ALTERATIONS OF DIGESTIVE FUNCTION IN CHILDREN


Disorders of the gastrointestinal tract, liver and pancreas in children include congenital anomalies with structural and functional alterations, enzyme deficiencies, and infections. These disorders lead to impairment of motility, digestion, nutrition and normal growth and development
DISORDERS OF THE GASTROINTESTINAL TRACT
Congenital Impairment of Motility
Cleft Lip and Cleft Palate
Cleft lip (harelip) and cleft palate are developmental anomalies of the first branchial arch (Figure 40-1). The incidence of cleft lip with or without cleft palate is estimated at 10.48 per 10,000 live births; the incidence of cleft palate only is 6.39 per 10,000 live births.1 Incidence is lower in black populations and higher in Asian populations.2 Cleft lip, with or without cleft palate, is more common in females. Both anomalies can be unilateral or bilateral, partial or complete and may also be associated with other malformations.3 Nonsyndromic cleft lip and/or palate is a malformation with an incomplete separation between nasal and oral cavities without any associated anomaly and is associated with a number of different genes.4
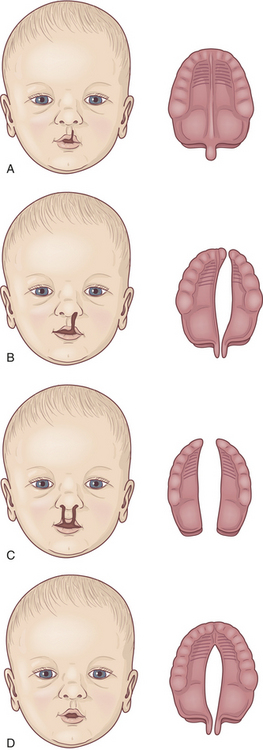
Figure 40-1 Variations in clefts of the lip and palate. A, Notch in vermilion border. B, Unilateral cleft lip and palate. C, Bilateral cleft lip and cleft palate. D, Cleft palate.
In most cases, cleft lip and cleft palate are caused by multiple gene-environmental interactions, including maternal deficiency of B vitamins (B6, folic acid, and B12), maternal tobacco5 and alcohol use, maternal diabetes mellitus, and genetic variations of several biomolecules including transforming growth factor, interferon regulatory factor-6, fibroblast growth factor, and other growth factors.6–9 (This phenomenon, called multifactorial inheritance, is discussed in Chapter 4.) Maternal hyperhomocysteinemia also may be a factor associated with orofacial clefts.10 Together these factors reduce the amount of neural crest mesenchyme that migrates into the area that will develop into the face of the embryo. If the amount is sufficiently reduced, clefting occurs.
Cleft Lip: Cleft lip is caused by the incomplete fusion of the nasomedial or intermaxillary process beginning during the fourth week of embryonic development,3 a period of very rapid fetal growth. The cleft causes structures of the face and mouth to develop without the normal restraints of encircling lip muscles. A characteristic depression or flattening of the infant’s midfacial contour may occur because normal antagonistic forces across the midline are absent, and growth of the involved facial segments is disturbed. The facial cleft may affect not only the lip but also the external nose, the nasal cartilages, the nasal septum, and the alveolar processes (bony ridge of maxilla that contains the tooth sockets).
The cleft is usually just beneath the center of one nostril. The defect may occur bilaterally and may be symmetric or asymmetric. The cleft can range in severity from a slight indentation of the lip to a fissure that extends to the nostril, causing a sagging and flattening of the nose. The failure of lip fusion by 35 days of gestation may impair closure of the palatal shelves. The more complete the cleft lip, the greater the chance that teeth in the line of the cleft will be missing or malformed.
Cleft Palate: Cleft palate is often associated with cleft lip but may occur without it. Cleft palate results from the failure of the primary palatal shelves, or processes, to fuse during the third month of gestation. The fissure may affect only the uvula and soft palate, or it may extend forward to the nostril and involve the hard palate and the maxillary alveolar ridge. It may be unilateral or bilateral, with the cleft occupying the midline posteriorly and as far forward as the alveolar process, where it deviates to the involved side. Clefts involving the palate only are usually but not necessarily in the midline. In some cases the vomer and nasal septum are partly or completely undeveloped. When these facial bones are involved, the nasal cavity may freely communicate with the oral cavity.
CLINICAL MANIFESTATIONS Feeding the infant with cleft lip usually presents no difficulty if the cleft lip is simple and the palate intact. Nursing at breast or bottle depends on suction developed by pressing the nipple against the hard palate with the tongue. Closure of the lips is not necessary, but the tongue must work harder if the lips cannot be pursed. A baby with cleft palate usually requires large, soft nipples with cross-cut openings. Although most infants with cleft palate can be successfully breast-fed, it may be impossible for some because of an unproductive suck.11,12 An orthodontic prosthesis for the roof of the mouth may facilitate sucking for some infants.13
EVALUATION AND TREATMENT Facial x-ray films confirm the extent of bone deformity. Soft tissue alterations are evaluated by history and physical examination. Prenatal ultrasound has aided early diagnosis of orofacial clefting.14
The nature and extent of the cleft, the infant’s condition, and the method of surgical correction proposed determine the course of treatment.15 The lip is united first. Although this can be done within a few weeks of birth, most surgeons prefer to wait until the infant is 3 months old to allow sufficient growth to occur. The initial repair may be revised when the child is 4 to 5 years old.
Repair of a cleft lip that is accompanied by bilateral cleft palate is technically more difficult, so the procedure is often performed in two steps. The lip is repaired when the infant is a few weeks old. Surgical correction of the cleft palate is often planned in stages with the soft palate closed first. Presurgical nasoalveolar molding can reduce the severity of the initial cleft.16 The palate is closed after the child is weaned from the nipple but before beginning to talk, usually at about 10 to 12 months of age. The aim of surgery is to obtain an airtight closure of the palatal cleft and to preserve the mobility and length of the soft palate. Even with early closure, the child may experience difficulty sealing off the nasopharynx from the buccal cavity during swallowing and while pronouncing certain consonants. Speech training and special attention by a prosthodontist and orthodontist are almost always required.3
Before and after surgery, children with cleft lip and palate tend to have recurrent infections of the paranasal sinuses and middle ear. Parents should be alerted to this increased risk so that otitis media can be detected and treated earlier to decrease the chance of long-term scarring and subsequent hearing loss.17 Breast-milk feedings have been associated with a lower incidence of otitis media in these infants.18 Hypertrophy of the tonsils and adenoids is common. Children with an orofacial cleft are at an increased risk of being infected by Streptococcus mutans and Lactobacillus at a very early age. Such colonization indicates a high risk for caries in the primary dentition.19,20 Displacement of the maxillary arches and malposition of the teeth usually require orthodontic correction.
Esophageal Malformations
Congenital malformations of the esophagus are rare and occur in 1 of 3000 to 5000 live births. Esophageal atresia is a condition in which the esophagus ends in a blind pouch. Esophageal atresia is usually accompanied by a fistula between the esophagus and the trachea. This connection is called a tracheoesophageal fistula (TEF). Either defect can occur alone (Figure 40-2).
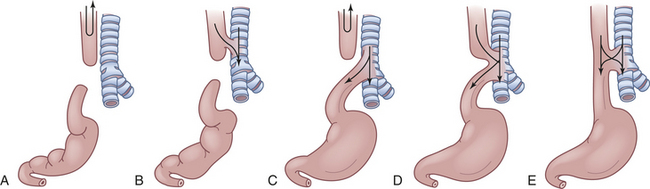
Figure 40-2 Five types of esophageal atresia and tracheoesophageal fistulae. A, Simple esophageal atresia. Proximal esophagus and distal esophagus end in blind pouches, and there is no tracheal communication. Nothing enters the stomach; regurgitated food and fluid may enter the lungs. B, Proximal and distal esophageal segments end in blind pouches, and a fistula connects the proximal esophagus to the trachea. Nothing enters the stomach; food and fluid enter the lungs. C, Proximal esophagus ends in a blind pouch, and a fistula connects the trachea to the distal esophagus. Air enters the stomach; regurgitated gastric secretions enter the lungs through the fistula. D, Fistula connects proximal and distal esophageal segments to the trachea. Air, food, and fluid enter the stomach and the lungs. E, Simple tracheoesophageal fistula between otherwise normal esophagus and trachea. Air, food, and fluid enter the stomach and the lungs. Between 85% and 90% of esophageal anomalies are type C; 6% to 8% are type A; 3% to 5% are type E; and less than 1% are type B or D. NOTE: Type F, esophageal stenosis is not shown.
PATHOPHYSIOLOGY The esophageal abnormalities are thought to arise from defective differentiation as the trachea separates from the esophagus during the fourth to sixth weeks of embryonic development. Defective growth of endodermal cells leads to atresia. Incomplete fusion of the lateral walls of the foregut leads to incomplete closure of the laryngotracheal tube and fistula formation.21
CLINICAL MANIFESTATIONS Polyhydramnios is reported to occur in 14% to 90% of mothers of affected infants.22 The blind end of the proximal esophagus has a capacity of only a few milliliters. As the infant with esophageal atresia swallows oral secretions, the pouch fills and overflows into the pharynx, resulting in drooling and occasionally in aspiration (see Figure 40-2, A and C).
If a fistula connects the trachea with the distal esophagus, the abdomen fills with air and becomes distended. The distention may be great enough to interfere with breathing (see Figure 40-2, C to E). If the fistula connects the proximal esophagus to the trachea, the first feeding after birth will be problematic (see Figure 40-2, B, D, and E). As the infant drinks, the blind end of the esophagus and the mouth fill with fluid. When the infant tries to take a breath, the fluid is aspirated into the lungs, which triggers protective cough and choke reflexes. Intermittent cyanosis may result. Plain water or glucose is recommended for the initial feeding to minimize the dangers associated with aspiration. If an abnormality of the esophagus is indicated, oral feedings are withheld until a diagnosis is confirmed.
Pulmonary complications are compounded by reflux of air and gastric secretions into the tracheobronchial tree through the fistula (see Figure 40-2, D and E), causing severe chemical irritation. The upper lobe of the right lung is most commonly involved because of its proximity to the tracheoesophageal fistula. Infants with esophageal atresia but no fistula have a scaphoid (boat-shaped), gasless abdomen. In fistula without atresia (see Figure 40-2, E), the usual symptoms are recurrent aspiration, pneumonia, and atelectasis that remains “silent” for days or even months. Late complication of esophageal atresia or tracheal esophageal fistula include stricture, reflux, and dysphagia.23
In at least 50% of infants with esophageal defects, other congenital anomalies are present as well. Cardiovascular anomalies are the most common, but other digestive tract, urinary, vertebral, and central nervous system defects can accompany esophageal atresia and tracheoesophageal fistula.
EVALUATION AND TREATMENT Esophageal atresia is usually diagnosed at birth, when attempts to pass a small-bore orogastric or nasogastric tube into the stomach fail.24 X-ray films show the catheter coiled in the upper esophageal pouch. Prenatal ultrasound reveals defects in some cases.25
Treatment is surgical and may include esophageal replacement for long-gap atresia. Esophageal continuity is restored, and the fistula is eliminated. Surgery is usually undertaken after birth, sometimes in stages. The child may continue to have problems with aspiration, gastroesophageal reflux, and esophagitis after surgical repair.21 The overall survival rate for infants with esophageal defects exceeds 90%.25
Pyloric Stenosis
Pyloric stenosis is an obstruction of the pyloric sphincter caused by hypertrophy of the sphincter muscle. It is one of the most common disorders of early infancy and affects infants between the ages of either 1 and 2 weeks or 3 and 4 months.26 The incidence of pyloric stenosis among males is approximately 5 in 1000, whereas among females it is only 1 in 1000. Whites are affected more often than blacks or Asians, and full-term infants are affected more often than premature infants.27 The cause is unknown but increased gastrin secretion by the mother in the last trimester of pregnancy increases the likelihood of pyloric stenosis in the infant. The overproduction of gastric secretions in the infant may be caused by stress-related factors in the mother. Exogenous administration of prostaglandin E is associated with an increased incidence of pyloric stenosis. There is an increased incidence of pyloric stenosis in children with Down syndrome; 6.9% of children have a parent who had pyloric stenosis, and 4.9% have a close relative that is affected.28,29 Pyloric stenosis occurs in approximately 20% of male and 10% of female descendants of mothers who had pyloric stenosis.30
PATHOPHYSIOLOGY The circular muscle of the pylorus is grossly enlarged because of an increase in cell size (hypertrophy) and an increase in cell number (hyperplasia).31 Research has shown that transforming growth factor-alpha plays a role in stimulating this increase in muscle mass.32 The mucosal lining of the pyloric opening is folded and the lumen is narrowed by the encroaching muscle. Because of the extra peristaltic effort necessary to force the gastric contents through the narrow pylorus, the muscle layers of the stomach may become hypertrophied as well.
CLINICAL MANIFESTATIONS Between 2 and 3 weeks after birth, an infant who has fed well and gained weight begins to vomit without apparent reason. The vomiting gradually becomes more forceful. In some cases, stomach contents may shoot out 3 or 4 feet. Food is often regurgitated through the nose. The forceful, or projectile, vomiting usually occurs immediately after eating, and the vomitus consists of the bulk of the feeding plus some food retained from previous feedings but is almost always free of bile. Usually infants are hungry and want to eat again after vomiting.33
Prolonged retention of food in the stomach is a characteristic feature of pyloric stenosis and food is present after 4 hours unless vomiting has occurred. Constipation is the rule because not much food reaches the intestine.
In severe untreated cases, increased gastric peristalsis and vomiting lead to severe fluid and electrolyte imbalances (hypochloremic metabolic alkalosis), chronic malnutrition, and weight loss that can be fatal within 4 to 6 weeks. Infants with pyloric stenosis are irritable because of hunger, and they may have esophageal discomfort caused by repeated vomiting and esophagitis. The vomitus may be blood streaked because of rupture of gastric and esophageal vessels.
EVALUATION AND TREATMENT Diagnosis is based on the history of clinical manifestations. Occasionally, gastric peristalsis is observable over the abdomen. A firm, small, movable mass, approximately the size of an olive, is felt in the right upper quadrant in 70% to 90% of infants with pyloric stenosis. A visible gastric peristaltic wave after eating is observed in some infants. Sonography is routinely done because this clearly shows the hypertrophied pyloric muscles and narrowed pyloric channel.34
The standard treatment for hypertrophic pyloric stenosis is a pyloromyotomy, in which the muscles of the pylorus are split and separated. The procedure can be completed with an open technique or with laparoscopy.35 The mortality rate associated with surgical correction is less than 0.5%.
Intestinal Malrotation
During the tenth week of embryonic development, the emerging ileum and cecum normally rotate, so that the cecum moves into the lower right quadrant of the abdomen and is fixed there by the mesentery. Intestinal malrotation is a condition in which rotation does not occur and the colon remains in the upper right quadrant, where an abnormal membrane may press on and obstruct the duodenum. The obstructing band over the duodenum, called a periduodenal band, is one of the most significant findings in malrotation. Associated abnormalities are seen in 30% to 62% of children in a reported series; 50% of children with duodenal atresia and 33% of those with jejunal atresia have associated malrotation.36
PATHOPHYSIOLOGY The small intestine lacks a normal posterior fixation in malrotation because it has only a rudimentary attachment near the origin of the superior mesenteric artery. Therefore, the entire mass can twist when the mobile loops of intestine from the duodenojejunal junction to the middle of the transverse colon twist on themselves. The twisting is termed volvulus. Intestinal twisting around the rudimentary mesentery angulates and obstructs the intestinal lumen and partly or completely occludes the superior mesenteric artery, causing infarction and necrosis of the entire midgut.
CLINICAL MANIFESTATIONS Although most cases of malrotation-associated volvulus and infarction develop during the neonatal period (50%) or infancy (85% are younger than 1 year), some develop during childhood or even adulthood.37 In infants the obstruction causes intermittent or persistent bile-stained vomiting after feedings. Abdominal distention is limited initially to the epigastrium because only the stomach and duodenum are dilated. The degree of distention depends on the pressure of swallowed air and the degree of obstruction caused by the volvulus. Dehydration and electrolyte imbalance may occur rapidly because large amounts of pancreatic juice, bile, and gastric secretions are lost through vomiting. Fever usually ensues. Pain, scanty stools, diarrhea, and bloody stools are associated with progressive volvulus, vascular compression, and infarction of the intestine in infants. Intermittent or partial volvulus may be seen in older children and adults. This may be asymptomatic (25% to 50% of the time) and discovered during unrelated abdominal surgery, or it may cause minor abdominal complaints, such as nausea after meals, recurrent episodes of vomiting, or abdominal pain.38
EVALUATION AND TREATMENT Diagnosis of malrotation with volvulus and infarction is based on a review of the clinical manifestations. X-ray films of the abdomen show gas bubbles and distention proximal to the site of obstruction.
Treatment consists of opening the abdomen and reducing the volvulus manually. Necrotic bowel is resected and a primary anastomosis performed. An enterostomy may be created. In cases of malrotation without duodenal obstruction, the operative survival rate is 80%. The operative survival rate is 40% to 50% in cases of malrotation complicated by obstruction caused by periduodenal bands or other intra-abdominal anomalies. Resection of large segments of the small intestine results in short-bowel syndrome and its long-term sequelae.
Meconium Ileus
Meconium is a substance that fills the entire intestine before birth. It consists of intestinal gland secretions and some amniotic fluid. Normally, meconium is passed from the rectum during the first 12 to 72 hours after birth.
Meconium ileus is intestinal obstruction caused by meconium formed in utero that is abnormally sticky and adheres firmly to the mucosa of the small intestine, resisting passage beyond the terminal ileum. The cause is usually a lack of digestive enzymes during fetal life. This meconium is also found to contain albumin, which is not normally found in meconium. The detection of albumin in meconium has been used as a screening test for cystic fibrosis.39 Neonatal meconium ileus occurs in 15% of newborns with cystic fibrosis.40 Partial aplasia of the pancreas is an associated factor, however, and one fifth of infants with meconium ileus are premature or have a history of maternal hydramnios (excessive amniotic fluid). After intestinal atresia and malrotation with volvulus, meconium ileus is the most common cause of small intestinal obstruction in newborns.
PATHOPHYSIOLOGY The terminal ileum is plugged with thick, viscous meconium resulting from the formation of an insoluble, calcium-glycoprotein compound in abnormal mucus. The segment of the ileum proximal to the obstruction is distended with liquid contents, and its walls may be hypertrophied. The segment distal to the obstruction is collapsed and filled with small pellets of pale-colored stool. Meconium in the obstructed segment has the consistency of thick syrup or glue. Peristalsis fails to propel this viscous material through the ileum, so it becomes impacted. Volvulus, atresia, or perforation of the bowel sometimes accompanies meconium ileus.
CLINICAL MANIFESTATIONS Abdominal distention usually develops during the first few days after birth. The infant does not pass meconium and begins to vomit within hours or days of birth. Infants with cystic fibrosis may have signs of pulmonary involvement, such as tachypnea, intercostal retractions, and grunting respirations. The distended abdomen shows patterns of dilated intestinal loops that feel doughlike when palpated. Some of the loops contain scattered, firm, movable masses. Despite hyperactive peristalsis, the rectal ampulla is empty.
EVALUATION AND TREATMENT Radiologic examination is used to confirm the presence of meconium ileus.41 The sweat test, which is accurate in 90% of infants, is performed to detect or rule out cystic fibrosis. The treatment of choice for cases not complicated by volvulus or perforation is a hyperosmolar enema done using fluoroscopy to evacuate the meconium. Although the success of this technique has not correlated with osmolality of the enema, the overall success rate is higher when metglutamine diatrizoate (Gastrografin) is used or when additives, such as polysorbate 80 (Tween 80) and acetylcysteine (Mucomyst), are used.42 Enterotomy and irrigation are reserved for complicated cases and enema failures.
Survival of infants with meconium ileus is improving, with a 97% to 98% survival rate at 1 year.43 The mortality rate increases to 70% if obstruction is complicated by peritonitis. After recovery from neonatal meconium ileus, the long-term outlook depends on the severity and progression of pulmonary disease. Recent research demonstrates a clear association of meconium ileus with poor long-term nutritional outcomes in children with cystic fibrosis related to surgical treatment for the ileus and poor essential fatty acid status.44
Distal Intestinal Obstruction Syndrome
Distal intestinal obstruction syndrome (DIOS), formerly called meconium ileus equivalent, affects approximately 15% of children and adults with cystic fibrosis.45 Intestinal contents may become abnormally thick and impact the intestinal lumen, particularly after episodes of dehydration or lack of pancreatic enzymes. Use of high-strength pancreatic enzymes has been implicated in formation of strictures of the ascending colon in children with cystic fibrosis and resultant chronic DIOS.46 The child displays signs and symptoms of intestinal obstruction. In most cases the obstruction is relieved by hypertonic enemas. Meconium ileus and DIOS have been shown to be risk factors for the development of liver disease and other abdominal manifestations in those with cystic fibrosis.47
Obstructions of the Duodenum, Jejunum, and Ileum
Congenital obstruction of the duodenum can be caused by intrinsic malformations or external pressure. Intrinsic obstruction is caused by failure of the duodenum to become patent. The obstruction may be partial or complete and usually is located at or near the major duodenal papilla. Extrinsic obstructions can be caused by peritoneal bands that constrict the duodenum. The duodenum can be obstructed by an annular pancreas—a defect in which the head of the pancreas surrounds part of the duodenum. Congenital obstructions of the jejunum and ileum can be attributable to atresia, stenosis, meconium ileus, megacolon (Hirschsprung disease), intussusception, Meckel diverticulum, intestinal duplication, or strangulated hernia.
In ileal atresia or jejunal atresia, the intestine ends blindly proximal and distal to an interruption in its continuity, with or without a gap in the mesentery. Stenosis (narrowing of the lumen) causes dilation proximal to the obstruction and luminal collapse distal to it.
Congenital Aganglionic Megacolon
Congenital aganglionic megacolon (Hirschsprung disease) is a functional obstruction of the colon caused by the absence of the enteric ganglia along a variable length of the colon with inadequate motility. The incidence is 1 in 5000 live births with an increased incidence in males, siblings of children with Hirschsprung disease, and children with Down syndrome.48 The exact cause is unknown but multiple interacting factors and a complex inheritance pattern that involves the RET proto-oncogene have been found.49
PATHOPHYSIOLOGY Congenital aganglionic megacolon is caused by a malformation of the parasympathetic nervous system. It is characterized by abnormalities of the basement membrane and extracellular matrix and absence of the intramural ganglion cells in the enteric nerve plexuses (Meissner and Auerbach plexuses) along variable lengths of the colon.50 Lacking neural stimulation, the muscle layers of the colon wall fail to propel feces through the colon, leading to functional obstruction. In 80% of cases the aganglionic segment is limited to the rectal end of the sigmoid colon; in 3% the entire colon lacks ganglion cells. The abnormally innervated colon impairs fecal movements, causing the proximal colon to become distended, hence the term megacolon (Figure 40-3).
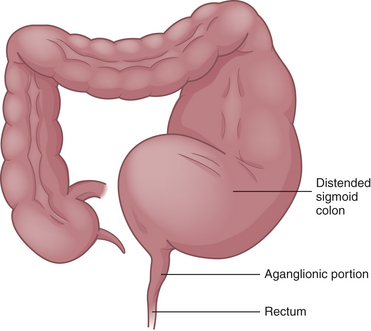
The ganglia normally develop from an advancing neural crest between the muscle layers (tunica muscularis) in the submucosal area (muscularis mucosae) of the intestinal wall. In cases of congenital megacolon, neurologic development is blocked and large, nonmyelinated fibers develop in place of these ganglion cells.50 The segment of colon that lacks ganglion cells has a relatively normal lumen caliber and wall thickness. In the segment of the colon proximal to it, the lumen is dilated and the muscle hypertrophied. Therefore, the abnormal portion of the colon appears to be normal and the normal portion appears to be diseased.
CLINICAL MANIFESTATIONS The extent of the aganglionic portion of the colon determines the severity of the symptoms of congenital aganglionic megacolon. The most distal part of the rectum is always involved. This is the extent of the aganglionic portion in some children, and the child is said to have “ultrashort-segment” Hirschsprung disease and generally has only mild constipation as a symptom. These individuals may not be diagnosed until adulthood.51 Symptoms of constipation, poor feeding, poor weight gain, and progressive abdominal distention increase in severity as the aganglionic portion extends proximally. Diarrhea may be the first sign, however, because only water can travel around the impacted feces.
The most serious complication in the neonatal period is enterocolitis related to fecal impaction. Bowel dilation stretches and partly occludes the encircling blood and lymphatic vessels, causing edema, ischemia, infarction of the mucosa, and significant outflow of fluid into the bowel lumen. Copious, liquid stools result. Infarction and destruction of the mucosa enable enteric microorganisms to penetrate the bowel wall. Frequently, gram-negative sepsis occurs, accompanied by fever and vomiting. Severe and rapid electrolyte changes may take place, causing collapse and rapid death.
EVALUATION AND TREATMENT Anorectal manometry and rectal suction biopsy are the most reliable screening tools for the diagnosis of Hirschsprung disease. Serial manometry measurements may be required in neonates.52 This test has uncovered ultrashort-segment Hirschsprung disease in older children with a history of constipation.53 The definitive diagnosis is made by rectal suction biopsy showing an absence of ganglion cells in the submucosa of the colon.54 X-ray films show dilated loops of colon, and contrast films show aganglionic areas. The infant usually cannot expel the barium.
The involved segment is resected within the first few months of life. For children with short-segment Hirschsprung disease, enemas are given to relieve impaction, and laxatives with a dietary and bowel training program are used in preference to surgical intervention.53 The child is not treated for diarrhea.
After surgery, enterocolitis sometimes recurs. If the postoperative enterocolitis is allowed to persist, pseudopolyps may appear. Because these are essentially identical to the lesions of ulcerative colitis, they have malignant potential. Therefore, a colectomy is indicated if pseudopolyps develop.
In general, the prognosis of congenital megacolon is satisfactory for children who undergo surgical treatment. Bowel training may be prolonged, but most children achieve bowel continence before puberty.55
Anorectal Malformations
Several congenital malformations of anorectal structures can obstruct the passage of feces. The incidence of minor abnormalities is approximately 1 in 500 and that of major anomalies is approximately 1 in 5000.
Congenital anorectal malformations range from mild anal stenosis, which is corrected by simple dilation, to complex deformities, such as anal or rectal agenesis, atresia, and rectourethral fistula (Figure 40-4). Deformities that cause complete obstruction are known collectively as imperforate anus.
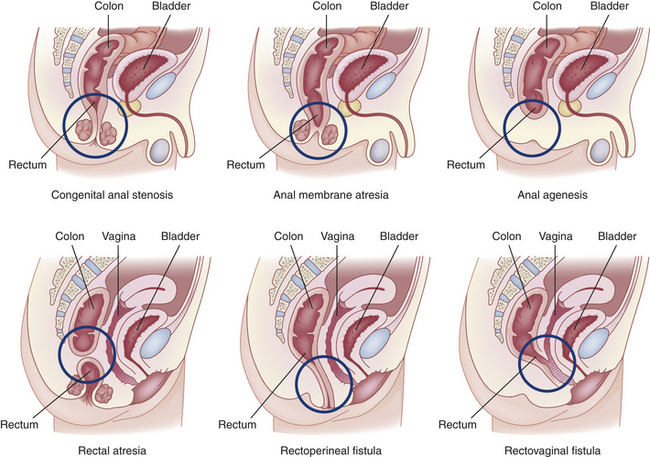
Approximately 40% of infants with anorectal malformations have other developmental anomalies as well. The most commonly associated major anomalies are Down syndrome, congenital heart disease, renal and urologic abnormalities, cryptorchidism, esophageal atresia, and malformations of the spine.56
Imperforate anus may not be obvious. It can be detected by gentle insertion of a rectal tube. X-ray films show dilations throughout the intestinal tract. Anal stenosis can be treated by dilations, but all other anorectal malformations require surgical correction. The overall mortality rate is approximately 10%. More than 90% of children with a low (anal) anomaly and intact sacrum achieve bowel continence; however, less than 30% of those with very high anomalies or anomalies associated with genitourinary fistulae achieve continence.57
Acquired Impairment of Motility
The most common cause of acquired intestinal obstruction in infants is intussusception. Intussusception is the telescoping or invagination of one portion of the intestine into another. Usually, the ileum invaginates the cecum and part of the ascending colon by collapsing through the ileocecal valve. Intussusception involving the ileum and colon (ileocolic intussusception) accounts for 80% to 90% of intestinal obstructions in infants and is two to three times more common in males than in females.58 Most intussusceptions occur before 1 year of age, most commonly between 4 and 7 months. Intussusception is rare in infants younger than 3 months and is infrequent after 36 months. Intussusception has occurred in children of all ages recovering from abdominal surgery; intussusception has been found in children with cystic fibrosis and symptoms of bowel obstruction that were initially misdiagnosed as having distal intestinal obstruction syndrome (meconium ileus equivalent).59,60
PATHOPHYSIOLOGY Most commonly, the proximal portion of the intestine, the intussusceptum, collapses into the distal portion, the intussuscipiens, in the direction of peristaltic flow (Figure 40-5). As it does so, the intussusceptum drags its mesentery into the enveloping lumen. Initially, the mesentery is constricted, obstructing venous return. Compression of the mesenteric vessels between the two layers of intestinal wall and at the U-shaped angle at either end of the intussusceptum leads within hours to venous stasis, engorgement, edema, exudation, and further vascular compression. Unless the intussusception is treated, bleeding and gangrene ensue. The tension of the mesentery on the intussusceptum tends to arch the bowel in a curve with its center at the mesenteric root. Edema and compression obstruct the flow of chyme through the intestine.
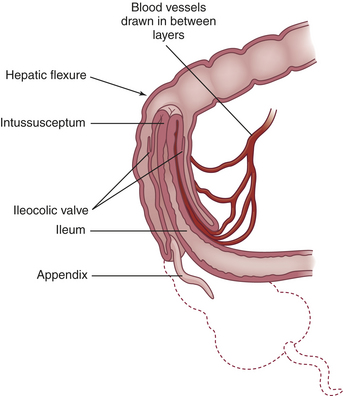
CLINICAL MANIFESTATIONS The affected infant suddenly develops abdominal pain, becomes irritable (colicky), and draws up the knees. Vomiting occurs soon afterward. A single normal stool may be passed, evacuating the colon distal to the apex of the intussusception. After that, 60% of infants pass “currant jelly” stools, which appear dark and gelatinous because of their blood and mucus content. In one study, less than one third of children had this clinical triad of vomiting, colicky abdominal pain, and bloody stools.61 Most infants have a tender, sausage-shaped abdominal mass. Abdominal tenderness and distention develop as intestinal obstruction becomes more acute.62
EVALUATION AND TREATMENT Diagnosis is based on clinical manifestations and onset of symptoms. Ultrasound of the abdomen, computed tomography (CT) and magnetic resonance imaging (MRI) are commonly completed for diagnosis.63 More than 82% of children have positive ultrasound results in ileocolic and jejunointestinal intussusception. Reduction is an emergency procedure involving hydrostatic pressure generated by an air or a barium enema given using fluoroscopic guidance.64 This technique is successful 45% to 90% of the time. A potential complication of enema reduction is bowel perforation, and for this reason the use of air rather than barium is favored.65 Surgical reduction is done on children who fail or are not candidates for hydrostatic reduction. Untreated intussusception in infants is nearly always fatal. Most infants recover if the intussusception is reduced within 24 hours.66 Spontaneous reduction of intussusception may occur in symptomatic or asymptomatic children and occurs more commonly than previously reported. These intussusceptions are usually short-segment, small-bowel intussusceptions with no recognizable lead point.67 Recurrent intussusception is more common after nonsurgical reduction than after surgical reduction. Risk of recurrence cannot be predicted by initial features or symptoms, and children with recurrent intussusception may exhibit fewer symptoms with a shorter interval to bowel necrosis and perforation.68
Gastroesophageal Reflux Disease
Gastroesophageal reflux disease (GERD) is common in children and involves dilation of the esophagus and intrusion of acid contents into it; GERD is believed to be related to relaxation or incompetence of the lower esophageal sphincter (see Chapter 39). In newborns, reflux is normal because neuromuscular control of the gastroesophageal sphincter is not fully developed. The frequency of reflux is highest in premature infants and decreases during the first 6 to 12 months of life.
GERD is thought to be a factor in the stimulation of reactive airway disease and otitis media with effusion in some children.69,70 Reflux also is thought to be a contributing cause in apparent life-threatening events and a significant problem for children with cerebral palsy and cystic fibrosis.71–74 The relationship between apnea of prematurity and GERD is controversial.75,76
PATHOPHYSIOLOGY Delayed maturation of the lower esophageal sphincter or impaired hormonal or neurotransmitter response mechanisms (i.e., vasoactive intestinal peptide and nitric oxide) are possible causes of inappropriate sphincter relaxation. Factors that maintain lower esophageal sphincter integrity in children include location of the gastroesophageal junction in a high-pressure zone within the abdomen, mucosal gathering within the sphincter, and the angle at which the esophagus is inserted into the stomach. Reflux persists if any of these pressure-maintaining factors is altered. Irritation of the mucosa by acidic gastric contents results in deterioration of the esophageal epithelium and stimulation of the vomiting reflex.77,78
Eosinophilic esophagitis also occurs in children (see Chapter 39). It is thought to be an atopic disease involving immediate as well as delayed hypersensitivity reactions to food ingestion. An eosinophilic infiltrate is associated with inflammation of the entire esophagus that is nonresponsive to acid-suppression therapy.79 Dysphagia, food impaction, and vomiting are common symptoms and other atopic diseases, such as asthma and eczema, may be present. Treatment includes elimination diets and corticosteroids.80 The eosinophilic inflammation may lead to progressive subepithelial fibrosis with esophageal strictures, narrowing, and dysphagia.81
CLINICAL MANIFESTATIONS The signs and symptoms of GERD are caused by exposure of the esophageal epithelium to refluxed gastric contents. Eighty-five percent of affected infants vomit excessively during the first week of life and usually have other symptoms by 6 weeks.
Vomiting may be forceful and must be differentiated from pyloric stenosis. Aspiration pneumonia develops in one third of infants with gastroesophageal reflux (GER). In cases that persist into childhood, chronic cough, wheezing, and recurrent pneumonia are common.82,83 Repeated vomiting leads to inadequate retention of nutrients, adversely affecting growth and weight gain. Esophagitis from exposure of the esophageal mucosa to acidic gastric contents is manifested by pain, bleeding, and eventually stricture formation and abnormal motility. Approximately 10% to 25% of children with GERD also have iron deficiency anemia caused by frank or occult blood loss.
EVALUATION AND TREATMENT The clinical manifestations are often adequate to confirm a diagnosis of GERD. Esophageal pH monitoring with a probe for 24 hours and endoscopy are routinely used for diagnosis.84
Mild GER resolves without treatment. Maintaining infants in a flat prone or a left lateral position, particularly during and for the first hour after a feeding, results in fewer or shorter episodes of GER.85 Infants with GER are excepted from the Academy of Pediatrics recommended sleep position for healthy infants to decrease the risk of sudden infant death syndrome (SIDS).86 Older infants and children achieve better results in an upright (sitting or standing) position while awake, with prone positioning used for sleeping.87 Thickened feedings may help some infants88; however, this has not been shown to be consistently helpful. For some infants, thickened feedings may actually worsen reflux by causing a delay in gastric emptying time. Small, frequent feedings and frequent burping are generally universally accepted strategies for managing reflux.29 Medications to increase motility, increase lower esophageal sphincter pressure, or decrease gastric acid production have been used to treat GERD. If no improvement is seen with medical management or the child has life-threatening events with reflux, an antireflux surgical procedure, including gastropexy and fundoplication, is performed. A fundoplication re-creates a valve by wrapping the fundus of the stomach around the lower esophagus.89
Impairment of Digestion, Absorption, and Nutrition
Cystic fibrosis (CF) of the pancreas, which is also called mucoviscidosis or fibrocystic disease of the pancreas, is a genetically transmitted disease (mutation of the long arm of chromosome 7) that involves many organs and systems and usually causes death in childhood or young adulthood. It is the most common cause of chronic suppurative lung disease in children and is the most common life-threatening inherited disease in the white population. This section focuses on the deficiency of pancreatic enzymes. (Chapter 34 discusses the pulmonary consequences of cystic fibrosis.)
PATHOPHYSIOLOGY The pathophysiologic triad that is the hallmark of CF includes (1) pancreatic enzyme deficiency, which causes maldigestion; (2) overproduction of mucus in the respiratory tract and inability to clear secretions, which cause progressive chronic obstructive pulmonary disease; and (3) abnormally elevated sodium and chloride concentrations in sweat. Exocrine secretions tend to be abnormally thick and precipitate in the glandular ducts, obstructing flow. Almost all clinical manifestations of CF are a result of overproduction of extremely viscous mucus and pancreatic enzyme deficiency. The full spectrum of involvement is summarized in Table 40-1.
Table 40-1
Pathophysiology, Clinical Manifestations, and Complications of Cystic Fibrosis

Data from Rudolph CD et al: Rudolph’s pediatrics, ed 21, New York, 2003, McGraw-Hill.
Pancreatic function may range from normal to completely ablated. Approximately 85% of patients have pancreatic insufficiency. Obstruction of the pancreatic ducts with thick mucus blocks the flow of pancreatic enzymes and causes degenerative and fibrotic changes in the pancreas. Pancreatic damage eventually can affect the beta cells, resulting in diabetes mellitus in some children. The incidence of diabetes mellitus and cirrhosis in this population has increased as larger numbers of people with cystic fibrosis have moved into young and middle adulthood. Severe problems with maldigestion of proteins, carbohydrates, and fats occur because of insufficient secretion of pancreatic enzymes.90
CLINICAL MANIFESTATIONS Clinical manifestations are presented in Table 40-1.
EVALUATION AND TREATMENT To determine the extent of pancreatic function 72-hour stool fat measurements are used. Stools also may be examined for absence of pancreatic enzymes, particularly fecal elastase, trypsin, and chymotrypsin. To optimize treatment, the carbon-13 (13C) mixed triglyceride breath test offers a simple, noninvasive way of assessing the need for pancreatic enzyme supplementation in children with cystic fibrosis.91 Pancreatic replacement enzymes are administered before or with meals and high-calorie, high-protein diets with frequent snacks and vitamin supplements are used to treat the malnutrition. However, anorexia is not uncommon in this group secondary to pulmonary disease and frequently large sputum output. To combat the worsening problem of growth failure in children with cystic fibrosis, nasogastric or gastrostomy tube feedings are used to supplement oral intake and promote weight gain. Monitoring of growth and body mass index is critical to treatment evaluation.92,93
Gluten-Sensitive Enteropathy
Gluten-sensitive enteropathy, formerly called celiac sprue or celiac disease, is an autoimmune disease that damages the small intestinal villous epithelium when there is ingestion of gluten (gliadin), the protein component of cereal grains. The gluten in wheat, rye, barley, and oats is toxic to the intestinal epithelial cells of genetically susceptible individuals.94 The disease occurs largely in whites and has been documented in Asians from India and Pakistan, but it is almost nonexistent in native Africans, Japanese, and Chinese. Prevalence rates in Europe range from 1 in 1000 to 1 in 3000. Recent data suggest that gluten-sensitive enteropathy, traditionally considered rare in the United States, occurs in about 1% of the population or 1 in 100 children.95 Other autoimmune diseases have been associated with gluten-sensitive enteropathy including type 1 diabetes mellitus and autoimmune thyroiditis.96
The pathogenesis appears to require interaction between a number of intrinsic factors (genetic susceptibility, activation of the immune system) and extrinsic factors (gluten and possibly other environmental factors). Cellular immunity as well as humoral immunity are implicated. There are increases in the percentages of T cells, immunoglobulin, and complement in the mucosa of active celiac disease. Immunoglobulin A (IgA) and immunoglobulin M (IgM) antigliadin antibodies have been found in jejunal fluid of persons with untreated disease.97 Autoantibody reactivity to transglutaminase-2 (TG-2) has been shown to closely correlate with the acute phase of the disease.98 Although gluten-sensitive enteropathy is widely perceived as a malabsorption syndrome of childhood, the diagnosis is increasingly being made for the first time in adult life.99
PATHOPHYSIOLOGY The major pathophysiologic characteristics of the disease are atrophy and flattening of villi in the upper small intestine and malabsorption of most nutrients in the presence of cereal gluten (Figures 40-6 and 40-7). The atrophy is caused by accelerated shedding of epithelial cells from the villi. To compensate for this loss, epithelial cell production increases, causing hypertrophy of the crypts of Lieberkühn.100 Increased cell production is not sufficient to keep pace with cell loss, however. Inflammation and edema develop around the enlarged cysts. The villi shorten and atrophy, and their surface cells are not mature enough to sustain absorptive functions. The microvilli and brush border disappear, leaving patches of bald mucosa. The loss of mucosal surface area and brush-border enzymes leads to severe malabsorption. The pathologic process is most pronounced in the duodenum and jejunum. The ileum may be spared. The severity of disease correlates with the length of the small intestinal mucosa involved.
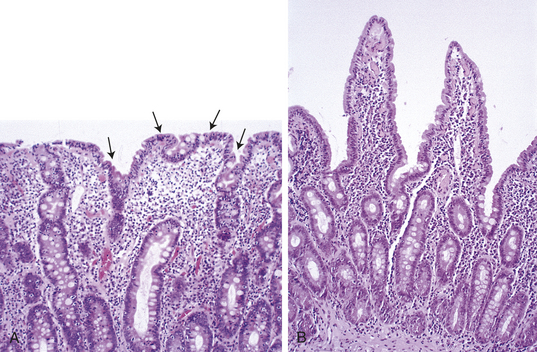
Figure 40-7 Gluten-sensitive enteropathy. A, Atrophy of villi and elongation of crypts that result in malabsorption (arrows)B, Recovery of normal villus structure after 6 months of gluten-free diet. (From Damjanov I, Linder J: Pathology: a color atlas, St Louis, 2000, Mosby.)
Damage to the mucosa of the duodenum and jejunum has secondary effects that exacerbate malabsorption. The secretion of intestinal hormones, such as secretin and cholecystokinin, may be diminished. Because these chemical messengers are scarce, secretion of pancreatic enzymes and expulsion of bile from the gallbladder decrease.
Destruction of mucosal cells causes inflammation, and water and electrolytes are secreted, leading to watery diarrhea. In addition, absorption that normally occurs by sodium-dependent active transport or facilitated diffusion is impaired. Carbohydrates, amino acids, dipeptides, water-soluble vitamins, bile salts, and cations are not absorbed from the intestinal lumen. Potassium loss, which is more severe than sodium loss, leads to muscle weakness. Magnesium and calcium malabsorption can cause seizures or tetany. Unabsorbed fatty acids combine with calcium, and secondary hyperparathyroidism increases phosphorus excretion, resulting in bone reabsorption. Calcium is no longer available to bind oxalate in the intestine and is absorbed, which causes hyperoxaluria. Gallbladder function may be abnormal, and bile salt conjugation may be decreased.
Fat malabsorption in the jejunum is the major cause of steatorrhea (fatty stools). Malabsorption may be mild early in the disease. Fecal nitrogen is elevated because peptidase deficiencies impair protein absorption. Pancreatic function is decreased, not only because of decreased hormonal levels but also because of malnutrition.
Deficiencies of fat-soluble vitamins are common in children with gluten-sensitive enteropathy. Vitamin K malabsorption leads to hypoprothrombinemia. In one third of cases, iron and folic acid malabsorption is manifested as cheilosis; anemia; and a smooth, red tongue. Vitamin B12 absorption is impaired in those with extensive ileal disease. Because the absorption of folate and iron is greatest in the proximal small intestine, deficiencies of these substances are common.
CLINICAL MANIFESTATIONS The onset of clinical manifestations of gluten-sensitive enteropathy depends on the age of the infant when gluten-containing substances are added to the diet. In 50% of affected children, onset occurs by 18 months of age, with latent intervals varying from months to years. Severity of symptoms can vary tremendously, and many children older than 3 years of age present with non-gastrointestinal symptoms.101
Diarrhea is an early sign in most infants accompanied by failure to thrive and anemia. The stools are pale, bulky, greasy, and foul smelling, and may contain oil droplets. Three to five such movements occur daily. As early as 3 or 4 months of age, growth failure, anorexia, and constipation can begin. In older children constipation is occasionally seen despite steatorrhea. Vomiting and abdominal pain are prominent in infants but unusual in older children. Anorexia is prevalent. The classic physical manifestations of organic failure to thrive, such as abdominal protuberance, wasted buttocks and limbs, and hypotonia, occur in less than 50% of infants with gluten-sensitive enteropathy. Growth is usually diminished.102
Manifestations of malabsorption, such as rickets, tetany, frank or occult bleeding, or anemia, may be obvious. Some children urinate more at night. The tongue is smooth and red, and the child may bruise and bleed easily. Hypomagnesemia and hypocalcemia cause irritability, tremor, convulsions, tetany, bone pain, osteomalacia, and dental abnormalities. If vitamin D deficiency is prolonged, rickets and clubbing of the terminal phalanges are likely. Eighty-six percent of older children have fingerprint changes (ridge atrophy). In older children, delayed puberty and infertility may be a manifestation of otherwise subtle gluten-sensitive enteropathy.103 Osteomalacia may be severe.104 Small intestinal lymphoma is also associated with gluten-sensitive enteropathy.105
A rare complication of gluten-sensitive enteropathy in infancy is celiac crisis. Celiac crisis is characterized by severe diarrhea, dehydration, and hypoproteinemia as a result of malabsorption and protein loss and requires aggressive treatment.
EVALUATION AND TREATMENT Diagnosis includes confirmation with serologic antiendomysial or antitransglutaminase IgA antibodies as well as an intestinal biopsy to detect the classic mucosal changes caused by gluten-sensitive enteropathy.106 The initial biopsy may be followed by a second intestinal biopsy to demonstrate regeneration of intestinal villi after treatment with a gluten-free diet. Most children with celiac disease remain undiagnosed.
Treatment consists of the immediate and permanent institution of a diet free of cereal grains (wheat, rye, barley, oats, malt). Lactose intolerance is presumed because of damage to the villi; therefore, lactose (milk sugar) is excluded from the diet. Tolerance to lactose improves with removal of gluten and healing of the mucosa and may be reintroduced at a later point.107 Infants are routinely given vitamin D, iron, and folic acid supplements to treat deficiencies. Breast-feeding at the time of gluten introduction in the diet delays the appearance of celiac disease.108
Approximately 25% of children experience recurrent relapses that interfere with growth. For most children, however, the long-term prognosis is excellent. There is an increased incidence of malignant disease, particularly T-cell lymphoma, in individuals who fail to respond to gluten-free diets.109
Protein Energy Malnutrition
Kwashiorkor and marasmus are the two most common types of malnutrition in children. These disorders are known collectively as protein energy malnutrition (PEM). Both are states of long-term starvation (see Chapter 39). Kwashiorkor is a severe protein deficiency, and marasmus is a severe deficiency of all nutrients. Kwashiorkor is a widespread nutritional problem among children in developing countries and economically destitute populations particularly when associated with human immunodeficiency virus (HIV) infection.110 The disease usually occurs in infants or children from 1 to 4 years of age who have been weaned from breast milk to a high-starch, protein-deficient diet.
Marasmus can occur at any age, but it is common in children younger than 1 year. In marasmus, starvation is attributable to lack of protein and carbohydrates. One third of the world’s children suffer from PEM, with the highest concentrations in, Asia, Africa, Latin America, and the Carribbean.111,112 The mortality risk for children in developing countries has been found to be inversely related to anthropometric indicators (height, weight, head circumference, skinfold thickness, midarm muscle circumference). There is elevated risk even in the mild to moderate range of malnutrition.113 Poor sanitation, early weaning of breast-fed infants, use of overdiluted commercial formulas, and infection (measles, malaria, pneumonia, HIV, and diarrheal disease) are major risk factors for PEM.
PEM is a complication of some diseases, such as chronic fever, tuberculosis, malignancy, digestive and malabsorptive disorders, and psychogenic illness. Radiation therapy and chemotherapy also can contribute to PEM. Acute and chronic malnutrition is common in hospitalized children in the United States and Europe. 114
PATHOPHYSIOLOGY In kwashiorkor, the deficit of dietary amino acids reduces protein synthesis in all tissues. Physical growth and mental growth are stunted, and maintenance of minimal life processes is in jeopardy. The lack of sufficient plasma proteins, particularly albumin, causes systemic pressure changes that result in generalized edema. The volume of total body water and extracellular fluid increases, causing a substantial loss of potassium. The liver swells with stored fat because no hepatic proteins are synthesized to form and release lipoproteins. Pancreatic atrophy and fibrosis may be present. Kwashiorkor also causes malabsorption, reduced bone density, and impaired renal function. If the condition is not reversed, the prognosis is very poor.
Because the intake of all dietary nutrients is reduced to a minimum in marasmus, metabolic processes, including liver function, are preserved but growth is severely retarded. Caloric intake is too low to support protein synthesis for growth or the storage of fat. If more protein is needed than is ingested, muscle wasting occurs. Fat wasting and anemia are common and can be severe. The volume of total body water is high. Serum triglyceride and phospholipid levels increase with increasing severity of malnutrition, but other serum values, such as cholesterol, are normal or slightly reduced. High fasting phospholipid and triglyceride concentrations are predictive of a poor prognosis for these children.115 Severe vitamin A deficiency contributes to blindness.116
CLINICAL MANIFESTATIONS Retarded physical, mental, and psychologic development; muscle wasting; diarrhea; dermatosis; low hemoglobin; and infection characterize marasmus. The presence of subcutaneous fat, hepatomegaly, and fatty liver distinguishes kwashiorkor from marasmus. These manifestations are missing in marasmus because caloric intake is not sufficient to support fat synthesis and storage.117
EVALUATION AND TREATMENT Evaluation of PEM is based on nutritional history and clinical manifestations. Providing deficient nutrients resolves clinical symptoms in 4 to 6 weeks. Physical and mental retardation may not be reversible, however. Nutritional rehabilitation with appropriate environmental stimulation for infants and young children resolves or improve cerebral shrinkage, physical growth, and psychomotor development.118,119 Advances are being made in the local preparation of ready-to-use therapeutic food for both home- and community-based malnutrition management.120
Failure to Thrive
Failure to thrive (FTT) is the inadequate physical development of an infant or a child. It is manifested as a deceleration in weight gain, a low weight/height ratio, or a low weight/height/head circumference ratio. FTT is a common problem and can present at any time in childhood.
PATHOPHYSIOLOGY Organic FTT has a pathophysiologic cause, such as GERD, pyloric stenosis, gastroenteritis, malabsorption syndromes, infection by intestinal parasites, congenital anomalies, very low birth weight, or chronic diseases of major body systems. All these factors either reduce the availability of nutrients for maintenance and growth or increase nutrient requirements, particularly when there is chronic infection. A chronic disease or congenital anomaly that causes weakness or reduced stature can create developmental, psychosocial, and emotional problems for the child.121
Nonorganic FTT occurs in the absence of any gastrointestinal, endocrine, or other chronic diseases. It is usually associated with psychosocial deprivation, although behavior problems may contribute to its occurrence in the absence of maternal pathologic findings. Behavioral and psychosocial problems may be compounded by inadequate economic resources and lack of knowledge. Generally the problem in nonorganic FTT is ineffective nurturing by parents and primary caregivers. A variety of parental stressors may be involved and include the following:
• Lack of nurturance in the parents’ own childhood
• Inability to bond with the infant because of health or other problems
• Postpartum or maternal depression
• Family crisis, such as a death or marital problems
The first few postnatal months appear to be a sensitive period in the relationship between growth and mental development, suggesting a critical need for early diagnosis and aggressive interventions.122,123
CLINICAL MANIFESTATIONS Clinical manifestations of organic FTT are restricted growth accompanied by manifestations of the underlying disease. Manifestations of nonorganic FTT are restricted growth plus reduced energy level, reduced responsiveness and interaction with the environment, social isolation, spasticity or rigidity when held or touched, inability to make eye contact or smile, refusal to eat, and rejection of foods. Weight loss and decelerated growth are accompanied by restricted development in many areas. Nonorganic FTT is a complex syndrome involving psychosocial, emotional, and parent-child problems that compound the pathophysiologic abnormalities.124,125 Children with primarily organic FTT have been found to have lower developmental skills, and their parents have been found to have higher emotional distress. Infant stress, vomiting, feeding disorders, and psychosocial factors have been noted in children with organic and inorganic FTT making the distinction between the two complex.126
EVALUATION AND TREATMENT Failure to thrive is suggested if a child falls below the third percentile on the growth curve or is falling off a previously established growth curve. Organic FTT is manifested in infancy by weight, height, and head circumference growth that may be parallel to but below the normal ranges. If no genetic, endocrine, or other systemic disorder is identified and if the physical and laboratory examinations show no abnormalities other than delayed growth, an environmental cause is indicated.
Hospital admission is recommended if the diagnosis is unclear or the child is in nutritional or emotional jeopardy. Eating patterns, food preferences, caloric intake, and family interactions can be assessed during the hospital stay. If the cause is environmental, the hospitalized child with FTT usually begins to gain weight.
If an organic problem has been identified, management of FTT consists of treating the cause. Management of nonorganic FTT involves the immediate total care of the child and measures to address (1) the psychosocial and emotional problems of the caregivers and (2) parent-child interactions. Counseling, parental modeling, and long-term family support are sometimes required.127,128 Clinical manifestations of organic FTT have inorganic components in the majority of children. The most successful interventions not only treat the underlying organic cause but also address assisting parents with feeding practices and management of psychosocial symptoms.129,130
Necrotizing Enterocolitis
Necrotizing enterocolitis (NEC) is an ischemic, inflammatory condition of the bowel that causes necrosis, perforation, and death if untreated. It is the most common gastrointestinal emergency in the newborn. The overall mortality rate is between 12% and 30% and is higher for infants requiring surgery.131 The incidence of NEC is increasing, causing 1500 to 2000 infant deaths every year in the United States.132 Premature very-low-birth-weight infants are the most likely victims; it rarely occurs in full-term infants, but when it does it usually is a result of other complications.133 The risk of NEC decreases as the gastrointestinal tract matures.
PATHOPHYSIOLOGY The exact etiology of NEC is unclear. Multiple factors probably contribute to the development of NEC including premature birth, immature immunity, immature intestinal motility and barrier function, abnormal bacterial colonization, infections, maternal age greater than 35 years, perinatal stress, effects of medications, feeding practices, and genetic predisposition.131,134 The intestinal mucosal barrier in premature infants is scanty, which delays digestion. Motility is slower, allowing for the accumulation of noxious substances that damage the intestine and increase the risk for infection. Immaturity of the intestinal mechanical and biochemical barrier contributes to increased permeability resulting in translocation of bacteria and other substances, causing injury, inflammation, and development of systemic inflammatory disease.135 Preterm infants also may have immature intestinal innate immunity and an unfavorable balance between commensal and pathogenic bacteria, promoting intestinal inflammation and release of proinflammatory mediators.136
Reduced mucosal blood flow leading to hypoxic injury to intestinal mucosa is thought to be a cause. Normally there is a balance between vasodilator and vasoconstrictor inputs with a tendency toward vasodilation because of the copious production of endothelium-derived nitric oxide. With endothelial injury the balance is shifted toward endothelin-1–mediated vasoconstriction leading to ischemia, injury, and inflammation.137 This injury allows bacterial invasion of the bowel wall and release of inflammatory mediators. Accumulation of gas in the mucosa and submucosa also can lead to ischemic inflammation and necrosis of intestinal segments. The terminal ileum and proximal colon are most often involved.138
CLINICAL MANIFESTATIONS Manifestations of NEC usually appear within 2 weeks of birth, with earlier symptoms in full-term infants (5.3 days compared with 15.3 days in premature infants).139 They range from mild abdominal distention to bowel perforation, sepsis, and death. Abdominal pain, unstable temperature, bradycardia, and apnea are nonspecific signs. Affected infants have abdominal distention, occult or grossly bloody stools, retained gastric contents, and septicemia with elevated white blood cell and falling platelet counts. The more premature the infant, the greater the incidence of NEC and related diseases of prematurity, such as respiratory distress syndrome and immunocompromise.
EVALUATION AND TREATMENT Diagnosis is based on clinical manifestations, laboratory results, and plain films of the abdomen that show gas accumulation in the intestine. Preventive strategies include breast milk feeding, judicious fluid management to prevent patent ductus arteriosus, administration of arginine and glutamine supplements and epidermal growth facor to support intestinal epithelial cell growth, and enteral probiotics to support commensal gut bacteria.140,141 Treatments include cessation of feeding, gastric suction to decompress the intestines, fluid and electrolyte maintenance, and administration of antibiotics to control sepsis.142 Surgical resection is the treatment of choice for intestinal perforation, and the mortality rate is high. For very ill infants weighing less than 1000 g, peritoneal drainage may used as an adjunct to laparotomy.143 Following treatment of NEC, infants treated by medical and surgical management are at risk for intestinal obstruction related to the development of strictures.144 Infants who have extensive resection of necrotic bowel may develop short-bowel syndrome, requiring chronic total parenteral nutrition. Intestinal lengthening procedures and intestinal transplantation are available as a lifesaving option for these children.145
Diarrhea
Diarrhea is a common gastrointestinal problem during infancy and early childhood, and infectious diarrhea is the leading cause of death worldwide, primarily in young children in developing countries. Severe diarrhea occurs one to three times during the first 3 years of life. Most episodes are self-limiting and resolve within 72 hours.
The pathophysiologic mechanisms of diarrhea in children are similar to those for adults described in Chapter 39. Prolonged diarrhea is more dangerous in children, however, because they have much smaller fluid reserves than adults. Therefore, dehydration can develop rapidly if any disturbance increases fluid secretion into the gastrointestinal lumen (secretory diarrhea), draws fluid into the lumen by osmosis (osmotic diarrhea), or prevents fluid absorption in the intestine.
Infant diarrhea is of special concern because its cause may be a congenital or metabolic anomaly. Infants have low fluid reserves and relatively rapid peristalsis and metabolism. Therefore, the danger of dehydration is great.
Common causes of acute diarrhea in infants include infections, congenital aganglionic megacolon, milk-protein allergies, and NEC. Less common causes are adrenogenital syndrome, impaired chloride-bicarbonate exchange, congenital lactase deficiency, glucose-galactose malabsorption, and sucrase-isomaltase deficiency.
Acute Diarrhea in Children
Infectious diarrhea in newborns is usually associated with nursery epidemics involving such pathogens as Escherichia coli, Klebsiella, staphylococci, Salmonella, and Shigella. Diarrhea caused by these agents has a rapid onset, and acidosis and shock can occur quickly. Clostridium difficile, often associated with previous antibiotic therapy, can cause acute, profuse, watery diarrhea and symptoms of colitis.146 Viral causes of diarrhea include rotaviruses, noroviruses, astroviruses, and certain types of adenoviruses. Causes of diarrhea in young children are unknown in about 40% of cases.147 True milk-protein allergy, which is uncommon, causes bloody, explosive stools after the introduction of milk into the diet.
Acute diarrhea in children is almost synonymous with acute viral or bacterial gastroenteritis. Viral gastroenteritis tends to be self-limiting. Bacterial gastroenteritis is treated with antibiotics if the causal pathogen can be identified. Other causes of acute diarrhea in the older child include antibiotic therapy, appendicitis, chemotherapy, inflammatory bowel disease, parasitic infestation, and ingestion of toxic substances.
Rotavirus, the leading cause of severe diarrhea in infants and young children, invades enterocytes of the intestinal mucosa and releases an enterotoxin that damages these cells. Damage decreases viable absorptive surface causing an imbalance of secretion and absorption and increases motility resulting in diarrhea and dehydration. Recovery from mucosal damage may take several weeks. Rotavirus is transmitted by the fecal-oral route among humans.148 Two new live, oral, attenuated rotavirus vaccines were licensed in 2006 and appear safe to use for prevention of rotavirus infection without any increased risk of intussusception as occurred with the first rotavirus vaccine.149 By 5 years of age most children have developed resistance to rotavirus infection.
Chronic Diarrhea in Children
Children with acute gastroenteritis often remain mildly symptomatic for up to 4 weeks; therefore, diarrhea that persists longer than 4 weeks is considered to be chronic. Children with chronic diarrhea can be divided into two groups: (1) otherwise well children whose growth is normal and (2) ill children whose growth is restricted. Causes of chronic diarrhea in the first group include abnormal colonic motility, lactose intolerance, encopresis, parasitic infestation, and antibiotic use.150,151 Chronic diarrhea in the second group is usually caused by a disease that impairs absorption.
Chronic Nonspecific Diarrhea: Chronic nonspecific diarrhea of childhood is a condition in which uncoordinated colonic motility causes forceful expulsion of feces in otherwise healthy children. It affects children between 1 and 5 years of age. Apparently, the lower sigmoid colon remains in a tonically contracted state. Defecation occurs when pressure in the upper sigmoid colon and distal descending colon becomes great enough to force feces through the nonmotile, contracted segment. Prostaglandin synthesis is increased in the jejunum, and there are bile salts in the stools that may act as secretagogues.
In some instances there is a family history of bowel complaints. As an infant the child is likely to have experienced colic and diarrhea associated with teething and immunizations. In more than 90% of cases, chronic nonspecific diarrhea resolves by 40 to 50 months of age. The cure often accompanies toilet training. Many children with chronic nonspecific diarrhea develop irritable bowel syndrome (which is also called mucous colitis) as adults.152 Children with chronic nonspecific diarrhea usually do well with normal food and fluid intake with a balance of fluid, fiber, fat, and fruit juices without high amounts of sorbitol (sorbitol content is higher in pear, apple, and prune juices and may cause diarrhea in children 1 to 3 years of age).
Primary Lactose Intolerance: Lactose intolerance is the inability to digest milk sugar. It is caused by inadequate production of lactase and is a common cause of diarrhea in children, particularly nonwhite children, younger than 7 years of age. The malabsorption of lactose results in osmotic diarrhea, in which fluids move by osmosis from the vascular compartment into the intestinal lumen. The undigested sugar is acted on by the colonic bacteria, and intestinal gas is produced. The diarrhea is accompanied by abdominal pain, bloating, and flatulence. Diagnosis includes elimination of dietary lactose or hydrogen breath testing. Treatment consists of lactase-treated dairy products, lactase supplements, or reducing dairy product consumption. Other sources of dietary calcium or supplements need to be provided if dairy products are eliminated. Some children can tolerate lactose in fermented forms, such as cheese and yogurt. Complete restriction of lactose-containing foods is rarely necessary in young infants.153
DISORDERS OF THE LIVER
Disorders of Biliary Metabolism and Transport
Neonatal Jaundice
Physiologic jaundice of the newborn is usually a transient, benign icterus that occurs during the first week of life in otherwise healthy full-term infants. Physiologic jaundice is caused by mild unconjugated (indirect-reacting) hyperbilirubinemia. Total serum bilirubin greater than 20 mg/dl or an indirect bilirubin greater than 15 mg/dl is considered pathologic jaundice. Risk factors include fetal-maternal blood type incompatibility (ABO and Rh incompatibility), prematurity, exclusive breast-feeding, maternal age greater than or equal to 25 years, male sex, delayed meconium passage, and excessive birth trauma such as bruising or cephalhematomas.154 Hyperbilirubinemia is a risk with early hospital discharge (within 48 hours) because hyperbilirubinemia may not be evident prior to this time.155
PATHOPHYSIOLOGY Physiologic jaundice results from the complex interaction of factors that cause (1) increased bilirubin production (e.g., hemolysis), (2) impaired hepatic uptake or excretion of unconjugated bilirubin, and (3) delayed maturation of liver conjugating mechanisms.155 Unconjugated bilirubin is lipid soluble, bound to albumin in the blood, and in the free form readily crosses the blood-brain barrier in infants. Chronic bilirubin encephalopathy (kernicterus) is caused by the deposition of toxic, unconjugated bilirubin in brain cells and usually does not occur in healthy full-term infants. Elevated conjugated bilirubin in a sign of underlying disease. A late rising indirect bilirubin level also may be a manifestation of glucose-6-phosphate dehydrogenase deficiency, a hereditary X-linked genetic defect.156,157 Elevated unconjugated bilirubin levels also can cause hemolysis, further increasing neonatal jaundice.158
CLINICAL MANIFESTATIONS Physiologic jaundice develops during the second or third day after birth and usually subsides in 1 to 2 weeks in full-term infants and 2 to 4 weeks in premature infants. After this, increasing bilirubin values and persistent jaundice indicate pathologic hyperbilirubinemia. Manifestations include yellowing skin, dark urine, light-colored stools, and weight loss. Premature infants with respiratory distress, acidosis, or sepsis are at greater risk for encephalopathy. The resulting disabilities include athetoid cerebral palsy and speech and hearing impairment.159,160
EVALUATION AND TREATMENT Total and direct (conjugated) bilirubin levels are monitored and the bilirubin/albumin ratio is being evaluated.161, 161a Pathologic jaundice should be suspected with serum bilirubin values that increase greater than 5 mg/dl per day, persistent jaundice (greater than 7 to 10 days in the full-term infant), or conjugated bilirubin greater than 2 mg/dl. Other causes of jaundice must be eliminated to confirm physiologic jaundice. Treatment depends on the degree of hyperbilirubinemia. Physiologic jaundice is usually treated by phototherapy (ultraviolet light) with good eye protection.162 Pathologic jaundice requires an exchange transfusion and treatment of the underlying cause.
Biliary Atresia
Biliary atresia is a rare congenital malformation characterized by the absence or obstruction of intrahepatic or extrahepatic bile ducts. Extrahepatic ducts may end in a blind pouch. The cause of the intrauterine injury to the ducts is not clear, but is thought to be related to a chromosomal abnormality or active agents, such as infection or drugs or an autoimmune response.163 The disease expression is a continuum in which the principal process is one of bile duct destruction. The points of destruction are influenced by the stage of intrauterine development in which injury occurs.161
The atresia or obstruction of the bile ducts leads to plugging, inflammation, and fibrosis of the bile canaliculi and extrahepatic biliary tree. Progressive obstruction may lead to biliary cirrhosis (see Chapter 39), portal hypertension, or liver failure.164
Jaundice is the primary clinical manifestation of biliary atresia. Other signs are hepatomegaly and acholic (clay-colored) stools. Fat absorption is impaired for lack of bile salts, and the infant may fail to gain weight. Cirrhosis and liver failure lead to death within 2 years if untreated.
Early diagnosis of biliary atresia is mandatory and is based on clinical manifestations and liver biopsy. Liver function test results are abnormal. Serum transaminase and alkaline phosphatase values are elevated, and conjugated (direct) serum bilirubin levels rise progressively.
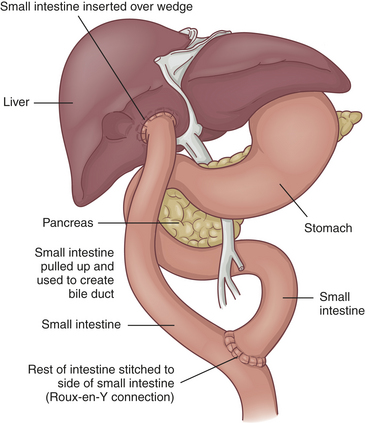
Extrahepatic atresia can be relieved by surgical drainage and correction in approximately 10% of cases. Some infants benefit from the Kasai procedure, in which a hepatic duct remnant is anastomosed to the jejunum or a jejunal segment is anastomosed to the porta hepatis if the patent hepatic duct remnant is not available (Figure 40-8). Even with initial restoration of bile flow, however, fibrosis and obliteration of intrahepatic bile ducts continue and cirrhosis results. Liver transplantation is the long-term therapy for biliary atresia. Approximately 40% of children with biliary atresia are immediate candidates for transplantation. Approximately 90% of children transplanted for biliary atresia become long-term survivors with good physical and mental development.165 The use of reduced and split livers from living, related donors has increased the number of children who survive after transplantation.166
Inflammatory Disorders
The pathophysiology of viral and fulminant hepatitis is described in Chapter 39.
Hepatitis A: Approximately one third to one half of the reported cases of hepatitis A virus (HAV) occur in children.167 Incidence is highest among young children of preschool age. Outbreaks tend to occur in daycare centers with large numbers of children who are not toilet trained and staff members who practice poor handwashing techniques.168 HAV replicates in the liver and is excreted through the biliary system into the stool. HAV in young children is usually mild and asymptomatic. Clinical manifestations, however, may include nausea, vomiting, and diarrhea. Because jaundice is absent, infected children appear to have the “flu.” Almost all children recover from hepatitis A without residual liver damage.169 Vaccination for hepatitis A should begin at 12 to 23 months of age.170
Hepatitis B: Infants of mothers who are chronic hepatitis B surface antigen (HBsAg) carriers, hemophiliacs who receive frequent blood transfusions, children who abuse parenteral drugs, and children who live in institutions for the mentally retarded are all at risk for hepatitis B virus (HBV) infection. Of newborns infected by their mothers, 90% develop chronic hepatitis and become carriers. The risk of chronic hepatitis is more than 25% to 90% for children younger than 6 years who contract HBV; HBV leads to chronic hepatitis in only 5% to 10% of cases among adults.171 Chronic hepatitis may develop more often in young children because of their immature immune systems. Infected infants and children are at risk for cirrhosis and hepatocellular carcinoma. The most serious consequence of HBV infection is fulminant hepatitis, which occurs in 1% of cases. Hepatitis D infection (HDV) depends on active infection with HBV. There is evidence that the risk of fulminant hepatitis is higher in individuals with combined infection of HBV and HDV than in those with HBV infection alone.172 The most effective approach to the treatment of hepatitis B is prevention. The American Academy of Pediatrics has added immunization for hepatitis B to those immunizations recommended for infants. Efforts are under way to immunize children and adolescents who did not receive this series as infants.
Hepatitis C: Hepatitis C virus (HCV) in children is most commonly transmitted vertically and is enhanced with maternal coinfection with HIV.173 It also is transmitted with blood transfusions. Between 10% and 50% of affected children develop chronic liver disease. Antivirals are effective in the treatment of both hepatitis B and hepatitis C.174
Chronic Hepatitis: The cause of chronic hepatitis is unknown in most cases, but an autoimmune mechanism is suggested because inflammatory findings are commonly seen in biopsy specimens of the liver. Manifestations of chronic hepatitis include malaise, anorexia, fever, gastrointestinal bleeding, hepatomegaly, edema, and transient joint pain. Serum transaminase and bilirubin levels are elevated. There may be evidence of impairment of synthetic functions of the liver: prolonged prothrombin time and hypoalbuminemia. Diagnosis is based on the clinical manifestations and liver biopsy.
Symptoms are often absent in children. Treatment has variable efficacy and differs by the causative pathogen. The goals are to suppress the virus and prevent cirrhosis and hepatocellular carcinoma. Treatment includes interferon-alpha, ribavirin, and lamivudine.175
Cirrhosis
Cirrhosis is the excessive formation of fibrous tissue in response to inflammation and tissue damage (see Chapter 39). Most forms of chronic liver diseases in children can progress to cirrhosis, but they seldom do. Nonalcoholic fatty liver disease (steatohepatitis) is increasing in children in correlation with an increase in childhood obesity and it can progress to cirrhosis and liver cancer176 (see What’s New? Childhood Obesity and Nonalcoholic Fatty Liver Disease).
The complications of cirrhosis in children are the same as those in adults: portal hypertension, the opening of collateral vessels between the portal and systemic veins, and varices. In addition, children with cirrhosis experience growth failure caused by nutritional deficits and developmental delay, particularly in gross motor function because of ascites and weakness. The cause of cirrhosis may influence its severity and course. Some types of cirrhosis can be stabilized if the cause is identified and treated early.
Portal Hypertension
The two basic causes of portal hypertension in children are (1) increased resistance to blood flow within the portal system and (2) increased volume of portal blood flow. Increased resistance to flow can occur anywhere in the portal circulatory system. Portal hypertension can accompany cirrhosis, intra-abdominal infections, portal vein thrombosis, congenital anomalies of the portal vein, and congenital hepatic fibrosis.
Types
Extrahepatic Portal Hypertension: Extrahepatic (prehepatic) portal venous obstruction causes 50% to 70% of extrahepatic portal hypertension in children. For at least half of these children, no specific cause can be found. Obstruction is almost always in the portal vein and is usually caused by thrombosis. Umbilical infection with or without a history of catheterization of the umbilical vein may be a cause in neonates. Portal vein thrombosis can occur as a complication of intra-abdominal infections, pancreatitis, and blunt abdominal trauma. It also has been associated
with neonatal dehydration, inflammatory bowel disease, and hypercoagulable states, such as protein C and protein S deficiencies.161 The liver is usually normal in cases of extrahepatic portal hypertension.
Intrahepatic Portal Hypertension: Cirrhosis is the primary cause of intrahepatic portal hypertension. The most common finding is fibrosis, which increases resistance to portal blood flow by constricting and reducing the compliance of the hepatic sinusoids.
CLINICAL MANIFESTATIONS The clinical manifestations of portal hypertension are splenomegaly, upper gastrointestinal variceal bleeding, ascites, and hepatic encephalopathy. Severe liver disease is characterized by hypoalbuminemia, prolonged prothrombin times, hyperbilirubinemia, electrolyte imbalance, and hypoglycemia. Splenomegaly is the most common sign of portal hypertension in children. The spleen may be firm or hard, depending on the duration of portal hypertension. Hematemesis, possibly associated with abdominal pain, often accompanies sudden pallor. Melena is observed, either at the time of hematemesis or soon afterward. In children most episodes of gastrointestinal bleeding are caused by rupture of esophageal varices. Clotting abnormalities caused by altered liver function promote the bleeding. If plasma volume is increased, esophageal varices readily rupture during activities, such as coughing, that increase blood pressure. Acetylsalicylic acid (aspirin), which should not be administered to children, can trigger bleeding, but its exact mechanisms of action are not known. Severe bleeding episodes can cause hypovolemic shock and death. Symptoms of ascites include weight gain, protruding abdomen, and reduced tidal volumes if the ascites is severe. Hepatic encephalopathy in children can be acute or chronic. Acute encephalopathy is characterized by major disorders of consciousness, which may progress to coma. This may follow an acute episode of variceal bleeding as the impaired liver attempts to metabolize the large protein (nitrogenous) load from the blood. Chronic, or minimal, encephalopathy is characterized by emotional or psychiatric disorders, decreased intellectual functioning, personality disorders caused by minimal brain dysfunction, and spatial disorientation.
EVALUATION AND TREATMENT Assessment of portal hypertension in children must be thorough because the cause dictates the management. The objectives of the clinical investigation are to locate the site of the venous block and identify the disease responsible for the portal hypertension. Thorough physical examination, laboratory tests of liver function, imaging procedures, and biopsy may be included in the diagnostic evaluation (see Chapter 38). Sclerotherapy is the initial treatment of choice for severe esophageal varices in children.177
The indications for surgical shunting include gastrointestinal hemorrhage not responsive to sclerotherapy. Surgical venous shunts rarely have been performed on small children because of the high failure rate secondary to vessel occlusion, but they may be an alternative in older children, with some success.161,178 Surgical shunts are no longer a contraindication for subsequent liver transplantation, although the shunt does make the transplantation technically more difficult. The transjugular intrahepatic portosystemic shunt (TIPS) procedure is performed for some children. This is a therapeutic option in which a stent is placed between the right hepatic vein and the right or left portal vein to allow shunting of blood without surgical intervention.179
The outcome of portal hypertension depends almost entirely on its cause. Children with extrahepatic disease are expected to recover with little morbidity. For children with intrahepatic disease, the prognosis varies.
Metabolic Disorders
More than 5000 genetically determined metabolic pathways have been identified in liver tissue. The earliest possible identification of metabolic disorders is essential because (1) early treatment may prevent permanent damage to vital organs, such as the liver or brain; (2) precise genetic counseling may be possible with prenatal diagnosis; and (3) complications can be minimized, even if cure is not possible. Galactosemia,180 fructosemia, and Wilson disease are rare treatable metabolic disorders that have hepatic clinical manifestations. These disorders are summarized in Table 40-2 and Wilson disease is discussed next.

Wilson Disease
Wilson disease (hepatolenticular degeneration) is an autosomal recessive defect of copper metabolism that causes toxic amounts of copper to accumulate in the liver, brain, kidneys, and corneas. The gene ATP7B is localized on chromosome 13 and encodes copper-transporting P-type adenosine triphosphatase (ATPase) membrane-spanning protein. It is highly expressed in the liver, kidney, and placenta and is expressed in lower levels in the brain, heart, muscle, and pancreas.181 This defect in the uptake and excretion of copper by hepatocytes is an important cause of progressive liver disease in children and young adults. Wilson disease is very rare, with an incidence of 1 in 30,000 live births worldwide.182 Between 1 in 200 and 1 in 500 persons are carriers.161
PATHOPHYSIOLOGY Two major abnormalities in copper metabolism have been identified: (1) diminished biliary excretion of copper and (2) failure to insert copper into ceruloplasmin (a glycoprotein that transports copper in the blood). A positive copper balance is present from birth in children with Wilson disease, despite increased excretion of copper in the urine. Copper toxicity with accumulation in the liver and brain is the major abnormality. Excesses of copper generate free radicals that disrupt cellular organelles, deoxyribonucleic acid (DNA), microtubules, enzymes, and proteins. Copper overload is related to impaired biliary excretion of copper and may be related to failure of hepatocyte lysosomes to eliminate copper by exocytosis.183
Early in the disease, intestinal absorption of copper is normal, as is hepatic clearance of albumin-bound absorbed copper. As copper-binding proteins in the liver become saturated, hepatic uptake of copper diminishes, with elevated serum copper levels and biochemical and clinical evidence of liver damage caused by copper accumulation. In later stages of the disease copper accumulates in extrahepatic tissues, including the eyes, brain, and kidneys.
When cerebral copper-binding proteins become saturated, a characteristic pattern of brain damage develops, particularly in the basal ganglia. Neural effects include intention tremor, unsteady gait, dystonia, and behavioral changes. Manifestations of renal tubular injury usually appear simultaneously. The uptake of copper by red blood cells is thought to cause hemolytic anemia, a condition sometimes seen early in the clinical course of Wilson disease.
CLINICAL MANIFESTATIONS The clinical manifestations of Wilson disease may begin as young as 4 years of age, when control mechanisms responsible for copper homeostasis and biliary excretion should have matured. The mean age at diagnosis in one large study was 15.5 years.184
The classic clinical presentation of Wilson disease is a triad of neuromuscular abnormalities, intention tremors, dysarthria (indistinct speech), and dystonia (disordered muscular tonicity): (1) Kayser-Fleischer rings (accumulation of copper in the limbus of the cornea, causing a greenish-yellow ring), (2) cirrhosis associated with elevated serum copper, and (3) low ceruloplasmin levels.185 Initial symptoms vary from malaise and abdominal pain to jaundice. Changes in mental and motor performance may develop at age 6 years or into adult life. The earliest signs of liver involvement include enlargement of the liver and spleen, jaundice, and anorexia. Edema and ascites may develop suddenly, or gastrointestinal hemorrhage may be the initial sign of the disease. Occasionally Wilson disease begins with a hemolytic crisis caused by the toxic effects of copper on the red blood cells. Cirrhosis develops in all untreated cases. Copper deposition in the kidneys causes a proximal renal tubular defect that results in losses of glucose, amino acids, phosphate, and uric acid in the urine and renal tubular acidosis. All untreated individuals will develop behavioral or psychiatric disorders.
EVALUATION AND TREATMENT Because Wilson disease is rare, it may not be diagnosed until clinical manifestations develop in older childhood or adulthood. Laboratory tests detect a serum ceruloplasmin concentration less than 30 mg/dl. Serum copper values may be normal or high, and urine copper values are elevated. Liver biopsy is used to assess structural changes and measure copper concentrations. The goal of therapy is to decrease copper accumulation by decreasing intestinal absorption and increasing renal excretion. Medical treatments include D-penicillamine, trientine, zinc, and ammonium tetrathiomolybdate.186 Copper intake is reduced by eliminating organ meats, nuts, legumes, shellfish, and chocolate from the diet. Physiotherapy may accelerate the recovery of gait and muscular coordination. Liver transplantation is the sole resolutive therapy for Wilson disease and is the treatment of choice for persons who develop fulminant hepatic failure or end-stage cirrhosis.187 Children with untreated Wilson disease die of neural, hepatic, renal, or hematologic complications.
Acute diarrhea 1530
Biliary atresia 1531
Chronic diarrhea 1530
Chronic hepatitis 1532
Chronic nonspecific diarrhea 1530
Cirrhosis 1532
Cleft lip (harelip) 1516
Cleft palate 1516
Congenital aganglionic megacolon (Hirschsprung disease) 1521
Cystic fibrosis (CF) 1524
Diarrhea 1530
Distal intestinal obstruction syndrome (DIOS) 1520
Eosinophilic esophagitis 1524
Esophageal atresia 1518
Extrahepatic (prehepatic) portal venous obstruction 1533
Failure to thrive (FTT) 1528
Gastroesophageal reflux disease (GERD) 1523
Glucose-6-phosphate dehydrogenase deficiency 1531
Gluten-sensitive enteropathy 1525
Hepatic encephalopathy 1533
Hepatitis A virus (HAV) 1532
Hepatitis B surface antigen (HBsAg) 1532
Hepatitis B virus (HBV) 1532
Hepatitis C virus (HCV) 1532
Imperforate anus 1522
Infant diarrhea 1530
Infectious diarrhea 1530
Intestinal malrotation 1519
Intrahepatic portal hypertension 1533
Intussusception 1522
Kernicterus 1531
Kwashiorkor 1528
Lactose intolerance 1531
Marasmus 1528
Meconium 1520
Meconium ileus 1520
Necrotizing enterocolitis (NEC) 1529
Nonorganic FTT 1528
Organic FTT 1528
Pathologic jaundice 1531
Periduodenal band 1519
Physiologic jaundice of the newborn 1531
Portal hypertension 1533
Protein energy malnutrition (PEM) 1528
Pyloric stenosis 1519
Rotavirus 1530
Splenomegaly 1533
Tracheoesophageal fistula (TEF) 1518
Wilson disease 1534
REFERENCES
1. Centers for Disease Control and Prevention. Improved national prevalence estimates for 18 selected major birth defects—United States, 1999-2001. MMWR. 2006;54(51,52):1301–1305.
2. Cooper, M.E., Tatay, J.S., Marazita, M.L. Asian oral-facial cleft birth prevalence. Cleft Palate Craniofac J. 2006;43(5):580–589.
3. Arosarena, O.A. Cleft lip and palate. Otolaryngol Clin North Am. 2007;4(1):27–60.
4. Carinci, F., et al. Human genetic factors in nonsyndromic cleft lip and palate: an update. Int J Pediatr Otorhinolaryngol. 2007;71(20):1509–1519.
5. Honein, M.A., et al. Maternal smoking, environmental tobacco smoke, and the risk of oral clefts. Epidemiology. 2007;18(2):226–330.
6. Shi, M., Wehby, G.L., Murray, J.C. Review on genetic variants and maternal smoking in the etiology or oral clefts and other birth defects. Birth Defects Res C Embryo Today. 2008;84(1):16–29.
7. Spilson, S.V., Kim, H.J., Chung, K.C. Association between maternal diabetes mellitus and newborn oral clefts. Ann Plast Surg. 2001;47(5):477–481.
8. Vieira, A.R. Unraveling human cleft lip and palate research. J Dent Res. 2008;87(2):119–125.
9. Zucchero, T.M., et al. Interferon regulating factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351(8):769–780.
10. Verkleij-Hagoort, A., et al. Hyperhomocysteinemia and MTHFR polymorphisms in association with orofacial clefts and congenital heart defects: a meta-analysis. Am J Med Genet A. 2007;143A(9):952–960.
11. da Silva Dalben, G., et al. Breastfeeding and sugar intake in babies with cleft lip and palate. Cleft Palate Craniofac J. 2003;40(1):84–87.
12. Redford-Badwal, D.A., Mabry, K., Frassinelli, J.D. Impact of cleft lip and/or palate on nutritional health and oral-motor development. Dent Clin North Am. 2003;47(2):305–317.
13. Turner, L., et al. The effects of lactation education and a prosthetic obturator appliance of feeding efficiency in infants with cleft lip and palate. Cleft Palate Craniofac J. 2001;38(5):519–524.
14. Ramos, G.A., et al. Diagnostic evaluation of the fetal face using 3-dimensional ultrasound. Ultrasound Q. 2008;24(4):215–253.
15. Weinfeld, A.B., et al. International trends in the treatment of cleft lip and palate. Clin Plas Surg. 2005;32(1):19–23.
16. Barillas, I., et al. Nasoalveolar molding improves long-term nasal symmetry in complete unilateral cleft lip-palate patients. Plast Reconst Surg. 2009;123(3):1002–1006.
17. Sheahan, P., et al. Incidence and outcome of middle ear disease in cleft lip and/or cleft palate. Int J Pediatr Otorhinolaryngol. 2003;67(7):785–793.
18. Aniansson, G., et al. Otitis media and feeding with breast milk of children with cleft palate. Scan J Plast Reconstr Surg Hand Surg. 2002;36(1):9–15.
19. Ahluwalia, M., et al. Dental caries, oral hygiene, and oral clearance in children with craniofacial disorders. J Dent Res. 2004;83(2):175–179.
20. Kirchberg, A., Treide, A., Hemprich, A. Investigation of caries prevalence in children with cleft lip, alveolus, and palate. J Craniomaxillofac Surg. 2004;32(4):216–219.
21. Achilidi, O., Grewal, H. Congenital anomalies of the esophagus. Otolaryngol Clin North Am. 2007;40(1):219–244. [viii].
22. Langer, J.C., et al. Prenatal diagnosis of esophageal atresia using sonography and magnetic resonance imaging. J Pediatr Surg. 2001;36(5):804–807.
23. Kovesi, T., Rubin, S. Long-term complications of congenital esophageal atreasia and/or tracheoesophageal fistula. Chest. 2004;126(3):915–925.
24. Spitz, L. Oesophageal atresia. Orphanet J Rare Dis. 2007;2:24.
25. Houben, C.H., Curry, H. Current status of prenatal diagnosis. operative management and outcome of esophageal atresia/tracheo-esophageal fistula, Prenat Diagn. 2008;28(7):667–675.
26. Hernanz-Schulman, M. Infantile hypertrophic pyloric stenosis. Radiology. 2003;227(2):319–331.
27. Phillips, J.D. Abdominal surgical emergencies. In Wyllie R., Hyams J., eds.: Pediatric gastrointestinal diseases, ed 2, Philadelphia: Saunders, 1999.
28. Armstrong, M. The child with a cognitive deficit. In McKinney E.S., et al, eds.: Maternal-child nursing, ed 2, St. Louis: Saunders, 2005.
29. Sams, C.A. The child with a gastrointestinal alteration. In McKinney E.S., et al, eds.: Maternal-child nursing, ed 2, St. Louis: Saunders, 2005.
30. Kliegman, R., et al. Nelson textbook of pediatrics, ed. 18. Philadelphia: Saunders; 2007.
31. Spinelli, C., et al. Muscle thickness in infantile hypertrophic pyloric stenosis. Pediatr Med Chir. 2003;25(2):148–150.
32. Shima, H., Puri, P. Increased expression of transforming growth factor-alpha in hypertrophic pyloric stenosis. Pediatr Surg Intern. 1999;15(3-4):198–200.
33. Blumer, S.L., et al. The vomiting neonate: a review of the ACR appropriateness criteria and ultrasound’s role in the work-up of such patients. Ultrasound Q. 2004;20(3):78–89.
34. Boneti, C., et al. Ultrasound as a diagnostic tool used by surgeons in pyloric stenosis. J Pediatr Surg. 2008;43(1):87091.
35. Aspelund, G., Langer, J.C. Current management of hypertrophic pyloric stenosis. Semin Pediatr Surg. 2007;16(1):27–33.
36. Sweeney, B., Surana, R., Puri, P. Jejunoileal atresia and associated malformations: correlation with the timing of in utero insult. J Pediatr Surg. 2001;36(5):774–776.
37. Durkin, E.T., et al. Age-related differences in diagnosis and morbidity of intestinal malrotation. J Am Coll Surg. 2008;206(4):658–663.
38. Cohen, Z., et al. How much of a misnomer is “asymptomatic” intestinal malrotation? Israel Med Assoc J. 2003;5(3):172–174.
39. Lai, H.C. Nutritional status of patients with cystic fibrosis with meconium ileus: a comparison with patients without meconium ileus and diagnosed early through neonatal screening. Pediatrics. 2000;105(1 Pt 1):53.
40. Blackman, S.M., et al. Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology. 2006;131(4):1030–1039.
41. McAlister, W.H., Kronemer, K.A. Emergency gastrointestinal radiology of the newborn. Radiol Clin North Am. 1996;34(4):819–844.
42. Kao, S.C., Franken, E.A., Jr. Nonoperative treatment of simple meconium ileus: a survey of the Society for Pediatric Radiology. Pediatr Radiol. 1995;25(2):97–100.
43. Evans, A.K., Fitzgerald, D.A., McKay, K.O. The impact of meconium ileus on the clinical course of children with cystic fibrosis. Eur Respir J. 2001;18(5):784–789.
44. Oliveira, M.C., et al. Effect of meconium ileus on the clinical prognosis of patients with cystic fibrosis. Braz J Med Biol Res. 2002;35(1):31–38.
45. Dray, X., et al. Distal intestinal obstruction syndrome in adults with cystic fibrosis. Clin Gastroenterol Hepatol. 2004;2(6):498–502.
46. Schibli, S., Dune, P.R., Tullis, E.D. Proper usage of pancreatic enzymes. Curr Opin Pulm Med. 2002;8(6):542–546.
47. Chaudry, G., et al. Abdominal manifestations of cystic fibrosis in children. Pediatr Radiol. 2006;36(3):233–240.
48. Hackam, D.J., et al. The influence of Down syndrome in the management and outcome of children with Hirschsprung disease. J Pediatr Surg. 2003;38(6):946–949.
49. Amiel, J., et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45(1):1–14.
50. Dasgupta, R., Langer, J.C. Hirschsprung disease. Curr Probl Surg. 2004;41(12):942–988.
51. Kessmann, J. Hirschsprung’s disease: diagnosis and management. Am Fam Physician. 2006;74(8):1319–1322.
52. de Lorijn, F., et al. Diagnostic test in Hirschsprung disease: a systematic review. J Pediatr Gastroenterol Nutr. 2006;42(5):496–505.
53. Khan, A.R., Vujanic, G.M., Huddart, S. The constipated child: how likely is Hirschsprung disease? Pediatr Surg Int. 2003;19(6):439–442.
54. de Lorijn, F., et al. Diagnostic tests in Hirschsprung disease: a systematic review. Pediatr Gastroenterol Nutr. 2006;42(5):496–505.
55. Bai, Y., et al. Long-term outcome and quality of life after the Swenson procedure for Hirschsprung disease. J Pediatr Surg. 2002;37(4):639–642.
56. Levitt, M.A., Pena, A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33.
57. Rintala, R.J. Fecal incontinence in anorectal malformations, neuropathy, and miscellaneous conditions. Semin Pediatr Surg. 2002;11(2):75–82.
58. Waseem, M., Rosenberg, H.K. Intussusception. Pediatr Emerg Care. 2008;24(11):793–800.
59. de Vries, S., Sleeboom, C., Aronson, D.C. Postoperative intussusception in children. Br J Surg. 1999;13(4):81–83.
60. Eggermont, E., De Boeck, K. Small-intestinal anomalies in cystic fibrosis patients. Eur J Pediatr. 1991;150(12):824–828.
61. Fischer, T.K., et al. Intussusception in early childhood: a cohort study of 1.7 million children. Pediatrics. 2004;114:782–785.
62. Blanch, A.J., Perel, S.B., Acworth, J.P. Paediatric intussusception: epidemiology and outcome. Emerg Med Australas. 2007;19(1):45–50.
63. Byrne, A.T., et al. The imaging of intussusception. Clin Radiol. 2005;60(3):412.
64. Kaiser, A.D., Applegate, K.E., Ladd, A.P. Current success in the treatment of intussusception in children. Surgery. 2007;142(4):469–477.
65. Navarro, O.M., Daneman, A., Chae, A. Intussusception: the use of delayed, repeated reduction attempts and the management of intussusception due to pathological lead points in pediatric patients. Am J Roentgenol. 2004;182(5):1169–1176.
66. Daneman, A., Navarro, O. Intussusception Part 1: a review of diagnostic approaches. Pediatr Radiol. 2003;33(2):79–85.
67. Kim, J.H. Ultrasound features of transient small bowel intussusception in pediatric patients. Korean J Radiol. 2004;5(3):178–184.
68. Bajaj, L., Roback, M.G. Postreduction management of intussusception in a children’s hospital emergency department. Pediatrics. 2003;112(6 Pt 1):1302–1307.
69. Crapko, M., et al. Role of extra-esophageal reflux in chronic otitis media with effusion. Laryngoscope. 2007;117(8):1419–1423.
70. Slocum, C., et al. Infant apnea and gastroesophageal reflux: a critical review and framework for further investigation. Curr Gastroenterol Rep. 2007;9(3):219–224.
71. Brodzicki, J., Trawinska-Bartnicka, M., Korzon, M. Frequency, consequences and pharmacological treatment of gastroesophageal reflux in children with cystic fibrosis. Med Sci Monit. 2002;8(7):529–537.
72. Campanozzi, A., et al. Impact of malnutrition on gastrointestinal disorders and gross motor abilities in children with cerebral palsy. Brain Dev. 2007;29(8):534.
73. Maggio, A.B., et al. Increased incidence of apparently life-threatening events due to supine position. Paediatr Perinat Epidemiol. 2006;20(6):491–496.
74. Tieder, J.S., et al. Variation in inpatient resource utilization and management of apparent life-threatening events. J Pediatr. 2008;152(5):629–635.
75. Bhat, R.Y., et al. Acid gastroesophageal reflux in convalescent preterm infants: effect of posture and relationship to apnea. Pediatr Res. 2007;62(5):620–623.
76. Magista, A.M., et al. Multichannel intraluminal impedance to detect relationship between gastroesophageal reflux and apnoea of prematurity. Dig Liver Dis. 2007;39(3):216–221.
77. Simanovsky, N., Buonomo, C., Nurko, S. The infant with chronic vomiting: the value of the upper GI series. Pediatr Radiol. 2002;32(8):549–550.
78. Spitz, L., McLeod, E. Gastroesophageal reflux. Semin Pediatr Surg. 2003;12(4):237–240.
79. Chang, F., Anderson, F. Clinical and pathological features of eosinophilic oesophagitis: a review. Pathology. 2008;40(1):3–8.
80. Ireland-Jenkin, K., et al. Oesophagitis in children: reflux or allergy? Pathology. 2008;40(2):188–195.
81. Chehade, M., et al. Esophageal subepithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2007;45(3):3119–3128.
82. Ciorba, A., et al. Gastroesophageal reflux and its possible role in the pathogenesis of upper aerodigestive tract disorders. Minerva Gastroenterol Dietol. 2007;53(2):171–180.
83. Rohen, R., Nurko, S. The importance of multichannel intraluminal impedence in the evaluation of children with persistent respiratory symptoms. Am J Gastroenterol. 2004;99(12):2452–2458.
84. Dalby, K., et al. Reproducibility of 24-hour combined multiple intraluminal impedance (MII) and pH measurements in infants and children. Evaluation of a diagnostic procedure for gastroesophageal reflux disease. Dig Dis Sci. 2007;52(9):219–265.
85. Omari, T. Gastroesophageal reflux in infants: can a simple left side positioning strategy help this diagnostic and therapeutic conundrum? Minerva Pediatr. 2008;60(2):193–200.
86. Rerksuppaphol, S., Barnes, G. Guidelines for the evaluation and treatment of gastroesophageal reflux in infants and children:recommendations of the North American Society for Pediatric Gastroenterology and Nutrition. J Pediatr Gastroenterol Nutr. 2002;35(4):583.
87. Arguin, A.L., Swartz, M.K. Gastroesophageal reflux in infants: a primary care perspective. Pediatr Nurs. 2004;30(1):45–51. [71].
88. Craig, W.R., et al. Metoclopramide, thickened feedings, and positioning for gastro-oesophageal reflux in children under two years. Cochrane Database Syst Rev. (4):2004. [CDC003502].
89. Lobe, T.E. The current role of laparoscopic surgery for gastroesophageal reflux disease in infants and children. Surg Endosc. 2007;21(2):167–174.
90. Konstan, M.W., et al. Ultrase MT12 and Ultrase MT20 in the treatment of exocrine pancreatic insufficiency in cystic fibrosis: safety and efficiency. Aliment Pharmacol Ther. 2004;20(11-12):1365–1371.
91. Borowitz, D. Update on the evaluation of pancreatic exocrine status in cystic fibrosis. Curr Opin Pulm Med. 2005;11(6):524–527.
92. Dodge, J.A., Turck, D. Cystic fibrosis: nutritional consequences and management. Best Pract Res Clin Gastroenterol. 2006;20(3):531–546.
93. Stallings, V.A., et al. Evidence-based recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008;108(5):832–839.
94. Losowsky, M.S. A history of celiac disease. Dig Dis. 2008;26(2):112–120.
95. Rodrigues, A.F., Jenkins, H.R. Investigation and management of celiac disease. Arch Dis Child. 2008;93(3):251–254.
96. Di Sabatino, A., Corazza, G.R. Coeliac disease. Lancet. 2009;373(9673):1480–1493.
97. Abdulkarim, A.S., Murray, J.A. Review article: the diagnosis of celiac disease. Aliment Pharmacol Ther. 2003;17(8):987–995.
98. Alaedini, A., Green, P.H. Autoantibodies in celiac disease. Autoimmunity. 2008;41(1):19–26.
99. Gasbarrini, G., et al. Celiac disease in the 21st century: issues of under- and over-diagnosis. Int J Immunopathol Pharmaco. 2009;22(1):1–7.
100. Rossi, T. Celiac disease. Adolesc Med Clin. 2004;15(1):91–103.
101. Telega, G., Bennet, T.R., Werlin, S. Emerging new clinical patterns in the presentation of celiac disease. Arch Pediatr Adolesc Med. 2008;162(2):164–168.
102. Catassi, C., Fasano, A. Celiac disease as a cause of growth retardation in children. Curr Opin Pediatr. 2004;16(4):445–449.
103. Eliakim, R., Sherer, D.M. Celiac disease: fertility and pregnancy. Gynecol Obstet Invest. 2001;51(1):3–7.
104. Mora, S. Celiac disease in children: impact on bone health. Rev Endocr Metab Disord. 2008;9(2):123–130.
105. Freeman, H.J. Free perforation due to intestinal lymphoma in biopsy-defined or suspected celiac disease. J Clin Gastroenterol. 2003;37(4):299–302.
106. Scherer, K.R. Celiac disease. Drugs Today (Barc). 2008;44(1):75–88.
107. Ojetti, V., et al. Regression of lactose malabsorption in celiac patients after receiving a gluten-free diet. Scand J Gastroenterol. 2008;43(2):174–177.
108. Guandalini, S. The influence of gluten: weaning recommendations for healthy children and children at risk for celiac disease. Nestle Nutr Workshop Ser Pediatr Program. 2007;60:139–151.
109. Bernardo, D., et al. Decreased circulating iNKT cell numbers in refractory celiac disease. Clin Immunol. 2008;126(2):172–179.
110. Saloojee, H., et al. What’s new: investigating risk factors for severe childhood malnutrition in a high HIV prevalence South African setting. Scand J Public Health Suppl. 2007;69:96–106.
111. Bern, C., et al. Assessment of potential indicators for protein-energy malnutrition in the algorithm for integrated management of childhood illness. Bull WHO. 1997;75(Suppl 1):87–96.
112. World Health Organization: Water related diseases: malnutrition. Available at www.who.int/water_sanitation_health/diseases/malnutrition/en.
113. Pelletier, D.L. The relationship between child anthropometry and mortality in developing countries: implications for policy, programs and future research. J Nutr. 1994;123(Suppl 10):2047S–2081S.
114. Joosten, K.F., Hulst, J.M. Prevalence of malnutrition in pediatric hospial patients. Curr Opin Pediatr. 2008;20(5):590–596.
115. Ogumkeye, O.O., Ighogboja, I.S. Increase in total serum triglyceride and phospholipids in kwashiorkor. Ann Trop Pediatr. 1992;12(4):463–466.
116. Maida, J.M., Mathers, K., Alley, C.L. Pediatric ophthalmology in the developing world. Curr Opin Ophthalmol. 2008;19(5):403–408.
117. Latham, M.C. The dermatosis of kwashiorkor in young children. Semin Dermatol. 1991;10(4):270–272.
118. Ahmed, T., et al. Management of severe malnutrition and diarrhea. Indian J Pediatr. 2001;68(1):45–51.
119. Greco, L., et al. Effect of a low-cost food on the recovery and death rate of malnourished children. J Pediatr Gastroenterol Nutr. 2006;43(4):512–517.
120. Linneman, Z., et al. A large-scale operational study of home-based therapy with ready to use therapeutic food in childhood malnutrition in Malawi. Matern Child Nutr. 2007;3(3):206–215.
121. Steward, D.K., Moser, D.K., Ryan-Wenger, N.A. Behavioral characteristics of infants with failure to thrive. J Pediatr Nurs. 2003;16(3):162–171.
122. Stewart, R.C. Maternal depression and infant growth: a review of recent evidence. Matern Child Nutr. 2007;3(2):94–107.
123. Krugman, S.D., Dubowitz, H. Failure to thrive. Am Fam Physician. 2003;68(5):879–884.
124. Piazza, C.C., et al. Functional analysis of inappropriate mealtime behaviors. J Appl Behav Anal. 2003;36(2):187–204.
125. Steward, D.K. Behavioral characteristics of infants with nonorganic failure to thrive during a play interaction. MCN Am J Matern Child Nurs. 2001;26(2):79–85.
126. Bergman, P., Graham, J. An approach to “failure to thrive,”. Aust Fam Physician. 2005;34(9):725–729.
127. Marino, R., Weinman, M.L., Soudelier, K. Social work intervention and failure to thrive in infants and children. Health Soc Work. 2001;26(2):90–97.
128. Robinson, J.R., Drotar, D., Boutry, M. Problem-solving abilities among mothers of infants with failure to thrive. J Pediatr Psychol. 2001;26(1):21–32.
129. Black, M.M., et al. Early intervention and recovery among children with failure to thrive: follow-up at age 8. Pediatrics. 2007;120(1):59–69.
130. Stewart, R.C. Maternal depression and infant growth: a review of recent evidence. Matern Child Nutr. 2007;3(2):94–107.
131. Lin, P.W., Stoll, B.J. Necrotizing enterocolitis. Lancet. 2006;368(9543):1271–1283.
132. Lee, J.S., Polin, R.A. Treatment and prevention of necrotizing enterocolitis. Semin Neonatol. 2003;8(6):449–459.
133. Lambert, D.K., et al. Necrotizing enterocolitis in term neonates: data from a multihospital health-care system. J Perinatol. 2007;27(7):437–443.
134. Hunter, C.J., et al. Understanding the susceptibility of the premature infant to necrotizing enterocolitis (NEC). Pediatr Res. 2008;63(2):117–123.
135. Anand, R.J., et al. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock. 2007;27(2):124–133.
136. Frost, B.L., Jilling, T., Caplan, M.S. The importance of pro-inflammatory signaling in neonatal necrotizing enterocolitis. Semin Perinatol. 2008;32(2):100–106.
137. Nankervis, C.A., Giannone, P.J., Reber, K.M. The neonatal intestinal vasculature: contributing factors to necrotizing enterocolitis. Semin Perinatol. 2008;32(2):83–91.
138. Chardot, C., et al. Surgical necrotizing enterocolitis: are intestinal lesions more severe in infants with low birth weight? J Pediatr Surg. 2003;38(2):167–172.
139. Kabeer, A., Gunnlaugsson, S., Coren, C. Neonatal necrotizing enterocolitis: a 12-year review at a county hospital. Dis Colon Rectum. 1995;38(8):866–872.
140. Alfaleh, K., Bassler, D. Probiotics for prevention of necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev. (1):2008. [CD005496].
141. Embleton, N.D., Yates, R. Probiotics and other preventative strategies for necrotizing enterocolitis. Semin Fetal Neonatal Med. 2008;13(1):35–43.
142. Srinivasan, P.S., Brandler, M.D., D’souza, A. Necrotizing enterocolitis. Clin Perinatol. 2008;35(1):251–272.
143. Nguyen, H., Lund, C.H. Exploratory laparotomy or peritoneal drain? Management of bowel perforation in the neonatal intensive care unit. Perinat Neonatal Nurs. 2007;21(1):50–60.
144. Butter, A., Flageole, H., Laberge, J.M. The changing face of surgical indications for necrotizing enterocolitis. J Pediatr Surg. 2002;37(3):496–499.
145. Nucci, A., et al. Interdisciplinary management of pediatric intestinal failure: a 10 year review of rehabilitation and transplantation. J Gastrointest Surg. 2008;12(3):429–435.
146. Bartlett, J.G., Gerding, D.N. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S12–S18.
147. Finkbeiner, S.R., et al. Metagenomic analysis of human diarrhea: vial detection and discovery. PLoS Pathol. 4(2), 2008. [e1000011].
148. Colbere-Garapin, F., et al. Prevention and treatment of enteric viral infections: possible benefits of probiotic bacteria. Microbes Infect. 2007;9(14-15):1623–1631.
149. Bernstein, D.I. Rotavirus overview. Pediatr Infect Dis J. 2009;28(3 Suppl):S50–S53.
150. Turck, D., et al. Incidence and risk factors of oral antibiotic-associated diarrhea in an outpatient pediatric population. J Pediatr Gastroenterol Nutr. 2003;37(1):22–26.
151. Lee, S.D., Surawicz, C.M. Infectious causes of chronic diarrhea. Gastroenterol Clin North Am. 2001;30(3):679–692.
152. Besedovsky, A., Li, B.U. Across the developmental continuum of irritable bowel syndrome: clinical and pathophysiologic considerations. Curr Gastroenterol Rep. 2004;6(3):247–253.
153. Heyman, M.B. Committee on Nutrition: lactose intolerance in infants, children and adolescents. Pediatrics. 2006;118(3):1279–1286.
154. Chou, S.C., et al. Management of hyperbilirubinemia in newborns: measuring performance by using a benchmarking model. Pediatrics. 2003;112(6):1264–1273.
155. Colletti, J.E., et al. An emergency medicine approach to neonatal hyperbilirubinemia. Emerg Med Clin North Am. 2007;25(4):1117–1135. [vii].
156. Cappellini, M.D., Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371(9606):64–74.
157. Iranpour, R., Akbar, M.R., Haghshenas, I. Glucose-6-phosphate dehydrogenase deficiency in neonates. Indian J Pediatr. 2003;70(1):855–857.
158. Alexandra Brito, M., Silva, R.F. Brites D: Bilirubin toxicity to human erythrocytes: a review. Clin Chim Acta. 2006;374(1-2):46–56.
159. Blackmon LR, Fanaroff AA, Raju TN: Research on prevention of bilirubin-induced brain injury and kernicterus: National Institute of Child Health and Human Development conference executive summary, Pediatrics 114(1):229-233, 2004.
160. Bhutani, V.K., Johnson, L.H. Newborn jaundice and kernicterus—health and societal perspectives. Indian J Pediatr. 2003;70(5):407–416.
161. Behrman, R.E., Kliegman, R., Jenson, H.B. Nelson textbook of pediatrics, ed 18. Philadelphia: Saunders; 2007.
161a. Hulzebos, C.V., et al. Usefulness of the bilirubin/albumin ratio for predicting bilirubin-induced neurotoxicity in premature infants. Arch Dis Child Fetal Neonatal Ed. 2008;93(5):F84–F88.
162. Moerschel, S.K., Cianciaruso, L.B., Tracy, L.R. A practical approach to neonatal jaundice. Am Fam Physician. 2008;77(9):1255–1262.
163. Mack, C.L. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27(3):233–242.
164. de Carvalho, E., Ivantes, C.A., Bezerra, J.A. Extrahepatic biliary atresia: current concepts and future directions. J Pediatr (Rio J). 2007;83(2):105–120.
165. Chardot, C. Biliary Atresia. Orphanet J Rare Dis. 2006;1:28.
166. Karakayali, H., et al. Liver transplantation for biliary atresia. Transplant Proc. 2008;40(1):231–233.
167. Armstrong, G.L., Bell, B.P. Hepatitis A virus infections in the United States: model-based estimates and implications for childhood immunizations. Pediatrics. 2002;109(5):839–845.
168. Muecke, C.J., et al. Hepatitis A seroprevalence and risk factors among day care educators. Clin Invest Med. 2004;27(5):259–264.
169. Koslap-Petraco, M.B., Shub, M., Judelsohn, R., Hepatitis, A. disease burden and current childhood vaccination strategies in the United States. J Pediatr Health Care. 2008;22(1):3–11.
170. Centers for Disease Control and Prevention. Prevention of hepatitis A through active or passive immunization: recommendations of the Advisory Committee on Immunizations Practices. MMWR. 2006;55(RR-7):1–23.
171. Chang, M.H. Hepatitis B virus infection. Semin Fetal Neonatal Med. 2007;12(3):160–167.
172. Shukla, N.B., Poles, M.A. Hepatitis B virus infection: co-infection with hepatitis C virus, hepatitis D virus, and human immunodeficiency virus. Clin Liver Dis. 2004;8(2):445–460.
173. Zein, N.N. Hepatitis C in children: recent advances. Curr Opin Pediatr. 2007;19(5):570–574.
174. Hsu, E.K., Murray, K.F. Hepatitis B and C in children. Nat Clin Pract Gastroenterol Hepatol. 2008;5(6):311–320.
175. Heller, S., Valencia-Mayoral, P. Treatment of viral hepatitis in children. Arch Med Res. 2007;38(6):702–710.
176. Roberts, E.A. Non-alcoholic steatohepatitis in children. Clin Liver Dis. 2007;11(1):155–172.
177. Sokucu, S., et al. Long-term outcomes after sclerotherapy with or without a beta blocker for variceal bleeding in children. Pediatr Int. 2003;45(4):388–394.
178. Ryckman, F.C., Alonso, M.H. Causes and management of portal hypertension in the pediatric population. Clin Liver Dis. 2001;5(3):789–818.
179. Rossle, M., Grandt, D. TIPS: an update. Best Pract Res Clin Gastroenterol. 2004;18(1):99–123.
180. Berry, G.T. Galactosemia and amenorrhea in the adolescent. Ann N Y Acad Sci. 2008;1135:112–117.
181. Ala, A., et al. Wilson’s disease. Lancet. 2008;369(9559):397–408.
182. Brewer, G.J. Recognition, diagnosis, and management of Wilson disease. Proc Soc Exp Biol Med. 2000;233(1):39–46.
183. Ferenci, P. Pathophysiology and clinical features of Wilson disease. Metab Brain Dis. 2004;19(3-4):229–239.
184. Stremmel, W., et al. Wilson disease: clinical presentation, treatment, and survival. Ann Intern Med. 1991;115(9):720–726.
185. Mak, C.M., Lam, C.W. Diagnosis of Wilson’s disease: a comprehensive review. Crit Rev Clin Lab Sci. 2008;45(3):263–290.
186. Medici, V., Rossaro, L., Sturniolo, G.C. Wilson disease—a practical approach to diagnosis, treatment and follow-up. Dig Liver Dis. 2007;39(7):601–609.
187. Sevmis, S., et al. Liver transplantation for Wilson’s disease. Transplant Proc. 2008;40(1):228–230.