toxins. To cause disease, most toxins must bind to surface molecules on the individual’s cells. Protective antibodies can bind to the toxins, prevent their interaction with cells, and neutralize their biologic effects. Detection of the presence of an antibody response against a specific toxin (antibodies referred to as antitoxins) can aid in the diagnosis of diseases. For example, laboratory tests that detect antistreptolysin O also can be very useful in diagnosing group A streptococcal infections. Antibodies that neutralize bacterial toxins can be induced to confer immunity against bacterial pathogens by means of immunization. To prevent harming the recipient of immunization, bacterial toxins are chemically inactivated so that they have lost most of their harmful properties but still retain their immunogenicity. These are referred to as toxoids. Examples of bacterial pathogens for which immunization with toxoids can provide immunologic protection include those that cause diphtheria and tetanus.
A new therapeutic application of antibodies uses their normal properties to treat human disease (see What’s New? Antibodies as Drugs).
Indirect Effects: Antibody can be protective by interacting with or activating components of nonspecific inflammation. Indirect effects are mediated by the Fc portion of the antibody molecule and include opsonic activity leading to enhanced phagocytosis and activation of the complement system that may lead to complement-mediated destruction of the pathogen or increased opsonic activity through deposition of C3b.44,45
In their role as opsonins, antibody and C3b make the pathogen more susceptible to phagocytosis through binding to Fc or C3b receptors on the phagocyte’s surface. Opsonization is often necessary for efficient bacterial clearance because many bacteria have an outer capsule that deters recognition by phagocytes unless it is coated with an antibody or complement protein. Bacterial surface molecules are usually complex and have multiple accessible antigenic determinants, enabling them to bind several different antibodies simultaneously. When an antigen reacts with the Fab regions of antibody, the Fc portion of that antibody is recognized and binds to Fc receptors on the surfaces of inflammatory cells. Engagement of Fc receptors results in their activation, making phagocytosis of the opsonized bacterium more efficient.46 The clustering of Fc regions has the added effect of more efficient complement activation.
Secretory Immune Response
The immune response that protects the entire body is produced by the systemic immune system. A distinct set of lymphoid tissues makes up another, partially independent, immune system at the external surfaces of the body. This system is called the secretory (mucosal) immune system (Figure 7-25). Most humoral immune responses occur when antibodies or B cells encounter antigens in the blood, but sometimes this encounter occurs in other body fluids. Antibodies are present in bodily secretions such as tears, sweat, saliva, mucus, and breast milk, where they can protect the body against antigens that have not yet penetrated the skin or mucous membranes.
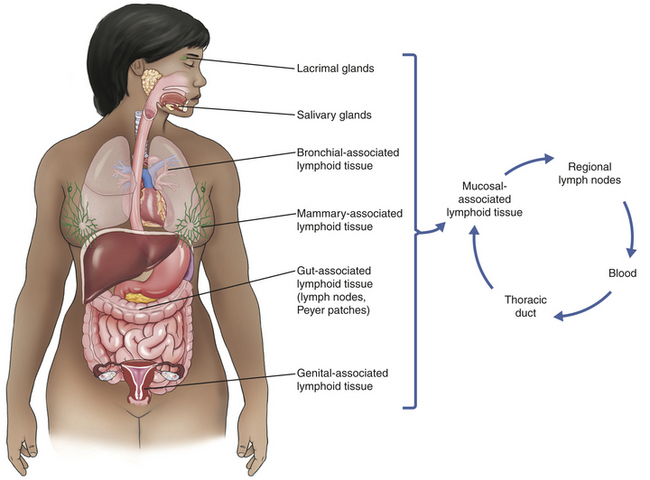
Figure 7-25 Secretory immune system. Lymphocytes from the mucosal-associated lymphoid tissues circulate throughout the body in a pattern separate from other lymphocytes. For example, lymphocytes from the gut-associated lymphoid tissue circulate through the regional lymph nodes, the thoracic duct, and the blood and return to other mucosal-associated lymphoid tissues rather than to lymphoid tissue of the systemic immune system.
Antibodies in secretions are produced by plasma cells of the secretory (mucosal) immune system.47 The B cells of these two systems follow a different pattern of migration after they leave the bone marrow. B lymphocytes of the systemic immune system travel through the spleen and most lymph nodes, whereas those of the secretory immune system travel through a different group of lymphoid tissues including the lacrimal and salivary glands and the lymphoid tissues of the breasts, bronchi, intestines, and genitourinary tract. Immunoglobulins that are secreted at these sites are called secretory immunoglobulins and act locally rather than systemically.
Local protection is necessary to combat antigens (chiefly infectious microorganisms) that are inhaled, swallowed, or otherwise come into contact with external body surfaces. Once they have taken up residence in the external layers of the body, harmful microorganisms can cause local disease or possibly penetrate the barriers described in Chapter 6 to cause systemic disease. Alternatively, the microorganisms may fail to cause disease in the individual, either because the microorganisms are passed out of the body without any ill effects or because the infection is thwarted by the systemic immune system. In the latter case the individual may continue to “carry” the infectious agent in the mucosal areas, thereby enabling its spread to other individuals. The major function of the secretory immune system is to halt viral and bacterial invasion before local or systemic disease can develop and to prevent a carrier state that may result in spread of the infection to others.
IgA is the dominant secretory immunoglobulin, although IgM and IgG also are present in secretions. The primary role of IgA is to prevent the attachment and invasion of pathogens through mucosal membranes, such as those of the gastrointestinal, pulmonary, and genitourinary tracts. To induce protective immunity against some pathogens that enter through these routes, local immunization seems to be preferable to inducing only systemic immunity. For instance, two different vaccines have been used against polio. The Sabin vaccine was administered orally as an attenuated (i.e., inactivated so as to render relatively harmless) live virus. This route caused a transient, limited infection and induced effective systemic immunity and secretory immunity, preventing both the disease and the establishment of a carrier state. The Salk vaccine, on the other hand, consisted of killed viruses that were administered intradermally. It induced adequate systemic protection but did not generally prevent an intestinal carrier state.
Because B lymphocytes of the secretory/mucosal immune system travel through breast-associated lymphoid tissue, most antigens to which the mother has been exposed gastrointestinally (e.g., poliovirus) induce secretion of specific IgAs, IgMs, and IgGs into the breast milk. Antibodies in the milk may provide protection against these infectious disease agents to the nursing newborn. Although colostral antibodies (i.e., found in colostrum of breast milk) provide the newborn with passive immunity against gastrointestinal infections, they do not provide systemic immunity because they do not cross the newborn’s gut into the bloodstream after the first 24 hours of life. Passive systemic immunity is provided by maternal antibodies that passed across the placenta into the fetus before birth.
The mechanisms and functions of antigen-antibody binding are the same in the secretory immune system as they are in the systemic immune systems; that is, binding neutralizes or opsonizes the antigen, preventing it from harming the host. The major differences between the two systems include (1) the order of utilization—the secretory immune response is part of the body’s first-line defense, whereas the systemic response is the body’s final defense; (2) the lymphocytes of each system follow different paths of migration and pass through different secondary lymphoid tissues; and (3) the secretory response occurs locally and externally (in body secretions), whereas the systemic response occurs systemically and internally (in blood and tissues).
IgE
IgE is a special class of antibody that is designed to help protect the individual from infection with large parasitic worms.48 However, when IgE is produced against relatively innocuous environmental antigens, it is also the primary cause of common allergies (e.g., hay fever, dust allergies, bee stings). The role of IgE in allergies is discussed in Chapter 8.
Large multicellular parasites usually invade mucosal tissues (Figure 7-26). In response to parasitic antigens, a variety of different antibody classes are produced with many B cells class-switching to IgE-secreting plasma cells under the direction of Th2 cells primarily producing IL-4 and IL-13. IgG, IgM, and IgA bind to the surface of parasites, activate complement, generate chemotactic factors for neutrophils and macrophages, and serve as opsonins for those phagocytic cells. The influx of neutrophils and macrophages progressively leads to development of a granulomatous response around the parasite.49 Unique to parasitic infections, the eosinophil is a primary cell in the granuloma. The influx of eosinophils results from IgE-triggered mast cell degranulation. Mast cells in the tissues have very high affinity Fc receptors for IgE, which rapidly bind IgE to the mast cell surface. Soluble macromolecules with multiple antigenic determinants are released from the parasite, react with the IgE-Fc receptors, and initiate mast cell degranulation (see Chapter 6). Eosinophil chemotactic factor of anaphylaxis (ECF-A) is released from mast cell granules and attracts eosinophils to the site of infection, as well as up-regulates surface receptors for IgG and complement component C3b. Eosinophil attachment to the parasite results in degranulation, releasing a variety of very toxic proteins that are at unusually high concentrations in eosinophilic granules, major basic protein (binds to heparin sulfate proteoglycans), eosinophil cationic protein (a member of the RNase A family), and others. These can cause extensive damage to the parasite if an adequate number of eosinophils are involved.
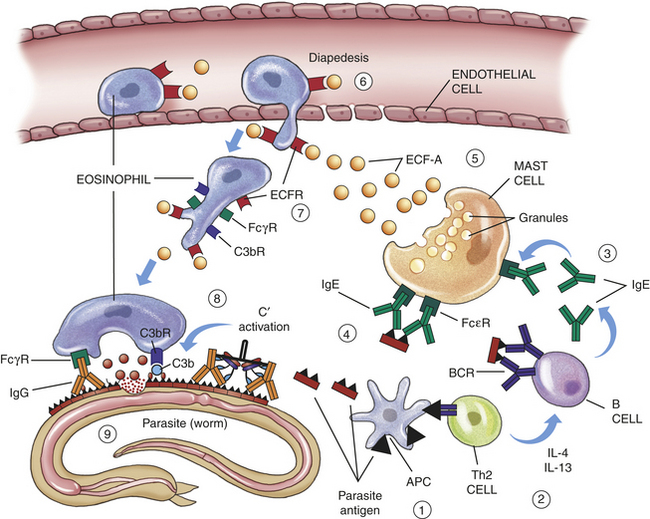
Figure 7-26 IgE function. Soluble antigens from a parasitic infection are processed by local antigen-presenting cells (APCs) and presented to Th2 cells (1), which respond by producing cytokines that favor class-switch to IgE production (2). B cells bind soluble parasite antigen, and some switch to producing IgG, whereas others switch to IgE. The secreted IgE molecules bind to IgE-specific receptors (FcεR) on the mast cell surface (3). Additional soluble parasite antigen crosslinks IgE-FcεR complexes on the mast cell surface (4), leading to mast cell degranulation and release of many proinflammatory products, including eosinophil chemotactic factor of anaphylaxis (ECF-A) (5). Eosinophils have receptors for ECF-A (ECFR) and are stimulated to increase adherence to the vessel walls and initiate diapedesis (6) and invasion of the surrounding tissue. The eosinophil also responds by increasing the density of surface receptors for IgG (FcγR) and complement component C3b (C3bR) (7). IgG had previously attached to the antigens on the parasite’s surface and activated the complement cascade (C′ activation) in a failed attempt to damage the parasite. The eosinophil attaches to the parasite’s surface through Fc and C3b receptors (8). Once bound to the parasite, the eosinophil releases its lysosomal enzymes onto the parasite, damaging its outer membrane (9).
T-Lymphocyte Function
Cytotoxic T Lymphocytes: Cytotoxic T lymphocytes (Tc cells or CTLs) are responsible for the cell-mediated destruction of such targets as tumor cells or cells infected with viruses.50 To perform this function, the Tc cell must directly adhere to the target cell through antigen presentation in association with MHC class I molecules and appropriate adhesion molecules (Figure 7-27). Most Tc-cell killing requires the αβ TCR complex and CD8 and is therefore class I restricted. Because of the cellular distribution of MHC class I molecules, Tc cells can recognize antigen on the surface of almost any type of cell that has been infected by a virus or has become cancerous.
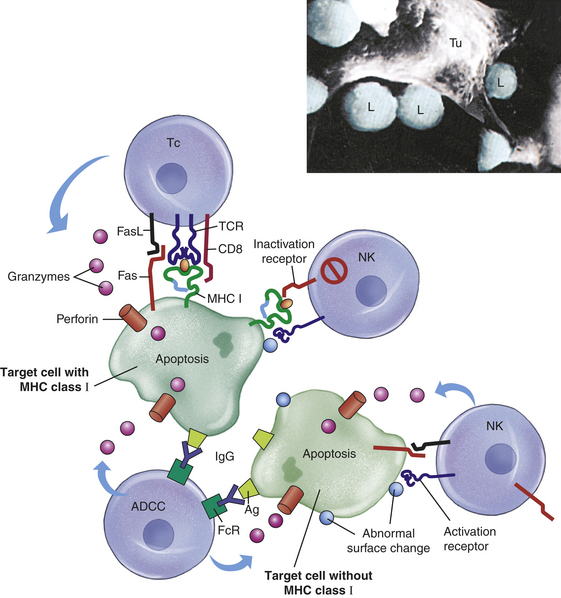
Figure 7-27 Cell killing mechanisms. Several cells have the capacity to kill abnormal (e.g., virally infected, cancerous) target cells. Cytotoxic T (Tc) cells recognized endogenous antigen presented by MHC class I molecules (cell on upper left). The intercellular interaction is enhanced through a variety of co-stimulatory adhesion molecules (not shown). The Tc cell mobilizes multiple killing mechanisms that induce apoptosis of the target cell, including the secretion of perforin that creates pores for the entrance of granzymes into the target cell and stimulation of Fas molecules on the target cell surface by Fas ligand (FasL) on the Tc cell. Natural killer (NK) cells (cells on right) use the same mechanisms to kill target cells through activation receptors that recognize “abnormal surface changes.” NK cells specifically kill targets that have down-regulated expression of surface MHC class I molecules. Targets expressing MHC class I molecules inactivate NK cells through a variety of inactivation receptors (cell on upper right). Several cells, including macrophages and NK cells, can kill by antibody-dependent cellular cytotoxicity (ADCC). IgG antibody binds to foreign antigen on the target cell. Cells involved in ADCC (cell on lower left) bind IgG through Fc receptors (FcRs) and initiate killing. The insert is a scanning electron microscopic view of Tc cells (L) attacking a much larger tumor cell (Tu). MHC, Major histocompatibility complex. (From Thibodeau GA, Patton KT: Anatomy & physiology, ed 5, St Louis, 2003, Mosby).
After attachment to a target cell, killing can occur by at least two different mechanisms that induce apoptosis: through the actions of perforin and granzyme or direct receptor interactions. Perforins and granzymes are contained in the Tc-cell lysosomal granules, which are released onto the surface of the target cell. Perforin acts in a fashion similar to C9 of the complement cascade and penetrates, polymerizes, and forms pores in the target cell’s plasma membrane. The granzymes enter the target cell through the perforin-lined pores and activate cellular enzymes (caspases) that are involved in apoptosis, resulting in death of the target. Additionally, target cell apoptosis can be induced directly through the stimulation of specific receptors on the cell surface. For instance, Tc cells express a surface molecule called Fas ligand, which is very similar to TNF-α and reacts with a protein called Fas (CD95) on the target cell surface. Activation of Fas signals the target cell to undergo apoptosis.
Other Cells That Kill Abnormal Cells: A variety of other cells kill targets in a fashion similar to Tc lymphocytes. Prominent among these cells are natural killer (NK) cells (see Chapter 6). In many ways, NK cells complement the effects of Tc cells.51 In some instances, a virally infected or cancerous cell will “protect” itself by down-regulating MHC class I molecule expression. Without surface MHC class I molecules, a cell becomes resistant to Tc-cell recognition and killing. NK cells are a special group of lymphoid cells that are similar to T cells but do not undergo maturation in the thymus and lack antigen-specific receptors.52 Instead, they express Fc receptors (CD16) for IgG and a variety of NK-specific cell surface receptors (similar to pattern recognition receptors, see Chapter 6) that identify protein changes on the surface of cells that have been infected or are in other ways abnormal. After attachment, the NK cell kills its target in a manner similar to that of Tc cells. However, NK cells also express another set of receptors, inhibitory receptors, that bind to MHC class I molecules.53 If the target cell continues to express MHC class I, the NK cell will bind to the class I molecule, and an inhibitory signal will result. Thus NK cells do not inadvertently kill MHC class I–bearing cells. If these cells are infected or malignant, yet still express MHC class I, they remain sensitive to Tc-cell killing. Thus Tc cells kill abnormal cells that continue to express MHC class I, whereas NK cells kill abnormal cells that have suppressed MHC class I expression.
NK cells, as well as some macrophages, can specifically kill targets through use of antibody. These cells express Fc receptors on their surface. If a pathogen or abnormal cell expresses a foreign antigen that elicits IgG antibody, which binds to the antigen, the NK cell can attach to the IgG through Fc receptors and activate its normal killing mechanisms. This is referred to as antibody-dependent cell-mediated cytotoxicity (ADCC) (see Figure 7-27).
Another population of NK-like cells has been identified, NK-T cells. NK-T cells are produced in the thymus and more closely resemble Tc cells. However, they express TCRs that have very limited variability and recognize antigens presented by CD1.
T Cells That Activate Macrophages
Under conditions of chronic inflammation, T cells produce cytokines that activate macrophages (see Chapter 6). Macrophage activation is usually accomplished by Th1 cells that recognize antigen and produce cytokines (particularly IFN-γ) that, in cooperation with microbial products (e.g., LPS), stimulate the macrophage to become a more efficient phagocyte and increase production of proteolytic enzymes and other antimicrobial substances (Figure 7-28).54,55 IFN-γ-induced macrophage activation also achieved by NK cells and CD8+ T cytotoxic cells. Additional signals (e.g., the CXC chemokine macrophage migration inhibitory factor) retain macrophages at inflammatory sites and increase intercellular adhesion between the Th1 cell (CD40L) and the macrophage (CD40).56
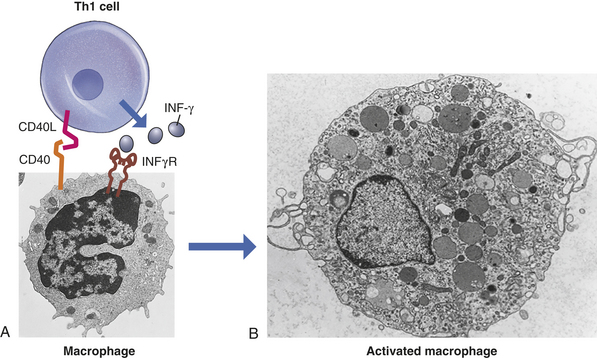
Figure 7-28 Activation of a macrophage by a T cell. A population of T cells that helps immune and inflammatory responses (helper T cells or Th1 cells) produces cytokines that activate macrophages. Optimal macrophage activation also requires close contact among the cells, which is mediated by a variety of adhesion molecules expressed on the surface of each cell (CD40L and CD40 shown here). CD40L, CD40 ligand; INFγ, interferon gamma; INFγR, receptor for interferon gamma. (Micrograph in A from Abbas AK, Lichtman AH: Cellular and molecular immunology, ed 5, Philadelphia, 2003, Saunders; B from Bloom W, Fawcett DW: A textbook of histology, ed 11, Philadelphia, 1986, Saunders.)
Regulatory T Lymphocytes
One form of peripheral tolerance to self-antigens occurs in Treg cells, a subpopulation of CD4+ T cells (see Figure 7-17).33,57–59 As with Th cells, Treg cells are activated by antigen presented in the context of class II MHC and differentiation under the control of specific cytokines, primarily TGF-β and IL-2, during which they express CD25 (the α-chain of the IL-2 receptor) and are frequently designated CD4+, CD25+ Treg cells. The role of Treg cells is to control or limit the immune response to protect the host’s own tissues against autoimmune reactions.60 Treg produce very high levels of TGF-β and IL-10, an immunosuppressive cytokine, which generally decrease Th1 and Th2 activity and suppress antigen recognition and Th cell proliferation. The role of Treg cells and other regulatory cells (e.g., CD25-cells, CD8+ regulatory cells, and Breg cells) is under intense investigation to determine the degree of their heterogeneity of derivation, function, and specificity.
Fetal and Neonatal Immune Function
The normal human infant is immunologically immature at birth. Although cell-mediated immunologic capabilities begin developing early in gestation and probably are completely functional at birth, antibody production is clearly deficient. In the last trimester, the fetus appears capable of producing a primary immune response (almost entirely IgM) to antigenic challenge in utero but is unable to produce a significant IgG response. Although some IgA can be detected, the capacity to produce IgA is underdeveloped.
To protect the child against infectious agents both in utero and during the first few postnatal months, a system of active transport facilitates the passage of maternal antibodies into the fetal circulation (Figure 7-29).61 In the placenta, maternal and fetal blood is separated by a layer of specialized cells termed trophoblasts. Immunoglobulins are too large to diffuse across this cellular layer so the trophoblastic cells actively transport immunoglobulins from the maternal to the fetal circulation. Active transport of maternal IgG is mediated by surface receptors that are specific for the Fc portion of free IgG but not for IgM, IgE, or IgA. Active transport sometimes results in higher antibody titers in umbilical cord blood than in maternal blood. (Active transport mechanisms are discussed in Chapter 1.)
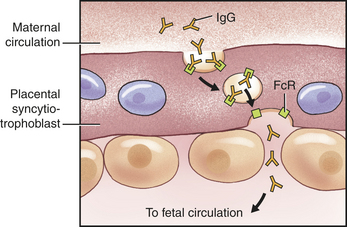
Figure 7-29 Transport of IgG across the syncytiotrophoblast. The human placenta is covered with a specialized multinucleated cell, the syncytiotrophoblast. Transport of maternal IgG across the syncytiotrophoblast and into the fetal circulation is an active process. Maternal IgG binds to Fc receptors on the surface of the syncytiotrophoblast and is internalized by the process of endocytosis. Receptors on the syncytiotrophoblast are specific for the Fc portion of IgG and do not bind other classes of immunoglobulins. Interaction of IgG with Fc receptors protects the antibody from lysosomal digestion during transport of the vacuole across the cell (i.e., transcytosis). On the fetal side of the syncytiotrophoblast, IgG is released by exocytosis (see Chapter 1).
At birth, total IgG levels in the umbilical cord are near adult levels (Figure 7-30). When the source of maternal antibodies is severed at birth, antibody titers in the newborn begin to drop as maternal antibody is catabolized. Thus antibody titers drop rapidly as the neonate’s production of IgG is beginning to rise. The rate of catabolism is usually more rapid than the rate of production so that the total immunoglobulin levels reach a minimum at 5 to 6 months in the normal child, occasionally causing transient hypogammaglobulinemia (insufficient quantities of circulating immunoglobulins). Many normal infants experience recurrent mild respiratory tract infections at this age.
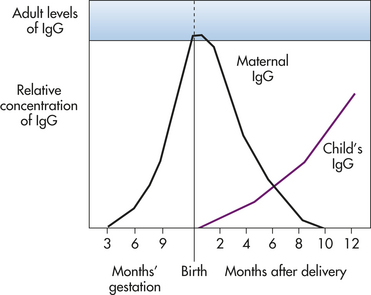
Figure 7-30 Antibody levels in umbilical cord blood and in neonatal circulation. Early in gestation maternal IgG begins crossing the placenta and enters the fetal circulation as shown in Figure 7-29. At birth, the fetal circulation may contain nearly adult levels of IgG, which is almost exclusively from the maternal source. The fetal immune system has the capacity to produce IgM and small amounts of IgA before birth (not shown). After delivery, maternal IgG is rapidly catabolized and neonatal IgG production increases.
Aging and Immune Function
Immune function decreases in old age as a result of changes in both lymphocyte function and relative lymphocyte populations. Individuals older than 60 years of age generally exhibit decreased T cell activity as demonstrated by laboratory assays of T-cell function,62 as well as in vivo reductions in cell-mediated responses to infections. The thymus, where T cells begin their development, reaches its maximum size at sexual maturity and then begins involuting until thymic size is only 15% of its maximum by middle age. Thymic capacity to mediate T-cell differentiation decreases with this atrophy. Although the total number of circulating T cells does not decrease with age, there is a shift in the populations of T-cell subtypes.
B-cell function is also altered with age as shown by decreases in specific antibody production in response to antigenic challenge, with concomitant increases in circulating immune complexes and in circulating autoantibodies (antibodies against self-antigens). A decrease in the number of circulating memory B cells is also observed.
Active acquired immunity (active immunity) 220
Adaptive (acquired) immunity (immune response) 217
Agglutination 244
Allergen 222
Antibody-dependent cell-mediated cytotoxicity (ADCC) 250
Antibody titer 244
Antigen-binding fragment (Fab) 224
Antigen presentation 226
Antigen-presenting cell (APC) 226
Antigen processing 236
Antigenic determinant 221
Attenuated virus 244
B lymphocyte (B cell) 218
B-cell receptor (BCR) 222
B-cell receptor (BCR) complex 226
Carrier 222
CD (cluster of differentiation) 221
Cellular immunity 220
Central tolerance 222
Class-switch 242
Clonal selection 230
Complementary-determining region (CDR) 225
Crystalline fragment (Fc) 224
Cytotoxic T lymphocyte (Tc cell or CTL) 247
Epitope 221
Framework region (FR) 225
Generation of clonal diversity 230
Haplotype 227
Hapten 222
Helper T cell (Th cell) 219, 237
Hinge region 224
High endothelial venule (HEV) 235
Human leukocyte antigen (HLA) 226
Humoral immunity 219
Immunity 217
Immunocompetent 218
Immunogen 221
Immunogenic 221
Immunoglobulin 218
Invariant chain 236
Isotype-switch 242
Lymphocyte 218
Lymphoid stem cell 230
Major histocompatibility complex (MHC) 226
MHC class I gene 226
MHC class II gene 226
Memory cell 220
Memory T cell 243
Neutralization 244
Opsonin 246
Opsonization 246
Paratope 221
Passive acquired immunity (passive immunity) 220
Plasma cell 240
Peripheral tolerance 222
Precipitation 244
Primary (central) lymphoid organ 230
Primary immune response 241
Secondary (anamnestic) immune response 241
Secondary (peripheral) lymphoid organ 230
Secretory (mucosal) immune system 246
Secretory immunoglobulin 247
Self-antigen 222
Somatic recombination 230
Superantigen (SAG) 244
Systemic immune system 246
T-cell receptor (TCR) 222
T-cell receptor (TCR) complex 226
T lymphocyte (T cell) 218
Th1 cell 239
Th2 cell 239
Th17 cell 239
T regulatory (Treg) cells 243
Tolerance 222
Valence 225
REFERENCES
1. Chaplin, D.D. Overview of the human immune response. J Allergy Clin Immunol. 2006;117(2 Suppl Mini-Primer):S430–S435.
2. Kalia, V., et al. Differentiation of memory B and T cells. Curr Opin Immunol. 2006;18(3):255–264.
3. Casadevall, A., Dadachova, E., Pirofski, L.A. Passive antibody therapy for infectious diseases. Nat Rev Microbiol. 2004;2(9):695–703.
4. Porter, R.R. The hydrolysis of rabbit γ-globulin and antibodies with crystalline papain. Biochem J. 1959;73:119–126.
5. Vargas-Madrazo, E., Paz-Garcia, E. An improved model of association for VH-VL immunoglobulin domains: asymmetries between VH and VL in the packing of some interface residues. J Mol Recognit. 2003;16(3):113–120.
6. Dal Porto, J.M., et al. B cell antigen receptor signaling 101. Mol Immunol. 2004;41(6-7):599–613.
7. Krogsgaard, M., Davis, M.M. How T cells “see” antigen. Nat Immunol. 2005;6(3):239–245.
8. Morris, C.R., et al. Association of intracellular proteins with folded major histocompatibility complex class I molecules. Immunol Res. 2004;30(2):171–179.
9. Immunogenetics database. Available at www.ebi.ac.uk/imgt/hla/stats.html. Accessed June 11, 2008.
10. Lawton, A.P., Kronenberg, M. The third way: progress on pathways of antigen processing and presentation by CD1. Immunol Cell Biol. 2004;82(3):295–306.
11. Barral, D.C., Brenner, M.B. CD1 antigen presentation: how it works. Nat Rev Immunol. 2007;7(12):929–941.
12. Metcalf, D. Hematopoietic cytokines. Blood. 2008;111(2):485–491.
13. Jerne, N.K. The natural-selection theory of antibody formation. Proc Natl Acad Sci U S A. 1955;41:849–857.
14. Burnet, F.M. The clonal selection theory of acquired immunity. London: Cambridge University Press; 1959.
15. Kyewski, B., Derbinski, J. Self-representation in the thymus: an extended view. Nat Rev Immunol. 2004;4(9):688–698.
16. von Boehmer, H. Selection of the T-cell repertoire: receptor-controlled checkpoints in T-cell development. Adv Immunol. 2004;84:201–238.
17. Bosselut, R. CD4/CD8-lineage differentiation in the thymus: from nuclear effectors to membrane signals. Nat Rev Immunol. 2004;4(7):529–540.
18. Ciofani, M., Zúñiga-Pflücker, J.C. A survival guide to early T cell development. Immunol Res. 2006;34(2):117–132.
19. Fuentes-Panana, E.M., Bannish, G., Monroe, J.G. Basal B-cell receptor signaling in B lymphocytes: mechanisms of regulation and role in positive selection, differentiation, and peripheral survival. Immunol Rev. 2004;197:26–40.
20. Chowdhury, D., Sen, R. Regulation of immunoglobulin heavy-chain gene rearrangements. Immunol Rev. 2004;200:182–196.
21. Verkoczy, L.K., Martensson, A.S., Nemazee, D. The scope of receptor editing and its association with autoimmunity. Curr Opin Immunol. 2004;16(6):808–814.
22. Pabst, O., et al. Elucidating the functional anatomy of secondary lymphoid organs. Curr Opin Immunol. 2004;16(4):394–399.
23. Klein, U., Dalla-Favera, R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8(1):22–33.
24. Trombetta, E.S., Mellman, I. Cell biology of antigen processing in vitro and in vivo. Ann Rev Immunol. 2005;23:975–1028.
25. Steinman, R.M. Dendritic cells: understanding immunogenicity. Eur J Immunol. 2007;37(Suppl 1):S53–S60.
26. Watts, C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol. 2004;5(7):685–692.
27. Savina, A., Amigorena, S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev. 2007;219:143–156.
28. Jiang, H., Chess, L. Regulation of immune responses by T cells. N Engl J Med. 2006;354(11):1166–1176.
29. Kidd, P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8(3):223–246.
30. Ochoa, J.B., Makarenkova, V. T lymphocytes. Crit Care Med. 2005;33(12 Suppl):S510–S513.
31. McGeachy, M.J., Cua, D.J. Th17 cell differentiation: the long and winding road. Immunity. 2008;28(4):445–453.
32. Ouyang, W., Kools, J.K., Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28(4):454–467.
33. Sigal, L.H. CD4+ T-cell subsets of probable clinical consequence. J Clin Rheum. 2007;13(4):229–233.
34. Dong, C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8(5):337–348.
35. Jenner, E. An inquiry into the causes and effects of the variolae vaccinae: a disease discovered in some of the western counties of England, particularly Gloucestershire, and known by the name of the cow pox. London: Sampson Low, 1798.
36. Eyler, J.M. Smallpox in history: the birth, death, and impact of a dread disease. J Lab Clin Med. 2003;142(4):216–220.
37. MacConmara, M., Lederer, J.A. B cells. Crit Care Med. 2005;33(12 Suppl):S514–S516.
38. Chaudhuri, J., Alt, F.W. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4(7):541–552.
39. Cerutti, A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8(6):421–434.
40. van der Merwe, P.A., Davis, S.J. Molecular interactions mediating T cell antigen recognition. Ann Rev Immunol. 2003;21:659–684.
41. Petersson, K., Forsberg, G., Walse, B. Interplay between superantigens and immunoreceptors. Scand J Immunol. 2004;59(4):345–355.
42. Baker, M.D., Acharya, K.R. Superantigens: structure-function relationships. Int J Med Microbiol. 2004;293(7-8):529–537.
43. Silverman, G.J., Goodyear, C.S. Confounding B cell defenses: lessons from a staphylococcal superantigen. Nat Rev Immunol. 2006;6(6):465–475.
44. Delves, P.J., Roitt, I.M. The immune system. I. N Engl J Med. 2000;343(1):37–49.
45. Delves, P.J., Roitt, I.M. The immune system. II. N Engl J Med. 2000;343(1):108–117.
46. Nimmerjahn, F., Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nature Rev Immunol. 2008;8(1):34–47.
47. Brandtzaeg, P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine. 2007;25(30):5467–5484.
48. Zacharia, B., Sherman, P. Atopy, helminths, and cancer. Med Hypotheses. 2003;60(1):1–5.
49. Anthony, R.M., et al. Protective immune mechanisms in helminth infection. Nat Rev Immunol. 2007;7(12):975–987.
50. Waterhouse, N.J., et al. Cytotoxic lymphocytes; instigators of dramatic target cell death. Biochem Pharmacol. 2004;68(6):1033–1040.
51. Bottino, C., et al. Learning how to discriminate between friends and enemies, a lesson from natural killer cells. Mol Immunol. 2004;41(6-7):569–575.
52. Vivier, E., et al. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510.
53. Lanier, L.L. NK cell recognition. Ann Rev Immunol. 2005;23:225–274.
54. Martinez, F.O., et al. Macrophage activation and polarization. Front Biosci. 2008;13:453–461.
55. Egen, J.G., et al. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28(2):271–284.
56. Bernhagen, J., et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and artherogenic cell recruitment. Nat Med. 2007;13(5):587–596.
57. Sakaguchi, S., Wing, K., Miyara, M. Regulatory T cells—a brief history and perspective. Eur J Immunol. 2007;37(Suppl 1):S116–S123.
58. Matarese, G., De Rosa, V., La Cava, A. Regulatory CD4 T cells: sensing the environment. Trends Immunol. 2008;29(1):12–17.
59. TretJiang, H., Chess, L. An integrated view of suppressor T cell subsets in immunoregulation. J Clin Invest. 2004;114(9):1198–1208.
60. D’Ambrosio, D. Regulatory T cells: how do they find their space in the immunological arena? Semin Cancer Biol. 2006;16(2):91–97.
61. Simister, N.E. Placental transport of immunoglobulin G. Vaccine. 2003;21(24):3365–3369.
62. Hakim, F.T., et al. Aging, immunity and cancer. Curr Opin Immunol. 2004;16(2):151–156.