ALTERATIONS OF RENAL AND URINARY TRACT FUNCTION IN CHILDREN


Renal and urinary disorders occur in children as well as adults. In childhood, however, the kidney and genitourinary structures are continuing to develop, so renal dysfunction may be associated with mechanisms and manifestations that are different from those in adults. In addition, some renal and urinary disorders are congenital and involve structural anomalies of the kidney and urinary drainage system.
STRUCTURE AND FUNCTION OF THE URINARY SYSTEM IN CHILDREN
Development of the Urinary System
The embryonic urinary system develops as three sets of sequentially replaced organs, the pronephros, mesonephros and metanephros. The pronephros is a nonfunctional structure that arises at the level of the cervical and upper thoracic regions during the third fetal week and connects the primitive Wolffian duct to the cloaca as the foundation for male sexual development (Figure 37-1). The development of the mesonephros and metanephros are described in Figure 37-1. The Wilms tumor 1 (WT1) gene plays an important role at all stages of kidney development and maintenance of kidney function.1 The wingless type signaling (WNT signaling) transduction pathway also is important for mesenchyme growth and differentiation.2
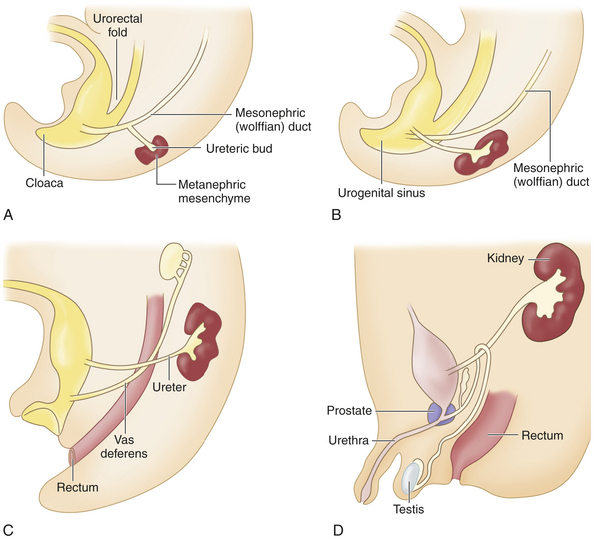
Figure 37-1 Embryonic development of the kidneys. The mesonephros begins development more caudally about the fourth fetal week and begins excretory function in the sixth week. Most of the mesonephros degenerates and disappears by the end of the embryonic period. The metanephros, the permanent kidney, arises distal to the bifurcation of the aorta and develops from two different sources: 1) the ureteric bud (metanephric duct) forms as an outgrowth of the mesonephritic (wolffian) duct and grows dorsocranially and starts subdividing to become the collecting system for the kidneys by forming the ureter, renal pelvis, and calyces; by the fifth fetal month it will have progressively branched into the collecting ducts, and 2) the metanephrogenic mesenchyme sits atop the terminal branches of the collecting ducts and develops into primitive glomeruli and uriniferous tubules (A). Genetic information from the metanephrogenic mesenchyme guides the development of the ureteric bud. Establishing the connection between the uriniferous tubules and the collecting ducts is a vital part of kidney development; errors in this stage can result in polycystic kidneys. As the embryo grows, the definitive kidneys migrate from the caudal position to the lumbar region and the ureters connect with the bladder (B, C, D). In the 8-week male embryo, the wolffian duct begins to give rise to the epididymis, the seminal vesicles, and the caudal part of the vas deferens (C). The external genitalia develop between 8 and 16 weeks, and testicular descent begins in month 7 of gestation (D). (From: Goldman L, Ausiello D: Cecil Medicine, ed 23, Philadelphia, 2008, Saunders.)
After glomeruli and tubules form, the tissues organize and progressively differentiate over approximately 30 days. Initial glomerular development is staggered, so there are glomeruli in various stages. In fact, a few of the first glomeruli formed degenerate and disappear during the later stages of fetal development. Progressive development continues into the ninth fetal month, when all metanephrogenic tissue then disappears.
As the embryo develops and the vertebral column straightens, the kidneys appear to ascend to the sacral area at about 6 weeks, to the third lumbar area by the third month, and to the first lumbar area at term. The kidneys rotate 90 degrees as they ascend so that renal tissue is lateral and the collecting system is medial.
While the kidneys mature, the cloaca becomes the urogenital sinus. It then differentiates into the vesicourethral canal, which forms the bladder and the upper urethra, and the urogenital sinus, which forms the main part of the urethra.
At birth the kidneys occupy a large portion of the posterior abdominal wall, and the ureters are proportionately shorter than those of an adult. All the nephrons are present at birth, and their number does not increase as the kidney grows and matures. The kidney reaches adult size by adolescence and, because of maturation of the tubular system, increases in weight 10-fold from the time of birth.
Urine formation and excretion begin by the third month of gestation, contributing to the amniotic fluid. In infancy the bladder lies close to the abdominal wall, making urinary bladder aspiration for diagnostic purposes a relatively simple procedure. The bladder descends into the pelvis with growth, changing from a cylindrical organ to the adult pyramidal shape. Although small amounts of urine are found in the bladder at birth, the newborn may not void for 12 to 24 hours. (The average daily urine output is shown in Table 37-1.)
Table 37-1
Average Daily Urine Output in Children

From Kempe CH, Silver KH, O’Brien D: Current pediatric diagnosis and treatment, Los Altos, CA, 1982, Lange Medical Publishers.
Immediately at birth the renal blood flow and glomerular filtration rate (GFR) increase because of a decrease in vascular resistance and the need to perform excretory functions no longer performed by the placenta. Renal vascular resistance remains higher in newborns and infants, however, which may be attributed to increased levels of circulating renin. The resistance progressively declines during the first year of development, with an increasing fraction of the cardiac output going to the kidney. The GFR continues to increase, becoming stable at 1 or 2 years, but retaining only 30% to 50% of adult levels until the end of the first year.
Fluid and Electrolyte Balance in Children
Because the kidney develops from the center toward the periphery, renal distribution of blood flow during the newborn period is primarily to the renal medulla. The result is a preferential flow to the medullary nephrons, which have comparatively short loops at this stage of development. The combination of higher blood flow and shorter loops produces a more dilute urine—approximately 600 to 700 mOsm (compared with 800 to 1200 mOsm in adults). The immature kidney is also less responsive to the actions of antidiuretic hormone (vasopressin).3 The dilute urine is accentuated by a low rate of urea excretion, which is necessary to establish the concentration gradient in the medulla. Urea excretion is low primarily because infants are in a high anabolic state and use their protein for growth.
Because of a high hydrogen ion concentration, limited ability to regulate the internal environment, and lowered osmotic pressure, the infant’s renal system has a narrow chemical safety margin. The immaturity and smaller surface area of the tubules also may diminish the water reabsorption response to antidiuretic hormone (ADH). An immature tubular transport capacity means that the ability to excrete a potassium load, reabsorb bicarbonate, or buffer hydrogen with ammonia does not become efficient until approximately 2 years of age. Consequently, any disturbance such as diarrhea, infection, fasting for diagnostic tests, or improper feeding can rapidly lead to severe acidosis and fluid imbalance because the infant can rapidly develop overhydration, or edema.4
After birth the proportion of total body water to body weight does not change markedly. Considerable change occurs, however, in the location of that body water as the child matures (see Chapter 3). The percentage of extracellular fluid volume of the newborn infant is nearly double that of an adult’s. Decrease in extracellular fluid volume occurs in two different periods of rapid growth—infancy and adolescence.
An infant has not only a greater content of extracellular fluid but also a greater rate of fluid exchange. The adult takes in and excretes approximately 2000 ml of water daily, representing 5% of the total body fluid and 14% of the extracellular fluid. In contrast, the infant’s daily exchange of 600 to 700 ml represents 290% of the total or nearly 50% of the extracellular volume, making control of dehydration and overhydration more difficult.
The composition of body fluids differs slightly with age. The total electrolyte concentration in extracellular fluids is greater in the newborn than in the adult. The concentration of sodium, chloride, phosphates, and organic acids is also greater. The concentration of bicarbonate ions is lower in the infant than in the older child, with a mild acidosis evidenced by a lowered pH. These variations, combined with a lowered plasma protein level, cause a reduced oncotic pressure of the vascular compartment and favor accumulation of fluid in the tissue spaces and an increased GFR. In the healthy child these differences remain for a few weeks or months. The premature infant and the normal newborn infant are usually in a state of well-compensated acidosis and potential edema.
ALTERATIONS IN RENAL and bladder FUNCTION IN CHILDREN
Congenital abnormalities of the kidney and urinary tract occur in about 1 out of 500 newborns.5 Structural abnormalities range from minor, nonpathologic, or easily correctable anomalies to those that are incompatible with life. For example, the kidneys may fail to ascend from the pelvis to the abdomen, causing ectopic kidneys—which usually function normally. The kidneys also may fuse in the midline as they ascend, causing a single U-shaped horseshoe kidney with an incidence of 1 per 600 births.6 Approximately one third of individuals with horseshoe kidneys are asymptomatic, and the most common problems are hydronephrosis, infection, and stone formation.7 Collectively, structural anomalies of the renal system account for approximately 45% of cases of renal failure in children.
Some anomalies are obvious at birth, whereas others remain latent. The following structural anomalies are commonly associated with urinary tract malformations8:
Chromosomal disorders, especially trisomy 13 (Patau syndrome) and trisomy 18
Absent abdominal muscles (prune-belly syndrome)
Anomalies of the spinal cord and lower extremities
Imperforate anus or genital deviation
Positive family history of renal disease (hereditary nephritis or cystic disease)
Hypospadias
Hypospadias is a congenital condition in which the urethral meatus is located on the ventral side or undersurface of the penis. The meatus can be located anywhere on the glans, the penile shaft, the base of the penis, the penoscrotal junction, or the perineum (Figure 37-2). This is the most common anomaly of the penis and occurs in about 1 in 300 infant boys. The etiology is multifactorial and related to disruptions in male hormones, including testosterone biosynthesis defects, 5alpha-reductase mutations, hormones administered for in vitro fertilization, advanced maternal age, and other environmental factors.9 Chordee, or penile torsion, may accompany hypospadias. In chordee a shortage of skin on the ventral surface causes the penis to bend or to “bow” ventrally (Figure 37-3). Penile torsion is a counterclockwise twist of the penile shaft. Partial absence of the foreskin, inguinal hernia and cryptorchidism (undescended testes, see Chapter 23) are associated with the anomaly.10
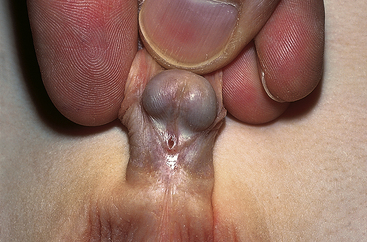
Figure 37-2 Hypospadias. (Courtesy H. Gil Rushton, MD, Children’s National Medical Center, Washington, DC; from Hockenberry MJ: Wong’s nursing care of infants and children, ed 7, St Louis, 2003, Mosby.)
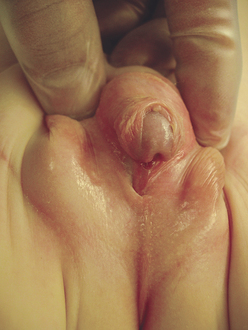
Figure 37-3 Perineal hypospadias with chordee and partial penoscrotal transposition. (From Kliegman RM et al, editors: Nelson textbook of pediatrics, ed 18, Philadelphia, 2007, Saunders.)
The goals for corrective surgery on the child with hypospadias are (1) a straight penis when erect to facilitate sexual intercourse as an adult, (2) a uniform urethra of adequate caliber to prevent spraying during urinations, (3) a cosmetic appearance satisfactory to the individual, and (4) repair completed in as few procedures as possible. Formerly performed in two or more stages, hypospadias repairs are now done in one stage. Improvements in microsurgical techniques have enhanced outcomes and decreased complications. Surgery is usually performed between 6 and 12 months of age.10
Epispadias
Epispadias and exstrophy of the bladder are the same congenital defect but expressed to a different degree. In male epispadias the urethral opening is on the dorsal surface of the penis. In females a cleft along the ventral urethra usually extends to the bladder neck. The incidence of epispadias is about 1 in 40,000 to 118,000 births. About twice as many boys as girls present with this defect.
In boys the urethral opening may be small and situated behind the glans (anterior epispadias), or a fissure may extend the entire length of the penis and into the bladder neck (posterior epispadias). Children with anterior epispadias can be continent with perhaps only stress incontinence, but those with posterior epispadias will experience constant dribbling of urine.11 Surgical repair provides good structural and functional outcomes.
Exstrophy of the Bladder
Exstrophy of the bladder is a rare extensive congenital anomaly in which the bladder opens directly onto the abdominal wall (Figure 37-4). The posterior portion of the bladder mucosa is exposed and appears bright red through a fissure in the abdominal wall. The incidence of exstrophy of the bladder is about 1 in 400,000 live births. Boys are predominant by a ratio of 5:1.12
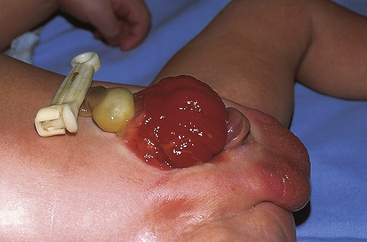
Figure 37-4 Exstrophy of bladder. (Courtesy H. Gil Rushton, MD, Children’s National Medical Center, Washington, DC; from Hockenberry MJ: Wong’s nursing care of infants and children, ed 7, St Louis, 2003, Mosby.)
Exstrophy of the bladder is caused by intrauterine failure of the abdominal wall and the mesoderm of the anterior bladder to fuse. Urine seeps onto the abdominal wall from the ureters, causing a constant odor of urine and excoriation of the surrounding skin. The rectus muscles below the umbilicus are separated, and the pubic rami (bony projections of the pubic bone) are not joined. The clitoris in girls is divided into two halves with the urethra between them. The penis in boys is epispadic. In addition, the posterior aspect of the pelvis is externally rotated, which retroverts the acetabula and causes external rotation of the feet. This causes a waddling gait when the child first learns to walk, but most children quickly learn to compensate and surgical correction is helpful.13
Because the exposed bladder mucosa becomes hyperemic and edematous, it bleeds easily and is painful. It should be covered with Silastic or a plastic dressing (e.g., kitchen plastic wrap) for protection from diaper irritation while permitting urine drainage. The unrepaired exstrophic bladder is cosmetically unacceptable and prone to cancerous changes as soon as 1 year after birth. Ideally the bladder and pubic defect should be closed before the infant is 48 hours old. Surgical reconstruction is performed usually within the first year as either a complete primary repair or as staged procedures. Staged procedures include bladder augmentation, bladder neck closure, or reconstruction of both bladder neck closures and reconstruction. Objectives of management include preservation of renal function, attainment of urinary control, prevention of infection, reconstructive repair of the defect, and improvement of sexual function and quality of life.
Cloacal exstrophy is the most rare and severe form of bladder exstrophy. The intestines, genitourinary tract, and spine may be involved, and reconstruction with restored urine and fecal control is difficult.14
Ureteropelvic Junction Obstruction
Ureteropelvic junction (UPJ) obstruction is a blockage of the tapered point where the renal pelvis transitions into the ureter.15 An intrinsic malformation of smooth muscle hypertrophy and fibrosis produces obstruction in 90% of cases.16 It is the most common cause of hydronephrosis in neonates.17 Diagnosis can be made by ultrasound, and treatment is a surgical pyeloplasty or endopyelotomy.18 During infancy or childhood, secondary ureteropelvic junction (UPJ) obstruction is caused by kinking or secondary scarring in the presence of high-grade vesicoureteral reflux. An increased risk of vesicoureteral reflux in children with UPJ obstruction affects the obstructed as well as contralateral kidneys. Other defects are sometimes associated with ureteral duplication including complete ureteral duplication (abnormal growth of two ureters and ureteral orifices draining a single kidney), incomplete duplication (bifurcation of the ureter terminates into one ureteral orifice and serves a single kidney), and ureterocele (cystic dilation of the intravesical ureter). Obstruction of the distant ureter causes dilation of the entire ureter, renal pelvis, and caliceal system.19 It occurs when a short acontractile segment of the ureter develops just above the ureterovesical junction.
Bladder Outlet Obstruction
Congenital causes of bladder outlet obstruction are rare and include urethral valves, urethral polyps, and urethral atresia. A urethral valve is a thin membrane of tissue that occludes the urethral lumen and obstructs urinary outflow in males and it is the most common cause of congenital lower urinary tract obstruction and renal failure. Most valves occur in the posterior urethra, although a few arise from the embryologically distinct anterior urethra.20 Urethral polyps rarely arise from the prostatic urethra. They often cause relatively severe obstruction and may impair renal embryogenesis and lead to urinary tract infection, vesicoureteric reflux, and renal failure.21 Urethral atresia is absence of the urethra and is rare.
Congenital urethral valves or polyps can be diagnosed with prenatal ultrasound and treated with prenatal bladder shunting or with resection during the first days of life.22 Infants with significant renal (and pulmonary) hypoplasia who are unable to undergo primary resection may be managed with a vesicostomy, a small opening created by pulling the bladder wall to the abdomen.23
Hypoplastic or Dysplastic Kidneys
During embryologic development the ureteric duct grows into the metanephric tissue, triggering the formation of the kidneys. If this growth does not occur, the kidney is absent—a condition called renal aplasia. Occasionally a hypoplastic kidney, a very small normal kidney, may develop. These aberrations may be unilateral or bilateral; the occurrence may be incidental or familial. Bilateral hypoplastic kidneys are a common cause of chronic renal failure in children. Segmental hypoplasia (the Ask-Upmark kidney) is not the result of developmental abnormalities but instead a deformity acquired secondary to vesicoureteral reflux.24
Renal dysplasia usually results from abnormal differentiation of the renal tissues; for example, primitive glomeruli and tubules, cysts, and nonrenal tissue (such as cartilage) are found in the dysplastic kidney. Dysplasia usually is associated also with a functional or organic obstruction of the collecting system. The obstruction may begin before birth, as in prune-belly syndrome (congenital absence of abdominal muscles), posterior urethral valves, or ureteroceles (dilation of the ureter where it enters the bladder).
Renal Agenesis
Renal agenesis (the absence of one or both kidneys) may be unilateral or bilateral, and it may occur randomly or be clearly hereditary. The condition may occur as an isolated entity or as a problem associated with anomalies in other organs.25
Unilateral renal agenesis occurs in approximately 1 of 1000 live births. Males are more often affected, and it is usually the left kidney that is absent. The single kidney is often completely normal so that the child can expect a normal, healthy life. The normal solitary kidney grows because of compensatory hypertrophy before and after birth, and by the time the child is several years older, the volume of this kidney may approach twice the normal size.26
In some instances the single kidney is abnormally formed and associated with abnormalities of its collecting system.27 Extrarenal congenital abnormalities are relatively more common with unilateral renal agenesis.
Bilateral renal agenesis (also called Potter syndrome) occurs in about 1 to 4 in 10,000 live births,28 and 75% of affected infants are male. Bilateral renal agenesis results from either an abnormal development of the normal progression from pronephros to mesonephros to metanephros or an isolated bilateral failure of development of the ureteral buds. The term Potter syndrome refers to the association with a specific group of facial anomalies (wide-set eyes, parrot-beak nose, low-set ears, and receding chin). Affected infants rarely live more than a few hours. Approximately 40% of affected infants are stillborn. Renal agenesis can be detected prenatally by ultrasound.
Polycystic Kidneys
Polycystic kidney disease (PKD) is an autosomal dominant inherited disorder that occurs in about 1 in 1000 live births. Mutations of two genes, PKD-1 (chromosome 16) and PKD-2 (chromosome 4) account for the disease most often found in adults. The gene products (polycystins) regulate growth and differentiation of the tubular epithelium.29 Defects in the formation of epithelial cells and their cilium result in cyst formation and obstruction accompanied by destruction of renal parenchyma, interstitial fibrosis, and loss of functional nephrons. Other organs also may have cysts, including the liver and pancreas, and hypertension, heart valve defects, and cerebral and aortic aneurysms may develop. Other signs include urinary tract infection, hematuria, and flank pain. Diagnosis is usually confirmed by ultrasound. Individuals may live for decades before developing symptoms; PKD is often an adult disease. Up to 10% require renal replacement therapy.30
In children there is an autosomal recessive form of PKD (ARPKD) with cystic changes in the kidney and liver. The gene mutation for ARPKD encodes a protein important to maintaining structural integrity and cellular function of the kidneys and liver.31
Glomerular Disorders
The most common glomerular disorders in children are glomerulonephritis, nephrotic syndrome, and hemolytic uremic syndrome. Most glomerular diseases are acquired and immunologically mediated. The disease can be acute or chronic; renal failure is rare.
Glomerulonephritis
Glomerulonephritis includes a number of renal disorders in which proliferation and inflammation of the glomeruli are secondary to an immune mechanism (Table 37-2). (The major glomerulopathies and their histologic characteristics can be reviewed in Chapter 36.) Chronic glomerulonephritis is the causative factor for 30% to 50% of renal failure in children and is the condition responsible for most school-age and teenage children requiring dialysis and kidney transplantation.
Acute Poststreptococcal Glomerulonephritis: Acute poststreptococcal glomerulonephritis (PSGN) is one of the most common postinfectious renal diseases in children ages 5 to 15 years. It occurs after a throat (pharyngitis) or skin (impetigo) infection with nephritogenic strains of group A beta-hemolytic streptococci and is characterized by a sudden onset of gross hematuria, edema, hypertension, and renal insufficiency.
The pathophysiology of PSGN in children is similar to that occurring in adults (see Chapter 36). Antigen-antibody complexes of immunoglobulin G (IgG), IgA, and C3 complement are deposited in the glomerulus or the antigen may be trapped within the glomerulus and immune complexes formed in situ. The exact mechanism of immune complex formation is unknown. The immune complexes initiate inflammation and glomerular injury. Immunofluorescence microscopy shows lumpy deposits of immunoglobulin and complement on the glomerular basement membrane (see Figure 36-8). Increased vascular permeability and loss of electrical negative charge along the glomerular vascular membrane leads to hematuria and proteinuria. Hypertension occurs with increased blood volume and release of endothelin-1, a potent vasoconstrictor.32
Typically a child is in good health until the onset of an upper respiratory or skin infection. Pharyngeal infections are most common during cold weather. Skin infections from impetigo, infected insect bites, or varicella sores usually occur during warm weather. One to 2 weeks later, mild proteinuria (less than 2 g per 4 hours), hematuria, and periorbital edema appear. The urine is usually smoky brown or cola colored because of the presence of red blood cells, and the volume is reduced. The onset of symptoms in the child is abrupt and consists of flank or midabdominal pain, irritability, general malaise, and fever. Acute hypertension may cause headache; vomiting; somnolence; and other central nervous system (CNS) manifestations, including seizures. Cardiovascular symptoms are related to circulatory overload and are compounded by hypertension. These include dyspnea, tachypnea, and an enlarged, tender liver. The most severely affected children develop acute renal failure with oliguria. As many as half the children affected are asymptomatic.
The disease is usually mild and runs its course in 1 month, but urine abnormalities may be found up to 1 year after the onset. Some children (less than 1%) become oliguric and develop rapidly progressive glomerulonephritis, whereas others slowly progress to chronic glomerulonephritis. Prolonged proteinuria and abnormal GFR indicate an unfavorable prognosis. More than 95% recover completely.
Acute glomerulonephritis (AGN) may be accompanied by a positive throat or skin culture for Streptococcus. Antistreptolysin-O titers confirm a recent streptococcal infection. Antihyaluronidase, antideoxyribonuclease B, and antistreptokinase antibody are other diagnostic markers. The urine usually contains red blood cells and proteins. Treatment is symptom specific. Because oliguria and hypertension are common, fluid, sodium, and potassium intakes are restricted. Antihypertensive medication and diuretic agents are indicated during the acute phase.
Immunoglobulin A Nephropathy: Immunoglobulin A (IgA) nephropathy (Berger nephropathy) is the most common type of childhood glomerulonephritis, occurs almost twice as often in males as in females, and is rare in blacks. It is characterized by deposition mainly of IgA, but also some IgM and complement proteins in the mesangium of the glomerular capillaries. There is no evidence of systemic immunologic disease such as systemic lupus erythematosus or anaphylactoid purpura (Henoch-Schönlein purpura). IgA may show decreased glycosylation that favors antibody formation and mesangial deposition.33 Binding of the IgA to mesangial cells stimulates them to proliferate, secrete extracellular matrix proteins, and release inflammatory cytokines and chemokines (interleukin-6, tumor necrosis factor-alpha, and transforming growth factor beta 1) that cause injury. The damage to the glomerulus can progress to glomerulosclerosis and tubular interstitial involvement, which is usually reversible.34
The classic presentation of the disease is recurrent gross hematuria, often after a respiratory infection. Most continue to have microscopic hematuria between the attacks of gross hematuria. Many children also have a mild proteinuria in spite of otherwise normal renal function and may report flank pain caused by renal swelling. The diagnosis may be missed and specific diagnosis is made by renal biopsy.35 Treatment is supportive with control of hypertension. Some studies report that tonsillectomy and use of corticosteroids reduces formation of IgA and reduces proteinuria.36 Long-term follow-up consists of evaluating blood pressure, urinalysis, proteinuria levels, and renal function every 6 to 12 months. Approximately 25% of affected children develop the progressive form of the disease with hypertension, proteinuria, and decreasing renal function that extends into adulthood.37 Hypertriglyceridemia and hyperuricemia are predictors of poor outcome.38 These children eventually require dialysis and transplantation.
Henoch-Schönlein Purpura Nephritis—IgA Nephropathy: Henoch-Schönlein purpura nephritis, also known as anaphylactoid purpura, is an IgA nephropathy that affects the glomerular blood vessels causing inflammation and damage to the vessel wall. The disease also involves small vessels in the skin and gut. The most typical renal lesion is focal segmental glomerulonephritis (see Table 36-7 in Chapter 36) with IgA deposits in the mesangium. Transient hematuria and mild proteinuria without functional impairment are more common in children. Children also may exhibit signs of intestinal colic and arthralgia.39 The development of interstitial fibrosis and crescent formation from subepithelial immune deposits along the glomeruli increases the risk of chronic renal failure.40 Most children recover with supportive care. Severe symptoms require steroids and other immunosuppressant drugs.41
Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) is an acute disorder characterized by microangiopathic hemolytic anemia and thrombocytopenia and is the most common cause of acute renal failure in young children.42 The etiology remains unknown, although an association between HUS and both bacterial and viral agents has been established. The disease also occurs with cancer and use of chemotherapeutic agents. Escherichia coli (E. coli) O157:H7, a shiga toxin-producing bacterium, is the most commonly associated microorganism in the United States, usually found in undercooked meat and unpasteurized milk.43 The disease occurs in infants and children younger than 4 years. The prognosis has improved dramatically in recent years, with more than 90% of children surviving and most regaining normal renal function.
PATHOPHYSIOLOGY In HUS, verotoxin from E. coli is absorbed from the intestines into the blood, binds to polymorphonuclear leukocytes, and is transported to the kidney, causing a cascade of effects including lysis of glomerular capillary endothelial cells, separation of endothelial cells from the basement membrane, activation and aggregation of platelets, and activation of the coagulation cascade. The glomerular arterioles becomes swollen and occluded with platelets and fibrin clots. There is decreased glomerular filtration, and the damaged glomerular membrane results in hematuria and proteinuria. Oliguria with renal failure occurs in up to 50% of children. Narrowed vessels damage erythrocytes as they pass through. These damaged red blood cells, identified as burr cells, helmet cells, and fragmented red blood cells, are removed by the spleen, causing acute hemolytic anemia. Fibrinolysis, the process of dissolution of a clot, acts on precipitated fibrin, causing the fibrin split products to appear in serum and urine. The platelet clustering within damaged vessels, combined with the damage and removal of platelets, produces thrombocytopenia. Fibrin-rich thrombi can be found throughout the microcirculation.44 Other tissues, including the brain, liver, heart, and intestines, are often involved, which portends a poorer prognosis.
CLINICAL MANIFESTATIONS Typical HUS is preceded by a prodromal gastrointestinal (GI) illness with diarrhea and is known as D+ HUS. Less frequently atypical or sporadic HUS is preceded by an unknown event or an upper respiratory infection and is known as D- HUS. A rare familial form is associated with mutations in complement proteins. The onset of D+ HUS occurs about 1 to 2 weeks after a GI illness with a symptom-free 1- to 5-day period. There is sudden onset of pallor, bruising or purpura, irritability, and oliguria. Slight fever, anorexia, vomiting, diarrhea (with the stool characteristically watery and blood stained—hemorrhagic diarrhea), abdominal pain, mild jaundice, and circulatory overload are accompanying symptoms. Seizures and lethargy indicate CNS involvement. Renal failure is apparent within 2 days to 2 weeks of onset. The renal failure causes metabolic acidosis, uremia, hyperkalemia, and often hypertension.
EVALUATION AND TREATMENT Clinical evaluation includes history of preexisting illness, presenting symptoms, and urine and blood analysis. Antibiotics are not used in the initial treatment because they increase shiga toxin release and increase the risk of HUS. Management consists of maintaining nutrition and hydration (to dilute toxins) and controlling hypertension, hyperkalemia, and seizures.45 When renal failure occurs, early and frequent dialysis is indicated. Blood transfusions with packed red blood cells are needed to maintain reasonable hemoglobin levels. The response to treatment is usually good and the disease is self-limiting. Death usually occurs from complications related to CNS or myocardial involvement.46 Preventing shiga toxin–producing bacterial infection (i.e., E. coli) prevents HUS.
Nephrotic Syndrome
Nephrotic syndrome is a term used to describe a symptom complex characterized by proteinuria, hypoproteinemia, hyperlipidemia, and edema. The syndrome is more common in children than adults. When no other identifiable causes are found the condition is termed primary (idiopathic) nephrotic syndrome. If it results from a systemic disease or other causes (e.g., drugs, toxins, diabetes mellitus, lupus nephritis) it is called secondary nephrotic syndrome. Primary nephrotic syndrome is usually described by histopathology (i.e., minimal change nephropathy [MCN], focal segmental glomerulosclerosis [FSGS], membraneous nephropathy [MN], or membranoproliferative glomerulonephritis [MPGN]) (see Table 36-7). Secondary nephrotic syndrome has the same patterns of histopathology but is associated with an underlying cause.
Approximately 95% of cases of nephrotic syndrome in children occur in the absence of systemic or preexisting renal disease. Primary nephrotic syndrome is found predominantly in preschool children, with a peak incidence of onset between 2 and 3 years of age. It is rare after 8 years of age. Boys are affected more often than girls. No prevalent racial or geographic distributions are evident. The incidence is approximately 3 per 100,000 children per year.
PATHOPHYSIOLOGY The cause of nephrotic syndrome in children is usually idiopathic and includes minimal change nephropathy (85%), FSGS (10%), and mesangial proliferative nephropathy (MPN) (5%).47 Secondary nephrotic syndrome may develop during the course of several different renal or systemic diseases. The pathophysiology (see Figure 36-11) and common clinical manifestations of nephrotic syndrome in adults are described in Chapter 36 (Table 36-8), and are similar in children. The most common causes of nephrotic syndrome in children are presented here.
Minimal change nephropathy (MCN), also known as lipoid nephrosis, is the most common cause of nephrotic syndrome in children. A systemic immune mechanism is a likely cause of the disease, but the true etiology is unknown. MCN is found in 85% of children with idiopathic nephrosis. The mechanism of increased glomerular permeability is unknown but is related, in part, to release of permeability factors from abnormal circulating T cells that injure the glomerular epithelial cells. The glomeruli appear normal, and immunoglobulin deposition is usually absent. The only change is fusion of epithelial cell podocyte foot processes.47 There are few other renal structural abnormalities. Loss of the electrical negative charge and increased permeability within the glomerular capillary wall leads to albuminuria.48,49 Hyperlipidemia leads to hyperlipiduria and primarily results from increased hepatic lipid synthesis and decreased plasma lipid catabolism.50
Focal segmental glomerulosclerosis (FSGS) is present in approximately 15% of children with nephrotic syndrome and is more common in blacks. The frequency of FSGS is increasing in children and adults.51 The primary injury is effacement (thinning or deletion) of epithelial podocytes, with a significant increase in pore size leading to impairment of size selectivity and proteinuria. Progressive disease results in proliferation of endothelial and mesangial cells with occlusion and sclerosis of glomerular capillaries. The more severe the proteinuria, the more likely that end-stage renal disease will occur.
Edema is the classic symptom of nephrotic syndrome. Several factors contribute to edema formation with hypoalbuminemia (decreased plasma oncotic pressure) and sodium retention as major contributors. The movement of fluid from the vascular to the interstitial space can decrease blood volume and increase activity of aldosterone and antidiuretic hormone (vasopressin), and decrease atrial natriuretic peptide, all of which promote fluid retention.52
Hyperlipidemia occurs in inverse proportion to the decrease in plasma proteins, particularly albumin. There are high concentrations of triglycerides, low-density lipoprotein (LDL), and very low-density lipoprotein (VLDL) cholesterol. High-density lipoprotein (HDL) cholesterol concentration is decreased. Hypoalbuminemia leads to a deficiency in the carrier protein for the transport of fatty acids, and they remain elevated in the serum. There is hepatic compensation for hypoalbuminemia with increased synthesis of lipoproteins to maintain plasma oncotic pressure. Hypoalbuminemia also leads to an increased hepatic stimulus for synthesis of LDL and VLDL cholesterol by the liver. Serum lipids may remain elevated from 1 to 3 months after remission of proteinuria.
Hypercoagulation with risk for arterial or venous thrombosis results from abnormalities in the coagulation pathways during nephrotic syndrome. Although rare, thrombosis can occur in the brain or lung. Family history and predisposing risk factors should be evaluated. Anticoagulants may be required.53
Congenital nephrotic syndrome (Finnish type) is caused by an autosomal recessive mutation of the NPHS1 gene that encodes an immunoglobulin-like protein, nephrin, at the podocyte slit membrane. Lack of nephrin causes heavy proteinuria.54 The disease usually manifests within the first 3 months of life, and these babies do not respond to steroid treatment.55
CLINICAL MANIFESTATIONS Onset of nephrotic syndrome is insidious, with periorbital edema as the first sign. The edema is most noticeable in the morning but subsides during the day as fluid shifts to the abdomen and lower extremities (Figure 37-5). Parents become alerted to an abnormality when they notice diminished “frothy” or “foamy” urine and when edema becomes pronounced with ascites, respiratory difficulty from pleural effusion, and labial or scrotal swelling.
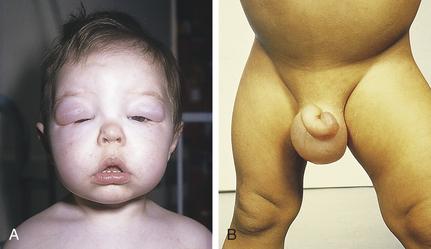
Figure 37-5 Nephrotic syndrome A, Facial edema. B, Gross edema of scrotum and legs with abdominal distention from ascites. (From Lissauer T, Clayden G: Illustrated textbook of pediatrics, St Louis, 2001, Mosby.)
Edema of the intestinal mucosa may cause diarrhea, anorexia, and poor absorption. Edema often masks the malnutrition caused by malabsorption and protein loss. Because of protein deficiency, changes in the quality of hair indicate a malnourished state. Pallor, with shiny skin and prominent veins, is also common. Blood pressure is usually normal or slightly decreased. The child has an increased susceptibility to infection, especially pneumonia, peritonitis, cellulitis, and septicemia. Irritability, fatigue, and lethargy are common. Infants born with congenital nephrotic syndrome have large fontanels, have separated cranial sutures, and may show gingival hyperplasia.56
EVALUATION AND TREATMENT The diagnosis of nephrotic syndrome is evident from the clinical presentation and findings of proteinuria, hyperlipidemia, and lipiduria. Several diagnostic tests, including kidney biopsy, may be required to determine whether the cause is an intrinsic renal disease or a consequence of systemic disease.
The goals of treatment are to reduce the excretion of protein and to maintain a protein-free urine. Prevention or treatment of infection, control of edema, establishment of a balanced nutritional state, and restoration of normal metabolic processes also are important in managing the disorder and avoiding adverse aspects of treatment. Basic management of nephrotic syndrome includes activity as tolerated; a low-sodium, well-balanced diet; diuretics (furosemide [Lasix]); angiotensin-converting enzyme (ACE) inhibitors (inhibits formation of angiotensin II and aldosterone resulting in decreased blood pressure and decreased renal sodium reabsorption); paracentesis (for ascites); and skin care. Corticosteroids are the primary therapeutic agents, and outcomes in children are often described according to their response to steroid therapy (Table 37-3). Steroid-sensitive nephrotic syndrome usually results in complete remission without serious adverse effects of therapy. Children who fail to respond to prednisone within 8 weeks are termed steroid resistant and may be treated with noncorticosteroid immunosuppressive agents or combinations of corticosteroids and noncorticosteroid immunosuppressives to prolong remission.57,58 Renal transplant is performed for those children who progress to renal failure; those with FSGS are at greatest risk.59
Table 37-3
Corticosteroid Treatment in Children with Nephrotic Syndrome
| Response to Corticosteroid | Incidence (%) | Outcomes |
| Steroid sensitive | >80 | Single course of therapy, low recurrence rate |
| Steroid dependent (frequently relapsing) | 7 | Intermittent exacerbations with remissions for several years |
| Steroid resistant | Rare | Resistance to steroids, eventual development of chronic renal failure |
Data from Kim JS et al: Kidney Int 68(3):1275-1281, 2005.
Renal Injury
Renal injury, either acute or chronic, is rare in children. The pathophysiology and management are similar to renal injury in adults (see Table 36-9). A modification of the RIFLE criteria (R = risk, I = injury, F = failure, L = loss, and E = end-stage kidney disease [ESKD]) that was proposed to standardize the definition of acute kidney injury in adults has been used in critically ill children (pRIFLE criteria) (Table 37-4).60
Table 37-4
Pediatric-Modified RIFLE (pRIFLE) Criteria

eCCl, Estimated creatinine clearance; GFR, glomerular filtration rate, pRIFLE, pediatric risk, injury, failure, loss, and end-stage renal disease.
Data from Akcan-Arikan A et al: Kidney Int 71(10):1028-1035, 2007.
The most common causes of prerenal acute renal injury are dehydration, hemorrhage, and sepsis. Glomerulonephritis, hemolytic uremic syndrome, and hypersensitivity reactions to drugs or infectious agents are the most common causes of intrinsic acute renal injury. Obstructive uropathies, such as posterior urethral valves and obstruction of the ureteropelvic junction, are associated with postrenal acute renal injury.61 Chronic renal failure in very young children is commonly associated with congenital renal structural abnormalities. In older children the most common cause is glomerulonephropathies.62,63 Renal transplants are successful in children.64,65 The use of growth hormone before and after transplant has contributed to normal growth and development particularly in prepubertal children.66
Bladder Disorders
Urinary tract infection (UTI) is the colonization of a pathogen anywhere along the urinary tract (urethra, bladder, ureter, kidney) and occurs commonly in children.67 The incidence of UTI is greater in boys up to 1 year of age, particularly among those not circumcised.68 Girls have a greater incidence after 1 year of age with an increasing incidence at adolescence. UTIs in girls occur as a result of perineal bacteria, especially E. coli, ascending the urethra. The incidence of UTI in neonates and infants is low, but the risk is high because of an immature immune system.
The pathophysiology of UTIs in children is similar to that of adults (see Chapter 36). However, UTIs in children are often clinically categorized as first or recurrent infection.69 Individual susceptibility, bacterial virulence, and the host’s anatomy (presence of reflux, obstruction, stasis, stones, or structural anomalies of the urinary tract) affect the severity of the disease. The recurrence rate is approximately 30% to 40%70 and is highest in females. Sexually active female adolescents are more likely to have a UTI. Similar to adult women, susceptibility is increased when genetically controlled blood group antigens (P1 and Lewis blood group nonsecretor) are present on surface uroepithelial cells and act as receptors for bacterial attachment.71
Cystitis, or infection of the bladder, results in mucosal inflammation and congestion. This causes detrusor muscle hyperactivity and a resulting decrease in the bladder capacity. It also can lead to transient reflux of urine up the ureters, sending bacteria all the way to the kidney, causing acute or chronic pyelonephritis and renal abscesses or scarring.
Symptoms of UTI in children are nonspecific, and differentiating whether an infection is in the bladder or kidneys is difficult based on symptoms alone. Infants usually develop nausea, vomiting, diarrhea, or jaundice. Infants and young children may present only with fever of undetermined origin and others may present with urinary tract symptoms of frequency; urgency; enuresis or incontinence in a previously dry child; abdominal, flank, or back pain; foul-smelling urine; and sometimes hematuria. Acute pyelonephritis usually causes chills, fever, and flank or abdominal pain along with enlarged kidney(s) caused by edema. Chronic pyelonephritis may be asymptomatic.
Diagnosis of UTIs is by urine culture of a pathogen prior to antimicrobial treatment. An accompanying urinalysis can show pyuria and microscopic hematuria. The presence of casts in the urine can indicate pyelonephritis. Ultrasound, voiding cystourethrography (VCUG) or radionuclide cystography, computed tomography (CT) scan, or voiding cystourethrogram may be necessary to rule out obstructions, abscesses, or reflux, particularly in young children that do not respond to antimicrobial therapy.72
With treatment, UTI symptoms are usually relieved in 1 to 2 days and the urine becomes sterile. A 2- to 4-day course of oral antibiotics is effective for uncomplicated UTI.73 More potent medications may be required if the child has a recurrent UTI, has a complicated UTI including congenital abnormalities of the urinary tract, or is immunosuppressed. About 3 to 6 weeks after treatment is completed, all children with a first UTI should have imaging done to rule out reflux and urinary tract abnormalities. Follow-up urine cultures should be done 2 to 3 weeks after the medication is completed and every 3 months for the next 1 to 2 years to monitor for recurrence, to prevent renal scarring, and for assessment of normal renal development and function, even if the child is asymptomatic.
Surgical correction of reflux or obstruction is necessary before the urinary tract can be sterilized. Children who develop frequent recurrences and who do not have surgically correctable anomalies may need prophylactic antibiotic therapy. These children also require regular cultures to rule out asymptomatic infections with resistant microorganisms.
Vesicoureteral Reflux
Vesicoureteral reflux (VUR) is the retrograde flow of bladder urine into the ureters. Reflux allows infected urine from the bladder to be repeatedly swept up into the kidneys. The reflux perpetuates infection by preventing complete emptying of the bladder, because infected, refluxed urine drains back into the bladder at the end of each voiding. In addition, the reflux allows the maximal intravesical pressure to be transmitted to the renal calyces and pyramids. The combination of reflux and infection is an important cause of pyelonephritis, especially in children younger than 5 years.
Vesicoureteral reflux occurs more often in girls by a ratio of 10:1 and is uncommon in blacks. Its incidence is approximately 1 in 1000 children, and siblings of those affected have up to a 50% chance of developing reflux.74 Although reflux is considered abnormal at any age, the shortness of the submucosal segment of the ureter during infancy and childhood renders the antireflux mechanism relatively inefficient and delicate. Thus reflux is seen commonly in association with infections during early childhood but rarely in older children and adults. (Among adults with UTIs, the incidence of reflux is approximately 5%.)
Reflux may be unilateral or bilateral, and it can be classified or graded (Figure 37-6) for comparative purposes:
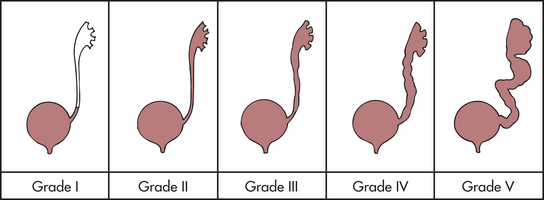
Figure 37-6 Grades of reflux. (From Retik A, Cukier J, editors: Pediatric urology, Baltimore, 1987, Williams & Wilkins.)
Grade I—Reflux into a nondilated distal ureter
Grade II—Reflux into the upper collecting system without dilation
Grade III—Reflux into dilated ureter or blunting of calyceal fornices
Grade IV—Reflux into a grossly dilated ureter
Grade V—Massive reflux with ureteral dilation and tortuosity and effacement of the calyceal details; occurs almost exclusively in male infants75
PATHOPHYSIOLOGY Primary reflux results from a congenitally abnormal or ectopic insertion of the ureter into the bladder. In some infants VUR may be related to inadequate relaxation of the external urethral sphincter.76 Occasionally the condition is hereditary. Secondary reflux is more serious and may be transient or persistent. It develops in association with infection, malformations of the ureterovesical (UV) junction, increased intravesical pressures, and surgery on the UV junction (Figure 37-7). Urinary tract infection associated with VUR may lead to permanent renal scarring, particularly when there is pyelonephritis.77 The actual cause of renal cell damage and scarring is unknown but contributing factors include inflammatory cytokines activated by bacterial virulence factors.78
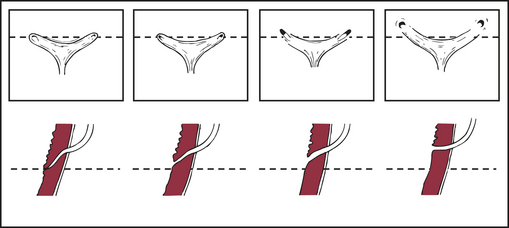
Figure 37-7 Normal and abnormal configuration of the ureteral orifices. Left to right: Progressive lateral displacement of the ureteral orifices and shortening of the intramural tunnels. Top row: Endoscopic appearance. Bottom row: Sagittal view through the intramural ureter. (From Behrman R et al, editors: Nelson textbook of pediatrics, ed 16, Philadelphia, 2000, Saunders.)
CLINICAL MANIFESTATIONS Children with reflux have recurrent UTIs or unexplained fever, poor growth and development, irritability, and feeding problems. Children and adults may have a family history of reflux or UTI, pain with voiding, and signs of urinary obstruction or nephropathy.
EVALUATION AND TREATMENT Early diagnosis of VUR in infants is critical to preventing renal scarring. Some infants may have renal scarring at birth from intrauterine damage caused by destruction or reflux.
In addition to the history of recurrent UTIs and other symptoms, imaging may be required for diagnosis and assessment of structural change, scarring, urinary tract function, and risk for future infection and renal damage.78 Most children with vesicoureteral reflux respond to nonoperative management aimed at prevention and treatment of infection. Spontaneous remission of grades I and II reflux may occur in 30% to 60% of children younger than 5 years. Children with grades III and IV reflux need long-term monitoring and prophylactic antibiotics, but there are concerns regarding antibiotic resistance.79 Endoscopic injection with biomaterials may be a treatment alternative.80 Recurrent infection may require surgical intervention. In cases of grade V reflux, early surgical intervention is indicated to prevent renal scarring, although spontaneous resolution can occur during the first year. Siblings of children
with vesicoureteral reflux should be monitored for risk of developing vesicoureteral reflux.81 Up to 50% have been found to have asymptomatic vesicoureteral reflux.82,83
Wilms Tumor
Wilms tumor is an embryonal tumor of the kidney arising from epigenetic and genetic changes that lead to abnormal proliferation of renal stem cells (metanephric blastema). It is also known by the histologic name of nephroblastoma and is the most common solid tumor occurring in children.
The incidence of Wilms tumor remains constant in the United States, with approximately 500 children diagnosed each year. Most children are between 1 and 5 years of age when they are diagnosed. The peak incidence occurs between 2 and 3 years of age. Wilms tumor is slightly more common in females and in black than in white children, and is less common in Asian children.84,85
Microscopically, Wilms tumor is composed of three cellular components: stromal, epithelial, and blastemic. This occurs because blastemic cells, which are primitive and undifferentiated, may have partially developed into epithelial or stromal tissue. With each of these three cellular components, varying stages of differentiation may be evident within the tumor.
PATHOGENESIS Wilms tumor has sporadic and inherited origins. The sporadic form occurs in children with no known genetic predisposition. Inherited cases, which are relatively rare (1% to 2% of cases), are transmitted in an autosomal dominant fashion.
The Wilms tumor suppressor gene (WT1) has been located at various chromosomal regions.86,87 The deletion or inactivation of the WT1 gene accounts for about 5% to 10% of Wilms tumor cases. The WTX gene located on the X chromosome is inactivated and found in 7% to 30% of cases; these tumors lack WTI mutations.88,89
The pathogenesis of the sporadic and inherited forms of Wilms tumor is similar to retinoblastoma (the “two hit” mutation) (see Figure 19-16). In the inherited form of the disease, the child inherits the loss of one copy of the Wilms tumor-suppressor gene (WT3) in all of the primitive metanephric blastemic (fetal renal) cells that normally differentiate into the renal tubules and glomeruli (“first hit”). All that is needed is the loss of the other copy of the gene for a Wilms tumor to develop (“second hit”). Because many cells are vulnerable to the loss of this second copy of the gene, bilateral presentation of Wilms tumor (tumor in both kidneys) occurs occasionally in the inherited form of the disease. In the sporadic form of Wilms tumor, both copies of the gene are lost during fetal development. Because normal renal development occurs during the eighth to the thirty-fourth week of gestation, gene loss likely occurs during this time.90
Approximately 10% of children who have Wilms tumor also have loss of other important genes and therefore have a number of congenital anomalies. These anomalies include aniridia (lack of an iris in the eye), hemihypertrophy (an asymmetry of the body), and genitourinary malformations (i.e., horseshoe kidneys, hypospadias, ureteral duplication, polycystic kidneys, uterine abnormalities).91,92 Children with congenital anomalies as well as Wilms tumor are more likely to have the inherited bilateral form of the disease.
CLINICAL MANIFESTATIONS Most Wilms tumors (90%) present as an enlarging asymptomatic upper abdominal mass in a healthy, thriving child. Other presenting complaints include vague abdominal pain, hematuria, fever, and hypertension.93
EVALUATION AND TREATMENT On physical examination the tumor feels firm, nontender, smooth, and generally is a solitary mass of varying size confined to one side of the abdomen. Once an abdominal mass is detected, diagnostic imaging demonstrates a solid intrarenal mass.
Diagnosis is based on surgical biopsy. Additional laboratory and radiologic studies are used to evaluate the presence or absence of metastasis. The most common sites of metastasis are regional lymph nodes and the lungs and less commonly liver, brain, and bone.
Staging systems for Wilms tumor have been developed and serve as guides to treatment. The most widely accepted system was developed by the National Wilms Tumor Study Group (Table 37-5). The system is based on surgical findings and the extent of disease at diagnosis.94 Children are further classified as either high or low risk according to favorable or unfavorable histology (anaplasia).
Table 37-5
| Stage | Tumor Characteristics |
| Stage I (40% to 45%) | Tumor limited to the kidney, completely resected |
| Stage II (20% to 25%) | Tumor ascending beyond the kidney or into vessels of renal sinus, but appearing to be totally resected |
| Stage III (20% to 25%) | Residual nonhematogenous tumor confined to the abdomen, positive lymph nodes in renal hila |
| Stage IV (10%) | Hematogenous metastases (e.g., lung, liver, bone, brain) |
| Stage V (5%) | Bilateral disease either at diagnosis or later, but need to stage each kidney |
NOTE: Staging system of the Third National Wilms Tumor Study Group (NWTS-3).
Data from American Cancer Society. Available at www.cancer.org/docroot/CRI/content/CRI_2_4_3X_How_is_Wilms_tumor_staged.asp.
Surgical exploration and resection begin the treatment of Wilms tumor. In bilateral disease, surgical intervention may include heminephrectomy of the less involved kidney and nephrectomy of the other. Radiation therapy has been found to be most effective if begun 1 to 3 days after surgery for stages III and IV disease and metastases. Chemotherapy is specific to histology and stage of disease.94
The overall cure rate is as high as 95% for children with stage I through stage III disease (Table 37-6). Prognosis is improving for children with metastases, and this is one of the few tumors for which lung metastases have been cured. Recurrent disease is treated aggressively in children with favorable histology.
Table 37-6
National Wilms Tumor Study 4-Year Survival Rates
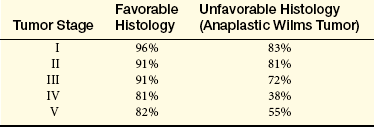
From National Wilms Tumor Studies, American Cancer Society. Available at www.cancer.org/docroot/CRI/content/CRI_2_4_3X_How_is_Wilms_tumor_staged.asp.
Enuresis
Enuresis refers to the voluntary or involuntary passage of urine by a child who is beyond the age when voluntary bladder control should have been acquired. Bladder control is accomplished by most children before the age of 4 years. Functional incontinence is urinary incontinence in which no structural or neurologic abnormality can be identified. The underlying mechanism may include disorders of both the storage and voiding phases of the bladder cycle.95
The incidence of enuresis is difficult to determine because it is not a problem that parents readily share with others and because definitions vary according to cultural norms and family practices. According to research data, the incidence of enuresis in children older than 5 years ranges from 15% to 20%. Boys are more enuretic than girls by a ratio of 3:2. Teenage and adult enuresis is usually a continuation of childhood bed-wetting in about 2% of individuals.96,97
PATHOGENESIS Multiple pathologic factors are likely responsible for enuresis. All or part of each one might be operating in a given child. A reasonable approach is to eliminate organic or physiologic causes for enuresis before exploring the psychologic ones.
Organic causes of enuresis account for 2% to 10% of cases. The causes include urinary tract infections; neurologic disturbances; congenital defects of the meatus, urethra, and bladder neck; allergies; or alteration in renal tubular ion and water transport related to prostaglandin secretion.98 Disorders that increase the normal output of urine, such as diabetes mellitus and diabetes insipidus, or disorders that impair the concentrating ability of the kidney must be considered in the evaluation of enuresis.
Enuresis in children is possibly caused by a maturational lag. Studies have demonstrated that the child with enuresis has a smaller functional bladder capacity than a nonenuretic child.99 A number of children show a general developmental delay along with elevated intravesical pressure and spikelike detrusor contractions during bladder filling. Enuresis may spontaneously disappear in these children as they get older. Other studies have shown that children with enuresis completely fill and empty their bladder several times each night because of a constant urine output and a stable level of ADH. Children who remain dry usually have elevated levels of ADH and thus a decreased urine output at night which may be combined with reduced bladder capacity.100 Still other studies indicated loss of diurnal variation in ADH.101
Genetic factors as a cause of enuresis are being investigated. Linkages have been proposed between nocturnal enuresis and chromosomes 8, 12, 13, and 22.102 Bed-wetting does occur with high frequency among parents, siblings, and other near relatives of symptomatic children. These observations are further supported by a high concordance rate in enuretic monozygotic twins.
Recent research studying sleep and nocturnal enuresis indicates that enuresis may be related to non–rapid eye movement (non-REM) sleep, and that those with enuresis may spend more time in stage 3 sleep and have a greater depth of sleep.103,104 Enuresis may be a symptom of obstructive sleep apnea. Inspiratory effort against a closed airway increases intrathoracic negative pressure causing cardiac distention and release of atrial natriuretic hormone and decreased vasopressin and renin-angiotensin-aldosterone complex. Children with nocturnal polyuria and enuresis should be evaluated for sleep-disordered breathing.105
A variety of psychosocial factors also have been postulated as explanations of enuresis. Enuresis has been associated with temper tantrums, fear reactions, excitability, low birth weight, and minimal brain dysfunction.106,107
CLINICAL MANIFESTATIONS Primary enuresis refers to a condition in which the child has never been continent. Secondary enuresis, or acquired enuresis, occurs when a child who has experienced a period of dryness of at least 3 to 6 months after toilet training becomes incontinent again. Secondary enuresis may be diurnal, nocturnal, or a combination of both. (Types of incontinence are defined in Table 37-7.) In 80% of children, enuresis that occurs at night only and more frequently than once a month is called nocturnal enuresis. Wetting during the day is called diurnal enuresis.
Table 37-7
Classification of Incontinence
| Types of Incontinence | Definition |
| Total incontinence | Inability to store any urine; indicates an anatomic or functional absence of urinary sphincters (e.g., epispadias, myelomeningocele) or a bypassing of urinary sphincters (e.g., vesicovaginal fistula) |
| Overflow incontinence | Frequent dribbling that relieves a constantly full bladder; occurs when urinary outlet is obstructed |
| Urge incontinence | Sudden and uncontrollable need to void that cannot be suppressed; suggests bladder irritation |
| Precipitate voiding | Voiding without a preceding urge to void; suggests neurologic origin |
| Stress incontinence | Uncontrollable voiding that occurs when intravesical pressure momentarily exceeds intravesical resistance, as in “giggle incontinence” |
| Paradoxical incontinence | Incontinence in spite of normal voiding; suggests an ectopic ureteral orifice outside the urinary sphincter mechanism (e.g., a girl who is constantly wet, yet voids normally) |
EVALUATION AND TREATMENT Evaluation of enuresis includes use of questionnaires, drinking and voiding charts, physical examination, and urinalysis. Underlying pathology, including kidney disease, vesicoureteral reflux, urinary tract infection, or neurogenic bladder, is excluded. Radiologic and urodynamic evaluation may be required.108
Behavioral interventions—such as self-awakening techniques, enuresis alarms, and motivational therapy—have good results in treating enuresis.96,97 Treatment with desmopressin (an antidiuretic hormone analog) and tricyclics has similar beneficial effects, but most children relapse when treatment is stopped.109,110 Desmopressin acetate nasal spray, a synthetic ADH, is best used when other treatments have not worked, but children must be evaluated for hyponatremia.111 Psychotherapy and behavior modification are recommended when enuresis is associated with psychologic stress. Stress is reduced when families and caregivers receive information and support.112
Acquired enuresis 1415
Acute poststreptococcal glomerulonephritis (PSGN) 1407
Acute pyelonephritis 1411
Anaphylactoid purpura 1408
Chordee 1405
Chronic pyelonephritis 1412
Cloacal exstrophy 1406
Congenital nephrotic syndrome (Finnish type) 1410
Cystitis 1411
Diurnal enuresis 1415
Edema 1410
Enuresis 1414
Epispadias 1405
Exstrophy of the bladder 1405
Focal segmental glomerulosclerosis (FSGS) 1409
Glomerulonephritis 1407
Hemolytic uremic syndrome 1408
Henoch-Schönlein purpura nephritis 1408
Horseshoe kidney 1404
Hypercoagulation 1410
Hyperlipidemia 1410
Hypoplastic kidney 1406
Hypospadias 1404
Immunoglobulin A (IgA) nephropathy (Berger nephropathy) 1408
Mesonephros 1402
Metanephros 1402
Minimal change nephropathy (MCN) 1409
Nephroblastoma 1413
Nephrotic syndrome 1409
Nocturnal enuresis 1415
Polycystic kidney disease (PKD) 1407
Potter syndrome 1407
Primary enuresis 1415
Primary (idiopathic) nephrotic syndrome 1409
Pronephros 1402
Renal agenesis 1407
Renal aplasia 1406
Renal dysplasia 1406
Secondary enuresis 1415
Secondary nephrotic syndrome 1409
Secondary uteropelvic junction (UPJ) obstruction 1406
Unilateral renal agenesis 1407
Urethral atresia 1406
Urethral polyp 1406
Urethral valve 1406
Urinary tract infection (UTI) 1411
Ureteropelvic junction (UPJ) obstruction 1406
Vesicoureteral reflux (VUR) 1412
Wilms tumor 1413
REFERENCES
1. Al, Menke, Schedl, A. WT1 and glomerular function. Semin Cell Dev Biol. 2003;14(4):233–240.
2. Merkel, C.E., Karner, C.M., Carroll, T.J. Molecular regulation of kidney development: is the answer blowing in the Wnt? Pediatr Nephrol. 2007;22(11):1825–1838.
3. Bonilla-Felix, M. Development of water transport in the collecting duct. Am J Physiol Renal Physiol. 2004;287(6):F1093–F1101.
4. Hartnoll, G. Basic principles and practical steps in the management of fluid balance in the newborn. Semin Neonatol. 2003;8(4):307–313.
5. Schedl, A. Renal abnormalities and their developmental origin. Nat Rev Genet. 2007;8(10):791–802.
6. Weizer, A.Z., et al. Determining the incidence of horseshoe kidney from radiographic data at a single institution. J Urol. 2003;170(5):1722–1726.
7. McAninch, J.W. Disorders of the kidneys. In: Taragho E.A., McAninch J.W., eds. Smith’s general urology. Norwalk, CT: Appleton & Lange, 1995.
8. Kelalis, P.P., Lowell, R.K., Bellmam, B.A. Clinical pediatric oncology, ed 3. Philadelphia: Saunders; 1992.
9. Nelson, C.P., et al. The increasing incidence of congenital penile anomalies in the United States,. J Urol. 2005;174(4 Pt 2):1573–1576.
10. Leung, A.K., Robson, W.L. Hypospadias: an update. Asian J Androl. 2007;9(1):16–22.
11. Grady, R.W., Mitchell, M.E. Management of epispadias. Urol Clin North Am. 2002;29(2):349–360. [vi].
12. Frimberger, D., Gearhart, J.P., Mathews, R. Female exstrophy: failure of initial reconstruction and its implications for continence. J Urol. 2003;170(6 PT 1):2428–2431.
13. Jones, D., Parkinson, S., Hosalkar, H.S. Oblique pelvic osteotomy in the exstrophy/epispadias complex. J Bone Joint Surg Br. 2006;88(6):799–806.
14. Thomas, J.C., et al. First stage approximation of the exstrophic bladder in patients with cloacal exstrophy—should this be the initial surgical approach in all patients? J Urol. 2007;178(4 Pt2):1632–1635.
15. Zhang, P.L., Peters, C.A., Rosen, S. Ureteropelvic junction obstruction: morphological and clinical studies. Pediatr Nephrol. 2000;14(8-9):820–826.
16. Rosen, S., et al. The kidney in congenital ureteropelvic junction obstruction: a spectrum from normal to nephrectomy,. J Urol. 2008;179(4):1257–1263.
17. Fefer, S., Ellsworth, P. Prenatal hydronephrosis. Pediatr Clin North Am. 2006;53(3):429–447.
18. Stein, R.J., Gill, I.S., Desai, M.M. Comparison of surgical approaches to ureteropelvic junction obstruction: endopyeloplasty versus endopyelotomy versus laparoscopic pyeloplasty. Curr Urol Rep. 2007;8(2):140–149.
19. Shokier, A.A., Nijman, R.J. Primary megaureter: current trends in diagnosis and treatment. BJU Int. 2000;86(7):861–868.
20. Krishnan, A., et al. The anatomy and embryology of posterior urethral valves,. J Urol. 2006;175(4):1214–1220.
21. Demircan, M., et al. Urethral polyps in children: a review of the literature and report of two cases. Int J Urol. 2006;13(6):841–843.
22. Baird, J.M., et al. Long-term outcomes in children treated by prenatal vesicoamniotic shunting for lower urinary tract obstruction. Obstet Gynecol. 2005;106(3):503–508.
23. Narasimhan, K.L., et al. Does mode of treatment affect the outcome of neonatal posterior urethral valves? J Urol. 2004;171(6 Pt 1):2423–2426.
24. Arant, B.S., Jr., Sotelo-Avila, C., Bernstein, J. Segmental “hypoplasia” of the kidney (ask-upmark). J Pediatr. 1979;95(6):931–939.
25. Dursan, H., et al. Associated anomalies in children with congenital solitary functioning kidney. Pediatr Surg Int. 2005;21(6):456–459.
26. Glazebrook, K.N., McGrath, F.P., Steele, B.T. Prenatal compensatory renal growth: documentation with US. Radiology. 1993;189(3):733.
27. Cascio, S., Paran, S., Puri, P. Associated urological anomalies in children with unilateral renal agenesis. J Urol. 1999;162(3 Pt 2):1081.
28. Parikh, C.R., et al. Congenital renal agenesis: case-control analysis of birth characteristics. Am J Kidney Dis. 2002;39:689–694.
29. Sutters, M. The pathogenesis of autosomal dominant polycystic kidney disease,. Nephron Exp Nephrol. 2006;103(4):e149–e155.
30. Chang, M.Y., Ong, A.C. Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and treatment. Nephron Physiol. 2008;108(1):1–7.
31. Al-Bhalal, L., Akhtar, M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD). Adv Anat Pathol. 2008;15(1):54–58.
32. Richter, C.M. Role of endothelin in chronic renal failure-developments in renal involvement. Rheumatology (Oxford). 2008;47(2):234–235.
33. Novak, J., et al. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28(1):78–87.
34. Moura, I.C., et al. The glomerular response to IgA deposition in IgA nephropathy,. Semin Nephrol. 2008;28(1):88–95.
35. Berthoux, F.C., Mohey, H., Afiani, A. Natural history of primary IgA nephropathy. Semin Nephrol. 2008;28(1):4–9.
36. Kawamura, T. treatment of IgA nephropathy: corticosteroids, tonsillectomy and mycophenolate mofetil. Contrib Nephrol. 157, 2007. [37-33].
37. Coppo, R. Pediatric IgA nephropathy: clinical and therapeutic perspectives. Semin Nephrol. 2008;28(1):18–26.
38. Syrhanen, J., Mustonen, J., Pasternack, A. Hypertriglyceridaemia and hyperuricaemia are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant. 2000;15(1):34.
39. Delos Santos, N.M., Wyatt, R.S. Pediatric IgA nephropathies: clinical aspects and therapeutic approaches,. Semin Nephrol. 2004;24(3):269–286.
40. Rieu, P., Noel, L.H. Henoch-Schönlein nephritis in children and adults. Morphological features and clinicopathological correlations. Ann Med Interne (Paris). 1999;150(2):151.
41. Zaffanello, M., Brugnara, M., Franchini, M. Therapy for children with Henoch-Schönlein purpura nephritis: a systematic review. Scientific World Journal. 2007;7:20–30.
42. Miller, D.P., et al. Incidence of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome. Epidemiology. 2004;15(2):208–215.
43. Serna, A., 4th., Boedeker, E.C. Pathogenesis and treatment of Shiga toxin-producing Escherichia coli infections. Curr Opin Gastroenterol. 2008;24(1):38–47.
44. Franchini, M., Zaffanello, M., Veneri, D. Advances in the pathogenesis, diagnosis and treatment of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Thromb Res. 2006;118(2):177–184.
45. Iijima, K., Kamioka, I., Nozu, K. Management of diarrhea-associated hemolytic uremic syndrome in children. Clin Exp Nephrol. 2008;12(1):16–19.
46. Repetto, H.A. Long-term course and mechanisms of progression of renal disease in hemolytic uremic syndrome. Kidney Int Suppl. 2005;8(97):S102–S106.
47. Behrman R.E., Kliegman R.M., Jenson H.B., eds. Nelson textbook of pediatrics, ed 17, Philadelphia: Saunders, 2004.
48. Salomon, R., et al. NF-kappa B p65 antagonizes IL-4 induction by c-maf in minimal change nephrotic syndrome. J Immunol. 2004;172(1):688–698.
49. Tain, Y.L., Chen, T.Y., Yang, K.D. Implications of serum TNF-beta and IL-13 in the treatment response of childhood nephrotic syndrome. Cytokine. 2003;21(3):155–159.
50. Saland, J.M., Ginsberg, H., Fisher, E.A. Dyslipidemia in pediatric renal disease: epidemiology, pathophysiology, and management. Curr Opin Pediatr. 2002;14(2):197–204.
51. Borges, et al. Is focal segmental glomerulosclerosis increasing in patients with nephritic syndrome? Pediatr Nephrol. 2007;22(9):1309–1313.
52. Kim, S.W., Frokiaer, J., Nielsen, S. Pathogenesis of oedema in nephrotic syndrome: role of epithelial sodium channel. Nephrology (Carlton). 2007;12(Suppl 3):8–10.
53. Zaffanello, M., Franchini, M. Thromboembolism in childhood nephrotic syndrome: a rare but serious complication. Hematology. 2007;12(1):69–73.
54. Simons, M., Huber, T.B. It’s not all about nephrin. Kidney Int. 2008;73(6):697–704.
55. Hinkes, B.G. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in four genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. 2007;119(4):Le907–Le919.
56. Mattoo, T.K. Gingival hyperplasia in congenital and infantile nephrotic syndrome. Pediatr Nephrol. 1997;11(3):388.
57. Hodson, E.M., Willis, N.S., Craig, J.C. Corticosteroid treatment for nephrotic syndrome in children. Cochrane Database Syst Rev. 17(4), 2007. [CD001533].
58. Hodson, E.M., Willis, N.S., Craig, J.C. Non-corticosteroid treatment for nephrotic syndrome in children. Cochrane Database Syst Rev. 23(1), 2008. [CD002290].
59. Crosson, J.T. Focal segmental glomerulosclerosis and renal transplantation. Transplant Proc. 2007;39(3):737–743.
60. Akcan-Arikan, A., et al. Modified RIFLE criteria in critically ill children with acute kidney injury. Kidney Int. 2007;71(10):1028–1035.
61. Andreoli, S.P. Acute renal failure in the newborn. Semin Perinatol. 2004;28(2):112–123.
62. Hari, P., et al. Chronic renal failure in children. Indian Pediatr. 2003;40(11):1035–1042.
63. Seikaly, M.G., et al. Acute renal failure in the newborn. Semin Perinatol. 2004;28(2):112–123.
64. Magee, J.C., et al. Pediatric transplantation. Am J Transplant. 2004;4(Suppl 9):54–71.
65. Magee, J.C., et al. Pediatric transplantation in the United States, 1997-2006. Am J Transplant. 2008;8(4 Pt 2):935–945.
66. Seikaly, M.G., et al. Use of rhGH in children with chronic kidney disease: lessons from NAPRTCS. Pediatr Nephrol. 2007;22(8):1195–1204.
67. Sedberry-Ross, S., Pohl, H.G. Urinary tract infections in children. Curr Urol Rep. 2008;9(2):165–171.
68. Kanellopoulos, T.A., et al. First urinary tract infection in neonates, infants and young children: a comparative study. Pediatr Nephrol. 2006;21(8):1131–1137.
69. Chang, S.L., Shortliffe, L.D. Pediatric urinary tract infections. Ped Clin North Am. 2006;53(3):379–400.
70. Le Saux, N., Pham, B., Moher, D. Evaluating the benefits of antimicrobial prophylaxis to prevent urinary tract infections in children: a systemic review. CMAJ. 2000;163(5):523.
71. Jantausch, B.A., et al. Association of Lewis blood group phenotypes with urinary tract infection in children. J Pediatr. 1994;124(6):863–868.
72. Bauer, R., Kogan, B.A. New developments in the diagnosis and management of pediatric UTIs. Urol Clin North Am. 2008;35(1):47–58.
73. Michael, M., et al. Short versus standard duration oral antibiotic therapy for acute urinary tract infection in children. Cochrane Database Syst Rev. (1):2003. [CD003966].
74. Mak, R.H., Kuo, H.J. Primary urethral reflux: emerging insights from molecular and genetic studies. Curr Opin Pediatr. 2003;15(2):181–185.
75. Sillen, U. Vesicoureteral reflux in infants. Pediatr Nephrol. 1999;13(4):355.
76. Chandra, M., Maddix, H. Urodynamic dysfunction in infants with vesicoureteral reflux. J Pediatr. 2000;136(6):754.
77. Jakobsson, G., Jacobson, S.H., Hjalmas, K. Vesico-ureteric reflux and other risk factors for renal damage: identification of high- and low-risk children. Acta Pediatr Suppl. 1999;88(431):31.
78. Smith, E.A. Pyelonephritis, renal scarring, and reflux nephropathy: a pediatric urologist’s perspective. Pediatr Radiol. 2008;38(Suppl 1):S76–S82.
79. Koyle, M.A., Caldamone, A.A. Part 4: considerations regarding the medical management of VUR: what have we really learned? Curr Med Res Opin. 2007;23(Suppl 4):S21–S25.
80. Hensle, T.W., et al. Part 2: examining pediatric vesicoureteral reflux: a real-world evaluation of treatment patterns and outcomes. Curr Med Res Opin. 2007;23(Suppl 4):S7–S13.
81. MacNeily, A.E., Afshar, K. Screening asymptomatic siblings for vesicoureteral reflux sound science or religious rhetoric? Can J Urol. 2006;13(6):3309–3316.
82. Chertin, B., Puri, P. Familial vesicoureteral reflux. J Urol. 2003;269(5):1804–1808.
83. Peeden, J.N., Noe, H.N. Is it practical to screen for familial vesicoureteral reflux within a private pediatric practice? Pediatrics. 1992;89(4):758.
84. Fukuzawa, R., Reeve, A.E. Molecular pathology and epidemiology of nephrogenic rests and Wilms tumors. J Pediatr Hematol Oncol. 2007;29(9):589–594.
85. National Cancer Institute: Wilms tumor and other childhood kidney tumors treatment (PDQ), 2009. Available from www.cancer.gov/cancertopics/pdq/treatment/wilms/HealthProfessional/page2
86. Brown KW, Malik KTA: The molecular biology of Wilms tumour, Exp Rev Mol Med. Available from www.expertreviews.org/010030227h.htm.
87. Peres, E.M., et al. Chromosome analyses of 16 cases of Wilms tumor: different pattern in unfavorable histology. Cancer Cenet Cytogenet. 2004;148(1):66–70.
88. Perotti, D., et al. Functional inactivation of the WTX gene is not a frequent event in Wilms tumors. Oncogene. 2008;27(33):4625–4632.
89. Rivera, M.N., et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315(5812):642–645.
90. Belasco, J., Chatten, J., D’Angio, G. Wilms tumor. In Sutow W., Fernbach D., Vietti T., eds.: Clinical pediatric oncology, ed 3, St Louis: Mosby, 1984.
91. Kurli, M., Finger, P.T. The kidney, cancer, and the eye: current concepts,. Surv Ophthalmol. 2005;50(6):507–518.
92. Nicholson, H.S., et al. Uterine anomalies in Wilms’ tumor survivors. Cancer. 1996;78(4):887.
93. Green, D.M. The diagnosis and management of Wilms’ tumor,. Pediatr Clin North Am. 1985;32:735.
94. Neville, H.L., Ritchey, M.L. Wilms’ tumor: overview of National Wilms’ Tumor Study Group results. Urol Clin North Am. 2000;27(3):435.
95. Djurhuus, J.C., Rittig, S. Nocturnal enuresis. Curr Opin Urol. 2002;12(4):317–320.
96. Glazener CM, Evans JH: Simple behavioral and physical interventions for nocturnal enuresis in children, Cocharane Database Syst Rev (2):CD003637.
97. Glazener CM, Evans JH, Peto RE: Alarm interventions for nocturnal enuresis in children, Cochrane Database Syst Rev (2):CD002911.
98. Kamperis, K., et al. Nocturnal polyuria in monosymptomatic nocturnal enuresis refractory to desmopressin treatment. Am J Physiol Renal Physiol. 2006;291(6):F1232–F1240.
99. Troup, C.W., Hodgson, N.B. Nocturnal functional bladder capacity in enuretic children. J Urol. 1971;105:129.
100. Stark, M. Assessment and management of the care of children with nocturnal enuresis: guidelines for primary care. Nurs Pract Forum. 1994;5(3):170.
101. Uygur, M.C., Ergen, A., Remzi, D. Enuresis nocturna: new concepts in pathophysiology. Int Urol Nephrol. 1995;27(4):439.
102. Gontard, A., et al. Molecular genetics of nocturnal enuresis: linkage to a locus on chromosome 22. Scand J Urol Nephrol Suppl. 1999;202:76.
103. Neveus, T. The role of sleep and arousal in nocturnal enuresis,. Acta Pediatr. 2003;92(10):1118–1123.
104. Neveus, T., et al. Sleep of children with enuresis: a polysomnographic study. Pediatrics. 1999;103(6 part 1):1193.
105. Firoozi, F., et al. Resolution of diurnal incontinence and nocturnal enuresis after adenotonsillectomy in children. J Urol. 2006;175(5):1885–1888.
106. Backes, M., et al. Cognitive and behavioral profile of fragile X boys: correlations to molecular data. Am J Med Genet. 2000;95(2):150–156.
107. Von Gontard, A., Hollmann, E. Comorbidity of functional urinary incontinence and encoporesis: somatic and behavioral associations. J Urol. 2004;171(6 Pt 2):2644–2647.
108. Feldman, A.S., Gauer, S.B. Diagnosis and management of dysfunctional voiding. Curr Opin Pediatr. 2006;18(2):139–147.
109. Cutting, D.A., Pallant, J.F., Cutting, F.M. Nocturnal enuresis: application of evidence-based medicine in community practice. J Paediatr Child Health. 2007;43(3):167–172.
110. Zaffanello, M., et al. Therapeutic options in childhood nocturnal enuresis. Minerva Urol Nefrol. 2007;59(2):199–205.
111. Robson, W.L., Leung, A.K., Norgaard, J.P. The comparative safety of oral versus intranasal desmopressin for the treatment of children with nocturnal enuresis,. J Urol. 2007;178(1):24–30.
112. Weaver, A., Dobson, P. Nocturnal enuresis in children. J Fam Health Care. 2007;17(5):159–161.