INFECTION



Modern healthcare has shown great progress in preventing and treating infectious diseases through the success of public health initiatives, vaccination programs, and the use of antibiotics. In developed countries death from infections is most common among those with debilitating diseases, nutritional deficiencies, or immunosuppression. Death from influenza and pneumonia and from sepsis were the eighth and tenth leading causes of death in the United States in 2006 estimates (see Table 30-1). Infectious disease remains a significant threat to life in many parts of the world, including India, Africa, and Southeast Asia (Table 9-1), although the advent of sanitary living conditions, clean water, uncontaminated food, vaccinations, and antimicrobials had improved the health of many. As a result of these initiatives, smallpox has been eradicated from the globe (the last reported case was in 1975 in Somalia), measles is almost eradicated in the Western Hemisphere, and many diseaes, such as tuberculosis and polio, are on the decline.
Table 9-1
Estimated Annual Number of Deaths by Cause Worldwide
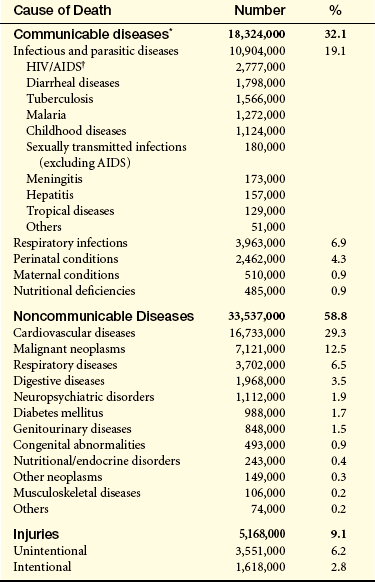
∗Communicable diseases, maternal and perinatal conditions and nutritional deficiencies
†Global data for human immunodeficiency syndrome/acquired immunodeficiency syndrome (HIV/AIDS) cases reported remain highly distorted for three reasons: (1) there are wide intercountry and interregional differences in the completeness of AIDS case detection and reporting, (2) reporting of AIDS cases to public health authorities and recognition of its importance have occurred in different countries at different times, and (3) pediatric AIDS remains substantially underrecognized and underreported.
From World Health Organization: The World Health Report 2004. Death by cause, sex, and mortality stratum in WHO regions, estimates for 2002, Annex Table 2. Available at www.who.int/whr/2004/annex/topic/en/annex_2_en.pdf.
Despite the widescale implementation of progressive public health and immunization policies infectious disease remains a significant cause of morbidity and mortality because of the emergence of previously unknown infections, the reemergence of old infections that were thought to be under control, and the development of infectious agents that are resistant to multiple antibiotics.1 The causes for these occurences are numerous and include the following:
• Vast and rapid urbanization in many areas of the world, resulting in a breakdown in public health programs and a more rapid spread of infection
• Poverty and social inequality
• Global travel, allowing more rapid spread of disease from isolated areas to virtually any point around the world in a few hours
• Globalization of the food supply
• Human encroachment into wilderness areas, resulting in contact with previously sequestered infectious agents
• Antibiotics that are prescribed excessively, that are not taken for a complete course of therapy, or that, even when appropriately used, result in the selection of antibiotic-resistant microorganisms
• Decreases in federal research budgets to study infectious disease
• Denial of a problem by governments, allowing infections to spread in an uncontrolled way
• Diminished use of effective insecticides
• Increased global warming, allowing insect vectors to spread into and breed in areas that were previously too cool for them
The emergence of previously unknown infections is not a new event in human history. However, the current rate may be unprecedented. Within one generation, more than 40 previously unknown infections have arisen and some examples are presented in Table 9-2. Several have extremely high mortality rates of more than 50% including severe acute respiratory syndrome (SARS) (in those older than 65 years), Ebola virus, Marburg virus, “mad cow” disease, Nipah virus (up to 75%), and acquired immunodeficiency syndrome (AIDS) (almost 100% in untreated persons). But most either spread very slowly (e.g., AIDS) or initially break out in relatively isolated areas and are effectively controlled by quarantine (e.g., Ebola virus). Although none of these infections has developed into the worldwide scourges portrayed in movies “Andromeda Strain” and “I Am Legend,” the potential of reversion to more rapidly spreading variants is a concen of public health agencies worldwide.

Concurrently, the incidence and spread of at least 20 previously known infections are increasing. A new strain of cholera that arose in Indonesia in 1961 has spread to Africa and in 1991 to South America. Malaria, dengue fever, and yellow fever are reemerging in areas where they had been eliminated or were unknown. The incidence of tuberculosis is increasing in countries that had reported declines and has risen by almost one third between the mid-1980s and early 1990s. Diphtheria has reemerged as a major health issue in Russia. In 1994 plague was reported in India after being dormant for a generation. War has led to outbreaks of Marburg hemorrhagic fever during civil war in Angola during 1975-2002 and cholera in the Democratic Republic of the Congo among Rwandan refugees in 1994; about 50,000 refugees died from a combination of cholera and shigella dysentery. The spread of cholera, yellow fever, and epidemic meningococcal disease has rebounded. Decreased insect control programs that were previously successful have led to spread of vector-borne diseases: African trypanosomiasis, dengue hemorrhagic fever, and malaria. Although the United States is relatively free of most of these diseases, the effects of global warming and relaxed control of vectors may result in resurgence. It should not be forgotten that in 1793 a yellow fever outbreak in Philadelphia killed 2000 of the city’s 55,000 inhabitants and forced the U.S. government to abandon the city until the outbreak ceased.
Many common and reemerging infections have become antibiotic and drug resistant. Streptococcus pneumoniae, a common cause of otitis media, pneumonia, and bacteremia, has been treated routinely and successfully with penicillin. Now at least 25% of isolates are penicillin resistant, and some are resistant to multiple antibiotics. Multiple antibiotic-resistant forms of Staphylococcus aureus, a primary cause of infections of wounds, surgical incisions, and catheters, are endemic in some hospitals. Some forms of this microorganism once were sensitive only to a single antibiotic, vancomycin, and now have become vancomycin-resistant. Antimicrobial resistance is now routinely observed in tuberculosis, diarrheal diseases, hospital-acquired infections, malaria, meningitis, respiratory tract infections, sexually transmitted infections (STIs), and human immunodeficiency virus (HIV).
Added to this collage of microbiologic dangers is the rising risk of bioterrorism. Agents such as smallpox, anthrax, and plague are continuing threats to public health and safety. All healthcare providers should have information about the characteristics and clinical manifestations of these biologic agents. The theoretical threat of bioterrorism became real in 2001 when letters containing anthrax were mailed; 22 individuals became infected and 5 died.
MICROORGANISMS AND HUMANS: A DYNAMIC RELATIONSHIP
For many microorganisms the human body is a hospitable site in which to grow and flourish because of its sufficient nutrients and appropriate conditions of temperature and humidity. In many cases a symbiotic relationship exists, in which both humans and microorganisms benefit (Box 9-1). These microorganisms make up the normal flora—the resident microorganisms found in different parts of the body, including the skin, mouth, gastrointestinal tract, respiratory tract, and genital tract.2 For instance, the normal bacterial flora of the human gut are provided with nutrients from ingested food and in exchange produce enzymes that facilitate the digestion and use of many of the more complex molecules found in the human diet, produce antibacterial factors (e.g., bacteriocins, colicins) that prevent colonization by pathogenic microorganisms, and produce usable metabolites (e.g., vitamin K, B vitamins). This beneficial homeostasis is normally maintained through the physical integrity of the gut and other mechanisms that sequester these microorganisms on the mucosal surface.
MICROORGANISMS AND INFECTIONS
The symbiotic relationship with the normal flora can be breached as a result of injury that compromises the physical protective barriers. Damage to the intestinal tract releases intestinal bacteria into the bloodstream, potentially leading to sepsis, shock, and death. Cuts in the skin may allow normally noninfectious bacteria (e.g., S. aureus) to cause local infections (e.g., abscesses, boils) and invade further and infect various organs. Symbiosis is also maintained by the immune and inflammatory systems. If those systems are compromised, many microorganisms will leave their normal sites and cause infection elsewhere in the body. Individuals with immune deficiencies easily become infected with opportunistic microorganisms, which normally would not cause disease but seize the opportunity to do so when a person’s defensive systems are weakened or suppressed (see Chapter 8).
Unlike opportunistic infectious agents, true pathogens have devised means to circumvent the individual’s defenses (discussed in Chapters 6 and 7) and directly cause infection. Successful infection with theses agents is usually dependent on adequate numbers of microorganisms rather than compromise of the host’s defenses.
From the perspective of the microorganisms that cause disease, the infectious process undergoes four separate stages of progression: colonization, invasion, multiplication, and spread.
Colonization
Infectious microorganisms usually exist in reservoirs, such as the environment (e.g., contaminated water, soil), animals, or another human who is infected or in a noninfectious state within the individual’s normal flora. An individual may obtain an infectious microorganism from a reservoir by several means.
Infections contracted from animal reservoirs (zoonotic infections) may be transmitted by direct contact (e.g., transmission of the rabies virus through bites) or indirectly by means of vectors (e.g., insects). Mechanical vectors (e.g., housefly) passively transfer microorganisms from a contaminated site to an individual.3 Biologic vectors (e.g., fleas, lice, mosquitoes, ticks) transmit infectious microorganisms through bites and stings. Individuals can also become infected by direct exposure to contaminated materials, such as fecal-oral transmission through food or water (e.g., salmonella food poisoning, cholera, hepatitis A infection, polio, rotavirus infection) or soil (e.g., tetanus).
Human-to-human transmission may occur through aerosolized microorganisms in droplets (e.g., produced by coughing or sneezing), which is the primary means of transmission for respiratory infections (e.g., agents that cause the common cold, influenza, streptococcal sore throat, bacterial meningitis). Other infectious agents require physical contact (e.g., sexual, blood transfusion, direct contact with contaminated clothes or bandages, entrance through wounds or openings in the skin). Direct contact is usually required for transmission of STIs, hepatitis B virus, cytomegalovirus (CMV), herpes simplex virus, or warts. Some microorganisms have the capacity to spread from mother to child across the placenta (e.g., Treponema pallidum, Listeria monocytogenes, cytomegalovirus [CMV], Toxoplasma gondii), ascending the birth canal or during delivery (e.g., group B Streptococcus, Escherichia coli, Chlamydia trachomatis, Neisseria gonorrhoeae, hepatitis B virus, HIV, Candida albicans), or through the breast milk (e.g., S. aureus). This is classified as vertical transmission, whereas spread from one person to another is horizontal transmission. In a very few instances, the infectious agent may develop airborne transmission.4 Effective airborne transmission may occur by prolonged presence of aerosolized droplets released by respiration, exercise, or other bodily activity and has been observed with SARS, tuberculosis, Norovirus, and smallpox.5
After deposition in receptive environments for colonization, the microorganism stabilizes the adherence to the tissue through specific surface receptors.6 For instance, infectious agents that cause respiratory infections specifically bind to molecules found on respiratory epithelium. Adherence helps protect the microorganism from removal by mechanical nonspecific forces, such as coughing of respiratory mucus. The specificity of adherence results in a particular microorganism being limited to where infections can occur (tissue tropism) such as the confinement of common cold viruses to producing respiratory infections.
Invasion
Once colonization has occurred the infectious agent can invade the surrounding tissue and, in many cases, other sites in the individual. Successful infectious agents have developed mechanisms to penetrate the tissues and evade the host’s nonspecific and specific defenses (inflammation and immunity). Many of these are discussed throughout this chapter.
Multiplication
Within the warm and nutrient-filled environment of human tissue most microorganisms undergo rapid multiplication with production of many new infectious progeny. Viral pathogens replicate within infected cells, and some bacteria are intracellular pathogens and replicate in macrophages and other cells. Many extracellular bacteria and fungi form multicellular masses called biofilms, which provides an optimal environment for growth.7
Spread
Many successful pathogens produce localized infections without spread to other regions of the body (e.g., Vibrio cholerae). Others, however, are highly invasive and may enter the lymphatics, blood, and internal organs. Successful spread relies on a variety of virulence factors, including adhesion molecules, toxins, and protection against the individual’s inflammatory and immune systems. Fungi, in particular, are opportunistic. In an individual with an intact immune system, the microorganism remains localized, whereas the infection may rapidly spread if the individual’s immune or inflammatory systems are compromised.
Clinical Infectious Disease
From the perspective of the individual who is infected, the clinical process occurs in four distinct stages:
• Incubation period—the period from initial exposure to the infectious agent and the onset of the first symptoms, during which the microorganism has entered the individual, undergone initial colonization, and begun multiplying, but is yet in insufficient numbers to cause symptoms; this period may last from several hours to years
• Prodromal stage—the occurrence of initial symptoms, which are often very mild and include a feeling of discomfort and tiredness
• Invasion period—the pathogen is multiplying rapidly, invading farther and affecting the tissues at the site of initial colonization as well as other areas; the immune and inflammatory responses are being triggered; development of symptoms specifically related to the pathogen and symptoms related to the ongoing protective inflammatory response
• Convalescence—in most instances, the individual’s immune and inflammatory systems have successfully removed the infectious agent, and symptoms decline; alternatively the disease may be fatal or may enter a latency phase with resolution of symptoms until reactivation at a later time
Clinical manifestations of infectious disease vary, depending on the pathogen, the organ system affected, and the severity. Effects of infection may be acute, chronic, secondary to the immune and inflammatory responses, or a consequence of bacterial toxins or viral injury. Manifestations can arise directly from the infecting microorganism or its products; however, the majority of manifestations result from the host’s inflammatory and immune responses. Infectious diseases typically begin with the nonspecific or general symptoms of fatigue, malaise, weakness, and loss of concentration. Generalized aching and loss of appetite are common complaints. However, the hallmark of most infectious diseases is fever.
Fever is not failure of the body to regulate temperature; rather, body temperature is being regulated at a higher level than normal. Body temperature is regulated by nervous system feedback to the hypothalamus, which functions as a central thermostat (see Chapter 15). A large number of agents (pyrogens) can produce fever. In current classification, those pyrogens derived from outside the host are termed exogenous pyrogens and those produced by the individual are termed endogenous pyrogens. There is little evidence that exogenous pyrogens cause fever directly. Such pyrogens indirectly affect the hypothalamus through endogenous pyrogens released by cells of the host. A number of cytokines have been identified as endogenous pyrogens. They are interleukins 1 and 6 (IL-1 and IL-6), interferon (IFN), tumor necrosis factor (TNF), and others (see Figure 15-8). These cytokines seem to raise the thermoregulatory set point through stimulation of prostaglandin synthesis and turnover in both thermoregulatory (brain) and non-thermoregulatory (peripheral) tissue. These mechanisms are discussed in detail in Chapter 15. Although it is generally believed that fever has a beneficial value in infection, the molecular mechanism behind the beneficial effects has not been established. Many investigators, however, consider fever as an adaptive host-defense response.
Several factors influence the capacity of a pathogen to cause disease.
• Communicability: The ability to spread from one individual to others and cause disease: measles and pertussis spread very easily, HIV is of lower communicability
• Immunogenicity: The ability of pathogens to induce an immune response
• Infectivity: The ability of the pathogen to invade and multiply in the host
• Mechanism of action: How the microorganism damages tissue
• Pathogenicity: The ability of an agent to produce disease—success depends on communicability, infectivity, extent of tissue damage, and virulence
• Portal of entry: Route by which a pathogenic microorganism infects the host: direct contact, inhalation, ingestion, or bites of an animal or insect
• Toxigenicity: The ability to produce soluble toxins or endotoxins, factors that greatly influence the pathogen’s degree of virulence
• Virulence: Capacity of a pathogen to cause severe disease—for example, measles virus is of low virulence; rabies virus is highly virulent
Infectious diseases are also classified by their prevalence and spread within the community.
• Endemic: Diseases with relatively high, but constant, rates of infection in a particular population
• Epidemic: The number of new infections in a particular population greatly exceeds the number usually observed
• Pandemic: An epidemic that spreads over a large area, such as a continent or worldwide (Table 9-3)

Classes of Infectious Microorganisms
Infectious disease can be caused by microorganisms that range in size from 20 nm (poliovirus) to 10 m (tapeworm). Classes of pathogenic microorganisms and their characteristics are summarized in Table 9-4 and discussed in detail in the following sections.

Bacterial Infection
Bacteria are prokaryotic unicellular microorganisms with no nuclei, mitochondria, or membrane-bound organelles. They are generally divided into several groups.
• “True bacteria” divide by binary fission and may have a variety of morphologies, including cocci (spherical), bacilli (rod shaped), vibrios (comma-shaped rods), or spirilla (twisted, rod shaped). Most disease-causing bacteria fall into this classification.
• Filamentous bacteria may have branching, mycelium-like structures that resemble fungi. Examples include the mycobacteria (Mycobacterium tuberculosis, Mycobacterium leprae) that cause tuberculosis and leprosy.
• Spirochetes are flexible spiral filaments that are motile. Most are anaerobic. Pertinent examples include Borrelia recurrentis (relapsing fever), T. pallidum (syphilis), and Borrelia burgdorferi (Lyme disease).
• Mycoplasma lack a rigid cell wall and are small and pleomorphic. They are the smallest and most simple members of the bacteria. Mycoplasma pneumoniae causes atypical pneumonia, and Mycoplasma genitalium is a suspected cause of urethritis and pelvic inflammatory disease.
• Rickettsia are strict intracellular parasites that can be rod-shaped, spherical, or pleomorphic. They are typically spread by insect vectors and cause Rocky Mountain spotted fever (Rickettsia rickettsii) and typhus (Rickettsia prowazekii).
• Chlamydia are also strict intracellular parasites, but with more complex intracellular life cycles. The primary chlamydial pathogen is Chlamydia trachomatis, which causes the most the most common bacterial sexual transmitted infection (pelvic inflammatory disease) and eye infections (conjunctivitis).
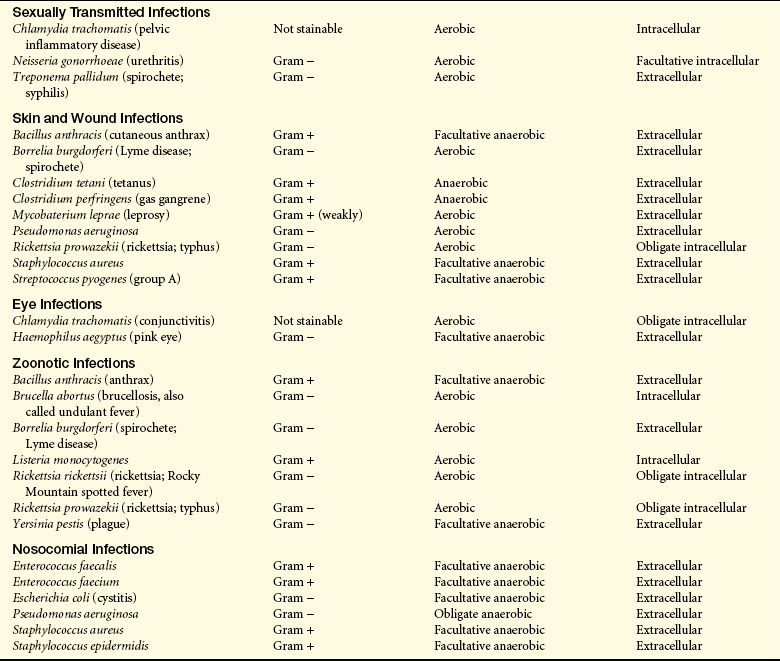
Bacteria are also categorized as gram-negative or gram-positive. Gram-negative bacteria do not retain crystal violet dye in the gram-staining process whereas gram-positive bacteria do retain crystal violet dye. Gram-negative bacteria also have a lipopolysaccharide (LPS) coat in the outer membrane which consists of lipid A, a core polysaccharide and O antigen. The LPS coat is also known as endotoxin (See Figure 9-1 and further discussion on page 301.)
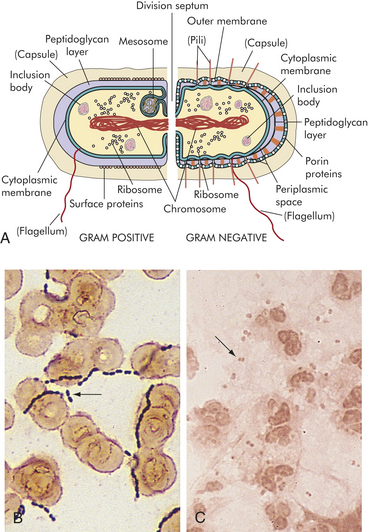
Figure 9-1 Gram-positive and gram-negative bacteria. A, The structure of the bacterial cell wall determines its staining characteristics with Gram stain. Gram-positive bacteria have a thick layer of peptidoglycan (left). Gram-negative bacteria have a thin peptidoglycan layer and an outer membrane of lipopolysaccharide (LPS) (right). B, Example of a gram-positive (darkly stained microorganisms, arrow) group A Streptococcus. This microorganism consists of cocci that frequently form chains. C, Example of a gram-negative (pink microorganisms, arrow) Neisseria meningitidis in cerebrospinal fluid. Neisseria form complexes of two cocci (diplococci).
Common bacterial pathogens are listed in Table 9-5.
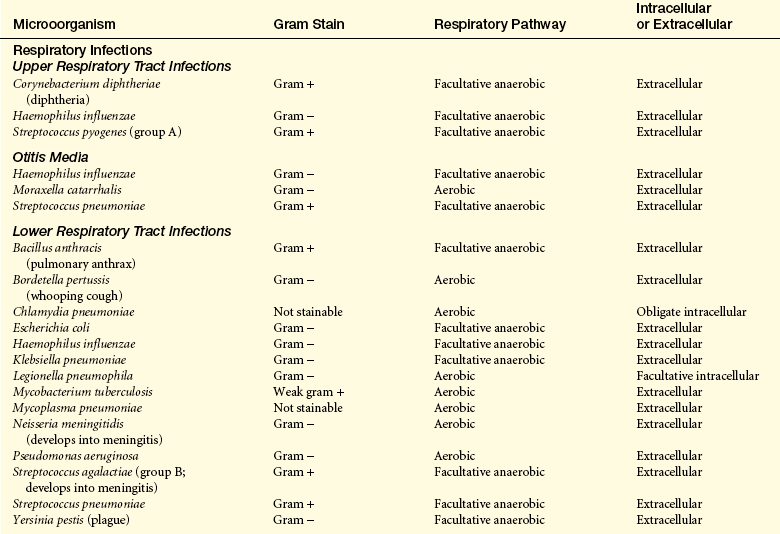
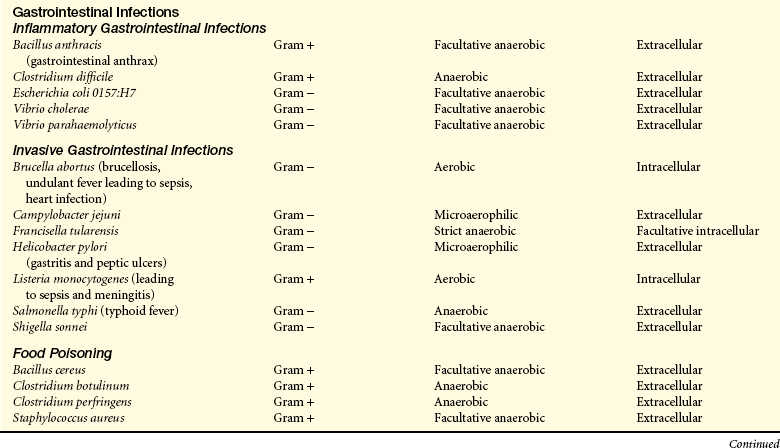
Transmission and Colonization: Transmission of bacterial infections occurs through the same routes generally described above. Many pathogenic bacteria normally reside in humans without causing disease. For instance, Streptococcus pyogenes (pharyngitis), S. pneumoniae (pneumonia, meningitis), Neisseria meningitidis (meningitis), and Haemophilus influenzae type b (meningitis). In most cases they are either present in inadequate numbers to cause disease or are effectively controlled by local protective mechanisms. Others reside in nature and cause infection after being ingested or entering wounds (e.g., V. cholerae [cholera] in contaminated water or Clostridium tetani [tetanus] in contaminated soil).
A large number require human-to-human contact, including several that are unstable on environmental surfaces (e.g., Bordetella pertussis [whooping cough], N. gonorrhoeae [gonorrhea], T. pallidum [syphilis]). Even the human-to-human spread of infectious bacteria contracted initially from environmental reservoirs can be facilitated by certain aspects of the disease, e.g., the explosive diarrhea of cholera. Chlamydia, which may be contracted as an STI, can spread from the mother to child during childbirth and may establish long-term intracellular persistence in the newborn.
The establishment of stable colonization requires adhesion.6 Many bacteria attach through pili (also called fimbriae), which are thin rod-like projections from the bacterial surface (see Figure 9-1 and Figure 9-2). Pathogenic strains of Escherichia coli involved in urinary tract infections have a variety of different specific pili-associated adhesion molecules, including mannose-binding protein that binds with glycoproteins specifically expressed on the bladder epithelium and PapG (pyelonephritis-associated protein) adhesin the binds to galactoses of the human P blood group. PapG is a specific adhesion for urinary tract epithelium, but PapG variants are specific for other cell types so that the particular PapG expressed will determine the preferred site of infection with E. coli. The particular adhesin may vary depending on growth conditions under which bacteria may undergo a “phase change” and shift from one type to another.
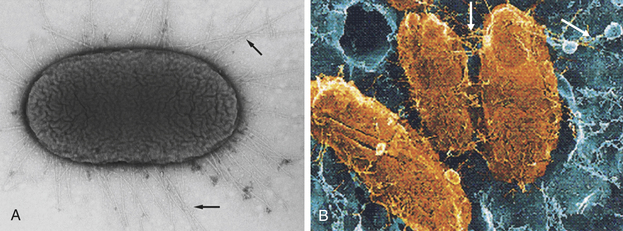
Figure 9-2 Attachment of Escherichia coli through pili. A, Transmission electron micrograph showing pili (arrows) of pathogenic E. coli. B, Scanning electron micrograph of E. coli (yellow) attached by pili (arrows) to bladder epithelium (blue). (A from Mandell G, Bennett J, Dolin R: Principles and practice of infectious diseases, ed 6, Philadelphia, 2005, Churchill Livingstone; B modified from Wein A et al: Campbell-Walsh urology, ed 9, Philadelphia, 2007, Saunders.)
Neisseria spp. bind to urinary tract epithelial cell membrane-associated cofactor protein (CD46), which is a receptor that regulates complement and protects cells by helping inactivate C3b and C4b. Several other microorganisms (e.g., B. pertussis, Legionella pneumophila, Mycobacterium tuberculosis) bind to the CR3 complement receptor (a receptor for C3b and C3b breakdown products) on the surface of monocytes/macrophages. B. pertussis expresses a surface hemagglutinin that recognizes CR3, whereas L. pneumophila and M. tuberculosis initially absorb inactivated C3b (C3bi) for use as an adhesin for CR3.
A variety of proteins and carbohydrates not associated with pili function as adhesion molecules. Flagella (used for motion) act as adhesions in V. cholerae. Hemagglutinins on B. pertussis, Salmonella spp., and Helicobacter pylori bind to erythrocyte surface molecules. Fibronectin is a common component of mucosal cell surfaces and is frequently used as a receptor. Several bacteria (S. pyogenes, S. aureus, T. pallidum) have developed specific adhesion molecules that recognize a particular amino acid sequence (Arg-Gly-Asp) in fibronectin. Many other bacteria have developed adhesion molecules that bind to collagen, laminin, and vitronectin, which are plentiful in connective tissue. A surface polysaccharide (poly-N-acetylglucosamine) is on nonencapsulated Staphylococcus spp., E. coli that infect the urinary system. Yersinia pestis, which causes plague, mediates adherence to the material in catheters and prosthetic devices, thus increasing the risk of infection of joint replacements and other implanted materials.
Invasion and Evasion: Invasion results in direct confrontation with the individual’s primary defense mechanisms against bacteria, which include the complement system, antibodies, and phagocytes, such as neutrophils and macrophages (see Chapters 6, 7, and 8). Bacterial survival and growth depend on the effectiveness of the body’s defenses and on the bacterium’s capacity to resist those defenses and obtain nutrients and multiply. Evasion of the body’s defense mechanisms may result in infectious microorganisms being transported in the blood (bacteremia) to infect other organs or even multiplying in the blood (sepsis).8
Efficient pathogens produce a variety of toxic molecules that may kill the individual’s cells, disrupt tissue, and protect against inflammation. Exotoxins are proteins released during bacterial growth. They are usually enzymes and have highly specific effects; they include cytotoxins, neurotoxins, pneumotoxins, enterotoxins, and hemolysins. Exotoxins can damage cell membranes, activate second messengers, and inhibit protein synthesis. For instance, a key component of invasion by N. meningitidis is a toxin that weakens intercellular adhesion between epithelial cells, thus allowing penetration into the underlying tissue. Pathogenic strains of streptococci and staphylococci produce hyaluronidase, lipases, and hemolysins that break down cells and intercellular matrix. Exotoxins are immunogenic and elicit the production of antibodies known as antitoxins. Consequently, vaccines are available for many of the exotoxins (i.e., tetanus, diphtheria, and pertussis).
Endotoxins are lipopolysaccharides (LPSs) contained in the cell walls of gram-negative bacteria and released during lysis (or destruction) of the bacteria (see Figure 9-1). The innermost part of the lipopolysaccharide, lipid A, is made of polysaccharides and fatty acids and is responsible for the substance’s toxic effects. Bacteria that produce endotoxins are called pyrogenic bacteria because they stimulate the release of inflammatory mediators, produce fever and the local and systemic effects of inflammation including septic shock (Figure 9-3).8 Endotoxin also may be released from the membrane of the bacteria, either during bacterial growth or during treatment with antibiotics. Therefore, antibiotics cannot prevent the toxic effects of the endotoxin.
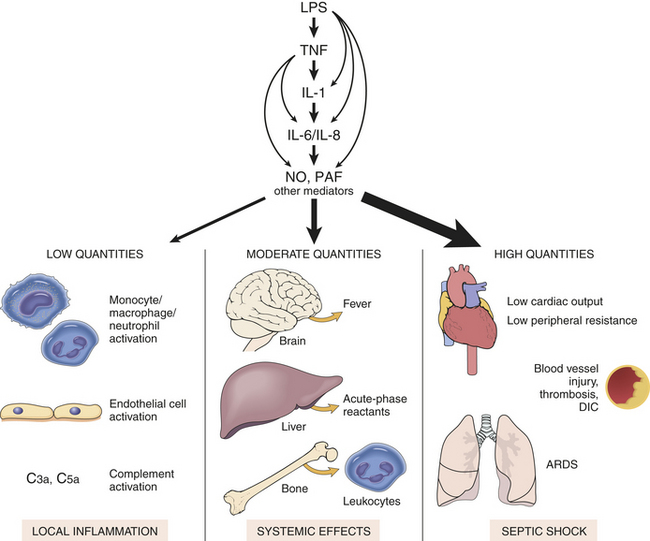
Figure 9-3 The many activities of lipopolysaccharide (LPS). Bacterial endotoxin (LPS) activates almost every aspect of inflammation. The release of LPS from gram-negative bacteria triggers successive waves of cytokine production, including tumor necrosis factor (TNF), interleukin-1 (IL-1), interleukin-6 (IL-6), and interleukin-8 (IL-8), and secondary mediators of inflammation, such as nitric oxide (NO) and platelet-activating factor (PAF). At low levels of LPS the effect is local. Moderate levels of LPS cause more systemic inflammatory responses. High levels of LPS may lead to septic shock and death. DIC, Disseminated intravascular coagulation; ARDS, acute respiratory distress syndrome. (From Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Evasion of the individual’s immune and inflammatory systems is multifaceted, and the most successful pathogens incorporate several mechanisms (Table 9-6).9


Rapid Division: Because the primary immune response may take 3 to 5 days to reach protective levels, some pathogens proliferate at rates that surpass the development of the immune system. Cholera causes severe vomiting and watery diarrhea, has a 60% mortality rate, and develops within 2 to 3 days of ingestion of the bacteria. Some strains of toxin-producing group A streptococci cause destructive skin infections and pneumonia that may kill an individual within 2 days. Group B streptococci from the maternal vagina may ascend the birth canal, penetrate fetal membranes, and infect the fluid surrounding the fetus. This microorganism may have already established an active infection of the child’s lungs by the time of birth, resulting in a pneumonia (50% mortality rate in newborns) that is too advanced to be treated successfully by antibiotics.
Intracellular Survival: Bacteria may hide from the immune response by growth in sites that are relatively poorly protected by immune cells, for example, V. cholerae in the gastrointestinal (GI) tract, and Salmonella typhi in the intestinal tract and biliary tract (gallbladder). Even asymptomatic people may undergo prolonged local colonization and shed infectious microorganisms in urine and feces, thus creating a carrier state.
Survival within cells (intracellular bacteria) affords a distinct advantage to some bacteria.10 Many intracellular bacteria can survive and even multiply in macrophages (Brucella, Listeria, M. leprae, M. tuberculosis) or in other cells (Yersinia, Shigella, Listeria, E. coli). Normally a macrophage would efficiently kill bacteria by fusing lysosomal granules with the phagosome to produce a phagolysosome and using oxygen-dependent and oxygen-independent mechanisms. Successful intracellular bacteria may block killing in several ways. Several microorganisms are resistant to killing and survive in the phagolysosome. Species of Salmonella secrete products that alter the environment of the phagolysosome. Production of catalase and superoxide dismutase (N. gonorrhoeae, Brucella abortus, S. aureus) destroys toxic oxygen products produced by the hexose-monophosphate-shunt. S. aureus also produces a cell-bound pigment (carotenoid) that “quenches” singlet oxygen. A second means of avoiding killing is escape from the phagosome. Several bacteria secrete lysins (e.g., hemolysin of Shigella, listeriolysin O and phospholipase C of L. monocytogenes, phospholipase A of Rickettsia spp.) that break down the phagosome membrane, and the bacteria are released into the cytoplasm, where they multiply.11 A third means of avoiding killing is by prevention of phagosome-lysosome fusion. M. tuberculosis and T. gondii produce toxins that prevent fusion so that the environment in the phagosome remains relatively nontoxic.
Protection Against Phagocytosis: Bacteria produce a large variety of toxins and extracellular enzymes, some of which kill phagocytic cells (e.g., Pseudomonas aeruginosa exotoxin A).12 Staphylococcal hemolysins and toxins from Bacillus anthracis and B. pertussis decrease phagocytic and chemotactic activities and may be toxic. Streptococcal products, such as streptolysin O, bind to cholesterol in the phagocyte’s plasma membrane and initiate destruction through the internal release of enzymes in lysosomal granules.13
Antiphagocytic capsules are expressed by most bacterial pathogens involved in pneumonia and meningitis (Figure 9-4). Capsules are mostly polysaccharide, although some very important proteins may be capsular components, which inhibit complement activation and phagocytosis. Such coatings inhibit phagocytosis and include the thick polysaccharide covering of the pneumococcus (S. pneumoniae), the waxy capsule surrounding the tubercle bacillus (M. tuberculosis), the polysaccharide “slime” capsule of P. aeruginosa, and the M protein of S. pyogenes. The M protein binds fibrinogen and fibrin and functions as an adhesin.
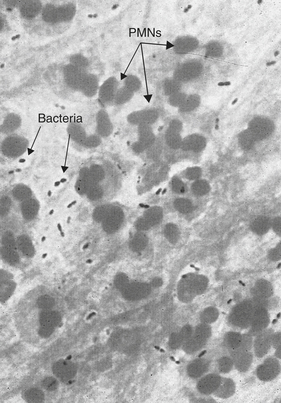
Figure 9-4 Bacterial capsule. Gram stain of a sputum sample from an individual with pneumococcal pneumonia (×1000 magnification). The sputum is rich in polymorphonuclear (PMN) cells (see arrow) and slightly elongated, gram-negative cocci (Streptococcus pneumoniae). Clear areas (arrows) around the bacteria indicate capsules. (Modified from Mandell G, Bennett J, Dolin R: Principles and practice of infectious diseases, ed 6, Philadelphia, 2005, Churchill Livingstone.)
Coating with Self-Protein: Some bacterial surface proteins (protein A of S. aureus, protein G of S. pyogenes) bind the Fc portion of the individual’s antibody, thus forming a protective coat of “self” protein. Binding through the Fc holds the antibody in an orientation that does not allow complement activation or phagocytosis. S. aureus produces a surface coagulase that induces fibrin clotting on the bacterial surface. T. pallidum coats itself with fibronectin. Capsules are produced that contain sialic acid (E. coli K-12), which closely resembles the sialic acid on the surface of most human cells, and hyaluronic acid (group A streptococci), which is the basic substance in connective tissue.
Antigenic Variation: Antigenic variation allows the pathogen to alter surface molecules that express antigens that are the targets of protective immune responses. Thus as the individual develops protective levels of antibodies, the pathogen responds by changing antigens and becoming resistant. The three primary mechanisms of antigenic variation are mutation, recombination, and gene switching. Antigenic variation can occur during the course of an infection in the host or during the spread of infection through the environment.
Neisseria use pili to adhere to epithelium, and antibody against these antigens can abrogate adherence. Neisseria undergoes phase shifts during which pilar antigens are changed by progressive silencing of one set of 10 or 11 available pili genes and activation of others that express different antigens. The spirochete B. recurrentis (relapsing fever) repeatedly relapses with spiking fevers as a result of as many as 10 episodes of antigenic changes over weeks or months. Many other bacteria have a large number of different antigenic types (serotypes) across the species. At least 80 different strains of group A S. pyogenes express different serotypes of the capsular M protein. S. pneumoniae has at least 100 different serotypes based on capsular polysaccharides. Multiple serotypes are also found in V. cholerae, S. aureus, E. coli, N. gonorrhoeae, and others. Although the particular serotype is stable on that bacterium, an immune response against one serotype will result in increased growth of a different serotype. Some gram-negative bacteria can revert to “rough” forms in which serotype-specific carbohydrates are deleted, thus becoming resistant to antibody and activation of complement.
Degradation of Immune Molecules: Protection is afforded by the breakdown of molecules of the immune or inflammatory system. An IgA protease produced by meningitis-causing microorganisms and other related bacteria (N. gonorrhoeae, N. meningitidis, H. influenzae, S. pneumoniae) cleaves IgA at the hinge region into ineffective Fc and Fab′2 regions. A staphylokinase produced by Staphylococcus spp. activates plasmin (resulting in the breakdown of clots) and degrades IgG and C3b of the complement system. Pseudomonas produces elastase (breakdown C3 of the complement system) and a 56-kDa protease (breaks down C5a).
Salmonella membrane protease degrades nonspecific antimicrobial molecules like defensins and cathelicidins. Salmonella can also alter surface lipid A by changing its fatty acid content to become resistant to small-molecular-weight antimicrobials.
Neutralization of Immune Molecules: Bacteria may spontaneously release surface molecules that bind to and neutralize antibody. These include endotoxin from gram-negative bacteria, capsular antigens from S. pneumoniae and N. meningitidis, and protein A from S. aureus. During infection the blood may contain high levels of complexes between antibody and bacterial antigen. At certain antigen-antibody ratios the complexes will deposit in the kidney, joints, and other target organs and initiate type III hypersensitivity reactions (Chapter 8).
Complement Evasion: Complement is a major component of the defense against bacterial infection through production of opsonin (C3b) and chemotactic factors (C3a, C5a) for neutrophils. Teichoic acid in the Gram-positive cell wall provides resistance against complement-mediated lysis.14 Staphylococcus produces proteins that inhibit complement activity, including C3 and C5 convertases, C5, C2, and the C5a receptor that mediates complement-induced chemotaxis.15 Bacterial regulatory proteins (e.g., Borrelia complement-regulator-acquiring protein, Neisseria porins, and members of the Streptococcus M protein family) affect complement activation, including destabilization of the C3 convertase complex or degradation of the opsonin C3b.
Immune Suppression: Although more prominent in viral infections, some bacterial pathogens can broadly suppress immune responses against their own antigens as well as other antigens unrelated to the infectious agent. Chronic bacterial infections, like leprosy (M. leprae) and tuberculosis (M. tuberculosis) induce anergy (suppressed response to multiple antigens) in infected hosts. H. pylori can release LPS that binds to dendritic cells and blocks development of Th1 cells, as well as produce toxins that block the T cell IL-2 receptor signaling pathway, thus inhibiting maturation of Th cells. N. gonorrhoeae pili antigens bind to adhesion molecules on CD4+ cells and prevent their activation.
Tissue Damage: Bacterial infections damage tissue either directly by means of bacterial products or indirectly as a result of inflammation.12 Many toxins break down tissue directly. These include a large variety of proteases, lipases, hyaluronidase, hemolysins, and many others discussed above. Some bacterial diseases arise completely from the systemic effects of toxins. Diphtheria is a respiratory tract infection (Corynebacterium diphtheriae) with systemic complications caused by toxin that inhibits messenger ribonucleic acid (mRNA) translation and causes low blood pressure, necrosis in the heart and liver, degeneration of myelin sheaths, and death. Gastrointestinal pathogens (V. cholerae, Salmonella, Shigella, E. coli) may produce enterotoxins that are cytotoxic and alter the permeability of intestinal cells leading to diarrheal diseases.
Clostridia are anaerobic bacteria that produce some of the most powerful toxins known. One type of food poisoning is caused by Clostridium botulinum that produces a paralytic neurotoxin (botulinum toxin) that blocks the release of acetylcholine at nerve-muscle synapses and results in flaccid paralysis. Infection of deep wounds with C. tetani results in release of a neurotoxin that causes severe muscle spasms, spastic paralysis of the voluntary muscles, and potentially death. C. perfringens produces multiple toxins that cause gas gangrene. One of these, alpha toxin, is a lecithinase that destroys the infected tissue by digesting cellular plasma membranes. Infection of the colon with Clostridium difficile results in watery diarrhea from toxins that damage the mucosa.
Bacterial superantigens are toxins that increase the adherence between major histocompatibilitiy complex (MHC) class II proteins on antigen-presenting cells and the T-cell receptor. Because the effect is independent of antigen, a large population of T cells is activated and overproduces proinflammatory cytokines, such as IL-1, IL-6, and TNF-α. Superantigens are responsible for several diseases, including food poisoning (enterotoxins of S. aureus and C. perfringens), toxic shock syndrome (toxic shock syndrome toxin [TSST] of S. aureus), and scarlet fever (erythrogenic toxin of S. pyogenes).
Inflammation is the body’s initial response to the presence of the bacteria.16 Vascular permeability is increased, allowing blood-borne substances (e.g., the complement system) involved in bacterial destruction to access the site of infection. The release of anaphylatoxins (C5a and C3a) of the complement cascade may increase the inflammatory response and lead to an increase in capillary permeability sufficient to permit the escape of large volumes of plasma, contributing to hypotension and, in severe cases, cardiovascular shock (see Chapter 46). Many persistent bacterial infections (e.g., M. tuberculosis) may lead to formation of granulomas, which diminishes the function of the affected organ (e.g., the lung).17
The release of a sufficient amount of endotoxin can lead to fatal endotoxic shock (septic shock), which is one of the leading causes of death in intensive care units.18 The usual cause is proliferation of gram-negative bacteria, although shock may be caused by a few gram-positive bacteria and fungi. Once in the blood, endotoxins cause the release of vasoactive peptides and cytokines that affect blood vessels, producing vasodilation, which reduces blood pressure, causes decreased oxygen delivery, and produces subsequent cardiovascular shock (see Figure 9-3 and Chapter 46). Endotoxin can activate the coagulation cascade, leading to the syndrome of disseminated (or diffuse) intravascular coagulation (see Chapter 27). Additionally, endotoxin induces the release of TNF-α (cachectin) by macrophages. TNF is a potent proinflammatory cytokine that is also called cachectin because of its role in promoting cachexia in individuals with cancer. (Cachexia is discussed in Chapter 11; cytokines are discussed in Chapters 6 and 7.)
Example of Bacterial Pathogenesis: S. aureus has become a major cause of hospital-acquired (nosocomial) infections.19 This microorganism is a common commensal inhabitant of normal skin and nasal passages (about 30% of individuals are nasal carriers) and can be transmitted by direct skin-to-skin contact or contact with shared items or surfaces that have become contaminated from someone else’s infection (e.g., towels, used bandages).
Skin infections may occur at sites of trauma, such as cuts and abrasions, and at areas of the body covered by hair (e.g., back of neck, groin, buttock, armpit, beard area of men). Most infections are relatively mild and localized, appearing as red and swollen pustules on the skin, containing pus or other drainage (Figure 9-5). They can develop into abscesses, boils, carbuncles, cellulitis, or furunculosis. Invasive disease may originate from wound infections (e.g., trauma, surgical wounds, indwelling medical devices, prosthetic joints) and lead to fatal septicemia and abscesses in internal organs (e.g., lungs, kidney, bones, skeletal muscle, meninges, or heart).
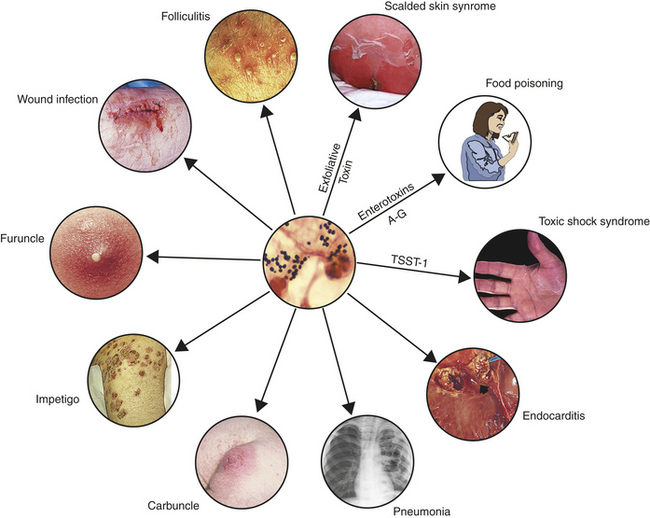
Figure 9-5 Staphylococcus aureus infections. Different strains of S. aureus (gram-positive cocci in sputum from an individual with pneumonia [center photograph]) cause a variety of infections. The particular infection may depend on the toxin produced: exfoliative toxin (scalded skin syndrome), enterotoxins A-G (food poisoning), or toxic-shock syndrome toxin-1 (TSST-1; Toxic shock syndrome, carbuncle, impetigo, and wound infection photos from Cohen J and Powderly WG: Infectious diseases, ed 2, St. Louis, 2004, Mosby. Folliculitis photo from Goldman L and Ausiello D: Cecil medicine, ed 23, Philadelphia, 2008, Saunders. Center photo and photos of food poisoning and endocarditis from Kumar V et al: Robbins & Cotran Pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders. Furuncle photo from Long S et al: Principles and practice of pediatric infectious diseases, ed 3, Philadelphia, 2009, Saunders. Scalded skin syndrome and pneumonia photos from Mandell G et al: Principles and practice of infectious diseases, ed 6, Philadelphia, 2005, Churchill Livingstone.)
Adherence to tissue is mediated by surface proteins that attach to connective tissue (laminin, fibrin, fibronectin) and endothelium. Attachment to collagen occurs in osteomyelitis and septic arthritis-causing strains, and capsular polysaccharide mediate attachment to prosthetic devices.
Staphylococci also produce very effective polysaccharide capsules that protect against phagocytosis as well as surface protein A that binds IgG by the Fc portion and masks Sbacterial antigens under a surface of self-proteins.20 Many pathogenic strains have increased resistance to intracellular oxidative killing when engulfed by a phagocyte.
Invasion is mediated by a variety of toxins, including membrane-damaging toxins (α-toxin, which forms pores in membranes; hemolysin, which destroys erythrocytes; β-toxin, which is a sphingomyelinase; δ-toxin, a detergent-like toxin; leukocidin, which lyses phagocytes), coagulase (causes clots), staphylokinase (breaks down of clots), exfoliatin toxins (causes separation of epidermis resulting in scalded skin syndrome), lipase (degrades lipids on skin surface; facilitates abscess formation), and a variety of enterotoxins.21 Many of the enterotoxins are superantigens that are responsible for staphylococcal food poisoning with diarrhea and vomiting and toxic shock syndrome. Each infectious strain of S. aureus produces a few of these toxins so that strains may differ in their capacities to cause particular diseases, thus different strains can cause purulent dermal infections, food poisoning, or toxic shock syndrome.
Antibiotic resistance has become a major problem with S. aureus.19 For several decades pathogenic strains have commonly produced β-lactamase, an enzyme that destroys penicillin. More recently staphylococci have developed resistance (methicillin-resistant Staphylococcus aureus [MRSA]) to broad spectrum antibiotics, including methicillin-like antibiotics, which were widely used to treat penicillin-resistant microorganisms.
Fungal Infections
Fungi are eukaryotic microorganisms with thick rigid cell walls and the capacity to form a variety of complex structures (Figure 9-6). Fungi may grow as a mold with branched filaments or a meshwork mycelium structure (e.g., Aspergillus spp., causing aspergillosis), yeast with ovoid or spherical shapes (C. albicans, which causes candidiasis), or dimorphic with a yeastlike appearance in tissue and mycelium in culture (e.g., Histoplasma capsulatum, which causes histoplasmosis, a systemic respiratory disease). The cell wall is composed of polysaccharides that differ from the peptidoglycans of bacteria and are thus are resistant to bacterial cell wall inhibitors such as penicillin and cephalosporin. In contrast to bacteria, the cytosol of fungi contains organelles: mitochondria, Golgi apparatus, microtubules, microvesicles, endoplasmic reticulum, and nuclei. Molds are aerobic, and yeasts are facultative anaerobes. Common pathologic fungi are summarized in Table 9-7.
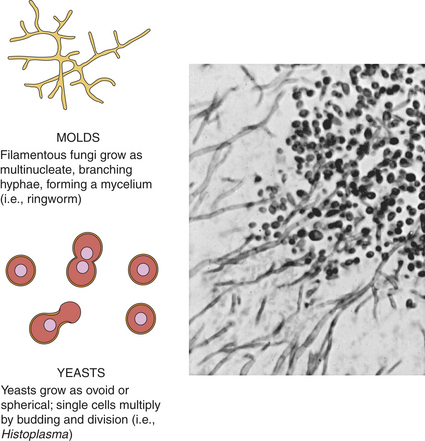
Figure 9-6 Morphology of fungi. Fungi may be either mold or yeast forms, or dimorphic. The photograph shows Candida albicans with both the mycelial and the yeast forms. (A from Mims CA et al: Medical microbiology, ed 3, London, 2004, Mosby; B from Townsend C et al: Sabiston textbook of surgery, ed 18, Philadelphia, 2008, Saunders.)
Fungi are diagnosed by microscopic observation of specimens treated with potassium hydroxide and stained to enhance visualization of spheres and filaments. Specimens also can be cultured. Skin tests are available for species of Aspergillus. Many of the antifungal drugs (e.g., amphotericin B, ketoconazole, fluconazole) used to treat deep or systemic infections are toxic to the host because the fungal cell composition is similar to the human cell.
Transmission and Colonization: Infection with a fungus is called mycosis. Most pathogenic fungi (e.g., H. capsulatum, Coccidioides immitis, Blastomyces dermatitidis) grow as saprophytes in the environment and are transmitted by inhalation or contamination of wounds. The majority of medically relevant fungi either exist as human commensals or cause relatively mild infections (superficial mycoses) of the skin, nails, hair, and mucous membranes of the mouth and vagina. These include dermatophytes (e.g., tineas, which refers to several skin mycoses including ringworm, athlete’s foot, and others) or yeasts (e.g., Candida, Aspergillus, Cryptococcus).
Human-to-human transmission is only a concern with dermatophyte infections. Systemic mycosis caused by pathogenic fungi generally results from inhalation of spores present in a contaminated environment and initially presents as a pulmonary infection. Infection that disseminates to other organs can be life threatening. Systemic mycosis caused by opportunistic fungi is usually secondary to immunosuppression caused by genetic defects, infections such as HIV, cancer, and drugs used to prevent transplant rejection.
Specific adherence to epithelium is provided by several polysaccharides on the fungal surface. Glucan, mannan, glycoprotein, and chitin molecules adhere with host receptors, including Toll-like receptors (TLRs), mannose receptors, dectin-1, and cadherins, respectively. A cell wall adhesion molecule, agglutinin-like sequence (ALS3), on several fungi (e.g., C. albicans) promotes adherence to epithelial cells as well as silicone, thus facilitating infection of implants and other medical devices.
In 2006, the opportunist pathogen Pneumocystis carinii was reclassified as a fungus, and the specific variant that infects humans was renamed Pneumocystis jiroveci.22 Despite the official change in nomenclature, much of the literature will continue to use P. carinii for some time. As with many fungi and protozoa, Pneumocystis has two life-cycle forms: a trophic form and the cyst form. Two surface proteins, glycoprotein A (gpA) and major surface glycoprotein (MSG), mediate attachment to alveolar epithelial cells.
Invasion and Evasion: The host defense against fungal infection includes the fungistatic properties of neutrophils and macrophages. T lymphocytes are crucial in limiting the extent of infection and producing cytokines to further activate macrophages. Human host defense against the inhaled spores begins with the mucous layer and the ciliary action in the respiratory tract, which then remove the fungus or facilitate phagocytosis by local macrophages. Pathogenic fungi have developed means to circumvent these mechanisms (see Table 9-6).
Intracellular Survival: Fungal virulence and resistance to the individual’s protective mechanisms is a complex process that requires the expression of multiple genes at different stages and different sites of infection. Pathologic fungi are generally dimorphic (e.g., H. capsulatum, B. dermatitidis, C. immitis) and readily adapt to the host environment, responding to temperature variations, low oxygen environment, more alkaline pH, and other conditions in the host tissue by undergoing changes in morphology and switching from avirulent mold forms to virulent yeast forms. Similar conditions also trigger yeasts (e.g., C. albicans) to switch from the yeast form to its more virulent hyphal form.
The more virulent morphologies allow fungi to survive in macrophages after phagocytosis. After phagocytosis by macrophages, the yeast form of Histoplasma replicates in phagosomes and phagolysosomes. Some yeast may produce proteins that inhibit the activity of lysosomal proteases. The macrophage may either be destroyed as a result of membrane modification by the fungus, or in the case of fungi like Histoplasma, harbor the fungus in granulomas. Breakdown of the granulomas over time may result in release of viable fungi and recurrence of the infection.
Protection Against Phagocytosis: Encapsulated yeast cells (e.g., Cryptococcus neoformans) are more resistant to phagocytosis than unencapsulated yeast. The cryptococcal polysaccharide capsule is antiphagocytic by blocking recognition by macrophages and may also be immunosuppressive by inhibiting migration of leukocytes into the site of fungal infection. Aspergillus fumigatus and many other fungi produce toxic metabolites (e.g., gliotoxin) that inhibit macrophage and neutrophil phagocytosis. Molecules like gliotoxin may also be immunosuppressive, including suppression of mast cell activation, degranulation, and secretion of leukotrienes and cytokines.
Antigenic Variation: Altered antigen expression affords protection against the developing immune responses, although this defense strategy is rarely used by fungal pathogens. Pneumocystis contains approximately 80 different gpA and MSG genes, only one of which is expressed at a time.23 Modulation of these antigens may provide resistance against immune destruction.
Immunosuppression: Several yeasts stimulate the production of immunosuppressive cytokines, resulting in down-regulation of some aspects of the host’s immune response. The yeast C. neoformans suppresses inflammation by inhibiting production of the proinflammatory cytokines TNF-α and IL-12 and inducing production of the anti-inflammatory cytokine IL-10. The overall result is suppression of macrophage function and protection against killing.
Tissue Damage: Fungal infections damage tissue directly by secretion of enzymes and indirectly by initiating an inflammatory response. Secreted enzymes, such as proteases, phospholipases, and elastases, damage cells and intercellular matrix, leading to necrosis. Many molds secrete mycotoxins when grown in environmental locations, such as on nuts, beans, and grains. Ingestion of this toxin affects muscle coordination, causes tremors, and may be fatal. Some fungal toxins may cause cancer; aflatoxins produced by some Aspergillus are especially carcinogenic.24
The typical immune and inflammatory reaction against fungal infections involves a cell-mediated response with infiltration of T cells and macrophages, as well as neutrophils.16 The inflammatory site is rich in proinflammatory cytokines. Fungal infections can be very difficult for these systems to eradicate, leading to progressively increasing production of cytokines. Thus, as the host’s response increases so do the destructive effects on surrounding healthy tissue. In cases of persistent infection, granulomas form and compromise the normal function of the infected tissue.
Example of Fungal Pathogenesis: Candida albicans is the most common cause of fungal infections in humans. It is an opportunistic yeast that is a commensal in the normal flora of many healthy individuals, residing in the skin, gastrointestinal tract, mouth (30% to 55% of healthy individuals), and vagina (20% of healthy women) and normally under the control of local defense mechanisms, including members of the bacterial flora that produce antifungal agents.25 In healthy individuals, particularly those whose normal flora has been disturbed by antibiotic therapy (e.g., diminished levels of Lactobacillus in the vaginal flora), Candida overgrowth may occur resulting in vaginitis or oropharyngeal infection (thrush). In those with an intact immune system the infection remains localized.
In immunocompromised individuals, particularly those with diminished levels of neutrophils (neutropenia), disseminated infection may occur.26 Candida is the most common fungal infection in people with cancer (particularly acute leukemia and other hematologic cancers), transplantation (bone marrow and solid organ), and HIV/AIDS. Almost 90% of people with AIDS have Candida at least one time during the disease, although infection is usually contained locally (thrush or vaginitis) because of adequate numbers of neutrophils (Figure 9-7). Invasive candidiasis may also be secondary to indwelling catheters, intravenous lines, or peritoneal dialysis, which provides direct entrance into the blood.
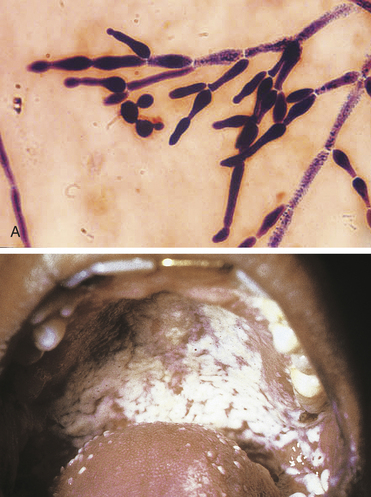
Figure 9-7 Candida albicans. Gram stain (upper photograph) of sputum showing chains of elongated budding yeasts of C. albicans producing pseudohyphae (×1000). Oral candidiasis (thrush) (lower photograph). (A from McPherson R, Pincus M: Henry’s clinical diagnosis and management by laboratory methods, ed 21, Philadelphia, 2006, Saunders; B from Mandell G, Bennett J, Dolin R: Principles and practice of infectious diseases, ed 6, Philadelphia, 2005, Churchill Livingstone.)
Disseminated candidiasis may involve several internal organs, including abscesses in the kidney, brain, liver, and heart, and is characterized by persistent or recurrent fever, gram-negative shocklike symptoms (hypotension, tachycardia), disseminated intravascular coagulation (DIC), and death.27 The mortality rates of sepsis or disseminated candidiasis are in the range of 30% to 40%.
Like most pathogenic yeasts, Candida undergoes morphologic changes from unicellular yeast to filamentous hyphal forms under conditions of neutral to alkaline pH, elevated temperature, and host serum factors. Switching of morphology results in altered profiles of surface antigens and increased resistance to immunologic destruction as well as altered tissue adhesion specificity.28 Candida expresses diverse surface adhesion molecules that permit adherence to materials in implants, epithelium, extracellular matrix (fibronectin, laminin, collagen), and leukocytes and facilitate invasion into tissue. One surface adherence molecule is a glycoprotein, INT1p (integrator complex subunit 1), which specifically binds to integrins. EAP1 (enhanced adhesion to plastic) is a glycoprotein that permits Candida to bind to epithelial cells and a variety of synthetic materials found in implants.
Candida secretes several enzymes that function as virulence factors and contribute to tissue destruction.27 Acid proteases and phospholipases damage the cell membrane and enhance invasive capacity. Secreted aspartic proteinases (Saps) increase the virulence of Candida by destroying cell membranes and removing surface molecules, including from cells of the immune and inflammatory systems.
Candida may suppress the host’s immune system. Production of the cytokine granulocyte-monocyte colony-stimulating factor (GM-CSF), suppresses monocyte/macrophage function, including the production of components of the complement cascade. Decreased production of C3 results in less opsonization (C3b) and production of chemotactic activity (C3a) for phagocytes.
Parasitic and Protozoan Infections
Parasitic organisms establish symbiosis with another species in which the parasite benefits at the expense of the other species. Parasites range from unicellular protozoan to large worms. Parasitic worms (helminths) include intestinal and tissue nematodes (e.g., hookworm, roundworm), flukes (e.g., liver fluke, lung fluke), and tapeworms. A protozoan is a eukaryotic, unicellular microorganism with a nucleus and cytoplasm. Pathogenic protozoa include malaria (Plasmodium), amoebae (e.g., Entamoeba histolytica, which causes amoebic dysentery), and flagellates (e.g., Giardia lamblia, which causes diarrhea; Trypanosoma, which causes sleeping sickness). Although less common in the United States, parasites and protozoa are common causes of infections worldwide, with a significant effect on the mortality and morbidity of individuals in developing countries. Important parasites of humans are listed in Table 9-8.
Transmission and Colonization: Parasitic and protozoal infections are rarely transmitted from human to human. The predominant means is through vectors in which the organism spends part of its life cycle. Examples include the transmission of malaria (Plasmodium spp.) by mosquitoes, trypanosomes (Trypanosoma cruzi, which causes Chagas disease in South America; Trypanosoma brucei, which causes sleeping sickness in Africa) by the tsetse fly, and Leishmania spp. by sand fleas. Many of the protozoal infectious agents (e.g., E. histolytica, G. lamblia) are encountered in contaminated water or food and transmission is by ingestion.
The initial attachment depends on whether the microorganism is injected into the bloodstream by a vector or whether entrance is through the gastrointestinal tract. Microorganisms in the bloodstream frequently have surface lectins that react with carbohydrates on specific cells. Malarial parasites attach to erythrocytes that express Duffy blood group antigens. Thus Duffy-negative individuals are resistant to malaria. T. cruzi can infect both CD4- and CD8-postive T cells through a T-cell surface receptor. Several parasites express surface glycoproteins that facilitate preferential entrance into monocytes/macrophages using various receptors, including the receptors for complement component C3b (Leishmania spp.). Leishmania expresses a surface glycoprotein (gp63) that is necessary for its entrance into macrophages. The gp63 molecule binds complement components (C3b, C3bi) and uses the macrophage complement receptors. E. histolytica expresses two surface proteins that react with the disaccharide galactose/N-acetylgalactosamine on epithelium.
Invasion and Evasion: An effective immune response varies depending on the particular parasite or protozoan. Intercellular pathogens are susceptible to cell-mediated immunity and activation of macrophages by T cells. Other organisms are more sensitive to antibody, particularly used in antibody-dependent cell-mediated cytotoxicity (ADCC) by macrophages and natural killer (NK) cells. Many helminth infections are sensitive to damage by eosinophils, which are attracted by IgE-mediated mast cell degranulation and release of eosinophil chemotactic factor of anaphylaxis (ECF-A). Evasion of the individual’s defenses is accomplished by several means (see Table 9-6).29
Intracellular Survival: Leishmania spp. are obligative intracellular parasites of monocytes/macrophages.30 They are protected from being killed in the phagosome and phagolysosome in which they multiply. Protection may be afforded by a surface protein that inhibits fusion of lysosomes to the phagosome, thus decreasing the amount of degradative enzymes within the phagolysosome. Toxoplasma spp. may be protected by entrance into a variety of cells, including macrophages.31 The probability of survival is increased by inhibiting fusion of lysosomes with the phagosome. Many of the intracellular organisms are sensitive to macrophage activation by T cells. T. cruzi bypasses the effects of macrophage activation by escaping from the phagosome and growing in the macrophage cytoplasm.
Protection Against Phagocytosis: Because many unicellular parasites survive within macrophages, the development of antiphagocytic capsules does not seem to be a major protective strategy. In some cases, a surface glycocalyx may function in a similar fashion as capsules to mask surface antigens from binding antibody. Some organisms produce toxins to protect themselves against phagocytosis; E. histolytica releases phospholipase and pore-forming proteins that disrupt the phagocyte’s plasma membrane.
Coating with Self-Proteins: Pathogens that coat themselves with human proteins may be disguised and “fool” the immune system. Schistosomes and trypanosomes mask their antigens by absorbing IgG by the Fc portion of the molecule.
Antigenic Variation: In general, those organisms that undergo part of their life cycle in humans or may assume multiple morphologic forms will also undergo antigenic changes related to the stage in the life cycle or morphology. Some protozoa have developed very complex alterations in surface antigens using gene switching. Infection with African trypanosomes may lead to fatal neurologic disease. During infection protective antibody is produced against surface antigens called variable surface glycoproteins (VSGs). An IgG or IgM response will successfully kill most of protozoa. However, a small percentage will undergo a gene shift that results in a change in VSG. The existing antibodies will not recognize the new VSG, and the level of schistosomes will rebound, causing recurrent disease. The schistosome has genetic information (about 10% of the genome) for hundreds of variations in VSG, which are expressed one at a time. Thus the organism undergoes periodic shifts in VSG expression, resulting in periodicity of parasites in the blood. A similar process occurs in some helminths (Trichinella spiralis).
Degradation of Immune Molecules: If a microorganism is sensitive to a particular component of the immune or inflammatory system, a survival advantage is obtained by production of enzymes that specifically degrade that component. Schistosomes secrete an enzyme that diminishes the effectiveness of IgG by specifically removing a critical peptide. E. histolytica secretes a protease that can degrade IgG and IgA.
Neutralization of Immune Molecules: Large parasitic organisms may release large amount of soluble antigen to neutralize antibody.
Complement Evasion: Complement activation is effective against several parasites.14 Some parasites (e.g., Echinococcus spp., Leishmania spp.) produce complement regulatory proteins that affect complement function by destabilizing C3 convertase or promoting degradation of C3b. Trypanosomes produce complement regulatory factors, including a surface gp63-like molecule, that confer resistance to complement. E. histolytica produces complement regulatory factors that inactivate C3a and C5a, thus inhibiting phagocyte chemotaxis activity.
Immune Suppression: Parasites may produce both pathogen-specific and nonspecific immune suppression.32 The release of large amounts of soluble antigen may induce specific tolerance to the pathogen as well as induce dysfunctional macrophage antigen processing. The function of immune cells is directly blocked by secretion of cytotoxic molecules (T. spiralis), selective inhibitors of T-cell function (schistosomes), or inducers of polyclonal B-cell activation (B-cell mitogen released by trypanosomes).
E. histolytica induces T-cell hyporesponsiveness by induction of IL-4 and IL-10 that down-regulate maturation of Th cells. The VSG molecules of African trypanosomes stimulate macrophages to overproduce TNF-α and CD8-positive cells to secrete high levels of IFN-γ, which impairs T-cell responses. Leishmania has developed an interesting approach to circumventing antigen processing by macrophages. This pathogen adsorbs IgG, which binds to the macrophage Fc receptor, resulting in the hyperproduction of IL-10 and suppression of IL-12 from infected macrophages. IL-10 prevents macrophage responses to IFN-γ, allowing the parasites to survive even in the immunologically intact individual.
Tissue Damage: Tissue damage may result directly by parasitic infestation in the tissue or be secondary to the individual’s immune and inflammatory responses. The particular process depends a great deal on burden of parasites infesting the site and sensitivity of the particular site to damage. Large infestations may lead to physical loss of function in a tissue or organ. For instance, a large number of intestinal parasites (e.g., the roundworm Ascaris lumbricoides, tapeworms, Giardia spp.) compete for and prevent uptake of nutrients, leading to various forms of malabsorption, blocked uptake of fats, or anemia from malabsorption of B12 or from large amounts of blood loss. Filarial parasites (e.g., Wuchereria bancrofti and Brugia malayi, which causes elephantiasis) block the lymphatics and cause accumulation of lymph in tissues. The larvae of tapeworms (e.g., Taenia solium) encyst in and prevent normal function of organs (e.g., muscle, liver, eye), which is particularly dangerous in the human brain.
Toxins released from the parasite may cause significant irreversible organ damage. Proteolytic enzymes from E. histolytica are very cytolytic leading to ulceration of intestinal walls, bloody diarrhea, amoebic dysentery, dehydration, and death in infants and young children. T. cruzi secretes a neurotoxin that affects the anatomic nervous system, a small-molecular-weight toxin that causes fever, and proteases and phospholipases, leading to tissue destruction.
The infected individual’s immune and inflammatory responses result in considerable histopathology.33 Schistosomiasis results in the deposition of eggs in organs (e.g., the liver), which leads to formation of granulomas and tissue destruction through fibrosis. Some parasitic products (T. brucei, malarial parasites) activate macrophages to overproduce cytokines, which leads to exacerbated inflammation.34 The IgE produced against parasitic worms is protective, but can also stimulate excessive degranulation of mast cells and even anaphylactic shock. Polyclonal B-cell activation leads to broad spectrum of autoantibodies that may precipitate autoimmune diseases. Overproduction of antigens by some pathogens (e.g., malaria, trypanosomiasis, schistosomiasis) may lead to excessive levels of pathogenic circulating immune complexes and initiation of type III hypersensitivity reactions (Chapter 8) in the kidneys and vasculature.
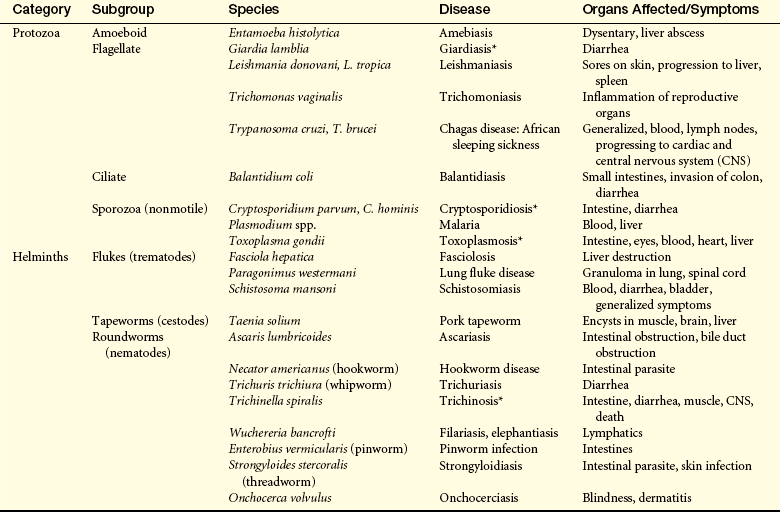
Example of Parasitic Pathogenesis: Malaria is one of the most common infections worldwide.35 Yearly an estimated 400 million cases occur, with about 1 million deaths. Four species infect humans: Plasmodium falciparum (accounts for most fatalities), Plasmodium vivax, Plasmodium ovale (both of which are more benign), and Plasmodium malariae. The four strains differ in disease severity and incidence of fever. Infection with P. falciparum, the most severe form, results in severe chills, high fever, sweating, headache, muscle pains, vomiting, severe anemia, pulmonary edema, and many other complications. Neurologic complications may result from infected red blood cells (RBCs) adhering to endothelium in capillaries of the brain. The individual may develop cardiovascular collapse, shock, coma, and death.
Transmission is through the bite of an infected female Anopheles mosquito, where the microorganism grows in the salivary gland. The infectious form enters the bloodstream, survives in the liver and invades parenchymal cells. After several rounds of division, the liver cell ruptures, and several thousand parasites enter the blood, where they infect red blood cells (Figure 9-8).36 Multiplication occurs in RBCs, resulting in the release of daughter parasites that reinfect other erythrocytes.
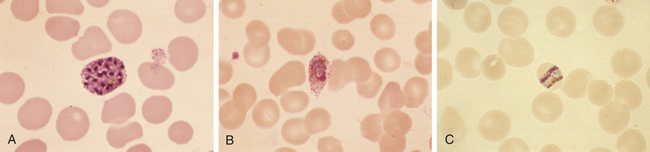
Figure 9-8 Malaria. Giemsa-stained smears. A, Plasmodium vivax schizont. B, Plasmodium ovale trophozoite. C, Characteristic band from trophozoite of Plasmodium malariae containing intracellular pigment hemozoin. (From Kliegman R et al: Nelson textbook of pediatrics, ed 18, St Louis, 2007, Saunders.)
P. vivax and P. ovale malaria can remain dormant in the liver for years, protected from the immune system by intracellular residence. The parasite in the blood avoids destruction by phagocytes in the spleen by expressing adhesion proteins that cause adherence and sequestration along the walls of the small vessels. Additionally, P. falciparum undergoes gene switching among about 60 different antigenic variants of antigens expressed on the surface of infected erythrocytes (P. falciparum erythrocyte membrane protein, PfEMP).37 Malaria induces a form of immune suppression. CD4 and CD8 T cells are diminished and lymphocytes from individuals infected with P. falciparum do not proliferate in the presence of PfEMP antigen in vitro.
Viral Infection and Injury
Viruses are extremely simple microorganisms and do not possess any of the metabolic organelles found in prokaryotes (e.g., bacteria) or eukaryotes (e.g., human cells). The basic viral structure (virion) consists of nucleic acid protected by a protein shell, the capsid. The capsid may take many characteristic shapes; helical, icosahedral, or large pleiomorphic (poxvirus) (Figure 9-9). Some viruses also have a protective envelope surrounding the capsid, which consists of the plasma membrane from the previously infected cell.
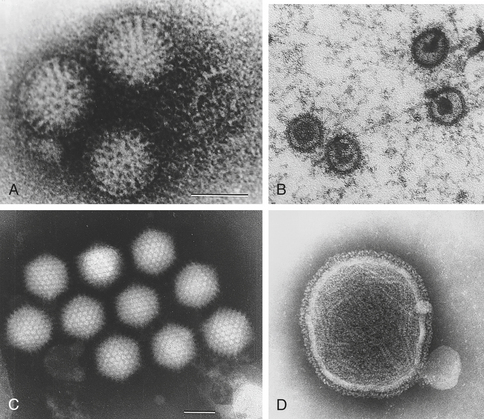
Figure 9-9 Electron micrographs of representative viral structures. A, Rotavirus: particles with a double shell and a characteristic “wheel-and-spoke” appearance. B, Epstein-Barr virus: icosahedral enveloped DNA virus. C, Adenovirus: particles with characteristic icosahedral structures. D, Paramyxovirus: spherical enveloped RNA virus. RNA is seen spilling out of the disrupted virus. (A and C from Kumar V et al: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders; B and D from Long S, Pickering L, Prober C: Principles and practice of pediatric infectious diseases, ed 3, Philadelphia, 2008, Churchill Livingstone.)
Viruses are classified by the format of nucleic acid in the virion, which may be RNA or deoxyribonucleic acid (DNA), and either single-stranded (ss) or double-stranded (ds), and whether the virus uses the enzyme reverse transcriptase (RT) for replication. Thus seven classifications are used: dsDNA (e.g., herpesvirus, smallpox virus), ssDNA (parvovirus), dsRNA (rotavirus), ssRNA +sense (+sense functions as mRNA) (e.g., hepatitis A and C viruses, SARS virus, poliovirus, rhinovirus), ssRNA −sense (e.g., Ebola virus, Marburg virus, Nipah, influenza virus, and viruses that cause measles, mumps, and rabies, hantavirus, Lassa virus), ssRNA +sense with RT (e.g., HIV), and dsDNA with RT (e.g., hepatitis B virus).
Viral diseases are the most common afflictions of humans and include the common cold, the “cold sore” of herpes simplex virus, several forms of hepatitis, HIV, and several types of cancer. Examples of human diseases caused by specific viruses are listed in Table 9-9.
Table 9-9
Human Diseases Caused by Specific Viruses
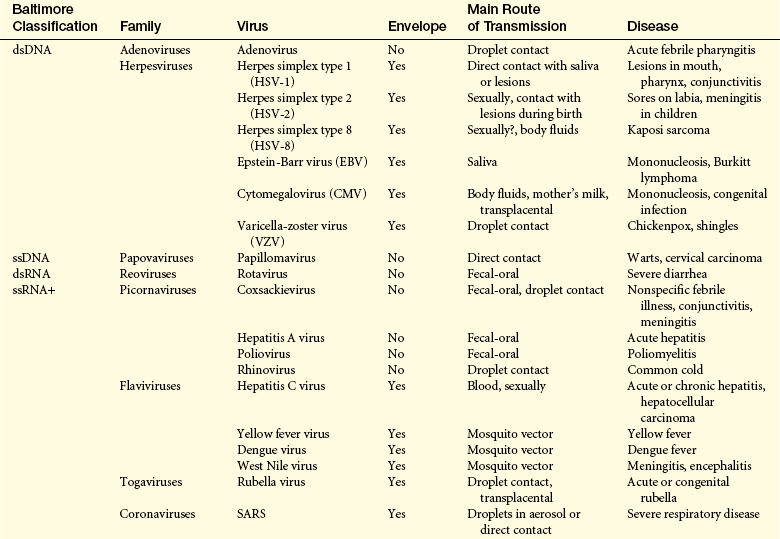

AIDS, Acquired immunodeficiency syndrome; DNA, deoxyribonucleic acid; ds, double-stranded; HIV, human immunodeficiency virus;
RNA, ribonucleic acid; RT, reverse transcriptase; SARS, severe acute respiratory syndrome; ss; single-stranded.
Transmission and Colonization: Viruses are obligatory intracellular parasites, thus transmission is usually from one infected individual to an uninfected individual or from an animal reservoir (zoonotic infection). Transmission may be direct or through a vector, such as mosquitoes. Human-to-human transmission may take many forms, including aerosols of respiratory fluids, contact with infected blood, or sexual contact.
The viral life cycle is completely intracellular and involves several steps: attachment to the target cell (determines host range and tropism), penetration (by endocytosis or membrane fusion), uncoating (release of viral nucleic acid from the viral capsid by viral or host enzymes), replication (synthesis of viral proteins and mRNA), assembly (formation of new virions), and release (by lysis, budding) (Figure 9-10).
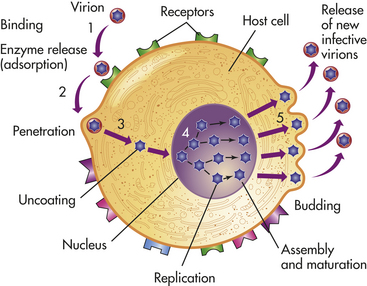
Attachment involves specific interactions between surface proteins on the virus and receptors on the cell to be infected.38 The specificity of the virus for these receptors and the distribution of receptors throughout the individual’s tissues dictate the range of host cells that a particular virus can infect. For example, HIV has a glycoprotein (gp120) that attaches to the CD4 molecule expressed on helper T cells, monocytes, and microglia. Rotavirus recognizes sialic acid and other constituents of epithelial junctions. Epstein-Barr virus (EBV, which causes mononucleosis) binds to complement receptor 2 (CR2) on B lymphocytes.
Once bound, the virion penetrates the plasma membrane by receptor-mediated endocytosis, by envelope fusion with the plasma membrane, or by directly crossing the plasma membrane.39 Within the cytoplasm the virus uncoats the protective nucleocapsid and releases viral genetic information. Most RNA viruses directly produce mRNA, which is translated into viral proteins, and genomic RNA, which is eventually packaged into new viruses. One particular family of viruses, retroviruses (e.g., HIV) carries an enzyme reverse transcriptase that creates a double-stranded DNA version of the virus. The DNA “provirus” enters the cell’s nucleus, where it becomes integrated into the host cell’s chromosomal DNA. DNA viruses also enter the nucleus and are transcribed into mRNA before protein translation. Some DNA viruses also may integrate into the infected cell’s chromosomal DNA.
The translation of viral-specific mRNA results in viral proteins that self-assemble. New virions are released from the cell for transmission of the viral infection to neighboring uninfected cells. Enveloped viruses are released through budding, in which shed viral particles are enveloped in the plasma membrane from the surface of the infected cell. Nonenveloped viruses commonly are released in large numbers concurrent with the destruction of the cell. Viral DNA that has become integrated with host DNA is transmitted to the daughter cells during mitosis. By this process, viral genes can become part of the genetic information of the cell and its progeny.
Invasion and Evasion: The primary defense mechanisms against viruses include antibody that prevents viral entrance into a cell and cellular immunity that recognizes antigenic changes on the surface of infected cells. Nonspecific defense includes production of α- and β-interferons that block intracellular viral replication. However, many viruses are highly successful pathogens and have developed a variety of mechanisms for bypassing immune rejection (see Table 9-6).9
Rapid Division: Some viruses, particularly those with small genomes, rapidly proliferate after the initial infection and by doing so produce a large number of virions more quickly than the immune system can develop. Norovirus virus and rotavirus (causes of severe diarrhea and vomiting) and Ebola virus, Marburg virus, and hantavirus (causes of hemorrhagic fever) have very short incubation periods. By the development of an effective adaptive immune response in 4 or 5 days, the virus has spread and caused severe clinical disease.
Intracellular Survival: As obligative intracellular pathogens, viruses hide within cells and away from normal inflammatory or immune responses. Viral agents that spread from cell to cell after the initial infection must encounter the immune response, which in most cases cures the infection. Thus most viral infections are self-limiting.
If a symbiotic relationship is maintained between the cell and the virus, persistent unapparent infection may result (latency). The infected cell will be functional, and the virus persists until it is activated to replicate (e.g., the recurrent coldsores of herpes simplex virus infection). Latent viruses usually possess latency-associated transcript (LAT) genes that control persistence indefinitely. Reactivation usually results from expression of lytic genes that lead to increased viral expression and destruction of the infected cell. Latency is characteristic of several chronic viral infections (e.g., HIV, hepatitis B and C viruses).
Coat with Self-Proteins: Enveloped viruses are the prime examples of how this mechanism may succeed. The viral capsid is completely surrounded by a cellular plasma membrane that is highly similar to that of an uninfected cell. Only a few critical differences exist. In order to infect another cell the viron expresses envelope proteins (SU and TM proteins) that are critical for the intercellular fusion process. These proteins are virus-specific and targets for protective immune responses.
Antigenic Variation: One of the classic examples of antigenic variation is influenza. This virus undergoes frequent antigen shifts and drifts, which are described in detail within the Example of Viral Pathogenesis section, later. Other viruses (e.g., HIV) add antigenic diversity by incorporating frequent functional translational errors. Some viral enzymes are designed to create small errors in reading mRNA leading to minor changes in the viral proteins. These changes are not at functionally critical sites, but may provide resistance to specific and nonspecific defense mechanisms.
In a manner similar to bacteria, some viruses have multiple stable antigenic serotypes. A person who recovers from an infection with one serotype may not have protective immunity against other serotypes of the same virus. At least 100 serotypes of rhinovirus can cause the “common cold,” which explains why individuals can catch many colds throughout their lives. HIV and hepatitis C virus also have multiple serotypes.
Neutralization of Immune Molecules: As with other pathogens, the secretion of large amounts of soluble viral antigen may lead to neutralization of antibody and formation of immune complexes. In infection such as hepatitis B significant levels of circulating immune complexes may form and be deposited in target tissues, such as the kidneys.
Some viruses also have the capacity to neutralize cytokines, such as IL-1 and TNF-α. Cells infected with vaccinia virus produce a protein that can bind to IL-1. These molecules are frequently called cytokine decoys.
Complement Evasion: Several viruses induce expression of regulators of complement activation.14 Cellular complement inhibitors are incorporated into the envelope of some viruses (e.g., HIV-1, vaccinia).15 Cytomegalovirus (CMV) induces complement inhibitors on the surface of infected cells. Herpes simplex virus expresses a cell surface protein (glycoprotein C-1) that binds and inhibits C3b, as well blocks the membrane attack complex.
Immune Suppression: HIV and other viruses have developed the capacity to infect and kill immune cells, thus protecting themselves, but also leading to a broad immunosuppression against other antigens. This process is discussed more thoroughly in the section on AIDS.
Some viruses have developed mechanisms for interfering with antigen processing and presentation by MHC class I molecules.40 Endogenous antigens, such as viral antigens, are normally degraded by proteasomes, transported by TAP proteins into the endoplasmic reticulum, and complexed with MHC class I molecules for expression on the cell surface and presentation to appropriate cells of the immune response (see Chapter 7). Many of the herpesviruses and retroviruses prevent steps in this process. For instance, Epstein-Barr virus inhibits degradation by the proteasomes. Herpes simplex virus can prevent binding of the antigenic peptide to MHC class I. Inhibition of antigen presentation by MHC class I prevents the generation of effective T-cell immune responses.
Human CMV has developed a unique modification of affecting MHC processing. NK cells are the principal defenders against tumor cells or virally infected cells that have down-regulated MHC expression and are therefore not recognized by T-cytotoxic cells. NK cell function is suppressed by targets with surface class I MHC molecules. CMV is capable of preventing antigen presentation by MHC class I and stimulates the expression of a MHC-like molecule that cannot present antigen but is recognized as a suppression signal by NK cells. Thus CMV-infected cells are protected from both T-cytotoxic cells and NK cell killing.
Tissue Damage: Once inside the infected cell, viruses may have many harmful effects, including the following:
• Cytopathic effects resulting from inhibition of cellular DNA, RNA, or protein synthesis, disruption of lysosomal membranes, resulting in release of “digestive” lysosomal enzymes that can kill the cell (cell lysis)
• Promotion of apoptosis of the cell
• Fusion of infected, adjacent cells, thereby producing multinucleated giant cells (herpesviruses and paramyxoviruses [measles virus, mumps virus, respiratory syncytial virus])
• Transformation of infected cell into cancerous cells, resulting in uninhibited and unregulated growth
• Alteration of the antigenic properties, or “identity,” of the infected cell, causing the immune system to attack the cell as if it were foreign16
Example of Viral Pathogenesis: Influenza is an ssRNA (−strand) with a segmented genome (seven or eight pieces of ssRNA) (Figure 9-11).4 It is transmitted through aerosols or body fluids and is highly infectious. The virions attach to respiratory epithelial cells and enter by endocytosis. Symptoms begin 1 to 4 days after infection and may include chills, fever, sore throat, muscle aches, severe headaches, coughing, weakness, generalized discomfort, nausea, and vomiting, and may lead to pneumonia; it can be fatal, particularly in young children and older adults.41 The normal rate of infectivity is about 5% to 15%, with a mortality rate of about 0.1%, and in most cases recovery occurs in 1 to 2 weeks. Yearly seasonal influenza outbreaks result in about 250,000 to 500,000 deaths worldwide.
Figure 9-11 Antigenic shifts in influenza virus. One theory proposes that antigenic shifts occur when a human influenza virus (blue) and an avian influenza virus (red) coinfect a species that is permissive for both. The 8 ssRNA strands are co-expressed in the same infected cell, resulting in mixing of the strands so that a hybrid virus can be produced. The hybrid virus indicated here contains all the genetic information of the original virus that infected humans, but contains a new hemagglutinin (HA)-containing stand from the avian virus. This virus expresses a new HA antigen and will be less susceptible to residual immunity that normally provides partial protection against yearly influenza infections.
The influenza virion expresses two surface proteins that are essential to virulence.41,42 The hemagglutinin (HA) protein is a glycoprotein that is necessary for entrance into cells by binding to glycan receptors, which are richly expressed on the surface of respiratory epithelium. The surface neuraminidase (NA) is an enzyme that is necessary for release of new virions from infected cells by cleaving cellular sialic acids (a common component of mammalian cell membranes).
Antibodies against the HA and NA antigens are responsible for protection against influenza infection. Infections are seasonal and protection gained from 1 year’s infection does not totally protect against influenza in the following year because the HA and NA antigens undergo yearly change.43 Usually antigenic variation is relatively minor (antigenic drift) and results from mutations. Individuals frequently have partial protection resulting from the previous year’s infection, which lessens the effects of the disease. Two groups of influenza virus, influenza A and influenza B, infect humans, and the yearly vaccine against it is a trivalent mixture of inactivated proteins from two influenza A subtypes and one influenza B subtype. Influenza B almost exclusively infects humans, mutates at a much lower rate than influenza A, and has reduced rate of antigenic change. Influenza B, however, has antigenically distinct subtypes based on HA (16 forms) and NA (9 forms) antigens and is infectious to birds and mammals. Currently subtypes H1N1, H1N2, and H3N2 are the primary causes of influenza worldwide.
Periodically influenza B undergoes major antigenic changes (antigenic shifts) (see Figure 9-11).41 Shifts occur in animals coinfected by a human and an avian strain of influenza. Because the genome is segmented, the segments can undergo recombination during which the human virus obtains a new HA or NA antigen. When such changes occur, previous protection may not exist, resulting in a major pandemic and much more severe disease. In 1918 an antigenic shift occurred in the western United States (first reported in Fort Riley, Kansas) resulting in an influenza A virus with an H1N1 serotype (called Spanish flu). The virus spread worldwide throughout 1918 and 1919. Symptoms were unusually severe, resulted from excessive production of cytokines, and included deaths from secondary bacterial pneumonia, massive hemorrhages, and pulmonary edema.44 Spanish flu possessed an extremely high rate of infectivity (up to 50%) and mortality (killed 20%
or more of those infected) and resulted in an estimated 40 million deaths worldwide. Strain variations due to antigenic drift occur within both HA and NA, and some may further affect virulence of that virus. The 1918 H1N1 serotype also possessed several mutations in the HA that resulted in a far greater virulence than the currently circulating H1N1 type. Studies have demonstrated that survivors of the 1918 pandemic still have protective levels of antibodies against that particular virus, confirming that protection against a particular influenza strain may be lifelong.
ACQUIRED IMMUNODEFICIENCY SYNDROME (AIDS)
The most notable form of secondary or acquired immune deficiency caused by an infectious agent is acquired immunodeficiency syndrome (AIDS).45 AIDS is a viral disease caused by the human immunodeficiency virus (HIV). HIV infects and depletes a portion of the immune system (Th cells), making individuals extremely susceptible to life-threatening infections and malignancies.
Despite major efforts by healthcare agencies around the world, HIV/AIDS remains the major cause of worldwide morbidity and mortality. Although aggressive antiretroviral therapy and public health campaigns have stabilized the number of new cases and deaths in the United States from HIV/AIDS, the number of cases and deaths continues to increase rapidly worldwide.46,47 The Centers for Disease Control and Prevention (CDC) surveillance report estimates that from 2002 through 2006 the number of new AIDS diagnoses in the United States was about 37,000 per year. Over the same period the estimated number of AIDS-related deaths was about 16,000 per year. Cumulatively since 1981 almost a million individuals have been diagnosed with AIDS in the United States, of which almost 550,000 have died as a result, and more than 400,000 are still living with AIDS. AIDS remains predominantly a disease of male homosexuals. In 2006, 50% of the newly diagnosed cases were attributed to male-to-male sexual contact, 33% to high-risk heterosexual contact, and 13% to injected drug abuse. About 26% of the new cases were diagnosed in females, primarily (74%) contracted through high-risk heterosexual contact.
Sub-Saharan Africa is still the epicenter of the AIDS pandemic. In 2007, the World Health Organization (WHO) estimated that 32% of newly diagnosed cases of HIV infection and AIDS-related deaths occurred in this region. In some areas AIDS is having a devastating effect. The United Nations (UN) AIDS program estimated that in 2006 from 4.9 million to 6.1 million people were HIV infected in South Africa, which has a total population of 48 million (national incidence of HIV infection is 10.8%). Women have the highest risk of infection: 13% of females are HIV infected, 90% of the newly diagnosed HIV infections are in 15- to 24-year-old women, and 29% of pregnant women are HIV infected. It is estimated that AIDS is responsible for almost 50% of all deaths in South Africa and 71% of deaths among individuals in the 15- to 40-year age group. As a result, the average life expectancy is now 54 years, whereas it would be at least 64 without AIDS. More than half of the current 15-year-olds are not expected to live to age 60.
The effects of AIDS are not limited to South Africa. In five countries in Africa the prevalence of HIV-infected adults exceeds 15%. In 2006, in Swaziland 26% of adults, 44% of men between the ages of 30 and 39, and 49% of pregnant women between the ages of 20 and 34 were HIV infected.
Transmission
HIV is a blood-borne pathogen present in body fluids (e.g., blood, vaginal fluid, semen, breast milk) with the typical routes of transmission: blood or blood products, intravenous drug abuse, heterosexual and homosexual activity, and maternal-child transmission before or during birth. As of the end of 2006, an estimated 8508 children in the United States developed AIDS after contracting HIV infection from their mothers across the placenta, through contact with infected blood during delivery, or through the milk during breast-feeding. Without treatment, symptoms usually develop within 6 months of life, and life expectancy is generally less than 3 years.
As with all blood-borne infections, healthcare providers are at increased risk of contracting the infection from an individual’s blood.48 The first reported case of occupational HIV infection was an emergency department nurse who became infected in 1986. The route of infection was probably through cuts in her hand that came into contact with contaminated blood through a gauze pad she was holding on a person’s open wound. Tens of thousands of healthcare workers have become infected with HIV, but as of January 2007, only 57 cases of confirmed HIV/AIDS and 140 possible cases were contracted by occupational exposure.49 Since initiation of widespread educational programs on universal precautions published by the CDC only 3 new cases have been reported. Nurses (42% of total cases) and clinical laboratory technicians (28% of cases) are by far the most commonly exposed healthcare workers.
Pathogenesis
HIV-1 was initially isolated by researchers at the Pasteur Institute as the lymphadenopathy/AIDS virus (LAV); a discovery for which they received the 2008 Nobel Prize in Medicine.50 A second major and less virulent variant, HIV-2, was identified later and is found mostly in western Africa. HIV is a member of the retrovirus family, which carries genetic information in the form of two copies of RNA (Figure 9-12). Retroviruses use a viral enzyme, reverse transcriptase, to convert RNA into double-stranded DNA (Figure 9-13). Using a second viral enzyme, an integrase, the new DNA is inserted into the infected cell’s genetic material, where it may remain dormant. If the cell is activated, translation of the viral information may be initiated, resulting in the formation of new virions, lysis and death of the infected cell, and shedding of infectious HIV particles. If, however, the cell remains relatively dormant, the viral genetic material may remain latent for years, and is probably present for the life of the individual.
Figure 9-12 The structure and genetic map of HIV-1. The HIV-1 virion consists of a core of two identical strands of viral RNA molecules of viral enzymes (reverse transcriptase [RT], protease [PR], integrase [IN]), encoated in a core capsid structure consisting primarily of the structural viral protein p24. The capsid is further encased in a matrix consisting primarily of a viral protein, p17. The outer surface is an envelope consisting of the plasma membrane of the cell from which the virus budded (lipid bilayer) and two viral glycoproteins: a transmembrane gp41 and a noncovalently attached surface protein, gp120. The HIV-1 genome contains regions that encode the structural proteins (gag), the viral enzymes (pol), and the envelope proteins (env). The gag region is translated into a large precursor (Pr55gag) that is cut by the HIV protease into smaller proteins that construct the capsid and matrix. The env region is translated into a gp160 precursor protein that is cut by a host-cell protease into the gp120 and gp41 envelope proteins. The genome of complex retroviruses, like HIV-1, often contain a variety of small regions that regulate expression of the virus. (Modified from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Figure 9-13 Life cycle and possible sites of therapeutic intervention of HIV-1. HIV infection begins when a virion, or virus particle, binds to CD4 and chemokine co-receptors on a susceptible cell. The HIV virion has viral proteins gp41 and gp120 on its surface (envelope), which interact with CD4 and the CCR5 co-receptor and initiate fusion between the viral envelope and the plasma membrane, resulting in the core of the virus being injected into the cytoplasm. Uncoating occurs in the cytoplasm, during which the core proteins are removed, and the viral RNA is released into the infected cell’s cytoplasm. The viral RNA is converted to a double-stranded DNA provirus by the action of the viral reverse transcriptase. The provirus migrates into the nucleus and is integrated into the cell’s own DNA. The provirus may remain latent. If the infected cell is activated (e.g., by cytokines), the provirus may be transcribed and translated into viral protein precursors. The precursor proteins are modified by viral (gag proteins) and cellular (env proteins) proteases into smaller proteins that are used to package the viral RNA into new virions that bud from the cell. The HIV-1 life cycle is susceptible to blockage at several sites. Some agents could block the attachment and entrance of the virus (entrance inhibitors). Reverse transcriptase inhibitors (e.g., azidothymidine [AZT]) prevent the reverse transcription of viral RNA into DNA. Drugs also may be able to inhibit the viral integrase (integrase inhibitors) and prevent insertion of the provirus into the host’s chromosomes. Protease inhibitors specifically inhibit the viral protease and prevent the processing of the gp160 into viral capsid and matrix proteins. (Modified from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
The primary surface receptor on HIV is the envelope glycoprotein gp120, which binds to the molecule CD4, found primarily on the surface of helper T cells (see Figure 9-13).51 Several other necessary co-receptors have been identified on the target cells, particularly the chemokine receptors CXCR4 and CCR5. Different strains of HIV-1 are selective for the CXCR4 or CCR5 co-receptors, which influences the tropism for different target cells. Strains that prefer the CXCR4 co-receptor tend to be T-cell tropic, usually found later in an infection, and cause infected cells to fuse and form a multinucleate syncytium. Strains that react better with the co-receptor CCR5 are macrophage-tropic, usually cause the primary HIV infection, and do not cause syncytium formation.
The primary cellular targets for HIV include the following:
• Dendritic cells (depending on the level of chemokine receptors the cell expresses)
• Macrophages (express low levels of CD4, but high amounts of heparin sulfate proteoglycans [syndecan] and other molecules [CCR5] that bind to gp120 and adsorb HIV)
• CD8-positive Tc cells (low rate of infection, but CD4 can be expressed by activated CD8-positive Tc cells)
• Double positive thymic cells (express CD4 and CD8 simultaneously)
• NK cells (some are CD4+, CCR5+)
• Neural cells of monocyte origin (macrophages and microglial cells)
Initially, the lymphoid areas of the mucosal surfaces are the primary sites of infection (Figure 9-14). Dendritic cells and mucosal T cells probably spread the infection to other peripheral lymphoid organs (especially follicular dendritic cells in the lymph nodes, which infect T cells). Infection also may involve the thymus and bone marrow, including the bone marrow stromal cells. Cells in the central nervous system (CNS) may act as a reservoir in which HIV can be relatively protected from antiviral drugs. The virus is also found in T cells and macrophages in semen and in the renal epithelium.
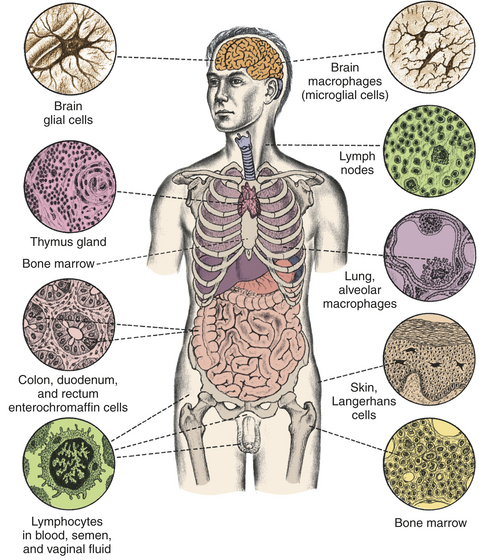
Figure 9-14 Distribution of tissues that can be infected by the HIV. Infection is closely linked to the presence of CD4 receptors or chemokine co-receptors on host tissue, particularly T cells and macrophages. (Modified from Weber JN, Weiss RA: HIV infection: the cellular picture, in the science of AIDS: readings from Scientific American, New York, 1989, Freeman.)
The major immunologic finding in AIDS is the striking decrease in the number of CD4+ Th cells (Figure 9-15). Individuals who are not HIV infected typically have 800 to 1000 CD4+ cells/μL of blood, with a range from 600 to 1200/μL. Numbers of CD8+ T cells are usually normal or slightly elevated. The decrease in CD4+ cell numbers results in a reversal of the normal CD4/CD8 T-cell ratio (normally about 1.9) to lower than 0.9 and often near zero.
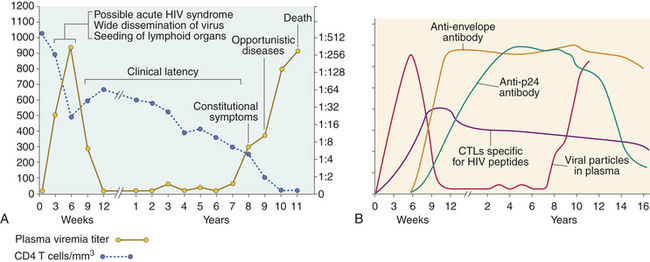
Figure 9-15 Typical course of progression from HIV infection to AIDS in untreated persons. A, Within weeks after infection, the person may experience symptoms of acute HIV syndrome. During this early period the virus progressively infects mucosal T cells and dendritic cells, propagates, and spreads to the lymphoid organs, with a sharp decrease in circulating CD4+ T cells. The resulting immune response usually induces a period of clinical latency, during which viral replication and T-cell destruction continue in the lymph nodes, although the individual is generally asymptomatic. As the disease progresses, the person may develop HIV-related disease (constitutional symptoms)—a variety of symptoms of acute viral infection that do not involve opportunistic infections or malignancies. When the number of CD4+ cells is critically suppressed, the person becomes susceptible to a variety of opportunistic infections and cancers. The length of time for progression from HIV infection to AIDS may vary considerably from person to person. B, Antibody and Tc cell (cytotoxic T lymphocytes [CTLs]) levels change during the progression to AIDS. During the initial phase antibodies against HIV-1 are not yet detectable (window period), but viral products, including p24 antigen, viral RNA, and infectious virus, may be detectable in the blood a few weeks after infection. Most antibodies produced against envelope proteins in the early phase are absorbed onto viral particles in the blood and are not detectable by most routine assays. During the latent phase of infection antibody levels against p24 and other viral proteins, as well as HIV-specific CTLs, generally increase, then remain constant until the development of AIDS. As the immune system becomes severely depressed and excess viral antigen is released into the blood, measurable antibody levels decrease. Disease progression usually ends in the death of the untreated individual. (A redrawn from Fauci AS, Lane HC: Human immunodeficiency virus disease: AIDS and related conditions. In Fauci AS et al, editors: Harrison’s principles of internal medicine, ed 14, New York, 1997, McGraw-Hill; B from Kumar V, Abbas A, Fausto N: Robbins and Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
HIV causes destruction of Th cells by a variety of means. Production of new HIV virions can be directly cytopathic to the infected cell, causing lysis (breakdown of the cell) or inducing apoptosis. Additionally, HIV-infected cells express new surface antigens and are targets for Tc-mediated lysis.
However, it is not unusual for a large majority (>98%) of peripheral CD4+ T cells to not be infected with HIV-1, although a significant amount (10% to 50%) show signs of apoptosis. Therefore, HIV-1 killing is probably an indirect rather than direct effect. HIV-infected cells shed soluble viral envelope protein, gp120, which can induce apoptotic cell death of uninfected T lymphocytes, neurons, and monocytes through interaction with cell surface receptors. The interaction between viral envelope protein on the surface of infected cells and its receptors on neighboring uninfected cells also can result in intercellular fusion and syncytium formation. The syncytia undergo apoptosis after a phase of latency. Envelope protein present on the surface of HIV-1–infected cells also can create partial fusion (hemifusion, membrane damage) that results in death of the uninfected cell. The presence of HIV virions and soluble viral antigen can result in a chronic activation of uninfected T cells with HIV-specific T-cell receptors (TCRs). Because activated T cells more efficiently support HIV replication, the most susceptible cells are those with TCRs against HIV, which undergo antigen-driven activation and infection. This observation may not bode well for successful vaccine development if the induced and supposedly protective CD4+ cells are also the most susceptible targets for HIV.
As a result of these processes the level of T cells decreases (particularly memory T cells, which seem more susceptible to HIV infection), thymic production of new T cells is decreased, and the secondary lymphoid organs (particularly the lymph nodes) are damaged.
Clinical Manifestations
Depletion of CD4+ cells has a profound effect on the immune system, causing a severely diminished response to a wide array of infectious pathogens and malignant tumors (Box 9-2).
At the time of diagnosis, the individual may manifest one of several different conditions: serologically negative (no detectable antibody), serologically positive (positive for antibody against HIV) but asymptomatic, early stages of HIV disease, or AIDS (see Figure 9-15).
The presence of circulating antibody against the HIV indicates infection by the virus, although many of these individuals are asymptomatic. Antibody appears rather rapidly after infection through blood products, usually within 4 to 7 weeks. After sexual transmission, however, the individual can be infected yet seronegative for 6 to 14 months or, in at least one case, for years. In addition, in the late stages of the disease, some individuals become seronegative because of a deficient immune system.
The period between infection and the appearance of antibody is referred to as the window period (see Figure 9-15). Although the individual may not have antibody, he or she may have virus growing, have virus in the blood and body fluids, and be infectious to others. Early symptoms are relatively nonspecific to HIV and include fatigue, fever, muscle aches, and headaches.
Those with the early stages of HIV disease (early stage disease or clinical latency) are usually asymptomatic. The early stage may last as long as 10 years in untreated people, during which viral load increases and numbers of CD4+ cells progressively decrease. Some estimates are that approximately 99% of untreated HIV-infected individuals would eventually progress to AIDS.
The currently accepted definition of AIDS relies on both laboratory tests and clinical symptoms. The most common laboratory test is for antibodies against HIV proteins (particularly p24). If the individual is seropositive, the diagnosis of AIDS is made in association with various clinical symptoms (see Box 9-2). The symptoms include atypical or opportunistic infections and cancers, as well as indications of debilitating chronic disease (e.g., wasting syndrome, recurrent fevers) (Figure 9-16). Most commonly, new cases of AIDS are diagnosed initially by decreased CD4+ T-cell numbers at or below 200 cells/μL.
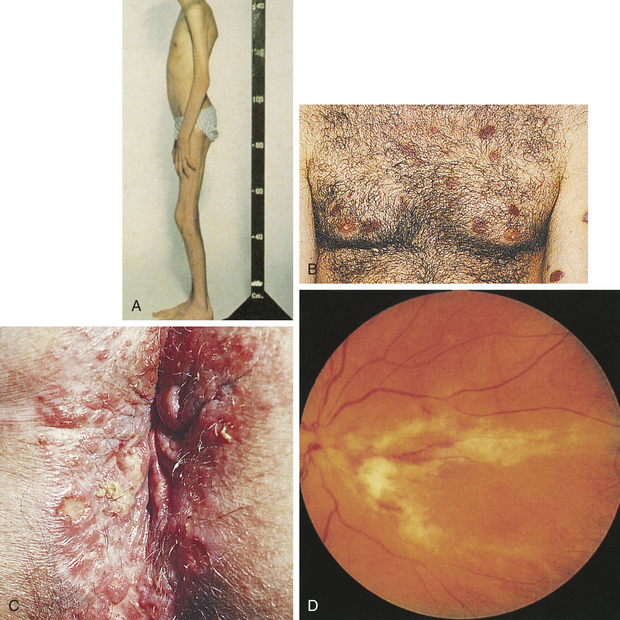
Figure 9-16 Clinical symptoms of AIDS. A, Severe weight loss and anorexia. B, Biopsy-proven Kaposi sarcoma lesions. C, Perianal vesicular and ulcerative lesions of herpes simplex infection. D, Deterioration of vision from cytomegalovirus retinitis leading to areas of infection; unless treated the progressive impairment will lead to blindness. (A and D from Taylor PK: Diagnostic picture tests in sexually transmitted diseases, London, 1995, Mosby; B and C from Morse SA et al editors: Atlas of sexually transmitted diseases and AIDS, ed 3, London, 2003, Mosby.)
Treatment and Prevention
The current regimen for treatment of HIV infection is a combination of drugs, termed highly active antiretroviral therapy (HAART).52 The combination includes at least three drugs from at least two classes of antiretroviral agents, including nucleoside reverse transcriptase inhibitors combined with a viral protease inhibitor or a non-nucleoside reverse transcriptase inhibitor (see Figure 9-13). Death from AIDS-related diseases has been reduced significantly since the introduction of HAART; without treatment an individual may live only 9 to 10 months after diagnosis of AIDS, whereas those who respond well to HAART may survive for several decades. However, many people do not respond to HAART therapy, those who do respond are not “cured”, and resistant variants to these drugs have been identified.53
Drug therapy for AIDS is difficult because, like most retroviruses, the AIDS virus incorporates into the genetic material of the host and may never be removed by antimicrobial therapy. Therefore, drug administration to control the virus may have to continue for the lifetime of the individual. Additionally, HIV may persist in regions where the antiviral drugs are not as effective, such as the CNS. Inhibitors of the initial viral entrance into the target cell (entrance inhibitors) may provide benefit for those who HAART does not benefit. Entrance inhibitors include the natural or modified ligands for the co-receptors (CXCR4 and CCR5) and can block infection and inhibit cell membrane fusion. Inhibitors of the viral integrase (integrase inhibitors) have undergone clinical trials and eventually may be added to the combination.
To date the development of an effective vaccine is the only hope for preventing the spread of HIV infection. Most common antiviral vaccines (e.g., rubella, mumps, influenza) induce protective antibodies that block the initial infection. Only one vaccine (rabies) is used after the infection has occurred. That approach is successful because the rabies virus proliferates and spreads very slowly. Whether an HIV vaccine would be effective in either preventing or treating HIV infection is problematic, and the results of a recent vaccine trial have for the moment dampened enthusiasm.
Pediatric AIDS and Central Nervous System Involvement
The clinical diagnosis of HIV in children is very often a difficult task. The CDC revised the classification in 1994 for HIV infection in children younger than 13 years (Box 9-3). The HIV infection may be identified by viral culture of blood or tissue, and the diagnosis is confirmed by the presence of specific antibodies to the virus. However, the presence of passive maternal antibody limits the use of HIV antibody testing in infants in the high-risk category up to 15 months of age.
WHAT’S NEW?
HIV Vaccine Trials
Very soon after the discovery in 1983 of the virus that causes AIDS, knowledgeable scientists and public officials predicted that an effective vaccine would be marketed within a few years. The prediction was not viewed as overly optimistic because vaccines had been produced against many other viral diseases, and the technology was available to swiftly purify, test, and manufacture viral antigens. In 2008, 25 years after the discovery of HIV, the chances of producing vaccine protection against AIDS appear more bleak than ever before.
BOX 9-3 Pediatric Human Immunodeficiency Virus (HIV) Classification
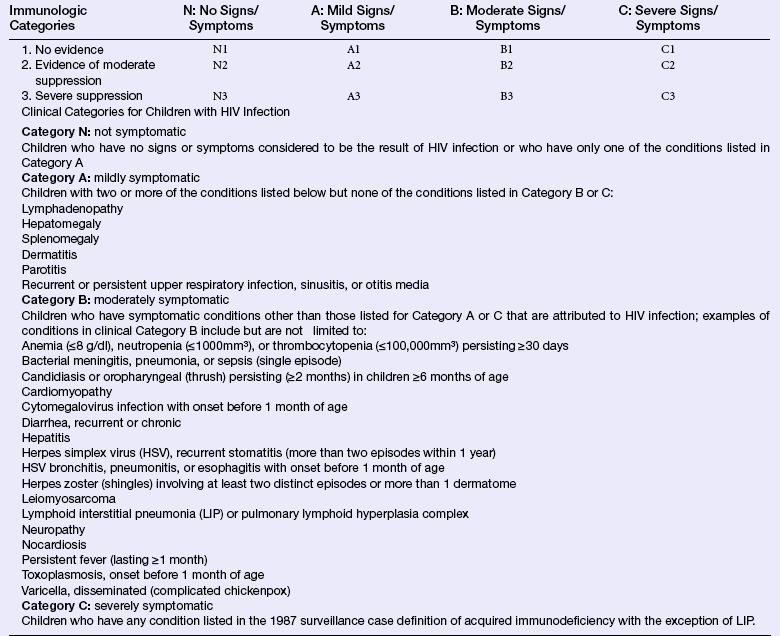
Data from Centers for Disease Control and Prevention: MMWR Morb Mortal Wkly Rep 43(12):1, 1994.
The goal of an AIDS vaccine is to either prevent the initial infection or enhance the immune response of an infected individual to reduce viral load, or both. There was a great deal of interest in a recent trial (the STEP Study) of vaccine V520, produced by Merck. V520 was designed to produce a T-cell response because a considerable amount of data supported the role of T cells in controlling HIV infection. The immunization protocol was very complex and elicited an immune response against several HIV antigens, including products of HIV env, gag, pol, and nef genes expressed in an altered adenovirus, which would normally cause a common cold. The trial was prematurely stopped in September of 2007 because the initial analysis indicated that the vaccine neither prevented HIV infection nor lowered virus in the blood of those who became infected. The vaccine was suspected of increasing the risk for HIV infection in some recipients. Of 741 individuals who received at least one dose of the vaccine, 24 became infected during the trial, compared with 21 who became infected of the 762 who received the sham control vaccine.
Since the first large scale vaccine trial that used recombinant HIV gp120 envelope protein, no trial has shown protection against HIV infection, although HIV-specific antibody or T cells have been induced. Despite optimistic predictions for vaccine success, several major caveats had been discussed. The scale of genetic diversity of HIV envelope proteins is more like influenza virus, which requires a new vaccine yearly, than other viral infections, such as measles, mumps, and others that express stable antigens. However, unlike influenza, which changes antigens yearly, HIV undergoes antigenic change during infection of each individual. HIV also remains latent within infected cells, and no vaccine has been developed to rid the individual of other latent viruses. Perhaps the most overlooked concern is that an antibody response to HIV may actually facilitate infection. T cells and macrophages express surface receptors for the Fc portion of IgG and for complement components, thus an HIV virion coated with IgG or C3b may rapidly enter the very target cells that support infection.
Thus, with the recurrent failure of AIDS vaccine trials, the only effective means to limit the spread of HIV are behavioral changes. In Africa in particular, the rate of HIV infection has been reduced by education about appropriate condom use, increased application of male circumcision, and reductions in multiple sexual partnerships. However, these approaches only diminish the spread of HIV; therefore, vaccination remains the only probable means of completely preventing transmission of HIV.
Data from Johnston MI, Fauci AS: N Engl J Med 356(20):2073-2081, 2007; Johnston MI, Fauci AS: N Engl J Med 359(9):888-890, 2008; Potts M et al: Science 320(5877):749-750, 2008; Steinbrook R: N Engl J Med 357(26):2653-2655, 2007; Walker BD, Burton DR: Science 320(5877):760-764, 2008; Patterson S et al: Handb Exp Pharmacol 188:275-293, 2009.
Therefore, two definitions of infection in children are necessary: one for prenatally exposed infants up to 15 months of age and one for older children.
HIV directly invades most major organ systems, including the CNS. Therefore, the clinical manifestations vary greatly from child to child. The initial signs and symptoms may be nonspecific and subtle, and they may progress slowly or rapidly to an acute, life-threatening condition. A definite diagnosis of HIV is made by personal history, viral culture, and clinical manifestations. Monitoring CD8+ T lymphocytes and monocytes, in addition to CD4+ T lymphocytes, has been suggested for predicting risk for progressive encephalopathy. Decreases in CD8+ T lymphocytes diminish defenses against viral infection and facilitate infected monocytes to cross the blood-brain barrier.
A particularly vulnerable site of HIV infection in infants and children is the CNS. HIV encephalopathy is more common in the advanced stages. Because survival of children with HIV has been prolonged with effective treatment, the incidence of progressive encephalopathy has increased. The 1994 classification from the CDC54 requires one of the following progressing findings to be present for at least 2 months, in the absence of a concurrent illness other than HIV that could explain the following findings:
• Failure to attain or loss of developmental milestones, or loss of intellectual ability, verified by standard developmental scale or neuropsychologic tests
• Impaired brain growth or acquired microcephaly demonstrated by head circumference measurements or brain atrophy demonstrated by computed tomography (CT) or magnetic resonance imaging (MRI) with serial imaging required in children less than 2 years of age
• Acquired symmetric motor deficits manifested by affecting a child 1 month of age or older
The onset of progressive encephalopathy may be a prognostic indicator of a poor outcome.
It may be difficult to completely differentiate the effect of HIV infection on the CNS from the effect of prenatal and perinatal exposure. In addition, other insults may accompany HIV in a young child and affect growth and development, such as drug exposure, prematurity, chronic illness, and a chaotic social atmosphere. The pathogenesis of HIV encephalopathy in children is poorly understood, but the presence of inflammatory mediators may be a contributing factor.
A growing number of investigational protocols are available for treatment of children with HIV. In general, treatment is focused on the preservation and maintenance of the immune system, aggressive response to opportunistic infections, and support and relief of symptomatic occurrences and administration of HAART.
COUNTERMEASURES AGAINST PATHOGENS
An extremely effective means of countering infectious microorganisms is rigorous use of environmental infection control measures, including control of insect vector populations, modern sanitation facilities, providing clean water and uncontaminated food supplies, and other measures. Additionally, prophylactic or interventive procedures have been developed to prevent pathogens from initiating disease (vaccines) or to subdue the pathogen once the disease process has started (antimicrobials). Vaccine development has focused successfully on preventing the most severe and common infections (Table 9-10). With the initial success of antibiotic therapy, there was no perceived need for vaccination against many common and non–life-threatening infections. The increasing problem of antibiotic-resistant pathogens, however, forced a reappraisal of that strategy, and a greater emphasis is being placed on the development of new vaccines.
Table 9-10
Reduction in Vaccine-Preventable Diseases in the United States
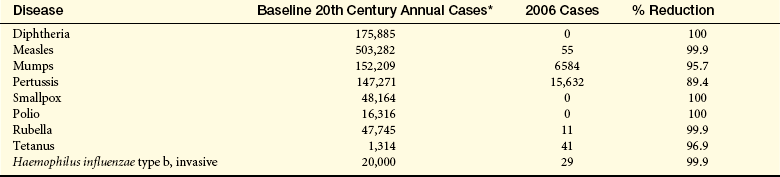
∗Average number of reported cases over multiple years before initiation of vaccine (Centers for Disease Control and Prevention: MMWR Morb Mortal Wkly Rep 48[12]:243-248, 1999, Morb Mortal Wkly Rep 57[11]:289-291, 2008.)
From National Institute of Allergy and Infectious Disease, National Institutes of Health: Vaccine, Vaccine Benefits available at www3.niaid.nih.gov/topics/vaccines/understanding/vaccineBenefits.htm. Accessed February 15, 2009.
Infection Control Measures
Although effective means of safeguarding populations from exposure to infectious disease are well known, lack of implementation or breakdowns in application of these initiatives has led to the reemergence of some infectious diseases.
John Snow, a British physician, is considered the “father of epidemiology.”55 Industrialization in England in the nineteenth century resulted in rapid expansion of London’s population that outgrew city services. An outbreak of cholera in the Soho area of London in 1854 resulted in more than 600 deaths. Snow discovered that the outbreak centered around one particular public water pump, which got water from a well that had been dug 3 feet from a cesspool (open sewage was common in London at that time). After the pump was closed, the rate of new cholera cases rapidly diminished. Snow’s observation stimulated the development of the construction of clean water supplies and sewers throughout the city.
Sewage removal and other public health initiatives (e.g., routine trash collection, disposal of garbage by incineration or in landfills) are now the expected norm in developed countries. The quality of water and food, as well as disposal of human and animal waste, remains poor in many developing countries. Rapid urbanization as well as other problems (e.g., poverty, overcrowding, mass relocation of people due to war, rapid destruction of forests) has put pressure on already inadequate systems.56 A 1991 outbreak of cholera in Peru resulted in an estimated 10,000 deaths and was attributed to inadequate sanitation.
Additionally, previously successful programs designed to control the breeding of insect vectors have been reversed. Despite an international emphasis on draining standing water that provides breeding grounds for mosquitoes, large unmanaged areas still abound. A very successful international mosquito eradication program resulted in decreasing incidence of mosquito-borne diseases. Some regions were declared “free” of mosquito-borne diseases. However, since the international ban on DDT for agricultural work and limitation of its use for control of disease vectors, mosquitoes and mosquito-related diseases have rebounded in previously disease-free areas. In northern India, heavy rains in the 1990s resulted in an increase in standing water that was not drained because of the lack of government-provided public health information and inadequate insecticides to control mosquitoes. Since 1996, recurrent and increasingly severe outbreaks of dengue fever (caused by dengue virus, which is related to yellow fever virus and West Nile virus) have occurred in this region. In the 2006 outbreak 10,344 cases were reported, with 162 deaths. This problem is exacerbated by the development of insecticide-resistant mosquitoes.
Antimicrobials
Since the first use of penicillin during World War II, antibiotics have had the greatest effect on controlling infection. Antibiotics are natural products of fungi, bacteria, and related microorganisms and kill or inhibit the growth of other microorganisms. Numerous chemicals or antimicrobials have been identified that either prevent the growth of microorganisms or directly destroy them. Antibiotics generally act by preventing the function of enzymes or cell structures that are unique to the infecting agent. Because viruses use the enzymes of the host’s cells, there has been far less success in developing antiviral antibiotics.
Immediately after antibiotics became widely used, microorganisms mutated and developed resistance.57 By 1944 an adequate supply of penicillin allowed its widespread use to treat infections. In 1946 a hospital in Britain reported that 14% of all S. aureus were penicillin resistant. By 1950 the same hospital reported an increase to 59% and to greater than 89% in the 1990s. Over the past few decades healthcare providers have observed increasing incidences of drug-resistant malaria, tuberculosis, gonorrhea, salmonellosis, shigellosis, and staphylococcal infections. S. pneumoniae, which causes pneumonia, meningitis, and acute otitis media (ear infections), was once routinely susceptible to penicillin. Since the 1980s, however, the incidence of penicillin-resistant microorganisms has risen to greater than 30% in some populations.
The goal of antibiotic therapy is elimination of the pathogenic microorganism. Some antibacterial antibiotics are bactericidal (kill the organism), whereas others are bacteriostatic (inhibit growth until the organism is destroyed by the individual’s own protective mechanisms). The mechanisms of action of most antibiotics are (1) inhibition of the function or production of the cell wall, (2) prevention of protein synthesis, (3) blockage of DNA replication, or (4) interference with folic acid metabolism.
Antibiotic resistance is usually a result of genetic mutations that can be transmitted directly to neighboring microorganisms by plasmid exchange. Microorganisms commonly develop the capacity to inactivate antibiotics. Penicillin resistance, for example, results from the production of an enzyme (β-lactamase) that breaks down the structure of the antibiotic. Other forms of resistance result from modification of the target molecule. Azidothymidine (AZT) is a family of antivirals that suppresses the enzymatic activity of reverse transcriptase,
a viral-specific enzyme responsible for the replication of viral RNA and production of a DNA copy. HIV frequently mutates and produces an AZT-resistant reverse transcriptase. A third mechanism of resistance is mediated by multidrug transporters in the microorganism’s membrane. These transporters affect the rate of intracellular accumulation of the antimicrobial by preventing entrance or, more commonly, increasing active efflux of the antibiotic. Antibiotic-resistant strains of M. tuberculosis are protected from aminoglycosides and tetracycline by the Tap protein (a mycobacterial multidrug efflux pump).
Many microorganisms are now resistant to multiple antibiotics. Methicillin-resistant Staphylococcus aureus (MRSA) incorporates several different mechanisms of resistance. MRSA produces a β-lactamase (penicillinase). Penicillinase-resistant antibiotics (e.g., methicillin) can be used as a substitute, but MRSA carrying the gene mecA, is also methicillin resistant because of a lower affinity for β-lactams. Resistance to the glycopeptide antibiotic vancomycin is controlled by the gene vanA, that results in alterations in peptidoglycans and loss of the vancomycin binding site.58
Why have multiple antibiotic–resistant microorganisms appeared? Overuse of antibiotics can lead to the destruction of the normal flora, allowing the selective overgrowth of antibiotic-resistant strains or pathogens that had previously been kept under control.59 For example, after treatment with the antibiotic clindamycin, the normal intestinal flora can become compromised, allowing the overgrowth of C. difficile and the development of pseudomembranous colitis. Also, lack of compliance concerning the necessity of completing the therapeutic regimen with antibiotics allows the selective resurgence of microorganisms that are more relatively resistant to the antibiotic.
With the development of multiple antibiotic–resistant strains, creativity in addressing this challenge must be rekindled. New antibiotics may solve a portion of the problem or may exacerbate it further as pathogens develop resistance to new antibiotics as well. Available antibiotics that prove to be no longer effective must be replaced with alternative forms of therapy, including the increased development and use of vaccines and other therapies.
Active Immunization: Vaccines
Contracting and surviving an infectious disease is the most effective means of developing lifelong immunity against a particular pathogen. However, some infections cause a great deal of morbidity and mortality. The purpose of vaccination is to induce active immunologic protection before exposure to the risks of infection. For each vaccine an initial immunization protocol is developed to produce large numbers of memory cells and a sustained protective secondary immune response in the greatest number of individuals. In general, vaccine-induced protection does not persist as long as infection-induced immunity, thus booster injections may be necessary to maintain protection throughout life.
Common vaccines used in the United States and their abbreviations, as noted in Tables 9-11 and 9-12, are as follows:
Table 9-11
Recommended Adult Immunization Schedule by Vaccine and Age Group—United States, October 2007 to September 2008
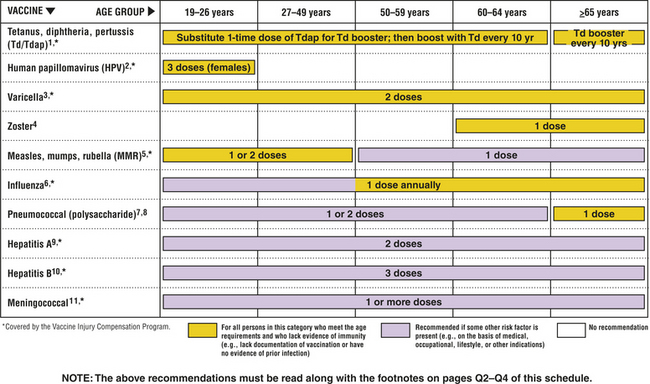
From the Centers for Disease Control and Prevention. Available at www.cdc.gov/vaccines/recs/schedules/default.htm.
Table 9-12
Recommended Immunization Schedule for Persons Ages 0 to 6 Years—United States 2008
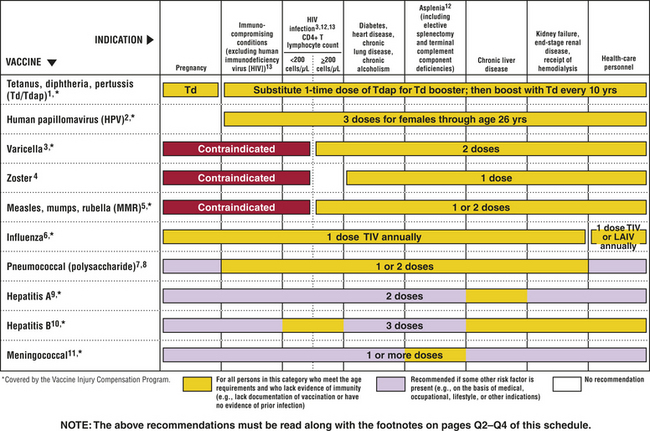
From the Centers for Disease Control and Prevention. Available at www.cdc.gov/vaccines/recs/schedules/default.htm.
• Diphtheria (D): A bacterial infection of the throat; produces a toxin that can lead to heart failure or paralysis; the vaccine is an inactivated form of diphtheria toxin (toxoid); part of DTaP combined vaccine
• H. influenzae type b (Hib): A bacterial infection that is commonly contracted by children younger than 5 years and infects the blood, joints, bones, and membrane covering the heart; most common cause of serious bacterial meningitis in children; the vaccine is an extracted bacterial antigen conjugated to a protein carrier
• Hepatitis A virus: A virus that causes liver disease; recommended in selected states and regions; vaccine is an inactivated virus
• Hepatitis B virus: Causes cirrhosis of the liver and liver cancer; the vaccine is a recombinant viral protein
• Influenza: a virus that causes severe upper respiratory tract infections; the most common vaccine is an injected inactivated virus
• Measles and mumps (MM): Viruses that cause fever and rash (measles) or inflammation of the salivary glands (mumps); mumps also may cause meningitis; both vaccines are attenuated live viruses; part of MMR combined vaccine
• Meningococcal: N. meningitidis is a major bacterial cause of meningitis, particularly in young adults; the vaccine contains four different extracted capsular polysaccharides
• Pertussis (acellular pertussis [aP]), or whooping cough: A bacterial infection that causes severe coughing in children younger than 5 years; the vaccine is an inactivated toxin and other bacterial antigens; part of DTaP combined vaccine
• Pneumococcal: S. pneumoniae causes pneumonia, particularly in older adults; the childhood vaccine is a protein conjugate with 7 different capsular antigens; the adult vaccine is a mixture of 10 extracted capsular polysaccharides
• Polio: Polio is a viral infection that causes paralysis and death; the vaccine is an inactivated virus
• Rotavirus: A viral infection that causes severe diarrhea primarily in babies and young children; the vaccine is an attenuated live virus
• Rubella (R), or German measles: A viral infection that causes fever and rash; may cause severe birth defects in pregnant women infected in the first trimester; part of MMR combined vaccine
• Tetanus (T): A bacterial infection that produces a toxin that attacks the nervous system and may cause death; the vaccine is an inactivated form of the tetanus toxin (toxoid); part of DTaP combined vaccine
• Varicella-zoster (varicella): A virus that causes chickenpox; vaccine is an attenuated live virus
Mass vaccination programs have led to major changes in the health of the world’s population. In the early 1950s an estimated 50 million cases of smallpox occurred each year, with about 15 million deaths. The World Health Organization (WHO) conducted a smallpox immunization campaign from 1967 to 1977 that resulted in the global eradication of smallpox by 1979. The 1988 immunization initiative against polio resulted in a 99% decrease in that disease. In 1960, 2525 cases of paralytic polio were reported in the United States, but no cases of naturally acquired polio have been reported since 1979. In 1994, polio was declared officially eradicated in all the Americas. The goal of the WHO is to eradicate polio worldwide in the next few years (Figure 9-17). Similar trends occurred for each disease against which an effective vaccine has been developed.
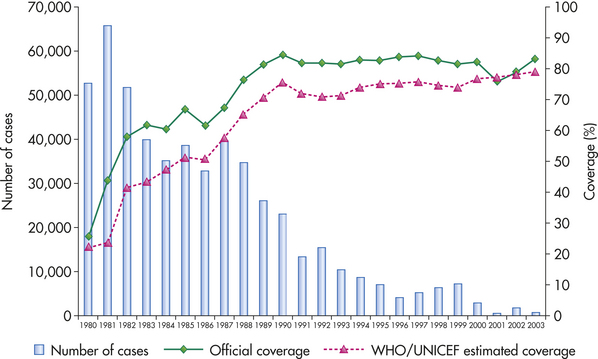
Figure 9-17 Effect of immunization on the global reported cases of polio, 1980 to 2003. The number of global cases of polio has progressively decreased in an inverse relationship to the extent of vaccination. The “official coverage” is the percent of the world’s population that has been reported to the World Health Organization (WHO) by 192 member countries as being immunized. The “WHO/UNICEF estimated coverage” indicates the best estimate of the real percentage of the population that has been immunized. (From World Health Organization: WHO vaccine-preventable diseases: monitoring system, 2003 global summary, Geneva, Switzerland, 2003. Additional information is available at www.who.int/vaccines-documents.)
Development of a successful vaccine depends on many factors. These include characterizing the desired protective immune response (e.g., antibody, T cell), identifying the appropriate antigen to induce that response (i.e., immune responses against some antigens on an infectious agent are ineffective or even increase the risk for infection), determining the most effective route of administration (e.g., injected, oral, inhaled), optimizing the number and timing of vaccine doses to induce protective immunity in a large proportion of the at-risk population, and deciding the most effective, yet safe, form in which to administer the vaccine. For instance, most vaccines against viral infections (e.g., measles, mumps, rubella, varicella [chickenpox], rotavirus) contain live viruses that are weakened (attenuated) to continue expressing the appropriate antigens but are unable to establish more than a limited and easily controlled infection. The limited proliferative capacity of attenuated live viruses appears to afford better long-term protection than using purified viral antigen. Two exceptions are the vaccines against hepatitis B, which uses a recombinant viral protein, and hepatitis A, which is an inactivated (killed) virus.
Even attenuated viruses can, however, establish life-threatening infections in vaccine recipients whose immune system is congenitally deficient or suppressed (see Chapter 8). Two different vaccines were developed against polio. The Sabin vaccine is an attenuated virus that is administered orally. It provides systemic protection and induces a secretory immune response to prevent growth of the poliovirus in the intestinal tract. The live attenuated vaccine causes polio in some children who have unsuspected immune deficiencies (about 1 case in 2.4 million doses). The Salk vaccine is a completely inactivated virus administered by injection. It induces protective systemic immunity but does not provide adequate secretory immunity. Therefore, even if the individual is protected from systemic infection the “wild-type” poliovirus can transiently infect the intestinal mucosa, be shed, and spread to others. When polio was epidemic, the live oral vaccine was preferred; however, about eight cases of paralytic polio per year in the United States resulted from the vaccine strain proliferating in individuals with inadequate immune systems. As a result, the CDC currently recommends vaccination with the killed virus.
Some common bacterial vaccines are killed microorganisms or extracts of bacterial antigens. The vaccine against pneumococcal pneumonia consists of a mixture of capsular polysaccharides from 10 strains of S. pneumoniae. Of the more than 90 known strains of this microorganism, only these 10 cause the most severe illnesses. However, the capsular vaccine is not very immunogenic in young children. A “conjugated” vaccine is available that contains capsular polysaccharides from 7 strains that are conjugated to carrier proteins in order to increase immunogenicity. A similar vaccine is available for Hib.
Some bacterial diseases are caused by potent toxins that act locally or systemically. These include diphtheria, cholera, and tetanus. Vaccination against the toxins is achieved using toxoids—purified toxins that have been chemically detoxified without loss of immunogenicity. Pertussis (whooping cough) vaccine was changed from a killed whole cell vaccine to an acellular vaccine that contained the pertussis toxin and additional bacterial antigens.
With so many available vaccines there has been an effort to mix vaccines in order to minimize the number of required injections. One of the first licensed vaccine mixtures was DPT, which now usually contains diphtheria (D) and tetanus (T) toxoids and acellular pertussis vaccine (aP). More recent mixtures include DTaP with inactivated poliovirus, with either Hib conjugate to tetanus toxoid or with hepatitis B vaccine.
A common problem is compliance of the susceptible population in vaccination programs.60 Depending on the microorganism, a certain percentage of the population (usually about 85%) should be immunized in order to achieve protection of the total population. This is referred to as herd immunity. If this level of immunization is not achieved, outbreaks of infection can occur. For instance, an effective measles (rubeola) vaccine was made available in 1963 and resulted in a dramatic decrease in the number of measles cases. Many parents became complacent and did not obtain measles vaccination for their preschool children. As a result, a large increase in the number of cases and deaths in 1989 and 1990 occurred, which initiated a reemphasis on complete immunization before children could start school. More recently resistance to immunization with measles has increased, and in early 2008 the number of measles cases in the United States increased by about fourfold. In several European countries immunization programs have been disrupted by anti-vaccine groups. As a result the incidence of pertussis (whooping cough) increased by 10 to 100 times compared with neighboring countries that maintained a high incidence of immunization.
The refusal to vaccinate has generally been based on potential vaccine dangers. As with any medicine, complications can arise. In the case of vaccines, these include pain and redness at the injection site, fever, allergic reactions to vaccine ingredients, infection associated with attenuated viruses in immunodeficient individuals, and others. For instance, a rotavirus vaccine approved a decade ago was found to increase the risk for a life-threatening bowel obstruction resulting from twisting of the intestines, and the vaccine was recalled.61 A commonly discussed fear is related to the presence of the preservative thimerosal in vaccines. Thimerosal is a mercury-containing compound that has been used as a preservative since the 1930s. Although no cases of mercury toxicity have been reported secondary to vaccination, thimerosal was removed from all vaccines in 2001, with the exception of inactivated influenza vaccines.62 In 2003, groups in northern Nigeria claimed that the oral polio vaccine was unsafe, which led to suspension of polio immunization in two states and reduction of immunization in other states. The result was a 36% increase in cases of polio and thousands of cases of paralysis, including in previously polio-free areas.
Passive Immunotherapy
Passive immunotherapy is a form of countermeasure against pathogens in which preformed antibodies are given to the individual. This form of therapy has been used for decades. Horse serum–containing antibodies were given to treat diphtheria, pneumococcal pneumonia, tetanus, and other diseases in the early twentieth century. However, because of foreign proteins in the serum, many individuals developed an immune reaction against the horse proteins and an immune complex–mediated serum sickness. Passive immunotherapy with human immunoglobulin has been approved for several infections, including hepatitis B and hepatitis A. Treatment of potential rabies infection after a bite combines passive and active immunization. The rabies virus proliferates very slowly. Individuals who have been bitten receive a onetime injection with human rabies immunoglobulin to slow down viral proliferation further, followed by multiple injections with a killed viral vaccine to induce greater protective immunity. For several diseases more specific therapy with monoclonal antibodies is being evaluated, and a monoclonal antibody against respiratory syncytial virus has been approved for therapy.
In the past, vaccines and therapeutic antibodies were developed for only the most deadly pathogens. With the increase in antibiotic-resistant microorganisms, the development and widespread use of new vaccines and antibodies against these microorganisms must be considered. For example, otitis media, a purulent ear infection, is caused primarily by Streptococcus, Haemophilus, and Staphylococcus. This infection routinely has been treated successfully with antibiotics; however, the increase in multiple antibiotic–resistant microorganisms may force a reevaluation of the use of childhood immunization as an alternative to preventing this disease. Other vaccines used now only to a limited degree may be used more widely in the future, and others may be developed soon, including vaccines against cholera, typhoid, malaria, West Nile virus, hantavirus, SARS, and several other diseases.
Acquired immunodeficiency syndrome (AIDS) 318
Antibiotic resistance 306
Antigenic drift 316
Antigenic shift 317
Antigenic variation 304
Antitoxin 301
Attenuated 331
Bacteremia 301
Bacterial superantigens 305
Bactericidal 327
Bacteriostatic 327
β-Lactamase 307
Biofilms 296
Capsule 304
Dimorphic 307
Endogenous pyrogen 297
Endotoxic shock (septic shock) 305
Endotoxin 301
Entrance inhibitor 324
Exogenous pyrogen 297
Exotoxin 301
Gene switching 311
Highly active antiretroviral therapy (HAART) 322
Human immunodeficiency virus (HIV) 318
Human immunoglobulin 332
IgA protease 304
Integrase 319
Integrase inhibitor 324
Lipopolysaccharide (LPS) 301
Methicillin-resistant Staphylococcus aureus (MRSA) 328
Mold 307
Multinucleated giant cell 316
Mutation 316
Mycosis 307
Passive immunotherapy 332
Pili (fimbriae) 298
Protease inhibitor 322
Recombination 317
Reverse transcriptase 319
Reverse transcriptase inhibitor 322
Sepsis 301
Syncytium 319
Tissue tropism 296
Toxoid 331
Window period 322
Yeast 307
Zoonotic infection 313
REFERENCES
1. Fauci, A.S., Touchette, N.A., Folkers, G.K. Emerging infectious diseases: a 10-year perspective from the National Institutes of Allergy and Infectious Disease. Emerg Infect Dis. 2005;11(4):519–525.
2. Hull, M.W., Chow, A.W. Indigenous microflora and innate immunity of the head and neck. Infect Dis Clin North Am. 2007;21(2):265–282.
3. Greger, M. The human/animal interface: emergence and resurgence of zoonotic infectious diseases. Crit Rev Microbiol. 2007;33(4):243–299.
4. Gillim-Ross, L., Subbarao, K. Emerging respiratory viruses: challenges and vaccine strategies. Clin Microbiol Rev. 2006;19(4):614–636.
5. Rothman, R.E., Hsieh, Y.H., Yang, S. Communicable respiratory threats in the ED: tuberculosis, SARS, and other aerosolized infections. Emerg Med Clin North Am. 2006;24(4):989–1017.
6. Pizarro-Cerda, J., Cossart, P. Bacterial adhesion and entry into host cells. Cell. 2006;124(4):715–727.
7. Otto, M. Staphylococcal biofilms. Curr Top Microbiol Immunol. 2008;322(1):207–228.
8. Kleinpell, R.M., Graves, B.T., Ackerman, M.H. Incidence, pathogenesis, and management of sepsis: an overview. AACN Adv Crit Care. 2006;17(4):385–393.
9. Finlay, B.B., McFadden, G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124(4):767–782.
10. Hybiske, K., Stephens, R.S. Exit strategies of intracellular pathogens. Nat Rev Microbiol. 2008;6(2):99–110.
11. Pizarro-Cerda, J., Cossart, P. Subversion of cellular functions by Listeria monocytogenes. J Pathol. 2006;208(2):215–223.
12. Labbe, K., Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008;15(9):1339–1349.
13. Kwinn, L.A., Nizer, V. How group A Streptococcus circumvents host phagocyte defenses. Future Med. 2007;2(1):75–84.
14. Lambris, J.D., Ricklin, D., Geisbrecht, B.V. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6(2):132–142.
15. Zipfel, P.F., Wurzner, R., Skerka, C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol. 2007;44(16):3850–3857.
16. Mizgerd, J.P. Acute lower respiratory tract infection. N Engl J Med. 2008;358(7):716–727.
17. Karin, M., Lawrence, T., Nizet, V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124(4):823–835.
18. Gao, H., Evans, T.W., Finney, S.J. Bench-to-bedside review: sepsis, severe sepsis, and septic shock—does the nature of the infecting organism matter? Crit Care. 2008;12(3):213.
19. Gordon, R.J., Lowy, F.D. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(Suppl 5):S350–S359.
20. Kraus, D., Peschel, A. Staphylococcus aureus evasion of innate antimicrobial defense. Future Microbiol. 2008;3(4):437–451.
21. Diep, B.A., Otto, M. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 2008;16(8):361–369.
22. D’Avignon, L.C., Schofield, C.M., Hospenthal, D.R. Pneumocystis pneumonia. Semin Respir Crit Care Med. 2008;29(2):132–140.
23. Stringer, J.R. Antigenic variation in Pneumocystis. J Eukaryot Microbiol. 2007;54(1):8–13.
24. Ferguson, L.R., Philpott, M. Nutrition and mutagenesis. Ann Rev Nutr. 2008;28(1):313–329.
25. Netea, M.G., et al. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol. 2008;6(1):67–78.
26. Marodi, L., Johnston, R.B., Jr. Invasive Candida species disease in infants and children: occurrence, risk factors, management, and innate host. Curr Opin Pediatr. 2007;19(6):693–697.
27. Mavor, A.L., Thewes, S., Hube, B. Systemic fungal infections caused by Candida species: epidemiology, infection process and virulence attributes. Curr Drug Targets. 2005;6(8):863–874.
28. Calderone, R.A., Fonzi, W.A. Virulence factors of Candida albicans. Trends Microbiol. 2001;9(7):327–335.
29. Zambrano-Villa, S., et al. How protozoan parasites evade the immune response. Trends Parasitol. 2002;18(6):272–278.
30. Mosser, D.M., Miles, S.A. Avoidance of innate immune mechanisms by the protozoan parasite, Leishmania spp. In: Denkers E., Gazzinelli R., eds. Protozoans in macrophages. Austin, TX: Landes Bioscience; 2007:118–129.
31. Alves, M.J., Colli, W. Trypanosoma cruzi: adhesion to the host cells and intracellular survival. IUBMB Life. 2007;59(4-5):274–279.
32. van Riet, E., Hartgers, F.C., Yazdanbakhsh, M. Chronic helminth infections induce immunomodulation: consequences and mechanisms. Immunobiol. 2007;212(6):475–490.
33. Raes, G., et al. Alternatively activated macrophages in protozoan infections. Curr Opin Immunol. 2007;19(4):454–459.
34. Stijlemans, B., et al. African trypanosomosis: from immune escape and immunopathology to immune intervention. Vet Parasitol. 2007;148(1):3–13.
35. Greenwood, B.M., et al. Malaria: progress, perils, and prospects for eradication. J Clin Invest. 2008;118(4):1266–1276.
36. Cowman, A.F., Crabb, B.S. Invasion of red blood cells by malaria parasites. Cell. 2006;124(4):755–766.
37. Dzikowski, R., Templeton, T.J., Deitsch, K. Variant antigen gene expression in malaria. Cell Microbiol. 2006;8(9):1371–1381.
38. Stewart, P.L., Nemerow, G.R. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 2007;15(11):500–507.
39. Marsh, M., Helenius, A. Virus entry: open sesame. Cell. 2006;124(4):729–740.
40. Loch, S., Tampe, R. Viral evasion of the MHC class I antigen-processing machinery. Eur J Physiol. 2005;451(3):409–417.
41. Taubenberger, J.K., Morens, D.M. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3(1):499–522.
42. Beigel, J.H. Influenza. Crit Care Med. 2008;36(9):2660–2666.
43. Boni, M.F. Vaccination and antigenic drift in influenza. Vaccine. 2008;26(Suppl 3):C8–C14.
44. Perrone, L.A., Tumpey, T.M. Reconstruction of the 1918 pandemic influenza virus. Infect Disord Drug Targets. 2007;7(4):294–303.
45. Chinen, J., Shearer, W.T. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol. 2008;121(2):S388–S392.
46. Merson, M.H., et al. The history and challenge of HIV prevention. Lancet. 2008;372(9637):475–488.
47. Padian, N.S., et al. Biomedical interventions to prevent HIV infection: evidence, challenges, and way forward. Lancet. 2008;372(9638):585–599.
48. Do, A.N., et al. Occupationally acquired human immunodeficiency virus (HIV) infection: national case surveillance data during 20 years of the HIV epidemic in the United States. Infect Control Hosp Epidemiol. 2003;24(2):86–96.
49. Centers for Disease Control and Prevention: Fact sheet: surveillance of occupationally acquired HIV/AIDS in healthcare personnel, as of December 2006. Available at www.cdc.gov/ncidod/dhqp/bp_hcp_w_hiv.html. Updated September, 2007.
50. Barre-Sinoussi, F., et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 1983;220(4599):868–871.
51. Goff, S.P. Host factors exploited by retroviruses. Nat Rev Microbiol. 2007;5(4):253–263.
52. Panel on Antiretroviral Guidelines for Adults and Adolescents: Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents, pp 1-139, Washington, DC, 2008 November 3, Department of Health and Human Services. Available at www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed November 14, 2008.
53. Daar, E.S. Emerging resistance profiles of newly approved antiretroviral drugs. Top HIV Med. 2008;16(4):110–116.
54. Caldwell, M.B., et al. revised classification system for human immunodeficiency virus infection in children less than 13 years of age. MMWR Morb Mortal Wkly Rep. 1994;43(RR-12):1–10. [1994].
55. Snow, S.J. John Snow: the making of a hero? Lancet. 2008;372(9632):22–23.
56. de Waal, A. On famine crimes and tragedies. Lancet. 2008;372(9649):1538–1539.
57. Hawkey, P.M. The growing burden of antimicrobial resistance. J Antimicrob Chemother. 2008;62(Suppl 1):i1–i9.
58. Werner, G., Strommerger, B., Witte, W. Acquired vancomycin resistance in clinically relevant pathogens. Future Micro. 2008;3(5):547–562.
59. Gould, I.M. Who’s winning the war? J Antimicrob Chemother. 2008;62(Suppl 3):iii3–iii6.
60. Feikin, D.R., et al. Individual and community risks of measles and pertussis associated with personal exemptions to immunization. JAMA. 2000;284(24):3145–3150.
61. Centers for Disease Control and Prevention (CDC). Intussusception among recipients of rotavirus vaccine—United States, 1998-1999. MMWR Morb Mortal Wkly Rep. 1999;48(27):577–581.
62. Schecter, R., Grether, J.K. Continuing increases in autism reported to California’s developmental services system: mercury in retrograde. Arch Gen Psych. 2008;65(1):19–24.