ALTERATIONS OF RENAL AND URINARY TRACT FUNCTION



Renal and urinary function can be affected by a variety of disorders. The most common type of urinary dysfunction is infection. Stones or tumors also can obstruct the urinary tract. Renal function can be impaired by disorders of the kidney itself or by many other systemic diseases and ultimately may result in renal insufficiency or renal failure. Because the kidney filters the blood, it is directly linked to every other organ system. Renal failure, whether acute or chronic, is therefore a life-threatening condition.
URINARY TRACT OBSTRUCTION
Urinary tract obstruction is an interference with the flow of urine at any site along the urinary tract (Figure 36-1). An obstruction may be anatomic or functional; it impedes flow proximal to the blockage, dilates of the urinary system, increases risk for infection, and compromises renal function. Anatomic changes in the urinary system caused by obstruction are referred to as obstructive uropathy. The severity of an obstructive uropathy is determined by (1) the location of the obstructive lesion, (2) whether one or both upper urinary tracts are involved, (3) the severity (completeness) of the blockage, (4) its duration, and (5) the nature of the obstructive lesion.1 Obstructions may be relieved or partially alleviated by correction of the obstruction, although permanent impairments occur if a complete or partial obstruction persists over weeks to months or longer.
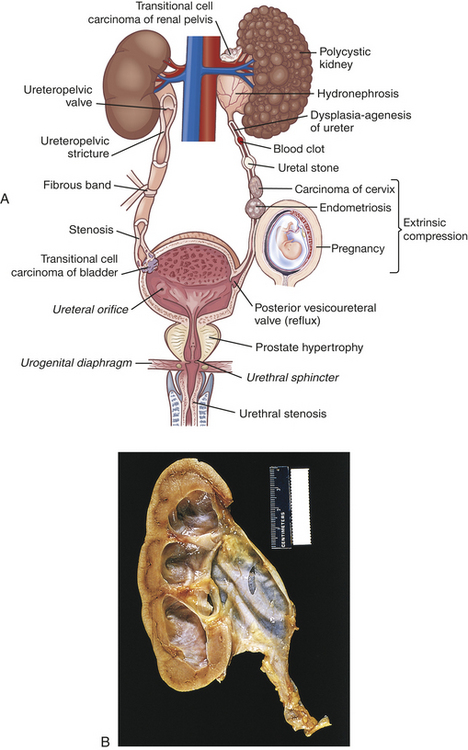
Figure 36-1 Urinary tract obstruction and hydronephrosis. A, Major sites of urinary tract obstruction. B, Hydronephrosis, marked dilation of renal pelvis and calyces with thinning of parenchyma.
Upper Urinary Tract Obstruction
Common causes of upper urinary tract obstruction include stricture or congenital compression of a calyx or the ureteropelvic or ureterovesical junction (i.e., stones [calculi]); compression from an aberrant vessel, tumor, or abdominal inflammation and scarring (retroperitoneal fibrosis); or ureteral blockage from stones or a malignancy of the renal pelvis or ureter.
Obstruction of the upper urinary tract causes dilation of the ureter, renal pelvis, calyces, and renal parenchyma proximal to the site of urinary blockage. Dilation of the ureter is referred to as hydroureter (accumulation of urine in the ureter), and dilation of the renal pelvis and calyces proximal to a blockage leads to hydronephrosis (enlargement of the renal pelvis and calyces) or ureterohydronephrosis (dilation of both the ureter and pelvicaliceal system) (Figure 36-2). Dilation of the upper urinary tract is an early response to obstruction and reflects smooth muscle hypertrophy and accumulation of urine above the level of blockage (urinary stasis/retention). The increased pressure is transmitted to the glomerulus, which decreases filtration. Unless the obstruction is relieved, this dilation leads to enlargement with tubulointerstitial fibrosis and apoptosis affecting the distal nephron and renal function. Tubulointerstitial fibrosis is the deposition of excessive amounts of extracellular matrix (collagen and other proteins). Deposition of extracellular matrix is a normal process of organ repair and maintenance, and the deposition of extracellular matrix is balanced by its breakdown under the influence of metalloproteinases. Multiple cytokines and growth factors have been implicated in the process of tubulointerstitial fibrosis and irreversible loss of kidney function, including transforming growth factor-beta-1 (TGF-β1), angiotensin II, and various tumor necrosis factors. Apoptosis is a normal process that the body uses to replace damaged or senescent cells with new ones, but the imbalance in growth factors provoked by obstruction leads to excess cellular destruction and death, ultimately resulting in loss of functioning nephrons and kidney damage.
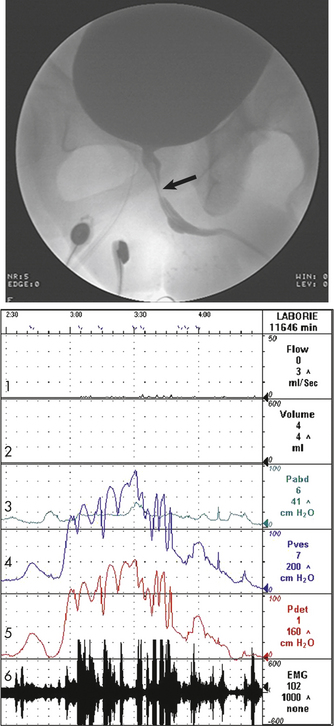
Figure 36-2 Neurogenic detrusor overactivity with vesicosphincter. The arrow indicates narrowing of the striated sphincter consistent with electromyographic activity (Line 6) noted on the urodynamic tracing. Note the characteristic poor flow pattern (Line 1) with elevated voiding pressures (Lines 4 and 5) indicating obstruction. Line 1 = Urine flow rate; Line 2 = urine volume; Line 3 = abdominal pressure (Pabd); Line 4 = intravesicular (inside) bladder) pressure (Pves); Line 5 = detrusor muscle pressure (Pdet); Line 6 = bladder electromyelogram (EMG).
Tubulointerstitial fibrosis and apoptosis result in detectable damage to the distal renal tubules within approximately 7 days. By 14 days, obstruction has adversely affected both distal and proximal aspects of the nephron. Within 28 days the glomeruli of the kidney have been damaged and the renal cortex and medulla are reduced in size (thinned). Distal tubular damage occurs initially and decreases the kidney’s ability to concentrate urine, causing an increase in urine volume despite a decrease in glomerular filtration rate (GFR). The affected kidney is unable to conserve sodium, bicarbonate, and water or to excrete hydrogen or potassium, leading to metabolic acidosis and dehydration. The magnitude of this damage, and the kidney’s ability to recover normal homeostatic function, is affected by the severity and duration of the obstruction. With complete obstruction, damage to the renal tubules and compression of the renal vasculature occurs in a matter of hours, and irreversible damage occurs within 3 to 4 weeks. Nevertheless, even in the face of a complete obstruction, the human kidney may recover at least partial homeostatic function provided the blockage is removed within 56 to 69 days.2 This recovery requires approximately 4 months. Partial obstruction, in the absence of renal infection, leads to subtler but ultimately permanent impairments including loss of the kidney’s ability to concentrate urine, reabsorb bicarbonate, excrete ammonia, or regulate metabolic acid-base balance. Complete bilateral obstruction causes anuria.
The body is able to partially counteract the negative consequences of unilateral obstruction by a process called compensatory hypertrophy and hyperfunction.3 The compensatory response is the result of two growth processes: obligatory growth occurs under the influence of somatomedins, and compensatory growth occurs under the influence of a yet-to-be-identified hormone or hormones. These processes cause the contralateral (unobstructed) kidney to increase the size of individual glomeruli and tubules but not the total number of functioning nephrons. The ability of the body to engage in compensatory hypertrophy and hyperfunction diminishes with age, and the process is reversible when relief of obstruction results in recovery of function by the obstructed kidney. Unilateral obstruction may remain silent for a long time.
Relief of bilateral, partial urinary tract obstruction, or complete obstruction of one kidney is usually followed by a brief period of diuresis (commonly called postobstructive diuresis).4 It is a physiologic response and is typically mild, representing a restoration of fluid and electrolyte imbalance caused by the obstructive uropathy. Alterations in tubular transport and water reabsorption and volume expansion contribute to the diuresis. Occasionally relief of obstruction will cause rapid excretion of large volumes of water, sodium, or other electrolytes, resulting in a urine output of 10 L/day or more. Rapid postobstructive diuresis causes dehydration and fluid and electrolyte imbalances if not promptly corrected. Risk factors for severe postobstructive diuresis include bilateral obstruction, impairment of one or both kidneys’ ability to concentrate urine or reabsorb sodium (nephrogenic diabetes insipidus), hypertension, edema and weight gain, congestive heart failure, and uremic encephalopathy.
Kidney Stones
Calculi, or urinary stones, are masses of crystals, protein, or other substances that are a common cause of urinary tract obstruction in adults. The prevalence of stones in the United States is approximately 6% in women and 15% in men.5 The recurrence rate is approximately 30% to 50% within 5 years.6 The risk of urinary calculi formation is influenced by a number of factors, including age, gender, race, geographic location, seasonal factors, fluid intake, diet, occupation, genetic predisposition and other conditions including urinary tract infection, hypertension, and obesity.7,8 Most persons develop their first stone before age 50 years. Geographic location influences the risk of stone formation because of indirect factors, including average temperature, humidity, and rain fall, and its influence on fluid and dietary patterns. Persons who regularly consume an adequate volume of water and those who are physically active are at reduced risk when compared with people who are inactive or consume lower volumes of fluid. Most renal stones are unilateral.
Urinary calculi can be classified according to the primary minerals (salts) that make up the stones. The most common stone types include calcium oxalate or phosphate (70% to 80%), struvite (magnesium, ammonium, and phosphate) (15%), and uric acid (7%). Cystine stones are rare, less than 1%. Less common stone elements include cystine, 2,8-dihydroxyadeninuria (a rare genetic disorder that increases risk of xanthine stones), triamterene (a diuretic), and indinavir (a protease inhibitor used in management of HIV infection).
PATHOPHYSIOLOGY The specific cascade of events that lead to stone formation is unknown. Stone formation requires (1) supersaturation of one or more salts in the urine, (2) precipitation of the salts from a liquid to a solid state, (3) growth through crystallization or agglomeration (sometimes called aggregation), and (4) the presence or absence of stone inhibitors.9 Supersaturation is the presence of a higher concentration of a salt within a fluid (in this case, the urine) than the volume is able to dissolve to maintain equilibrium.
Human urine contains many positively and negatively charged ions capable of precipitating from solution and forming a variety of salts. The salts form crystals that are retained and grow into stones. Crystallization is the process by which crystals grow from a small nidus or nucleus to larger stones in the presence of supersaturated urine. Although supersaturation is essential for stone formation, the urine need not remain continuously supersaturated for a calculus to grow once its nidus has precipitated from solution. Intermittent periods of supersaturation after the ingestion of a meal or during times of dehydration are sufficient for stone growth in many individuals. In addition, the renal tubules and papillae have many surfaces that may attract a crystalline nidus and add biologic material (matrix) to the forming stone. Matrix is an organic material that is formed in the presence of urea-splitting pathogens and is high in stones associated with infection.9
Temperature and pH of the urine also influence the risk of precipitation and calculus formation—pH is more important. An alkaline urinary pH significantly increases the risk of calcium phosphate stone formation, whereas acidic urine increases the risk of a uric acid stone. Cystine and xanthine precipitate more readily in acidic urine.
Stone or crystal growth inhibiting substances, including Tamm-Horsfall protein, potassium citrate, pyrophosphate, and magnesium, are capable of crystal growth inhibition, thereby reducing the risk of calcium phosphate or calcium oxalate precipitation in the urine and preventing subsequent stone formation.
Retention of crystal particles occurs primarily at the papillary collecting ducts. Although most crystals are flushed from the tract through antegrade urine flow, urinary stasis, anatomic abnormalities, or inflamed epithelium within the urinary tract may prevent prompt flushing of crystals from the system, thus increasing the risk of calculus formation.
The size of a stone determines the likelihood that it will pass through the urinary tract and be excreted through micturition.10 A stone that is smaller than 5 mm has about a 50% chance of spontaneous passage, whereas a stone that is 1 cm has almost no chance of spontaneous passage. Nevertheless, the person with ureteral dilation from the previous passage of a stone may be able to excrete larger stones when compared with the person experiencing an initial obstructing calculus.
Calcium stones (calcium phosphate or calcium oxalate) account for 70% to 80% of all stones requiring treatment. Most of these individuals have idiopathic calcium urolithiasis (ICU), a condition whose exact etiology has not yet been defined. However, hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, mild renal tubular acidosis, or crystal growth inhibitor deficiencies are associated with calcium stones.11 Hypercalciuria is usually attributable to intestinal hyperabsorption of dietary calcium and less commonly to a defect in renal calcium reabsorption. Hyperparathyroidism and bone demineralization associated with prolonged immobilization are also known to cause hypercalciuria. An alkaline urine also promotes calcium stone formation.12 Although oxalate in the diet influences the risk of calcium stones, primary hyperoxaluria is a rare, inherited disorder.
Struvite stones primarily contain magnesium-ammonium-phosphate as well as varying levels of matrix. Matrix forms in an alkaline urine and during infection with a urease-producing bacterial pathogen, such as a Proteus, Klebsiella, or Pseudomonas. Struvite calculi may grow quite large and branch into a staghorn configuration (staghorn calculus) that approximates the pelvicaliceal collecting system.13 Women are at greater risk for struvite stones because they have an increased incidence of urinary tract infection.
Uric acid is primarily a product of biosynthesis of endogenous purines and is secondarily affected by consumption of purines in the diet. Persons who excrete excessive uric acid in the urine, such as those with gouty arthritis, are at particular risk for uric acid stones. A consistently acidic urine greatly increases this risk. Cystine and xanthine are amino acids that precipitate more readily in acidic urine. Cystinuria and xanthinuria are genetic disorders of amino acid metabolism, and their excess in urine can cause cystinuric, or xanthine, stone formation in the presence of a low urine pH of 5.5 or less.
CLINICAL MANIFESTATIONS Renal colic, described as moderate to severe pain often originating in the flank and radiating to the groin, usually indicates obstruction of the renal pelvis or proximal ureter.14 Colic that radiates to the lateral flank or lower abdomen typically indicates obstruction in the midureter, and bothersome lower urinary tract symptoms (urgency, frequent voiding, urge incontinence) indicate obstruction of the lower ureter or ureterovesical junction. The pain can be severe and incapacitating and may be accompanied by nausea and vomiting. Gross or microscopic hematuria may be present.
EVALUATION AND TREATMENT The evaluation and diagnosis of urinary calculi are based on presenting symptoms and history combined with a focused physical assessment, imaging studies, and possibly a functional study of renal pelvic and ureteral pressures.15 The history also queries dietary habits, the age of the first stone episode, stone analysis, and presence of complicating factors including hyperparathyroidism or recent gastrointestinal or genitourinary surgery. Urinalysis (including pH) is obtained and a 24-hour urine is completed to identify calcium oxalate, citrate, and other significant constituents. In addition, every effort is made to retrieve and analyze calculi that are passed spontaneously or retrieved through aggressive intervention. Additional tests are obtained in selected individuals, such as those with suspected hyperparathyroidism or cystine or uric acid stones, in order to diagnose and manage underlying metabolic disorders. An x-ray film of the kidneys, ureters, and bladder (KUB radiograph) is obtained to evaluate radiopaque stones (comprising more than 90% of all stones), and an ultrasound, intravenous pyelogram (IVP), or computed tomography (CT) scan or ultrasonography is obtained to determine the location of the calculi, the severity of obstruction, and associated obstructive uropathy.16 CT urography is used for pre- and postoperative evaluation and fluoroscopy guides intraoperative imaging.17
The goals of treatment are to manage acute pain, promote stone passage, reduce the size of stones already formed, and prevent new stone formation. The components of treatment include (1) parenteral and/or oral analgesics for acute pain, (2) medical therapy promoting stone passage, (3) reducing the concentration of stone-forming substances by increasing urine flow rate with high fluid intake, (4) decreasing the amount of stone-forming substances in the urine by decreasing dietary intake or endogenous production or by altering urine pH,18 and (5) removing stones using percutaneous nephrolithotomy, ureteroscopy, or ultrasonic or laser lithotripsy to fragment stones for excretion in the urine.19,20
Lower Urinary Tract Obstruction
Obstructive disorders of the lower urinary tract (LUT) are primarily related to storage of urine in the bladder or emptying of urine through the bladder outlet. The causes of the obstruction include neurogenic and anatomic alterations or, in some instances, a combination of both. Incontinence is a common symptom and types of incontinence are reviewed in Table 36-1.
Table 36-1

Data from Agency for Healthcare Research and Quality: Overview: urinary incontinence in adults, clinical practice guideline update, Rockville, MD, 1996. Available at www.ahrq.gov/clinic/uiovervw.htm.
Neurogenic Bladder
Neurogenic bladder is a general term for bladder dysfunction caused by neurologic disorders. The types of dysfunction are related to the sites in the nervous system that control sensory and motor bladder function (Figure 36-3). Lesions that develop in upper motor neurons of the brain and spinal cord result in dyssynergia (loss of coordinated neuromuscular contraction) and overactive or hyperreflexive bladder function. Lesions in the sacral area of the spinal cord or peripheral nerves result in underactive, hypotonic, or atonic (flaccid) bladder function, often with loss of bladder sensation.
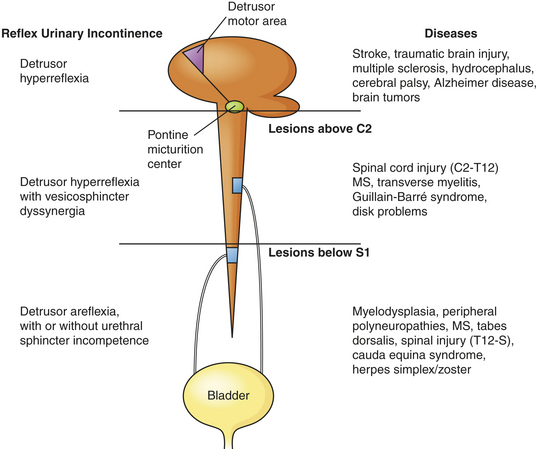
Figure 36-3 Causes of neurogenic bladder and reflex incontinence. (Adapted from Doughty DB: Urinary and fecal incontinence. In Doughty DB, editor: Urinary and fecal incontinence: nursing management, ed 2, St Louis, 2000, Mosby.)
Neurologic disorders that develop above the pontine micturition center result in detrusor hyperreflexia, also known as an uninhibited or reflex bladder. This is an upper motor neuron disorder in which the bladder empties automatically when it becomes full and the external sphincter functions normally. Because the pontine micturition center remains intact, there is coordination between detrusor muscle contraction and the relaxation of the urethral sphincter. Stroke, traumatic brain injury, dementia, and brain tumors are examples of disorders that result in detrusor hyperreflexia. Symptoms include urine leakage and incontinence.
Neurologic lesions that occur below the pontine micturition center but above the sacral micturition center (between C2 and S1) are also upper motor neuron lesions and result in detrusor hyperreflexia with vescicosphincter dyssynergia. There is loss of pontine coordination of detrusor muscle contraction and external sphincter relaxation, so both the bladder and the sphincter are contracting at the same time, causing a functional obstruction of the bladder outlet.21 Spinal cord injury, multiple sclerosis, Guillain-Barré syndrome, and intervertebral disk problems are causes of this disorder. There is diminished bladder relaxation during storage with small urine volumes and high intravesicular (inside the bladder) pressures. This results in an overactive bladder syndrome with symptoms of frequency, urgency, and urge incontinence and increased risk for urethral turbulence and urinary tract infection.
Lesions that involve the sacral micturition center (below S1; may also be termed cauda equina syndrome) or peripheral nerve lesions result in detrusor areflexia (acontractile detrusor), a lower motor neuron disorder. The result is an acontractile detrusor or atonic bladder with retention of urine and distention. If the sensory innervation of the bladder is intact, the full bladder will be sensed but the detrusor may not contract. This is an underactive bladder syndrome and may have symptoms of stress and overflow incontinence. Myelodysplasia, multiple sclerosis, tabes dorsalis, and peripheral polyneuropathies are associated with this disorder.
Overactive Bladder Syndrome
Overactive bladder syndrome (OAB) is a syndrome of detrusor overactivity characterized by urgency with involuntary detrusor contractions during the bladder filling phase that may be spontaneous or provoked.22 There is coordination between the contracting bladder and the external sphincter, but the detrusor is too weak to empty the bladder, resulting in urinary retention with overflow or stress incontinence. Overactive bladder as defined by the International Continence Society as a symptom syndrome of urgency, with or without urge incontinence and usually associated with frequency and nocturia.23 Overactive bladder syndrome affects millions of men, women, and children; adults are often reluctant to discuss this syndrome with their healthcare provider. Sexual dysfunction and bowel problems often accompany OAB.24 Diagnosis is usually made by evaluation of symptoms. Urodynamic evaluation confirms the diagnosis. Antimuscarinics are the most common treatment and in intractable cases, surgery is recommended.25 When left untreated, OAB is costly, impairs health and quality of life, causes depression, and leads to social isolation; in older adults it may cause risk for falls and urinary tract infection.26
Obstructions to Urine Flow
Anatomic causes of resistance to urine flow include urethral stricture, prostatic enlargement in men, and pelvic organ prolapse in women. Symptoms of obstruction are more common in men and include (1) frequent daytime voiding (urination more than every 2 hours while awake); (2) nocturia (awakening more than once each night to urinate for adults less than 65 years of age or more than twice for older adults); (3) poor force of stream; (4) intermittency of urinary stream; (5) bothersome urinary urgency, often combined with hesitancy; and (6) feelings of incomplete bladder emptying despite micturition.
A urethral stricture is a narrowing of its lumen. It occurs when infection, injury, or surgical manipulation produces a scar that reduces the caliber of the urethra.27 The vast majority of urethral strictures occur in men; they are rare in women.28 The severity of obstruction is influenced by its location within the urethra, its length, and the minimum caliber of urethral lumen within the stricture. Specifically, proximal urethral strictures cause more severe obstruction than do strictures of the distal urethra, longer strictures tend to be more obstructive, and the magnitude of blockage is in reverse proportion to the urethral caliber.
Prostate enlargement is caused by acute inflammation, benign prostatic hyperplasia, or prostate cancer (see Chapter 23). Each of these disorders can cause encroachment on the urethra with obstruction to urine flow and the symptoms summarized previously.
Severe pelvic organ prolapse (see Chapter 23) in a woman causes bladder outlet obstruction when a cystocele (the downward protrusion of the bladder into the vagina) descends below the level of the urethral outlet. Cystoceles that reach or protrude beyond the vaginal introitus create the greatest risk for obstruction, particularly if the bladder neck has been surgically repaired without simultaneous repair of the cystocele.29 In men the bladder may rarely herniate into the scrotum, causing a similar type of obstruction.
Partial obstruction of the bladder outlet or urethra initially causes an increase in the force of detrusor contraction. If the blockage persists, afferent nerves within the bladder wall are adversely affected, leading to urinary urgency and, in some cases, overactive detrusor contractions (a myogenic cause of overactive bladder). When obstruction persists, there is an increased deposition of collagen within the smooth muscle bundles of the detrusor muscle (trabeculation), possibly in an attempt to increase the force of its contraction strength. Ultimately, the bladder wall loses its ability to stretch and accommodate urine, a condition called low bladder wall compliance, and the detrusor loses its ability to contract efficiently. Low bladder wall compliance chronically elevates intravesicular pressure, greatly increasing the problems of hydroureter, hydronephrosis, and impaired renal function.
EVALUATION AND TREATMENT Although the history and physical examination are critical to the evaluation of lower urinary tract disorders, it must be remembered that no symptom or cluster of symptoms has been identified that accurately differentiates the various causes of these disorders. For example, symptoms such as urgency, urge incontinence, frequent urination, and nocturia may develop because of overactive bladder or either increased or decreased bladder outlet resistance. Reduced resistance is associated with the symptom of stress incontinence (incontinence with coughing or sneezing) and symptoms of increased resistance are similar to bladder outlet obstruction, including poor force of urinary stream, hesitancy, and feelings of incomplete bladder emptying.
Various diagnostic tests assist with evaluation. The postvoid urine is measured by catheterization within 5 to 15 minutes of urination or through a bladder ultrasound machine that measures bladder height and width to provide an approximation of urine within the vesicle. This measurement may be combined with uroflowmetry, a graphic representation of the force of the urinary stream expressed as milliliters voided per second. Each of these measurements assesses the lower urinary tract’s efficiency in evacuating urine through micturition but neither differentiates poor detrusor contraction strength from obstruction as a cause of urinary retention. Instead, multichannel urodynamic testing is used to identify obstruction, quantify its severity, and measure detrusor contraction strength (see Figure 36-2). Video-urodynamic recordings can also demonstrate overactive bladder and detrusor sphincter dyssynergia. An evaluation of renal function, including functional imaging studies and serum creatinine, is completed particularly when obstruction is severe and associated with elevated residuals or urinary tract infection.
Because the bladder neck consists of circular smooth muscle with adrenergic innervation, detrusor sphincter dyssynergia may be managed by α-adrenergic blocking (antimuscarinic) medications. Obstruction that is not adequately managed by pharmacotherapy may require bladder neck incision. Detrusor sphincter dyssynergia may be managed by intermittent catheterization in combination with higher dose antimuscarinic drugs to prevent overactive detrusor contractions and associated dyssynergia while ensuring regular, complete bladder evacuation through catheterization. Alternatively, men with dyssynergia may be managed by condom catheter containment, supplemented by an α-adrenergic blocking drug or transurethral sphincterotomy (surgical incision of the striated sphincter) in order to relieve obstruction. Low bladder wall compliance may be managed by antimuscarinic drugs and intermittent catheterization; however, more severe cases may require augmentation enterocystoplasty (enlargement of the low compliant bladder wall using a detubularized piece of small bowel), urinary diversion, or long-term indwelling catheterization.
Prostate enlargement is managed by treating the underlying cause of the prostate enlargement with medication or surgery. Acute prostatitis is initially managed by broad-spectrum antibiotics until the results of a urine culture are obtained. Urinary retention may require transient placement of a suprapubic catheter. The management of benign prostatic hyperplasia and treatment options for prostate cancer are presented in Chapter 23.
Urethral stricture is treated with urethral dilation accomplished by using a steel instrument shaped like a catheter (urethral sound) or a series of incrementally increasing catheter-like tubes (filiforms and followers). Long, dense strictures typically require surgical repair to prevent recurrence.
A pessary (rubber or silicone device designed to compensate for vaginal wall prolapse) may be inserted to mechanically reverse severe pelvic organ (bladder, uterus, or rectum) prolapse. Depending on the device, the woman may be able to remove, cleanse, and replace the pessary, or it may be changed during a clinic visit. Intravaginal hormone replacement therapy and regular follow-up are critical to the long-term success of a pessary. Alternatively, pelvic organ prolapse may be repaired surgically; the procedure may be combined with a urethral suspension to correct stress urinary incontinence or rectocele repair.
Tumors
Renal tumors account for about 57,760 (3.8%) new cancer cases and 12,980 (2.3%) deaths each year.30 There are a number of different types of kidney tumors. Renal adenomas (benign tumors) are uncommon but are increasing in number. The tumors are solid, encapsulated and are usually located near the cortex of the kidney. Because they can become malignant, they are usually surgically removed. Renal cell carcinoma (RCC) is the most common renal neoplasm (85% of all renal neoplasms) and represents about 2% of cancer deaths (Figure 36-4). Renal cell carcinoma usually occurs in men (two times more often than in women) between 50 and 60 years of age. Risk factors include cigarette smoking, obesity and hypertension.31 Five-year survival is 96% for stage I cancer and about 23% for stage V cancer.30 Black Americans have shorter survival than their white counterparts and this may be related to extent of comorbid conditions and lower rate of surgical treatment.32
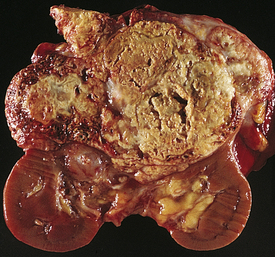
Figure 36-4 Renal cell carcinoma. Renal cell carcinomas usually are spheroidal masses composed of yellow tissue mottled with hemorrhage, necrosis, and fibrosis. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
PATHOGENESIS Renal cell carcinomas are adenocarcinomas that usually arise from tubular epithelium commonly in the renal cortex. The etiology is unknown. They are classified according to cell type and extent of metastasis. Clear cell tumors, the most common, present a better prognosis than granular cell or spindle tumors. Confinement within the renal capsule, together with treatment, is associated with a better survival rate. The tumors usually occur unilaterally (see Figure 36-4). About 25% to 30% of individuals with RCC present with metastasis.33
CLINICAL MANIFSTATIONS The classic clinical manifestations of renal tumors are hematuria, dull and aching flank pain, and palpable flank mass in thinner individuals. Systemic manifestations usually represent an advanced stage of disease and include weight loss, fatigue, intermittent fever from tumor toxins, anemia from hematuria and lack of erythropoietin or polycythemia from tumor secretion of erythropoietin, hypertension from elevated renin levels, and alterations in liver function tests. All of these symptoms occur in less than 10% of cases. Further, they represent an advanced stage of disease, whereas earlier stages are often silent. The most common sites of distant metastasis are the lung, lymph nodes, liver, bone, thyroid, and central nervous system.34
EVALUATION AND TREATMENT Diagnosis is based on the clinical symptoms, plain x-ray films of the abdomen, intravenous pyelography, renal angiography, and CT. (Staging of renal cell carcinoma is presented in Table 36-2.) Staging systems using molecular tumor markers are rapidly advancing.35 Treatment for localized disease is surgical removal of the affected kidney (radical nephrectomy) with combined use of chemotherapeutic agents. Smaller tumors may be removed by nephron-sparing surgery (partial nephrectomy).36 Radiation therapy also may be used and new techniques using radiofrequency ablation, cryoablation and laparoscopy are promising.37 Immunotherapy (i.e., interferon-alpha and interleukin-2) is promising in selected cases, and new targeted therapies are being developed.38 Tumor obstruction is relieved by placing ureteral catheters, placement of nephrostomy tubes, or completion of urinary diversion procedures. Survival is related to tumor grade, tumor cell type, and extent of metastasis.
Table 36-2
Staging of Renal Cell Carcinoma (TNM System)
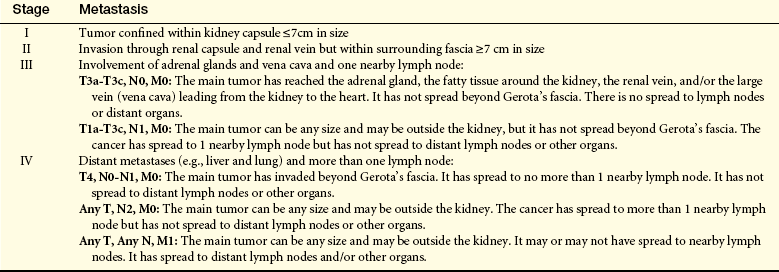
T, Tumor; N, node; M, metastasis.
Adapted from American Cancer Society: Detailed guide: kidney cancer. How is kidney cancer (renal cell carcinoma) staged? Available at www.cancer.org/docroot/CRI/content/CRI_2_4_3X_How_is_kidney_cancer_staged_22.asp (accessed April 2008).
Bladder Tumors
Bladder tumors represent about 3% of all malignant tumors and are the fourth most common malignancy in men.30,39 Approximately 70,980 (4.8%) people develop bladder cancer each year, and 14,430 (2.5%) die of it.30,39 The development of bladder cancer is most common in men older than 60 years. Urothelial carcinoma previously known as transitional cell carcinoma is the most common bladder malignancy, appearing usually as a superficial tumor.
PATHOGENESIS The risk of primary bladder cancer is greater among people who smoke or are exposed to metabolites of aniline dyes or other aromatic amines or chemicals and with heavy consumption of phenacetin.40 Oncogenes of the ras gene family and tumor-suppressor genes including TP53 mutations and inactivation of retinoblastoma gene (pRb) are implicated in bladder cancer. The tumor is usually composed of uroepithelial cells (cells lining the bladder, ureters, urethra and renal pelvis) and most have a papillary growth pattern (a tuftlike lesion attached to a stalk). (Figure 36-5). Nonpapillary tumors (sessile or nodular represent 10% to 30% of bladder tumors) are not as common as papillary tumors, but they tend to be more invasive and have a poorer prognosis. Carcinoma in situ can present with papillary tumors. World- wide squamous cell carcinoma is the most prevalent. Metastasis is usually to lymph nodes, liver, bones, or lungs. Staging for bladder carcinoma is presented in Table 36-3. Secondary bladder cancer develops by invasion of cancer from bordering organs, such as cervical carcinoma in women or prostatic carcinoma in men.
Table 36-3
Staging of Bladder Carcinoma (TNM∗ System)
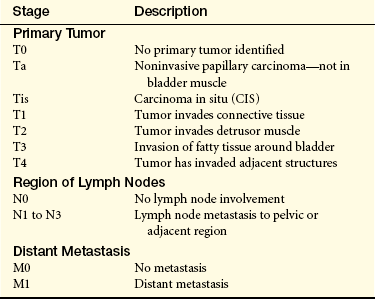
∗T, Tumor; N, node; M, metastasis.
Adapted from American Cancer Society: Detailed guide: bladder cancer. How is bladder cancer staged? 2006. Available at www.cancer.org/docroot/CRI_2_4_3X_How_is_bladder_cancer_staged_22.asp?sitearea= (accessed April, 2007).
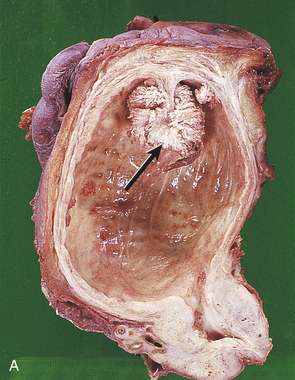
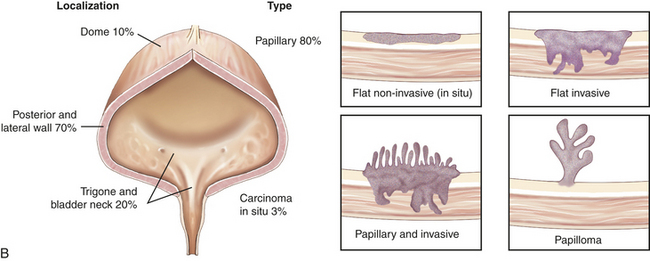
Figure 36-5 Carcinoma of the bladder. A, Bladder cancer with morphologic patterns of most common tumors. B, Papillary transitional cell carcinoma arising in the dome of the bladder as a cauliflower-like lesion (arrow). (A from Stevens A, Lowe J, editors: Pathology, ed 2, St Louis, 2008, Mosby; B from Kissane JM, editor: Anderson’s pathology, ed 9, St Louis, 1990, Mosby.)
CLINICAL MANIFESTATIONS Gross painless hematuria is the archetypal clinical manifestation of bladder cancer. Episodes of hematuria tend to recur, and they are often accompanied by bothersome lower urinary tract symptoms including daytime voiding frequency, nocturia, urgency, and urge urinary incontinence. Flank pain may occur if tumor growth obstructs one or both ureterovesical junctions. Bothersome lower urinary tract symptoms are particularly intense in individuals with carcinoma in situ. Metastasis is the cause of death from bladder cancer.41
EVALUATION AND TREATMENT Urinalysis for evidence of hematuria in the absence of infection provides a useful screening tool for high-risk patients. Several bladder tumor antigen-testing systems have been developed for screening, but they have proved more useful in monitoring patients with known cancer as compared to being used for primary screening. Urine cytology (pathologic analysis of sloughed cells within the urine) is completed in individuals with evidence of hematuria from unknown causes; cystoscopy or fluorescence cystoscopy with tissue biopsy can confirm the diagnosis. Biologic markers for the diagnosis of bladder cancer are under investigation.42 Transurethral resection or laser ablation, combined with intravesical chemotherapy or immunotherapy, is effective for superficial tumors.43 Radical cystectomy with urinary diversion and adjuvant chemotherapy is required for locally invasive tumors, and radiation therapy may be used to support palliative or adjuvant treatment for muscle invasive tumors.44
URINARY TRACT INFECTION
Causes of Urinary Tract Infection
A urinary tract infection (UTI) is an inflammation of the urinary epithelium usually caused by bacteria from gut flora. A UTI can occur anywhere along the urinary tract including the urethra, prostate, bladder, ureter, or kidney. At risk are premature newborns; prepubertal children; sexually active and pregnant women; women treated with antibiotics that disrupt vaginal flora; spermicide users; estrogen-deficient postmenopausal women; individuals with indwelling catheters; and persons with diabetes mellitus, neurogenic bladder, or urinary tract obstruction. UTIs are commonly classified by their location or complicating factors: cystitis (bladder inflammation), pyelonephritis (inflammation of upper urinary tract), uncomplicated UTI (occur in a normally functioning urinary system) and complicated UTI (occur with defects in the urinary system or in individuals with other health problems).
Cystitis is more common in women because of the shorter urethra and the closeness of the urethra to the anus (increasing the possibility of bacterial contamination). Up to 10% of women have an acute uncomplicated UTI in a year and up to 60% of women may have a lower UTI at some time in their life.45 Several factors normally combine to protect against UTIs. Most bacteria are washed out of the urethra during micturition. The low pH and high osmolality of urea, the presence of Tamm-Horsfall protein, and secretions from the uroepithelium provide a bactericidal effect. The ureterovesical junction closes during bladder contraction, preventing reflux of urine to the ureters and kidneys. Both the longer urethra and prostatic secretions decrease the risk of infection in men.
Types of Urinary Tract Infection
Cystitis: Acute cystitis is an inflammation of the bladder and is the most common site of UTI. The morphologic appearance of the bladder through cystoscopy describes different types of cystitis. With mild inflammation, the mucosa is hyperemic (red). More advanced cases may show diffuse hemorrhage (termed hemorrhagic cystitis), pus formation, or suppurative exudates (termed suppurative cystitis) on the epithelial surface of the bladder. Prolonged infection may lead to sloughing of the bladder mucosa with ulcer formation (termed ulcerative cystitis). The most severe infections may cause necrosis of the bladder wall (termed gangrenous cystitis). Generally infections are mild, without complications, and occur in individuals with a normal urinary tract. Acute cystitis may occur alone or in association with pyelonephritis or prostatitis.
PATHOPHYSIOLOGY The most common infecting microorganisms are uropathic strains of Escherichia coli (E. coli) (80% to 85%) and the second most common is Staphylococcus saprophyticus (10%). Less common microorganisms include Klebsiella, Proteus, Pseudomonas, fungi, viruses, parasites, or tubercular bacilli. Bacterial contamination of the normally sterile urine usually occurs by retrograde movement of gram-negative bacilli into the urethra and bladder and then to the ureter and kidney.46 Some women may be genetically susceptible to certain strains of E. coli attachment.47
Fungal infections are comparatively uncommon. The most common pathogen is Candida, but multiple fungal species may colonize the urinary tract or urinary catheters and produce symptomatic UTI particularly in those who are immunosuppressed.48
Hematogenous infections are uncommon and often preceded by septicemia. Infection initiates an inflammatory response and the symptoms of cystitis. The inflammatory edema in the bladder wall stimulates discharge of stretch receptors, initiating symptoms of bladder fullness with small volumes of urine and producing the urgency and frequency of urination associated with cystitis.
Schistosomiasis is the most common cause of parasitic invasion of the urinary tract on a global basis; it infects more than 200 million people.49 Although rare among people living in the United States, the parasite dwells in waters of the various rivers of fresh water bodies in Africa, South America, and Pacific Rim countries. It usually enters the human by swimming up the urethra while the host swims or is partly submerged in an infected body of water. The parasite burrows into the walls of the urinary tract, causing inflammation and scarring of the urinary tract and an increased risk for urothelial malignancies.50
Two factors account for the presence of a UTI: the efficiency of defense mechanisms within the host (individual) and the virulence of the pathogen (bacterium, fungus, or parasite). In the healthy individual, host defense mechanisms maintain a sterile posterior urethra and bladder. Even if bacteria manage to enter the bladder, these defense mechanisms prevent it from clinging to the walls of the bladder or ascending to the upper urinary tracts.51 Most people are able to rapidly rid the urinary tract of invading bacteria, but some show evidence of bacteria in the urine that does not provoke an infection. This condition is called asymptomatic bacteriuria and does not harm urinary function or require intervention except in pregnant women.52 A UTI occurs when a pathogen circumvents or overwhelms the host’s defense mechanisms and rapidly reproduces.
Virulence of Uropathogens
Virulence is a pathogen’s ability to evade or overwhelm the host defense mechanisms and cause disease in a host (see Chapter 9). Several factors contribute to bacterial virulence within the urinary tract,53 including the ability of uropathic bacteria to adhere (attach) to the uroepithelium.54 Uropathic strains of E. coli have type-1 fimbriae that bind to latex catheters and to receptors on uroepithelium resisting flushing during normal micturition. Additionally they have pyelonephritis-associated fimbriae (P fimbriae) that bind to the uroepithelial P-blood group antigen that is present in most of the human population and readily ascend the urinary tract.55 Strains of E. coli also produce siderophores for acquiring nutrient iron, are resistant to bactericidal effects of complement, and express toxins including cytotoxin necrotizing factor-1 and hemolysins. Certain bacterial species also enhance their virulence by acting together to form a biofilm that enhances colonization and resists efficiency of innate host defense mechanisms and antimicrobial therapy.56,57
Host Defense Mechanisms: Opposing bacterial virulence are multiple host defense mechanisms including the immune system and the uroepithelium. Periurethral mucus-secreting glands surround the distal two thirds of the female urethra. Mucus from these glands traps bacteria before it can ascend from proximal urethra to the bladder. In men, the length of the male urethra and secretions from the prostate and accessory periurethral glands combine to form a protective barrier against infection. In addition, the urethral sphincter mechanism acts as a mechanical barrier to bacterial ascent from the distal urethra.
Bacteria that successfully ascend the urethra face detection and destruction by components of the body’s immune system provided they come into contact with the bladder wall. Unfortunately, time is required for the immune system to respond to the potential threat, and this period may provide adequate time for bacteria or other pathogens to reproduce several times.
The efficiency of the bladder’s defenses is also influenced by the person’s Lewis blood group.58 This taxonomy is based on recognition of inherited antigens associated with the ABO blood factors. Individuals with certain Lewis blood groups are more prone to UTI because they secrete fewer antigens capable of resisting bacterial adherence by pili formation.
The urine itself may contain components that enhance resistance to UTI. These include hydrogen ion concentration (pH), osmolarity (concentration of salts within the urine), glucose content, urea, and glycoproteins. Ideally, the urine should have a slightly acidic pH (6 or less), moderate to high urea concentration, and abundant glycoproteins (slimy substances that interfere with bacterial adherence). Dilute urine washes out bacteria, and urine with higher urea concentrations (high osmolarity) is more bacteriostatic. In contrast, glucose in the urine, a higher (alkaline) pH, or urine with high osmolarity but low urea concentration is less bacteriostatic.
CLINICAL MANIFESTATIONS Many individuals with bacteriuria are asymptomatic. Clinical manifestations of cystitis, however, usually include frequency, urgency, dysuria (painful urination), and suprapubic and low back pain. Hematuria, cloudy and foul-smelling urine, and flank pain are more serious symptoms. Approximately 10% of individuals with bacteriuria have no symptoms, and 30% of individuals with symptoms are abacteriuric. Older adults with cystitis may be asymptomatic or demonstrate confusion or vague abdominal discomfort. Older adults with recurrent UTI and other concurrent illness have a higher risk of mortality.59
EVALUATION AND TREATMENT Infections are diagnosed by urine culture of specific microorganisms with counts of 10,000/ml or more from freshly voided urine. Dipstick urinalysis and microscopy are adequate to diagnose an uncomplicated UTI, but urine culture is critical for complicated infections. Risk factors, such as a urinary tract obstruction, which are associated with a complicated UTI, should be identified and treated. Increased fluid intake and urinary analgesics can relieve symptoms. Evidence of bacteria from urine culture and antibiotic sensitivity warrants treatment with a microorganism-specific antibiotic to eradicate the underlying pathogen. A single large dose of antibiotic or a 3-day course may be effective when symptoms are of short duration and there are no complications. Three to 7 days of treatment is most common depending on antimicrobial selection; older adults with obstructive disorders may require 7 to 14 days of treatment. Frequent, recurrent, acute uncomplicated UTI requires low-dose antimicrobial therapy from 6 months to 2 years.46 Follow-up urine cultures should be obtained 1 week after initiation of treatment and at monthly intervals for 3 months. Clinical symptoms are frequently relieved, but bacteriuria may still be present, particularly with the development of antimicrobial-resistant bacterial strains. Repeat cultures should be obtained every 3 to 4 months until 1 year after treatment for evaluation of recurrent infection.60 Urosepsis and septic shock are medical emergencies that usually demand parenteral, broad-spectrum antibiotic therapy and may require hospitalization. A UTI caused by
Schistosomiasis is treated with praziquantel, and vaccines are under development.61
Painful Bladder Syndrome/Interstitial Cystitis: Painful bladder syndrome/interstitial cystitis (PBS/IC) is a condition that includes nonbacterial infectious cystitis (viral, mycobacterial, chlamydial, fungal), and noninfectious cystitis (radiation, chemical, autoimmune, hypersensitivity).62 It occurs most commonly in women ages 20 to 30 years who have symptoms of cystitis, such as frequency, urgency, dysuria, and nocturia, but with negative urine cultures and no other known etiology. Nonbacterial infectious cystitis is most common among those who are immunocompromised. Noninfectious cystitis is associated with radiation or chemotherapy treatment for pelvic and urogenital cancers.
The cause of PBS/IC is unknown, but an autoimmune reaction may be responsible for the inflammatory response, which includes mast cell activation, altered epithelial permeability, and increased sensory nerve sensitivity.63 The inflammation is associated with a derangement of the bladder mucosa that makes it more susceptible to penetration by bacteria and noxious urinary solutes.64 Inflammation and fibrosis of the bladder wall are accompanied by the presence of hemorrhagic ulcers (Hunner ulcers), and bladder volume may decrease as a result of fibrosis. More recently, the identification of antiproliferative factor (APF), a protein expressed by the bladder uroepithelium in those with IC, is important. APF appears to block the normal growth of cells that line the inside wall of the bladder and indirectly increases bladder sensation.65 Characteristic symptoms of IC include bladder fullness, frequency (including nocturia), small urine volume, and chronic pelvic pain with symptoms lasting longer than 9 months. Diagnosis of IC requires the exclusion of other diagnoses, and extensive evaluations are completed.66 No single treatment is effective, and different approaches are used for symptom relief.67
Acute Pyelonephritis: Pyelonephritis is an infection of one or both upper urinary tracts (ureter, renal pelvis, and interstitium). Common causes are summarized in Table 36-4. Urinary obstruction and reflux of urine from the bladder (vesicoureteral reflux) are the most common underlying risk factors. One or both kidneys may be involved. Most cases occur in women. The responsible microorganism is usually E. coli, Proteus, or Pseudomonas. The latter two microorganisms are more commonly associated with infections after urethral instrumentation or urinary tract surgery. These microorganisms also split urea into ammonia, making alkaline urine that increases the risk of stone formation.
Table 36-4
Common Causes of Pyelonephritis
| Predisposing Factors | Pathologic Mechanisms |
| Kidney stones | Obstruction and stasis of urine contributing to bacteriuria and hydronephrosis; irritation of epithelial lining with entrapment of bacteria |
| Vesicoureteral reflux | Chronic reflux of urine up the ureter and into kidney during micturition contributing to bacterial infection |
| Pregnancy | Dilation and relaxation of ureter with hydroureter and hydronephrosis; partly caused by obstruction from enlarged uterus and partly from ureteral relaxation caused by higher progesterone levels |
| Neurogenic bladder | Neurologic impairment interfering with normal bladder and urethral sphincter contraction with residual urine and ascending infection |
| Instrumentation | Introduction of organisms into urethra and bladder by catheters and endoscopes introduced into the urinary tract for diagnostic purposes |
| Female sexual trauma | Movement of organisms from the urethra into the bladder with infection and retrograde spread to kidney |
PATHOPHYSIOLOGY The infection is probably spread by ascending uropathic microorganisms along the ureters, but spread also may occur by way of the bloodstream. The inflammatory process is usually focal and irregular, primarily affecting the pelvis, calyces, and medulla. The infection causes medullary infiltration of white blood cells with renal inflammation, renal edema, and purulent urine. In severe infections, localized abscesses may form in the medulla and extend to the cortex. Primarily affected are the tubules; the glomeruli usually are spared. Necrosis of renal papillae can develop. After the acute phase, healing occurs with deposition of scar tissue, fibrosis and atrophy of affected tubules (Figure 36-6). Acute pyelonephritis rarely causes renal failure.68
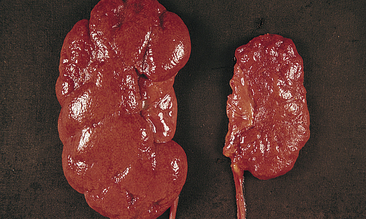
Figure 36-6 Pyelonephritis. Right: Small, shrunken, irregularly scarred kidney of an individual with chronic pyelonephritis. Left: Kidney is of normal size but also shows scarring on the upper pole. (From Damjanov I: Pathology for the health professions, ed 3, St Louis, 2006, Saunders.)
CLINICAL MANIFESTATIONS The onset of symptoms is usually acute, with fever, chills, and flank or groin pain. Symptoms characteristic of a UTI, including frequency, dysuria, and costovertebral tenderness, may precede systemic signs and symptoms. Older adults may have nonspecific symptoms, such as low-grade fever and malaise.
EVALUATION AND TREATMENT Differentiating symptoms of cystitis from those of pyelonephritis by clinical assessment alone is difficult. The specific diagnosis is established by urine culture, urinalysis, and clinical signs and symptoms. White blood cell casts indicate pyelonephritis, but they are not always present in the urine. Complicated pyelonephritis requires blood cultures and urinary tract imaging.69,70
Uncomplicated acute pyelonephritis responds well to 2 to 3 weeks of microorganism-specific antibiotic therapy. Follow-up urine cultures are obtained at 1 and 4 weeks after treatment if symptoms recur. Antibiotic-resistant microorganisms or reinfection may occur in cases of urinary tract obstruction or reflux. Intravenous pyelography and voiding cystourethrography identify surgically correctable lesions.
Chronic Pyelonephritis: Chronic pyelonephritis is a persistent or recurrent infection of the kidney leading to scarring of the kidney. One or both kidneys may be involved. The specific cause of chronic pyelonephritis may be unknown (idiopathic) or associated with versicoureteral reflux or renal stones. Recurrent infections from acute pyelonephritis may be associated with chronic pyelonephritis. Causes other than chronic pyelonephritis include drug toxicity from analgesics such as nonsteroidal anti-inflammatory drugs, ischemia, irradiation, and immune-complex diseases.
PATHOPHYSIOLOGY Chronic urinary tract obstruction prevents elimination of bacteria and starts a process of progressive inflammation, altered renal pelvis and calyces, destruction of the tubules, atrophy or dilation and diffuse scarring, and finally impaired urine-concentrating ability, leading to chronic kidney failure. The lesions of chronic pyelonephritis are sometimes termed chronic interstitial nephritis because the inflammation and fibrosis are located in the interstitial spaces between the tubules (see Figure 36-6).
CLINICAL MANIFESTATIONS The early symptoms of chronic pyelonephritis are often minimal and may include hypertension, frequency, dysuria, and flank pain. With loss of tubular function is an inability to conserve sodium, and development of hyperkalemia and metabolic acidosis. Risk for dehydration must be considered if there is loss of ability to concentrate the urine. Progression of disease leads to renal failure, particularly in the presence of other risk factors (i.e., obstructive uropathy or diabetes mellitus).71
EVALUATION AND TREATMENT Urinalysis, intravenous pyelography, and ultrasound are used diagnostically. Treatment is related to the underlying cause. Obstruction must be relieved. Antibiotics may be given, with prolonged antibiotic therapy for recurrent infection.
GLOMERULAR DISORDERS
Glomerular disease can be caused by primary injury within the glomulus or secondarily as a result of systemic disease including metabolic disorders such as diabetes mellitus, bacterial or viral infectious disease, or systemic immune disease such as systemic lupus erythematosus (lupus nephritis). Most primary and many secondary glomerular diseases are the result of immune injury (Figure 36-7). Immune injury includes (1) deposition of circulating antigen-antibody immune complexes in the glomulus (type III hypersensitivity reaction); (2) antibodies reacting in situ against planted antigens within the glomerulus (type III hypersensitivity); (3) action of antibodies directed against the glomerular capillary wall (antiglomerular basement membrane antibodies), the least common and most severe form of immune injury (type II hypersensitivity); and (4) cell-mediated immune injury (type IV hypersensitivity) (see Chapter 8). The severity of glomerular damage and decline in glomerular function are related to the size, number, location (focal or diffuse), duration of exposure, and type of antigen-antibody complexes.
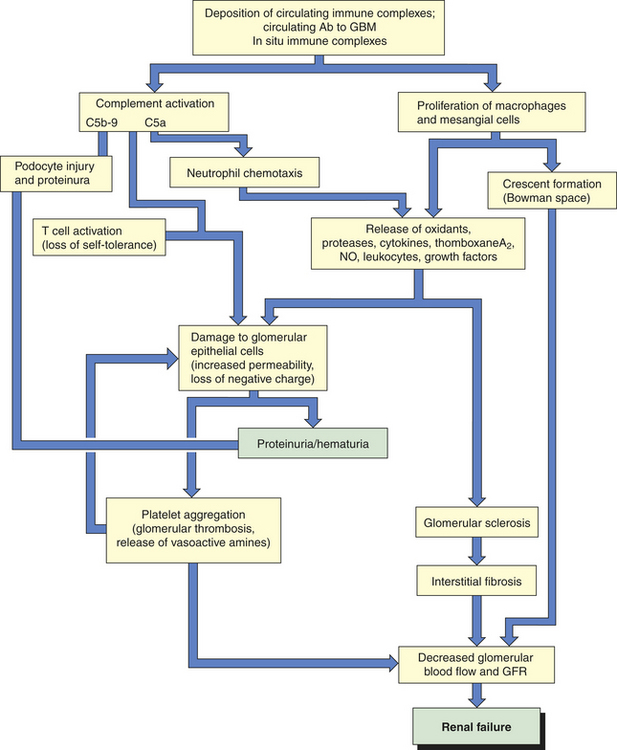
Figure 36-7 Mechanisms of glomerular injury. Ab, antibody; GBM, glomerular basement membrane; GFR, glomerular filtration rate; NO, nitric oxide.
Patterns of antigen-antibody complex deposition or formation within the glomerular capillary filtration membrane have been established using light, electron, and immunofluorescent microscopy for different disease processes (Table 36-5). Electron microscopy differentiates morphologic changes within the glomerular capillary wall. Staining with fluorescein identifies different antibodies (i.e., immunoglobulin G [IgG] or IgA) and their configurations when viewed under ultraviolet light with a microscope (see Figure 36-8, C, p. 1382).
Table 36-5
Immunologic Pathogenesis of Glomerulonephritis
| Glomerular Injury | Mechanism |
| Soluble immune-complex glomerulonephritis (90%) | Formation of antibodies stimulated by the presence of endogenous or exogenous antigens results in circulating soluble antigen-antibody complexes, which are deposited in glomerular capillaries, or the in situ formation of immune complexes to planted antigens or to structural components within the glomerulus; glomerular injury occurring with complement deposition and activation and release of immunologic substances that lyse cells and increase membrane permeability; immune deposits with a microscopic appearance that fluoresce in a granular pattern when stained with fluorescein and viewed under ultraviolet light; severity of glomerular injury related to the number of complexes formed; a type III hypersensitivity |
| Antiglomerular basement membrane glomerulonephritis (5%) | Antibodies are formed and act directly against the glomerular basement membrane; immune response that causes crescent formation and a linear pattern of immunofluorescence; generally associated with rapidly progressive renal failure such as Goodpasture syndrome (type II hypersensitivity) |
| Alternative complement pathway | A relatively obscure mechanism associated with low levels of complement and membranoproliferative glomerulonephritis |
| Cell-mediated immunity | A delayed hypersensitivity response that damages the glomerulus; actual cellular mechanism not clearly understood but may involve cytokine secretion, activation of effector cells such as macrophages or by inducing autoantibodies or immune complexes. Cytotoxic CD8+ T-cell responses and failure of regulatory T cells may represent two additional types of antirenal hypersensitivity∗ |
∗Data from Kurts C et al: Role of T cells and dendritic cells in glomerular immunopathology, Semin Immunopathol 29(4):317-335, 2007.
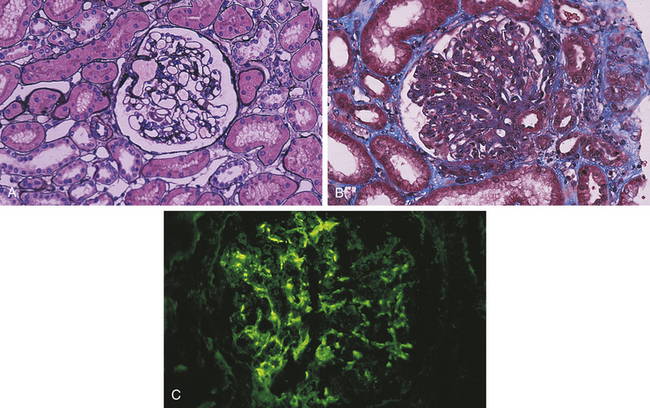
Figure 36-8 Glomerulonephritis. A, Normal glomerulus; note single-contoured walls, patent capillaries, inconspicuous mesangium, and degree of cellularity. (Periodic acid–methenamine silver stain.) B, Acute postinfectious glomerulonephritis. There is considerable increase in cellularity, mainly because of accumulation of numerous polymorphonuclear leukocytes in capillary lumina. Note numerous subepithelial hump-shaped fuchsinophilic deposits in many capillary walls. Protein precipitates (hyalinization) are in the arteriole. (Masson trichrome stain.) C, Postinfectious glomerulonephritis. Irregular mesangial and capillary wall immunostaining for C3. (From Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
Immune injury is caused by activation of mediators of inflammation (complement, leukocytes, fibrin) and begins after the antibody or antigen-antibody complexes have localized in the glomerular capillary wall. Complement is deposited with the antibodies, and its activation can cause cell lysis or serve as a chemotactic stimulus for attraction of neutrophils and monocytes.72 These phagocytes further the inflammatory reaction by releasing lysosomal enzymes, reactive oxygen species, and cytokines, which damage glomerular cell walls and contribute to swelling and proliferation of mesangial cells and expansion of the extracellular matrix causing a decrease in glomerular blood flow, decreased GFR, and hypoxic injury.73
The inflammatory processes alter membrane permeability with injury to epithelial cells, the glomerular basement membrane (GBM) and endothelial cells (podocytes). Loss of the negative electrical charge across the glomerular filtration membrane enhances filtration of proteins into the urine, which are normally repelled because they also have a negative charge. Membrane damage can lead to platelet aggregation and degranulation, whereby platelets release substances that increase glomerular permeability, permitting the passage of protein molecules or red blood cells into the urine and causing proteinuria (excess protein in the urine, usually albumin), hematuria (blood in the urine), or both. The coagulation system also may be activated and lead to fibrin deposition in Bowman space, contributing to crescent formation (deposition of substances in the Bowman space forming the shape of a crescent moon).74 Renal blood flow decreases, and glomerular filtration is reduced. Hematuria results from increased glomerular permeability or bleeding along the nephron.
Different causes of injury may cause more than one type of glomerular lesion, so lesions are not necessarily disease-specific (Table 36-6). The onset of glomerular disease may be sudden or insidious and significant loss of nephron function can occur before symptoms develop. Glomerular disease may be silent, mild, moderate, or severe in symptom presentation. Severe or progressive glomerular disease causes oliguria, hypertension, and renal failure.
Table 36-6
Classification of Glomerular Lesions
| Lesion | Distribution When Many Glomeruli Considered |
| Diffuse | Relatively uniform involvement of most (>50%) or all glomeruli; most common form of glomerulonephritis |
| Focal | Changes in only some glomeruli (>50%), whereas others are normal |
| Lesion | Distribution When Single Glomeruli Considered |
| Global | A lesion involving the entire glomerulus |
| Segmental-local | Changes in one part of the glomerulus with other parts unaffected |
| Lesion | Lesion Characteristics |
| Mesangial | Deposits of immunoglobulins in the mesangial matrix; mesangial cell proliferation |
| Membranous | Thickening of the glomerular capillary wall with immune deposits (i.e., IgG and C3) |
| Proliferative | Increase in the number of glomerular cells: endothelial, epithelial, mesangial |
| Sclerotic | Glomerular scarring from previous glomerular injury |
| Crescentic | Accumulation of proliferating cells within Bowman space, making the appearance of a crescent |
| Interstitial fibrosis | Scarring between the glomerulus and the tubules |
Different types of glomerular disease may be associated with patterns of urinary sediment (Table 36-7). Urine in diseases associated with nephrotic sediment contains massive amounts of protein and lipids and either a microscopic amount of blood or no blood. Urine in diseases associated with nephritic sediment is characterized by the presence of blood in the urine with red cell casts, white cell casts, and varying degrees of protein, which usually is not severe. The sediment of chronic glomerular disease has waxy casts, granular casts, and less protein and blood than does nephrotic or nephritic sediment.
Table 36-7
Features of the Common Types of Glomerulonephritis
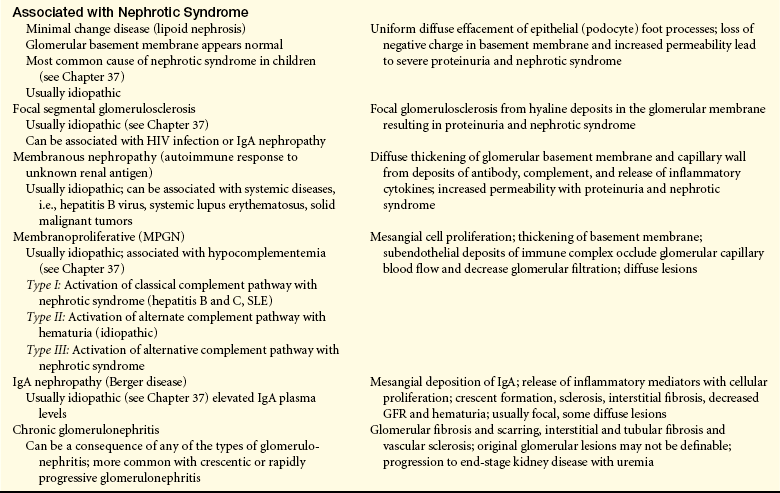
GBM, Glomerular basement membrane; GFR, glomerular filtration rate; HIV, human immunodeficiency virus; IgA, immunoglobulin A; IgG, immunoglobulin G; SLE, systemic lupus erythematosus.
Reduced GFR during glomerular disease is evidenced by elevated plasma urea, creatinine concentration, or reduced renal creatinine clearance (see Chapter 35). Edema, caused by excessive sodium and water retention, may require the use of diuretics or dialysis. The volume expansion that accompanies salt and water retention leads to hypertension. During the first few weeks, the major life-threatening problems are acute renal insufficiency with fluid, electrolyte, and acid-base imbalances; acute hypertension that may cause hypertensive encephalopathy; circulatory failure; and pulmonary edema.
Glomerulonephritis
Glomerulonephritis is an inflammation of the glomerulus caused by numerous factors, including infection, immunologic abnormalities (the most common cause), ischemia, free radicals, drugs, toxins, vascular disorders, and systemic diseases, including diabetes mellitus and lupus erythematosus. Glomerular disease is the most common cause of chronic kidney disease and end-stage renal failure.75
The classification of glomerulonephritis can be described according to cause, pathologic lesions, disease progression (acute, rapidly progressive, chronic), or clinical presentation (nephrotic syndrome, nephritic syndrome, acute or chronic kidney failure). In nearly all types of glomerulonephritis, the epithelial or podocyte layer of the glomerular capillary membrane is disturbed with loss of negative charges and changes in membrane permeability; the mesangial matrix may be expanded or the basement membrane thickened (see Figure 36-7). Many types of glomerulonephritis occur most often in children or young adults. Minimal change disease, focal segmental glomerulosclerosis, and membranoproliferative glomerulonephritis are discussed in Chapter 37.
Acute Postinfectious Glomerulonephritis
Acute postinfectious glomerulonephritis (PIGN) usually involves an immunologic mechanism that activates inflammation with damage to the glomerular basement membrane, capillary endothelium, and mesangium. Acute postinfectious glomerulonephritis is most often is associated with a streptococcal infection (acute poststreptococcal glomerulonephritis). The disease begins abruptly and usually occurs 7 to 10 days after a streptococcal infection of the skin (impetigo) or of the throat (pharyngitis) and commonly affects children (see Chapter 37). Sporadic occurrences have been observed after bacterial endocarditis, which may be associated with streptococcal or staphylococcal microorganisms, or after viral diseases such as varicella and hepatitis B and C. Glomerular injury is immune mediated with streptococcal antigen-antibody complexes either depositing in the GBM or forming in situ against planted antigens. The antigen-antibody complex activates complement and the release of inflammatory mediators that damage endothelial and epithelial cells lying on the basement membrane and causes altered permeability and cellular proliferation.76
Symptoms may be insidious or sudden and usually occur 10 to 21 days after infection and include hematuria, red blood cell casts, proteinuria, decreased GFR, oliguria, hypertension, edema around the eyes or feet and ankles, and, occasionally, ascites or pleural effusions. Blood urea nitrogen (BUN) is elevated. Immunofluorescent findings from renal biopsy indicate immune complex deposits in the glomerulus (complement C3 and IgG), neutrophil and macrophage recruitment and activation, with diffuse mesangial cell and capillary endothelial cell proliferation of the entire glomeruli77 (Figure 36-8). The thickened glomerular membrane contributes to the decreased GFR. Activated complement, inflammatory cytokines, oxidants, proteases, and growth factors attack epithelial cells, alter membrane permeability, and cause proteinuria. More severe renal disease is observed after a prolonged infection and before antibiotic therapy. In acute poststreptococcal glomerulonephritis the streptococcal exoenzymes are elevated, such as antistreptolysin-O and antistreptokinase. Serum complement is decreased, and serum creatinine concentration and blood urea nitrogen are elevated.
There is no specific treatment for glomerulonephritis. Most individuals, especially children, recover without significant loss of renal function or recurrence of the disease. During the first few weeks the major life-threatening problems are acute renal insufficiency with fluid, electrolyte, and acid-base imbalances; acute hypertension may cause hypertensive encephalopathy. Death occurs in about 1% of all persons with PIGN, mostly in developing countries.78
Lupus Nephritis
Lupus nephritis is an inflammatory complication of the chronic autoimmune syndrome, systemic lupus erythematosus (see Chapter 8). The renal component of the disease is one of the more severe complications and is associated with autoantibodies against double-stranded deoxyribonucleic acid (dsDNA) and nucleosomes.79 Complexes of these autoantibodies and complement accumulate in the glomerulus, causing cell proliferation, inflammation, and injury. Different glomerular lesion patterns are identifiable on biopsy including membranous, mesangial, membranoproliferative, and diffuse proliferative glomerulonephritis (see Table 36-6).80 Symptom presentation is variable depending on lesion involvement and can include proteinuria, edema, and other signs of nephrotic syndrome (p. 1384). Disease progression may be silent or may progress to end-stage kidney failure over a period of years. Long-term management includes corticosteroids, immunosuppressants, and renal replacement therapy.
IgA Nephropathy
IgA nephropathy (Berger disease) is the most common form of acute glomerulonephritis in developed countries, especially Asia. The cause is unknown and more commonly affects adults ages 20 to 30 years. Henoch-Schönlein purpura is a milder systemic form of the disease that presents with hematuria and occurs more often in children (see Chapter 37). Abnormal glycosylated IgA-1 (galactose-deficient IgA-1) produced by the bone marrow and complement molecules binds to glomerular mesangial cells, stimulating them to proliferate and release oxidants and proteases, thereby contributing to diffuse mesangioproliferative glomerular injury and glomerulosclerosis.81 The disease manifests with gross or microscopic (30% to 40%) hematuria 24 to 48 hours after an upper respiratory or gastrointestinal viral infection. Proteinuria, edema, and hypertension are less common. Diagnosis is made by renal biopsy. Treatment may include angiotensin-converting enzyme (ACE) inhibitors, glucocorticoids, and cyclophosphamide. The prognosis is variable, with 14% to 39% of cases progressing to renal failure over a period of years.82
Crescentic or Rapidly Progressive Glomerulonephritis
Rapidly progressive (crescentic) glomerulonephritis (RPGN) is also known as subacute or extracapillary glomerulonephritis and develops over days to weeks. The disease affects primarily adults in their 50s and 60s and may be idiopathic or associated with a proliferative glomerular disease (diffuse proliferation of extracapillary cells), such as lupus or poststreptococcal glomerulonephritis. Antiglomerular basement membrane antibodies and antineutrophil cytoplasmic antibodies are associated with glomerular injury.83 There is extensive proliferation of cells into the Bowman space with crescent formation (the shape of the Bowman capsule). Typically the glomerular injury is accompanied by a rapid decline in glomerular function, progressing to renal failure in a few weeks or months.84 Hematuria is common and may or may not be accompanied by proteinuria, edema, or hypertension. There are three types of RPGN:
Type I: Antiglomerular basement membrane disease (Goodpasture syndrome) is a type of RPGN. The disease is rare and associated with IgG antibody formation against pulmonary capillary and glomerular basement membranes, with activation of complement and neutrophils that damage the basement membrane. The disease occurs most often in men 20 to 30 years of age, often accompanied by pulmonary hemorrhage and renal failure.
Type II: Immune complex deposition involves deposition of immune complexes in the mesangium and is often associated with lupus nephritis and PIGN.
Type III: Pauci immune glomerulonephritis involves the presence of serum antineutrophil cytoplasmic antibodies without immune complex deposition. It is usually idiopathic.
RPGN has a relatively poor prognosis if not diagnosed and treated early. Anticoagulants may be of some benefit in reducing the fibrin component of crescent formation. Plasmapheresis is usually combined with steroids and immunosuppression therapy, including plasma exchange. Dialysis or transplantation is required when failure is irreversible.85
Mesangial Proliferative Glomerulonephritis
Mesangial proliferative glomerulonephritis involves deposits of immune complex in the mesangium with mesangial cell proliferation. Mesangial expansion reduces blood flow and alters filtration membrane permeability with development of hematuria, proteinuria, hypertension, and uremia (nephritic syndrome).
Membranous Nephropathy
Membranous nephropathy, also known as membranous glomerulonephritis, is usually caused by deposition of circulating antibodies or antibodies formed in situ to antigens located in the glomerular basement membrane. The antigen-antibody complexes activate C5b-C9 fragments of complement (the membrane attack complex) on glomerular epithelial cells with injury and release of inflammatory mediators by mesangial and epithelial cells resulting in increased membrane permeability, thickening of the glomerular membrane, and ultimately glomerular sclerosis. Proteinuria and nephrotic syndrome are common manifestations.
Membranoproliferative Glomerulonephritis
Membranoproliferative glomerulonephritis (MPGN) is usually idiopathic, involves proliferation of mesangial cells, and the formation of crescents related to the deposition of complement. This disease occurs more commonly in children and young adults. Hypocomplementemia is associated with all types of MPGN. Immune complexes are deposited in the mesangium and subendothelial spaces in type I MPGN, activating complement and release of inflammatory cytokines that leads to mesangial and endothelial cell proliferation. In type II MPGN there are dense deposits that are also present in other organs, so it is a systemic disease. There are no circulating immune complexes. Type III MPGN can be familial and involves deposition and activation of complement in the capillary wall with subepithelial and subendothelial deposits. Injury to the glomerular capillary wall in all types of MPGN can cause proteinuria, hematuria, nephrotic syndrome, and acute or chronic kidney failure.
Chronic Glomerulonephritis
Chronic glomerulonephritis encompasses glomerular diseases with a progressive course leading to chronic kidney disease (see p. 1389). Immune injury may cause progressive glomerular destruction but in many cases the cause is unknown and there may be no history of renal disease before the diagnosis. Hypercholesterolemia and proteinuria have been associated with progressive glomerulosclerosis, tubulointerstitial fibrosis and tubular atrophy.86 The primary cause may be difficult to establish because advanced pathologic changes may obscure specific disease characteristics (Figures 36-9 and 36-10). Diabetes mellitus and systemic lupus erythematosus are secondary causes of chronic glomerular injury.87
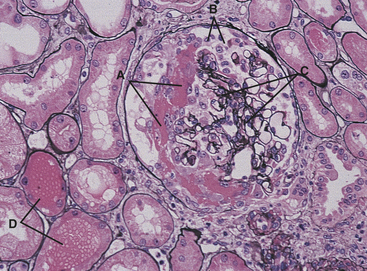
Figure 36-9 Antiglomerular basement membrane nephritis. Glomerulus with a fresh crescent consisting of fibrin and cells in the Bowman space (A). There is disruption of the basement membrane of the Bowman capsule, with migration of cells from the interstitium into the Bowman space (B). The capillary tufts (C) are distorted and compressed because of the crescent. Note the free erythrocytes in tubular lumina (D). The interstitium is mildly edematous. (Periodic acid–methenamine silver stain.) (Modified from Damjanov I, Linder J, editors: Anderson’s pathology, ed 10, St Louis, 1996, Mosby.)
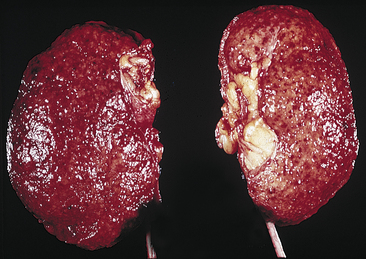
Figure 36-10 Chronic glomerulonephritis. The kidneys appear small, are uniformly shrunken, and have a finely granular external surface (From Damjanov I: Pathology for the health professions, ed 3, St Louis, 2006, Saunders.)
CLINICAL MANIFESTATIONS Injury to the glomeruli causes various signs and symptoms consequent to changes in glomerular capillary wall structure and GFR. Two major changes distinctive of more severe glomerulonephritis are (1) hematuria with red blood cell casts and (2) proteinuria exceeding 3 to 5 g/day with albumin as the major protein. Gross proteinuria is associated with nephrotic syndrome and a decrease in urine output accompanies a decreased GFR with elevations in plasma creatinine (see Chapter 35).
Several disorders may produce hematuria because bleeding can occur anywhere along the urinary tract. The characteristics of hematuria from red blood cells escaping through the glomerular membrane include a smoky brown–tinged urine, red blood cell casts, and accompanying proteinuria. Bleeding from sites lower in the urinary tract may produce a pink or red-tinged urine. Glomerular bleeding provides prolonged contact with the acidic urine and transforms hemoglobin to methemoglobin, which is brownish and has no blood clots. The history and physical examination may disclose findings that differentiate glomerular disease from another source of urinary tract bleeding.
The immune-mediated inflammatory response with cellular infiltration decreases GFR, which leads to fluid retention. Salt and water are also reabsorbed, contributing to fluid volume expansion, edema, and hypertension.
Microscopic proteinuria and hematuria may occur during the early years of the disease. Blood pressure may be normal. After 5 to 20 years, renal insufficiency usually begins to develop, followed by nephrotic syndrome and an accelerated progression to end-stage renal failure. Symptom patterns vary depending on the underlying cause. Biopsy may reveal the underlying glomerular lesion (see Table 36-6).
EVALUATION AND TREATMENT The diagnosis of glomerular disease is confirmed by the progressive development of clinical manifestations and laboratory findings of abnormal urinalysis with proteinuria, red blood cells, white blood cells, and casts. Microscopic evaluation from renal biopsy provides a specific determination of renal injury and type of pathologic condition (see Tables 36-6 and 36-7).
Management principles for treating glomerulonephritis are related to treating the primary disease, preventing or minimizing immune responses, and correcting accompanying problems, such as edema, hypertension, hyperlipidemia, hyperkalemia, and hyperglycemia in those with diabetes mellitus. Specific treatment regimens are necessary for particular types of glomerulonephritis. Antibiotic therapy is essential for the management of underlying infections that may be contributing to ongoing antigen-antibody responses. Corticosteroids may decrease antibody synthesis and suppress inflammatory responses. Cytotoxic agents (i.e., cyclophosphamide) may be used to suppress the immune response. Anticoagulants may be useful for controlling fibrin crescent formation in RPGN. Dialysis or kidney transplantation ultimately may be needed.
Nephrotic Syndrome
Nephrotic syndrome is the excretion of 3.5 g or more of protein in the urine per day and is characteristic of glomerular injury. Lipoid nephrosis (minimal change disease), membranous glomerulonephritis, and focal glomerulosclerosis are directly related to nephrotic syndrome, although these conditions can occur with other types of glomerular disease.88 Nephrotic syndrome is more common in children than adults (see Chapter 37). Secondary forms of nephrotic syndrome occur in systemic diseases, including diabetes mellitus, amyloidosis, systemic lupus erythematosus, and Henoch-Schönlein purpura. Nephrotic syndrome is also seen with certain drugs, infections, malignancies, and vascular disorders. Familial forms of nephrotic syndrome result from genetic defects that affect the function and composition of the glomerular capillary wall (i.e., Alport syndrome with alterations in basement membrane type IV collagen).89 Nephrotic syndrome often signifies a more serious prognosis when present as a secondary complication.
PATHOPHYSIOLOGY Disturbances in the GBM and podocyte injury lead to increased permeability to protein and loss of electrical negative charge. Loss of plasma proteins, particularly albumin and some immunoglobulins, occurs across the injured glomerular filtration membrane (Figure 36-11).88 Sustained proteinuria can result in the release of inflammatory mediators and cytokines by tubular cells with influx of leukocytes resulting in progressive glomerulosclerosis and renal fibrosis.90 Hypoalbuminemia results from urinary loss of albumin combined with a diminished synthesis of replacement albumin by the liver. Albumin is lost in the greatest quantity because of its high plasma concentration and low molecular weight. Decreased dietary intake of protein from anorexia or malnutrition or accompanying liver disease may also contribute to lower levels of plasma albumin. Loss of albumin stimulates lipoprotein synthesis by the liver and hyperlipidemia. Loss of immunoglobulins may increase susceptibility to infections. Sodium retention also is associated with nephrotic syndrome contributing to the development of edema and ascites. The exact mechanism is unknown but the site of retention is the distal nephron tubules and collecting ducts.91
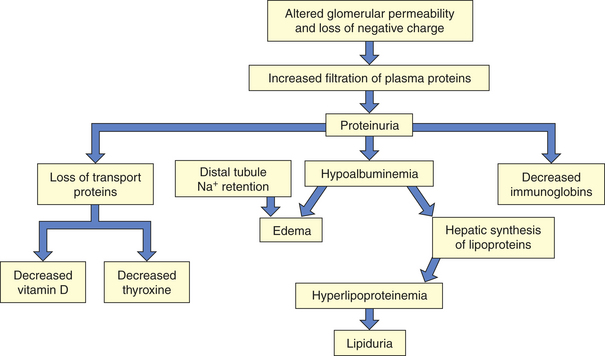
CLINICAL MANIFESTATIONS Many clinical manifestations of nephrotic syndrome are related to loss of serum proteins (Table 36-8) and sodium retention. They include edema, hyperlipidemia, lipiduria, vitamin D deficiency, and hypothyroidism.92,93 Vitamin D deficiency is related to loss of serum transport proteins and decreased vitamin D activation by the kidney. Hypothyroidism can result from urinary loss of thyroid-binding protein and thyroxine. Alterations in coagulation factors cause hypercoagulability and may lead to thromboembolic events, particularly in young adults.94

EVALUATION AND TREATMENT Nephrotic syndrome is diagnosed when the protein level in a 24-hour urine collection is greater than 3.5 g. Serum albumin decreases (to less than 3 g/dl), and serum cholesterol, phospholipids, and triglycerides increase. Fat bodies may be present in the urine. The specific pathologic condition is identified by renal biopsy.
Nephrotic syndrome is commonly treated with a normal-protein (i.e., 1 g/kg body weight/day) low-fat diet, salt restriction, diuretics, immunosuppression, and heparinoids. When diuretics are used, care must be taken to observe for hypovolemia and hypokalemia or potassium toxicity in the presence of renal insufficiency. Aldactone may be combined with loop diuretics to suppress aldosterone activity to conserve potassium. Steroids may be particularly effective for the initial treatment of nephrotic syndrome in children.95
ACUTE KIDNEY INJURY
Classification of Kidney Dysfunction
Kidney injury may be acute and rapidly progressive (within hours), and the process may be reversible. Kidney failure also can be chronic, progressing to end-stage kidney failure over a period of months or years. The terms renal insufficiency, renal failure, uremia, and azotemia are associated with decreasing renal function but are not specific in relation to kidney function. Often they are used synonymously, although with some distinctions. Generally, renal insufficiency refers to a decline in renal function to about 25% of normal or a GFR of 25 to 30 ml/minute. Levels of serum creatinine and urea are mildly elevated. Renal failure refers to significant loss of renal function. When less than 10% of renal function remains, this is termed end-stage renal failure (ESRF). Specific criteria for acute renal dysfunction are discussed in the next section. Uremia is a syndrome of renal failure and includes elevated blood urea and creatinine levels accompanied by fatigue, anorexia, nausea, vomiting, pruritus, and neurologic changes. Uremia represents the numerous consequences related to renal failure, including retention of toxic wastes, deficiency states, and electrolyte disorders. Azotemia means increased serum urea levels and frequently increased creatinine levels as well. Renal insufficiency or renal failure causes azotemia. Both azotemia and uremia indicate an accumulation of nitrogenous waste products in the blood, a common characteristic that explains the overlap in definitions of terms.
Acute Kidney Injury
Acute kidney injury (AKI) is a sudden decline in kidney function with a decrease in glomerular filtration and accumulation of nitrogenous waste products in the blood as demonstrated by an elevation in plasma creatinine and blood urea nitrogen. The term acute kidney injury is preferred to the term acute renal failure as it captures the diverse nature of this syndrome ranging from minimal or subtle changes in renal function to complete renal failure requiring renal replacement therapy. Classification criteria have been developed to guide the diagnosis of renal injury described by the acronym RIFLE (R = risk, I = injury, F = failure, L = loss, and E = end-stage kidney disease [ESKD]) representing three levels of renal dysfunction of increasing severity (Table 36-9).96
Table 36-9
RIFLE Criteria for Acute Renal Dysfunction/Failure
| Category | GFR Criteria | Urine Output (UO) Criteria |
| Risk | Increased creatinine × 1.5 or GFR decrease >25% | UO <0.5ml/kg/hr × 6 hr |
| Injury | Increased creatinine × 2 or GFR decrease >50% | UO <0.5ml/kg/hr × 12 hr |
| Failure | Increased creatinine × 3 or GFR decrease >75% | UO <0.3ml/kg/hr × 24 hr or anuria |
| Loss | Persistent ARF = complete loss of kidney function >4 weeks | |
| ESKD | End-stage kidney disease (>3 months) |
ARF, Acute renal failure; GFR, glomerular filtration rate.
Adapted from Bellomo R et al: Curr Opin Crit Care 8(6):505-508, 2002; Bellomo R et al: Crit Care 8(4):R204-R212, 2004. Available at www.medicalcriteria.com/criteria/neph_rifle.htm (accessed October, 2008).
PATHOPHYSIOLOGY AKI commonly results from extracellular volume depletion, decreased renal blood flow, or toxic/inflammatory injury to kidney cells that results in alterations in renal function that may be minimal or severe, subtle, or dramatic. Even small changes in renal function may be associated with significant morbidity and mortality. The pathophysiology of AKI can be described considering three categories of injury: (1) renal hypoperfusion (prerenal AKI); (2) disorders involving the renal parenchymal or interstitial tissue (intrarenal AKI); and (3) disorders associated with acute urinary tract obstruction (postrenal AKI) (Table 36-10 and Figure 36-12). Most types of acute renal failure are reversible if diagnosed and treated early.

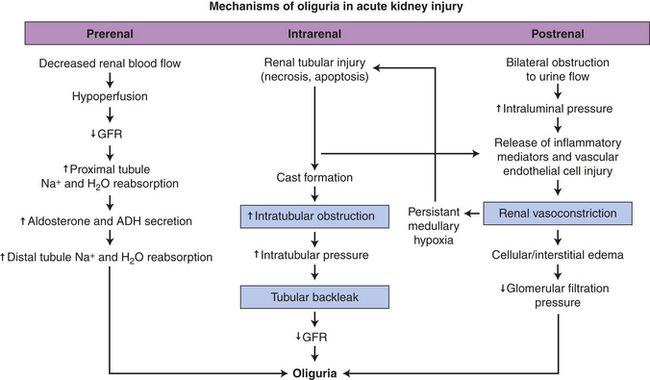
Figure 36-12 Acute kidney injury and mechanisms of oliguria. ADH, Antidiuretic hormone; GFR, glomerular filtration rate.
Prerenal acute kidney injury is the most common cause of AKI and is caused by renal hypoperfusion that occurs rapidly over a period of hours with elevation of BUN and plasma creatinine levels. During the early phases of hypoperfusion protective autoregulatory mechanisms maintain GFR at a relatively constant level through afferent arteriolar dilation and efferent arteriolar vasoconstriction (mediated by angiotensin II). Tubuloglomerular feedback mechanisms also maintain GFR and distal tubular nephron flow (see Chapter 35). The GFR ultimately declines because of the decrease in filtration pressure. Poor perfusion can result from renal vasoconstriction, hypotension, hypovolemia, hemorrhage, or inadequate cardiac output. AKI may occur during chronic kidney failure if a sudden stress is imposed on already marginally functioning kidneys. Failure to restore blood volume or blood pressure and oxygen delivery can cause cell injury and acute tubular necrosis or acute interstitial necrosis, a more severe form of AKI.
Intrarenal (intrinsic) acute kidney injury (AKI) may result from ischemic acute tubular necrosis (ATN), nephrotoxic ATN (i.e., exposure to radiocontrast media), acute glomerulonephritis, vascular disease (malignant hypertension, disseminated intravascular coagulation, and renal vasculitis), allograft rejection, or interstitial disease (drug allergy, infection, tumor growth). Acute tubular necrosis (ATN) caused by ischemia is the most common cause of intrarenal AKI. It occurs most often after surgery (40% to 50% of cases) but is also associated with severe sepsis, obstetric complications, and severe trauma, including severe burns. A combination of events and predisposing factors leads to the greatest risk for acute renal failure. The terms acute tubular necrosis and acute renal failure are sometimes used interchangeably, but the conditions are not the same because acute renal failure can occur without ATN. ATN is generally described as postischemic or nephrotoxic or it can be a combination of both.97 Postischemic ATN involves persistent hypotension, hypoperfusion, and hypoxemia producing ischemia, reduced ATP, and generate toxic oxygen-free radicals with loss of antioxidant protection that causes cell swelling, injury, and necrosis. The release of inflammatory cytokines contributes to tubular injury and increases neutrophil adhesion.98 Transport of sodium and other molecules is disrupted with damage to the tubular epithelium and shedding of the brush border. Ischemic necrosis tends to be patchy and may be distributed along any part of the nephron tubules. Injury is most severe in the outer medulla with scattered necrosis in the cortex and loss of cells along the tubular epithelium. Severe disease of the glomeruli (i.e., acute or rapidly progressive glomerulonephritis) or renal microvascular disorders can also cause intrinsic kidney injury. Oliguria is common (urine output less than 30 ml/hour) with intrarenal AKI, but anuria is rare. Acute kidney failure associated with sepsis may involve different mechanisms of tubular injury including apoptosis (does not result in inflammation because there is no release of intracellular contents into the extracellular space) (see Chapter 2 for apoptosis) and tubular cell dysfunction. Urinalysis in septic renal injury may not reflect renal injury because elevated creatinine and oliguria may be physiologic responses to hypovolemia and decreased renal blood flow.99,100
Nephrotoxic ATN can be produced by numerous antibiotics, but the aminoglycosides (neomycin, gentamicin, tobramycin) are the major culprits. The drugs tend to accumulate in the renal cortex and may not cause renal failure until after treatment is complete. Radiocontrast media (x-ray media) and cisplatin also may be nephrotoxic. Dehydration, advanced age, concurrent renal insufficiency, and diabetes mellitus tend to enhance nephrotoxicity from either aminoglycosides or radiocontrast media. Other substances such as excessive myoglobin (oxygen-transporting substance in muscles), carbon tetrachloride, heavy metals (mercury, arsenic), methoxyflurane anesthesia, or bacterial toxins may promote renal failure. Necrosis and tubular cell apoptosis caused by nephrotoxins is usually uniform and limited to the proximal tubules. The high surface area of the brush border of the proximal tubular cells makes them more vulnerable to toxic injury.101
Postrenal acute kidney injury is rare and usually occurs with urinary tract obstruction that affects the kidneys bilaterally (e.g., bilateral ureteral obstruction, bladder outlet obstruction–prostatic hypertrophy, tumors or neurogenic bladder, and urethral obstruction). The obstruction causes an increase in intraluminal pressure upstream from the site of obstruction with a gradual fall in GFR. A pattern of several hours of anuria with flank pain followed by polyuria is a characteristic finding. This type of AKI can occur after diagnostic catheterization of the ureters, a procedure that may cause edema of the tubular lumen.
Oliguria can occur in AKI, and three mechanisms have been proposed to account for the decrease in urine output. All three mechanisms probably contribute to oliguria in varying combinations and degrees throughout the course of the disease (Figure 36-12). These mechanisms are as follows102:
1. Alterations in renal blood flow. Efferent arteriolar vasoconstriction may be produced by intrarenal release of angiotensin II or by redistribution of blood flow from the cortex to the medulla. Autoregulation of blood flow may be impaired, resulting in decreased GFR. Changes in glomerular permeability and decreased GFR also may result from the ischemia.
2. Tubular obstruction. Advanced injury with necrosis of the tubules causes sloughing of cells, cast formation, or ischemic edema that results in tubular obstruction, which in turn causes a retrograde increase in pressure and reduces the GFR. Renal failure can occur within 24 hours.
3. Backleak. Glomerular filtration remains normal, but tubular reabsorption or “leak” of filtrate is accelerated as a result of permeability caused by ischemia and increased tubular pressure from obstruction.
CLINICAL MANIFESTATIONS The clinical progression of acute renal failure, particularly acute tubular necrosis, occurs in three phases: the initiation phase, maintenance phase, and recovery phase. The initiation phase is the phase of reduced perfusion or toxicity in which renal injury is evolving. Prevention of injury is possible during this phase. The maintenance phase is the period of established renal injury and dysfunction after the initiating event has been resolved and may last from weeks to months. Urine output is lowest during this phase, and serum creatinine and blood urea nitrogen increase. The recovery phase is the interval when renal injury is repaired and normal renal function is reestablished. Diuresis is common during this phase, with a decline in serum creatinine and urea and an increase in creatinine clearance.
Oliguria begins within 1 day after a hypotensive event and lasts for 1 to 3 weeks, but it may regress in several hours or extend for several weeks, depending on the duration of ischemia or severity of toxic injury and the initiation of treatment. Anuria (urine output less than 50 ml/day) is uncommon in ATN, and 10% to 20% of cases have nonoliguric failure (decreased urine output but greater than 30 ml/hour). Anuria involves both kidneys and suggests bilateral renal artery occlusion, obstructive uropathy, or acute cortical necrosis. Nonoliguric failure usually represents less severe injury. The urine output may vary in volume, but the BUN and plasma creatinine concentrations increase (plasma creatinine is inversely proportional to the GFR). Urine sediment should be evaluated for casts, cells, and crystals.
Other early manifestations depend on the underlying cause of renal failure. Individuals who have experienced trauma or surgery or people in a catabolic state may have more rapid elevations in BUN. They are prone to hyperkalemia and hyperphosphatemia from cellular breakdown. Fluid retention may cause edema. Symptoms of congestive heart failure develop in persons with cardiac disease. Nausea, vomiting, and fatigue accompany uremia and electrolyte imbalances. Wound healing is delayed, and the risk of infection, particularly pneumonia, is greater. Nonoliguric renal failure generally has a better prognosis because of fewer complications. Oliguric patients may require maintenance dialysis to attenuate symptoms of renal failure.
As renal function improves during the recovery phase, increase in urine volume (diuresis) is progressive. During the early diuretic phase the tubules are still recovering secretory and reabsorptive function. Sodium and potassium are lost in the urine, and the risk for hypokalemia is greater. Volume depletion may ensue, with fluid losses of 3 to 4 L/day. Fluid and electrolyte balance must be carefully monitored and excessive urinary losses replaced. Return to normal status may take 3 to 12 months, and approximately 30% of individuals do not have full recovery of a normal GFR or tubular function.
EVALUATION AND TREATMENT The diagnosis of AKI is related to the cause of the disease. A history of surgery, trauma, or cardiovascular disorders is common, and exposure to nephrotoxins and obstructive uropathies (i.e., an enlarged prostate) must be considered. The diagnostic challenge is to differentiate prerenal acute renal injury from acute tubular necrosis. Urine composition may provide helpful diagnostic clues to changes in tubular function (Table 36-11).
Table 36-11
Urine Characteristics of Prerenal and Intrinsic Acute Kidney Injury
| Diagnostic Index | Prerenal | ATN |
| Urine volume | <400 ml | <400 ml |
| Urine specificity | 1.016-1.020 | 1.010-1.012 |
| Urine osmolality | >500 mOsm | <300 mOsm |
| Urine sodium | <10 mEq/L | >30 mEq/L |
| BUN/plasma | >15:1 | <15:1 creatinine |
| FENa | <1% (also seen in acute glomerulonephritis | >1% (also seen in urinary tract obstruction and renal parenchymal disease) |
| Urine sediment | Usually no cells, some hyaline casts | Brown granular casts, epithelial cells |
ATN, Acute tubular necrosis; BUN, blood urea nitrogen; FENa, fractional excretion of sodium.
The ratios of the BUN to plasma creatinine concentration and fractional excretion of sodium (the ratio of filtered sodium to excreted sodium) are helpful diagnostic indicators because the tests reflect renal tubular reabsorption ability. In prerenal AKI, tubular function is maintained and salt, water, and urea are reabsorbed. With ATN, reabsorption and urinary concentration abilities are compromised. Other causes of renal failure also may exhibit similar clinical findings. Serial measurements of plasma creatinine provide an index of renal function during the recovery phase. Cystatin C, a serum protein constantly produced by nucleated cells, is freely filtered by the glomerulus, and its concentration can serve as a measure of GFR and may be useful for detecting early changes in glomerular filtration rate.103 Biomarkers of AKI that detect injury very early are needed to prevent delay in initiating therapy. New diagnostic tests are being developed to provide more sensitive markers of AKI.104,105
Prevention of acute renal injury and maintenance of renal perfusion are major treatment factors and involve maintenance of fluid volume before and after surgery or diagnostic procedures or when nephrotoxic drugs or contrast agents are in use. The primary goal of therapy is to maintain the individual’s life until renal function has recovered. There is no specific treatment for acute renal failure. Management principles directly related to physiologic alterations generally include (1) correcting fluid and electrolyte disturbances, (2) managing blood pressure, (3) treating infections, (4) maintaining nutrition, and (5) remembering that drugs or their metabolites are not excreted. Fluid and electrolyte replacement must be carefully calculated with consideration of urine losses, insensible losses (up to 1000 ml/day), and production of endogenous water by oxidation (450 ml/day). Overhydration of patients dilutes their plasma sodium concentration. Metabolic acidosis is usually not treated until serum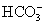 is less than 15 mEq/L.106
is less than 15 mEq/L.106
Hyperkalemia can be managed by restricting dietary sources of potassium, using non–potassium-sparing diuretics, or using cation-ion exchange resins, which may be administered orally or rectally. These resins exchange potassium for another cation, such as sodium in the bowel, and the potassium then is excreted attached to the resin. With severe hyperkalemia (more than 6.5 mEq/L), dialysis may be required or potassium can be driven back temporarily into the cells by administering glucose and insulin or by infusing sodium bicarbonate or albuterol. Glucose metabolism causes potassium to move to the intracellular fluid, and insulin infusions therefore can be effective in shifting potassium from the extracellular to intracellular space, along with the transport of glucose, within 30 minutes. (Glucose metabolism is discussed in Chapter 1 and Chapter 21.) Using sodium bicarbonate to cause alkalemia also shifts potassium into cells in exchange for hydrogen ions.
Careful monitoring of the electrocardiogram for peaking T waves is essential for individuals with hyperkalemia. Intravenous infusion of calcium is the most rapid method of treating cardiac effects of hyperkalemia. Calcium decreases the threshold potential and reduces the membrane excitability caused by hyperkalemia (see Chapter 3). Calcium should be used only in emergencies, however, because hypercalcemia also may cause cardiac arrest.
Azotemia is generally controlled and nutrition maintained with a low-protein, high-carbohydrate diet. Essential amino acid replacement can be given orally or parenterally. Adequate carbohydrate intake slows protein catabolism and helps prevent hyperkalemia. Because sepsis is a common serious or fatal complication of renal failure, observation for signs of infection and early treatment with antibiotics are necessary. Drug dosage levels may require adjustment if they are metabolized or excreted by the kidneys. Recovery may take up to 1 year.
Continuous renal replacement therapy (hemodialysis) (mechanical removal of water, electrolytes, and toxins from the blood) is indicated for uncontrollable hyperkalemia or acidosis or severe fluid overload. Continuous renal replacement therapy is particularly promising in critically ill patients with multiple organ dysfunction.107
CHRONIC KIDNEY DISEASE
Chronic kidney disease (CKD) is the progressive loss of renal function associated with systemic diseases such as hypertension and diabetes mellitus or intrinsic kidney disease including chronic glomerulonephritis, chronic pyelonephritis, obstructive uropathies or vascular disorders. The National Kidney Foundation (www.kidney.org/professionals/) defines kidney damage as a GFR less than 60 mL/min/1.73 m2 for 3 months or more, irrespective of cause. Chronic kidney disease is the preferred terminology and is referenced to declining GFR. The terms renal insufficiency and chronic renal failure are still often used to describe declining renal function, but they do not have the specificity of the stages recommended by the National Kidney Foundation (Table 36-12). CKD decreases GFR and tubular functions with changes manifested throughout all organ systems (Table 36-13).108
Table 36-12
Stages of Chronic Kidney Disease
Normal glomerular filtration rate in a 70-kg male is about 120 ml/min.
GFR, Glomerular filtration rate; PTH, parathyroid hormone.
Adapted from National Kidney Foundation, Chronic Kidney Disease 2006: A Guide to Select NKF KDOQI Guidelines and Recommendations, 2006. Available at http://www.kidney.org/professionals/kls/pdf/Pharmacist_CPG.pdf
Table 36-13
Systemic Effects of Chronic Kidney Disease and Uremia

With data from Keane WF: Kidney Int Suppl 75:S27-S31, 2000; Uribarri J: Semin Dial 13(4):232-234, 2000.
PATHOPHYSIOLOGY The kidneys have a remarkable ability to adapt to loss of nephron mass.109 Symptomatic changes resulting from increased creatinine, urea,
potassium, and alterations in salt and water balance usually do not become apparent until renal function declines to less than 25% of normal when adaptive renal reserves have been exhausted.
Different theories have been proposed to account for the adaptation to loss of renal function. The intact nephron hypothesis proposes that loss of nephron mass with progressive kidney damage causes the surviving nephrons to sustain normal kidney function. These nephrons are capable of a compensatory hypertrophy and expansion or hyperfunction in their rates of filtration, reabsorption, and secretion and can maintain a constant rate of excretion in the presence of overall declining GFR. The intact nephron hypothesis explains adaptive changes in solute and water regulation that occur with advancing renal failure. Although the urine of an individual with chronic kidney failure may contain abnormal amounts of protein and red and white blood cells or casts, the major end products of excretion are similar to those of normally functioning kidneys until the advanced stages of renal failure when there is a significant reduction of functioning nephrons.110,111 However, the continued loss of functioning nephrons and the adaptive hyperfiltration (increased glomerular pressure) can result in glomerulosclerosis contributing to uremia and end-stage renal failure.111 This is known as the trade-off hypothesis.
The particular location of kidney damage can also influence loss of kidney function. For example, tubular interstitial diseases damage primarily the tubular or medullary parts of the nephron, producing problems such as renal tubular acidosis, salt wasting, and difficulty diluting or concentrating the urine. When the damage is primarily vascular or glomerular, proteinuria, hematuria, and nephrotic syndrome are more prominent. This theory is useful for planning treatment in early stages of renal failure when symptomatic differences in renal disease may be distinct. A summary of factors involved in the progression of chronic kidney disease is outlined in Table 36-14.
Table 36-14
Factors Representing Progression of Chronic Kidney Disease
| Factor | Characteristics |
| Proteinuria | Glomerular hyperfiltration of protein contributes to tubular intestinal injury by accumulating in the interstitial space and promoting inflammation and progressive fibrosis. |
| Creatine and urea clearance | In chronic renal failure, the GFR falls and the plasma creatinine concentration increases by a reciprocal amount; because there is no regulatory adjustment for creatinine, plasma levels continue to rise and serve as an index of changing glomerular function. |
| As GFR declines, urea clearance increases. (note: Urea is filtered and reabsorbed and varies with the state of hydration.) | |
| Sodium and water balance | In chronic renal failure, sodium load delivered to nephrons exceeds normal, so excretion must increase, thus less is reabsorbed. Obligatory loss occurs, leading to sodium deficits and volume depletion. As GFR is reduced, ability to concentrate and dilute urine diminishes. |
| Phosphate and calcium balance | Changes in acid-base balance affect phosphate and calcium balance. The major disorders associated with chronic renal failure are reduced renal phosphate excretion, decreased renal synthesis of 1,25-(OH)2 vitamin D3, and hypocalcemia. |
| Hypocalcemia leads to secondary hyperparathyroidism, GFR falls, and progressive hyperphosphatemia, hypocalcemia, and dissolution of bone result. | |
| Hematocrit | Lack of erythropoietin and anemia accompanies chronic renal failure. Lethargy, dizziness, and low hematocrit are common. |
| Potassium balance | In chronic renal failure, tubular secretion of potassium increases until oliguria develops. Use of potassium-sparing diuretics also may precipitate elevated serum potassium levels. As disease progresses, total body potassium levels can rise to life-threatening levels and dialysis is required. |
| Acid-base balance | In early renal insufficiency, acid excretion and bicarbonate reabsorption are increased to maintain normal pH. Metabolic acidosis begins when GFR reaches 30% to 40%. Metabolic acidosis and hyperkalemia may be severe enough to require dialysis when end-stage renal failure develops. |
| Dyslipidemia | Chronic hyperlipidemia may induce glomerular and tubulointerstitial injury contributing to the progression of chronic kidney disease. |
Progression of chronic kidney disease is thought to be associated with common pathogenic processes regardless of the initial disease (Figure 36-13).112 These processes include the following:
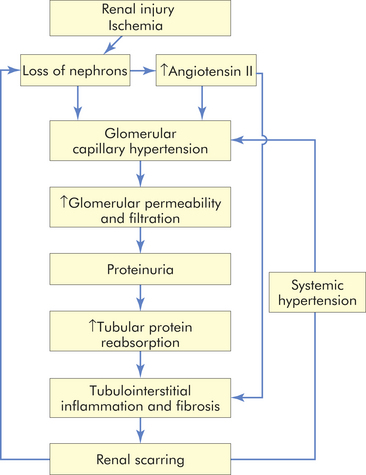
The factors that contribute to the pathogenesis of chronic kidney disease are complex and involve the interaction of many cells, cytokines, and structural alterations. Two factors that have consistently been recognized to advance renal disease are proteinuria and angiotensin II.113,114 Glomerular hyperfiltration and increased glomerular capillary permeability lead to proteinuria. Proteinuria contributes to tubulointerstitial injury by accumulating in the interstitial space and activating complement proteins and other mediators and cells, such as macrophages, that promote inflammation and progressive fibrosis. Angiotensin II activity is elevated with progressive nephron injury. Angiotension II promotes glomerular hypertension and hyperfiltration caused by efferent arteriolar vasoconstriction and also promotes systemic hypertension. The chronically high intraglomerular pressure increases glomerular capillary permeability contributing to proteinuria. Angiotensin II also may promote the activity of inflammatory cells and growth factors that participate in tubulointerstitial fibrosis and scarring.
CLINICAL MANIFESTATIONS The clinical manifestations of chronic kidney disease are often described using the terms azotemia and uremia. Azotemia is increased levels of serum urea, serum creatinine and other nitrogenous compounds related to decreasing kidney function. Uremic syndrome is the systemic manifestations associated with the accumulation of urea and other nitrogenous compounds and toxins caused by decline in renal function. Sources of toxins include the accumulation of end products of protein metabolism, alterations in fluid and electrolytes, metabolic acidosis, intestinal absorption of toxins produced by gut bacteria and results of altered renal hormone synthesis (i.e., anemia, hyperphosphatemia, and hypocalcemia). Uremia represents a proinflammatory state with many systemic effects.115 Generally, the symptoms include hypertension, anorexia, nausea, vomiting, diarrhea or constipation, weight loss, pruritus, edema, anemia, and neurologic, cardiovascular, and skeletal changes. The many systemic manifestations associated with uremia are summarized in Table 36-13, p. 1391 and Figure 36-14.
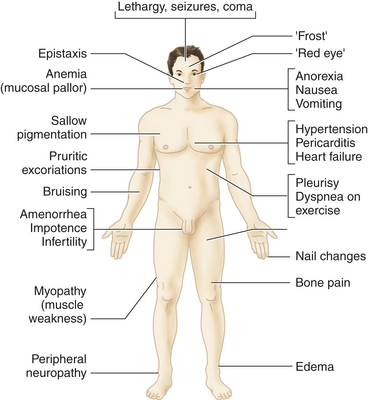
Figure 36-14 Common signs and symptoms of kidney failure (see text for reference site). (From Goldman L, Ausiello D: Cecil medicine, ed 23, Philadelphia, 2008, Saunders.)
Creatinine and Urea Clearance
Creatinine is constantly released from muscle and excreted primarily by glomerular filtration. In CKD, as the GFR declines, the plasma creatinine level increases by a reciprocal amount to maintain a constant rate of excretion (Figure 36-15). Because no significant tubular adjustment occurs for creatinine (i.e., tubular secretion), the plasma levels continue to increase as the GFR decreases. Therefore, measures of plasma creatinine can serve as an index of changing glomerular function. The clearance of urea follows a similar pattern, but urea is filtered as well as reabsorbed and varies with the state of hydration and it is not a good index of GFR. However, as the GFR decreases, plasma urea concentration also increases.
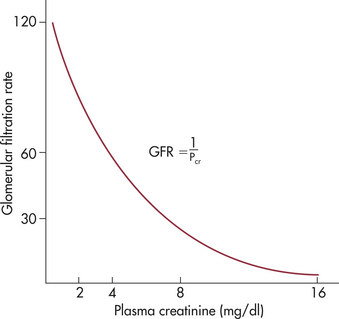
Fluid and Electrolyte Balance
Fluid and electrolyte and acid-base balance are significantly disturbed with chronic kidney disease. A summary of electrolyte and acid-base balance alterations is presented in Table 36-15.

Levels of sodium must be regulated within narrow limits because sodium is the major extracellular solute. In CKD sodium and water balance is maintained very close to normal until the development of stage V ESKD. This occurs because of the increased fractional excretion of sodium, particularly in the distal nephron, in relation to decreasing GFR. Hormones including aldosterone, prostaglandins, and natriuretic peptides also modulate sodium excretion and they are elevated with progressive renal failure. Individual variation in the underlying pathology of CKD must be considered in the management of sodium intake or restriction. Sodium wasting may be present with tubulointerstitial causes of CKD and there may also be extra renal losses of sodium from vomiting, diarrhea, or fever. Sodium retention is more likely in ESKD particularly in the presence of nephrotic syndrome or heart failure. Sodium retention contributes to hypertension, edema, and heart failure. Management of salt and water balance requires individual assessment, and low dietary salt intake is usually recommended.116,117
The regulation of water balance and osmolality is normally achieved by urinary concentration mediated by ADH. As GFR is reduced, ability to concentrate and dilute the urine diminishes. In earlier stages of renal failure, this may be caused by osmotic diuresis produced by increased fractional excretion of solutes by the remaining nephrons or by a decreased tubular response to ADH. Individual nephrons can maintain water balance until severe renal failure occurs and GFR declines to 15% to 20% of normal with extensive loss of nephron and tubular function. At this stage the urinary concentration becomes fixed and approaches that of the plasma at 285 mOsm/L with a specific gravity of about 1.010.
Urinary excretion of potassium is related primarily to distal tubular secretion mediated by aldosterone and sodium-potassium adenosine triphosphatase (see Chapter 3). In renal failure there is increased tubular secretion that provides effective regulation until the onset of oliguria. Larger amounts of potassium are also lost through the bowel.118 Although nonoliguric patients can maintain potassium excretion with normal dietary intake, they are more prone to develop hyperkalemia with increased loading (i.e., use of salt substitutes). Use of potassium-sparing diuretics, such as spironolactone (aldactone), volume depletion, acute infection, severe acidosis, or marked hyperglycemia also may precipitate elevated levels of serum potassium. With progression of disease to ESRF, total body potassium can increase to life-threatening levels and must be controlled by dietary restriction, loop diuretics, cation exchange resins, and dialysis.119 Severe acute hyperkalemia is treated with intravenous calcium, intravenous glucose, and insulin and nebulized albuterol (sympathetic beta2-agonist, promotes Na+, K+-ATPase pump and intracellular movement of potassium).120
The intake of a normal diet produces 50 to 100 mEq of hydrogen per day. These ions are secreted from the renal tubules and excreted in the urine combined with phosphate and ammonia buffers (buffering is described in Chapter 3). Metabolic acidosis develops when GFR decreases to less than 20% to 25% of normal.121 The causes of acidosis are primarily related to decreased hydrogen ion elimination and decreased bicarbonate reabsorption. With end-stage renal failure, metabolic acidosis may be severe enough to require alkali therapy and dialysis.122
Calcium, Phosphate, and Bone
Bone and skeletal changes develop with alterations in calcium and phosphate metabolism (Table 36-16). These changes begin when GFR decreases to 25% or less. Hypocalcemia is accelerated by impaired renal synthesis of 1,25-vitamin D3 (calcitriol) with decreased intestinal absorption of calcium. Renal phosphate excretion also decreases and the increased serum phosphate binds calcium, further contributing to hypocalcemia. Acidosis also contributes to a negative calcium balance. Decreased serum calcium stimulates parathyroid hormone secretion with mobilization of calcium from bone and may cause calcium levels to approach normal. The combined effect of hyperparathyroidism and vitamin D deficiency can result in renal osteodystrophies (i.e., osteomalacia and osteitis fibrosa with increased risk for fractures.123 Other consequences of secondary hyperparathyroidism include soft tissue and vascular calcification, cardiovascular disease, and less commonly, calcific uremic arteriolopathy.124
Table 36-16
Calcium and Phosphate Metabolism in Chronic Kidney Failure
| Kidney | Plasma | Bone |
| Decreased renal production of vitamin D3 | Decreased calcium absorption from gut | Decreased calcium deposition |
| Decreased ionized calcium | ||
| Increased PTH secretion (secondary hyperparathyroidism) | ||
| Decreased phosphate excretion | Elevated phosphate | Release of calcium and phosphate |
| Formation of CaHPO4 | Osteitis fibrosa, osteomalacia, calcium deposits in soft tissue (occurs when kidney fails to respond to PTH secretion because of loss of renal mass and calcium and phosphate continues to be absorbed from bone) |
Protein, Carbohydrate, and Fat Metabolism
Protein, carbohydrate, and fat metabolism are altered in chronic renal failure (CRF). Proteinuria and a catabolic state contribute to a negative nitrogen balance. Serum proteins diminish, including albumin, complement, and transferrin, and there is loss of muscle mass. Proteinuria may independently cause renal damage by promoting tubular inflammation and fibrosis.125 The amount of proteinuria is also related to the extent of renal injury and predicts disease progression.126 The effectiveness of a low-protein diet on progression of chronic kidney disease is inconclusive.127,128
Hyperinsulinemia and glucose intolerance related to insulin resistance are common and may be related to alterations in adipokines (high leptin and low adiponectin) that interfere with insulin action and oxidative stress that contribute to renal tubular and vascular injury in both nondiabetic and diabetic CKD.129 Hyperparathyroidism also decreases insulin sensitivity and impairs glucose tolerance.
Dyslipidemia is common among individuals with CKD. There is a high ratio of low-density lipoprotein (LDL) to high-density lipoprotein (HDL), high triglycerides, accumulation of LDL particles with accelerated atherosclerosis and vascular calcification.130 Uremia causes a deficiency in lipoprotein lipase and decreased hepatic triglyceride lipase. Decreased lipolytic activity results in a reduction in HDL. Apolipoprotein B is also elevated, thereby accelerating atherogenesis.131
Cardiovascular System
Cardiovascular disease is a major cause of morbidity and mortality in CKD. Proinflammatory mediators, oxidative stress, and metabolic derangements are significant contributors. Elevated renin stimulates the secretion of aldosterone, increasing sodium reabsorption. Hypertension is the result of excess sodium and fluid volume. Dyslipidemia occurs early in CKD. Arterial wall thickness increases with decreased elastic fibers and increased extracellular matrix. Atheromatous plaque and calcium deposits contribute to loss of vessel elasticity and obstruction and are accelerated by the oxidative stress of CKD.132 Macrovascular disease is responsible for increased risk for ischemic heart disease, left ventricular hypertrophy, congestive heart failure, stroke, and peripheral vascular disease in individuals with uremia.
Endothelial cell dysfunction and calcium deposits lead to a loss of vessel elasticity and vascular calcification. The resulting vascular disease increases the risk for ischemic heart disease, left ventricular hypertrophy, congestive heart failure, stroke, and peripheral vascular disease in individuals with uremia. Declining erythropoietin production causes anemia, thereby increasing demands for cardiac output and adding to cardiac workload. Pericarditis can develop from inflammation caused by the presence of uremic toxins. Accumulation of fluid in the pericardial space can compromise ventricular filling and cardiac output.
Pulmonary System
Pulmonary complications are associated with fluid overload and congestive heart failure. Pulmonary edema develops and metabolic acidosis can cause Kussmaul respirations. Dyspnea is common in ESKD.
Hematologic System
Hematologic alterations include normochromic normocytic anemia, impaired platelet function, and hypercoagulability. Inadequate production of erythropoietin decreases red blood cell production and is the most significant factor in contributing to anemia. Chronic inflammation, iron deficiency, and decreased half-life of erythrocytes are also contributing factors. Anemia contributes to decreased tissue oxygenation and contributes to progression of kidney disease. Lethargy, dizziness, low hematocrit, and increased cardiac workload and increased risk for congestive heart failure are common findings. Treatment of anemia includes erythropoiesis stimulating agents (i.e., recombinant human erythropoietin) and intravenous iron.
Disorders of hemostasis in CKD and uremia are primarily related to defective platelet aggregation and impaired adhesion of platelets to the vascular endothelium. The consequence is an increased bleeding tendency and an increased risk for bruising, epistaxis, gastrointestinal bleeding, or cerebrovascular hemorrhage and cutaneous or submucosal hemorrhage.133 Adequate dialysis improves platelet function. Alterations of individual clotting factors, fibrin, thrombin and fibrinolysis contribute to alterations of blood coagulation and can promote a hypercoagulable state and thrombosis with increased risk for myocardial infarction and stroke. 134,135
Immune System
Immune system dysregulation with immune suppression, deficient response to vaccination, and increased risk for infection develops with CRF.136 Chemotaxis, phagocytosis, antibody production, and cell-mediated immune responses are suppressed. Malnutrition, metabolic acidosis, hyperglycemia, or effects of hemodialysis may amplify immunosuppression.
Neurologic System
Neurologic symptoms are common and progressive with CRF and are related to uremic toxicity, chronic hyperkalemic depolarization, and anemia.137 Symptoms may include headache, drowsiness, pain, sleep disorders, impaired concentration, memory loss, and impaired judgment. Neuromuscular irritation can cause hiccups, muscle cramps, and muscle twitching. In advanced stages of renal failure, symptoms may progress to seizures and coma. Peripheral neuropathies also develop with impaired sensations decreased tendon reflexes, muscle weakness and muscle atrophy, most commonly in the lower extremities.
Gastrointestinal System
Gastrointestinal complications are common in individuals with CRF. Uremic gastroenteritis can cause bleeding ulcer and significant blood loss. Nonspecific symptoms include anorexia, nausea, vomiting, and constipation or diarrhea. Uremic fetor is a form of bad breath caused by the breakdown of urea by salivary enzymes. Malnutrition is common.
Endocrine and Reproductive Systems
Endocrine and reproductive alterations develop with progression of CKD. Males and females have a decrease in circulating sex steroids. Males often experience a reduction in testosterone levels and may be impotent. Oligospermia and germinal cell dysplasia can result in infertility. Females have reduced estrogen levels, amenorrhea, and difficulty maintaining a pregnancy to term. A decrease in libido occurs in both genders.138,139
Insulin resistance is common in uremia. Low-grade systemic inflammation and oxidative stress may be contributing factors with increased risk for cardiovascular disease.140 As CKD progresses the ability of the kidney to degrade insulin is reduced, and the half-life of insulin is prolonged. Individuals with diabetes mellitus and CKD need to carefully manage their insulin dosages. Low-protein diets and renal replacement therapy improve insulin sensitivity.141
CRF also causes alterations in thyroid hormone metabolism and low thyroid hormone levels and is known as nonthyroidal illness syndrome (euthyroid sick syndrome). Low-grade inflammation and oxidative stress may be contributing factors.142 Uremia also reduces conversion of T3 to T4.143 A low-protein, low-phosphorus diet may improve thyroid hormone function.144
Integumentary System
Skin changes are associated with other complications that develop with CKD. Anemia can cause pallor and bleeding into the skin and results in hematomas and ecchymosis. Retained urochromes manifest as a sallow skin color. Hyperparathyroidism and uremic skin residues (known as uremic frost) are associated with irritation and pruritus with scratching, excoriation, and increased risk for infection.145
EVALUATION AND TREATMENT Early screening and evaluation of chronic kidney disease are based on risk factors, history, presenting signs and symptoms, and diagnostic testing. Prediction equations are used for estimating GFR from serum creatinine values or creatinine clearance may be completed. Markers of kidney damage include urine protein, particularly albumin and examination of urine sediment. Ultrasound, CT scan, or plain x-ray films will show small kidney size. Renal biopsy confirms the diagnosis.
Management involves dietary control, including phosphate restriction, vitamin D supplementation, sodium and fluid maintenance, potassium restriction, adequate caloric intake, management of dyslipidemias, and erythropoietin as needed. ACE inhibitors or receptor blockers are often used to control systemic hypertension and provide renoprotection.146 ESKD related to diabetic nephropathy can be significantly reduced with control of hyperglycemia by intense insulin therapy.147 ESKD is treated with dialysis, supportive therapy, and renal transplantation.
Acute cystitis 1374
Acute kidney injury (AKI) 1386
Acute postinfectious glomerulonephritis (PIGN) 1380
Acute renal failure 1386
Acute tubular necrosis (ATN) 1387
Angiotensin II 1393
Antiglomerular basement membrane disease (Goodpasture syndrome) 1383
Anuria 1388
Apoptosis 1367
Asymptomatic bacteriuria 1375
Azotemia 1386
Calcium metabolism 1394
Calcium stone 1368
Calculus (pl., calculi) (urinary stone) 1368
Chronic glomerulonephritis 1383
Chronic kidney disease (CKD) 1389
Chronic pyelonephritis 1377
Compensatory growth 1367
Compensatory hypertrophy 1367
Continuous renal replacement therapy (hemodialysis) 1389
Cystinuric (xanthine) stone 1369
Detrusor areflexia 1370
Detrusor hyperreflexia 1369
Detrusor hyperreflexia with vesicosphincter dyssynergia 1370
Dyssynergia 1369
End-stage renal failure (ESRF) 1386
Glomerulonephritis 1379
Hematuria 1378
Hydronephrosis 1365
Hydroureter 1365
Hyperfunction 1367
IgA nephropathy (Berger disease) 1382
Immune complex deposition 1383
Intrarenal (intrinsic) acute kidney injury (AKI) 1387
Low bladder wall compliance 1371
Lupus nephritis 1382
Membranoproliferative glomerulonephritis (MPGN) 1383
Membranous nephropathy (membranous glomerulonephritis) 1383
Mesangial proliferative glomerulonephritis 1383
Nephritic sediment 1378
Nephrotic sediment 1378
Nephrotic syndrome 1384
Neurogenic bladder 1369
Nonbacterial infectious cystitis 1376
Noninfectious cystitis 1376
Obligatory growth 1367
Obstructive uropathy 1365
Oliguria 1388
Overactive bladder syndrome (OAB) 1370
Painful bladder syndrome/interstitial cystitis (PBS/IC) 1376
Partial obstruction of the bladder outlet or urethra 1371
Pauci immune glomerulonephritis 1383
Pelvic organ prolapse 1371
Phosphate metabolism 1394
Postobstructive diuresis 1367
Postrenal acute kidney injury 1388
Prerenal acute kidney injury 1386
Prostate enlargement 1371
Proteinuria 1378
Pyelonephritis 1377
Rapidly progressive (crescentic) glomerulonephritis (RPGN) 1383
Renal adenoma 1372
Renal cell carcinoma (RCC) 1372
Renal colic 1369
Renal insufficiency 1386
Renal failure 1386
Staghorn calculus (pl., calculi) 1368
Struvite stone 1368
Tubulointerstitial fibrosis 1366
Uremia 1386
Uremic syndrome 1393
Ureterohydronephrosis 1366
Urethral stricture 1371
Uric acid stone 1369
Urinary tract infection (UTI) 1373
Virulence 1375
REFERENCES
1. Selius, B.A., Subedi, R. Urinary retention in adults: diagnosis and initial management. Am Fam Physician. 2008;77(5):643–650.
2. Gillenwater, J.Y. Hydronephrosis. In Gillenwater J.Y., et al, eds.: Adult and pediatric urology, ed 4, Philadelphia: Lippincott Williams & Wilkins, 2002.
3. Maarten, T.W., Brenner, B.M., Adaptation to nephron loss. Brenner B.M., ed. Brenner and Rector’s the kidney. ed 8. 2008:783.
4. Frokiaer, J., Zeidel, M.L., Urinary tract obstruction. Brenner B.M., ed. Brenner and Rector’s the kidney. ed 8. 2008:1260.
5. Porena, M., Guggi, P., Micheli, C. Prevention of stone disease. Urol Int. 2007;79(Suppl 1):37–46.
6. Chandhoke, P.S. Evaluation of the recurrent stone former. Urol Clin North Am. 2007;34(3):315–322.
7. Miano, R., Germani, S., Vespasiani, G. Stones and urinary tract infection. Urol Int. 2007;79(Suppl 1):32–36.
8. Obligado, S.H., Goldfarb, D.S. The association of nephrolithiasis with hypertension and obesity: a review. Am J Hypertens. 2008;21(3):257–264.
9. Miller, N.L., Evan, A.P., Lingeman, J.E. Pathogenesis of renal calculi. Urol Clin North Am. 2007;34(3):295–313.
10. Lingeman, J.E., Lifshitz, D.A., Evan, A.P. Surgical management of urinary lithiasis. In Walsh P.C., et al, eds.: Campbell’s urology, ed 8, Philadelphia: Saunders, 2002.
11. Trinchieri, A., et al. Calcium stone disease: a multiform reality. Urol Res. 2005;33(3):194–198.
12. Park, S., Pearle, M.S. Pathophysiology and management of calcium stones. Urol Clin North Am. 2007;34(3):323–334.
13. Healy, K.A., Ogan, K. Pathophysiology and management of infectious staghorn calculi. Urol Clin North Am. 2007;34(3):363–374.
14. Ahmed, H.U., et al. Diagnosis and management of renal (ureteric) colic. Br J Hosp Med (Lond). 2006;67(9):465–469.
15. Pietrow, P.K., Karellas, M.E. Medical management of common urinary calculi. Am Fam Physician. 2006;74(1):86–94.
16. Asplin, J.R. Evaluation of the kidney stone patient. Semin Nephrol. 2008;28(2):99–110.
17. Park, S., Pearle, M.S. Imaging for percutaneous renal access and management of renal calculi. Urol Clin North Am. 2006;33(3):353–364.
18. Taylor, E.N., Curhan, G.C. Diet and fluid prescription in stone disease. Kidney Int. 2006;70(5):835–839.
19. Steggall, M.J., Omara, M. Urinary tract stones: types, nursing care and treatment options. Br J Nurs. 2008;17(9):520–523.
20. Wen, C.C., Nakada, S.Y. Treatment selection and outcomes: renal calculi. Urol Clin North Am. 2007;34(3):409–419.
21. Karsenty, G., et al. Understanding detrusor sphincter dyssynergia—significance of chronology. Urology. 2005;66(4):763–768.
22. Chu, F.M., Dmochowski, R. Pathophysiology of overactive bladder. Am J Med. 2006;199(3 Suppl 1):3–8.
23. Abrams, P., et al. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Am J Obstet Gynecol. 2002;187(1):116–126.
24. Franco, I. Overactive bladder in children: Part 1: pathophysiology. J Urol. 2007;178(3 Pt 1):761–768.
25. Srikrishna, S., et al. Management of overactive bladder syndrome. Postgrad Med J. 2007;83(981):481–486.
26. Staskin, D.R., MacDiarmid, S.A. Pharmacologic management of overactive bladder: practical options for the primary care physician. Am J Med. 2006;119(3 Suppl 1):24–28.
27. Mundy, A.R. Management of urethral strictures. Postgrad Med J. 2006;82(970):489–493.
28. Valchanov, K., et al. An unusual cause of acute renal failure: urethral stricture in a female. Nephron. 2001;87(1):89–90.
29. Anger, J.U., Raz, S., Rodriguez, L.V. Severe cystocele: optimizing results. Curr Urol Rep. 2007;8(5):394–398.
30. American Cancer Society, Inc., S urveillance and Health Policy Research: Estimated New Cancer Cases and Deaths by Sex, U.S . 2009 available at www.cancer.org/downloads/stt/CFF2009_EstCD_3.pdf.asp
31. Setoawan, V.W., et al. Risk factors for renal cell cancer: the multiethnic cohort. Am J Epidemiol. 2007;166(8):932–940.
32. Berndt, S.I., et al. Disparities in treatment and outcome for renal cell cancer among older black and white patients. J Clin Oncol. 2007;24(25):3589–3595.
33. Gupta, K., et al. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008;34(3):193–205.
34. Corgna, E., et al. Renal cancer. Crit Rev Oncol Hematol. 2007;64(3):247–262.
35. George, S., Burkowski, R.M. Biomarkers in clear cell renal cell carcinoma. Expert Rev Anticancer Ther. 2007;7(12):1737–1747.
36. Pahernik, S., et al. Elective nephron sparing surgery for renal cell carcinoma larger than 4 cm. J Urol. 2008;179(1):71–74.
37. Lehman, D.S., Landman, J. Cryoablation and radiofrequency for kidney tumor. Curr Urol Rep. 2008;9(2):128–134.
38. Costa, L.J., Drabkin, H.A. Renal cell carcinoma: new developments in molecular biology and potential for targeted therapies. Oncologist. 2007;12(12):1404–1415.
39. Pelucchi, C., et al. Mechanisms of disease: the epidemiology of bladder cancer. Nat Clin Pract Urol. 2006;3(6):327–340.
40. Murta-Nascimento, C., et al. Epidemiology of urinary bladder cancer: from tumor development to patient’s death. World J Urol. 2007;25(3):285–295.
41. Madeb, R., et al. Current state of screening for bladder cancer. Expert Rev Anticancer Ther. 2007;7(7):981–987.
42. Mohammed, A., et al. Biological markers in the diagnosis of recurrent bladder cancer: an overview. Expert Rev Mol Diag. 2008;8(1):63–72.
43. Dalbagni, G. The management of superficial bladder cancer. Nat Clin Pract Urol. 2007;4(5):254–260.
44. Barocas, D.A., Clark, P.E. Bladder cancer. Curr Opin Oncol. 2008;20(3):307–314.
45. Foxman, B., et al. Urinary tract infection: self-reported incidence and associated costs. Ann Epidemiol. 2000;10:509–515.
46. Nicole, L.E. Uncomplicated urinary tract infection in adults including uncomplicated pyelonephritis. Urol Clin North Am. 2008;35(1):1–12.
47. Bouckaert, J., et al. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesion. Mol Microbiol. 2005;55(2):441–455.
48. Malani, A.N., Kaufman, C.A. Candida urinary tract infections: treatment options. Expert Rev Anti Infect Ther. 2007;5(2):277–284.
49. Barsoum, R.S. Schistosomiasis and the kidney. Semin Nephrol. 2003;23(1):24–46.
50. Vauhkonen, H., et al. Can bladder adenocarcinomas be distinguished from schistosomiasis-associated bladder cancers by using array comparative genomic hybridization analysis? Cancer Genet Cytogenet. 2007;177(2):153–157.
51. Hladunewich, M., Rosenthal, M.H. Pathophysiology and management of renal insufficiency in the perioperative and critically ill patient. Anesthesiol Clin North Am. 2000;18(4):773–789.
52. Smaill, F. Asymptomatic bacteriuria in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2007;21(3):439–450.
53. Yamamoto, S. Molecular epidemiology of uropathogenic Escherichia coli. J Infect Chemother. 2007;13(2):68–73.
54. Bricker, N.S., Morrin, P.A., Kime, S.W., Jr. The pathologic physiology of chronic Bright’s disease: an exposition of the “intact nephron hypothesis,”. J Am Soc Nephrol. 1997;8(9):1470–1476.
55. Neal, D.E. Complicated urinary tract infections. Urol Clin North Am. 2008;35(1):13–22.
56. Hancock, V., Ferrieres, L., Klemm, P. Biofilm formation by asymptomatic and virulent urinary tract infectious Escherichia coli strains. FEMS Microbiol Lett. 2007;267(1):30–37.
57. Jacobsen, S.M., et al. Complicated catheter-associated urinary tract infections due to Escherichia coli and Proteus mirabilis. Clin Microbiol Rev. 2008;21(1):26–59.
58. Sakallioglu, O., Sakallioglu, A.E. The effect of ABO-Rh blood group determinants on urinary tract infections. Int Urol Nephrol. 2007;39(2):577–579.
59. Juthani-Mehta, M. Asymptomatic bacteriuria and urinary tract infection in older adults. Clin Geriatr Med. 2007;23(3):585–594.
60. Czaja, C.A., Hooton, T.M. Update on acute uncomplicated urinary tract infection in women. Postgrad Med. 2006;119(1):39–45.
61. McManus, D.P., Loukas, A. Current status of vaccines for schistosomiasis. Clin Microbiol Rev. 2008;21(1):225–242.
62. van de Merwe, J.P., et al. Diagnostic criteria, classification, and nomenclature for painful bladder syndrome/intersitial cystitis: an ISSIC proposal. Eur Urol. 2008;53(1):60–67.
63. Nazif, O., Teichman, J.M., Bebhart, G.F. Neural upregulation in interstitial cystitis. Urology. 2007;60(4 Suppl):24–30.
64. Teichman, J.M., Moldwin, R. The role of the bladder surface in interstitial cystitis/painful bladder syndrome. Can J Urol. 2007;14(4):3599–3607.
65. Graham, E., Chai, T.C. Dysfunction of bladder urothelium and bladder urothelial cells in interstitial cystitis. Curr Urol Rep. 2006;7(6):440–446.
66. Teichman, J.M., Parsons, C.L. Contemporary clinical presentation of interstitial cystitis. Urology. 2007;69(4 Suppl):41–47.
67. Dell, J.R. Interstitial cystitis/painful bladder syndrome: appropriate diagnosis and management. J Womens Health (Larchmt). 2007;16(8):1181–1187.
68. Funfstuck, R., Ott, U., Naber, K.G. The interaction of urinary tract infection and renal insufficiency. Int J Antimicrob Agents. 2006;28(Suppl 1):S72–S77.
69. Craig, W.D., Wagner, B.J., Travis, M.D. Pyelonephritis: radiologic-pathologic review. Radiographics. 2008;28(1):255–277.
70. Hsu, C.Y., et al. The clinical impact of bacteremia in complicated acute phylonephritis. Am J Med Sci. 2006;332(4):175–180.
71. Funfstuck, R., Ott, U., Naber, K.G. The interaction of urinary tract infection and renal insufficiency. Int J Antimicrob Agents. 2006;28(Suppl 1):S72–S77.
72. Berger, S.P., Daha, M.R. Complement in glomerular injury. Semin Immunopathol. 2007;29(4):375–384.
73. Norman, J.T., Fine, L.G. Intrarenal oxygenation in chronic renal failure. Clin Exp Pharmacol Physiol. 2006;33(10):989–996.
74. Lizakowski, S., et al. Plasma tissue factor and tissue factor pathway inhibitor in patients with primarily glomerulonephritis. Scand J Urol Nephrol. 2007;41(3):237–242.
75. Cattran, D.C. Outcomes research in glomerulonephritis. Semin Nephrol. 2003;23(4):340–354.
76. Naicker, S., et al. Infection and glomerulonephritis. Semin Immunopathol. 2007;29(4):397–414.
77. El-Husseini, A.A., et al. Acute postinfectious crescentic glomerulonephritis: clinicopathologic presentation and risk factors. Int Urol Nephrol. 2005;37(3):603–609.
78. Carapetis, J.R., et al. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5(11):685–694.
79. Mortensen, E.S., Fenton, K.A., Rekvig, O.P. Lupus nephritis: the central role of nucleosomes revealed. Am J Pathol. 2008;172(2):275–283.
80. Liapis, H., Tsokos, G.C. Pathology and immunology of lupus glomerulonephritis: can we bridge the two? Int Urol Nephrol. 2007;39(1):223–231.
81. Novak, J., et al. IgA glycoslylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28(1):78–87.
82. Berthoux, F.C., Mohey, H., Afiani, A. Natural history of primary IgA nephropathy. Semin Nephrol. 2008;28(1):4–9.
83. Lionaki, S., Jeannette, J.C., Falk, R.J. Anti-neutrophil cytoplasmic (ANCA) and anti-glomerular basement membrane (GBM) autoantibodies in necrotizing and crescentic glomerulonephritis. Semin Immunopathol. 2007;29(4):459–474.
84. Little, M.A., Pusey, C.D. Rapidly progressive glomerulonephritis: current and evolving treatment strategies. J Nephrol. 2004;17(Suppl 8):S10–S19.
85. Papiris, S.A., et al. Bench-to-bedside review: pulmonary-renal syndromes-an update for the intensivist. Crit Care. 2007;11(3):213.
86. Wolf, G., Ziyadeh, F.N. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol. 2007;106(2):26–31.
87. Johnson, D.W. Evidence-based guide to slowing the progression of early renal insufficiency. Intern Med J. 2004;34(1-2):50–57.
88. Cho, M.H., et al. Pathophysiology of minimal change nephritic syndrome and focal segmental glomerulosclerosis. Nephrology (Carlton). 2007;12(Suppl 3):S11–S14.
89. Glubler, M.C. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol. 2008;4(1):24–37.
90. Camici, M. The nephrotic syndrome is an immunoinflammatory disorder. Med Hypothesis. 2007;68(4):900–905.
91. Kim, S.W., Frøkiaer, J., Nielsen, S. Pathogenesis of oedema in nephritic syndrome: role of epithelial sodium channel. Nephrology (Carlton). 2007;12(Suppl 3):S8–S10.
92. Crew, R.J., Radhakrishnan, J., Appel, G. Complications of the nephrotic syndrome and their treatment. Clin Nephrol. 2004;62(4):245–259.
93. Fehally J., Floege J., Johnson R., eds. Comprehensive clinical nephrology. St Louis: Mosby, 2007.
94. Zaffanello, M., Franchini, M. Thromboembolism in childhood nephritic syndrome: a rare but serious complication. Hematology. 2007;12(1):69–73.
95. Hodson, E.M., Willis, N.S., Craig, J.C. Corticosteroid therapy for nephritic syndrome in children. Cochrane Database Syst Rev. (4):2007. [CD001533].
96. Bellomo R et al: Acute Dialysis Quality Initiative workgroup. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group, Crit Care 8(4):R204-R212, 2004.
97. Santos, W.J., et al. Patients with ischaemic, mixed and nephrotoxic acute tubular necrosis in the intensive care unit—a homogeneous population? Crit Care. 2006;10(2):R68.
98. Bonventre, J.V. Pathophysiology of acute kidney injury: roles of potential inhibitors of inflammation. Contrib Nephrol. 2007;156:39–46. [review].
99. Kellum, J.A. Acute kidney injury. Crit Care Med. 2008;36(4 Suppl):S141–S145.
100. Wan, L., et al. Pathophysiology of septic acute kidney injury: what do we really know? Crit Care Med. 2008;36(4 Suppl):S198–S203.
101. Ortiz, A., et al. Targeting apoptosis in acute tubular injury. Biochem Pharmacol. 2003;66(8):1589–1594.
102. Clarkson, M.R., et al. Acute kidney injury. In Brenner B.M., ed.: Brenner and Rector’s the kidney, ed 8, Philadelphia: Saunders, 2007.
103. Bagshaw, S.M., Gibney, R.T. Convention markers of kidney function. Crit Care Med. 2008;36(4 Suppl):S152–S158.
104. Endre, Z.H., Westhuzyen, J. Early detection of acute kidney injury: emerging new biomarkers. Nephrology (Carlton). 2008;13(2):91–98.
105. Parikh, C.R., Devarajan, P. New biomarkers of acute kidney injury. Crit Care Med. 2008;36(4 Suppl):S159–S165.
106. Kraut, J.A., Kurtz, I. Metabolic acidosis of CKD: diagnosis, clinical characteristics, and treatment. Am J Kidney Dis. 2005;45(6):978–993.
107. Ronco, C., Cruz, D., Bellomo, R. Continuous renal replacement in critical illness. Contrib Nephrol. 2007;156:309–319.
108. Snyder, S., Pendergraph, B. Detection and evaluation of chronic kidney disease. Am Fam Physician. 2005;72(9):1723–1732.
109. Mene, P., Polci, R., Festuccia, F. Mechanisms of repair after kidney injury. J Nephrol. 2003;16(2):186–195.
110. Bricker, N.S., Morrin, P.A., Kime, S.W., Jr. The pathologic physiology of chronic Bright’s disease: an exposition of the “intact nephron hypothesis,”. J Am Soc Nephrol. 1997;8(9):1470–1476.
111. Tall, M.W., Luychx, V.A., Brenner, B.M., Adaptation to nephron loss. Brenner B.M., ed. Brenner and Rector’s the kidney. ed 7. 2004:1954.
112. Schieppati, A., Pisoni, R., Remuzzi, G. Pathophysiology and management of chronic kidney disease. In: Greenberg A., ed. Primer on kidney diseases. ed 4. St Louis: Saunders; 2005:445–447.
113. Chang, S.S. Albuminuria and diabetic nephropathy. Pediatr Endocrinol Rev. 2008;5(Suppl 4):974–979.
114. Nangaku, M., Jujita, T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens Res. 2008;31(2):175–184.
115. Vanholder, R., et al. Uremic toxins: do we know enough to explain uremia? Blood Purif. 2008;26(1):77–81.
116. Thijssen, S., Kitzler, T.M., Levin, N.W. Salt: its role in chronic kidney disease. J Ren Nutr. 2008;18(1):18–26.
117. Weir, M.R., Fink, J.C. Salt intake and progression of chronic kidney disease: an overlooked modifiable exposure: a commentary. Am J Kidney Dis. 2005;45(1):176–188. [review].
118. Kupin, W.L., Narins, R.G. The hyperkalemia of renal failure: pathophysiology, diagnosis, and therapy. In: Bourke E., Mallick N.P., Pollak B.E., eds. Moving points in nephrology. Basel: Karger, 1993.
119. Giovannetti, S., Cupisti, A., Barsotti, G. The mentabolic acidosis of chronic renal failure: pathophysiology and treatment. In: Berlyn G.M., ed. The kidney today. Basel: Karger, 1992.
120. Putcha, N., Allon, M. Management of hyperkalemia in dialysis patients. Semin Dial. 2007;20(5):431–439.
121. Kraut, J.A., Kurtz, J. Metabolic acidosis of CKD: diagnosis, clinical characteristics, and treatment. Am J Kidney Dis. 2005;45(6):978–993.
122. Fehally, J., Floege, J., Johnson, R. Comprehensive clinical nephrology. St Louis: Mosby; 2007.
123. Ferreira, A. Development of renal bone disease. Eur J Clin Invest. 2006;36(Suppl 2):2–12.
124. Brancaccio, D., et al. Management of secondary hyperparathyroidism in uremic patients: the role of the new vitamin D analogs. J Nephrol. 2007;20(1):3–9.
125. Abbate, M., Zoja, C., Remuzzi, G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17(11):2974–2984.
126. Bakris, G.L. Slowing nephropathy progression: focus on proteinuria reduction. Clin J Am Soc Nephrol. 2008;3(Suppl 1):S3–S10.
127. Chaturvedi, S., Jones, C. Protein restriction for children with chronic renal disease. Cochrane Database Syst Rev. (4):2007. [CD002181].
128. Levey, A.S., et al. Effect of dietary protein restriction on the progression of kidney disease: long-term follow-up of Modification of Diet in Renal Disease (MDRD) study. Am J Kidney Dis. 2006;48(6):879–888.
129. Ikee, R., et al. Glucose metabolism, insulin resistance and renal pathology in non-diabetic chronic kidney disease. Nephron Clin Pract. 2008;108(2):c163–c168.
130. Chan, D.T., et al. Dyslipidaemia and cardiorenal disease: mechanisms, therapeutic opportunities and clinical trials. Atherosclerosis. 2008;196(2):823–834.
131. Krane, V., Wanner, C. Dyslipidaemia in chronic kidney disease. Minerva Urol Nefrol. 2007;59(3):299–316.
132. Zanetti, M., et al. Vascular sources of oxidative stress: implications for uremia-related cardiovascular disease. J Ren Nutr. 2007;17(1):53–56.
133. Kaw, D., Malhotra, D. Platelet dysfunction and end-stage renal disease. Semin Dial. 2006;19(4):317–322.
134. Adams, M.J., et al. Hypercoagulability in chronic kidney disease is associated with coagulation activation but not endothelial function. Thromb Res. 2008;123(2):374–380.
135. Molino, D., et al. Coagulation disorders in uremia. Semin Nephrol. 2006;26:46–51.
136. Chonchol, M. Neutrophil dysfunction and infection risk in end-stage renal disease. Semin Dial. 2006;19(4):291–296.
137. Krishnan, A.V., Kiernan, M.C. Uremic neuropathy: clinical features and new pathophysiological insights. Muscle Nerve. 2007;35(3):273–290.
138. Bellinghieri, G., Savica, V., Santoro, D. Vascular erectile dysfunction in chronic renal failure. Semin Nephrol. 2006;26(1):42–45.
139. Kettas, E., et al. Sexual dysfunction and associated risk factors in women with end-stage renal disease. J Sex Med. 2007;5(4):872–877.
140. Zanetti, M., Barazzoni, R., Guarnieri, G. Inflammation and insulin resistance in uremia. J Ren Nut. 2008;18(1):70–75.
141. Rigalleau, V., Gin, H. Carbohydrate metabolism in uraemia. Curr Opin Clin Nutr Metab Care. 2005;8(4):463–469.
142. Carrero, J.J., et al. Clinical and biochemical implication of low thyroid hormone levels (total and free forms) in euthyroid patients with chronic kidney disease. J Intern Med. 2007;262(6):690–701.
143. Adler, S., Wartofsky, L. The nonthyroidal illness syndrome. Endocrinol Metab Clin North Am. 2007;36(3):657–672.
144. Rosolowska-Huszcz, D., Kozlowska, L., Rydzewski, A. Influence of low protein diet on nonthyroidal illness syndrome in chronic renal failure. Endocrine. 2005;27(3):283–288.
145. Patel, T.S., Freedman, B.I., Yosipovitch, G. An update on pruritus associated with CKD. Am J Kidney Dis. 2007;50(1):11–20.
146. Kunz, R., et al. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008;148(1):30–48.
147. Schrijvers, B.F., De Vriese, A.S. Novel insights in the treatment of diabetic nephropathy. Acta Clin Belg. 2007;62(5):278–290.