Oxygen and Life
Learning objectives
After reading this chapter you should be able to:
 Identify the major reactive oxygen species (ROS) and their sources in the cell.
Identify the major reactive oxygen species (ROS) and their sources in the cell.
Identify targets of reactive oxygen damage and describe the effects of ROS on biomolecules.
Identify the major antioxidant enzymes, vitamins, and biomolecules that provide protection against ROS.
Describe the role of reactive oxygen in regulatory biology and immunologic defenses.
Explain the role of ROS in development of chronic and inflammatory diseases.
Describe the functions of the antioxidant response element in protection against nucleophiles and ROS.
Introduction
At body temperature, oxygen is a relatively sluggish oxidant
The element oxygen (O2) is essential for the life of aerobic organisms. Although it is highly reactive in combustion reactions at high temperature, oxygen is relatively inert at body temperature; it has a high activation energy for oxidation reactions. This is fortunate, otherwise we might spontaneously combust. About 90% of our O2 usage is committed to oxidative phosphorylation. Enzymes that use O2 for hydroxylation and oxygenation reactions consume another 10%, and a residual fraction, <1%, is converted to reactive oxygen species (ROS), such as superoxide and hydrogen peroxide, which are reactive forms of oxygen. ROS are important in metabolism – some enzymes use H2O2 as a substrate. ROS also play a role in regulation of metabolism and in immunologic defenses against infection. However, ROS are also a source of chronic damage to tissue biomolecules. One of the risks of harnessing O2 as a substrate for energy metabolism is that we may, and do, get burned. For this reason, we have a range of antioxidant defenses that protect us against ROS.
This chapter will deal with the biochemistry of reactive oxygen, the mechanisms of formation and detoxification of ROS, and their role in human health and disease.
The inertness of oxygen
In most textbooks, oxygen is shown as a diatomic molecule with two bonds between the oxygen atoms. This is an attractive presentation from the viewpoint of electron dot structures and electron pairing to form chemical bonds, but it is incorrect. In fact, at body temperature, O2 is a biradical, a molecule with two unpaired electrons (Fig. 37.1). These electrons have parallel spins and are unpaired. Since most organic oxidation reactions, e.g. the oxidation of an alkane to an alcohol or an aldehyde to an acid, are two-electron oxidation reactions, O2 is generally not very reactive in these reactions. In fact, it is completely stable, even in the presence of a strong reducing agent such as H2. When enough heat (activation energy) is applied, one of the unpaired electrons flips to form an electron pair, which then participates in the combustion reaction. Once started, the combustion provides the heat needed to propagate the reaction, sometimes explosively.
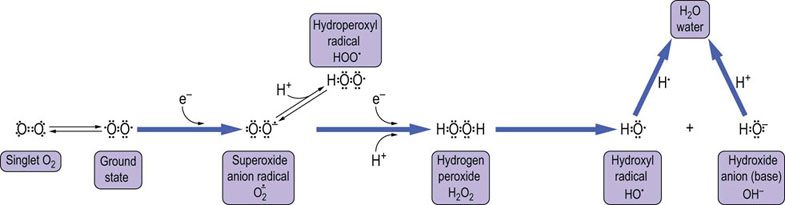
Fig. 37.1 Structure of oxygen and reactive oxygen species (ROS).
Oxygen is shown at the far left as the incorrect double-bonded diatomic form. This form, known as singlet oxygen, exists to a significant extent only at high temperature or in response to irradiation. The diradical is the natural, ground-state form of O2 at body temperature. ROS are partially reduced, reactive forms of oxygen. The first reduction product is the anion radical, superoxide ( ), which is in equilibrium with the weak acid, hydroperoxyl radical (pKa,~4.5). Reduction of superoxide yields hydroperoxide O2–2, in the form of H2O2. Reduction of H2O2 causes a hemolytic cleavage reaction that releases hydroxyl radical (OH•) and hydroxide ion (OH–). Water is the end product of complete reduction of O2.
), which is in equilibrium with the weak acid, hydroperoxyl radical (pKa,~4.5). Reduction of superoxide yields hydroperoxide O2–2, in the form of H2O2. Reduction of H2O2 causes a hemolytic cleavage reaction that releases hydroxyl radical (OH•) and hydroxide ion (OH–). Water is the end product of complete reduction of O2.
Oxygen is activated by transition metal ions, such as iron or copper, in the active of metalloenzymes
Metabolic reactions are conducted at body temperature, far below the temperature required to activate free oxygen. In biological redox reactions involving O2, the oxygen is always activated by redox active metal ions, such as iron and copper; these metals also have unpaired electrons and form reactive metal–oxo complexes. All enzymes that use O2 in vivo are metalloenzymes and, in fact, even the oxygen transport proteins, hemoglobin and myoglobin, contain iron in the form of heme (Chapter 5). These metal ions provide one electron at a time to oxygen, activating O2 for metabolism. Because iron and copper, and sometimes manganese and other ions, activate oxygen, these redox-active metal ions are kept at very low (sub-micromolar) free concentrations in vivo. Normally, they are tightly sequestered (compartmentalized) in storage or transport proteins, and they are locally activated at the active sites of enzymes where oxidation chemistry can be contained and focused on a specific substrate. Free redox-active metal ions are dangerous in biological systems because, in free form, they activate O2, and ROS formed in these reactions cause oxidative damage to biomolecules. Damage to proteins is often site specific, occurring at sites of metal binding to proteins, indicating that metal–oxo complexes participate in ROS-mediated damage in vivo.
 Clinical box Iron overload increases risk for diabetes and cardiomyopathy
Clinical box Iron overload increases risk for diabetes and cardiomyopathy
Patients with hematologic disorders such as hereditary hemochromatosis, thalassemias and sickle cell disease, or who receive frequent blood transfusions, gradually develop iron overload, a condition that increases the risk for development of cardiomyopathy and diabetes. The heart and β-cells are rich in mitochondria. The development of secondary disease in iron overload is considered the result of iron-mediated enhancement of mitochondrial ROS production in these tissues. Mutations in the mitochondrial genome may lead to progressive mitochondrial dysfunction, compromising cardiac and β-cell function.
Reactive oxygen species and oxidative stress
ROS are reactive, strongly oxidizing forms of oxygen
Oxidative stress is defined as a condition in which the rate of generation of ROS exceeds our ability to protect ourselves against them, resulting in an increase in oxidative damage to biomolecules (Fig. 37.2). Oxidative stress is a characteristic feature of inflammatory diseases in which cells of the immune system produce ROS in response to challenge. Oxidative stress may be localized, for instance in the joints in arthritis or in the vascular wall in atherosclerosis, or can be systemic, e.g. in systemic lupus erythematosus (SLE) or diabetes.
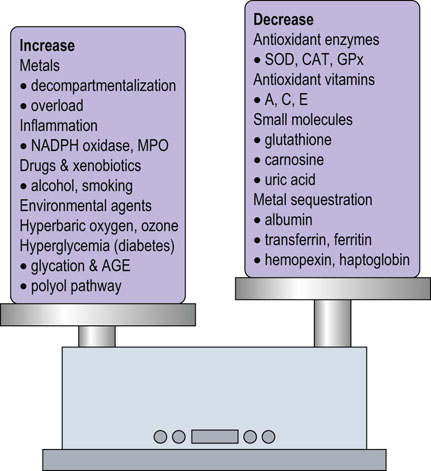
Fig. 37.2 Oxidative stress: an imbalance between prooxidant and antioxidant systems.
As described in this chapter, numerous factors contribute to the enhancement and inhibition of oxidative stress. AGE, advanced glycation end product; CAT, catalase; GPx, glutathione peroxidase; MPO, myeloperoxidase; SOD, superoxide dismutase.
Among the ROS, H2O2 is present at highest concentration in blood and tissues, albeit at micromolar or lower concentrations. H2O2 is relatively stable; it can be stored in the laboratory or medicine cabinet for years but decomposes in the presence of redox-active metal ions. The hydroxyl radical (OH•) is the most reactive and damaging species; its half-life, measured in nanoseconds, is diffusion limited, i.e. determined by the time to collision with a target biomolecule. Superoxide (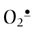 ) is intermediate in stability and may actually serve as either an oxidizing or reducing agent, forming H2O2 or O2, respectively. At physiologic pH, the hydroperoxyl radical (HOO•, pKa < 4.5), the protonated form of superoxide (see Fig. 37.1), represents only a small fraction of total
) is intermediate in stability and may actually serve as either an oxidizing or reducing agent, forming H2O2 or O2, respectively. At physiologic pH, the hydroperoxyl radical (HOO•, pKa < 4.5), the protonated form of superoxide (see Fig. 37.1), represents only a small fraction of total 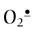 (about 0.1%), but this radical is intermediate in reactivity, between
(about 0.1%), but this radical is intermediate in reactivity, between 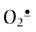 and OH•. HOO• and H2O2 are small, uncharged molecules and readily diffuse through cell membranes.
and OH•. HOO• and H2O2 are small, uncharged molecules and readily diffuse through cell membranes.
ROS are formed by three major mechanisms in vivo: by reaction of oxygen with decompartmentalized metal ions (Fig. 37.3); as a side reaction of mitochondrial electron transport (Fig. 37.4); or by normal enzymatic reactions, e.g. formation of H2O2 by fatty acid oxidases in the peroxisome (Chapter 15). Secondary ROS are also formed by enzymatic reactions, e.g. myeloperoxidase in the macrophage catalyzes the reaction of H2O2 with Cl– to produce another ROS, hypochlorous acid (HOCl). HOCl, which is the major oxidizing species in chlorine-based bleaches, is also part of the bactericidal machinery of the macrophage (see below).
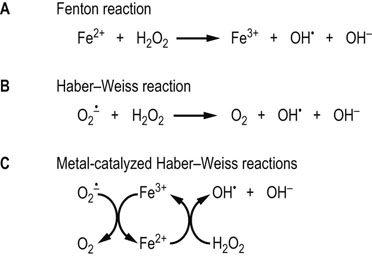
Fig. 37.3 Formation of ROS by the Fenton and Haber–Weiss reactions.
(A) Fenton first described the oxidizing power of solutions of Fe2+ and H2O2. This reaction generates the strong oxidant OH•. Cu+ catalyzes the same reaction. (B) The Haber–Weiss reaction describes the production of OH• from 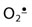 and H2O2. (C) Under physiologic conditions, the Haber–Weiss reaction is catalyzed by redox-active metal ions.
and H2O2. (C) Under physiologic conditions, the Haber–Weiss reaction is catalyzed by redox-active metal ions.
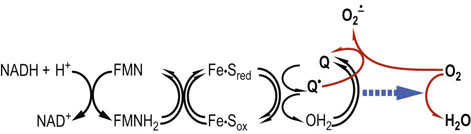
Fig. 37.4 Formation of superoxide by mitochondria.
After the oxidation of NADH, the electron transport chain catalyzes single-electron redox reactions. The semiquinone radical, an intermediate in the reduction of Q to QH2 by Complex I or II, is sensitive to oxidation by molecular oxygen and is considered a major source of superoxide radicals in the cell. This reaction is enhanced at high oxygen tension and also at low oxygen tension: e.g. during ischemia-reperfusion injury (p. 489) when NADH is increased and the electron transport chain is saturated with electrons and the membrane potential is high.
 Advanced concept box Radiotherapy: medical application of reactive oxygen
Advanced concept box Radiotherapy: medical application of reactive oxygen
Radiation therapy uses a focused beam of high-energy electrons or γ-rays from an X-ray or cobalt-60 source to destroy tumor tissue. The radiation produces a flux of hydroxyl radicals (from water) and organic radicals at the site of the tumor. The localized oxidative stress causes damage to all biomolecules in the tumor cell, but the damage to DNA is critical – it prevents tumor cell replication, inhibiting tumor growth. Irradiation is also used as a method of sterilization of food, destroying viral or bacterial contaminants or insect infestations and preserving food products during long-term storage.
Exposure to ionizing radiation from nuclear explosions or accidents, or breathing or ingestion of radioactive elements, such as radon gas or strontium-90, also causes oxidative damage to DNA. Cells that survive the damage may have mutations in DNA that eventually lead to development of cancers. Leukemias are particularly prominent because of the rapid division of bone marrow cells.
Clinical box Toxicity of hyperoxia
Supplemental oxygen therapy may be used for treatment of patients with hypoxemia, respiratory distress, or following exposure to carbon monoxide. Under normobaric conditions, the fraction of oxygen in air can be increased to nearly 100% using a facial mask or nasal cannula. However, patients develop chest pain, cough and alveolar damage within a few hours of exposure to 100% oxygen. Edema gradually develops and compromises pulmonary function. The damage results from overproduction of ROS in the lung. Rats can be protected from oxygen toxicity by gradually increasing the oxygen tension over a period of several days. During this time, antioxidant enzymes, such as superoxide dismutase, are induced in the lung and provide increased protection against oxygen toxicity.
The lung is not the only tissue affected by hyperoxia. Premature infants, especially those with acute respiratory distress syndrome, often require supplemental oxygen for survival. During the 1950s, it was recognized that the high oxygen tension used in incubators for premature infants increased the risk for blindness, resulting from retinopathy of prematurity (retrolental fibroplasia).
Clinical box Ischemia/reperfusion injury: a patient with myocardial infarction
A patient suffered a severe myocardial infarction, which was treated with tissue plasminogen activator, a clot-dissolving (thrombolytic) enzyme. During the days following hospitalization, the patient experienced palpitations, irregular rapid heartbeat, associated with weakness and faintness. The patient was treated with antiarrhythmic agents.
Ischemia, meaning limited blood flow, is a condition in which a tissue is deprived of oxygen and nutrients. Damage to heart tissue during a myocardial infarction occurs not during the hypoxic or ischemic phase, but during reoxygenation of the tissue. This type of damage also occurs following transplantation, and cardiovascular surgery. ROS are thought to play a major role in reperfusion injury. When cells are deprived of oxygen, they must rely on anaerobic glycolysis and glycogen stores for ATP synthesis. NADH and lactate accumulate, and all the components of the mitochondrial electron transport system are saturated with electrons (reduced), because the electrons cannot be transferred to oxygen. The mitochondrial membrane potential is increased (hyperpolarized), and when oxygen is reintroduced, great quantities of ROS are rapidly produced, overwhelming antioxidant defenses. ROS flood throughout the cell, damaging membrane lipids, DNA and other vital cellular constituents, leading to necrosis. Antioxidant supplements and drugs are being evaluated for use during recovery from myocardial infarction and stroke, during surgery, and for protection of tissues prior to transplantation.
Reactive nitrogen species and nitrosative stress
Peroxynitrite is a strongly oxidizing reactive nitrogen species
Nitric oxide synthases (NOS) catalyze the production of the free radical nitric oxide (NO•) from the amino acid L-arginine. There are three isoforms of NOS: nNOS in neuronal tissue, where NO• serves a neurotransmitter function; iNOS in the immune system, where it is involved in regulation of the immune response; and eNOS in endothelial cells, where NO•, known as endothelium-derived relaxation factor (EDRF), has a role in the regulation of vascular tone.
In a side reaction at sites of inflammation, NO• reacts with 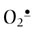 to form the strong oxidant, peroxynitrite (ONOO–), a reactive nitrogen species (RNS). Like ROS, which produce oxidative stress, RNS produce nitrosative stress by reaction with biomolecules. ONOOH has many of the strong oxidizing properties of OH• but has a longer biological half-life. It is also a potent nitrating agent, producing nitrotyrosine in proteins, nitrated phospholipids in membranes and nucleotides in DNA. Simultaneous production of NO• and
to form the strong oxidant, peroxynitrite (ONOO–), a reactive nitrogen species (RNS). Like ROS, which produce oxidative stress, RNS produce nitrosative stress by reaction with biomolecules. ONOOH has many of the strong oxidizing properties of OH• but has a longer biological half-life. It is also a potent nitrating agent, producing nitrotyrosine in proteins, nitrated phospholipids in membranes and nucleotides in DNA. Simultaneous production of NO• and 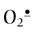 , with the concomitant increase in ONOO– and a decrease in NO•, is thought to limit vasodilatation and exacerbate hypoxia and oxidative stress in the vascular wall during ischemia-reperfusion injury, setting the stage for vascular disease. ONOOH degrades, in part, by homolytic cleavage to produce two very reactive species, OH• and NO2•. NO2• is also formed by eosinophil peroxidase or myeloperoxidase-catalyzed oxidation of NO• by H2O2.
, with the concomitant increase in ONOO– and a decrease in NO•, is thought to limit vasodilatation and exacerbate hypoxia and oxidative stress in the vascular wall during ischemia-reperfusion injury, setting the stage for vascular disease. ONOOH degrades, in part, by homolytic cleavage to produce two very reactive species, OH• and NO2•. NO2• is also formed by eosinophil peroxidase or myeloperoxidase-catalyzed oxidation of NO• by H2O2.
The nature of oxygen radical damage
The hydroxyl radical is the most reactive and damaging ROS
The reaction of ROS with biomolecules produces characteristic products, described as biomarkers of oxidative stress. These compounds may be formed directly in the oxidation reaction with the ROS, or by secondary reactions between oxidation products and other biomolecules. The hydroxyl radical reacts with biomolecules primarily by hydrogen abstraction and addition reactions. One of the most sensitive sites of free radical damage are cell membranes, which are rich in readily oxidized polyunsaturated fatty acids (PUFA). Peroxidative damage to the plasma membrane affects the integrity and function of the membrane, compromising the cell's ability to maintain ion gradients and membrane phospholipid asymmetry. As shown in Figure 37.5, when OH• abstracts a hydrogen atom from a PUFA, it initiates a chain of lipid peroxidation reaction, producing secondary oxidation products, lipid peroxides and lipid peroxyl radicals. The lipid oxidation products formed in this reaction degrade to form reactive compounds, such as malondialdehyde (MDA) and hydroxynonenal (HNE). These compounds react with proteins to form adducts and crosslinks, known as advanced lipoxidation end products (ALE). Increased levels of MDA and HNE adducts to lysine residues have been measured in lipoproteins in plasma and the vascular wall in atherosclerosis and in amyloid plaque in Alzheimer's disease, implicating oxidative stress and damage in the pathogenesis of these diseases.
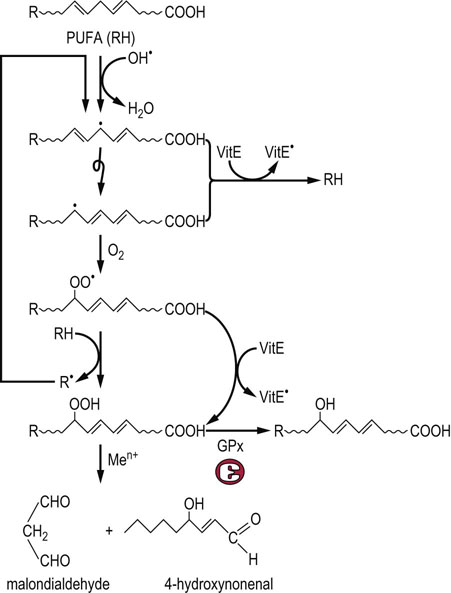
Fig. 37.5 Pathway of lipid peroxidation.
OH• attacks PUFA, forming a carbon-centered lipid radical. The radical rearranges to form a conjugated dienyl radical. This radical reacts with ambient O2, forming a hydroperoxyl radical, which then abstracts a hydrogen from a neighboring lipid, forming a lipid peroxide and R•, which starts a chain reaction. This reaction continues until the supply of PUFA is exhausted, unless a termination reaction occurs. Vitamin E (below) is the major chain-terminating antioxidant in membranes; it reduces both the conjugated dienyl and hydroperoxyl radicals, quenching the chain or cycle of lipid peroxidation reactions. Lipid peroxides may also be reduced by glutathione peroxidase (GPx), forming inert lipid alcohols Otherwise, they decompose to form a range of ‘reactive carbonyl species’ such as malondialdehyde and hydroxynonenal, which react with protein to form advanced lipoxidation end products (ALE), which are biomarkers of oxidative stress. The reaction scheme shown here for PUFA also occurs with intact phospholipids and cholesterol esters in lipoproteins and cell membranes.
Hydroxyl radicals also react by addition to phenylalanine, tyrosine and nucleic acid bases to form hydroxylated derivatives and crosslinks (Fig. 37.6). Other ROS and RNS leave tell-tale tracks, such as nitro- and chlorotyrosine, formed from ONOOH and HOCl, respectively, and methionine sulfoxide, formed by reaction of H2O2 or HOCl with methionine residues in proteins (see Fig. 37.6). Nitrotyrosine, like ALEs, is increased in atherosclerotic and Alzheimer's plaques.
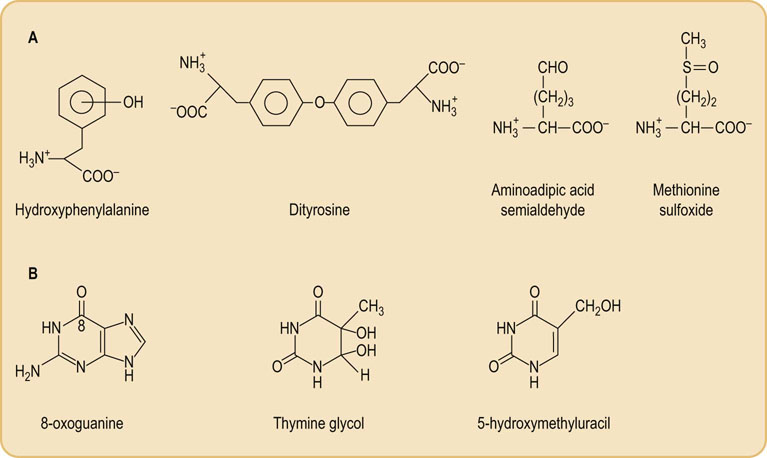
Fig. 37.6 Products of hydroxyl radical damage to biomolecules.
(A) Amino acid oxidation products: o-, m- and p-tyrosine and dityrosine from phenylalanine; amino adipic acid semialdehyde from lysine; methionine sulfoxide. Other products include chlorotyrosine (from HOCl), nitrotyrosine (from ONOO– and NO2•), dihydroxyphenylalanine produced by hydroxylation of tyrosine, and aliphatic amino acid hydroperoxides, such as leucine hydroperoxide. (B) Nucleic acid oxidation products: 8-oxoguanine is the most commonly measured indicator of DNA damage.
ROS also react with carbohydrates to form reactive carbonyl compounds that react with protein to form crosslinks and adducts, known as advanced glycation end products (AGE). AGEs are increased in tissue proteins in diabetes as a result of hyperglycemia and oxidative stress, and the increase in chemical modification of proteins by AGEs and ALEs is implicated in the development of diabetic vascular, renal, and retinal complications (see Chapter 21).
Antioxidant defenses
There are several levels of protection against oxidative damage
ROS damage to lipids and proteins is repaired largely by degradation and resynthesis. Oxidized proteins, for example, are preferred targets for proteasomal degradation, and damaged DNA is repaired by a number of excision-repair mechanisms. The process is not perfect. Some proteins, such as collagens and crystallins, turn over slowly, so that damage accumulates and function may be impaired, e.g. age-dependent browning of lens proteins, crosslinking of collagen and elastin, and loss of elasticity or changes in permeability of the vascular wall and renal basement membrane (see Chapter 43). The association between chronic inflammation and cancer indicates that chronic exposure to ROS causes cumulative damage to the genome in the form of nonlethal mutations in DNA.
Advanced concept box Sentinel function of methionine
Methionine (Met) residues in protein may be oxidized to methionine sulfoxide (MetSO) by H2O2, HOCl or lipid peroxides. Met is generally on the surface of proteins and rarely has a role in the active site or mechanism of action of enzymes. However, there is evidence that it serves as an ‘antioxidant pawn’, protecting the active site of enzymes. Half of the Met residues of glutamine synthetase can be oxidized without affecting the enzyme's specific activity. These residues are physically arranged in an array that ‘guards’ the entrance to the active site, protecting the enzyme from inactivation by ROS. MetSO can be reduced back to methionine by methionine sulfoxide reductases, providing a catalytic amplification of the antioxidant potential of each methionine residue.
Clinical box Methionine oxidation and emphysema
α1-Antitrypsin (A1AT) is a plasma protein, synthesized and secreted by liver. It is a potent inhibitor of elastase and protects tissues from damage by the neutrophil enzyme secreted during inflammation. Deficiency of this protein (about 1 in 4000 worldwide) is commonly associated with emphysema, progressive lung disease, and also hepatic damage resulting from accumulation of protein aggregates. The lung damage is attributed to failure of A1AT to inhibit elastase released by alveolar macrophages during phagocytosis of airborne particulate matter.
Therapy includes replacement therapy by weekly intravenous infusion of a purified plasma concentrate or recombinant protein. Cigarette smoking and exposure to mineral dust (coal, silica) exacerbate pathology in patients with A1AT deficiency, but are also independent risk factors for emphysema and pulmonary fibrosis. Cigarette smoke and microparticulate materials activate lung macrophages, leading to release of proteolytic enzymes and increased production of ROS as a result of inflammation. The ROS cause oxidation of a specific Met residue in A1AT, irreversibly inhibiting the anti-elastase activity of this protein. Increased levels of inactive, Met(O)-containing A1AT are present in plasma of chronic smokers.
Advanced concept box Selenium, an antioxidant micronutrient
Selenocysteine is an unusual amino acid in proteins, found in only 25 proteins in the human proteome. It is encoded by UGA, which is normally a STOP codon, under direction of a SElenoCysteine Insertion Sequence (SECIS), a 50-nucleotide stem-loop structure in the mRNA. The 25-member selenoproteome includes five glutathione peroxidase isozymes, three thioredoxin reductases, methionine sulfoxide reductase, one of three enzymes that reduces methionine sulfoxide back to methionine, and three iodothyronine deiodinases. About a third of the selenoproteins have no known function, but appear to be involved in antioxidant defense mechanisms. Selenium is essential for life, in part because of severe hypothyroidism and oxidative stress in its absence. Selenium deficiency in adults is associated with cardiomyopathy in Keshan disease, osteoarthropathy (cartilage degeneration) in Kashin–Beck disease, and with symptoms of hypothyroidism, including chronic fatigue and goiter.
Our first line of defense against oxidative damage is sequestration or chelation of redox-active metal ions
Advanced concept box The antioxidant response element
Cells adapt to oxidative stress by induction of antioxidant enzymes. Many of these are controlled by the antioxidant response element (ARE), also known as the electrophile response element. The central regulator of the ARE is the transcription factor Nrf2, which is retained in inactive form in the cytoplasmic compartment by binding to a cysteine-rich protein, Keap1. Under normal conditions, Keap1 directs ubiquitination and proteasomal degradation of Nrf2. During oxidative stress, modification of the sulfhydryl groups of Keap1 by nucleophiles, such as the lipid peroxidation products, hydroxynonenal or acrolein, causes dissociation of Nrf2 from Keap1. Nrf2 then translocates to the nucleus and activates ARE-dependent genes. Keap1 also reacts with exogenous nucleophiles, including carcinogens that would otherwise react with DNA.
ARE-dependent enzymes include catalase (CAT) and superoxide dismutase (SOD), and enzymes that catalyze the oxidation and conjugation of carcinogens and oxidants for excretion. One of these enzymes, hepatic glutathione S-transferase, catalyzes conjugation with GSH. The conjugates are then excreted in urine as mercapturic acid, which are S-substituted N-acetyl-cysteine derivatives.
Endogenous chelators include a number of metal-binding proteins that sequester iron and copper in inactive form, such as transferrin and ferritin, the transport and storage forms of iron. The plasma protein haptoglobin binds to hemoglobin from ruptured red cells, and delivers the hemoglobin molecule to the liver for catabolism. Plasma hemopexin binds heme, the lipid-soluble form of iron, which catalyzes ROS formation in lipid environments; it delivers the heme to the liver for catabolism. Albumin, the major plasma protein, has a strong binding site for copper and effectively inhibits copper-catalyzed oxidation reactions in plasma. Carnosine (β-alanyl-L-histidine) and related peptides are present in muscle and brain at millimolar concentrations; they are potent copper chelators and may have a role in intracellular antioxidant protection.
Despite these manifold and potent metal chelation systems, ROS are formed continuously in the body, both by enzymes and spontaneous metal-catalyzed reactions. In these cases, there are a group of enzymes that act to detoxify ROS and their precursors. These include superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) (Fig. 37.7). SOD converts 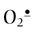 to the less toxic H2O2. There are two classes of SOD: an MnSOD isozyme which is found in mitochondria, and CuZnSOD isozyme which is widely distributed throughout the cell. An extracellular, secreted glycoprotein isoform of CuZnSOD (EC-SOD) binds to proteoglycans in the vascular wall and is thought to protect against
to the less toxic H2O2. There are two classes of SOD: an MnSOD isozyme which is found in mitochondria, and CuZnSOD isozyme which is widely distributed throughout the cell. An extracellular, secreted glycoprotein isoform of CuZnSOD (EC-SOD) binds to proteoglycans in the vascular wall and is thought to protect against 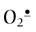 and ONOO– injury. CAT, which inactivates H2O2, is found largely in peroxisomes, the major site of H2O2 generation in the cell.
and ONOO– injury. CAT, which inactivates H2O2, is found largely in peroxisomes, the major site of H2O2 generation in the cell.
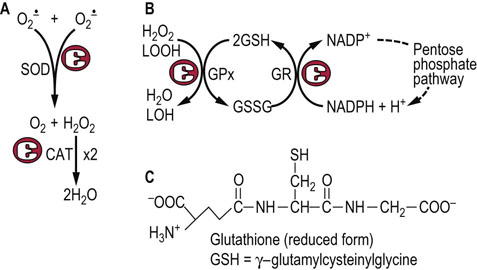
Fig. 37.7 Enzymatic defenses against ROS.
(A) Superoxide dismutase (SOD) and catalase (CAT) are dismutases, catalyzing oxidation and reduction of two separate substrate (H2O2) molecules; both are highly specific for their substrates, 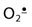 and H2O2, respectively. (B) Glutathione peroxidase (GPx) reduces H2O2 and lipid peroxides (LOOH), using GSH as a co-substrate. The GSH is recycled by glutathione reductase (GR) using NADPH from the pentose phosphate pathway. (C) Structure of GSH.
and H2O2, respectively. (B) Glutathione peroxidase (GPx) reduces H2O2 and lipid peroxides (LOOH), using GSH as a co-substrate. The GSH is recycled by glutathione reductase (GR) using NADPH from the pentose phosphate pathway. (C) Structure of GSH.
GPx is widely distributed in the cytosol, in mitochondria and the nucleus. It reduces H2O2 and lipid hydroperoxides to water and a lipid alcohol, respectively, using reduced glutathione (GSH) as a co-substrate. GSH is a tripeptide (Chapter 2) that is present at 1–5 mM concentration in all cells. The GSH is recycled by an NADPH-dependent enzyme, GSH reductase. The NADPH, provided by the pentose phosphate pathway, maintains about a 100 : 1 ratio of GSH : GSSG in the cell. GPx is actually a family of selenium-containing isozymes; a phospholipid hydroperoxide glutathione peroxidase will reduce lipid hydroperoxides in phospholipids in lipoproteins and membranes, while other isozymes are specific for free fatty acid or cholesterol ester hydroperoxides. There is also an isoform of GPx in intestinal epithelial cells, which is thought to have a role in detoxification of dietary hydroperoxides, e.g. in fried foods.
Vitamin C is the outstanding antioxidant in biological systems
Three antioxidant vitamins, A, C and E, provide the third line of defense against oxidative damage. These vitamins, primarily vitamin C (ascorbate) (Fig. 37.8) in the aqueous phase and vitamin E (α- and γ-tocopherol; Fig. 37.9) in the lipid phase, act as chain-breaking antioxidants (see Fig. 37.5). They act as reducing agents, donating a hydrogen atom (H•) and quenching organic radicals formed by reaction of ROS with biomolecules. The vitamin C and E radicals produced in this reaction are unreactive, resonance-stabilized species; they do not propagate radical damage and are enzymatically recycled, e.g. by dehydroascorbate reductase (see Fig. 37.8). Vitamin C reduces superoxide and lipid peroxyl radicals, but also has a special role in reduction and recycling of vitamin E. In response to severe oxidative stress. vitamin C recycles vitamin E, so that vitamin E is maintained at constant concentration in the lipid phase until all the vitamin C is consumed (see Fig. 37.9). These antioxidants work together to inhibit lipid peroxidation reactions in plasma lipoproteins and membranes. Vitamin A (carotene; Chapter 11) is also a lipophilic antioxidant. Although best understood for its role in vision, it is a potent singlet oxygen scavenger and protects against damage from sunlight in the retina and skin.
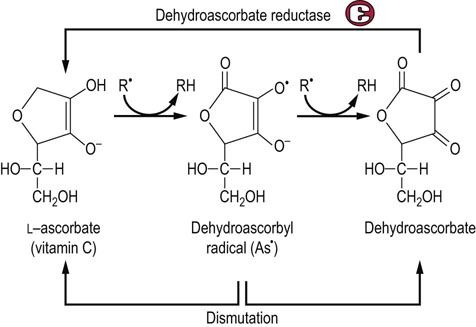
Fig. 37.8 Antioxidant activity of ascorbate.
Vitamin C exists as the enolate anion at physiologic pH. The enolate anion spontaneously reduces superoxide, organic (R•) and vitamin E radicals, forming a dehydroascorbyl radical (As•). Dehydroascorbyl radical may dismutate to ascorbate and dehydroascorbate. Dehydroascorbate is also recycled by dehydroascorbate reductase, a GSH-dependent enzyme present in all cells.
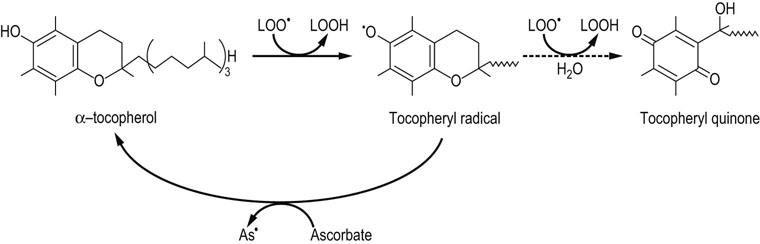
Fig. 37.9 Antioxidant activity of vitamin E.
The term vitamin E refers to a family of related tocopherol and tocotrienol isomers with potent lipophilic antioxidant and membrane-stabilizing activity. Tocopherols reduce lipid hydroperoxyl radicals and also inactivate singlet oxygen. α-Tocopherol is the most effective form in humans and the major form of vitamin E in the diet. It consists of a chromanol ring structure with a polyisoprenoid side chain which helps to anchor the vitamin in membranes; the isoprene units are unsaturated in tocotrienol. The α, β, γ, and δ Isomers differ in the pattern of methyl groups on the benzene ring (see Figure 11.3). The major commercial form of vitamin E is α-tocopherol acetate, which is more stable than free tocopherol during storage. The tocopheryl radical, the major product formed during antioxidant action of vitamin E, is recycled by ascorbate. Tocopheryl quinone is also formed in small quantities.
Glutathionylation of proteins – protection against ROS under stress
Despite the multiplicity of defensive mechanisms, there is always some evidence of ongoing oxidative damage in tissues. Under physiologic conditions, when proteins are exposed to O2, their sulfhydryl groups gradually oxidize to form disulfides, either intramolecularly or intermolecularly with other proteins. This is multi-step process. First, a protein sulfhydryl group is oxidized to a sulfenic acid (PrSOH) by an ROS, such as H2O2 or HOCl; then the sulfenic acid reacts with another PrSH to form a crosslinked protein PrS-SPr. These crosslinked proteins are reduced by glutathione to form oxidized glutathione and regenerate the native protein with free sulfhydryl groups. The reaction sequence is
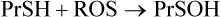
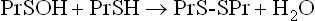
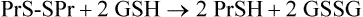
During oxidative stress, there is a significant increase in S-glutathionylated proteins (PrS-SG) in the cell. In this case, the reaction sequence is
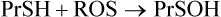
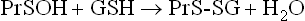
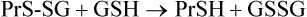
S-Glutathionylation is reversed by nonenzymatic reduction by GSH (above) or by enzymes using thiol protein cofactors (thioredoxin, glutaredoxin). This pathway inhibits the formation of crosslinked protein aggregates, such as Heinz bodies, which are hemoglobin precipitates that develop in red cells in glucose-6-phosphate dehydrogenase deficiency, characterized by decreased levels of GSH (Chapter 12). S-glutathionylation is thought to have a dual role, not only in protecting cysteine against irreversible oxidation to the sulfinic or sulfonic acid during oxidative and/or nitrosative stress but also in modulating cellular metabolism (redox regulation). Target proteins include a wide range of enzymes with active site or regulatory –SH groups, such as glyceraldehyde-3-phosphate dehydrogenase in glycolysis and protein kinases in signaling cascades, as well as chaperones and transport proteins. S-glutathionylation also appears to protect proteins from ubiquitin-mediated proteasomal degradation during oxidative stress.
The beneficial effects of reactive oxygen species
ROS are essential for many metabolic and signaling pathways
 Clinical test box Peroxidase activity for detection of occult blood
Clinical test box Peroxidase activity for detection of occult blood
Peroxidases, such as the glutathione peroxidase (GPx), are enzymes that catalyze the oxidation of a substrate using H2O2. Hemoglobin and heme have a pseudoperoxidase activity in vitro. In the guaiac-based test for occult fecal blood, a stool sample is applied to a small card containing guaiac acid. Hemoglobin in a stool specimen oxidizes phenolic compounds in guaiac acid to quinones. A positive test is indicated by a blue stain along the edge of the fecal smear. Incompletely digested hemoglobin and myoglobin from animal meat and some plant peroxidases may cause false positives. Similar peroxidase-dependent assays are used to identify bloodstains at crime scenes.
Advanced concept box The glyoxalase pathway: a special role for glutathione
A small fraction of triose phosphates produced during metabolism spontaneously degrades to methylglyoxal (MGO), a reactive dicarbonyl sugar. MGO is also formed during metabolism of glycine and threonine, and as a product of nonenzymatic oxidation of carbohydrates and lipids – it is a significant precursor of advanced glycation and lipoxidation end products (AGE/ALEs) (see Chapters 21 and 43). MGO reacts primarily with arginine residues in proteins, but also with lysine, histidine and cysteine, leading to enzyme inactivation and protein crosslinking.
MGO is inactivated by enzymes of the glyoxalase pathway, a GSH-dependent system found in all cells in the body. The glyoxalase pathway (Fig. 37.10) consists of two enzymes that catalyze an internal redox reaction in which carbon-1 of MGO is oxidized from an aldehyde to a carboxylic acid group and carbon-2 is reduced from a ketone to a secondary alcohol. The end product, D-lactate, does not react with proteins; D-lactate is distinct from L-lactate, the product of glycolysis, but may be converted into L-lactate for further metabolism.
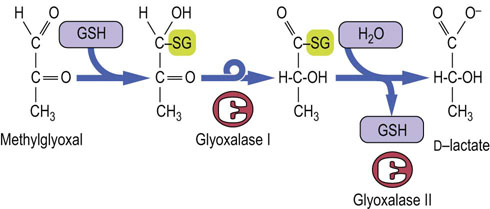
Fig. 37.10 The glyoxalase system.
Glyoxalase I catalyzes the formation of a thiohemiacetal adduct between GSH and MGO and its rearrangement to a thioester. Glyoxalase II catalyzes the hydrolysis of the thioester, forming D-lactate and regenerating GSH. Unlike GPx, this pathway does not consume GSH.
Levels of MGO and D-lactate are increased in blood of diabetic patients, because levels of glucose and glycolytic intermediates, including triose phosphates, are increased intracellularly in diabetes. The glyoxalase system also inactivates glyoxal, and other dicarbonyl sugars produced during nonenzymatic oxidation of carbohydrates and lipids. Glyoxalase inhibitors are being evaluated for chemotherapy because cancer cells appear to be more sensitive to glyoxalase inhibitors, perhaps because of their increased reliance on glycolysis.
Advanced concept box The respiratory burst in macrophages
As outlined in Fig. 37.11, macrophages launch a sequence of ROS-producing reactions during the burst of oxygen consumption accompanying phagocytosis. NADPH oxidase in the macrophage plasma membrane is activated to produce  , which is then converted to H2O2 by superoxide dismutase. The H2O2 is used by another macrophage enzyme, myeloperoxidase (MPO), to oxidize chloride ion, ubiquitous in body fluids, to hypochlorous acid (HOCl). H2O2 and HOCl mediate bactericidal activity by oxidative degradation of microbial lipids, proteins, and DNA. The macrophage has a high intracellular concentration of antioxidants, especially ascorbate, to protect itself during ROS production, but its relatively short life span, 2–4 months, suggests that it is not immune to oxidative damage.
, which is then converted to H2O2 by superoxide dismutase. The H2O2 is used by another macrophage enzyme, myeloperoxidase (MPO), to oxidize chloride ion, ubiquitous in body fluids, to hypochlorous acid (HOCl). H2O2 and HOCl mediate bactericidal activity by oxidative degradation of microbial lipids, proteins, and DNA. The macrophage has a high intracellular concentration of antioxidants, especially ascorbate, to protect itself during ROS production, but its relatively short life span, 2–4 months, suggests that it is not immune to oxidative damage.
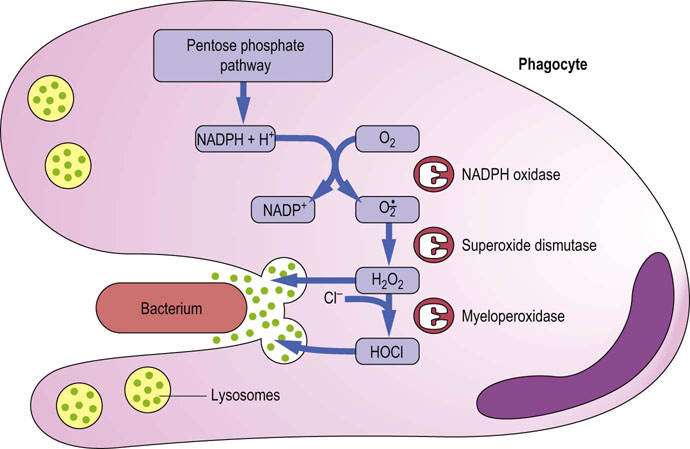
Fig. 37.11 Generation and release of ROS during phagocytosis.
A cascade of reactions generating ROS is initiated during phagocytosis to kill invading organisms. Hydrolytic enzymes are also released from lysosomes to assist in degradation of microbial debris.
The consumption of O2 by NADPH oxidase is responsible for the ‘respiratory burst’, the sharp increase in O2 consumption for production of ROS, which accompanies phagocytosis. One of the end products of this reaction sequence, HOCl, is also the active oxidizer in chlorine-containing laundry bleaches. Intravenous infusion of dilute HOCl solutions was actually used for treatment of bacterial sepsis in battlefield hospitals during World War I, before the advent of penicillin and other antibiotics. Chronic granulomatous disease (CGD) is an inherited disease resulting from a genetic defect in NADPH oxidase. The inability to produce superoxide leads to chronic life-threatening bacterial and fungal infections.
Advanced concept box Antioxidant defenses in the red blood cell (RBC)
The RBC does not use oxygen for metabolism, nor is it involved in phagocytosis. However, because of the high O2 tension in arterial blood and the heme iron content of RBCs, ROS are formed continuously in the RBC. Hb spontaneously produces superoxide (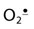 ) in a minor side reaction associated with binding of O2. The occasional reduction of O2 to
) in a minor side reaction associated with binding of O2. The occasional reduction of O2 to  is accompanied by oxidation of normal (ferro) Hb to methemoglobin (ferrihemoglobin), a rust-brown protein that does not bind or transport O2. Methemoglobin may release heme, which reacts with
is accompanied by oxidation of normal (ferro) Hb to methemoglobin (ferrihemoglobin), a rust-brown protein that does not bind or transport O2. Methemoglobin may release heme, which reacts with 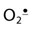 and H2O2 in Fenton-type reactions to produce hydroxyl radical (OH•) and reactive iron-oxo species. These ROS initiate lipid peroxidation reactions that can lead to loss of membrane integrity and cell death.
and H2O2 in Fenton-type reactions to produce hydroxyl radical (OH•) and reactive iron-oxo species. These ROS initiate lipid peroxidation reactions that can lead to loss of membrane integrity and cell death.
The RBC is well fortified with antioxidant defenses to protect itself against oxidative stress. These include catalase (CAT), superoxide dismutase (SOD) and glutathione peroxidase (GPx), as well as a methemoglobin reductase activity that reduces methemoglobin back to normal ferrohemoglobin. Normally, less than 1% of Hb is present as methemoglobin. However, persons with congenital methemoglobinemia, resulting from methemoglobin reductase deficiency, typically have a dark and cyanotic appearance. Treatment with large doses of ascorbate (vitamin C) is used to reduce their methemoglobin to functional hemoglobin.
GSH, present at ~2 mmol/L in the RBC, not only supports antioxidant defenses but also is an important sulfhydryl buffer, maintaining –SH groups in hemoglobin and enzymes in the reduced state.
While this chapter has focused thus far on the dangerous aspects of reactive oxygen, it is worth closing with some recognition of the beneficial effects of ROS. Among these are the regulatory functions of NO, the role of ROS in activation of the ARE, the requirement for ROS in the bactericidal activity of macrophages, and the use of ROS as substrates for enzymes, e.g. H2O2 for the hemeperoxidases involved in iodination of thyroid hormone. There is also increasing evidence that ROS, particularly H2O2, are important signaling molecules involved in regulation of metabolism. The tissue concentration of H2O2 is estimated to be in the submicromolar range; estimates vary widely, from 1 to 700 nmol/L. However, significant changes in H2O2 concentration occur in response to cytokines, growth factors and biomechanical stimulation. The fact that these signaling events are inhibited by peroxide scavengers or by overexpression of catalase implicates H2O2 in the signaling cascade. Insulin signaling, for example, appears to involve H2O2 as part of the mechanism for reversible inactivation of some protein tyrosine phosphatases, at the same time that protein tyrosine kinases are activated through the insulin receptor (Chapter 21). As the evidence for the signaling role of H2O2 has become convincing, there is increasing interest in research on the regulatory role of superoxide.
Summary
Reactive oxygen species (ROS) are the sparks produced by oxidative metabolism, and oxidative stress may be viewed as the price we pay for using oxygen for metabolism.
ROS and RNS, such as superoxide, peroxide, hydroxyl radical, and peroxynitrite, are reactive and toxic, sometimes difficult to contain, but their production is important for regulation of metabolism, turnover of biomolecules and protection against microbial infection.
ROS and RNS cause oxidative damage to all classes of biomolecules: proteins, lipids and DNA.
There are a number of protective antioxidant mechanisms, including sequestration of redox-active metal ions, enzymatic inactivation of major ROS, inactivation of organic radicals by small molecules, such as GSH and vitamins, and, when all else fails, repair and/or turnover, and, in extremis, apoptosis.
Biomarkers of oxidative stress are readily detected in tissues in inflammation, and oxidative stress is increasingly implicated in the pathogenesis of age-related, chronic disease.
Despite their damaging actions, ROS are also essential for the normal functions of the immune system, and for many enzymes and cell signaling pathways.
Burgoyne, JR, Oka, SI, Ale-Agha, N, et al. Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system. Antioxid Redox Signal. 2013; 18:1042–1052.
Ferrari, CK, Souto, PC, França, EL, et al. Oxidative and nitrosative stress on phagocytes’ function: from effective defense to immunity evasion mechanisms. Arch Immunol Ther Exp. 2011; 59:441–448.
Greenough, MA, Camakaris, J, Bush, AI. Metal dyshomeostasis and oxidative stress in Alzheimer's disease. Neurochem Int. 2013; 62:540–555.
Riccioni, G, D'Orazio, N, Salvatore, C, et al. Carotenoids and vitamins C and E in the prevention of cardiovascular disease. Int J Vitam Nutr Res. 2012; 82:15–26.
Tkachev, VO, Menshchikova, EB, Zenkov, NK. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Mosc). 2011; 76:407–422.
Zhang, H, Forman, HJ. Glutathione synthesis and its role in redox signaling. Semin Cell Dev Biol. 2012; 23:722–728.
Zhang, Y, Tocchetti, CG, Krieg, T, et al. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic Biol Med. 2012; 53:1531–1540.
Antioxidants and cancer. www.cancer.gov/cancertopics/factsheet/antioxidantsprevention.
Free radical lectures. http://web.mst.edu/~nercal/documents/chem464/lectures/Lec01_FreeRadicals.pdf.
Oxidative stress and disease. www.oxidativestressresource.org/.
Reactive oxygen species and antioxidant vitamins. http://lpi.oregonstate.edu/f-w97/reactive.html.
Virtual Free Radical Schoo. www.sfrbm.org/sections/education/frs-presentations