Oxygen Transport
Learning objectives
After reading this chapter you should be able to:
 Describe the mechanism of oxygen binding to myoglobin and hemoglobin.
Describe the mechanism of oxygen binding to myoglobin and hemoglobin.
Describe conformational differences between deoxygenated and oxygenated hemoglobins.
Define the concept of cooperativity in oxygen binding to hemoglobin.
Describe the Bohr effect and its role in modulating the binding of oxygen to hemoglobin.
Explain how 2,3-bisphosphoglycerate interacts with hemoglobin and influences oxygen binding.
Summarize the processes by which carbon dioxide is transported from peripheral tissues to the lungs.
Introduction
Vertebrates are aerobic organisms
They have a closed circulatory system and a mechanism for extraction of O2 from air (or water), and release of carbon dioxide (CO2) in waste products. Inspired O2 leads to an efficient utilization of metabolic fuels, such as glucose and fatty acids; expired CO2 is a major product of cellular metabolism. This utilization of O2 as a metabolic substrate is accompanied by the generation of reactive oxygen species (ROS) that are capable of damaging virtually all biological macromolecules (Chapter 37). Organisms protect themselves from radical damage in several ways: sequestering O2, limiting production of ROS, and detoxifying them. Heme proteins, interestingly, participate in these protective mechanisms by sequestering and transporting oxygen. The major heme proteins in mammals are myoglobin (Mb) and hemoglobin (Hb). Mb is found primarily in skeletal and striated muscle, and serves to store O2 in the cytoplasm and deliver it on demand to the mitochondrion. Hb is restricted to erythrocytes where it facilitates the transport of O2 and CO2 between the lungs and peripheral tissues. This chapter presents the molecular features of Mb and Hb, the biochemical and physiologic relationships between the structures of Mb and Hb and their interaction with O2 and other small molecules, and the pathologic aspects of selected Hb mutations.
Properties of oxygen
Most oxygen in the body is bound to a carrier protein containing heme
Photosynthetic organisms release diatomic oxygen into the earth's atmosphere during energy production, contributing to the current level of 21% oxygen in air. In mixtures of gases. Each component makes a specific contribution, known as its partial pressure (Dalton's Law), that is directly proportional to its concentration. It is also customary to use the partial pressure of a gas as a measure of its concentration in physiologic fluids. For atmospheric O2 at a barometric pressure (sea level) of 760 mmHg or torr (101.3 kP or kPa; 1 atmosphere absolute or ATA), the partial pressure of oxygen, pO2, is 150–160 mmHg. The amount of O2 in solution is, in turn, directly proportional to its partial pressure. Thus, in the arterial blood (37°C, pH 7.4) the pO2 is 100 mmHg, which produces a concentration of dissolved O2 of 0.13 mmol/L. This level of dissolved O2, however, is inadequate to support efficient aerobic metabolism.
Rather, the major fraction of O2 transported in blood and stored in muscle is complexed with the iron (ferrous, Fe2+) proteins Hb and Mb, respectively. Hb is a tetrameric protein with four O2-binding sites (heme groups). In arterial blood with a Hb concentration of 150 g/L (2.3 mmol/L) and O2 saturation of 97.4%, the contribution of protein-bound O2 is about 8.7 mmol/L. This concentration represents a dramatic 67-fold increase over physically dissolved O2. The total oxygen-carrying capacity of the arterial blood, in dissolved and protein-bound forms, is 8.8 mmol/L – almost 200 mL of dissolved oxygen per liter of blood.
Characteristics of mammalian globin proteins
Globins constitute an ancient family of soluble metalloproteins
Globins are ubiquitous family of proteins, found in microorganisms, plants, invertebrates, and vertebrates. Present-day globins, with their spectacular diversity of function, are most likely derived from a single ancestral globin. While the extent of amino acid identity among invertebrate and vertebrate globins varies widely and can often appear random, two features are noteworthy: the invariant residues PheCD1 and HisF8 and the characteristic patterns of hydrophobic residues in helical segments (Fig. 5.1). Human Mb consists of a single globin polypeptide (153 amino acid residues, 17,053 Da). Human Hb is a tetrameric assembly of two α-globin polypeptides (141 residues, 15,868 Da) and two β-globin polypeptides (146 residues, 15,126 Da). A single heme prosthetic group is noncovalently associated with each globin apoprotein.
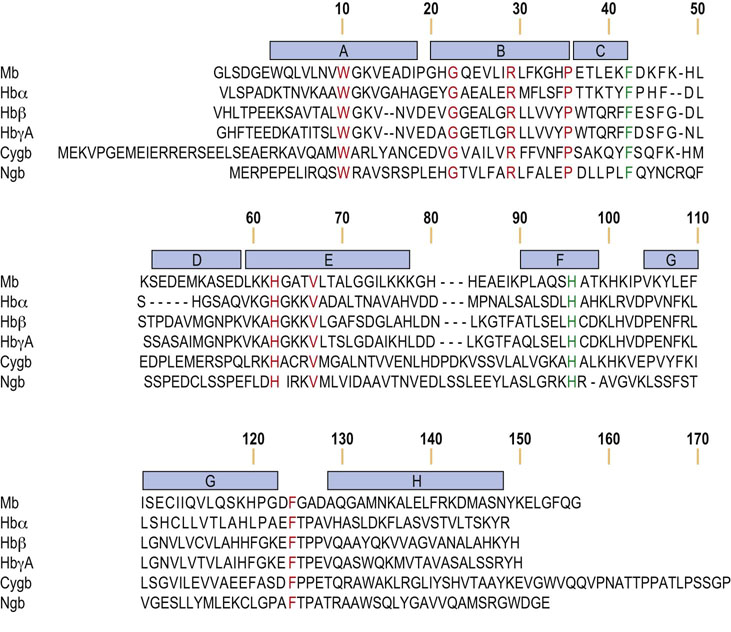
Fig. 5.1 Human globin amino acid sequences are highly conserved.
An alignment of human globins is depicted, with identical amino acid residues in orange. The two residues in green, PheCD1 (F) and HisF8 (H), are absolutely conserved in all metazoan globins. Helical segments in myoglobin are identified by the blue bars. Mb, myoglobin; Hbα, α-globin; Hbβ, β-globin; HbγA, γA-globin; Cygb, cytoglobin; Ngb, neuroglobin.
The secondary structure of mammalian globins is dominated by a high proportion of α-helix, with over 75% of the amino acids associated with eight helical segments. These α-helices are organized into a tightly packed, nearly spherical, tertiary structure, designated the globin fold (Fig. 5.2). So universal is this overall tertiary structure among all globins that the conventional nomenclature for globin residues follows that defined initially for sperm whale Mb, namely helices A, B, C, etc., starting at the N-terminus, separated by corners AB, BC, etc., with residues numbered within each helix and corner. For example, residue A14, an amino acid that participates in electrostatic stabilization between helix A and the GH corner, corresponds to Lys15 in insect Hb, Lys16 in Mb and α-globin, and Lys17 in β-globin (arrow, lower center, Fig. 5.2).
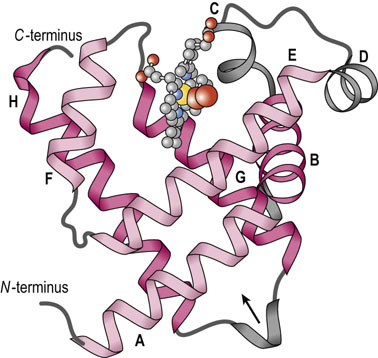
Fig. 5.2 Myoglobin is a compact globular protein.
In this drawing of mammalian Mb only the globin polypeptide backbone is shown, with emphasis on the high proportion of secondary structure (exclusively α-helix). The two-layer, three-over-three arrangement of α-helices is highlighted by the light and dark shades of red. The heme group is illustrated as a gray ‘ball-and-stick’ structure, with iron (yellow) and a bound oxygen molecule (red).
Polar amino acids are located almost exclusively on the exterior surface of globin polypeptides and contribute to the remarkably high solubility of these proteins (e.g. 370 g/L (37% protein solution; 5.7 mmol/L) Hb in the erythrocyte). Amino acids that are both polar and hydrophobic, such as threonine, tyrosine and tryptophan, are oriented with their polar functions toward the protein's exterior. Hydrophobic residues are buried within the interior, where they stabilize the folding of the polypeptide and form a pocket that accommodates the heme prosthetic group. Notable exceptions to this general distribution of amino acid residues in globins are the two histidines that play indispensable roles deep within the heme pocket (Fig. 5.3). Their side chains are oriented perpendicular to and lay on either side of the heme prosthetic group. One of the side chain imidazole nitrogens of the invariant proximal histidine (HisF8) is close enough to bond directly to the pentacoordinate Fe2+ atom. On the opposite side of the heme plane the distal histidine (HisE7), which is too far from the heme iron for direct bonding, functions to stabilize bound O2 by hydrogen bonding.
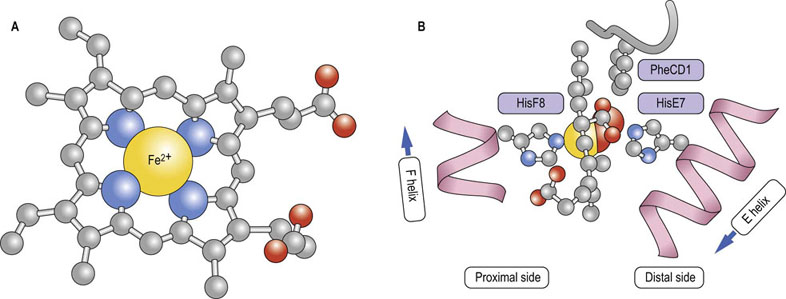
Fig. 5.3 Heme is a complex of porphyrin and iron.
(A) In this view the carbon framework of protoporphyrin IX, a conjugated tetrapyrrole ring, is shown in gray; O2 molecules are red. Iron (yellow sphere) prefers six ligands in an octahedral coordination geometry; pyrrole nitrogen atoms (blue spheres) provide four of these. PheCD1 makes critical hydrophobic and electrostatic stacking interactions with the porphyrin ring. (B) In the oxygenated globin structure, the planar heme is positioned between the proximal and distal histidines (HisF8 and HisE7, respectively); only HisF8 has an imidazole nitrogen (blue sphere) close enough to bond with iron. The α-helices that contain these histidines are shown in pink. In deoxygenated globins, the sixth position remains vacant, leaving a pentacoordinated iron. In the oxygenated state, O2 occupies the sixth position. Both porphyrin propionate moieties participate in hydrogen and electrostatic bonding interactions with globin side chains and solvent.
Structure of the heme prosthetic group
Heme, the O2-binding moiety common to Mb and Hb, is a porphyrin molecule to which an iron atom (Fe2+) is coordinated
The Fe-porphyrin prosthetic group of heme is planar and hydrophobic, with the exception of two propionate groups which are exposed to solvent. Heme becomes an integral component of the holoprotein during polypeptide synthesis; it is heme that gives globins, their characteristic purple-red color – purple in the deoxygenated state in venous blood, red in the oxygenated state in arterial blood.
Globins increase the aqueous solubility of the otherwise poorly soluble, hydrophobic heme prosthetic group. Once sequestered inside a hydrophobic pocket created by the folded globin polypeptide, heme is in a protective environment that minimizes the spontaneous oxidation of Fe2+ (ferrous) to Fe3+ (ferric: rusting) in the presence of O2. Such an environment is also essential for globins to bind and release O2. Should the iron atom become oxidized to the ferric state, heme can no longer interact reversibly with O2, compromising its function in O2 storage and transport.
Myoglobin: an oxygen storage protein
Mb binds O2 that has been released from Hb in tissue capillaries and subsequently diffused into tissues
Located in the cytosol of skeletal, cardiac and some smooth muscle cells, Mb stores a supply of O2 that is readily available to cellular organelles, particularly the mitochondrion, that carry out oxidative metabolism. With its single ligand-binding site, the reversible reaction of Mb with O2:
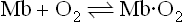
may be described by the following equations:
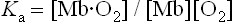
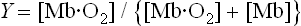
where Ka is an affinity or equilibrium constant, and Y is the fractional O2 saturation. Combining these two equations, expressing the concentration of O2 in terms of its partial pressure (pO2), and substituting the term P50 for 1/Ka yields the equation for the O2 saturation curve of Mb:
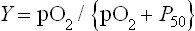
By definition, the constant P50 is the value of pO2 at which Y = 0.5 or half the ligand sites are occupied by O2. In a plot of Y versus pO2, the equation for ligand binding by Mb describes a hyperbola (Fig. 5.4) with a P50 of 4 mmHg. The low value of P50 reflects a high affinity for O2. In the capillary beds of muscle tissues, pO2 values are in the range of 20–40 mmHg. Predictably, working muscles exhibit lower pO2 values than muscles at rest. With its high affinity for O2, myocyte Mb readily becomes saturated with O2 that has entered from the blood. As O2 is consumed during aerobic metabolism, it dissociates through mass action from Mb and diffuses into mitochondria, the power plants of the muscle cell. Whales and other diving mammals have unusually high concentrations of Mb in their muscle tissue; the Mb is thought to function as a large oxygen reservoir for prolonged periods under water.

Fig. 5.4 Oxygen saturation curves of myoglobin and hemoglobin.
Mb and Hb have different O2 saturation curves. The fractional saturation (Y) of O2-binding sites is plotted against the concentration of O2 (pO2 (mmHg) ). Curves are shown for Mb, fetal Hb (HbF), and adult Hb (HbA). Also indicated, by arrows and shading, are the normal levels of O2 measured in various adult and fetal blood samples.
 Clinical box Hyperbaric O2 therapy for acute carbon monoxide poisoning
Clinical box Hyperbaric O2 therapy for acute carbon monoxide poisoning
A 22-year-old pregnant woman, carrying a fetus of 31 weeks gestational age, was brought to the maternity clinic of a hospital for suspected CO poisoning.
The patient was experiencing headache, nausea, and visual abnormalities. She stated that her workplace had been undergoing repairs to the heating and ventilation systems during the past 2 weeks, and on the day of her hospital visit the fire department had evacuated the building after detecting a high level of CO (200 ppm), compared to a typical urban street level of 10 ppm. Her blood pressure was 116/68 mmHg, pulse rate of 100, and respiratory rate of 24/min. Noteworthy in the patient's evaluation was a carboxy-Hb (COHb) component of 15% of total Hb at time of admission (normal = 3%, but may exceed 10% in heavy smokers). Fetal monitoring indicated a fetal heart rate of 135, with occasional, moderate irregularities. Uterine contractions were occurring every 3–5 min.
The patient was treated in the hospital's hyperbaric O2 chamber: 30 min at 2.5 ATA, then 60 min at 2.0 ATA. She also received magnesium sulfate intravenously to resolve the premature contractions. The patient was discharged 2 days later. She delivered a healthy female infant at 38 weeks of gestational age who, on examination at birth and at 6 weeks of age, exhibited no apparent sequelae to her in utero exposure to CO or 100% O2.
Carbon monoxide is a normal product of heme catabolism and has a range of physiologic activities in vascular, neuronal and immunologic systems. Like O2, CO also binds to heme prosthetic groups. Because the affinity of globin-bound heme for CO is about 250 times that for O2, prolonged exposure of hemoglobin to exogenous CO would be virtually irreversible (t1/2 for reversal in blood, 4–8 h) and lead to highly toxic levels of carboxy-Hb. Hyperbaric O2 is the treatment of choice for severe or complicated CO poisoning.
The administration of 100% O2 at 2–3 ATA creates arterial and tissue pO2 values of 2000 mmHg and 400 mmHg, respectively (20 times normal). The immediate result is a reduction in the t1/2 of carboxy-Hb to less than 30 min. Hyperbaric O2 is also used in the treatment of decompression sickness, arterial gas embolism, radiation-induced or ischemic tissue injury, and severe hemorrhage.
Hemoglobin: an oxygen transport protein
Hb is the principal O2-transporting protein in human blood; it is localized exclusively in erythrocytes
Adult Hb (HbA) is a tetrahedral array of two identical α-globin, and two identical β-globin subunits, a geometry that predicts several types of subunit–subunit interactions in the quaternary structure (Fig. 5.5). Importantly, within the Hb tetrahedron each subunit is in contact with the other three. Experimental analysis of the quaternary structure indicates multiple noncovalent interactions (hydrogen bonds and electrostatic bonds) between each pair of dissimilar subunits, i.e. at the α–β interfaces. In contrast, there are fewer and predominantly hydrophobic interactions between identical subunits, at the α1–α2 or β1–β2 interfaces. The actual number and nature of contacts differ in the presence or absence of O2. Strong associations within each αβ heterodimer and at the interface between the two heterodimers (see Fig. 5.5) are now recognized as major factors determining O2 binding and release. Thus, Hb is more appropriately considered a dimer of heterodimers, (αβ)2, rather than an α2β2 tetramer. Although a solution of HbA is theoretically a dynamic mixture of heterodimers and tetramers, under physiologic conditions (high Hb and neutral pH) the equilibria greatly favor the tetramer: 99% for oxygenated Hb, and 99.9% for deoxygenated Hb.
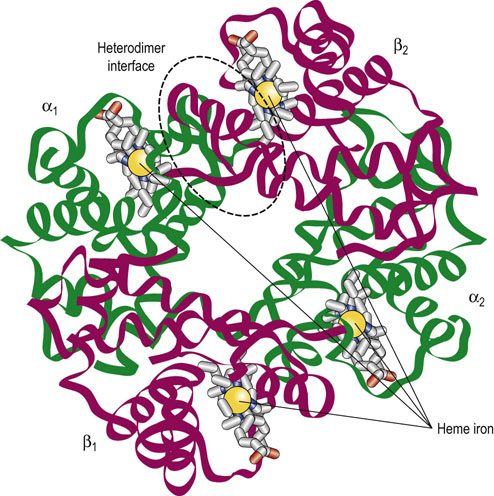
Fig. 5.5 Hemoglobin is a tetramer of four globin subunits.
Hb is a tetrahedral complex of two identical α-globins (α1 and α2, greens) and two identical β-globins (β1 and β2, reds). With this geometry each globin subunit contacts the other three subunits, creating the interfaces and interactions that define cooperativity. One of the heterodimer interfaces is outlined in a dashed oval.
Interactions of hemoglobin with oxygen
Hb binds oxygen cooperatively, with a Hill coefficient of ~2.7
As a gas delivery vehicle, Hb must be able to bind O2 efficiently as it enters the lung alveoli during respiration and to release O2 to the extracellular environment with similar efficiency as erythrocytes circulate through tissue capillaries. This remarkable duality of function is achieved by cooperative interactions among globin subunits. When deoxygenated Hb becomes oxygenated, significant structural changes extend throughout the protein molecule. In the heme pocket, as a consequence of O2 coordination to iron and a new orientation of atoms in the heme structure, the proximal histidine and helix F to which it belongs, shift their positions (see Fig. 5.3). This subtle conformational change triggers major structural realignments elsewhere within that globin subunit. In turn, these tertiary structural changes are transmitted, even amplified, in the overall quaternary structure, such that a 12–15° rotation and a 0.10 nm displacement of the α1β1 dimer relative to the α2β2 dimer take place. Because of the inherent asymmetry of the α2β2 tetramer, these combined motions result in quite dramatic changes within and, more importantly, between the αβ heterodimers. Because of structural changes in hemoglobin as a result of binding of oxygen and other effectors, the binding affinity for subsequent molecules of oxygen may be increased (positive cooperativity) or decreased (negative cooperativity).
Hb can bind up to four molecules of O2 in a cooperative manner
With its multiple ligand-binding sites and structural changes in response to binding, the oxygen affinity and the fractional saturation of Hb are more complex functions than those of Mb. Consequently, the equation for the fractional O2 saturation curve must be modified to:
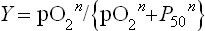
where n is the Hill coefficient. In a plot of Y versus pO2 when n > 1, the equation for ligand binding describes a sigmoid (S-shaped) curve (see Fig. 5.4). The Hill coefficient, determined experimentally, is a measure of cooperativity among ligand-binding sites, i.e. the extent to which the binding of O2 to one subunit influences the affinity of O2 to other subunits. For fully cooperative binding, n is equal to the number of sites (four in Hb), an indication that binding at one site maximally enhances binding at other sites in the same molecule. The normal Hill coefficient for adult Hb (n = 2.7) reflects strongly cooperative ligand binding. Hb has a considerably lower affinity for O2, reflected in a P50 of 27 ± 2 mmHg, compared to myoglobin (P50 = 4 mmHg). In the absence of cooperativity, even with multiple sites, the Hill coefficient would be 1, i.e. binding of one molecule of O2 would not influence the binding of other molecules. Decreased or absent cooperativity is observed for Hb mutants that have lost functional subunit–subunit contacts (Table 5.1). The steepest slope of the saturation curve for Hb lies in a range of pO2 that is found in most tissues (see Fig. 5.4). Thus, relatively small changes in pO2 will result in considerably larger changes in the interaction of Hb with O2. Accordingly, slight shifts of the curve in either direction will also dramatically influence O2 affinity.
Table 5.1
Classification and examples of hemoglobinopathies
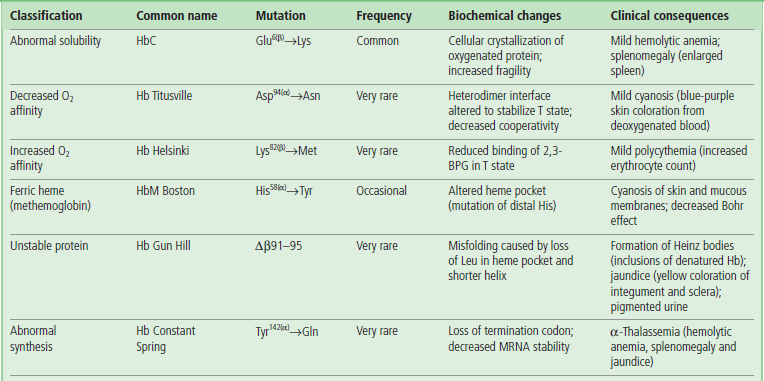
Hemoglobinopathies are usually classified according to the most prominent change to the protein's structure, function, or regulation. Initial identification of a mutation often involves electrophoretic or chromatographic analysis, as shown in Figure 5.9 for HbSC, a double heterozygous genotype associated with a sickle cell disease-like phenotype. Δ = deletion mutant.
Hemoglobin subunits may assume two different conformations that differ in O2 affinity
The mechanism underlying the cooperativity in oxygen binding by hemoglobin involves a shift between two conformational states of the hemoglobin molecule which differ in oxygen affinity. These two quaternary conformations are known as the T (tense) and R (relaxed) states, respectively. In the T state, interactions between the heterodimers are stronger; in the R state, these noncovalent bonds are, in summation, weaker. O2 affinity is lower in the T state and higher in the R state. Transition between these states is accompanied by the breaking of existing noncovalent bonds and formation of new ones at the heterodimer interfaces (Fig. 5.6). Contact between the two αβ heterodimers (see Fig. 5.5) is stabilized by a mixture of hydrogen and electrostatic bonds. Approximately 30 amino acids participate in the noncovalent interactions that characterize the deoxygenated and oxygenated Hb conformations.
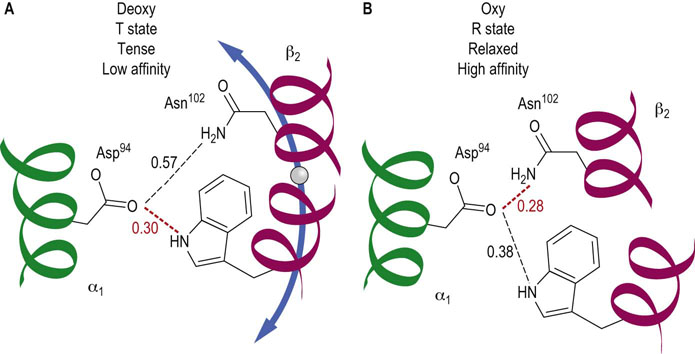
Fig. 5.6 Noncovalent bonds differ in deoxygenated and oxygenated hemoglobin.
In the middle of the interface between the two αβ heterodimers are the residues Asp94(α) on the α1-globin of one heterodimer and Trp37(β) and Asn102(β) on the β2-globin of the other heterodimer (see dashed oval in Fig. 5.5). Each has side chain atoms capable of noncovalent interactions. (A) In the deoxygenated T state the distance between the Asp and Trp residues favors a hydrogen bond, whereas the distance between Asp and Asn is too great. (B) As a result of the conformational changes that accompany the transition to the oxygenated R state, the distance between Asp and Trp is now too large, but that between Asp and Asn is compatible with formation of a new hydrogen bond. Elsewhere along this interface, other bonds are created and broken. An identical alignment of residues and noncovalent interactions is found between the α2- and β1-globin monomers. Distances are shown in nm. Hydrogen bonds are commonly 0.27–0.31 nm in length.
Several models have been developed to describe the transition between the T and R states of Hb. At one extreme is a model in which each Hb subunit sequentially responds to O2 binding with a conformational change, thereby permitting hybrid intermediates of the T and R states. At the opposite extreme is a model in which all four subunits switch concertedly; hybrid states are forbidden, and binding of O2 to one subunit shifts the equilibrium of all subunits from the T to the R state simultaneously. The molecular structures of deoxygenated and partially and fully ligated Hb have been studied extensively by a broad range of thermodynamic and kinetic techniques. Yet, progress toward reconciling inconsistencies among classic and more recent models has been slow. These different viewpoints on conformational changes in multi-subunit proteins are discussed further in the section on allosteric enzymes in Chapter 6.
 Clinical test box Pulse oximetry
Clinical test box Pulse oximetry
Pulse oximetry (‘pulse-ox’) is a noninvasive method of estimating the oxygen saturation of arterial Hb. Two physical principles are involved: first, the visible and infrared spectral characteristics of oxy- and deoxy-Hb are different; second, arterial blood flow has a pulsatile component that results from volume changes with each heartbeat. Transmission or reflectance measurements are made in a translucent tissue site with reasonable blood flow, commonly a finger, toe or ear of adults and children, or a foot or hand of infants. The photodetector and microprocessor of the pulse oximeter permit a calculation of oxygen saturation (SpO2 = saturation of peripheral oxygen) that typically correlates within 4–6% of the value found by arterial blood gas analysis.
Pulse oximetry is used to monitor the cardiopulmonary status during local and general anesthesia, in intensive care and neonatal units, and during patient transport. Body movement, radiated ambient light, elevated bilirubin, artificial or painted fingernails can interfere with pulse oximetry.
Conventional, two-wavelength instruments ‘assume’ that the optical measurements are associated with oxygenated and deoxygenated hemoglobins; they cannot discriminate among oxy-, carboxy- and metHb. Newer technologies, however, utilize six or eight wavelengths and permit multiple Hb species discrimination with an accuracy of ±2.0% and precision of ±1.0%.
Allosteric Modulation of the Oxygen Affinity of Hemoglobin
Allosteric proteins and effectors
Hb is an allosteric protein; its affinity for O2 is regulated by small molecules
Hb is one of the best studied examples of an allosteric protein, These small bind to proteins at sites that are spatially distinct from the ligand-binding sites, thus their designation as allosteric (other site) effectors. Through long-range conformational effects, they alter the ligand or substrate binding affinity of the protein. Allosteric proteins are typically multi-subunit proteins. The O2 binding affinity of Hb is affected positively by O2, as well as by a number of chemically diverse allosteric effectors, including H+, CO2, and 2,3-bisphosphoglycerate (2,3-BPG) (Fig. 5.7). When an allosteric effector affects its own binding to the protein (at other sites), the process is termed homotropic, e.g. the effect of binding of O2 at one site on Hb enhances the affinity for binding of O2 to other sites on Hb. When the allosteric effector is different from the ligand whose binding is altered, the process is termed heterotropic, e.g. the effect of H+ (pH) on the P50 for oxygen binding to Hb. These interactions lead to horizontal shifts in the O2 binding curves (see Fig. 5.7).
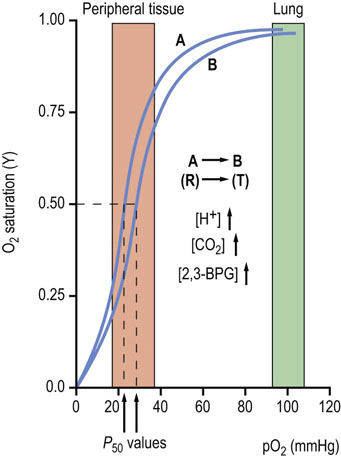
Fig. 5.7 Allosteric effectors decrease the oxygen affinity of hemoglobin.
O2 interaction with Hb is regulated by allosteric effectors. Under physiologic conditions HbA exhibits a highly cooperative O2 saturation curve. With an increase in the erythrocyte concentration of any of three allosteric effectors, H+, CO2 or 2,3-bisphosphoglycerate (2,3-BPG), the curve shifts to the right (position B), indicating a decreased affinity for O2 (increase in P50 value). Actions of the effectors that modulate O2 affinity appear to be additive. Conversely, a decrease in any of the allosteric effectors shifts the curve to the left (position A). Increasing temperature will also shift the curve to the right. The sensitivity of O2 saturation to H+ is known as the Bohr effect. Normal ranges of O2 measured in pulmonary and peripheral tissue capillaries are indicated by shaded areas.
Bohr effect
Acidic pH (protons) decreases the O2 affinity of Hb
The O2 affinity of Hb is exquisitely sensitive to pH, a phenomenon known as the Bohr effect. The Bohr effect is most readily described as a right shift in the O2 saturation curve with decreasing pH. Thus, an increased concentration of H+ (decreased pH) favors an increased P50 (lower affinity) for O2 binding to Hb, equivalent to an H+-dependent shift of Hb from the R to the T state.
To understand the Bohr effect at the level of protein structure and to appreciate the role of H+ as a heterotropic allosteric effector, it is important to recall that Hb is a highly charged molecule. The residues that participate in the Bohr effect include the N-terminal Val amino group of α-globin and the C-terminal His side chain of β-globin. The pKa values of these weak acids differ sufficiently between the deoxygenated and oxygenated forms of Hb to cause the uptake of 1.2–2.4 protons by the deoxygenated, compared to oxygenated, tetramer.
Identification of specific amino acid residues of the α- and β-globins that participate in the Bohr effect is complicated by differential interactions of other charged solutes with deoxy- and oxy-Hb. Thus, a preferential binding of a given anion, i.e. Cl− and/or organic phosphates, to deoxygenated Hb involves the alteration of the pKs of some cationic groups, thereby contributing to the overall observed Bohr effect. For example, there is compelling evidence showing that Val1(α) is relevant to the Bohr effect only in the presence of Cl−. The pKa of this group shifts from 8.0 in deoxygenated Hb to 7.25 in oxygenated Hb in the presence of physiologic concentration of Cl− (≈100 mmol/L). Further, the participation of the Val1(α) groups in the chloride-dependent Bohr effect is strongly modulated by CO2 because of the formation of CO2 (carbamino) adducts of Hb (described below).
As Hb binds O2, protons dissociate from selected weak acid functions. Conversely, in acidic media, protonation of the conjugate bases inhibits O2 binding
During their circulation between pulmonary alveoli and peripheral tissue capillaries, erythrocytes encounter markedly different conditions of pO2 and pH. The high pO2 in the lungs promotes ligand saturation and forces protons from the Hb molecule to stabilize the R state. In the capillary bed, particularly in metabolically active tissues, the pH is slightly lower, due to the production of acidic metabolites, such as lactate. Oxygenated Hb, upon entering this environment, will acquire some ‘excess’ protons and shift toward the T state, promoting release of O2 for uptake by tissues for aerobic metabolism.
Effects of CO2 and temperature
Like H+, CO2 is is increased in venous capillaries and is a negative allosteric effector of the O2 affinity of Hb
Closely related to the Bohr effect is the ability of CO2 to alter the O2 affinity of Hb. The increase in pCO2 in venous capillaries decreases the affinity of Hb for O2. Accordingly, a right shift in the ligand saturation curve occurs as pCO2 increases. It should be emphasized that the allosteric effector is, in fact, CO2, not HCO3−: CO2 reacts reversibly with the unprotonated N-terminal amino groups of the globin polypeptides to form carbamino adducts:
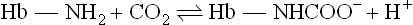
This transient covalent chemical modification of Hb is not only a specialized example of allosteric control, resulting in a stabilization of deoxygenated Hb; it also represents one form of transport of CO2 to the lungs for clearance from the body. Between 5% and 10% of the total CO2 content of blood exists as carbamino adducts.
There is a strong physiologic correlation between pCO2 and the O2 affinity of Hb. CO2 is a major product of mitochondrial oxidation and, like H+, is particularly abundant in metabolically active tissues. Upon diffusing into blood, CO2 can react with oxygenated Hb, shift the equilibrium toward the T state, and thereby promote the dissociation of bound O2 (see Fig. 5.7). The vast majority of peripheral tissue CO2, however, is hydrated by erythrocyte carbonic anhydrase to carbonic acid (H2CO3), a weak acid that dissociates partially to H+ and HCO3−:
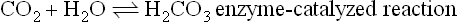
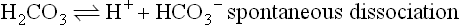
Interestingly, from both carbamino adduct formation and hydration/dissociation reactions involving CO2, an additional pool of protons is generated. These are protons that become available to participate in the Bohr effect and facilitate O2–CO2 exchange. During its return to the lungs, blood transports two forms of CO2: carbamino-Hb and the H2CO3/HCO3− acid–conjugate base pair. Blood and Hb are now exposed to a low pCO2, and through mass action the carbamino adduct formation is reversed and binding of O2 is again favored. Similarly, in the pulmonary capillaries, erythrocyte carbonic anhydrase converts H2CO3 to CO2 and H2O, which are expired into the atmosphere (see Chapter 25).
 Advanced concept box Artificial hemoglobins
Advanced concept box Artificial hemoglobins
The supply-and-demand curves for whole blood and packed red cell availability and utilization point to an impending crisis and the need to develop alternatives. Red cell substitutes are transfusion alternatives that are potentially useful during major surgical procedures and hemorrhagic shock emergencies.
Three types of artificial O2 carriers have been investigated: Hb-based oxygen carrier (HBOC), liposome- or nanoparticle-encapsulated Hb, and perfluorocarbon emulsions. HBOCs are hemoglobins derived from allogeneic, xenogeneic or recombinant sources that have been modified by polymerization, crosslinkage or conjugation. These modifications facilitate purification and sterilization, and minimize toxicity and immunogenicity. They are also necessary to stabilize the extracellular Hb tetramers; otherwise the hemoglobin dissociates into dimers and monomers in plasma and is excreted in urine. The artificial forms have O2 affinity (P50) in the range 16–38 mmHg, but usually have diminished cooperativity (n = 1.3–2.1) and Bohr effects.
Several HBOCs have progressed through extensive clinical evaluation and some are used in medical procedures in some countries. Adverse effects are not uncommon with HBOCs. Increased vasoconstriction with subsequent hypertension occurs, a result of increased binding of nitric oxide (NO), an endogenous regulator of vasodilation, by extracellular Hb. Other problems include increased heme oxidation to metHb, elevated iron deposition in tissues, gastrointestinal distress, neurotoxicity, and interference with diagnostic measurements. Molecular engineering of human Hb, now underway in a number of laboratories, seeks to improve O2 binding, allosteric properties, and side effects of HBOCs. Packaging of hemoglobin in liposomes or nanocapsules, producing artificial red blood cells, is also a promising technology since this limits the escape of Hb into extravascular spaces.
Working muscles not only produce the allosteric effectors H+ and CO2 as byproducts of aerobic metabolism but also liberate heat. Because the binding of O2 to heme is an exothermic process, the O2 affinity of Hb decreases with increasing temperature. Thus, the microenvironment of an exercising muscle profoundly favors a more efficient release of Hb-bound O2 to the surrounding tissue.
Effect of 2,3-bisphosphoglycerate
2-3-Bisphoglycerate (2,3-BPG), an intermediate in carbohydrate metabolism, is an important allosteric effector of Hb
2,3-BPG is synthesized in human erythrocytes in a one-step shunt off the glycolytic pathway (Chapter 12). Like H+ and CO2, 2,3-BPG is an indispensable negative allosteric effector that, when bound to Hb, causes a marked increase in P50 (see Fig. 5.7). Were it not for the high erythrocyte concentration of 2,3-BPG (4.1 mmol/L, nearly equal to that of Hb), the O2 saturation curve of Hb would approach that of Mb!
At one end of the twofold symmetry axis within the quaternary structure of Hb there is a shallow cleft defined by cationic amino acids of the juxtaposed β-globin subunits (Fig. 5.8). A single molecule of 2,3-BPG binds to this site. A critical consequence of the conformational differences between the T and R states is that deoxygenated Hb preferentially interacts with the negatively charged 2,3-BPG. Multiple electrostatic interactions stabilize the complex between the polyanionic effector and deoxygenated Hb. The cleft is too narrow in fully oxygenated Hb to accommodate 2,3-BPG.
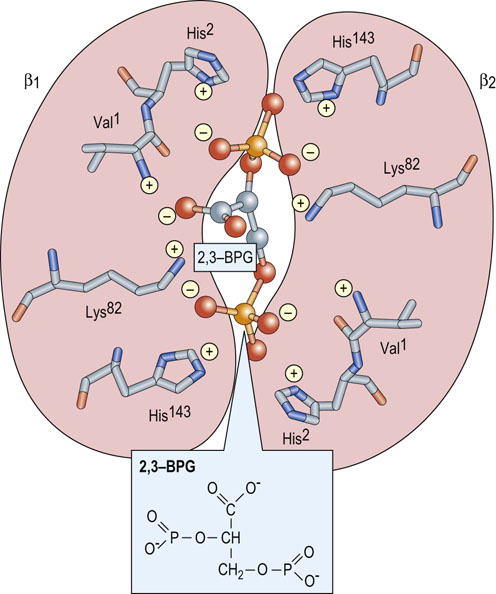
Fig. 5.8 2,3-Bisphosphoglycerate binds preferentially to deoxygenated hemoglobin.
On the surface of the deoxygenated Hb tetramer where the two β-globins (purple) interact, there is a cleft formed by the N-terminal amino acid residue (Val1(β) ) and the side chains of His2(β), Lys82(β), and His143(β) (stick models). This site consists of eight cationic groups, sufficient to bind with high affinity one molecule of 2,3-BPG (ball-and-stick model; phosphorus, orange), a molecule with five anionic groups at physiologic pH. This array of positive charges does not exist in oxygenated Hb. In fetal Hb (HbF) His143(β) is replaced by a Ser residue.
The importance of 2,3-BPG as an allosteric effector is underscored by observations that its concentration in the erythrocyte changes in response to various physiologic and pathologic conditions. During chronic hypoxia (decreased pO2) secondary to pulmonary disease, anemia or shock, the level of 2,3-BPG increases. Such compensatory increases have also been described in cigarette smokers and on adaptation to high altitudes. The net result is a greater stabilization of the deoxygenated, low-affinity T state and a further shift of the saturation curve to the right, thereby facilitating release of more O2 to tissues. Under most circumstances, the rightward shift has an insignificant effect on the O2 saturation of Hb in the lungs.
Selected Topics
Interaction of hemoglobin with nitric oxide
Nitric oxide, a potent vasodilator, is stored on Hb as S-nitrosoHb (SNO-Hb)
Nitric oxide (NO) is a gaseous free radical capable of oxidative modification (nitration, nitrosation, nitrosylation) of biological macromolecules. Yet this highly reactive molecule, also known as endothelium-derived relaxing factor (EDRF), is synthesized in endothelial cells and participates in normal vascular physiology, including vasodilation (smooth muscle), hemostasis (platelet), and adhesion molecule expression (endothelial cell). Erythrocytes are the largest intravascular reservoir of bioactive NO, and Hb is indispensable for its formation, storage, and release. SNO-Hb is the product of S-nitrosylation of the Cys93β side chains of Hb. These Cys thiol groups can accept NO by transfer from intracellular S-nitrosoglutathione or from heme-bound NO (nitrosyl-Hb). NO is released by exchange from SNO-Hb to Cys side chains of anion exchanger 1, an erythrocyte membrane protein that can then deliver NO to the plasma. The formation and breakdown of SNO-Hb are sensitive to pO; NO is released from Hb in response to hypoxia or on conversion to the T state, e.g. in venous capillaries.
Another remarkable process within the erythrocyte is the allosterically regulated conversion of nitrite (NO2−) to NO, a reaction performed by deoxygenated Hb. This intrinsic ‘nitrite reductase’ activity takes advantage of the moderate NO2− concentration in the erythrocyte (up to 0.3 µmol/L). While the chemistry is complex, the reaction is thought to yield a labile intermediate nitrosyl-metHb(ferric) that can readily transfer NO to Cys93(β) on oxygenated Hb.
Advanced Concept Box Acute mountain sickness – too high, too fast
Acute mountain sickness (AMS) develops in individuals who ascend rapidly to ambient conditions of hypobaric hypoxia. Symptoms include shortness of breath, rapid heart rate, headache, nausea, anorexia, and sleep disturbance. These can develop at altitudes of 2000 m (25% incidence) and higher to 4000 m or more (50% incidence). The most severe form is high-altitude cerebral edema (2% incidence), a potentially fatal condition characterized by ataxia and other neuromuscular and neurologic problems.
At 4000 m the barometric pressure is 460 mmHg, leading to an ambient partial pressure of O2 of 96 mmHg (sea level, 160). Physiologic calculations yield values of a tracheal pO2 of 86 mmHg (sea level, 149), an alveolar pO2 of 50 mmHg (sea level, 105), and an arterial pO2 of 45 mmHg (sea level, 100). At this arterial partial pressure of O2, Hb saturation is only 81% (see Fig. 5.4). Consequently, the O2-carrying capacity of arterial blood decreases to 160 mL/L (sea level, 198). Hypoxia can also lead to overperfusion of vascular beds, endothelial leakage, and edema.
Humans adapt to high altitude (acclimatization) by several mechanisms. Hyperventilation is a critical short-term response that serves to decrease alveolar pCO2 and, in turn, increase alveolar pO2. Arterial pH is also increased during hyperventilation, leading to a higher affinity of Hb for O2. A gradual increase in 2,3-BPG typically occurs in response to chronic hypoxia. Another important adaptive mechanism is polycythemia, an increase in erythrocyte concentration that results from erythropoietin stimulation of bone marrow cells. Within one week of acclimatization the Hb concentration can increase by as much as 20% to provide near-normal arterial O2 content.
Neuroglobin and cytoglobin: minor mammalian hemoglobins
Two other globins have recently been identified in humans
Neuroglobin (Ngb) is expressed primarily in the central nervous system and some endocrine tissues; cytoglobin (Cygb) is ubiquitously expressed, primarily in cells of fibroblast origin. Tissue concentrations of both are <1 mmol/L. The Ngb polypeptide has 151 amino acid residues (16,933 Da), whereas Cygb contains 190 residues (21,405 Da), with ‘extensions’ of 20 amino acids at both the N- and C-termini (see Fig. 5.1). Both human proteins share only about 25% sequence identity with Mb and Hb. Yet all key elements of the globin fold are present: the three-over-three α-helix sandwich; the proximal and distal His residues; and a hydrophobic, heme-containing pocket.
In contrast to Mb and Hb, Ngb and Cygb contain hexacoordinate hemes for both the Fe2+ and Fe3+ valency states. The distal HisE7, serving as the sixth ligand, must be displaced to permit binding of O2. Yet the O2 affinities of Ngb and Cygb are surprisingly high, with P50 values in the range 1–7.5 mmHg and 0.7–1.8 mmHg, respectively, compared to a P50<27 mmHg for Hb. Binding of O2 to the dimeric Cygb is cooperative (Hill coefficient = 1.2–1.7) but independent of pH. On the other hand, monomeric Ngb exhibits a pH-dependent O2 affinity. The functions of these minor globins remain elusive. Ngb appears to be comparable to Mb, mediating the delivery of O2 to retina mitochondria. Cygb is thought to function as an enzyme cofactor, supplying O2 for the hydroxylation of Pro and Lys side chains in some proteins.
Clinical box A student with hyperventilation numbness, and dizziness
A college student with severe muscle spasms in her arms, numbness in her extremities, some dizziness, and respiratory difficulty was brought to the student health center. The patient had been vigorously exercising in an attempt to relieve the stress of forthcoming examinations when she suddenly began to experience forced, rapid breathing. Suspecting hyperventilation, a health care worker began to reassure the student and helped her recover by getting her to breathe into a paper bag. After 20 minutes the spasms ceased, feeling returned to her fingers, and the lightheadedness resolved.
Alveolar hyperventilation is an abnormally rapid, deep, and prolonged breathing pattern that leads to respiratory alkalosis, i.e. a profound decrease in pCO2 and an increase in blood pH that can be attributed to an increased loss of CO2 from the body. With decreased [CO2] and [H+], two allosteric effectors of O2 binding and release, the affinity of Hb for O2 increases sufficiently to reduce the efficiency of delivery of O2 to peripheral tissues, including the central nervous system. Another characteristic of alkalosis is a decreased level of ionized calcium in plasma, a situation that contributes to muscle spasms and cramps. In general, hyperventilation may be triggered by hypoxemia, pulmonary and cardiac diseases, metabolic disorders, pharmacologic agents, and anxiety. See also Chapter 25.
Hemoglobin variants
Over 95% of the Hb found in adult humans is HbA, with the α2β2 globin subunit composition. HbA2 accounts for 2–3% of the total and has an α2δ2 polypeptide composition. HbA2 is elevated in β-thalassemia, a disease characterized by a deficiency in β-globin biosynthesis. Functionally, these two adult hemoglobins are indistinguishable. Not surprisingly, mutations of the gene encoding δ-globin are without clinical consequence.
Another minor Hb is fetal Hb, HbF; its subunits are α-globin and γ-globin. While it accounts for no more than 1% of adult Hb, HbF predominates in the fetus during the second and third trimesters of gestation and in the neonate. Gene switching on chromosome 11 causes HbF to decrease shortly after birth. The most striking functional difference between HbF and HbA is its decreased sensitivity to 2,3-BPG. Comparison of the primary structures of the β- and γ- polypeptides reveals a replacement of His143β by Ser in γ-globin (see Fig. 5.1). Consequently, two of the cationic groups that participate in the binding of the anionic allosteric effector are no longer available (see Fig. 5.8). Predictably, the interaction of 2,3-BPG with HbF is weaker, resulting in an increased affinity for O2 (P50 of 19 mmHg for HbF compared to 27 mmHg for HbA) and a greater stabilization of the oxygenated R state. The direct benefit of this structural and functional change in the HbF isoform is a more efficient transfer of O2 from maternal HbA to fetal HbF (see Fig. 5.4). Separation of these and other Hb variants in the clinical laboratory is performed by electrophoretic and chromatographic analysis (Fig. 5.9).
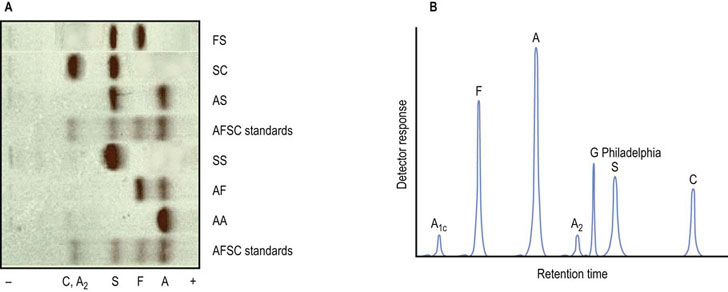
Fig. 5.9 Normal and abnormal hemoglobins can be separated by electrophoretic and chromatographic methods.
(A) This panel shows cellulose acetate electrophoresis (pH 8.4) of blood samples obtained for neonatal screening. This rapid technique will tentatively identify HbS and HbC, two common mutant hemoglobins in the African-American population. Additional tests are required for a definitive diagnosis. FS, newborn with sickle cell disease; SC, double heterozygote child with sickle cell-like disease; AS, child with sickle cell trait; AF, normal neonate. (B) This trace illustrates high-pressure liquid chromatography (HPLC) with a cation-exchanger solid phase, a technique capable of separating and quantifying more than 40 hemoglobins. HPLC may also be used to measure HbA1c, a glycated protein that is measured clinically as an index of mean blood glucose concentration in diabetes mellitus. Also shown is the elution profile of Hb G Philadelphia (Asn68(α)→Lys), a common but benign variant that co-migrates with HbS on electrophoresis.
Sickle cell disease, a common hemoglobinopathy
In sickle cell disease (SCD), distortion of erythrocyte structure (sickling) limits capillary blood flow
Clinically, an individual with SCD presents with intermittent episodes of hemolytic anemia, resulting from chronic lysis of red cells, and painful vaso-occlusive crises. Common features also include impaired growth, increased susceptibility to infections, and multiple organ damage. In the African-American population in the US, SCD affects 90,000–100,000 individuals, a frequency of 0.2%. Heterozygous, mostly asymptomatic, carriers, number 8% in this same population.
SCD is caused by an inherited, single point mutation in the gene encoding β-globin, leading to the expression of the Hb variant HbS. Indeed, HbS has been studied biochemically, biophysically, and genetically for over 50 years, making SCD the paradigm of a molecular disease. The mutation is Glu6(β)→Val: a surface-localized charged amino acid is replaced by a hydrophobic residue. Valine on the mutant β-globin subunit fits into a complementary pocket (sometimes called a ‘sticky patch’) formed on the β-globin subunit of a deoxygenated Hb molecule, a pocket that becomes exposed only upon the release of bound O2 in tissue capillaries.
HbA remains a true solute at rather high concentrations, largely as a result of a polar exterior surface that is compatible and nonreactive with nearby Hb molecules. In contrast, HbS, when deoxygenated, is less soluble. It forms long, filamentous polymers that readily precipitate, distorting erythrocyte morphology to the characteristic sickle shape. In the homozygous individual with SCD (HbS/HbS), the complex process of nucleation and polymerization occurs rapidly, producing about 10% of circulating erythrocytes that are sickled. In the heterozygous individual (HbA/HbS, sickle cell trait), the kinetics of sickling are decreased by at least a factor of 1000, thereby accounting for the asymptomatic nature of this genotype. In dilute solution, HbS has interactions with O2 (P50 value, Hill coefficient) that are similar to those for HbA. However, the Bohr effect on concentrated HbS is more pronounced, leading to greater release of O2 in the capillaries and increased propensity for sickling.
Clinical box Acquired methemoglobinemia
In a rural region of the state, a 4-month-old infant was seen at the local emergency room for episodes of seizures, breathing difficulty, and vomiting. The infant's skin and mucous membranes were bluish, indicating cyanosis. Analysis of arterial blood revealed a chocolate brown color, a normal pO2, an O2 saturation of 60%, and a metHb (ferric-heme) level of 35%.
The tentative cause of the acute toxic methemoglobinemia was found to be well water contaminated by a nitrate/nitrite concentration of 34 mg/L. The infant was treated successfully by intravenous administration of methylene blue (1–2 mg/kg) that serves to accelerate indirectly the enzymatic reduction of metHb to normal (ferrous) Hb by NADPH metHb reductase, which is normally a minor pathway for conversion of metHb to Hb.
MetHb is formed when the ferrous iron of heme is oxidized to ferric iron; it is produced spontaneously at a low rate and more rapidly in the presence of certain drugs, nitrites, and aniline dyes. In genetic forms of methemoglobinemia, mutation of either the proximal or distal His to Tyr makes the heme iron more susceptible to oxidation (see Table 5.1). The extent of oxidation in Hb tetramers can range from one heme group to all four. Erythrocytes contain an NADH-cytochrome b5 reductase, or NADH diaphorase, that is responsible for the majority of metHb reduction. Infants are particularly vulnerable to methemoglobinemia because their level of NADH-cytochrome b5 reductase is half that of adults. Moreover, their higher level of HbF is more sensitive to oxidants compared to HbA.
Sickled erythrocytes exhibit less deformability. They no longer move freely through the microvasculature and often block blood flow, especially in the spleen and joints. Moreover, these cells lose water, become fragile, and have a considerably shorter life span, leading to hemolysis and anemia. Except during extreme physical exertion, the heterozygous individual appears normal. For reasons that remain to be elucidated, heterozygosity is associated with an increased resistance to malaria, specifically growth of the infectious agent Plasmodium falciparum in the erythrocyte. This observation represents an example of a selective advantage that the HbA/HbS heterozygote exhibits over either the HbA/HbA normal or the HbS/HbS homozygote and probably offers an explanation for the persistence of HbS in the gene pool.
Clinical Box Analgesic treatment of sickle cell vasoocclusive crises
Acute vaso-occlusive crises are the most common problem reported by individuals with SCD; they are also the most frequent reason for emergency room treatment and hospital admission. Episodes of vaso-occlusive pain are unpredictable and are often excruciating and incapacitating. The origin of this progressive pain involves altered rheologic and hematologic properties of erythrocytes attributable to HbS polymerization and aggregation. Microvascular dysfunction is precipitated by an inflammatory response, indicated by elevation of plasma acute-phase proteins. Ultimately, impaired vasomotor responses in arterioles and adhesive interactions between sickled erythrocytes and endothelial cells in postcapillary venules restrict blood flow to tissues throughout the body.
Epidemiologic data indicate that 5% of patients with SCD can expect to experience 3–10 episodes of severe pain annually. Typically, the pain crisis resolves within 5–7 days, but a severe crisis may cause pain that persists for weeks. To provide relief to the patient, nonnarcotic, narcotic, and adjuvant analgesics are used alone or in combination.
The severity and duration of the pain dictate the most appropriate analgesic regimen. Parenterally administered opioids (morphine, hydromorphone, meperidine) are frequently used for treatment of severe pain in vasoocclusive crises. Several recent studies suggest additional options for the patient and physician: continuous intravenous infusion of a nonsteroidal antiinflammatory drug (ketorolac) and continuous epidural administration of local anesthetic (lidocaine) and opioid analgesic (fentanyl) effectively decreased pain that was unresponsive to conventional measures. In addition to analgesia, oxygen therapy and fluid management are also initiated.
Other hemoglobinopathies
More than 1000 mutations in the genes encoding the α- and β-globin polypeptides have been documented
As with most mutational events, most lead to few, if any, clinical problems. There are, however, several hundred mutations that give rise to abnormal Hb and pathologic phenotypes. Hb mutants or hemoglobinopathies are usually named after the location (hospital, city or geographical region) in which the abnormal protein was first identified. They are classified according to the type of structural change and altered function and the resulting clinical characteristics (see Tables 5.1, 5.2). While many of these mutants have predictable phenotypes, others are surprisingly pleiotropic in their impact on multiple properties of the Hb molecule. With few exceptions, Hb variants are inherited as autosomal recessive traits. Occasionally, double heterozygotes are identified, e.g. HbSC (Fig. 5.9).
Table 5.2
| Parameter | Patient (male) | Reference value (SI units)* |
| White blood cell count, WBC | 6.82 × 109/L | 4.0–11.0 × 109/L |
| Red cell count, RBC | 4.78 × 1012/L | 4.0–5.2 × 1012/L (F); 4.5–5.9 × 1012/L (M) |
| Hemoglobin, Hb | 6.1 mmol/L | 7.4–9.9 mmol/L (F); 8.4–10.9 mmol/L (M) |
| Hematocrit, HCT | 33.4% | 41–46% (F); 37–49% (M) |
| Mean corpuscular volume, MCV | 71.9 fL | 80–96 fL |
| Mean corpuscular hemoglobin, MCH | 21.3 pg/cell | 26–34 pg/cell |
| Mean corpuscular hemoglobin concentration, MCHC | 296 g/L | 320–360 g/L |
| Red cell distribution width, RDW | 17.7% | 11.5–14.5% |
| Platelet count, PLT | 274 × 109/L | 150–350 × 109/L |
| Mean platelet volume, MPV | 8.6 fL | 6.4–11.0 fL |
*F, female; M, male; fL, 10−15 L; pg, 10−12 g. To convert mmol Hb/L to g Hb/dL, multiply by 0.01611. Automated laboratory evaluation of blood provides invaluable information for the diagnosis and monitoring of health problems. The complete blood count, performed on a sample of whole blood, includes counts of red cells (erythrocytes), white cells (leukocytes), and platelets and quantitative indices of the red cells (MCV, MCH, MCHC, and RDW). The results describe the hematopoietic status of the bone marrow and the presence of anemia and its possible cause. Data presented are characteristic of an individual with iron deficiency anemia: low HGB, low MCV (microcytosis), and low MCH (hypochromia). See also reference values in the Appendix 1.
Clinical test box Separation of hemoglobin variants and mutants; diagnosis of hemoglobinopathies
The mobility of a protein during electrophoresis or chromatography is determined by its charge and interaction with the matrix. Three commonly used techniques provide sufficient resolution to separate Hb variants differing in a single charge from HbA: electrophoresis, isoelectric focusing, and ion exchange chromatography. Electrophoretic and chromatographic separations of Hb are illustrated in Figure 5.9.
The volume of hemolysate required (<100 µL) makes these techniques suitable for neonatal and adult blood samples. Quantification is performed by scanning densitometry or absorption spectrometry. Indications of abnormalities in screening tests are followed by complete blood count (Table 5.2), additional protein analysis, and DNA analysis to identify specific mutations to the globin genes.
Clinical test box Complete blood count
A complete blood count (CBC) provides information on blood cell populations and their characteristics. Data are obtained from whole blood samples by automated hematology analysis. Some instruments also provide leukocyte differentials, reticulocyte count, and red cell morphology. A typical printout of the results for one individual and the reference range is shown in Table 5.2.
Summary
This chapter describes two important proteins that reversibly interact with O2: myoglobin (Mb), a tissue oxygen storage protein, and hemoglobin (Hb), a blood oxygen transport protein. Both use an ancient heme-containing polypeptide domain motif to sequester O2 and increase its solubility.
As a tetramer of globins, Hb is one of the best characterized examples of cooperativity in ligand interactions.
With its wide variety of effector molecules, Hb is also a prototype of an allosteric proteins and enzymes.
2,3-Bisphosphoglycerate is an important allosteric effector of Hb, decreasing the oxygen affinity of hemoglobin; this is an important adaptation to high altitude and in pulmonary disease. Protons, through the Bohr effect, and CO2 also promote the release of oxygen from hemoglobin in peripheral tissue. Conformational changes in both the tertiary and quaternary structures characterize the transition between deoxygenated and oxygenated states.
Mutations to globin genes lead to a spectrum of structural and functional variants, some of which are pathogenic, such as HbS, which causes sickle cell disease.
Allen, BW, Stamler, JS, Piantadosi, CA. Hemoglobin, nitric oxide and molecular mechanisms of hypoxic vasodilation. Trends Mol Med. 2009; 15:452–460.
Bauer, I, Pannen, BH. Bench-to-bedside review: Carbon monoxide – from mitochondrial poisoning to therapeutic use. Crit Care. 2009; 13:220.
Jahr, JS, Akha, AS, Holtby, RJ. Crosslinked, polymerized, and PEG-conjugated hemoglobin-based oxygen carriers: clinical safety and efficacy of recent and current products. Curr Drug Discov Technol. 2012; 9:158–165.
Kutlar, F. Diagnostic approach to hemoglobinopathies. Hemoglobin. 2007; 31:243–250.
Malowany, JI, Butany, J. Pathology of sickle cell disease. Semin Diagn Pathol. 2012; 29:49–55.
Skold, A, Cosco, DL, Klein, R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J. 2011; 104:757–761.
Thein, SL. Milestones in the history of hemoglobin research. Hemoglobin. 2011; 35:450–462.
Vogt, M, Hoppeler, H. Is hypoxia training good for muscles and exercise performance? Prog Cardiovasc Dis. 2010; 52:525–533.
Winslow, RM. Oxygen: the poison is in the dose. Transfusion. 2012.
The Red Cell and Anemia (detailed five-part presentation by pathologist E Uthman): Blood cells and the CBC. http://web2.airmail.net/uthman/blood_cells.html.
Anemia: Pathophysiologic Consequences, Classification, and Clinical Investigation. http://web2.airmail.net/uthman/anemia/anemia.html.
Nutritional Anemias and Anemia of Chronic Disease. http://web2.airmail.net/uthman/nutritional_anemia/nutritional_anemia.html.
Hemolytic Anemias. http://web2.airmail.net/uthman/hemolytic_anemia/hemolytic_anemia.html.
Hemoglobinopathies and Thalassemias. http://web2.airmail.net/uthman/hemoglobinopathy/hemoglobinopathy.html.
Sickle Cell Information Center (comprehensive site for both patients and professionals). www.scinfo.org/.
Interactive examination (Jmol format) of hemoglobin and heme structures, binding of oxygen, and impact of sickle cell disease mutation. Molecular Visualization Resources, University of Massachusetts. www.umass.edu/molvis/tutorials/hemoglobin.
Teaching Cases, American Society of Hematology. teachingcases.hematology.org.